 Sen Zhang
Sen Zhang Qinglu Wang
Qinglu Wang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 21 November 2022
Sec. Inflammation
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1049936
This article is part of the Research Topic Adipose Tissue Inflammation and Metabolic Disease View all 8 articles
Ferroptosis requires not only the accumulation of iron ions, but also changes in many ferroptosis-related regulators, including a decrease in GPX4 and inhibition of SLC7A11 for classical ferroptosis, a deletion of FSP1 or GCH1. Surprisingly, adipose tissue (AT) in the obesity conditions is also accompanied by iron buildup, decreased GSH, and increased ROS. On the neurological side, the pro-inflammatory factor released by AT may have first caused ferroptosis in the vagus nerve by inhibiting of the NRF2-GPX4 pathway, resulting in disorders of the autonomic nervous system. On the immune side, obesity may cause M2 macrophages ferroptosis due to damage to iron-rich ATMs (MFehi) and antioxidant ATMs (Mox), and lead to Treg cells ferroptosis through reductions in NRF2, GPX4, and GCH1 levels. At the same time, the reduction in GPX4 may also trigger the ferroptosis of B1 cells. In addition, some studies have also found the role of GPX4 in neutrophil autophagy, which is also worth pondering whether there is a connection with ferroptosis. In conclusion, this review summarizes the associations between neuroimmune regulation associated with obesity and ferroptosis, and on the basis of this, highlights their potential molecular mechanisms, proposing that ferroptosis in one or more cells in a multicellular tissue changes the fate of that tissue.
Iron, which has the highest content of all essential trace elements in the human body, plays a critical role as a biologically essential component of every living organism (1), which is closely related to the development of the human body and the occurrence and development of diseases. Thus, a constant balance requires to be maintained between iron uptake, transport, storage, and usage to maintain iron homeostasis (2). Iron homeostasis involves the action of multiple cell types, including red blood cells, intestinal cells, hepatocytes, and macrophages (3). When the iron steady state is broken, its basic chemical properties lay the foundation for potential toxicity via the production of reactive oxygen species (ROS), including hydrogen peroxide (H2O2), hydroxyl radicals (·HO), superoxide anions (O2−), and other reaction intermediates with free radical properties (4, 5). During ATP production in mitochondria, some of the oxygen consumption will be reduced through the monovalent route (5), resulting in the production of the above reactive intermediates. Moreover, because of its redox properties, iron can just have a “Fenton reaction” with H2O2 in it, thereby generating extremely reactive ·HO. Furthermore, when ROS exceeds a certain threshold, it leads to oxidative stress (6, 7); therefore, too much iron can negatively impact the body. Increasing studies have proved that iron overload is the key cause of ferroptosis in cells, and the area of occurrence in the human body is extremely extensive (8). Additionally, long-term intake of a high-fat diet can lead to iron accumulation, which may exacerbate the mechanism of onset (9). The diseases associated with ferroptosis include neurodegenerative diseases (such as Alzheimer’s disease and Parkinson’s disease), ischemic reperfusion damage to the heart, brain, lungs, and kidneys, and stomach cancer and breast cancer (10–17).
Ferroptosis, unlike apoptosis, autophagy, and cell necrosis, is a regulatory form of cell death that depends on iron and ROS. This form of death was first reported in Brent R Stock well’s lab in 2012 (18). The mechanism of ferroptosis involves the highly expressed polyunsaturated fatty acids (PUFAs; such as docosahexaenoic acid and arachidonic acid) on the cell membrane, which are driven by acyl-Coenzyme A synthase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3). As a result, these PUFAs are modified to long chains and re-esterify them into phospholipids that infiltrate into lipids and membranes, forming PUFA-PL with carbon-centered phospholipid radicals (PL•) (19–22) (Figure 1A). Subsequently, non-enzymatic self-oxidation occurs with molecular oxygen under the catalysis of the Fenton reaction, which is initiated by iron and H2O2 to produce phospholipid peroxyl radicals (PLOO•), which are generated by removing hydrogen from another PUFA to generate PLOOH (6, 22). Simultaneously, with the participation of iron, PUFA-PL undergoes an enzymatic reaction to generate PLOOH under the direct or indirect catalysis of lipoxygenase (LOX) and cytochrome P450 oxidoreductase (POR) (23, 24) (Figure 1A). When PLOOH is not rapidly neutralized as an PLOH by glutathione peroxidase 4 (GPX4) in time, PLOOH further reacts with ferrous ions, and the resulting lipid radicals, such as alkoxyl phospholipid radicals (PLO•) and PLOO•, react with PUFA-PLs (8). By continuing the non-enzymatic reaction catalyzed by the previous removal of hydrogen atoms and the Fenton reaction with molecular oxygen, the production of phospholipid hydroperoxides (PLOOHs), known as ferroptosis executioners, continues. This ultimately generates numerous secondary products and various cascade peroxide chain reactions, as well as a large accumulation of lipid peroxides (such as malonaldehyde and 4-hydroxynenoic acid); these actions have the potential to destroy the integrity of the membrane, eventually leading to rupture or organelle membrane and cell death (25). During this process, O2− produced by the mitochondrial tricarboxylic acid (TCA) cycle and the reduction of nitrogen oxides by NADPH produce H2O2 catalyzed by superoxide dismutase (SOD) and participate in the Fenton reaction of iron (26) (Figure 1A). Simultaneously, O2− is also involved in the recovery of iron; PUFA removal of hydrogen can reduce trivalent iron ions to divalent iron ions, and trivalent iron ions can be reduced to divalent iron ions under the action of O2− to generate molecular oxygen for PUFA-PL oxidation. The recovery between iron and ferrous is crucial for the Fenton reaction and lipid peroxidation process (24, 27).
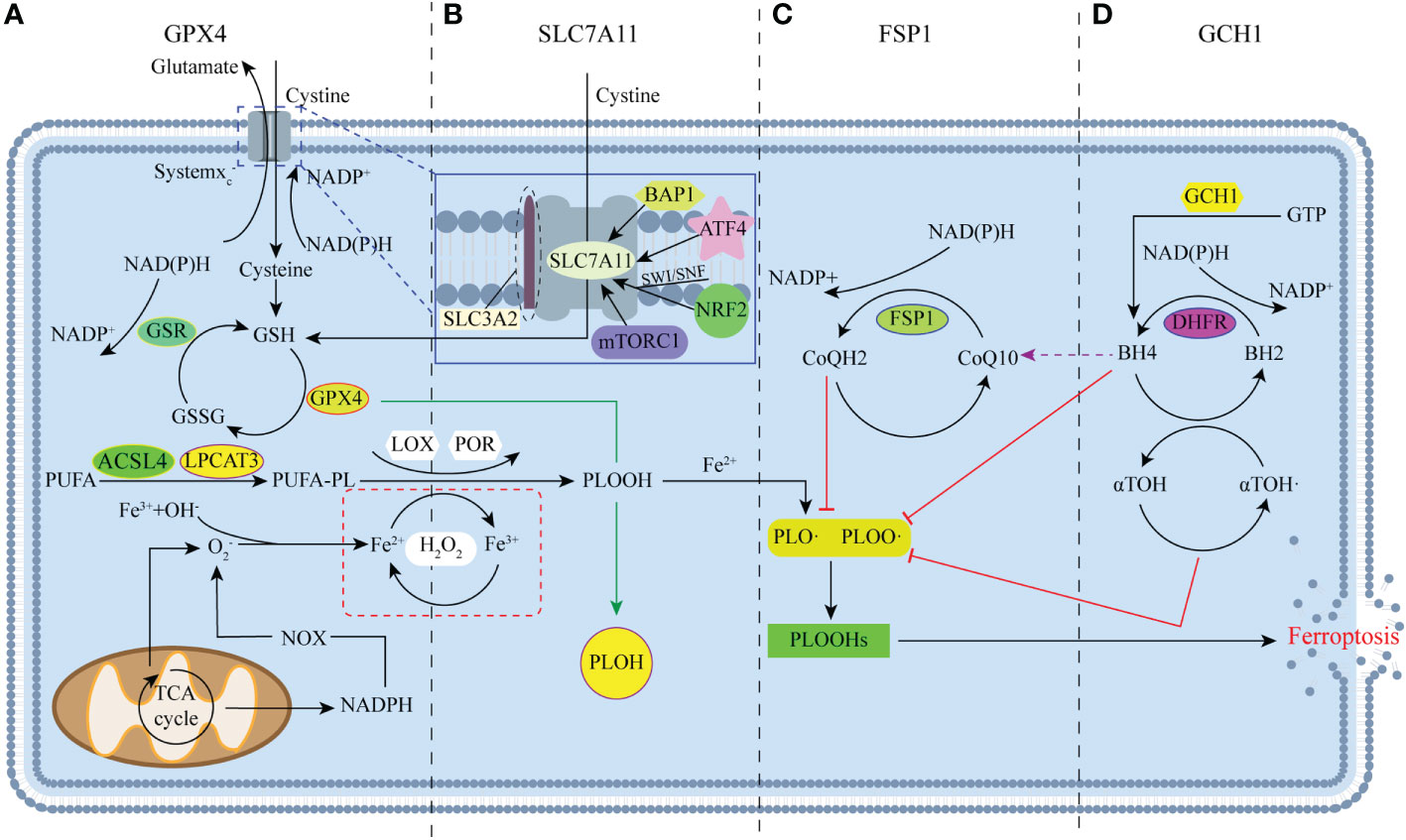
Figure 1 Major regulators of ferroptosis. (A) Glutathione peroxidase 4 (GPX4) is the key regulator of classical ferroptosis, and is mainly involved in the antioxidant axis system xc--GSH-GPX4. (B) Many studies have focused on system xc-, with SLC7A11 acting as a mediator of system xc-. (C) FSP1 protects cells from ferroptosis through the NAD (P)H/FSP1/CoQ10 system as a result of genetic deletion or pharmacological inhibition of GPX4. (D) GTP cyclase hydrolase-1 (GCH1) protects cells from ferroptosis mainly through the GCH1/BH4/DHFR pathway.
This mechanism of death differs from that of other types of morphology, genetics, and biochemistry. Morphologically, ferroptosis is marked by a decrease or disappearance of mitochondrial crests, rupture and contraction of the outer mitochondrial membrane, and shrinkage and darkening of the volume and color of the mitochondria, but the morphological changes in the nucleus are not obvious (28, 29). In terms of genetics, ferroptosis leads to changes in iron transport pathways, RAS/MAPK/Raf pathways, and cysteine transport pathways, such as those including cyclooxygenase-2 (Cox2), ACSL4, and NADPH oxidases 1 (NOX1) genes, while also downregulating GPX4, solute carrier family 7 member 11 (SLC7A11), and Ferritin (30). In terms of biochemistry, ferroptosis occurs due a series of preparations, mainly manifested in the reduction of the antioxidant capacity of cells (18, 29, 31). For example, in the main inhibitory pathway of iron apoptosis, the system xc–GSH-GPX4 antioxidant axis, the upstream regulator system xc- is inhibited, and the cystine transport pathway is blocked, resulting in a reduction in cystine transfer and cysteine reduced by NADPH (8, 25). As the rate-limiting precursor of glutathione (GSH), cysteine can cause a decrease in the activity of GPX4 and a large accumulation of lipid peroxides when deficient, which eventually leads to ferroptosis.
Since Scott J. Dixon first discovered and named ferroptosis, the critical role of GPX4 in the inhibition of ferroptosis in his study has received widespread attention, mainly in the antioxidant axis system xc--GSH-GPX4 (18). Due to GPX4’s excellent antioxidant capacity, it can detoxify intracellular lipid and cholesterol hydroperoxide product build-up by using GSH as a cofactor (32, 33). Furthermore, increasing studies have proven that GPX4 is the core regulator of ferroptosis (8). Indeed, as long as part of the GPX4 activity is maintained, cells lacking other selenoproteins can still survive and proliferate. However, the loss of GPX4 leads to the lethality of early embryos in mice. For example, in CAMKIIalpha-driven GPX4 knockout (KO) mice, mitochondrial dysfunction and ferroptosis due to chelated lipid peroxidation caused mice to display spinal motor neuron degeneration, paralysis, and death within 8 days of knockout (34), suggesting that cellular ferroptosis can be affected by regulating GPX4. GPX4 is also regulated in terms of activity and stability levels. GSH, as a cofactor is a direct factor affecting GPX4 (35), is derived from the tripeptides of glutamic acid, glycine, and cysteine, and is the most abundant reducing agent in mammalian cells (36). Notably, independent of the antioxidant properties of GSH, it also can regulate the NO steady state, forming nitrosyl mercaptans (37). In summary, GSH is important for the biological occurrence of iron-sulfur clusters, preventing oxidative damage, and directly regulating glucose homeostasis. When system xc- is inhibited, obstruction of the cystine transport pathway can decrease GSH and GPX4, which can lead to cell death due to ongoing oxidative stress (Figure 1A).
In relation to obesity, genetic variants in GPX4 have been linked to obesity and inflammation in humans (38, 39), suggesting a role for GPX4 in metabolic disorders. And studies have described that impaired GPX4 activity is associated with obesity, although the biological consequences have not yet been determined (32). It is certain that obesity leads to the expansion of adipocytes, causing an inadequate supply of oxygen within AT and promoting the secretion of pro-inflammatory cytokines and ROS (40–42). The presence of the antioxidant GPX4 has also been shown to neutralize the ROS produced by the oxidative stress of AT and control AT inflammation by preventing lipid peroxidation (43). Furthermore, the same study also revealed that the activity of adipocytes GPX4 in obese patients is reduced, but the consequences seem to more of effect on undifferentiated adipocytes, but not as obvious for mature adipocytes (32, 43, 44). As a substance necessary for adipocytes differentiation, the reduction in GPX4 may affect AT remodeling, which is manifested by the finding that Gpx4−/−AT mice have larger adipocytes and a considerably increased body weight compared to wild-type mice; additionally, GPX4 can inhibits the phospholipid peroxidation of adipocytes, although the specific mechanism is unclear (43). Additionally, increased fasting blood glucose and insulin resistance were also observed in Gpx4−/− AT mice, and spontaneously produced impaired insulin signaling in the liver, resulting in systemic inflammation and disorders of glucose metabolism (43). In conclusion, there is a strong association between GPX4 and obesity, and GPX4-centered ferroptosis may be the key link between GPX4 and obesity
Ferroptosis-suppressor-protein 1 (FSP1) is a recently discovered non-GPX4-dependent ferroptosis inhibitory factor, which exhibits a different pathway of action from GPX4 in the process of ferroptosis inhibition, mainly regulated by the NAD (P)H/FSP1/CoQ10 system (45) (Figure 1C). As a consequence, both ultimately act on lipid radicals to reduce cellular damage caused by their excessive accumulation, and the loss of FSP1 can also increase PLOOHs. Apparently, FSP1 provides another important pathway for ferroptosis that is not limited to mitochondria (46, 47). Indeed, the sensitivity of different cancer cells to GPX4 inhibitors is highly variable, and some are unaffected by GPX4 (48). Therefore, the emergence of FSP1 may compensate for some of the shortcomings of GPX4. However, whether there is an FSP1-based pathway at play in adipocytes without ferroptosis when GPX4 is reduced requires further investigation. The available results suggest that FSP1 can play an important role in predicting the efficacy of ferroptosis-inducing drugs in cancer, and FSP1 inhibitors are considered to have great potential to overcome ferroptosis resistance.
To identify other ways to regulate ferroptosis sensitivity, a new set of ferroptosis inhibition genes was identified by genome-wide activation library screening. As a result, a new ferroptosis inhibition axis with GTP cyclohydrolase-1 (GCH1) as the core was finally determined, which was found to be mainly regulated by the GCH1/BH4/DHFR pathway, which also protected cells from ferroptosis in a GPX4-independent manner (49, 50) (Figure 1D). However, studies only points to the key role of BH4 in protecting cells when GPX4 is inhibited, and its influence on lipid peroxidation is unrelated to its cofactors, but the specific mechanism remains unclear (50). Currently, the GCH1-based ferroptosis inhibition pathway only focuses on the therapeutic feasibility of tumors and cancer cells, with no obesity-related studies as of yet (49). However, as a polymorphism site for human and chronic diseases, including diabetes and hypertension, GCH1 can play a role in adipocytes and their related regulatory pathways.
With the deepening of ferroptosis research, studies have strengthened the strategy of using the Cystine/Glutamate reverse transporter SLC7A11 (xCT) pathway to regulate ferroptosis, the principle of which is also based on the antioxidant axis system xc--GSH-GPX4, but the focus falls on the key precursor system of the system xc- (51). The system xc- is actually a sodium-independent antiporter, consisting of the heavy chain subunit solute carrier family 3 member 2 (SLC3A2) and the light chain subunit SLC7A11 (52, 53). SLC3A2 can anchor SLC7A11 to the plasma membrane and maintain the stability of its protein (54), while SLC7A11 can mediate the activity of the reverse transporter of system xc- (55, 56) (Figure 1B). The two work together to transfer extracellular cystine and intracellular glutamate out in a 1:1 ratio, thereby driving the efficient operation of the GPX4 antioxidant axis and protecting cells from oxidative stress damage (57, 58). However, because the function of SLC3A2 is not limited to cystine transport pathways, studies mainly focus on regulating SLC7A11 to inhibit the ferroptosis resistance of cancer cells (56). In addition, the activity and expression of SLC7A11 is strictly regulated by multiple mechanisms, such as transcription factor 4 (ATF4), nuclear factor erythroid 2–related factor 2 (NRF2), ubiquitin BRCA1 (Breast cancer gene 1)-associated protein-1 (BAP1), the SWI/SNF complex, and rapamycin complex 1 (mTORC1) (51), through which the expression of SLC7A11 can be upregulated to enhance the antioxidant defense of cells and inhibit the occurrence of ferroptosis (Figure 1B).
In general, there are four main mechanisms, GPX4, FSP1, GCH1 and SLC7A11, to help the body resist oxidative stress. Their operation is also precisely controlled by many regulators, but even under the conditions of so many pathways, more and more studies still regard SLC7A11 as the rate-limiting precursors and GPX4 as the core antioxidant defense to against ferroptosis (8).
Studies found that obesity is closely related to iron metabolism disorders, which are mainly reflected as an excessive iron level (59). Ferroptosis caused by iron accumulation is accompanied by elevated ROS and inflammatory response, insulin resistance and mitochondrial dysfunction, leading to metabolic disorders and the development of obesity (60). As a result, the link between obesity and ferroptosis in different aspects has gradually become clear.
At the iron level, a previous study found that iron accumulation occurred in the epididymal adipose tissue (eAT) of polygenic obese and diabetic mice, but did not occur in the subcutaneous and brown adipose tissue (61). The available data are sufficient to show that iron accumulation is more obvious in the pancreas, liver, and heart, all of which are traditional tissues with excessive iron. Adipose tissue (AT) iron overload is also closely related to macrophages, mainly reflected in the important role of macrophages in controlling iron metabolism (62). Studies have shown that obesity induces increased M1 macrophages (M1) polarization in mice, which may further promote iron deposition, according to in vitro studies (63). Interestingly, studies have identified adipose tissue macrophages (ATMs) with iron-processing phenotypes, noting that a high-fat diet (HFD) promotes iron entry into adipocytes and inhibits the ability of ATMs to process iron (64). These studies also further illustrate the role of ATMs in the connection between iron and adipocytes. In addition, another study in mice with high-speed rail diet intervention also yielded an interesting result, showing that eAT produced robust remodeling, which was accompanied by AT weight reduction and a significant increase in iron content. This further suggests that processes associated with ferroptosis may play roles in AT remodeling (61). In a word, these findings further embody the relevant metabolic regulatory network in complex biological systems, with ferroptosis and obesity caused by iron accumulation as the connection point, and the parallel occurrence of the inflammatory response and insulin resistance.
Under an inflammatory environment and oxidative stress, as obesity progresses, the increased secretion of pro-inflammatory cytokines and chemokines, such as monocyte chemotin-1, tumor necrosis factor (TNF) α, and interleukin (IL)-6, lead to a low-grade inflammatory response throughout the body (65). This is similar to the environmental changes that accompany ferroptosis. Moreover, the increased iron is also accompanied by oxidative stress in the pancreas, liver, and eAT. In polygenic obese mice, iron chelating agents inhibit inflammatory factors and the production of oxidative stress, as well as the infiltration of ATMs to improve the hypertrophy of adipocytes (66, 67), which further indicates that ferroptosis may occur in AT. Indeed, the development of obesity and ferroptosis indicates the weakening of the body’s antioxidant capacity, which in turn leads to a series of metabolic disorders.
Regarding insulin resistance, studies have shown that omental adipocytes size and insulin resistance are positively correlated, and long-term obesity induces systemic inflammation and insulin resistance (68, 69). Moreover, an increase in inflammatory factors and a decrease in adiponectin may also lead to the interruption of insulin signaling pathways, reducing insulin sensitivity (70, 71). An increase in iron storage also causes insulin resistance, and insulin sensitivity increases after reducing serum iron levels. During insulin therapy, iron uptake can be increased by increasing the transferrin receptor 1 (TfR1) in adipocytes (72).
Regarding mitochondrial function, obesity can lead to mitochondrial dysfunction, an abnormality that mostly occurs in the liver, muscles, and AT that are associated with high energy, nutrient, and lipid overload (73). Furthermore, the morphology and number of mitochondria in obese patients have also changed. For example, the mitochondria in their skeletal muscle have become smaller and shorter, while those of white adipose tissue (WAT) are small and elongated, with an irregular crest, and the larger the adipocytes, the less mitochondria are present (74–76). Additionally, mitochondrial dysfunction in AT under obese conditions can significantly increase respiration and biogenesis, promote fatty acid oxidation and alter metabolism, all of which lead to the production of excess acetyl-CoA (77, 78). This is due to the fact that obesity enhances the production of ROS in mitochondrial, which can damage the lipid membranes, proteins, and enzymes of the respiratory chain in the mitochondria, as well as DNA (77). In summary, obesity leads to decreased mitochondrial function and increased insulin resistance. It is thought that there is a close link between the two given the known mitochondrial dysfunction caused by iron accumulation in adipocytes.
The special link between adipocytes and ferroptosis. Although there are substances, genes, and environmental changes within AT that are required for ferroptosis. However, there seems to be a contradictory result between adipocytes and ferroptosis, as shown by the proliferation of adipocytes despite being under ferroptosis conditions (79). Therefore, to demonstrate whether adipocytes can undergo apoptosis in response to oxidative stress, one study treated mature adipocytes with H2O2 and found that an increase in ROS enhanced adipocyte apoptosis (80). These results suggest that not only AT remodeling but also differentiated adipocytes can still be affected by ferroptosis. So how do adipocytes in AT defend against ferroptosis? One speculation is that although adipocytes have reduced GPX4, adipocytes may be similar to some tumor cells, which still have a high level of GSH in their cells, so that although adipocytes are in an overall oxidative stress condition, the remaining GPX4 from GSH is still needed to neutralize large amounts of ROS and remove some of the lipid peroxides, resulting in differentiated adipocytes that are not or less affected by ferroptosis (81). Compared to the former speculation, another study found that adipocytes can secrete specific fatty acids to induce ferroptosis resistance in breast cancer cells, and this process was dependent on the fatty acid synthetase ACSL3, confirming that breast cancer cells co-cultured with peri-tumor adipocytes showed resistance to ferroptosis (82). In addition, the team verified the protection of adipocytes against ferroptosis in triple-negative breast cancer through animal models. These results provide a new idea as to why adipocytes in obese AT do not undergo ferroptosis and continue to proliferate.
In conclusion, on the basis of these studies, this review speculated that there may be two mechanisms of ferroptosis leading to obesity: (i) In the autonomic nervous system, vagus and parasympathetic nerves in some organs or tissues may be affected by ferroptosis and become dysfunctional, while the inflammatory factors released from adipocytes lead to excessive sympathetic activation and hypothalamic neuronal ferroptosis which promotes the development of obesity through a series of processes; (ii) Ferroptosis may have occurred in certain anti-inflammatory immune cells in AT, which allows a large increase in pro-inflammatory immune cells, further causing systemic inflammation and insulin resistance, and ultimately leading to the development of obesity. Unfortunately, although ferroptosis, a novel mode of cell death, has received widespread attention from researchers since its discovery, there are still few studies on AT related to ferroptosis. Most of the studies are still mainly focused on tumor cells (50), and a certain theoretical system has not yet been formed, resulting in many specific mechanisms related to it are not very clear.
At present, the rapidly rising prevalence of obesity has become a global problem that needs to be solved. The discovery that AT can be affected by the process of ferroptosis provides us with another idea to treat obesity. Therefore, this review will investigate whether the neurological and immune systems can regulate the ferroptosis process in AT. Finding out the role of ferroptosis in AT, and thus provide a new therapeutic target for the treatment of obesity, which is important for the clinical treatment of human obesity.
The nervous system is known to play a leading role in the regulation of physiological functional activities, but is also closely related to AT as a result of the dynamic crosstalk between adipocytes and other types of cells in the highly neurologically innervated and vascularized tissue matrix (83). Therefore, a large number of studies in recent years have actively explored the innervation of AT, and the results have confirmed the presence of only sensory innervation and sympathetic innervation within AT (83). Among them, the fat sensory nerve will convey the metabolic signal in AT to the central nervous system, and then instruct AT through the sympathetic nerve (84–86), so that it can carry out the corresponding lipolysis and thermogenesis according to the instruction (87, 88) (Figure 2). However, under obese conditions, the number of nerve fibers within the AT appears to be drastically reduced, which not only leads one to ponder whether there is some association with ferroptosis. Besides, the exact mechanism leading to the reduction of nerve fibers remains unclear. What is known is that the expansion of adipocytes causes tissue inflammation and diminished sympathetic signaling (83, 89). This leads to dysregulation of sympathetic neurotransmitter levels of NPY, ATP and NE, resulting in a chronic over-activation of sympathetic nerves, but with reduced sensitivity of β-adrenergic receptors, resulting in a weakened regulation of AT and a proliferation of adipocytes. Moreover, cytokines released from adipocytes such as leptin stimulate AT sensory nerve fibers and may lead to their hyperactivation and release of inflammatory factors, further inflammatory stimulation and oxidative stress may inhibit normal hypothalamic and nervous system function (89–91). In addition to the innervation within the AT, the autonomic nervous system is known to populate all organs and tissues of the body. As a whole, there is also a network of alternating influences between these autonomic nervous systems and the AT. For example, in the vagus nerve, although postganglionic neurons of the vagus nerve may be located outside of AT, studies have found that some preganglionic axons of the vagus nerve terminate on neurons of unknown phenotype in the adrenal, celiac, and superior mesenteric ganglia. Since neurons in these ganglia may contribute to sympathetic innervation of at least the intraperitoneal AT, this could also be a potential pathway for vagal innervation. Not only that, but studies has found ferroptosis in the nervous system (92). The reason may be related to the high activity of brain nerves. The high metabolic rate and high oxygen consumption of brain parenchyma will inevitably lead to the production of a large number of by-products ROS, and may further cause oxidative stress (93). At the same time, the brain is relatively limited in antioxidant factors, but it is also rich in iron, which can react with endogenous H2O2 to promote lipid peroxidation (94). And membranes with high PUFA-PL content should be particularly susceptible to peroxidation, as has been demonstrated in neurons (93). In addition to this, the iron accumulation seen in the liver of obese patients is an interesting echo of hepatic parasympathetic dysfunction. In conclusion, the systemic inflammation induced by obesity may accelerate the process of ferroptosis in certain neuronal cells, and this could potentially further promote the development of obesity (95).
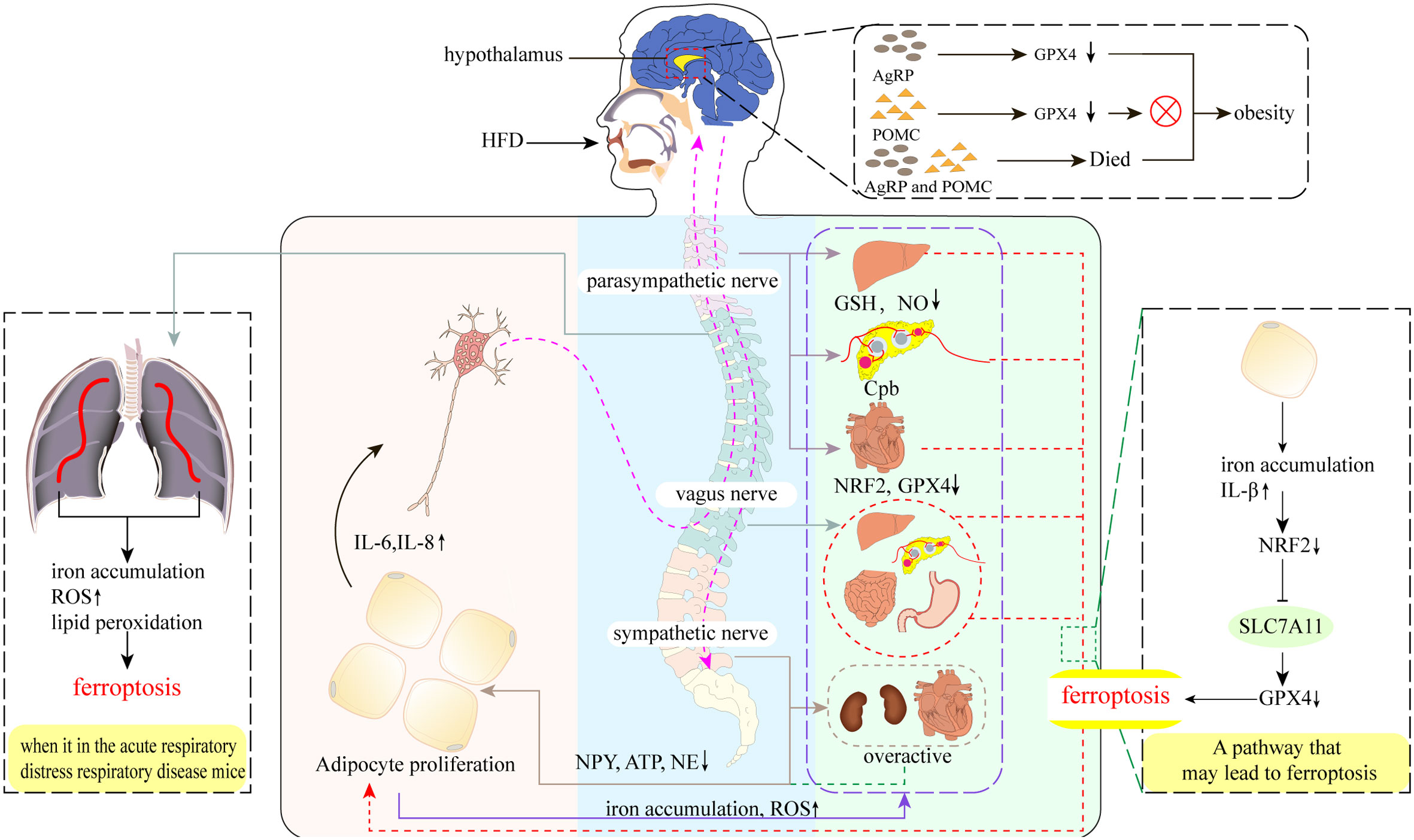
Figure 2 Neural regulation of obesity and ferroptosis. A high-fat diet (HFD) leads to increased secretion of pro-inflammatory cytokines such as IL-6 and IL-8 in adipose tissue (AT), which stimulates AT sensory nerves and subsequent vagal-parasympathetic transmission to the nociceptors, which then transmit regulatory information directly to AT via sympathetic nerves, as well as by targeting tissues and organs elsewhere in the body to promote AT lipolysis and thermogenesis. However, the emergence of obesity disrupts these transmission pathways. Parasympathetic: obesity decreases hepatic GSH, NO levels, pancreatic cholinergic pathway block (Cpd), and cardiac NRF2 and GPX4 levels, which may cause ferroptosis in hepatic, pancreatic, and cardiac parasympathetic nerves, thus promoting AT amplification. Vagus nerve: part of the parasympathetic nerve. The presence of obesity may cause hepatic vagal ferroptosis, which further leads to possible ferroptosis of islet and intestinal and gastric vagal nerves. Sympathetic nerves: obesity leads to over-activation of sympathetic nerves, including cardiac and renal sympathetic nerves, and reduced levels of the sympathetic transmitters NPY, ATP, and NE, which also reduce their direct innervation of AT. Hypothalamus: AgRP neurons do not undergo ferroptosis after the knockdown of GPX4, do not undergo ferroptosis after knockdown, yet still resulted in the appearance of obesity, which was not observed in POMC neurons. Interestingly, AgRP and POMC neurons also led to obesity after inactivation.
The vagus nerve is the main nerve of the parasympathetic branch (96), which can participate in taste sensation and innervate most organs, including the liver, stomach, and pancreas, and regulate circulation, breathing, and digestion by conducting sensory impulses in organs, particularly those controlling the activity of the heart muscle, glands, and smooth muscles (97, 98). In terms of the relationship with obesity, studies have found that in obese humans and rodent models, autonomic imbalances include excessive activation of the vagus nerve (99, 100), and a decrease in the mass of WAT is induced after vagus nerve resection (100). However, this idea, while demonstrating the ameliorative effect of vagus nerve excision on the association with obesity (101), is limited by the absence of significant changes in some lean animals and some populations (98), as well as the increased occurrence of secondary trauma and mortality after excision (102). As an important component of regulating metabolism and immune homeostasis, the vagus nerve can regulate WAT content through the central nervous system to control sympathetic peripheral muscle bundles, and can also regulate the release of GSH and NO to enhance antioxidant capacity and prevent ferroptosis (36). Thus, the vagus nerve may be involved in maintaining oxidative homeostasis and responding to external stimuli or injury. For example, in acute respiratory distress, ferroptosis has been found in mouse lung tissue, together with iron accumulation, increased ROS, and lipid peroxidation (103). The use of electro-acupuncture to stimulate the Zusanli (ST36) acupoint can activate the α7 nicotinic acetylcholine receptor in the lung tissue through the cervical vagus nerve and sciatic nerve to protect the ferroptosis in the lung tissue, which also verifies the importance of the vagus nerve in protecting the oxidative damage of cells (104) (Figure 2). The vagus nerve can also inhibit the release of pro-inflammatory cytokines in such a way that mediates selective cholinergic activation in the inflammatory response, thereby improving inflammation and obesity (97, 98, 105). Surprisingly, the emergence of obesity seems to break the protective ability of the vagus nerve, not only the oxidative damage of organs and tissues (i.e., the liver and pancreas), but also in terms of transmission signal interruption and decreased plasticity (98) (Figure 2). Indeed, some studies have pointed out that this phenomenon may be an important mechanism for developing obesity (106). Overall, autonomic imbalances caused by obesity may be associated with the first occurrence of ferroptosis in the vagus nerve, which impairs its ability to transmit information collected by the sensory nerve to the nerve center, as well as its ability to mobilize GSH and NO to prevent ferroptosis.
As another major component of the autonomic nervous system that neutralizes sympathetic activity, and has a complete mediated pathway to the pancreas, the parasympathetic nervous system plays an important role in maintaining metabolic homeostasis, such as maintaining blood sugar (107). Parasympathetic efferent nerves can transmit neuropeptides and acetylcholine to the islet liver, increasing insulin release and liver NO production through muscarinic receptors and lowering blood sugar. In addition, despite a lack of evidence of the parasympathetic innervation of WAT (108, 109), it can still affect WAT through multiple pathways, and studies have suggested that reduced parasympathetic activity is associated with obesity (110). Correspondingly, the link between parasympathetic injury and ferroptosis has gradually became clear. Diabetes mellitus, which is a complication of obesity, can activate satellite glial cells of the supracervical ganglia through P2Y receptors, resulting in increased expression of pro-inflammatory cytokines such as IL-1β, and causing oxidative damage to the autonomic nerves such as via an inflammatory response (111, 112) (Figure 2). Among them, oxidative damage to the autonomic nervous system due to iron accumulation associated with diabetes may be due to changes in the NRF2-GPX4 pathway in ferroptosis (113, 114), the transcription factor that regulates SLC7A11 (51) can effectively prevent oxidative damage in various pathological conditions upon NRF2 activation (115, 116). Furthermore, both obesity and diabetes are accompanied by significant increases in ROS and decreases in NRF2 and GPX4 levels, indicating a weakening of antioxidant capacity and activation of the ferroptosis process. Simultaneously, ferroptosis may be involved in the progression of cardiac autonomic neuropathy (DCAN) through the P2Y14 receptor (a new member of the metabolic receptor family), while P2Y14 targeting naringin can alleviate DCAN by promoting the NRF2-GPX4 pathway to eliminate ROS and inhibit ferroptosis (117) (Figure 2). Surprisingly, another study showed that diabetes-related cardiovascular autonomic damage is first manifested in the vagus nerve (106), which may involve a ferroptosis-mediated overall decrease in parasympathetic tone, ultimately leading to over-activation of the sympathetic nerve and the worsening development of metabolic complications. In conclusion, these studies elucidate the role of obesity and diabetes in promoting autonomic nervous system imbalances and metabolic disorders through the ferroptosis process (106).
The sympathetic nervous system plays a key role in regulating metabolic control as an important hub that connects the brain to most organs, glands, and tissues, including the heart. Particularly in regulating AT, which is only innervated by the sympathetic nerves, while the liver, skeletal muscle, and pancreas are under the control of the parasympathetic nervous system (118). Studies have shown that sympathetic hyperactivation is strongly associated with hyperinsulinemia, impaired baroreceptors, angiotensin II, apnea syndrome, and adipokines (119). Furthermore, studies have highlighted that ROS can activate the sympathetic nervous system (120), suggesting that the sympathetic nervous system may be less inhibited under high oxidative stress. However, it has also been shown that sympathetic neurons undergo lipid peroxidation following cleavage of the p75 neurotrophic factor receptor, causing degeneration and cell death (121). Considered another way, sympathetic hyperactivation indicates that the inhibitory effects of the vagus and parasympathetic nerves have weakened, which may also be an important mechanism for developing obesity. In obese conditions, the vagus and parasympathetic nerves may be damaged due to ferroptosis, which also promotes further sympathetic activity and autonomic nervous system imbalance. Furthermore, studies have found that antioxidants can reverse sympathetic dysfunction, oxidative stress, and hypertension in male obese rats following intervention with antioxidants (122). It has also been shown that impaired baroreceptor reflexes are caused by reduced inhibition of the vagus and parasympathetic nerves (123). In conclusion, these data show that the overactivation of the sympathetic nerve under obese conditions is closely related to ferroptosis of the vagus nerve and the parasympathetic nervous system. Additionally, the sympathetic nervous system may have a complex mechanism that inhibits the effects of oxidative stress represented by ferroptosis.
In addition to the vegetative nervous system described above, traces of ferroptosis have been found in neurons in some specific areas associated with obesity. For example, HFD has increased secretion of free fatty acids and pro-inflammatory cytokines. When these pro-inflammatory mediators reach the hypothalamus, they increased the levels of oxidative stress and neuronal inflammation in the hypothalamus through processes such as microglia proliferation (124, 125). Insufficient GSH, the only antioxidant in the hypothalamus, may cause ferroptosis in some neurons, leading to further metabolic disorders. Leptin-activated hypothalamic proopiomelanocortin (POMC) and agouti-related protein (AgRP) neurons located in the arcuate nucleus are typical (126, 127). The former increases appetite and the latter suppresses appetite, both of which are critical for the metabolic homeostasis of the body (128). Studies have shown that a high-fat, high-sucrose (HFHS) diet decreased GPX4 expression and activity in the hypothalamus of mice associated with elevated GPX4 levels in WAT (129). Surprisingly, the decrease in GPX4 failed to damage the integrity of both neurons, and mice that only lost GPX4 in AgRP neurons experienced weight gain, while POMC neurons required for ROS buffering and metabolic homeostasis were unaffected by GPX4 deletion (129) (Figure 2). This phenomenon is indeed elusive, but it is speculated that there may be other antioxidant pathways that inhibit this process. In contrast, another study that specifically induced the death of AgRP and POMC neurons, profound metabolic disorders and obesity emerged (129) (Figure 2). In summary, although the two neurons did not die of GPX4 reduction, the study focused on only one point of GPX4 and did not delve into other regulators, indicating that there are other pathways affecting the activity of POMC neurons under obesity conditions (129). However, this phenomenon also confirms that the increase in high levels of plasma under previous obesity conditions can cause POMC neuron stimulation, which will further induce sympathetic activation. Additionally, these studies continue to demonstrate that GPX4 deletion can cause oxidative damage to membranes and exacerbate neuroinflammation. Therefore, improving obesity by improving oxidative stress caused by ferroptosis in the hypothalamus and nervous system is an essential consideration.
With the continuous in-depth study of obesity, the importance of AT immune cells as a key hub for regulating the AT environment has become recognized. AT immune cells not only promote AT homeostasis, but also change phenotype and increase their number to promote the maintenance and development of local AT inflammation. AT immune cells also secrete pro-inflammatory cytokines and other pro-inflammatory products into the systemic circulation, which affect the entire body, thus representing a key factor in the transition from simple obesity to cardiovascular disease, type II diabetes, and related metabolic disorder complications (130). It should be clarified that although the contribution of immune cells to metabolic diseases is mainly concentrated in AT, an effective source of such inflammation also exists in the liver, intestines, and other metabolic regulatory tissues, promoting the production of inflammation and obesity throughout the body through the circulation (131). In addition, AT remains the most commonly studied source of immune-mediated inflammation in obesity. In the case of eWAT, an increase in endogenous fatty acids following adherence to a HFD activates two sensors within adipocytes, Toll-like receptor 4(TLR4) and inflammasomes, which causes an influx of neutrophils to enter the eWAT, leading to increased levels of chemokine and cytokine expression (132). Among them, the most typical is the chemokine Leukotriene B4 (LTB4), the increased expression of which causes recruitment of neutrophils, ATMs, T cells, and B cells, which increase expression of pro-inflammatory cytokines, eventually leading to the development of eWAT inflammation and obesity (132) (Figure 3). Therefore, in response to iron accumulation and high levels of ROS within AT, certain anti-inflammatory immune cells may also be affected by the ferroptosis process, resulting in their inability to neutralize the inflammatory response caused by pro-inflammatory cytokines in a timely manner. Currently, known immune cells present within AT include ATMs, T cells (CD4+, CD8+, Tregs), natural killer T cells, B cells, neutrophils, eosinophils, and dendritic and mast cells (130, 133, 134). Based on current research progress and the association of these immune cells with obesity and oxidative stress, this review identified macrophages, T cells, B cells, and neutrophils, which are closely related to oxidative stress and metabolic disorders, and sorted out their changes in obesity and these immune cells complication-related pathways to identify ferroptosis traces.
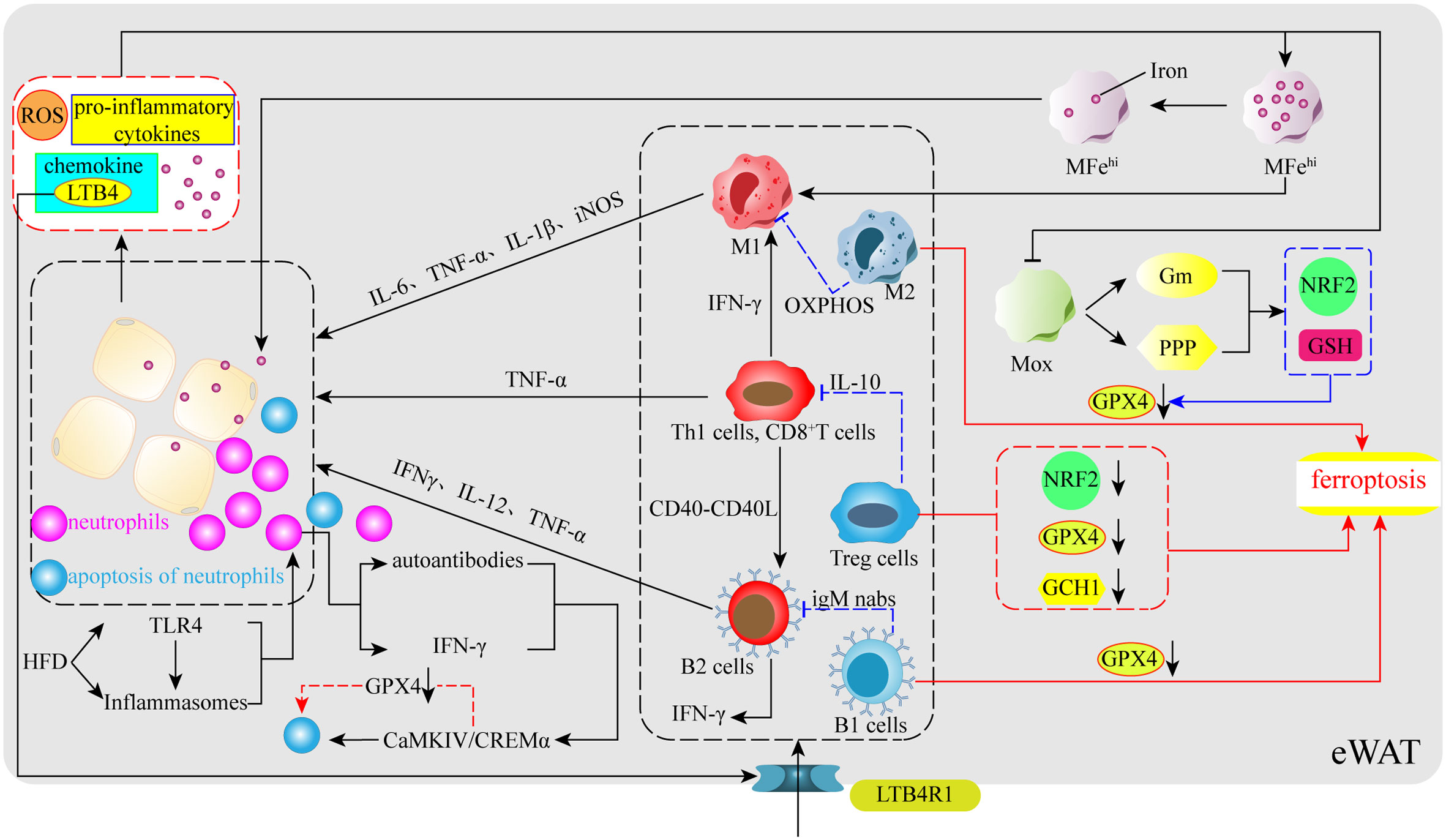
Figure 3 Immunoregulation of obesity and ferroptosis. In the early stages of obesity, HFD stimulates TLR4, a sensor within VAT, and inflammatory vesicles to release certain signals to promote neutrophil entry into VAT, followed by the release of chemokines (LTB4) by neutrophils to further promote the aggregation of ATMs, T cells, and B cells in VAT. Simultaneously, their pro-inflammatory phenotype releases numerous pro-inflammatory cytokines, which increases the levels of ROS, iron, pro-inflammatory cytokines, adipocyte-secreted chemokines, and leads to increased inflammation due to ferroptosis in some types of anti-inflammatory immune cells. ATMs: obesity impairs the iron reserve capacity of MFehi macrophages and promotes their polarization toward the M1 phenotype. At the same time, a large number of pro-inflammatory cytokines also inhibit the upregulation of the NRF2 gene and GSH levels through glucose metabolism and the pentose phosphate pathway in Mox macrophages, which have antioxidant capacity, resulting in reduced GPX4 levels and ferroptosis in M2 macrophages. T cells: Obesity causes T cells to release IFN-γ and CD40-CD40L to promote M1 and B2 production. Tregs undergo ferroptosis due to reduced levels of NRF2, GPX4, and GCH1. B cells: obesity promotes the entry of B cells into VAT via the LTB4R1 receptor, whereas B1 cells may also undergo ferroptosis due to reduced GPX4. Neutrophils: neutrophils activate the CaMKIV/CREMα axis to induce apoptosis spontaneously, which may involve ferroptosis-related processes.
ATMs are central players in obesity-related inflammatory and metabolic diseases and can be involved in the regulation of adipocytes energy metabolism and mitochondrial function (133), thereby promoting post-inflammatory or post-injury repair, as well as tissue remodeling (135). ATMs have two main phenotypes: M1 macrophages (M1) is considered the culprit of AT inflammation, can participate in glycolysis to rapidly produce ATP, and plays a key role in the initiation and maintenance of systemic inflammation and insulin resistance; and M2 macrophages (M2) uses the effective coupling of respiration in the electron transport chain to enhance oxidative phosphorylation (OXPHOS), reduces ROS production, and promotes improved obesity and systemic glucose homeostasis by inhibiting adipocytes proliferation (136, 137). Additionally, studies have identified two new ATMs that are closely related to iron metabolism and oxidative stress: iron-rich ATMs (MFehi) and antioxidant ATMs (Mox) (138). Impaired MFehi affects iron distribution, leading to AT dysfunction and adipocyte iron overload in obese patients; Mox inhibits oxidative damage by upregulating NRF2-dependent antioxidant enzymes and producing GSH, which has been shown to be the main ATMs for lean AT (64, 138).
At present, the interaction between ATMs and ferroptosis is gradually becoming clear, studies have shown that ATMs secrete IL-6, TNF-α, IL-1β, and iNOS, which can induce or inhibit the process of ferroptosis through different routes (139–142). Notably, IL-6 not only modulates hepcidin to affect the release of stored iron from ATMs and intestinal absorption, but also reduces transferrin expression and aggravates iron accumulation by promoting hepcidin production. IL-6 can even induce the polarization of ATMs to M1 or M2 (143). IL-1β can also activate hepcidin to promote transferrin degradation and iron accumulation, which is also related to the occurrence of ferroptosis (144). In addition, the ROS produced by ATMs first affect their recruitment and polarization, especially in cases such as diabetes, ROS induces ATMs to polarize towards M1 and the release of inflammatory factors (145, 146). Based on these results, an interesting association emerged, in that obesity leads to the accumulation of ATMs; indeed, in most obese individuals, up to half of the cells in the fat pool are ATMs (147). However, iron accumulation still occurs in AT in obese patients, which indicates that IL-6 and IL-1β in AT may cause induce damage to MFehi and Mox, resulting in high levels of oxidative stress and lipid peroxidation of AT (64) (Figure 3). Furthermore, as HFD mice show a decrease in M2 marker expression, does the inflammatory environment and iron accumulation cause M2 ferroptosis? Recent reports have found that GPX4 deletion induces iron apoptosis in M2, while M1 does not, largely because iNOS is a negative regulator of iron apoptosis and promotes M1 survival (148). In addition, another study showed that ATMs taken from the bone marrow died of iron in M0 in the absence of GPX4 (148). As the original source of ATMs, this phenomenon also provides a certain basis for the possibility of ferroptosis of ATMs. In short, AT under obese conditions due to the presence of many pro-inflammatory factors and iron accumulation, which may trigger the occurrence of ferroptosis and inflammation in M2, eventually leads to high rates of adipocytes proliferation and further development of obesity.
Following the identification of macrophages as the culprit of most inflammatory events, studies have found that when the obese phenotype is activated, T cells are involved in the inflammatory response (149, 150). However, more T cells located in visceral adipose tissue (VAT) play a role compared to those in subcutaneous fat; this is because VAT contains more immune cells compared to subcutaneous fat and the main immune cell types in VAT are ATMs and T cells, which play a more critical role in immune metabolic homeostasis (151). Similar to ATMs, T cells have both pro- and anti-inflammatory action types; indeed, Th1 cells have pro-inflammatory effects (a CD4+ T cell subsets) as do CD8+ T cells, while Th2 cells and regulatory T cells (Tregs) have anti-inflammatory effects (152). Tregs are mainly derived from the thymus gland or peripheral gland and serve to regulate local and systemic inflammation and metabolism. However, studies have shown that Tregs in HFD mice VAT decrease with age, and Treg cells are also significantly reduced in obese patients (153, 154). Interestingly, another study mentioned the key role of GPX4 in preventing ferroptosis in activated T cells (155). Thus, under obese conditions, inhibition of antioxidant enzyme pathways may cause ferroptosis in activated Treg cells, decreasing their number and increasing inflammation and metabolic disorders (156). Surprisingly, according to the previous system, the obesity-induced oxidative stress should lead to ferroptosis of traditional T cells, but the number of CD4+ T cells with pro-inflammatory effects and CD8+ T cells has been found to increased significantly (152). However, there are currently fewer mechanisms for regulating ROS in T cells, so it is still difficult to explain how the altered systemic metabolic environment after obesity affects T cells in the context of the immune response. However, another study demonstrated that NRF2 is a key factor in facilitating Treg resistance to ROS (157, 158). HFD inhibits the transcription of key antioxidant genes by triggering NRF2 translocation into obese mouse Tregs nucleus, causing a rapid decline in HO-1 expression in Tregs, leading to enhanced oxidative stress and possible ferroptosis, and causing an intensification of obesity and inflammatory responses (156) (Figure 3). Additionally, studies have pointed to the effect of BH4 inhibiting ferroptosis in T cells, and when the rate-limiting enzyme GCH1 of BH4 is deficient, it will inhibit the proliferation of T cells (159) (Figure 3). In summary, a large decrease in Tregs under obese conditions may be greatly related to ferroptosis, which may also fall on the classic antioxidant axis xc−-GSH-GPX4, serving to induce ferroptosis in activated Tregs in this antioxidant pathway. Simultaneously, GPX4 is essential for the maintenance, immunity, and proliferation of regular T cells.
Recently, studies have shown that B cells are also involved in the regulation of obesity-induced AT inflammation and insulin resistance (131, 160–162). As effector cells that produce specific antibodies, B cells can be mainly divided into B1 and B2 cells; among which, B2 cells have a pro-inflammatory effect. In an obese environment, many B2 cells are recruited into VAT through the action of the chemokine leukotriene B4 (LTB4) and its receptor LTB4R1, leading to tissue inflammation and the development of insulin resistance (163) (Figure 3). For B1 cells, B-1b cell subsets and the anti-inflammatory IgM natural antibodies alleviate dietary glucose intolerance and obesity metabolic dysfunction (164). However, among the pro-inflammatory and anti-inflammatory B cells, obesity is largely characterized by B2 cell accumulation and B1 cell damage within VAT (163, 165). Interestingly, GPX4 prevents ferroptosis due to lipid peroxidation in B1 cells, but is optional for maintenance and development in B2 cells (166). Therefore, it is important to determine whether the high concentration of ROS and the decline in antioxidant capacity caused by obesity causes ferroptosis in B1 cells. This is currently uncertain, with many unexplained mechanisms between AT, GPX4, and immune cells. It is certain that while B1 cells from the spleen did not die of iron during GPX4 deficiency, B1 cells taken from the abdominal cavity of mice experienced a higher degree of ferroptosis at the time of GPX4 defect (166) (Figure 3). It is worth pondering whether B1 cells in the obesity eAT will also undergo similar changes.
New research data suggest that neutrophils are also involved in the development of AT inflammation and insulin resistance, for instance, by infiltrating AT early in obesity and producing chemokines and cytokines that promote ATM infiltration (167). Neutrophil-derived proteolytic enzymes (i.e., myeloperoxidase and elastase) may also be involved in the initiation of AT inflammation (132). On this basis, some studies have indicated that an HFD caused by the NF-kB pathway increases the specific expression of IL-1b in eWAT neutrophils, which far exceeds the contribution of ATMs (132). Taken together, these results support the idea that neutrophils penetrate AT, causing high numbers of ATMs and IL-1b levels to drive obesity and insulin resistance (132). Surprisingly, to prevent activated neutrophils from releasing their highly cytotoxic inflammatory mediators into the tissue environment and damaging surrounding tissues along the bloodstream, neutrophils spontaneously undergo apoptosis. Although the mechanism that triggers this apoptosis remains unclear, the apoptosis process plays a key role in maintaining immune homeostasis and eliminating inflammation (168). Furthermore, neutrophil apoptosis is accompanied by a lack of GSH, and GSH may play an important role in this apoptosis process. Correspondingly, treatment with exogenous GSH and LPS was found to delay apoptosis and reduce the level of the pro-apoptotic protein caspase-3 (169). Although the relevant data do not prove the effect of ferroptosis in this process, the effect of GSH infers an association with ferroptosis. Therefore, to explore whether there is an association between neutrophils and ferroptosis, another study explored the role of GPX4 in neutrophil ferroptosis and identified two key factors, namely serum autoantibodies and IFN-α (170), both of which induce iron apoptosis in neutrophils by activating the CaMKIV/CREMα axis, leading to SLE neutropenia. An increase in the nuclear translocation of CaMKIV/CREMα decreases GPX4, which is accompanied by an increase in intracellular lipid-ROS, eventually leading to neutrophil iron apoptosis (170) (Figure 3). Although the focus of the study was on systemic lupus erythematosus, the data demonstrate that ferroptosis can occur in neutrophils (170). Overall, these data suggest a strong link between neutrophils and ferroptosis. The accumulation of many neutrophils in obese AT shows that neutrophils are unaffected by ferroptosis, which may be similar to other pro-inflammatory immune cells. However, the role of neutrophil ferroptosis in immunodeficient diseases is worth considering, particularly whether there are shared pathways that mediate the process of ferroptosis.
This review summarizes the characteristics of iron metabolism in humans, the association between obesity and ferroptosis, and the neuroimmune regulation associated with obesity and ferroptosis. While many of these mechanisms are still unknown, there is growing evidence that ferroptosis plays a key role in obesity and its complications, including type II diabetes, hypertension, nonalcoholic steatohepatitis, atherosclerosis, and obese cardiomyopathy (171). Moreover, ferroptosis may not only play a direct role in AT, but also indirectly promote the development of obesity through the inflammatory response and insulin resistance caused by liver autonomic and immunomodulatory disorders(Figure 4). Indeed, ferroptosis occurs through the intersection of multiple pathways to form an intricate regulatory network with obesity, so that one or more cells in the AT ferroptosis has the potential to change the fate of the entire tissue development. Therefore, research related to obesity with different ferroptosis targets is constantly emerging, and its specific mechanisms are gradually becoming clear. In addition to this, research into cuprotosis, which is similar to ferroptosis, has also appeared in recent years. As the name suggests, this is a form of cell death caused by copper, in which copper ions directly bind to protein lipoylation components in the tricarboxylic acid circulation pathway, causing abnormal aggregation of lipoylated proteins and loss of iron-sulfur cluster protein, resulting in protein toxic stress responses and cell death (172). Interestingly, the cuprotosis approach to cell death caused by the tricarboxylic acid cycle is similar to ferroptosis, in that ferroptosis also involves mitochondrial dysfunction and the production of excess acetyl-CoA. Thus, it is worth exploring whether the development of cuprotosis is indirectly related to ferroptosis. Ultimately, the current research on both ferroptosis and cuprotosis is still in its infancy, and it is not yet possible to confirm their specific mechanisms. However, the performance of nerve and immune cells involved in their action process still proposes novel therapeutic targets for obesity and related metabolic diseases, as well as cancer. With the continuation of research, the mechanism of ferroptosis and obesity metabolic disorders will likely be deciphered in the near future, opening up the option to reasonably mediate cellular ferroptosis through accurate scientific targets for treating obesity.
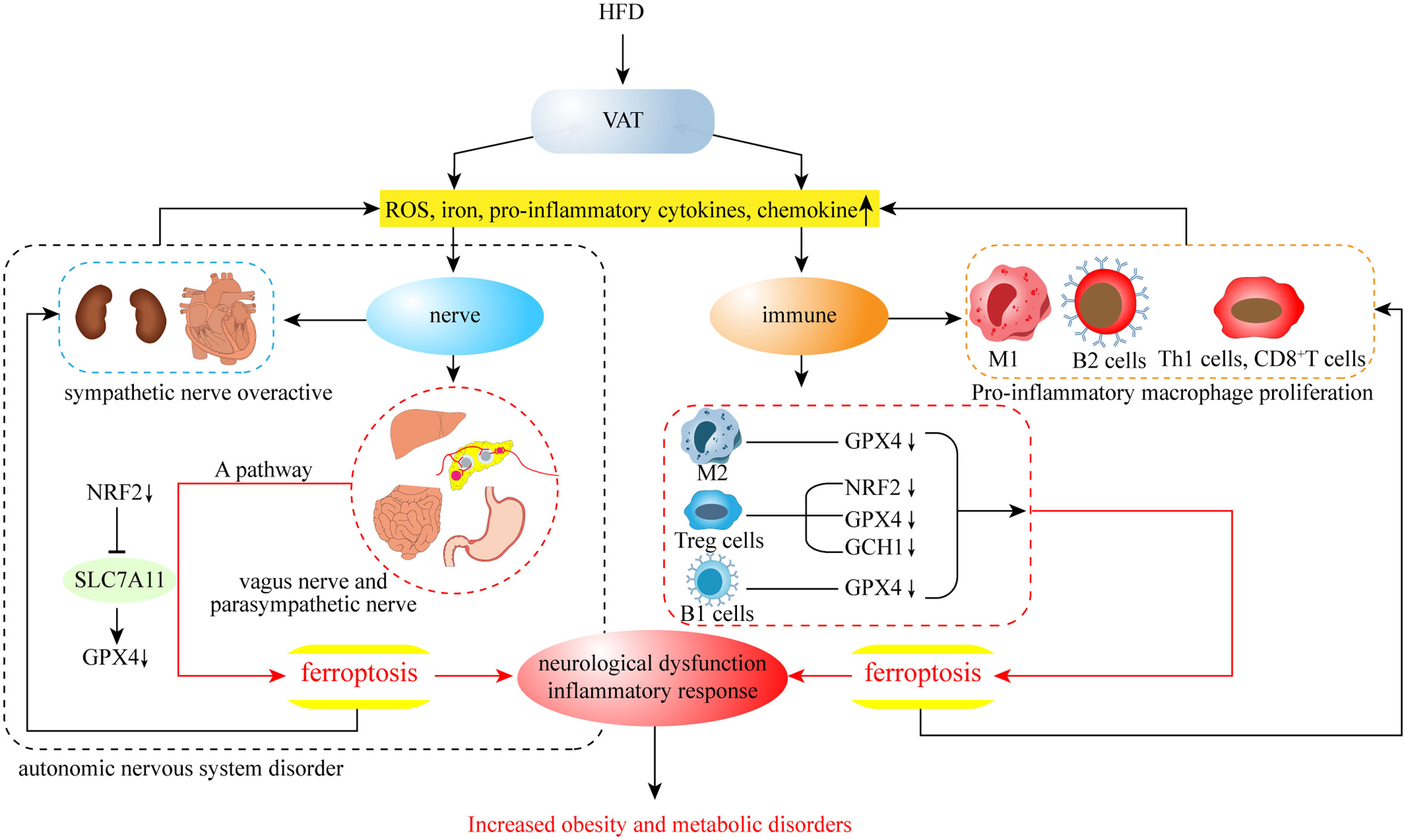
Figure 4 In VAT, HFD leads to an increase in ferroptosis-related factors, which may further contribute to the development of obesity from both neurological and immune aspects.
SZ read the literature related to the topic and participated in drafting the manuscript. ZS, XJ and ZL participated in searching and archiving the literature related to the topic and discussed the contents of the manuscript. LD, CL, QW and XT revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the Taishan Scholars Program of Shandong Province (tsqn201909148), the central government guides local science and technology development funds (YDZX2022091), and the technology research and integrated application of scientific fitness smart chip and cloud service platform(2020CXGC010902).
We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Aisen P, Enns C, Wessling-Resnick M. Chemistry and biology of eukaryotic iron metabolism. Int J Biochem Cell Biol (2001) 33:940–59. doi: 10.1016/S1357-2725(01)00063-2
2. Lieu PT, Heiskala M, Peterson PA, Yang Y. The roles of iron in health and disease. Mol Aspects Med (2001) 22:1–87. doi: 10.1016/S0098-2997(00)00006-6
3. Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science (2004) 306:2090–3. doi: 10.1126/science.1104742
4. Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev (1979) 59:527–605. doi: 10.1152/physrev.1979.59.3.527
5. Cadenas E. Mitochondrial free radical production and cell signaling. Mol Aspects Med (2004) 25:17–26. doi: 10.1016/j.mam.2004.02.005
6. Galaris D, Pantopoulos K. Oxidative stress and iron homeostasis: mechanistic and health aspects. Crit Rev Clin Lab Sci (2008) 45:1–23. doi: 10.1080/10408360701713104
7. Merkofer M, Kissner R, Hider RC, Brunk UT, Koppenol WH. Fenton chemistry and iron chelation under physiologically relevant conditions: Electrochemistry and kinetics. Chem Res Toxicol (2006) 19:1263–9. doi: 10.1021/tx060101w
8. Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol (2021) 22:266–82. doi: 10.1038/s41580-020-00324-8
10. Jakaria M, Belaidi AA, Bush AI, Ayton S. Ferroptosis as a mechanism of neurodegeneration in alzheimer's disease. J Neurochem (2021) 159:804–25. doi: 10.1111/jnc.15519
11. Wang ZL, Yuan L, Li W, Li JY. Ferroptosis in parkinson's disease: glia-neuron crosstalk. Trends Mol Med (2022) 28:258–69. doi: 10.1016/j.molmed.2022.02.003
12. Tu H, Zhou YJ, Tang LJ, Xiong XM, Zhang XJ, Ali Sheikh MS, et al. Combination of ponatinib with deferoxamine synergistically mitigates ischemic heart injury via simultaneous prevention of necroptosis and ferroptosis. Eur J Pharmacol (2021) 898:173999. doi: 10.1016/j.ejphar.2021.173999
13. Xie BS, Wang YQ, Lin Y, Mao Q, Feng JF, Gao GY, et al. Inhibition of ferroptosis attenuates tissue damage and improves long-term outcomes after traumatic brain injury in mice. CNS Neurosci Ther (2019) 25:465–75. doi: 10.1111/cns.13069
14. Wang Y, Dong Z, Zhang Z, Wang Y, Yang K, Li X. Postconditioning with irisin attenuates lung Ischemia/Reperfusion injury by suppressing ferroptosis via induction of the Nrf2/HO-1 signal axis. Oxid Med Cell Longev (2022) 2022:9911167. doi: 10.1155/2022/9911167
15. Huang LL, Liao XH, Sun H, Jiang X, Liu Q, Zhang L. Augmenter of liver regeneration protects the kidney from ischaemia-reperfusion injury in ferroptosis. J Cell Mol Med (2019) 23:4153–64. doi: 10.1111/jcmm.14302
16. Sun X, Yang S, Feng X, Zheng Y, Zhou J, Wang H, et al. The modification of ferroptosis and abnormal lipometabolism through overexpression and knockdown of potential prognostic biomarker perilipin2 in gastric carcinoma. Gastric Cancer (2020) 23:241–59. doi: 10.1007/s10120-019-01004-z
17. Sui S, Xu S, Pang D. Emerging role of ferroptosis in breast cancer: New dawn for overcoming tumor progression. Pharmacol Ther (2022) 232:107992. doi: 10.1016/j.pharmthera.2021.107992
18. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
19. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol (2017) 13:91–8. doi: 10.1038/nchembio.2239
20. Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol (2017) 13:81–90. doi: 10.1038/nchembio.2238
21. Kim JH, Lewin TM, Coleman RA. Expression and characterization of recombinant rat acyl-CoA synthetases 1, 4, and 5. selective inhibition by triacsin c and thiazolidinediones. J Biol Chem (2001) 276:24667–73. doi: 10.1074/jbc.M010793200
22. Conrad M, Pratt DA. The chemical basis of ferroptosis. Nat Chem Biol (2019) 15:1137–47. doi: 10.1038/s41589-019-0408-1
23. Kuhn H, Banthiya S, van Leyen K. Mammalian lipoxygenases and their biological relevance. Biochim Biophys Acta (2015) 1851:308–30. doi: 10.1016/j.bbalip.2014.10.002
24. Zou Y, Li H, Graham ET, Deik AA, Eaton JK, Wang W, et al. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol (2020) 16:302–09. doi: 10.1038/s41589-020-0472-6
25. Zheng J, Conrad M. The metabolic underpinnings of ferroptosis. Cell Metab (2020) 32:920–37. doi: 10.1016/j.cmet.2020.10.011
26. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell (2015) 59:298–308. doi: 10.1016/j.molcel.2015.06.011
27. Ghosh MK, Mukhopadhyay M, Chatterjee IB. NADPH-initiated cytochrome P450-dependent free iron-independent microsomal lipid peroxidation: Specific prevention by ascorbic acid. Mol Cell Biochem (1997) 166:35–44. doi: 10.1023/A:1006841228483
28. Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol (2014) 16:1180–91. doi: 10.1038/ncb3064
29. Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature (2007) 447:864–8. doi: 10.1038/nature05859
30. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell (2017) 171:273–85. doi: 10.1016/j.cell.2017.09.021
31. Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, et al. Ferroptosis: process and function. Cell Death Differ (2016) 23:369–79. doi: 10.1038/cdd.2015.158
32. Kobayashi H, Matsuda M, Fukuhara A, Komuro R, Shimomura I. Dysregulated glutathione metabolism links to impaired insulin action in adipocytes. Am J Physiol Endocrinol Metab (2009) 296:E1326–34. doi: 10.1152/ajpendo.90921.2008
33. Seiler A, Schneider M, Forster H, Roth S, Wirth EK, Culmsee C, et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab (2008) 8:237–48. doi: 10.1016/j.cmet.2008.07.005
34. Chen L, Hambright WS, Na R, Ran Q. Ablation of the ferroptosis inhibitor glutathione peroxidase 4 in neurons results in rapid motor neuron degeneration and paralysis. J Biol Chem (2015) 290:28097–106. doi: 10.1074/jbc.M115.680090
35. Wei Y, Lv H, Shaikh AB, Han W, Hou H, Zhang Z, et al. Directly targeting glutathione peroxidase 4 may be more effective than disrupting glutathione on ferroptosis-based cancer therapy. Biochim Biophys Acta Gen Subj (2020) 1864:129539. doi: 10.1016/j.bbagen.2020.129539
36. Macedo MP, Lima IS, Gaspar JM, Afonso RA, Patarrao RS, Kim YB, et al. Risk of postprandial insulin resistance: the liver/vagus rapport. Rev Endocr Metab Disord (2014) 15:67–77. doi: 10.1007/s11154-013-9281-5
37. Hogg N. The biochemistry and physiology of s-nitrosothiols. Annu Rev Pharmacol Toxicol (2002) 42:585–600. doi: 10.1146/annurev.pharmtox.42.092501.104328
38. Villette S, Kyle JA, Brown KM, Pickard K, Milne JS, Nicol F, et al. A novel single nucleotide polymorphism in the 3' untranslated region of human glutathione peroxidase 4 influences lipoxygenase metabolism. Blood Cells Mol Dis (2002) 29:174–8. doi: 10.1006/bcmd.2002.0556
39. Ruperez AI, Olza J, Gil-Campos M, Leis R, Mesa MD, Tojo R, et al. Association of genetic polymorphisms for glutathione peroxidase genes with obesity in Spanish children. J Nutrigenet Nutrigenomics (2014) 7:130–42. doi: 10.1159/000368833
40. Suganami T, Nishida J, Ogawa Y. A paracrine loop between adipocytes and macrophages aggravates inflammatory changes: role of free fatty acids and tumor necrosis factor alpha. Arterioscler Thromb Vasc Biol (2005) 25:2062–8. doi: 10.1161/01.ATV.0000183883.72263.13
41. Arner E, Westermark PO, Spalding KL, Britton T, Ryden M, Frisen J, et al. Adipocyte turnover: Relevance to human adipose tissue morphology. Diabetes (2010) 59:105–9. doi: 10.2337/db09-0942
42. Li B, Leung JCK, Chan LYY, Yiu WH, Tang SCW. A global perspective on the crosstalk between saturated fatty acids and toll-like receptor 4 in the etiology of inflammation and insulin resistance. Prog Lipid Res (2020) 77:101020. doi: 10.1016/j.plipres.2019.101020
43. Schwarzler J, Mayr L, Radlinger B, Grabherr F, Philipp M, Texler B, et al. Adipocyte GPX4 protects against inflammation, hepatic insulin resistance and metabolic dysregulation. Int J Obes (Lond) (2022) 46:951–59. doi: 10.1038/s41366-022-01064-9
44. Long EK, Olson DM, Bernlohr DA. High-fat diet induces changes in adipose tissue trans-4-oxo-2-nonenal and trans-4-hydroxy-2-nonenal levels in a depot-specific manner. Free Radic Biol Med (2013) 63:390–8. doi: 10.1016/j.freeradbiomed.2013.05.030
45. Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature (2019) 575:693–98. doi: 10.1038/s41586-019-1707-0
46. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature (2019) 575:688–92. doi: 10.1038/s41586-019-1705-2
47. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell (2014) 156:317–31. doi: 10.1016/j.cell.2013.12.010
48. Zou Y, Palte MJ, Deik AA, Li H, Eaton JK, Wang W, et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat Commun (2019) 10:1617. doi: 10.1038/s41467-019-09277-9
49. Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Muller C, Zandkarimi F, et al. GTP cyclohydrolase 1/Tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent Sci (2020) 6:41–53. doi: 10.1021/acscentsci.9b01063
50. Soula M, Weber RA, Zilka O, Alwaseem H, La K, Yen F, et al. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat Chem Biol (2020) 16:1351–60. doi: 10.1038/s41589-020-0613-y
51. Koppula P, Zhuang L, Gan B. Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell (2021) 12:599–620. doi: 10.1007/s13238-020-00789-5
52. Lee N, Carlisle AE, Peppers A, Park SJ, Doshi MB, Spears ME, et al. xCT-driven expression of GPX4 determines sensitivity of breast cancer cells to ferroptosis inducers. Antioxidants (Basel) (2021) 10:317. doi: 10.3390/antiox10020317
53. Yan R, Xie E, Li Y, Li J, Zhang Y, Chi X, et al. The structure of erastin-bound xCT-4F2hc complex reveals molecular mechanisms underlying erastin-induced ferroptosis. Cell Res (2022) 32:687–90. doi: 10.1038/s41422-022-00642-w
54. Nakamura E, Sato M, Yang H, Miyagawa F, Harasaki M, Tomita K, et al. 4F2 (CD98) heavy chain is associated covalently with an amino acid transporter and controls intracellular trafficking and membrane topology of 4F2 heterodimer. J Biol Chem (1999) 274:3009–16. doi: 10.1074/jbc.274.5.3009
55. Sato H, Tamba M, Ishii T, Bannai S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem (1999) 274:11455–8. doi: 10.1074/jbc.274.17.11455
56. Koppula P, Zhang Y, Zhuang L, Gan B. Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Cancer Commun (Lond) (2018) 38:12. doi: 10.1186/s40880-018-0288-x
57. Bannai S. Exchange of cystine and glutamate across plasma membrane of human fibroblasts. J Biol Chem (1986) 261:2256–63. doi: 10.1016/S0021-9258(17)35926-4
58. Conrad M, Sato H. The oxidative stress-inducible cystine/glutamate antiporter, system x (c) (-) : Cystine supplier and beyond. Amino Acids (2012) 42:231–46. doi: 10.1007/s00726-011-0867-5
59. Gonzalez-Dominguez A, Visiedo-Garcia FM, Dominguez-Riscart J, Gonzalez-Dominguez R, Mateos RM, Lechuga-Sancho AM. Iron metabolism in obesity and metabolic syndrome. Int J Mol Sci (2020) 21:5529. doi: 10.3390/ijms21155529
60. Abbaspour N, Hurrell R, Kelishadi R. Review on iron and its importance for human health. J Res Med Sci (2014) 19:164–74.
61. Ma X, Pham VT, Mori H, MacDougald OA, Shah YM, Bodary PF. Iron elevation and adipose tissue remodeling in the epididymal depot of a mouse model of polygenic obesity. PloS One (2017) 12:e0179889. doi: 10.1371/journal.pone.0179889
62. Ganz T. Macrophages and systemic iron homeostasis. J Innate Immun (2012) 4:446–53. doi: 10.1159/000336423
63. Corna G, Campana L, Pignatti E, Castiglioni A, Tagliafico E, Bosurgi L, et al. Polarization dictates iron handling by inflammatory and alternatively activated macrophages. Haematologica (2010) 95:1814–22. doi: 10.3324/haematol.2010.023879
64. Orr JS, Kennedy A, Anderson-Baucum EK, Webb CD, Fordahl SC, Erikson KM, et al. Obesity alters adipose tissue macrophage iron content and tissue iron distribution. Diabetes (2014) 63:421–32. doi: 10.2337/db13-0213
65. Hotamisligil GS. Inflammation, metaflammation and immunometabolic disorders. Nature (2017) 542:177–85. doi: 10.1038/nature21363
66. Forouhi NG, Harding AH, Allison M, Sandhu MS, Welch A, Luben R, et al. Elevated serum ferritin levels predict new-onset type 2 diabetes: results from the EPIC-Norfolk prospective study. Diabetologia (2007) 50:949–56. doi: 10.1007/s00125-007-0604-5
67. Tajima S, Ikeda Y, Sawada K, Yamano N, Horinouchi Y, Kihira Y, et al. Iron reduction by deferoxamine leads to amelioration of adiposity via the regulation of oxidative stress and inflammation in obese and type 2 diabetes KKAy mice. Am J Physiol Endocrinol Metab (2012) 302:E77–86. doi: 10.1152/ajpendo.00033.2011
68. Arner P, Andersson DP, Thorne A, Wiren M, Hoffstedt J, Naslund E, et al. Variations in the size of the major omentum are primarily determined by fat cell number. J Clin Endocrinol Metab (2013) 98:E897–901. doi: 10.1210/jc.2012-4106
69. Ahmed B, Sultana R, Greene MW. Adipose tissue and insulin resistance in obese. BioMed Pharmacother (2021) 137:111315. doi: 10.1016/j.biopha.2021.111315
70. Hotamisligil GS. Inflammatory pathways and insulin action. Int J Obes Relat Metab Disord (2003) 27 Suppl 3:S53–5. doi: 10.1038/sj.ijo.0802502
71. Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest (1995) 95:2409–15. doi: 10.1172/JCI117936
72. Tanner LI, Lienhard GE. Insulin elicits a redistribution of transferrin receptors in 3T3-L1 adipocytes through an increase in the rate constant for receptor externalization. J Biol Chem (1987) 262:8975–80. doi: 10.1016/S0021-9258(18)48032-5
73. de Mello AH, Costa AB, Engel JDG, Rezin GT. Mitochondrial dysfunction in obesity. Life Sci (2018) 192:26–32. doi: 10.1016/j.lfs.2017.11.019
74. Putti R, Sica R, Migliaccio V, Lionetti L. Diet impact on mitochondrial bioenergetics and dynamics. Front Physiol (2015) 6:109. doi: 10.3389/fphys.2015.00109
75. Zorzano A, Liesa M, Palacin M. Role of mitochondrial dynamics proteins in the pathophysiology of obesity and type 2 diabetes. Int J Biochem Cell Biol (2009) 41:1846–54. doi: 10.1016/j.biocel.2009.02.004
76. Jheng HF, Tsai PJ, Guo SM, Kuo LH, Chang CS, Su IJ, et al. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol Cell Biol (2012) 32:309–19. doi: 10.1128/MCB.05603-11
77. Prasun P. Mitochondrial dysfunction in metabolic syndrome. Biochim Biophys Acta Mol Basis Dis (2020) 1866:165838. doi: 10.1016/j.bbadis.2020.165838
78. Bohm A, Keuper M, Meile T, Zdichavsky M, Fritsche A, Haring HU, et al. Increased mitochondrial respiration of adipocytes from metabolically unhealthy obese compared to healthy obese individuals. Sci Rep (2020) 10:12407. doi: 10.1038/s41598-020-69016-9
79. Cao Y, Wang H, Wang Q, Han X, Zeng W. Three-dimensional volume fluorescence-imaging of vascular plasticity in adipose tissues. Mol Metab (2018) 14:71–81. doi: 10.1016/j.molmet.2018.06.004
80. Cao W, Li M, Wu T, Feng F, Feng T, Xu Y, et al. alphaMSH prevents ROS-induced apoptosis by inhibiting Foxo1/mTORC2 in mice adipose tissue. Oncotarget (2017) 8:40872–84. doi: 10.18632/oncotarget.16606
81. Luo C, Sun J, Liu D, Sun B, Miao L, Musetti S, et al. Self-assembled redox dual-responsive prodrug-nanosystem formed by single thioether-bridged paclitaxel-fatty acid conjugate for cancer chemotherapy. Nano Lett (2016) 16:5401–8. doi: 10.1021/acs.nanolett.6b01632
82. Xie Y, Wang B, Zhao Y, Tao Z, Wang Y, Chen G, et al. Mammary adipocytes protect triple-negative breast cancer cells from ferroptosis. J Hematol Oncol (2022) 15:72. doi: 10.1186/s13045-022-01297-1
83. Guilherme A, Henriques F, Bedard AH, Czech MP. Molecular pathways linking adipose innervation to insulin action in obesity and diabetes mellitus. Nat Rev Endocrinol (2019) 15:207–25. doi: 10.1038/s41574-019-0165-y
84. Ryu V, Garretson JT, Liu Y, Vaughan CH, Bartness TJ. Brown adipose tissue has sympathetic-sensory feedback circuits. J Neurosci (2015) 35:2181–90. doi: 10.1523/JNEUROSCI.3306-14.2015
85. Garretson JT, Szymanski LA, Schwartz GJ, Xue B, Ryu V, Bartness TJ. Lipolysis sensation by white fat afferent nerves triggers brown fat thermogenesis. Mol Metab (2016) 5:626–34. doi: 10.1016/j.molmet.2016.06.013
86. Harris RBS. Denervation as a tool for testing sympathetic control of white adipose tissue. Physiol Behav (2018) 190:3–10. doi: 10.1016/j.physbeh.2017.07.008
87. Imai J, Katagiri H. Regulation of systemic metabolism by the autonomic nervous system consisting of afferent and efferent innervation. Int Immunol (2022) 34:67–79. doi: 10.1093/intimm/dxab023
88. Collins S. Beta-adrenoceptor signaling networks in adipocytes for recruiting stored fat and energy expenditure. Front Endocrinol (Lausanne) (2011) 2:102. doi: 10.3389/fendo.2011.00102
89. Kuo LE, Kitlinska JB, Tilan JU, Li L, Baker SB, Johnson MD, et al. Neuropeptide y acts directly in the periphery on fat tissue and mediates stress-induced obesity and metabolic syndrome. Nat Med (2007) 13:803–11. doi: 10.1038/nm1611
90. Pandolfi J, Ferraro A, Lerner M, Serrano JR, Dueck A, Fainboim L, et al. Purinergic signaling modulates human visceral adipose inflammatory responses: implications in metabolically unhealthy obesity. J Leukoc Biol (2015) 97:941–49. doi: 10.1189/jlb.3A1214-626R
91. Eltzschig HK, Sitkovsky MV, Robson SC. Purinergic signaling during inflammation. N Engl J Med (2012) 367:2322–33. doi: 10.1056/NEJMra1205750
92. Hu X, Xu Y, Xu H, Jin C, Zhang H, Su H, et al. Progress in understanding ferroptosis and its targeting for therapeutic benefits in traumatic brain and spinal cord injuries. Front Cell Dev Biol (2021) 9:705786. doi: 10.3389/fcell.2021.705786
93. Cobley JN, Fiorello ML, Bailey DM. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol (2018) 15:490–503. doi: 10.1016/j.redox.2018.01.008
94. Halliwell B. Reactive oxygen species and the central nervous system. J Neurochem (1992) 59:1609–23. doi: 10.1111/j.1471-4159.1992.tb10990.x
95. Bazinet RP, Laye S. Polyunsaturated fatty acids and their metabolites in brain function and disease. Nat Rev Neurosci (2014) 15:771–85. doi: 10.1038/nrn3820
96. Balcioglu AS, Muderrisoglu H. Diabetes and cardiac autonomic neuropathy: Clinical manifestations, cardiovascular consequences, diagnosis and treatment. World J Diabetes (2015) 6:80–91. doi: 10.4239/wjd.v6.i1.80
97. Browning KN, Verheijden S, Boeckxstaens GE. The vagus nerve in appetite regulation, mood, and intestinal inflammation. Gastroenterology (2017) 152:730–44. doi: 10.1053/j.gastro.2016.10.046
98. de Lartigue G. Role of the vagus nerve in the development and treatment of diet-induced obesity. J Physiol (2016) 594:5791–815. doi: 10.1113/JP271538
99. Cohen M, Syme C, McCrindle BW, Hamilton J. Autonomic nervous system balance in children and adolescents with craniopharyngioma and hypothalamic obesity. Eur J Endocrinol (2013) 168:845–52. doi: 10.1530/EJE-12-1082
100. Balbo SL, Ribeiro RA, Mendes MC, Lubaczeuski C, Maller AC, Carneiro EM, et al. Vagotomy diminishes obesity in cafeteria rats by decreasing cholinergic potentiation of insulin release. J Physiol Biochem (2016) 72:625–33. doi: 10.1007/s13105-016-0501-9
101. Kuchler JC, Siqueira BS, Ceglarek VM, Chasko FV, Moura IC, Sczepanhak BF, et al. The vagus nerve and spleen: Influence on white adipose mass and histology of obese and non-obese rats. Front Physiol (2021) 12:672027. doi: 10.3389/fphys.2021.672027
102. Peterson CY, Krzyzaniak M, Coimbra R, Chang DC. Vagus nerve and postinjury inflammatory response. Arch Surg (2012) 147:76–80. doi: 10.1001/archsurg.2011.237
103. Xia H, Liu Z, Liang W, Zeng X, Yang Y, Chen P, et al. Vagus nerve stimulation alleviates hepatic ischemia and reperfusion injury by regulating glutathione production and transformation. Oxid Med Cell Longev (2020) 2020:1079129. doi: 10.1155/2020/1079129
104. Zhang Y, Zheng L, Deng H, Feng D, Hu S, Zhu L, et al. Electroacupuncture alleviates LPS-induced ARDS through alpha7 nicotinic acetylcholine receptor-mediated inhibition of ferroptosis. Front Immunol (2022) 13:832432. doi: 10.3389/fimmu.2022.832432
105. Pavlov VA, Tracey KJ. The vagus nerve and the inflammatory reflex–linking immunity and metabolism. Nat Rev Endocrinol (2012) 8:743–54. doi: 10.1038/nrendo.2012.189
106. Agashe S, Petak S. Cardiac autonomic neuropathy in diabetes mellitus. Methodist Debakey Cardiovasc J (2018) 14:251–56. doi: 10.14797/mdcj-14-4-251
107. Hampton RF, Jimenez-Gonzalez M, Stanley SA. Unravelling innervation of pancreatic islets. Diabetologia (2022) 65:1069–84. doi: 10.1007/s00125-022-05691-9
108. Giordano A, Song CK, Bowers RR, Ehlen JC, Frontini A, Cinti S, et al. White adipose tissue lacks significant vagal innervation and immunohistochemical evidence of parasympathetic innervation. Am J Physiol Regul Integr Comp Physiol (2006) 291:R1243–55. doi: 10.1152/ajpregu.00679.2005
109. Berthoud HR, Fox EA, Neuhuber WL. Vagaries of adipose tissue innervation. Am J Physiol Regul Integr Comp Physiol (2006) 291:R1240–2. doi: 10.1152/ajpregu.00428.2006
110. Afonso RA, Lautt WW, Schafer J, Legare DJ, Oliveira AG, Macedo MP. High-fat diet results in postprandial insulin resistance that involves parasympathetic dysfunction. Br J Nutr (2010) 104:1450–9. doi: 10.1017/S0007114510002400
111. Hanani M. Satellite glial cells in sensory ganglia: from form to function. Brain Res Brain Res Rev (2005) 48:457–76. doi: 10.1016/j.brainresrev.2004.09.001
112. Hanani M. Satellite glial cells in sympathetic and parasympathetic ganglia: in search of function. Brain Res Rev (2010) 64:304–27. doi: 10.1016/j.brainresrev.2010.04.009
113. Roman-Pintos LM, Villegas-Rivera G, Rodriguez-Carrizalez AD, Miranda-Diaz AG, Cardona-Munoz EG. Diabetic polyneuropathy in type 2 diabetes mellitus: Inflammation, oxidative stress, and mitochondrial function. J Diabetes Res (2016) 2016:3425617. doi: 10.1155/2016/3425617
114. de MBS, da Fonseca LJ, da SGG, Rabelo LA, Goulart MO, Vasconcelos SM. Oxidative stress as an underlying contributor in the development of chronic complications in diabetes mellitus. Int J Mol Sci (2013) 14:3265–84. doi: 10.3390/ijms14023265
115. Lin J, Liu F, Zhang YY, Song N, Liu MK, Fang XY, et al. P2Y14 receptor is functionally expressed in satellite glial cells and mediates interleukin-1beta and chemokine CCL2 secretion. J Cell Physiol (2019) 234:21199–210. doi: 10.1002/jcp.28726
116. Song X, Long D. Nrf2 and ferroptosis: A new research direction for neurodegenerative diseases. Front Neurosci (2020) 14:267. doi: 10.3389/fnins.2020.00267
117. Tang G, Pi L, Guo H, Hu Z, Zhou C, Hu Q, et al. Naringin relieves diabetic cardiac autonomic neuropathy mediated by P2Y14 receptor in superior cervical ganglion. Front Pharmacol (2022) 13:873090. doi: 10.3389/fphar.2022.873090
118. Thorp AA, Schlaich MP. Relevance of sympathetic nervous system activation in obesity and metabolic syndrome. J Diabetes Res (2015) 2015:341583. doi: 10.1155/2015/341583
119. Hall JE, da Silva AA, do Carmo JM, Dubinion J, Hamza S, Munusamy S, et al. Obesity-induced hypertension: role of sympathetic nervous system, leptin, and melanocortins. J Biol Chem (2010) 285:17271–6. doi: 10.1074/jbc.R110.113175
120. Hirooka Y, Kishi T, Sakai K, Takeshita A, Sunagawa K. Imbalance of central nitric oxide and reactive oxygen species in the regulation of sympathetic activity and neural mechanisms of hypertension. Am J Physiol Regul Integr Comp Physiol (2011) 300:R818–26. doi: 10.1152/ajpregu.00426.2010
121. Metzger JM, Matsoff HN, Zinnen AD, Fleddermann RA, Bondarenko V, Simmons HA, et al. Post mortem evaluation of inflammation, oxidative stress, and PPARgamma activation in a nonhuman primate model of cardiac sympathetic neurodegeneration. PloS One (2020) 15:e0226999. doi: 10.1371/journal.pone.0226999
122. Lopes FNC, da Cunha NV, de Campos BH, Fattori V, Panis C, Cecchini R, et al. Antioxidant therapy reverses sympathetic dysfunction, oxidative stress, and hypertension in male hyperadipose rats. Life Sci (2022) 295:120405. doi: 10.1016/j.lfs.2022.120405
123. Reyes del Paso GA. A biofeedback system of baroreceptor cardiac reflex sensitivity. Appl Psychophysiol Biofeedback (1999) 24:67–77. doi: 10.1023/A:1022899115220
124. Milanski M, Degasperi G, Coope A, Morari J, Denis R, Cintra DE, et al. Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: Implications for the pathogenesis of obesity. J Neurosci (2009) 29:359–70. doi: 10.1523/JNEUROSCI.2760-08.2009
125. Sears B, Perry M. The role of fatty acids in insulin resistance. Lipids Health Dis (2015) 14:121. doi: 10.1186/s12944-015-0123-1
126. Belgardt BF, Okamura T, Bruning JC. Hormone and glucose signalling in POMC and AgRP neurons. J Physiol (2009) 587:5305–14. doi: 10.1113/jphysiol.2009.179192
127. Denis RG, Joly-Amado A, Cansell C, Castel J, Martinez S, Delbes AS, et al. Central orchestration of peripheral nutrient partitioning and substrate utilization: Implications for the metabolic syndrome. Diabetes Metab (2014) 40:191–7. doi: 10.1016/j.diabet.2013.11.002
128. Jiang J, Morgan DA, Cui H, Rahmouni K. Activation of hypothalamic AgRP and POMC neurons evokes disparate sympathetic and cardiovascular responses. Am J Physiol Heart Circ Physiol (2020) 319:H1069–H77. doi: 10.1152/ajpheart.00411.2020
129. Schriever SC, Zimprich A, Pfuhlmann K, Baumann P, Giesert F, Klaus V, et al. Alterations in neuronal control of body weight and anxiety behavior by glutathione peroxidase 4 deficiency. Neuroscience (2017) 357:241–54. doi: 10.1016/j.neuroscience.2017.05.050
130. Cinkajzlova A, Mraz M, Haluzik M. Adipose tissue immune cells in obesity, type 2 diabetes mellitus and cardiovascular diseases. J Endocrinol (2021) 252:R1–22. doi: 10.1530/JOE-21-0159
131. Liu R, Nikolajczyk BS. Tissue immune cells fuel obesity-associated inflammation in adipose tissue and beyond. Front Immunol (2019) 10:1587. doi: 10.3389/fimmu.2019.01587
132. Watanabe Y, Nagai Y, Honda H, Okamoto N, Yanagibashi T, Ogasawara M, et al. Bidirectional crosstalk between neutrophils and adipocytes promotes adipose tissue inflammation. FASEB J (2019) 33:11821–35. doi: 10.1096/fj.201900477RR
133. Cinkajzlova A, Mraz M, Haluzik M. Lymphocytes and macrophages in adipose tissue in obesity: markers or makers of subclinical inflammation? Protoplasma (2017) 254:1219–32. doi: 10.1007/s00709-017-1082-3
134. Caslin HL, Bhanot M, Bolus WR, Hasty AH. Adipose tissue macrophages: Unique polarization and bioenergetics in obesity. Immunol Rev (2020) 295:101–13. doi: 10.1111/imr.12853
135. Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature (2013) 496:445–55. doi: 10.1038/nature12034
136. Li Y, Yun K, Mu R. A review on the biology and properties of adipose tissue macrophages involved in adipose tissue physiological and pathophysiological processes. Lipids Health Dis (2020) 19:164. doi: 10.1186/s12944-020-01342-3
137. O'Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol (2016) 16:553–65. doi: 10.1038/nri.2016.70
138. Serbulea V, Upchurch CM, Ahern KW, Bories G, Voigt P, DeWeese DE, et al. Macrophages sensing oxidized DAMPs reprogram their metabolism to support redox homeostasis and inflammation through a TLR2-syk-ceramide dependent mechanism. Mol Metab (2018) 7:23–34. doi: 10.1016/j.molmet.2017.11.002
139. Han F, Li S, Yang Y, Bai Z. Interleukin-6 promotes ferroptosis in bronchial epithelial cells by inducing reactive oxygen species-dependent lipid peroxidation and disrupting iron homeostasis. Bioengineered (2021) 12:5279–88. doi: 10.1080/21655979.2021.1964158
140. Wang CY, Babitt JL. Hepcidin regulation in the anemia of inflammation. Curr Opin Hematol (2016) 23:189–97. doi: 10.1097/MOH.0000000000000236
141. Jung HS, Shimizu-Albergine M, Shen X, Kramer F, Shao D, Vivekanandan-Giri A, et al. TNF-alpha induces acyl-CoA synthetase 3 to promote lipid droplet formation in human endothelial cells. J Lipid Res (2020) 61:33–44. doi: 10.1194/jlr.RA119000256
142. Wen Y, Chen H, Zhang L, Wu M, Zhang F, Yang D, et al. Glycyrrhetinic acid induces oxidative/nitrative stress and drives ferroptosis through activating NADPH oxidases and iNOS, and depriving glutathione in triple-negative breast cancer cells. Free Radic Biol Med (2021) 173:41–51. doi: 10.1016/j.freeradbiomed.2021.07.019
143. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol (2018) 233:6425–40. doi: 10.1002/jcp.26429
144. Kanamori Y, Murakami M, Sugiyama M, Hashimoto O, Matsui T, Funaba M. Hepcidin and IL-1beta. Vitam Horm (2019) 110:143–56. doi: 10.1016/bs.vh.2019.01.007
145. Yuan Y, Chen Y, Peng T, Li L, Zhu W, Liu F, et al. Mitochondrial ROS-induced lysosomal dysfunction impairs autophagic flux and contributes to M1 macrophage polarization in a diabetic condition. Clin Sci (Lond) (2019) 133:1759–77. doi: 10.1042/CS20190672
146. Zhang B, Yang Y, Yi J, Zhao Z, Ye R. Hyperglycemia modulates M1/M2 macrophage polarization via reactive oxygen species overproduction in ligature-induced periodontitis. J Periodontal Res (2021) 56:991–1005. doi: 10.1111/jre.12912
147. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest (2003) 112:1796–808. doi: 10.1172/JCI200319246
148. Piattini F, Matsushita M, Muri J, Bretscher P, Feng X, Freigang S, et al. Differential sensitivity of inflammatory macrophages and alternatively activated macrophages to ferroptosis. Eur J Immunol (2021) 51:2417–29. doi: 10.1002/eji.202049114
149. Zhou H, Liu F. Regulation, communication, and functional roles of adipose tissue-resident CD4 T cells in the control of metabolic homeostasis. Front Immunol (2018) 9:1961. doi: 10.3389/fimmu.2018.01961
150. Wang Q, Wu H. T Cells in adipose tissue: Critical players in immunometabolism. Front Immunol (2018) 9:2509. doi: 10.3389/fimmu.2018.02509
151. Nosalski R, Guzik TJ. Perivascular adipose tissue inflammation in vascular disease. Br J Pharmacol (2017) 174:3496–513. doi: 10.1111/bph.13705
152. Wang Q, Wang Y, Xu D. The roles of T cells in obese adipose tissue inflammation. Adipocyte (2021) 10:435–45. doi: 10.1080/21623945.2021.1965314
153. Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med (2009) 15:930–9. doi: 10.1038/nm.2002
154. Dorneles GP, da Silva I, Boeira MC, Valentini D, Fonseca SG, Dal Lago P, et al. Cardiorespiratory fitness modulates the proportions of monocytes and T helper subsets in lean and obese men. Scand J Med Sci Sports (2019) 29:1755–65. doi: 10.1111/sms.13506
155. Xu C, Sun S, Johnson T, Qi R, Zhang S, Zhang J, et al. The glutathione peroxidase Gpx4 prevents lipid peroxidation and ferroptosis to sustain treg cell activation and suppression of antitumor immunity. Cell Rep (2021) 35:109235. doi: 10.1016/j.celrep.2021.109235
156. Wang B, Sun J, Ma Y, Wu G, Shi Y, Le G. Increased oxidative stress and the apoptosis of regulatory T cells in obese mice but not resistant mice in response to a high-fat diet. Cell Immunol (2014) 288:39–46. doi: 10.1016/j.cellimm.2014.02.003
157. Muri J, Kopf M. Redox regulation of immunometabolism. Nat Rev Immunol (2021) 21:363–81. doi: 10.1038/s41577-020-00478-8
158. Maj T, Wang W, Crespo J, Zhang H, Wang W, Wei S, et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat Immunol (2017) 18:1332–41. doi: 10.1038/ni.3868
159. Cronin SJF, Seehus C, Weidinger A, Talbot S, Reissig S, Seifert M, et al. The metabolite BH4 controls T cell proliferation in autoimmunity and cancer. Nature (2018) 563:564–68. doi: 10.1038/s41586-018-0701-2
160. Winer DA, Winer S, Shen L, Wadia PP, Yantha J, Paltser G, et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med (2011) 17:610–7. doi: 10.1038/nm.2353
161. DeFuria J, Belkina AC, Jagannathan-Bogdan M, Snyder-Cappione J, Carr JD, Nersesova YR, et al. B cells promote inflammation in obesity and type 2 diabetes through regulation of T-cell function and an inflammatory cytokine profile. Proc Natl Acad Sci U S A. (2013) 110:5133–8. doi: 10.1073/pnas.1215840110
162. Ying W, Tseng A, Chang RC, Wang H, Lin YL, Kanameni S, et al. miR-150 regulates obesity-associated insulin resistance by controlling b cell functions. Sci Rep (2016) 6:20176. doi: 10.1038/srep20176
163. Ying W, Wollam J, Ofrecio JM, Bandyopadhyay G, El Ouarrat D, Lee YS, et al. Adipose tissue B2 cells promote insulin resistance through leukotriene LTB4/LTB4R1 signaling. J Clin Invest (2017) 127:1019–30. doi: 10.1172/JCI90350
164. Harmon DB, Srikakulapu P, Kaplan JL, Oldham SN, McSkimming C, Garmey JC, et al. Protective role for b-1b b cells and IgM in obesity-associated inflammation, glucose intolerance, and insulin resistance. Arterioscler Thromb Vasc Biol (2016) 36:682–91. doi: 10.1161/ATVBAHA.116.307166
165. Kosaraju R, Guesdon W, Crouch MJ, Teague HL, Sullivan EM, Karlsson EA, et al. B cell activity is impaired in human and mouse obesity and is responsive to an essential fatty acid upon murine influenza infection. J Immunol (2017) 198:4738–52. doi: 10.4049/jimmunol.1601031
166. Muri J, Thut H, Bornkamm GW, Kopf M. B1 and marginal zone b cells but not follicular B2 cells require Gpx4 to prevent lipid peroxidation and ferroptosis. Cell Rep (2019) 29:2731–44 e4. doi: 10.1016/j.celrep.2019.10.070
167. Talukdar S, Oh DY, Bandyopadhyay G, Li D, Xu J, McNelis J, et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med (2012) 18:1407–12. doi: 10.1038/nm.2885
168. Witko-Sarsat V, Pederzoli-Ribeil M, Hirsch E, Sozzani S, Cassatella MA. Regulating neutrophil apoptosis: New players enter the game. Trends Immunol (2011) 32:117–24. doi: 10.1016/j.it.2011.01.001
169. Yuyun X, Fan Y, Weiping W, Qing Y, Bingwei S. Metabolomic analysis of spontaneous neutrophil apoptosis reveals the potential involvement of glutathione depletion. Innate Immun (2021) 27:31–40. doi: 10.1177/1753425920951985
170. Li P, Jiang M, Li K, Li H, Zhou Y, Xiao X, et al. Glutathione peroxidase 4-regulated neutrophil ferroptosis induces systemic autoimmunity. Nat Immunol (2021) 22:1107–17. doi: 10.1038/s41590-021-00993-3
171. Du H, Ren X, Bai J, Yang W, Gao Y, Yan S. Research progress of ferroptosis in adiposity-based chronic disease (ABCD). Oxid Med Cell Longev (2022) 2022:1052699. doi: 10.1155/2022/1052699
Keywords: ferroptosis pathway, obesity, neurological dysfunction, inflammatory response, metabolic disorders
Citation: Zhang S, Sun Z, Jiang X, Lu Z, Ding L, Li C, Tian X and Wang Q (2022) Ferroptosis increases obesity: Crosstalk between adipocytes and the neuroimmune system. Front. Immunol. 13:1049936. doi: 10.3389/fimmu.2022.1049936
Received: 21 September 2022; Accepted: 07 November 2022;
Published: 21 November 2022.
Edited by:
Zhihao Jia, Purdue University, United StatesReviewed by:
Jingbo Pang, University of Illinois at Chicago, United StatesCopyright © 2022 Zhang, Sun, Jiang, Lu, Ding, Li, Tian and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xuewen Tian, eHVld2VudGlhbjE5NzhAMTYzLmNvbQ==; Qinglu Wang, d3FsX3pjcUAxMjYuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.