 Kohsuke Shirakawa
Kohsuke Shirakawa Motoaki Sano
Motoaki Sano
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 04 January 2023
Sec. T Cell Biology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1044737
This article is part of the Research TopicAdaptive Immunity in Local TissuesView all 12 articles
Obesity has a pronounced effect on the immune response in systemic organs that results in not only insulin resistance but also altered immune responses to infectious diseases and malignant tumors. Obesity-associated microenvironmental changes alter transcriptional expression and metabolism in T cells, leading to alterations in T-cell differentiation, proliferation, function, and survival. Adipokines, cytokines, and lipids derived from obese visceral adipose tissue (VAT) may also contribute to the systemic T-cell phenotype, resulting in obesity-specific pathogenesis. VAT T cells, which have multiple roles in regulating homeostasis and energy utilization and defending against pathogens, are most susceptible to obesity. In particular, many studies have shown that CD4 T cells are deeply involved in the homeostasis of VAT endocrine and metabolic functions and in obesity-related chronic inflammation. In obesity, macrophages and adipocytes in VAT function as antigen-presenting cells and contribute to the obesity-specific CD4 T-cell response by inducing CD4 T-cell proliferation and differentiation into inflammatory effectors via interactions between major histocompatibility complex class II and T-cell receptors. When obesity persists, prolonged stimulation by leptin and circulating free fatty acids, repetitive antigen stimulation, activating stress responses, and hypoxia induce exhaustion of CD4 T cells in VAT. T-cell exhaustion is characterized by restricted effector function, persistent expression of inhibitory receptors, and a transcriptional state distinct from functional effector and memory T cells. Moreover, obesity causes thymic regression, which may result in homeostatic proliferation of obesity-specific T-cell subsets due to changes in T-cell metabolism and gene expression in VAT. In addition to causing T-cell exhaustion, obesity also accelerates cellular senescence of CD4 T cells. Senescent CD4 T cells secrete osteopontin, which causes further VAT inflammation. The obesity-associated transformation of CD4 T cells remains a negative legacy even after weight loss, causing treatment resistance of obesity-related conditions. This review discusses the marked transformation of CD4 T cells in VAT and systemic organs as a consequence of obesity-related microenvironmental changes.
The start of the obesity epidemic in the US in the early 1970s led to much research on adipose tissue, which regulates metabolic and nutritional homeostasis (1). Then, in the mid-1990s, Hotamisligil et al. proposed the intriguing hypothesis that immune cells are activated in response to a state of excess energy, which induces inflammation and, consequently, insulin resistance (2). Compared with subcutaneous adipose tissue, visceral adipose tissue (VAT) is composed of more diverse cell populations and is highly vascularized and contains numerous sympathetic and sensory nerves. Furthermore, during the development of obesity, a more complex diversity of immune cells arises in VAT than in subcutaneous adipose tissue. Since 2003, when Weisberg et al. and Xu et al. independently reported that macrophages accumulate in obese VAT, macrophages—which express high levels of inflammatory cytokines—have been considered the central players in VAT inflammation. However, subsequent studies revealed that VAT inflammation involves a greater variety of immune cells than previously thought and that not only the innate but also the acquired immune system is activated (3, 4). Obese VAT has a higher proportion of CD4 T cells, which have been recognized as central regulators of chronic VAT inflammation. Even in the lean state, VAT CD4 T cells continue to be weakly activated against an autoantigen specific to VAT. Interestingly, a much larger amount of regulatory T cells (Tregs) are present in VAT than in secondary lymphoid tissues, indicating that local immunometabolic homeostasis is maintained within VAT by enhanced immune tolerance (5).
During the development of obesity, strong activation of effector T cells initiates VAT inflammation (6). Furthermore, in chronic obesity, acute inflammation transitions to chronic inflammation and T cells become dysfunctional (because of T-cell exhaustion and senescence) (7–11). Once obesity has formed, the memory of obesity is imprinted on T cells and does not disappear with weight loss (12, 13); for this reason, weight rebound after weight loss induces more inflammation in the VAT than was present before weight loss (13).
VAT dynamically functions as not only a lipid storage organ but also an endocrine organ that produces a variety of soluble mediators, such as adipokines, cytokines, and lipids. The drastic changes in the VAT microenvironment in obesity affect the phenotype of not only VAT T cells but also systemic ones.
In this review, we summarize what is known about how T-cell activation, differentiation, and function change in response to obesity-associated factors.
In general, T-cell activation (survival, proliferation, differentiation, and functional enhancement) requires three positive signals: a T-cell receptor (TCR) signal, co-stimulation signal (CD28 signal), and cytokine signal. The TCR-mediated signal primarily activates the ZAP70-PLCγ pathway; the co-stimulation signal, the phosphatidylinositol-3-kinase-protein kinase B pathway; and the cytokine signal, the Janus family of kinases-signal transducer and activator of transcription (JAK-STAT) pathway (14). To suppress excessive responses, T cells are equipped with brakes on each of these three signals. TCR-mediated signaling is suppressed by programmed cell death 1 (PD-1) (15); PD-1 has an immunoreceptor tyrosine-based inhibitory motif, and upon stimulation by the ligands PD-L1 and PD-L2, it recruits Src homology 2 domain-containing protein tyrosine phosphatase 1, which dephosphorylates and inactivates ZAP7 (16, 17). Co-stimulation is suppressed by cytotoxic T-lymphocyte associated protein 4 (CTLA4), which has an extracellular domain similar to that of costimulatory receptor CD28 and competes with CD28 for binding to its ligand CD80/86; CTLA4 has higher affinity for CD80/86 than CD28 does, but it does not generate a signal (18) and thus physically blocks CD28-mediated signaling (19). And the cytokine signaling JAK-STAT pathway is suppressed by the suppressor of cytokine signaling family of molecules (20).
In addition to known T cell responses, obesity-associated insulin resistance and changes in adipokine secretion profiles all affect T-cell activation, function, and survival, not only in VAT but also in systemic organs (21). Obesity-associated adipocyte hypertrophy and hyperplasia cause VAT microenvironment remodeling, including impaired angiogenesis, deposition of extracellular matrix protein, and hypoxia-induced pyroptosis (22), which may result in changes in T-cell phenotype Obese VAT cells also regulate adipose T-cell activity through antigen presentation and co-stimulatory or co-inhibitory receptor signaling. Obesity-associated disruption of T-cell homeostasis may contribute to the development of an inflammatory state, which is followed by disruption of tissue homeostasis.
The finding that T cells with a restricted TCR repertoire accumulate in the VAT of diet-induced obese (DIO) mice (23) suggests that in obesity, T cells recognize some kind of antigen. Antigen-presenting cells process an antigen and present it to T cells in the form of an antigen-peptide major histocompatibility complex (MHC). The MHC is recognized by the TCR on the T cell, and the T cell is activated. In addition, antigen-presenting cells highly express co-stimulatory molecules, which pair with co-stimulator receptors on the T-cell surface to modulate T-cell activation (24). Activation of T cells by antigen-presenting cells also plays an important role in triggering the VAT inflammation induced by a high-fat diet (HFD). In VAT T cells, MHC class II (MHCII)-T cell receptor interaction upregulates the expression of inflammatory Th1 marker genes, including Tbx21 and Ifng. Deletion of MHCII, which plays a role in presenting antigen-derived peptides to CD4 T cells, reduces VAT CD4 T helper type 1 (Th1) cell activity and macrophage accumulation within VAT (23, 25–27).
In VAT, besides the classical antigen-presenting cells, i.e., dendritic cells (DCs) (28), macrophages (27, 29), and B cells (30), adipocytes (26) also play an important role in the formation of T-cell responses as antigen-presenting cells (31). Adipocytes express MHCII molecules and the costimulatory molecules CD80/86 and thus act as antigen-presenting cells and promote CD4 T cell activation (26).Interestingly, in obesity, adipocytes have higher expression levels of MHCII molecules and costimulatory molecules (25).The above findings indicate that during the progression of obesity, antigens presented on MHCII molecules induce T-cell proliferation and differentiation into specific subclasses of inflammatory effectors and that this process is the basis for the initiation and persistence of inflammation in VAT. Knowledge about obesity-related antigens could potentially lead to the development of vaccines and treatments to prevent chronic inflammation of VAT, but unfortunately, such antigens have not yet been identified.
Immune checkpoint molecules maintain immune homeostasis by suppressing self-immune responses and excessive activation of T cells. Co-inhibitory receptors such as CTLA-4 and PD-1 are co-expressed on effector T cells and are involved in immune response homeostasis (19). T-cell exhaustion is characterized by restricted effector function, persistent expression of inhibitory receptors, and a transcriptional state distinct from functional effector and memory T cells. In general, PD-1 is expressed in T cells in response to most immune challenges; however, it is rapidly downregulated during the acute phase of the immune response, allowing for normal immune responses (15). On the other hand, PD-1 expression remains high in chronically stimulated antigen-specific T cells, so the immune response to additional stimulation is impaired (32–35).
T-cell exhaustion has also been identified in VAT in mice and humans with obesity (7, 9, 36).
In fact, in HFD-induced obese mice and patients with type 2 diabetes, adipose T cells are less able than lean adipose T cells to produce cytokines such as IL-2 and IFN-γ (9). HFD-induced obesity increases the PD-1 expression level in adipose T cells, and T cells with high PD-1 expression are a subset of T cells that acquire the exhaustion phenotype (7–9). Various obesity-associated environmental factors lead to heterogeneous T-cell exhaustion profiles, suggesting that obesity may not promote classical T-cell exhaustion.The transient elevation in the expression of immune checkpoint molecules during the priming process, in which native T cells are sensitized and activated by antigen-presenting cells, not only inhibits excessive activation but is also involved in determining the polarity of the effector T cells (37, 38). PD-L1 is expressed in large amounts on DCs in VAT of HFD-fed mice. DC-specific PD-L1 deficiency shifts the polarity of T cells in the VAT of HFD-fed mice toward Th1, exacerbating weight gain and abnormal glucose metabolism (39). PD-L1 expression on DCs is an important factor in suppression of Th1, Th17, and cytotoxic T cells in antitumor responses and autoimmune diseases (40, 41) and appears to play a similar role in the pathogenesis of obesity.
Upregulation of PD-L1 in human adipose tissue is positively correlated with body mass index, but not with type 2 diabetes (42). On the other hand, a negative correlation was reported between body mass index and PD-L1 expression (43). The pathology of human obesity is highly heterogeneous, with multifactorial contributions by dietary quantity and quality, physical inactivity, and genetic factors. Therefore, despite the common phenotype of accumulation of adipose tissue, the number and qualitative changes of immune cells involved in chronic inflammation of VAT are thought also to be diverse. These differences may stem from the different roles of immune cells: They are involved in the initiation, amplification, and/or suppression of chronic inflammation of VAT, and their roles differ greatly depending on the phase of obesity. The role of PD-1/PD-L1 signaling in human VAT T cells remains unclear and requires further elucidation.
Differentiation of CD4 T cells into functional subsets is supported by complex metabolic programs (44). Th1, Th17, and Th2 effector cells generate energy by aerobic glycolysis rather than oxidative phosphorylation, whereas Tregs rely on fatty acid oxidation-fueled oxidative phosphorylation rather than glycolysis (45).
Obesity-related alterations in environmental signaling in VAT alter cellular metabolism and contribute to obesity-specific T-cell responses (46, 47). However, it is difficult to measure the metabolic state of VAT T cells in mice because of the complexity of the isolation process, so splenic T cell metabolism has mainly been studied.
Glucose uptake and oxygen consumption are increased in splenic CD4 T cells from obese mice compared with those from lean mice (46). Furthermore, β3-adrenergic receptor stimulation mimics T-cell metabolism in DIO mice and reduces expression of the mitochondrial-localized chaperone protein disulfide bond A oxidoreductase like protein (DsbA-L) in T cells. Although alterations in mitochondrial respiration are an important mechanism controlling cytokine production, loss of DsbA-L in T cells reduces mitochondrial oxidative phosphorylation capacity in both CD4 and CD8 T cells. Mice with T-cell–specific knockout of DsbA-L have reduced IFN-γ–producing Th1 cells in brown adipocytes, enhanced brown adipocyte thermogenic signaling, and less obesity and insulin resistance when fed a HFD (48). Thus, the obesity-related changes in T cell metabolism greatly contribute to the pathogenesis of obesity, including insulin resistance; however, many aspects of the relationship between metabolism and function specific to VAT T cells remain unclear.
Adipocytes secrete a variety of bioactive substances, collectively referred to as “adipokines.” Changes in adipokines associated with the progression of obesity affect T-cell proliferation, differentiation, and function (49). Adipocyte-derived lipids also affect T-cell phenotypes in obesity (Table 1).

Table 1 Effects of humoral factors on CD4 T cells in obesity.
Leptin stimulates the satiety center in the hypothalamus. As body fat mass increases, adipocytes produce more leptin, and the serum leptin concentration rises. In normal-weight individuals, the higher serum leptin concentration stimulates the satiety center, which suppresses eating behavior, allowing body fat mass to return to its base level. In the periphery, leptin promotes fatty acid oxidation and glucose uptake in skeletal muscle (64).
Leptin has been reported to have various physiological activities in both normal weight and obese conditions. It also acts on the immune system by promoting T-cell formation by the thymus. Conversely, leptin deficiency results in thymic atrophy and decreased numbers of circulating T cells (50). Administration of leptin to young leptin mutant (ob/ob) and normal mice increases CD4 single-positive thymocytes in the thymus and CD4 T cells in the periphery (51). The long-chain leptin receptor ObRb is expressed on double-negative, double-positive, and CD4 single-positive thymocyte subsets, but not on CD8 single-positive thymocytes (52). Among other things, leptin may promote differentiation from double-positive into CD4 single-positive cells (52).
In obesity, thymus function is reduced despite hyperleptinemia (65). Individuals with obesity are known to have high blood levels of leptin, but their appetite is not suppressed and they are in a leptin-resistant state. The question whether the leptin resistance observed in the hypothalamus is also observed in the thymus or whether other factors, such as adipogenesis of thymic tissue, are involved in obesity-related thymic hypofunction needs further clarification.
Leptin is also involved in the differentiation and proliferation of CD4 T cells in the periphery: TCR stimulation upregulates the expression of leptin receptors on T cells (66), which require leptin for metabolic reprogramming in which activated effector T cells upregulate the glucose transporter Glut1 and enhance the glycolytic system (50). Leptin also promotes differentiation of T cells into Th17 cells via T-cell–like leptin receptors (53–57).
In VAT, leptin gene expression in adipocytes begins to increase within 1 week after HFD loading, indicating that leptin may be an initiator of the adipose inflammatory cascade. Leptin promotes differentiation of VAT T cells into Th1 cells and secretion of IFN-γ (26). On the other hand, ob/ob and db/db mice also develop VAT inflammation and severe insulin resistance, suggesting that humoral factors other than leptin may contribute to the initiation of VAT inflammation (67). In addition, in the pathogenesis of chronic obesity, leptin signaling contributes to T-cell exhaustion by activating homeostatic STAT3 signaling and thus inducing PD-1 expression (8).
In VAT, in which adipocytes are the major constituent cells, T cells may be exposed to high concentrations of leptin. In addition, the degree and duration of obesity, which determine the amount of exposure to leptin, greatly affect T-cell phenotypic changes. Leptin is one of the initiators of VAT inflammation that promotes Th1 differentiation of VAT T cells, and long-term exposure to leptin induces immune exhaustion. Thus, leptin is a key molecule in obesity-induced T cell phenotypic changes.
Adiponectin would be expected to prevent obesity and obesity-related diseases by promoting insulin sensitivity and fatty acid oxidation and exerting anti-inflammatory effects (68). However, serum levels of adiponectin are negatively correlated with obesity, and obesity decreases adiponectin receptor expression (68).
In general, most T cells store the adiponectin receptors AdipoR1 and AdipoR2 in intracellular compartments. After antigen-specific stimulation, AdipoR1 and AdipoR2 are transported to the cell surface and expressed along with the receptor CTLA-A and other receptors. In various in vivo inflammatory models, adiponectin acts as a negative regulator of effector T cells by suppressing T-cell proliferation and cytokine production and promoting apoptosis (58–60).
Adiponectin also appears to affect the mechanism by which naïve T cells are activated by DCs, and adiponectin treatment reduces the expression of MHCII, CD80, and CD86 on DCs and suppresses production of IL-12p40 (21). Furthermore, PD-L1 expression is increased in adiponectin-treated DCs. Co-culture of adiponectin-treated DCs with allogeneic T cells in vitro decreases T-cell proliferation and IL-2 production, and this phenomenon can be partially reversed by blocking the PD-1/PD-L1 pathway (61).
Although the question whether the obesity-related decrease in adiponectin releases the brake on the inflammatory response of VAT T cells remains unanswered, in vitro research suggests that adiponectin may suppress the inflammatory response of obesity-activated T cells. In fact, obesity enhances the glycolysis pathway in splenic Th1 and Th17 cells, and adiponectin inhibits glycolysis in both an AMPK-dependent and -independent manner, resulting in the amelioration of inflammation (62). In obese patients, adiponectin expression is reduced not only in adipose tissue but also in serum (69). The reduction in VAT-derived adiponectin may affect systemic immune function and contribute to the development of obesity-specific inflammatory conditions.
During the progression of obesity, lipids released from adipocytes are also involved in the enrichment of IFN-γ–producing CD4 T cells in VAT (63). The differentiation of TCR-stimulated IFN-γ–producing effector T cells is enhanced by co-culture with adipocytes (62). Among the soluble factors, fatty acids were shown to be the strongest modulators of differentiation into Th1 (63). Patients with obesity have high serum fatty acid concentrations, showing that the bias for differentiation of naïve T cells into IFN-γ–producing Th1 cells is not limited to VAT. Indeed, upon antigen presentation in secondary lymphoid tissues, T cells primed with excess saturated fatty acids have been shown to undergo biased differentiation into pro-inflammatory effector memories that tend to cluster in pro-inflammatory nonlymphoid tissues such as obese adipose tissue and atherosclerotic lesions (70).
Because VAT CD4 T cells appear to acquire unique characteristics that differ from those of CD4 T cells in other organs, including blood and spleen, it is unclear whether fatty acids have the same effects on CD4 T cells in VAT as they do on those in blood. However, VAT CD4 T cells are exposed to higher concentrations of fatty acids, which may result in higher production of IFN-γ and development of VAT inflammation.
From an immunological perspective, VAT is a unique environment. In the physiological (non-obese) state, T cells are kept weakly activated against a self-antigen specific to VAT, and Tregs that are highly reactive to self-antigens are also assembled. In this way, peripheral immune tolerance is enhanced to suppress inflammation in VAT and maintain immunometabolic homeostasis. During the development of obesity, the storage of excess fat leads to further enhancement of T-cell activation signals. However, in the chronic phase of obesity, T cells in VAT become dysfunctional. In this section, we describe how the immune-tolerant state of VAT is breached in obesity, how T-cell activation affects adipocyte function, and how T-cell transformation is involved in chronic VAT inflammation.
Endogenous tissue Tregs differentiate and mature in response to tissue-specific environmental signals and play an important role in organ homeostasis (5). VAT Tregs are functionally specialized tissue-resident cells that prevent obesity-associated inflammation and maintain insulin sensitivity and glucose tolerance (71). Under homeostatic conditions in a lean state, VAT has a large population of Tregs, and research showed that as non-obese mice age, Tregs accumulate in the VAT (72). Indeed, studies in mice found that at 20 to 30 weeks of age, VAT Tregs represent a surprisingly high percentage (50%-80%) of the CD4 T-cell compartment (73, 74). For reference, the proportion of Tregs in the CD4 T-cell compartment in spleen and lymphoid tissues is about 5% to 15%.
VAT is thought have such a high proportion of CD4 T cells because it requires Tregs for immunological homeostasis. Within VAT, T cells continue to be loosely activated by autoantigens in adipose tissue. Tissue-resident Tregs play an important role in preventing the activation of effector T cells and the accompanying dysfunction of adipocytes and in maintaining systemic insulin sensitivity.
There are two types of Tregs: Tregs that are generated in the thymus (referred to as naturally occurring or thymic Tregs) and Tregs that differentiate from naïve CD4 T cells in the periphery (peripheral Tregs) (75).
The intestinal lamina propria has a large population of Tregs to prevent excessive immune responses to dietary components and intestinal bacteria; the population comprises peripheral Tregs, which increase locally depending on intestinal bacteria, and thymic Tregs (76). When all germs were experimentally removed from the intestinal mucosa, the population of Tregs in the intestinal mucosa decreased to the level seen in lymphoid tissues (61). Among intestinal bacteria, Clostridium species are known to be a strong inducer of peripheral Tregs (76). In VAT, most Tregs are thymic Tregs. CD4 T cells that undergo negative selection in the thymus and do not die express the lineage-determining transcription factor Foxp3 to become Tregs, which are self-recognizing and thus inherently more likely to invade self-tissue. Compared with Tregs in lymphoid tissue, VAT Tregs have a unique TCR repertoire that exhibits specific antigen recognition (77), meaning that VAT Tregs react to certain antigens specific to VAT.
Foxp3 and the signal-dependent transcription factor peroxisome proliferator-activated receptor gamma (PPAR-γ), as well as signaling by the cytokine IL-33 through the IL-33 receptor ST2, are important for the proliferation and functional maturation of Tregs in VAT (78). VAT Tregs express insulin receptors, which are rarely expressed on Tregs in lymphoid tissues, and highly express PPAR-γ and the ST2 receptor in an insulin signaling-dependent manner (79). VAT Tregs have transcripts driven by PPARγ that differ from lymphoid-organ and other nonlymphoid-tissue Treg populations (80). Mice lacking Treg-specific PPARγ have greatly reduced VAT Tregs.
In mice, the VAT Treg population was found to show sex differences and IL-33 was found to be particularly important for the maintenance of Tregs in adipose tissue of males (81). Under homeostatic conditions in a lean state, the Treg population is larger in male than in female mice. In female mice, adipose tissue inflammation is suppressed by estrogen, and the expression of inflammatory cytokines such as IL-6, C-C motif chemokine ligand 2, and IL-1b in VAT is more pronounced in males than in females (81). Consistent with this finding, male mice are more insulin resistant than females. In males, the environment of higher inflammation in the VAT causes more Tregs to be recruited from the spleen. Androgens regulate the differentiation of IL-33–producing stromal cell populations specific to VAT in males (81).
In male mice, VAT Tregs upregulate expression of the ST2 receptor in a manner dependent on transcription factor Blimp1, which is also highly expressed in males; the upregulated expression leads to local expansion of VAT Tregs in an IL-33 signaling-dependent manner (81).
VAT Tregs also enhance IL-10 production in a Blimp1-dependent manner. IL-10 produced by VAT Tregs suppresses not only effector T activity but also white adipose tissue browning (82). Because female mice have smaller VAT Treg populations, they have lower IL-10 levels and are thus more prone to white adipose tissue browning; lower IL-10 may also be one of the reasons why female mice are protected from white adipose tissue accumulation and glucose intolerance compared with age-matched male mice.
As obesity progresses, VAT Tregs suppress inflammation by regulating effector T cells, DCs, and macrophage activity directly or via IL-10 production (74). Tregs, which constantly express CD25 (also known as IL-2 receptor alpha chain), preferentially bind to IL-2 to inhibit proliferation of effector T cells (74). Tregs express the adhesion factors lymphocyte function-associated antigen-1 (LFA-1) and CTLA4, and the former enables them to strongly adhere to DCs (83). In addition, Tregs downregulate the expression of CD80/86 on DCs in both a CTLA-4- and LFA-1-dependent manner (83). IL-10 acts mainly on DCs and macrophages to suppress production of IL-12 and tumor necrosis factor alpha (TNFα) and the expression of CD86, MHCII, and CD40 by strongly activating STAT3 (84).
In severe obesity, the Treg cell population is reduced to 10% to 20% of the CD4 T-cell compartment (73, 74). One of the reported mechanisms for this decrease is the presence of ST2 soluble isoforms, which are secreted by adipocytes in obesity and function as decoy receptors for the ST2 receptor to weaken IL-33 signaling (85, 86).
The immune compartments of adipose tissue are markedly different in mice, humans, and primates: Lean male mice have a high proportion of Tregs in VAT, but the proportion is much lower in female mice, humans, and cynomolgus macaques (87). In healthy lean humans, the proportion of Tregs is significantly lower in VAT than in peripheral blood; however, OX40-expressing Tregs, which have potent suppressive activity and high proliferative capacity, are selectively distributed in VAT in all individuals and further increase with obesity. Upregulation of OX40 Tregs is thought to be a protective mechanism that suppresses excessive inflammation (88).
Although the actions of immune cells in the maintenance of VAT homeostasis appear to differ greatly between species, Tregs play an important role in regulating VAT inflammatory across species.
The main players in VAT inflammation in obesity are macrophages. In the lean state, macrophages localize diffusely in the VAT, but in obesity, they explode in number and become intensively localized as crown-like structures, which comprise clusters of macrophages surrounding dead adipocytes (89). A wide variety of T cells modulate monocyte recruitment to the VAT and macrophage proliferation and differentiation in the crown-like structures (90).
CD8 T cells were first noticed as initiators of VAT inflammation. CD8 T-cell infiltration into VAT precedes accumulation of macrophages, and genetic deletion of CD8 T cells reduces VAT inflammation and ameliorates systemic insulin resistance. Adoptive transfer of CD8 T cells into CD8-deficient mice exacerbates VAT inflammation (91).
CD4 T cells also regulate VAT inflammation. Macrophage-driven inflammation is induced by the Th1 response. In VAT, this response is induced by a HFD. Th1 cells produce TNFα, which activates the vascular endothelium and promotes monocyte invasion, and IFN-γ, which induces polarization of proinflammatory M1 macrophages (92).
Thus, Th1 cells contribute to the recruitment of monocytes into the VAT and their differentiation into M1 macrophages (26). IFN-γ suppresses insulin signaling in mature adipocytes, which attenuates insulin-dependent glucose uptake and lipid storage, and also inhibits differentiation of pre-adipocytes to mature adipocytes (93). In sum, Th1 cells play a central role in the induction of early adipose inflammation and adipocyte dysfunction associated with a HFD. Th1 cells are elevated in VAT in patients with type 2 diabetes and correlate with obesity-induced inflammation and insulin resistance (94). In human adipocytes, IFN-γ sustains activation of the JAK/STAT1 pathway, attenuates lipid storage and insulin signaling, and suppresses differentiation (93). Indeed, obese IFN-γ knockout mice have smaller adipocytes and less accumulation of VAT inflammatory cells, resulting in improved insulin sensitivity (95).
In HFD-induced obese mice, the Th17 cell population in the spleen and circulating levels of IL-17 are increased in an IL-6–dependent manner, and IL-17 inhibits insulin signaling in hepatocytes and glucose uptake in skeletal muscle (96). IL-17 has a modulatory effect on adipocytes, inhibiting lipid uptake and glucose uptake by insulin and suppressing adipogenesis (97). Obese insulin-resistant patients have more Th17 cells in their abdominal subcutaneous visceral fat than insulin-sensitive obese patients (98). Furthermore, in obese and metabolically unhealthy individuals, Th17 cells infiltrate obese adipose tissue and Th17 cytokines promote TNF-α production and induce inflammation (99).
Th2 cytokines, such as IL-4 and IL-13, have complex effects in VAT, i.e., they do more than antagonize Th1 cytokines and exert anti-inflammatory effects. In healthy adipose tissues, metabolic homeostasis is maintained by local IL-4 secretion by VAT eosinophils and maintenance of alternatively activated macrophages. In the absence of eosinophils, the numbers of alternatively activated macrophages in VAT are greatly reduced (100). In obese humans, the number of Th2 cells is decreased in subcutaneous and visceral fat and peripheral blood and Th2 frequency is inversely correlated with insulin resistance and serum levels of C-reactive protein, a marker of systemic inflammation (101). Th2 cytokines are abundant in the crown-like structures of obese VAT, where macrophages actively proliferate in a manner dependent on the IL-4 receptor α-chain (IL-4Rα), a molecule essential for Th2 signaling. In these structures, IL-6 functions as a major driver of proliferation of VAT macrophages in obesity by upregulating IL-4Rα (102). The role of the Th2 cytokines IL-4 and IL-13 in VAT inflammation caused by a HFD load is supported by the finding that inflammation is reduced in mice with myeloid cell-specific knockout of IL-4Rα (103). Thus, in the obese environment, Th2 cytokines play an important role in the maintenance of fast-proliferating macrophages within the crown-like structures (102). In the physiological state, innate lymphoid cells (ILC2s) are important producers of type 2 cytokines, which are critical for maintenance of alternatively activated or M2-like adipose tissue macrophages and glucose homeostasis (104). In obese VAT, ILC2s regulates saturated fatty acid absorption, resulting in the amelioration of chronic inflammation (105); however, in obesity the number of ILC2s decreases in both mouse and humans (104). The sources and roles of type 2 cytokines appear to be significantly different between VAT homeostasis and obesity-induced exacerbation of chronic inflammation, and further research is needed.
Overeating activates the effector function of T cells in VAT and initiates inflammation of the tissue. However, the mechanism by which this inflammation becomes chronic is still being elucidated. Recent studies have revealed that T-cell dysfunction due to both T-cell exhaustion and aging is involved in the chronicity of VAT inflammation, and these two aspects are discussed below.
As a result of chronic inflammation, obese T cells exhibit an exhausted phenotype that includes long-term antigen-stimulation, stress responses, and hypoxia (106). Given that a significant proportion of VAT CD4 T cells are not recruited from the circulation and proliferate in situ (107), T-cell exhaustion can be expected to significantly interfere with antigen-specific and memory responses. Soluble factors from the obese stromal vascular fraction inhibit activation of VAT T cells, suggesting that the microenvironment of obese VAT may trigger T-cell exhaustion (9). In mice, chronic HFD intake attenuates the inflammatory capacity of effector T cells in VAT such that they lose their ability to respond to TCR-specific stimulation (9). Strikingly, VAT T-cell dysfunction has been suggested to occur early, i.e., before macrophage infiltration into the VAT (9).
VAT T cells from DIO mice fed a HFD for 18 weeks fail to upregulate CD25 or secrete T cell effector cytokines such as IFN-γ and IL-2 (9). CD25 is required for formation of TCR-stimulated, high-affinity IL-2 receptors. The same is true for VAT T cells in humans with obesity. On the other hand, the ability of Tregs to suppress effector T cells is reduced by downregulation of CD25.
The dysfunction of VAT T cells in DIO mice involves persistent antigen presentation from antigen-presenting cells. Although many T cells in the VAT of DIO mice express high levels of PD-1, blocking PD-1 in the VAT is not able to restore the ability to produce cytokines upon TCR stimulation (9).
In addition to persistent antigen presentation by antigen-presenting cells, some soluble factors in obese VAT are involved in T-cell dysfunction. One candidate for soluble factors is leptin (see also section 5.2). The high levels of leptin associated with obesity lead to T-cell exhaustion by inducing PD-1 expression through strong activation of STAT3 (8, 108). However, adipose tissue T cells from db/db mice also cause T-cell exhaustion (9), suggesting that it involves other humoral factors besides leptin.
Obese VAT is in a state of insulin resistance, in which glucose uptake and fatty acid synthesis are suppressed in the presence of high insulin levels and, conversely, triglycerides are broken down and fatty acids and glycerol are released (109). Impairments in angiogenic capacity also occur in obese VAT. Inhibition of angiogenesis leads to rarefaction of capillaries, impaired proliferation of multipotent progenitor cells, and adipocyte hypertrophy (110). These changes in the environment surrounding T cells may affect the intracellular metabolic system and induce T-cell exhaustion.
Aerobic glycolysis is activated when TCRs are stimulated and suppression of the glycolytic system induces elevated PD-1 expression and T-cell exhaustion, which decreases IFN-γ production. One group proposed a mechanism by which, when the glycolytic system is suppressed, glyceraldehyde 3-phosphate dehydrogenase binds to the 3’UTR of IFN-γ mRNA and inhibits its translation, resulting in decreased IFN-γ production (111).
In the cancer microenvironment, glucose deprivation by cancer cells results in reduced T-cell glycolytic flux, decreased IFN-γ production, and increased PD-1 expression (111). In vitro co-culture experiments have shown that in the cancer microenvironment, metabolic constraints, rather than chronic antigen stimulation per se, may be responsible for T-cell exhaustion (112). In the obese environment, a reduction in the glycolytic flux of VAT T cells may also accelerate T-cell exhaustion.
PD-1 is one of the indicators of T-cell exhaustion, and inhibitors of the PD-1/PD-L1 axis, which restores T cells from exhaustion, have provided major clinical breakthroughs in cancer (16, 113–115). PD-1 expression on T cells is elevated in people with obesity (8). Highly exhausted T cells are susceptible to PD-1 blockade, and obesity is positively correlated with the efficacy of PD-1/PD-L1 inhibitors in patients and mice with cancer (8, 116, 117), together suggesting that obesity induces T-cell exhaustion.
Obesity also induces CD4 T-cell exhaustion in obese VAT, and exhausted CD4 T cells highly express PD-1 (7). However, the finding that PD-1 blockade does not improve VAT CD4 T-cell exhaustion indicates that VAT CD4 T cells may have acquired a unique exhaustion phenotype independent of PD-1/PD-L1 signaling (9). CD8 T cells also appear to acquire diverse exhaustion traits in obese VAT. VAT CD8 T cells from obese mice highly express T-cell exhaustion markers such as Pdcd1, Tox, Entpd1, Tigit, and Lag3 and have an exhaustion profile similar to T cells chronically infected with Lymphocytic choriomeningitis virus (36). On the other hand, obese VAT CD8 T cells do not have increased immune checkpoints such as TIM-3, LAG-3, TIGIT, and EB4 or exhaustion markers such as TOX, TCF-7, and Eomes; instead, VAT CD8 T cells upregulate the co-inhibitory receptor B and T Lymphocyte Attenuator (Btla), Nlrc3, and Dicer1 genes, which suppress TCR signaling by a mechanism different from that of PD1 signaling (118).
Differences in T-cell exhaustion profiles in VAT may result from the intensity and duration of exposure to specific antigens associated with HFDs and obesity. Immunosuppressive signals of VAT are also considered to be a defense mechanism that suppresses excessive obesity-related inflammation of VAT. However, VAT is an important reservoir of immune cells in obesity, and exhaustion of VAT T cells may be disadvantageous in case of various pathologies, such as infectious diseases and cancer.
To sum up, in developing obesity, a HFD load causes acute inflammation by inducing excessive activation of VAT T cells. On the other hand, in the chronic phase of obesity, inhibitory signals suppress the normal VAT T cell response. The fact that chronic inflammation in VAT persists suggests that the presence of not only T-cell exhaustion but also cell-intrinsic functional changes in VAT T cells, which more actively sustain inflammation.
TCR spectratyping analyses show that diet-induced obesity causes thymic regression and limits TCR diversity. Obesity due to melanocortin-4 receptor deficiency, the most common genetic cause of human obesity, also reduces T-cell repertoire diversity (65). A key factor in obesity-induced thymic regression is the conversion of thymic fibroblasts to adipocytes as a result of lipid accumulation (119). Adipocyte proliferation leads to an increase in leukemia inhibitory factor, oncostatin M, and IL-6, which inhibits thymic function and induces thymocyte apoptosis, resulting in a compromised pool of T-cell progenitor cells (120). After thymic degeneration, peripheral T cells are maintained by homeostatic proliferation. In humans, homeostatic proliferation of circulating CD4 T cells is accelerated in individuals with obesity (121).
VAT T cells have significantly less TCR diversity than splenic T cells, and obesity exacerbates this difference (23). Repeated antigen stimulation, the effects of antigen-presenting cells themselves, and homeostatic proliferation may contribute to the repertoire restriction of T cells.
After thymic degeneration, peripheral T cells are maintained by homeostatic proliferation, but a significant proportion of VAT CD4 T cells are not recruited from the circulation (107). Furthermore, VAT contains a huge pool of T cells in obesity (122). These findings suggest that to compensate for the decrease in VAT T cells, VAT T cells are activated and maintained by repeated antigen stimulation rather than by homeostatic proliferation. Studies showed HLA- and MHC2-mediated activation of T cells by antigen-presenting cells in obese humans and mice (25–27, 123, 124), suggesting that repeated stimulation with some kind of antigen may result in TCR repertoire restriction. Differences in the expression of various co-stimulatory and inhibitory receptors expressed on antigen-presenting cells also influence T-cell phenotypic changes (24–27, 29, 31), so antigen-presenting cells may also contribute to T-cell repertoire restriction.
On the other hand, a specific subset of T cells that emerges with obesity may be maintained through homeostatic proliferation because studies showed that during the physiological aging process, T cells are maintained through homeostatic proliferation triggered by homeostatic cytokines and MHC associated with self-peptides (123, 124) and we previously found that obesity accelerates T-cell senescence and that senescent T cells have similar characteristics to T cells created by homeostatic proliferation during aging (7). Although no studies have directly demonstrated homeostatic proliferation of T cells in VAT, we hypothesize that obesity-associated senescent T cells may be maintained by homeostatic proliferation in the same way as aging-associated senescent T cells.
Cellular senescence is triggered by DNA damage, telomere dysfunction, inflammation, and metabolic dysfunction and is accompanied by irreversible cell cycle arrest and the acquisition of the senescence-associated secretory phenotype (125). Oxidative stress, inflammation, and repeated antigenic stimulation associated with obesity may induce shortening of telomere length and accelerate cellular aging (111, 126, 127). Obesity is also associated with leukocyte DNA methylation changes that can lead to immune dysfunction (128). However, the effects of long-term exposure to obesity on gene expression and metabolic status in T cells remain to be elucidated.
In mice fed a HFD, the absolute number of CD4 T cells per gram of VAT continues to increase as obesity progresses. One study attributed this increase in the absolute number of VAT CD4 T cells to an increase in antigen-stimulated activated CD44hiCD62Llo cells (7). In this study, a unique population of CD44hi CD62Llo CD4 T cells that constitutively express CD153 and PD-1 was found to exhibit cellular senescence properties. A CD153+ PD-1hi subset expressing T-bet, the master transcription factor of Th1, had high senescence-associated beta-galactosidase activity and were positive for the DNA damage marker γH2AX, indicating characteristics of cellular senescence. The authors concluded that CD153+ PD-1hi subset continues to secrete abundant osteopontin (OPN) without PD-1–mediated negative signaling inhibition at the cost of normal function and causes VAT inflammation (7). In fact, OPN is elevated in the circulation of patients with obesity and enhances VAT inflammation, leading to the development of diabetes (129, 130).
OPN functions as a potent chemoattractant for macrophages. Chronic inflammation is characterized by macrophage retention, and OPN is a particularly important molecule in promoting macrophage migration and retention. OPN also regulates cytokine production by macrophages and acts on T cells to promote IL-12 production while inhibiting IL-10 production and promoting Th1 cell-mediated responses (131).
A portion of Th1 effector cells terminally differentiates into OPN-producing senescent T cells, and increased numbers of senescent T cells produce large amounts of OPN and maintain high levels of OPN in VAT (7). In turn, high OPN levels cause persistent accumulation and activation of macrophages and induce a Th1 response. These experimental results suggest that, at least in mice, VAT CD4 T cells that have deviated from immune checkpoint mechanisms and acquired a senescent phenotype continue to produce OPN independently of classical TCR stimulation and that this process is closely related to the maintenance of chronic inflammation in obesity. Recently, vaccination targeting CD153 was shown to reduce senescent cells in visceral fat of obese mice fed a HFD for 10 to 11 weeks and to improve VAT inflammation and insulin resistance (132). Senescent T cells were suggested as a potential therapeutic target for obesity-associated immunometabolic disorders.
CD153+ PD-1hi CD4 T cells were originally identified as a characteristic T-cell subset that emerges with aging and were named senescence-associated T cells (124, 133). Noteworthy is that the same cells also appear in the VAT microenvironment in obesity and are involved in chronic local inflammation, making T cell senescence a common underlying pathology for both obesity-related and aging-related diseases.
VAT CD4 T cells play an important role in regulating inflammation and metabolism in obesity, but the underlying mechanisms are largely unknown. Recently, DIO was shown to increase p38 activity in VAT T cells and promote obesity-associated adipose tissue senescence. p38α, an essential subunit of p38, promotes T-cell glycolysis through a mechanistic target of rapamycin signaling, resulting in enhanced Th1 differentiation. T cell-specific deletion of p38α protected mice from HFD-induced obesity, fatty liver, adipose tissue inflammation, and insulin resistance (134).
To sum up, the negative linkage between obesity-accelerating T-cell senescence and T-cell activation-accelerating VAT senescence plays an important role in the development of chronic VAT inflammation (Figure 1).
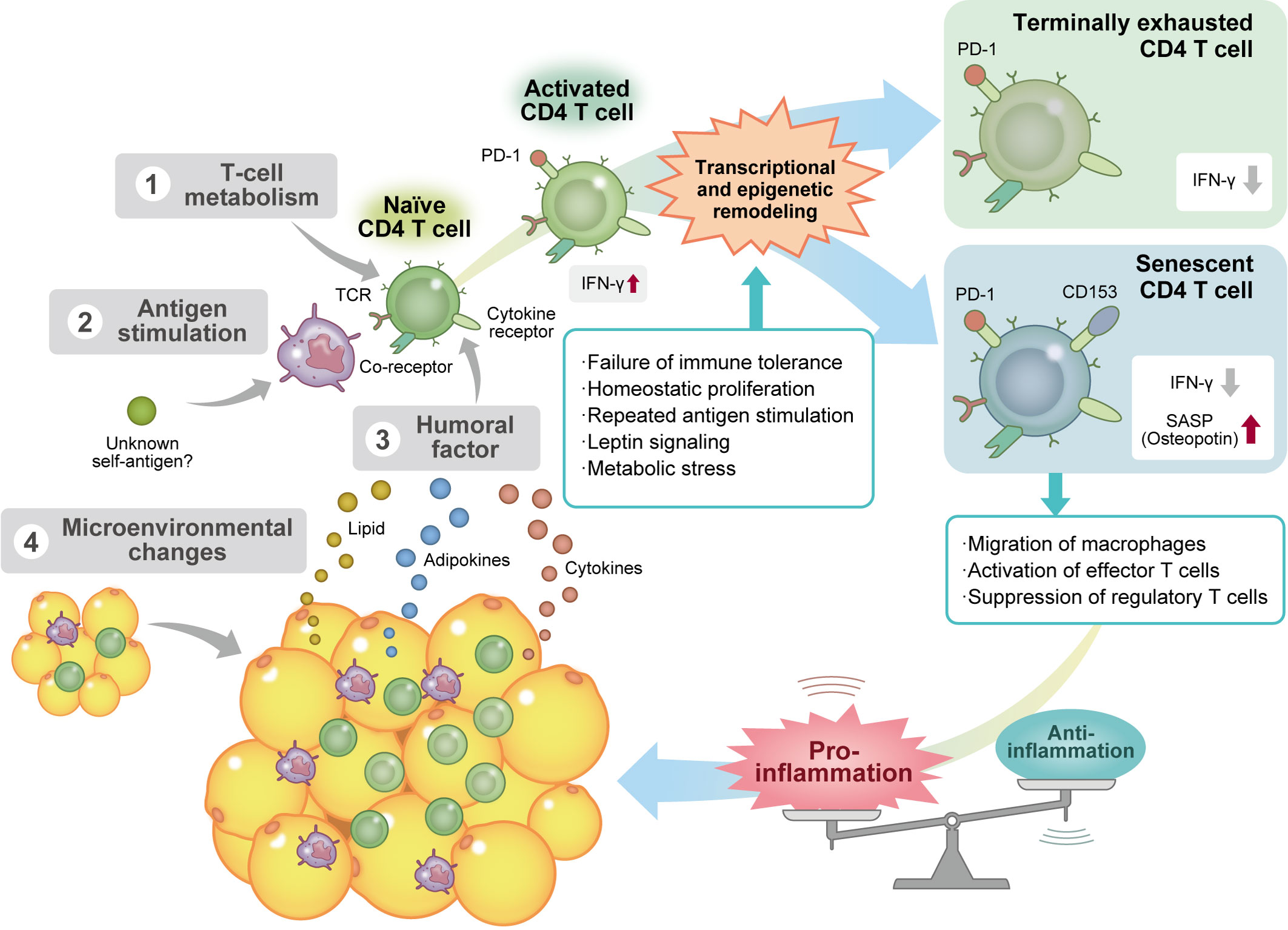
Figure 1 Obesity induces CD4 T-cell senescence via several mechanisms. Factors affecting the differentiation and maturation process of visceral adipose tissue (VAT) CD4 T cells in obesity include 1) alteration of T-cell metabolism; 2) repeated antigen stimulation by antigen-presenting cells, such as macrophages and dendritic cells; 3) various humoral factors, such as cytokines, adipokines, and lipids; and 4) changes in the VAT microenvironment. These stimuli activate T helper 1 (Th1) CD4 T cells and induce various types of transcriptional and epigenetic remodeling. Chronic activation of T cells induces immune exhaustion and reduces the secretion of proinflammatory cytokines, including interferon γ. On the other hand, some Th1 effector T cells deviate from the immune checkpoint mechanism and acquire a cellular senescence phenotype without anergy because of repeated antigenic stimulation. Senescent CD4 T cells that produce a large amount of osteopontin accelerate macrophage migration and effector T-cell activation and suppress regulatory T cell function, resulting in the maintenance of chronic VAT inflammation. Ifn-γ, interferon γ; PD-1, programmed cell death protein 1; SASP, senescence-associated secretory phenotype; TCR, T-cell receptor.
Obesity can be corrected by weight loss. However, weight loss through caloric restriction and increased physical activity is not as easy as it sounds, and even if weight is lost, it may be regained quickly. Traditionally, bariatric surgery has been performed in severely obese patients who are refractory to medical therapy because of a combination of factors such as dietary environment and unique personality traits. With the recent launch of a glucagon-like peptide 1 receptor agonist and dual glucose-dependent insulinotropic polypeptide/glucagon-like peptide 1 receptor agonist, long-term maintenance of successful lost weight by medication has finally become feasible (135, 136). However, studies on the long-term effects of weight reduction on cardiovascular events and life expectancy in patients with obesity and type 2 diabetes have yielded conflicting results. A meta-analysis showed that weight reduction is associated with reduced mortality in unhealthy individuals with obesity (137), and bariatric surgery reduced the incidence of myocardial infarction in patients with obesity and type 2 diabetes over a mean follow-up period of 13.3 years (138). However, a prospective randomized study found that an intensive lifestyle intervention focused on weight reduction was not associated with reduced cardiovascular disease and mortality in adults with overweight or obesity and type 2 diabetes at almost 10 years of follow-up (139).
The phenotypic changes in T cells upon weight reduction in obesity appear to be very complex. In obese mice, weight reduction induces the accumulation of CD4 T cells in VAT, resulting in the recruitment and retainment of pro-inflammatory macrophages in VAT despite normalization of body weight (13, 140). In mice with weight reduction after being switched from a HFD to a control diet, body weight and visceral fat decreased to the same level as lean mice fed the control diet; however, the VAT of the weight-loss mice showed dense infiltration of macrophages, which formed more crown-like structures than those in HFD-fed obese mice. Mechanistically, CD153+ PD-1hi CD4 T cells are long-lived and not easily eliminated after weight loss, and the continued presence of senescent T cells is associated with the production of large amounts of OPN, creating a chronic inflammatory loop (Figure 2). Thus, senescent CD4 T cells are suggested to be a negative legacy effect of obesity (13).
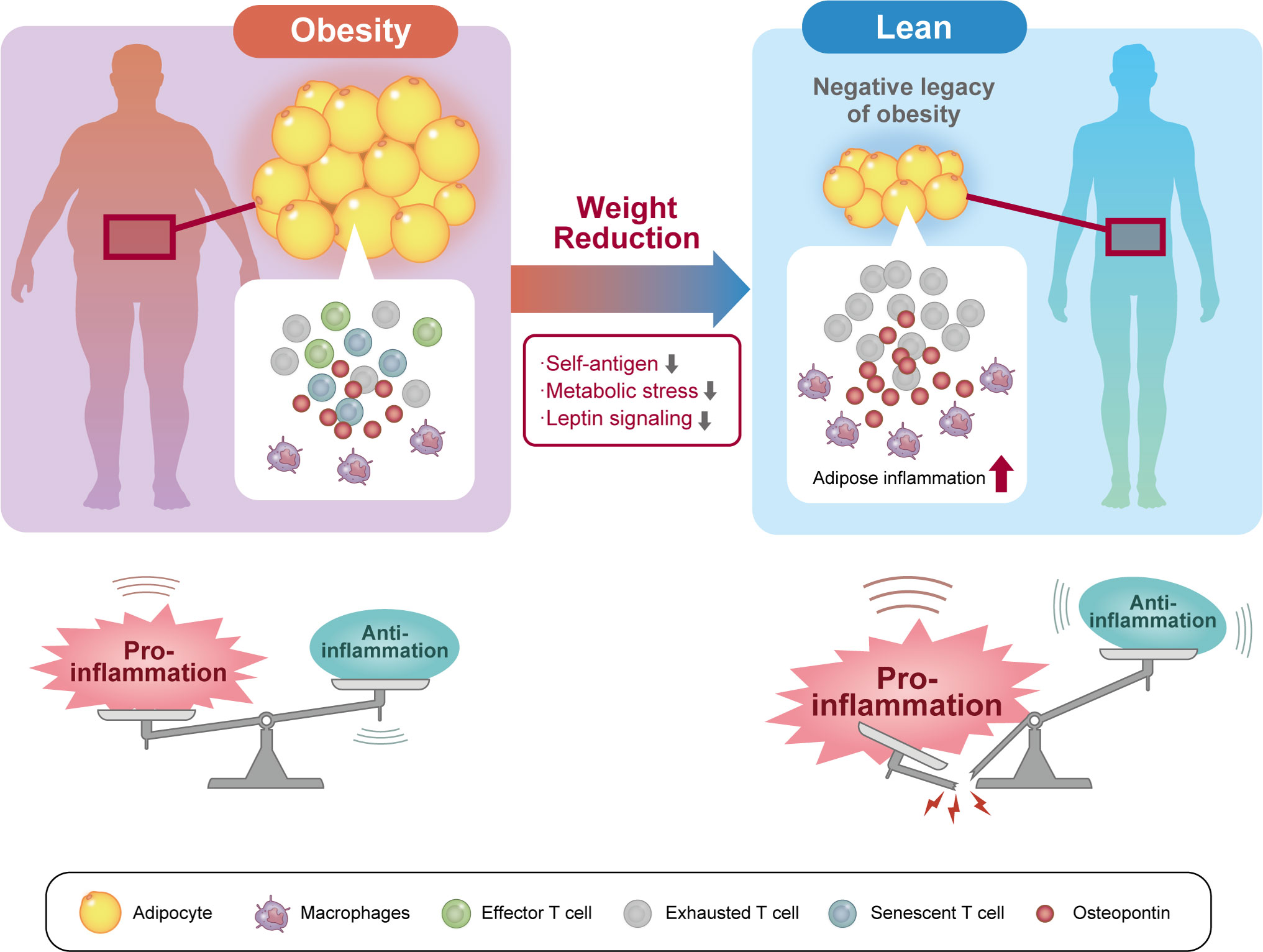
Figure 2 Senescent CD4 T cells are a negative legacy of obesity. In the pathophysiology of obesity, not only effector and exhausted T cells but also senescent T cells accumulate in visceral adipose tissue (VAT). VAT inflammation induced by these VAT T cells induces systemic insulin resistance and contributes to the pathogenesis of diabetes. Weight reduction reduces exposure to self-antigens, metabolic stress, and humoral factors in the VAT microenvironment. However, long-lived senescent CD4 T cells concentrate in VAT after weight loss and continue to secrete proinflammatory osteopontin. As a result, VAT inflammation persists after weight reduction and may represent a residual risk factor for cardiovascular disease.
Interestingly, in a weight gain-loss-regain model, mice with a history of obesity regain weight very quickly. Although the molecular mechanism for this obesity memory is unknown, CD4 effector T cells were shown to be involved (12). Furthermore, one study reported that glucose tolerance was worse in DIO weight-loss-regain mice than in normal DIO mice and that this metabolic dysfunction was associated with increased T-cell accumulation in VAT (36).
The above experimental results explain the body’s propensity to repeatedly gain and lose weight, which makes it difficult to maintain weight loss and increases the risk of developing diabetes. Noteworthy is that the background imprinting of an obesity-related immune phenotype is present in VAT.
VAT is not only a storage organ for lipids, but also an endocrine organ that regulates energy balance in the body by secreting various bioactive substances called adipokines, which are involved in food intake and insulin resistance. Dysregulation of adipokine function and production in obese VAT plays a major role in the development and progression of metabolic syndrome. Furthermore, it is becoming clear that various immunocompetent cells, including macrophages, increasingly infiltrate obese VAT and that the accompanying mild systemic chronic inflammatory response is the underlying pathogenesis of lifestyle-related diseases such as metabolic syndrome.
In this review, we sought to summarize the role of CD4 T cells in maintaining VAT homeostasis, inflammation caused by excessive fat accumulation in VAT, and the chronicity of this inflammation. In addition to TCR signals, co-stimulation signals, and cytokine signals from antigen-presenting cells, T cells undergo tissue-specific differentiation in response to organ- and disease-specific environmental signals.
To date, most of the findings on chronic inflammation of VAT associated with obesity are from analyses of DIO mice fed a HFD rich in saturated fatty acids. Therefore, future research needs to carefully examine the extent to which findings from DIO mice can be extrapolated to humans. Epidemiological evidence indicates that obese VAT increases the risk of developing cardiovascular disease. However, the mediators that affect remote organs via immunological changes in VAT remain unclear. In obesity, ectopic fat accumulates around the heart and blood vessels, and development of cardiovascular disease may be more strongly influenced by this neighboring ectopic fat. Obesity also affects systemic immune function. When considering the relationships between obesity and infectious diseases, cancer, and autoimmune diseases, the hypothesis is attractive that immunological abnormalities in obese VAT can also affect systemic immune function. In fact, some research suggests such an association, but proving a causal relationship remains a challenge for the future. Nevertheless, an understanding of the qualitative and quantitative changes of immune cells within the microenvironment in VAT will undoubtedly help to elucidate the mechanisms by which obesity causes various diseases, including lifestyle-related ones, via excessive local or systemic activation of the immune system. In the future, tailored therapeutic strategies based on an understanding of immune cell trait changes may contribute to improving the prognosis of obesity-related diseases.
Both KS and MS contributed to conceptualization, writing (original draft preparation, review, and editing), and funding acquisition. All authors contributed to the article and approved the submitted version.
The following funding was received: JSPS Grant-in-Aid for Young Scientists JP21K16096 (2021–2022), JSPS Grant-in-Aid for Scientific Research (B) 18H02812 (2018-2020), Grant for The Japan Research Foundation for Healthy Aging, Grant for Japan Foundation for Applied Enzymology, Grant for The Japanese Heart Failure Society, Grant for Takeda Science Foundation, and The Vehicle Racing Commemorative Foundation. The funders had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; or decision to submit the manuscript for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Rosen ED, Spiegelman BM. What we talk about when we talk about fat. Cell (2014) 156(1-2):20–44. doi: 10.1016/j.cell.2013.12.012
2. Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science (1993) 259(5091):87–91. doi: 10.1126/science.7678183
3. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. (2003) 112(12):1796–808. doi: 10.1172/JCI200319246
4. Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. (2003) 112(12):1821–30. doi: 10.1172/JCI200319451
5. Panduro M, Benoist C, Mathis D. Tissue tregs. Annu Rev Immunol (2016) 34:609–33. doi: 10.1146/annurev-immunol-032712-095948
6. Wu H, Ghosh S, Perrard XD, Feng L, Garcia GE, Perrard JL, et al. T-Cell accumulation and regulated on activation, normal T cell expressed and secreted upregulation in adipose tissue in obesity. Circulation (2007) 115(8):1029–38. doi: 10.1161/CIRCULATIONAHA.106.638379
7. Shirakawa K, Yan X, Shinmura K, Endo J, Kataoka M, Katsumata Y, et al. Obesity accelerates T cell senescence in murine visceral adipose tissue. J Clin Invest. (2016) 126(12):4626–39. doi: 10.1172/JCI88606
8. Wang Z, Aguilar EG, Luna JI, Dunai C, Khuat LT, Le CT, et al. Paradoxical effects of obesity on T cell function during tumor progression and PD-1 checkpoint blockade. Nat Med (2019) 25(1):141–51. doi: 10.1038/s41591-018-0221-5
9. Porsche CE, Delproposto JB, Geletka L, O'Rourke R, Lumeng CN. Obesity results in adipose tissue T cell exhaustion. JCI Insight (2021) 6(8):e139793. doi: 10.1172/jci.insight.139793
10. Frebel H, Richter K, Oxenius A. How chronic viral infections impact on antigen-specific T-cell responses. Eur J Immunol (2010) 40(3):654–63. doi: 10.1002/eji.200940102
11. Klenerman P, Hill A. T Cells and viral persistence: lessons from diverse infections. Nat Immunol (2005) 6(9):873–9. doi: 10.1038/ni1241
12. Zou J, Lai B, Zheng M, Chen Q, Jiang S, Song A, et al. CD4+ T cells memorize obesity and promote weight regain. Cell Mol Immunol (2018) 15(6):630–9. doi: 10.1038/cmi.2017.36
13. Shirakawa K, Endo J, Katsumata Y, Yamamoto T, Kataoka M, Isobe S, et al. Negative legacy of obesity. PloS One (2017) 12(10):e0186303. doi: 10.1371/journal.pone.0186303
14. Wilcox RA. A three-signal model of T-cell lymphoma pathogenesis. Am J Hematol (2016) 91(1):113–22. doi: 10.1002/ajh.24203
15. Riley JL. PD-1 signaling in primary T cells. Immunol Rev (2009) 229(1):114–25. doi: 10.1111/j.1600-065X.2009.00767.x
16. Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med (2012) 209(6):1201–17. doi: 10.1084/jem.20112741
17. Sheppard KA, Fitz LJ, Lee JM, Benander C, George JA, Wooters J, et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett (2004) 574(1-3):37–41. doi: 10.1016/j.febslet.2004.07.083
18. Yokosuka T, Kobayashi W, Takamatsu M, Sakata-Sogawa K, Zeng H, Hashimoto-Tane A, et al. Spatiotemporal basis of CTLA-4 costimulatory molecule-mediated negative regulation of T cell activation. Immunity (2010) 33(3):326–39. doi: 10.1016/j.immuni.2010.09.006
19. Brunner-Weinzierl MC, Rudd CE. CTLA-4 and PD-1 control of T-cell motility and migration: Implications for tumor immunotherapy. Front Immunol (2018) 9:2737. doi: 10.3389/fimmu.2018.02737
20. Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol (2007) 7(6):454–65. doi: 10.1038/nri2093
21. Procaccini C, De Rosa V, Galgani M, Carbone F, La Rocca C, Formisano L, et al. Role of adipokines signaling in the modulation of T cells function. Front Immunol (2013) 4:332. doi: 10.3389/fimmu.2013.00332
22. Giordano A, Murano I, Mondini E, Perugini J, Smorlesi A, Severi I, et al. Obese adipocytes show ultrastructural features of stressed cells and die of pyroptosis. J Lipid Res (2013) 54(9):2423–36. doi: 10.1194/jlr.M038638
23. Yang H, Youm YH, Vandanmagsar B, Ravussin A, Gimble JM, Greenway F, et al. Obesity increases the production of proinflammatory mediators from adipose tissue T cells and compromises TCR repertoire diversity: implications for systemic inflammation and insulin resistance. J Immunol (2010) 185(3):1836–45. doi: 10.4049/jimmunol.1000021
24. La Gruta NL, Gras S, Daley SR, Thomas PG, Rossjohn J. Understanding the drivers of MHC restriction of T cell receptors. Nat Rev Immunol (2018) 18(7):467–78. doi: 10.1038/s41577-018-0007-5
25. Xiao L, Yang X, Lin Y, Li S, Jiang J, Qian S, et al. Large Adipocytes function as antigen-presenting cells to activate CD4(+) T cells via upregulating MHCII in obesity. Int J Obes (Lond). (2016) 40(1):112–20. doi: 10.1038/ijo.2015.145
26. Deng T, Lyon CJ, Minze LJ, Lin J, Zou J, Liu JZ, et al. Class II major histocompatibility complex plays an essential role in obesity-induced adipose inflammation. Cell Metab (2013) 17(3):411–22. doi: 10.1016/j.cmet.2013.02.009
27. Cho KW, Morris DL, DelProposto JL, Geletka L, Zamarron B, Martinez-Santibanez G, et al. An MHC II-dependent activation loop between adipose tissue macrophages and CD4+ T cells controls obesity-induced inflammation. Cell Rep (2014) 9(2):605–17. doi: 10.1016/j.celrep.2014.09.004
28. Porsche CE, Delproposto JB, Patrick E, Zamarron BF, Lumeng CN. Adipose tissue dendritic cell signals are required to maintain T cell homeostasis and obesity-induced expansion. Mol Cell Endocrinol (2020) 505:110740. doi: 10.1016/j.mce.2020.110740
29. Morris DL, Cho KW, Delproposto JL, Oatmen KE, Geletka LM, Martinez-Santibanez G, et al. Adipose tissue macrophages function as antigen-presenting cells and regulate adipose tissue CD4+ T cells in mice. Diabetes (2013) 62(8):2762–72. doi: 10.2337/db12-1404
30. Winer DA, Winer S, Shen L, Wadia PP, Yantha J, Paltser G, et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med (2011) 17(5):610–7. doi: 10.1038/nm.2353
31. Song J, Deng T. The adipocyte and adaptive immunity. Front Immunol (2020) 11:593058. doi: 10.3389/fimmu.2020.593058
32. Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, Yagita H, et al. Expression of the PD-1 antigen on the surface of stimulated mouse T and b lymphocytes. Int Immunol (1996) 8(5):765–72. doi: 10.1093/intimm/8.5.765
33. Carter L, Fouser LA, Jussif J, Fitz L, Deng B, Wood CR, et al. PD-1:PD-L inhibitory pathway affects both CD4(+) and CD8(+) T cells and is overcome by IL-2. Eur J Immunol (2002) 32(3):634–43. doi: 10.1002/1521-4141(200203)32:3<634::AID-IMMU634>3.0.CO;2-9
34. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol (2008) 26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331
35. Sharpe AH, Pauken KE. The diverse functions of the PD1 inhibitory pathway. Nat Rev Immunol (2018) 18(3):153–67. doi: 10.1038/nri.2017.108
36. Cottam MA, Caslin HL, Winn NC, Hasty AH. Multiomics reveals persistence of obesity-associated immune cell phenotypes in adipose tissue during weight loss and weight regain in mice. Nat Commun (2022) 13(1):2950. doi: 10.1038/s41467-022-30646-4
37. Odorizzi PM, Pauken KE, Paley MA, Sharpe A, Wherry EJ. Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J Exp Med (2015) 212(7):1125–37. doi: 10.1084/jem.20142237
38. Ahn E, Araki K, Hashimoto M, Li W, Riley JL, Cheung J, et al. Role of PD-1 during effector CD8 T cell differentiation. Proc Natl Acad Sci U S A. (2018) 115(18):4749–54. doi: 10.1073/pnas.1718217115
39. Schwartz C, Schmidt V, Deinzer A, Hawerkamp HC, Hams E, Bayerlein J, et al. Innate PD-L1 limits T cell-mediated adipose tissue inflammation and ameliorates diet-induced obesity. Sci Transl Med (2022) 14(635):eabj6879. doi: 10.1126/scitranslmed.abj6879
40. Chang CB, Lee SP, Chen WM, Wang CM, Song YC, Chan MW, et al. Dendritic cell upregulation of programmed death ligand-1 via DNA demethylation inhibits experimental autoimmune encephalomyelitis. J Autoimmun (2020) 107:102362. doi: 10.1016/j.jaut.2019.102362
41. Peng Q, Qiu X, Zhang Z, Zhang S, Zhang Y, Liang Y, et al. PD-L1 on dendritic cells attenuates T cell activation and regulates response to immune checkpoint blockade. Nat Commun (2020) 11(1):4835. doi: 10.1038/s41467-020-18570-x
42. MacLaren RE, Cui W, Lu H, Simard S, Cianflone K. Association of adipocyte genes with ASP expression: a microarray analysis of subcutaneous and omental adipose tissue in morbidly obese subjects. BMC Med Genomics (2010) 3:3. doi: 10.1186/1755-8794-3-3
43. Wu B, Chiang HC, Sun X, Yuan B, Mitra P, Hu Y, et al. Genetic ablation of adipocyte PD-L1 reduces tumor growth but accentuates obesity-associated inflammation. J Immunother Cancer (2020) 8(2):e000964. doi: 10.1136/jitc-2020-000964
44. Dumitru C, Kabat AM, Maloy KJ. Metabolic adaptations of CD4(+) T cells in inflammatory disease. Front Immunol (2018) 9:540. doi: 10.3389/fimmu.2018.00540
45. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol (2011) 186(6):3299–303. doi: 10.4049/jimmunol.1003613
46. Alwarawrah Y, Nichols AG, Green WD, Eisner W, Kiernan K, Warren J, et al. Targeting T-cell oxidative metabolism to improve influenza survival in a mouse model of obesity. Int J Obes (Lond). (2020) 44(12):2419–29. doi: 10.1038/s41366-020-00692-3
47. Turbitt WJ, Buchta Rosean C, Weber KS, Norian LA. Obesity and CD8 T cell metabolism: Implications for anti-tumor immunity and cancer immunotherapy outcomes. Immunol Rev (2020) 295(1):203–19. doi: 10.1111/imr.12849
48. Zhou H, Liu F. T Cell metabolism in obesity and beyond: comments on 'DsbA-l deficiency in T cells promotes diet-induced thermogenesis through suppressing IFN-γ production'. J Mol Cell Biol (2021) 13(5):389–91. doi: 10.1093/jmcb/mjab008
49. Saltiel AR, Olefsky JM. Inflammatory mechanisms linking obesity and metabolic disease. J Clin Invest. (2017) 127(1):1–4. doi: 10.1172/JCI92035
50. Saucillo DC, Gerriets VA, Sheng J, Rathmell JC, Maciver NJ. Leptin metabolically licenses T cells for activation to link nutrition and immunity. J Immunol (2014) 192(1):136–44. doi: 10.4049/jimmunol.1301158
51. Howard JK, Lord GM, Matarese G, Vendetti S, Ghatei MA, Ritter MA, et al. Leptin protects mice from starvation-induced lymphoid atrophy and increases thymic cellularity in ob/ob mice. J Clin Invest. (1999) 104(8):1051–9. doi: 10.1172/JCI6762
52. Kim SY, Lim JH, Choi SW, Kim M, Kim ST, Kim MS, et al. Preferential effects of leptin on CD4 T cells in central and peripheral immune system are critically linked to the expression of leptin receptor. Biochem Biophys Res Commun (2010) 394(3):562–8. doi: 10.1016/j.bbrc.2010.03.019
53. Fildes A, Charlton J, Rudisill C, Littlejohns P, Prevost AT, Gulliford MC. Probability of an obese person attaining normal body weight: Cohort study using electronic health records. Am J Public Health (2015) 105(9):e54–9. doi: 10.2105/AJPH.2015.302773
54. Gerriets VA, Danzaki K, Kishton RJ, Eisner W, Nichols AG, Saucillo DC, et al. Leptin directly promotes T-cell glycolytic metabolism to drive effector T-cell differentiation in a mouse model of autoimmunity. Eur J Immunol (2016) 46(8):1970–83. doi: 10.1002/eji.201545861
55. Lee GR. The balance of Th17 versus treg cells in autoimmunity. Int J Mol Sci (2018) 19(3):730. doi: 10.3390/ijms19030730
56. Reis BS, Lee K, Fanok MH, Mascaraque C, Amoury M, Cohn LB, et al. Leptin receptor signaling in T cells is required for Th17 differentiation. J Immunol (2015) 194(11):5253–60. doi: 10.4049/jimmunol.1402996
57. Yu Y, Liu Y, Shi FD, Zou H, Matarese G, La Cava A. Cutting edge: Leptin-induced RORγt expression in CD4+ T cells promotes Th17 responses in systemic lupus erythematosus. J Immunol (2013) 190(7):3054–8. doi: 10.4049/jimmunol.1203275
58. Shibata S, Tada Y, Hau CS, Mitsui A, Kamata M, Asano Y, et al. Adiponectin regulates psoriasiform skin inflammation by suppressing IL-17 production from γδ-T cells. Nat Commun (2015) 6:7687. doi: 10.1038/ncomms8687
59. Wilk S, Scheibenbogen C, Bauer S, Jenke A, Rother M, Guerreiro M, et al. Adiponectin is a negative regulator of antigen-activated T cells. Eur J Immunol (2011) 41(8):2323–32. doi: 10.1002/eji.201041349
60. Zhang K, Guo Y, Ge Z, Zhang Z, Da Y, Li W, et al. Adiponectin suppresses T helper 17 cell differentiation and limits autoimmune CNS inflammation via the SIRT1/PPARγ/RORγt pathway. Mol Neurobiol (2017) 54(7):4908–20. doi: 10.1007/s12035-016-0036-7
61. Tsang JY, Li D, Ho D, Peng J, Xu A, Lamb J, et al. Novel immunomodulatory effects of adiponectin on dendritic cell functions. Int Immunopharmacol. (2011) 11(5):604–9. doi: 10.1016/j.intimp.2010.11.009
62. Surendar J, Frohberger SJ, Karunakaran I, Schmitt V, Stamminger W, Neumann AL, et al. Adiponectin limits IFN-γ and IL-17 producing CD4 T cells in obesity by restraining cell intrinsic glycolysis. Front Immunol (2019) 10:2555. doi: 10.3389/fimmu.2019.02555
63. Ioan-Facsinay A, Kwekkeboom JC, Westhoff S, Giera M, Rombouts Y, van Harmelen V, et al. Adipocyte-derived lipids modulate CD4+ T-cell function. Eur J Immunol (2013) 43(6):1578–87. doi: 10.1002/eji.201243096
64. Minokoshi Y, Toda C, Okamoto S. Regulatory role of leptin in glucose and lipid metabolism in skeletal muscle. Indian J Endocrinol Metab (2012) 16(Suppl 3):S562–8. doi: 10.4103/2230-8210.105573
65. Yang H, Youm YH, Vandanmagsar B, Rood J, Kumar KG, Butler AA, et al. Obesity accelerates thymic aging. Blood (2009) 114(18):3803–12. doi: 10.1182/blood-2009-03-213595
66. Procaccini C, La Rocca C, Carbone F, De Rosa V, Galgani M, Matarese G. Leptin as immune mediator: Interaction between neuroendocrine and immune system. Dev Comp Immunol (2017) 66:120–9. doi: 10.1016/j.dci.2016.06.006
67. Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature (1998) 395(6704):763–70. doi: 10.1038/27376
68. Achari AE, Jain SK. Adiponectin, a therapeutic target for obesity, diabetes, and endothelial dysfunction. Int J Mol Sci (2017) 18(6):1321. doi: 10.3390/ijms18061321
69. Kern PA, Di Gregorio GB, Lu T, Rassouli N, Ranganathan G. Adiponectin expression from human adipose tissue: relation to obesity, insulin resistance, and tumor necrosis factor-alpha expression. Diabetes (2003) 52(7):1779–85. doi: 10.2337/diabetes.52.7.1779
70. Mauro C, Smith J, Cucchi D, Coe D, Fu H, Bonacina F, et al. Obesity-induced metabolic stress leads to biased effector memory CD4(+) T cell differentiation via PI3K p110δ-Akt-Mediated signals. Cell Metab (2017) 25(3):593–609. doi: 10.1016/j.cmet.2017.01.008
71. Li C, Spallanzani RG, Mathis D. Visceral adipose tissue tregs and the cells that nurture them. Immunol Rev (2020) 295(1):114–25. doi: 10.1111/imr.12850
72. Becker M, Levings MK, Daniel C. Adipose-tissue regulatory T cells: Critical players in adipose-immune crosstalk. Eur J Immunol (2017) 47(11):1867–74. doi: 10.1002/eji.201646739
73. Deiuliis J, Shah Z, Shah N, Needleman B, Mikami D, Narula V, et al. Visceral adipose inflammation in obesity is associated with critical alterations in tregulatory cell numbers. PloS One (2011) 6(1):e16376. doi: 10.1371/journal.pone.0016376
74. Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med (2009) 15(8):930–9. doi: 10.1038/nm.2002
75. Ohkura N, Sakaguchi S. Transcriptional and epigenetic basis of treg cell development and function: its genetic anomalies or variations in autoimmune diseases. Cell Res (2020) 30(6):465–74. doi: 10.1038/s41422-020-0324-7
76. Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, et al. Induction of colonic regulatory T cells by indigenous clostridium species. Science (2011) 331(6015):337–41. doi: 10.1126/science.1198469
77. Kolodin D, van Panhuys N, Li C, Magnuson AM, Cipolletta D, Miller CM, et al. Antigen- and cytokine-driven accumulation of regulatory T cells in visceral adipose tissue of lean mice. Cell Metab (2015) 21(4):543–57. doi: 10.1016/j.cmet.2015.03.005
78. Vasanthakumar A, Moro K, Xin A, Liao Y, Gloury R, Kawamoto S, et al. The transcriptional regulators IRF4, BATF and IL-33 orchestrate development and maintenance of adipose tissue-resident regulatory T cells. Nat Immunol (2015) 16(3):276–85. doi: 10.1038/ni.3085
79. Li Y, Lu Y, Lin SH, Li N, Han Y, Huang Q, et al. Insulin signaling establishes a developmental trajectory of adipose regulatory T cells. Nat Immunol (2021) 22(9):1175–85. doi: 10.1038/s41590-021-01010-3
80. Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, et al. PPAR-γ is a major driver of the accumulation and phenotype of adipose tissue treg cells. Nature (2012) 486(7404):549–53. doi: 10.1038/nature11132
81. Vasanthakumar A, Chisanga D, Blume J, Gloury R, Britt K, Henstridge DC, et al. Sex-specific adipose tissue imprinting of regulatory T cells. Nature (2020) 579(7800):581–5. doi: 10.1038/s41586-020-2040-3
82. Beppu LY, Mooli RGR, Qu X, Marrero GJ, Finley CA, Fooks AN, et al. Tregs facilitate obesity and insulin resistance via a blimp-1/IL-10 axis. JCI Insight (2021) 6(3):e140644. doi: 10.1172/jci.insight.140644
83. Onishi Y, Fehervari Z, Yamaguchi T, Sakaguchi S. Foxp3+ natural regulatory T cells preferentially form aggregates on dendritic cells in vitro and actively inhibit their maturation. Proc Natl Acad Sci U S A. (2008) 105(29):10113–8. doi: 10.1073/pnas.0711106105
84. Williams LM, Ricchetti G, Sarma U, Smallie T, Foxwell BM. Interleukin-10 suppression of myeloid cell activation–a continuing puzzle. Immunology (2004) 113(3):281–92. doi: 10.1111/j.1365-2567.2004.01988.x
85. Han JM, Wu D, Denroche HC, Yao Y, Verchere CB, Levings MK. IL-33 reverses an obesity-induced deficit in visceral adipose tissue ST2+ T regulatory cells and ameliorates adipose tissue inflammation and insulin resistance. J Immunol (2015) 194(10):4777–83. doi: 10.4049/jimmunol.1500020
86. Zhao XY, Zhou L, Chen Z, Ji Y, Peng X, Qi L, et al. The obesity-induced adipokine sST2 exacerbates adipose t(reg) and ILC2 depletion and promotes insulin resistance. Sci Adv (2020) 6(20):eaay6191. doi: 10.1126/sciadv.aay6191
87. Laparra A, Tricot S, Le Van M, Damouche A, Gorwood J, Vaslin B, et al. The frequencies of immunosuppressive cells in adipose tissue differ in human, non-human primate, and mouse models. Front Immunol (2019) 10:117. doi: 10.3389/fimmu.2019.00117
88. Donninelli G, Del Cornò M, Pierdominici M, Scazzocchio B, Varì R, Varano B, et al. Distinct blood and visceral adipose tissue regulatory T cell and innate lymphocyte profiles characterize obesity and colorectal cancer. Front Immunol (2017) 8:643. doi: 10.3389/fimmu.2017.00643
89. Murano I, Barbatelli G, Parisani V, Latini C, Muzzonigro G, Castellucci M, et al. Dead adipocytes, detected as crown-like structures, are prevalent in visceral fat depots of genetically obese mice. J Lipid Res (2008) 49(7):1562–8. doi: 10.1194/jlr.M800019-JLR200
90. Wang Q, Wu H. T Cells in adipose tissue: Critical players in immunometabolism. Front Immunol (2018) 9:2509. doi: 10.3389/fimmu.2018.02509
91. Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med (2009) 15(8):914–20. doi: 10.1038/nm.1964
92. Rocha VZ, Folco EJ, Sukhova G, Shimizu K, Gotsman I, Vernon AH, et al. Interferon-gamma, a Th1 cytokine, regulates fat inflammation: a role for adaptive immunity in obesity. Circ Res (2008) 103(5):467–76. doi: 10.1161/CIRCRESAHA.108.177105
93. McGillicuddy FC, Chiquoine EH, Hinkle CC, Kim RJ, Shah R, Roche HM, et al. Interferon gamma attenuates insulin signaling, lipid storage, and differentiation in human adipocytes via activation of the JAK/STAT pathway. J Biol Chem (2009) 284(46):31936–44. doi: 10.1074/jbc.M109.061655
94. Zhao Y, Lin L, Li J, Xiao Z, Chen B, Wan L, et al. CD4(+) T cells in obesity and obesity-associated diseases. Cell Immunol (2018) 332:1–6. doi: 10.1016/j.cellimm.2018.08.013
95. O'Rourke RW, White AE, Metcalf MD, Winters BR, Diggs BS, Zhu X, et al. Systemic inflammation and insulin sensitivity in obese IFN-γ knockout mice. Metabolism (2012) 61(8):1152–61. doi: 10.1016/j.metabol.2012.01.018
96. Winer S, Paltser G, Chan Y, Tsui H, Engleman E, Winer D, et al. Obesity predisposes to Th17 bias. Eur J Immunol (2009) 39(9):2629–35. doi: 10.1002/eji.200838893
97. Zúñiga LA, Shen WJ, Joyce-Shaikh B, Pyatnova EA, Richards AG, Thom C, et al. IL-17 regulates adipogenesis, glucose homeostasis, and obesity. J Immunol (2010) 185(11):6947–59. doi: 10.4049/jimmunol.1001269
98. Fabbrini E, Cella M, McCartney SA, Fuchs A, Abumrad NA, Pietka TA, et al. Association between specific adipose tissue CD4+ T-cell populations and insulin resistance in obese individuals. Gastroenterology (2013) 145(2):366–74.e1-3. doi: 10.1053/j.gastro.2013.04.010
99. Ip B, Cilfone NA, Belkina AC, DeFuria J, Jagannathan-Bogdan M, Zhu M, et al. Th17 cytokines differentiate obesity from obesity-associated type 2 diabetes and promote TNFα production. Obes (Silver Spring). (2016) 24(1):102–12. doi: 10.1002/oby.21243
100. Wu D, Molofsky AB, Liang HE, Ricardo-Gonzalez RR, Jouihan HA, Bando JK, et al. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science (2011) 332(6026):243–7. doi: 10.1126/science.1201475
101. McLaughlin T, Ackerman SE, Shen L, Engleman E. Role of innate and adaptive immunity in obesity-associated metabolic disease. J Clin Invest. (2017) 127(1):5–13. doi: 10.1172/JCI88876
102. Braune J, Weyer U, Hobusch C, Mauer J, Brüning JC, Bechmann I, et al. IL-6 regulates M2 polarization and local proliferation of adipose tissue macrophages in obesity. J Immunol (2017) 198(7):2927–34. doi: 10.4049/jimmunol.1600476
103. Ackermann J, Arndt L, Kirstein M, Hobusch C, Brinker G, Klöting N, et al. Myeloid cell-specific IL-4 receptor knockout partially protects from adipose tissue inflammation. J Immunol (2021) 207(12):3081–9. doi: 10.4049/jimmunol.2100699
104. Bénézech C, Jackson-Jones LH. ILC2 orchestration of local immune function in adipose tissue. Front Immunol (2019) 10:171. doi: 10.3389/fimmu.2019.00171
105. Okamura T, Hashimoto Y, Mori J, Yamaguchi M, Majima S, Senmaru T, et al. ILC2s improve glucose metabolism through the control of saturated fatty acid absorption within visceral fat. Front Immunol (2021) 12:669629. doi: 10.3389/fimmu.2021.669629
106. Lu J, Zhao J, Meng H, Zhang X. Adipose tissue-resident immune cells in obesity and type 2 diabetes. Front Immunol (2019) 10:1173. doi: 10.3389/fimmu.2019.01173
107. Han SJ, Glatman Zaretsky A, Andrade-Oliveira V, Collins N, Dzutsev A, Shaik J, et al. White adipose tissue is a reservoir for memory T cells and promotes protective memory responses to infection. Immunity (2017) 47(6):1154–68.e6. doi: 10.1016/j.immuni.2017.11.009
108. Bally AP, Austin JW, Boss JM. Genetic and epigenetic regulation of PD-1 expression. J Immunol (2016) 196(6):2431–7. doi: 10.4049/jimmunol.1502643
109. Gastaldelli A, Gaggini M, DeFronzo RA. Role of adipose tissue insulin resistance in the natural history of type 2 diabetes: Results from the San Antonio metabolism study. Diabetes (2017) 66(4):815–22. doi: 10.2337/db16-1167
110. Corvera S, Solivan-Rivera J, Yang Loureiro Z. Angiogenesis in adipose tissue and obesity. Angiogenesis (2022) 25(4):439–53. doi: 10.1007/s10456-022-09848-3
111. Chang CH, Curtis JD, Maggi LB Jr., Faubert B, Villarino AV, O'Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell (2013) 153(6):1239–51. doi: 10.1016/j.cell.2013.05.016
112. Lim AR, Rathmell WK, Rathmell JC. The tumor microenvironment as a metabolic barrier to effector T cells and immunotherapy. Elife (2020) 9:e55185. doi: 10.7554/eLife.55185
113. Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med (2015) 372(21):2018–28. doi: 10.1056/NEJMoa1501824
114. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med (2015) 373(1):23–34. doi: 10.1056/NEJMoa1504030
115. Liu J, Chen Z, Li Y, Zhao W, Wu J, Zhang Z. PD-1/PD-L1 checkpoint inhibitors in tumor immunotherapy. Front Pharmacol (2021) 12:731798. doi: 10.3389/fphar.2021.731798
116. Cortellini A, Bersanelli M, Buti S, Cannita K, Santini D, Perrone F, et al. A multicenter study of body mass index in cancer patients treated with anti-PD-1/PD-L1 immune checkpoint inhibitors: when overweight becomes favorable. J Immunother Cancer. (2019) 7(1):57. doi: 10.1186/s40425-019-0527-y
117. McQuade JL, Daniel CR, Hess KR, Mak C, Wang DY, Rai RR, et al. Association of body-mass index and outcomes in patients with metastatic melanoma treated with targeted therapy, immunotherapy, or chemotherapy: a retrospective, multicohort analysis. Lancet Oncol (2018) 19(3):310–22. doi: 10.1016/S1470-2045(18)30078-0
118. Xu X, Hou B, Fulzele A, Masubuchi T, Zhao Y, Wu Z, et al. PD-1 and BTLA regulate T cell signaling differentially and only partially through SHP1 and SHP2. J Cell Biol (2020) 219(6):e201905085. doi: 10.1083/jcb.201905085
119. Dixit VD. Thymic fatness and approaches to enhance thymopoietic fitness in aging. Curr Opin Immunol (2010) 22(4):521–8. doi: 10.1016/j.coi.2010.06.010
120. Sempowski GD, Hale LP, Sundy JS, Massey JM, Koup RA, Douek DC, et al. Leukemia inhibitory factor, oncostatin m, IL-6, and stem cell factor mRNA expression in human thymus increases with age and is associated with thymic atrophy. J Immunol (2000) 164(4):2180–7. doi: 10.4049/jimmunol.164.4.2180
121. van der Weerd K, Dik WA, Schrijver B, Schweitzer DH, Langerak AW, Drexhage HA, et al. Morbidly obese human subjects have increased peripheral blood CD4+ T cells with skewing toward a treg- and Th2-dominated phenotype. Diabetes (2012) 61(2):401–8. doi: 10.2337/db11-1065
122. Grant RW, Dixit VD. Adipose tissue as an immunological organ. Obes (Silver Spring). (2015) 23(3):512–8. doi: 10.1002/oby.21003
123. Minato N, Hattori M, Hamazaki Y. Physiology and pathology of T-cell aging. Int Immunol (2020) 32(4):223–31. doi: 10.1093/intimm/dxaa006
124. Shimatani K, Nakashima Y, Hattori M, Hamazaki Y, Minato N. PD-1+ memory phenotype CD4+ T cells expressing C/EBPalpha underlie T cell immunodepression in senescence and leukemia. Proc Natl Acad Sci U S A. (2009) 106(37):15807–12. doi: 10.1073/pnas.0908805106
125. Di Micco R, Krizhanovsky V, Baker D, d'Adda di Fagagna F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol (2021) 22(2):75–95. doi: 10.1038/s41580-020-00314-w
126. Igarashi H, Sakaguchi N. Telomerase activity is induced by the stimulation to antigen receptor in human peripheral lymphocytes. Biochem Biophys Res Commun (1996) 219(2):649–55. doi: 10.1006/bbrc.1996.0288
127. Mundstock E, Sarria EE, Zatti H, Mattos Louzada F, Kich Grun L, Herbert Jones M, et al. Effect of obesity on telomere length: Systematic review and meta-analysis. Obes (Silver Spring). (2015) 23(11):2165–74. doi: 10.1002/oby.21183
128. Wang X, Zhu H, Snieder H, Su S, Munn D, Harshfield G, et al. Obesity related methylation changes in DNA of peripheral blood leukocytes. BMC Med (2010) 8:87. doi: 10.1186/1741-7015-8-87
129. Gordin D, Forsblom C, Panduru NM, Thomas MC, Bjerre M, Soro-Paavonen A, et al. Osteopontin is a strong predictor of incipient diabetic nephropathy, cardiovascular disease, and all-cause mortality in patients with type 1 diabetes. Diabetes Care (2014) 37(9):2593–600. doi: 10.2337/dc14-0065
130. Bertola A, Deveaux V, Bonnafous S, Rousseau D, Anty R, Wakkach A, et al. Elevated expression of osteopontin may be related to adipose tissue macrophage accumulation and liver steatosis in morbid obesity. Diabetes (2009) 58(1):125–33. doi: 10.2337/db08-0400
131. Lund SA, Giachelli CM, Scatena M. The role of osteopontin in inflammatory processes. J Cell Commun Signal (2009) 3(3-4):311–22. doi: 10.1007/s12079-009-0068-0
132. Yoshida S, Nakagami H, Hayashi H, Ikeda Y, Sun J, Tenma A, et al. The CD153 vaccine is a senotherapeutic option for preventing the accumulation of senescent T cells in mice. Nat Commun (2020) 11(1):2482. doi: 10.1038/s41467-020-16347-w
133. Tahir S, Fukushima Y, Sakamoto K, Sato K, Fujita H, Inoue J, et al. A CD153+CD4+ T follicular cell population with cell-senescence features plays a crucial role in lupus pathogenesis via osteopontin production. J Immunol (2015) 194(12):5725–35. doi: 10.4049/jimmunol.1500319
134. Meng D, Zhang B, Wang Y, Zheng T, Hu R, Wang B, et al. p38α deficiency in T cells ameliorates diet-induced obesity, insulin resistance, and adipose tissue senescence. Diabetes (2022) 71(6):1205–17. doi: 10.2337/db21-0653
135. Neeland IJ, Marso SP, Ayers CR, Lewis B, Oslica R, Francis W, et al. Effects of liraglutide on visceral and ectopic fat in adults with overweight and obesity at high cardiovascular risk: a randomised, double-blind, placebo-controlled, clinical trial. Lancet Diabetes Endocrinol (2021) 9(9):595–605. doi: 10.1016/S2213-8587(21)00179-0
136. Jastreboff AM, Aronne LJ, Ahmad NN, Wharton S, Connery L, Alves B, et al. Tirzepatide once weekly for the treatment of obesity. N Engl J Med (2022) 387(3):205–16. doi: 10.1056/NEJMoa2206038
137. Harrington M, Gibson S, Cottrell RC. A review and meta-analysis of the effect of weight loss on all-cause mortality risk. Nutr Res Rev (2009) 22(1):93–108. doi: 10.1017/S0954422409990035
138. Romeo S, Maglio C, Burza MA, Pirazzi C, Sjöholm K, Jacobson P, et al. Cardiovascular events after bariatric surgery in obese subjects with type 2 diabetes. Diabetes Care (2012) 35(12):2613–7. doi: 10.2337/dc12-0193
139. Wing RR, Bolin P, Brancati FL, Bray GA, Clark JM, Coday M, et al. Cardiovascular effects of intensive lifestyle intervention in type 2 diabetes. N Engl J Med (2013) 369(2):145–54. doi: 10.1056/NEJMoa1212914
Keywords: CD4 T cells, obesity, adipose tissue, osteopontin, immunosenescence
Citation: Shirakawa K and Sano M (2023) Drastic transformation of visceral adipose tissue and peripheral CD4 T cells in obesity. Front. Immunol. 13:1044737. doi: 10.3389/fimmu.2022.1044737
Received: 15 September 2022; Accepted: 12 December 2022;
Published: 04 January 2023.
Edited by:
Yuekang Xu, Anhui Normal University, ChinaReviewed by:
Yusuke Endo, Kazusa DNA Research Institute, JapanCopyright © 2023 Shirakawa and Sano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Motoaki Sano, bXNhbm9AYTgua2Vpby5qcA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.