 Shengwei Tao
Shengwei Tao Shuhang Liang
Shuhang Liang Taofei Zeng
Taofei Zeng Dalong Yin
Dalong Yin
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 04 January 2023
Sec. Cancer Immunity and Immunotherapy
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1043667
This article is part of the Research Topic Epigenetic Regulation in Cancer Immunity View all 12 articles
Hepatocellular carcinoma (HCC) constitutes most primary liver cancers and is one of the most lethal and life-threatening malignancies globally. Unfortunately, a substantial proportion of HCC patients are identified at an advanced stage that is unavailable for curative surgery. Thus, palliative therapies represented by multi-tyrosine kinase inhibitors (TKIs) sorafenib remained the front-line treatment over the past decades. Recently, the application of immune checkpoint inhibitors (ICIs), especially targeting the PD-1/PD-L1/CTLA-4 axis, has achieved an inspiring clinical breakthrough for treating unresectable solid tumors. However, many HCC patients with poor responses lead to limited benefits in clinical applications, which has quickly drawn researchers’ attention to the regulatory mechanisms of immune checkpoints in HCC immune evasion. Evasion of immune surveillance by cancer is attributed to intricate reprogramming modulation in the tumor microenvironment. Currently, more and more studies have found that epigenetic modifications, such as chromatin structure remodeling, DNA methylation, histone post-translational modifications, and non-coding RNA levels, may contribute significantly to remodeling the tumor microenvironment to avoid immune clearance, affecting the efficacy of immunotherapy for HCC. This review summarizes the rapidly emerging progress of epigenetic-related changes during HCC resistance to ICIs and discusses the mechanisms of underlying epigenetic therapies available for surmounting immune resistance. Finally, we summarize the clinical advances in combining epigenetic therapies with immunotherapy, aiming to promote the formation of immune combination therapy strategies.
Primary liver cancer is the sixth most commonly diagnosed malignancy and the third leading reason for cancer mortality worldwide, accompanied by an extremely high number of new and fatal cases (1).
Hepatocellular carcinoma (HCC) constitutes 75%-85% of primary liver cancer as the commonest subtype. Potential Curative treatments, comprising surgical resection, liver transplantation, and local ablation, are amenable to early-stage HCC patients, which account for less than 20% of all HCC cases (2–4). However, most HCC patients are identified in the unresectable stage given its rapid progression, high malignancy, and inconspicuous early symptoms, resulting in palliative or symptomatic treatment, such as transarterial chemoembolisation (TACE) and systemic therapies (4, 5). Therefore, the identification of appropriate systemic therapy for advanced HCC has been an area of intense interest.
In recent years, with the advancement of precision cancer management, therapeutic strategies for HCC have emerged, including precision surgical resection, targeted molecular therapy based on different subtypes, and immunotherapy. Herein, the current landscape of systemic therapy for HCC treatment strategies is shown in Figure 1A. The application of multityrosine kinase inhibitor (TKIs) sorafenib as first-line therapy represented a milestone in systematic therapy (6). Despite the SHARPE trial and the ORIENTAL trial demonstrating improved median overall survival (OS) with sorafenib of 2.8 (10.7 vs 7.9)/2.3 (6.5 vs 4.2) months compared with placebo, respectively, anticancer efficacy remained suboptimal with a median survival of less than one year (6, 7). Notably, the therapeutic landscape has vigorously evolved after 2016 despite sorafenib remaining the pillar option for HCC patients during the past decade (5). Lenvatinib was approved for first-line treatment of advanced HCC due to its proven superior progression-free survival (PFS), and overall response rate (ORR) compared to sorafenib (8). In addition, regorafenib and cabozantinib were applied as second-line treatment since it was proven to prolong survival in progressed patients after suffering sorafenib treatment (9, 10). The biomarker-driven REACH-2 trial confirmed the efficacy of ramucirumab, especially in cases with baseline AFP >400ng/ml (11). Despite the promising outlook shown by these phase III studies, the OS benefits remain unsatisfactory.
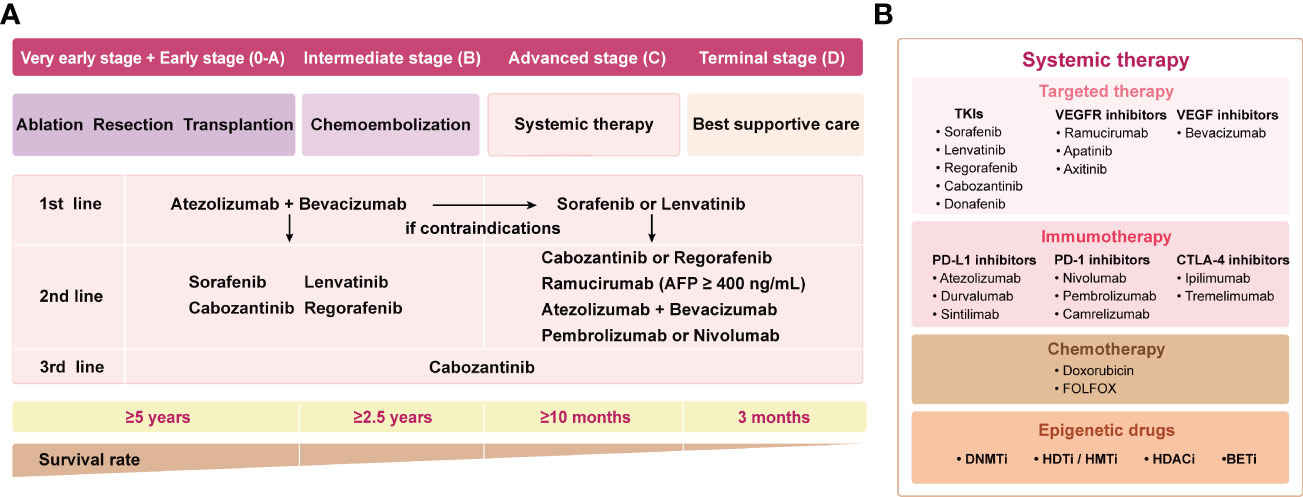
Figure 1 The landscape of systemic therapy for HCC treatment strategies. (A) The guidelines are sorted out and summarized by 2018 ASCO, 2018 AASLD, and 2020 EASL. The clinical stages of the patients are defined according to BCLC. (B) Main drugs for systemic therapy. TKI, Multi-tyrosine kinase inhibitor; VEGFR, vascular endothelial growth factor receptor; VEGF, vascular endothelial growth factor; ASCO, American Society of Clinical Oncology; AASLD, American Association for the Study of Liver Diseases; EASL, European Association for the Study of the Liver; BCLC, Barcelona Clinic Liver Cancer.
During past decades, the application of T-cell immune checkpoint inhibitors (ICIs) such as cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) and programmed cell death protein 1 (PD-1)/programmed cell death-ligand 1 (PD-L1) monoclonal antibodies have completely opened the door to tumor immunotherapy (12–16). Currently, immunotherapy has achieved compelling efficacy in treating several solid and hematologic tumors, and promising clinical breakthroughs in the immune-based treatment of HCC are emerging (17–22). However, it is undeniable that a considerable number of patients experienced primary resistance to ICIs, despite ICIs offering prognostic improvement in certain groups (23). In addition, even patients who initially benefit may ultimately develop resistance, which means only a minority of patients have a long-lasting response to these treatments (19, 20, 24). Many hypotheses have been suggested to be responsible for primary nonresponse or acquired resistance to ICIs, focusing on the tumor-intrinsic factors or the tumor microenvironment (TME), such as lack of immunogenic epitopes, immunosuppressive cell populations, inflammatory phenotypes, and T-cell exhaustion (25–29). Therefore, discovering and identifying the causes of tumor immune escape and screening the best beneficiary population guided by predictive markers may lead to further breakthroughs in managing advanced HCC.
Nowadays, accumulating evidence has found that epigenetic modifications may act essentially in remodeling TME to avoid immune surveillance, affecting the efficacy of immunotherapy. This review mainly concentrates on the epigenetic changes in tumor cells, and the impact of TME reprogramming can be consulted in other reviews (30, 31). The history and advances in immunotherapy for HCC were introduced first, followed by a discussion of the mechanisms of HCC resistance to immunotherapy. Then, we summarize the rapidly emerging progress of epigenetic-related changes during HCC resistance to ICIs and discuss underlying epigenetic therapies available for surmounting immune resistance. Finally, this review summarizes the clinical advances in combining epigenetic therapies with immunotherapy, aiming to promote the formation of immune combination therapy strategies.
Systemic therapy based on TKIs and chemotherapy constituted the dominant treatment for advanced HCC before 2017. Notably, immunotherapies have been regarded as a breakthrough that has changed the treatment landscape for advanced HCC since 2017. Among immunotherapies, the success of ICIs therapy targeting PD-1, PD- L1, and CTLA-4 in solid and hematological tumors has shifted more attention to its potential in advanced HCC (Table 1 and Figure 2).
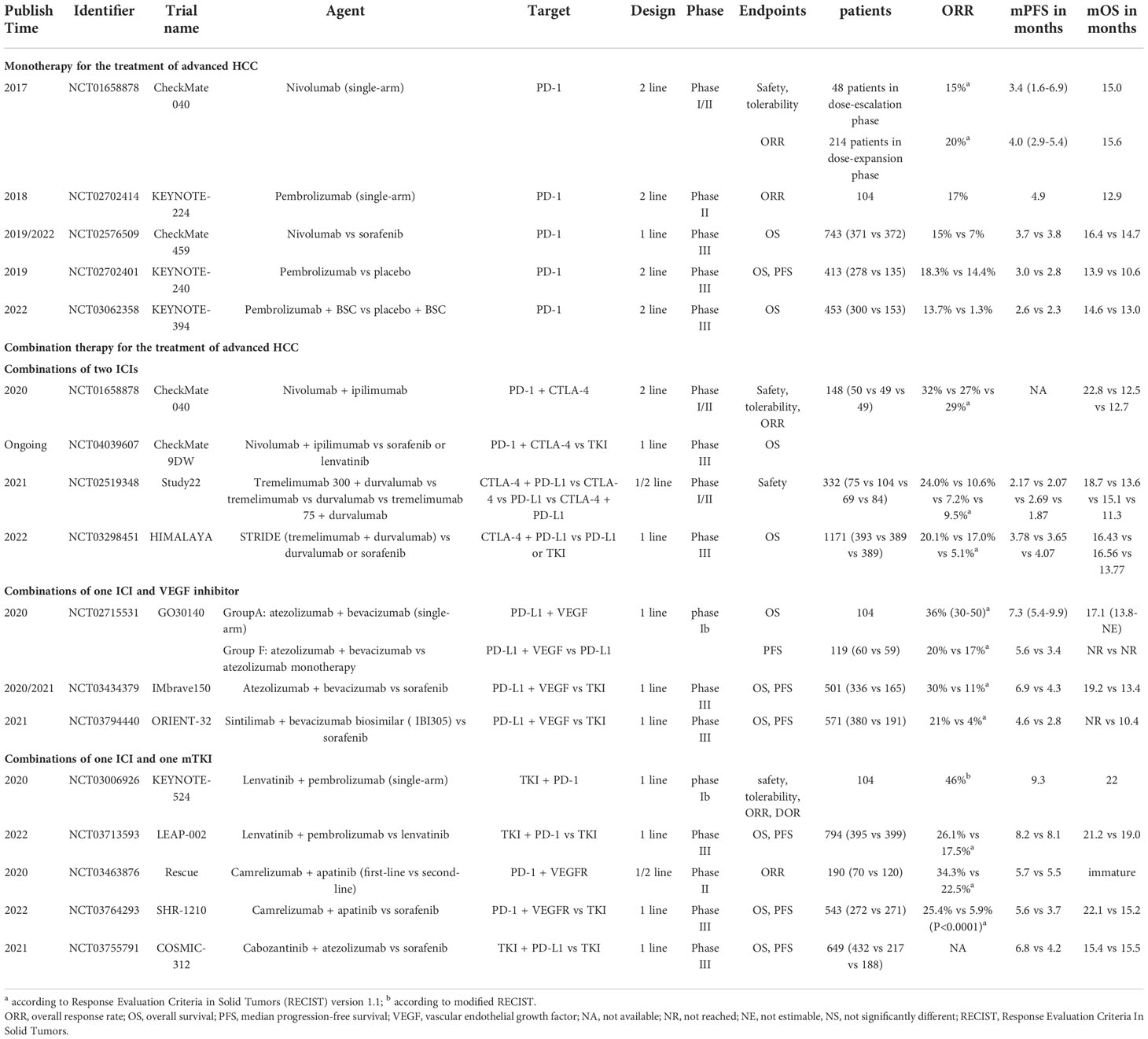
Table 1 Representative clinical trials for the systemic treatment of HCC.
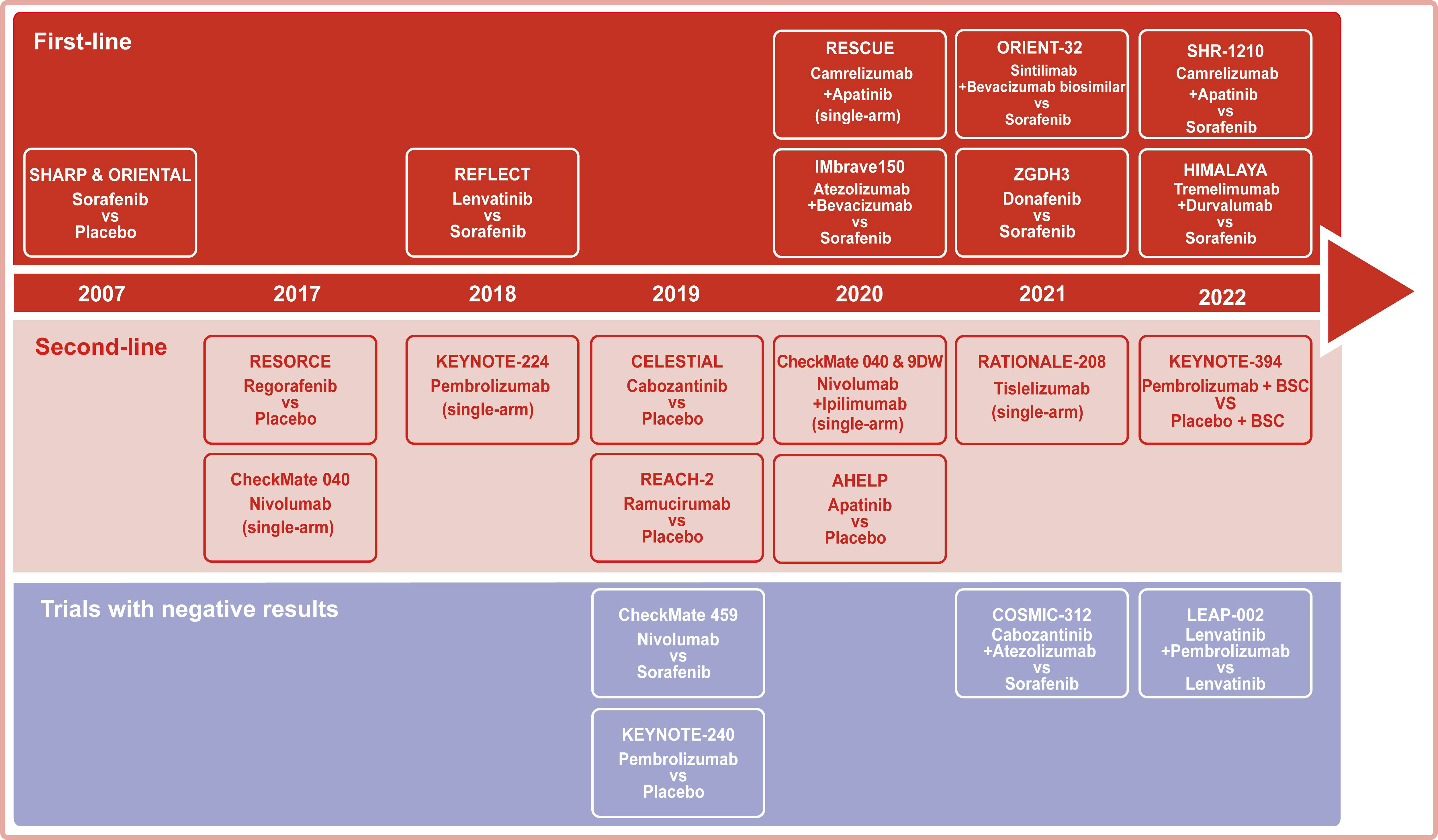
Figure 2 Representative clinical trials for the systemic treatment of HCC. Background color: red for trials with first-line settings, pink for trials with second-line settings, and purple for trials with negative results.
Monotherapy employing antibodies targeting PD-1, PD-L, and CTLA-4 for the treatment of HCC, as outlined in Figure 1B. Well-known CheckMate 040 trial (NCT01658878) and KEYNOTE-224 trial (NCT02702414) were the first phase II ICIs trials to have proven promising results, confirming the significant efficacy of nivolumab and pembrolizumab in patients with advanced HCC (19, 20). CheckMate 040 was an open-label, non-comparative, phase I/II dose escalation and expansion trial, enrolling 262 patients with or without sorafenib exposure, demonstrating an ORR of 15% and 20% for nivolumab in the dose-expansion phase and dose-escalation phase, respectively (19). The median OS was 15.6 months in patients treated with nivolumab as second-line therapy, whereas the median OS was 28.6 months with nivolumab in the first-line setting without prior sorafenib therapy. Similarly, another phase II trial KEYNOTE-224, which enrolled 104 patients to evaluate the potential of pembrolizumab as second-line therapy, demonstrated an ORR of 17%, and a median OS of 12.9 months (20). Subsequently, the exciting results of these two trials led to accelerated approvals by the United States Food and Drug Administration (FDA) for nivolumab and pembrolizumab monotherapy as second-line therapy after sorafenib failure for progressive HCC.
Based on positive results from the uncontrolled single-arm trials, some randomized phase III trials were performed to validate the efficiency of monotherapy. CheckMate 459 (NCT02576509) is an open-label, randomized phase III trial that evaluated the efficacy of nivolumab and sorafenib as first-line treatment for advanced HCC in 743 patients (32, 33). The results showed that while patients treated with nivolumab showed a survival benefit of 1.7 months more than those treated with sorafenib, the trial did not meet the statistical significance of its predefined OS endpoint (median 16.4 months versus 14.7 months, HR=0.85; P=0.075). Furthermore, another randomized phase III trial KEYNOTE-240 (NCT02702401) compared the efficiency of pembrolizumab versus placebo as second-line therapy for 413 patients who progressed on previous sorafenib (34). Although the results showed significantly higher median OS and ORR in the pembrolizumab group than in the placebo group, the predefined OS and PFS statistical threshold were still not reached. In spite of demonstrated antitumor activity for nivolumab in the first-line setting and pembrolizumab in the second-line setting, the results of two phase III trials failed, which can be attributed to treatment with new drugs or ICIs after disease progression in the control group. Rewardingly, a recent phase III trial KEYNOTE-394 (NCT03062358) enrolled 453 Asian patients who failed in first-line treatment with sorafenib or oxaliplatin, confirming the remarkable benefits of second-line therapy with pembrolizumab in OS, PFS, and ORR (35). To date, other ICI monotherapies are currently being explored, and detailed results are presented in Table 1.
The dilemma of ICIs monotherapy suggests that combining with other drugs to potentiate the efficacy of ICIs may be a promising direction. Several combination strategies are exhibiting outstanding results, including the combined application of two ICIs, one ICI plus vascular endothelial growth factor (VEGF) inhibitor, and one ICI plus one TKI. (Table 1).
The combination of nivolumab with ipilimumab was first evaluated by the CheckMate 040 trial, validating an ORR of 32% and a 2-year OS rate of 48% in child-Pugh class A patients previously treated with sorafenib (36). Positive data from this strategy led to accelerated FDA approval in 2020 for second-line treatment, and arm A was selected in an ongoing phase III study CheckMate 9DW (NCT04039607) for comparison with sorafenib or lenvatinib in first-line treatment.
Another regimen, termed STRIDE (Single Tremelimumab Regular Interval Durvalumab), has been shown to have clinical activity and manageable safety in phase I/II trial Study 22 (NCT02519348) (37). More recently, results from this regimen’s Phase III clinical trial HIMALAYA (NCT03298451) were also announced, demonstrating significantly improved ORR and three-year OS rates compared to sorafenib (38). In addition, durvalumab monotherapy was shown to be non-inferior to sorafenib in the first-line treatment. Hence, the STRIDE regimen was approved by FDA in October 2022 for the first-line treatment of unresectable HCC patients.
The combination of atezolizumab and bevacizumab was explored in a phase Ib trial GO30140 (NCT02715531) that demonstrated an ORR of 36% and a median PFS of 7. 3 months (39). Given such results, a global, open-label, phase III randomized trial IMbrave150 (NCT03434379) was performed to validate the safety and efficiency of this strategy (40, 41). According to the updated study, median OS was prolonged by 5.8 months with atezolizumab plus bevacizumab compared to sorafenib, establishing its superior first-line status in the treatment of advanced HCC over sorafenib (41). This pioneering advance demonstrated the mechanistic synergy and efficacy of the combination of anti-vascular therapy and immunotherapy, inspiring subsequent clinical trials with ICIs in combination with TKIs or anti-VEGF monoclonal antibodies. For instance, a randomized, open-label, phase II/III trial ORIENT-32 (NCT03794440) enrolled 571 Chinese patients with unresectable HBV-associated HCC (42). The results showed that sintilimab plus bevacizumab biosimilar (IBI305) showed significant OS and PFS benefits in first-line therapy with a manageable safety profile compared to sorafenib.
A preclinical study revealed that the combination of lenvatinib and PD-1 inhibitors had a synergistic effect and increased efficacy (43). Therefore, a phase Ib clinical trial KEYNOTE-524 (NCT03006926) was performed to verify the safety and efficacy of lenvatinib plus pembrolizumab in patients with unresectable HCC (44). Based on the favorable safety and efficacy demonstrated by this regimen, a phase III trial LEAP-002 (NCT03713593) was subsequently conducted (45). Unexpectedly, although lenvatinib plus pembrolizumab was found to have elevated benefits in OS and PFS, it did not reach the predefined statistically significant difference. Nevertheless, the subgroup analysis of OS suggested that patients with portal vein invasion/extrahepatic metastases, HBV-related HCC, and APF >400ng/mL all benefited more from lenvatinib in combination with pembrolizumab. The regimen of Camrelizumab plus Apatinib has also been shown in Rescue (NCT03463876) and SHR-1210 (NCT03764293) to have positive efficacy and a manageable safety profile in first-line setting (46, 47). Another promising regimen, cabozantinib in combination with atezolizumab, was assessed in advanced renal cell carcinoma (48). However, the phase III trial COSMIC-312 (NCT0375579) of cabozantinib plus atezolizumab for the first-line setting of advanced HCC showed that the combination therapy significantly improved median PFS compared with sorafenib, but failed to improve median OS (49).
In the past few years, despite the failure of some clinical trials, several studies have shown exciting survival benefits, shifting to first-line treatment options (Figure 2). Meanwhile, the combination regimens also showed hidden dangers in tolerability and safety that need attention. Honestly, the efficiency of immunotherapy for HCC remains inadequate compared to other tumors with favorable responses. In fact, only a small percentage of patients with advanced HCC derive significant benefits from ICIs treatment. To address this need, efforts are required to focus on identifying molecular biomarkers that predict response to TKIs or ICIs and exploring novel drug mechanisms in combination with ICIs.
The hallmark of developing cancer is a dynamic immunoediting process that interacts with the immune system. This hypothesis explains the ability of immune cells to eliminate the tumor (immune surveillance) while shaping the tumor immunogenicity to produce an environment that contributes to tumor growth and progression (immune tolerance) (Figures 3, 4). In the battle between the immune system and the tumor, there are three phases in sequence: immune elimination (early cancerous cells are recognized and eliminated by the immune system), immune equilibrium (sporadic tumor cells that survive immune elimination are not visible and cannot grow excessively), and immune escape (tumors gradually grow with distinct clinical features and establish an immunosuppressive microenvironment to evade killing by the immune system) (50, 51). For immunotherapy to be successful, three essential criteria need to be met: firstly, tumor antigen-specific T cell responses need to be activated; secondly, T cells need to infiltrate the TME; and finally, activated T cells need to trigger tumor cell killing mechanisms. Taken together, ICIs treatment failure can be caused by a defect in the above steps.
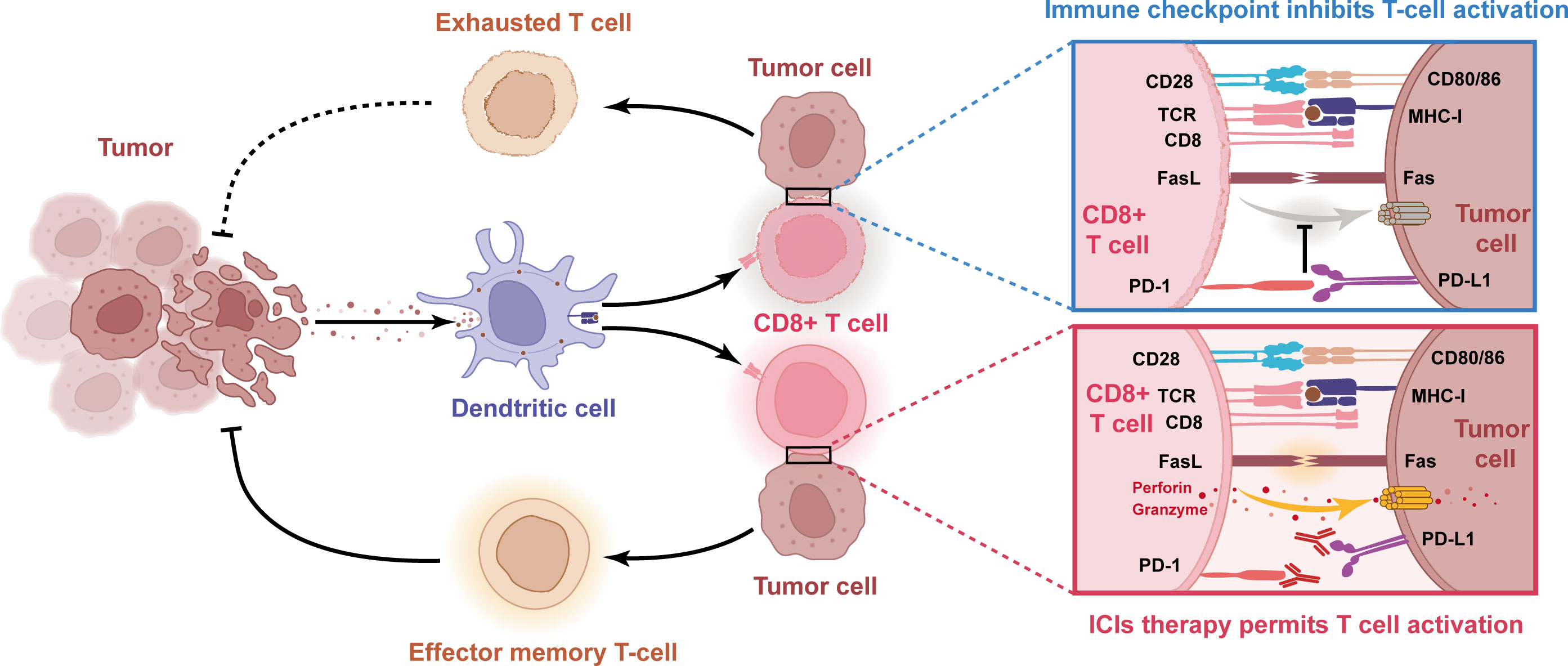
Figure 3 Schematic diagram of the molecular mechanism of impaired anti-tumor immunity caused by immune checkpoints and reactivation of T cells with PD-1/PD-L1 blocking antibody. When tumor cell PD-L1 binds to T cell PD-1, this interaction leads to T cell dysfunction and lack of anti-tumor activity. Thus, blocking the interaction between PD-1 and PD-L1 with anti-PD-1 or PD-L1 antibodies can reactivate T cells and release their anti-tumor activity.
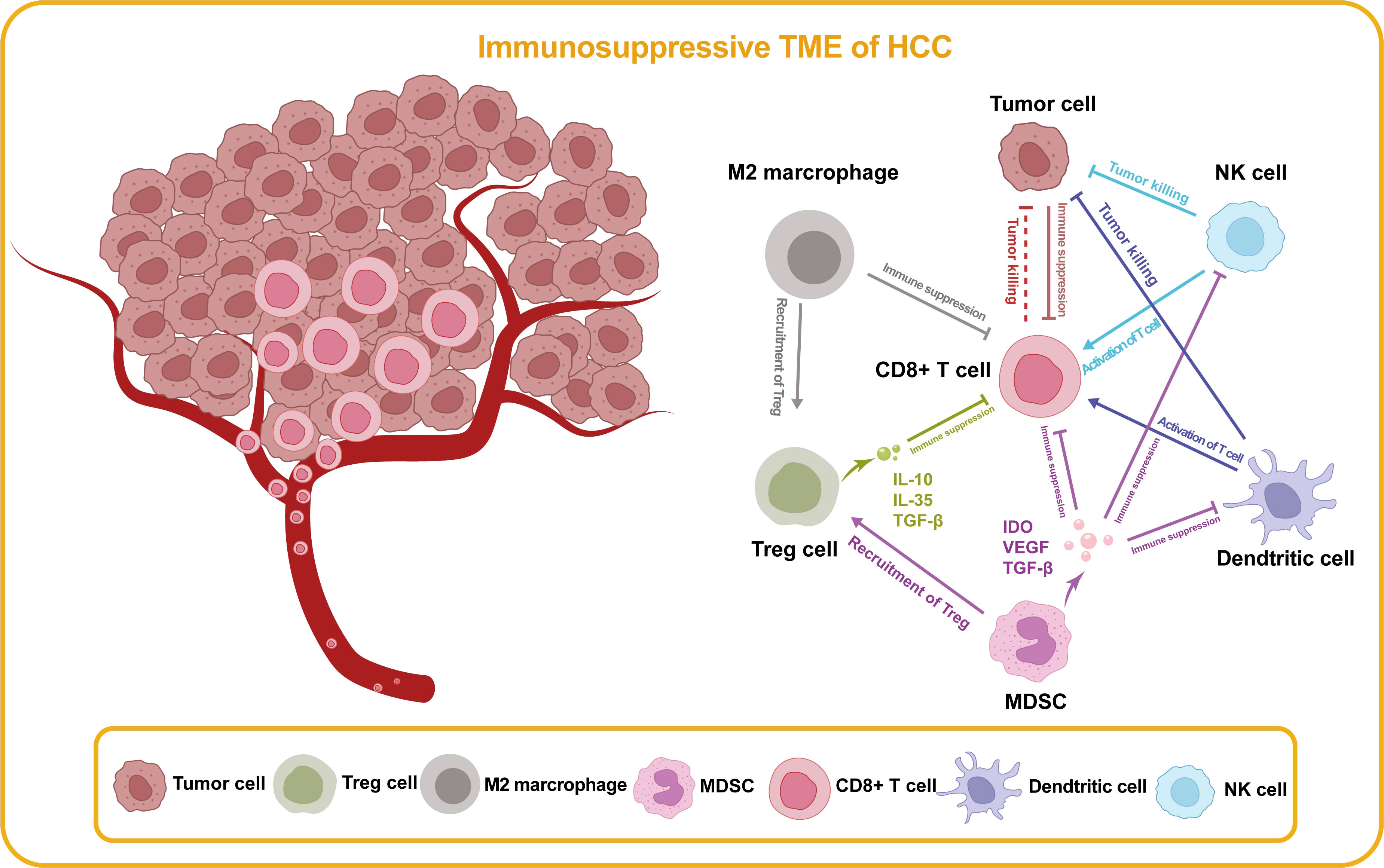
Figure 4 Immunosuppressive TME of HCC. In the TME of HCC, there are cell types that promote anti-tumor immunity and cell types that impede effective immune surveillance, which are illustrated in this figure. Treg cell, regulatory T cell; MDSC, myeloid-derived suppressor cell; NK, natural killer; IDO, indoleamine 2,3-dioxygenase; VEFG, vascular endothelial growth factor; TGF-β, transforming growth factor-β.
It is growingly recognized that epigenetic modifications occurring in tumor cells and immune cells within the TME represent the essential factors of cell growth, immune evasion, and drug resistance. Critical factors of primary response resistance to immune checkpoint blockade have been increasingly recognized, including tumor-intrinsic factors (tumor neoantigen burden and activation of oncogenic signaling pathways), TME (low tumor-infiltrating lymphocytes, exhausted CD8+ T cells, immunosuppressive cells and epigenetic silencing of chemokine), host immune components and microbiomes. Epigenetic modification-induced silencing of gene expression and loss of mutation-associated antigens, which impairs immunogenicity and immune recognition, have been implicated as one of the mechanisms of acquired drug resistance. For example, hypermethylation regulated by DNMTs and histone deacetylation regulated by HDACs contribute to the loss of function of tumor suppressor genes or immune presenting genes (e.g., MHC class-I expression), leading to tumor antigen presentation dysfunction and immune evasion (52). Moreover, activating mutations in CTNNB1 were correlated with a low response to ICIs monotherapy in advanced HCC patients (53). Despite multiple factors that might explain the therapeutic outcomes of ICIs, acquired resistance to HCC may be primarily attributable to the reprogramming of TME, which impedes the infiltration of lymphocytes (54–57).
Epigenetics is a mechanism that changes biological phenotypes without involving DNA sequence changes, and such changes can be passed on to offspring. Specifically, epigenetic modifications include chromatin structure remodeling, DNA methylation, histone post-translational modifications, and non-coding RNA levels (Figure 5).
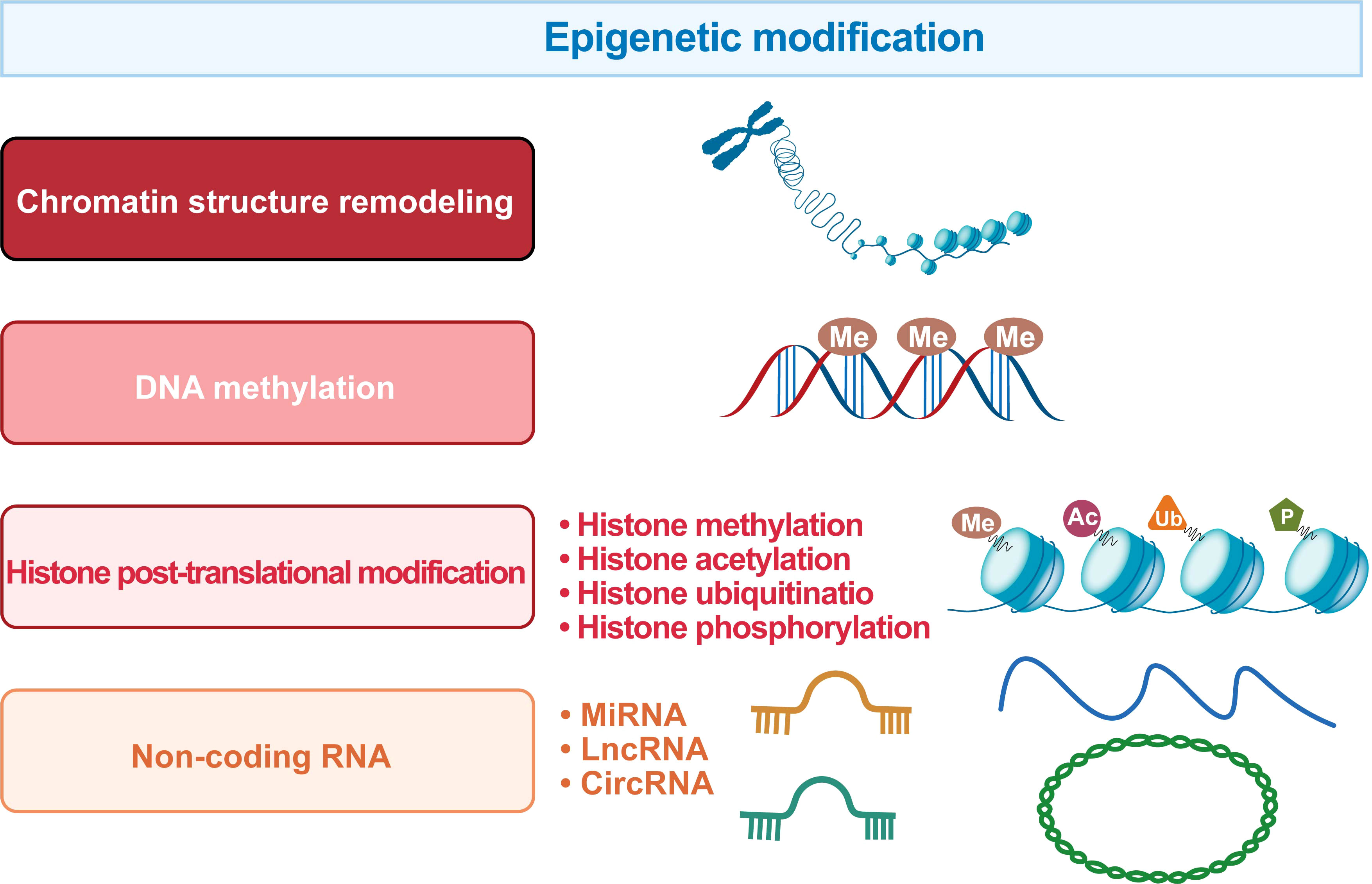
Figure 5 The regulatory systems involved in the epigenetic landscape of HCC. The epigenetic marks in HCC include chromatin structure remodeling, DNA methylation, histone post-translational modification and non-coding RNA.
Chromatin is a dynamic structure composed of DNA and histones, consisting of H2A, H2B, H3, H4, and DNA (58, 59). Chromatin conformation is essentially controlled by DNA modifications and histones subjected to post-translational modifications, leading to altered transcriptional activity (60, 61). The mating-type switch/sucrose non-fermenting (SWI/SNF) complex regulates chromatin accessibility in an adenosine triphosphate (ATP)-dependent nucleosome remodeling manner, which is the most intensively studied chromatin remodeling complex. Hence, its gene-encoding region is frequently mutated in tumors (62). The SWI/SNF complex is a macromolecular complex of 12-15 subunits, including a catalytic ATPase subunit, SWI/SNF related, matrix associated, actin-dependent regulator of chromatin, subfamily a, member 4 (SMARCA4), and several subunits, such as AT-rich interaction domain 1A (ARID1A) and ARID1B or polybromo 1 (PBRM1) and ARID2 (63). SWI/SNF subunit inactivating mutations, such as ARID1A, ARID1B, ARID2, PBRM1, and SMARCA4, are frequently detected in HCC (62).
ARID1A encodes one subunit of the SWI/SNF complex and regulates many processes requiring DNA access (such as transcription, DNA damage repair, and replication) by remodeling chromatin structure. ARID1A mutation was found to be associated with larger HCC and highly or moderately differentiated HCC (64). Moreover, He et al. found that ARID1A downregulation was associated with metastasis and poor prognosis in HCC, possibly due to the downregulation of E-cadherin (65). In addition, ARID1A mutations have also been reported to cause angiogenesis by upregulating angiopoietin-2 (Ang2) via H3K27ac modification (66). An in vivo experiment revealed that ARID1A knockdown could lead to mouse hepatocarcinogenesis, accompanied by macrophage and neutrophil infiltration and activation of STAT3 and NF-κB pathways (67). Notably, mutations in the SWI/SNF complex have been found to be involved in resistance to ICIs. ARID1A-deficient tumor-bearing mice exhibited increased mutational load as well as better lymphocytic tumor infiltration accompanied by increased PD- L1 expression in ovarian cancer. And ARID1A-deficient mice combined with anti-PD-L1 treatment were significantly more effective than mice with ARID1A wild-type ovarian tumors (68). Furthermore, inhibition of PBRM1, another SWI/SNF complex, was found to enhance immunotherapeutic response by increasing tumor immunogenicity (69). These results suggest that the aberrant SWI/SNF complex may engage in the TME of HCC and inhibition of the SWI/SNF complex may offer a combined effect with ICIs. More in-depth studies are needed to elucidate the mechanism of mutated SWI/SNF complexes shaping the TME.
DNA methylation is achieved by adding a methyl group to cytosines at CpG sequences in gene-promoter regions by DNA methyl transferases (DNMT), resulting in gene silencing. The overall depletion of methylation at repetitive element regions that preserve genomic stability while exhibiting hypermethylation at promoter regions of tumor-suppressor genes is prevalent in cancer (70–73). In HCC, significantly elevated levels of DNA hypermethylation were observed in the promoters of genes associated with TP53, cAMP, serine proteases, and NADH regulation compared to normal tissues (74). Analysis of the cohort of human HCC versus normal tissues revealed the signature of tumor suppressor gene hypermethylation as detected by methylation-specific PCR (75). Moreover, genes regulated by methylation sites in non-tumor tissues of hepatitis cirrhosis potentially drive tumorigenesis and recurrence and carry significant prognostic value (76–78). During the progression of cirrhosis to advanced HCC, ascending DNA methylation can distinguish the normal liver from the diseased tissue (74, 79–81).
The mechanisms contributing to hepatocarcinogenesis and drug resistance due to dysregulated DNA methylation may be diversified. For example, the expression of DNMT and TET is altered during hepatocarcinogenesis (82, 83). DNMT3 is overexpressed in HCC and correlates with hypermethylation of promoters controlling 22 oncogenes (83, 84). High Dnmt3b expression in HCC is regulated by the IL-6/STAT3 signaling pathway, contributing to resistance to sorafenib and poor prognosis (85). DNA methylation in HCC may also alter genes involved in immune surveillance (86–89). In sorafenib-resistant HCC cells, overexpression of DNIMT1 is accompanied by PD-L1 expression, causing poor prognosis (90). PD-L1knockdown or drug interference reverses sorafenib resistance in HCC by restoring the expression of CDH1, an intercellular adhesion molecule that inhibits HCC metastasis, which is silenced by DNMT1 methylation (90). Subsequently, it was shown that the DNMT inhibitor 5-azacytidine combined with anti-PD-L1 therapy led to tumor regression accompanied by increased cytotoxic T-lymphocyte infiltration in mouse models compared to monotherapy (91). This mechanism is synergistic with immunotherapy by inducing upregulation of the T helper 1 (Th1)-type chemokines CXCL9 and CXCL10, causing effective T cell traffic to the TME. Furthermore, SGI-110 (Guadecitabine), a second-generation DNMT1 inhibitor, exhibited anti-carcinogenic and anti-angiogenic activity in a xenograft HepG2 model (75, 92). These studies suggest that inhibition of epigenetic modifiers may collaborate with ICIs by strengthening immunogenicity, remodeling effective T cell function, and modifying the immunosuppressive TME, making more exploration needed.
Histone modifications, including histone methylation, acetylation, ubiquitination, sumoylation, and phosphorylation, have been intensively studied in different cancers and are considered crucial factors for disease progression and immunotherapy resistance (93–96). The histone tails are modified to reversibly modulate chromatin compaction to promote or constrain accessibility to genes and further activate or silence gene transcription processes (97). Dysregulation of epigenetic modifiers of histones is recognized to exert an essential influence on hepatocarcinogenesis and immune escape (Figure 6).
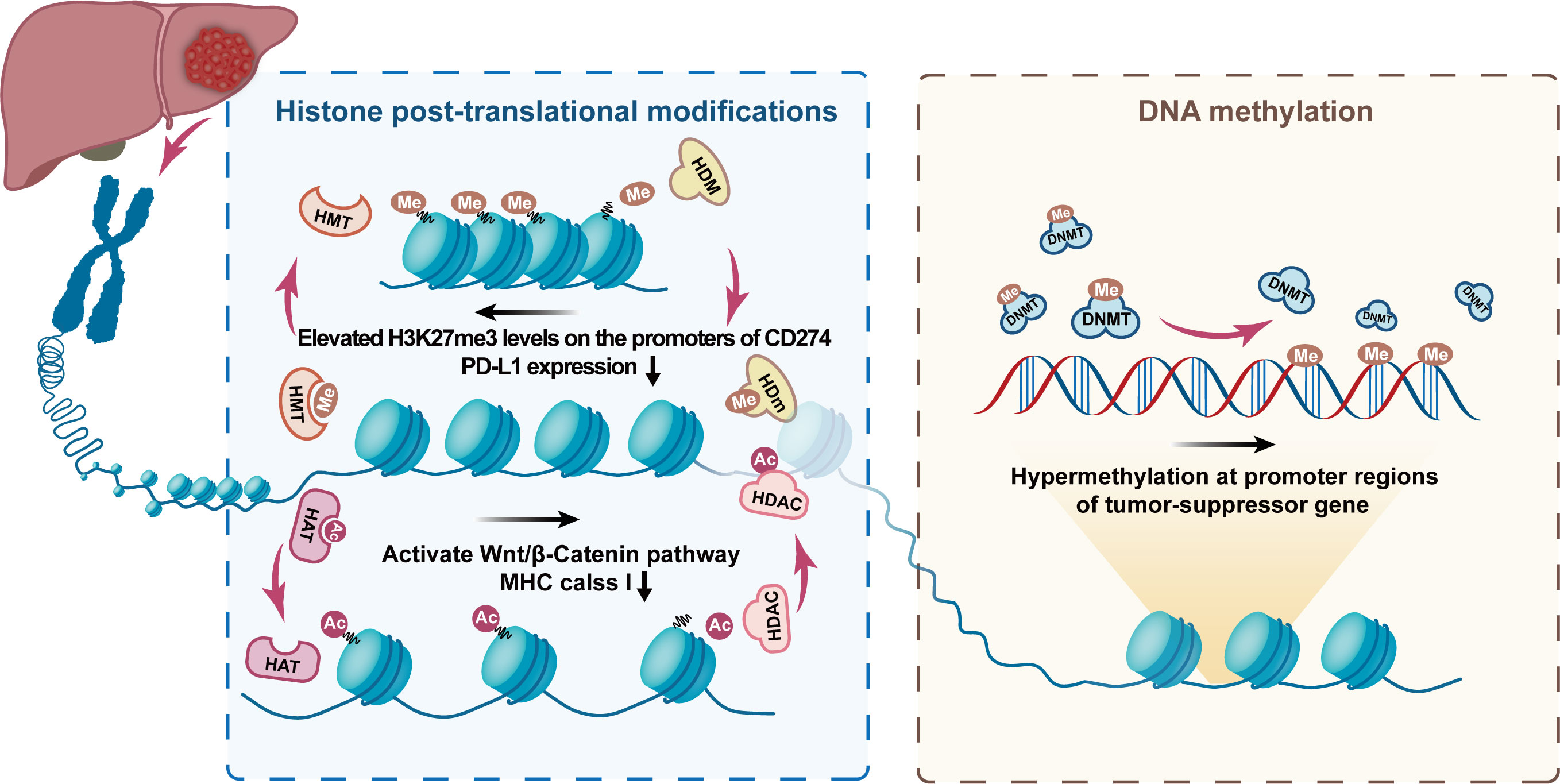
Figure 6 Typical histone post-translational modifications and DNA methylation mechanism. The figure shows the role of epigenetic regulation of chromatin by DNA methylation and histone post-translational modifications in the occurrence and development of HCC. The figure highlights the role of histone demethylase (HDM), histone methyltransferase (HMT), histone acetyltransferases (HAT), histone deacetylase (HDAC), and DNA methyltransferase (DNMT) in the formation of epigenetic characteristics of HCC. Ac, acetylation; Me, Methylation. The figure shows that EZH2 overexpression leads to elevated H3K27me3 levels on the promoters of CD274 and interferon regulatory factor 1 (IRF1), impeding PD-L1 expression. In, addition, the alteration caused by HDAC8 overexpression activates the Wnt/β-Catenin pathway, which in turn impairs antitumor immunity of antigen-specific T cells resulting in ICI resistance.
The methylation of lysines on histones is mainly modulated by histone methyltransferases (HMTs) and histone demethylase (HDMs). In contrast to transcriptional activation due to histone acetylation, the effects of histone methylation are dictated by the location and number of residue methylation. For instance, the monomethylation of lysine 9 and 27 of histone H3 (H3K9me1 and H3K27me1) is correlated with an active chromatin state. In contrast, the trimethylations on lysine 9 and 27 of histone H3 (H3K9me3 and H3K27me3) are associated with a repressive chromatin (98).
Available evidence suggests that aberrant alterations in histone-modifying enzymes and anomalous histone modifications of genes involved in angiogenesis, cell cycle regulation, and cell adhesion are common in the progression of hepatocarcinogenesis. For example, H3K9me2 and H3K9me3 were overexpressed in HCC and correlated with a bad prognosis (99). Similarly, another study has also emphasized the critical role that high levels of H3K9me3 and its HMT SUV39H1 perform in the development and recurrence of HCC (100). The identical effect was also substantiated in other HMTs, such as SETDB1, G9a, and EHMT2 (101–105). Among the HMTs family, enhancer of zeste homolog 1 and 2 (EZH2), a catalytic subunit of the polycomb repressive complex 2 (PRC2), has recently captured much discussions. EZH2 is the crucial molecule of PRC2 and catalyses the trimethylation of H3 (H3K27me3) lysine residues to mediate the silencing of target genes (106, 107). A high association has been established between hyper-expressed EZH2 and HCC, indicating that EZH2 is tightly related to an aggressive phenotype and unfavorable prognosis (108, 109). Moreover, both EZH2 knockdown and drug blockade reduced the level of H3K27me3, resulting in reduced tumorigenesis (110).
The benefits of EZH2 inhibitors in combination with targeted therapy have begun to emerge. Kusakabe et al. observed overexpression of EZH2 and H3K27me3 in sorafenib-resistant cells, while the combination of sorafenib with an EZH2 inhibitor reversed sorafenib resistance resulting in a synergistic antitumor effect (111). Recently, it has been proposed that EZH2 can influence lymphocyte subpopulation differentiation and function to reshape the TME, and thus inhibition of EZH2 may be a novel strategy to improve anti-tumor immunity in certain cancers (112–114). Two H3K4me3-specific HMT, MLL1, and WDR5 have been proven to block immune escape and improve anti-PD-1 immunotherapy in pancreatic cancer (115, 116). Xiao et al. found that EZH2 potentiated H3K27me3 levels on the promoters of CD274 and interferon regulatory factor 1 (IRF1) to impede PD-L1 expression, suggesting that EZH2 inhibition combined with ICIs may provide accessible benefit (117). A study by Bugide et al. came to the identical conclusion, demonstrating that genetic or pharmacological inhibition of EZH2 induces re-expression of chemokine CXCL10 in HCC and therefore promotes migration and infiltration of NK cells into the tumor (118). The scope for clinical application of EZH2 inhibitors is being actively explored (119). Furthermore, given the outstanding inhibition of tumor growth demonstrated by EZH2 inhibitors in combination with anti-PD-1 therapy, Tazemetostat has been approved as the most widely studied EZH2 inhibitor as a first-line treatment option for epithelioid sarcoma (112, 114).
The acetylation of lysines on histones is mainly controlled by histone acetyltransferases (HATs) and histone deacetylases (HDACs), which are responsible for mediating chromatin structural sparing and transcriptional activation of genes (120). Numerous studies have established a robust correlation between HCC and the dysregulation of HDAC. For example, Ler et al. revealed that HDAC1 and HDAC2 expression was upregulated in most HCC tissues, and HDAC1 expression is related to the degree of malignancy (121, 122). HDAC3 has been reported to be involved in DNA damage and repair processes, adjusting the balance between DNA damage and protumorigenic transcription (123, 124). hMOF, the histone acetyltransferase responsible for H4K16 acetylation, was found to have a dual effect of inhibiting growth and promoting vascular invasion in HCC, the exact mechanism of which remains unclear (125, 126).
Based on the currently available evidence, many efforts have focused on the function of HDAC in TME. Several studies have suggested that, in addition to altering the intrinsic phenotype of tumor cells, epigenetic therapies exhibit the potential to reverse primary or acquired resistance to ICIs. For example, Aberrant epigenetic modification by HDAC8 overexpression was demonstrated to play a crucial role in resistance to ICI (127). The alteration caused by HDAC8 overexpression activates the Wnt/β-Catenin pathway, which in turn impairs antitumor immunity of antigen-specific T cells resulting in ICI resistance (127, 128). Moreover, HDAC10 was found to recruit EZH2 enabling modification of the CXCL10 promoter region H3K27me3, producing CXCL10 transcriptional repression and therefore inhibiting NK cell migration and infiltration towards HCC (129). Therefore, inhibitors against HDACs and HATS are expected to be new targets for HCC management and require validation of effects verified in preclinical and clinical trials.
Non-coding RNAs (ncRNAs) are not responsible for the translation into proteins and perform roles in regulating DNA methylation, histone modification, and gene silencing. Aberrations of ncRNAs have been reported extensively in HCC, and detailed information on these reports can be found in the following reviews (130–134). Given the immune-related focus of this review, representative ncRNAs have been selected for elaboration.
The expression of many microRNAs were closely associated with the degree of differentiation, and tumor metastasis of HCC, such as miR-497, miR-1246, and miR-378a-3p (135, 136). In addition, miR-378a-3p was also proved to directly regulate PD-L1 and STAT3 signaling to inhibit HCC, which may be a potential target in the future (137). Moreover, liver-derived exosome miR-92a-3p was identified as a potential biomarker for predicting HCC metastasis. Mechanistically, exosomal miR-92a-3p plays a key role in epithelial-mesenchymal transition (EMT) progression and promotion of metastasis through inhibition of PTEN and activation of the Akt / Snail signaling pathway (138). MiR-1 was found to be induced by NRF-2, promoting upregulation of PD-L1 expression and maintaining HCC resistance to sorafenib (139). MiR-200c inhibits PD-L1 expression by binding to the 3’ untranslated region (3’ UTR) of CD274 in HCC (140). Similarly, MIR-570 can also affect PD-L1 mRNA by binding its 3'UTR in HCC (141). The miR-144/451a cluster was also found to promote macrophage M1 polarisation and activity in a hepatocyte growth factor (HGF) and macrophage migration inhibitory factor (MIF)-dependent manner to inhibit HCC development (142). MiR-144/451a was also revealed to promote macrophage M1 polarisation in a specific cytokine-dependent manner to inhibit HCC development.
In terms of long non-coding RNAs (lncRNAs), KCNQ1 overlapping transcript 1 (KCNQ1OT1) was found to promote sorafenib resistance and PD-L1-mediated immune escape by sponging miR-506 and miR-146a-5p (143, 144). Cancer susceptibility candidate 11 (CASC11) was found to stabilize E2F transcription factor 1 (E2F1) mRNA by recruiting eukaryotic translation initiation factor 4A3 (EIF4A3), which in turn affects the activation of NF-KB signaling pathway and Pl3K/AKT/mTOR pathway, and further regulated PD-L1 expression (145). The above researches suggest that selected ncRNAs therapies combined with ICIs may be promising treatment candidates. It is encouraging that OTX-2002, the first mRNA therapeutic to downregulate MYC expression pre-transcriptionally through epigenetic regulation, has been approved for application in HCC patients. Currently, MYCHELANGELO I (NCT05497453) is evaluating the potential of OTX-2002 as a single agent or in combination with TKIs or ICIs.
Unlike genetic mutations, epigenetic alterations are amenable to pharmacological interventions due to their flexible and variable interactions, rendering them a promising target for reversing ICIs resistance (146). Actually, epigenetic drugs not only exert a direct effect on tumors but also have the potential to remodel the suppressed TME and synergistically improve ICI efficacy (147–149). Currently, there is a wealth of research demonstrating the effectiveness of epigenetic modifiers drugs in many cancer models and pre-clinical applications (150, 151). In addition, some of these drugs have been intensively investigated in treating HCC. Therefore, it is reasonable to expect that coupling epigenetic drugs with ICIs may offer a desirable prospect for ICIs-resistant patients (Figure 7). Epigenetic modifications within TME were elaborated on in a previous review, and this review focuses on the epigenetic alterations occurring in HCC (30, 152). The current exploration of epigenetic drugs combined with ICIs in the clinic is summarized in Table 2.
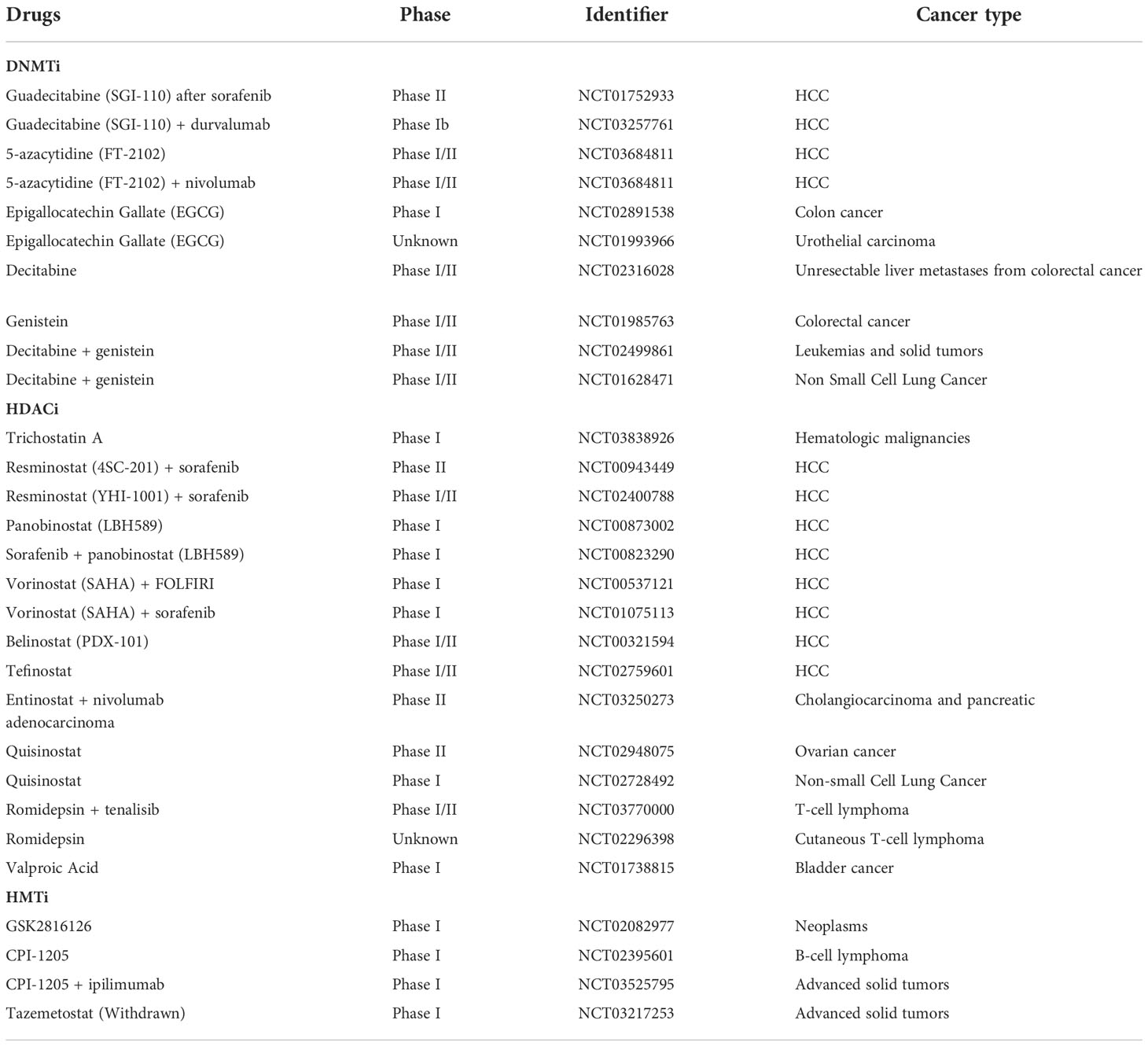
Table 2 Epigenetics drugs in clinical trial.
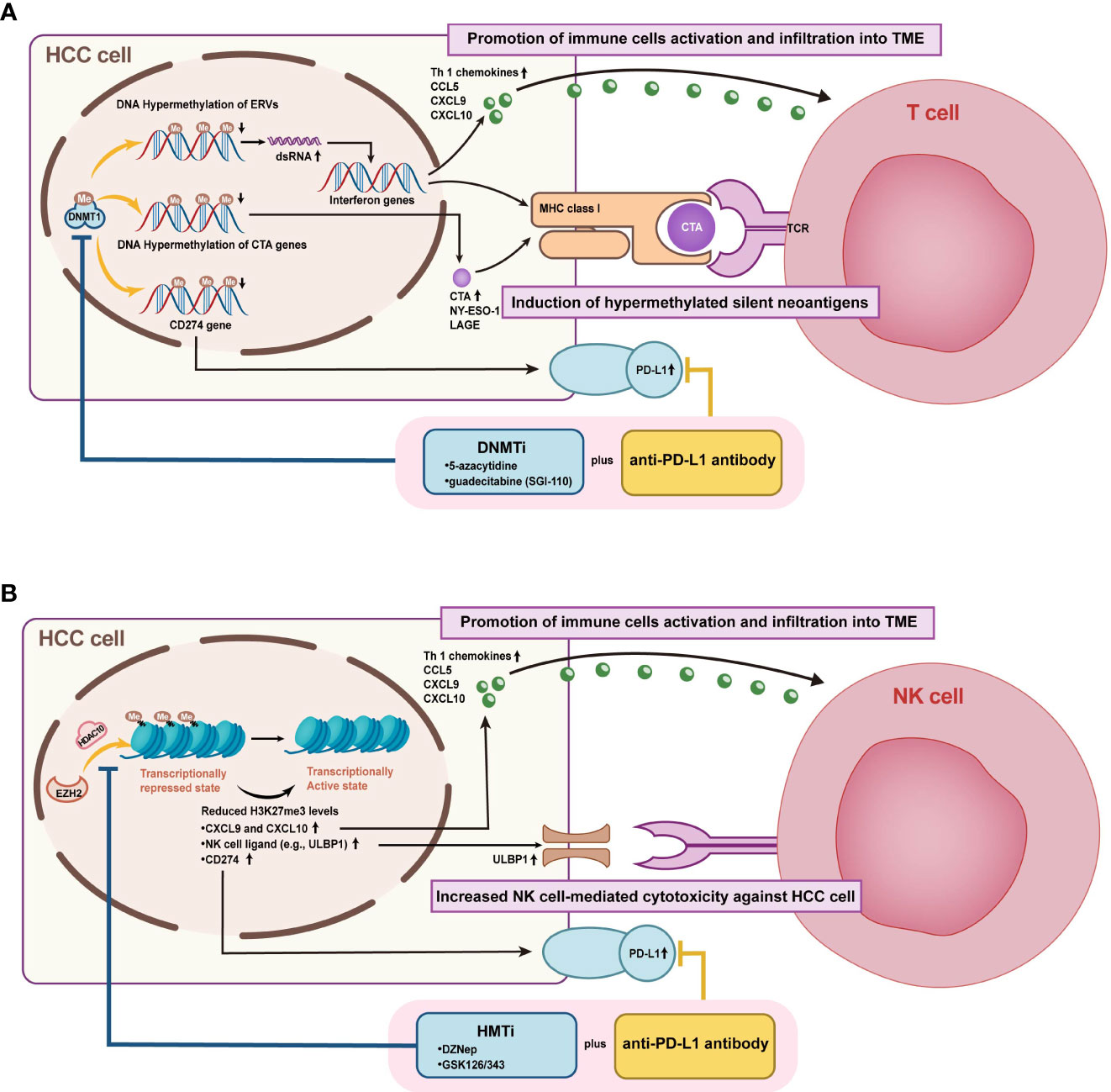
Figure 7 Mechanisms of combined epigenetic and immunotherapeutic strategies for HCC treatment. (A) Figure A illustrates the enhanced efficacy of DNMTi combined with ICIs by promoting immune cell activation and infiltration into TME and inducing hypermethylated silenced neoantigen expression. (B) Figure B demonstrates that HMTi combined with ICIs promotes immune cell activation and infiltration to TME and enhances NK cell-mediated HCC killing by upregulating the expression of chemokines, PD- L1, and NK cell ligand, which are inhibited by high histone methylation.
DNMTi was shown to enable the demethylation of cancer-testis genes and repetitive sequences, leading to improved host immune surveillance by increasing immunogenicity (153). Currently, the inhibitors of DNMT include 5-azacytidine, decitabine, zebularine (ZEB), and guadecitabine (SGI-110). Some studies have also highlighted the association between DNA methylation and immune checkpoints or lymphocytic infiltration. In mouse models, the DNMT inhibitor 5-azacytidine reactivated the expression of TH1-type chemokines (CXCL9 and CXCL10), increased infiltration of cytotoxic T-lymphocyte infiltration, and caused tumor regression in combination with anti-PD-L1 (91). Besides resensitizing sorafenib-resistant cells, decitabine also brought PFS and OS benefits clinically (90, 154). SGI-110, a novel DNMTi, has already been demonstrated in a preclinical study to inhibit HCC growth and enhance the antitumor effects of sorafenib (75). Moreover, Liu et al. proposed that SGI-110 has the potential to combine immune checkpoint therapy by reactivating endogenous retroviral elements (ERVs) to stimulate immune response pathways (92). More clinical trials combining SGI-110 and immunotherapy are ongoing (NCT01752933, NCT03257761).
However, DNMTi does not seem to perform a favorable function necessarily. A previous study has reported that brain-expressed X-linked protein 1 (BEX1) methylation mediated by DNMT1 inhibited HCC stemness and tumorigenicity, while DNMT1i ZEB promoted self-renewal and invasiveness in the high cancer stem cell (CSC) score HCC group (155). They found that the promoter region of the BEX1 gene was highly methylated, and the activation of BEX expression by ZEB treatment aberrantly triggered the Wnt/β-catenin signaling pathway, causing the proliferation of tumor hepatocytes. Moreover, BEX overexpression was also found to lead to resistance to sorafenib, and knockdown treatment restored its sensitivity. Considering the differential effects of DNMTI, its exploration in different patient subtypes should be emphasized in subsequent studies to ensure a better survival benefit.
The potential role of HMT and HDM inhibitors in managing HCC is also being actively pursued. For example, EZH2 negatively regulates PD-L1 in an IFN γ-dependent manner and might provide a promising target for combination immunotherapy (117). While previous studies demonstrate that EZH2 inhibitor GSK126 enhances transcription of NK cell ligands to promote natural killer cell-mediated HCC cell death, additional studies ought to be conducted to validate the benefit of EZH2 inhibitors in conjunction with ICIs (118). Furthermore, upregulation of another HMT, G9a, was defined to be significantly associated with HCC progression and aggressive clinicopathological features. Thus, inhibition of G9a may lead to novel approaches (104). Interestingly, the combination therapy of DNMTi and HMTi/HDTi also yielded therapeutic benefits. As an example, improved antitumor effects of ICIs combining 5-aza and 3-deazaneplanocin A were verified in a subcutaneous transplanted hepatoma cell model (91). CM-272, a dual inhibitor of G9a and DNMT1, exhibits anticancer efficacy in vitro and in vivo by restoring the differentiation phenotype of HCC cells (101). While CM-272 was proven to potentiate ICIs therapy in liver fibrosis and bladder cancer, the propensity in HCC remains to be established both at a fundamental and clinical phase (156, 157).
HDACi is clinically approved for treating hematologic malignancies that warrant continuous evaluation of its value in HCC. HDAC inhibitors such as TSA, panobinostat, valproic acid, and ITF2357 can inhibit HCC cells and may provide a combined effect with immunotherapy (158–161). TSA has been shown to improve the anticancer effects in combination with sorafenib (162). The pan-deacetylase inhibitor panobinostat demonstrated inhibitory effects by affecting the expression of angiogenic and epithelial-mesenchymal transition markers in the HCC model (163, 164). In addition, low expression of another HDAC, SIRT7, is proposed to induce global H3K18 acetylation and reactivate key metabolic and immune regulators, affecting tumorigenicity in vitro and in vivo (165). And it has been demonstrated that SIRT7 blockade stimulates PD-L1 expression, which provides a foundation for combining SIRT7 inhibitors with ICIs (166). Moreover, HDACi combined with DNMTi could provide stronger anti-proliferative effects compared to single agents in a xenograft HCC model (167).
Accumulating evidence has demonstrated that HDAC inhibition reprograms the TME to convert cold tumors into hot ones. HDAC inhibitor Belinostat was previously tested for its potential to treat advanced unresectable liver cancer in a multicenter phase I/II study (168). Later, a preclinical study demonstrated that belinostat has immune-mediated antitumor effects and may enhance the effectiveness of immunotherapy. In a subcutaneous Hepa129 mouse HCC model, Belinostat was observed to enhance anti-CTLA-4 antitumor activity by promoting early infiltration of M1 macrophages and suppressing regulatory T cells (169). Yang et al. observed that the inhibition of HDAC8 relieved T-cell hypo-infiltration by reversing H3K27 hypoacetylation occurring in the metabolic and immunomodulatory factor genome and activating T-cell transport chemokine expression in HCC (128). In a syngeneic and orthotopic C57BL/6 mouse Hepa1-6 hepatoma model, selective HDAC8 inhibitor PCI -34051 combined with anti-PD-L1 therapy improved tumor-infiltrating CD8+ T cells, eliciting an effective and up to 15-month tumor-free response to ICB. Comparably, HDAC2 inhibitors were also shown to block the transcription of immune checkpoint genes mediated by nuclear translocation of PD-L1, causing increased infiltration of CD8+ cytotoxic T cells in HCC (170). Inhibition of HDAC6 also specifically triggers TH17 cell activation and strengthens the anti-tumor immune response (171). Considering the prominent regulatory effects of HDAC in hepatocarcinogenesis and TME, it is conceivable that the combination of HDACi and immunotherapy will result in a better survival benefit.
Satisfactory results were also obtained for targeted epigenetic readers. The expression of acetylated H3 and H4 reader BRD4 is augmented in HCC, and the BRD4 inhibitor JQ-1 was proven to suppress HCC proliferation (172–174). Importantly, targeting BRD4 also showed enhanced efficacy of ICI in the experimental HCC model. BET bromodomain inhibitor molibresib combined with anti-PD-L1 shows enhanced anti-tumor effects by decreasing Monocytic-MDSC and increasing tumor-infiltrating lymphocytes (TILs) in fibrotic HCC mouse models (112).
Taken together, several therapeutic approaches targeting epigenetic mechanisms can modify tumor progression and response to treatment, supporting great promise in combining epigenetic strategies with ICIs. However, efforts must also be devoted to further understanding the complex epigenome and its regulation, discovering and exploiting novel epigenetic mechanisms, and assessing the effectiveness and safety of these approaches. The clinical application of epigenetic modulators is far from being realized, and subsequent implements for evaluating safer and more effective combined ICIs in larger populations warrant adequate attention. As the results of these trials are reported, distribution patterns based on biomarkers will likely provide the greatest benefit to patients.
Despite remarkable advances in etiological prevention, diagnostic techniques, and treatment strategies, the 5-year survival rate for all stages of HCC is still only 18%, prompting clinicians and scientists to scout for bio-predictive marker models for ICIs efficacy based on tumor and TME (118, 119). Different immunocompetent subtypes respond differently to ICIs treatment. Through in-depth characterization of the high-resolution HCC immune landscape, better prognostic enhancement strategies may be developed for specific immunocompetent subtypes. Therefore, identifying molecular biomarkers predicting response to TKIs or ICIs remains a valuable area of research to be actively explored.
Although previous studies have attributed positive immune responses to PD-L1 staining, tumor mutational burden (TMB), and microsatellite instability (MSI) in many tumors, it does not apply to HCC (175–178). A clinical study found no significant differences in OS and relapse-free survival (RFS) between PD-L1 high- and low-expressing subgroups in 2979 HCC patients. On the flip side, CheckMate017 and OAK also found that non-small-cell lung cancer patients with negative PD-L1 expression could benefit from immunotherapy (179, 180). Meanwhile, PD-L1 expression levels did not associate with immune response in HCC, according to the CheckMate040 and Keynote224 studies (19, 20, 181). ICIs response does not correlate with mutational burden in HCC, as revealed by next-generation sequencing (NGS) (53, 177). Analysis of RNA-seq data in the TCGA database also revealed consistent results (182). The incidence of MSI-high or mismatch repair defects (dMMR) in HCC is estimated to be low (~3%); patients with high microsatellite instability did not indicate a high response rate (183, 184). Nonetheless, two recent studies showed that higher intra-tumoral frequency of PD-1high CD8+ T cells and CD38+ CD68+ macrophages were correlated with better response to ICIs detected by flow cytometry and multiplex IHC (185, 186). Currently, the prognostic relationship between PD-L1 expression on tumor cells and TILs remains controversial (187, 188). Therefore, the importance of spatial heterogeneity of the HCC TME in the evaluation of ICIs efficacy markers deserves further evaluation.
Recently, a clinical study showed that Wnt/β-Catenin pathway mutations frequently occur in HCC patients resistant to ICIs therapy (189). Mechanically, activation of Wnt/β-Catenin leads to ICIs resistance by impairing antigen-specific T cell-mediated antitumor immunity, which is demonstrated by constructing tail vein injections of a transposon-based vector expressing MYC; p53−/− (190, 191). There is much evidence that cytokine and immune cell infiltration in TME may influence the outcome of ICIs. For instance, serum CD137 concentration and M1 macrophage infiltration were potential predictors for HCC patients treated with immune-combined anti-vascular therapy. Moreover, several studies have also found that TGF β attenuates the ICIs by limiting TIL within the tumor (192, 193). In addition, the etiology of HCC may also be responsible for the heterogeneity of patient response to immunotherapy. For example, the HBV-associated HCC microenvironment is more immunosuppressive and exhaustive than the non-viral-associated HCC. The high enrichment of PD-1high Tregs and PD-1+ CD8+ resident memory T cells in HBV-associated HCC implies an advantage of anti-PD-1therapy (194–196). Notably, a previous study found that the gut microbiome regulates chemokine-mediated immune cell accumulation via bile acids, affecting immune surveillance in HCC (197). Two recent clinical studies have demonstrated robust correlations between the gut microbiome and bile acids and the efficacy of ICIs therapy in HCC (198, 199).
Although several predictors have been identified, any single predictive biomarker has limitations and cannot effectively identify the beneficiary population. Using combined assays or building effective predictive models may improve predictive sensitivity and effectively capture the immune status of tumor patients. In the future, analyzing tumor and microenvironment characteristics through large samples and building multivariate models for immunotherapy efficacy prediction using machine learning and artificial intelligence will help develop a new paradigm for precision tumor therapy.
HCC is a malignancy that severely threatens human health, and most patients will progress to the advanced stage with a poor prognosis. Treatment for advanced HCC has been lacking effective means, and with the development of immunotherapy, HCC treatment is at the dawn of a new era. However, immune resistance and disappointingly low patient response rates are critical reasons plaguing efficacy improvement. As introduced by this review, epigenetic changes may be essential biomarkers for identifying immunotherapy responders, as well as promising targets for overcoming resistance to ICIs. The mechanisms that operate to drive drug resistance remain to be further elucidated.
Pretreatment of the microenvironment with epigenetic reagents before immunotherapy may help reprogram immune cells toward subtypes that are effective against cancer. The development of predictive biomarkers can help reveal the mechanism underlying ICIs resistance and the interaction mechanism within tumors and TME, which is crucial for individualized immunotherapy. Although our knowledge is still constrained, the current evidence indicates that epigenetic therapies exhibit sufficient potential. With the exploration of combination therapies (such as combining ICIs with TKIs or Epigenetic drugs) and immunotherapy guided by practical predictive markers to screen for optimal benefit populations, there may be further breakthroughs in managing advanced HCC.
ST and DY contributed to designing, drafting, revising, and approving the final version of the manuscript. SL revised and reviewed the manuscripts. TZ provided help in software support and figure drawing. All authors contributed to the article and approved the submitted version.
We would like to thank the Anhui Provincial Key Laboratory of Hepatopancreatobiliary Surgery for the help in writing the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2021) 71(3):209–49. doi: 10.3322/caac.21660
2. Davila JA, Duan Z, McGlynn KA, El-Serag HB. Utilization and outcomes of palliative therapy for hepatocellular carcinoma: A population-based study in the united states. J Clin Gastroenterol (2012) 46(1):71–7. doi: 10.1097/MCG.0b013e318224d669
3. Clark HP, Carson WF, Kavanagh PV, Ho CP, Shen P, Zagoria RJ. Staging and current treatment of hepatocellular carcinoma. Radiographics (2005) 25 Suppl 1:S3–23. doi: 10.1148/rg.25si055507
4. Liver EAftSot. Easl clinical practice guidelines: Management of hepatocellular carcinoma. J Hepatol (2018) 69(1):182–236. doi: 10.1016/j.jhep.2018.03.019
5. Yang JD, Hainaut P, Gores GJ, Amadou A, Plymoth A, Roberts LR. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol (2019) 16(10):589–604. doi: 10.1038/s41575-019-0186-y
6. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med (2008) 359(4):378–90. doi: 10.1056/NEJMoa0708857
7. Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S, Kim JS, et al. Efficacy and safety of sorafenib in patients in the Asia-pacific region with advanced hepatocellular carcinoma: A phase iii randomised, double-blind, placebo-controlled trial. Lancet Oncol (2009) 10(1):25–34. doi: 10.1016/s1470-2045(08)70285-7
8. Kudo M, Finn RS, Qin S, Han KH, Ikeda K, Piscaglia F, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet (2018) 391(10126):1163–73. doi: 10.1016/s0140-6736(18)30207-1
9. Bruix J, Qin S, Merle P, Granito A, Huang YH, Bodoky G, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (Resorce): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet (2017) 389(10064):56–66. doi: 10.1016/s0140-6736(16)32453-9
10. Abou-Alfa GK, Meyer T, Cheng AL, El-Khoueiry AB, Rimassa L, Ryoo BY, et al. Cabozantinib in patients with advanced and progressing hepatocellular carcinoma. N Engl J Med (2018) 379(1):54–63. doi: 10.1056/NEJMoa1717002
11. Zhu AX, Kang YK, Yen CJ, Finn RS, Galle PR, Llovet JM, et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased A-fetoprotein concentrations (Reach-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol (2019) 20(2):282–96. doi: 10.1016/s1470-2045(18)30937-9
12. Fehrenbacher L, Spira A, Ballinger M, Kowanetz M, Vansteenkiste J, Mazieres J, et al. Atezolizumab versus docetaxel for patients with previously treated non-Small-Cell lung cancer (Poplar): A multicentre, open-label, phase 2 randomised controlled trial. Lancet (2016) 387(10030):1837–46. doi: 10.1016/s0140-6736(16)00587-0
13. Herbst RS, Baas P, Kim DW, Felip E, Pérez-Gracia JL, Han JY, et al. Pembrolizumab versus docetaxel for previously treated, pd-L1-Positive, advanced non-Small-Cell lung cancer (Keynote-010): A randomised controlled trial. Lancet (2016) 387(10027):1540–50. doi: 10.1016/s0140-6736(15)01281-7
14. Sharon E, Streicher H, Goncalves P, Chen HX. Immune checkpoint inhibitors in clinical trials. Chin J Cancer (2014) 33(9):434–44. doi: 10.5732/cjc.014.10122
15. Carlino MS, Larkin J, Long GV. Immune checkpoint inhibitors in melanoma. Lancet (2021) 398(10304):1002–14. doi: 10.1016/s0140-6736(21)01206-x
16. Lipson EJ, Drake CG. Ipilimumab: An anti-Ctla-4 antibody for metastatic melanoma. Clin Cancer Res (2011) 17(22):6958–62. doi: 10.1158/1078-0432.Ccr-11-1595
17. Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat Rev Immunol (2020) 20(11):651–68. doi: 10.1038/s41577-020-0306-5
18. Qin S, Ren Z, Meng Z, Chen Z, Chai X, Xiong J, et al. Camrelizumab in patients with previously treated advanced hepatocellular carcinoma: A multicentre, open-label, parallel-group, randomised, phase 2 trial. Lancet Oncol (2020) 21(4):571–80. doi: 10.1016/s1470-2045(20)30011-5
19. El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, et al. Nivolumab in patients with advanced hepatocellular carcinoma (Checkmate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet (2017) 389(10088):2492–502. doi: 10.1016/s0140-6736(17)31046-2
20. Zhu AX, Finn RS, Edeline J, Cattan S, Ogasawara S, Palmer D, et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (Keynote-224): A non-randomised, open-label phase 2 trial. Lancet Oncol (2018) 19(7):940–52. doi: 10.1016/s1470-2045(18)30351-6
21. Finn RS, Ryoo B-Y, Merle P, Kudo M, Bouattour M, Lim H-Y, et al. Results of keynote-240: Phase 3 study of pembrolizumab (Pembro) vs best supportive care (Bsc) for second line therapy in advanced hepatocellular carcinoma (Hcc). J Clin Oncol (2019) 37(15):4004–4. doi: 10.1200/JCO.2019.37.15_suppl.4004
22. Sangro B, Park JW, Dela Cruz CM, Anderson J, Lang LX, Neely J, et al. A randomized, multicenter, phase 3 study of nivolumab vs sorafenib as first-line treatment in patients (Pts) with advanced hepatocellular carcinoma (Hcc): Checkmate-459. J Clin Oncol (2016) 34(15):2. doi: 10.1200/JCO.2016.34.15_suppl.TPS4147
23. Llovet JM, Kelley RK, Villanueva A, Singal AG, Pikarsky E, Roayaie S, et al. Hepatocellular carcinoma. Nat Rev Dis Primers (2021) 7(1):6. doi: 10.1038/s41572-020-00240-3
24. Vasan N, Baselga J, Hyman DM. A view on drug resistance in cancer. Nature (2019) 575(7782):299–309. doi: 10.1038/s41586-019-1730-1
25. Perrier A, Didelot A, Laurent-Puig P, Blons H, Garinet S. Epigenetic mechanisms of resistance to immune checkpoint inhibitors. Biomolecules (2020) 10(7):1061. doi: 10.3390/biom10071061
26. Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and transcriptomic features of response to anti-Pd-1 therapy in metastatic melanoma. Cell (2016) 165(1):35–44. doi: 10.1016/j.cell.2016.02.065
27. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic B-catenin signalling prevents anti-tumour immunity. Nature (2015) 523(7559):231–5. doi: 10.1038/nature14404
28. Chen PL, Roh W, Reuben A, Cooper ZA, Spencer CN, Prieto PA, et al. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discovery (2016) 6(8):827–37. doi: 10.1158/2159-8290.Cd-15-1545
29. Rieth J, Subramanian S. Mechanisms of intrinsic tumor resistance to immunotherapy. Int J Mol Sci (2018) 19(5):1391. doi: 10.3390/ijms19051340
30. Wang F, Malnassy G, Qiu W. The epigenetic regulation of microenvironment in hepatocellular carcinoma. Front Oncol (2021) 11:653037. doi: 10.3389/fonc.2021.653037
31. Maes K, Mondino A, Lasarte JJ, Agirre X, Vanderkerken K, Prosper F, et al. Epigenetic modifiers: Anti-neoplastic drugs with immunomodulating potential. Front Immunol (2021) 12:652160. doi: 10.3389/fimmu.2021.652160
32. Yau T, Park JW, Finn RS, Cheng AL, Mathurin P, Edeline J, et al. Nivolumab versus sorafenib in advanced hepatocellular carcinoma (Checkmate 459): A randomised, multicentre, open-label, phase 3 trial. Lancet Oncol (2022) 23(1):77–90. doi: 10.1016/s1470-2045(21)00604-5
33. Yau T, Park JW, Finn RS, Cheng A, Mathurin P, Edeline J, et al. Checkmate 459: A randomized, multi-center phase 3 study of nivolumab (Nivo) vs sorafenib (Sor) as first-line (1l) treatment in patients (Pts) with advanced hepatocellular carcinoma (Ahcc). Ann Oncol (2019) 30(suppl_5):v851–934. doi: 10.1093/annonc/mdz394.029
34. Finn RS, Ryoo BY, Merle P, Kudo M, Bouattour M, Lim HY, et al. Pembrolizumab as second-line therapy in patients with advanced hepatocellular carcinoma in keynote-240: A randomized, double-blind, phase iii trial. J Clin Oncol (2020) 38(3):193–202. doi: 10.1200/jco.19.01307
35. Qin S, Chen Z, Fang W, Ren Z, Xu R, Ryoo B-Y, et al. Pembrolizumab plus best supportive care versus placebo plus best supportive care as second-line therapy in patients in Asia with advanced hepatocellular carcinoma (Hcc): Phase 3 keynote-394 study. J Clin Oncol (2022) 40(4_suppl):383–. doi: 10.1200/JCO.2022.40.4_suppl.383
36. Yau T, Kang YK, Kim TY, El-Khoueiry AB, Santoro A, Sangro B, et al. Efficacy and safety of nivolumab plus ipilimumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib: The checkmate 040 randomized clinical trial. JAMA Oncol (2020) 6(11):e204564. doi: 10.1001/jamaoncol.2020.4564
37. Kelley RK, Sangro B, Harris W, Ikeda M, Okusaka T, Kang Y-K, et al. Safety, efficacy, and pharmacodynamics of tremelimumab plus durvalumab for patients with unresectable hepatocellular carcinoma: Randomized expansion of a phase I/Ii study. J Clin Oncol (2021) 39(27):2991–3001. doi: 10.1200/jco.20.03555
38. Abou-Alfa GK, Lau G, Kudo M, Chan SL, Kelley RK, Furuse J, et al. Tremelimumab plus durvalumab in unresectable hepatocellular carcinoma. NEJM Evidence (2022) 1(8):EVIDoa2100070. doi: 10.1056/EVIDoa2100070
39. Lee MS, Ryoo BY, Hsu CH, Numata K, Stein S, Verret W, et al. Atezolizumab with or without bevacizumab in unresectable hepatocellular carcinoma (Go30140): An open-label, multicentre, phase 1b study. Lancet Oncol (2020) 21(6):808–20. doi: 10.1016/s1470-2045(20)30156-x
40. Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med (2020) 382(20):1894–905. doi: 10.1056/NEJMoa1915745
41. Cheng AL, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, et al. Updated efficacy and safety data from Imbrave150: Atezolizumab plus bevacizumab vs. sorafenib for unresectable hepatocellular carcinoma. J Hepatol (2022) 76(4):862–73. doi: 10.1016/j.jhep.2021.11.030
42. Ren Z, Xu J, Bai Y, Xu A, Cang S, Du C, et al. Sintilimab plus a bevacizumab biosimilar (Ibi305) versus sorafenib in unresectable hepatocellular carcinoma (Orient-32): A randomised, open-label, phase 2-3 study. Lancet Oncol (2021) 22(7):977–90. doi: 10.1016/s1470-2045(21)00252-7
43. Kimura T, Kato Y, Ozawa Y, Kodama K, Ito J, Ichikawa K, et al. Immunomodulatory activity of lenvatinib contributes to antitumor activity in the Hepa1-6 hepatocellular carcinoma model. Cancer Sci (2018) 109(12):3993–4002. doi: 10.1111/cas.13806
44. Finn RS, Ikeda M, Zhu AX, Sung MW, Baron AD, Kudo M, et al. Phase ib study of lenvatinib plus pembrolizumab in patients with unresectable hepatocellular carcinoma. J Clin Oncol (2020) 38(26):2960–70. doi: 10.1200/jco.20.00808
45. Finn RS, Kudo M, Merle P, Meyer T, Qin S, Ikeda M, et al. Primary results from the phase iii leap-002 study: Lenvatinib plus pembrolizumab versus lenvatinib as first-line (1l) therapy for advanced hepatocellular carcinoma (Ahcc). Ann Oncol (2022) 33(suppl_7):S808–S69. doi: 10.1016/annonc/annonc1089
46. Xu J, Shen J, Gu S, Zhang Y, Wu L, Wu J, et al. Camrelizumab in combination with apatinib in patients with advanced hepatocellular carcinoma (Rescue): A nonrandomized, open-label, phase ii trial. Clin Cancer Res (2021) 27(4):1003–11. doi: 10.1158/1078-0432.Ccr-20-2571
47. Qin S, Chan LS, Gu S, Bai Y, Ren Z, Lin X, et al. Camrelizumab (C) plus rivoceranib (R) vs. sorafenib (S) as first-line therapy for unresectable hepatocellular carcinoma (Uhcc): A randomized, phase iii trial. Ann Oncol (2022) 33(suppl_7):S808–S69. doi: 10.1016/annonc/annonc1089
48. Pal SK, McGregor B, Suárez C, Tsao C-K, Kelly W, Vaishampayan U, et al. Cabozantinib in combination with atezolizumab for advanced renal cell carcinoma: Results from the cosmic-021 study. J Clin Oncol (2021) 39(33):3725–36. doi: 10.1200/jco.21.00939
49. Kelley RK, Rimassa L, Cheng AL, Kaseb A, Qin S, Zhu AX, et al. Cabozantinib plus atezolizumab versus sorafenib for advanced hepatocellular carcinoma (Cosmic-312): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol (2022) 23(8):995–1008. doi: 10.1016/s1470-2045(22)00326-6
50. O’Donnell JS, Teng MWL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol (2019) 16(3):151–67. doi: 10.1038/s41571-018-0142-8
51. Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol (2004) 22:329–60. doi: 10.1146/annurev.immunol.22.012703.104803
52. Nebbioso A, Tambaro FP, Dell’Aversana C, Altucci L. Cancer epigenetics: Moving forward. PloS Genet (2018) 14(6):e1007362. doi: 10.1371/journal.pgen.1007362
53. Harding JJ, Nandakumar S, Armenia J, Khalil DN, Albano M, Ly M, et al. Prospective genotyping of hepatocellular carcinoma: Clinical implications of next-generation sequencing for matching patients to targeted and immune therapies. Clin Cancer Res (2019) 25(7):2116–26. doi: 10.1158/1078-0432.Ccr-18-2293
54. Pitt JM, Vétizou M, Daillère R, Roberti MP, Yamazaki T, Routy B, et al. Resistance mechanisms to immune-checkpoint blockade in cancer: Tumor-intrinsic and -extrinsic factors. Immunity (2016) 44(6):1255–69. doi: 10.1016/j.immuni.2016.06.001
55. Syn NL, Teng MWL, Mok TSK, Soo RA. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol (2017) 18(12):e731–e41. doi: 10.1016/s1470-2045(17)30607-1
56. Prieto J, Melero I, Sangro B. Immunological landscape and immunotherapy of hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol (2015) 12(12):681–700. doi: 10.1038/nrgastro.2015.173
57. Zheng C, Zheng L, Yoo JK, Guo H, Zhang Y, Guo X, et al. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell (2017) 169(7):1342–56.e16. doi: 10.1016/j.cell.2017.05.035
58. Kornberg RD, Thomas JO. Chromatin structure; oligomers of the histones. Science (1974) 184(4139):865–8. doi: 10.1126/science.184.4139.865
59. Venkatesh S, Workman JL. Histone exchange, chromatin structure and the regulation of transcription. Nat Rev Mol Cell Biol (2015) 16(3):178–89. doi: 10.1038/nrm3941
60. Hauer MH, Gasser SM. Chromatin and nucleosome dynamics in DNA damage and repair. Genes Dev (2017) 31(22):2204–21. doi: 10.1101/gad.307702.117
61. Cutter AR, Hayes JJ. A brief review of nucleosome structure. FEBS Lett (2015) 589(20 Pt A):2914–22. doi: 10.1016/j.febslet.2015.05.016
62. Hu B, Lin JZ, Yang XB, Sang XT. The roles of mutated Swi/Snf complexes in the initiation and development of hepatocellular carcinoma and its regulatory effect on the immune system: A review. Cell Prolif (2020) 53(4):e12791. doi: 10.1111/cpr.12791
63. Centore RC, Sandoval GJ, Soares LMM, Kadoch C, Chan HM. Mammalian Swi/Snf chromatin remodeling complexes: Emerging mechanisms and therapeutic strategies. Trends Genet (2020) 36(12):936–50. doi: 10.1016/j.tig.2020.07.011
64. Abe H, Hayashi A, Kunita A, Sakamoto Y, Hasegawa K, Shibahara J, et al. Altered expression of at-rich interactive domain 1a in hepatocellular carcinoma. Int J Clin Exp Pathol (2015) 8(3):2763–70.
65. He F, Li J, Xu J, Zhang S, Xu Y, Zhao W, et al. Decreased expression of Arid1a associates with poor prognosis and promotes metastases of hepatocellular carcinoma. J Exp Clin Cancer Res (2015) 34(1):47. doi: 10.1186/s13046-015-0164-3
66. Hu C, Li W, Tian F, Jiang K, Liu X, Cen J, et al. Arid1a regulates response to anti-angiogenic therapy in advanced hepatocellular carcinoma. J Hepatol (2018) 68(3):465–75. doi: 10.1016/j.jhep.2017.10.028
67. Fang JZ, Li C, Liu XY, Hu TT, Fan ZS, Han ZG. Hepatocyte-specific Arid1a deficiency initiates mouse steatohepatitis and hepatocellular carcinoma. PloS One (2015) 10(11):e0143042. doi: 10.1371/journal.pone.0143042
68. Shen J, Ju Z, Zhao W, Wang L, Peng Y, Ge Z, et al. Arid1a deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat Med (2018) 24(5):556–62. doi: 10.1038/s41591-018-0012-z
69. Pan D, Kobayashi A, Jiang P, Ferrari de Andrade L, Tay RE, Luoma AM, et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science (2018) 359(6377):770–5. doi: 10.1126/science.aao1710
70. Baylin SB, Jones PA. Epigenetic determinants of cancer. Cold Spring Harb Perspect Biol (2016) 8(9):1–35. doi: 10.1101/cshperspect.a019505
71. Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature (1983) 301(5895):89–92. doi: 10.1038/301089a0
72. Easwaran H, Tsai HC, Baylin SB. Cancer epigenetics: Tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol Cell (2014) 54(5):716–27. doi: 10.1016/j.molcel.2014.05.015
73. Arechederra M, Bazai SK, Abdouni A, Sequera C, Mead TJ, Richelme S, et al. Adamtsl5 is an epigenetically activated gene underlying tumorigenesis and drug resistance in hepatocellular carcinoma. J Hepatol (2021) 74(4):893–906. doi: 10.1016/j.jhep.2020.11.008
74. Hernandez-Meza G, von Felden J, Gonzalez-Kozlova EE, Garcia-Lezana T, Peix J, Portela A, et al. DNA Methylation profiling of human hepatocarcinogenesis. Hepatology (2021) 74(1):183–99. doi: 10.1002/hep.31659
75. Jueliger S, Lyons J, Cannito S, Pata I, Pata P, Shkolnaya M, et al. Efficacy and epigenetic interactions of novel DNA hypomethylating agent guadecitabine (Sgi-110) in preclinical models of hepatocellular carcinoma. Epigenetics (2016) 11(10):709–20. doi: 10.1080/15592294.2016.1214781
76. Hlady RA, Zhao X, Pan X, Yang JD, Ahmed F, Antwi SO, et al. Genome-wide discovery and validation of diagnostic DNA methylation-based biomarkers for hepatocellular cancer detection in circulating cell free DNA. Theranostics (2019) 9(24):7239–50. doi: 10.7150/thno.35573
77. Zheng Y, Huang Q, Ding Z, Liu T, Xue C, Sang X, et al. Genome-wide DNA methylation analysis identifies candidate epigenetic markers and drivers of hepatocellular carcinoma. Brief Bioinform (2018) 19(1):101–8. doi: 10.1093/bib/bbw094
78. Li GX, Ding ZY, Wang YW, Liu TT, Chen WX, Wu JJ, et al. Integrative analysis of DNA methylation and gene expression identify a six epigenetic driver signature for predicting prognosis in hepatocellular carcinoma. J Cell Physiol (2019) 234(7):11942–50. doi: 10.1002/jcp.27882
79. Um TH, Kim H, Oh BK, Kim MS, Kim KS, Jung G, et al. Aberrant cpg island hypermethylation in dysplastic nodules and early hcc of hepatitis b virus-related human multistep hepatocarcinogenesis. J Hepatol (2011) 54(5):939–47. doi: 10.1016/j.jhep.2010.08.021
80. Nishida N, Kudo M, Nagasaka T, Ikai I, Goel A. Characteristic patterns of altered DNA methylation predict emergence of human hepatocellular carcinoma. Hepatology (2012) 56(3):994–1003. doi: 10.1002/hep.25706
81. Hlady RA, Tiedemann RL, Puszyk W, Zendejas I, Roberts LR, Choi JH, et al. Epigenetic signatures of alcohol abuse and hepatitis infection during human hepatocarcinogenesis. Oncotarget (2014) 5(19):9425–43. doi: 10.18632/oncotarget.2444
82. Park HJ, Yu E, Shim YH. DNA Methyltransferase expression and DNA hypermethylation in human hepatocellular carcinoma. Cancer Lett (2006) 233(2):271–8. doi: 10.1016/j.canlet.2005.03.017
83. Oh BK, Kim H, Park HJ, Shim YH, Choi J, Park C, et al. DNA Methyltransferase expression and DNA methylation in human hepatocellular carcinoma and their clinicopathological correlation. Int J Mol Med (2007) 20(1):65–73.
84. Li H, Rauch T, Chen ZX, Szabó PE, Riggs AD, Pfeifer GP. The histone methyltransferase Setdb1 and the DNA methyltransferase Dnmt3a interact directly and localize to promoters silenced in cancer cells. J Biol Chem (2006) 281(28):19489–500. doi: 10.1074/jbc.M513249200
85. Lai SC, Su YT, Chi CC, Kuo YC, Lee KF, Wu YC, et al. Dnmt3b/Oct4 expression confers sorafenib resistance and poor prognosis of hepatocellular carcinoma through il-6/Stat3 regulation. J Exp Clin Cancer Res (2019) 38(1):474. doi: 10.1186/s13046-019-1442-2
86. Shi R, Zhao H, Zhao S, Yuan H. Molecular subtypes, prognostic and immunotherapeutic relevant gene signatures mediated by DNA methylation regulators in hepatocellular carcinoma. Aging (Albany NY) (2022) 14(12):5271–91. doi: 10.18632/aging.204155
87. Wang X, Chen M, Liang X, Bai Y, Zeng J, Xu X, et al. Rnf135 promoter methylation is associated with immune infiltration and prognosis in hepatocellular carcinoma. Front Oncol (2021) 11:752511. doi: 10.3389/fonc.2021.752511
88. Lin Y, Yao Y, Wang Y, Wang L, Cui H. Pd-L1 and immune infiltration of M(6)a rna methylation regulators and its mirna regulators in hepatocellular carcinoma. BioMed Res Int (2021) 2021:5516100. doi: 10.1155/2021/5516100
89. Xu Y, He X, Deng J, Xiong L, Li Y, Zhang X, et al. Comprehensive analysis of the immune infiltrates and pd-L1 of M(6)a rna methylation regulators in hepatocellular carcinoma. Front Cell Dev Biol (2021) 9:681745. doi: 10.3389/fcell.2021.681745
90. Liu J, Liu Y, Meng L, Liu K, Ji B. Targeting the pd-L1/Dnmt1 axis in acquired resistance to sorafenib in human hepatocellular carcinoma. Oncol Rep (2017) 38(2):899–907. doi: 10.3892/or.2017.5722
91. Hong YK, Li Y, Pandit H, Li S, Pulliam Z, Zheng Q, et al. Epigenetic modulation enhances immunotherapy for hepatocellular carcinoma. Cell Immunol (2019) 336:66–74. doi: 10.1016/j.cellimm.2018.12.010
92. Liu M, Zhang L, Li H, Hinoue T, Zhou W, Ohtani H, et al. Integrative epigenetic analysis reveals therapeutic targets to the DNA methyltransferase inhibitor guadecitabine (Sgi-110) in hepatocellular carcinoma. Hepatology (2018) 68(4):1412–28. doi: 10.1002/hep.30091
93. Audia JE, Campbell RM. Histone modifications and cancer. Cold Spring Harb Perspect Biol (2016) 8(4):a019521. doi: 10.1101/cshperspect.a019521
94. Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res (2011) 21(3):381–95. doi: 10.1038/cr.2011.22
95. Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet (2007) 8(4):286–98. doi: 10.1038/nrg2005
96. Lawrence M, Daujat S, Schneider R. Lateral thinking: How histone modifications regulate gene expression. Trends Genet (2016) 32(1):42–56. doi: 10.1016/j.tig.2015.10.007
97. Ruthenburg AJ, Li H, Patel DJ, Allis CD. Multivalent engagement of chromatin modifications by linked binding modules. Nat Rev Mol Cell Biol (2007) 8(12):983–94. doi: 10.1038/nrm2298
98. Li Y, Chen X, Lu C. The interplay between DNA and histone methylation: Molecular mechanisms and disease implications. EMBO Rep (2021) 22(5):e51803. doi: 10.15252/embr.202051803
99. Qian Y, Li Y, Zheng C, Lu T, Sun R, Mao Y, et al. High methylation levels of histone H3 lysine 9 associated with activation of hypoxia-inducible factor 1α (Hif-1α) predict patients’ worse prognosis in human hepatocellular carcinomas. Cancer Genet (2020) 245:17–26. doi: 10.1016/j.cancergen.2020.04.077
100. Chiba T, Saito T, Yuki K, Zen Y, Koide S, Kanogawa N, et al. Histone lysine methyltransferase Suv39h1 is a potent target for epigenetic therapy of hepatocellular carcinoma. Int J Cancer (2015) 136(2):289–98. doi: 10.1002/ijc.28985
101. Bárcena-Varela M, Caruso S, Llerena S, Álvarez-Sola G, Uriarte I, Latasa MU, et al. Dual targeting of histone methyltransferase G9a and DNA-methyltransferase 1 for the treatment of experimental hepatocellular carcinoma. Hepatology (2019) 69(2):587–603. doi: 10.1002/hep.30168
102. Fan DN, Tsang FH, Tam AH, Au SL, Wong CC, Wei L, et al. Histone lysine methyltransferase, suppressor of variegation 3-9 homolog 1, promotes hepatocellular carcinoma progression and is negatively regulated by microrna-125b. Hepatology (2013) 57(2):637–47. doi: 10.1002/hep.26083
103. Wong CM, Wei L, Law CT, Ho DW, Tsang FH, Au SL, et al. Up-regulation of histone methyltransferase Setdb1 by multiple mechanisms in hepatocellular carcinoma promotes cancer metastasis. Hepatology (2016) 63(2):474–87. doi: 10.1002/hep.28304
104. Wei L, Chiu DK, Tsang FH, Law CT, Cheng CL, Au SL, et al. Histone methyltransferase G9a promotes liver cancer development by epigenetic silencing of tumor suppressor gene Rarres3. J Hepatol (2017) 67(4):758–69. doi: 10.1016/j.jhep.2017.05.015
105. Zheng Y, Tang L, Chen G, Liu Z. Comprehensive bioinformatics analysis of key methyltransferases and demethylases for histone lysines in hepatocellular carcinoma. Technol Cancer Res Treat (2020) 19:1533033820983284. doi: 10.1177/1533033820983284
106. Valk-Lingbeek ME, Bruggeman SW, van Lohuizen M. Stem cells and cancer; the polycomb connection. Cell (2004) 118(4):409–18. doi: 10.1016/j.cell.2004.08.005
107. Au SL, Ng IO, Wong CM. Epigenetic dysregulation in hepatocellular carcinoma: Focus on polycomb group proteins. Front Med (2013) 7(2):231–41. doi: 10.1007/s11684-013-0253-7
108. Yonemitsu Y, Imazeki F, Chiba T, Fukai K, Nagai Y, Miyagi S, et al. Distinct expression of polycomb group proteins Ezh2 and Bmi1 in hepatocellular carcinoma. Hum Pathol (2009) 40(9):1304–11. doi: 10.1016/j.humpath.2009.01.017
109. Chen Z, Lin X, Wan Z, Xiao M, Ding C, Wan P, et al. High expression of Ezh2 mediated by ncrnas correlates with poor prognosis and tumor immune infiltration of hepatocellular carcinoma. Genes (Basel) (2022) 13(5):1–20. doi: 10.3390/genes13050876
110. Chiba T, Suzuki E, Negishi M, Saraya A, Miyagi S, Konuma T, et al. 3-deazaneplanocin a is a promising therapeutic agent for the eradication of tumor-initiating hepatocellular carcinoma cells. Int J Cancer (2012) 130(11):2557–67. doi: 10.1002/ijc.26264
111. Kusakabe Y, Chiba T, Oshima M, Koide S, Rizq O, Aoyama K, et al. Ezh1/2 inhibition augments the anti-tumor effects of sorafenib in hepatocellular carcinoma. Sci Rep (2021) 11(1):21396. doi: 10.1038/s41598-021-00889-0
112. Kim HJ, Cantor H, Cosmopoulos K. Overcoming immune checkpoint blockade resistance Via Ezh2 inhibition. Trends Immunol (2020) 41(10):948–63. doi: 10.1016/j.it.2020.08.010
113. Eich ML, Athar M, Ferguson JE 3rd, Varambally S. Ezh2-targeted therapies in cancer: Hype or a reality. Cancer Res (2020) 80(24):5449–58. doi: 10.1158/0008-5472.Can-20-2147
114. Kang N, Eccleston M, Clermont PL, Latarani M, Male DK, Wang Y, et al. Ezh2 inhibition: A promising strategy to prevent cancer immune editing. Epigenomics (2020) 12(16):1457–76. doi: 10.2217/epi-2020-0186
115. Lu C, Paschall AV, Shi H, Savage N, Waller JL, Sabbatini ME, et al. The Mll1-H3k4me3 axis-mediated pd-L1 expression and pancreatic cancer immune evasion. J Natl Cancer Inst (2017) 109(6):1–12. doi: 10.1093/jnci/djw283
116. Lu C, Liu Z, Klement JD, Yang D, Merting AD, Poschel D, et al. Wdr5-H3k4me3 epigenetic axis regulates opn expression to compensate pd-L1 function to promote pancreatic cancer immune escape. J Immunother Cancer (2021) 9(7):1–14. doi: 10.1136/jitc-2021-002624
117. Xiao G, Jin LL, Liu CQ, Wang YC, Meng YM, Zhou ZG, et al. Ezh2 negatively regulates pd-L1 expression in hepatocellular carcinoma. J Immunother Cancer (2019) 7(1):300. doi: 10.1186/s40425-019-0784-9
118. Bugide S, Green MR, Wajapeyee N. Inhibition of enhancer of zeste homolog 2 (Ezh2) induces natural killer cell-mediated eradication of hepatocellular carcinoma cells. Proc Natl Acad Sci U.S.A. (2018) 115(15):E3509–e18. doi: 10.1073/pnas.1802691115
119. Huang X, Yan J, Zhang M, Wang Y, Chen Y, Fu X, et al. Targeting epigenetic crosstalk as a therapeutic strategy for Ezh2-aberrant solid tumors. Cell (2018) 175(1):186–99.e19. doi: 10.1016/j.cell.2018.08.058
120. Zhang L, Lu Q, Chang C. Epigenetics in health and disease. Adv Exp Med Biol (2020) 1253:3–55. doi: 10.1007/978-981-15-3449-2_1
121. Ler SY, Leung CH, Khin LW, Lu GD, Salto-Tellez M, Hartman M, et al. Hdac1 and Hdac2 independently predict mortality in hepatocellular carcinoma by a competing risk regression model in a southeast Asian population. Oncol Rep (2015) 34(5):2238–50. doi: 10.3892/or.2015.4263
122. Jin X, Tian S, Li P. Histone acetyltransferase 1 promotes cell proliferation and induces cisplatin resistance in hepatocellular carcinoma. Oncol Res (2017) 25(6):939–46. doi: 10.3727/096504016x14809827856524
123. Wu H, Yang TY, Li Y, Ye WL, Liu F, He XS, et al. Tumor necrosis factor receptor-associated factor 6 promotes hepatocarcinogenesis by interacting with histone deacetylase 3 to enhance c-myc gene expression and protein stability. Hepatology (2020) 71(1):148–63. doi: 10.1002/hep.30801
124. Ji H, Zhou Y, Zhuang X, Zhu Y, Wu Z, Lu Y, et al. Hdac3 deficiency promotes liver cancer through a defect in H3k9ac/H3k9me3 transition. Cancer Res (2019) 79(14):3676–88. doi: 10.1158/0008-5472.Can-18-3767
125. Zhang J, Liu H, Pan H, Yang Y, Huang G, Yang Y, et al. The histone acetyltransferase hmof suppresses hepatocellular carcinoma growth. Biochem Biophys Res Commun (2014) 452(3):575–80. doi: 10.1016/j.bbrc.2014.08.122
126. Poté N, Cros J, Laouirem S, Raffenne J, Negrão M, Albuquerque M, et al. The histone acetyltransferase hmof promotes vascular invasion in hepatocellular carcinoma. Liver Int (2020) 40(4):956–67. doi: 10.1111/liv.14381
127. Tian Y, Wong VW, Wong GL, Yang W, Sun H, Shen J, et al. Histone deacetylase Hdac8 promotes insulin resistance and B-catenin activation in nafld-associated hepatocellular carcinoma. Cancer Res (2015) 75(22):4803–16. doi: 10.1158/0008-5472.Can-14-3786
128. Yang W, Feng Y, Zhou J, Cheung OK, Cao J, Wang J, et al. A selective Hdac8 inhibitor potentiates antitumor immunity and efficacy of immune checkpoint blockade in hepatocellular carcinoma. Sci Transl Med (2021) 13(588):1–14. doi: 10.1126/scitranslmed.aaz6804
129. Bugide S, Gupta R, Green MR, Wajapeyee N. Ezh2 inhibits nk cell-mediated antitumor immunity by suppressing Cxcl10 expression in an Hdac10-dependent manner. Proc Natl Acad Sci U.S.A. (2021) 118(30):1–12. doi: 10.1073/pnas.2102718118
130. Manna D, Sarkar D. Non-coding rnas: regulating disease progression and therapy resistance in hepatocellular carcinoma. Cancers (Basel) (2020) 12(5). doi: 10.3390/cancers12051243
131. Peng L, Yuan XQ, Zhang CY, Peng JY, Zhang YQ, Pan X, et al. The emergence of long non-coding rnas in hepatocellular carcinoma: an update. J Cancer (2018) 9(14):2549–58. doi: 10.7150/jca.24560
132. Niu ZS, Niu XJ, Wang WH. Long non-coding rnas in hepatocellular carcinoma: potential roles and clinical implications. World J Gastroenterol (2017) 23(32):5860–74. doi: 10.3748/wjg.v23.i32.5860
133. Hu X, Jiang J, Xu Q, Ni C, Yang L, Huang D. A systematic review of long noncoding rnas in hepatocellular carcinoma: molecular mechanism and clinical implications. BioMed Res Int (2018) 2018:8126208. doi: 10.1155/2018/8126208
134. Han TS, Hur K, Cho HS, Ban HS. Cancers (Basel) (2020) 12(9). Epub 2020/09/18. doi: 10.3390/cancers12092622
135. Chen S, Fu Z, Wen S, Yang X, Yu C, Zhou W, et al. Expression and diagnostic value of mir-497 and mir-1246 in hepatocellular carcinoma. Front Genet (2021) 12:666306. doi: 10.3389/fgene.2021.666306
136. Qian F, Wang J, Wang Y, Gao Q, Yan W, Lin Y, et al. Mir-378a-3p as a putative biomarker for hepatocellular carcinoma diagnosis and prognosis: computational screening with experimental validation. Clin Transl Med (2021) 11(2):e307. doi: 10.1002/ctm2.307
137. Li Y, Zhou T, Cheng X, Li D, Zhao M, Zheng WV. Microrna-378a-3p regulates the progression of hepatocellular carcinoma by regulating pd-l1 and stat3. Bioengineered (2022) 13(3):4730–43. doi: 10.1080/21655979.2022.2031408
138. Yang B, Feng X, Liu H, Tong R, Wu J, Li C, et al. High-metastatic cancer cells derived exosomal mir92a-3p promotes epithelial-mesenchymal transition and metastasis of low-metastatic cancer cells by regulating pten/akt pathway in hepatocellular carcinoma. Oncogene (2020) 39(42):6529–43. doi: 10.1038/s41388-020-01450-5
139. Li D, Sun FF, Wang D, Wang T, Peng JJ, Feng JQ, et al. Programmed death ligand-1 (pd-l1) regulated by nrf-2/microrna-1 regulatory axis enhances drug resistance and promotes tumorigenic properties in sorafenib-resistant hepatoma cells. Oncol Res (2020) 28(5):467–81. doi: 10.3727/096504020x15925659763817
140. Sun C, Lan P, Han Q, Huang M, Zhang Z, Xu G, et al. Oncofetal gene sall4 reactivation by hepatitis b virus counteracts mir-200c in pd-l1-induced t cell exhaustion. Nat Commun (2018) 9(1):1241. doi: 10.1038/s41467-018-03584-3
141. Guo W, Tan W, Liu S, Huang X, Lin J, Liang R, et al. Mir-570 inhibited the cell proliferation and invasion through directly targeting b7-h1 in hepatocellular carcinoma. Tumour Biol (2015) 36(11):9049–57. doi: 10.1007/s13277-015-3644-3
142. Zhao J, Li H, Zhao S, Wang E, Zhu J, Feng D, et al. Epigenetic silencing of mir-144/451a cluster contributes to hcc progression via paracrine hgf/mif-mediated tam remodeling. Mol Cancer (2021) 20(1):46. doi: 10.1186/s12943-021-01343-5
143. Yang G, Zhou L, Xu Q, Meng F, Wan Y, Meng X, et al. Lncrna kcnq1ot1 inhibits the radiosensitivity and promotes the tumorigenesis of hepatocellular carcinoma via the mir-146a-5p/acer3 axis. Cell Cycle (2020) 19(19):2519–29. doi: 10.1080/15384101.2020.1809259
144. Zhang J, Zhao X, Ma X, Yuan Z, Hu M. Kcnq1ot1 contributes to sorafenib resistance and programmed death−ligand−1−mediated immune escape via sponging mir−506 in hepatocellular carcinoma cells. Int J Mol Med (2020) 46(5):1794–804. doi: 10.3892/ijmm.2020.4710
145. Song H, Liu Y, Li X, Chen S, Xie R, Chen D, et al. Long noncoding rna casc11 promotes hepatocarcinogenesis and hcc progression through eif4a3-mediated e2f1 activation. Clin Transl Med (2020) 10(7):e220. doi: 10.1002/ctm2.220
146. Cossío FP, Esteller M, Berdasco M. Towards a more precise therapy in cancer: exploring epigenetic complexity. Curr Opin Chem Biol (2020) 57:41–9. doi: 10.1016/j.cbpa.2020.04.008
147. Saleh MH, Wang L, Goldberg MS. Improving cancer immunotherapy with dna methyltransferase inhibitors. Cancer Immunol Immunother (2016) 65(7):787–96. doi: 10.1007/s00262-015-1776-3
148. Zhu S, Denman CJ, Cobanoglu ZS, Kiany S, Lau CC, Gottschalk SM, et al. The narrow-spectrum hdac inhibitor entinostat enhances nkg2d expression without nk cell toxicity, leading to enhanced recognition of cancer cells. Pharm Res (2015) 32(3):779–92. doi: 10.1007/s11095-013-1231-0
149. Costantini B, Kordasti SY, Kulasekararaj AG, Jiang J, Seidl T, Abellan PP, et al. The effects of 5-azacytidine on the function and number of regulatory t cells and t-effectors in myelodysplastic syndrome. Haematologica (2013) 98(8):1196–205. doi: 10.3324/haematol.2012.074823
150. Dhanak D, Jackson P. Development and classes of epigenetic drugs for cancer. Biochem Biophys Res Commun (2014) 455(1-2):58–69. doi: 10.1016/j.bbrc.2014.07.006
151. Di Costanzo A, Del Gaudio N, Migliaccio A, Altucci L. Epigenetic drugs against cancer: An evolving landscape. Arch Toxicol (2014) 88(9):1651–68. doi: 10.1007/s00204-014-1315-6
152. Wu ZH, Yang DL, Wang L, Liu J. Epigenetic and immune-cell infiltration changes in the tumor microenvironment in hepatocellular carcinoma. Front Immunol (2021) 12:793343. doi: 10.3389/fimmu.2021.793343
153. Jones PA, Ohtani H, Chakravarthy A, De Carvalho DD. Epigenetic therapy in immune-oncology. Nat Rev Cancer (2019) 19(3):151–61. doi: 10.1038/s41568-019-0109-9
154. Mei Q, Chen M, Lu X, Li X, Duan F, Wang M, et al. An open-label, single-arm, phase i/ii study of lower-dose decitabine based therapy in patients with advanced hepatocellular carcinoma. Oncotarget (2015) 6(18):16698–711. doi: 10.18632/oncotarget.3677
155. Wang Q, Liang N, Yang T, Li Y, Li J, Huang Q, et al. Dnmt1-mediated methylation of bex1 regulates stemness and tumorigenicity in liver cancer. J Hepatol (2021) 75(5):1142–53. doi: 10.1016/j.jhep.2021.06.025
156. Barcena-Varela M, Paish H, Alvarez L, Uriarte I, Latasa MU, Santamaria E, et al. Epigenetic mechanisms and metabolic reprogramming in fibrogenesis: dual targeting of g9a and dnmt1 for the inhibition of liver fibrosis. Gut (2021) 70(2):388–400. doi: 10.1136/gutjnl-2019-320205
157. Segovia C, San José-Enériz E, Munera-Maravilla E, Martínez-Fernández M, Garate L, Miranda E, et al. Inhibition of a g9a/dnmt network triggers immune-mediated bladder cancer regression. Nat Med (2019) 25(7):1073–81. doi: 10.1038/s41591-019-0499-y
158. Lachenmayer A, Toffanin S, Cabellos L, Alsinet C, Hoshida Y, Villanueva A, et al. Combination therapy for hepatocellular carcinoma: additive preclinical efficacy of the hdac inhibitor panobinostat with sorafenib. J Hepatol (2012) 56(6):1343–50. doi: 10.1016/j.jhep.2012.01.009
159. Buurman R, Gürlevik E, Schäffer V, Eilers M, Sandbothe M, Kreipe H, et al. Histone deacetylases activate hepatocyte growth factor signaling by repressing microrna-449 in hepatocellular carcinoma cells. Gastroenterology (2012) 143(3):811–20.e15. doi: 10.1053/j.gastro.2012.05.033
160. Pathil A, Armeanu S, Venturelli S, Mascagni P, Weiss TS, Gregor M, et al. Hdac inhibitor treatment of hepatoma cells induces both trail-independent apoptosis and restoration of sensitivity to trail. Hepatology (2006) 43(3):425–34. doi: 10.1002/hep.21054
161. Armeanu S, Pathil A, Venturelli S, Mascagni P, Weiss TS, Göttlicher M, et al. Apoptosis on hepatoma cells but not on primary hepatocytes by histone deacetylase inhibitors valproate and itf2357. J Hepatol (2005) 42(2):210–7. doi: 10.1016/j.jhep.2004.10.020
162. Chen JC, Chuang HY, Liao YJ, Hsu FT, Chen YC, Wang WH, et al. Enhanced cytotoxicity of human hepatocellular carcinoma cells following pretreatment with sorafenib combined with trichostatin a. Oncol Lett (2019) 17(1):638–45. doi: 10.3892/ol.2018.9582
163. Gahr S, Mayr C, Kiesslich T, Illig R, Neureiter D, Alinger B, et al. The pan-deacetylase inhibitor panobinostat affects angiogenesis in hepatocellular carcinoma models via modulation of ctgf expression. Int J Oncol (2015) 47(3):963–70. doi: 10.3892/ijo.2015.3087
164. DIF P, Montalbano R, Quint K, Alinger B, Kemmerling R, Kiesslich T, et al. The pan-deacetylase inhibitor panobinostat modulates the expression of epithelial-mesenchymal transition markers in hepatocellular carcinoma models. Oncol Lett (2013) 5(1):127–34. doi: 10.3892/ol.2012.951
165. Wu F, Xu L, Tu Y, Cheung OK, Szeto LL, Mok MT, et al. Sirtuin 7 super-enhancer drives epigenomic reprogramming in hepatocarcinogenesis. Cancer Lett (2022) 525:115–30. doi: 10.1016/j.canlet.2021.10.039
166. Xiang J, Zhang N, Sun H, Su L, Zhang C, Xu H, et al. Disruption of sirt7 increases the efficacy of checkpoint inhibitor via mef2d regulation of programmed cell death 1 ligand 1 in hepatocellular carcinoma cells. Gastroenterology (2020) 158(3):664–78.e24. doi: 10.1053/j.gastro.2019.10.025
167. Venturelli S, Armeanu S, Pathil A, Hsieh CJ, Weiss TS, Vonthein R, et al. Epigenetic combination therapy as a tumor-selective treatment approach for hepatocellular carcinoma. Cancer (2007) 109(10):2132–41. doi: 10.1002/cncr.22652
168. Yeo W, Chung HC, Chan SL, Wang LZ, Lim R, Picus J, et al. Epigenetic therapy using belinostat for patients with unresectable hepatocellular carcinoma: a multicenter phase i/ii study with biomarker and pharmacokinetic analysis of tumors from patients in the mayo phase ii consortium and the cancer therapeutics research group. J Clin Oncol (2012) 30(27):3361–7. doi: 10.1200/jco.2011.41.2395
169. Llopiz D, Ruiz M, Villanueva L, Iglesias T, Silva L, Egea J, et al. Enhanced anti-tumor efficacy of checkpoint inhibitors in combination with the histone deacetylase inhibitor belinostat in a murine hepatocellular carcinoma model. Cancer Immunol Immunother (2019) 68(3):379–93. doi: 10.1007/s00262-018-2283-0
170. Gao Y, Nihira NT, Bu X, Chu C, Zhang J, Kolodziejczyk A, et al. Acetylation-dependent regulation of pd-l1 nuclear translocation dictates the efficacy of anti-pd-1 immunotherapy. Nat Cell Biol (2020) 22(9):1064–75. doi: 10.1038/s41556-020-0562-4
171. Qiu W, Wang B, Gao Y, Tian Y, Tian M, Chen Y, et al. Targeting histone deacetylase 6 reprograms interleukin-17-producing helper t cell pathogenicity and facilitates immunotherapies for hepatocellular carcinoma. Hepatology (2020) 71(6):1967–87. doi: 10.1002/hep.30960
172. Tsang FH, Law CT, Tang TC, Cheng CL, Chin DW, Tam WV, et al. Aberrant super-enhancer landscape in human hepatocellular carcinoma. Hepatology (2019) 69(6):2502–17. doi: 10.1002/hep.30544
173. Bayo J, Fiore EJ, Dominguez LM, Real A, Malvicini M, Rizzo M, et al. A comprehensive study of epigenetic alterations in hepatocellular carcinoma identifies potential therapeutic targets. J Hepatol (2019) 71(1):78–90. doi: 10.1016/j.jhep.2019.03.007
174. Jühling F, Hamdane N, Crouchet E, Li S, El Saghire H, Mukherji A, et al. Targeting clinical epigenetic reprogramming for chemoprevention of metabolic and viral hepatocellular carcinoma. Gut (2021) 70(1):157–69. doi: 10.1136/gutjnl-2019-318918
175. Davis AA, Patel VG. The role of pd-l1 expression as a predictive biomarker: an analysis of all us food and drug administration (fda) approvals of immune checkpoint inhibitors. J Immunother Cancer (2019) 7(1):278. doi: 10.1186/s40425-019-0768-9
176. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. mutational landscape determines sensitivity to pd-1 blockade in non-small cell lung cancer. Science (2015) 348(6230):124–8. doi: 10.1126/science.aaa1348
177. Ang C, Klempner SJ, Ali SM, Madison R, Ross JS, Severson EA, et al. Prevalence of established and emerging biomarkers of immune checkpoint inhibitor response in advanced hepatocellular carcinoma. Oncotarget (2019) 10(40):4018–25. doi: 10.18632/oncotarget.26998
178. Shrestha R, Prithviraj P, Anaka M, Bridle KR, Crawford DHG, Dhungel B, et al. Monitoring immune checkpoint regulators as predictive biomarkers in hepatocellular carcinoma. Front Oncol (2018) 8:269. doi: 10.3389/fonc.2018.00269
179. Rittmeyer A, Barlesi F, Waterkamp D, Park K, Ciardiello F, von Pawel J, et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (oak): a phase 3, open-label, multicentre randomised controlled trial. Lancet (2017) 389(10066):255–65. doi: 10.1016/s0140-6736(16)32517-x
180. Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WE, Poddubskaya E, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med (2015) 373(2):123–35. doi: 10.1056/NEJMoa1504627
181. Yau T, Hsu C, Kim TY, Choo SP, Kang YK, Hou MM, et al. Nivolumab in advanced hepatocellular carcinoma: sorafenib-experienced asian cohort analysis. J Hepatol (2019) 71(3):543–52. doi: 10.1016/j.jhep.2019.05.014
182. Mauriello A, Zeuli R, Cavalluzzo B, Petrizzo A, Tornesello ML, Buonaguro FM, et al. High somatic mutation and neoantigen burden do not correlate with decreased progression-free survival in hcc patients not undergoing immunotherapy. Cancers (Basel) (2019) 11(12). doi: 10.3390/cancers11121824
183. First tissue-agnostic drug approval issued. Cancer Discovery (2017) 7(7):656. doi: 10.1158/2159-8290.Cd-nb2017-078
184. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to pd-1 blockade. Science (2017) 357(6349):409–13. doi: 10.1126/science.aan6733
185. Macek Jilkova Z, Aspord C, Kurma K, Granon A, Sengel C, Sturm N, et al. Immunologic features of patients with advanced hepatocellular carcinoma before and during sorafenib or anti-programmed death-1/programmed death-l1 treatment. Clin Transl Gastroenterol (2019) 10(7):e00058. doi: 10.14309/ctg.0000000000000058
186. Ng HHM, Lee RY, Goh S, Tay ISY, Lim X, Lee B, et al. Immunohistochemical scoring of cd38 in the tumor microenvironment predicts responsiveness to anti-pd-1/pd-l1 immunotherapy in hepatocellular carcinoma. J Immunother Cancer (2020) 8(2). doi: 10.1136/jitc-2020-000987
187. Kurebayashi Y, Ojima H, Tsujikawa H, Kubota N, Maehara J, Abe Y, et al. Landscape of immune microenvironment in hepatocellular carcinoma and its additional impact on histological and molecular classification. Hepatology (2018) 68(3):1025–41. doi: 10.1002/hep.29904
188. Itoh S, Yoshizumi T, Yugawa K, Imai D, Yoshiya S, Takeishi K, et al. Impact of immune response on outcomes in hepatocellular carcinoma: association with vascular formation. Hepatology (2020) 72(6):1987–99. doi: 10.1002/hep.31206
189. Pinyol R, Sia D, Llovet JM. Immune exclusion-wnt/ctnnb1 class predicts resistance to immunotherapies in hcc. Clin Cancer Res (2019) 25(7):2021–3. doi: 10.1158/1078-0432.Ccr-18-3778
190. Ruiz de Galarreta M, Bresnahan E, Molina-Sánchez P, Lindblad KE, Maier B, Sia D, et al. β-catenin activation promotes immune escape and resistance to anti-pd-1 therapy in hepatocellular carcinoma. Cancer Discovery (2019) 9(8):1124–41. doi: 10.1158/2159-8290.Cd-19-0074
191. Tian Y, Mok MT, Yang P, Cheng AS. Epigenetic activation of wnt/β-catenin signaling in nafld-associated hepatocarcinogenesis. Cancers (Basel) (2016) 8(8). doi: 10.3390/cancers8080076
192. Okrah K, Tarighat S, Liu B, Koeppen H, Wagle MC, Cheng G, et al. Transcriptomic analysis of hepatocellular carcinoma reveals molecular features of disease progression and tumor immune biology. NPJ Precis Oncol (2018) 2:25. doi: 10.1038/s41698-018-0068-8
193. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. Tgfβ attenuates tumour response to pd-l1 blockade by contributing to exclusion of t cells. Nature (2018) 554(7693):544–8. doi: 10.1038/nature25501
194. Lim CJ, Lee YH, Pan L, Lai L, Chua C, Wasser M, et al. Multidimensional analyses reveal distinct immune microenvironment in hepatitis b virus-related hepatocellular carcinoma. Gut (2019) 68(5):916–27. doi: 10.1136/gutjnl-2018-316510
195. Inada Y, Mizukoshi E, Seike T, Tamai T, Iida N, Kitahara M, et al. Characteristics of immune response to tumor-associated antigens and immune cell profile in patients with hepatocellular carcinoma. Hepatology (2019) 69(2):653–65. doi: 10.1002/hep.30212
196. Duan M, Hao J, Cui S, Worthley DL, Zhang S, Wang Z, et al. Diverse modes of clonal evolution in hbv-related hepatocellular carcinoma revealed by single-cell genome sequencing. Cell Res (2018) 28(3):359–73. doi: 10.1038/cr.2018.11
197. Ma C, Han M, Heinrich B, Fu Q, Zhang Q, Sandhu M, et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via nkt cells. Science (2018) 360(6391). doi: 10.1126/science.aan5931
198. Lee PC, Wu CJ, Hung YW, Lee CJ, Chi CT, Lee IC, et al. Gut microbiota and metabolites associate with outcomes of immune checkpoint inhibitor-treated unresectable hepatocellular carcinoma. J Immunother Cancer (2022) 10(6). doi: 10.1136/jitc-2022-004779
Keywords: tumor immunotherapy, immune checkpoint inhibitors, hepatocellular carcinoma (HCC), epigenetic modification, immune resistance mechanisms
Citation: Tao S, Liang S, Zeng T and Yin D (2023) Epigenetic modification-related mechanisms of hepatocellular carcinoma resistance to immune checkpoint inhibition. Front. Immunol. 13:1043667. doi: 10.3389/fimmu.2022.1043667
Received: 13 September 2022; Accepted: 28 November 2022;
Published: 04 January 2023.
Edited by:
Hailin Tang, Sun Yat-sen University Cancer Center (SYSUCC), ChinaCopyright © 2023 Tao, Liang, Zeng and Yin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dalong Yin, ZG9jdG9yeWluQHVzdGMuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.