 Swati Sharma
Swati Sharma Tarun Tyagi
Tarun Tyagi Silvio Antoniak
Silvio Antoniak- 1UNC Blood Research Center, School of Medicine, University of North Carolina at Chapel Hill, Chapel Hill, NC, United States
- 2Yale Cardiovascular Research Center, Yale School of Medicine, New Haven, CT, United States
In the broad range of human diseases, thrombo-inflammation appears as a clinical manifestation. Clinically, it is well characterized in context of superficial thrombophlebitis that is recognized as thrombosis and inflammation of superficial veins. However, it is more hazardous when developed in the microvasculature of injured/inflamed/infected tissues and organs. Several diseases like sepsis and ischemia-reperfusion can cause formation of microvascular thrombosis subsequently leading to thrombo-inflammation. Thrombo-inflammation can also occur in cases of antiphospholipid syndrome, preeclampsia, sickle cell disease, bacterial and viral infection. One of the major contributors to thrombo-inflammation is the loss of normal anti-thrombotic and anti-inflammatory potential of the endothelial cells of vasculature. This manifest itself in the form of dysregulation of the coagulation pathway and complement system, pathologic platelet activation, and increased recruitment of leukocyte within the microvasculature. The role of platelets in hemostasis and formation of thrombi under pathologic and non-pathologic conditions is well established. Platelets are anucleate cells known for their essential role in primary hemostasis and the coagulation pathway. In recent years, studies provide strong evidence for the critical involvement of platelets in inflammatory processes like acute ischemic stroke, and viral infections like Coronavirus disease 2019 (COVID-19). This has encouraged the researchers to investigate the contribution of platelets in the pathology of various thrombo-inflammatory diseases. The inhibition of platelet surface receptors or their intracellular signaling which mediate initial platelet activation and adhesion might prove to be suitable targets in thrombo-inflammatory disorders. Thus, the present review summarizes the concept and mechanism of platelet signaling and briefly discuss their role in sterile and non-sterile thrombo-inflammation, with the emphasis on role of platelets in COVID-19 induced thrombo-inflammation. The aim of this review is to summarize the recent developments in deciphering the role of the platelets in thrombo-inflammation and discuss their potential as pharmaceutical targets.
1 Introduction
Platelets originate from megakaryocytes as anucleate cells equipped with their own survival machinery to function in circulation. The main hemostatic role of platelets is to form the platelet plug at the site of vascular lesion or injury, to limit the blood loss and thus maintain hemostasis. Importantly, under pathological conditions, the dysregulated intravascular activation and aggregation of platelets along with activation of local coagulation factors lead to formation of vessel-occluding thrombi. This series of events may result in ischemia and infarction of affected tissues. Increasing number of reports revealing the complexity associated with platelet signaling pathways have encouraged researchers to dig deeper into exploring the role of platelets beyond hemostasis. Prior to last few decades, platelets were not studied for their role in the pathogenesis of infectious diseases. Their key role was thought to be limited as primary effectors of hemostasis and thrombosis, with very few studies exploring platelets in host immune response during various infectious diseases.
In the past two decades, platelets have been extensively explored for their involvement in different physiological processes including inflammation, vascular permeability, and tissue repair (1–5). There is a growing number of evidence from literature that emphasize on the role of platelets as main sentinel and effector cells involved in bridging hemostasis, inflammation, and the immune system. The role of platelets now extends beyond their traditional role as marginal mediators of hemostasis and thrombosis, as the contributors to diverse immunological processes. The recent investigations provide substantial evidence for their role against microbial threats, modulating antigen presentation, enhancement of adaptive immune responses, recruitment and promotion of innate effector cell functions. Platelets until last decade were underappreciated orchestrator of the immune system but now emerged as crucial players during infectious diseases too, as recent studies document the role of platelets in inflammation, hemostasis, and maintenance of vascular integrity during infection (3, 6–10). As platelets through different receptors can respond to both PAMPs (Pathogen associated molecular patterns) and DAMPs (Damage associated molecular patterns) released during non-sterile (infection) and sterile inflammation, it becomes pertinent to investigate their role and mechanism of action in both kind of inflammation. Investigating the role of platelets in both sterile and non-sterile inflammation will not only shed the light on shared molecular mechanism but can also reveal novel therapeutic targets.
The platelets are highly sensitive to environmental cues. These environmental stimuli tightly regulate not only hemostatic and immunomodulatory functions of platelets but also their interaction with the vasculature (endothelium) and components of innate immunity. Platelets can sense intravascular invasion of pathogen, whether its parasitic, bacterial, or viral infection, either directly through receptors namely integrin receptors and pattern recognition receptors (PRRs), or through pathogen: immunoglobulin complexes via Fc and complement receptors. Platelets can also respond to pathogens indirectly via their interactions with leukocytes and the vascular endothelium at the area of local injury or inflammation. Recognition of the pathogen by platelets leads to their activation. The activated platelets through various possible mechanisms can directly kill or sequester the pathogen or orchestrate pathogen clearance by activating neutrophils and macrophages. Platelet neutrophil interaction promotes the formation of neutrophil extracellular traps (NETs) as part of the innate immune response. However, NETs can enhance platelet activation, platelet aggregation and formation of microthrombi. Intravascular platelet activation can occur as an aberrant response and if unchecked may lead to the exacerbation of inflammation, promoting the endothelial dysfunction and thrombosis. Consequently, these events can be injurious to the host and cause thrombo‐inflammation (11). The term thrombo-inflammation broadly describes a phenomenon that exhibit thrombus formation as the result of crosstalk between inflammation including immune cells and coagulation, which encompasses an array of diseases (deep vein thrombosis, stroke, atherosclerosis, and infectious diseases). The concept of thrombo‐inflammation was also used to define the role of the platelets in inflammatory response followed by cerebral ischemia‐reperfusion injury (12).
With the increase in the breadth of our knowledge regarding the complexity of the platelets and their signaling during the last decade, it is well established now that there is a crosstalk between hemostasis, thrombosis, and inflammation and they are tightly interconnected with each other. Importantly, platelets are also the key effector cells that bridge and link these three processes. Thus, this narrative review attempts to address the role of platelets in both sterile and non-sterile thrombo-inflammation with major focus on role of platelets signaling in viral infection induced thrombo-inflammation including Coronavirus disease 2019 (COVID-19). Figure 1 summaries the theme of the present review. The last section also summaries the recent developments in deciphering the role of the main platelet receptors in thrombo-inflammation and discuss their potential as potential pharmacotherapeutic targets. As thromo-inflammation is common manifestation of both sterile (deep vein thrombosis [DVT], atherosclerosis and infectious disease [viral and bacterial infections]), it is important to study how platelets differentially regulates these processes.
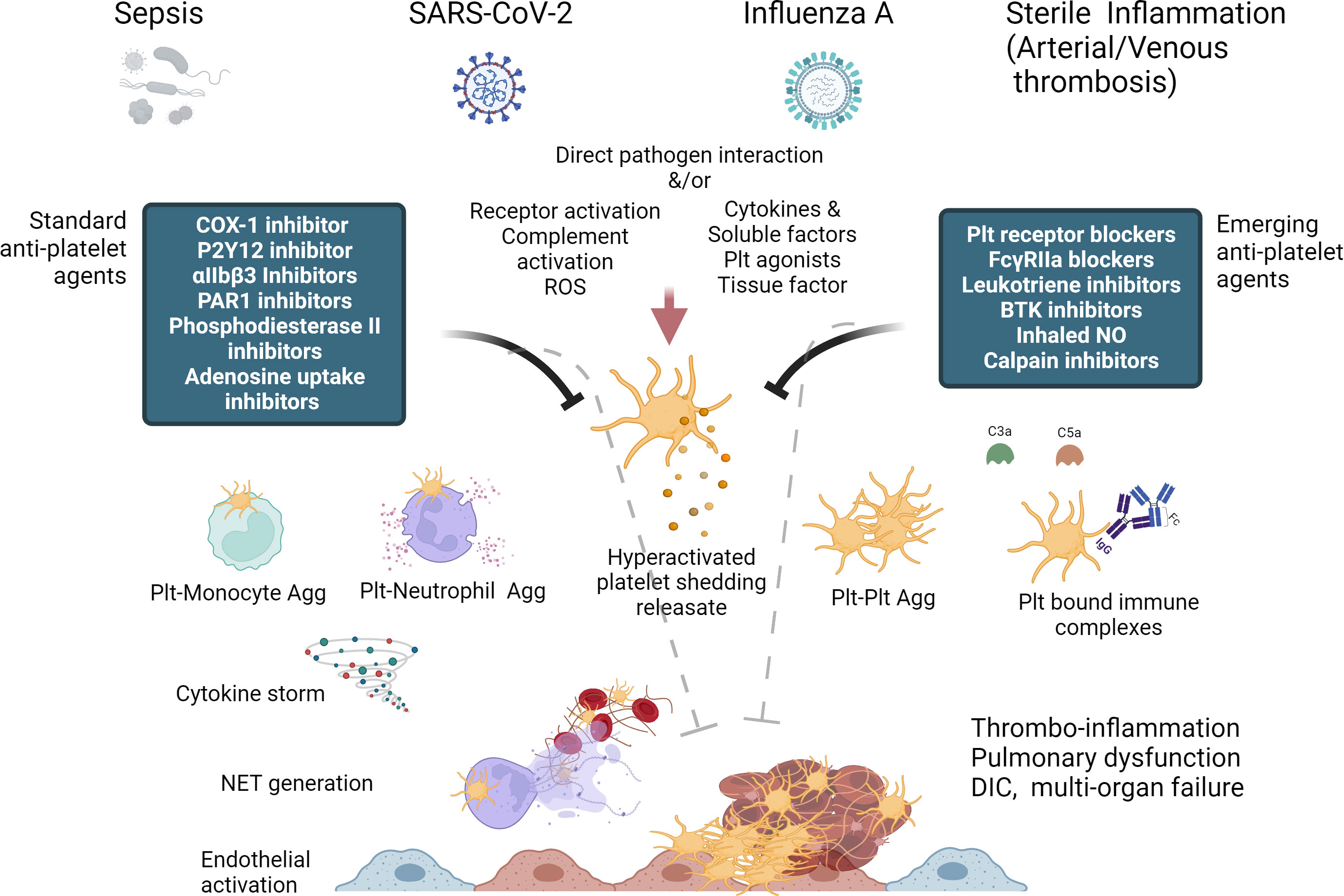
Figure 1 Diagrammatic summary of Platelet hyperactivation and dysfunction in thrombo-inflammation and potential of standard and emerging anti-platelet therapies, some of which can act as both anti-thrombotic and anti-inflammatory in their action. (Created with BioRender.com).
The versatile functions performed by platelets can be attributed to various receptors present on them. Platelets have receptors like glycoprotein (GP) Ib-V-IX, GPVI and α2β1 that aids in adhesion to the damaged vasculature. Platelet surface receptors that respond to soluble agonists are P2Y12, thromboxane receptor and protease-activated receptor (PAR) 1 and 4 (13). Together, they belong to the G-protein coupled receptor family and mediate the second wave of platelet activation. Pathogen binding and the cellular activation of platelets is mediated by receptors like integrins, lectins and toll-like receptors (TLRs) (14, 15). Immune complexes are recognized by Fc receptor FcγRIIA present on human platelets. Infection-associated thrombocytopenia is due to an increase in platelet activation (16, 17). Activated platelets bind to leukocytes including neutrophils, monocytes, eosinophils and lymphocytes which forms heterotypic cell aggregates. Activated platelets also express P-selectin (CD62P) on their surface and released on extracellular vesicles (EVs) and harbor an array of cytokines, such as IL-1β and chemokines, such as RANTES (CCL5) (18).
2 Role of platelets in sterile and non-sterile inflammation
2.1 Sterile inflammation
2.1.1 Atherosclerosis
In patients with atherosclerosis, which is a thrombo‐inflammatory disorder, the inflammatory and immune response to the endothelial dysfunction and oxidized lipids leads to formation of atherosclerotic plaques. There is accumulation of platelets and leukocytes at the site of atherosclerosis, that promotes plaque growth and progression. This eventually leads to destabilization of the underlying endothelial cell (EC) layer resulting in plaque instability/rupture and exposure of extracellular matrix (ECM) and procoagulant factors including tissue factor (TF) (19–22). Intravascular exposure of TF leads to activation of coagulation and thrombin generation. Platelets respond to ECM exposure and thrombin (primary activation) generation. In addition, platelets are also believed to be the initial regulators in the development of atherosclerotic lesions. They bind to activated ECs, leukocytes and initiate the transformation of the monocytes into macrophages. Platelets promote foam cell formation and internalize oxidized phospholipids. They also recruit progenitor cells that, depending on conditions, can differentiate into foam cells or ECs to the site of lesion. Hence, platelets not only promote progression but also regulate initiation and development of the atherosclerotic lesions (23). The platelets are recruited through GPVI‐laminin interaction, on the intact plaque promoting atheroprogression (24). Plaque rupture at the site of fissured lesions triggers the recruitment of platelets via GPVI‐collagen interaction. GPVI is the essential platelet collagen receptor in atherothrombosis, thus inhibition of either its extracellular domain or the downstream signaling can inhibit thrombus formation on atherosclerotic plaque. Janina et al, showed that anti-GPVI antibodies can inhibit atherosclerotic plaque-induced platelet aggregation under flow and static conditions, thus, targeting GPVI-collagen interaction along with other antiplatelet therapies in patients with spontaneous plaque rupture or intervention-associated plaque injury can be beneficial (25).
The activation of the platelets can also contribute to the pathogenesis of chronic-vascular inflammation and atherosclerosis independently of atherothrombosis. Platelets promote the uptake of oxidated low‐density lipoproteins (OxLDLs) by monocytes and macrophages, thereby increasing the monocyte recruitment and their adhesion to the atherosclerotic or inflamed endothelium. They also secrete different cytokines/chemokines contributing systemic inflammatory responses (26). Monocyte recruitment by activated platelets is mediated through interaction of P‐selectin with P-selectin glycoprotein ligand-1 (PSGL‐1) and CD40L–MAC‐1, and via deposition of platelet factor 4 (PF4, CXCL4) and RANTES on ECs and monocytes (27–31). Moreover, there are reports which show that PF4 downregulates the athero-protective genes in human macrophages (32). It is also reported PF4 increases OxLDL uptake by macrophages, thus exacerbating atherosclerosis (33). Studies with hyperlipidemic mice and mouse model of stroke showed that platelet PF4 forms heterodimers with RANTES, causing increased monocyte adhesion to ECs. Disruption of this interaction can be an approach to reduce atherosclerotic plaque formation (34, 35). Also, the monocyte recruitment and activation via platelets promote plaque instability, partly by increasing matrix metallopeptidase 9 production by monocytes (36). Factors released from activated platelets might also enhance the endothelial permeability, thus facilitating the accumulation of lipids within the vessel wall. Patients with atherosclerotic vascular disease exhibit increased number of platelet-leukocyte aggregates (PLAs). The elevated number of heterotypic PLAs in circulation is suggestive of their proinflammatory role in atherosclerosis, as it is directly corelated with higher risk of cardiovascular and cerebrovascular diseases associated with increased ECs activation (37). Due to the crucial role played by the platelets in thrombo-inflammatory events, the classical antiplatelet drugs are useful in treating thrombotic complications in arterial cerebrovascular and cardiovascular thrombosis including atherosclerosis (38, 39). P2Y12 receptor antagonists are widely used antiplatelet agents and have additional anti‐inflammatory properties that are associated with a decrease in platelet P‐selectin, circulatory PLAs, and soluble CD40L and RANTES (37). Despite having anti-thrombotic and anti‐inflammatory effects, these drugs fail to prevent the progression of already established atherosclerosis in patients. Therefore, more recently, the therapies that disrupt the PLAs have gained interest, this includes the inhibitors of P‐selectin, PSGL‐1, CD40L, and GPIb. The review by Rainger et al. summarizes the harmful and beneficial outcomes of using above mentioned inhibitors in atherosclerosis (38). Soluble CD40L is reported to plays an important role in regulating platelet-dependent thrombotic and inflammatory responses (40, 41). It mediates stimulation-induced platelet release of reactive oxygen species (ROS) through activation of Akt and p38 mitogen activated protein kinase (MAPK) signaling pathways that directs stimulation of nuclear factor kappaB and enhanced synthesis of CD40L and MCP1 (42). Increased levels of CD40L and MCP1 leads to adherence of CD40-positive cells, like platelets, to the vasculature modulating atherothrombosis (43). Along with atherosclerosis, various cardiovascular diseases (CVDs) are associated with platelet oxidative stress, thus various cardiovascular therapeutics explore altering oxidative-dependent platelet function as preventive or therapeutic measure. It has been shown that both pravastatin and aspirin inhibit expression of lectin-like oxidized LDL receptor 1 (LOX-1) on platelets by favorably affecting release of ROS from activated platelets (4). Dipyridamole at therapeutically relevant concentrations, suppresses the formation of ROS in endothelial cells and platelets thereby improving the cellular redox status (43). Polymeric flavonoids, alter platelet release of reactive oxygen intermediates with increased NO release and attenuated superoxide production, thereby leading to an immediate attenuation of release of sCD40L an important inflammatory mediator (44), The polyphenols catechin and quercetin act synergistically to reduce platelet recruitment via inhibition of PKC-dependent NAD(P)H oxidase activation, resulting in downregulation of NO-mediated platelet GPIIb/IIIa (45).
The use of these inhibitors is presently debatable as some of them have shown promising results in pre-clinical models, but clinical trials have been disappointing, and several studies are still on going. Also, heterogeneity and multifactorial nature of the atherosclerosis along with engagement of multiple receptors through disease progression can be held responsible for the differences observed between patients and experimental models (46).
2.1.2 Deep vein thrombosis
DVT is a multifactorial disease, dictated by presence of various genetic and environmental risk factors. It is characterized by reduction of blood flow, endothelial and stromal cell activation, and coagulation dysregulation that eventually leads to thrombus formation in veins. DVT is most prevalent in the lower proximities i.e., legs under the muscular fascia of the limbs or in the central deep veins. Vascular hemostasis and thrombosis are mediated by two major mechanisms that depends on vascular damage or vessel structure (47). These two mechanisms are intrinsic and extrinsic pathways of coagulation mediated by collagen and TF, respectively. The damage to EC layer during normal hemostasis may occur which exposes the collagen from the subendothelial space. Platelets interact with the exposed collagen and von Willebrand factor (vWF) through their GPVI and GPIb/V/IX and act as mediators of primary hemostasis. The collagen exposure results in platelet adhesion and formation of the platelet monolayer at the site of vascular injury. The activated platelets result in recruitment of other circulating platelets via secreting various aggregatory mediators including thromboxane A2, ADP, ultra-large vWF (ulvWF) multimers. Coagulation activation leads to thrombin generation and subsequently platelet PAR activation. Also, platelet form a 3D structure through aggregating by their activated GPIIb/IIIa (αIIbβ3) integrins (48). In murine DVT models, initiated by the ligation of inferior vena cava (IVC), it was reported that the platelets and leukocytes are recruited to vessel wall prior to thrombus formation (49, 50). Platelets are either recruited as small aggregates or as single cells at the site of ligation and bind to the vasculature forming heterotypic PLAs by their GPIbα receptor. The monocytes assist the DVT progression predominantly by expressing TF that triggers the extrinsic coagulation pathway The neutrophils on the other hand aid thrombus formation by interacting with platelets and releasing NETs (51). The platelet‐derived high mobility group box 1 (HMGB‐1) is proposed to be responsible for triggering NETosis in the sterile environment inside the blood vessel wall. There are studies that show that the effect of HMGB‐1 is potentiated via the P‐selectin–PSGL‐1 axis (52), as P-selectin deficient mice have better protection against DVT (53, 54). Platelet‐originated HMGB‐1 is known to increase the local inflammation as the result of increased increases neutrophil and monocyte sequestration at venous wall, while also promoting NETosis. The recent studies encompassing in vivo murine models of DVT show that complement activation also regulate the development of DVT with the components of complement displaying distinct role in thrombus formation. The activation of C3 results in platelet and fibrin deposition, whereas C5 activation results in increased TF expression on monocytes. It also promotes thrombo-inflammation by precipitating fibrin generation (55). Clinical data show increased PLAs in venous thrombosis patients (56). The high levels of platelet-neutrophil aggregates represent a risk factor for VT, as patients with DVT show correlation between platelet activation and increase in circulating platelet-neutrophil aggregates (57). Increase in platelet-monocyte aggregates were found in surgery-associated VTE (58), which correlated with increased plasma C-reactive protein levels, which itself is an important systemic proinflammatory marker (59). Anticoagulants have been keystone of contemporary therapy for thrombosis, although mechanical thrombectomy, thrombolysis and angioplasty have been also explored and used. With the emerging role of platelets in DVT, the antiplatelet drugs are also being explored for their therapeutic use to treat the disease. In particular acetylsalicylic acid (ASA), when used after initial anticoagulant treatment as a long-term secondary preventive strategy in patients with VTE, were shown to reduce risk of primary thromboembolism as well as recurrence of secondary VTE (60–62). The cardioprotective effect of ASA are mediated through irreversible inhibition of platelet cyclooxygenase 1 and blockade of the production of thromboxane A2 (TXA2). The anti-platelet effect of ASA is through reduced production of TXA2 (63).
In conclusion, initial driver of venous thrombosis is the activation of coagulation system, with significant contribution of platelets interacting with the innate immune system. These directly contribute, both systemically (in circulation) and locally (vein vasculature), to the initiation and progression of DVT and associated thrombo-inflammatory response.
2.2 Non-sterile inflammation driven by infection
2.2.1 Sepsis
In the past decade, platelets have emerged as one of the crucial players in mediating the response to infectious disease. The ability of platelets to sense and respond to various exogenous and endogenous infectious and inflammatory signals to initiate an immune response is attributed to the wide range of complement and PRRs expressed by them including TLRs and Fc receptors (64). In response to pathogenic challenge, the activated platelets secrete their granules which contain variety of immunomodulatory and antimicrobial molecules that either aids in immune cell differentiation and activation or can directly kill pathogens. In murine models of sepsis, the platelet depletion and thrombocytopenia are associated with more severe response to disease, highlighting the protective role of platelets in sepsis (65). Early platelet transfusion has been reported to be protective in the mouse model of bacterial peritonitis (66). Platelet transfusion to reduce thrombocytopenia, was found to decrease the plasma levels of various inflammatory cytokines such as IL-6 and TNF‐α, and thus improving survival (66). The proposed underlying mechanism is macrophage polarization towards the pro‐inflammatory phenotype and subsequent increase in survival of septic mice (67). The plausible mechanism of bacterial clearance by platelets is thought to be via GPIb-CD11b interaction. In addition, platelet transfusion also dampens the systemic inflammation reaction by sequestering cytokines released from activation of immune cells (67). Lipopolysaccharide (LPS)-induced endotoxemia has sepsis-like features. LPS activate platelet TLR4 which enhances the release of NETs (68). NET release is mediated by P-selectins (52), release of HMGB‐1 (69) and β1‐defensins (70). Platelets also release IL‐1β-rich EVs in response to LPS-dependent TLR4 activation, which enhances ECs activation and propagation of the inflammatory response (71).
NETs and platelets are also reported to induce disseminated intravascular coagulation (DIC) which deteriorates the organ function (71). Additionally, platelets also contribute to the innate immune surveillance system of the liver as they transiently interact with the Kupffer cells that lines the walls of sinusoids in the liver through GPIbα‐vWF interaction (72). The interaction of Kupffer cells and platelets is stabilised via GPIIb-IIIa‐mediated platelet adhesion in presence of bacteria like Bacillus cereus and methicillin‐resistant Staphylococcus aureus. This stable adhesion leads to increased infiltration of neutrophils to the liver sinusoids and evoke host response against the pathogen (73).
Studies show that, platelets through TLR4 act as inflammatory sentinels and thus surrounds and isolate an infection, along with modulating release of proinflammatory cytokine (74). Thus, therapeutics that affect platelet-TLR signaling are being explored. Eritoran, is one such example. It competitively binds and antagonize TLR4 as mimics lipid A portion of LPS and acts as a synthetic analogue (75, 76). Therefore, it has been extensively explored as a therapeutic intervention for rampant endotoxin-mediated inflammation.
During infection, the platelets’ interactions with neutrophils or macrophages are mediated through the platelet immunoreceptor tyrosine-based activation motif (ITAM) receptors C-type lectin-like receptor 2 (CLEC‐2) and GPVI (77–79). Platelet receptors involved in PLA formation during sepsis depends on the time course of infection and the organs involved. The increase in PLAs in septic patients is inversely correlated with survival, and patients develop multiple organ failure most likely due to an increase in sequestration (80). Though during the early phases of sepsis there are elevated levels of platelet-neutrophil aggregates, but they decrease significantly with the disease progression (80). Also, the association of platelet‐monocyte aggregates with mortality depends upon age of patients, with higher mortality in older patients but not in young patients (81). Moreover, platelet depletion resulted in markedly reduced leukocyte infiltration into tissues in animal models of sepsis or ischemia reperfusion (IR) (81). Thus, in septic patients, whether targeting the platelet-immune cell interaction is beneficial or detrimental, it depends upon various other factors like disease stage and associated comorbidities.
The data from various preclinical studies support the hypothesis that the antiplatelet agents, integrin αIIbβ3 antagonists or ASA, may reduce sepsis-associated mortality (82). The αIIbβ3 antagonists were shown to be beneficial in animal models of sepsis as they inhibit platelet aggregation by blocking fibrinogen binding to integrin αIIbβ3. In LPS-induced rat sepsis model the administration of abciximab, a Fab fragment which inhibitsαIIbβ3 activation, reduced tissue edema and vascular leakage (83, 84). The use of αIIbβ3 antagonists also reduced EC damage and mortality in septic mice (85). Lastly, the blockade of integrin αIIbβ3 preserved red blood cell (RBC) and white blood cell (WBC) count and resulted in delayed thrombocytopenia and reduced renal damage in a baboon model of sepsis (86). Even with the success in animal models, these inhibitors have not been tested in sepsis patients. The crucial role of platelets in accelerating the septic complications is further emphasized by the study of triggering receptor expressed in myeloid cells (TREM) gene cluster. TREM-like transcript-1 (TLT-1) receptor is highly expressed in platelets and remain sequestered within platelet alpha-granules in resting platelets and is translocated to the surface after activation such as with LPS or thrombin (87, 88). Platelet TLT-1 binds to fibrinogen and facilitate platelet aggregation, and the soluble form of TLT-1 (sTLT-1) acts as a potent endogenous regulator of sepsis-associated inflammation (89). Sepsis patients exhibit elevated levels of sTLT-1, that strongly correlates with DIC score and high levels of D-dimer (89), suggestive of role of platelet-derived TLT-1 in pathology of diseases manifested as acute lung injury and acute respiratory distress syndrome (ARDS) (89).
Considering all the studies stated above, platelets play a crucial role at various stages of sepsis contributing to the thrombo-inflammatory pathophysiology and the potential use of platelet activation inhibitors as potential therapeutic approach should be explored more.
2.2.2 Influenza A virus infection
Seasonal influenza infection is one of the leading causes of death by infectious disease in the United States (90). IAV is a common respiratory tract infection, that during pandemic of 1918-1919, caused 50-100 million deaths and still infects 10-20% population every year (91). In addition, IAV can cause reoccurring pandemic as seen in 2009 (92). Though adaptive immunity is required for later clearance of IAV, innate immunity plays a key role in recognition of the viral infection and initiation of immune response. It is important for viral control and clearance, but aberrant immune response can also lead to collateral damage and subsequent organ dysfunction/failure (92, 93). Severe cases of IAV infection are marked by tissue pathology and excessive inflammation and coagulation activation within the lungs (94–96). Platelets play a significant role in host defense against influenza viruses as they express various immune receptors and immune effectors upon activation by the viral invasion (97, 98). Human platelets can endocytose IAV in vitro and IAV viral particles have been seen within platelets in infected patients (99). In the murine model of pulmonary viral infection, a direct correlation of platelet accumulation in the lungs with the progression of disease has been reported (100, 101). Soon after IAV infection, platelets form aggregates with monocytes (102). Platelets also form PLAs with neutrophils and these PLAs transmigrate from circulation to the lung airspace of IAV infected mice (100). During viral infections, platelet-neutrophil interactions are often needed for neutrophil recruitment to site of infection, and studies demonstrates that these interactions were crucial for NET releases (101). Koupenova et al. recently showed that IAV engulfment via platelets causes TLR7-dependent release of complement factor C3 and subsequent activation of neutrophils and their NETosis (99). Though several recent studies validate that platelet and neutrophil recruitment to lungs drive pathogenesis of IAV, but the specific mechanisms involved have not been deciphered. The release of NETs along with their constituent neutrophil proteases, histone proteins and DNA act as primary driving forces in initiation of both thrombosis and fibrinolysis simultaneously, which form the hallmarks of associated DIC pathology (103, 104). NET generation can also act as part of antiviral immune response (105). These findings are suggestive of a vicious cycle including NET release, formation microvascular thrombosis and thrombin-induced platelet activation that together can further leads to platelet hyperactivation amplifying thrombo-inflammation and tissue damage. The recent findings by Kim et al. showed that inhibition of platelet aggregation in CD41-deficient mice or platelet-neutrophil interactions by using CD18 blocking antibodies prevents the formation of NETs and subsequent lung tissue damage in IAV infected mice (105). After IAV infection, thrombin activates platelets that promotes coagulation along with the release of inflammatory cytokines from platelets (106). The early thrombin signal generated just after the severe IAV infection is speculated to link coagulation and inflammation, further enhancing platelet-mediated thrombo-inflammatory responses (106).
Different PRRs present on platelets can bind IAV virions, further illustrating the importance of platelets in host response to influenza. These PRRs include TLR7 and TLR3 which induce prothrombotic responses by altering platelet-leukocyte interactions independent of aggregation mediated by thrombin (107). Stimulation of TLR7 on the platelet leads to surface expression of P-selectin and CD154, causing α-granule release. However, TLR3 upon stimulation, potentiates arachidonic acid-, ADP- or collagen-mediated platelet aggregation at high concentration of agonist that possibly represent late stages of infection (108, 109).
In addition, PAR1 and PAR4 are reported to be involved in progression of IAV pathogenesis (102, 110, 111). Mouse platelets lack PAR1, therefore, thrombin-mediated platelet activation occurs through PAR4 in mice (112, 113). The work by Tatsumi et al. (114) showed that global deficiency of PAR4 in mice worsens the disease outcome in the context of mild to moderate IAV infection suggestive of protective role of PAR4 during influenza infection. However, the authors were not able to separate the contribution of PAR4 on the different cell-types including platelets to IAV pathology. In line with the findings by Tatsumi et al (114),, a more recent study showed that PAR4 inhibition in wildtype mice caused more IAV-induced pathology during mild infection (105). However, during severe IAV infection PAR4 inhibition in wildtype mice seemed to mediate protection (105), which was suggested before by observations of Riteau’s group (101). The dual infection severity-dependent role of platelet PAR4 in at least IAV infection is intriguing and needs further investigation.
These studies, therefore, suggest that platelets contribute to lung injury in IAV infection likely by increasing inflammation, neutrophil recruitment/activation, and NET formation by various mechanisms. Therefore, platelet receptors can be explored for identifying reliable therapeutic targets. P2Y12 receptor antagonists are commonly used as antiplatelet agents that reduce platelet–leukocyte interactions and alter inflammatory biomarkers, associated with improved lung function in mouse models of pneumonia (80, 115). Studies showed that P2Y12 inhibition as well as ASA treatment reduced IAV pathology in mice (101, 116). Preliminary findings with P2Y12 receptor antagonists also exhibited improved lung function in humans with pneumonia (117–119).
The non-coding miR-223 regulate platelet expression of the P2Y12 receptor (120) and has been also implicated in negatively regulating TLR-NFκB signaling (121). The studies show that overexpression of miR-223 remarkably decreases pro-inflammatory cytokine production in TLR-stimulated macrophages (122, 123) and reduces neutrophilic inflammation (124). In human studies, lower plasma levels of miR-223 in patients with both stable (125) and acute (123, 126) CVD are associated with high platelet reactivity (126). In addition, ASA treatment was shown to reduce ARDS onset but did not reduce ARDS severity which may be due to the small patient numbers in those trials. These preliminary but underpowered observations underscore the need of larger multicentral clinical trials to finally test the usefulness of anti-platelet drugs in viral ARDS. The leap in knowledge for defining new pathways accelerating thrombo-inflammation warrants the study of platelet receptors belonging to the ITAM family as potential alternative therapeutic targets. For instance, the role of the ITAM receptors in IAV are not fully understood. While Fc gamma receptor (FcγRIIA) involvement in platelet activation during IAV infection was shown, the contribution of other receptors such as CLEC-2 or GPVI was unclear until recently, Boilard et al. showed Influenza virus activates platelets through FcγRIIA signaling and thrombin generation (107). Deficiency of either receptor not posing a major bleeding risk, make them potential attractive therapeutic targets in severe IAV infection (107).
Thus, the platelet-neutrophil interaction promotes acute thrombo-inflammatory responses. Also, formation of platelet-neutrophil aggregates leads to microvascular obstruction and inflammation in various thrombo-inflammatory disorders, including the acute coronary syndromes, lung injury as in infections, and ischemic stroke (119, 127, 128). The receptors involved in these interactions can be explored for their therapeutic potential. Preclinical studies have confirmed that targeting adhesion molecules on platelets like P-selectin, GPIb, αIIbβ3 and on neutrophils like PSGL-1, Mac-1, inhibits the formation of neutrophil–platelet aggregates and thereby improves inflammation response and curtails microvascular dysfunction. Inclacumab, a monoclonal antibody against P-selectin in phase 1 trial has confirmed the safety and dosing as it does not increase bleeding time (128).
As the studies with new age anti-platelet drugs are still in clinical trials, it remains unclear whether emerging strategies, that primarily have cytoprotective and anti-inflammatory properties with less impact on hemostasis, will be effective. This is because previous therapeutic approaches that do not alter coagulation and tissue perfusion have not successfully improved survival or reduced organ injury. Therefore, effective, and safe therapies based on targeting platelet receptors, remains a challenge for researchers.
2.2.3 SARS-corona Virus-2 infection
Initial COVID-19 patients’ autopsy studies had confirmed thrombosis occurring particularly within small vessels (platelet rich thrombotic microangiopathy) in lungs, with or without hemorrhage, which is a feature of the typical ARDS pathology of diffusely edematous lungs (129–131). As pandemic progressed, COVID-19 associated thrombo-embolic episodes included both microvascular and macrovascular thrombotic events including in situ-pulmonary thrombosis, and in liver and other sites (132–134), were associated with mortality. This indicated a hypercoagulable state in COVID-19 patients which involved endothelial and platelet dysfunction (135). The COVID-19 associated platelet dysfunction included reports of platelet hyperreactivity, platelet destruction and platelet-immune complex formation in patients regardless in cases of mild or severe disease progression.
3 Multidimensional effects of SARS-CoV-2 on platelets
The platelets can be affected by SARS-CoV-2 infection directly or indirectly. As platelets are known to have ability to interact with viruses, the reports about SARS-CoV-2 (a single stranded RNA virus) interaction with platelets were not surprising. The known receptors for SARS-CoV-2 entry in human cells include angiotensin-converting enzyme 2 (ACE-2) and transmembrane protease serine 2 (TMPRSS-2). The conflicting reports on ACE-2 expression in platelets imply that ACE-2 presence in platelets is arguable and may only be present, if at all, in very low amounts in a fraction of patients (136). TMPRSS2 is another receptor, which has also been suggested to be possible mediator for SARS-CoV-2 interaction with platelets (137). Besides ACE-2, SARS-CoV-2 spike protein can interact with integrin α5β1, the fibronectin receptor, on ECs inducing a proinflammatory phenotype (138). Moreover, α5β1 mediates SARS-CoV-2 infection in an ACE2-independent way (139). Importantly, platelets express α5β1 and a direct interaction of SARS-CoV-2 spike protein with platelet α5β1 is therefore possible. Indeed, the α5β1 binding peptide ATN-161 can block SAR-CoV-2 infection and was further showed to reduce platelet activation (140, 141). While mechanism of entry of SARS-CoV-2 in platelets may not be confirmed, its presence (Viral RNA traces, proteins, and whole virus) in patient platelets has been reported by several studies (136, 142). A remarkable study demonstrated the SARS-CoV-2 induced programmed cell death and necroptosis of platelets in which the virions were shown by transmission electron microscopy, to get internalized in platelets by being bound to EVs (120).
Besides, directly binding to viral particles, In COVID-19, platelets may also be influenced indirectly. As one of the hallmarks of COVID-19 pathology is ‘cytokine storm’ or ‘hypercytokinemia’ characterised by immune dysfunction involving systemic inflammation which spreads to multiple organs of patient and can lead to fatal multi organ failure (142). Such a systemic inflammation milieu resulting from a disproportionate immune system activation can activate platelet either through individual cytokines or by immune complexes formed in COVID-19 patients. Presence of autoantibodies in COVID-19 patients can also trigger platelet dysfunction. COVID-19 platelets have exhibited heparin-induced thrombocytopenia (HIT)-like features which are thought to be because of anti-PF4-heparin complex antibody generation (143, 144). However, such HIT-like features were also seen in patients without exposure to heparin (145), which indicates a complex mechanism behind this observation Other pathological factors such as hypoxemia have also been proposed to promote platelet hyperreactivity in COVID-19, as hypoxia has been known to induce platelet hyperactivation (146). Very recently, platelets were found partly desensitised along with presence of autophagosomes and even with visible traces of viral particles in COVID-19 patient samples (147). Interestingly, a platelet lipidome study found altered lipid composition of platelets in COVID-19 patients and linked it to platelet activation (148). Thus, platelets are affected at multiple levels in COVID-19.
4 Consequences of COVID-19 associated platelet dysfunction
The dysfunctional platelets in COVID-19, whether its hyperactivation of platelets or related to platelet death, consumption, or all of these, can complicate clinical management of COVID-19 patient in form of increased risk of thrombo-embolism, DIC or haemorrhage. Emerging platelet studies have provided some insights into intricacies of platelet dysfunction in COVID-19.
In context of haemostatic roles, several studies found platelets to be hyperreactive in COVID-19 in response to agonists. An altered transcriptional profile with enrichment in the pathways of antigen presentation, and mitochondrial dysfunction was observed by RNA sequencing of platelets in COVID-19 patients (142). Platelet surface markers for activation were also increased in a unique pattern in COVID-19 patients (149) along with platelet proteomic changes (150). Apart from themselves being hyperreactive, they were also reported to induce TF expression in monocytes from patients (151). Platelets’ contribution to plasma vWF, fibrinogen and FXII was also found to be higher in COVID-19 patients (152). More recently, a large-scale single cell platelet imaging by intelligent platelet morphometry, the investigators analyzed whole blood samples to identify platelet aggregates in nearly 90% of all COVID-19 patients (153). Interestingly, in this study, the platelet aggregates were also observed in some patients even after full recovery from COVID-19 symptoms, which indicate a long-lasting effect of COVID-19 on platelet physiology. As a large fraction of COVID-19 patients also show mild thrombocytopenia, platelet apoptosis was studied. The patients’ platelets showed more signs of apoptosis than healthy donors and even SARS-CoV-2 was demonstrated to directly cause apoptosis and necroptosis ex vivo (136, 150). COVID-19 associated platelet apoptosis was also found mediated by circulating immune complexes in patient’s sera (154). There are only limited data available from animal studies with SARS-CoV-2 or the related murine coronavirus (MHV, murine hepatitis virus) which can cause SARS-like symptoms and its effect on platelets. The recent study by Andrade et al. found that MHV infection caused early thrombocytopenia, hypercytokinemia and multiorgan failure and thereby mirroring some clinical features observed in moderate and severe cases of COVID-19 (155). Importantly, local lung and systemic changes trigged by the MHV infection could be prevented by inhibition of the TNF signaling pathway (156). Lack of TNFR1 or the use of the TNF inhibitor etanercept reduced thrombocytopenia, cytokine expression, lung injury and improved survival of MHV-infected mice. In support of the murine study, etanercept also reduced SARS-CoV-2 replication in human lung epithelial cells (156, 157).
Activated or dysfunctional platelets can release multiple cytokines including IL-1β, IL-7, IL-8, MCP-1, MIP-1α, HGF, MCP-3 (158, 159). An initial study on platelets from mild and severe disease COVID-19 patients, found platelets prone to release certain cytokines including IL-1β and CD40L, while many others including IFN-α, TNF-α and TNF-β, Eotaxin, and others were reduced. Interestingly, the same study also reported increase in EVs from COVID-19 platelets in non-severe cases but reduction in severe disease patients (160). However, thrombin induced platelet aggregation was found enhanced in both non-severe and severe disease (160). According to one report, the sCD40L was found to increase in early phase in COVID-19 patients while it decreased in the later phase of disease while sP-Selectin, the other platelet soluble marker showed opposite pattern (161). This suggests platelet response may be very dynamic as COVID-19 progresses.
In addition to cytokine release, platelet-leukocyte crosstalk has also been seen in COVID-19. The neutrophil recruitment to the pulmonary vasculature is an important event shared by ARDS and COVID-19 pathologies (162). NETs can trigger thrombo-inflammation leading to vascular thrombosis and significant mortality (163). The secondary capture’ event defined by activated platelet-neutrophil binding and rolling on endothelium, plays a key role in initiating immune-thrombosis (119). The secondary capture facilitates their transmigration to alveolar lumen and contributes to formation of edematous lungs, which can cause further platelet activation in turn. Platelets from COVID-19 patients have been reported by multiple studies to form increased PLAs which includes neutrophils or monocytes (151, 152). Platelet-Monocyte aggregates were present in higher numbers in critically ill COVID-19 patients than in those with non-severe symptoms (151). Thus, besides promoting increased prothrombotic tendency in COVID-19, platelets can also contribute to immune dysregulation in COVID-19 through soluble factors and direct cellular crosstalk with other immune cells.
5 Emerging therapeutic targets in COVID-19 associated platelet dysfunction
The COVID-19 patients were reported to contain increased titters of neutralizing antibodies, against SARS-CoV-2 components, the levels of which rise with severity of disease. The sera and IgG fractions from COVID-19 patients was able to induce procoagulant platelets. The study of these immune complexes revealed that aberrant glycosylation of Fc domain of these antibodies could activate platelets (164). The afucosylated IgG were higher in severe disease patients and found to activate platelets through platelet surface FcRIIA (165). In another study, the signalling mediators behind this activation was proposed to be PI3K-AKT pathway downstream of activated FcγRIIA receptor on platelet surface (166). Blocking the FcγRIIA could prevent IgG mediated platelet activation in COVID-19 platelets by preventing AKT and PI3K phosphorylation. The investigators also demonstrated that the pharmacological inhibition of AKT and PI3K phosphorylation by BAY1125976 or BYL719 could revert the induction of procoagulant platelets (166). PI3K-AKT signalling pathway has been shown earlier to mediate platelet activation by alpha-IIb mediated induction by agonists (167).
Another extensive study of FcγRIIA and complement pathway in platelets recently uncovered novel anti-platelet drug targets in COVID-19. The FcγRIIA signalling is also mediated by a protein tyrosine kinase Syk, whose inhibition by a pharmacological inhibitor drug fostamatinib could rescue FcγRIIA mediated platelet hyperactivation ex vivo (168). Fostamatinib is an FDA approved drug for first-line treatment for chronic ITP (169), and thus present a potentially safer therapeutic candidate against COVID-19 induced platelet dysfunction. Platelets express complement receptors C3aR (170) and C5aR (171) and complement components such as anaphylatoxins. Complement components have role in inflammatory cascade during COVID-19 infection (172, 173). A murine study with SARS-CoV indicated C3 as driver of disease. In line with this, blockade of C5a and/or C3a by neutralizing antibodies was reduce COVID-19 plasma induced platelet activation in vitro; the effect was more robust when each or both were combined with FcγRIIA blockade (168). As substantial complement activation is seen in autopsies studies in lung and kidney from COVID-19 patients (174, 175), the complement blockade strategy could offer a dual advantage of anti-platelet as well as immunotherapeutic against COVID-19 infection.
A related form of pathological platelet activation is also seen in small fraction of vaccinated COVID-19 patients who manifest Vaccinated Immune Thrombotic Thrombocytopenia (VITT). VITT is also seen to present features of HIT where autoantibodies against PF4 or viral spike protein can trigger platelet activation (176). The platelet immune complex formation in response to VITT antibodies respond in similar manner as that in case of HIT associated anti-PF4 antibodies (177). VITT associated platelet dysfunction also involves platelet FcγRIIA activation (178, 179). Intravenous IVIG could was able to limit the VITT (180). Platelet activation induced by plasma from VITT patients was also reverted by action of antiplatelet drugs – indomethacin and ticagrelor, and tyrosine kinase inhibitors, in the ex vivo settings (181).
The drug screening for in search of effective therapeutic against SARS-CoV-2 infection led to identification of Mpro inhibitors. Mpro is a cysteine protease in host cells which is also used by invading viruses including SARS-CoV-2 for its multiplication and propagation. One of the agents which inhibited Mpro was a Calpain VI inhibitor and Calpain II and XII inhibitors. One of the calpain inhibitors has been shown previously to inhibit the previous coronavirus SARS-CoV replication (182). Calpain is also important for platelets, where it acts by proteolysing structural platelet proteins and thus facilitating both early and late events of platelet activation and spreading (183). Calpain is also involved in endocytosis and apoptosis (146). Calpain inhibition has also been shown to restrict venous thrombosis in rat models (184). A recently manufactured A‐705053, small molecule calpain inhibitor can be orally administrated with enhanced pharmacokinetics (185). BLD-2660 is a new, artificial, orally active, small-molecule taken selective inhibitor of Calpain 1, 2, and 9 which is metabolically stable and permeable. It has been developed for the therapy of COVID-19 and is under trial (ClinicalTrials.gov Identifier: NCT04334460). Such a drug, if successfully tested for both anti-platelet and anti-viral functions and safety, can become one dual action drug against COVID-19 to treat both viral growth and associated platelet dysfunction.
Another drug target which can serve dual purpose of anti-platelet and anti-inflammatory actions in COVID-19 are leukotrienes. LTE4, a cysteinyl leukotriene has recently been found to induce platelet activation in vitro (186). The targeting of LTE4 by an approved clinical use drug – Monteleukast, has been shown to have anti-platelet activity. The mechanism was proposed to be preventing the surface expression of TF and P-selectin, reducing the formation of circulating monocyte– and granulocyte–platelet aggregates, and, finally, in completely inhibiting the release of TF positive-circulating EVs (186). The antiplatelet mechanism can also be indirect as this LTE4 antagonist reduces IL6 levels or other inflammatory cytokines which thereby may cause reduction of platelet activation. Monteleukast is in clinical use against bronchoconstriction and other inflammatory conditions which makes this a safer drug candidate for testing as anti-platelet and anti-inflammatory drug in COVID-19 (187). Among approved drugs, Nitric oxide (NO) promote vascular smooth muscle cell relaxation (vasodilation), inhibition of platelet activation, and reducing leukocyte adhesion and inflammation (188). Thus, inhaled NO may be a promising strategy for treatment of COVID-19 and an anti-platelet agent, and multiple trials of inhaled NO are in progress (189). Very recently, platelet GPIIb/IIIa receptor blockers eptifibatide or tirofiban have been reported to have reduced thrombus formation in blood from COVID-19 patients, in response to ex vivo shear stress (190).
Another possible antiplatelet target in COVID-19 could be platelet ITAM signaling including podoplanin-CLEC-2 axis. Podoplanin is expressed by lung epithelial cells and on tissue macrophages and could be released from the injured lung. The tyrosine kinase linked receptor CLEC-2 is expressed on platelets and has been reported play a critical role in thrombo-inflammation in mice (88). Recently, it is shown that targeting a non-receptor signaling kinase – Btk which is downstream of CLEC-2 has very effective anti-platelet effect in mice (191). In humans, Btk inhibitors - ibrutinib and dasatinib have recently been approved for use in B-cell malignancies and chronic myeloid leukemia/prostate cancer respectively (191, 192). Post-hoc analysis of these trials has shown that both ibrutinib and dasatinib significantly reduce venous thrombosis and that ibrutinib also reduces arterial thrombosis in B-cell lymphoma patients (193). Btk inhibitors including Ibrutinib have also been proposed to inhibit GPVI dependent platelet activation and thrombosis (194). Apart from its anti-thrombotic action, Btk inhibition has also been shown to reduce hyperinflammation based on myeloid cell cytokine release as demonstrated in mice model of brain ischemia/reperfusion injury (195). Thus, Btk inihibitors can also be potential drug candidates which can meet the need of new anti and anti-inflammatory therapies against COVID-19. Table 1 summaries detail of emerging major pharmacological agents/antibodies (and their molecular targets) having anti-platelet/anti-thrombotic action to combat thrombo-inflammation being tested in COVID-19.
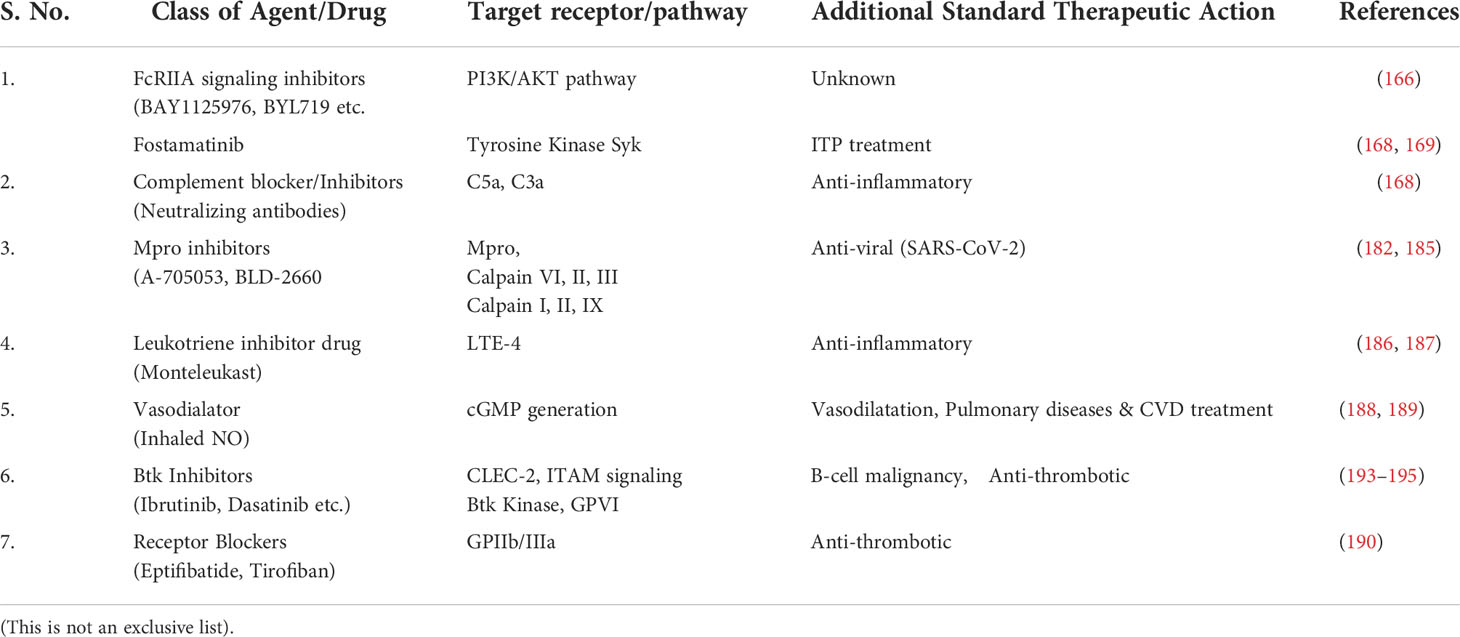
Table 1 Summarized detail of emerging major pharmacological agents/antibodies (and their molecular targets) having anti-platelet/anti-thrombotic action to combat thrombo-inflammation being tested in Covid-19.
Therefore, SARS-CoV-2 infection associated platelet dysfunction and thrombo-inflammation are tightly interlinked. The growing knowledge of platelet signaling and their interactions with immune cells has helped to understand platelet dysfunction and thrombo-inflammation in COVID-19. Understanding the complex multiple layers of COVID-19 associated effects on platelets and their response to affect hemostatic and immune events is the key towards development of novel effective and safer therapies.
6 Conclusion and future directions
The major therapeutic challenge in the field of cardiovascular and associated diseases is to reduce the deleterious impact of microvascular thrombosis and inflammation. Even with the advent progress in therapeutics and better understanding of disease, the successful clinical management remains a formidable challenge in these disorders. This is mainly because of multifactorial and complex nature of innate immunity and hemostatic responses that directs various stages of thrombo-inflammatory process. Therefore, it is difficult to get a single “Holy Grail” that can be used to address and treat the thrombo-inflammatory complications. There are various factors that are critical for optimizing the treatment including diverse genetic background of patients, environmental cues, pathogenic heterogeneity, time of therapeutic interventions etc. Platelets are effective sentinels for sensing and responding to infection by pathogens thereby bridging hemostatic, inflammatory, and immune continuums (196, 197). The activated platelet express integrin αIIbβ3, P-selectin and other receptors on the surface which play key roles in platelet activation. The activation of platelets also leads to formation of platelet aggregates and micro-thrombi, their adherence to the endothelium causing endothelial damage, increased interactions with macrophages and neutrophils, increased NETs formation, and ultimately release of cytokines. All these bridging factors added to the evolution of concept of thrombo-inflammation in various diseases. Though, studies have expanded the armamentarium of platelet functions during infectious diseases, there is lot more to be done. Specially, exploring the platelet receptors and downstream signaling for safer and effective therapeutic development. The various anti-platelet drugs depending upon the receptor they act on are summarized in Figure 2 along with the different diseases they can prevent/manage/treat. Even with the emerging role of platelets in infectious diseases, most of clinically approved anti-platelet drugs are approved for CVDs and related complications only. In future, emerging technological innovations are likely to facilitate use of these receptors as either biomarker or therapeutic targets for infectious-inflammatory diseases as well. Developing the antiplatelet therapies which can serve dual purpose of treating unwanted hyperinflammation and thrombosis would be most effective in clinical management of thrombo-inflammatory diseases such as arterial and venous thrombosis, sepsis, influenza and COVID-19.
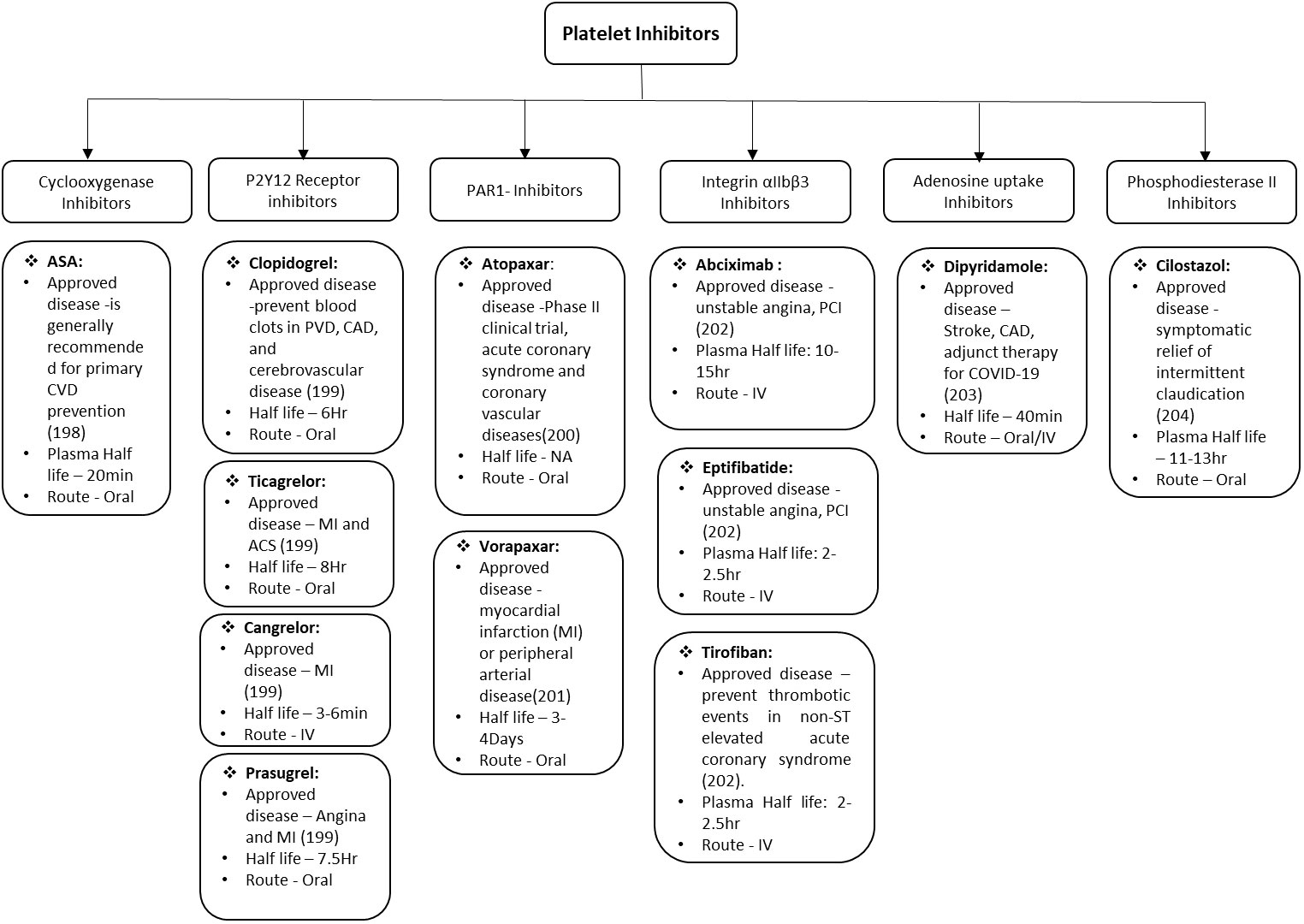
Figure 2 Schematic representation of Anti-platelet drugs in use and under development. ASA, acetylsalicylic acid; CVD, cardiovascular disease; PVD, pulmonary vascular disease; CAD, coronary artery disease; MI, myocardial infarction; ACS, acute coronary syndrome; IV, intra veinous; PCI percutaneous coronary intervention.
Though, the incredible progress in the understanding of anucleate platelet function, beyond just primary hemostasis, has been exciting journey, further discoveries will not only continue decipher role of platelets in infectious diseases but open new doors for better management and treatment of thrombo-inflammatory diseases.
Author contributions
SS, TT and SA conceptualised the idea and wrote the manuscript. SA edited the manuscript and provided funding. All authors contributed to the article and approved the submitted version.
Funding
The presented study was supported by grants HL142799 and HL148432 from the NHLBI to SA.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Deppermann C, Kubes P. Platelets and infection. Semin Immunol (2016) 28:536–45. doi: 10.1016/j.smim.2016.10.005
2. Thomas MR, Storey RF. The role of platelets in inflammation. Thromb Haemost (2015) 114:449–58. doi: 10.1160/TH14-12-1067
3. Rayes J, Watson SP, Nieswandt B. Functional significance of the platelet immune receptors GPVI and CLEC-2. J Clin Invest (2019) 129:12–23. doi: 10.1172/JCI122955
4. Gawaz M, Vogel S. Platelets in tissue repair: control of apoptosis and interactions with regenerative cells. Blood (2013) 122:2550–4. doi: 10.1182/blood-2013-05-468694
5. Li JL, Zarbock A, Hidalgo A. Platelets as autonomous drones for hemostatic and immune surveillance. J Exp Med (2017) 214(8):2193–204. doi: 10.1084/jem.20170879
6. Boulaftali Y, Hess PR, Getz TM, Cholka A, Stolla M, Mackman N, et al. Platelet ITAM signaling is critical for vascular integrity in inflammation. J Clin Invest (2013) 123:908–16. doi: 10.1161/ATVBAHA.118.310224
7. Goerge T, Ho-Tin-Noe B, Carbo C, Benarafa C, Remold-O'Donnell E, Zhao B-Q, et al. Inflammation induces hemorrhage in thrombocytopenia. Blood (2008) 111:4958–64. doi: 10.1113/jphysiol.2011.212886
8. Rayes J, Jadoui S, Lax S, Gros A, Wichaiyo S, Ollivier V, et al. The contribution of platelet glycoprotein receptors to inflammatory bleeding prevention is stimulus and organ dependent. Haematologica (2018) 103:e256–8. doi: 10.1038/s41573-020-0061-0
9. Gros A, Syvannarath V, Lamrani L, Ollivier V, Loyau S, Goerge T, et al. Single platelets seal neutrophil-induced vascular breaches via GPVI during immune-complex-mediated inflammation in mice. Blood (2015) 126:1017–26. doi: 10.1111/jth.12782
10. Ho-Tin-Noe B, Boulaftali Y, Camerer E. Platelets and vascular integrity: how platelets prevent bleeding in inflammation. Blood (2018) 131:277–88. doi: 10.1186/s12865-015-0092-1
11. Grover SP, Bergmeier W, Mackman N. Platelet signaling pathways and new inhibitors. Arterioscler Thromb Vasc Biol (2018) 38(4):e28–35. doi: 10.1007/s00018-009-0209-x
12. Nieswandt B, Kleinschnitz C, Stoll G. Ischaemic stroke: A thrombo-inflammatory disease? J Physiol (2011) 589:4115–23. doi: 10.3389/fimmu.2014.00649
13. Mackman N, Bergmeier W, Stouffer GA and Weitz JI. Therapeutic strategies for thrombosis: new targets and approaches. Nat Rev Drug Discov (2020) 19:333–52. doi: 10.1161/CIRCRESAHA.117.310795
14. Negrotto S, Jaquenod de Giusti C, Rivadeneyra L, Ure AE, Mena HA, Schattner M and Gomez RM. Platelets interact with coxsackieviruses b and have a critical role in the pathogenesis of virusinduced myocarditis. J Thromb haemostasis: JTH (2015) 13:271–82. doi: 10.1016/0002-8703(95)90266-X
15. Chabert A, Hamzeh-Cognasse H, Pozzetto B, Cognasse F, Schattner M, Gomez RM and Garraud O. Human platelets and their capacity of binding viruses: meaning and challenges? BMC Immunol (2015) 16:26. doi: 10.1084/jem.20012044
16. Flaujac C, Boukour S, Cramer-Borde E. Platelets and viruses: An ambivalent relationship. Cell Mol Life sciences: CMLS (2010) 67:545–56. doi: 10.1161/CIRCULATIONAHA.110.961714
17. Assinger A. Platelets and infection - an emerging role of platelets in viral infection. Front Immunol (2014) 5:649. doi: 10.3389/fimmu.2014.00649
18. Koupenova M, Clancy L, Corkrey HA and Freedman JE. Circulating platelets as mediators of immunity, inflammation, and thrombosis. Circ Res (2018) 122:337–51. doi: 10.1111/j.1538-7836.2007.02517.x
19. Merhi Y, Guidoin R, Provost P, Leung TK, Lam JY. Increase of neutrophil adhesion and vasoconstriction with platelet deposition after deep arterial injury by angioplasty. Am Heart J (1995) 129:445–51. doi: 10.1016/j.yjmcc.2010.04.009
20. Massberg S, Brand K, Grüner S, Page S, Müller E, Müller I, et al. A critical role of platelet adhesion in the initiation of atherosclerotic lesion formation. J Exp Med (2002) 196:887–96. doi: 10.1016/j.jacc.2015.03.573
21. Drechsler M, Megens RT, van Zandvoort M, Weber C, Soehnlein O. Hyperlipidemia-triggered neutrophilia promotes early atherosclerosis. Circulation (2010) 122:1837–45. doi: 10.1161/ATVBAHA.113.302919
22. Ylitalo R, Oksala O, Yla-Herttuala S, Ylitalo P. Effects of clodronate (dichloromethylene bisphosphonate) on the development of experimental atherosclerosis in rabbits. J Lab Clin Med (1994) 123:769–76. doi: 10.1161/CIRCRESAHA.111.247684
23. Lindemann S, Krämer B, Seizer P, Gawaz M. Platelets, inflammation and atherosclerosis. J Thromb Haemost (2007) 5(Suppl 1):203–11. doi: 10.1111/j.1538-7836.2007.02517.x
24. Bültmann A, Li Z, Wagner S, Peluso M, Schönberger T, Weis C, et al. Impact of glycoprotein VI and platelet adhesion on atherosclerosis–a possible role of fibronectin. J Mol Cell Cardiol (2010) 49:532–42. doi: 10.1182/blood-2010-01-261206
25. Jamasbi J, Megens R, Bianchini M, Münch G, Ungerer M, Faussner A, et al. Differential inhibition of human atherosclerotic plaque-induced platelet activation by dimeric GPVI-fc and anti-GPVI antibodies: functional and imaging studies. J Am Coll Cardiol (2015) 65:2404–15. doi: 10.1038/28204
26. Badrnya S, Schrottmaier WC, Kral JB, Yaiw K-C, Volf I, Schabbauer G, et al. Platelets mediate oxidized low-density lipoprotein-induced monocyte extravasation and foam cell formation. Arterioscler Thromb Vasc Biol (2014) 34:571–80. doi: 10.1161/01.ATV.0000170133.43608.37
27. Wolf D, Hohmann J-D, Wiedemann A, Bledzka K, Blankenbach H, Marchini T, et al. Binding of CD40L to mac-1's I-domain involves the EQLKKSKTL motif and mediates leukocyte recruitment and atherosclerosis–but does not affect immunity and thrombosis in mice. Circ Res (2011) 109:1269–79. doi: 10.1074/jbc.M208894200
28. Donners M, Beckers L, Lievens D, Munnix I, Heemskerk J, Janssen BJ, et al. The CD40-TRAF6 axis is the key regulator of the CD40/CD40L system in neointima formation and arterial remodeling. Blood (2008) 111:4596–604. doi: 10.3389/fphys.2012.00001
29. Lievens D, Zernecke A, Seijkens T, Soehnlein O, Beckers L, Munnix I, et al. Platelet CD40L mediates thrombotic and inflammatory processes in atherosclerosis. Blood (2010) 116:4317–27. doi: 10.1038/nm.1898
30. Mach F, Schonbeck U, Sukhova GK, Atkinson E, Libby P. Reduction of atherosclerosis in mice by inhibition of CD40 signalling. Nature (1998) 394:200–3. doi: 10.1161/JAHA.116.003615
31. Mause SF, von Hundelshausen P, Zernecke A, Koenen RR, Weber C. Platelet microparticles: A transcellular delivery system for RANTES promoting monocyte recruitment on endothelium. Arterioscler Thromb Vasc Biol (2005) 25:1512–8. doi: 10.1161/hh1801.096339
32. Nassar T, Sachais BS, Sa'ed A, MA K, Bdeir K, Leitersdorf E, et al. Platelet factor 4 enhances the binding of oxidized low-density lipoprotein to vascular wall cells. J Biol Chem (2003) 278:6187–93. doi: 10.1067/mcp.2003.13
33. Gleissner CA. Macrophage phenotype modulation by CXCL4 in atherosclerosis. Front Physiol (2012) 3:1. doi: 10.3109/09537104.2015.1064881
34. Koenen RR, von Hundelshausen P, Nesmelova IV, Zernecke A, Liehn EA, Sarabi A, et al. Disrupting functional interactions between platelet chemokines inhibits atherosclerosis in hyperlipidemic mice. Nat Med (2009) 15:97–103. doi: 10.1161/ATVBAHA.107.159178
35. Fan Y, Xiong X, Zhang Y, Yan D, Jian Z, Xu B, et al. A peptide inhibitor of CXCL4-CCL5 heterodimer formation, protects against stroke in mice. J Am Heart Assoc (2016) 5(9):e003615. doi: 10.1161/01.ATV.0000184765.59207.f3
36. Galt SW, Lindemann S, Medd D, Allen LL, Kraiss LW, Harris ES, et al. Differential regulation of matrix metalloproteinase-9 by monocytes adherent to collagen and platelets. Circ Res (2001) 89:509–16. doi: 10.1074/jbc.M700211200
37. Klinkhardt U, Bauersachs R, Adams J, Graff J, Lindhoff-Last E, Harder S. Clopidogrel but not aspirin reduces p-selectin expression and formation of platelet-leukocyte aggregates in patients with atherosclerotic vascular disease. Clin Pharmacol Ther (2003) 73:232–41. doi: 10.1067/mcp.2003.13
38. Ed Rainger G, Chimen M, Harrison MJ, Yates CM, Harrison P, Watson SP, et al. The role of platelets in the recruitment of leukocytes during vascular disease. Platelets (2015) 26(6):507–20. doi: 10.1124/jpet.105.089987
39. Freedman JE. Oxidative stress and platelets. Arterioscler Thromb Vasc Biol (2008) 28(3):s11–6. doi: 10.1161/ATVBAHA.107.159178
40. Chakrabarti S, Varghese S, Vitseva O, Tanriverdi K, Freedman JE. CD40 ligand influences platelet release of reactive oxygen intermediates. Arterioscler Thromb Vasc Biol (2005) 25:2428–34. doi: 10.1096/fj.05-5269com
41. Chakrabarti S, Blair P, Freedman JE. CD40-40L signaling in vascular inflammation. J Biol Chem (2007) 282:18307–17. doi: 10.1093/eurheartj/ehr373
42. Marwali MR, Hu C, Mohandas B, Dandapat A, Deonikar P, Chen J, et al. Modulation of ADP-induced platelet activation by aspirin and pravastatin: Role of lectin-like oxidized low-density lipoprotein receptor-1, nitric oxide, oxidative stress, and inside-out integrin signaling. J Pharmacol Exp Ther. (2007) 322(3):1324–32. doi: 10.1124/jpet.107.12285
43. Chakrabarti S, Vitseva O, Iyu D, Varghese S, Freedman JE. The effect of dipyridamole on vascular cell-derived reactive oxygen species. J Pharmacol Exp Ther (2005) 315:494–500. doi: 10.1124/jpet.105.089987
44. Vitseva O, Varghese S, Chakrabarti S, Folts JD, Freedman JE. Grape seed and skin extracts inhibit platelet function and release of reactive oxygen intermediates. J Cardiovasc Pharmacol (2005) 46:445–51. doi: 10.1084/jem.20112322
45. Pignatelli P, Di Santo S, Buchetti B, Sanguigni V, Brunelli A, Violi F. Polyphenols enhance platelet nitric oxide by inhibiting protein kinase c-dependent NADPH oxidase activation: effect on platelet recruitment. FASEB J (2006) 20:1082–9. doi: 10.1182/blood-2010-05-287623
46. Patrono C, Andreotti F, Arnesen H, Badimon L, Baigent C, Collet J-P, et al. Antiplatelet agents for the treatment and prevention of atherothrombosis. Eur Heart J (2011) 32:2922–32. doi: 10.1084/jem.20112322
47. Furie B, Furie BC. Mechanisms of thrombus formation. N Engl J Med (2008) 359:938–49. doi: 10.1182/blood-2010-05-287623
48. Koupenova M, Kehrel BE, Corkrey HA, Freedman JE. Thrombosis and platelets: An update. Eur Heart J (2017) 38:785–91. doi: 10.1111/j.1538-7836.2011.04544.x
49. von Brühl M-L, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med (2012) 209:819–35. doi: 10.1182/blood-2015-01-624023
50. Brill A, Fuchs TA, Chauhan AK, Yang JJ, De Meyer SF, Kollnberger M, et al. Von willebrand factor-mediated platelet adhesion is critical for deep vein thrombosis in mouse models. Blood (2011) 117:1400–7. doi: 10.1182/blood-2016-04-710632
51. Brill A, Fuchs TA, Savchenko AS, Thomas GM, Martinod K, De meyer SF, et al. Neutrophil extracellular traps promote deep vein thrombosis in mice. J Thromb Haemost (2012) 10:136–44. doi: 10.1016/j.jacc.2004.12.075
52. Etulain J, Martinod K, Wong SL, Cifuni SM, Schattner M, Wagner DD. P-selectin promotes neutrophil extracellular trap formation in mice. Blood (2015) 126:242–6. doi: 10.1515/cclm-2018-0909
53. Stark K, Philippi V, Stockhausen S, Busse J, Antonelli A, Miller M, et al. Disulfide HMGB1 derived from platelets coordinates venous thrombosis in mice. Blood (2016) 128:2435–49. doi: 10.1160/TH13-02-0113
54. Dyer MR, Chen Q, Haldeman S, Yazdani H, Hoffman R, Loughran P, et al. Deep vein thrombosis in mice is regulated by platelet HMGB1 through release of neutrophil-extracellular traps and DNA. Sci Rep (2018) 8:2068. doi: 10.1038/srep27478
55. Subramaniam S, Jurk K, Hobohm L, Jäckel S, Saffarzadeh M, Schwierczek K, et al. Distinct contributions of complement factors to platelet activation and fibrin formation in venous thrombus development. Blood (2017) 129:2291–302. doi: 10.1056/NEJMoa1114238
56. Chirinos JA, Heresi GA, Velasquez H, Jy W, Jimenez JJ, Ahn E, et al. Elevation of endothelial microparticles, platelets, and leukocyte activation in patients with venous thromboembolism. J Am Coll Cardiol (2005) 45:1467–71. doi: 10.1056/NEJMoa1700518
57. Zhou J, Xu E, Shao K, Shen W, Gu YI, Li M, et al. Circulating platelet-neutrophil aggregates as risk factor for deep venous thrombosis. Clin Chem Lab Med (2019) 57:707–15. doi: 10.1136/bmj.f5133
58. Seizer P, May AE. Platelets and matrix metalloproteinases. Thromb Haemost (2013) 110:903–9. doi: 10.1160/TH13-02-0113
59. Shih L, Kaplan D, Kraiss LW, Casper TC, Pendleton RC, Peters CL, et al. Platelet-monocyte aggregates and c-reactive protein are associated with VTE in older surgical patients. Sci Rep (2016) 6:27478. doi: 10.3389/fimmu.2019.01687
60. Becattini C, Agnelli G, Schenone A, Eichinger S, Bucherini E, Silingardi M, et al. Aspirin for preventing the recurrence of venous thromboembolism. N Engl J Med (2012) 366:1959–67. doi: 10.1038/ncomms3657
61. Weitz JI, Lensing A, Prins MH, Bauersachs R, Beyer-Westendorf J, Bounameaux H, et al. Rivaroxaban or aspirin for extended treatment of venous thromboembolism. N Engl JmMed (2017) 376:1211–22. doi: 10.1016/j.celrep.2019.06.062
62. Castellucci LA, Cameron C, Le Gal G, Rodger MA, Coyle D, Wells PS, et al. Efficacy and safety outcomes of oral anticoagulants and antiplatelet drugs in the secondary prevention of venous thromboembolism: systematic review and network meta-analysis. BMJ (2013) 347:f5133. doi: 10.1038/nm1565
63. Diep R, Garcia D. Does aspirin prevent venous thromboembolism? Hematol Am Soc Hematol Educ Program (2020) 2020(1):634–41. doi: 10.1182/hematology.2020000150
64. Assinger A, Schrottmaier WC, Salzmann M, Rayes J. Platelets in sepsis: An update on experimental models and clinical data. Front Immunol (2019) 10:1687. doi: 10.3389/fimmu.2019.01687
65. Xiang B, Zhang G, Guo L, Li X-A, Morris AJ, Daugherty A, et al. Platelets protect from septic shock by inhibiting macrophage-dependent inflammation via the cyclooxygenase 1 signalling pathway. Nat Commun (2013) 4:2657. doi: 10.4049/jimmunol.1001623
66. Carestia A, Mena HA, Olexen CM, Ortiz Wilczyñski JM, Negrotto S, Errasti AE, et al. Platelets promote macrophage polarization toward pro-inflammatory phenotype and increase survival of septic mice. Cell Rep (2019) 28:e5. doi: 10.1182/blood-2016-09-741298
67. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med (2007) 13:463–9. doi: 10.1038/ni.2631
68. Tadie J-M, Bae H-B, Jiang S, Park DW, Bell CP, Yang H, et al. HMGB1 promotes neutrophil extracellular trap formation through interactions with toll-like receptor 4. Am J Physiol Lung Cell Mol Physiol (2013) 304:L342–9. doi: 10.1152/ajplung.00023.2017
69. Kraemer BF, Campbell RA, Schwertz H, Cody MJ, Franks Z, Tolley ND, et al. Novel anti-bacterial activities of beta-defensin 1 in human platelets: suppression of pathogen growth and signaling of neutrophil extracellular trap formation. PLoS Pathog (2011) 7:e1002355. doi: 10.1016/j.thromres.2009.11.004
70. Brown GT, McIntyre TM. Lipopolysaccharide signaling without a nucleus: kinase cascades stimulate platelet shedding of proinflammatory IL-1beta-rich microparticles. J Immunol (2011) 186:5489–96. doi: 10.1124/jpet.102.044487
71. McDonald B, Davis RP, Kim S-J, Tse M, Esmon CT, Kolaczkowska E, et al. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood (2017) 129:1357–67. doi: 10.1001/jama.2013.2194
72. Wong CH, Jenne CN, Petri B, Chrobok NL, Kubes P. Nucleation of platelets with blood-borne pathogens on kupffer cells precedes other innate immunity and contributes to bacterial clearance. Nat Immunol (2013) 14:785–92. doi: 10.1172/JCI79070
73. Lax S, Rayes J, Wichaiyo S, Haining EJ, Lowe K, Grygielska B, et al. Platelet CLEC-2 protects against lung injury via effects of its ligand podoplanin on inflammatory alveolar macrophages in the mouse. Am J Physiol Lung Cell Mol Physiol (2017) 313:L1016–29. doi: 10.1038/s41467-017-02402-6
74. Beaulieu LM, Freedman JE. The role of inflammation in regulating platelet production and function: Toll-like receptors in platelets and megakaryocytes. Thromb Res (2010) 125(3):205–9. doi: 10.1111/j.1365-2362.1995.tb01694.x
75. Mullarkey M, Rose JR, Bristol J, Kawata T, Kimura A, Kobayashi S, et al. Inhibition of endotoxin response by e5564, a novel toll-like receptor 4-directed endotoxin antagonist. J Pharm Exp (2003) 304:1093–102. doi: 10.1124/jpet.102.044487
76. Opal SM, Laterre P-F, Francois B, LaRosa SP, Angus DC, Mira J-P, et al. Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: The ACCESS randomized trial. JAMA (2013) 309:1154–62. doi: 10.1001/jama.2013.2194
77. Hitchcock JR, Cook CN, Bobat S, Ross EA, Flores-Langarica A, Lowe KL, et al. Inflammation drives thrombosis after salmonella infection via CLEC-2 on platelets. J Clin Invest (2015) 125:4429–46. doi: 10.1016/j.thromres.2013.07.002
78. Rayes J, Lax S, Wichaiyo S, Watson SK, Di Y, Lombard S, et al. The podoplanin-CLEC-2 axis inhibits inflammation in sepsis. Nat Commun (2017) 8:2239. doi: 10.1080/10739680490266216
79. Gawaz M, Fateh-Moghadam S, Pilz G, Gurland HJ, Werdan K. Platelet activation and interaction with leucocytes in patients with sepsis or multiple organ failure. Eur J Clin Invest (1995) 25:843–51. doi: 10.1097/00003246-200106000-00019
80. Gremmel T, Michelson AD, Frelinger AL III, Bhatt DL. Novel aspects of antiplatelet therapy in cardiovascular disease. Res Pract Thromb Haemost (2018) 2:439–49. doi: 10.1182/blood.V89.11.4078
81. Slaba I, Wang J, Kolaczkowska E, McDonald B, Lee WY, Kubes P. Imaging the dynamic platelet-neutrophil response in sterile liver injury and repair in mice. Hepatology (2015) 62(5):1593–605. doi: 10.1371/journal.pone.0041549
82. Akinosoglou K, Alexopoulos D. Use of antiplatelet agents in sepsis: A glimpse into the future. Thromb Res (2014) 133(2):131–8. doi: 10.1182/blood-2004-01-0315
83. Walther A, Czabanka M, Gebhard MM, Martin E. Glycoprotein IIB/IIIA-inhibition and microcirculatory alterations during experimental endotoxemia–an intravital microscopic study in the rat. Microcirculation (2004) 11(1):79–88. doi: 10.4049/jimmunol.1102674
84. Pu Q, Wiel E, Corseaux D, Bordet R, Azrin MA, Ezekowitz MD, et al. Beneficial effect of glycoprotein IIb/IIIa inhibitor (AZ-1) on endothelium in escherichia coli endotoxin-induced shock. Crit Care Med (2001) 29(6):1181–8. doi: 10.1182/blood-2018-03-841593
85. Taylor FB, Coller BS, Chang AC, Peer G, Jordan R, Engellener W, et al. 7E3 F(ab9)2, a monoclonal antibody to the platelet GPIIb/IIIa receptor, protects against microangiopathic hemolytic anemia and microvascular thrombotic renal failure in baboons treated with C4b binding protein and a sublethal infusion of escherichia coli. Blood (1997) 89(11):4078–84. doi: 10.1542/peds.2006-3226
86. Sharron M, Hoptay CE, Wiles AA, Garvin LM, Geha M, Benton AS, et al. Platelets induce apoptosis during sepsis in a contact-dependent manner that is inhibited by GPIIb/IIIa blockade. PLoS One (2012) 7(7):e41549. doi: 10.1016/j.vaccine.2008.06.087
87. Washington AV, Schubert RL, Quigley L, Disipio T, Feltz R, Cho EH, et al. A TREM family member, TLT-1, is found exclusively in the alpha-granules of megakaryocytes and platelets. Blood (2004) 104(4):1042–7. doi: 10.1016/j.bj.2018.12.009
88. Derive M, Bouazza Y, Sennoun N, Marchionni S, Quigley L, Washington V, et al. Soluble TREM-like transcript-1 regulates leukocyte activation and controls microbial sepsis. J Immunol (2012) 188(11):5585–92. doi: 10.1177/1753425913508992
89. Morales-Ortíz J, Deal V, Reyes F, Maldonado-Martínez G, Ledesma N, Staback F, et al. Platelet-derived TLT-1 is a prognostic indicator in ALI/ARDS and prevents tissue damage in the lungs in a mouse model. Blood (2018) 132(23):2495–505. doi: 10.1016/j.jiph.2020.07.003
90. Hamilton BE, Minino AM, Martin JA, Kochanek KD, Strobino DM, Guyer B. Annual summary of vital statistics: 2005. Pediatrics (2007) 119:345–60. doi: 10.1542/peds.2006-3226
91. Schanzer DL, Langley JM, Tam TWS. Co-Morbidities associated with influenza-attributed mortality, 1994–2000, Canada. Vaccine (2008) 26:4697–703. doi: 10.1016/j.vaccine.2008.06.087
92. Biondo C, Lentini G, Beninati C, Teti G. The dual role of innate immunity during influenza. BioMed J (2019) 42(1):8–18. doi: 10.1016/j.bj.2018.12.009
93. Tripathi S, White MR, Hartshorn KL. The amazing innate immune response to influenza a virus infection. Innate Immun (2015) 21(1):73–98. doi: 10.1177/1753425913508992
94. Manna S, Baindara P, Mandal SM. Molecular pathogenesis of secondary bacterial infection associated to viral infections including SARS-CoV-2. J Infect Public Health (2020) 13(13):1397–404. doi: 10.1016/j.jiph.2020.07.003
95. Sharma-Chawla N, Sender V, Kershaw O, Gruber AD, Volckmar J, Henriques-Normark B, et al. Influenza a virus infection predisposes hosts to secondary infection with different streptococcus pneumoniae serotypes with similar outcome but serotype-specific manifestation. Infect Immun (2016) 84(12):3445–57. doi: 10.1128/IAI.00422-16
96. Antoniak S. The coagulation system in host defense. Res Pract Thromb Haemost (2018) 2:549–57. doi: 10.1002/rth2.12109
97. Semple JW, Freedman J. Platelets and innate immunity. Cell MolLife Sci (2010) 67:499–511. doi: 10.1007/s00018-009-0205-1
98. Antoniak S, Mackman N. Platelets and viruses. Platelets (2021) 32(3):325–30. doi: 10.1080/09537104.2021.1887842
99. Koupenova M, Corkrey HA, Vitseva O, Manni G, Pang CJ, Clancy L, et al. The role of platelets in mediating a response to human influenza infection. Nat Commun (2019) 10:1780. doi: 10.1038/s41467-019-09607-x
100. Pulavendran S, Rudd JM, Maram P, Thomas PG, Akhilesh R, Malayer JR, et al. Combination therapy targeting platelet activation and virus replication protects mice against lethal influenza pneumonia. Am J Respir Cell Mol Biol (2019) 61:689–701. doi: 10.3389/fimmu.2021.772859
101. Le VB, Schneider JG, Boergeling Y, Berri F, Ducatez M, Guerin JL, et al. Platelet activation and aggregation promote lung inflammation and influenza virus pathogenesis. Am J Respir Crit Care Med (2015) 191:804–19. doi: 10.1164/rccm.201406-1031OC
102. Rondina MT, Brewster B, Grissom CK, Zimmerman GA, Kastendieck DH, Harris ES and Weyrich AS. In vivo platelet activation in critically ill patients with primary 2009 influenza A(H1N1). Chest (2012) 141:1490–5. doi: 10.1182/blood-2013-11-536003
103. McDonald B, Urrutia R, Yipp BG, Jenne CN, Kubes P. Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe (2012) 12:324–33. doi: 10.1111/jth.12842
104. Fuchs TA, Bhandari AA, Wagner DD. Histones induce rapid and profound thrombocytopenia in mice. Blood (2011) 118:3708–14. doi: 10.1182/blood2011-01-332676
105. Kim S-J, Carestia A, McDonald B, Zucoloto AZ, Grosjean H, Davis RP, et al. Platelet-mediated NET release amplifies coagulopathy and drives lung pathology during severe influenza infection. Front Immunol (2021) 12:772859. doi: 10.3389/fimmu.2021.772859
106. Boilard E, Pare G, Rousseau M, Cloutier N, Dubuc I, Levesque T, et al. Influenza virus H1N1 activates platelets through fc RIIA signaling and thrombin generation. Blood (2014) 123:2854–63. doi: 10.1182/blood-2013-07-515536
107. Koupenova M, Vitseva O, MacKay CR, Beaulieu LM, Benjamin EJ, Mick E, et al. Platelet-tlr7 mediates host survival and platelet count during viral infection in the absence of platelet-dependent thrombosis. Blood (2014) 124:791–802. doi: 10.1111/j.1538-7836.2005.01377.x
108. D'Atri LP, Etulain J, Rivadeneyra L, Lapponi MJ, Centurion M, Cheng K, et al. Expression and functionality of toll-like receptor 3 in the megakaryocytic lineage. J Thromb Haemost (2015) 13:839–50. doi: 10.1038/35025229
109. Anabel AS, Eduardo PC, Pedro Antonio HC, Carlos SM, Juana NM, Honorio TA, et al. Human platelets express toll-like receptor 3 and respond to poly i:C. Hum Immunol (2014) 75:1244–51. doi: 10.1016/j.cellimm.2019.103949
110. Antoniak S, Owens APIII, Baunacke M, Williams JC, Lee RD, Weithauser A, et al. PAR-1 contributes to the innate immune response during viral infection. J Clin Invest (2013) 123:1310–22. doi: 10.1172/JCI66125
111. Khoufache K, Berri F, Nacken W, Vogel AB, Delenne M, Camerer E, et al. PAR1 contributes to influenza a virus pathogenicity in mice. J Clin Invest (2013) 123:206–14. doi: 10.1172/JCI61667
112. Coughlin SR. Protease-activated receptors in hemostasis, thrombosis and vascular biology. J Thromb Haemost (2005) 3:1800–14. doi: 10.1111/j.1538-7836.2005.01377.x65
113. Coughlin SR. Thrombin signalling and protease-activated receptors. Nature (2000) 407:258–64. doi: 10.1038/35025229
114. Tatsumi K, Schmedes CM, Houston ER, Butler E, Mackman N, Antoniak S. Protease-activated receptor 4 protects mice from coxsackievirus B3 and H1N1 influenza a virus infection. Cell Immunol (2019) 344:1039–49. doi: 10.1016/j.cellimm.2019.103949
115. Liverani E, Rico MC, Tsygankov AY, Kilpatrick LE, Kunapuli SP. P2Y12 receptor modulates sepsis-induced inflammation. Arterioscler Thromb Vasc Biol (2016) 36(5):961–71. doi: 10.1038/nsmb.1651
116. Gross AK, Dunn SP, Feola DJ, Martin CA, Charnigo R, Li Z, et al. Clopidogrel treatment and the incidence and severity of community acquired pneumonia in a cohort study and meta-analysis of antiplatelet therapy in pneumonia and critical illness. J Thromb Thrombolysis (2013) 35(2):147–54. doi: 10.1016/j.celrep.2018.01.058
117. Sexton TR, Zhang G, Macaulay TE, Callahan LA, Charnigo R, Vsevolozhskaya OA, et al. Ticagrelor reduces thromboinflammatory markers in patients with pneumonia. JACC Basic Transl Sci (2018) 3(4):435–49. doi: 10.1016/j.jacbts.2018.05.005
118. Zarbock A, Polanowska-Grabowska RK, Ley K. Platelet-neutrophil-interactions: linking hemostasis and inflammation. Blood Rev (2007) 21(2):99–111. doi: 10.1016/j.molimm.2017.08.026
119. Ott I, Neumann FJ, Gawaz M, Schmitt M, Schomig A. Increased neutrophil-platelet ¨ adhesion in patients with unstable angina. Circulation (1996) 94(6):1239–46. doi: 10.1002/hep.29153
120. Landry P, Plante I, Ouellet DL, Perron MP, Rousseau G, Provost P. Existence of a microRNA pathway in anucleate platelets. Nat Struct Mol Biol (2009) 16:961–6. doi: 10.1038/nsmb.1651
121. Zhou W, Pal AS, Hsu AY-H, Gurol T, Zhu X, Wirbisky-Hershberger SE, et al. MicroRNA-223 suppresses the canonical NF-κB pathway in basal keratinocytes to dampen neutrophilic inflammation. Cell Rep (2018) 22:1810–23. doi: 10.1016/j.thromres.2019.12.016
122. Chen Q, Wang H, Liu Y, Song Y, Lai L, Han Q, et al. Inducible microRNA-223 down-regulation promotes TLR-triggered IL-6 and IL-1β production in macrophages by targeting STAT3. PLoS One (2012) 7:e42971. doi: 10.1371/journal.pone.0042971
123. Zhang N, Fu L, Bu Y, Yao Y, Wang Y. Downregulated expression of miR-223 promotes toll-like receptor-activated inflammatory responses in macrophages by targeting RhoB. Mol Immunol (2017) 91:42–8. doi: 10.1016/j.molimm.2017.08.026
124. He Y, Feng D, Li M, Gao Y, Ramirez T, Cao H, et al. Hepatic mitochondrial DNA/Toll-like receptor 9/MicroRNA-223 forms a negative feedback loop to limit neutrophil overactivation and acetaminophen hepatotoxicity in mice. Hepatology (2017) 66:220–34. doi: 10.1056/NEJMoa2015432
125. Chyrchel B, Totoń-Żurańska J, Kruszelnicka O, Chyrchel M, Mielecki W, Kołton-Wróż M, et al. Association of plasma miR-223 and platelet reactivity in patients with coronary artery disease on dual antiplatelet therapy: A preliminary report. Platelets (2015) 26:593–7. doi: 10.1093/ajcp/aqaa062
126. Liu J, Qin L, Wang Z, Peng L, Liu J, Wang X, et al. Platelet-derived miRNAs as determinants of the antiplatelet response in clopidogrel-treated patients with ACS. Thromb Res (2020) 186:71–4. doi: 10.1101/2020.04.06.20050575
127. Zarbock A, Ley K. The role of platelets in acute lung injury (ALI). Front Biosci (Landmark Ed) (2009) 14:150–8. doi: 10.1016/j.thromres.2020.05.025
128. Schmitt C, Abt M, Ciorciaro C, Kling D, Jamois C, Schick E, et al. First-inman study with inclacumab, a human monoclonal antibody against p-selectin. J Cardiovasc Pharmacol (2015) 65(6):611–9. doi: 10.1001/jamanetworkopen.2020.10478
129. Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in covid-19. N Engl J Med (2020) 383:120–8. doi: 10.1055/s-0042-1744185
130. Barton LM, Duval EJ, Stroberg E, Ghosh S, Mukhopadhyay S. COVID-19 autopsies, Oklahoma, USA. Am J Clin Pathol (2020) 153(6):725–33. doi: 10.1038/s41569-020-00469-1
131. Pulmonary and cardiac pathology in covid-19: The first autopsy series from new Orleans. medRxiV (2020) 8(7):681–6. doi: 10.1161/CIRCRESAHA.121.319117
132. Kashi M, Jacquin A, Dakhil B, Zaimi R, Mahé E, Tella E, et al. Severe arterial thrombosis associated with covid-19 infection. Thromb Res (2020) 192:75–7. doi: 10.1016/j.thromres.2020.05.025
133. Nahum J, Morichau-Beauchant T, Daviaud F, Echegut P, Fichet J, Maillet J-M, et al. Venous thrombosis among critically ill patients with coronavirus disease 2019 (COVID-19). JAMA Netw Open (2020) 3:e2010478. doi: 10.1001/jamanetworkopen.2020.10478
134. Sharma S, Mishra A, Ashraf Z. COVID-19 induced coagulopathy (CIC): Thrombotic manifestations of viral infection. TH Open (2022) 6(1):e70–9. doi: 10.1055/s-0042-1744185
135. Gu S, Tyagi T, Jain K, Gu VW, Lee SH, Hwa JM, et al. Thrombocytopathy and endotheliopathy are key contributors to COVID-19 thromboinflammation. Nat Rev Cardio (2021) 18(3):194–209. doi: 10.1016/j.jacbts.2020.10.003
136. Koupenova M, Corkrey HA, Vitseva O, Tanriverdi K, Somasundaran M, Liu P, et al. SARS-CoV-2 initiates programmed cell death in platelets. Circ Res (2021) 129:631–46. doi: 10.1161/CIRCRESAHA.121.319117
137. Zaid Y, Guessous F. The ongoing enigma of SARS-CoV-2 and platelet interaction. Res Pract Thromb Haemost (2022) 6(1):e12642. doi: 10.1002/rth2.12642
138. Robles JP, Zamora M, Adan-Castro E, Siqueiros-Marquez L, Martinez de la Escalera G, Clapp C. The spike protein of SARS-CoV-2 induces endothelial inflammation through integrin α5β1 and NF-κB signaling. J Biol Chem (2022) 298(3):101695. doi: 10.1016/j.jbc.2022.101695
139. Liu J, Lu F, Chen Y, Plow E, Qin J. Integrin mediates cell entry of the SARS-CoV-2 virus independent of cellular receptor ACE2. J Biol Chem (2022) 298(3):101710. doi: 10.1016/j.jbc.2022.101710
140. Beddingfield BJ, Iwanaga N, Chapagain PP, Zheng W, Roy CJ, Hu TY, et al. The integrin binding peptide, ATN-161, as a novel therapy for SARS-CoV-2 infection. JACC Basic Transl Sci (2021) 6(1):1–8. doi: 10.1016/j.jacbts.2020.10.003
141. Senchenkova EY, Russell J, Vital SA, Yildirim A, Orr AW, Granger DN, et al. A critical role for both CD40 and VLA5 in angiotensin II-mediated thrombosis and inflammation. FASEB J (2018) 32(6):3448–56. doi: 10.1096/fj.201701068R
142. Manne BK, Denorme F, Middleton EA, Portier I, Rowley JW, Stubben C, et al. Platelet gene expression and function in patients with COVID-19. Blood (2020) 136:1317–29. doi: 10.1182/blood.2020007214
143. Ye Q, Wang B, Mao J. The pathogenesis and treatment of the `Cytokine storm' in COVID-19. J Infect (2020) 80(6):607–13. doi: 10.1055/s-0042-1749438
144. Warkentin TE, Kaatz S. COVID-19 versus HIT hypercoagulability. Thromb Res (2020) 196:38–51. doi: 10.1016/j.thromres.2020.08.017
145. Brodard J, Kremer Hovinga JA, Fontana P, Studt J-D, Gruel Y, Greinacher A. COVID-19 patients often show high-titer non-Platelet-Activating anti-PF4/heparin IgG antibodies. J Thromb Haemost JTH (2021) 19:1294–8. doi: 10.1111/jth.15262
146. Tyagi T, Ahmad S, Gupta N, Sahu A, Ahmad Y, Nair V, et al. Altered expression of platelet p roteins and calpain activity mediate hypoxia-induced prothrombotic phenotype. Blood (2014) 123(8):1250–60. doi: 10.1182/blood.2020007252
147. Garcia C, Au Duong J, Poëtte M, Ribes A, Payre B, Mémier V, et al. Platelet activation and partial desensitization are associated with viral xenophagy in patients with severe COVID-19. Blood Adv (2022) 6(13):3884–98. doi: 10.1161/ATVBAHA.120.315175
148. Schuurman AR, Léopold V, Pereverzeva L, Chouchane O, Reijnders TDY, Brabander J, et al. The platelet lipidome is altered in patients with COVID-19 and correlates with platelet reactivity. Thromb Haemost (2022) 122(10):1683–92. doi: 10.1101/2021.04.29.21256354
149. Puhm F, Allaeys I, Lacasse E, Dubuc I, Galipeau Y, Zaid Y, et al. Platelet activation by SARS-CoV-2 implicates the release of active tissue factor by infected cells. Blood Adv (2021) 6(12):3593–605. doi: 10.1182/bloodadvances.2022007444
150. Trugilho MRO, Azevedo-Quintanilha IG, Gesto JSM, Moraes ECS, Mandacaru SC, Campos MM, et al. Platelet proteome reveals features of cell death, antiviral response and viral replication in covid-19. Cell Death Discov (2022) 8:324. doi: 10.1038/s41420-022-01122-1
151. Hottz ED, Azevedo-Quintanilha IG, Palhinha L, Teixeira L, Barreto EA, Pão CR, et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood (2020) 136:1330–41. doi: 10.1182/blood.2020007252
152. Taus F, Salvagno G, Canè S, Fava C, Mazzaferri F, Carrara E, et al. Platelets promote thromboinflammation in SARS-CoV-2 pneumonia. Arterioscler Thromb Vasc Biol (2020) 40(12):2975–89. doi: 10.1161/ATVBAHA.120.315175
153. Kanno H, Zhou Y, Nishikawa M, Xiao T-H, Suzuki T, Ibayashi Y, et al. Understanding microvascular thrombosis in COVID-19 via massive single-cell imaging of circulating platelets. medRxiv (2021). doi: 10.1101/2021.04.29.21256354
154. Althaus K, Marini I, Zlamal J, Pelzl L, Singh A, Häberle H, et al. Antibody-induced procoagulant platelets in severe COVID-19 infection. Blood (2021) 137:1061–71. doi: 10.1182/blood.2020008762
155. Andrade ACDSP, Campolina-Silva GH, Queiroz-Junior CM, de Oliveira LC, Lacerda LSB, Gaggino JCP, et al. A biosafety level 2 mouse model for studying betacoronavirus-induced acute lung damage and systemic manifestations. J Virol (2021) 95(22):e0127621. doi: 10.1182/blood.2020007008
156. Gryzia A, Volkmann T, Brechling A, Hoeke V, Schneider L, Kuepper K, et al. Crystallographic order and decomposition of [MnIII6CrIII]3+ single-molecule magnets deposited in submonolayers and monolayers on HOPG studied by means of molecular resolved atomic force microscopy (AFM) and kelvin probe force microscopy in UHV. Nanoscale Res Lett (2014) 9(1):60. doi: 10.1186/1556-276X-9-60
157. Fitch-Tewfik JL, Flaumenhaft R. Platelet granule exocytosis: A comparison with chromaffin cells. Front Endocrinol (Lausanne) (2013) 4:77. doi: 10.1016/S2352-3026(20)30109-5
158. Sut C, Tariket S, Aubron C, Aloui C, Hamzeh-Cognasse H, Berthelot P, et al. The non-hemostatic aspects of transfused platelets. Front Med (Lausanne) (2018) 5:42. doi: 10.3389/fmed.2018.00042
159. Zaid Y, Puhm F, Allaeys I, Naya A, Oudghiri M, Khalki L, et al. Platelets can associate with SARS-CoV-2 RNA and are hyperactivated in COVID-19. Circ Res (2020)127(11):1404–18. doi: 10.1161/CIRCRESAHA.120.317703
160. Middleton EA, He XY, Denorme F, Campbell RA, Ng D, Salvatore SP, et al. Neutrophil extracellular traps (NETs) contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood (2020) 136(10):1169–79. doi: 10.1111/jth.15587
161. Hamzeh-Cognasse H, Mansour A, Reizine F, Mismetti P, Gouin-Thibault I, Cognasse F. Platelet-derived sCD40L: specific inflammatory marker for early-stage severe acute respiratory syndrome coronavirus 2 infection. Virol J (2021) 18(1):211. doi: 10.1186/s12985-021-01680-3
162. Wang T, et al. Attention should be paid to venous thromboembolism prophylaxis in the management of COVID-19. Lancet Haematol (2020) 7(5):e362–3. doi: 10.1101/2021.05.01.442279
163. Larsen MD, de Graaf EL, Sonneveld ME, Plomp HR, Nouta J, Hoepel W, et al. Afucosylated IgG characterizes enveloped viral responses and correlates with COVID-19 severity. Science (2021) 371(6532):eabc8378. doi: 10.1126/science.abc8378
164. Bye AP, Hoepel W, Mitchell JL, Jégouic S, Loureiro S, Sage T, et al. Aberrant glycosylation of anti-SARS-CoV-2 spike IgG is a prothrombotic stimulus for platelets. Blood (2021) 138:1481–9. doi: 10.1182/blood.2021011871
165. Pelzl L, Singh A, Funk J, Witzemann A, Marini I, Zlamal J, et al. Antibody-mediated procoagulant platelet formation in COVID-19 is AKT dependent. J Thromb Haemost (2022) 20:387–98. doi: 10.1111/jth.15587
166. Niu H, Chen X, Gruppo RA, Li D, Wang Y, Zhang L, et al. Integrin alphaIIb-mediated PI3K/Akt activation in platelets. PLoS One (2012) 7:e47356. doi: 10.1016/j.cell.2020.10.005
167. Apostolidis SA, Sarkar A, Giannini HM, Goel RR, Mathew D, Suzuki A, et al. Signaling through FcγRIIA and the C5a-C5aR pathway mediate platelet hyperactivation in COVID-19. Front Immunol (2021) 13:834988. doi: 10.3389/fimmu.2022.834988
168. Bussel J, Arnold DM, Grossbard E, Mayer J, Treliński J, Homenda W, et al. Fostamatinib for the treatment of adult persistent and chronic immune thrombocytopenia: Results of two phase 3, randomized, placebo-controlled trials. Am J Hematol (2018) 93:921–30. doi: 10.1016/S2666-5247(20)30144-0
169. Mezger M, Nording H, Sauter R, Graf T, Heim C, von Bubnoff N, et al. Platelets and immune responses during thromboinflammation front. Immunol (2019) 10:1731. doi: 10.1038/s41467-021-22781-1
170. Patzelt J, Mueller KAL, Breuning S, Karathanos A, Schleicher R, Seizer P, et al. Expression of anaphylatoxin receptors on platelets in patients with coronary heart disease. Atherosclerosis (2015) 238:289–95. doi: 10.1038/s41586-021-03744-4
171. Aid M, Busman-Sahay K, Vidal SJ, Maliga Z, Bondoc S, Starke C, et al. Vascular disease and thrombosis in SARS-CoV-2-Infected rhesus macaques. Cell (2020) 183:1354–66.e13. doi: 10.1126/sciadv.abl8213
172. Skendros P, Mitsios A, Chrysanthopoulou A, Mastellos DC, Metallidis S, Rafailidis P, et al. Complement and tissue factor–enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J Clin Invest (2020) 130:6151–7. doi: 10.1111/jth.15862
173. Schurink B, Roos E, Radonic T, Barbe E, Bouman CSC, de Boer HH, et al. Viral presence and immunopathology in patients with lethal COVID-19: A prospective autopsy cohort study. Lancet Microbe (2020) 1:e290–9. doi: 10.1016/j.jaut.2021.102662
174. Diao B, Wang C, Wang R, Feng Z, Zhang J, Yang H, et al. Human kidney is a target for novel severe acute respiratory syndrome coronavirus 2 infection. Nat Commun (2021) 12:2506. doi: 10.1182/blood.2021012819
175. Huynh A, Kelton JG, Arnold DM, Daka M, Nazy I. Antibody epitopes in vaccine-induced immune thrombotic thrombocytopaenia. Nature (2021) 596:565–9. doi: 10.1038/s41586-021-03744-4
176. Baker AT, Boyd RJ, Sarkar D, Teijeira-Crespo A, Chan CK, Bates E, et al. ChAdOx1 interacts with CAR and PF4 with implications for thrombosis with thrombocytopenia syndrome. Sci Adv (2021) 7:eabl8213. doi: 10.1126/sciadv.abl8213
177. Schönborn L, Thiele T, Esefeld M, El Debuch K, Wesche J, Seck SE, et al. Quantitative interpretation of PF4/heparin-EIA optical densities in predicting platelet-activating VITT antibodies. J Thromb Haemost (2022) 20(11):2579–86. doi: 10.1111/jth.15862
178. McGonagle D, de MG, Bridgewood C. Mechanisms of immunothrombosis in vaccine-induced thrombotic thrombocytopenia (VITT) compared to natural SARS-CoV-2 infection. J Autoimmun (2021) 121:102662. doi: 10.1016/j.jaut.2021.102662
180. Smith CW, Montague SJ, Kardeby C, Di Y, Lowe GC, Lester WA, et al. Antiplatelet drugs block platelet activation by VITT patient serum. Blood (2021) 138:2733–40. doi: 10.1182/blood.2021012277
181. Abhithaj J, Francis D, Sharanya CS, Arun KG, Sadasivan C, Variyar EJ. Repurposing simeprevir, calpain inhibitor IV and a cathepsin f inhibitor against SARS-CoV-2 and insights into their interactions with mpro. J Biomol Struct Dyn (2020) 40(1):325–36. doi: 10.3389/fmolb.2020.610132
182. Croce K, Flaumenhaft R, Rivers M, Furie B, Furie BC, Herman IM, et al. Inhibition of calpain blocks platelet secretion, aggregation, and spreading. J Biol Chem (1999) 274(51):36321–7. doi: 10.3389/fphar.2020.01344
183. Wingrave JM, Schaecher KE, Sribnick EA, Wilford GG, Ray SK, Hazen-Martin DJ, et al. Early induction of secondary injury factors causing activation of calpain and mitochondria-mediated neuronal apoptosis following spinal cord injury in rats. J Neurosci Res (2003) 73(1):95–104. doi: 10.1161/CIRCULATIONAHA.105.602532
184. Mahaman YAR, Huang F, Kessete Afewerky H, Maibouge TMS, Ghose B, Wang X. Involvement of calpain in the neuropathogenesis of alzheimer’s disease. Med Res Rev (2019) 39(2):608–30. doi: 10.1101/2020.03.10.20033522
185. Camera M, Canzano P, Brambilla M, Rovati GE. Montelukast inhibits platelet activation induced by plasma from COVID-19 patients. Front Pharmacol (2022) 13:784214. doi: 10.3389/fphar.2022.784214
186. Aigner L, Pietrantonio F, Bessa de Sousa DM, Michael J, Schuster D, Reitsamer HA, et al. The leukotriene receptor antagonist montelukast as a potential COVID-19 therapeutic. Front Mol Biosci (2020) 7:610132. doi: 10.3389/fmolb.2020.610132
187. Barré J, Sabatier J-M, Annweiler C. Montelukast drug may improve COVID-19 prognosis: A review of evidence. Front Pharmacol (2020) 11:1344. doi: 10.1016/S0140-6736(15)00667-4
188. Forstermann U, Munzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation (2006) 113:1708–14. doi: 10.1111/bjh.16209
189. Lei C, Su B, Dong H, Fakhr BS, Grassi LG, Di Fenza R, et al. Protocol for a randomized controlled trial testing inhaled nitric oxide therapy in spontaneously breathing patients with COVID-19. medRxiv (2020). doi: 10.1101/2020.03.10.20033522
190. Nicolson PLR, Nock SH, Hinds J, Garcia-Quintanilla L, Smith CW, Campos J, et al. Low-dose btk inhibitors selectively block platelet activation by CLEC-2. Haematologica (2021) 106(1):208–19. doi: 10.3324/haematol.2019.218545
191. Barr PM, Robak T, Owen C, Tedeschi A, Bairey O, Bartlett NL, et al. Sustained efficacy and detailed clinical follow-up of first-line ibrutinib treatment in older patients with chronic lymphocytic leukemia: extended phase 3 results from RESONATE-2. Haematologica (2018) 103(9):1502–10. doi: 10.3324/haematol.2018.192328
192. Dreyling M, Jurczak W, Jerkeman M, Silva RS, Rusconi C, Trneny M, et al. Ibrutinib versus temsirolimus in patients with relapsed or refractory mantle-cell lymphoma: An international, randomised, open-label, phase 3 study. Lancet (2016) 387(10020):770–8. doi: 10.1016/S0140-6736(15)00667-4
193. Kander EM, Zhao Q, Bhat SA, Hirsch J, Byrd JC, Ooka L, et al. Venous and arterial thrombosis in patients with haematological malignancy during treatment with ibrutinib. Br J Haematol (2019) 187(3):399–402. doi: 10.1111/bjh.16209
194. Levade M, David E, Garcia C, Laurent PA, Cadot S, Michallet AS, et al. Ibrutinib treatment affects collagen and von willebrand factor-dependent platelet functions. Blood (2014) 124(26):3991–5. doi: 10.1182/blood-2014-06-583294
195. Ito M, Shichita T, Okada M, Komine R, Noguchi Y, Yoshimura A, et al. Bruton's tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury. Nat Commun (2015) 6:7360. doi: 10.1038/ncomms8360
196. Semple JW, Italiano JE, Freedman J, Italiano JE, Freedman J. Platelets and the immune continuum. Nat Rev Immunol (2011) 11:264–74. doi: 10.1038/nri2956
197. Weiss LJ, Drayss M, Manukjan G, Zeitlhöefler MJ, Kleiss J, Weigel ML, et al. Uncoupling of platelet granule release and integrin activation suggests GPIIb/IIIa as therapeutic target in COVID-19. Blood Adv (2022). doi: 10.1182/bloodadvances.2022008666
198. Altman R, Luciardi HL, Muntaner J, Herrera RN. The antithrombotic profile of aspirin. aspirin resistance, or simply failure? Thromb J (2004) 2:1. doi: 10.1186/1477-9560-2-1
199. Pradhan A, Tiwari A, Caminiti G, Salimei C, Muscoli S, Sethi R, et al. Ideal P2Y12 inhibitor in acute coronary syndrome: A review and current status. Int J Environ Res Public Health (2022) 19(15):8977. doi: 10.3390/ijerph19158977
200. O'Donoghue ML, Bhatt DL, Flather MD, Goto S, Angiolillo DJ, Goodman SG, et al. Atopaxar and its effects on markers of platelet activation and inflammation: results from the LANCELOT CAD program. J Thromb Thrombolysis (2012) 34(1):36–43. doi: 10.1007/s11239-012-0750-6
201. Chaudhary R, Mohananey A, Sharma SP, Singh S, Singh A, Kondur A. Improving outcomes in cardiovascular diseases: A review on vorapaxar. Cardiol Rev (2022) 30(5):241–6. doi: 10.1097/CRD.0000000000000390
202. Muñiz-Lozano A, Rollini F, Franchi F, Angiolillo DJ. Update on platelet glycoprotein IIb/IIIa inhibitors: recommendations for clinical practice. Ther Adv Cardiovasc Dis (2013) 7(4):197–213. doi: 10.1177/1753944713487781
203. Aliter KF, Al-Horani RA. Potential therapeutic benefits of dipyridamole in COVID-19 patients. Curr Pharm Des (2021) 27(6):866–75. doi: 10.2174/1381612826666201001125604
Keywords: platelets, hemostasis, thrombo-inflammation, signaling, infections
Citation: Sharma S, Tyagi T and Antoniak S (2022) Platelet in thrombo-inflammation: Unraveling new therapeutic targets. Front. Immunol. 13:1039843. doi: 10.3389/fimmu.2022.1039843
Received: 08 September 2022; Accepted: 18 October 2022;
Published: 14 November 2022.
Edited by:
Ilaria Canobbio, University of Pavia, ItalyReviewed by:
Cristina Nocella, Sapienza University of Rome, ItalyFabrice Cognasse, INSERM U1059 Santé INgéniérie BIOlogie, France
Copyright © 2022 Sharma, Tyagi and Antoniak. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Silvio Antoniak, c2lsdmlvX2FudG9uaWFrQG1lZC51bmMuZWR1