 Pamela A. McCombe
Pamela A. McCombe Todd A. Hardy2,3
Todd A. Hardy2,3 Judith M. Greer
Judith M. Greer
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 09 December 2022
Sec. Autoimmune and Autoinflammatory Disorders : Autoimmune Disorders
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1038411
This article is part of the Research Topic Sexual Dimorphism in Autoimmune and Immune-dysregulated Diseases View all 7 articles
Guillain Barré syndrome (GBS) and its variants, and chronic inflammatory demyelinating polyradiculoneuropathy (CIDP and its variants, are regarded as immune mediated neuropathies. Unlike in many autoimmune disorders, GBS and CIDP are more common in males than females. Sex is not a clear predictor of outcome. Experimental autoimmune neuritis (EAN) is an animal model of these diseases, but there are no studies of the effects of sex in EAN. The pathogenesis of GBS and CIDP involves immune response to non-protein antigens, antigen presentation through non-conventional T cells and, in CIDP with nodopathy, IgG4 antibody responses to antigens. There are some reported sex differences in some of these elements of the immune system and we speculate that these sex differences could contribute to the male predominance of these diseases, and suggest that sex differences in peripheral nerves is a topic worthy of further study.
The Guillain Barré syndrome (GBS) and its variants, and chronic inflammatory demyelinating polyradiculoneuropathy (CIDP and its variants, are regarded as immune mediated neuropathies, due to their pathological findings and their response to immune therapy (1–3). Because there is sexual dimorphism of the immune system (4) it could be that the sex of the subject might affect the features of these diseases. This review summarizes knowledge of the effects of sex on GBS and CIDP and discusses possible mechanisms. Firstly, however, we provide a brief overview of GBS and CIDP, and also of their animal model, experimental autoimmune neuritis (EAN).
The term GBS arises from the initial description by Guillain, Barre and Strohl of a syndrome of ascending weakness, associated with loss of deep tendon reflexes, and early recovery, in two soldiers (5). However, the earliest report is likely to be that that of Landry who, in 1859, described a patient who died of respiratory failure after developing ascending weakness (6). The modern use of the term “the Guillain Barre syndrome” is attributed to Haymaker and Kernohan (7). The history of GBS has been written at different times and these reports show evolution of our understanding and conception of the syndrome and its causes (8–10).
Although at one stage GBS was well-known as a demyelinating disease, in modern times it is recognized that there are variants with axonal pathology; it is also known that there are focal variants, such as the Fisher syndrome, first described in 1954 (11). There are clinical and neurophysiological criteria for the diagnosis of GBS (12–14). The “Brighton criteria” (15) are widely accepted for the diagnosis of GBS and Fisher syndrome and are shown in Table 1. The prevalence of GBS increases with age up to 70–75 years, then declines (16–18).

Table 1 The Brighton Criteria for diagnosis of GBS and Fisher syndrome.
Based on clinical findings and electrodiagnostic studies, GBS can be divided into subtypes (19). These include acute inflammatory demyelinating polyradiculoneuropathy (AIDP), in which there is inflammation in nerves and demyelination of nerve fibers, acute motor axonal neuropathy (AMAN), and acute motor and sensory neuropathy (AMSAN) which cause generalized weakness with demyelinating or axonal features. The relationship between the demyelinating and axonal disorders is unclear in that there appear to be conditions where there is primary damage to axons, and conditions where severe demyelinating disease leads on to axonal damage (20). There can be difficulty in distinguishing between conduction block and axonal degeneration (21, 22). There are other GBS variants with specific restricted phenotypes; as well as Fisher syndrome (FS), a syndrome of ophthalmoplegia, ataxia and areflexia (23), there is a phenotype of isolated facial diplegia (24) and a pharyngeal-cervical-brachial variant (25).
GBS often follows a preceding illness or event such as an infection. In an analysis of the first 1000 patients enrolled in the International GBS Outcome Study, 72% of patients reported an antecedent infection (26). In that study, the organisms that lead to the preceding infections included Campylobacter jejuni (30%), Mycoplasma pneumoniae (10%) and cytomegalovirus (4%) (26, 27) and more than one preceding infection was identified in 6% of patients. In our own study, we found that 75% of subjects had a preceding infection (16). This association with a prior infection has led to the view that the immune abnormalities in GBS are triggered in many by exposure to an infectious agent.
However, it must be noted that these infections do not lead to GBS in most people, which suggests that humans vary in their response to infection and in the tendency to develop GBS. It is likely that this is due to genetically mediated variation in the function of the immune system, but it could also be due to “target organ resistance” to immune attack.
There is also increasing recognition that GBS can follow trauma or surgery. The first report of GBS after surgery was in 1968 (28), and since then there have been numbers of case reports. More detailed studies have attempted to quantify the risk. A study from Switzerland found that the relative risk of developing GBS in the 6 weeks after surgery was 13.1 times that of the non-surgically exposed population (29). A French nationwide case-crossover study found that among 8364 cases of GBS, 175 had undergone surgery within the preceding 60 days which increased the risk by greater than 1.5 times compared those who had undergone surgery in the preceding 336-425 days (30). While any recent surgery was a risk, the risk was stronger with bone and digestive organ surgery. Furthermore, a systematic review of 136 cases and 6 cases series of trauma-related GBS found that 89% of patients developed GBS following injury or surgery, with spontaneous intracranial haemorrhage making up nearly 10% of the remaining cases (31). In this study, trauma or surgery to brain and spine were the most likely to pre-date GBS. At present the mechanism for “trauma-related GBS” is speculative and warrants further research.
In AIDP, the inflammatory demyelinating form of GBS, the pathological findings include many macrophages and small numbers of T cells in the nerves (32, 33). Both CD4+ and CD8+ T cells have been found in nerve biopsies from AIDP patients, as have γδ-T cells and NK T cells (34). Autopsy studies show that pathological changes are prominent in the nerve roots (32). This infiltration is followed by segmental demyelination, meaning demyelination of internodes, which are produced by a single Schwann cell. Demyelination occurs by stripping of myelin by macrophages (35–37). Ultrastructural studies indicate the presence of vesicular degeneration in the outer myelin layer can predate macrophage infiltration (32). Secondary axonal degeneration, thought to occur with severe inflammation, can be seen in spinal roots and in motor and sensory nerves (38). In GBS there are reports of deposition of complement in peripheral nerves (32, 33), including on the outer surface of Schwann cells. The process of recovery from GBS involves remyelination with the production of thinly myelinated fibers with short internodes (37).
In AMAN there is axonal injury but macrophage-activated demyelination and inflammatory infiltrates are not characteristic (39). Activated complement deposition occurs at the nodes of Ranvier with formation of the membrane attack complex and, in addition, macrophages infiltrate along the periaxonal space leading to myelin detachment (39, 40). The resulting injury leads to nodal lengthening and axonal degeneration (39, 41). The pathology of FS, on the other hand, is not well elucidated as it is rarely fatal, and post-mortem studies are rare but the available pathology indicates demyelination of the extraocular nerves (42).
There are many reports of immune abnormalities in GBS and its variants. There have been reports of T cell and antibody reactivity to myelin antigens (eg P0 and P2 proteins) (43), but these were found only in a few patients. These antigens were of interest because immunization of experimental animals with such antigens and adjuvant led to experimental autoimmune neuritis, which shares clinical and histological features with GBS. More recently, immunity to nodal antigens (44) has been demonstrated in GBS.
However, there is now extensive evidence to suggest that glycolipid antigens are the target of a humoral and cell-mediated immune response in some patients with GBS and its variants; this is seen predominantly in AMAN and FS rather than in AIDP and AMSAN (45, 46). The glycolipid targets are mainly gangliosides which are found widely in cell membranes and are important in the biology of the nervous system (47). As well as reactivity to structures found on individual gangliosides there is also reactivity to ganglioside complexes (48). This immune response is thought to be triggered with cross-reactivity between glycolipids on infectious agents and on peripheral nerve. This is known as molecular mimicry (49, 50) and GBS is an excellent example (51). It must be noted that although the gangliosides that are the targets of these antibodies are expressed in other tissues such as the brain and the kidney, the pathological process in GBS is generally confined to the peripheral nerve, suggesting that there is some factor that makes the peripheral nerves vulnerable. However, there are exceptions; GBS can be associated with nephrotic syndrome (52) and with papilloedema (53).
It is notable that the immune response to glycolipid antigens requires antigen presentation by major histocompatibility complex (MHC)-like molecules. These include cluster of differentiation 1 (CD1) and possibly Major Histocompatibility Complex, Class I-Related (MR1) protein (54–57). The T cells that respond to such molecules presented by CD1 or MR1 are sometimes referred to as “unconventional T cells” (58, 59) and include natural killer T (NKT) cells, γδ T cells, and MR1-expressing T cells (MR1T), a subset of which are the mucosal associated invariant T (MAIT) cells (60). Antigen presentation by MAIT cells is potentially relevant to GBS after C.jejuni infection since it is a gut infection. Activation of antibody producing cells by NKT cells can occur independently of lymphoid follicles and can lead to a transient activation of plasmablasts (61); this would be consistent with the acute self-limited nature of GBS.
Immune abnormalities are reported in the blood of patients with GBS. These include alterations in levels of T cells The percentage of CD8+ cells in the blood is increased (62) and the proportion of Treg cells is decreased (62, 63). There are increased levels of activated T cells (62, 63).
There are elevated circulating levels of cytokines such as tumor necrosis factor (TNF), interferon (IFN)-γ, interleukin (IL)-1β, IL-6 and IL-17 (64), whereas levels of other circulating factors such as brain derived neurotrophic factor (BDNF) are reduced (65). Stimulation of peripheral blood mononuclear cells (PBMCs) with C. jejuni results in a γδT cell response in patients with GBS, and patients with GBS including those with prior C. jejuni infection have been found to have increased numbers of circulating γδT cells (66).
In the early stages of GBS infiltrating T cells produce TNFα which can induce demyelination by a direct cytotoxic effect on myelin and by altering myelin protein and glycolipid synthesis (67). Another cytokine of importance is the proinflammatory cytokine IFNα. Th1 cells produce IFNα which activates endothelial cells and enhances expression of major histocompatibility complex (MHC) II leading to an increase in antigen presentation by macrophages. IFNα also effects T cells causing a switch to a Th1 phenotype and stimulating T cell apoptosis. IFNα facilitates TNFα`, IL1β and IL6 production and B cell class switching. Other cytokines implicated in the immunopathogenesis of GBS include IL-17 and IL-23 (68). There is also dysregulation of the IL33/ST2 immune axis (69). A role for Th17 cells is supported by a study showing elevated levels of cytokines of the Th17 pathway (IL17, IL-6 and Il-22) in the cerebrospinal fluid (CSF) of patients with GBS compared to controls (70).
GBS is not thought to be an inherited disease. Some genes have been associated with GBS, but there is no clear HLA association (71). Genetic evidence supports that the killer immunoglobulin receptor (KIR) system is involved in GBS (72); this indicates that the innate immune system is involved in pathogenesis, since KIR molecules are expressed on NK cells that are part of this system (73, 74).
In summary GBS appears to be a monophasic immune mediated disease, associated with antibodies to gangliosides. GBS can be subdivided into groups according to clinical features. GBS patients can also be subdivided according to the serum ganglioside antibody associated with their illness.
CIDP is a term that describes a chronic or relapsing illness, often characterized by demyelination of peripheral nerves and nerve roots. In some patients, CIDP is predominantly a motor neuropathy that causes chronic or recurrent episodes of weakness (75). However, there are variants of CIDP that can show mainly sensory features or focal features (76) and variants that are characterized by axonal degeneration rather than demyelination. These variants include multifocal acquired demyelinating sensory and motor neuropathy (MADSAM), that is characterized by multifocal onset and proximal inflammation of nerve roots and plexuses (77, 78) and distal acquired demyelatinating symmetric (DADS) neuropathy that is characterized by distal but not proximal weakness (77, 79).
Different diagnostic criteria have been developed for the diagnosis of CIDP, with different sensitivity and specificity (76, 80, 81). One challenging issue is the distinction between GBS with fluctuations, and CIDP presenting with an acute deterioration (82, 83). The widely accepted EFNS/PNS criteria (81) provide electrodiagnostic criteria as shown in Table 2, and state that:

Table 2 EFNS/PNS electrodiagnostic criteria for CIDP.
“CIDP should be considered in any patient with a progressive symmetrical or asymmetrical polyradiculoneuropathy in whom the clinical course is relapsing and remitting or progresses for more than 2 months, especially if there are positive sensory symptoms, proximal weakness, areflexia without wasting, or preferential loss of vibration or joint position sense.”
It must be noted that there are other chronic immune-mediated neuropathies, such as multifocal motor neuropathy (84), neuropathy associated with antibodies to myelin-associated glycoprotein (MAG) (85) and Polyneuropathy, Organomegaly, Endocrinopathy, Monoclonal Gammopathy, and Skin Changes Syndrome (POEMS) (86) that are not regarded as CIDP. It is well-known that some neuropathies are associated with circulating paraproteins (87); these include the aforementioned anti-MAG neuropathy and POEMS as well as other neuropathies associated with haematological conditions (88). However, some patients with CIDP also have circulating paraproteins (89). For further information, the reader is referred to more detailed studies of the classification and frequency of CIDP and its variants (90, 91).
In contrast to GBS, there is seldom a history of an infectious illness before the onset of CIDP. A study from Italy found that 15.5% of 435 patients had preceding infections or vaccinations before the onset of disease, and patients with antecedent infections were more likely to have acute onset CIDP with cranial nerve involvement (92). Another large series of 268 patients found preceding infection in 10.4% of subjects (93). Other series reported a frequency of preceding infections ranging from 9.7% of 294 patients (80) to 32% of 92 patients (75).
Findings from sural nerve biopsies indicate that typical CIDP is characterized by paranodal interstitial oedema, and endoneurial cell infiltrates with prominent macrophage invasion causing demyelination (80, 94, 95). Macrophage-mediated damage could facilitate inflammation by exposing new autoantigenic epitopes leading to epitope spreading (96).
In CIDP, in a minority of patients, there have been reports of T cell and antibody reactivity to myelin antigens (43, 97–100). Involvement of cell-mediated immunity in the pathogenesis of CIDP is supported by evidence that activated T cells cross the blood-nerve barrier and that various T cell associated cytokines such as TNFα, IFNα and IL-2 are expressed in abundance in the perineurium, endoneurium and endoneurial blood vessels in patients with CIDP (101). T cells are also found in bopsy samples of nerves from patients with CIDP, and evidence of restricted clonality of CD8+ cells suggests that these cells play a role (96). Both CD4+ and CD8+ T cells have been found in nerve biopsies from CIDP patients, as have γδ-T cells and NK T cells (34). One study showed that CD8+ cells were more common than CD4+ cells (102). There is also deposition of complement in nerves (103).
In a small minority of patients there are reports of circulating antibodies to periperal nerve, peripheral nerve myelin and peripheral nerve proteins (104). There are rare instances of antibodies to gangliosides GM1 (105) and also LM1, a lacto series ganglioside, in a minority of patients with CIDP (106).
More recently there have been reports of immune reactivity to nodal antigens in a subset of patients with CIDP (44, 107–109). Antibodies targeting these nodal antigens are predominantly of the IgG4 type (107) and the associated syndromes are characterized by poor response to treatment with intravenous immunoglobulin (IVIG). The specific antibody targets in these patients include neurofascin 155, a glial cell paranodal protein, and NF140 and NF186, proteins expressed at nodes and the initial segments of axons (110), and contactin antigens such as contactin-1 (CNTN1) and contactin-associated protein 1 (CASPR1) (111). A comprehensive study of the presence of antibodies in 65 CIDP patients found that 8 had antibodies to nodal/paranodal proteins, 11 had ganglioside antibodies and one had antibody to myelin P2 protein (112). The mechanism of nerve damage from antibodies to nodal/paranodal antigens could involve disruption of axonal-glial junctions (113).
Pathological findings in patients with nodal antibodies are different from those of classical CIDP, without prominent macrophage-mediated demyelination or axonal damage, and this might indicate that nodal/paranodal antibody associated forms of disease have a different pathogenesis to that of classical CIDP. Patients with antibodies to nodal/paranodal antigens are resistant to treatment with IVIG (treatment-resistant CIDP), likely because these antibodies are of the IgG4 type, and are best regarded as having a different disease, called autoimmune nodopathy (AN) (114, 115).
There are some reports of associations of different genes with CIDP, and some reports of human leukocyte antigen (HLA) associations but these have not been replicated in large series (71). There is one report that associations with HLA DR2 are sex dependent, being present in females (116). As with GBS, genetic studies indicated that NK cells of the innate immune system are involved in pathogenesis (117).
In summary CIDP is a chronic or chronic relapsing disease that is immune mediated and associated with a variety of antibodies including gangliosides and nodal and paranodal proteins. Patients with CIDP can be divided into different groups on clinical grounds and also be subdivided according to the antibody they carry.
Animal models of autoimmune disease have been developed since the first description of experimental allergic encephalitis (an animal model of multiple sclerosis) by inoculation with central nervous system tissue and later with the addition of adjuvant, which stimulates the innate immune system (118, 119). The first description of experimental allergic neuritis (EAN), as an animal model of GBS, was provided by Waksman and Adams who set out to produce a disease where inflammation was confined to the peripheral nervous system, and achieved this in rabbits, guinea pigs and mice by inoculation with homogenized nerve (119, 120). EAN was also induced in chickens by inoculation with sciatic nerve (121). EAN can now be induced in many different animal strains with a variety of purified antigens including peripheral myelin proteins, with the use of adjuvants. Table 3 shows the first descriptions of these models, showing the evolution of types of active EAN and these are discussed below.
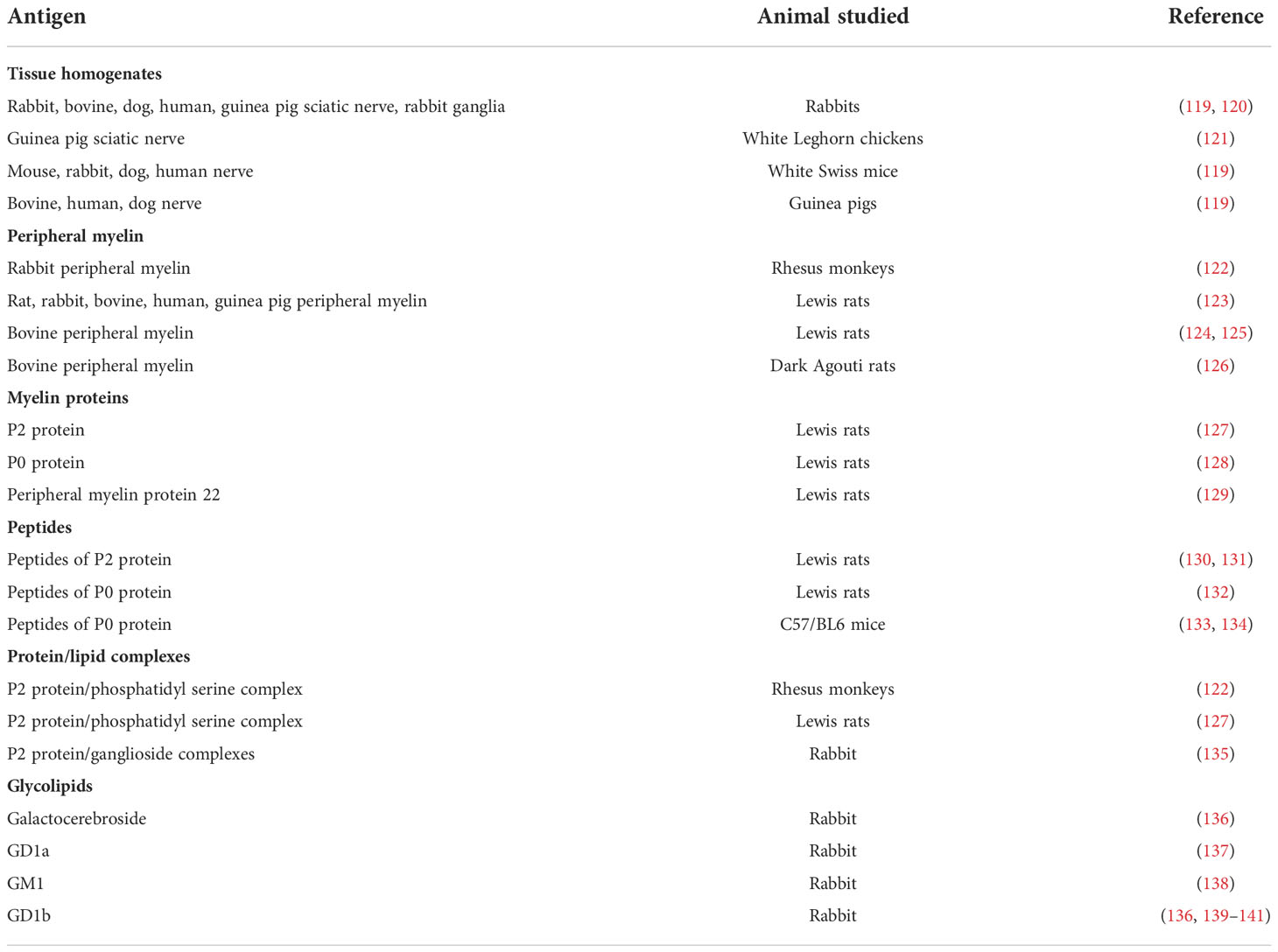
Table 3 Development of models of actively induced EAN.
After induction of EAN by inoculation with peripheral nerve tissue, studies were performed to investigate whether sensitization to myelin could produce disease. EAN was induced with peripheral nerve myelin in rats and monkeys (122, 123, 126) and with bovine peripheral myelin in SJL/J mice (142). These studies were followed by attempts to identify the “ neuritogen” — the component of nerve or myelin that caused the sensitization that led to disease. Myelin P2 protein was the first purified “neuritogen” to be identified (127, 143). Myelin P0 protein can also induce EAN in Lewis rats (128). EAN has also been induced in Lewis rats by inoculation with peripheral myelin protein 22 (PMP22) (129).
Once myelin proteins has been identified as neuritogenic, studies were performed to identify the epitopes of the proteins that caused disease. EAN can be induced by inoculation of P2 peptides in Lewis rats (130, 131). EAN can also be induced in Lewis rats and C57/BL6 mice by inoculation with myelin P0 peptides (132–134). EAN can also be passively transferred by sensitized T cells (144, 145).
These are generally acute forms of EAN. After recovery, animals are resistant to re-induction of disease by inoculation with the same antigen. This acquired tolerance is related to active tolerance by Treg cells. It has been shown that transfer of CD4+CD25+ Treg cells from animals that have recovered from EAN inhibits the induction of EAN (146).
The pathology of EAN is of inflammation and demyelination similar to that of GBS with myelin removal by macrophages (147, 148). There is also deposition of antibody and complement in peripheral nerves (149, 150). Studies of passively transferred EAE suggest a role for T cells in the pathogenesis of disease.
There are few models of CIDP, however immune suppression can convert monophasic EAN induced by peripheral nerve myelin into a chronic form of disease suggesting that in this model, active immune suppression controls the immunity to nerves (151, 152). Chronic EAN can also be induced in Lewis rats by high doses of P0 peptides (132). It can also be induced in Lewis rats by palmitoylated P0 peptides (153).
There is also a model of spontaneous EAN in non-obese diabetic (NOD) mice that are deficient in the co-stimulatory B7-2 molecule (154). In this model there is inflammation and demyelination of peripheral nerves, with a chronic course. However, C57/BL6 mice that overexpress B7-2 also develop spontaneous autoimmune polyneuropathy with a chronic course (155).
Lipids are also major components of myelin. Early studies focused on the possibility that a combination of lipid and protein was required to produce EAN and found that EAN could be induced in rabbits and Lewis rats by inoculation with a complex of lipids and P2 protein (122, 127, 135). Stimulus to finding animal models of disease induced by glycolipids was increased by the finding of antibodies to gangliosides in patients with neuropathy (see above). The first such disease was produced in New Zealand albino rabbits by inoculation with galactocerebroside C (GalC), and is a demyelinating disease (136). Disease induced by GD1a was a flaccid paralysis (137). The disease induced in rabbits by inoculation with GM1 is a severe motor axonal neuropathy (138). The experimental disease produced by GD1b in rabbits is a usually sensory/ataxic neuropathy (136, 139–141) and is associated with apoptosis of cells in the dorsal root ganglion (141).
We now present our views on the pathogenesis of these diseases; we have previously published our views on GBS (156). These diseases are associated with immune changes in peripheral nerve and we take the view that the pathogenesis involves antigen-specific autoimmunity, even though the target antigen is unknown in many cases.
In GBS and CIDP these immune changes can arise after a preceding event, such as an infection, or after a stressful event such as surgery, or can arise without apparent trigger. In the case of the animal models, the immune process is initiated by inoculation. It is likely that host factors play a role in the susceptibility to disease, since not all patients develop GBS after infection, since there are some limited suggestions of genetic associations with GBS and CIDP, and since animal strains vary in susceptibility. It is also notable that there is heterogeneity of GBS and CIDP, both in clinical features and pathology.
It is likely that there is a phase of initiation of disease, when the innate immune system is responsible for activation of the adaptive immune system that leads to an antigen specific process in peripheral nerves and especially the nerve roots. During this phase there is considerable heterogeneity. In GBS after C. jejuni infection, the site of initation of innate immunity would be the gut, after respiratory infections antigen would interact with cells in the respiratory mucosa and in EAN, the antigen would be engulfed by dendritic cells in the skin. In EAN, activation of the innate immune system is achieved by the use of adjuvant.
In all cases it is expected that these antigen presenting cells would travel to the regional lymph nodes. In the lymph nodes there would be activation of T cells and B cells. For protein antigens, the interaction between antigen presenting cells and effector cells would require antigen presentation in association with MHC class I and class II molecules (157). For glycolipid antigens, antigen presentation would require CD1 molecules (158). In most cases, activation of T cells and B cells would occur in association with lymph node follicles, but we speculate that in GBS, unconventional extra-follicular activation could occur, since this leads to short term, self-limited immune activation.
The activated cells and antibody then travel though the blood and through the tissues of the body as part of normal immune surveillance and accumulate at the sites where their target antigen is expressed. Antibody also binds to target epitopes and in some cases triggers complement activation.
In the effector phase of disease, demyelination is caused by myelin stripping and other mechanisms other mechanisms. In the axonal disorders, the mechanism of axonal loss includes other mechanisms. There seems to be a role for complement in GBS, CIDP and EAN since complement deposition is seen in biopsies of patients. The role of complement could be to form the membrane attack complex, resulting in damage.
In GBS and acute EAN here is termination of the immune attack- either because the activation was itself limited, such as the transient activation that can occur after infection (159) or because of development of tolerance mechanisms such as Treg cells. In CIDP there is ongoing inflammation/immune processes due to failure of tolerance mechanisms. In the recovery stage of GBS and EAN and during periods of remission of CIDP there is remyelination (seen as thinly myelinated fibers). There is limited capacity for axonal regeneration in GBS and CIDP.
Differences in prevalence: There is evidence that GBS is more common in males than females, and that the incidence increases with age. A systematic review of 63 papers found the incidence of GBS increased with age and was estimated to be between 1.1/100,000 per year to 1.8 per 100,000 per year (17).
A meta-analysis found that the incidence of GBS increases with age and ranges from 0.90 per 100,000 person years in age 20-29 years to 2.66 per 100,000 person years in ages 80-89 years and that at all ages the incidence was greater in males (160); the authors acknowledged that “the reason for the higher risk of GBS in males is unknown”. A large survey of 150,095 GBS patients, part of the global burden of disease (GBD) 2019 project, conducted by the Institute for Health Metrics and Evaluation (IHME), found that the prevalence increased with age until age 75 years and then declined (18) and the prevalence was greater in males than females at all ages up to 75 years.
Table 4 lists a selection of studies of GBS, since 2010, with more than 100 patients, that show the percentage of male patients. In most reports, the number of males was greater than the number of females. Figure 1 shows a meta-analysis of the data from Table 4. The overall odds ratio was 2.7; the overall relative risk was 1.7.
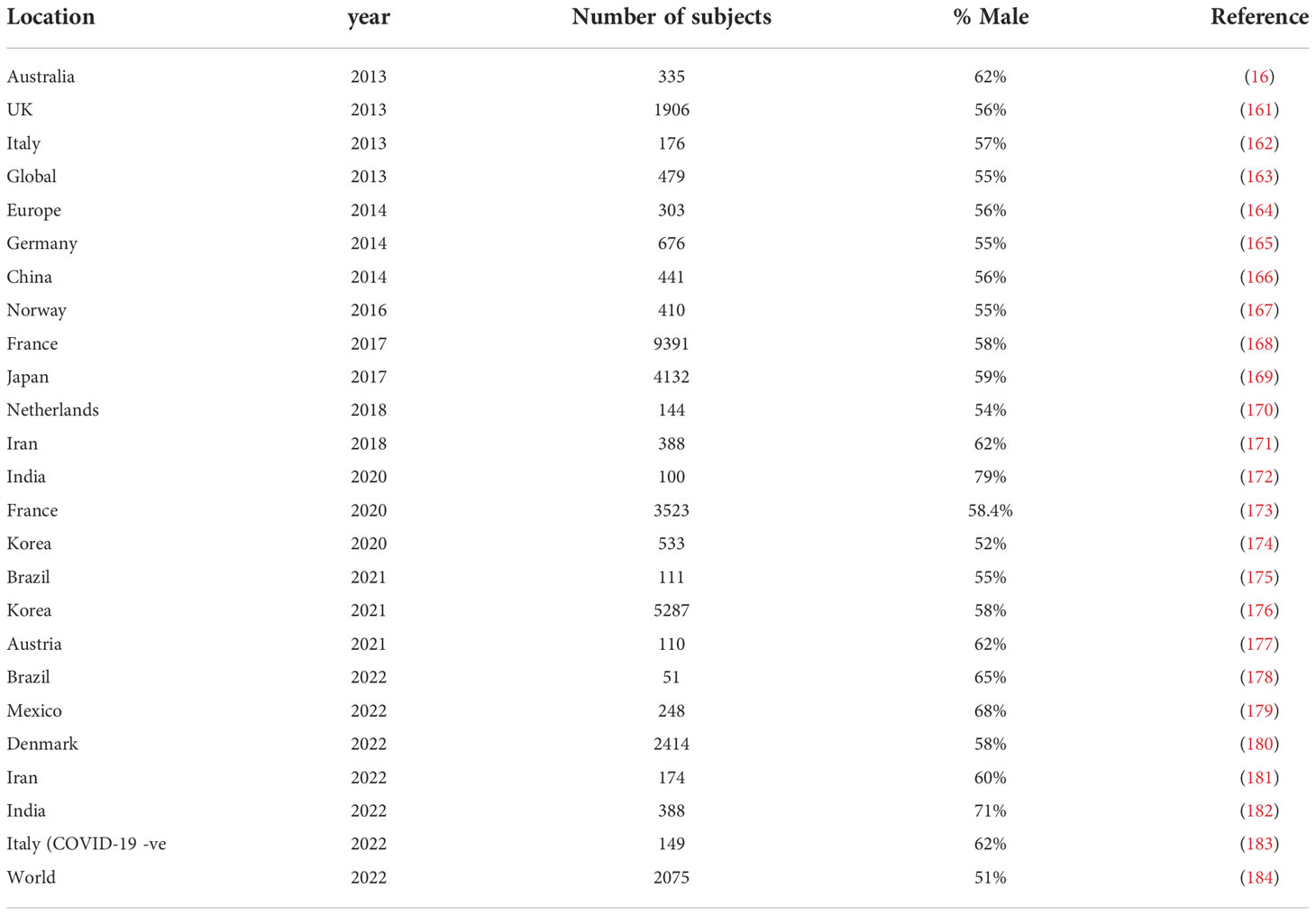
Table 4 Percentage of males in GBS series with 100 or more cases since 2010.
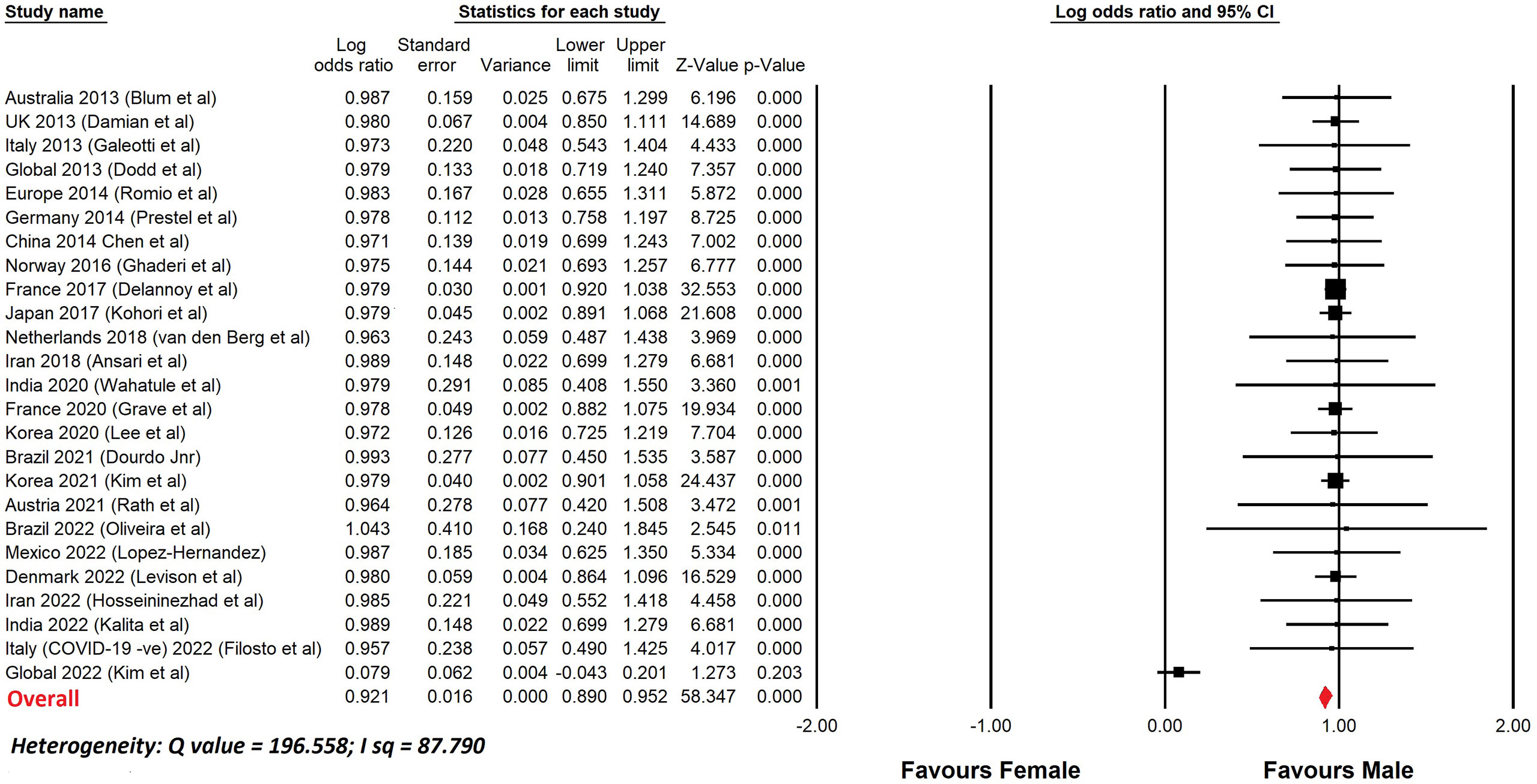
Figure 1 Meta-analysis of sex differences in GBS. This shows a meta-analysis of the data from Table 4. Comprehensive Meta-analysis software v 3.31 (Biotech, USA, 2022) was used. Sex differences are displayed using a log odds ratio. The overall odds ratio was 2.7. The overall relative risk was 1.6.
There have been reports of childhood GBS and a selection of these with more than 20 cases are shown in Table 5. In most cases the upper age limit was 18 years, so these series included pre- and post-pubertal subjects. This is important because sex differences due to the effects of gonadal hormones become apparent after puberty. Figure 2 shows a meta-analysis of the data in Table 5. The overall odds ratio was 1.7; the overall relative risk was 1.3.
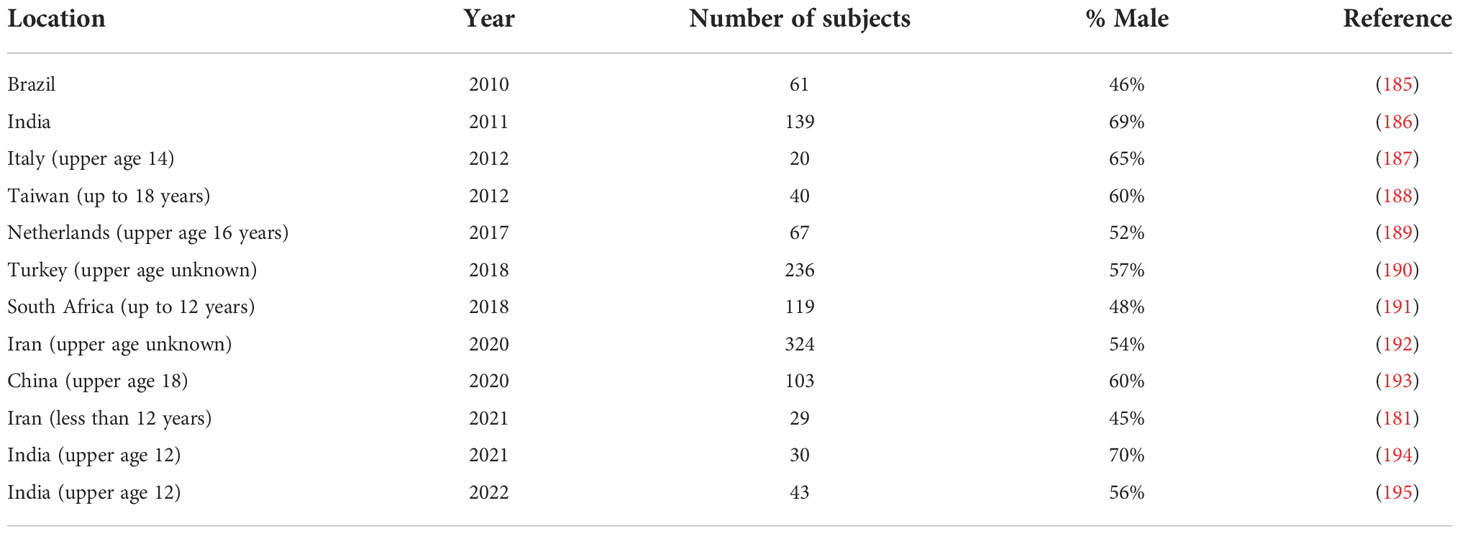
Table 5 Percentage of males in childhood GBS series of 20 or more cases since 2010.
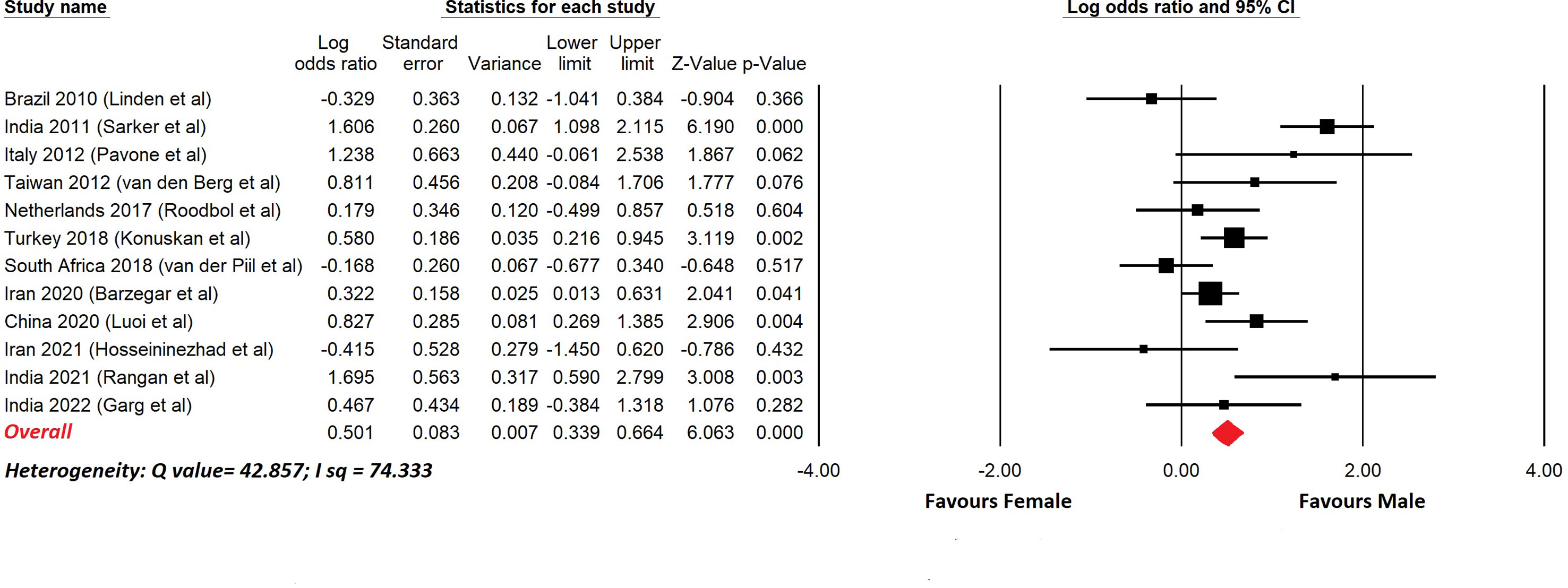
Figure 2 Meta-analysis of sex differences in childhood GBS. This shows a meta-analysis of the data from Table 5. Comprehensive Meta-analysis software v 3.31 (Biotech, USA, 2022) was used. Sex differences are displayed using a log odds ratio. The overall odds ratio was 1.6. The overall relative risk was 1.3.
Sex differences in outcome: The usual treatment for GBS is intravenous infusion of immunoglobulin (IVIG). IVIG was shown to be at least as effective as (if not superior to) plasma exchange in a randomized controlled trial (196). Plasma exchange also appears to be effective, and may be used as an alternative, but tends to be less well tolerated to completion by patients, and is less straightforward to administer (197). Corticosteroids are no of clear benefit and may even be harmful in GBS (198). In GBS, criteria for admission to the intensive care unit include rapidly progressive respiratory muscle weakness, respiratory distress, severe dysautonomia or dysphagia, or a score >4 on the Erasmus GBS respiratory insufficiency score (EGRIS) (199). EGRIS is a composite score that incorporates time from symptom onset to hospitalization, facial and bulbar weakness, and the severity of muscle weakness at hospital admission (200).
A retrospective study of 121 patients from Austria extending back 20 years calculated that sex was not a predictor of prognosis (201). In addition, there was no difference in the odds of receiving more treatment for GBS according to sex. In a large Japanese study of 4132 patients, multivariate regression indicated that males with GBS were less likely than females to require mechanical ventilation (OR 0.76) (169). Once intubated, male patients with GBS tend to have a poorer outcome than females due to ICU-related complications including pneumonia (202).
Difference in Prevalence: An early study found the prevalence of CIDP to be 1/100,000 in South East England (203). In Australia, the prevalence was 1.9 per 100,000 (204)[80]. A higher prevalence of 7.7 per 100,000 was found in Norway (205)[81]. Later studies found prevalence of 4.77 per 100,000 using EFNS/PNS criteria and 1.97 per 100,000 using AAN criteria (206).
As in GBS, the incidence and prevalence of CIDP appears to increase with older age. In a Japanese study, the crude incidence rate of CIDP was 0.06 per 100 000 person years in the age group 0-15 years; 0.40 per 100 000 person years in the age group 15-55 years and 0.73 40 per 100 000 person years among those older than 55 years (207). In a Dutch study, incidence was 17 times as high in those with ages greater than 50 years compared to less than 50 years (208). A systematic review and meta-analysis showed that the age specific prevalence increased steadily from childhood through to old age across 5 different studies (209).
Table 6 lists studies of CIDP that show the prevalence in males and females, with nearly all studies showing a male predominance. Figure 3 shows a meta-analysis of the data in Table 6. The overall odds ratio was 2.9; the overall relative risk was 1.7.
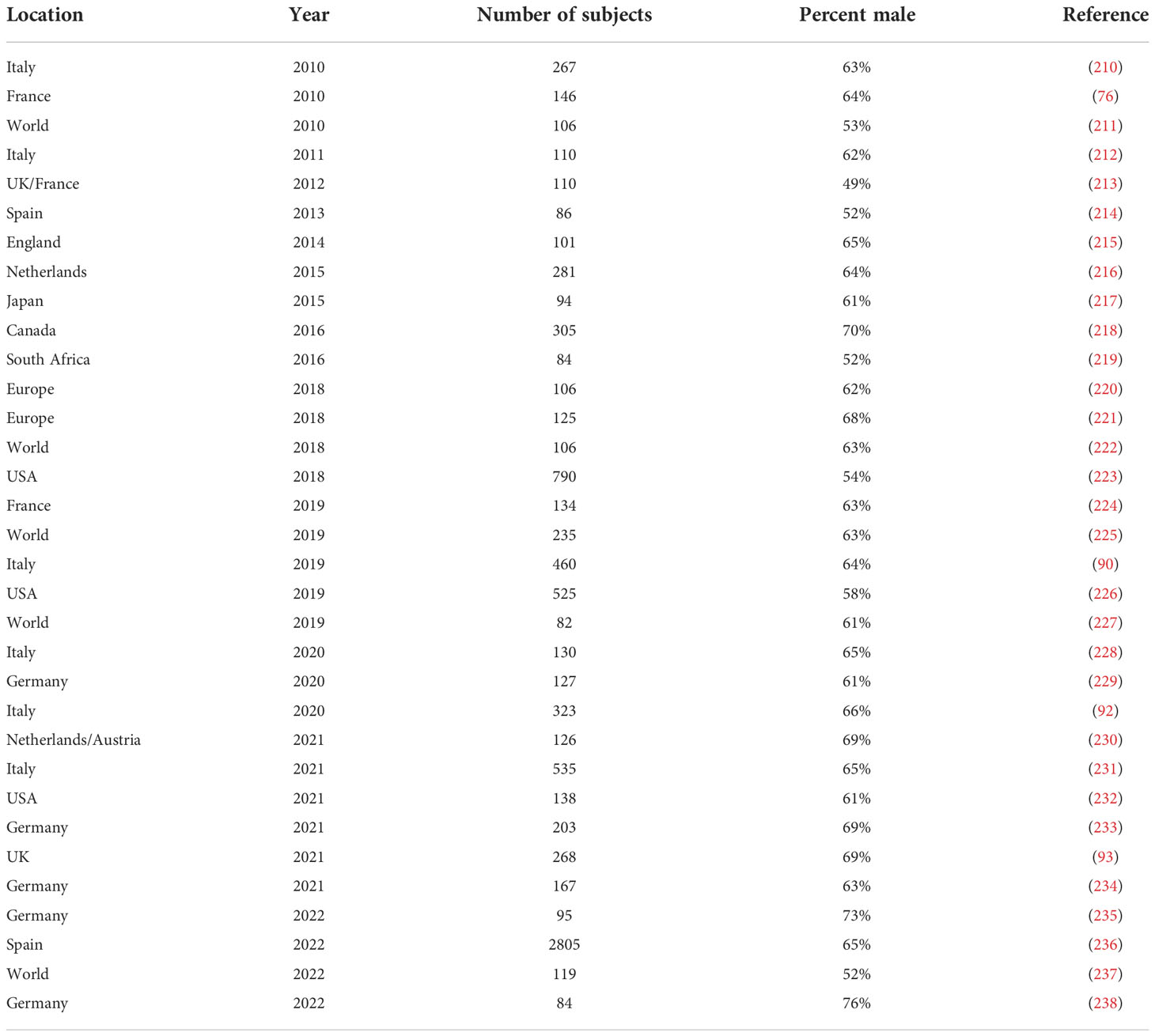
Table 6 Percentage of males in a selection of studies of CIDP of 80 or more cases since 2010.
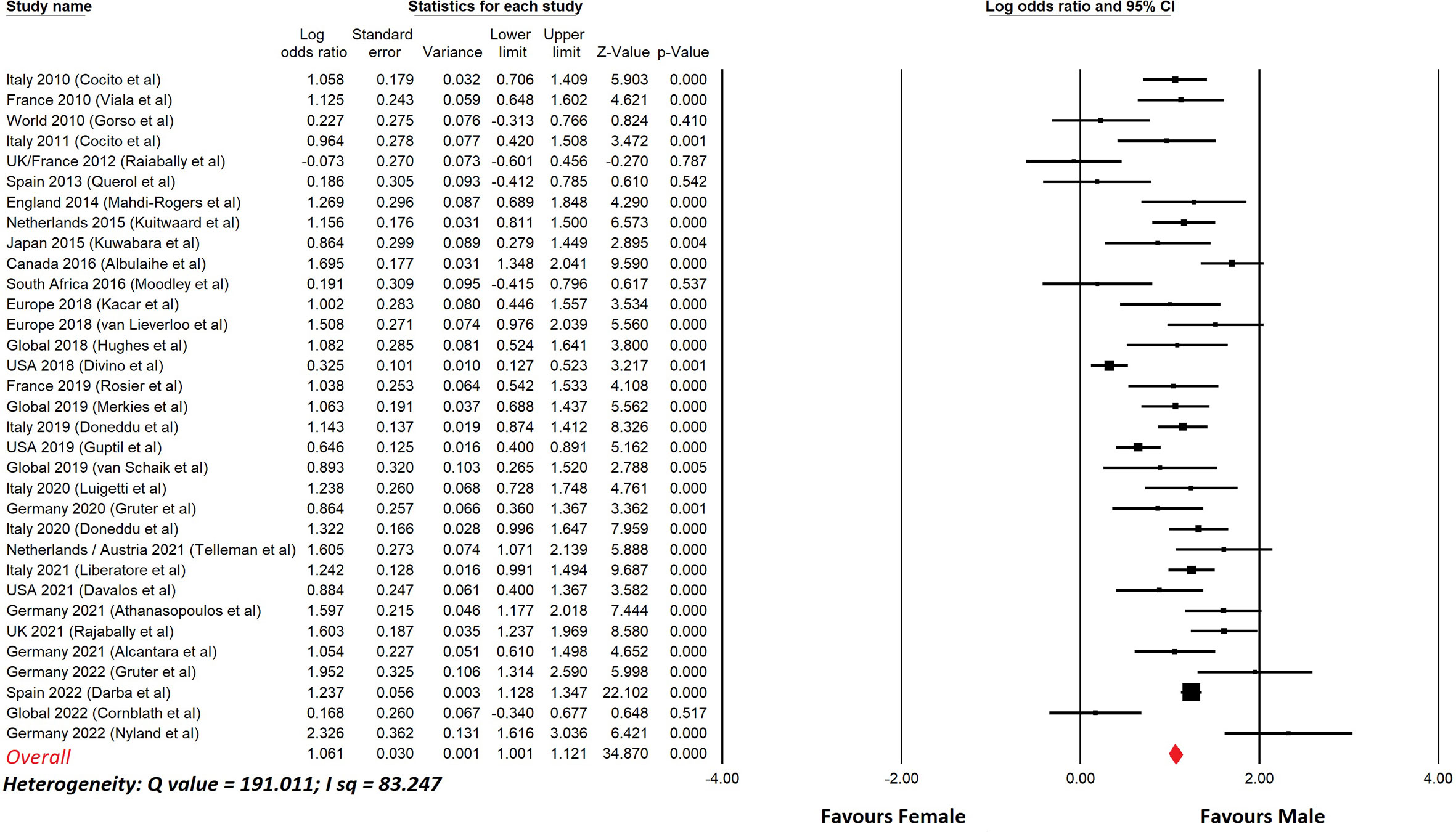
Figure 3 Meta-analysis of sex differences in CIDP.This shows a meta-analysis of the data from Table 6. Comprehensive Meta-analysis software v 3.31 (Biotech, USA, 2022) was used. Sex differences are displayed using a log odds ratio. The overall odds ratio was 2.9. The overall relative risk was 1.7.
In childhood CIDP, a review of a series of patients combined with a review of previous series found no sex differences (239).
Sex difference in outcome: Patients with CIDP can be treated with oral corticosteroids, IVIG or plasma exchange (240). Chronic immune suppressing therapies, such as azathioprine, cyclophosphamide and rituximab are also widely used as part of a combination immune intervention strategy, but quality evidence for efficacy is lacking (241). In the randomized, controlled FORCIDP trial, fingolimod was not of benefit in CIDP (222).
Currently there are trials underway investigating agents that block the neonatal Fc receptor (FcRn) and therefore lead to a decrease in circulating levels of immunoglobulin (Ig) and Ig immune complexes. The results of the first of these trials, which was a phase 2 placebo-controlled trial of rozanoliximab, a monoclonal antibody that antagonizes the FcRn, were disappointing, being negative for an improvement in a patient reported scale of activity and social participation (242).
Sex was not found to be a predictor of severe disability in a multivariate analysis in 165 patients with CIDP (80). A Japanese study found that factors which predict poor responsiveness to IVIG in CIDP patients include male sex, longer disease duration, and slower progression (243).
The role of sex differences has not been a focus of studies of EAN. In early studies there was little recognition of the possible importance of sex differences, and the sex of the animal was not reported, only one sex was used or there was no study of sex differences. Table 3 shows the sex of the animals used in the initial studies of various forms of EAN.
In the original paper, female rabbits were inoculated with CNS tissue and adjuvants (120). In an early paper about EAN in Lewis rats, both sexes were used for inoculation with myelin from guinea pig, frog, rat and rabbit and there was no reported difference between the sexes (123). In another early paper that reported passive transfer of EAN to rabbits, the sex of the donors and the recipients was not stated (244). EAN could be induced in SJL mice by inoculation with myelin but the sex of the mice was not stated (142). In a study of EAN induced in chickens with guinea pig sciatic nerve, both sexes were studied (121). In studies of inoculation of Lewis rats with bovine myelin, male (124) and female Lewis (125) rats have been used.
In the early studies of disease induced in Lewis rats with P2 protein, males were used (127). EAN can be induced in male (130) and female (130, 131) Lewis rats by peptides of myelin P2 protein. Recently there are numbers of studies of EAE induced in female Lewis rats (245–250) by P2 peptides, and occasional studies of males (251) but no studies comparing the sexes.
EAN was induced by myelin P0 protein in male Lewis rats (128). EAN was induced by peptides of P0 protein in male Lewis rats (132). With this model, males have been used in recent studies (252). EAN was induced also with peptides of P0 and in male (133) and female C57/BL6 mice (134), and recent studies have used male Lewis rats (253). PMP22 EAN has been induced in male Lewis rats (129) but there are no recent studies.
Similarly, there is little information about the diseases induced by glycolipids. EAN after GalC sensitization was produced in male New Zealand rabbits (136). GM1 disease and GD1a disease was first induced in male Japanese white rabbits (138, 254, 255). The sex of the rabbits used for induction of GD1 disease was not specified (140).
It must be noted that in experimental autoimmune encephalomyelitis, which has been more thoroughly studied than EAN, and where sex differences have been explored, the effects of sex are complex and vary with animal species/strain and with the inoculating antigen (256).
There are sex differences (sexual dimorphism) in many aspects of physiology and pathophysiology. These have developed since the evolution of the sex chromosomes and sexual reproduction (257–259). Sex differences can be attributed to the effects of the sex chromosomes and to the effects of gonadal hormones. There are widespread examples of sexual dimorphism in morphology, physiology and biochemistry (260). There is marked sexual dimorphism of the immune system (261, 262). Across the animal kingdom, females have a more active immune system (sometimes referred to as having greater immunocompetence) than males (263, 264). Tables 7, 8 show some sex differences in the immune system.

Table 7 Sexual dimorphism in immune processes.

Table 8 Sex differences in immune cell numbers and functions.
In this review we have studied sex differences in GBS and CIDP. GBS and CIDP are consistently more common in males than females. The sex association was less obvious in childhood GBS, where some of the subjects would have been pre-pubertal. Sex differences that appear after puberty can be due to the effects of gonadal hormones. It can be noted that in multiple sclerosis, where there is a strong influence of sex on the prevalence of disease in adults, there is no sex difference in childhood before puberty (286). There is little evidence of an effect of sex on the outcome of disease. It must be noted that the majority of studies were cohort studies rather than population based studies, which is a limitation.
The male predominance would be unusual if GBS and CIDP were like other autoimmune diseases, which are usually more common in females (260, 287). For these other autoimmune diseases, the female predominance is often attributed to the stronger immune system of females, Tables 7, 8 provide lists of the effects of sex on different immune cells and functions, and shows that, for many functions, the female immune system has stronger responses. Females have greater resistance to infection, greater resistance to the induction of immune tolerance, greater in vitro response to mitogens, greater response to vaccination, greater IgM levels and greater Th1 responses (262). The stronger immune responses are thought to have arisen through evolutionary mechanisms allowing females to live longer to protect their offspring (264).
We have considered what is known about the pathogenesis of GBS and CIDP and now speculate on some possible reasons for the sex differences. The pathogenesis involves macrophages, monocytes, CD4+ T cells, B cells, Th17 cells, antibody, complement and cytokines. As outlined in Tables 7, 8, for most of these elements of the immune system, females are thought to have stonger responses. Exceptions are CD8+ cells and NK cells which have increased levels in males and have a possible role in GBS and CIDP (72, 117, 288). Therefore, we have looked for other explanations for the sex differences in GBS and CIDP.
We consider that GBS and CIDP are autoimmune diseases, likely to be due to an antigen-specific immune response, athough for many patients the antigen remains unknown. However, GBS and CIDP have some features that are unusual for autoimmune diseases. Criteria for accepting an autoimmune aetiology for disease were first put forward in 1957 (289) and later updated in 1993 (290). These criteria include direct proof (transmission to another human or experimental animal), indirect proof (induction of disease by autoantigen, pathological features) and circumstantial evidence (MHC association). GBS and CIDP have features that differ from these criteria. There is no clear target antigen – although it could be argued that each of the syndromes associated with the known targets of autoantibodies could be regarded as separate diseases. For some of the known antigen, namely the glycolipid antigens, disease transfer has been more difficult than with more classical autoimmune diseases. Finally, there is no clear HLA association (71); this could be related primarily to the fact that, in GBS, and some CIDP patients, as opposed to the case in well-recognized autoimmune diseases, the target antigens are not proteins but glycolipids, which are not presented by HLA molecules. There is more limited polymorphism in the CD1 and MR1 molecules than for HLA molecules, but nothing is currently known about whether there are any sex-related effects in their expression.
GBS differs from autoimmune diseases in being self-limited. This suggests a transient activation of the immune system. This can occur through activation of plasmablasts by pathways that do not include the lymphoid follicle or through non-canonical pathways of activation (291). There are some suggestions that sex influences the activation of plasmablasts and plasma cells (292). Further exploration of the role of plasmablast activation could be useful in understanding GBS and exploring sex differences.
Genome wide gene expression analysis of peripheral leukocytes has indicated differences between male and female patients with GBS. In one study, male GBS patients were enriched for twenty genes involved in a range of immunological processes, including macrophage and leukocyte migration, and female GBS patients were enriched for 62 genes including those for viral infection and defense (293). Genes involved in the production of matrix metaloproteinase-9 (MMP9), which has previously been shown to be associated with disease severity in GBS, were highly expressed in males implicating MMP9 as being potentially relevant to the higher prevalence of GBS in males.
In most cases of GBS, and some patients with CIDP, gangliosides are the target antigen. Gangliosides are small glycolipid molecules that react with unconventional T cells including γδ T cells, NKT cells and MR1T/MAIT cells (58–60) after antigen presentation in association with CD1 (158). Little is known about sex differences in these pathways.
However, one study measured the numbers of unconventional T cells and showed females had more iNKT cells, fewer γδ T cells and the same number of MAIT cells as males (294). Another study showed that there are sex differences in the function of NKT cells, with male cells having a Th1 bias (295). However, a third study found that women have greater numbers of NKT cells than men, and that stimulation with alpha-galactosylceramide leads to higher production of IFNγ, IL-4, IL-17 and TNF by CD4+ and double negative NKT cells (296). Further studies of the mechanisms of development of reactivity to gangliosides could also be helpful in the understanding of GBS and CIDP and possible sex differences.
In CIDP, studies suggest a role for CD8 cells, that interact with MHC class I. A recent study found that HLA-associated shaping of T cell receptor B variable (TCRBV) usage in CD8+ T cells differed between the sexes, with male cells showing greater expansion of TCRBV usage than females (297); this is evidence that there could be sex differences in the capacity of these cells to interact with self antigens. It remains to be determined if this occurs in CIDP.
Patients with “treatment resistant CIDP” can be placed in a separate group of autoimmune nodopathies (AN), which are emediated by IgG4 antibodies. There are other autoimmune diseases associated with IgG4 antibodies, and some of these are neurological, including anti-muscle specific tyrosine kinease (MUSK) myasthenia gravis, leucine-rich glioma-inactivated (LGI)-1 and CASPR2 autoimmune syndromes, and anti-immunoglobulin LSAMP, OBCAM, Neurotrimin 5 (IgLON5) disorder disorder (298). It must be noted, that in contrast to the findings in CIDP, MUSK MG is reported to be more common in females. A switch to IgG4 antibody production is triggered by chronic antigen exposure, and an environment enriched by T cell-produced cytokines, in particular IL-4, IL-10, IL-12, IL-13 and IL-21 (299).There are many sex differences in T cells, for example the ratio of Th1 to Th2 cells is greater for females than males (300)and there are sex differences in the regulation of TfH cells, which are needed for antibody production (301). Interestingly, a group of systemic immune-mediated disorders that are all characterized by infiltration of IgG4-expressing plasma cells into involved organs have now been consolidated into a grouping known as IgG4-related disease, and one of the predominant clinical features is the male predominance of these disorders (302).
In CIDP, another distinguishing feature that could suggest unusual immunological features, is the lack of response to therapies that were expected to be of benefit. This could be due to the acknowledged challenges of trials in CIDP, for example the difficulty in identifying patients with active disease who deteriorate after withdrawal of IVIG. However, some of the pathological features of CIDP are similar to those of multiple sclerosis (MS), but fingolimod, which is of benefit in MS, was not helpful in CIDP. In antibody mediated diseases, such as myasthenia gravis (MG), blockade of the neonatal Fc receptor is of benefit. However, the results of the first trial in CIDP showed no benefit (242). There is no information about what these failures reveal about the underlying pathogenesis of CIDP. However, this could be a further indicator that CIDP differs from other immune and inflammatory diseases.
The sex differences in GBS and CIDP could relate to sex differences in the underlying immunopathogenetic mechanisms. However, our understandng of the pathogenesis of GBS and CIDP is incomplete. Going forward, it will be beneficial to perform further studies of the immunological basis of GBS and CIDP, with attention to the heterogeneity of these diseases. There is marked clinical heterogeneity and also heterogeneity in the immunological targets that have been identified. Once the immunological basis of the subtypes of disease is clear, then further attention can be given to the sex differences in these mechanisms.
It must be noted that the prevalence of GBS and CIDP increases with age. In myasthenia gravis, which shows a late peak of disease (late-onset MG), studies, including our own, have also shown that there is an increased proportion of males in the late onset disease (303, 304). This could suggest that older males are more susceptible to immune disease and this could contribute to the male predominance. It is known that there are changes in the immune system with aging (immunosenescence) (305). We have shown sex differences in immunosenescence, with males showing greater decline (265). Above we have speculated on a role for unconventional T cells; there is no information about the effect of ageing on these cells, or any sex differences in these cells with aging. It is possible that the sex differences in immunosenescence could make males more susceptible- possibly by waning of immunoregulation.
Another possible issue is sex differences in the susceptibility of nerves to immunological attack. In autoimmunity, this can be referred to as “target organ resistance”. Little has been written about this, but some of the earliest studies come from the model of autoimmune thyroiditis that arises in obese strain chickens, and is a model of human Hashimoto’s thyroiditis (306). In this disorder, inherited alterations of thyroid function predispose to spontaneous autoimmune thyroiditis (307, 308). This has been related to viral infection and aberrant expression of MHC class II molecules (309). In experimental autoimmune oophoritis, a model of autoimmune ovarian failure (310), it has been shown that after recovery from oophoritis, animals are resistant to further episodes of disease and that this resistance is a property of the recovered ovaries. When normal ovarian tissue was transplanted under the capsule of recovered ovaries, disease developed only in the transplanted tissue. In experimental autoimmune encephalomyelitis, lack of apoptosis in target tissue leads to increased EAE (311). We have previously suggested that sex differences in target organ resistance could contribute to sex differences in autoimmunity (260).
There is some evidence that alterations in peripheral nerve myelin can lead to inflammation of nerves. This is seen in the reports of inflammation typical of CIDP, or the onset of clinical features of CIDP in people with various types of Charcot Marie Tooth (CMT) disease (312–314). This is highly suggestive that variation in the structure of nerves could possibly lead to vulnerability to inflammation.
The underlying mechanism for putative sex differences in target organ resistance of peripheral nerves would be expected to be due to the biological properties of nerves. There are some reports of sex differences in the peripheral nerves and in the response of peripheral nerve to injury. Gene expression profiles from the dorsal root ganglia (DRG) of rats show that 6% of expressed genes differ according to sex with more genes involved in immunological mechanisms expressed in females than males, and that after injury to the DRG, neural pathways linked to pain also show considerable differences between males and females (315). Furthermore, following sciatic nerve axotomy in mice, males exhibited greater expression of structural cytoskeletal proteins in the regenerating nerve than females. In addition there were sex differences in coding and non-coding mRNAs responsible for other neurotrophic, metabolic and sex chromosome-linked molecular programs (316). It is possible that these sex differences in the biology of peripheral nerves could also contribute to the differences in susceptibility to GBS and CIDP. However, there is still much that needs to be determined.
Our conclusion is that there is evidence that unlike other autoimmune diseases, GBS and CIDP are more common in males. This difference is not likely to have any single cause, and could relate to differnces in the immune system or in the target organ. There are no studies of sex differences in EAN and in future it would be very helpful if such studies were to be performed, although we note that in experimental autoimmune encephalomyelitis (EAE), there are many sex differences that are specific for the strain of animal and the type of disease that is studied (256). Therefore it will be necessary to report all these details. It will also be necessary to report the age of the animal, given that sex differences change over time- becoming more apparent with puberty and showing differences in older age. It has also been found that, in some experiments, the sex of the researcher can influence the results of the study (317, 318), so this should also be recorded.
Going forward we suggest that there is a need to investigate sex differences in GBS and CIDP. For males compared to females the odds ratio for developing GBS and CIDP are 2.7 and 2. 9, respectively. This is a substantial increase in risk, and understanding the mechanisms for this could help understand what is important in pathogenesis. We recommend that all epidemiological studies stratify for age and sex, and that all investigations into the biology of GBS and CIDP also take account of the sex of the subjects. We also recommened that all clinical trials should be stratified according to sex, in case there are sex differences in the response to therapy.
There are some challenges. Studies will need to be increased in size, to provide sufficient power to detect sex differences, and investigators need to consider the effects of sex at different stages of investigation such as recruitment, randomization and analysis (319). However, despite the challenges, research into sex differences has the potential to be rewarding. Sex differences are pervasive in all aspects of biology, and the finding that GBS and CIDP are more common in males deserves further enquiry.
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Kieseier BC, Mathey EK, Sommer C, Hartung HP. Immune-mediated neuropathies. Nat Rev Dis Primers (2018) 4(1):31. doi: 10.1038/s41572-018-0027-2
2. Kuwabara S, Misawa S. Chronic inflammatory demyelinating polyneuropathy. Adv Exp Med Biol (2019) 1190:333–43. doi: 10.1007/978-981-32-9636-7_21
3. Yuki N, Hartung HP. Guillain-Barré Syndrome. New Engl J Med (2012) 366(24):2294–304. doi: 10.1056/NEJMra1114525
4. Klein SL, Flanagan KL. Sex differences in immune responses. Nat Rev Immunol (2016) 16(10):626–38. doi: 10.1038/nri.2016.90
5. Guillain G, Barré JA, Strohl A. Sur un syndrome de radiculonévrite avec hyperalbuminose Du liquide cephalo-rachidien sans reaction cellulaire. remarques sur Les caractères cliniques et graphiques des réflexes tendineux. Bull Soc Med Hop Paris (1916) 40:1462–70.
7. Haymaker W, Kernohan JW. The landry-Guillain-Barré syndrome. a clinicopathological report of fifty fatal cases and a critique of the literature. Med Baltimore (1949) 28:59–141. doi: 10.1097/00005792-194902010-00003
8. Wiederholt WC, Mulder DW, Lambert EH. The landry-Guillain-Barré-Strohl syndrome or polyradiculoneuropathy: Historical review, report on 97 patients, and present concepts. Mayo Clin Proc (1964) 39:427–51.
9. Asbury AK. Guillain-Barré Syndrome: Historical aspects. Ann Neurol (1990) 27(Supplement):S2–6. doi: 10.1002/ana.410270703
10. Goodfellow JA, Willison HJ. Guillain-Barré Syndrome: A century of progress. Nat Rev Neurol (2016) 12(12):723–31. doi: 10.1038/nrneurol.2016.172
11. Fisher M. An unusual variant of acute idiopathic polyneuritis (Syndrome of ophthalmoplegia, ataxia and areflexia). New Engl J Med (1956) 255(2):57–65. doi: 10.1056/nejm195607122550201
12. Asbury AK, Cornblath DR. Assessment of current diagnostic criteria for Guillain-Barré syndrome [See comments]. Ann Neurol (1990) 27 Suppl:S21–S4. doi: 10.1002/ana.410270707
13. Hadden RD, Cornblath DR, Hughes RA, Zielasek J, Hartung HP, Toyka KV, et al. Electrophysiological classification of Guillain-barre syndrome: Clinical associations and outcome. Plasma Exchange/Sandoglobulin Guillain-Barre Syndrome Trial Group Ann Neurol (1998) 44(5):780–8. doi: 10.1002/ana.410440512
14. Fokke C, van den Berg B, Drenthen J, Walgaard C, van Doorn PA, Jacobs BC. Diagnosis of Guillain-Barré syndrome and validation of Brighton criteria. Brain (2014) 137(Pt 1):33–43. doi: 10.1093/brain/awt285
15. Sejvar JJ, Kohl KS, Gidudu J, Amato A, Bakshi N, Baxter R, et al. Guillain-Barré Syndrome and Fisher syndrome: Case definitions and guidelines for collection, analysis, and presentation of immunization safety data. Vaccine (2011) 29(3):599–612. doi: 10.1016/j.vaccine.2010.06.003
16. Blum S, Reddel S, Spies J, McCombe P. Clinical features of patients with Guillain-barre syndrome at seven hospitals on the East coast of Australia. J Peripher Nerv Syst (2013) 18(4):316–20. doi: 10.1111/jns5.12045
17. McGrogan A, Madle GC, Seaman HE, de Vries CS. The epidemiology of Guillain-barre syndrome worldwide. A Systematic Literature Review. Neuroepidemiology (2009) 32(2):150–63. doi: 10.1159/000184748
18. Bragazzi NL, Kolahi AA, Nejadghaderi SA, Lochner P, Brigo F, Naldi A, et al. Global, regional, and national burden of Guillain-Barré syndrome and its underlying causes from 1990 to 2019. J Neuroinflamm (2021) 18(1):264. doi: 10.1186/s12974-021-02319-4
19. Arends S, Drenthen J, Van den Bergh PYK, Hadden RDM, Shahrizaila N, Dimachkie MM, et al. Electrodiagnostic subtyping in Guillain-Barré syndrome: Use of criteria in practice based on a survey study in IGOS. J peripheral nervous system JPNS (2022) 27(3):107-205. doi: 10.1111/jns.12504
20. Berciano J. Axonal degeneration in Guillain-Barré syndrome: A reappraisal. J Neurol (2021) 268(10):3728–43. doi: 10.1007/s00415-020-10034-y
21. Uncini A, Manzoli C, Notturno F, Capasso M. Pitfalls in electrodiagnosis of Guillain-barre syndrome subtypes. J Neurol Neurosurg Psychiatry (2010) 81(10):1157–63. doi: 10.1136/jnnp.2010.208538
22. Uncini A, Yuki N. Electrophysiologic and immunopathologic correlates in Guillain-barre syndrome subtypes. Expert Rev Neurother (2009) 9(6):869–84. doi: 10.1586/ern.09.43
23. Hughes RA, Cornblath DR. Guillain-Barre syndrome. Lancet (2005) 366(9497):1653–66. doi: 10.1016/S0140-6736(05)67665-9
24. Wakerley BR, Yuki N. Isolated facial diplegia in Guillain-Barré syndrome: Bifacial weakness with paresthesias. Muscle Nerve (2015) 52(6):927–32. doi: 10.1002/mus.24887
25. Rousseff RT, Khuraibet AJ, Neubauer D. The "Child in the barrel syndrome"–severe pharyngeal-Cervical-Brachial variant of Guillain-barre syndrome in a toddler. Neuropediatrics (2008) 39(6):354–6. doi: 10.1055/s-0029-1202768
26. Leonhard SE, van der Eijk AA, Andersen H, Antonini G, Arends S, Attarian S, et al. An international perspective on preceding infections in Guillain-Barré syndrome: The IGOS-1000 cohort. Neurology (2022) 99(12):e1299–313. doi: 10.1212/wnl.0000000000200885
27. Sinha S, Prasad KN, Jain D, Pandey CM, Jha S, Pradhan S. Preceding infections and anti-ganglioside antibodies in patients with Guillain-Barré syndrome: A single centre prospective case-control study. Clin Microbiol infection (2007) 13(3):334–7. doi: 10.1111/j.1469-0691.2006.01636.x
28. Arnason BG, Asbury AK. Idiopathic polyneuritis after surgery. Arch Neurol (1968) 18(5):500–7. doi: 10.1001/archneur.1968.00470350058005
29. Gensicke H, Datta AN, Dill P, Schindler C, Fischer D. Increased incidence of Guillain-Barré syndrome after surgery. Eur J Neurol (2012) 19(9):1239–44. doi: 10.1111/j.1468-1331.2012.03730.x
30. Rudant J, Dupont A, Mikaeloff Y, Bolgert F, Coste J, Weill A. Surgery and risk of Guillain-Barré syndrome: A French nationwide epidemiologic study. Neurology (2018) 91(13):e1220–e7. doi: 10.1212/wnl.0000000000006246
31. Huang C, Zhang Y, Deng S, Ren Y, Lu W. Trauma-related Guillain-Barré syndrome: Systematic review of an emerging concept. Front Neurol (2020) 11:588290. doi: 10.3389/fneur.2020.588290
32. Hafer-Macko CE, Sheikh KA, Li CY, Ho TW, Cornblath DR, McKhann GM, et al. Immune attack on the schwann cell surface in acute inflammatory demyelinating polyneuropathy. Ann Neurol (1996) 39(5):625–35. doi: 10.1002/ana.410390512
33. Wanschitz J, Maier H, Lassmann H, Budka H, Berger T. Distinct time pattern of complement activation and cytotoxic T cell response in Guillain-barre syndrome. Brain (2003) 126(Pt 9):2034–42. doi: 10.1093/brain/awg207
34. Winer J, Hughes S, Cooper J, Ben Smith A, Savage C. Gamma delta T cells infiltrating sensory nerve biopsies from patients with inflammatory neuropathy. J Neurol (2002) 249(5):616–21. doi: 10.1007/s004150200072
35. Prineas JW. Pathology of the Guillain-Barré syndrome. Ann Neurol (1981) 9(supplement):6–19. doi: 10.1002/ana.410090704
36. Prineas JW. Acute idiopathic polyneuritis. Electron microscope study. Lab Invest (1972) 26:133–47.
37. Prineas JW. Demyelination and remyelination in recurrent idiopathic polyneuropathy. an electron microscope study. Acta Neuropathol (1971) 18(1):34–57. doi: 10.1007/bf00684474
38. Asbury AK, Arnason BG, Adams RD. The inflammatory lesion in idiopathic polyneuritis. its role in pathogenesis. Med Baltimore (1969) 48:173–215. doi: 10.1097/00005792-196905000-00001
39. Hafer-Macko C, Hsieh ST, Li CY, Ho TW, Sheikh K, Cornblath DR, et al. Acute motor axonal neuropathy: An antibody-mediated attack on axolemma. Ann Neurol (1996) 40(4):635–44. doi: 10.1002/ana.410400414
40. Kuwabara S, Yuki N. Axonal Guillain-Barré syndrome: Concepts and controversies. Lancet Neurol (2013) 12(12):1180–8. doi: 10.1016/s1474-4422(13)70215-1
41. Griffin JW, Li CY, Macko C, Ho TW, Hsieh ST, Xue P, et al. Early nodal changes in the acute motor axonal neuropathy pattern of the Guillain-Barré syndrome. J Neurocytol (1996) 25(1):33–51. doi: 10.1007/bf02284784
42. Dehaene I, Martin JJ, Geens K, Cras P. Guillain-Barre syndrome with ophthalmoplegia: Clinicopathologic study of the central and peripheral nervous systems, including the oculomotor nerves. Neurology (1986) 36:851–4. doi: 10.1212/wnl.36.6.851
43. Csurhes PA, Sullivan AA, Green K, Pender MP, McCombe PA. T Cell reactivity to P0, P2, pmp-22, and myelin basic protein in patients with Guillain-barre syndrome and chronic inflammatory demyelinating polyradiculoneuropathy. J Neurol Neurosurg Psychiatry (2005) 76(10):1431–9. doi: 10.1136/jnnp.2004.052282
44. Devaux JJ, Odaka M, Yuki N. Nodal proteins are target antigens in Guillain-barre syndrome. J Peripher Nerv Syst (2012) 17(1):62–71. doi: 10.1111/j.1529-8027.2012.00372.x
45. Kusunoki S, Willison HJ, Jacobs BC. Antiglycolipid antibodies in Guillain-Barré and Fisher syndromes: Discovery, current status and future perspective. J neurology neurosurgery Psychiatry (2021) 92(3):311–8. doi: 10.1136/jnnp-2020-325053
46. Csurhes PA, Sullivan AA, Green K, Greer JM, Pender MP, McCombe PA. Increased circulating T cell reactivity to Gm1 ganglioside in patients with Guillain-barre syndrome. J Clin Neurosci (2005) 12(4):409–15. doi: 10.1016/j.jocn.2004.04.006
47. Vasques JF, de Jesus Gonçalves RG, da Silva-Junior AJ, Martins RS, Gubert F, Mendez-Otero R. Gangliosides in nervous system development, regeneration, and pathologies. Neural Regener Res (2023) 18(1):81–6. doi: 10.4103/1673-5374.343890
48. Kaida K, Morita D, Kanzaki M, Kamakura K, Motoyoshi K, Hirakawa M, et al. Ganglioside complexes as new target antigens in Guillain-Barré syndrome. Ann Neurol (2004) 56(4):567–71. doi: 10.1002/ana.20222
49. Laman JD, Huizinga R, Boons GJ, Jacobs BC. Guillain-Barré Syndrome: Expanding the concept of molecular mimicry. Trends Immunol (2022) 43(4):296–308. doi: 10.1016/j.it.2022.02.003
50. Damian RT. Molecular mimicry: Antigen sharing by parasite and host and its consequences. Am Nat (1964) 98(900):129–49. doi: 10.1086/282313
51. Ang CW, Jacobs BC, Laman JD. The Guillain-barre syndrome: A true case of molecular mimicry. Trends Immunol (2004) 25(2):61–6. doi: 10.1016/j.it.2003.12.004
52. Kitamura H, Nakano T, Kakihara M, Nishino M, Isshiki K, Kawano K, et al. A case of Guillain-Barré syndrome developed minimal change nephrotic syndrome simultaneously. Am J Nephrol (1998) 18(2):151–4. doi: 10.1159/000013325
53. Zhao PP, Ji QK, Sui RB, Zhang R, Zhang LJ, Xu ZX, et al. Increased intracranial pressure in Guillain-Barré syndrome: A case report. Med (Baltimore) (2018) 97(30):e11584. doi: 10.1097/md.0000000000011584
54. Corbett AJ, Eckle SB, Birkinshaw RW, Liu L, Patel O, Mahony J, et al. T-Cell activation by transitory neo-antigens derived from distinct microbial pathways. Nature (2014) 509(7500):361–5. doi: 10.1038/nature13160
55. Van Rhijn I, Godfrey DI, Rossjohn J, Moody DB. Lipid and small-molecule display by Cd1 and Mr1. Nat Rev Immunol (2015) 15(10):643–54. doi: 10.1038/nri3889
56. Mori L, Lepore M, De Libero G. The immunology of Cd1- and Mr1-restricted T cells. Annu Rev Immunol (2016) 34:479–510. doi: 10.1146/annurev-immunol-032414-112008
57. Shimamura M, Huang YY, Okamoto N, Suzuki N, Yasuoka J, Morita K, et al. Modulation of Valpha19 nkt cell immune responses by alpha-mannosyl ceramide derivatives consisting of a series of modified sphingosines. Eur J Immunol (2007) 37(7):1836–44. doi: 10.1002/eji.200636689
58. Lepore M, Mori L, De Libero G. The conventional nature of non-Mhc-Restricted T cells. Front Immunol (2018) 9:1365. doi: 10.3389/fimmu.2018.01365
59. Wo J, Zhang F, Li Z, Sun C, Zhang W, Sun G. The role of gamma-delta T cells in diseases of the central nervous system. Front Immunol (2020) 11:580304. doi: 10.3389/fimmu.2020.580304
60. Harriff MJ, McMurtrey C, Froyd CA, Jin H, Cansler M, Null M, et al. Mr1 displays the microbial metabolome driving selective Mr1-restricted T cell receptor usage. Sci Immunol (2018) 3(25) eaao2556. doi: 10.1126/sciimmunol.aao2556
61. Gálvez NMS, Bohmwald K, Pacheco GA, Andrade CA, Carreño LJ, Kalergis AM. Type I natural killer T cells as key regulators of the immune response to infectious diseases. Clin Microbiol Rev (2021) 34(2) e00232-20. doi: 10.1128/cmr.00232-20
62. Dahle C, Vrethem M, Ernerudh J. T Lymphocyte subset abnormalities in peripheral blood from patients with the Guillain-barre syndrome. J Neuroimmunol (1994) 53(2):219–25. doi: 10.1016/0165-5728(94)90032-9
63. Harness J, McCombe PA. Increased levels of activated T-cells and reduced levels of Cd4/Cd25+ cells in peripheral blood of Guillain-barre syndrome patients compared to controls. J Clin Neurosci (2008) 15(9):1031–5. doi: 10.1016/j.jocn.2007.09.016
64. Sun T, Chen X, Shi S, Liu Q, Cheng Y. Peripheral blood and cerebrospinal fluid cytokine levels in Guillain Barré syndrome: A systematic review and meta-analysis. Front Neurosci (2019) 13:717. doi: 10.3389/fnins.2019.00717
65. Wu CL, Chao CH, Lin SW, Chien YY, Huang WY, Weng WC, et al. Case report: Plasma biomarkers reflect immune mechanisms of Guillain-Barré syndrome. Front Neurol (2021) 12:720794. doi: 10.3389/fneur.2021.720794
66. Battistini L, Borsellino G, Sawicki G, Poccia F, Salvetti M, Ristori G, et al. Phenotypic and cytokine analysis of human peripheral blood gamma delta T cells expressing nk cell receptors. J Immunol (1997) 159(8):3723–30.
67. Wang Y, Zhang J, Luo P, Zhu J, Feng J, Zhang HL. Tumor necrosis factor-A in Guillain-Barré syndrome, friend or foe? Expert Opin Ther Targets (2017) 21(1):103–12. doi: 10.1080/14728222.2017.1258402
68. Debnath M, Nagappa M, Murari G, Taly AB. Il-23/Il-17 immune axis in Guillain Barré syndrome: Exploring newer vistas for understanding pathobiology and therapeutic implications. Cytokine (2018) 103:77–82. doi: 10.1016/j.cyto.2017.12.029
69. Sharma PP, Seshagiri DV, Nagappa M, Mullapudi T, Sreenivas N, Dey S, et al. Role of altered il-33/St2 immune axis in the immunobiology of Guillain-Barré syndrome. Eur J Neurol (2022) 29(7):2074–83. doi: 10.1111/ene.15334
70. Debnath M, Nagappa M, Dutta D, Talukdar PM, Subbanna M, Shivakumar V, et al. Evidence of altered Th17 pathway signatures in the cerebrospinal fluid of patients with Guillain Barré syndrome. J Clin (2020) 75:176–80. doi: 10.1016/j.jocn.2020.03.010
71. Blum S, McCombe PA. Genetics of Guillain-barre syndrome (Gbs) and chronic inflammatory demyelinating polyradiculoneuropathy (Cidp): Current knowledge and future directions. J Peripher Nerv Syst (2014) 19(2):88–103. doi: 10.1111/jns5.12074
72. Blum S, Csurhes P, Reddel S, Spies J, McCombe P. Killer immunoglobulin-like receptor and their hla ligands in Guillain-barre syndrome. J Neuroimmunol (2013) 267:92–6. doi: 10.1016/j.jneuroim.2013.12.007
73. Thielens A, Vivier E, Romagné F. Nk cell mhc class I specific receptors (Kir): From biology to clinical intervention. Curr Opin Immunol (2012) 24(2):239–45. doi: 10.1016/j.coi.2012.01.001
74. Dębska-Zielkowska J, Moszkowska G, Zieliński M, Zielińska H, Dukat-Mazurek A, Trzonkowski P, et al. Kir receptors as key regulators of nk cells activity in health and disease. Cells (2021) 10(7):1777. doi: 10.3390/cells10071777
75. McCombe PA, McManis PG, Frith JA, Pollard JD, McLeod JG. Chronic inflammatory demyelinating polyradiculoneuropathy associated with pregnancy. Ann Neurol (1987) 21:102–4. doi: 10.1002/ana.410210120
76. Viala K, Maisonobe T, Stojkovic T, Koutlidis R, Ayrignac X, Musset L, et al. A current view of the diagnosis, clinical variants, response to treatment and prognosis of chronic inflammatory demyelinating polyradiculoneuropathy. J peripheral nervous system JPNS (2010) 15(1):50–6. doi: 10.1111/j.1529-8027.2010.00251.x
77. Shibuya K, Tsuneyama A, Misawa S, Sekiguchi Y, Beppu M, Suichi T, et al. Different distribution of demyelination in chronic inflammatory demyelinating polyneuropathy subtypes. J Neuroimmunol (2020) 341:577170. doi: 10.1016/j.jneuroim.2020.577170
78. Koike H, Katsuno M. Pathophysiology of chronic inflammatory demyelinating polyneuropathy: Insights into classification and therapeutic strategy. Neurol Ther (2020) 9(2):213–27. doi: 10.1007/s40120-020-00190-8
79. Katz JS, Saperstein DS, Gronseth G, Amato AA, Barohn RJ. Distal acquired demyelinating symmetric neuropathy. Neurology (2000) 54(3):615–20. doi: 10.1212/wnl.54.3.615
80. Chiò A, Cocito D, Bottacchi E, Buffa C, Leone M, Plano F, et al. Idiopathic chronic inflammatory demyelinating polyneuropathy: An epidemiological study in Italy. J neurology neurosurgery Psychiatry (2007) 78(12):1349–53. doi: 10.1136/jnnp.2007.114868
81. Hughes RA, Bouche P, Cornblath DR, Evers E, Hadden RD, Hahn A, et al. European Federation of neurological Societies/Peripheral nerve society guideline on management of chronic inflammatory demyelinating polyradiculoneuropathy: Report of a joint task force of the European federation of neurological societies and the peripheral nerve society. Eur J Neurol (2006) 13(4):326–32. doi: 10.1111/j.1468-1331.2006.01278.x
82. Dalakas MC. Advances in the diagnosis, pathogenesis and treatment of cidp. Nat Rev Neurol (2011) 7(9):507–17. doi: 10.1038/nrneurol.2011.121
83. Ruts L, Drenthen J, Jacobs BC, van Doorn PA. Distinguishing acute-onset cidp from fluctuating Guillain-barre syndrome: A prospective study. Neurology (2010) 74(21):1680–6. doi: 10.1212/WNL.0b013e3181e07d14
84. van Schaik IN, Leger J-M, Nobile-Orazio E, Cornblath DR, Hadden RDM, Koski C, et al. European Federation of neurological Societies/Peripheral nerve society guideline on management of multifocal motor neuropathy. report of a joint task force of the European federation of neurological societies and the peripheral nerve society–first revision. J peripheral nervous system JPNS (2010) 15(4):295–301. doi: 10.1111/j.1529-8027.2010.00290.x
85. Steck AJ. Anti-mag neuropathy: From biology to clinical management. J Neuroimmunol (2021) 361:577725. doi: 10.1016/j.jneuroim.2021.577725
86. Khouri J, Nakashima M, Wong S. Update on the diagnosis and treatment of poems (Polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes) syndrome: A review. JAMA Oncol (2021) 7(9):1383–91. doi: 10.1001/jamaoncol.2021.0586
87. Vallat JM, Duchesne M, Corcia P, Richard L, Ghorab K, Magy L, et al. The wide spectrum of pathophysiologic mechanisms of paraproteinemic neuropathy. Neurology (2021) 96(5):214–25. doi: 10.1212/wnl.0000000000011324
88. Koike H, Katsuno M. Paraproteinemia and neuropathy. Neurological Sci (2021) 42(11):4489–501. doi: 10.1007/s10072-021-05583-7
89. Vital A, Lagueny A, Julien J, Ferrer X, Barat M, Hermosilla E, et al. Chronic inflammatory demyelinating polyneuropathy associated with dysglobulinemia: A peripheral nerve biopsy study in 18 cases. Acta Neuropathol (2000) 100(1):63–8. doi: 10.1007/s004010051193
90. Doneddu PE, Cocito D, Manganelli F, Fazio R, Briani C, Filosto M, et al. Atypical cidp: Diagnostic criteria, progression and treatment response. Data Ital Cidp Database. J neurology neurosurgery Psychiatry (2019) 90(2):125–32. doi: 10.1136/jnnp-2018-318714
91. Nobile-Orazio E. Chronic inflammatory demyelinating polyradiculoneuropathy and variants: Where we are and where we should go. J peripheral nervous system JPNS (2014) 19(1):2–13. doi: 10.1111/jns5.12053
92. Doneddu PE, Bianchi E, Cocito D, Manganelli F, Fazio R, Filosto M, et al. Risk factors for chronic inflammatory demyelinating polyradiculoneuropathy (Cidp): Antecedent events, lifestyle and dietary habits. Data Ital Cidp Database. Eur J Neurol (2020) 27(1):136–43. doi: 10.1111/ene.14044
93. Rajabally YA, Peric S, Bozovic I, Loo LK, Kalac A, Palibrk A, et al. Antecedent infections and vaccinations in chronic inflammatory demyelinating polyneuropathy: A European collaborative study. Muscle Nerve (2021) 64(6):657–61. doi: 10.1002/mus.27374
94. Dyck PJ, Lais AC, Ohta M, Bastron JA, Okazaki H, Groover RV. Chronic inflammatory polyradiculoneuropathy. Mayo Clin Proc (1975) 50:621–37.
95. Krendel DA, Parks HP, Anthony DC, St.Clair MB, Graham DG. Sural nerve biopsy in chronic inflammatory demyelinating polyradiculoneuropathy. Muscle Nerve (1989) 12:257–64. doi: 10.1002/mus.880120402
96. Schneider-Hohendorf T, Schwab N, Uceyler N, Gobel K, Sommer C, Wiendl H. Cd8+ T-cell immunity in chronic inflammatory demyelinating polyradiculoneuropathy. Neurology (2012) 78(6):402–8. doi: 10.1212/WNL.0b013e318245d250
97. Yan WX, Archelos JJ, Hartung HP, Pollard JD. P0 protein is a target antigen in chronic inflammatory demyelinating polyradiculoneuropathy. Ann Neurol (2001) 50(3):286–92. doi: 10.1002/ana.1129
98. Allen D, Giannopoulos K, Gray I, Gregson N, Makowska A, Pritchard J, et al. Antibodies to peripheral nerve myelin proteins in chronic inflammatory demyelinating polyradiculoneuropathy. J Peripher Nerv Syst (2005) 10(2):174–80. doi: 10.1111/j.1085-9489.2005.0010207.x
99. Inglis HR, Csurhes PA, McCombe PA. Antibody responses to peptides of peripheral nerve myelin proteins P0 and P2 in patients with inflammatory demyelinating neuropathy. J Neurol Neurosurg Psychiatry (2007) 78(4):419–22. doi: 10.1136/jnnp.2006.106617
100. Sanvito L, Makowska A, Mahdi-Rogers M, Hadden RD, Peakman M, Gregson N, et al. Humoral and cellular immune responses to myelin protein peptides in chronic inflammatory demyelinating polyradiculoneuropathy. J neurology neurosurgery Psychiatry (2009) 80(3):333–8. doi: 10.1136/jnnp.2008.159798
101. Mathey EK, Pollard JD, Armati PJ. Tnf alpha, ifn gamma and il-2 mrna expression in cidp sural nerve biopsies. J Neurol Sci (1999) 163(1):47–52. doi: 10.1016/s0022-510x(99)00009-x
102. Matsumuro K, Izumo S, Umehara F, Osame M. Chronic inflammatory demyelinating polyneuropathy: Histological and immunopathological studies on biopsied sural nerves. J Neurol Sci (1994) 127(2):170–8. doi: 10.1016/0022-510x(94)90070-1
103. Querol LA, Hartung HP, Lewis RA, van Doorn PA, Hammond TR, Atassi N, et al. The role of the complement system in chronic inflammatory demyelinating polyneuropathy: Implications for complement-targeted therapies. Neurotherapeutics (2022) 19(3):864–73. doi: 10.1007/s13311-022-01221-y
104. Nyland H, Aarli JA. Guillain-Barré Syndrome: Demonstration of antibodies to peripheral nerve tissue. Acta Neurol Scand (1978) 58:35–43. doi: 10.1111/j.1600-0404.1978.tb02857.x
105. McCombe PA, Wilson R, Prentice RL. Antiganglioside antibodies in peripheral neuropathy. Clin Exp Neurol (1992) 29:182–8.
106. Kuwahara M, Suzuki H, Samukawa M, Hamada Y, Takada K, Kusunoki S. Clinical features of cidp with Lm1-associated antibodies. J neurology neurosurgery Psychiatry (2013) 84(5):573–5. doi: 10.1136/jnnp-2012-303440
107. Cortese A, Lombardi R, Briani C, Callegari I, Benedetti L, Manganelli F, et al. Antibodies to neurofascin, contactin-1, and contactin-associated protein 1 in cidp: Clinical relevance of igg isotype. Neurology(R) neuroimmunology Neuroinflamm (2020) 7(1) e639. doi: 10.1212/nxi.0000000000000639
108. Moritz CP, Tholance Y, Stoevesandt O, Ferraud K, Camdessanché JP, Antoine JC. Cidp antibodies target junction proteins and identify patient subgroups: An autoantigenomic approach. Neurology(R) neuroimmunology Neuroinflamm (2021) 8(2) e944. doi: 10.1212/nxi.0000000000000944
109. Pascual-Goñi E, Fehmi J, Lleixà C, Martín-Aguilar L, Devaux J, Höftberger R, et al. Antibodies to the Caspr1/Contactin-1 complex in chronic inflammatory demyelinating polyradiculoneuropathy. Brain (2021) 144(4):1183–96. doi: 10.1093/brain/awab014
110. Delmont E, Manso C, Querol L, Cortese A, Berardinelli A, Lozza A, et al. Autoantibodies to nodal isoforms of neurofascin in chronic inflammatory demyelinating polyneuropathy. Brain (2017) 140(7):1851–8. doi: 10.1093/brain/awx124
111. Doppler K, Appeltshauser L, Wilhelmi K, Villmann C, Dib-Hajj SD, Waxman SG, et al. Destruction of paranodal architecture in inflammatory neuropathy with anti-Contactin-1 autoantibodies. J neurology neurosurgery Psychiatry (2015) 86(7):720–8. doi: 10.1136/jnnp-2014-309916
112. Querol L, Siles AM, Alba-Rovira R, Jáuregui A, Devaux J, Faivre-Sarrailh C, et al. Antibodies against peripheral nerve antigens in chronic inflammatory demyelinating polyradiculoneuropathy. Sci Rep (2017) 7(1):14411. doi: 10.1038/s41598-017-14853-4
113. Koike H, Kadoya M, Kaida KI, Ikeda S, Kawagashira Y, Iijima M, et al. Paranodal dissection in chronic inflammatory demyelinating polyneuropathy with anti-Neurofascin-155 and anti-Contactin-1 antibodies. J neurology neurosurgery Psychiatry (2017) 88(6):465–73. doi: 10.1136/jnnp-2016-314895
114. Uncini A, Susuki K, Yuki N. Nodo-paranodopathy: Beyond the demyelinating and axonal classification in anti-ganglioside antibody-mediated neuropathies. Clin Neurophysiol (2013) 124(10):1928–34. doi: 10.1016/j.clinph.2013.03.025
115. Uncini A, Kuwabara S. Nodopathies of the peripheral nerve: An emerging concept. J neurology neurosurgery Psychiatry (2015) 86(11):1186–95. doi: 10.1136/jnnp-2014-310097
116. McCombe PA, Csurhes PA, Greer JM. Studies of hla associations in Male and female patients with Guillain-barre syndrome (Gbs) and chronic inflammatory demyelinating polyradiculoneuropathy (Cidp). J Neuroimmunol (2006) 180(1-2):172–7. doi: 10.1016/j.jneuroim.2006.07.017
117. Blum S, Csurhes P, McCombe P. The frequencies of killer immunoglobulin-like receptors and their hla ligands in chronic inflammatory demyelinating polyradiculoneuropathy are similar to those in guillian barre syndrome but differ from those of controls, suggesting a role for nk cells in pathogenesis. J Neuroimmunol (2015) 285:53–6. doi: 10.1016/j.jneuroim.2015.05.017
118. Rivers TM, Sprunt DM, Berry GP. Observations on attempts to produce acute disseminated encephalomyelitis in monkeys. J Exp Med (1933) 58:39–53. doi: 10.1084/jem.58.1.39
119. Waksman BH, Adams RD. A comparative study of experimental allergic neuritis in the rabbit, Guinea pig, and mouse. J Neuropathol Exp Neurol (1956) 15:293–332. doi: 10.1097/00005072-195607000-00005
120. Waksman BH, Adams RD. Allergic neuritis: An experimental disease of rabbits induced by the injection of peripheral nervous tissue and adjuvants. J Exp Med (1955) 102:213–35. doi: 10.1084/jem.102.2.213
121. Petek M, Quaglio GL. Experimental allergic neuritis in the chicken. Pathol Vet (1967) 4:464–76. doi: 10.1177/030098586700400503
122. Eylar EH, Toro Goyco E, Kessler MJ, Szymanska I. Induction of allergic neuritis in rhesus monkeys. J Neuroimmunol (1982) 3:91–8. doi: 10.1016/0165-5728(82)90043-1
123. Smith ME, Forno LS, Hofmann WW. Experimental allergic neuritis in the Lewis rat. J Neuropathol Exp Neurol (1979) 38:377–91. doi: 10.1097/00005072-197907000-00003
124. Hartung HP, Schafer B, van der Meide PH, Fierz W, Heininger K, Toyka KV. The role of interferon-gamma in the pathogenesis of experimental autoimmune disease of the peripheral nervous system. Ann Neurol (1990) 27:247–57. doi: 10.1002/ana.410270306
125. Du T, Yang CL, Ge MR, Liu Y, Zhang P, Li H, et al. M1 macrophage derived exosomes aggravate experimental autoimmune neuritis Via modulating Th1 response. Front Immunol (2020) 11:1603. doi: 10.3389/fimmu.2020.01603
126. Jung S, Gaupp S, Korn T, Köllner G, Hartung HP, Toyka KV. Biphasic form of experimental autoimmune neuritis in dark agouti rats and its oral therapy by antigen-specific tolerization. J Neurosci Res (2004) 75(4):524–35. doi: 10.1002/jnr.10879
127. Rostami A, Brown MJ, Lisak RP, Sumner AJ, Zweiman B, Pleasure DE. The role of myelin P2 protein in the production of experimental allergic neuritis. Ann Neurol (1984) 16:680–5. doi: 10.1002/ana.410160610
128. Milner P, Lovelidge CA, Taylor WA, Hughes RA. P0 myelin protein produces experimental allergic neuritis in Lewis rats. J Neurol Sci (1987) 79:275–85. doi: 10.1016/0022-510x(87)90235-8
129. Gabriel CM, Hughes RA, Moore SE, Smith KJ, Walsh FS. Induction of experimental autoimmune neuritis with peripheral myelin protein-22. Brain (1998) 121(Pt 10):1895–902. doi: 10.1093/brain/121.10.1895
130. Shin HC, McFarlane EF, Pollard JD, Watson EG. Induction of experimental allergic neuritis with synthetic peptides from myelin P2 protein. Neurosci Lett (1989) 102:309–12. doi: 10.1016/0304-3940(89)90097-9
131. Rostami A, Gregorian SK, Brown MJ, Pleasure DE. Induction of severe experimental autoimmune neuritis with a synthetic peptide corresponding to the 53-78 amino acid sequence of the myelin P2 protein. J Neuroimmunol (1990) 30:145–51. doi: 10.1016/0165-5728(90)90098-8
132. Zhu J, Pelidou SH, Deretzi G, Levi M, Mix E, van der Meide P, et al. P0 glycoprotein peptides 56-71 and 180-199 dose-dependently induce acute and chronic experimental autoimmune neuritis in Lewis rats associated with epitope spreading. J Neuroimmunol (2001) 114(1-2):99–106. doi: 10.1016/s0165-5728(01)00245-4
133. Gonsalvez DG, Yoo S, Craig GA, Wood RJ, Fletcher JL, Murray SS, et al. Myelin protein Zero(180-199) peptide induced experimental autoimmune neuritis in C57bl/6 mice. Methods Mol Biol (Clifton NJ) (2018) 1791:243–50. doi: 10.1007/978-1-4939-7862-5_19
134. Zhao Y, Liu B, Wang Y, Xiao B. Effect of fasudil on experimental autoimmune neuritis and its mechanisms of action. Braz J Med Biol Res = Rev Bras pesquisas medicas e biologicas (2020) 53(1):e8669. doi: 10.1590/1414-431x20198669
135. Takeda S, Ikuta F, Nagai Y. Neuropathological comparative studies on experimental allergic neuritis (Ean) induced in rabbits by P2 protein-ganglioside complexes. Jpn J Exp Med (1980) 50:453–62.
136. Saida T, Saida K, Dorfman SH, Silberberg DH, Sumner AJ, Manning MC, et al. Experimental allergic neuritis induced by sensitization with galactocerebroside. Science (1979) 204:1103–6. doi: 10.1126/science.451555
137. Nagai Y, Momoi T, Saito M, Mitsuzawa E, Ohtani S. Ganglioside syndrome, a new autoimmune neurologica disorder, experimentally induced with brain gangliosides. Neurosci Lett (1976) 2:107–11. doi: 10.1016/0304-3940(76)90033-1
138. Yuki N, Yamada M, Koga M, Odaka M, Susuki K, Tagawa Y, et al. Animal model of axonal Guillain-Barré syndrome induced by sensitization with Gm1 ganglioside. Ann Neurol (2001) 49(6):712–20. doi: 10.1002/ana.1012
139. Kusunoki S, Chiba A, Hitoshi S, Takizawa H, Kanazawa I. Anti-Gal-C antibody in autoimmune neuropathies subsequent to mycoplasma infection. Muscle Nerve (1995) 18(4):409–13. doi: 10.1002/mus.880180407
140. Kusunoki S, Shimizu J, Chiba A, Ugawa Y, Hitoshi S, Kanazawa I. Experimental sensory neuropathy induced by sensitization with ganglioside Gd1b. Ann Neurol (1996) 39(4):424–31. doi: 10.1002/ana.410390404
141. Takada K, Shimizu J, Kusunoki S. Apoptosis of primary sensory neurons in Gd1b-induced sensory ataxic neuropathy. Exp Neurol (2008) 209(1):279–83. doi: 10.1016/j.expneurol.2007.09.010
142. Rostami A. Comparative study of experimental autoimmune neuritis in sjl mice and Lewis rats. Immunol Res (1990) 9:314–9. doi: 10.1007/BF02935530
143. Suzuki M, Kitamura K, Uyemura K, Ogawa Y, Ishihara Y, Matsuyama H. Neuritogenic activity of peripheral nerve myelin proteins in Lewis rats. Neurosci Lett (1980) 19(3):353–8. doi: 10.1016/0304-3940(80)90287-6
144. Rostami A, Burns JB, Brown MJ, Rosen J, Zweiman B, Lisak RP, et al. Transfer of experimental allergic neuritis with P2-reactive T- cell lines. Cell Immunol (1985) 91:354–61. doi: 10.1016/0008-8749(85)90233-3
145. Linington C, Izumo S, Suzuki M, Uyemura K, Meyermann R, Wekerle H. A permanent rat T cell line that mediates experimental allergic neuritis in the Lewis rat in vivo. J Immunol (1984) 133:1946–50.
146. Tran GT, Hodgkinson SJ, Carter N, Verma ND, Robinson CM, Plain KM, et al. Autoantigen specific il-2 activated Cd4(+)Cd25(+)T regulatory cells inhibit induction of experimental autoimmune neuritis. J Neuroimmunol (2020) 341:577186. doi: 10.1016/j.jneuroim.2020.577186
147. Rosen JL, Brown MJ, Hickey WF, Rostami A. Early myelin lesions in experimental allergic neuritis. Muscle Nerve (1990) 13:629–36. doi: 10.1002/mus.880130712
148. Hartung HP, Schafer B, Heininger K, Stoll G, Toyka KV. The role of macrophages and eicosanoids in the pathogenesis of experimental allergic neuritis. serial clinical, electrophysiological, biochemical and morphological observations. Brain (1988) 111:1039–59. doi: 10.1093/brain/111.5.1039
149. Olsson T, Holmdahl R, Klareskog L, Forsum U. Ia-expressing cells and T lymphocytes of different subsets in peripheral nerve tissue during experimental allergic neuritis in Lewis rats. Scand J Immunol (1983) 18:339–43. doi: 10.1111/j.1365-3083.1983.tb01805.x
150. Stoll G, Schmidt B, Jander S, Toyka KV, Hartung HP. Presence of the terminal complement complex (C5b-9) precedes myelin degradation in immune-mediated demyelination of the rat peripheral nervous system. Ann Neurol (1991) 30:147–55. doi: 10.1002/ana.410300205
151. McCombe PA, van der Kreek SA, Pender MP. Neuropathological findings in chronic relapsing experimental allergic neuritis induced in the Lewis rat by inoculation with intradural root myelin and treatment with low dose cyclosporin a. Neuropathology Appl Neurobiol (1992) 18(2):171–87. doi: 10.1111/j.1365-2990.1992.tb00778.x
152. McCombe PA, van der Kreek SA, Pender MP. The effects of prophylactic cyclosporin a on experimental allergic neuritis (Ean) in the Lewis rat. induction of relapsing ean using low dose cyclosporin a. J Neuroimmunol (1990) 28(2):131–40. doi: 10.1016/0165-5728(90)90027-k
153. de Sèze J, Kremer L, Alves do Rego C, Taleb O, Lam D, Beiano W, et al. Chronic inflammatory demyelinating polyradiculoneuropathy: A new animal model for new therapeutic targets. Rev neurologique (2016) 172(12):767–9. doi: 10.1016/j.neurol.2016.05.006
154. Salomon B, Rhee L, Bour-Jordan H, Hsin H, Montag A, Soliven B, et al. Development of spontaneous autoimmune peripheral polyneuropathy in B7-2-Deficient nod mice. J Exp Med (2001) 194(5):677–84. doi: 10.1084/jem.194.5.677
155. Brisebois M, Zehntner SP, Estrada J, Owens T, Fournier S. A pathogenic role for Cd8+ T cells in a spontaneous model of demyelinating disease. J Immunol (2006) 177(4):2403–11. doi: 10.4049/jimmunol.177.4.2403
156. Hardy TA, Blum S, McCombe PA, Reddel SW. Guillain-Barre syndrome: Modern theories of etiology. Curr Allergy Asthma Rep (2011) 11(3):197–204. doi: 10.1007/s11882-011-0190-y
157. Yaneva R, Schneeweiss C, Zacharias M, Springer S. Peptide binding to mhc class I and ii proteins: New avenues from new methods. Mol Immunol (2010) 47(4):649–57. doi: 10.1016/j.molimm.2009.10.008
158. Cohen NR, Garg S, Brenner MB. Antigen presentation by Cd1 lipids, T cells, and nkt cells in microbial immunity. Adv Immunol (2009) 102:1–94. doi: 10.1016/s0065-2776(09)01201-2
159. Courey-Ghaouzi AD, Kleberg L, Sundling C. Alternative b cell differentiation during infection and inflammation. Front Immunol (2022) 13:908034. doi: 10.3389/fimmu.2022.908034
160. Sejvar JJ, Baughman AL, Wise M, Morgan OW. Population incidence of Guillain-barre syndrome: A systematic review and meta-analysis. Neuroepidemiology (2011) 36(2):123–33. doi: 10.1159/000324710
161. Damian MS, Ben-Shlomo Y, Howard R, Bellotti T, Harrison D, Griggs K, et al. The effect of secular trends and specialist neurocritical care on mortality for patients with intracerebral haemorrhage, myasthenia gravis and Guillain-Barré syndrome admitted to critical care : An analysis of the intensive care national audit & research centre (Icnarc) national united kingdom database. Intensive Care Med (2013) 39(8):1405–12. doi: 10.1007/s00134-013-2960-6
162. Galeotti F, Massari M, D'Alessandro R, Beghi E, Chiò A, Logroscino G, et al. Risk of Guillain-Barré syndrome after 2010-2011 influenza vaccination. Eur J Epidemiol (2013) 28(5):433–44. doi: 10.1007/s10654-013-9797-8
163. Dodd CN, Romio SA, Black S, Vellozzi C, Andrews N, Sturkenboom M, et al. International collaboration to assess the risk of Guillain Barré syndrome following influenza a (H1n1) 2009 monovalent vaccines. Vaccine (2013) 31(40):4448–58. doi: 10.1016/j.vaccine.2013.06.032
164. Romio S, Weibel D, Dieleman JP, Olberg HK, de Vries CS, Sammon C, et al. Guillain-Barré Syndrome and adjuvanted pandemic influenza a (H1n1) 2009 vaccines: A multinational self-controlled case series in Europe. PloS One (2014) 9(1):e82222. doi: 10.1371/journal.pone.0082222
165. Prestel J, Volkers P, Mentzer D, Lehmann HC, Hartung HP, Keller-Stanislawski B. Risk of Guillain-Barré syndrome following pandemic influenza a(H1n1) 2009 vaccination in Germany. Pharmacoepidemiol Drug Saf (2014) 23(11):1192–204. doi: 10.1002/pds.3638
166. Chen Y, Ma F, Zhang J, Chu X, Xu Y. Population incidence of Guillain-barre syndrome in parts of China: Three Large populations in jiangsu province, 2008-2010. Eur J Neurol (2014) 21(1):124–9. doi: 10.1111/ene.12265
167. Ghaderi S, Gunnes N, Bakken IJ, Magnus P, Trogstad L, Håberg SE. Risk of Guillain-Barré syndrome after exposure to pandemic influenza a(H1n1)Pdm09 vaccination or infection: A Norwegian population-based cohort study. Eur J Epidemiol (2016) 31(1):67–72. doi: 10.1007/s10654-015-0047-0
168. Delannoy A, Rudant J, Chaignot C, Bolgert F, Mikaeloff Y, Weill A. Guillain-Barré Syndrome in France: A nationwide epidemiological analysis based on hospital discharge data (2008-2013). J peripheral nervous system JPNS (2017) 22(1):51–8. doi: 10.1111/jns.12202
169. Kobori S, Kubo T, Otani M, Muramatsu K, Fujino Y, Adachi H, et al. Coexisting infectious diseases on admission as a risk factor for mechanical ventilation in patients with Guillain-Barré syndrome. J Epidemiol (2017) 27(7):311–6. doi: 10.1016/j.je.2016.07.003
170. van den Berg B, Storm EF, Garssen MJP, Blomkwist-Markens PH, Jacobs BC. Clinical outcome of Guillain-Barré syndrome after prolonged mechanical ventilation. J neurology neurosurgery Psychiatry (2018) 89(9):949–54. doi: 10.1136/jnnp-2018-317968
171. Ansari B, Basiri K, Derakhshan Y, Kadkhodaei F, Okhovat AA. Epidemiology and clinical features of Guillain-barre syndrome in isfahan, Iran. Adv BioMed Res (2018) 7:87. doi: 10.4103/abr.abr_50_17
172. Wahatule R, Dutta D, Debnath M, Nagappa M, Mahadevan A, Sinha S, et al. Ganglioside complex antibodies in an Indian cohort of Guillain-Barré syndrome. Muscle Nerve (2020) 62(6):728–34. doi: 10.1002/mus.27071
173. Grave C, Boucheron P, Rudant J, Mikaeloff Y, Tubert-Bitter P, Escolano S, et al. Seasonal influenza vaccine and Guillain-Barré syndrome: A self-controlled case series study. Neurology (2020) 94(20):e2168–e79. doi: 10.1212/wnl.0000000000009180
174. Lee H, Kang HY, Jung SY, Lee YM. Incidence of Guillain-Barré syndrome is not associated with influenza vaccination in the elderly. Vaccines (Basel) (2020) 8(3). doi: 10.3390/vaccines8030431
175. Dourado Junior MET, Sousa BF, Costa N, Jeronimo SMB. Cytomegalovirus infection in Guillain-Barré syndrome: A retrospective study in Brazil. Arq Neuropsiquiatr (2021) 79(7):607–11. doi: 10.1590/0004-282x-anp-2020-0464
176. Kim AY, Lee H, Lee YM, Kang HY. Epidemiological features and economic burden of Guillain-Barré syndrome in south Korea: A nationwide population-based study. J Clin Neurol (2021) 17(2):257–64. doi: 10.3988/jcn.2021.17.2.257
177. Rath J, Zulehner G, Schober B, Grisold A, Krenn M, Cetin H, et al. Cerebrospinal fluid analysis in Guillain-Barré syndrome: Value of albumin quotients. J Neurol (2021) 268(9):3294–300. doi: 10.1007/s00415-021-10479-9
178. Oliveira AFM, Silva A, Manga Aridjae U, Bastos MM, Wachira VK, Gallo LG. Characterization of Guillain-Barré syndrome in the integrated development region of the federal district and surrounding areas (Ride), Brazil, between 2017 and 2019. Acta Trop (2022) 229:106366. doi: 10.1016/j.actatropica.2022.106366
179. López-Hernández JC, Galnares-Olalde JA, Gutiérrez A, Estrada SA, García-Grimshaw M, Vargas-Cañas ES. Guillain-Barre syndrome in Mexico: Clinical features and validation of Brighton collaboration group criteria. Rev Neurol (2022) 74(8):258–64. doi: 10.33588/rn.7408.2021437
180. Levison LS, Thomsen RW, Andersen H. Increased mortality following Guillain-Barré syndrome: A population-based cohort study. Eur J Neurol (2022) 29(4):1145–54. doi: 10.1111/ene.15204
181. Hosseininezhad M, Khatami SS, Saadat S, Asghari M, Ghovvati Choshal H, Hooshmand Marvasti A, et al. Ten years evaluation of epidemiology- and mortality-related factors in adults and children with Guillain-Barré syndrome in the north of Iran. Neurological Sci (2022) 43(3):1929–38. doi: 10.1007/s10072-021-05562-y
182. Kalita J, Misra UK, Chaudhary SK, Das M, Mishra A, Ranjan A, et al. Outcome of Guillain-Barré syndrome following intravenous immunoglobulin compared to natural course. Eur J Neurol (2022). doi: 10.1111/ene.15500
183. Filosto M, Cotti Piccinelli S, Gazzina S, Foresti C, Frigeni B, Servalli MC, et al. Guillain-Barré Syndrome and covid-19: A 1-year observational multicenter study. Eur J Neurol (2022). doi: 10.1111/ene.15497
184. Kim JE, Park J, Min YG, Hong YH, Song TJ. Associations of Guillain-Barré syndrome with coronavirus disease 2019 vaccination: Disproportionality analysis using the world health organization pharmacovigilance database. J peripheral nervous system (2022). doi: 10.1111/jns.12507
185. Linden V, da Paz JA, Casella EB, Marques-Dias MJ. Guillain-Barré Syndrome in children: Clinic, laboratorial and epidemiologic study of 61 patients. Arq Neuropsiquiatr (2010) 68(1):12–7. doi: 10.1590/s0004-282x2010000100004
186. Sarkar UK, Menon L, Sarbapalli D, Pal R, Zaman FA, Kar S, et al. Spectrum of Guillain-Barré syndrome in tertiary care hospital at kolkata. J Nat Sci Biol Med (2011) 2(2):211–5. doi: 10.4103/0976-9668.92320
187. Pavone P, Praticò AD, Ruggieri M, Verrotti A, Castellano-Chiodo D, Greco F, et al. Acquired peripheral neuropathy: A report on 20 children. Int J Immunopathol Pharmacol (2012) 25(2):513–7. doi: 10.1177/039463201202500222
188. Hu MH, Chen CM, Lin KL, Wang HS, Hsia SH, Chou ML, et al. Risk factors of respiratory failure in children with Guillain-Barré syndrome. Pediatr Neonatol (2012) 53(5):295–9. doi: 10.1016/j.pedneo.2012.07.003
189. Roodbol J, de Wit MY, van den Berg B, Kahlmann V, Drenthen J, Catsman-Berrevoets CE, et al. Diagnosis of Guillain-Barré syndrome in children and validation of the Brighton criteria. J Neurol (2017) 264(5):856–61. doi: 10.1007/s00415-017-8429-8
190. Konuşkan B, Okuyaz Ç, Taşdelen B, Kurul SH, Anlar B. Electrophysiological subtypes and prognostic factors of childhood Guillain-Barré syndrome. Noro Psikiyatr Ars (2018) 55(3):199–204. doi: 10.5152/npa.2017.16996
191. van der Pijl J, Wilmshurst JM, van Dijk M, Argent A, Booth J, Zampoli M. Acute flaccid paralysis in south African children: Causes, respiratory complications and neurological outcome. J Paediatr Child Health (2018) 54(3):247–53. doi: 10.1111/jpc.13709
192. Barzegar M, Toopchizadeh V, Golalizadeh D, Pirani A, Jahanjoo F. A predictive model for respiratory failure and determining the risk factors of prolonged mechanical ventilation in children with Guillain-barre syndrome. Iran J Child Neurol (2020) 14(3):33–46.
193. Luo H, Hong S, Li M, Wang L, Jiang L. Risk factors for mechanical ventilation in children with Guillain-Barré syndrome. Muscle Nerve (2020) 62(2):214–8. doi: 10.1002/mus.26905
194. Rangan RS, Tullu MS, Deshmukh CT, Mondkar SA, Agrawal M. Clinical profile and outcome of Guillain-barre syndrome in pediatric patients admitted to a tertiary care centre: A retrospective study. Neurol India (2021) 69(1):81–4. doi: 10.4103/0028-3886.310112
195. Garg D, Dhamija RK, Choudhary A, Shree R, Kumar S, Samal P, et al. Impact of the covid-19 pandemic on the frequency, clinical spectrum and outcomes of pediatric Guillain-Barré syndrome in India: A multicentric ambispective cohort study. Ann Indian Acad Neurol (2022) 25(1):60–7. doi: 10.4103/aian.aian_392_21
196. van der Meche FG, Schmitz PI, Dutch Guillain-Barré Study Group. A randomized trial comparing intravenous immune globulin and plasma exchange in Guillain-Barré syndrome. . N Engl J Med (1992) 326:1123–9.
197. Chevret S, Hughes RA, Annane D. Plasma exchange for Guillain-Barré syndrome. Cochrane Database systematic Rev (2017) 2(2):Cd001798. doi: 10.1002/14651858.CD001798.pub3
198. Hughes RA, Brassington R, Gunn AA, van Doorn PA. Corticosteroids for Guillain-Barré syndrome. Cochrane Database systematic Rev (2016) 10(10):Cd001446. doi: 10.1002/14651858.CD001446.pub5
199. Leonhard SE, Mandarakas MR, Gondim FAA, Bateman K, Ferreira MLB, Cornblath DR, et al. Diagnosis and management of Guillain-Barré syndrome in ten steps. Nat Rev Neurol (2019) 15(11):671–83. doi: 10.1038/s41582-019-0250-9
200. Walgaard C, Lingsma HF, Ruts L, Drenthen J, van Koningsveld R, Garssen MJ, et al. Prediction of respiratory insufficiency in Guillain-Barré syndrome. Ann Neurol (2010) 67(6):781–7. doi: 10.1002/ana.21976
201. Rath J, Zulehner G, Schober B, Grisold A, Krenn M, Cetin H, et al. Real-world treatment of adult patients with Guillain-Barré syndrome over the last two decades. Sci Rep (2021) 11(1):19170. doi: 10.1038/s41598-021-98501-y
202. Dhar R, Stitt L, Hahn AF. The morbidity and outcome of patients with Guillain-Barré syndrome admitted to the intensive care unit. J Neurol Sci (2008) 264(1-2):121–8. doi: 10.1016/j.jns.2007.08.005
203. Lunn MP, Manji H, Choudhary PP, Hughes RA, Thomas PK. Chronic inflammatory demyelinating polyradiculoneuropathy: A prevalence study in south East England. J Neurol Neurosurg Psychiatry (1999) 66(5):677–80. doi: 10.1136/jnnp.66.5.677
204. McLeod JG, Pollard JD, Macaskill P, Mohamed A, Spring P, Khurana V. Prevalence of chronic inflammatory demyelinating polyneuropathy in new south Wales, Australia. Ann Neurol (1999) 46(6):910–3. doi: 10.1002/1531-8249(199912)46:6<910::AID-ANA14>3.0.CO;2-2
205. Mygland A, Monstad P. Chronic polyneuropathies in vest-agder, Norway. Eur J Neurol (2001) 8(2):157–65. doi: 10.1046/j.1468-1331.2001.00187.x
206. Rajabally YA, Nicolas G, Pieret F, Bouche P, Van den Bergh PY. Validity of diagnostic criteria for chronic inflammatory demyelinating polyneuropathy: A multicentre European study. J neurology neurosurgery Psychiatry (2009) 80(12):1364–8. doi: 10.1136/jnnp.2009.179358
207. Iijima M, Koike H, Hattori N, Tamakoshi A, Katsuno M, Tanaka F, et al. Prevalence and incidence rates of chronic inflammatory demyelinating polyneuropathy in the Japanese population. J Neurol Neurosurg Psychiatry (2008) 79(9):1040–3. doi: 10.1136/jnnp.2007.128132
208. Broers MC, de Wilde M, Lingsma HF, van der Lei J, Verhamme KMC, Jacobs BC. Epidemiology of chronic inflammatory demyelinating polyradiculoneuropathy in the Netherlands. J peripheral nervous system JPNS (2022) 27(3):182–8. doi: 10.1111/jns.12502
209. Broers MC, Bunschoten C, Nieboer D, Lingsma HF, Jacobs BC. Incidence and prevalence of chronic inflammatory demyelinating polyradiculoneuropathy: A systematic review and meta-analysis. Neuroepidemiology (2019) 52(3-4):161–72. doi: 10.1159/000494291
210. Cocito D, Paolasso I, Antonini G, Benedetti L, Briani C, Comi C, et al. A nationwide retrospective analysis on the effect of immune therapies in patients with chronic inflammatory demyelinating polyradiculoneuropathy. Eur J Neurol (2010) 17(2):289–94. doi: 10.1111/j.1468-1331.2009.02802.x
211. Gorson KC, van Schaik IN, Merkies IS, Lewis RA, Barohn RJ, Koski CL, et al. Chronic inflammatory demyelinating polyneuropathy disease activity status: Recommendations for clinical research standards and use in clinical practice. J peripheral nervous system JPNS (2010) 15(4):326–33. doi: 10.1111/j.1529-8027.2010.00284.x
212. Cocito D, Grimaldi S, Paolasso I, Falcone Y, Antonini G, Benedetti L, et al. Immunosuppressive treatment in refractory chronic inflammatory demyelinating polyradiculoneuropathy. a nationwide retrospective analysis. Eur J Neurol (2011) 18(12):1417–21. doi: 10.1111/j.1468-1331.2011.03495.x
213. Rajabally YA, Lagarde J, Cassereau J, Viala K, Fournier E, Nicolas G. A European multicentre reappraisal of distal compound muscle action potential duration in chronic inflammatory demyelinating polyneuropathy. Eur J Neurol (2012) 19(4):638–42. doi: 10.1111/j.1468-1331.2011.03605.x
214. Querol L, Rojas-Garcia R, Casasnovas C, Sedano MJ, Muñoz-Blanco JL, Alberti MA, et al. Long-term outcome in chronic inflammatory demyelinating polyneuropathy patients treated with intravenous immunoglobulin: A retrospective study. Muscle Nerve (2013) 48(6):870–6. doi: 10.1002/mus.23843
215. Mahdi-Rogers M, Hughes RA. Epidemiology of chronic inflammatory neuropathies in southeast England. Eur J Neurol (2013). doi: 10.1111/ene.12190
216. Kuitwaard K, Hahn AF, Vermeulen M, Venance SL, van Doorn PA. Intravenous immunoglobulin response in treatment-naïve chronic inflammatory demyelinating polyradiculoneuropathy. J neurology neurosurgery Psychiatry (2015) 86(12):1331–6. doi: 10.1136/jnnp-2014-309042
217. Kuwabara S, Isose S, Mori M, Mitsuma S, Sawai S, Beppu M, et al. Different electrophysiological profiles and treatment response in 'Typical' and 'Atypical' chronic inflammatory demyelinating polyneuropathy. J neurology neurosurgery Psychiatry (2015) 86(10):1054–9. doi: 10.1136/jnnp-2014-308452
218. Albulaihe H, Alabdali M, Alsulaiman A, Abraham A, Breiner A, Barnett C, et al. Disease activity in chronic inflammatory demyelinating polyneuropathy. J Neurol Sci (2016) 369:204–9. doi: 10.1016/j.jns.2016.08.034
219. Moodley K, Bill PL, Patel VB. A comparative study of cidp in a cohort of hiv-infected and hiv-uninfected patients. Neurology(R) neuroimmunology Neuroinflamm (2017) 4(2):e315. doi: 10.1212/nxi.0000000000000315
220. Kacar A, Bjelica B, Bozovic I, Peric S, Nikolic A, Cobeljic M, et al. Neuromuscular disease-specific questionnaire to assess quality of life in patients with chronic inflammatory demyelinating polyradiculoneuropathy. J peripheral nervous system JPNS (2018) 23(1):11–6. doi: 10.1111/jns.12251
221. van Lieverloo GGA, Peric S, Doneddu PE, Gallia F, Nikolic A, Wieske L, et al. Corticosteroids in chronic inflammatory demyelinating polyneuropathy : A retrospective, multicentre study, comparing efficacy and safety of daily prednisolone, pulsed dexamethasone, and pulsed intravenous methylprednisolone. J Neurol (2018) 265(9):2052–9. doi: 10.1007/s00415-018-8948-y
222. Hughes R, Dalakas MC, Merkies I, Latov N, Léger JM, Nobile-Orazio E, et al. Oral fingolimod for chronic inflammatory demyelinating polyradiculoneuropathy (Forcidp trial): A double-blind, multicentre, randomised controlled trial. Lancet Neurol (2018) 17(8):689–98. doi: 10.1016/s1474-4422(18)30202-3
223. Divino V, Mallick R, DeKoven M, Krishnarajah G. The economic burden of cidp in the united states: A case-control study. PloS One (2018) 13(10):e0206205. doi: 10.1371/journal.pone.0206205
224. Rosier C, Graveline N, Lacour A, Antoine JC, Camdessanché JP. Intravenous immunoglobulin for treatment of chronic inflammatory demyelinating polyneuropathy and multifocal motor neuropathy in France: Are daily practices in accordance with guidelines? Eur J Neurol (2019) 26(4):575–80. doi: 10.1111/ene.13841
225. Merkies ISJ, van Schaik IN, Léger JM, Bril V, van Geloven N, Hartung HP, et al. Efficacy and safety of ivig in cidp: Combined data of the prima and path studies. J peripheral nervous system JPNS (2019) 24(1):48–55. doi: 10.1111/jns.12302
226. Guptill JT, Runken MC, Eaddy M, Lunacsek O, Fuldeore RM. Treatment patterns and costs of chronic inflammatory demyelinating polyneuropathy: A claims database analysis. Am Health Drug Benefits (2019) 12(3):127–35.
227. van Schaik IN, Mielke O, Bril V, van Geloven N, Hartung HP, Lewis RA, et al. Long-term safety and efficacy of subcutaneous immunoglobulin Igpro20 in cidp: Path extension study. Neurology(R) neuroimmunology Neuroinflamm (2019) 6(5):e590. doi: 10.1212/nxi.0000000000000590
228. Luigetti M, Romano A, Di Paolantonio A, Bisogni G, Rossi S, Conte A, et al. Pathological findings in chronic inflammatory demyelinating polyradiculoneuropathy: A single-center experience. Brain Sci (2020) 10(6). doi: 10.3390/brainsci10060383
229. Grüter T, Motte J, Fisse AL, Bulut Y, Köse N, Athanasopoulos D, et al. Pathological spontaneous activity as a prognostic marker in chronic inflammatory demyelinating polyneuropathy. Eur J Neurol (2020) 27(12):2595–603. doi: 10.1111/ene.14476
230. Telleman JA, Herraets IJT, Goedee HS, van Eijk RPA, Verhamme C, Eftimov F, et al. Prognostic value of nerve ultrasonography: A prospective multicenter study on the natural history of chronic inflammatory neuropathies. Eur J Neurol (2021) 28(7):2327–38. doi: 10.1111/ene.14885
231. Liberatore G, Manganelli F, Doneddu PE, Cocito D, Fazio R, Briani C, et al. Chronic inflammatory demyelinating polyradiculoneuropathy: Can a diagnosis be made in patients not fulfilling electrodiagnostic criteria? Eur J Neurol (2021) 28(2):620–9. doi: 10.1111/ene.14545
232. Davalos L, London ZN, Stino AM, Gallagher GW, Callaghan BC, Wieland CM, et al. Patients who meet electrodiagnostic criteria for cidp rarely present with a sensory predominant dsp phenotype. Muscle Nerve (2021) 63(6):881–4. doi: 10.1002/mus.27235
233. Athanasopoulos D, Motte J, Grüter T, Köse N, Yoon MS, Otto S, et al. Evaluation of the Efns/Pns diagnostic criteria in a cohort of cidp patients. Ann Clin Trans Neurol (2021) 8(5):1110–21. doi: 10.1002/acn3.51357
234. Alcantara M, Hartung HP, Lawo JP, Durn BL, Mielke O, Bril V. Electrophysiological predictors of response to subcutaneous immunoglobulin therapy in chronic inflammatory demyelinating polyneuropathy. Clin Neurophysiol Off J Int Fed Clin Neurophysiol (2021) 132(9):2184–90. doi: 10.1016/j.clinph.2021.05.018
235. Grüter T, Motte J, Bulut Y, Kordes A, Athanasopoulos D, Fels M, et al. Axonal damage determines clinical disability in chronic inflammatory demyelinating polyradiculoneuropathy (Cidp): A prospective cohort study of different cidp subtypes and disease stages. Eur J Neurol (2022) 29(2):583–92. doi: 10.1111/ene.15156
236. Darbà J, Marsà A. Chronic inflammatory demyelinating polyneuropathy in Spain: A retrospective analysis of hospital incidence and medical costs. Expert Rev Pharmacoecon Outcomes Res (2022) 22(4):665–70. doi: 10.1080/14737167.2022.2000862
237. Cornblath DR, van Doorn PA, Hartung HP, Merkies ISJ, Katzberg HD, Hinterberger D, et al. Randomized trial of three ivig doses for treating chronic inflammatory demyelinating polyneuropathy. Brain (2022) 145(3):887–96. doi: 10.1093/brain/awab422
238. Nyland H, Matre R, Mork S. Immunological characterization of sural nerve biopsies from patients with Guillain-Barré syndrome. Ann Neurol (1981) 9(supplement):80–6. doi: 10.1002/ana.410090713
239. McMillan HJ, Kang PB, Jones HR, Darras BT. Childhood chronic inflammatory demyelinating polyradiculoneuropathy: Combined analysis of a Large cohort and eleven published series. Neuromuscular Disord NMD (2013) 23(2):103–11. doi: 10.1016/j.nmd.2012.09.008
240. Van den Bergh PY, Hadden RD, Bouche P, Cornblath DR, Hahn A, Illa I, et al. European Federation of neurological Societies/Peripheral nerve society guideline on management of chronic inflammatory demyelinating polyradiculoneuropathy: Report of a joint task force of the European federation of neurological societies and the peripheral nerve society - first revision. Eur J Neurol (2010) 17(3):356–63. doi: 10.1111/j.1468-1331.2009.02930.x
241. Mahdi-Rogers M, Brassington R, Gunn AA, van Doorn PA, Hughes RA. Immunomodulatory treatment other than corticosteroids, immunoglobulin and plasma exchange for chronic inflammatory demyelinating polyradiculoneuropathy. Cochrane Database systematic Rev (2017) 5(5):Cd003280. doi: 10.1002/14651858.CD003280.pub5
242. Querol L, DeSeze LD,T, Rao H, Rivner M, Hartung PK,P, Shimzu S, et al. Rosanolixizumab in chronic inflammatroy demyelinating polyradiculoneuropathy; a randomized , subject-blind, investigator-blind, placebo controlled phase 2a trial. J Peripheral Nervous system (2022) 27(53):S110–S1.
243. Iijima M, Yamamoto M, Hirayama M, Tanaka F, Katsuno M, Mori K, et al. Clinical and electrophysiologic correlates of ivig responsiveness in cidp. Neurology (2005) 64(8):1471–5. doi: 10.1212/01.Wnl.0000158680.89323.F8
244. Astrom K-E, Waksman BH. The passive transfer of experimental allergic encephalomyelitis and neuritis with living lymphoid cells. J Pathol Bacteriol (1962) 83:89–106. doi: 10.1002/path.1700830112
245. Mao M, Fan W, Zheng Y, Qi P, Xi M, Yao Y. Upregulation of n-type voltage-gated calcium channels induces neuropathic pain in experimental autoimmune neuritis. Evid Based Complement Alternat Med (2022) 2022:8547095. doi: 10.1155/2022/8547095
246. Szepanowski F, Winkelhausen M, Steubing RD, Mausberg AK, Kleinschnitz C, Stettner M. Lpa(1) signaling drives schwann cell dedifferentiation in experimental autoimmune neuritis. J Neuroinflamm (2021) 18(1):293. doi: 10.1186/s12974-021-02350-5
247. Klimas R, Sgodzai M, Motte J, Mohamad N, Renk P, Blusch A, et al. Dose-dependent immunomodulatory effects of bortezomib in experimental autoimmune neuritis. Brain Commun (2021) 3(4):fcab238. doi: 10.1093/braincomms/fcab238
248. Pitarokoili K, Bachir H, Sgodzai M, Grüter T, Haupeltshofer S, Duscha A, et al. Induction of regulatory properties in the intestinal immune system by dimethyl fumarate in Lewis rat experimental autoimmune neuritis. Front Immunol (2019) 10:2132. doi: 10.3389/fimmu.2019.02132
249. Liu S, Liu Y, Xiao Z, Pan S, Gong Q, Lu Z. Th17 cells and their cytokines serve as potential therapeutic target in experimental autoimmune neuritis. Brain Behav (2019) 9(12):e01478. doi: 10.1002/brb3.1478
250. Xue Y, Yin P, Li G, Zhong D. Transcriptomes in rat sciatic nerves at different stages of experimental autoimmune neuritis determined by rna sequencing. Clin Exp Immunol (2019) 198(2):184–97. doi: 10.1111/cei.13354
251. Xu L, Li L, Zhang CY, Schluesener H, Zhang ZY. Natural diterpenoid oridonin ameliorates experimental autoimmune neuritis by promoting anti-inflammatory macrophages through blocking notch pathway. Front Neurosci (2019) 13:272. doi: 10.3389/fnins.2019.00272
252. Jin T, Yu H, Wang D, Zhang H, Zhang B, Quezada HC, et al. Bowman-birk inhibitor concentrate suppresses experimental autoimmune neuritis Via shifting macrophages from M1 to M2 subtype. Immunol Lett (2016) 171:15–25. doi: 10.1016/j.imlet.2016.01.004
253. Shen D, Chu F, Lang Y, Zheng C, Li C, Liu K, et al. Nuclear factor kappa b inhibitor suppresses experimental autoimmune neuritis in mice Via declining macrophages polarization to M1 type. Clin Exp Immunol (2021) 206(1):110–7. doi: 10.1111/cei.13637
254. Moyano AL, Comín R, Lardone RD, Alaniz ME, Theaux R, Irazoqui FJ, et al. Validation of a rabbit model of neuropathy induced by immunization with gangliosides. J Neurol Sci (2008) 272(1-2):110–4. doi: 10.1016/j.jns.2008.05.006
255. Aoyagi T, Wada T, Ishikawa Y, Kojima F, Nagai M, Osanai T, et al. Role of intramuscular enzymatic changes in the development of muscular weakness in rats with experimental allergic neuritis. Exp Neurol (1984) 84:326–37. doi: 10.1016/0014-4886(84)90229-2
256. McCombe PAG JM. Effects of biological sex and pregnancy in experimental autoimmune encephalomyelitis: It’s complicated. Front Immunol (2022). doi: 10.3389/fimmu.2022.1059833
257. Mank JE. Sex chromosomes and the evolution of sexual dimorphism: Lessons from the genome. Am Nat (2009) 173(2):141–50. doi: 10.1086/595754
258. Torgrimson BN, Minson CT. Sex and gender: What is the difference? J Appl Physiol (Bethesda Md 1985) (2005) 99(3):785–7. doi: 10.1152/japplphysiol.00376.2005
259. Fairbairn DJ. Sexual dimorphism. In: Kliman R, editor. The encyclopedia of evolutionary biology. San Diego: Elsevier Science and Technology (2016). p. 105–13.
260. McCombe PA, Greer JM, Mackay IR. Sexual dimorphism in autoimmune disease. Curr Mol Med (2009) 9(9):1058–79. doi: 10.2174/156652409789839116
261. Gal-Oz ST, Maier B, Yoshida H, Seddu K, Elbaz N, Czysz C, et al. Immgen report: Sexual dimorphism in the immune system transcriptome. Nat Commun (2019) 10(1):4295. doi: 10.1038/s41467-019-12348-6
262. McCombe PAG JM. Sexual dimorphism in the immune system. In: Rose NRM ,IR, editor. The autoimmune diseases. London: Academic Press (2020). p. 419–28.
263. Kelly CD, Stoehr AM, Nunn C, Smyth KN, Prokop ZM. Sexual dimorphism in immunity across animals: A meta-analysis. Ecol Lett (2018) 21(12):1885–94. doi: 10.1111/ele.13164
264. Nunn CL, Lindenfors P, Pursall ER, Rolff J. On sexual dimorphism in immune function. Philos Trans R Soc London Ser B Biol Sci (2009) 364(1513):61–9. doi: 10.1098/rstb.2008.0148
265. Yan J, Greer JM, Hull R, O'sullivan JD, Henderson RD, Read SJ, et al. The effect of ageing on human lymphocyte subsets: Comparison of males and females. Immun Ageing (2010) 7:4. doi: 10.1186/1742-4933-7-4
266. Butterworth M, McClellan B, Allansmith M. Influence of sex in immunoglobulin levels. Nature (1967) 214(5094):1224–5. doi: 10.1038/2141224a0
267. Lichtman MA, Vaughan JH, Hames CG. The distribution of serum immunoglobulins, anti-Gamma-G globulins ("Rheumatoid factors") and antinuclear antibodies in white and Negro subjects in Evans county, Georgia. Arthritis Rheum (1967) 10(3):204–15. doi: 10.1002/art.1780100306
268. Kongshavn PA, Bliss JQ. Sex differences in survival of h-2 incompatible skin grafts in mice treated with antithymocyte serum. Nature (1970) 226(5244):451. doi: 10.1038/226451a0
269. Santoli D, Trinchieri G, Zmijewski CM, Koprowski H. HLA-related control of spontaneous and antibody-dependent cell-mediated cytotoxic activity in humans. J Immunol (1976) 117(3):765–70.
270. Weinstein Y, Ran S, Segal S. Sex-associated differences in the regulation of immune responses controlled by the mhc of the mouse. J Immunol (1984) 132(2):656–61.
271. Bebo BF Jr., Adlard K, Schuster JC, Unsicker L, Vandenbark AA, Offner H. Gender differences in protection from eae induced by oral tolerance with a peptide analogue of mbp-Ac1-11. J Neurosci Res (1999) 55(4):432–40. doi: 10.1002/(SICI)1097-4547(19990215)55:4<432::AID-JNR4>3.0.CO;2-2
272. Lotter H, Jacobs T, Gaworski I, Tannich E. Sexual dimorphism in the control of amebic liver abscess in a mouse model of disease. Infect Immun (2006) 74(1):118–24. doi: 10.1128/IAI.74.1.118-124.2006
273. dos Santos CD, Toldo MP, do Prado Junior JC. Trypanosoma cruzi: The effects of dehydroepiandrosterone (Dhea) treatment during experimental infection. Acta Trop (2005) 95(2):109–15. doi: 10.1016/j.actatropica.2005.05.005
274. Kleinschnitz C, Schwab N, Kraft P, Hagedorn I, Dreykluft A, Schwarz T, et al. Early detrimental T-cell effects in experimental cerebral ischemia are neither related to adaptive immunity nor thrombus formation. Blood (2010) 115(18):3835–42. doi: 10.1182/blood-2009-10-249078
275. Huygen K, Palfliet K. Strain variation in interferon gamma production of bcg-sensitized mice challenged with ppd ii. importance of one major autosomal locus and additional sexual influences. Cell Immunol (1984) 85(1):75–81.
276. Spitzer JA. Gender differences in some host defense mechanisms. Lupus (1999) 8(5):380–3. doi: 10.1177/096120339900800510
277. Bouman A, Schipper M, Heineman MJ, Faas MM. Gender difference in the non-specific and specific immune response in humans. Am J Reprod Immunol (2004) 52(1):19–26. doi: 10.1111/j.1600-0897.2004.00177.x
278. Wikby A, Månsson IA, Johansson B, Strindhall J, Nilsson SE. The immune risk profile is associated with age and gender: Findings from three Swedish population studies of individuals 20-100 years of age. Biogerontology (2008) 9(5):299–308. doi: 10.1007/s10522-008-9138-6
279. Das BR, Bhanushali AA, Khadapkar R, Jeswani KD, Bhavsar M, Dasgupta A. Reference ranges for lymphocyte subsets in adults from Western India: Influence of sex, age and method of enumeration. Indian J Med Sci (2008) 62(10):397–406. doi: 10.4103/0019-5359.42725
280. Lee BW, Yap HK, Chew FT, Quah TC, Prabhakaran K, Chan GS, et al. Age- and sex-related changes in lymphocyte subpopulations of healthy Asian subjects: From birth to adulthood. Cytometry (1996) 26(1):8–15. doi: 10.1002/(sici)1097-0320(19960315)26:1<8::Aid-cyto2>3.0.Co;2-e
281. Abdullah M, Chai PS, Chong MY, Tohit ER, Ramasamy R, Pei CP, et al. Gender effect on in vitro lymphocyte subset levels of healthy individuals. Cell Immunol (2012) 272(2):214–9. doi: 10.1016/j.cellimm.2011.10.009
282. Gupta S, Nakabo S, Blanco LP, O'Neil LJ, Wigerblad G, Goel RR, et al. Sex differences in neutrophil biology modulate response to type I interferons and immunometabolism. Proc Natl Acad Sci United States America (2020) 117(28):16481–91. doi: 10.1073/pnas.2003603117
283. Becerra-Díaz M, Lerner AD, Yu DH, Thiboutot JP, Liu MC, Yarmus LB, et al. Sex differences in M2 polarization, chemokine and il-4 receptors in monocytes and macrophages from asthmatics. Cell Immunol (2021) 360:104252. doi: 10.1016/j.cellimm.2020.104252
284. So J, Tai AK, Lichtenstein AH, Wu D, Lamon-Fava S. Sexual dimorphism of monocyte transcriptome in individuals with chronic low-grade inflammation. Biol sex Dif (2021) 12(1):43. doi: 10.1186/s13293-021-00387-y
285. Robinson ,J, Peckham H, Butler G, Pineda-Torra I, Ciurtin C, Jury EC. Investigating sex differences in T regulatory cells from cisgender and transgender healthy individuals and patients with autoimmune inflammatory disease; a cross-sectional study. Lancet Rheumatol (2022) 4:e710–34. doi: 10.1016/S2665-9913(22)00198-9
286. McCombe PA. The role of sex and pregnancy in multiple sclerosis: What do we know and what should we do? Expert Rev Neurother (2022) 22(5):377–92. doi: 10.1080/14737175.2022.2060079
287. Ngo ST, Steyn FJ, McCombe PA. Gender differences in autoimmune disease. Front Neuroendocrinol (2014) 35(3):347–69. doi: 10.1016/j.yfrne.2014.04.004
288. Yang M, Peyret C, Shi XQ, Siron N, Jang JH, Wu S, et al. Evidence from human and animal studies: Pathological roles of Cd8(+) T cells in autoimmune peripheral neuropathies. Front Immunol (2015) 6:532. doi: 10.3389/fimmu.2015.00532
289. Witebsky E, Rose NR, Terplan K, Paine JR, Egan RW. Chronic thyroiditis and autoimmunization. J Am Med Assoc (1957) 164(13):1439–47. doi: 10.1001/jama.1957.02980130015004
290. Rose NR, Bona C. Defining criteria for autoimmune diseases (Witebsky's postulates revisited). Immunol Today (1993) 14(9):426–30. doi: 10.1016/0167-5699(93)90244-F
291. Cerutti A, Cols M, Puga I. Activation of b cells by non-canonical helper signals. EMBO Rep (2012) 13(9):798–810. doi: 10.1038/embor.2012.111
292. Pioli PC,M, Kornas M, Renner P, Pioli K-A. Sex dictates organ-specific differences in the production and activation of plasmablasts and plasma cells. J Immunol (2021) 206(98).
293. Yin PQ, Sun YY, Chen HP, Li GZ, Zhong D. Genome-wide gene expression analysis of peripheral leukocytes in relation to the Male predominance of Guillain-barre syndrome: Differential gene expression between Male and female patients. Int J Neurosci (2016) 126(6):531–41. doi: 10.3109/00207454.2015.1044088
294. Singh P, Szaraz-Szeles M, Mezei Z, Barath S, Hevessy Z. Gender-dependent frequency of unconventional T cells in a healthy adult Caucasian population: A combinational study of invariant nkt cells, Γδ T cells, and mucosa-associated invariant T cells. J leukocyte Biol (2022). doi: 10.1002/jlb.5a1121-583rr
295. Snyder-Cappione JE, Tincati C, Eccles-James IG, Cappione AJ, Ndhlovu LC, Koth LL, et al. A comprehensive ex vivo functional analysis of human nkt cells reveals production of Mip1-A and Mip1-B, a lack of il-17, and a Th1-bias in males. PloS One (2010) 5(11):e15412. doi: 10.1371/journal.pone.0015412
296. Bernin H, Fehling H, Marggraff C, Tannich E, Lotter H. The cytokine profile of human nkt cells and pbmcs is dependent on donor sex and stimulus. Med Microbiol Immunol (2016) 205(4):321–32. doi: 10.1007/s00430-016-0449-y
297. Schneider-Hohendorf T, Görlich D, Savola P, Kelkka T, Mustjoki S, Gross CC, et al. Sex bias in mhc I-associated shaping of the adaptive immune system. Proc Natl Acad Sci United States America (2018) 115(9):2168–73. doi: 10.1073/pnas.1716146115
298. Dalakas MC. Autoimmune neurological disorders with Igg4 antibodies: A distinct disease spectrum with unique Igg4 functions responding to anti-b cell therapies. Neurotherapeutics (2022). doi: 10.1007/s13311-022-01210-1
299. Koneczny I, Tzartos J, Mané-Damas M, Yilmaz V, Huijbers MG, Lazaridis K, et al. Igg4 autoantibodies in organ-specific autoimmunopathies: Reviewing class switching, antibody-producing cells, and specific immunotherapies. Front Immunol (2022) 13:834342. doi: 10.3389/fimmu.2022.834342
300. Niu HQ, Zhao XC, Li W, Xie JF, Liu XQ, Luo J, et al. Characteristics and reference ranges of Cd4(+)T cell subpopulations among healthy adult han Chinese in shanxi province, north China. BMC Immunol (2020) 21(1):44. doi: 10.1186/s12865-020-00374-9
301. Park HJ, Park HS, Lee JU, Bothwell AL, Choi JM. Gender-specific differences in pparγ regulation of follicular helper T cell responses with estrogen. Sci Rep (2016) 6:28495. doi: 10.1038/srep28495
302. Brito-Zerón P, Ramos-Casals M, Bosch X, Stone JH. The clinical spectrum of Igg4-related disease. Autoimmun Rev (2014) 13(12):1203–10. doi: 10.1016/j.autrev.2014.08.013
303. Blum S, Lee D, Gillis D, McEniery DF, Reddel S, McCombe P. Clinical features and impact of myasthenia gravis disease in Australian patients. J Clin Neurosci (2015) 22(7):1164–9. doi: 10.1016/j.jocn.2015.01.022
304. Yildiz Celik S, Durmus H, Yilmaz V, Saruhan Direskeneli G, Gulsen Parman Y, Serdaroglu Oflazer P, et al. Late-onset generalized myasthenia gravis: Clinical features, treatment, and outcome. Acta neurologica Belgica (2020) 120(1):133–40. doi: 10.1007/s13760-019-01252-x
305. Pawelec G. Age and immunity: What is "Immunosenescence"? Exp Gerontol (2018) 105:4–9. doi: 10.1016/j.exger.2017.10.024
306. Wick G, Boyd R, Hala K, Thunold S, Kofler H. Pathogenesis of spontaneous autoimmune thyroiditis in obese strain (Os) chickens. Clin Exp Immunol (1982) 47(1):1–18.
307. Wick G, Hala K, Wolf H, Ziemiecki A, Sundick RS, Stoffler-Meilicke M, et al. The role of genetically-determined primary alterations of the target organ in the development of spontaneous autoimmune thyroiditis in obese strain (Os) chickens. Immunol Rev (1986) 94:113–36. doi: 10.1111/j.1600-065x.1986.tb01167.x
308. Wick G. The role of the target organ in the development of autoimmune diseases exemplified in the obese strain (Os) chicken model for human hashimoto disease. Exp Clin Endocrinol Diabetes (1996) 104 Suppl 3:1–4. doi: 10.1055/s-0029-1211667
309. Kuhr T, Hala K, Dietrich H, Herold M, Wick G. Genetically determined target organ susceptibility in the pathogenesis of spontaneous autoimmune thyroiditis: Aberrant expression of mhc-class ii antigens and the possible role of virus. J Autoimmun (1994) 7(1):13–25. doi: 10.1006/jaut.1994.1002
310. Nelson LM. Autoimmune ovarian failure: Comparing the mouse model and the human disease. J Soc Gynecol Investig (2001) 8(1 Suppl Proceedings):S55–S7. doi: 10.1016/s1071-5576(00)00110-6
311. Lukic ML, Mensah-Brown E, Galadari S, Shahin A. Lack of apoptosis of infiltrating cells as the mechanism of high susceptibility to eae in da rats. Dev Immunol (2001) 8(3-4):193–200. doi: 10.1155/2001/32636
312. Houlden H, Laura M, Ginsberg L, Jungbluth H, Robb SA, Blake J, et al. The phenotype of charcot-Marie-Tooth disease type 4c due to Sh3tc2 mutations and possible predisposition to an inflammatory neuropathy. Neuromuscular Disord NMD (2009) 19(4):264–9. doi: 10.1016/j.nmd.2009.01.006
313. Fernandez-Lizarbe S, Civera-Tregón A, Cantarero L, Herrer I, Juarez P, Hoenicka J, et al. Neuroinflammation in the pathogenesis of axonal charcot-Marie-Tooth disease caused by lack of Gdap1. Exp Neurol (2019) 320:113004. doi: 10.1016/j.expneurol.2019.113004
314. Cardellini D, Zanette G, Taioli F, Bertolasi L, Ferrari S, Cavallaro T, et al. Cidp, Cmt1b, or Cmt1b plus cidp? Neurological Sci (2021) 42(3):1127–30. doi: 10.1007/s10072-020-04789-5
315. Stephens KE, Zhou W, Ji Z, Chen Z, He S, Ji H, et al. Sex differences in gene regulation in the dorsal root ganglion after nerve injury. BMC Genomics (2019) 20(1):147. doi: 10.1186/s12864-019-5512-9
316. Chernov AV, Shubayev VI. Sexually dimorphic transcriptional programs of early-phase response in regenerating peripheral nerves. Front Mol Neurosci (2022) 15:958568. doi: 10.3389/fnmol.2022.958568
317. Georgiou P, Zanos P, Mou TM, An X, Gerhard DM, Dryanovski DI, et al. Experimenters' sex modulates mouse behaviors and neural responses to ketamine Via corticotropin releasing factor. Nat Neurosci (2022) 25(9):1191–200. doi: 10.1038/s41593-022-01146-x
318. Sorge RE, Martin LJ, Isbester KA, Sotocinal SG, Rosen S, Tuttle AH, et al. Olfactory exposure to males, including men, causes stress and related analgesia in rodents. Nat Methods (2014) 11(6):629–32. doi: 10.1038/nmeth.2935
Keywords: Guillain Barré syndrome, chronic inflammatory demyelinating polyradiculoneuropathy, sex differences, gangliosides, IgG4 antibodies, experimental autoimmune neuritis
Citation: McCombe PA, Hardy TA, Nona RJ and Greer JM (2022) Sex differences in Guillain Barré syndrome, chronic inflammatory demyelinating polyradiculoneuropathy and experimental autoimmune neuritis. Front. Immunol. 13:1038411. doi: 10.3389/fimmu.2022.1038411
Received: 07 September 2022; Accepted: 24 November 2022;
Published: 09 December 2022.
Edited by:
Trine N. Jorgensen, Case Western Reserve University, United StatesReviewed by:
Bruce V. Taylor, University of Tasmania, AustraliaCopyright © 2022 McCombe, Hardy, Nona and Greer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pamela A. McCombe, UGFtZWxhLk1jQ29tYmVAdXEuZWR1LmF1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.