 Melissa Martins Oliveira1
Melissa Martins Oliveira1 Camila Ramalho Bonturi2
Camila Ramalho Bonturi2 Bruno Ramos Salu2
Bruno Ramos Salu2 Maria Luiza Vilela Oliva2
Maria Luiza Vilela Oliva2 Renato Arruda Mortara1
Renato Arruda Mortara1 Cristina Mary Orikaza1*
Cristina Mary Orikaza1*- 1¹Microbiology, Immunology and Parasitology Department, Escola Paulista de Medicina, Federal University of São Paulo - UNIFESP, São Paulo, Brazil
- 2²Biochemistry Department, Escola Paulista de Medicina, Federal University of São Paulo - UNIFESP, São Paulo, Brazil
Trypanosoma cruzi is the causative protozoan of Chagas’ Disease, a neglected tropical disease that affects 6−7 million people worldwide. Interaction of the parasite with the host immune system is a key factor in disease progression and chronic symptoms. Although the human immune system is capable of controlling the disease, the parasite has numerous evasion mechanisms that aim to maintain intracellular persistence and survival. Due to the pronounced genetic variability of T. cruzi, co-infections or mixed infections with more than one parasite strain have been reported in the literature. The intermodulation in such cases is unclear. This study aimed to evaluate the co-infection of T. cruzi strains G and CL compared to their individual infections in human macrophages derived from THP-1 cells activated by classical or alternative pathways. Flow cytometry analysis demonstrated that trypomastigotes were more infective than extracellular amastigotes (EAs) and that strain G could infect more macrophages than strain CL. Classically activated macrophages showed lower number of infected cells and IL-4-stimulated cells displayed increased CL-infected macrophages. However, co-infection was a rare event. CL EAs decreased the production of reactive oxygen species (ROS), whereas G trypomastigotes displayed increased ROS detection in classically activated cells. Co-infection did not affect ROS production. Monoinfection by strain G or CL mainly induced an anti-inflammatory cytokine profile by decreasing inflammatory cytokines (IFN-γ, TNF-α, IL-1β) and/or increasing IL-4, IL-10, and TGF-β. Co-infection led to a predominant inflammatory milieu, with reduced IL-10 and TGF-β, and/or promotion of IFN-γ and IL-1β release. Infection by strain G reduced activation of intracellular signal transducer and activator of transcription (STAT) factors. In EAs, monoinfections impaired STAT-1 activity and promoted phosphorylation of STAT-3, both changes may prolong cell survival. Coinfected macrophages displayed pronounced activation of all STATs examined. These activations likely promoted parasite persistence and survival of infected cells. The collective results demonstrate that although macrophages respond to both strains, T. cruzi can modulate the intracellular environment, inducing different responses depending on the strain, parasite infective form, and co-infection or monoinfection. The modulation influences parasite persistence and survival of infected cells.
Introduction
Chagas’ Disease (CD) is an anthropozoonosis caused by Trypanosoma cruzi protozoan. CD is endemic in 21 Latin American countries with an estimated 12,000−14,000 deaths annually. An estimated 6 million people are infected worldwide (1ߝ4–6).
T. cruzi is a digenetic parasite with a life cycle that comprises three distinct evolutionary forms: epimastigotes, metacyclic or blood trypomastigotes, and amastigotes (1, 7, 8). Trypomastigotes are the classic infective forms. However, extracellular differentiation of trypomastigotes or early cell rupture can release extracellular amastigotes (EAs), which can also infect host cells and sustain the parasite life cycle (revised in 9). T. cruzi is currently categorized into seven discrete typing units (4, 10). This pronounced genetic variability allows reinfections by different strains, also known as co-infections, polyparasitism, or mixed infections. Previous studies have described polyparasitism in insect vectors (11, 12), human patients (13–16), experimental infection and treatment effects in animal models (17–20). At endemic areas, the occurrence of co-infections is estimated between 9 and 57% (14-16, 21). This broad range of mixed infections reflects the great abundance of strains, besides, some factors may underestimate the polyparasitism numbers, like the difficulty of parasite detection in the host, especially at chronic phase of infection, and socioeconomic conditions intrinsically associated with neglected tropical diseases, such as CD. It is not clear whether the correlation of these strain groups and clinical symptoms are related (22) and whether they modulate disease progression in the host. If so, it would at least partially explain the wide range of outcomes in patients with CD.
Besides parasite intrinsic virulence genetic factors, mechanisms of the host cell response are also related to the control or aggravation of disease. Macrophages are the first defense cells to encounter the parasite (23, 24) and these cells are highly responsive to the surrounding milieu and develop specific and complex profiles, which are classically activated macrophages (M1) and alternatively activated macrophages (M2) (25–27; revised in 28). The latter can be subdivided into M2a, M2b, M2c, and M2d (27).
M1 macrophages have high phagocytic capacity and can be generated in vitro by monocytes stimulated by interferon-gamma (IFN-γ) and/or lipopolysaccharide (LPS) (29). M2 macrophages are commonly obtained by the polarization of monocytes with IL-4 and/or IL-13, which are linked to tissue repair and angiogenesis mechanisms (reviewed by 30). Generally, in the acute phase of infection, proinflammatory cytokines that include IFN-γ, tumor necrosis factor-alpha (TNF-α), IL-1β, and IL-12 are related to parasite control, and anti-inflammatory cytokines (IL-10, IL-4, and transforming growth factor-beta [TGF-β]) result in opposing actions (3, 31). During the chronic phase of infection, the imbalance of few anti-inflammatory stimuli with an exacerbated inflammatory response appears to lead to tissue lesions, as seen in chronic chagasic cardiomyopathy (32–34). Reactive oxygen species (ROS) and reactive nitrogen species, such as nitric oxide (NO), are important molecules implicated in killing several intracellular pathogens. This is despite the presence of T. cruzi antioxidant enzymes that can inactivate these molecules and favor parasite persistence inside the host cell (35, 36; revised in 37).
Cytokines can induce the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway, in which STATs dimerize and translocate to the nucleus and promote or repress DNA transcription factors of genes related to immunological responses (38–40). STAT-1 can be phosphorylated in response to IFN-γ. Stahl et al. (41) and Kulkarni et al. (42) demonstrated the essential role of the IFN-γ/STAT-1 axis in the control of T. cruzi infection. Infection of cardiomyocytes with the Tulahuén strain of T. cruzi induced an anti-apoptotic effect in an IL-6/STAT-3-dependent manner (43). In contrast, de Souza etal. (44) reported that T. cruzi Dm28c provoked apoptosis in peritoneal macrophages, but the authors discussed that apoptosis may not be induced by T. cruzi depending on the parasite strain and host cell tissue. IL-6 is a cytokine associated with macrophage polarization to the M2 profile, specifically M2d (45), and downstream phosphorylation of STAT-3 that is linked to the promotion of cellular proliferation and reduction of apoptosis (46). IL-4 can cause STAT-6 activation that promotes an anti-inflammatory response that is important to counteract the excess of inflammatory stimuli but is also related to parasite persistence and spread (47, 48).
Considering the important role of macrophages in T. cruzi infection response and the paucity of studies involving co-infection with different strains, the present study evaluated the activation of STAT-1, -3, and -6 in polarized macrophages derived from THP-1 cells. The results of co-infection with G (TcI) and/or CL (TcVI) strains were compared to those with monoinfections by each strain.
Methods
Ethics statement
This study was approved by the Ethical Research Committee of Federal University of São Paulo - UNIFESP (number 8152030419) in April 2019.
THP-1 cells, macrophages, and cytokine stimuli
THP-1 human monoblasts were kindly provided by Dr. Fatima Ribeiro-Dias, Universidade Federal de Goiás, Goiânia, Brazil. The cells were cultivated in RMPI 1640 (Vitrocell, Brazil) supplemented with 10 mM HEPES (Thermo Fisher Scientific, USA), 20 mM sodium bicarbonate, 2 mM L-glutamine, 25 mM glucose, 1 mM sodium pyruvate, 5 mM mercaptoethanol, 2 mM penicillin, 0.15 mM streptomycin (all from Sigma-Aldrich, USA), and 10% fetal calf serum (FCS, Vitrocell). Cells were maintained in T25 culture flasks (TPP, Swiss) at a cell density of 5 × 104 to 1 × 106 cells/mL. To obtain macrophages derived from THP-1 cells, the cells were plated (2 × 105 cells/mL) in 6-, 12-, or 96-well plates (Corning, USA) and incubated for 48 h in complete medium supplemented with 100 ng/mL phorbol 12-myristate 13-acetate (PMA, Sigma-Aldrich) at 37°C in an atmosphere of 5% CO2. The wells were washed twice with Hanks Balanced Salt Solution (Sigma-Aldrich) supplemented with 2 mM sodium bicarbonate to remove PMA and unattached cells. The plates were further incubated for 48 h in complete medium at 37°C and 5% CO2. Macrophages were then activated with cytokines for 24 h as follows using: IFN-γ (20 ng/mL) + LPS (100 ng/mL), IL-4 (25 ng/mL), or IL-6 (50 ng/mL). One group was not exposed to cytokines (M0 phenotype, representing the basal control).
Trypanosoma cruzi
T. cruzi G (49) and CL (50) strains were used in its wild type form (WT) for ROS assay or transfected with fluorescent plasmids harboring green fluorescent protein (GFP) (G-pTREX-GFP; 51) or DsRed (CL-pTREX-DsRed; Ferreira et al., 2016). Tissue culture-derived trypomastigotes (TCTs) of T. cruzi were obtained from the supernatant of Vero cells (Instituto Adolfo Lutz, São Paulo, Brazil). TCTs were cultured in RPMI 1640 supplemented with 2.5% FCS, 2 mM penicillin, 0.2 mM streptomycin, and 84 µM gentamicin (all from Sigma-Aldrich) at 37°C and 5% CO2. EAs were obtained as previously described (52–54). Briefly, TCTs were cultured in liver infusion tryptose medium (pH 5.8) for 14−16 h, followed by 1 h in RPMI 1640 supplemented with 10% FCS. Four groups were used for every experiment: non-infected cells (NI), cells infected with G-GFP (G), cells infected with CL-DsRed (CL), and cells coinfected with both T. cruzi strains (COI). WT strains were only used in the ROS assay, due to fluorescence interference on measurements, since the infectivity of transfected and WT parasites was similar, as previously described (51). Multiplicity of infection (MOI) for TCTs and EAs was 2:1 for G and 20:1 for CL. T. cruzi was incubated with the cells for 3 h, the wells were washed three to seven times to remove parasites that were not internalized and maintained for 48 h at 37 °C and 5% CO2 until analyses were performed.
Analyses of infected cells and cell viabilities
Cells (2 × 105) were seeded in 12-well plates and stimulated and/or infected as described above and detached using 0.2% trypsin and 0.02% EDTA. Aliquots were obtained to assess the cell viability of each group using Fixable Viability Dye eFluor 780 (eBiosciences, USA) according to the manufacturer’s instructions. The suspended cells were fixed for 15 min in 4% paraformaldehyde (PFA) in phosphate buffered saline (PBS), washed in MACS buffer (PBS containing 0.05% bovine serum albumin and 2 mM EDTA), and suspended in 300 µL of MACS buffer. Cell suspensions were analyzed by flow cytometry using a LSRFortessa flow cytometer (BD Biosciences, USA), acquiring 5000 events/sample, with the gating strategy illustrated in Supplementary Figure 1S. For compensation controls of fluorescence, monoinfected cells were used. Analyses were performed using BD FACSDiva 6.2 and FlowJo 10.8.1 software (BD Biosciences).
Measurements of nitric oxide and cytokines
THP-1 cells were plated as described above 48 h post-infection (hpi). The medium was filtered through a 0.22 µm pore size filter (Millipore, USA) and frozen at –20°C until measurements were made. NO was analyzed using a model 280i NO analyzer (Zysense). For the measurement of cytokines, the samples were stored in 10 mM EDTA, 9 μM aprotinin, and 10 μM E-64 (all from Sigma-Aldrich) at –80°C. IFN-γ, TNF-α, IL-1β, IL-4, IL-6, IL-10, and IL-12p70 were analyzed using the MILLIPLEX®MAP Human Cytokine/Chemokin Magnetic Bead Panel (Merck Millipore). TGF-β was analyzed with the MILLIPLEX®MAP TGF-β1,2,3 Magnetic Bead Kit (Merck Millipore). All analyses used the MAGPIX® system (Merck Millipore), according to the manufacturer’s instructions.
Measurement of ROS
Cells (1 × 104) were seeded in black 96-well plates and cultivated as previously described. We used the 2’,7’-dichlorofluorescein diacetate/2’,7’-dichlorodihydrofluorescein diacetate (DCFDA/H2DCFDA) − Cellular ROS Assay Kit (Abcam, UK) according to the manufacturer’s instructions. Measurements were performed at 3 and 24 hpi by fluorescence quantification of each cell using ImageJ v.1.53m software (NIH) from at least four images per group (40× magnification) using an Olympus IX70 inverted microscope. Data are presented as corrected total cell fluorescence (CTCF) calculated as integrated density – (area of selected cell × mean fluorescence of background readings). Analyses were performed using the ratio of cell CTCF and M0 mean CTCF.
Evaluation of STATs
THP-1 cells (5.5 × 105) were seeded into 6-well plates and stimulated as previously described. After 48 hpi, cells were detached with 300 µL of trypsin solution, inactivated with complete medium, and washed with MACS. Aliquots were reserved to test cell viability as described above. Cells were fixed with 4% PFA in PBS, incubated with 5% autologous serum, and permeabilized with PermBufferIII (BD Biosciences). The samples were incubated with combinations of two antibodies: anti-STAT-1 pY701 Pacific Blue and anti-STAT-1 pS727 AlexaFluor 647, or anti-STAT-3 pY705 Pacific Blue and anti-STAT-6 pY641 AlexaFluor 647 (all from BD Biosciences). As compensation controls, we used beads for STATs antibodies and monoinfected cells for G-GFP and CL-DsRed. A gating strategy (Supplementary Figure 1S) consisted of the selection of single cells, followed by macrophages, and then GFP × DsRed. Thus, each quadrant represents one type of infected cell (Q1 by G, Q2 coinfected, Q3 by CL, and Q4 NI cells). Within each quadrant, we analyzed the presence of phosphorylated STATs. Acquisition of 20.000 events/sample was performed with BD FACSDiva version 6.2 in a BD LSRFortessa flow cytometer. Median fluorescence intensity (MFI) of STATs were analyzed with FlowJo software version 10.8.1.
Statistical analyses
All experiments were performed in duplicate or triplicate, with two or three biological replicates. Data are expressed as the mean ± standard deviation for all parameters except ROS; the data of the latter are expressed as standard error of the mean. Differences were considered significant at a p-value < 0.05. The determinations involved two-way ANOVA with Tukey’s post-test when samples presented a normal distribution and otherwise by Kruskal−Wallis with Dunn’s post-test calculated with GraphPad Prism 8.3.0. Data of the phosphorylated STATs are presented as heatmaps with the ratio of MFI in each condition in relation to M0 basal MFI.
Results
Macrophage infection is greater with T. cruzi G strain, but IL-4 favors infection by CL strain
THP-1 derived macrophages were cultivated and stimulated with cytokines as described above. None of these treatments significantly interfered with cell viability in all groups, as evidenced by the cell viability rate >90% (Supplementary Figure 2S). T. cruzi strains G and CL, and both TCTs and EAs were capable of infecting macrophages, independent of cytokine stimulus (Figures 1A−C). However, infection by TCTs presented higher infection rates at 48 hpi than by EAs. Macrophages stimulated with IFN-γ + LPS [M(IFN-γ)] led to lower numbers of infected cells in all conditions. Macrophages stimulated with IL-4 or IL-6 [M(IL-4) and M(IL-6), respectively] tended to augment CL-infected cells, mainly when infected with TCTs. Consequently, infected cells also increased in the co-infection group. This effect was more evident in M(IL-4). Among the infected cells in the co-infection condition, we could separate into G-only, CL-only, or cohabited cells, as shown in Figures 1D, E. We observed that only G cells were predominant in M0 and M(IFN-γ), and that CL macrophages appeared in almost the same proportion as G macrophages in M(IL-4). Despite this variance, cohabiting cells were rare under all conditions tested.
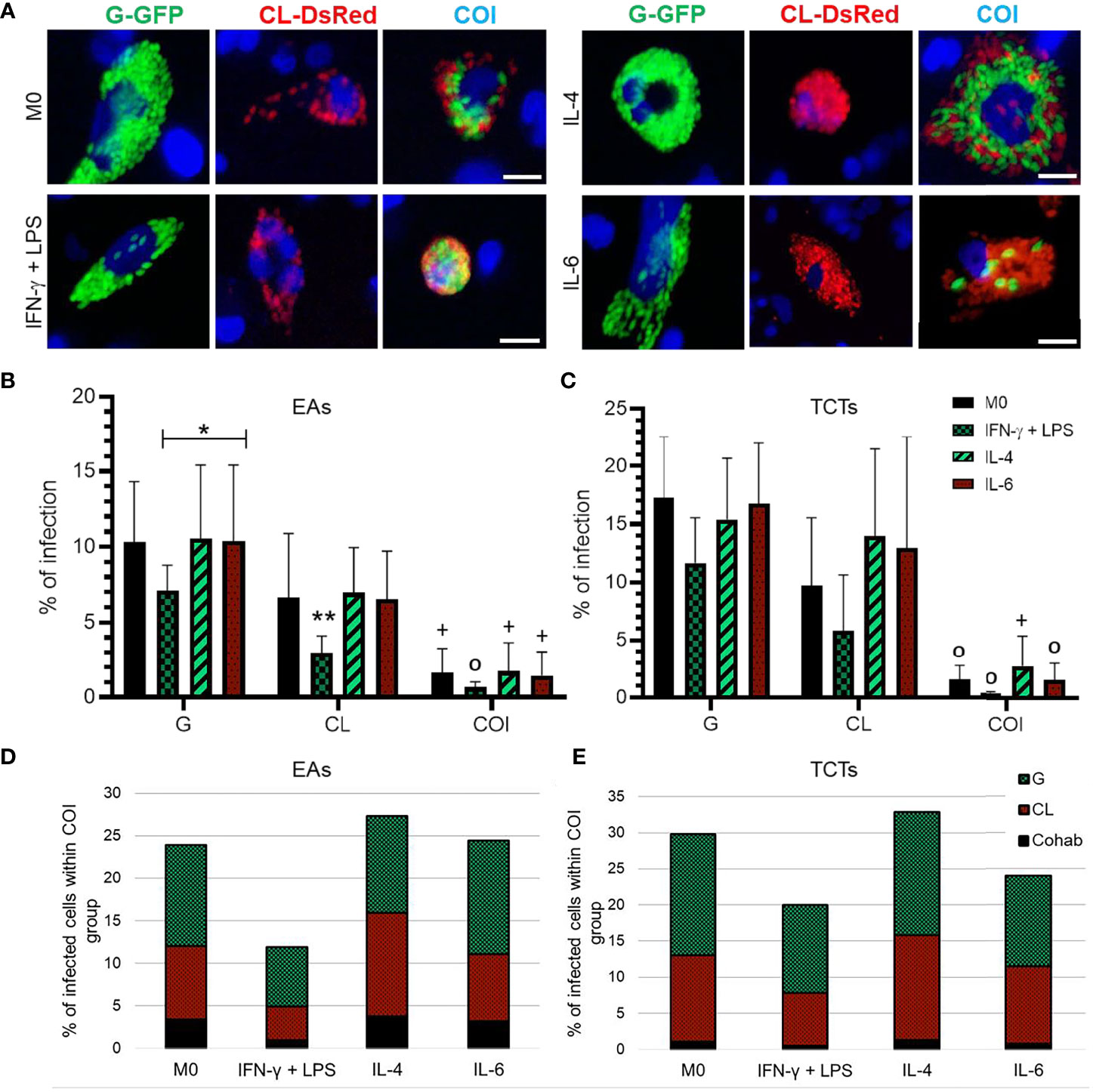
Figure 1 IFN-γ + LPS treatment reduced the number of infected macrophages by both strains, while IL-4 and IL-6 augmented CL infected cell numbers. (A) Representative images of Trypanosoma cruzi amastigotes in THP-1 derived macrophages at 72 h post-infection (hpi); Green = G-GFP; red = CL-DsRed; blue = cell nuclei stained with Hoechst 33342. COI = co-infection. Bar = 20 μm. Percentage of infected cells at 48 hpi analyzed by flow cytometry when cells were infected by extracellular amastigotes (EAs) (B), or by tissue culture-derived trypomastigote forms (TCTs) (C). Graphs represent mean and standard deviation of three independent experiments. *p < 0.05; **p < 0.01; +p < 0.05 COI vs. monoinfections of both strains; 0p ≤ 0.01 COI vs. G monoinfection; determined by two-way ANOVA with Tukey’s post hoc. Distribution of strains within co-infection group, cells only with the G strain were predominant, while CL strain was more present in the groups stimulated with IL-4 or IL-6 and cohabited cells (Cohab) presented lower numbers in the infection either by EAs (D) or TCTs (E).
NO levels vary in infection by CL, but not by G strain, otherwise G strain infection increased ROS in classically activated macrophages
According to the results presented above, TCTs had higher infection rates. Thus, we evaluated NO production by these cells at 48 and 72 hpi (Figures 2A, B). M0 and M(IFN-γ) groups showed no differences in NO detection patterns at 48 or 72 hpi. CL monoinfection diminished NO levels in M0 and M(IFN-γ) at 48 hpi, while at 72 hpi the levels were higher compared to their NI control, including M(IL-6). G monoinfection seemed to maintain stable levels of NO, even in M(IL-4) cells, which decreased NO in the NI group. After 72 hpi, COI decreased NO in M0 and M(IFN-γ) and increased in the M(IL-4) group.
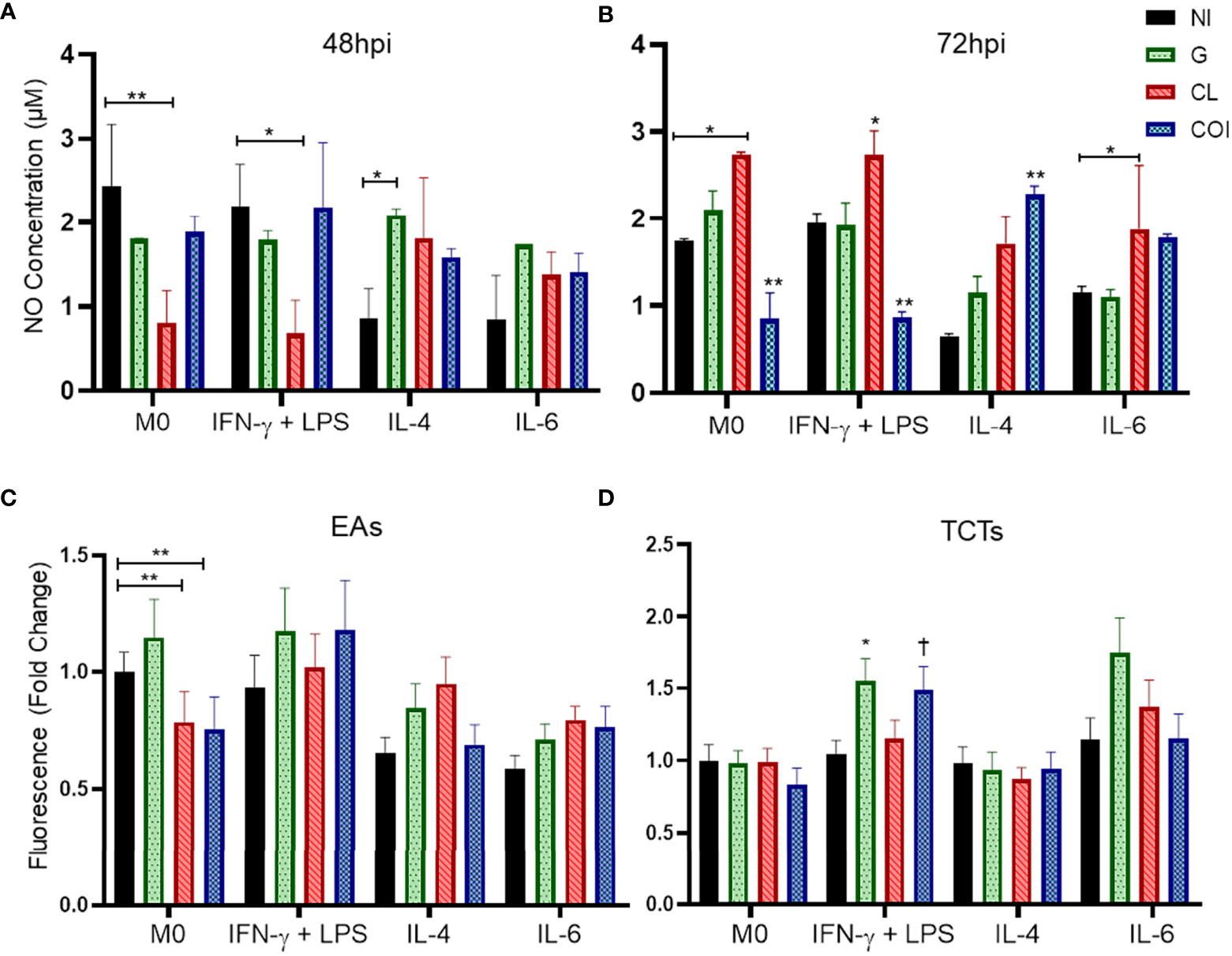
Figure 2 Nitric oxide (NO) concentration remains at stable levels when macrophages are infected by strain G, but not CL or co-infection (COI); otherwise, G infection promoted increase of reactive oxygen species (ROS) in cells stimulated with IFN-γ+LPS. CL monoinfection and co-infection showed an opposite pattern of NO production by non-polarized macrophages (M0) and cells stimulated with IFN-γ + LPS at the two evaluated times (48 and 72 hours post infection – hpi). Graphs represent the mean and standard deviation of duplicates from two independent experiments at 48 hpi (A) and 72 hpi (B). Statistical differences determined by the two-way ANOVA test with Tukey’s post hoc, *p < 0.05; **p < 0.01. Extracellular amastigotes (EAs) of CL strain infection and co-infection in M0 produced less ROS than the uninfected macrophage (C). Regarding TCTs, macrophages stimulated with IFN-γ + LPS displayed the highest production of ROS with G and COI (D). Graphs summarize the observations of two experiments performed in duplicate, quantified at 3 hpi. Statistical differences determined by Kruskal−Wallis test with Dunn’s post hoc. *p < 0.05; **p < 0.01; †p < 0.05 vs. M0 COI.
ROS were evaluated at 3 and 24 hpi. No significant differences were observed at 24 hpi (Supplementary Figure 3S). At 3 hpi (Figures 2C, D), CL EAs alone or with strain G reduced ROS in M0. Otherwise, TCTs in M(IFN-γ) COI and G were higher than the same infection at M0 or M(IL-4), respectively.
Monoinfections predominantly promoted decrease and co-infections increase of inflammatory cytokines release
Another way to evaluate macrophage activation is through the secreted cytokines. Since M(IFN-γ) did not elevate NO levels as expected, we analyzed IFN-γ, TNF-α, IL-1β, IL-4, IL-6, IL-10, IL-12, and TGF-β. Unlike the NO measurements, we observed a higher release of proinflammatory cytokines (IFN-γ, IL-1β, and TNF-α) in the M(IFN-γ) group (Figure 3) and this was likely due to cell activation.
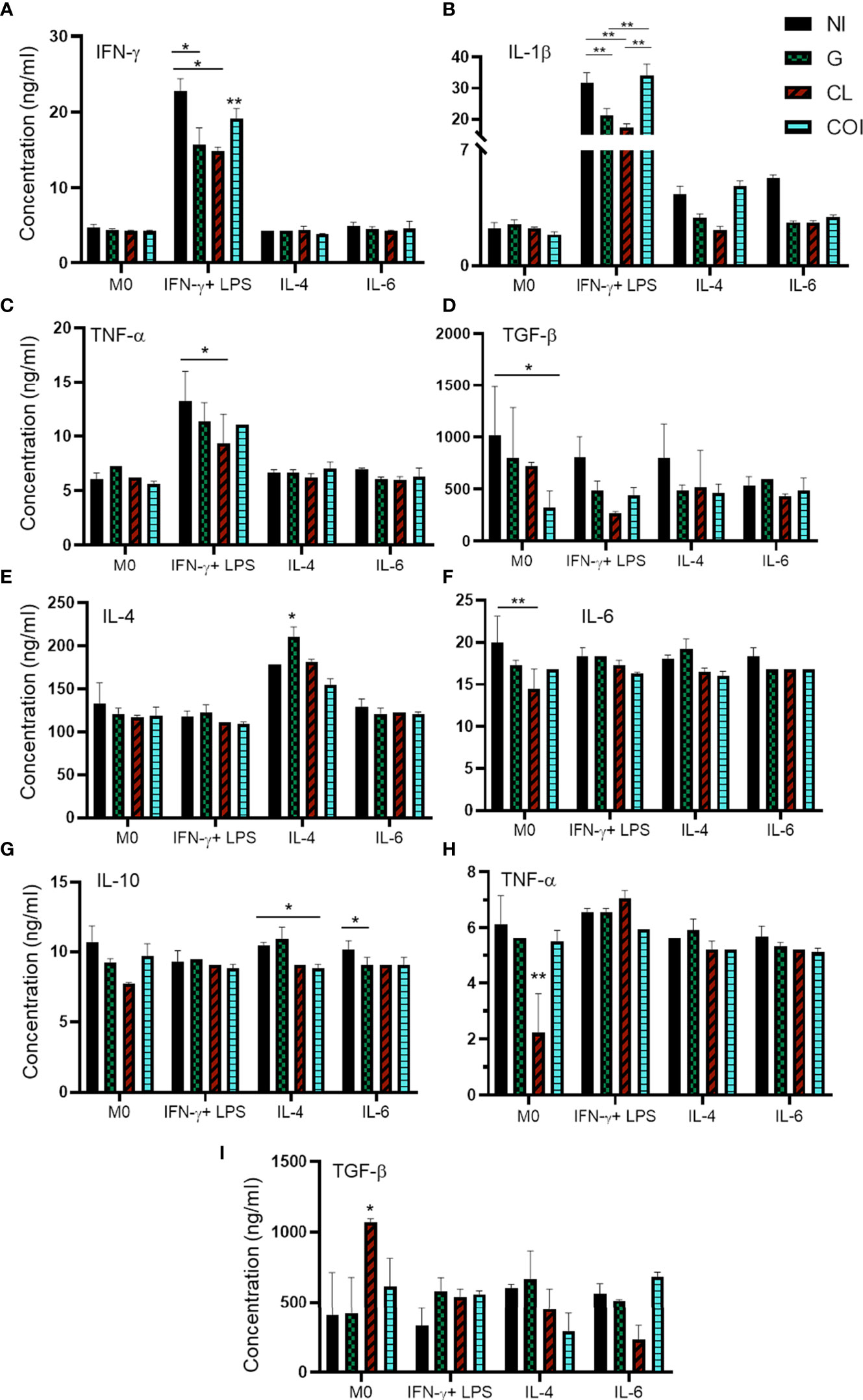
Figure 3 Quantification of cytokines in the supernatant of macrophages showing predominant decreased levels of proinflammatory cytokines in monoinfections and increased levels of inflammatory cytokines in co-infection. Macrophages were polarized and/or infected by extracellular amastigotes (EAs) (A–D) or by tissue-culture trypomastigotes (TCTs) (E–I), and quantification was measured 48 h after infection. Graphs represent the mean and standard deviation of concentration values (ng/mL) from two measurements of each sample in duplicate, statistical differences determined by the two-way ANOVA test with Tukey’s post hoc test. *p < 0.05; **p < 0.01.
Monoinfection of EAs with G or CL reduced the levels of IFN-γ and IL-1β compared to those in NI cells among M(IFN-γ). Co-infection elevated the levels of both cytokines, especially IL-1β. Monoinfection with CL also diminished TNF-α levels in the same IFN-stimulated group. Co-infection significantly reduced TGF-β levels in the M0 group (Figures 3A−D). The other conditions did not produce significant differences in EAs infection (Supplementary Figures 4S).
Infection of TCTs led to different responses. CL monoinfection reduced TNF-α and IL-6 in addition to enhanced TGF-β levels in M0 cells. M(IL-4) resulted in increased IL-4 when infected with only the G strain and reduced IL-10 during co-infection. IL-10 was also reduced in the monoinfection of G when cells were stimulated with IL-6 (Figures 3E−I). The other conditions and cytokines did not show significant differences during infection of TCTs (Supplementary Figure 5S).
Analysis of STATs activation
JAK-STAT is an important intracellular pathway activated by cytokines. Accordingly, we evaluated four domains of the three STATs at 48 hpi. The basal MFI of the M0 NI group was used as a reference (ratio 1, yellow) to evaluate the increase (>1, red) or decrease (<1, green) of the other MFIs (Figure 4). We observed that the activation effect due to the examined cytokine stimuli was still detectable, as observed on the NI line in M(IFN-γ) on STAT-1, M(IL-6) on STAT-3, and M(IL-4) on STAT-6. These results were obtained even though we could observe distinct activation patterns in each infection, depending on the infective form of the parasite.
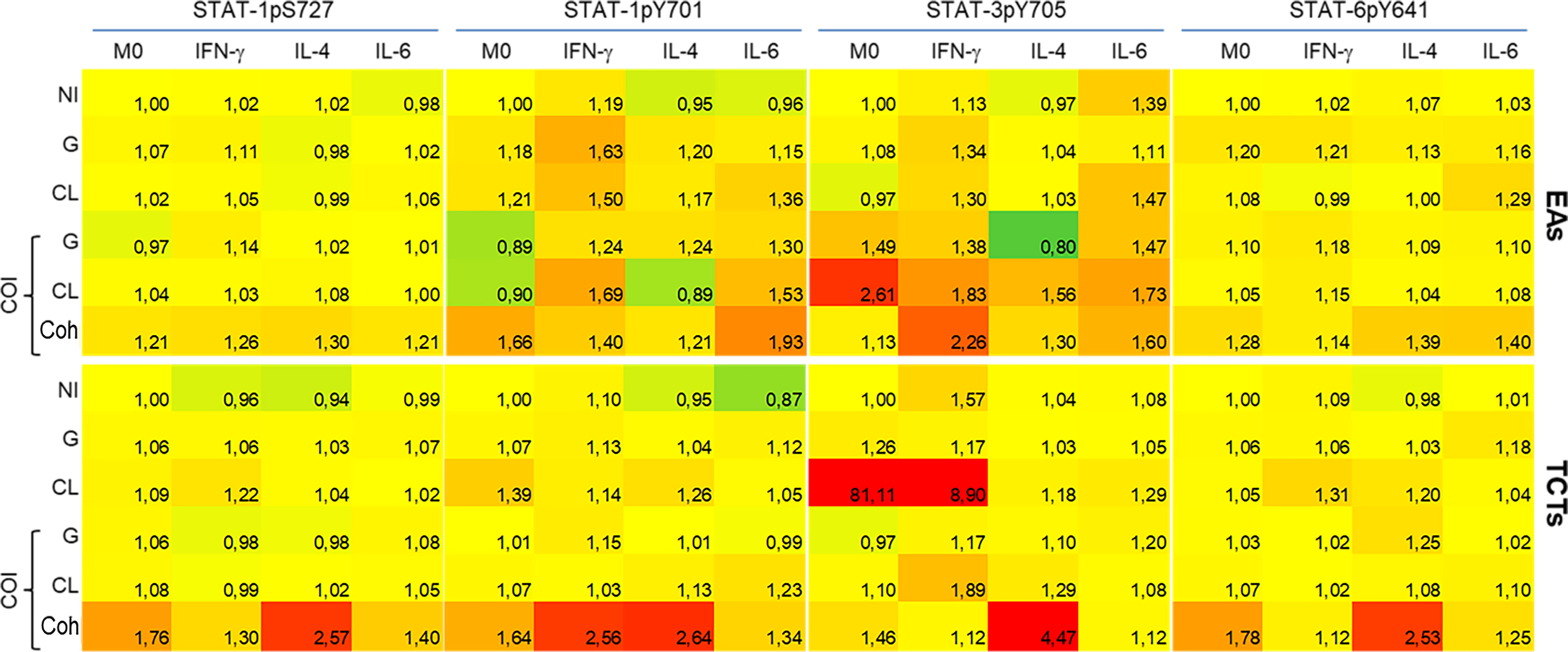
Figure 4 Coinfected cells induced increased phosphorylation of all STATs residues evaluated when compared to monoinfections at 48 h post infection. Activation of STATs were assessed by flow cytometry, with detection of phosphorylated Y701 (pY701) and pS727 of STAT-1, pY705 of STAT-3 and pY641 of STAT-6. Rows indicate infection groups: NI uninfected; G infection; CL infection; and subgroups within co-infection (COI) inside the brace. Coh: cohabited cells, presence of both strains in a cell. Top rows represent infection by extracellular amastigotes (EAs), and bottom rows by tissue-culture trypomastigotes (TCTs). Columns indicate the STATs residue evaluated and macrophage stimuli. The comparative activation is presented as green-red scale heatmap with the ratio of median fluorescence intensity (MFI) of each condition in relation to uninfected M0 basal MFI (ratio 1, yellow; <1 less phosphorylation = green; >1 more phosphorylation = red).
The heatmap graphic enables two main comparisons: between lines (effect of infection within cytokine-treated group) or between columns (effect of cytokine stimuli within each infection). For our purposes, we made comparisons between lines, as our aim was to observe differences among mono- or co-infection. STAT-1pS727 and STAT-6pY641 were generally near basal levels, except for the EAs and TCTs infected with both parasites. Infected EAs displayed elevated activation of tyrosine residues of STAT-1 and STAT-3 in the presence of either parasite in M(IFN-γ) or M(IL-6) cells; the latter did not show this increase with G monoinfection. Infection of TCTs seemed to cause fewer alterations, but stronger effects on phosphorylation; CL monoinfection greatly increased STAT-3 activation, mainly in M0 and M(IFN-γ) groups. EAs and TCTs harboring both strains displayed the highest phosphorylation among the studied residues under almost all conditions.
We also analyzed the uninfected cells within each infection condition, as illustrated by the Q4 quadrant in Supplementary Figures 1S–C. These results are presented in Figure 5, taking as reference the M0 NI condition and comparisons as mentioned in the previous figure. We observed minor effects that mostly tended to be dephosphorylation of STATs-1 in M(IL-4) or M(IL-6) cells. The exception was NI COI with EAs, which displayed higher activation of STAT-3 in M(IFN-γ) or M(IL-6) cells.
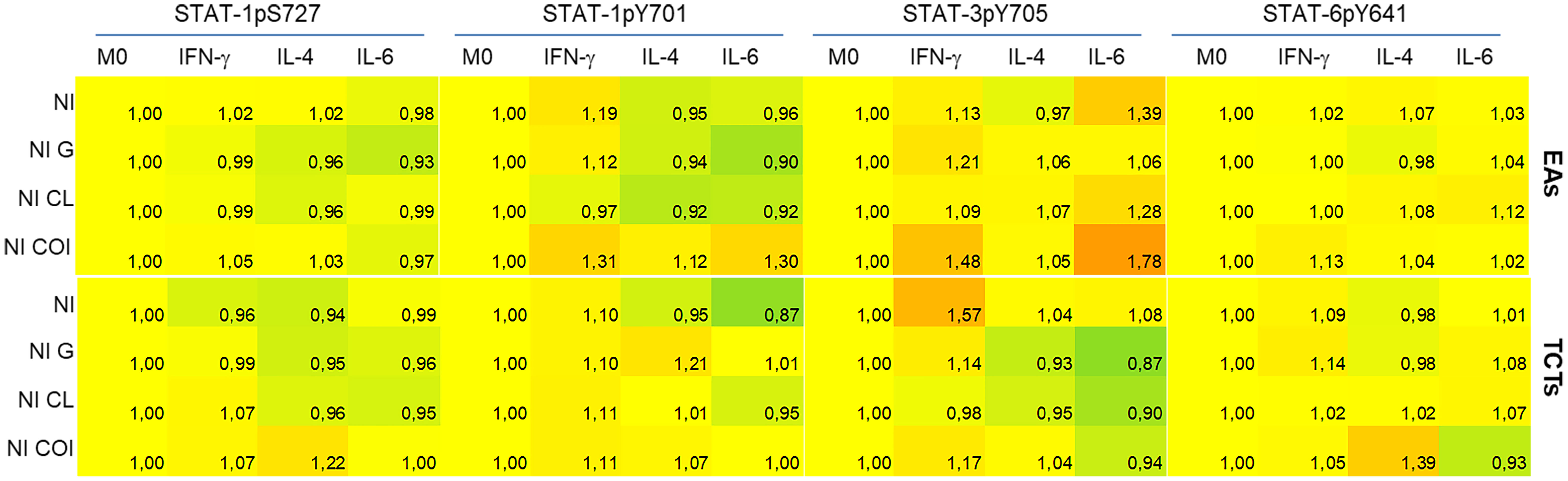
Figure 5 Uninfected cells (NI) demonstrated dephosphorylation of STATs, mainly in monoinfections, except for STAT-3 evaluated in cells infected by extracellular amastigotes (EAs). Activation of STATs were assessed by flow cytometry at 48 h post-infection, with detection of phosphorylated Y701 (pY701) and pS727 of STAT-1, pY705 of STAT-3 and pY641 of STAT-6 residues. Rows indicate the condition group: NI - uninfected; NI G - uninfected cells within G infected group; NI CL - uninfected cells within CL infected group; and NI COI uninfected cells within co-infected group. Top rows represent infection by extracellular amastigotes (EAs), and bottom rows by tissue-culture trypomastigotes (TCTs). Columns indicate the STATs residue evaluated and macrophage stimuli. The comparative activation is presented as green-red scale heatmaps with the ratio of each condition’s median fluorescence intensity (MFI) in relation to M0 basal MFI (ratio 1, yellow; <1 less phosphorylation = green; >1 more phosphorylation = red).
Discussion
T. cruzi strains and vector diversity in endemic zones provide multiple contacts between humans and triatomines. The latter also have the chance to feed on the blood of numerous infected individuals (55, 56). This environment has a direct impact on evolution and parasite genetics. Reinfections in one individual can result in co-infection or mixed infection with more than one strain interacting in the same host. These events can influence symptoms, disease progression, and even treatment effectiveness (18).
Our observations demonstrate that both G and CL strains could infect macrophages, whether they were activated or not, independent of the infective form, either EAs or TCTs. The G strain is characterized by low infectivity of trypomastigotes while displaying highly infective EAs in vitro. CL metacyclic trypomastigotes present high infection rates and low infectivity as EAs in epithelial cell cultures (49; reviewed by 57). Interestingly, our results in a model of professional phagocytic cells showed that TCTs were more infective than EAs independent of strain, and strain G was able to infect a higher number of cells than strain CL. Even when using an MOI that was ten times higher, strain CL resulted in low numbers of infected THP-1-derived macrophages, except when cells were stimulated with IL-4 or IL-6, then, both strains presented similar infection rates (Figure 1). Owing to the multiclonal structure of T. cruzi, different isolates can exhibit symbiotic behavior. However, environmental stressors can also promote competition among them (55, 56), this fact may at least partially explain our low co-infection numbers.
The main differences between infected and NI cells at 48 hpi are shown in Figure 6. Infection with strain G caused less intracellular reaction than infection with CL. This may contribute to the thriving of G in this cell model. We observed that in the M(IL-4) condition, which favored CL infection, no or little intracellular disturbance was evident, this data corroborates that of Vaena de Avalos et al. (58). The latter authors concluded that T. cruzi infection in susceptible cells induced few transcriptional changes at early stages. Finally, we observed that co-infection resulted in a different pattern of responses from the cells with the activation of more STATs, independent of cytokine stimuli.
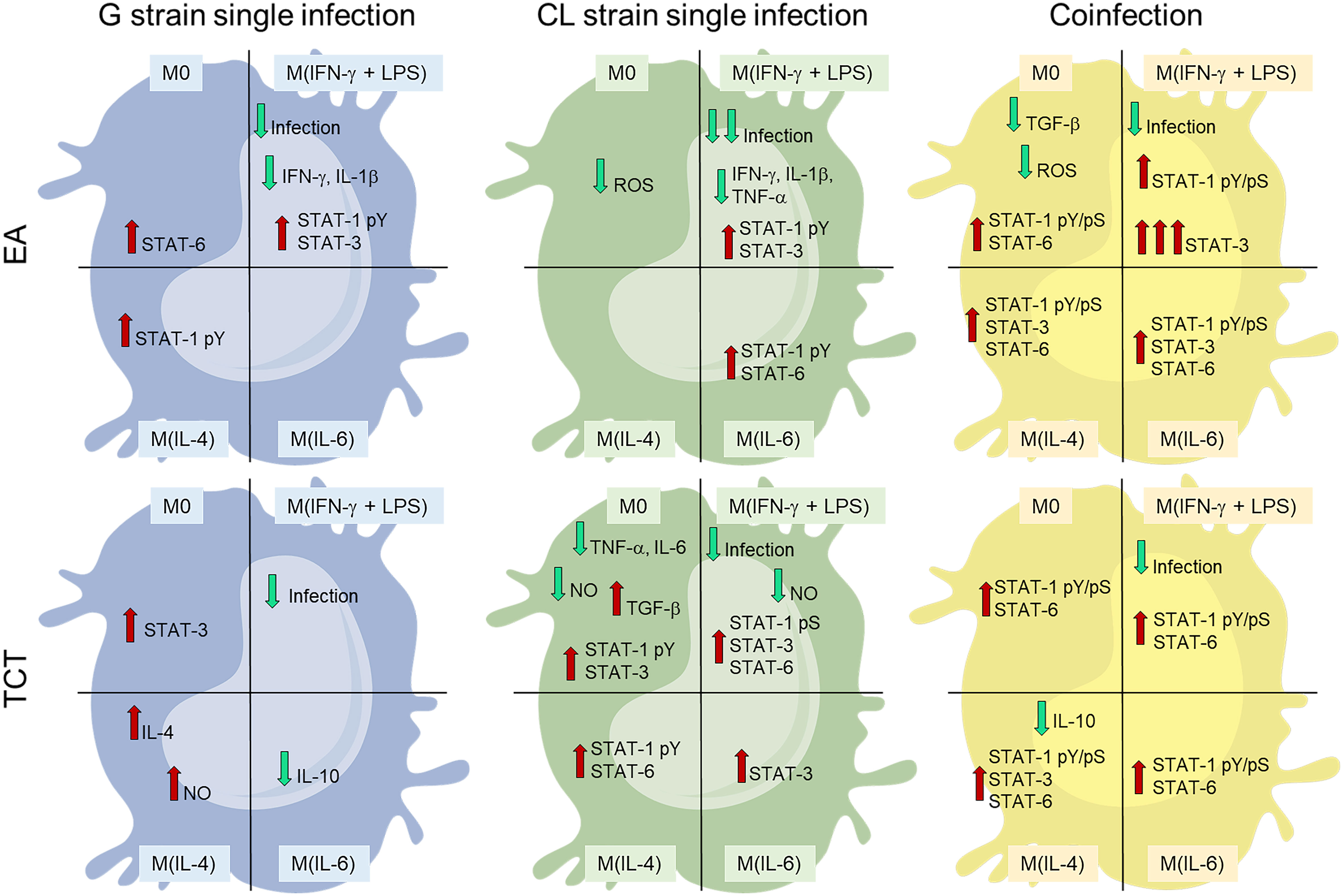
Figure 6 Monoinfection conditions of higher infected cell numbers [strain G or strain CL M(IL4)] demonstrated less host cell responses than co-infection (COI). COI promoted important activation of STATs and resulted in low infection rates. Cells are divided in four categories, representing the four stimuli states: M0 – without cytokine; M(IFN- γ+LPS) – stimulated with 20 ng/mL and 100 ng/mL, respectively; M(IL-4) – stimulated with 25 ng/mL of IL-4; or M(IL-6) – stimulated with 50 ng/mL of IL-6. The top cells represent infection by extracellular amastigotes (EAs). The bottom cells depict tissue cultured trypomastigotes (TCTs). Blue cells are infection by G strain, green cells by CL strain, and yellow cells by both strains. Green and red arrows denote decrease and increase, respectively.
The number of infected cells decreased in M(IFN-γ) macrophages under all infection conditions, demonstrating the effectiveness of IFN-γ in hindering parasite entry. IFN-γ is a key cytokine in T. cruzi control. Its release is increased in the acute phase of infection, and this provides an inflammatory environment that elicits a Th1 response, these events are essential for the reduction of parasitemia and disease counteraction (24, 34, 59–62). An effector pathway that can be activated by IFN-γ is inducible NO synthase (iNOS). Subsequent NO production and other cytotoxic radicals are detrimental to T. cruzi and to the host (63, 64). However, some studies have also discussed the role of NO, which may need synergistic cytokine effects or cause tissue toxicity, or at low concentrations can promote growth and survival of intracellular pathogens (64–67). Nevertheless, we did not observe any increase in NO due to IFN-γ stimulation, even using a more sensible technique (chemiluminescent method) (Figure 2). However, recent studies argued that THP-1 cells are not good NO producers because macrophage differentiation with PMA treatment can exhaust NO production capacity (68–71). We observed that NO varied over time when infected by CL (low to high) or by both strains (high to low) with opposite patterns, whereas G maintained stable NO levels in M0 and M(IFN-γ) (Figure 2). It is possible that the intracellular amastigotes of CL interfere with the NO pathway, and CL growth in COI cells is delayed compared to that in infection alone. Zalloum etal. (72) demonstrated that T. cruzi could suppress NO release in BALB/c peritoneal macrophages. Depending on the strain, this effect was only seen in cells in contact with trypomastigotes on supernatants, so the release of trypomastigotes by an infected cell could cause downstream signaling to reduce NO production. de Castro Neto etal. (37) also reviewed several parasite antioxidant factors that can counteract the oxidative stress induced by T. cruzi infection. The expression of these proteins and enzymes can be specific to each parasite cycle stage, as it has to adapt its energetic requirements and immune response evasion.
Monoinfections seemed to induce an anti-inflammatory profile of cytokines, with decreased levels of IFN-γ, IL-6, IL-1β, and TNF-α, and increased levels of TGF-β and IL-4. These findings demonstrated that the establishment of infection, although with variations according to stimuli, induced a favorable environment for parasite persistence. Co-infection tended to promote inflammation with heightened IFN-γ and IL-1β and diminished IL-10 levels when compared to monoinfections.
Full activation of STAT-1 requires phosphorylation of both serine 727 and tyrosine 701 to enhance the transcription of innate immunity-related genes (41, 73). The only condition in which we detected both phosphorylated residues was on COI, independent of previous stimuli. Phosphorylation of Y701 residue was detected in almost all infected M(IFN-γ). This activation is essential for dimerization, stability, nuclear translocation, and enhanced transcriptional effects of STATs (74, 75). Phosphorylation of S727 is required to fully activate transcriptional effects and for pathogen control (73, 76, 77). STAT-1 has also been described as an important transcriptional factor for procaspases (76), thus, impairment of its full active state can inhibit intrinsic signaling for apoptosis.
STAT-3 can have opposite effects of STAT-1 on the cell cycle and apoptosis. The constitutive activation of STAT-3 has been described in various cancer cells, including downregulation of apoptotic gene transcription and upregulation of proto-oncogene expression (78–82). In this context, as STAT-1pS (phosphorylation of serine residue) is required to induce apoptosis in cardiomyocytes after stress, and intracellular T. cruzi amastigotes inhibited this domain activation (41, 83) plus the increased STAT-3 activation at M(IFN-γ) infected with EAs of G or CL strains, we suggest that these two distinct mechanisms act synergistically to increase the survival of infected cells. T. cruzi induces the activation of STAT-3 in different cell types in response to IL-6. However, TGF-β can also phosphorylate STAT-3 and cause positive feedback in the transcription of both TGF-β and itself (43, 84–88), consistent with our results in M0 infected with CL TCTs (Figure 6).
Stahl et al. (85) demonstrated that although Y strain-infected cardiomyocytes had higher pSTAT-3 levels, cells were more apoptotic compared to uninfected controls. The authors discussed that the interaction and equilibrium of STAT-1 and STAT-3 activation can determine cell survival or death in this model of T. cruzi infection. Indeed, activated STAT-3 can promote SOCS3 (suppressor of cytokine signaling 3) which can inhibit IFN-γ/STAT-1 signaling in T. cruzi infection (84), and STATs-1 and -3 can form heterodimers that can target different DNA biding sites than their homodimers (82). Only some co-infection conditions resulted in fully activated STAT-1 and phosphorylated STAT-3.
IL-4 is a critical cytokine that controls antibody production, inflammation, and allergy. IL-4 has a main role in Th2 reprogramming and alternatively activated macrophages, demonstrating some opposing actions compared to IFN-stimulated cells, which generate Th1 and classically activated macrophages (reviewed by 89). IL-4 promotes the resistance of macrophages to T. cruzi (Tulahuén strain), while in vivo reduced resistance to infection has been described by the same strain (90, 91). The findings highlight the complex interplay of cytokines and cells in parasite control.
STAT-6 activated by IL-4 and TGF-β induce T cells to produce IL-9 from naïve or Th2 cells (89). IL-9 has recently been described as an important cytokine that controls fibrosis in cardiac tissues of mice infected with T. cruzi (92). Tarleton et al. (47) demonstrated that STAT-6 knockout mice were more resistant to T. cruzi chronic infection. The mice displayed less inflammation in cardiac or muscle tissues and no detection of parasites in situ. These findings indicate the essential role of STAT-6 in stimulating the Th2 response, which contributes to parasite persistence and disease severity. STAT-6 phosphorylation was observed in THP-1 derived macrophages infected with Toxoplasma gondii, with an anti-apoptotic effect on the cells (93).
The altered activation state of STATs seen in uninfected cells could be caused by vesicles or proteins directly secreted from the parasite, or extracellular vesicles (EVs) generated by infected macrophages (94–97). Factors secreted by T. cruzi can modulate uninfected cell responses in a paracrine manner to parasite invasion. EVs also can have a proinflammatory effect via the binding of Toll-like receptor-2 (95, 98, 99). However, different strains have surface variations. Thus, EVs can be very heterogeneous with altered quantity or content, which can interfere with parasite virulence and infectivity (99, 100). Interestingly, Cronemberger-Andrade et al. (95) evaluated EVs from THP-1 derived macrophages infected with T. cruzi strain Y and observed a reduction in STAT-1 and STAT-3 transcripts. These findings are consistent with our findings of reduced activation of these proteins in neighboring uninfected cells (Figure 5).
The role of STATs has been studied in several conditions, including other neglected tropical diseases (67, 76, 93, 101–103). The present study demonstrates that the activation state of macrophages elicits different interactions and responses from the host cells to T. cruzi. Both infective forms induce phosphorylation of the STAT residues evaluated within 48 hpi to prolong the survival of infected cells. The present data increase the understanding of T. cruzi infection, especially polyparasitism. The latter has not been extensively studied and related data can help to understand the variability in prognoses, symptoms, and effectiveness of treatments available for patients with CD.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Author contributions
MeO, CO, and RM conceived the study and designed the experiments. MeO, CO, CB, BS, and MaO performed the experiments. MeO, CO, and RM interpreted results and wrote the manuscript. All the authors contributed to the study and approved the submitted version of the manuscript.
Funding
This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, grants 2016/15000-4 and 2019/08933-2) Coordenação de Aperfeiçoamento Pessoal de Nível Superior (CAPES) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq).
Acknowledgments
We would like to thank Gabriela Dantas de Oliveira Vieira, who began this study, Dr. Milena Brunialti and Dr. Reinaldo Salomão for their kind support with flow cytometry equipment, and Wiley Editing Services, for the careful and thorough review of this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.1038332/full#supplementary-material
References
1. Brener Z. Biology of trypanosoma cruzi. Annu Rev Microbiol (1973) 27:347–82. doi: 10.1146/annurev.mi.27.100173.002023
2. Pérez-Molina JA, Molina I. Chagas disease. Lancet (Lond Engl) (2018) 391(10115):82–94. doi: 10.1016/S0140-6736(17)31612-4
3. Petray PB, Rottenberg ME, Bertot G, Corral RS, Diaz A, Orn A, et al. Effect of anti-gamma-interferon and anti-interleukin-4 administration on the resistance of mice against infection with reticulotropic and myotropic strains of trypanosoma cruzi. Immunol Lett (1993) 35(1):77–80. doi: 10.1016/0165-2478(93)90151-q
4. Zingales B, Andrade SG, Briones MR, Campbell DA, Chiari E, Fernandes O, et al. A new consensus for trypanosoma cruzi intraspecific nomenclature: Second revision meeting recommends TcI to TcVI. Memorias do Instituto Oswaldo Cruz (2009) 104(7):1051–4. doi: 10.1590/s0074-02762009000700021
5. Pan American Health Organization. Chagas disease (2020). Available at: https://www.paho.org/en/topics/chagas-disease (Accessed June, 2022).
6. World Health Organization. Neglected tropical diseases (2022). Available at: https://www.who.int/health-topics/neglected-tropical-diseases (Accessed June, 2022).
7. De Souza W. Basic cell biology of Trypanosoma cruzi. Curr Pharm design (2002) 8(4):269–85. doi: 10.2174/1381612023396276
8. Lima FM, Oliveira P, Mortara RA, Silveira JF, Bahia D. The challenge of chagas' disease: Has the human pathogen, Trypanosoma cruzi, learned how to modulate signaling events to subvert host cells? New Biotechnol (2010) 27(6):837–43. doi: 10.1016/j.nbt.2010.02.003
9. Ferreira ER, Bonfim-Melo A, Mortara RA, Bahia D. Trypanosoma cruzi extracellular amastigotes and host cell signaling: More pieces to the puzzle. Front Immunol (2012) 3:363. doi: 10.3389/fimmu.2012.00363
10. Zingales B. Trypanosoma cruzi genetic diversity: Something new for something known about chagas disease manifestations, serodiagnosis and drug sensitivity. Acta tropica (2018) 184:38–52. doi: 10.1016/j.actatropica.2017.09.017
11. Bosseno MF, Telleria J, Vargas F, Yaksic N, Noireau F, Morin A, et al. Trypanosoma cruzi: Study of the distribution of two widespread clonal genotypes in Bolivian triatoma infestans vectors shows a high frequency of mixed infections. Exp Parasitol (1996) 83(3):275–82. doi: 10.1006/expr.1996.0075
12. Araújo CA, Waniek PJ, Jansen AM. TcI/TcII co-infection can enhance trypanosoma cruzi growth in rhodnius prolixus. Parasites Vectors (2014) 7:94. doi: 10.1186/1756-3305-7-94
13. Steindel M, Kramer Pacheco L, Scholl D, Soares M, de Moraes MH, Eger I, et al. Characterization of trypanosoma cruzi isolated from humans, vectors, and animal reservoirs following an outbreak of acute human chagas disease in Santa catarina state, Brazil. Diagn Microbiol Infect Dis (2008) 60(1):25–32. doi: 10.1016/j.diagmicrobio.2007.07.016
14. Monje-Rumi M M, Brandán C P, Ragone P G, Tomasini N, Lauthier J J, Alberti D'Amato A M, et al. Trypanosoma cruzi diversity in the Gran Chaco: mixed infections and differential host distribution of TcV and TcVI. Infection, genetics and evolution : journal of molecular epidemiology and evolutionary genetics in infectious diseases, (2015) 29, 53–59. doi: 10.1016/j.meegid.2014.11.001
15. Martinez-Perez A, Poveda C, Ramírez JD, Norman F, Gironés N, Guhl F, et al. Prevalence of trypanosoma cruzi's discrete typing units in a cohort of Latin American migrants in Spain. Acta Tropica (2016) 157:145–50. doi: 10.1016/j.actatropica.2016.01.032
16. Prescilla-Ledezma A, Blandon R, Schijman AG, Benatar A, Saldaña A, Osuna A, et al. Mixed infections by different Trypanosoma cruzi discrete typing units among Chagas disease patients in an endemic community in Panama. PLoS ONE, (2020) 15(11): e0241921. doi: 10.1371/journal.pone.0241921
17. Sales-Campos H, Kappel HB, Andrade CP, Lima TP, de Castilho A, Giraldo LE, et al. Trypanosoma cruzi DTU TcII presents higher blood parasitism than DTU TcI in an experimental model of mixed infection. Acta Parasitologica (2015) 60(3):435–41. doi: 10.1515/ap-2015-0060
18. Perez CJ, Thompson R, Keatley SK, Walsh AL, Lymbery AJ. The effect of reinfection and mixed trypanosoma cruzi infections on disease progression in mice. Acta tropica (2018) 178:107–14. doi: 10.1016/j.actatropica.2017.11.002
19. Mateus J, Nocua P, Lasso P, López MC, Thomas MC, Egui A, et al. CD8+ T cell response quality is related to parasite control in an animal model of single and mixed chronic Trypanosoma cruzi infections. Front Cell Infect Microbiol (2021) 11:723121. doi: 10.3389/fcimb.2021.723121
20. Strauss M, Lo Presti MS, Ramírez J C, Bazán P C, Velázquez López D A, Báez A L, et al. Differential tissue distribution of discrete typing units after drug combination therapy in experimental Trypanosoma cruzi mixed infection. Parasitology (2021) 148(13):1595–601. doi: 10.1017/S0031182021001281
21. Tomasini N, Ragone PG, Gourbière S, Aparicio JP, Diosque P. Epidemiological modeling of trypanosoma cruzi: Low stercorarian transmission and failure of host adaptive immunity explain the frequency of mixed infections in humans. PloS Comput Biol (2017) 13(5):e1005532. doi: 10.1371/journal.pcbi.1005532
22. Macedo AM, Segatto M. Implications of trypanosoma cruzi intraspecific diversity in the pathogenesis of chagas disease. Am Trypanosomiasis (2010), 489–522. doi: 10.1016/B978-0-12-384876-5.00020-4
23. De Araújo-Jorge TC. The biology of trypanosoma cruzi-macrophage interaction. Memorias do Instituto Oswaldo Cruz (1989) 84(4):441–62. doi: 10.1590/s0074-02761989000400001
24. Ribeiro-Gomes FL, Lopes MF, DosReis GA. Negative signaling and modulation of macrophage function in trypanosoma cruzi infection in madame curie bioscience database. (Austin TX: Landes Bioscience) (2013). Bookshelf ID: NBK6246, and URL https://www.ncbi.nlm.nih.gov/books/NBK6246/.
25. Cassetta L, Cassol E, Poli G. Macrophage polarization in health and disease. TheScientificWorldJournal (2011) 11:2391–402. doi: 10.1100/2011/213962
26. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000prime Rep (2014) 6:13. doi: 10.12703/P6-13
27. Atri C, Guerfali FZ, Laouini D. Role of human macrophage polarization in inflammation during infectious diseases. Int J Mol Sci (2018) 19(6):1801. doi: 10.3390/ijms19061801
28. Locati M, Curtale G, Mantovani A. Diversity, mechanisms, and significance of macrophage plasticity. Annu Rev Pathol (2020) 15:123–47. doi: 10.1146/annurev-pathmechdis-012418-012718
29. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol (2008) 8(12):958–69. doi: 10.1038/nri2448
30. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol (2002) 23(11):549–55. doi: 10.1016/s1471-4906(02)02302-5
31. Abrahamsohn IA, Coffman RL. Trypanosoma cruzi: IL-10, TNF, IFN-gamma, and IL-12 regulate innate and acquired immunity to infection. Exp Parasitol (1996) 84(2):231–44. doi: 10.1006/expr.1996.0109
32. Abrahamsohn IA, da Silva AP, Coffman RL. Effects of interleukin-4 deprivation and treatment on resistance to trypanosoma cruzi. Infection Immun (2000) 68(4):1975–9. doi: 10.1128/IAI.68.4.1975-1979.2000
33. Cunha-Neto E, Nogueira LG, Teixeira PC, Ramasawmy R, Drigo SA, Goldberg AC, et al. Immunological and non-immunological effects of cytokines and chemokines in the pathogenesis of chronic chagas disease cardiomyopathy. Memorias do Instituto Oswaldo Cruz (2009) 104(Suppl 1):252–8. doi: 10.1590/s0074-02762009000900032
34. Rodrigues AA, Saosa JS, da Silva GK, Martins FA, da Silva AA, Souza Neto CP, et al. IFN-γ plays a unique role in protection against low virulent trypanosoma cruzi strain. PloS Negl Trop Dis (2012) 6(4):e1598. doi: 10.1371/journal.pntd.0001598
35. Piacenza L, Peluffo G, Alvarez MN, Martínez A, Radi R. Trypanosoma cruzi antioxidant enzymes as virulence factors in chagas disease. Antioxidants Redox Signaling (2013) 19(7):723–34. doi: 10.1089/ars.2012.4618
36. Mesías AC, Garg NJ, Zago MP. Redox balance keepers and possible cell functions managed by redox homeostasis in Trypanosoma cruzi. Front Cell Infect Microbiol (2019) 9:435. doi: 10.3389/fcimb.2019.00435
37. de Castro Neto AL, da Silveira JF, Mortara RA. Comparative analysis of virulence mechanisms of trypanosomatids pathogenic to humans. Front Cell infection Microbiol (2021) 11:669079. doi: 10.3389/fcimb.2021.669079
38. Rawlings JS, Rosler KM, Harrison DA. The JAK/STAT signaling pathway. J Cell Sci (2004) 117(Pt 8):1281–3. doi: 10.1242/jcs.00963
39. Villarino AV, Kanno Y, O'Shea JJ. Mechanisms and consequences of jak-STAT signaling in the immune system. Nat Immunol (2017) 18(4):374–84. doi: 10.1038/ni.3691
40. Seif F, Khoshmirsafa M, Aazami H, Mohsenzadegan M, Sedighi G, Bahar M. The role of JAK-STAT signaling pathway and its regulators in the fate of T helper cells. Cell communication Signaling CCS (2017) 15(1):23. doi: 10.1186/s12964-017-0177-y
41. Stahl P, Ruppert V, Schwarz RT, Meyer T. Trypanosoma cruzi evades the protective role of interferon-gamma-signaling in parasite-infected cells. PloS One (2014) 9(10):e110512. doi: 10.1371/journal.pone.0110512
42. Kulkarni MM, Varikuti S, Terrazas C, Kimble JL, Satoskar AR, McGwire BS. Signal transducer and activator of transcription 1 (STAT-1) plays a critical role in control of trypanosoma cruzi infection. Immunology (2015) 145(2):225–31. doi: 10.1111/imm.12438
43. Ponce NE, Carrera-Silva EA, Pellegrini AV, Cazorla SI, Malchiodi EL, Lima AP, et al. Trypanosoma cruzi, the causative agent of chagas disease, modulates interleukin-6-induced STAT3 phosphorylation via gp130 cleavage in different host cells. Biochim Biophys Acta (2013) 1832(3):485–94. doi: 10.1016/j.bbadis.2012.12.003
44. de Souza EM, Araújo-Jorge TC, Bailly C, Lansiaux A, Batista MM, Oliveira GM, et al. Host and parasite apoptosis following trypanosoma cruzi infection in in vitro and in vivo models. Cell Tissue Res (2003) 314(2):223–35. doi: 10.1007/s00441-003-0782-5
45. Wang Q, Ni H, Lan L, Wei X, Xiang R, Wang Y. Fra-1 protooncogene regulates IL-6 expression in macrophages and promotes the generation of M2d macrophages. Cell Res (2010) 20(6):701–12. doi: 10.1038/cr.2010.52
46. Forbes LR, Milner J, Haddad E. Signal transducer and activator of transcription 3: a year in review. Current opinion in hematology, (2016) 23(1): 23–27. doi: 10.1097/MOH.0000000000000206
47. Tarleton RL, Grusby MJ, Zhang L. Increased susceptibility of Stat4-deficient and enhanced resistance in Stat6-deficient mice to infection with trypanosoma cruzi. J Immunol (Baltimore Md. 1950) (2000) 165(3):1520–5. doi: 10.4049/jimmunol.165.3.1520
48. Goenka S, Kaplan MH. Transcriptional regulation by STAT6. Immunologic Res (2011) 50(1):87–96. doi: 10.1007/s12026-011-8205-2
49. Yoshida N. Surface antigens of metacyclic trypomastigotes of trypanosoma cruzi. Infection Immun (1983) 40(2):836–9. doi: 10.1128/iai.40.2.836-839.1983
50. Brener Z, Chiari E. Variações morfológicas observadas em diferentes amostras de trypanosoma cruzi. Rev Inst Med Trop São Paulo (1963) 5:220–4. Available at: https://www.imt.usp.br/wp-content/uploads/revista/vol05/220-224.pdf.
51. Cruz MC, Souza-Melo N, da Silva CV, Darocha WD, Bahia D, Araújo PR, et al. Trypanosoma cruzi: Role of δ-amastin on extracellular amastigote cell invasion and differentiation. PloS One (2012) 7(12):e51804. doi: 10.1371/journal.pone.0051804
52. Ley V, Andrews NW, Robbins ES, Nussenzweig V. Amastigotes of trypanosoma cruzi sustain an infective cycle in mammalian cells. J Exp Med (1988) 168(2):649–59. doi: 10.1084/jem.168.2.649
53. Mortara RA. Trypanosoma cruzi: amastigotes and trypomastigotes interact with different structures on the surface of HeLa cells. Exp Parasitol (1991) 73(1):1–14. doi: 10.1016/0014-4894(91)90002-e
54. Tomlinson S, Vandekerckhove F, Frevert U, Nussenzweig V. The induction of trypanosoma cruzi trypomastigote to amastigote transformation by low pH. Parasitology (1995) 110(Pt 5):547–54. doi: 10.1017/s0031182000065264
55. Macedo AM, Pena SD. Genetic variability of trypanosoma cruzi:Implications for the pathogenesis of chagas disease. Parasitol Today (Personal ed.) (1998) 14(3):119–24. doi: 10.1016/s0169-4758(97)01179-4
56. Manoel-Caetano F, Silva AE. Implications of genetic variability of trypanosoma cruzi for the pathogenesis of chagas disease. Cadernos saude publica (2007) 23(10):2263–74. doi: 10.1590/s0102-311x2007001000002
57. Mortara RA, Andreoli WK, Taniwaki NN, Fernandes AB, Silva CV, Fernandes MC, et al. Mammalian cell invasion and intracellular trafficking by trypanosoma cruzi infective forms. Anais da Academia Bras Cienc (2005) 77(1):77–94. doi: 10.1590/s0001-37652005000100006
58. Vaena de Avalos S, Blader I.J, Fisher M, Boothroyd JC, Burleigh BA. Immediate/early response to trypanosoma cruzi infection involves minimal modulation of host cell transcription. J Biol Chem (2002) 277(1):639–44. doi: 10.1074/jbc.M109037200
59. Abrahamsohn IA. Cytokines in innate and acquired immunity to trypanosoma cruzi infection. Braz J Med Biol Res = Rev Bras pesquisas medicas e biologicas (1998) 31(1):117–21. doi: 10.1590/s0100-879x1998000100015
60. Hölscher C, Köhler G, Müller U, Mossmann H, Schaub GA, Brombacher F. Defective nitric oxide effector functions lead to extreme susceptibility of trypanosoma cruzi-infected mice deficient in gamma interferon receptor or inducible nitric oxide synthase. Infection Immun (1998) 66(3):1208–15. doi: 10.1128/IAI.66.3.1208-1215.1998
61. Laucella SA, Postan M, Martin D, Hubby Fralish B, Albareda MC, Alvarez MG, et al. Frequency of interferon- gamma -producing T cells specific for trypanosoma cruzi inversely correlates with disease severity in chronic human chagas disease. J Infect Dis (2004) 189(5):909–18. doi: 10.1086/381682
62. Boscardin SB, Torrecilhas AC, Manarin R, Revelli S, Rey EG, Tonelli RR, et al. Chagas' disease: An update on immune mechanisms and therapeutic strategies. J Cell Mol Med (2010) 14(6B):1373–84. doi: 10.1111/j.1582-4934.2010.01007.x
63. MacMicking J, Xie QW, Nathan C. Nitric oxide and macrophage function. Annu Rev Immunol (1997) 15:323–50. doi: 10.1146/annurev.immunol.15.1.323
64. Gutierrez FR, Mineo TW, Pavanelli WR, Guedes PM, Silva JS. The effects of nitric oxide on the immune system during trypanosoma cruzi infection. Memorias do Instituto Oswaldo Cruz (2009) 104(Suppl 1):236–4510.1590/s0074–02762009000900030. doi: 10.1590/S0074-02762009000900030
65. Machado FS, Martins GA, Aliberti JC, Mestriner FL, Cunha FQ, Silva JS. Trypanosoma cruzi-infected cardiomyocytes produce chemokines and cytokines that trigger potent nitric oxide-dependent trypanocidal activity. Circulation (2000) 102(24):3003–8. doi: 10.1161/01.cir.102.24.3003
66. Cummings KL, Tarleton RL. Inducible nitric oxide synthase is not essential for control of trypanosoma cruzi infection in mice. Infection Immun (2004) 72(7):4081–9. doi: 10.1128/IAI.72.7.4081-4089.2004
67. El Kasmi KC, Qualls JE, Pesce JT, Smith AM, Thompson RW, Henao-Tamayo M, et al. Toll-like receptor-induced arginase 1 in macrophages thwarts effective immunity against intracellular pathogens. Nat Immunol (2008) 9(12):1399–406. doi: 10.1038/ni.1671
68. Park EK, Jung HS, Yang HI, Yoo MC, Kim C, Kim KS. Optimized THP-1 differentiation is required for the detection of responses to weak stimuli. Inflammation Res Off J Eur Histamine Res Soc (2007) 56(1):45–50. doi: 10.1007/s00011-007-6115-5
69. van den Bogaart E, Mens PF, Adams ER, Grobusch MP, Schallig H. Phagocytosis of hemozoin by RAW 264.7 cells, but not THP-1 cells, promotes infection by leishmania donovani with a nitric oxide-independent mechanism. Parasitol Int (2017) 66(2):196–206. doi: 10.1016/j.parint.2016.09.003
70. Chang YY, Lu CW, Jean WH, Shieh JS, Lin TY. Phorbol myristate acetate induces differentiation of THP-1 cells in a nitric oxide-dependent manner. Nitric oxide: Biol Chem (2021) 109-110:33–41. doi: 10.1016/j.niox.2021.02.002
71. Ozleyen A, Yilmaz YB, Tumer TB. Dataset on the differentiation of THP-1 monocytes to LPS inducible adherent macrophages and their capacity for NO/iNOS signaling. Data Brief (2021) 35:106786. doi: 10.1016/j.dib.2021.106786
72. Zalloum L, Lala ER, Moreira NM, Silveira TG, Dalálio MM, Toledo MJ, et al. Induction of phagocytic activity and nitric-oxide production in natural populations of trypanosoma cruzi I and II from the state of paraná, Brazil. Rev do Instituto Medicina Trop Sao Paulo (2011) 53(5):247–53. doi: 10.1590/s0036-46652011000500002
73. Varinou L, Ramsauer K, Karaghiosoff M, Kolbe T, Pfeffer K, Müller M, et al. Phosphorylation of the Stat1 transactivation domain is required for full-fledged IFN-gamma-dependent innate immunity. Immunity (2003) 19(6):793–802. doi: 10.1016/s1074-7613(03)00322-4
74. Sadzak I, Schiff M, Gattermeier I, Glinitzer R, Sauer I, Saalmüller A, et al. Recruitment of Stat1 to chromatin is required for interferon-induced serine phosphorylation of Stat1 transactivation domain. Proc Natl Acad Sci United States America (2008) 105(26):8944–9. doi: 10.1073/pnas.0801794105
75. Wenta N, Strauss H, Meyer S, Vinkemeier U. Tyrosine phosphorylation regulates the partitioning of STAT1 between different dimer conformations. Proc Natl Acad Sci United States America (2008) 105(27):9238–43. doi: 10.1073/pnas.0802130105
76. Najjar I, Fagard R. STAT1 and pathogens, not a friendly relationship. Biochimie (2010) 92(5):425–44. doi: 10.1016/j.biochi.2010.02.009
77. Majoros A, Platanitis E, Szappanos D, Cheon H, Vogl C, Shukla P, et al. Response to interferons and antibacterial innate immunity in the absence of tyrosine-phosphorylated STAT1. EMBO reports, (2016) 17(3): 367–382. doi: 10.15252/embr.201540726
78. Grandis JR, Drenning SD, Zeng Q, Watkins SC, Melhem MF, Endo S, et al. Constitutive activation of Stat3 signaling abrogates apoptosis in squamous cell carcinogenesis in vivo. Proc Natl Acad Sci USA (2000) 97(8):4227–32. doi: 10.1073/pnas.97.8.4227
79. Huang M, Page C, Reynolds RK, Lin J. Constitutive activation of stat 3 oncogene product in human ovarian carcinoma cells. Gynecologic Oncol (2000) 79(1):67–73. doi: 10.1006/gyno.2000.5931
80. Horiguchi A, Oya M, Marumo K, Murai M. STAT3, but not ERKs, mediates the IL-6-induced proliferation of renal cancer cells, ACHN and 769P. Kidney Int (2002) 61(3):926–38. doi: 10.1046/j.1523-1755.2002.00206.x
81. Stephanou A, Latchman DS. Opposing actions of STAT-1 and STAT-3. Growth factors (Chur Switzerland) (2005) 23(3):177–82. doi: 10.1080/08977190500178745
82. You L, Wang Z, Li H, Shou J, Jing Z, Xie J, et al. The role of STAT3 in autophagy. Autophagy (2015) 11(5):729–39. doi: 10.1080/15548627.2015.1017192
83. Stephanou A, Scarabelli TM, Brar BK, Nakanishi Y, Matsumura M, Knight RA, et al. Induction of apoptosis and fas receptor/Fas ligand expression by ischemia/reperfusion in cardiac myocytes requires serine 727 of the STAT-1 transcription factor but not tyrosine 701. J Biol Chem (2001) 276(30):28340–7. doi: 10.1074/jbc.M101177200
84. Hovsepian E, Penas F, Siffo S, Mirkin GA, Goren N.B, N. B. IL-10 inhibits the NF-κB and ERK/MAPK-mediated production of pro-inflammatory mediators by up-regulation of SOCS-3 in trypanosoma cruzi-infected cardiomyocytes. PloS One (2013) 8(11):e79445. doi: 10.1371/journal.pone.0079445
85. Stahl P, Ruppert V, Meyer T, Schmidt J, Campos MA, Gazzinelli RT, et al. Trypomastigotes and amastigotes of trypanosoma cruzi induce apoptosis and STAT3 activation in cardiomyocytes in vitro. Apoptosis an Int J programmed Cell Death (2013) 18(6):653–63. doi: 10.1007/s10495-013-0822-x
86. Pedroza M, To S, Assassi S, Wu M, Tweardy D, Agarwal SK. Role of STAT3 in skin fibrosis and transforming growth factor beta signalling. Rheumatol (Oxford England) (2018) 57(10):1838–50. doi: 10.1093/rheumatology/kex347
87. Hoffman KA, Villar MJ, Poveda C, Bottazzi ME, Hotez PJ, Tweardy DJ, et al. Signal transducer and activator of transcription-3 modulation of cardiac pathology in chronic chagasic cardiomyopathy. Front Cell Infect Microbiol (2021) 11:708325. doi: 10.3389/fcimb.2021.708325
88. Lattanzi R, Maftei D, Fullone MR, Miele R. Trypanosoma cruzi trans-sialidase induces STAT3 and ERK activation by prokineticin receptor 2 binding. Cell Biochem Funct (2021) 39(2):326–34. doi: 10.1002/cbf.3586
89. Keegan AD, Leonard WJ, Zhu J. Recent advances in understanding the role of IL-4 signaling. Faculty reviews, (2021) 10: 71. doi: 10.12703/r/10-71
90. Wirth JJ, Kierszenbaum F, Zlotnik A. Effects of IL-4 on macrophage functions: increased uptake and killing of a protozoan parasite (Trypanosoma cruzi). Immunology (1989) 66(2):296–301.
91. Hiyama K, Hamano S, Nakamura T, Nomoto K, Tada I. IL-4 reduces resistance of mice to trypanosoma cruzi infection. Parasitol Res (2001) 87(4):269–74. doi: 10.1007/pl00008577
92. Silva N, Orikaza CM, de Santana FR, Dos Santos LA, Salu BR, Oliva M, et al. Interleukin-9 in immunopathology of Trypanosoma cruzi experimental infection. Front Cell infection Microbiol (2021) 11:756521. doi: 10.3389/fcimb.2021.756521
93. Song KJ, Ahn HJ, Nam HW. Anti-apoptotic effects of SERPIN B3 and B4 via STAT6 activation in macrophages after infection with toxoplasma gondii. Korean J Parasitol (2012) 50(1):1–6. doi: 10.3347/kjp.2012.50.1.1
94. da Silva CV, Kawashita SY, Probst CM, Dallagiovanna B, Cruz MC, da Silva EA, et al. Characterization of a 21kDa protein from trypanosoma cruzi associated with mammalian cell invasion. Microbes Infect (2009) 11(5):563–70. doi: 10.1016/j.micinf.2009.03.007
95. Cronemberger-Andrade A, Xander P, Soares RP, Pessoa NL, Campos MA, Ellis CC, et al. Trypanosoma cruzi-infected human macrophages shed proinflammatory extracellular vesicles that enhance host-cell invasion via toll-like receptor 2. Front Cell Infect Microbiol (2020) 10:99. doi: 10.3389/fcimb.2020.00099
96. Torrecilhas AC, Soares RP, Schenkman S, Fernández-Prada C, Olivier M. Extracellular vesicles in trypanosomatids: Host cell communication. Front Cell infection Microbiol (2020) 10:602502. doi: 10.3389/fcimb.2020.602502
97. D'Avila H, de Souza NP, Albertoni A, Campos LC, Rampinelli PG, Correa JR, et al. Impact of the extracellular vesicles derived from Trypanosoma cruzi: A paradox in host response and lipid metabolism modulation. Front Cell Infect Microbiol (2021) 11:768124. doi: 10.3389/fcimb.2021.768124
98. de Pablos Torró LM, Retana Moreira L, Osuna A. Extracellular vesicles in chagas disease: A new passenger for an old disease. Front Microbiol (2018) 9:1190. doi: 10.3389/fmicb.2018.01190
99. Florentino P, Real F, Orikaza CM, da Cunha J, Vitorino F, Cordero EM, et al. A carbohydrate moiety of secreted stage-specific glycoprotein 4 participates in host cell invasion by Trypanosoma cruzi extracellular amastigotes. Front Microbiol (2018) 9:693. doi: 10.3389/fmicb.2018.00693
100. Ribeiro KS, Vasconcellos CI, Soares RP, Mendes MT, Ellis CC, Aguilera-Flores M, et al. Proteomic analysis reveals different composition of extracellular vesicles released by two Trypanosoma cruzi strains associated with their distinct interaction with host cells. J extracellular vesicles (2018) 7(1):1463779. doi: 10.1080/20013078.2018.1463779
101. Osorio EY, Zhao W, Espitia C, Saldarriaga O, Hawel L, Byus CV, et al. Progressive visceral leishmaniasis is driven by dominant parasite-induced STAT6 activation and STAT6-dependent host arginase 1 expression. PloS Pathog (2012) 8(1):e1002417. doi: 10.1371/journal.ppat.1002417
102. Faz-López B, Ledesma-Soto Y, Romero-Sánchez Y, Calleja E, Martínez-Labat P, Terrazas LI. Signal transducer and activator of transcription factor 6 signaling contributes to control host lung pathology but favors susceptibility against toxocara canis infection. BioMed Res Int (2013), 2013: 696343. doi: 10.1155/2013/696343
Keywords: STAT-1, STAT-3, STAT-6, Trypanosoma cruzi, THP-1, macrophages, co-infection
Citation: Oliveira MM, Bonturi CR, Salu BR, Oliva MLV, Mortara RA and Orikaza CM (2022) Modulation of STAT-1, STAT-3, and STAT-6 activities in THP-1 derived macrophages infected with two Trypanosoma cruzi strains. Front. Immunol. 13:1038332. doi: 10.3389/fimmu.2022.1038332
Received: 06 September 2022; Accepted: 04 October 2022;
Published: 26 October 2022.
Edited by:
Diana Bahia, Federal University of Minas Gerais, BrazilReviewed by:
Gonzalo Cabrera, University of Chile, ChileRossella Miele, Sapienza University of Rome, Italy
Copyright © 2022 Oliveira, Bonturi, Salu, Oliva, Mortara and Orikaza. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cristina Mary Orikaza, Y20ub3Jpa2F6YUB1bmlmZXNwLmJy