 Lalitha Palanivelu
Lalitha Palanivelu Ching-Hsuan Liu2,3,4†
Ching-Hsuan Liu2,3,4† Liang-Tzung Lin
Liang-Tzung Lin- 1International Ph.D. Program in Medicine, College of Medicine, Taipei Medical University, Taipei, Taiwan
- 2Department of Microbiology and Immunology, School of Medicine, College of Medicine, Taipei Medical University, Taipei, Taiwan
- 3Graduate Institute of Medical Sciences, College of Medicine, Taipei Medical University, Taipei, Taiwan
- 4Department of Microbiology & Immunology, Dalhousie University, Halifax, NS, Canada
According to the World Health Organization, cancer is one of the leading global health concerns, causing nearly 10 million deaths in 2020. While classical chemotherapeutics produce strong cytotoxicity on cancer cells, they carry limitations of drug resistance and off-target effects and sometimes fail to elicit adequate antitumor protection against tumor relapse. Additionally, most cancer cells have developed various ways to escape immune surveillance. Nevertheless, novel anticancer strategies such as oncolytic viro-immunotherapy can trigger immunogenic cell death (ICD), which can quickly grasp the attention of the host defense machinery, resulting in an ensuing antitumor immune response. Specifically, oncolytic viruses (OVs) can infect and destroy targeted cancer cells and stimulate the immune system by exposing pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) to promote inflammatory reactions, and concomitantly prime and induce antitumor immunity by the release of neoantigens from the damaged cancer cells. Thus, OVs can serve as a novel system to sensitize tumor cells for promising immunotherapies. This review discusses the concept of ICD in cancer, centralizing ICD-associated danger signals and their consequence in antitumor responses and ICD induced by OVs. We also shed light on the potential strategies to enhance the immunogenicity of OVs, including the use of genetically modified OVs and their combination with ICD-enhancing agents, which are helpful as forthcoming anticancer regimens.
1 Introduction
Cancers are the derivative of the genetic and epigenetic transformations leading to cell immortality and the generation of neoantigens. Neoantigens are newly formed proteins sourced by the mutated tumor-specific genes or tumor-associated viruses integrated into the genome, which previously have not been recognized by the host immune machinery (1). Furthermore, previous studies illustrated that the antitumor immune response provoked by tumor cell death could lead to prolonged therapeutic effects (2). The induced antitumor immunity is attributed to the alarming cellular molecules released by the dying cells in the tumor microenvironment (TME), and such danger cues are broadly referred to as “damage-associated molecular patterns” (DAMPs) (2, 3). Collectively, the immunogenicity conferred by tumor cell death depends on both antigenicity induced by neoantigen epitopes and adjuvanticity produced by specific DAMPs (4). This recipe for cell demise is recognized as “immunogenic cell death” (ICD).
Since the birth of the concept of ICD, the mode of viewing cancer therapy has drastically deviated in a different direction. ICD gained recognition as a host immunogenicity enhancer and initiator of lasting tumor-specific response as it recruits dendritic cells (DCs) and DC precursors. For successful immunogenicity, the danger signals’ production and exposure are critical for mediating cellular stress. However, cancer cells have designed various strategies to abrogate danger signals and dodge immune surveillance through loss of antigenicity or immunogenicity and via creating an immunosuppressive TME. Nonetheless, such immune evasion mechanisms mediated by cancer cells can be reversed by the evolving ICD inducers. Anticancer treatment strategies such as certain chemotherapeutics (e.g. the anthracycline family, oxaliplatin, and bortezomib) (5), radiotherapy (6), and photodynamic therapy (7) have been demonstrated to trigger ICD. A new category of ICD inducers—OVs can induce ICD specifically in targeted tumors. Microbial components from OVs can serve as pathogen-associated molecular patterns (PAMPs), which specific receptors can recognize in immune cells (8). Consequently, the discovery of OVs helped approach the viruses with a distinct perspective rather than viewing them as infectious disease-causing agents. With the advent of recombinant viruses, it is possible to engineer and develop attenuated viruses that retain their oncolytic ability while improving their tumor specificity (9, 10). Furthermore, the OVs combinatory approach with potent therapeutics has led to synergistic augmentations in ICD-induction and antitumor responses in several cancer types in vitro and in vivo. This review sheds light on the concept of ICD and summarizes the rationale and insights on preclinical research that led to the clinical trials of ICD-inducing OVs in cancer treatment.
2 Canonical mechanism of ICD: danger signaling and its consequence
During the ICD, dying cells secrete and express a broad panel of signature molecules on the cell surface in a defined temporal sequence (3). Recognition of these molecules, i.e., ICD-associated DAMPs, by specific pattern recognition receptors (PRRs) expressed on DCs ultimately results in the activation of both innate and adaptive immune responses (11).
2.1 Calreticulin
Calreticulin (CRT) is one of the early DAMPs that translocate from the ER to cell surface (ecto-CRT) during the pre-apoptotic stage by complexing with the disulfide isomerase ERp57 (12). Ecto-CRT facilitates the phagocytosis of antigens by DCs, thus triggering immune responses (13). The receptor for CRT is ambiguous; however, studies show it might act on CD91, a low-density lipoprotein receptor-related protein 1 (LRP1), scavenger receptors SREC-1 and SR-A, CD40 ligand, TRAIL, and Fas ligand present on innate immune cells, such as APCs (14–17). On the other hand, the phagocytosis mediating ability of CRT is counterbalanced by the robust expression of CD47, a CRT antagonist which serves as a ‘do not eat me signal’ (18). Besides, reconstituting CRT-deficient cancer cells with exogenous recombinant CRT could revive their susceptibility to phagocyte clearance and reverse their resistance to ICD inducers (19). Furthermore, in non-small cell lung cancer patients, neoplastic cells manifesting high CRT levels showed a favorable prognosis and enhanced accumulation of immune cells (20). Therefore, CRT is an essential antitumor immune booster as it plays a vital part in promoting a tumor-specific immune response.
2.2 Adenosine-triphosphate
The release of ATP from dying cells is one of the major hallmarks of ICD. Multiple pathways are required for ICD-associated ATP secretion, including autophagy, lysosomal exocytosis, apoptosis, membrane blebbing, and plasma membrane permeabilization (21). ATP released from apoptotic cells binds to the P2Y2 purinergic receptor on monocytes and macrophages to facilitate clearance of apoptotic cells (22). The released ATP also binds to the P2X7 purinergic receptor on DCs, triggering NALP3-ASC-inflammasome formation and discharge of IL-1β, which is essential for T cell priming (23). Of note, human breast cancer patients with the P2RX7 loss-of-function allele were shown to have shorter metastatic disease-free survival (23), suggesting the important role of ATP secretion in ICD induction.
2.3 High mobility group box 1
When cells are undergoing ICD, high mobility group box 1 (HMGB1) is freed from the nucleus and translocated to the extracellular space through the permeabilization of both the nuclear lamina and the plasma membrane (11). The extracellular HMGB1 triggers DC maturation by activating TLR4 expressed on immature DCs. Once TLR recognizes HMGB1, it induces the transcription of the adaptor molecule myeloid differentiation response gene (MyD88). Reports suggest that the TLR4/MyD88 cascade inhibits lysosome and phagosome fusion which causes the phagocytic vesicle processing in DCs and expedites the uptake of tumor antigens by DCs (24–27). HMGB1, in its redox state, can elicit diverse immunomodulatory activities, such as a chemoattractant (reduced HMGB1) and a proinflammatory cytokine-mediator (oxidized HMGB1 possessing disulfide bond) (28, 29). However, some former studies proposed that HMBG1 may bind to T cell immunoglobulin and mucin domain-containing protein 3 (TIM-3) on DCs to mediate immunosuppressive function (30) or augment the immunosuppressive activities of myeloid-derived suppressor cells (MDSCs) (31). Therefore, its implication in cancer treatment is yet to be fully understood.
2.4 Heat shock proteins
Heat shock proteins (HSPs) are molecular chaperones generally located in the cytosol, ER, mitochondria, and nucleus and are crucial for their role in protein folding, refolding, and degeneration (32). Some HSPs including HSP70 and HSP90 were found to be released after cell death and act as alert stress signals to prime the immune cells (33). Extracellular HSPs serve as DAMPs recognized by the pattern recognition receptors (PPRs), such as TLR2 or TLR4, and stimulate APCs by enhanced expression of costimulatory molecules, MHC molecules, and proinflammatory and Th-1 cytokines (34, 35). In addition, HSPs can bind to distinct DC receptors and encourage the cross-presentation of the processed antigenic peptides. Typically, they act via scavenger receptors, activating intrinsic proinflammatory cascades like NF-kB, mitogen-activated protein kinases (MAPK/ERK), or associate with TLRs (36).
2.5 Annexin A1
Annexin A1 (ANXA1) belongs to the Ca2+-dependent phospholipid-binding protein family and plays a crucial part in resolving inflammatory responses (37). During ICD, secreted or surface-exposed ANXA1 can assist DC activity. Vacchelli et al. demonstrated that the interaction between ANXA1 and its receptor, formyl peptide receptor 1 (FPR1), on DCs is essential to direct DCs to the dying cancer cells. Tumors lacking ANXA1 or hosts lacking FPR1 showed loss of immunotherapeutic outcome in anthracycline and oxaliplatin-treated mice (38). Besides, a loss-of-function polymorphic mutation in FPR1 reduced the overall survival and progression-free survival in breast and colorectal cancer patients receiving anthracycline or oxaliplatin-based chemotherapy (38). These results suggested that ANXA1 in tumor cells and the presence of its receptor (FPR1) in hosts are essential to initiate antitumor T cell immunity.
2.6 Cellular nucleic acids, cytokines, chemokines, and translation factors
Cellular RNA, such as mRNA, released from damaged cells can promote the production of type I interferons (IFNs) and proinflammatory cytokines via the TLR3 signaling cascade (39). Therefore, uptake and recognition of extracellular self RNA in the endosomal compartment is crucial for the TLR3 signaling in tumor cells undergoing ICD (40). On the other hand, when recognized by cytosolic dsDNA sensor (CDS), dsDNA can mediate type I IFNs secretion via stimulator of interferon genes (STING) activation for an anticancer immune response (41). Type I IFNs aid in chemotherapy-induced ICD mainly through autocrine or paracrine signaling cascade, elicit immunostimulatory effect via IFNAR signaling in immune cells, and trigger CXCL10 secretion by tumor cells (40, 42). Notably, CXCL10 secreted due to anthracycline-induced ICD of tumor cells can recruit T cells to the TME (40). In addition, growing evidence supports that determining the phosphorylation levels of eukaryotic translation initiation factor 2α (eIF2α) can be a potential marker for auditing the ICD process, since eIF2α phosphorylation is mandatory for the pre-apoptotic surface exposure of ecto-CRT (43).
Overall interactions between DAMPs with purinergic receptors, phagocytic receptors, and PRRs in host cells are crucial in stimulating DCs to actively display antigens to major histocompatibility complex (MHC) I/MHC II molecules on T cells for a tumor antigen-specific antitumor response. Therefore, immunogenic dying cells could mediate the phagocytic engulfing of dying tumor cells and target the leftover malignant cells (Figure 1).
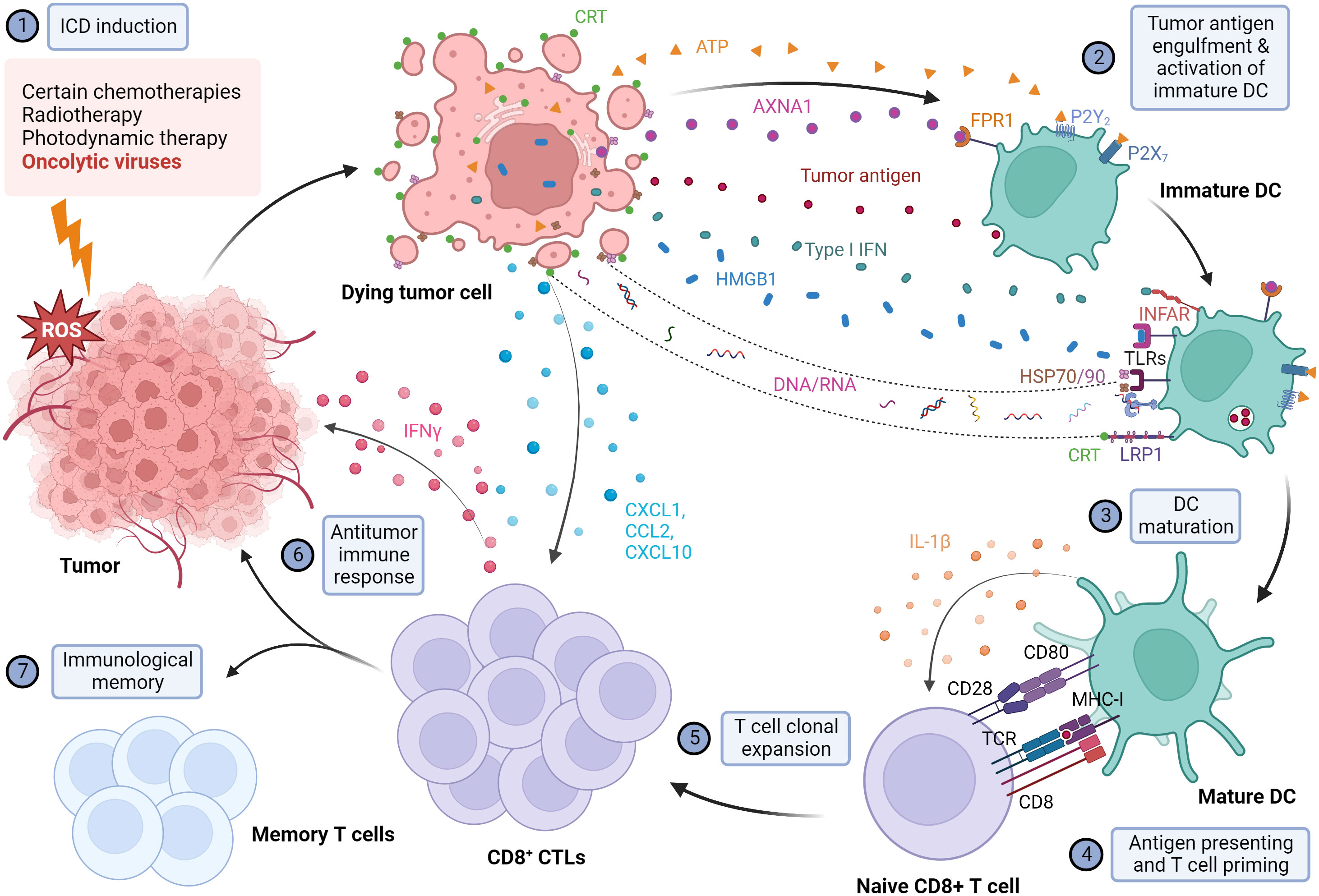
Figure 1 Illustration of the mechanism of immunogenic cell death and the subsequent activation of the antitumor immune response. When exposed to different immunogenic cell death (ICD) inducers, including OVs, cancer cells are under extreme ER stress and undergo ICD. Dying cancer cells express various damage-associated molecular patterns (DAMPs), including the release of high mobility group box 1 (HMGB1) from the nucleus, translocation and cell surface exposure of calreticulin (ecto-CRT) and heat shock proteins HSP70/90, and extracellular secretion of ATP, Annexin A1 (AXNA1), cytokines, chemokines, and nucleic acids. Exposure to DAMPs serves as a "find me" signal which recruits immature DC to TME and induces the maturation of DC. Ecto-CRT provides a pro-phagocytic signal that promotes the phagocytosis of antigens by DC. In addition, HMGB1 and HSP70/90 assist in promoting the processing of phagocytic cargo by binding to toll-like receptors (TLRs), thereby escalating antigen engulfment, processing, and presentation to T cells to mediate tumor-specific immune response and protective immunological memory. Created with BioRender.com.
3 Oncolytic viruses — an emerging anticancer modality over conventional chemotherapies
Traditional cytotoxic chemotherapeutic drugs can restrict the rapidly growing tumor cells by inducing DNA damage, disrupting DNA synthesis or repair, or targeting the basic functions of cell division (44) but can face significant obstacles when cancer stem cells develop drug resistance. A noticeable example is the expression of specific ATP-binding cassette (ABC) transport protein, also known as breast cancer resistance protein (BCRP)/ATP-binding cassette superfamily G member 2 (ABCG2) protein in hematopoietic stem cells and cancerous cells (45, 46). BCRP-ABCG2 protein effluxes the intracellular toxic-chemical agents to the extracellular space by utilizing the energy liberated by ATP hydrolysis (47). In acute myeloid leukemia (AML), BCRP-ABCG2 is predominantly expressed in stem cell subpopulations to mediate energy-driven clearance of toxic agents (48). Other drug resistance mechanisms may also be acquired, such as by the elevated expression of detoxifying enzyme aldehyde dehydrogenase (ALDH) against alkylating agents (e.g., cyclophosphamide) (49). Additionally, chemotherapy is often associated with patient toxicities including immunosuppression with reduced level of immune cells, increasing the chance of therapy intolerance (44).
On the contrary, OVs can selectively target and lyse cancer cells. Some OVs exhibit natural tropism in cancerous tissues, while some OVs are genetically modified to detect specific targets, replicate in cancer cells, and deliver certain genes. The unique feature of OVs is their ability to distinguish tumor cells from healthy cells, resulting in more tumor-specific toxicity, unlike conventional chemotherapy which flows to every region in the systemic circulation and damages the fast-growing normal cells. Above all, the OVs can activate the immune system against cancer cells to elicit potent antitumor responses. Several OVs can trigger ICD via the expression of danger signals (i.e., DAMPs), thereby marking the dying tumor cells for recognition by the innate immune cells. Apart from this, OVs elicit a specific antiviral immune response following ICD induction for virus clearance to prevent possible virus-induced toxicity. PRRs including toll-like receptors (TLRs), RIG-I-like receptors (RLRs) and cytoplasmic DNA receptors can recognize viral PAMPs, which activate the antiviral innate immune responses that induce the expression of the type I IFNs and pro-inflammatory cytokines (50). Type I IFNs can also trigger the adaptive immune response by priming T helper cells and cytotoxic T cells, leading to the induction of antigen-specific responses (50). In addition, OVs can serve as a vector to deliver the desired therapeutic agent to target cells. Several studies also demonstrated that combining OV with traditional therapies showed synergistic tumor killing, implying that OV’s tumor-specificity and antitumor feature may be a booster for adequate antitumor protection in combination therapy (51). The features above make it ideal to employ ICD-inducing OVs as a novel treatment alternative in cancer therapy.
4 Immunogenic and multimodal cell death mediated by OVs
OVs can induce ICD with various features, such as causing ER stress, HMGB1 and ATP release, and elevated surface expression of CRT during infection. A recent study illustrated that parapoxvirus ovis (ORFV) induced extracellular secretion of HMGB1 and ATP in lung cancer cells (52). Squamous cell carcinoma (SCC) cells infected with herpes simplex virus type 1 (HSV-1) manifested extracellular ATP and HMGB1 release, as well as CRT translocation to the membrane (53). Similarly, Adenovirus (Ad), Semliki Forest virus (SFV4), and Vaccinia virus (VACV) mediated CRT exposure, the release of HMGB1 and HSP90, as well as ATP release in lung cancer and bone cancer cells (54). The attenuated poliovirus (PV)-based vector PVSRIPO (55, 56), which has its Internal Ribosomal Entry Site (IRES) substituted with IRES from rhinovirus type 2 (HRV2) to eliminate PV’s neurovirulence, was shown to induce HSPs, HMGB1, viral dsRNA, and tumor antigen release from melanoma and breast cancer cells (57). Besides, some related OVs may induce differential ICD hallmarks in distinct tumors. For example, Coxsackievirus B3 (CVB3) mediated cytotoxicity in lung cancer cells with high CRT exposure, HMGB1 release, and ATP secretion (58), whereas CVA21 mediated cytotoxicity in bladder cancer cells positively correlated with high CRT exposure and HMGB1 release but not ATP release (59). In parallel, the Newcastle disease virus (NDV) Hitchner B1 strain induced surface translocation of CRT and HMGB1 secretion in glioma cells, which produced a long-term, tumor-specific immunological memory response in an orthotopic glioma model (60). NDV/FMV strain-infected melanoma, lung cancer, and prostate cancer cells also displayed HSP70/90 and ATP secretion in addition to CRT exposure and HMGB1 release (61–63). As for measles virus (MV), the Edmonston strain mediated the release of type I IFNs and HMGB1 in human melanoma (64) and CRT exposure, ATP, and HMGB1 release in hepatocellular carcinoma (HCC) (65). MV-Schwarz strain mediated the release of HSP70 in mesothelioma cells (66). Examples of ICD-inducing OVs are summarized in Figure 2 and Table 1.
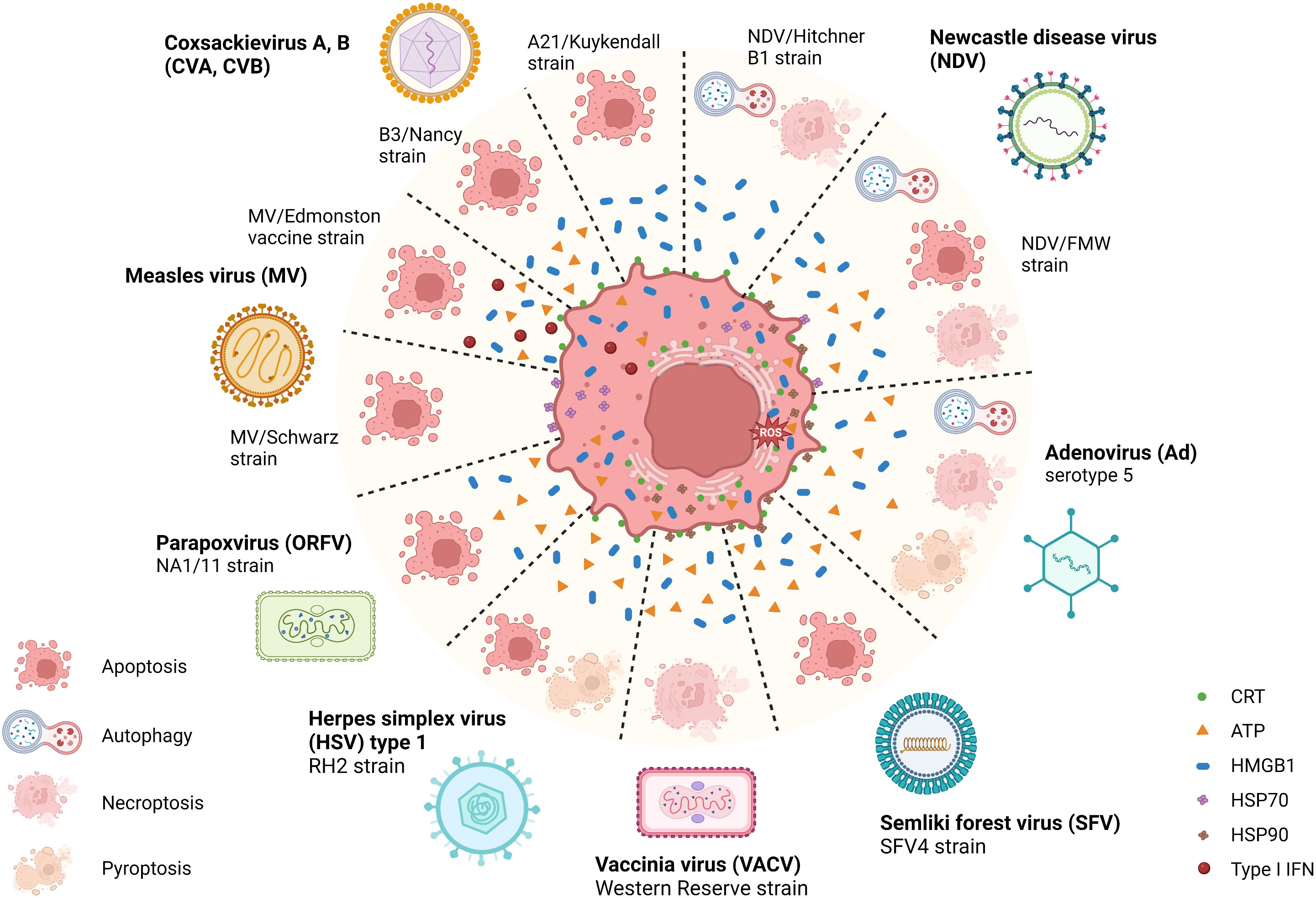
Figure 2 Illustration of the immunogenic cell death related pathways induced by natural OVs. Several natural oncolytic viruses (OVs), including coxsackievirus A, B (CVA, CVB), Newcastle disease virus (NDV), adenovirus (Ad), semliki forest virus (SFV), measles virus (MV), herpes simplex virus (HSV) and poxviruses, such as vaccinia virus (VACV) and parapoxvirus (ORFV) can induce a distinct mode of immunogenic cell death (ICD). For example, CVA, CVB, MV, ORFV, and SFV induce immunogenic apoptosis, and VACV induces necroptosis. While some OVs, such as HSV, NDV, and Ad, induce multimodal cell death, mediating chronic exposure of DAMPs in dying cancer cells. During this phase, the dying tumor cells exposing multiple damage-associated molecular patterns (DAMPs) recruit innate immune responders, thus connecting the bridge between TME and host innate immune machinery, eliciting a potent antitumor immune response. Created with BioRender.com.
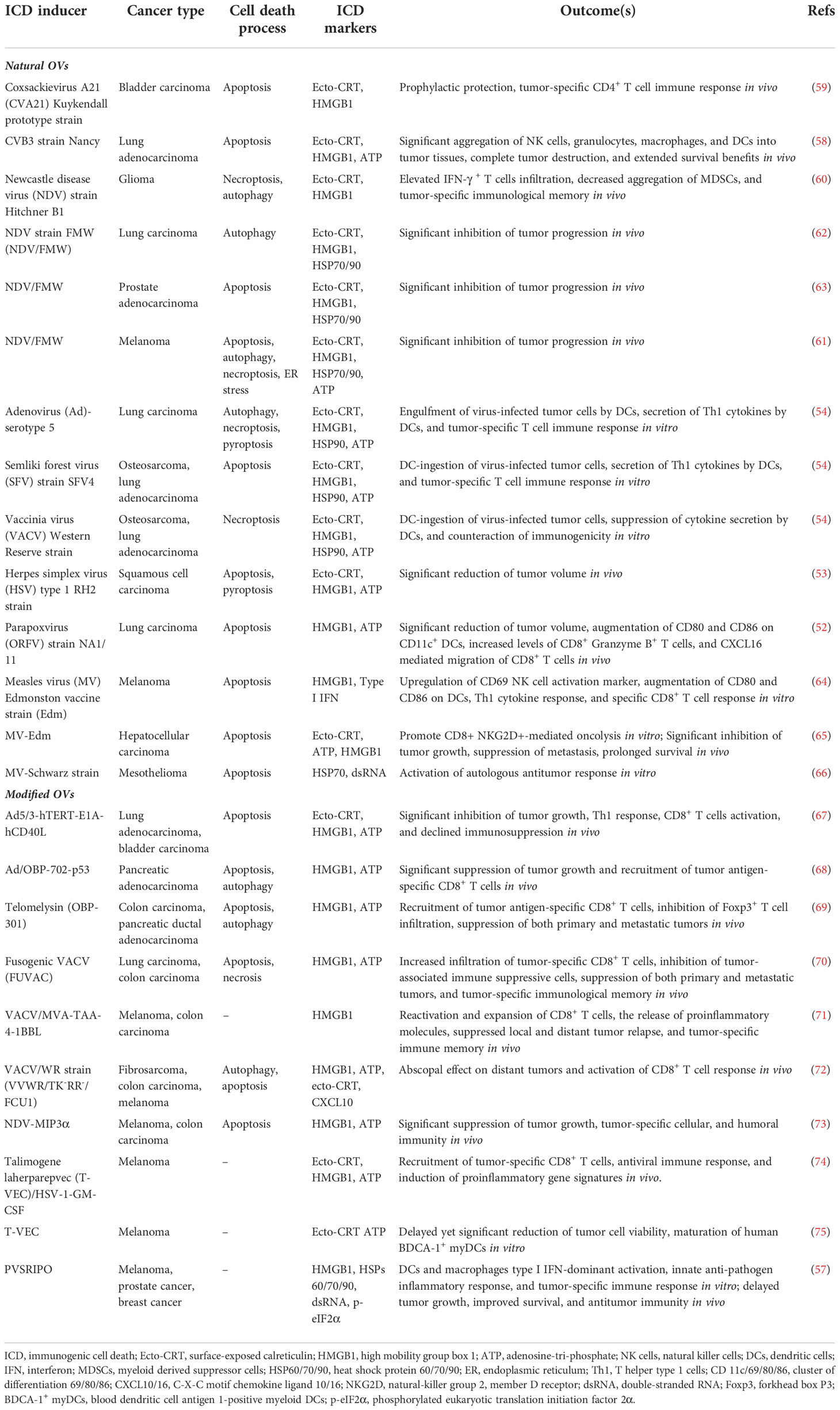
Table 1 ICD hallmarks induced by OVs.
The fate of tumor cells undergoing ICD establishes tumor-specific immune effects by cross-priming tumor antigens to T cells via APCs. For instance, the innate immune cells are recruited in TME to uptake, process, and present tumor-specific antigens in human non-small cell lung cancer (58). For the application in oncolytic viro-immunotherapy, OVs must cause the proper ways of cell death that elicit robust antitumor immunity. Initial investigations of ICD were associated only with the concept of “immunogenic apoptosis,” but over a decade of research shed light on multimodal cell death related pathways, including necrosis, necroptosis, pyroptosis, and autophagy (76). Various tumors and associated endothelial cells killed by OVs showed distinct types of cell death, characterized by the drastic fine-structural transition of dying cells. Predominantly, immunogenic apoptosis is the most common mode of cell death induced by OVs. SFV4 replication displayed rapid cell lysis coupled with induction of an intrinsic caspase-3/7-mediated apoptotic pathway (54). CVA21, CVB3, and ORFV induced immunogenic apoptosis of bladder cancer cells and lung cancer cells, respectively, while ORFV enhanced DC activation in infected cancer cells (52, 58, 59). In addition, MV-infected melanoma, HCC and mesothelioma cells, and NDV-infected prostate cancer cells were also susceptible to immunogenic apoptosis (63–66).
Nonetheless, many studies have emphasized the significance of necroptosis, a programmed necrotic cell death process in a caspase-independent fashion, in overcoming apoptotic resistance and may induce or augment antitumor immunity. Many key mediators in necroptotic signaling cascades are less expressed in various tumors, implying that malignant cells may dodge necroptotic hook for survival (77, 78). For example, VACV infection in lung cancer cells showed decreased caspase-8 activity with a marked increase in phosphorylated mixed lineage kinase domain-like (MLKL), a known obligate effector of necroptosis (54). Some OVs also trigger multiple cell death processes in the target cancer cells. For example, wild-type Ad-serotype 5 induced autophagy and activated necroptotic and pyroptotic cell death pathways (54). The immunogenic killing of HSV-1 in SCC was reduced by pan-caspase and caspase-1 inhibitors, demonstrating the involvement of apoptosis and pyroptosis (53). In the case of NDV, NDV/Hitchner B1 was suggested to induce necroptosis rather than apoptosis in glioma cells (60), whereas NDV/FMW induced autophagy-dependent ICD (not affected by apoptosis and necroptosis inhibitors) in lung cancer cells (62) and multiple cell death processes including autophagy, apoptosis, and necroptosis in melanoma cells (61). These results suggest that OVs may trigger various cell death modalities in cancer cells, and the types of cell death induced may depend not only on the virus but also on the cancer type.
5 Strategies to modulate the immunogenic efficacy of OVs
Although OVs have selective tumor lysis potential, antiviral immunity may restrict OVs’ replication and spread, leading to premature OV clearance (79). In addition, numerous preclinical and clinical research investigated the antitumor efficacy of “non-armed” OVs; however, their immune-provoking ability does not always produce the anticipated antitumor response. Therefore, strategies to modulate the immunogenicity of OVs may be beneficial to enhance their therapeutic potential in cancer immunotherapy.
5.1 Genetic engineering of OVs to improve oncolysis, innate and adaptive immune responses
Viruses can be engineered at the gene level to optimize antitumor immunity by delivering various immuno-modulatory transgenes, most prominently DC and T cell activating cytokines (80).
5.1.1 Cell death-inducing factors for enhanced oncolysis
Researchers have introduced cell death-inducing factors in OV vectors for more efficient immune-oncolytic therapeutic outcomes. For example, Van Hoecke et al. found that intratumoral delivery of mRNA encoding MLKL, a necroptosis-inducing factor, induced tumor lysis and mediated antitumor immunity in mouse models (81). In a follow-up study, they employed wild-type VACV for targeted intratumoral delivery of MLKL, which led to the activation of necroptosis-like tumor cell death in vitro (82). Furthermore, MLKL expressing VACV vectors induced a noticeable antitumor activity coupled with potent intrinsic antitumor immunity against neo-epitopes (82). On the other hand, Ad OBP-702 expressing p53, a tumor suppressor protein, substantially enhanced ICD via HMGB1 and ATP release by favoring apoptotic and autophagic cancer cell death (68). Ad/OBP-702-p53 thus promoted significant CD8+ T cell infiltration in pancreatic ductal adenocarcinoma (PDAC) tumors to achieve efficient antitumor response (68).
5.1.2 Immuno-modulatory transgenes to optimize APCs activation
The first U.S. FDA-approved oncolytic virus, talimogene laherparepvec (T-VEC), is an attenuated HSV-1 armed with two copies of human granulocyte-macrophage colony stimulating factor (GM-CSF) gene to promote DC infiltration and maturation (74). T-VEC was shown to induce apoptosis in melanoma cells (83) and trigger ICD hallmarks including ecto-CRT, HMGB1, and ATP release (74, 75), resulting in the recruitment of tumor-specific CD8+ T cells and induction of proinflammatory responses in both injected and non-injected tumor sites (74). On the other hand, expressing CD40 ligand (CD40L) to the CD40 receptor on APCs leads to cancer cell apoptosis and Th-1 immune reaction, followed by cytotoxic T cell activation and nullified immunosuppression. Chimeric Ad5/3-hTERT-E1A-hCD40L, coding for CD40L, substantially reduced tumor development by oncolytic and apoptotic properties in vivo (67). (Ad5/3-hTERT-E1A-hCD40L)-induced oncolysis culminated in ATP and HMGB1 release and augmented CRT exposure implying immunogenicity. In vivo therapeutic intervention of immunocompetent mice with Ad5/3-hTERT-E1A-hCD40L showed macrophages’ recruitment and activation, which induced Th-1 cell immune response via IL-2 release. This effect was coupled with a cytotoxic T cell response via CCL5/RANTES, IFN-γ, and TNF-α cytokines, suggesting the activation of a systemic T cell immune response (67). Recombinant NDV NDV-MIP3α expressing the macrophage inflammatory protein-3α (MIP3α), a chemokine of DCs, exhibited tumor killing and ICD initiation, same as the wild-type NDV via HMGB1 and ATP release (73). Moreover, NDV-MIP3α successfully promoted the maturation and activation of DCs with strong expression of CD80 and CD86 costimulatory markers and IFN-γ and TNF-α active cytokines. Ultimately, significant reversion of TME and tumor-specific cellular and humoral responses are observed in melanoma and colorectal murine models (73).
5.1.3 Immuno-modulatory transgenes to optimize T cell immune responses
T cell activation demands a minimum of two signals, including a TAA-specific signal dispatched via a T cell receptor and a costimulatory signal attributed via designated molecules. Previous studies have shown that intratumoral delivery of immunomodulatory genes such as B7-1 and B7-2 could augment antitumor responses (84, 85). The inoculation of recombinant VACV expressing the murine B7-1 or B7-2 genes (rVACV-B7-1 or rVACV-B7-2) into immunocompetent mouse models appeared to halt the tumor growth (84). Similarly, Todo et al. developed a defective HSV-1 vector incorporated with B7-1-immunoglobulin (B7-1-Ig) fusion transgene (dvB7Ig/G207) (85). Intraneoplastic inoculation of dvB7Ig/G207 in the neuroblastoma syngeneic mouse model inhibited the cancer progression and substantially increased CD4+ and CD8+ T cell infiltration, and the mice cured by dvB7Ig/G207 treatment were protected against tumor rechallenge (85). Furthermore, lack of adequate stimulation and sufficient tumor infiltration due to immune-hostile TME often downgrades the naturally primed CD8+ T cell response. However, intratumoral delivery of the VACV/MVA vaccine strain armed with 4-1BBL (a T cell immunostimulatory molecule) led to reactivation and increased expansion of tumor-specific CD8+ T cells (71). In addition, MVA-TAA-4-IBBL induced strong ICD with HMGB1 exposure and generated tumor-specific immune memory that eliminated local and distant tumor relapse (71).
5.1.4 Recombination of OVs to enhance immunogenicity
Recombination of OVs has been explored to enhance the oncolytic activity and immunogenicity of viral vectors. For example, the reovirus p14 fusion-associated small transmembrane (FAST) protein, a viral fusogen that induces cell-cell fusion and syncytia formation, has been recombined with the attenuated vesicular stomatitis virus vector VSVΔM51 to form the VSV-p14 chimera (86). VSV-p14 infected breast cancer spheroids more efficiently and enhanced immune cell activation in syngeneic breast cancer mouse model compared to VSVΔM51 encoding green fluorescence protein (VSV-GFP) (86). Likewise, recombinant VSV-NDV (rVSV-NDV) chimera is produced by retaining the highly replicative VSV backbone and replacing its glycoprotein with the hemagglutinin-neuraminidase (HN) and modified fusion (F) envelope protein of fusogenic NDV to improve the efficacy and safety of VSV (87). rVSV-NDV induced tumor-specific syncytia formation, followed by dynamic cell-to-cell virus transmission for rapid onset of ICD in HCC, as observed by ATP, HMGB1, Hsp70, and Hsp90 release and CRT expression (87). Systemic administration of rVSV-NDV substantially increased the overall survival of the orthotopic HCC-bearing mice (87). Therefore, combining the valuable traits of two or more OVs may pave an attractive and beneficial platform for the clinical safety and efficacy of oncolytic viro-immunotherapy.
5.2 Combination of OVs and ICD-enhancing agents for enhanced immunotherapeutic efficacies
This approach uses OVs with ICD-enhancing chemical drugs, some of which are clinically available, to break immune tolerance and induce a long-lasting tumor-specific immune response.
5.2.1 Enzyme inhibitors
Bortezomib is a novel peptide-based proteasome inhibitor. The ICD-inducing property comes from its ability to cause reactive oxygen species (ROS) generation and mitochondrial dysfunction, which leads to cellular stress and DAMP release (88, 89). In HCC, ICD induced by Ad expressing human telomerase reverse-transcriptase (hTert-Ad) infection is enhanced with bortezomib treatment. Proteasome inhibition during hTert-Ad infection triggered the complementary ER stress pathways by negatively disrupting unfolded protein response (UPR) and augmented the caspase-dependent activation of antitumor immunity (90). Furthermore, bortezomib repressed the antiviral immune response in immunocompetent mice, which improved oncolysis of hTert-Ad (90). In the immunocompetent HCC set-up, adenovirotherapy-induced CD8+ T cell immunity suppressed the primary tumor and the offshoot of non-infected lung metastases (90). In another study, VSV and bortezomib showed an antagonistic effect against myeloma cells in vitro, but in contrast, a synergistic anti-myeloma effect was observed in vivo. This effect is explained by the bystander immune cells in host TME absent in vitro (91), suggesting the possible ER stress-induced ICD upon proteasome inhibition.
5.2.2 STAT3 inhibitors
Several signaling pathways are aberrantly hyperactivated in diverse cancers, among which activation of transcription factor STAT3 accounts for immunosuppression. Hence, hindering STAT3 may provide new strategies for anticancer immunotherapies. Wang et al. demonstrated that impairing STAT3 signaling cascade with small-molecule STAT3 inhibitor, C188-9, which binds to the phosphotyrosyl peptide binding site within STAT3, has significantly reduced the tyrosine phosphorylation status of STAT3 in prostate cancer cells (63). This C188-9 pre-treatment, followed by NDV/FMW infection, profoundly enhanced HMGB1 and HSP70/90 release and increased CRT exposure. To eliminate possible off-target effects of C188-9, stable knock-down of STAT3 with lentivirus-based shRNA has significantly elevated NDV/FMW-induced tumor growth inhibition, ICD, and substantially augmented all three ICD hallmarks as in C188-9 pre-treatment (63). Furthermore, the supernatant from NDV/FMV-infected prostate cancer cells significantly repressed tumor growth in vivo (63). On the contrary, the same group showed that STAT3 is crucial in the NDV/FMW-caused ICD process in melanoma cells (61). Pharmacological inhibition or shRNA-mediated depletion of STAT3 suppressed the NDV/FMW-caused ICD determinants, including HMGB1 and HSP70/90 release, CRT exposure, and ATP secretion (61), suggesting plausible mechanisms to fine-tune NDV-induced ICD in diverse cancer types.
5.2.3 Immune checkpoint inhibitors
A class of cancer immunotherapy, immune checkpoint inhibitors (ICIs), has recently caused a paradigm deviation that dramatically enhanced the clinical output in malignant tumor therapies, such as gastric cancer, melanoma, Hodgkin’s lymphoma, and non-small cell lung cancer (92–95). Among these, blocking antibodies against programmed cell death-1 (PD-1) combined with potential OVs synergized the OVs-induced ICD, thus favoring the combination therapy. A telomerase-specific oncolytic Ad (Telomelysin, OBP-502) caused the release of ICD determinants, such as ATP and HMGB1, and chemokines such as RANTES and CXCL10/IP-1, resulting in CD8+ T cell recruitment and blockade of intratumoral penetration of Foxp3+ T cells (69). Combining intratumoral OBP-502 administration and anti-PD-1 Ab systemic administration in bilateral subcutaneous tumor models has elicited an abscopal effect by gradually repressing both OBP-502-treated tumors and OBP-502-non-treated tumors by active recruitment of CD8+ T cells (69). In addition, the combination efficacies are evaluated in a more clinically relatable orthotopic rectal tumor model with liver metastasis, which significantly prolonged survival and gravely suppressed both primary and metastatic tumors (69). Moreover, a novel fusogenic oncolytic vaccinia virus (FUVAC) displayed fusogenic cytopathic effects resulting in cell-cell fusion, apoptosis, necrosis, and ICD via HMGB1 and ATP release, which increased CD8+ T cell infiltration and decreased tumor-associated immune repressive cells (70). Strikingly, combining FUVAC and anti-PD-1 Ab offered synergistic effects by achieving complete response (CR), abscopal effect against non-treated distant tumors, and antitumor immunological memory against tumor re-implantation (70). Most importantly, coupling ICD-inducing OVs with anti-PD-1 monotherapy may have positive outcomes in areas of significant unmet needs. For instance, the VACV/GLV-1H68 strain induced ICD with the substantial secretion of ATP, HMGB1, and surface exposure of CRT (96). Direct delivery of VACV/GLV-1H68 using isolated limb perfusion (ILP) and subsequent PD-1 blockade showed no local and distant relapse with significant survival in neoadjuvant models (96). Likewise, VACV/WR strain (VVWR/TK-RR-/FCU1) mediated an abscopal effect on distant tumors by activating CD8+ T cell response (72). This systemic efficacy of VACV/WR was further augmented when combined with anti-CTLA-4 and anti-PD-1 ICIs, thereby displaying a higher survival benefit (72).
5.2.4 Vitamins
Novel insights into the pharmacological action of vitamin C (Vit-C) suggest that high-dose Vit-C can trigger the accumulation of large quantities of ROS through multiple pathways, which ultimately damages cancer cells (97). High-dose Vit-C was also shown to enhance ICIs (97). Recently, Ma et al. demonstrated that combination therapy of high-dose Vit-C and oncolytic Ad could mediate synergistic antitumor effect by augmenting ICD (98). This combination resulted in significant infiltration of T cells in TME, activation of CD8+ T cell-dependent antitumor effects, and decreased proportion of regulatory T cells (98). Hence, OVs and Vit-C combination regimens may offer new dimensions in cancer immunotherapy and deserves further exploration.
6 Conclusions and prospects
Cancers are known to hijack routine cell regulatory cascades, escape immune surveillance, and adapt to various anticancer treatment strategies, thereby diminishing therapeutic efficacies. Nonetheless, OVs identified for their abilities to selectively kill cancer cells and induce systemic antitumor immune response through ICD are arising as a new class of potent anticancer agents. Furthermore, recent progress has demonstrated that genetic engineering of OVs with transgenes and combining OVs with ICD-enhancing agents can potentiate ICD and establish a robust antitumor response with immunological memory. These aspects underscore ICD as a key player and essential cornerstone of oncolytic viro-immunotherapy. Future development of advanced OV regimens will need to consider how to achieve optimal and balanced oncolytic activity in inducing ICD, promoting tumor clearance, and eliciting effective long-lasting antitumor protection, which are vital for a successful oncolytic viro-immunotherapy as a next-generation anticancer modality.
Author contributions
Conceptualization: LP, C-HL, and L-TL. Writing—Original Draft: LP and C-HL. Writing—Review and Editing: LP, C-HL, and L-TL. Supervision: L-TL. Funding Acquisition: L-TL. All authors contributed to the article and approved the submitted version.
Funding
L-TL is supported by funding from the Ministry of Science and Technology of Taiwan (MOST110-2320-B-038-041-MY3).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Zhang Z, Lu M, Qin Y, Gao W, Tao L, Su and Zhong W J, et al. Neoantigen: A new breakthrough in tumor immunotherapy. Front Immunol (2021) 12:672356. doi: 10.3389/fimmu.2021.672356
2. Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P, et al. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer (2012). doi: 10.1038/nrc3380
3. Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol (2013) 31:51–72. doi: 10.1146/annurev-immunol-032712-100008
4. Broz P, Monack DM. Newly described pattern recognition receptors team up against intracellular pathogens. Nat Rev Immunol (2013) 13(8):551–65. doi: 10.1038/nri3479
5. Vanmeerbeek I, Sprooten J, De Ruysscher D, Tejpar S, Vandenberghe P, Fucikova J, et al. Trial watch: Chemotherapy-induced immunogenic cell death in immuno-oncology. Oncoimmunology (2020) 9(1):1703449. doi: 10.1080/2162402X.2019.1703449
6. Golden EB, Apetoh L. Radiotherapy and immunogenic cell death. Semin Radiat Oncol (2015) 25(1):11–7. doi: 10.1016/j.semradonc.2014.07.005
7. Alzeibak R, Mishchenko TA, Shilyagina NY, Balalaeva IV, Vedunova MV, Krysko DV, et al. Targeting immunogenic cancer cell death by photodynamic therapy: Past, present and future. J Immunother Cancer (2021) 9(1):e001926. doi: 10.1136/jitc-2020-001926
8. Jiang H, Fueyo J. Healing after death: antitumor immunity induced by oncolytic adenoviral therapy. Oncoimmunology (2014) 3(7):e947872. doi: 10.4161/21624011.2014.947872
9. Fukuhara H, Ino Y, Todo T. Oncolytic virus therapy: A new era of cancer treatment at dawn. Cancer Sci (2016) 107(10):1373–9. doi: 10.1111/cas.13027
10. Chulpanova DS, Solovyeva VV, Kitaeva KV, Dunham SP, Khaiboullina SF, Rizvanov AA, et al. Recombinant viruses for cancer therapy. Biomedicines (2018) 6(4):94. doi: 10.3390/biomedicines6040094
11. Fucikova J, Kepp O, Kasikova L, Petroni G, Yamazaki T, Liu P, et al. Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis (2020) 11(11):1013. doi: 10.1038/s41419-020-03221-2
12. Panaretakis T, Joza N, Modjtahedi N, Tesniere A, Vitale I, Durchschlag M, et al. The co-translocation of ERp57 and calreticulin determines the immunogenicity of cell death. Cell Death Diff (2008) 15(9):1499–509. doi: 10.1038/cdd.2008.67
13. Feng M, Jiang W, Kim BYS, Zhang CC, Fu YX, Weissman IL, et al. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat Rev Cancer (2019) 19(10):568–86. doi: 10.1038/s41568-019-0183-z
14. Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, et al. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell (2005) 123(2):321–34. doi: 10.1016/j.cell.2005.08.032
15. Berwin B, Delneste Y, Lovingood RV, Post SR, Pizzo SV. SREC-I, a type f scavenger receptor, is an endocytic receptor for calreticulin. J Biol Chem (2004) 279(49):51250–7. doi: 10.1074/jbc.M406202200
16. Berwin B, Hart JP, Rice S, Gass C, Pizzo SV, Post SR, et al. Scavenger receptor-a mediates gp96/GRP94 and calreticulin internalization by antigen-presenting cells. EMBO J (2003) 22(22):6127–36. doi: 10.1093/emboj/cdg572
17. Duus K, Pagh RT, Holmskov U, Højrup P, Skov S, Houen G, et al. Interaction of calreticulin with CD40 ligand, TRAIL and fas ligand. Scandinavian J Immunol (2007) 66(5):501–7. doi: 10.1111/j.1365-3083.2007.01999.x
18. Chao MP, Jaiswal S, Weissman-Tsukamoto R, Alizadeh AA, Gentles AJ, Volkmer J, et al. Calreticulin is the dominant pro-phagocytic signal on multiple human cancers and is counterbalanced by CD47. Sci Transl Med (2010) 2(63):63ra94. doi: 10.1126/scitranslmed.3001375
19. Garg AD, Elsen S, Krysko DV, Vandenabeele P, de Witte P, Agostinis P, et al. Resistance to anticancer vaccination effect is controlled by a cancer cell-autonomous phenotype that disrupts immunogenic phagocytic removal. Oncotarget (2015) 6(29):26841–60. doi: 10.18632/oncotarget.4754
20. Fucikova J, Becht E, Iribarren K, Goc J, Remark R, Damotte D, et al. Calreticulin expression in human non-small cell lung cancers correlates with increased accumulation of antitumor immune cells and favorable prognosis. Cancer Res (2016) 76(7):1746–56. doi: 10.1158/0008-5472.CAN-15-1142
21. Martins I, Wang Y, Michaud M, Ma Y, Sukkurwala AQ, Shen S, et al. Molecular mechanisms of ATP secretion during immunogenic cell death. Cell Death Diff (2014) 21(1):79–91. doi: 10.1038/cdd.2013.75
22. Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, et al. Nucleotides released by apoptotic cells act as a find-me signal. Nature (2010) 461(7261):282–6. doi: 10.1038/nature08296
23. Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1β-dependent adaptive immunity against tumors. Nat Med (2009) 15(10):1170–8. doi: 10.1038/nm.2028
24. Yu M, Wang H, Ding A, Golenbock DT, Latz E, Czura CJ, et al. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock (2006) 26(2):174–9. doi: 10.1097/01.shk.0000225404.51320.82
25. Apetoh L, Ghiringhelli F, Tesniere A, Criollo A, Ortiz C, Lidereau R, et al. The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol Rev (2007) 220:47–59. doi: 10.1111/j.1600-065X.2007.00573.x
26. Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol (2010).
27. Manfredi AA, Capobianco A, Esposito A, De Cobelli F, Canu T, Monno A, et al. Maturing dendritic cells depend on RAGE for in vivo homing to lymph nodes. J Immunol (2008) 180(4):2270–5. doi: 10.4049/jimmunol.180.4.2270
28. Venereau E, Casalgrandi M, Schiraldi M, Antoine DJ, Cattaneo A, De Marchis F, et al. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J Exp Med (2012) 209(9):1519–28. doi: 10.1084/jem.20120189
29. Yang H, Hreggvidsdottir HS, Palmblad K, Wang H, Ochani M, Li J, et al. A critical cysteine is required for HMGB1 binding to toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci United States America (2010) 107(26):11942–7. doi: 10.1073/pnas.1003893107
30. Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol (2012) 13(9):832–42. doi: 10.1038/ni.2376
31. Parker KH, Sinha P, Horn LA, Clements VK, Yang H, Li J, et al. HMGB1 enhances immune suppression by facilitating the differentiation and suppressive activity of myeloid-derived suppressor cells. Cancer Res (2014) 74(20):5723–33. doi: 10.1158/0008-5472.CAN-13-2347
32. Whitley D, Goldberg SP, Jordan WD. Heat shock proteins: A review of the molecular chaperones. J Vasc Surg (1999) 29(4):748–51. doi: 10.1016/S0741-5214(99)70329-0
33. De Maio A, Vazquez D. Extracellular heat shock proteins: A new location, a new function. Shock (2013) 40(4):239–46. doi: 10.1097/SHK.0b013e3182a185ab
34. Murshid A, Gong J, Calderwood SK. The role of heat shock proteins in antigen cross presentation. Front Immunol (2012) 3:63. doi: 10.3389/fimmu.2012.00063
35. Graner MW. HSP90 and immune modulation in cancer. Adv Cancer Res (2016) 129:191–224. doi: 10.1016/bs.acr.2015.10.001
36. Binder RJ. Functions of heat shock proteins in pathways of the innate and adaptive immune system. J Immunol (2014) 193(12):5765–71. doi: 10.4049/jimmunol.1401417
37. Perretti M, D'Acquisto F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat Rev Immunol (2009) 9(1):62–70. doi: 10.1038/nri2470
38. Vacchelli E, Ma Y, Baracco EE, Sistigu A, Enot DP, Pietrocola F, et al. Chemotherapy-induced antitumor immunity requires formyl peptide receptor 1. Science (2015) 350(6263):972–8. doi: 10.1126/science.aad0779
39. Karikó K, Ni H, Capodici J, Lamphier M, Weissman D. mRNA is an endogenous ligand for toll-like receptor 3. J Biol Chem (2004) 279(13):12542–50. doi: 10.1074/jbc.M310175200
40. Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, et al. Cancer cell–autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med (2014) 20(11):1301–9. doi: 10.1038/nm.3708
41. Barber GN. STING-dependent cytosolic DNA sensing pathways. Trends Immunol (2014) 35(2):88–93. doi: 10.1016/j.it.2013.10.010
42. McNab F, Mayer-Barber K, Sher A, Wack A, O'Garra A. Type I interferons in infectious disease. Nat Rev Immunol (2015) 15(2):87–103. doi: 10.1038/nri3787
43. Kepp O, Semeraro M, Bravo-San Pedro JM, Bloy N, Buqué A, Huang X, et al. eIF2α phosphorylation as a biomarker of immunogenic cell death. Semin Cancer Biol (2015) 33:86–92. doi: 10.1016/j.semcancer.2015.02.004
44. Fabian KP, Wolfson B, Hodge JW. From immunogenic cell death to immunogenic modulation: Select chemotherapy regimens induce a spectrum of immune-enhancing activities in the tumor microenvironment. Front Oncol (2021) 11:728018. doi: 10.3389/fonc.2021.728018
45. Abbott BL. ABCG2 (BCRP) expression in normal and malignant hematopoietic cells. Hematol Oncol (2003) 21(3):115–30. doi: 10.1002/hon.714
46. Austin Doyle L, Yang W, Abruzzo LV, Krogmann T, Gao Y, Rishi AK, et al. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc Natl Acad Sci United States America (1998) 95(26):15665–70. doi: 10.1073/pnas.95.26.15665
47. Mao Q, Unadkat JD. Role of the breast cancer resistance protein (BCRP/ABCG2) in drug transport–an update. AAPS J (2015) 17(1):65–82. doi: 10.1208/s12248-014-9668-6
48. Raaijmakers MHGP, de Grouw EP, Heuver LH, van der Reijden BA, Jansen JH, Scheper RJ, et al. Breast cancer resistance protein in drug resistance of primitive CD34+38- cells in acute myeloid leukemia. Clin Cancer Res (2005) 11(6):2436–44. doi: 10.1158/1078-0432.CCR-04-0212
49. Gasparetto M, Pei S, Minhajuddin M, Pollyea DA, Vasiliou V, Humphries RK, et al. Aldehyde dehydrogenases play a role in acute myeloid leukemia and have prognostic and therapeutic significance. Blood (2014) 124(21):2238. doi: 10.1182/blood.V124.21.2238.2238
50. Bowie AG, Unterholzner L. Viral evasion and subversion of pattern-recognition receptor signalling. Nat Rev Immunol (2008) 8(12):911–22. doi: 10.1038/nri2436
51. Lan Q, Xia S, Wang Q, Xu W, Huang H, Jiang S, et al. Development of oncolytic virotherapy: From genetic modification to combination therapy. Front Med (2020) 14(2):160–84. doi: 10.1007/s11684-020-0750-4
52. Wang R, Mo J, Luo X, Zhang G, Liu F, Luo S, et al. ORFV infection enhances CXCL16 secretion and causes oncolysis of lung cancer cells through immunogenic apoptosis. Front Cell Infect Microbiol (2022) 12:910466. doi: 10.3389/fcimb.2022.910466
53. Takasu A, Masui A, Hamada M, Imai T, Iwai S, Yura Y, et al. Immunogenic cell death by oncolytic herpes simplex virus type 1 in squamous cell carcinoma cells. Cancer Gene Ther (2016) 23(4):107–13. doi: 10.1038/cgt.2016.8
54. Ma J, Ramachandran M, Jin C, Quijano-Rubio C, Martikainen M, Yu D, et al. Characterization of virus-mediated immunogenic cancer cell death and the consequences for oncolytic virus-based immunotherapy of cancer. Cell Death Dis (2020) 11(1):48. doi: 10.1038/s41419-020-2236-3
55. Gromeier M, Alexander L, Wimmer E. Internal ribosomal entry site substitution eliminates neurovirulence in intergeneric poliovirus recombinants. Proc Natl Acad Sci United States America (1996) 93(6):2370–5. doi: 10.1073/pnas.93.6.2370
56. Denniston E, Crewdson H, Rucinsky N, Stegman A, Remenar D, Moio K, et al. The practical consideration of poliovirus as an oncolytic virotherapy. Am J Virol (2016) 5(1):1–7. doi: 10.3844/ajvsp.2016.1.7
57. Brown MC, Holl EK, Boczkowski D, Dobrikova E, Mosaheb M, Chandramohan V, et al. Cancer immunotherapy with recombinant poliovirus induces IFN-dominant activation of dendritic cells and tumor antigen-specific CTLs. Sci Transl Med (2017) 9(408):eaan4220. doi: 10.1126/scitranslmed.aan4220
58. Miyamoto S, Inoue H, Nakamura T, Yamada M, Sakamoto C, Urata Y, et al. Coxsackievirus B3 is an oncolytic virus with immunostimulatory properties that is active against lung adenocarcinoma. Cancer Res (2012) 72(10):2609–21. doi: 10.1158/0008-5472.CAN-11-3185
59. Annels NE, Arif M, Simpson GR, Denyer M, Moller-Levet C, Mansfield D, et al. Oncolytic immunotherapy for bladder cancer using coxsackie A21 virus. Mol Ther Oncolytics (2018) 9:1–12. doi: 10.1016/j.omto.2018.02.001
60. Koks CA, Garg AD, Ehrhardt M, Riva M, Vandenberk L, Boon L, et al. Newcastle Disease virotherapy induces long-term survival and tumor-specific immune memory in orthotopic glioma through the induction of immunogenic cell death. Int J Cancer (2015) 136(5):E313–25. doi: 10.1002/ijc.29202
61. Shao X, Wang X, Guo X, Jiang K, Ye T, Chen J, et al. STAT3 contributes to oncolytic Newcastle disease virus-induced immunogenic cell death in melanoma cells. Front Oncol (2019) 9:436. doi: 10.3389/fonc.2019.00436
62. Ye T, Jiang K, Wei L, Barr MP, Xu Q, Zhang G, et al. Oncolytic Newcastle disease virus induces autophagy-dependent immunogenic cell death in lung cancer cells. Am J Cancer Res (2018) 8(8):1514–27.
63. Wang X, Shao X, Gu L, Jiang K, Wang S, Chen J, et al. Targeting STAT3 enhances NDV-induced immunogenic cell death in prostate cancer cells. J Cell Mol Med (2020) 24(7):4286–97. doi: 10.1111/jcmm.15089
64. Donnelly OG, Errington-Mais F, Steele L, Hadac E, Jennings V, Scott K, et al. Measles virus causes immunogenic cell death in human melanoma. Gene Ther (2013) 20(1):7–15. doi: 10.1038/gt.2011.205
65. Chen A, Zhang Y, Meng G, Jiang D, Zhang H, Zheng M, et al. Oncolytic measles virus enhances antitumour responses of adoptive CD8+NKG2D+ cells in hepatocellular carcinoma treatment. Sci Rep (2017) 7(1):5170. doi: 10.1038/s41598-017-00654-2
66. Gauvrit A, Brandler S, Sapede-Peroz C, Boisgerault N, Tangy F, Gregoire M, et al. Measles virus induces oncolysis of mesothelioma cells and allows dendritic cells to cross-prime tumor-specific CD8 response. Cancer Res (2008) 68(12):4882–92. doi: 10.1158/0008-5472.CAN-07-6265
67. Diaconu I, Cerullo V, Hirvinen ML, Escutenaire S, Ugolini M, Pesonen SK, et al. Immune response is an important aspect of the antitumor effect produced by a CD40L-encoding oncolytic adenovirus. Cancer Res (2012) 72(9):2327–38. doi: 10.1158/0008-5472.CAN-11-2975
68. Araki H, Tazawa H, Kanaya N, Kajiwara Y, Yamada M, Hashimoto M, et al. Oncolytic virus-mediated p53 overexpression promotes immunogenic cell death and efficacy of PD-1 blockade in pancreatic cancer. Mol Ther Oncolytics (2022) 27:3–13. doi: 10.1016/j.omto.2022.09.003
69. Kanaya N, Kuroda S, Kakiuchi Y, Kumon K, Tsumura T, Hashimoto M, et al. Immune modulation by telomerase-specific oncolytic adenovirus synergistically enhances antitumor efficacy with anti-PD1 antibody. Mol Ther (2020) 28(3):794–804. doi: 10.1016/j.ymthe.2020.01.003
70. Nakatake M, Kuwano N, Kaitsurumaru E, Kurosaki H, Nakamura T. Fusogenic oncolytic vaccinia virus enhances systemic antitumor immune response by modulating the tumor microenvironment. Mol Ther (2021) 29(5):1782–93. doi: 10.1016/j.ymthe.2020.12.024
71. Hinterberger M, Giessel R, Fiore G, Graebnitz F, Bathke B, Wennier S, et al. Intratumoral virotherapy with 4-1BBL armed modified vaccinia Ankara eradicates solid tumors and promotes protective immune memory. J Immunother Cancer (2021) 9(2):e001586. doi: 10.1136/jitc-2020-001586
72. Fend L, Yamazaki T, Remy C, Fahrner C, Gantzer M, Nourtier V, et al. Immune checkpoint blockade, immunogenic chemotherapy or IFN-α blockade boost the local and abscopal effects of oncolytic virotherapy. Cancer Res (2017) 77(15):4146–57. doi: 10.1158/0008-5472.CAN-16-2165
73. Huang FY, Wang JY, Dai SZ, Lin YY, Sun Y, Zhang L, et al. A recombinant oncolytic Newcastle virus expressing MIP-3α promotes systemic antitumor immunity. J Immunother Cancer (2020) 8(2):e000330. doi: 10.1136/jitc-2019-000330
74. Bommareddy PK, Zloza A, Rabkin SD, Kaufman HL. Oncolytic virus immunotherapy induces immunogenic cell death and overcomes STING deficiency in melanoma. Oncoimmunology (2019) 8(7):1591875–1591875. doi: 10.1080/2162402X.2019.1591875
75. Kalus P, De Munck J, Vanbellingen S, Carreer L, Laeremans T, Broos K, et al. Oncolytic herpes simplex virus type 1 induces immunogenic cell death resulting in maturation of BDCA-1(+) myeloid dendritic cells. Int J Mol Sci (2022) 23(9):4865. doi: 10.3390/ijms23094865
76. Inoue H, Tani K. Multimodal immunogenic cancer cell death as a consequence of anticancer cytotoxic treatments. Cell Death Diff (2014) 21(1):39–49. doi: 10.1038/cdd.2013.84
77. Koo GB, Morgan MJ, Lee DG, Kim WJ, Yoon JH, Koo J, et al. Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res (2015) 25(6):707–25.
78. Geserick P, Wang J, Schilling R, Horn S, Harris PA, Bertin J, et al. Absence of RIPK3 predicts necroptosis resistance in malignant melanoma. Cell Death Dis (2015) 6(9):e1884. doi: 10.1038/cddis.2015.240
79. Lemos de Matos A, Franco LS, McFadden G. Oncolytic viruses and the immune system: The dynamic duo. Mol Ther Methods Clin Dev (2020) 17:349–58. doi: 10.1016/j.omtm.2020.01.001
80. de Graaf JF, de Vor L, Fouchier RAM, van den Hoogen BG. Armed oncolytic viruses: A kick-start for anti-tumor immunity. Cytokine Growth Factor Rev (2018) 41:28–39. doi: 10.1016/j.cytogfr.2018.03.006
81. Van Hoecke L, Van Lint S, Roose K, Van Parys A, Vandenabeele P, Grooten J, et al. Treatment with mRNA coding for the necroptosis mediator MLKL induces antitumor immunity directed against neo-epitopes. Nat Commun (2018) 9(1). doi: 10.1038/s41467-018-05979-8
82. Van Hoecke L, Riederer S, Saelens X, Sutter G, Rojas JJ. Recombinant viruses delivering the necroptosis mediator MLKL induce a potent antitumor immunity in mice. Oncoimmunology (2020) 9(1):1802968.
83. Bommareddy PK, Aspromonte S, Zloza A, Rabkin SD, Kaufman HL. MEK inhibition enhances oncolytic virus immunotherapy through increased tumor cell killing and T cell activation. Sci Transl Med (2018) 10(471). doi: 10.1126/scitranslmed.aau0417
84. Hodge JW, Abrams S, Schlom J, Kantor JA. Induction of antitumor immunity by recombinant vaccinia viruses expressing B7-1 or B7-2 costimulatory molecules. Cancer Res (1994) 54(21):5552–5.
85. Todo T, Martuza RL, Dallman MJ, Rabkin SD. In situ expression of soluble B7-1 in the context of oncolytic herpes simplex virus induces potent antitumor immunity. Cancer Res (2001) 61(1):153–61.
86. Le Boeuf F, Gebremeskel S, McMullen N, He H, Greenshields AL, Hoskin DW, et al. Reovirus FAST protein enhances vesicular stomatitis virus oncolytic virotherapy in primary and metastatic tumor models. Mol Ther Oncolytics (2017) 6:80–9. doi: 10.1016/j.omto.2017.08.001
87. Abdullahi S, Jäkel M, Behrend SJ, Steiger K, Topping G, Krabbe T, et al. A novel chimeric oncolytic virus vector for improved safety and efficacy as a platform for the treatment of hepatocellular carcinoma. J Virol (2018) 92(23). doi: 10.1128/JVI.01386-18
88. Ling YH, Liebes L, Zou Y, Perez-Soler R. Reactive oxygen species generation and mitochondrial dysfunction in the apoptotic response to bortezomib, a novel proteasome inhibitor, in human H460 non-small cell lung cancer cells. J Biol Chem (2003) 278(36):33714–23. doi: 10.1074/jbc.M302559200
89. Yoo JY, Hurwitz BS, Bolyard C, Yu JG, Zhang J, Selvendiran K, et al. Bortezomib-induced unfolded protein response increases oncolytic HSV-1 replication resulting in synergistic antitumor effects. Clin Cancer Res (2014) 20(14):3787–98. doi: 10.1158/1078-0432.CCR-14-0553
90. Boozari B, Mundt B, Woller N, Strüver N, Gürlevik E, Schache P, et al. Antitumoural immunity by virus-mediated immunogenic apoptosis inhibits metastatic growth of hepatocellular carcinoma. Gut (2010) 59(10):1416–26. doi: 10.1136/gut.2009.196519
91. Yarde DN, Nace RA, Russell SJ. Oncolytic vesicular stomatitis virus and bortezomib are antagonistic against myeloma cells invitro but have additive anti-myeloma activity invivo. Exp Hematol (2013) 41(12):1038–49. doi: 10.1016/j.exphem.2013.09.005
92. Kang Y-K, Boku N, Satoh T, Ryu MH, Chao Y, Kato K, et al. Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538-12, ATTRACTION-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet (2017) 390(10111):2461–71. doi: 10.1016/S0140-6736(17)31827-5
93. Weber JS, D'Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol (2015) 16(4):375–84. doi: 10.1016/S1470-2045(15)70076-8
94. Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WE, Poddubskaya E, et al. Nivolumab versus docetaxel in advanced squamous-cell non-Small-Cell lung cancer. N Engl J Med (2015) 373(2):123–35. doi: 10.1056/NEJMoa1504627
95. Horn L, Spigel DR, Vokes EE, Holgado E, Ready N, Steins M, et al. Nivolumab versus docetaxel in previously treated patients with advanced non-Small-Cell lung cancer: Two-year outcomes from two randomized, open-label, phase III trials (CheckMate 017 and CheckMate 057). J Clin Oncol (2017) 35(35):3924–33. doi: 10.1200/JCO.2017.74.3062
96. Smith HG, Mansfield D, Roulstone V, Kyula-Currie JN, McLaughlin M, Patel RR, et al. PD-1 blockade following isolated limb perfusion with vaccinia virus prevents local and distant relapse of soft-tissue sarcoma. Clin Cancer Res (2019) 25(11):3443–54. doi: 10.1158/1078-0432.CCR-18-3767
97. Mussa A, Mohd Idris RA, Ahmed N, Ahmad S, Murtadha AH, Tengku Din TADAA, et al. High-dose vitamin c for cancer therapy. Pharmaceuticals (2022) 15(6). doi: 10.3390/ph15060711
Keywords: immunogenic cell death (ICD), oncolytic virus (OV), anticancer immunotherapy, pathogen-associated molecular patterns (PAMPs), damage-associated molecular patterns (DAMPs), viral engineering, combination therapy
Citation: Palanivelu L, Liu C-H and Lin L-T (2023) Immunogenic cell death: The cornerstone of oncolytic viro-immunotherapy. Front. Immunol. 13:1038226. doi: 10.3389/fimmu.2022.1038226
Received: 06 September 2022; Accepted: 28 November 2022;
Published: 23 January 2023.
Edited by:
Hongwei Zhang, Department of Neurosurgery, Capital Medical University, ChinaReviewed by:
Masmudur Mohammed Rahman, Arizona State University, United StatesVolker Schirrmacher, German Cancer Research Center (DKFZ), Germany
Copyright © 2023 Palanivelu, Liu and Lin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liang-Tzung Lin, bHRsaW5AdG11LmVkdS50dw==
†These authors have contributed equally to this work