 Sean M. Morris
Sean M. Morris Abhishek Chauhan
Abhishek Chauhan
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 27 October 2022
Sec. Cytokines and Soluble Mediators in Immunity
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1037645
This article is part of the Research TopicPlatelet Signaling rediscovered in the Era of ThromboinflammationView all 3 articles
Acute liver injuries have wide and varied etiologies and they occur both in patients with and without pre-existent chronic liver disease. Whilst the pathophysiological mechanisms remain distinct, both acute and acute-on-chronic liver injury is typified by deranged serum transaminase levels and if severe or persistent can result in liver failure manifest by a combination of jaundice, coagulopathy and encephalopathy. It is well established that platelets exhibit diverse functions as immune cells and are active participants in inflammation through processes including immunothrombosis or thromboinflammation. Growing evidence suggests platelets play a dualistic role in liver inflammation, shaping the immune response through direct interactions and release of soluble mediators modulating function of liver sinusoidal endothelial cells, stromal cells as well as migrating and tissue-resident leucocytes. Elucidating the pathways involved in initiation, propagation and resolution of the immune response are of interest to identify therapeutic targets. In this review the provocative role of platelets is outlined, highlighting beneficial and detrimental effects in a spatial, temporal and disease-specific manner.
Acute liver inflammation as a result of hepatic injury can occur both in patients with and without pre-existent liver disease. Acute failure (ALF) occurs in the former, specifically when a patient with no antecedent history of liver disease develops significant liver dysfunction due to an acute hepatocellular insult (1). ALF is characterized by deranged liver biochemistry, jaundice, coagulopathy and encephalopathy (1). Viral hepatitides are the leading cause of ALF in the Asia and Africa whilst in the West the majority of ALF due to drug-induced liver injury [acetaminophen (APAP) overdose] (1). Treatments options for APAP-induced ALF are limited (2) and treatment is therefore based largely around supportive care often in an intensive care setting; ultimately liver transplantation may be required (1). Ten percent of liver transplants in the West are due to acute liver failure (1). Dilemmas surrounding timing of liver transplantation, prognosis and donor shortage highlight the need for disease modifying treatments in patients with ALF (3).
Acute-on-chronic liver failure (ACLF) on the other hand occurs in patients with established chronic liver disease or cirrhosis. Multiple stages of cirrhosis are recognized; in the compensated cirrhosis phase symptoms are modest and mortality low. Acute liver injuries result in hepatic and systemic inflammation in cirrhotic patients which then drive hepatic decompensation as manifest by the development of one or a combination of ascites, portal hypertensive gastrointestinal bleeding and or encephalopathy (4). The one year mortality increases from 3% in patients with compensated cirrhosis to 57% with acute decompensation; median patient survival falls from 12 years to only 2 years (4). There is a subgroup of patients that after acutely decompensating develop progressive extrahepatic organ failures on a background of severe systemic inflammation (5), this syndrome is referred to as ACLF (6) and has a short term mortality as high as 80% at 28 days (7); ACLF in fact represents the leading cause of mortality amongst patients with decompensated cirrhosis (7). Precipitating events for decompensation and eventually ACLF in patients with established cirrhosis include drugs, infections, bleeds and flares of the underlying disease (4).
There are differences and similarities in the pathophysiological responses that underpin the development of ALF and ACLF, both involve a complex interplay between damaged hepatocytes, liver sinusoidal endothelial cells (LSECs), resident and circulating immune cells that initiate and potentiate inflammation but also determine resolution. Platelets are key protagonists in a number of these processes. Known historically for their hemostatic function at sites of vascular injury, it is now well established that platelets participate actively in the immune response intimately linking thrombosis and inflammation, in a process described as thromboinflammation (8–11). Whilst platelets are clearly involved in acute liver inflammation (12, 13), the involvement is likely to be stage and location specific varying in patients with and without pre-existent liver disease (14, 15). Of particular interest in the treatment of liver diseases is defining coagulopathy sparing platelet activation pathways (12, 16). Here we discuss the dualistic roles of platelets in the initiation, amplification and resolution of acute liver injury and how this drives the development of ALF and ACLF.
Sterile inflammation is a response to host cell damage in the absence of pathogens, a key step in restoration of homeostasis, however, becomes a pathological process in several causes of ALF including drug-induced liver injury, alcoholic hepatitis, non-alcoholic steatohepatitis and ischemia-reperfusion injury (17, 18). Central to this condition is the release and recognition of damage-associated molecular patterns (DAMPs) via pattern recognition receptors (PRRs). DAMPs are self-molecules with an ability to activate inflammation (19) including a number of different proteins, nucleic acids and mitochondrial components (20). They are released in inflammatory cell death, pyroptosis, necrosis and necroptosis (18, 21), in addition to non-inflammatory cell death during secondary necrosis of apoptotic bodies (22). PRRs are highly conserved receptors, originally discovered for their role in responding to pathogen associated molecular patterns (PAMPs), of which Toll-like receptors (TLRs) are best studied, responsible for promoting inflammation through cytokine production, chemokine production and expression of ligands involved in leucocyte adhesion and activation (17). In the liver, immune surveillance is performed by a number of resident and circulating leucocytes. Kupffer cells are particularly important sentinel macrophages of the liver participating in the immune response through detection of injury, leucocyte recruitment and mediate tissue repair (23). Importantly it has been demonstrated that platelets are the first cells to accumulate at sites of injury within the liver, thus generating interest into their role in the disease process (24).
In ACLF the main driver of widespread tissue injury is a systemic hyperinflammatory response (25), arising from massive release of inflammatory mediators including cytokines, chemokines, growth factors and bioactive lipid mediators. This leads to immune cell activation and subsequent immune-mediated tissue damage (26). Little is known about exact triggers however is likely to involve recognition of both PAMPs and DAMPs via PRRs. Approximately one-third of cases involve bacterial infections (27), attributed to bacterial translocation across the intestinal lumen (28). Inflammatory cell death, necroptosis and pyroptosis, is common in advanced liver disease triggering release of DAMPs propagating the immune response (29). The hyperinflammatory response often co-exists with innate immune dysfunction at humoral, physical and cell-mediated level (26). The condition is characterised by increased pro- and anti-inflammatory mediators (30), with the prevailing phenotype temporally and spatially dependent, although immunodeficiency has greater importance in advanced disease (31). Bernsmeier et al. (32), proposed a model whereby exaggerated inflammatory responses to DAMPs and PAMPs in cirrhosis promotes polarization of monocytes/macrophages to immunoregulatory phenotypes. In the presence of endothelial dysfunction, reverse migration of these regulatory cells leads to population expansion in distant organs and global immunosuppression. One might hypothesize that populations of exhausted leucocytes predispose patients to sepsis, driving expansion and activation of naïve innate immune cells potentiating cell damage. Dissecting these pathways, and elucidating the role of platelets, if any, is key to identify suitable therapeutic targets.
Platelets possess a range of receptors and a diverse proteasome facilitating interactions with endothelial cells, immune cells, the extracellular matrix and other platelets (Figure 1) (9, 10, 33). Activation leads to platelet degranulation, through classical glycoprotein (GP) pathways at sites of vascular injury (9, 10) and more recently discovered alternative pathways (16), releasing cytokines, chemokines, vasoactive substances, growth factors and platelet-derived extracellular vesicles (PEVs) (containing microvesicles, exosomes and apoptotic bodies) (10, 34). Platelets exhibit dual roles in inflammation with pro- and anti-inflammatory effector functions, which are likely to be disease, organ and time-specific (10). They promote leucocyte recruitment and modulate effector functions through direct interactions (P-selectin-P-selectin glycoprotein ligand 1 (PSGL-1), GPIbα-macrophage-1 antigen (MAC-1, a complement receptor), GPIIbIIIa-MAC-1 through fibrinogen and CD40-CD40L pairings (35, 36)), chemokine/cytokine secretion and increasing vascular permeability (9, 10). They provide a link between innate and adaptive immunity, supporting antigen presentation and lymphocyte function (37). At sites of inflammation platelets also limit bleeding (38) through physical sealing and tightening of endothelial junctions (39). During injury resolution, platelets promote regeneration and homeostasis through chemokine, angiogenic factor and growth factor release (9). The interaction between platelets and leucocytes is bidirectional – leucocytes also promote platelet activation and, in turn, the coagulation cascade; this process is termed immunothrombosis (9). Initially recognized as a means to potentially limit pathogen spread and enhance clearance; there is now increasing recognition of the role microthrombi can play in liver pathobiology by inducing endothelial dysfunction and organ damage (13, 40).
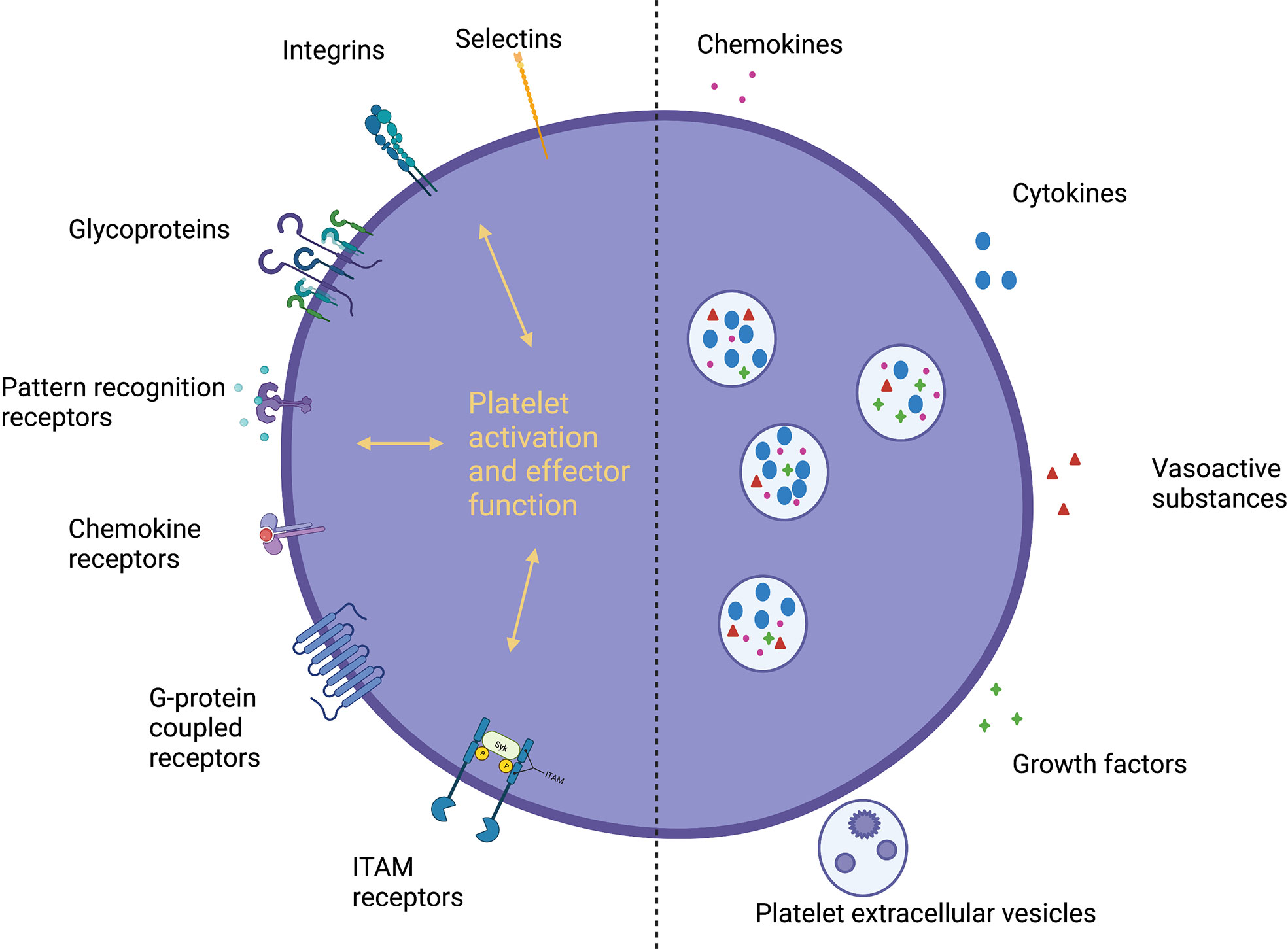
Figure 1 Platelet receptors and secreted mediators. Platelets express a diverse range of receptors and a large proteasome, secreting a variety of soluble mediators, facilitating direct and indirect interactions with immune cells, the endothelium and the extracellular matrix. Pattern recognition receptors, PRRs; G-protein coupled receptors, GPCRs; Immunoreceptor tyrosine-based activation motif, ITAM; Platelet-derived extracellular vesicles, PEVs.
The role for thromboinflammation is evident in conditions traditionally associated with thrombosis including atherosclerosis (41), deep vein thrombosis (42) and reperfusion injury after ischemic stroke (43). There is also increasing evidence for a protective role in sepsis, limiting tissue injury and promoting pathogen clearance (37, 44–48). Clinically, thrombocytopenia is associated with poor outcomes in sepsis (44, 46) and platelet transfusion improves bacterial clearance through macrophage recruitment (48). With a wide array of PRRs platelets aid with pathogen clearance during viral infection, but also exacerbate tissue injury in response to DAMPs (49). Interestingly platelets demonstrate plasticity in function, as evidenced in major trauma, emphasizing temporally and spatially diverse roles in the immune response (50). In the acute phase of injury platelets are poorly responsive to activation ex vivo, contributing to coagulopathy, followed by hyper-responsiveness exhibiting a pro-inflammatory and prothrombotic phenotype resulting in secondary organ damage (50). Pathways modulating platelet immune functions without impairing normal hemostasis are attractive therapeutic targets. Whilst this is particularly relevant in acute liver injury where patients can have an unpredictable coagulopathy, the bleeding diathesis in ALF is arguably an overstated concern but beyond the scope of this current review (51, 52). Immunoreceptor tyrosine-based activation motif receptors C-type lectin-like receptor 2 (CLEC2) and GPVI share similar downstream pathways involved in platelet activation (Figure 2) (16). CLEC2, activated by endogenous ligand podoplanin, is important in hemostasis although, promisingly, blockade does not produce a hemorrhagic phenotype (53). On the other hand, the podoplanin-CLEC2 axis appears to be vital in several models of thromboinflammation (16). In infection these receptors have both beneficial and detrimental roles (13, 45–47, 54). GPVI promotes neutrophil recruitment during pneumonia (54), whilst inflammatory macrophages promote platelet aggregation via CLEC2 leading to pathogenic thrombosis in peritoneal sepsis (13). In other mouse models targeting the podoplanin-CLEC2 axis may have a role in limiting immune activation (45–47).
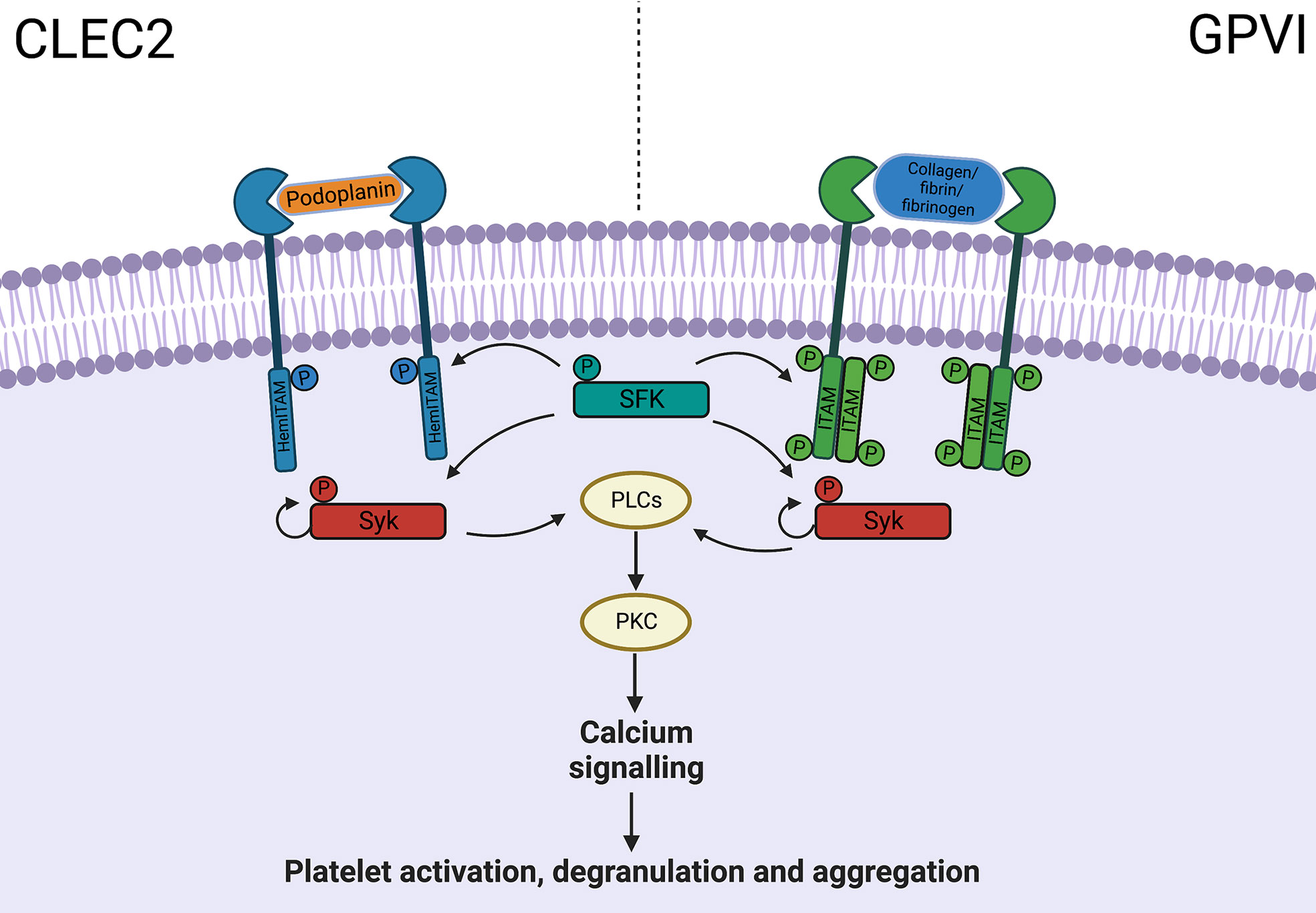
Figure 2 ITAM receptor downstream signalling pathway. CLEC-2 and GPVI share common downstream pathways, with activation leading to platelet aggregation and degranulation, through PLC, PKC and calcium signalling. C-type lectin-like receptor 2, CLEC2; Glycoprotein VI, GPVI; Immunoreceptor tyrosine-based activation motif, ITAM; Phosphate, P; Src family kinase, SFK; Phospholipase C, PLC; Protein kinase C, PKC.
In the liver, platelets promote hemostasis, fine tune the immune response through direct and indirect interactions and serve as a reservoir of biologically active substances (15). Unsurprisingly, platelet effector functions are complex and dualistic – often disease and stage specific (14).
LSECs constitute a unique vascular bed with a powerful scavenger system and potent endocytic capacity, aptly placed to respond appropriately to an array of antigens balancing immune activation and tolerance (55). LSECs orchestrate the immune response but also interact with hepatocytes and hepatic stellate cells in the process of regeneration or fibrosis (55). The hepatic sinusoids are classically narrow, characterised by low shear stress and thus initial recruitment is often selectin independent. The sinusoids express minimal levels of selectins in vivo (56, 57). Platelet sequestration is observed within hepatic sinusoids in numerous models of inflammation including APAP-induced (12), non-alcoholic steatohepatitis (58), ischemia-reperfusion injury (59, 60) and viral hepatitis (61).
Platelet-endothelial interactions are also bidirectional and vary between type of injury encountered. Due to the diverse range of receptors, surface ligands and ability to secrete soluble mediators, both cell types participate in recruitment, adhesion, and activation of immune cells (Figure 3A). In vitro studies have demonstrated platelet adhesion is, in part, integrin mediated (through GPIIbIIIa and αVβ3) (62). Platelet adhesion triggers CXCL8 and CCL2 production by endothelial cells, promoting leucocyte recruitment. In a model of liver ischemia-reperfusion injury, LSECs are highly susceptible during cold preservation (63). Depletion of adenosine triphosphate (ATP) impairs transmembrane active ion transport leading to cellular swelling and mitochondrial dysfunction (64). On reperfusion reactive oxygen species production depletes free radical scavengers, leading to activation with increased expression of P-selectin (60). Upregulation of P-selectin promotes platelet adhesion, activation and LSEC apoptosis (65). In a bile duct (BDL) model of cholestatic liver injury of mice, platelet accumulation promotes leucocyte sequestration and hepatocyte damage in a partially p-selectin dependent manner (66). In this model, the role of LSEC podoplanin expression has been explored (67). Podoplanin expression is increased in BDL-treated mice and those pre-treated with anti-CLEC2 antibodies had reduced hepatic inflammation and subsequent fibrosis. The authors hypothesized platelet-derived serotonin (5-HT) released on activation of CLEC2 was responsible for reduced injury and promoting regeneration. Plasma serotonin was found to be raised in untreated BDL-mice. PEVs have been shown to induce endothelial cell apoptosis in models of sepsis (68). In a study of patients presenting with ALF (50% APAP-induced), increased levels of circulating microparticles were associated with presence of systemic inflammatory response syndrome, high-grade hepatic encephalopathy and death or requirement for liver transplantation (69). This may implicate PEVs in driving inflammation in ALF or simply highlight their utility as a marker of systemic inflammation. In ischemia-reperfusion injury, PEV levels increase rapidly at initiation of injury (70) and inhibition of microparticle release has been shown to reduce degree of injury (71). Endothelial cells interact with resident and migrating leucocytes to promote platelet adhesion and activation. In a model of ischemia-reperfusion injury, migrating CD4 T cells were shown to interact with endothelial cells to promote platelet adhesion (72) and Kupffer cells produced a similar effect via tumor necrosis factor (TNF)-α secretion (73). Bidirectional communication promotes a self-perpetuating cycle of inflammation leading to significant immune-mediated damage. These murine and human data reveal how platelets modulate the inflammatory landscape in acute liver injury in non-fibrotic livers to potentially drive ALF.
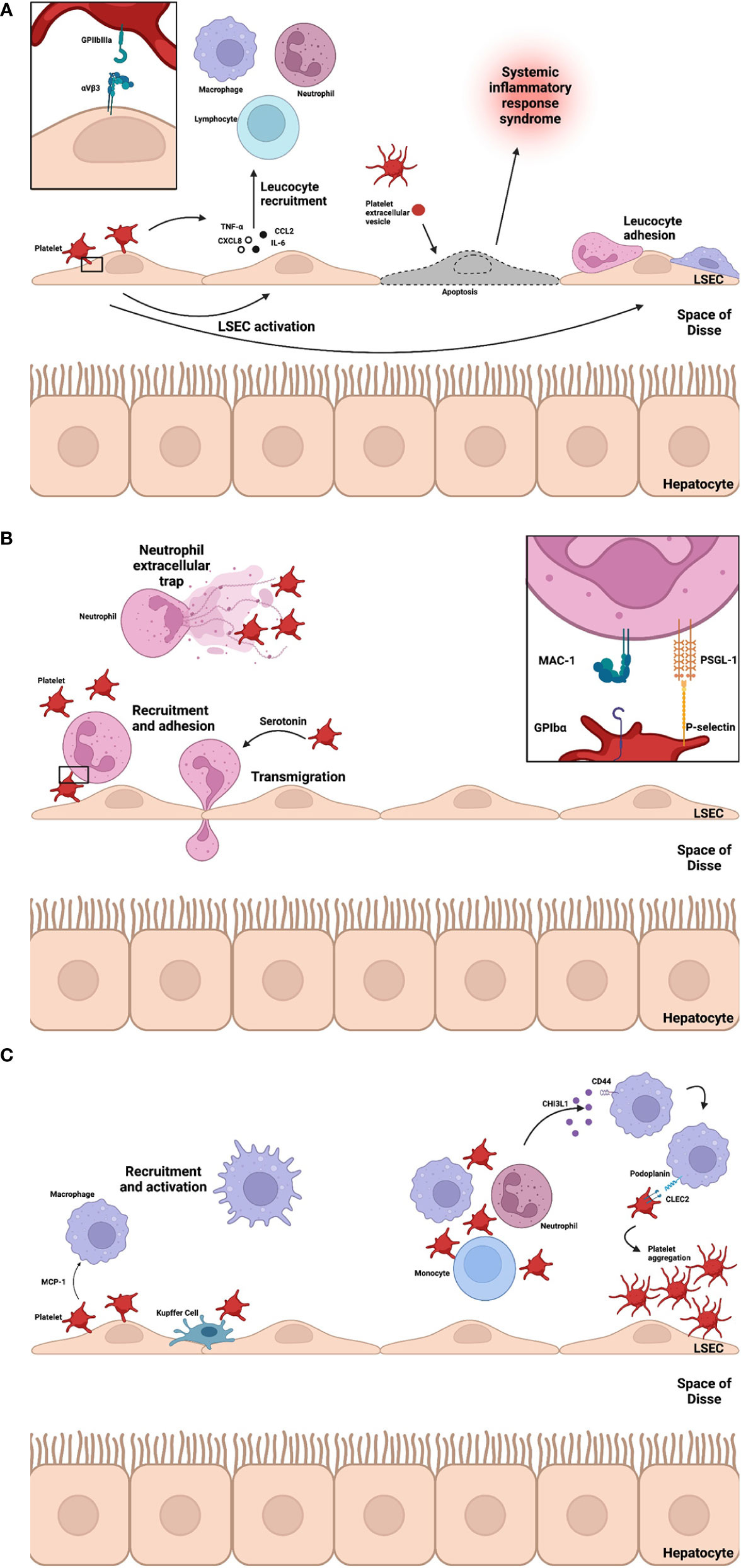
Figure 3 (A-C) Platelet interactions drive liver inflammation. At sites of inflammation platelets interact with LSECs leading to chemokine and cytokine (CXCL8, CCL2, IL-6 and TNF-α) secretion to facilitate recruitment of leucocytes. Platelet-LSEC interaction is partially integrin mediated (GPIIbIIIa and αVbeta3). PEVs rise rapidly at these sites and may induce apoptosis, driving systemic inflammation. Adherent platelets recruit neutrophils via P-selectin-PSGL-1 interactions and MAC-1 via GPIbα and platelet-derived 5-HT mediates neutrophil transmigration. The bidirectional relationship between platelet activation, aggregation and NETosis, a key effector function of neutrophils, is a key component of immunothrombosis and a driver of inflammation in ALF. During homeostasis KCs and platelets interact transiently through GPIb-vWF – A process of immune surveillance. Monocytes and macrophages are recruited to liver sinusoids through chemoattractant MCP-1. Recently, CHI3L1 release in models of ALF trigger podoplanin expression on macrophages which promote platelet aggregation and drive inflammation via CLEC2. Liver sinusoidal endothelial cell, LSEC; Glycoprotein, GP; Tumour necrosis factor alpha, TNF-α; CXC motif chemokine ligand 8, CXCL8; interleukin 6, IL-6; CC ligand 2, CCL2; Systemic inflammatory response syndrome, SIRS; Platelet-extracellular vesicle, PEV; Neutrophil extracellular trap, NET; Serotonin, 5-HT; Macrophage-1 antigen, MAC-1; P-selectin glycoprotein ligand-1, PSGL-1; Acute liver failure, ALF; Kupffer cell, KC; Von Willebrand Factor, vWF; Monocyte chemoattractant protein-1, MCP-1; chitinase-3 like protein-1, CHI3L1; C-type lectin-like receptor 2, CLEC2.
Evidence for thromboinflammation mediated liver damage in acute liver injury has clearly been demonstrated in viral models of murine hepatitis. Activated platelets in these models reduce sinusoidal blood flow through the secretion of vasoactive mediators including 5-HT, which then drives the development of inflammatory intrahepatic microthrombosis. Thromboinflammation thus directly delays effector cell recruitment reducing viral clearance and enhancing liver damage (74). During homeostasis, surface expression of CD39 on LSECs cleave ATP and adenosine diphosphate to adenosine monophosphate limiting platelet activation (75). In reperfusion injury, for example during ischemic hepatitis, CD39 expression is reduced promoting platelet activation via ATP. Injury is again potentiated through increased vascular tone and reduced blood flow from platelet-derived thromboxane A2 and 5-HT (59). Historic models reveal that during acute liver injury in rats endothelial damage causes the deposition of platelet rich thrombi within hepatic sinusoids (76). These models demonstrate that thromboinflammation disrupts hepatic microcirculation (77); reduced sinusoidal blood flow exacerbates hepatocyte dysfunction, increases DAMP expression and amplifies inflammation. These data provide a cogent explanation to how platelet driven thromboinflammation can drive both de novo acute liver failure for instance in virus induced ALF but also decompensation and ACLF in patients who contract a viral infection or suffer an ischemic hepatitis on a background of established liver cirrhosis.
Non-selective beta blockers are used in cirrhosis to reduce portal pressure. They also exert beneficial non-hemodynamic effects in relation to bacterial translocation, particularly reducing incidence of ACLF secondary to bacteria-induced systemic inflammation (CANONIC study) (78). Recently reduced von Willebrand Factor (vWF) levels, as a marker of endothelial dysfunction, were identified as a possible marker of non-hemodynamic non-selective beta blocker effect (79). This may illustrate a link between endothelial dysfunction, platelets and bacteria-induced systemic inflammation in ACLF. More recently, examination into predictors of ACLF development during acute decompensation identified severity of inflammation as a key determinant of the development of portal venous thrombosis; again highlighting the link between inflammation and thrombosis in the context of liver cirrhosis (80). Further evidence for the role of immunothrombosis is observed in severity of decompensated cirrhosis (81). Here, portal and hepatic vein sampling was performed in patients undergoing transjugular intrahepatic portal systemic shunt insertion and demonstrated higher markers of platelet activation, lipopolysaccharide levels and inducible nitric oxide synthetase in the portal vein. This correlated with clinical disease severity, linking bacterial translocation and platelet activation with progression of disease. The specific molecular endothelial triggers for platelet activation within the liver and how this varies when comparing cirrhotic to non-cirrhotic livers merits further investigation.
Neutrophils are one of the key effector cells in the innate immune system responsible for driving inflammation and tissue injury in the liver (82–86). They are essential to host defense (87) but can also contribute to indiscriminate tissue injury and are responsible for cell damage in many diseases (88, 89). Neutrophil recruitment and activation is evident in a multitude of acute liver injuries which can drive acute liver failure including drug-induced liver injury (82), and ischemia-reperfusion injury (86) but also ACLF including viral hepatitis (83), alcoholic hepatitis (84), and non-alcoholic steatohepatitis (85). Neutrophils infiltrate the site of liver injury within minutes to hours via stimulated LSECs involves a process of selectin-mediated rolling, integrin-mediated firm adhesion followed by transendothelial migration in a well-described recruitment cascade (89). Recruitment to the liver during inflammation is influenced by resident Kupffer cells (90–92), LSECs (93) and stromal cells (94). Necrotic cells release necrotaxis signals including mitochondrial formylated peptides to facilitate precise homing (91). Neutrophils migrate through intravascular channels and exhibit swarming behaviour at the site of injury (91). In addition to initiating and propagating inflammation, neutrophils promote resolution and a return to homeostasis in focal thermal (24), APAP- (12) and carbon tetrachloride-induced (95) liver injury through clearance of cellular debris, production of extracellular matrix and normal revascularization. During ACLF neutrophils exhibit an exhausted phenotype – characterised by high levels of activation (96) but impaired core functions including phagocytosis, reactive oxygen specifies production and degranulation (97). Lower CXCR1/2 expression on neutrophils, a key chemotactic receptor, predicted poor outcome in hepatitis B-virus (HBV) related ACLF (98). On the other hand, neutrophil-to-lymphocyte ratio has been shown to be an independent predictor of prognosis in HBV ACLF (99). Interestingly, patients with ratios ≥3 had lower mortality, but those with >6 were at greater risk of mortality in ACLF.
Platelets contribute to neutrophil recruitment and activation through ligand-receptor interactions and chemokine secretion (Figure 3B) (10, 100, 101). Initial interactions between platelet P-selectin and neutrophil PSGL-1 are critical for recruitment, activation of MAC-1 and LFA-1 (lymphocyte function-associated antigen 1, an integrin expressed on leucocytes) and release of neutrophil extracellular traps (NETs) (102, 103). The importance of platelet-neutrophil interactions via p-selectin is illustrated by improved survival of p-selectin deficient mice in an ischemia-reperfusion injury model of acute liver injury (104). MAC-1 allows direct binding with platelets via GPIbα and indirectly to GPIIbIIIa via fibrinogen (105). Recruitment is further amplified through secretion of cytokines, chemokines and growth factors including platelet factor 4 (PF4), interleukin(IL)-1, RANTES, beta-thromboglobulin, platelet-derived growth factor (PDGF), platelet-activating factor, CXCL7, migration inhibiting factor, thromboxane A2 and 5-HT (10). The interaction between platelets and neutrophils in the liver has been observed in models of sterile injury (24). Using intravital confocal microscopy, platelets were observed to line the endothelium at sites of focal injury facilitating neutrophil rolling through GPIIbIIIa dependent mechanisms.
ACLF, often triggered by sepsis, rapidly leads to multiorgan dysfunction (6), and platelets may contribute to development of the systemic inflammatory response. In a mouse model of endotoxemia-mediated acute lung injury, P-selectin-PSGL-1 interactions were investigated (106). Administration of PSGL-1 blocking antibody reduced recruitment of neutrophils, platelet-neutrophil aggregates, lung injury and survival. It may be of interest to assess the role of this interaction in models of ALF and ACLF. Whilst platelet-neutrophil interactions may exacerbate injury, recent work may highlight a beneficial role for platelet-derived 5-HT in neutrophil recruitment to sites of inflammation. Giovanni et al. (107), demonstrate 5-HIAA, a metabolite of platelet-derived 5-HT, mediates neutrophil recruitment and transmigration to inflamed tissues via GPR35. Here, treatment with serotonin inhibitors diminished recruitment of neutrophils and clearance of peritoneal bacteria.
How platelets influence neutrophil recruitment and final phenotype within the damaged liver and whether parallels from data in other organs can be extrapolated to the liver is key to develop rational antiplatelet therapies in liver disease.
Neutrophils release NETs as an antimicrobial effector mechanism (108, 109). These consist of a fibrous mesh of decondensed DNA mixed with a number of nuclear and granular proteins capturing and neutralizing microbes in an attempt to prevent dissemination (110). In the liver, NETs are released in response to infection, ischemia and sterile inflammation (111). Whilst providing an important role in pathogen defense, NETs are cytotoxic towards host cells (112). The neutrophil-platelet-NET axis is a complex interaction whereby platelet-mediated recruitment and activation of neutrophils at sites of inflammation triggers NET release (113). NET release in response to platelet activation is integrin- (114) and potentially selectin-mediated (115). Histone and polyphosphate entities within the NET matrix subsequently activate platelets through TLRs and directly activate coagulation within the bloodstream (116, 117). The neutrophil-platelet-NET axis illustrates the important role for link between infection, inflammation and thrombosis enhancing host immunity (118). In models of sepsis (116, 119), and recently in a galactosamine hydrochloride and lipopolysaccharide model of ALF (120), NETs contribute to tissue damage. Importantly, inhibition of NET generation or DNAse to breakdown NET reduces observed collateral tissue damage (116, 119). Cell-free DNA, often referred to as a NET marker (albeit somewhat non-specific) is associated with mortality in ACLF though the link with the more specific myeloperoxidase-DNA was not established (121). In a study by von Meijenfeldt et al (122), 676 patients with ALF were recruited from the U.S ALF Study group. Forty-six percent had APAP-induced ALF. Cell-free DNA and myeloperoxidase-DNA complexes were measured in comparison to healthy controls and tissue obtained at liver transplantation was stained for NETs in 20 patients. Levels of cell-free DNA and myeloperoxidase-DNA complexes were 7.1-fold and 2.5-fold higher than healthy controls respectively. High cell-free DNA was not associated with mortality. Myeloperoxidase-DNA levels were 30% higher in patients with ALF who died or required urgent liver transplant. The observed differences between ALF and ACLF may be explained, in part, by innate immune cell dysfunction in ACLF. NETs may represent an attractive therapeutic target to reduce immunothrombosis and cytotoxic cell damage, however this needs to be carefully balanced with loss of beneficial immune function. In animal models of sepsis, disruption of NET formation reduced liver injury and microcirculation thrombosis without impairing bacterial clearance (123–125). More research is required to accurately measure NET formation (108) and their impact in different models of liver injury.
Monocytes and macrophages have a critical role in homeostatic immune mechanisms, immune-mediated liver injury, fibrosis and regeneration (126). Infiltrating the site of injury 24-48hrs after neutrophils, monocytes perform diverse functions during ALF including inflammatory mediator release, clearance of dead cells, stimulation of the extracellular matrix and parenchymal regeneration (18). Distinct subsets of monocytes possess predominately inflammatory or anti-inflammatory phenotypes (Table 1) (18). Differentiation into macrophages is also an important function (127, 128). Platelet interactions are evident in immune surveillance functions of the liver, initiation of inflammation, recruitment of monocytes and polarisation to a pro-inflammatory macrophage profile (Figure 3C) (14, 15). Monocytes, sharing common regulatory and effector properties with neutrophils, are recruited to inflamed endothelium by platelets in a similar fashion (15). Efficient monocyte recruitment requires specific stimuli, namely monocyte chemoattractant protein 1 (MCP-1) (10).

Table 1 Monocyte subsets.
The interaction between Kupffer cells and platelets is an important step in initial pathogen detection. Platelets survey macrophages through transient GPIb-vWF interactions during homeostasis (129). In the presence of blood-borne bacteria, sustained platelet-macrophage interactions are observed through vWF-GPIIb/IIIa encasing the bacterium and facilitating clearance. Increasing evidence is suggesting that the podoplanin-CLEC2 axis is central to platelet interactions with macrophages. Recently, Shan et al. (130), identified a novel interaction between platelets and macrophages potentiating APAP-induced liver injury through chitinase-3 like protein-1 (CHI3L1). CHI3L1 is a soluble protein released by multiple immune cells and found to be raised in a range of liver disease (131). In this study, CHI3L1 interaction via CD44 on macrophages upregulated podoplanin expression and subsequent platelet aggregation via CLEC2. In this model, disruption of the pathway at the level of CHI3L1 and podoplanin-CLEC2 greatly inhibited liver injury after APAP administration. In the model of APAP-induced liver injury (132), platelet depletion greatly reduced tissue damage and the study by Shan et al (130), highlights one potential pathway underlying this – its role in the other models of liver injury are yet to be determined.
Monocytes participate in early stages of the innate immune response to acute liver injury through cytokine production, antigen presentation and polarisation to inflammatory macrophages (30). Plasticity in response to the local microenvironment is demonstrated with anti-inflammatory monocytes appearing 12-24 hours later promoting resolution via IL-10, transforming growth factor beta (TGF-β) and vascular endothelial growth factor (VEGF) (133–136). This arises from recruitment and in situ reprogramming (136). Apart from monocyte differentiation, specific macrophage populations resident to the peritoneal cavity, characterised by GATA6 expression, are recruited to the inflamed liver to assist in liver repair (137–140). This appears to be mediated by ATP release and exposed hyaluronan (137, 138). Recently, Jin et al. (139), demonstrated through dual recombinase mediated genetic GATA6+ lineage tracing, macrophages are only recruited to surface of liver during carbon tetrachloride-induced liver injury, questioning a potential role in ALF.
In ACLF, there is evidence of dampened function, characterised by reduced human leukocyte antigen (HLA)-DR expression, correlated with high mortality rates and increased prothrombin time (31). Underlying immunoparesis in ACLF, there is also expansion of several monocyte populations including MERTK+ (32), monocytic myeloid-derived suppressor cells (141) and intermediate CD14++CD16+ (142) with classical monocytes also exhibiting impaired function, characterised by reduced TLR2/4 expression, phagocytic activity and upregulation of genes related to dampened immune response (142). Monocyte/macrophage polarisation and plasticity is influenced by platelet activity. In vitro studies by Lee et al. (143), demonstrated adenosine diphosphate-activated platelets induced CD16 expression on CD14+CD16- monocytes from platelet-derived TGF-β and monocyte-derived IL-6. These monocytes preferentially differentiated towards M2 macrophages expressing CD163 and MerTK. It may be postulated that in platelet-monocyte interactions in advanced cirrhosis contributes to immunoparesis through expansion of MerTK macrophages precipitating widespread inflammation in ACLF. In contrast, in vitro lipopolysaccharide-treated monocytes co-incubated with platelets are skewed from an M2 towards a pro-inflammatory M1 phenotype demonstrating increased TNF-α expression, improved bacterial phagocytic activity and reduced healing capability (48). In vivo platelet transfusion increased inducible nitrous oxide synthase-expressing macrophages, improving bacterial clearance and survival in septic mice. In both of these settings, blockade of CD11b-GPIb interaction abolished the effect. The effect of platelet-macrophage interactions in liver disease is not limited to the hepatic environment as in a murine model of acute liver injury monocyte-platelet aggregates modulated microglial activation and drove the development of sickness behaviors in TLR4-dependent pathways (140).
Platelet interactions with monocytes and macrophages are complex and diverse. Given the distinct cellular niches that exist within fibrotic livers (144), studying spatio-temporal platelet driven immunothrombosis in acute and chronic liver disease and how this influences macrophage phenotype and function in liver inflammation remains an exciting avenue to study.
Platelets play a provocative role in the resolution of inflammation with the ability to potentiate immune-mediated damage, promote regeneration and drive fibrosis (Figure 4). This complex relationship may reflect limitations in models used to study liver inflammation, represent gaps in our knowledge or identify roles for platelets that vary throughout and in different types of injury.
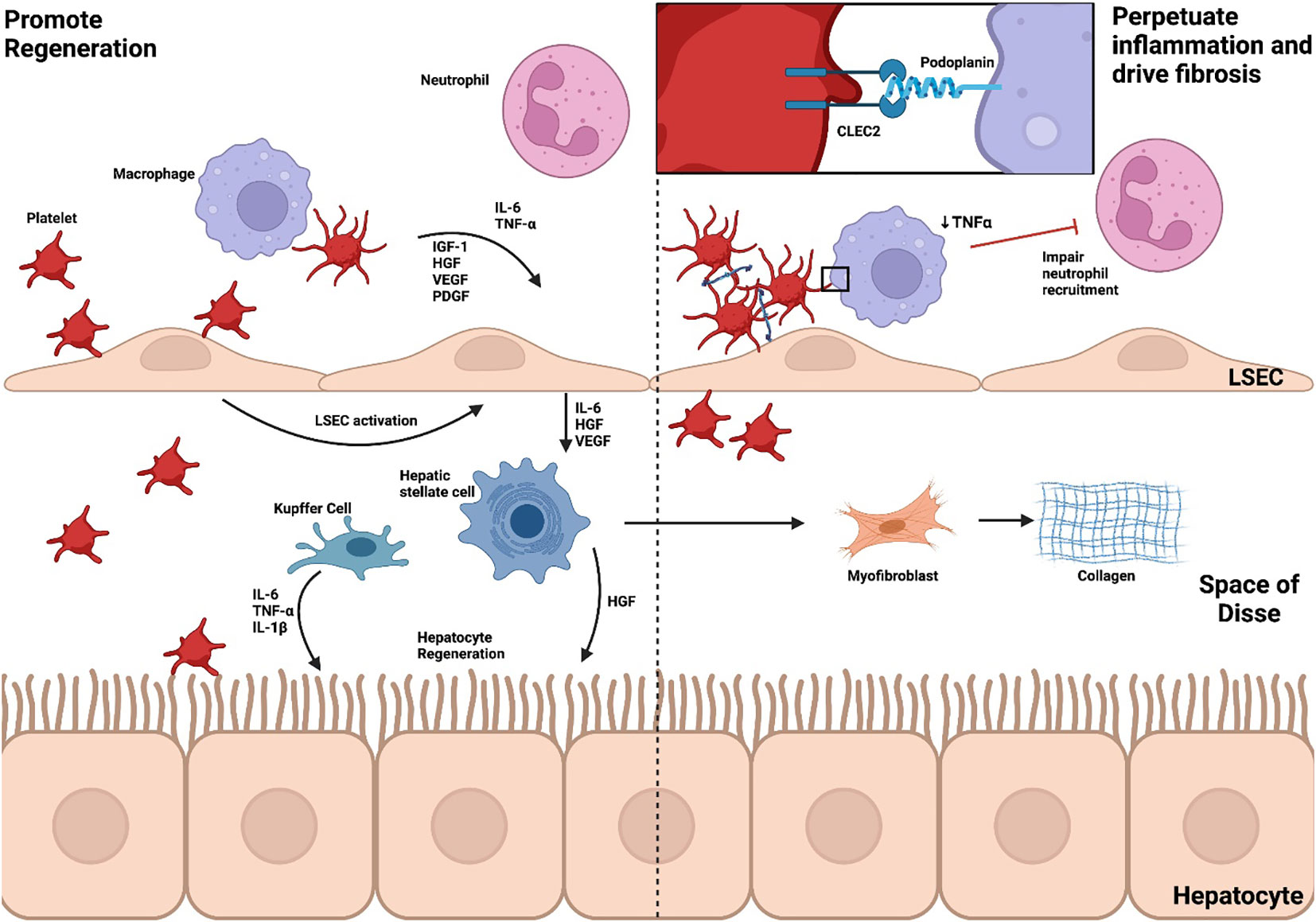
Figure 4 Platelets promote regeneration, perpetuate inflammation and drive fibrosis. The role of platelets in the resolution of inflammation is dualistic, potentiating immune-mediated damage, promoting regeneration and driving fibrosis. Through CLEC2, platelets activate LSECs to produce IL-6, TNF-α, HGF and VEGF which, in turn, triggers HGF release from HSCs to stimulate hepatocyte regeneration. HSC activation also leads to myofibroblast differentiation and collagen synthesis. Neutrophils, critical to resolution of inflammation, are also recruited through cytokine and chemokine release. In an APAP model of ALF, however, platelet-macrophage interactions through CLEC2 reduce neutrophil recruitment and perpetuate inflammation. Liver sinusoidal endothelial cell, LSEC; Kupffer cell, KC; Hepatic stellate cell, HSC; interleukin, IL; Tumour necrosis factor alpha, TNF-α; Insulin-like growth factor-1, IGF-1; Hepatocyte growth factor, HGF; Vascular endothelial growth factor, VEGF; Platelet-derived growth factor, PDGF; C-type lectin-like receptor 2, CLEC2; Acetaminophen, APAP; Acute liver failure, ALF.
During liver regeneration, hepatocyte proliferation is controlled by a multitude of extracellular signals including cytokine, growth factor and metabolic pathways (15). TNF-α and IL-6 are cytokines central to regulation of liver regeneration whilst growth factors such as hepatocyte growth factor, endothelial growth factor, Insulin-like growth factor-1 and PDGF drive cell cycle progression (145). Platelets accumulate within the space of Disse rapidly after partial hepatectomy and have an active role in hepatic regeneration (146–148). Such a role for platelets is also appreciated in the clinical setting in a recent meta-analysis of 3966 patients (149). In this study preoperative thrombocytopenia constituted a significant risk factor for post-hepatectomy liver failure. Platelets mediate regeneration through interactions with LSECs, Kupffer cells and hepatocytes. These interactions are facilitated by direct contact and platelet-derived soluble mediators including hepatocyte growth factor, insulin-like growth factor 1, PDGF, VEGF, 5-HT, adenosine diphosphate and ATP (150). Downstream cascades key to these pathways include TNF-α/NF-kB, Il-6/STAT3, phosphatidylinositol 3-kinase (PI3K)/Akt and ERK1/2 (15, 151).
LSECs produce mitotic substances, specifically IL-6, hepatocyte growth factor and VEGF (152). Direct adhesion between platelets and LSECs induces IL-6 release, in turn, leading to hepatocyte growth factor secretion from hepatic stellate cells promoting hepatocyte regeneration in vitro (153). This is likely controlled through podoplanin-CLEC2 signalling (154) and expression of vWF (155). Soluble mediators released by activated platelets including TGF–β1 (156) and sphingosine-1-phosphate (157) are also sufficient to support IL-6 production by LSECs.
Kupffer cells are an important source of regenerative cytokines TNF-α, IL-6 and IL-1β (126). Depletion of Kupffer cells impairs hepatocyte proliferation during liver regeneration in cholestatic injury (158), alcohol-induced injury (159) and partial hepatectomy (147). Platelets contribute to regeneration through promoting TNF-α production by Kupffer cells, however utilize alternate pathways in Kupffer cell depleted environments (147).
Platelet-derived 5-HT is an important mediator of liver regeneration, most likely through production of growth factors at the site of injury (160, 161). Recently a multi-center trial has demonstrated that perioperative use of selective serotonin reuptake inhibitors and serotonin noradrenaline reuptake inhibitors is associated with adverse outcomes after hepatic resection further supporting a role for 5-HT (162).
Within the space of Disse, platelets are also able to interact with hepatocytes directly. Internalization of platelets and platelet-like particles followed by horizontal transfer of mRNA has also been demonstrated to contribute to hepatocyte proliferation (163). Linking coagulation and inflammation, liver-specific tissue factor release promotes platelet accumulation through fibrin(ogen) deposition, facilitating resolution after partial hepatectomy (164).
Whilst it is generally accepted that platelets are able to promote liver regeneration through a variety of mechanisms, there is emerging evidence implicating them in delaying resolution (12, 77). Neutrophils are not only central drivers of inflammation but also promote resolution during sterile injury (24). Recently our group demonstrated signalling via podoplanin-CLEC2 between platelets and inflammatory macrophages reduced TNF-α secretion and subsequent neutrophil recruitment to facilitate resolution of inflammation in APAP toxicity (12). Moreover, increased vWF deposition and impaired clearance has been attributed to persisting platelet accumulation in APAP-induced liver disease (77). In this model, deficiency or inhibition of vWF accelerated resolution. These results add a further layer of complexity to our understanding highlighting that targeting platelets may be temporally sensitive but also vary in different models of liver injury (12, 77, 155). Partial hepatectomy is the most commonly studied model of liver regeneration, however pathways involved in injury and resolution vary in other models (165). Thus, further investigation is required in different models of ALF and ACLF to elucidate specific roles of platelets and identify potential therapeutic targets.
Platelets play a vital role at all stages of acute liver injury through direct and indirect interactions with immune cells, stromal cells and the endothelium. Their involvement is both beneficial and detrimental. On one hand, they can intelligently sense and appropriately respond to pathogens, recruit leucocytes and promote regeneration at the resolution of inflammation. On the other, they exaggerate immune-mediated tissue injury, worsen hepatocyte dysfunction through microthrombi formation and delay mechanisms of resolution. Pathways controlling platelet effector function, such as the podoplanin-CLEC2 axis, represent an attractive therapeutic target however the disease-specific and temporal roles of platelets need to be carefully dissected in order to develop effective disease modifying treatments.
SM – conceptualization, writing of original draft and editing. AC – overall supervision of article. All authors contributed to the article and approved the submitted version.
All figures created with BioRender.com
Author AC is recipient of a Wellcome trust clinical fellowship and an NIHR clinical lectureship. AC has received consultation fees from Principia biopharma now part of Sanofi.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Bernal W, Wendon J. Acute liver failure. N Engl J Med (2013) 369(26):2525–34. doi: 10.1056/nejmra1208937
2. Chiew AL, Gluud C, Brok J, Buckley NA. Interventions for paracetamol (acetaminophen) overdose. Cochrane Database Syst Rev (2018) 2(2):CD003328. doi: 10.1002/14651858.CD003328.pub3
3. Kumar R, Anand U, Priyadarshi RN. Liver transplantation in acute liver failure: Dilemmas and challenges. World J Transplant (2021) 11(6):187–202. doi: 10.5500/wjt.v11.i6.187
4. Abbas N, Rajoriya N, Elsharkawy AM, Chauhan A. Acute-on-chronic liver failure (ACLF) in 2022: have novel treatment paradigms already arrived? (2022). Available at: https://www.tandfonline.com/action/journalInformation?journalCode=ierh20.
5. Gustot T, Stadlbauer V, Laleman W, Alessandria C, Thursz M. Transition to decompensation and acute-on-chronic liver failure: Role of predisposing factors and precipitating events. J Hepatol (2021) 75:S36–48. doi: 10.1016/j.jhep.2020.12.005
6. Moreau R, Jalan R, Gines P, Pavesi M, Angeli P, Cordoba J, et al. Acute-on-chronic liver failure is a distinct syndrome that develops in patients with acute decompensation of cirrhosis. Gastroenterology (2013) 144(7):1426–1437.e9. doi: 10.1053/j.gastro.2013.02.042
7. Hernaez R, Solà E, Moreau R, Ginès P. Acute-on-chronic liver failure: an update. Gut (2017) 66(3):541–53. doi: 10.1136/gutjnl-2016-312670
8. Garraud O, Cognasse F. Are platelets cells? and if yes, are they immune cells? Front Immunol (2015) 6(FEB):70. doi: 10.3389/fimmu.2015.00070
9. Mezger M, Nording H, Sauter R, Graf T, Heim C, von Bubnoff N, et al. Platelets and immune responses during thromboinflammation. Front Immunol (2019) 10:1731. doi: 10.3389/fimmu.2019.01731
10. Rayes J, Bourne JH, Brill A, Watson SP. The dual role of platelet-innate immune cell interactions in thrombo-inflammation. Res Pract Thromb Haemost (2020) 4(1):23–35. doi: 10.1002/rth2.12266
11. Mandel J, Casari M, Stepanyan M, Martyanov A, Deppermann C. Beyond hemostasis: Platelet innate immune interactions and thromboinflammation. Int J Mol Sci (2022) 23(7):3868. doi: 10.3390/ijms23073868
12. Chauhan A, Sheriff L, Hussain MT, Webb GJ, Patten DA, Shepherd EL, et al. The platelet receptor CLEC-2 blocks neutrophil mediated hepatic recovery in acetaminophen induced acute liver failure. Nat Commun (2020) 11(1):1–12. doi: 10.1038/s41467-020-15584-3
13. Hitchcock JR, Cook CN, Bobat S, Ross EA, Flores-Langarica A, Lowe KL, et al. Inflammation drives thrombosis after salmonella infection via CLEC-2 on platelets. J Clin Invest (2015) 125(12):4429. doi: 10.1172/JCI79070
14. Chauhan A, Adams DH, Watson SP, Lalor PF. Platelets: No longer bystanders in liver disease. Hepatology (2016) 64(5):1774–84. doi: 10.1002/hep.28526
15. Mussbacher M, Brunnthaler L, Panhuber A, Starlinger P, Assinger A. Till death do us part–the multifaceted role of platelets in liver diseases. Int J Mol Sci (2021) 22(6):3113. doi: 10.3390/ijms22063113
16. Meng D, Luo M, Liu B. The role of CLEC-2 and its ligands in thromboinflammation. Front Immunol (2021) 12:688643. doi: 10.3389/fimmu.2021.688643
17. Kubes P, Mehal WZ. Sterile inflammation in the liver. Gastroenterology (2012) 143(5):1158–72. doi: 10.1053/j.gastro.2012.09.008
18. Mcdonald B, Kubes P. Innate immune cell trafficking and function during sterile inflammation of the liver. Gastroenterology (2016) 151(6):1087–95. doi: 10.1053/j.gastro.2016.09.048
19. Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol (1994) 12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015
20. Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol (2008) 8(4):279–89. doi: 10.1038/nri2215
21. Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol (2010) 11(10):700–14. doi: 10.1038/nrm2970
22. Silva MT. Secondary necrosis: the natural outcome of the complete apoptotic program. FEBS Lett (2010) 584(22):4491–9. doi: 10.1016/j.febslet.2010.10.046
23. Crispe IN. The liver as a lymphoid organ. Annu Rev Immunol (2009) 27:147–63. doi: 10.1146/annurev.immunol.021908.132629
24. Slaba I, Wang J, Kolaczkowska E, Mcdonald B, Lee WY, Kubes P. Imaging the dynamic platelet-neutrophil response in sterile liver injury and repair in mice. Hepatology (2015) 62(5):1593–605. doi: 10.1002/hep.28003
25. Clària J, Stauber RE, Coenraad MJ, Moreau R, Jalan R, Pavesi M, et al. Systemic inflammation in decompensated cirrhosis: Characterization and role in acute-on-chronic liver failure. Hepatology (2016) 64(4):1249–64. doi: 10.1002/hep.28740
26. Casulleras M, Zhang IW, López-Vicario C, Clària J. Leukocytes, systemic inflammation and immunopathology in acute-on-Chronic liver failure. Cells (2020) 9(12):2632. doi: 10.3390/cells9122632
27. Fernández J, Acevedo J, Wiest R, Gustot T, Amoros A, Deulofeu C, et al. Bacterial and fungal infections in acute-on-chronic liver failure: prevalence, characteristics and impact on prognosis. Gut (2018) 67(10):1870–80. doi: 10.1136/gutjnl-2017-314240
28. Wiest R, Lawson M, Geuking M. Pathological bacterial translocation in liver cirrhosis. J Hepatol (2014) 60(1):197–209. doi: 10.1016/j.jhep.2013.07.044
29. Aizawa S, Brar G, Tsukamoto H. Cell death and liver disease. Gut Liver (2020) 14(1):20–9. doi: 10.5009/gnl18486
30. Khanam A, Kottilil S. Abnormal innate immunity in acute-on-Chronic liver failure: Immunotargets for therapeutics. Front Immunol (2020) 11:2013. doi: 10.3389/fimmu.2020.02013
31. Xing T, Li L, Cao H, Huang J. Altered immune function of monocytes in different stages of patients with acute on chronic liver failure. Clin Exp Immunol (2007) 147(1):184–8. doi: 10.1111/j.1365-2249.2006.03259.x
32. Bernsmeier C, Pop OT, Singanayagam A, Triantafyllou E, Patel VC, Weston CJ, et al. Patients with acute-on-Chronic liver failure have increased numbers of regulatory immune cells expressing the receptor tyrosine kinase MERTK. Gastroenterology (2015) 148(3):603–615.e14. doi: 10.1053/j.gastro.2014.11.045
33. Fong KP, Barry C, Tran AN, Traxler EA, Wannemacher KM, Tang HY, et al. Deciphering the human platelet sheddome. Blood (2011) 117(1):e15–26. doi: 10.1182/blood-2010-05-283838
34. Balaphas A, Meyer J, Sadoul K, Fontana P, Morel P, Gonelle-Gispert C, et al. Platelets and platelet-derived extracellular vesicles in liver physiology and disease. Hepatol Commun (2019) 3(7):855–66. doi: 10.1002/hep4.1358
35. Ed Rainger G, Chimen M, Harrison MJ, Yates CM, Harrison P, Watson SP, et al. The role of platelets in the recruitment of leukocytes during vascular disease. Platelets (2015) 26(6):507–20. doi: 10.3109/09537104.2015.1064881
36. Rossaint J, Margraf A, Zarbock A. Role of platelets in leukocyte recruitment and resolution of inflammation. Front Immunol (2018) 9(NOV):2712. doi: 10.3389/fimmu.2018.02712
37. Marcoux G, Laroche A, Espinoza Romero J, Boilard E. Role of platelets and megakaryocytes in adaptive immunity. Platelets (2021) 32(3):340–51. doi: 10.1080/09537104.2020.1786043
38. Hillgruber C, Pöppelmann B, Weishaupt C, Steingräber AK, Wessel F, Berdel WE, et al. Blocking neutrophil diapedesis prevents hemorrhage during thrombocytopenia. J Exp Med (2015) 212(8):1255–66. doi: 10.1084/jem.20142076
39. Gros A, Syvannarath V, Lamrani L, Ollivier V, Loyau S, Goerge T, et al. Single platelets seal neutrophil-induced vascular breaches via GPVI during immune-complex-mediated inflammation in mice. Blood (2015) 126(8):1017–26. doi: 10.1182/blood-2014-12-617159
40. Mai SHC, Khan M, Dwivedi DJ, Ross CA, Zhou J, Gould TJ, et al. Delayed but not early treatment with DNase reduces organ damage and improves outcome in a murine model of sepsis. Shock (2015) 44(2):166–72. doi: 10.1097/SHK.0000000000000396
41. Stark K, Massberg S. Interplay between inflammation and thrombosis in cardiovascular pathology. Nat Rev Cardiol (2021) 189:2021. doi: 10.1038/s41569-021-00552-1
42. Brill A, Fuchs TA, Chauhan AK, Yang JJ, De Meyer SF, Köllnberger M, et al. Von willebrand factor-mediated platelet adhesion is critical for deep vein thrombosis in mouse models. Blood (2011) 117(4):1400–7. doi: 10.1182/blood-2010-05-287623
43. Stoll G, Nieswandt B. Thrombo-inflammation in acute ischaemic stroke — implications for treatment. Nat Rev Neurol (2019) 158:2019. doi: 10.1038/s41582-019-0221-1
44. De Stoppelaar SF, Van ‘t Veer C, Claushuis TAM, Albersen BJA, Roelofs JJTH, van der Poll T. Thrombocytopenia impairs host defense in gram-negative pneumonia-derived sepsis in mice. Blood (2014) 124(25):3781–90. doi: 10.1182/blood-2014-05-573915
45. Lax S, Rayes J, Wichaiyo S, Haining EJ, Lowe K, Grygielska B, et al. Platelet CLEC-2 protects against lung injury via effects of its ligand podoplanin on inflammatory alveolar macrophages in the mouse. Am J Physiol Lung Cell Mol Physiol (2017) 313(6):L1016–29. doi: 10.1152/ajplung.00023.2017
46. Rayes J, Lax S, Wichaiyo S, Watson SK, Di Y, Lombard S, et al. The podoplanin-CLEC-2 axis inhibits inflammation in sepsis. Nat Commun (2017) 8(1):1–14. doi: 10.1038/s41467-017-02402-6
47. Lax S, Rayes J, Thickett DR, Watson SP. Effect of anti-podoplanin antibody administration during lipopolysaccharide-induced lung injury in mice. BMJ Open Respir Res (2017) 4(1):e000257. doi: 10.1136/bmjresp-2017-000257
48. Carestia A, Mena HA, Olexen CM, Ortiz Wilczyñski JM, Negrotto S, Errasti AE, et al. Platelets promote macrophage polarization toward pro-inflammatory phenotype and increase survival of septic mice. Cell Rep (2019) 28(4):896–908.e5. doi: 10.1016/j.celrep.2019.06.062
49. Antoniak S, Mackman N. Platelets and viruses. Platelets (2021) 32(3):325–30. doi: 10.1080/09537104.2021.1887842
50. Vulliamy P, Kornblith LZ, Kutcher ME, Cohen MJ, Brohi K, Neal MD. Alterations in platelet behavior after major trauma: adaptive or maladaptive? Platelets (2021) 32(3):295. doi: 10.1080/09537104.2020.1718633
51. Tripodi A, Mannucci PM. The coagulopathy of chronic liver disease. N Engl J Med (2011) 365(2):147–56. doi: 10.1056/NEJMra1011170
52. Wendon J, Cordoba J, Dhawan A, Larsen FS, Manns M, Nevens F, et al. EASL clinical practical guidelines on the management of acute (fulminant) liver failure. J Hepatol (2017) 66(5):1047–81. doi: 10.1016/j.jhep.2016.12.003
53. Bender M, May F, Lorenz V, Thielmann I, Hagedorn I, Finney BA, et al. Combined in vivo depletion of glycoprotein VI and c-type lectin-like receptor 2 severely compromises hemostasis and abrogates arterial thrombosis in mice. Arterioscler Thromb Vasc Biol (2013) 33(5):926–34. doi: 10.1161/ATVBAHA.112.300672
54. Claushuis TAM, de Vos AF, Nieswandt B, Boon L, Roelofs JJTH, de Boer OJ, et al. Platelet glycoprotein VI aids in local immunity during pneumonia-derived sepsis caused by gram-negative bacteria. Blood (2018) 131(8):864–76. doi: 10.1182/blood-2017-06-788067
55. Shetty S, Lalor PF, Adams DH. Liver sinusoidal endothelial cells — gatekeepers of hepatic immunity. Nat Rev Gastroenterol Hepatol (2018) 15(9):555–67. doi: 10.1038/s41575-018-0020-y
56. Adams DH, Hubscher SG, Fisher NC, Williams A, Robinson M. Expression of e-selectin and e-selectin ligands in human liver inflammation. Hepatology (1996) 24(3):533–8. doi: 10.1002/hep.510240311
57. Campbell JJ, Qin S, Bacon KB, Mackay CR, Butcher EC. Biology of chemokine and classical chemoattractant receptors: differential requirements for adhesion-triggering versus chemotactic responses in lymphoid cells. J Cell Biol (1996) 134(1):255–66. doi: 10.1083/jcb.134.1.255
58. Malehmir M, Pfister D, Gallage S, Szydlowska M, Inverso D, Kotsiliti E, et al. Platelet GPIbα is a mediator and potential interventional target for NASH and subsequent liver cancer. Nat Med (2019) 25(4):641–55. doi: 10.1038/s41591-019-0379-5
59. Miyashita T, Nakanuma S, Ahmed AK, Makino I, Hayashi H, Oyama K, et al. Ischemia reperfusion-facilitated sinusoidal endothelial cell injury in liver transplantation and the resulting impact of extravasated platelet aggregation. Eur Surg (2016) 48(2):92. doi: 10.1007/s10353-015-0363-3
60. Massberg S, Enders G, Leiderer R, Eisenmenger S, Vestweber D, Krombach F, et al. Platelet-endothelial cell interactions during Ischemia/Reperfusion: The role of p-selectin. Blood (1998) 92(2):507–15. doi: 10.1182/blood.V92.2.507
61. Iannacone M, Sitia G, Isogawa M, Marchese P, Castro MG, Lowenstein PR, et al. Platelets mediate cytotoxic T lymphocyte–induced liver damage. Nat Med (2005) 11(11):1167–9. doi: 10.1038/nm1317
62. Lalor PF, Herbert J, Bicknell R, Adams DH. Hepatic sinusoidal endothelium avidly binds platelets in an integrin-dependent manner, leading to platelet and endothelial activation and leukocyte recruitment. Am J Physiol - Gastrointest Liver Physiol (2013) 304(5):G469. doi: 10.1152/ajpgi.00407.2012
63. Russo L, Gracia-Sancho J, García-Calderó H, Marrone G, García-Pagán JC, García-Cardeña G, et al. Addition of simvastatin to cold storage solution prevents endothelial dysfunction in explanted rat livers. Hepatology (2012) 55(3):921–30. doi: 10.1002/hep.24755
64. Upadhya GA, Topp SA, Hotchkiss RS, Anagli J, Strasberg SM. Effect of cold preservation on intracellular calcium concentration and calpain activity in rat sinusoidal endothelial cells. Hepatology (2003) 37(2):313–23. doi: 10.1053/jhep.2003.50069
65. Sindram D, Porte RJ, Hoffman MR, Bentley RC, Clavien PA. Platelets induce sinusoidal endothelial cell apoptosis upon reperfusion of the cold ischemic rat liver. Gastroenterology (2000) 118(1):183–91. doi: 10.1016/S0016-5085(00)70427-6
66. Laschke MW, Dold S, Menger MD, Jeppsson B, Thorlacius H. Platelet-dependent accumulation of leukocytes in sinusoids mediates hepatocellular damage in bile duct ligation-induced cholestasis. Br J Pharmacol (2008) 153(1):148–56. doi: 10.1038/sj.bjp.0707578
67. Maruyama S, Kono H, Furuya S, Shimizu H, Saito R, Shoda K, et al. Platelet c-type lectin-like receptor 2 reduces cholestatic liver injury in mice. Am J Pathol (2020) 190(9):1833–42. doi: 10.1016/j.ajpath.2020.05.009
68. Gambim MH, de Oliveira do Carmo A, Marti L, Veríssimo-Filho S, Lopes LR, Janiszewski M. Platelet-derived exosomes induce endothelial cell apoptosis through peroxynitrite generation: experimental evidence for a novel mechanism of septic vascular dysfunction. Crit Care (2007) 11(5):R107. doi: 10.1186/cc6133
69. Stravitz RT, Bowling R, Bradford RL, Key NS, Glover S, Thacker LR, et al. Role of procoagulant microparticles in mediating complications and outcome of acute liver injury/acute liver failure. Hepatology (2013) 58(1):304–13. doi: 10.1002/hep.26307
70. Freeman CM, Quillin RC, Wilson GC, Nojima H, Johnson BL, Sutton JM, et al. Characterization of microparticles after hepatic ischemia-reperfusion injury. PloS One (2014) 9(5):e97945. doi: 10.1371/journal.pone.0097945
71. Teoh NC, Ajamieh H, Wong HJ, Croft K, Mori T, Allison AC, et al. Microparticles mediate hepatic ischemia-reperfusion injury and are the targets of diannexin (ASP8597). PloS One (2014) 9(9):e104376. doi: 10.1371/journal.pone.0104376
72. Khandoga A, Hanschen M, Kessler JS, Krombach F. CD4+ T cells contribute to postischemic liver injury in mice by interacting with sinusoidal endothelium and platelets. Hepatology (2006) 43(2):306–15. doi: 10.1002/hep.21017
73. Peralta C, Fernández L, Panés J, Prats N, Sans M, Piqué JM, et al. Preconditioning protects against systemic disorders associated with hepatic ischemia-reperfusion through blockade of tumor necrosis factor-induced p-selectin up-regulation in the rat. Hepatology (2001) 33(1):100–13. doi: 10.1053/jhep.2001.20529
74. Lang PA, Contaldo C, Georgiev P, El-Badry AM, Recher M, Kurrer M, et al. Aggravation of viral hepatitis by platelet-derived serotonin. Nat Med (2008) 14(7):756–61. doi: 10.1038/nm1780
75. Enjyoji K, Sévigny J, Lin Y, Frenette PS, Christie PD, Am Esch JS, et al. Targeted disruption of cd39/ATP diphosphohydrolase results in disordered hemostasis and thromboregulation. Nat Med (1999) 5(9):1010–7. doi: 10.1038/12447
76. Hirata K, Ogata I, Ohta Y, Fujiwara K. Hepatic sinusoidal cell destruction in the development of intravascular coagulation in acute liver failure of rats. J Pathol (1989) 158(2):157–65. doi: 10.1002/path.1711580211
77. Groeneveld D, Cline-Fedewa H, Baker KS, Williams KJ, Roth RA, Mittermeier K, et al. Von Willebrand factor delays liver repair after acetaminophen-induced acute liver injury in mice. J Hepatol (2020) 72(1):146–55. doi: 10.1016/j.jhep.2019.09.030
78. Mookerjee RP, Pavesi M, Thomsen KL, Mehta G, MacNaughtan J, Bendtsen F, et al. Treatment with non-selective beta blockers is associated with reduced severity of systemic inflammation and improved survival of patients with acute-on-chronic liver failure. J Hepatol (2016) 64(3):574–82. doi: 10.1016/j.jhep.2015.10.018
79. Jachs M, Hartl L, Simbrunner B, Bauer D, Paternostro R, Scheiner B, et al. Decreasing von willebrand factor levels upon nonselective beta blocker therapy indicate a decreased risk of further decompensation, acute-on-chronic liver failure, and death. Clin Gastroenterol Hepatol (2022) 20(6):1362–1373.e6. doi: 10.1016/j.cgh.2021.07.012
80. Zanetto A, Pelizzaro F, Campello E, Bulato C, Balcar L, Gu W, et al. Severity of systemic inflammation is the main predictor of ACLF and bleeding in patients with acutely decompensated cirrhosis. J Hepatol (2022) S0168-8278(22):03110–5. doi: 10.1016/j.jhep.2022.09.005
81. Queck A, Carnevale R, Uschner FE, Schierwagen R, Klein S, Jansen C, et al. Role of portal venous platelet activation in patients with decompensated cirrhosis and TIPS. Gut (2020) 69(8):1535–6. doi: 10.1136/gutjnl-2019-319044
82. Liu ZX, Han D, Gunawan B, Kaplowitz N. Neutrophil depletion protects against murine acetaminophen hepatotoxicity. Hepatology (2006) 43(6):1220–30. doi: 10.1002/hep.21175
83. Takai S, Kimura K, Nagaki M, Satake S, Kakimi K, Moriwaki H. Blockade of neutrophil elastase attenuates severe liver injury in hepatitis b transgenic mice. J Virol (2005) 79(24):15142–50. doi: 10.1128/JVI.79.24.15142-15150.2005
84. Jaeschke H. Neutrophil-mediated tissue injury in alcoholic hepatitis. Alcohol (2002) 27(1):23–7. doi: 10.1016/S0741-8329(02)00200-8
85. Rensen SS, Bieghs V, Xanthoulea S, Arfianti E, Bakker JA, Shiri-Sverdlov R, et al. Neutrophil-derived myeloperoxidase aggravates non-alcoholic steatohepatitis in low-density lipoprotein receptor-deficient mice. PloS One (2012) 7(12):e52411. doi: 10.1371/journal.pone.0052411
86. De Oliveira THC, Marques PE, Proost P, Teixeira MMM. Neutrophils: a cornerstone of liver ischemia and reperfusion injury. Lab Invest (2018) 98(1):51–62. doi: 10.1038/labinvest.2017.90
87. Lekstrom-Himes JA, Gallin JI. Immunodeficiency diseases caused by defects in phagocytes. N Engl J Med (2000) 343(23):1703–14. doi: 10.1056/NEJM200012073432307
88. Ramaiah SK, Jaeschke H. Role of neutrophils in the pathogenesis of acute inflammatory liver injury. Toxicol Pathol (2007) 35(6):757–66. doi: 10.1080/01926230701584163
89. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol (2013) 13(3):159–75. doi: 10.1038/nri3399
90. Kono H, Karmarkar D, Iwakura Y, Rock KL. Identification of the cellular sensor that stimulates the inflammatory response to sterile cell death. J Immunol (2010) 184(8):4470–8. doi: 10.4049/jimmunol.0902485
91. McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CCM, et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science (2010) 330(6002):362–6. doi: 10.1126/science.1195491
92. Soehnlein O, Lindbom L. Phagocyte partnership during the onset and resolution of inflammation. Nat Rev Immunol (2010) 10(6):427–39. doi: 10.1038/nri2779
93. Imaeda AB, Watanabe A, Sohail MA, Mahmood S, Mohamadnejad M, Sutterwala FS, et al. Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J Clin Invest (2009) 119(2):305–14. doi: 10.1172/JCI35958
94. Tsung A, Klune JR, Zhang X, Jeyabalan G, Cao Z, Peng X, et al. HMGB1 release induced by liver ischemia involves toll-like receptor 4 dependent reactive oxygen species production and calcium-mediated signaling. J Exp Med (2007) 204(12):2913–23. doi: 10.1084/jem.20070247
95. Kwon HJ, Won YS, Park O, Feng D, Gao B. Opposing effects of prednisolone treatment on T/NKT cell- and hepatotoxin-mediated hepatitis in mice. Hepatology (2014) 59(3):1094–106. doi: 10.1002/hep.26748
96. Huang CH, Jeng WJ, Ho YP, Teng W, Hsieh YC, Chen WT, et al. Increased EMR2 expression on neutrophils correlates with disease severity and predicts overall mortality in cirrhotic patients. Sci Rep (2016) 6(1):1–10. doi: 10.1038/srep38250
97. Boussif A, Rolas L, Weiss E, Bouriche H, Moreau R, Périanin A. Impaired intracellular signaling, myeloperoxidase release and bactericidal activity of neutrophils from patients with alcoholic cirrhosis. J Hepatol (2016) 64(5):1041–8. doi: 10.1016/j.jhep.2015.12.005
98. Xu R, Bao C, Huang H, Lin F, Yuan Y, Wang S, et al. Low expression of CXCR1/2 on neutrophils predicts poor survival in patients with hepatitis b virus-related acute-on-chronic liver failure. Sci Rep (2016) 6:38714. doi: 10.1038/srep38714
99. Liu H, Zhang H, Wan G, Sang Y, Chang Y, Wang X, et al. Neutrophil-lymphocyte ratio: a novel predictor for short-term prognosis in acute-on-chronic hepatitis b liver failure. J Viral Hepat (2014) 21(7):499–507. doi: 10.1111/jvh.12160
100. Pittman K, Kubes P. Damage-associated molecular patterns control neutrophil recruitment. J Innate Immun (2013) 5(4):315–23. doi: 10.1159/000347132
101. Zarbock A, Polanowska-Grabowska RK, Ley K. Platelet-neutrophil-interactions: Linking hemostasis and inflammation. Blood Rev (2007) 21(2):99–111. doi: 10.1016/j.blre.2006.06.001
102. Lisman T. Platelet-neutrophil interactions as drivers of inflammatory and thrombotic disease. Cell Tissue Res (2018) 371(3):567–76. doi: 10.1007/s00441-017-2727-4
103. Sreeramkumar V, Adrover JM, Ballesteros I, Cuartero MI, Rossaint J, Bilbao I, et al. Neutrophils scan for activated platelets to initiate inflammation. Science (2014) 346(6214):1234–8. doi: 10.1126/science.1256478
104. Yadav SS, Howell DN, Steeber DA, Harland RC, Tedder TF, Clavien PA. P-selectin mediates reperfusion injury through neutrophil and platelet sequestration in the warm ischemic mouse liver. Hepatology (1999) 29(5):1494–502. doi: 10.1002/hep.510290505
105. Weber C, Springer TA. Neutrophil accumulation on activated, surface-adherent platelets in flow is mediated by interaction of mac-1 with fibrinogen bound to alphaIIbbeta3 and stimulated by platelet-activating factor. J Clin Invest (1997) 100(8):2085–93. doi: 10.1172/JCI119742
106. Wang X, Deng Hf, Tan C, Xiao Zh, Liu Md, Liu K, et al. The role of PSGL-1 in pathogenesis of systemic inflammatory response and coagulopathy in endotoxemic mice. Thromb Res (2019) 182:56–63. doi: 10.1016/j.thromres.2019.08.019
107. De Giovanni M, Tam H, Valet C, Xu Y, Looney MR, Cyster JG. GPR35 promotes neutrophil recruitment in response to serotonin metabolite 5-HIAA. Cell (2022) 185(5):815–830.e19. doi: 10.1016/j.cell.2022.01.010
108. Meijenfeldt FAV, Jenne CN. Netting liver disease: Neutrophil extracellular traps in the initiation and exacerbation of liver pathology. Semin Thromb Hemost (2020) 46(6):724–34. doi: 10.1055/s-0040-1715474
109. Gómez RM, López Ortiz AO, Schattner M. Platelets and extracellular traps in infections. Platelets (2021) 32(3):305–13. doi: 10.1080/09537104.2020.1718631
110. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science (2004) 303(5663):1532–5. doi: 10.1126/science.1092385
111. Huang H, Tohme S, Al-Khafaji AB, Tai S, Loughran P, Chen L, et al. Damage-associated molecular pattern-activated neutrophil extracellular trap exacerbates sterile inflammatory liver injury. Hepatology (2015) 62(2):600–14. doi: 10.1002/hep.27841
112. Gupta AK, Joshi MB, Philippova M, Erne P, Hasler P, Hahn S, et al. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Lett (2010) 584(14):3193–7. doi: 10.1016/j.febslet.2010.06.006
113. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med (2007) 13(4):463–9. doi: 10.1038/nm1565
114. Carestia A, Kaufman T, Rivadeneyra L, Landoni VI, Pozner RG, Negrotto S, et al. Mediators and molecular pathways involved in the regulation of neutrophil extracellular trap formation mediated by activated platelets. J Leukoc Biol (2016) 99(1):153–62. doi: 10.1189/jlb.3A0415-161R
115. Etulain J, Martinod K, Wong SL, Cifuni SM, Schattner M, Wagner DD. P-selectin promotes neutrophil extracellular trap formation in mice. Blood (2015) 126(2):242–6. doi: 10.1182/blood-2015-01-624023
116. McDonald B, Davis RP, Kim SJ, Tse M, Esmon CT, Kolaczkowska E, et al. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood (2017) 129(10):1357–67. doi: 10.1182/blood-2016-09-741298
117. Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood (2011) 118(7):1952–61. doi: 10.1182/blood-2011-03-343061
118. Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol (2012) 13(1):34–45. doi: 10.1038/nri3345
119. Jiménez-Alcázar M, Rangaswamy C, Panda R, Bitterling J, Simsek YJ, Long AT, et al. Host DNases prevent vascular occlusion by neutrophil extracellular traps. Sci (80- ) (2017) 358(6367):1202–6. doi: 10.1126/science.aam8897
120. Ye D, Yao J, Du W, Chen C, Yang Y, Yan K, et al. Neutrophil extracellular traps mediate acute liver failure in regulation of miR-223/Neutrophil elastase signaling in mice. Cell Mol Gastroenterol Hepatol (2022) 14(3):587–607. doi: 10.1016/j.jcmgh.2022.05.012
121. Blasi A, Patel VC, Adelmeijer J, Azarian S, Aziz F, Fernández J, et al. Plasma levels of circulating DNA are associated with outcome, but not with activation of coagulation in decompensated cirrhosis and ACLF. JHEP Rep Innov Hepatol (2019) 1(3):179–87. doi: 10.1016/j.jhepr.2019.06.002
122. von Meijenfeldt FA, Stravitz RT, Zhang J, Adelmeijer J, Zen Y, Durkalski V, et al. Generation of neutrophil extracellular traps in patients with acute liver failure is associated with poor outcome. Hepatology (2022) 75(3):623–33. doi: 10.1002/hep.32174
123. Zucoloto AZ, Jenne CN. Platelet-neutrophil interplay: Insights into neutrophil extracellular trap (NET)-driven coagulation in infection. Front Cardiovasc Med (2019) 6. doi: 10.3389/fcvm.2019.00085
124. Carestia A, Davis RP, Grosjean H, Lau MW, Jenne CN. Acetylsalicylic acid inhibits intravascular coagulation during staphylococcus aureus-induced sepsis in mice. Blood (2020) 135(15):1281–6. doi: 10.1182/blood.2019002783
125. Carestia A, Davis RP, Davis L, Jenne CN. Inhibition of immunothrombosis does not affect pathogen capture and does not promote bacterial dissemination in a mouse model of sepsis. Platelets (2020) 31(7):925–31. doi: 10.1080/09537104.2019.1704711
126. Wen Y, Lambrecht J, Ju C, Tacke F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell Mol Immunol (2020) 18(1):45–56. doi: 10.1038/s41423-020-00558-8
127. Mossanen JC, Krenkel O, Ergen C, Govaere O, Liepelt A, Puengel T, et al. Chemokine (C-c motif) receptor 2–positive monocytes aggravate the early phase of acetaminophen-induced acute liver injury. Hepatology (2016) 64(5):1667–82. doi: 10.1002/hep.28682
128. Crane MJ, Daley JM, Van Houtte O, Brancato SK, Henry WL, Albina JE. The monocyte to macrophage transition in the murine sterile wound. PloS One (2014) 9(1):e86660. doi: 10.1371/journal.pone.0086660
129. Wong CHY, Jenne CN, Petri B, Chrobok NL, Kubes P. Nucleation of platelets with blood-borne pathogens on kupffer cells precedes other innate immunity and contributes to bacterial clearance. Nat Immunol (2013) 14(8):785–92. doi: 10.1038/ni.2631
130. Shan Z, Li L, Atkins CL, Wang M, Wen Y, Jeong J, et al. Chitinase 3-like-1 contributes to acetaminophen-induced liver injury by promoting hepatic platelet recruitment. Elife (2021) 10:e68571. doi: 10.7554/eLife.68571.sa2
131. Wang S, Hu M, Qian Y, Jiang Z, Shen L, Fu L, et al. CHI3L1 in the pathophysiology and diagnosis of liver diseases. BioMed Pharmacother (2020) 131:110680. doi: 10.1016/j.biopha.2020.110680
132. Miyakawa K, Joshi N, Sullivan BP, Albee R, Brandenberger C, Jaeschke H, et al. Platelets and protease-activated receptor-4 contribute to acetaminophen-induced liver injury in mice. Blood (2015) 126(15):1835–45. doi: 10.1182/blood-2014-09-598656
133. Nahrendorf M, Pittet MJ, Swirski FK. Monocytes: protagonists of infarct inflammation and repair after myocardial infarction. Circulation (2010) 121(22):2437–45. doi: 10.1161/CIRCULATIONAHA.109.916346
134. Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science (2010) 327(5966):656–61. doi: 10.1126/science.1178331
135. Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med (2007) 204(12):3037–47. doi: 10.1084/jem.20070885
136. Dal-Secco D, Wang J, Zeng Z, Kolaczkowska E, Wong CHY, Petri B, et al. A dynamic spectrum of monocytes arising from the in situ reprogramming of CCR2+ monocytes at a site of sterile injury. J Exp Med (2015) 212(4):447–56. doi: 10.1084/jem.20141539
137. Wang J, Kubes P. A reservoir of mature cavity macrophages that can rapidly invade visceral organs to affect tissue repair. Cell (2016) 165(3):668–78. doi: 10.1016/j.cell.2016.03.009
138. Honda M, Kadohisa M, Yoshii D, Komohara Y, Hibi T. Directly recruited GATA6 + peritoneal cavity macrophages contribute to the repair of intestinal serosal injury. Nat Commun (2021) 12(1):7294. doi: 10.1038/s41467-021-27614-9
139. Jin H, Liu K, Tang J, Huang X, Wang H, Zhang Q, et al. Genetic fate-mapping reveals surface accumulation but not deep organ invasion of pleural and peritoneal cavity macrophages following injury. Nat Commun (2021) 12(1):1–15. doi: 10.1038/s41467-021-23197-7
140. Zindel J, Peiseler M, Hossain M, Deppermann C, Lee WY, Haenni B, et al. Primordial GATA6 macrophages function as extravascular platelets in sterile injury. Sci (80- ) (2021) 371(6533):eabe0595. doi: 10.1126/science.abe0595
141. Bernsmeier C, Triantafyllou E, Brenig R, Lebosse FJ, Singanayagam A, Patel VC, et al. CD14 + CD15 - HLA-DR - myeloid-derived suppressor cells impair antimicrobial responses in patients with acute-on-chronic liver failure. Gut (2018) 67(6):1155–67. doi: 10.1136/gutjnl-2017-314184
142. Korf H, Du Plessis J, Van Pelt J, De Groote S, Cassiman D, Verbeke L, et al. Inhibition of glutamine synthetase in monocytes from patients with acute-on-chronic liver failure resuscitates their antibacterial and inflammatory capacity. Gut (2019) 68(10):1872–83. doi: 10.1136/gutjnl-2018-316888
143. Lee SJ, Yoon BR, Kim HY, Yoo SJ, Kang SW, Lee WW. Activated platelets convert CD14+CD16- into CD14+CD16+ monocytes with enhanced FcγR-mediated phagocytosis and skewed M2 polarization. Front Immunol (2021) 11:3363. doi: 10.3389/fimmu.2020.611133
144. Ramachandran P, Dobie R, Wilson-Kanamori JR, Dora EF, Henderson BEP, Luu NT, et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature (2019) 575(7783):512–8. doi: 10.1038/s41586-019-1631-3
145. Michalopoulos GK, Khan Z. Liver regeneration, growth factors, and amphiregulin. Gastroenterology (2005) 128(2):503. doi: 10.1053/j.gastro.2004.12.039
146. Murata S, Ohkohchi N, Matsuo R, Ikeda O, Myronovych A, Hoshi R. Platelets promote liver regeneration in early period after hepatectomy in mice. World J Surg (2007) 31(4):808–16. doi: 10.1007/s00268-006-0772-3
147. Murata S, Matsuo R, Ikeda O, Myronovych A, Watanabe M, Hisakura K, et al. Platelets promote liver regeneration under conditions of kupffer cell depletion after hepatectomy in mice. World J Surg (2008) 32(6):1088–96. doi: 10.1007/s00268-008-9493-0
148. Myronovych A, Murata S, Chiba M, Matsuo R, Ikeda O, Watanabe M, et al. Role of platelets on liver regeneration after 90% hepatectomy in mice. J Hepatol (2008) 49(3):363–72. doi: 10.1016/j.jhep.2008.04.019
149. Meyer J, Balaphas A, Combescure C, Morel P, Gonelle-Gispert C, Bühler L. Systematic review and meta-analysis of thrombocytopenia as a predictor of post-hepatectomy liver failure. HPB (Oxford) (2019) 21(11):1419–26. doi: 10.1016/j.hpb.2019.01.016
150. Yadav S, Storrie B--. The cellular basis of platelet secretion: Emerging structure/function relationships. Platelets (2017) 28(2):108–18. doi: 10.1080/09537104.2016.1257786
151. Liang C, Takahashi K, Furuya K, Ohkohchi N, Oda T. Dualistic role of platelets in living donor liver transplantation: Are they harmful? World J Gastroenterol (2022) 28(9):897. doi: 10.3748/wjg.v28.i9.897
152. De Rudder M, Dili A, Stärkel P, Leclercq IA. Critical role of LSEC in post-hepatectomy liver regeneration and failure. Int J Mol Sci (2021) 22(15):8053. doi: 10.3390/ijms22158053
153. Meyer J, Balaphas A, Fontana P, Morel P, Robson SC, Sadoul K, et al. Platelet interactions with liver sinusoidal endothelial cells and hepatic stellate cells lead to hepatocyte proliferation. Cells (2020) 9(5):1243. doi: 10.3390/cells9051243
154. Kono H, Fujii H, Suzuki-Inoue K, Inoue O, Furuya S, Hirayama K, et al. The platelet-activating receptor c-type lectin receptor-2 plays an essential role in liver regeneration after partial hepatectomy in mice. J Thromb Haemost (2017) 15(5):998–1008. doi: 10.1111/jth.13672
155. Kirschbaum M, Jenne CN, Veldhuis ZJ, Sjollema KA, Lenting PJ, Giepmans BNG, et al. Transient von willebrand factor-mediated platelet influx stimulates liver regeneration after partial hepatectomy in mice. Liver Int (2017) 37(11):1731–7. doi: 10.1111/liv.13386
156. Balaphas A, Meyer J, Perozzo R, Zeisser-Labouebe M, Berndt S, Turzi A, et al. Platelet transforming growth factor-β1 induces liver sinusoidal endothelial cells to secrete interleukin-6. Cells (2020) 9(5):1311. doi: 10.3390/cells9051311
157. Nowatari T, Murata S, Nakayama K, Sano N, Maruyama T, Nozaki R, et al. Sphingosine 1-phosphate has anti-apoptotic effect on liver sinusoidal endothelial cells and proliferative effect on hepatocytes in a paracrine manner in human. Hepatol Res (2015) 45(11):1136–45. doi: 10.1111/hepr.12446
158. Osawa Y, Seki E, Adachi M, Suetsugu A, Ito H, Moriwaki H, et al. Role of acid sphingomyelinase of kupffer cells in cholestatic liver injury in mice. Hepatology (2010) 51(1):237–45. doi: 10.1002/hep.23262
159. Owumi SE, Corthals SM, Uwaifo AO, Kamendulis LM, Klaunig JE. Depletion of kupffer cells modulates ethanol-induced hepatocyte DNA synthesis in C57Bl/6 mice. Environ Toxicol (2014) 29(8):867–75. doi: 10.1002/tox.21814
160. Lesurtel M, Graf R, Aleil B, Walther DJ, Tian Y, Jochum W, et al. Platelet-derived serotonin mediates liver regeneration. Sci (80- ) (2006) 312(5770):104–7. doi: 10.1126/science.1123842
161. Sulaiman P, Joseph B, Kaimal SB, Paulose CS. Decreased hepatic 5-HT1A receptors during liver regeneration and neoplasia in rats. Neurochem Res (2008) 33(3):444–9. doi: 10.1007/s11064-007-9452-4
162. Starlinger P, Pereyra D, Hackl H, Ortmayr G, Braunwarth E, Santol J, et al. Consequences of perioperative serotonin reuptake inhibitor treatment during hepatic surgery. Hepatology (2021) 73(5):1956–66. doi: 10.1002/hep.31601
163. Kirschbaum M, Karimian G, Adelmeijer J, Giepmans BNG, Porte RJ, Lisman T. Horizontal RNA transfer mediates platelet-induced hepatocyte proliferation. Blood (2015) 126(6):798–806. doi: 10.1182/blood-2014-09-600312
164. Groeneveld D, Pereyra D, Veldhuis Z, Adelmeijer J, Ottens P, Kopec AK, et al. Intrahepatic fibrin(ogen) deposition drives liver regeneration after partial hepatectomy in mice and humans. Blood (2019) 133(11):1245–56. doi: 10.1182/blood-2018-08-869057
Keywords: platelet, innate immunity, liver failure- therapy, acute on chronic liver failure (ACLF), ALF, liver, thromboinflammation, , immunothrombosis
Citation: Morris SM and Chauhan A (2022) The role of platelet mediated thromboinflammation in acute liver injury. Front. Immunol. 13:1037645. doi: 10.3389/fimmu.2022.1037645
Received: 06 September 2022; Accepted: 14 October 2022;
Published: 27 October 2022.
Edited by:
Lucia Stefanini, Sapienza University of Rome, ItalyReviewed by:
Erica Sparkenbaugh, University of North Carolina at Chapel Hill, United StatesCopyright © 2022 Morris and Chauhan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Abhishek Chauhan, YS5jaGF1aGFuLjFAYmhhbS5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.