
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 01 December 2022
Sec. Viral Immunology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1034968
This article is part of the Research TopicInterferon and its Antiviral Effect in Response to HBV InfectionView all 5 articles
 Qirong Li1,2†
Qirong Li1,2† Baozhen Sun3†Yue Zhuo4†
Baozhen Sun3†Yue Zhuo4† Ziping Jiang5Rong Li2Chao Lin6
Ziping Jiang5Rong Li2Chao Lin6 Ye Jin7
Ye Jin7 Yongjian Gao1*
Yongjian Gao1* Dongxu Wang2*
Dongxu Wang2*Human hepatitis B virus (HBV) is a small enveloped DNA virus with a complex life cycle. It is the causative agent of acute and chronic hepatitis. HBV can resist immune system responses and often causes persistent chronic infections. HBV is the leading cause of liver cancer and cirrhosis. Interferons (IFNs) are cytokines with antiviral, immunomodulatory, and antitumor properties. IFNs are glycoproteins with a strong antiviral activity that plays an important role in adaptive and innate immune responses. They are classified into three categories (type I, II, and III) based on the structure of their cell-surface receptors. As an effective drug for controlling chronic viral infections, Type I IFNs are approved to be clinically used for the treatment of HBV infection. The therapeutic effect of interferon will be enhanced when combined with other drugs. IFNs play a biological function by inducing the expression of hundreds of IFN-stimulated genes (ISGs) in the host cells, which are responsible for the inhibiting of HBV replication, transcription, and other important processes. Animal models of HBV, such as chimpanzees, are also important tools for studying IFN treatment and ISG regulation. In the present review, we summarized the recent progress in IFN-HBV treatment and focused on its mechanism through the interaction between HBV and ISGs.
Hepatitis B virus (HBV) infection and its related diseases is an important medical problem in China and all over the world. In addition to causing chronic hepatitis B (CHB), it is a major cause of advanced liver disease and hepatocellular carcinoma (HCC) (1). HBV is a non-cytopathic DNA virus, belonging to the hepatophilic DNA virus family (2). Chronic HBV infection can cause persistent low-grade hepatic inflammation in patients, accompanied by transient episodes of high hepatic inflammation and the development of fibrotic processes, which results in liver fibrosis, cirrhosis, and ultimately decompensated liver disease or HCC in 25–40% of patients (3). CHB is characterized by the persistence of free covalently closed circular DNA (cccDNA) of the HBV genome as a stable miniature chromosome in the nucleus of infected hepatocytes (4). After treatment discontinuation or loss of immune defense, HBV cccDNA multiples in hepatocytes and can reactivate viral replication to produce an intact virus (5). Therefore, complete elimination of cccDNA from infected hepatocytes is important to achieve complete elimination of HBV. However, the presently available therapies can only control HBV infection or replication and cannot cure it completely. Previous studies have divided HBV cures into “functional” and “complete” (6). Functional cure refers to serum clearance of hepatitis B surface antigen (HBsAg), which is sometimes accompanied by serum DNA and continuously transcribed inactive cccDNA. Complete cure refers to the complete elimination of cccDNA (6).
HBV infection is generally controlled by reverse transcriptase inhibitors (nucleosides or nucleotide analogs [NAs]) and interferon (IFN) therapy (7). Presently, antiviral drugs approved for CHB treatment can be divided into two major groups. One is pegylated IFN-α (PEG-IFN-α), which inhibits viral replication in about 25% of patients (8). The other is the new generation of NAs that have high antiviral potency and resistance barriers and produce strong viral suppression in many patients (9). IFNs are a group of cytokines first discovered and explored in 1957. It is a key regulator of the immune response process against various viruses and cancers and also one of the first lines of defense for host cells against viruses (10). The following three types of IFNs are found: I (α, β, 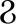 , κ, and ε), II (γ), and III (λ). IFN complexes can activate the Janus-activated kinase (JAK)-signal transducer and the activator of the transcription (STAT) pathway, which leads to the expression of IFN-stimulated genes (ISGs). These genes can further regulate viral replication and immune response as downstream effectors (11). The proteins encoded by ISGs inhibit the proliferation of viruses by inhibiting their transcription, translation, and replication, which promotes the degradation of viral nucleic acid, and changes the cellular lipid metabolism level (12).
, κ, and ε), II (γ), and III (λ). IFN complexes can activate the Janus-activated kinase (JAK)-signal transducer and the activator of the transcription (STAT) pathway, which leads to the expression of IFN-stimulated genes (ISGs). These genes can further regulate viral replication and immune response as downstream effectors (11). The proteins encoded by ISGs inhibit the proliferation of viruses by inhibiting their transcription, translation, and replication, which promotes the degradation of viral nucleic acid, and changes the cellular lipid metabolism level (12).
Studies have shown that ISG expression is associated with HBV infection and treatment (13). IFNs can regulate almost 10% of genes in the human genome. The proteins encoded by ISGs can individually or collectively play a role in inducing the intrinsic antiviral proliferation activity of cells and activating adaptive immunity for antiviral defense (14). In this review, we mainly focus on the mechanism underlying the treatment of IFNs, emphasizing the regulation of ISGs. Elucidating the regulatory mechanism underlying ISGs is helpful to understand their future impact better on antiviral therapy and pave the way for research of long-term HBV control therapy and the identification of new therapeutic targets.
CHB is prevalent in Africa, Asia, and parts of Central and Eastern Europe. Nearly 1 million people die every year due to complications of persistent HBV infection, cirrhosis, and HCC, with 250 million people affected by CHB globally (15). The present research has reported the gene expression and replication mechanisms underlying the HBV life cycle. Viral and host determinants influence whether the virus can successfully infect (16). Studies have reported that HBV naturally infects humans, chimpanzees, and some primates to a lesser extent. The parenchymal cells in the liver are the only sites where HBV can multiply (17).
HBV can be transmitted through infected bodily fluids such as blood and semen, which can be caused immune-mediated liver disease (7). HBV does not directly damage cells. The inflammation and necrosis of liver tissue are mainly due to the host’s recognition of invading antigens and the activation of its own immune system, which targets and destroys infected hepatocytes. Liver injury caused by excessive immune activation can further contribute to liver fibrotic disease and HCC during chronic HBV infection (18). HBV is highly effective in invading recognition by the innate immune system owing to its unique replication strategies, such as the use of capped and polyglandulated transcripts similar to host-derived mRNAs or the restriction of RNA/DNA genomes produced by replication to nucleocapsid particles in the cytoplasm (19). The HBV genotypes can be classified based on their genome sequences from A to J with many subtypes (20). The pathogenicity, virulence, clinical outcome, and response of HBV to type I IFN treatment are associated with its genotype. HBV DNA levels and hepatitis B E antigen (HBeAg) seroconversion rates were lower in patients infected with HBV genotypes C or D than those with HBV genotypes A or B (21). HBeAg seroconversion rate refers to patients who no longer express HBeAg and produce anti-HBeAg antibodies (22). HBV infection can be divided into the following four stages: immune tolerance, HBeAg positive immune active, HBeAg negative immune inactive (CHB loss or low replication), and HBeAg negative immune active (23). Serological markers should be discovered to determine the disease stage. General serum markers can diagnose CHB and help in distinguishing between acute and chronic infections. Common serological tests can detect HBV surface antigen (HBS), HBeAg, HBV surface antibody (anti-HBS), hepatitis B core antibody (anti-HBC), HBV envelope antibody (anti-HBE), and HBV DNA (24).
During the HBV life cycle, HBV DNA is transformed into a highly stable double-stranded circular DNA structure called cccDNA, which is an important stage in the nucleus of the liver cells. During this stage, cccDNA is integrated into the host genome as a template for viral RNA transcription, and cccDNA hides in the nuclei of the liver cell nuclei and serves as a template for viral replication (25) (Figure 1). HBV infectious virions are enveloped nucleocapsids that selectively enter hepatocytes and deliver incomplete circular DNA genomes, which initiates multiple viral replication processes (26). The circulating virions are initially attached to heparan sulfate proteoglycans (HSPG) (27), then viral surface proteins facilitate their entry into host hepatocytes. The preS1 domain is a crucial structure for mediating large surface proteins (28). HBV can enter the hepatocytes, which is co-mediated by surface molecules called sodium taurocholate cotransport polypeptides (NTCPs) (29). After the entry of the virus, the HBV nucleocapsid containing relaxed circular DNA (rcDNA) is delivered into the nucleus, where host enzymes transform the viral genome into cccDNA (30). Human RNA polymerase II mediates cccDNA transcription to produce pregenomic RNA (pgRNA). PgRNAs are mRNAs of core proteins and polymerases that serve as templates for HBV DNA replication (31). PgRNA is reverse transcribed to form incomplete rcDNA, wherein the HBV capsid is coated with HBsAg to become mature virus particles (32). Capsid-containing rcDNAs are transported back to the nucleus to increase the cccDNA content or enter multivesicular bodies. They come into contact with viral envelope proteins and exit hepatocytes to circulate in the blood as infectious virions (33).
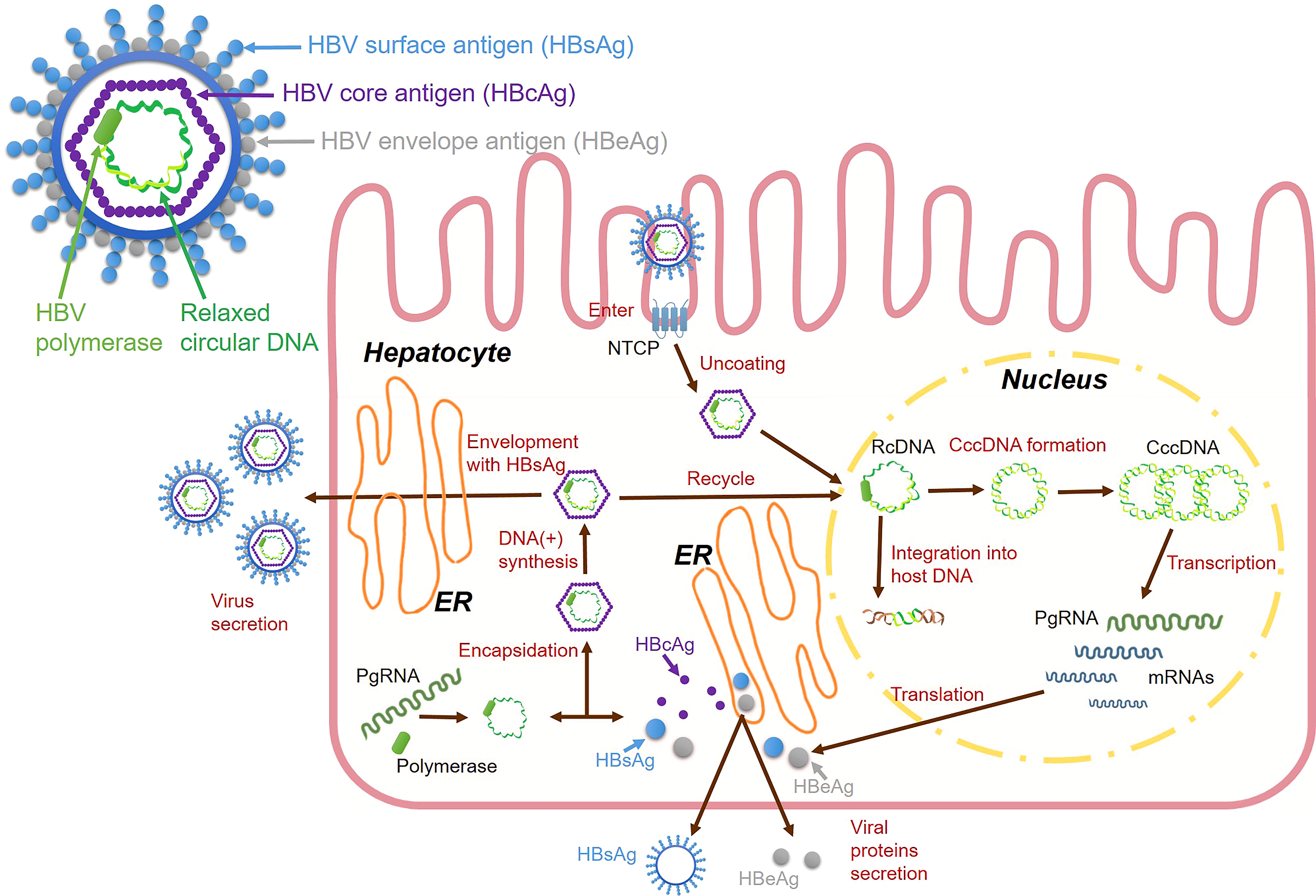
Figure 1 HBV virions and HBV life cycle.
Establishing animal HBV infection models is important for elucidating the mechanism underlying the immune response to HBV infection, which leads to hepatitis and the progression of liver injury and repair. Establishing relevant animal models has facilitated the development of methods to control chronic HBV infection and the study of ISG regulatory pathways. Mice have good immune system characteristics and are easy to handle. However, they cannot naturally be infected with HBV. Therefore, many studies have established various HBV infection models in transgenic mice using gene editing and humanized liver technologies (34). Past studies have found that sterile alpha motif domain-containing 4A (SAMD4A) is an important anti-HBV ISG by overexpressing or knocking down ISGs in HBV transgenic mice (35). Besides, interferon alpha-inducible protein 27 (IFI27) as ISG can inhibit HBV gene expression and DNA replication in mouse models (36). Other studies have shown that the steady-state level of HBV DNA in ubiquitin specific peptidase 18 (USP18) (UBP43) deficient mice is significantly reduced (37). Moreover, some studies have used the human liver chimeric mouse model and shown that HBV/HDV infection significantly induced ISG expression (38). Chimpanzees are the only immunocompetent animals that are naturally susceptible to HBV, and they are the main animal model for studying HBV infection (39). However, their HBV-related studies were limited because of ethical issues. Other animals, such as woodchucks, are naturally infected with hepatitis viruses similar to HBV (40). Woodchucks can be infected with woodchucks hepatitis virus (WHV), and ducks can be infected with duck hepatitis virus (41). These viruses have characteristics similar to HBV infection in humans. Some studies have investigated the changes in ISG expression after HBV infection using custom woodchuck microarray platforms (42). Moreover, another HBV-like virus, woolly monkey HBV (WMHBV), can infect its natural host, woolly monkeys, and was investigated for antiviral therapies for HBV infection (43). Other smaller non-human primate models are also being developed, such as tupaias, cynomolgus monkeys, and rhesus monkeys. The development of these animal models is crucial for studying HBV infection (44) (Table 1). HBV has a high species specificity. However, recent advances in transgenic mice, humanized mice, and strategies to make macaques more susceptible to HBV infection are gradually improving our ability to study HBV in a more suitable in vivo environment (47).
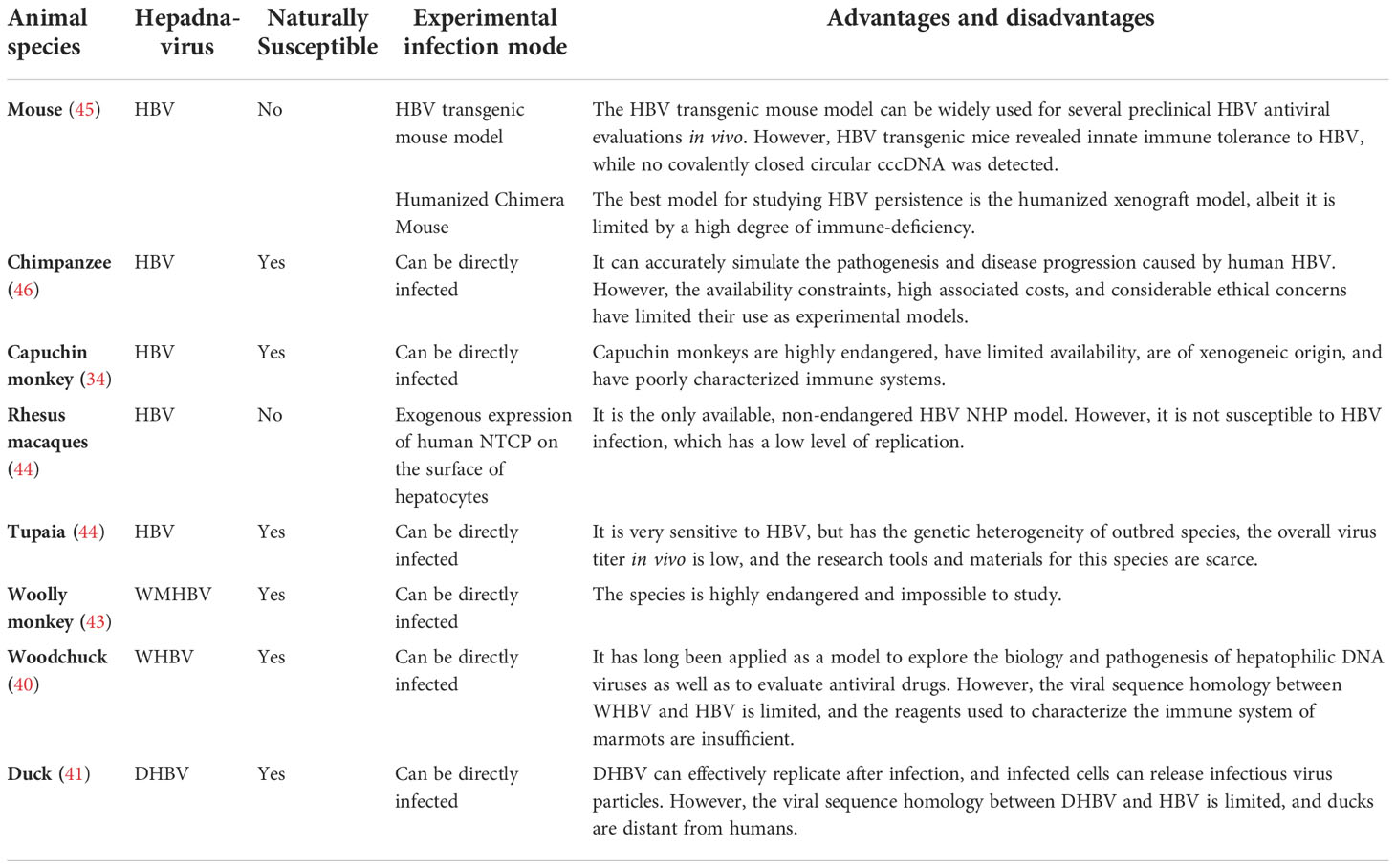
Table 1 Animal models for HBV researches.
The prophylactic vaccine for HBV is adopted in all developed countries. It is a common and crucial measure for preventing and controlling HBV (48). However, this vaccine does not affect patients with prolonged infections. Currently, treatment for these patients is limited to immunomodulators, including many direct-acting antivirals (DAAs), known as third-generation nuclear analogs (NUCs), such as entecavir, tenofovir, and tenofovir alanine or regular and pegylated type I IFNs (7). Induction of IFN expression occurs in response to viral or bacterial infection. With the development of recombinant IFNs, IFNs have been increasingly applied in HBV treatment, and have become a more popular treatment option (49).
When the HBV viral load is low, it can induce a type I IFN response and stimulate HBV gene expression and replication (50). However, type I IFNs inhibits HBV replication when the viral load is high. IFN-α and IFN-γ can interfere with the synthesis of negative-strand DNA virus by inducing apolipoprotein B mRNA editing enzyme catalytic subunit 3G (APOBEC3G) expression and binding to viral DNA polymerase (51). Therefore, type I IFNs can promote or inhibit HBV infection depending on the viral expression.
IFN-α induces genes encoding intracellular or secreted proteins (ISGs) that promote immune cell activation. They have direct or indirect antiviral activity (52). Human IFN-α can reduce HBV DNA, HBeAg, and HBsAg levels in hepatocytes (53). Furthermore, IFN-α14 can be the most effective IFN subtype for inhibiting HBV cccDNA transcription and HBeAg/HBsAg production. IFN-α14 can activate IFN-α and IFN-γ signaling and induce the expression of many potent antiviral effectors, synergistically limiting HBV replication (54). The anti-HBV activity of IFN-α is regulated by a complex mode of action, which includes natural killer (NK) T cell activation (55). They decrease pgRNA and subgenomic RNA transcription in HBV cccDNA microsomes and decrease signal transducer and activator of transcription 1 (STAT1) and 2 (STAT2) transcription factor binding to active cccDNA, which collectively inhibit HBV replication (56). IFN-α can be used to treat HBV by degrading cccDNA via APOBEC3A activation in infected cells (56). Furthermore, IFN-α treatment significantly upregulated the expression of the host gene ubiquitin-conjugating enzyme E2 L3 (UBE2L3), whereas UBE2L3 silencing increased the antiviral activity of IFN-α against HBV RNA, cccDNA, and DNA (57). IFN-α can also transfer antiviral molecules from cell to cell through exosomes, which contributes to its antiviral response to HBV in mice (58). Cross-linking IFN-α with apolipoprotein A-I produces a molecule with different antiviral and immune-stimulating activities that decrease IFN-α hematologic toxicity and have HBV therapeutic effects (59). Moreover, IFNs inhibit HBV secretion by inducing the protein Tetherin, which is the potential anti-HBV response mechanism triggered by IFNs (60).
PEG-IFN-α is added to some therapeutic agents that are pegylated by partially incorporating polyethylene into the active product. PEG-IFN-α molecules are mainly used to increase the pharmacokinetic properties of unmodified IFN-α (61). The binding of pegylated molecules to IFNs increases its half-life more than that by IFN-α alone. This reduces its rates of absorption and renal and cellular clearance. Moreover, PEG-IFN-α requires less frequent administration than IFN-α and produced more durable viral inhibitory effects in clinical trials (62). A recent study created and evaluated two pegylated IFN preparations (PEG IFN-α-2a and PEG IFN-α-2b) with different molecular sizes and structures, in vivo and in vitro properties, and half-lives (63, 64). The immunomodulatory function of PEG-IFN-α, especially NK cell activation, plays a key role in response to HBV treatment (65). Furthermore, PEG-IFN-α-2b improved the resistance of CHB patients to HBV by increasing the number of HBV-specific CD8+ T cells and regulating the expression of Th1 and Th2 cytokines (66). PEG-IFN-α treatment upregulates exosomal microRNAs (miRNAs) miR-193a-5p, miR-25-5p, and miR-574-5p, with exosomes secreted by macrophages transferring IFN-α-related miRNA into HBV-infected hepatocytes, which inhibits HBV replication and transcription (67).
Systematic reviews and meta-analyses of the role of conventional IFN-α in patients with HBeAg-positive CHB have found that it can improve their biological, serological, and virological responses. Treatment with higher doses of IFN-α and a longer duration of continuous administration can have a better therapeutic effect; however, it can also lead to side effects and increased treatment costs (68). IFN-α is presently the first choice of antiviral therapy for children with CHB older than one year, whereas PEG-IFN-α-2a is the recommended treatment for children with CHB older than three years. The results showed that antiviral monotherapy with IFNα-2B or PEG-IFNα-2a was well tolerated and effective in CHB children compared with adults with higher HBeAg seroconversion rates and HBsAg clearance rates (69). Many studies have shown that standard IFN-α has a specific role in anti-HBV infection; however, pure IFN-α is not commonly given as a therapy in clinical trials (70). Some studies have shown that IFN-α treatment is ineffective in most patients with HBV infection possibly because HBV prevents the induction of IFN-α signaling and interferes with ISG transcription in hepatocytes by inhibiting STAT1 nuclear translocation, which results in a low IFN-α therapeutic effectiveness (71). Overall, its antiviral effects in patients with CHB are modest for unknown reasons but may include inadequate delivery to the infected liver, tolerance of infected hepatocytes to IFN-α signaling, or other mechanisms (72).
Clinical results showed that PEG-IFN-α-2b was effective in treating HBeAg-positive CHB (73). In addition, PEG-IFN-α monotherapy was effective in 298 Chinese inactive HBV carriers, with good tolerability and safety (74). Using PEG-IFN-α in treating HBeAg-positive patients with CHB could inhibit viral production to some extent in 10%–40% of patients, and the HBeAg serum conversion rate of patients was about 25%–30%. Loss of HBsAg expression was observed in approximately 5% of patients six months after treatment discontinuation (75). Treatment regimens with PEG-IFN-α should be determined based on host-related factors and viral predictive markers, such as age, alanine transaminase (ALT) levels, viral load, and HBV genotype (76). Moreover, hepatitis B core-related antigen and HBsAb levels at the end of treatment can help determine the curative effect of PEG-IFN-α-based treatment in patients with CHB (77).
Combining IFN-α and PEG-IFN-α with other drugs is currently an attractive approach. The co-administration of ribavirin and IFN-α may be effective in treating viremic anti-HBE-positive patients with CHB who have not responded well to previous IFN treatment (78). Another clinical trial showed that sequential combination therapy with lamivudine and IFN-α induced a sustained virological response, including HBS seroconversion, in patients with CHB who were unresponsive to IFN-α alone. This observation suggests that this treatment regimen needs to be further evaluated in clinical trials (79). NVR3-778 is one of the core protein allosteric modulators (CpAMs), which has been shown to reactivate the host innate immune response by inducing the expression of ISGs (80, 81). Clinical studies have shown that combining PEG-IFN-α and NVR3-778 exerts a good antiviral effect in vivo (82). In addition, combining entecavir or tenofovir with PEG-IFN-α can reduce HBsAg levels consistently (83). Additional treatment with PEG-IFN-α results in higher serological response rates than monotherapy and may facilitate NAs discontinuation (84).
Furthermore, current regimens that may be of more interest include combining IFNs with traditional Chinese medicine (TCM) (85). Many studies have reported that TCM and related active compounds extracted from TCM have a potential anti-HBV activity, including Salvia miltiorrhiza, Astragalus, Oxymatrine, Artemisinin, and Vogoning. TCM preparations have better safety than IFN-α regarding dose-dependent side effects and drug resistance and are potential candidates for anti-HBV therapies (86). TCM preparations combined with IFNs considerably decreased serum HBeAg, increased serum HBV DNA clearance rates, and improved serum ALT normalization compared with IFNs alone (87). Moreover, a polysaccharide from Radix isatidis (Isatis indigotica Fortune) can exert an antiviral effect by activating the IFN-α-dependent JAK/STAT signaling pathway and increasing anti-HBV protein levels (88). Despite many IFN-related clinical trials, stronger evidence and more detailed experiments are needed to evaluate the safety and efficacy of combination therapy. In addition, more studies are needed to develop more convenient and effective IFN-α-based HBV treatment strategies.
After HBV infection, the host can induce many ISGs, the core components of intracellular antiviral innate immunity (89). ISGs can regulate IFN signaling and even directly inhibit viral infection. Studies on ISG mechanisms can show how IFN-induced signaling reprograms and primes cells to enhance viral detection, achieve effective viral defense, and return cells to normal functions. In addition, some studies have shown that ISGs are related to treating HBV using IFN-α (90). In the present study, we focused on the role of ISGs in treating HBV by regulating type I IFNs.
All type I IFNs, including IFN-α and IFN-β, are regulated by the IFN-α/β receptor (IFNAR) complex, which contains two subunits, IFNAR1 and IFNAR2 (91). However, type I IFN binding to IFNAR can induce ISG expression and activate the JAK/STAT signaling pathway (92). The heterotrimeric ISG factor 3 (ISGF3) transcription factor complex comprises phosphorylated STAT1/STAT2 and interferon regulatory factor 9, and type I IFNs can activate ISGF3 expression via the JAK/STAT signaling pathway (14). Activated ISGF3 binds to ISG upstream promoter regions in the nucleus in response to IFN stimulation (Figure 2). Furthermore, studies have shown that the increased interaction between STAT1 methylation and STAT1- protein inhibitor of activated STAT-1 is involved in IFN-α HBV antagonism, and the antiviral effect of IFN-α can be enhanced by increasing the expression of methylated STAT1 and S-adenosyl methionine (93). In addition, the unbiased high-throughput RNA interference technology was used to screen cells that showed HBV inhibition after IFN-α treatment. Among 711 epigenetic modifiers, SET domain containing 2-mediated K525 STAT1 methylation is an important antiviral signaling mechanism (94). Activating the JAK signaling pathway further induces alternative signaling pathways such as mitogen-activated kinase-like protein, phosphatidylinositol 3-kinase, and nuclear factor Kappa-light chain enhancer of activated B cells (NF-κB), amplifying the strength and magnitude of type I IFN signaling (49). Though previous studies considered ISGs as IFN-induced protein-coding mRNAs, recent studies have shown that IFNs also mediate changes in the expression of many non-coding RNAs, including long non-coding RNAs and miRNAs (95).
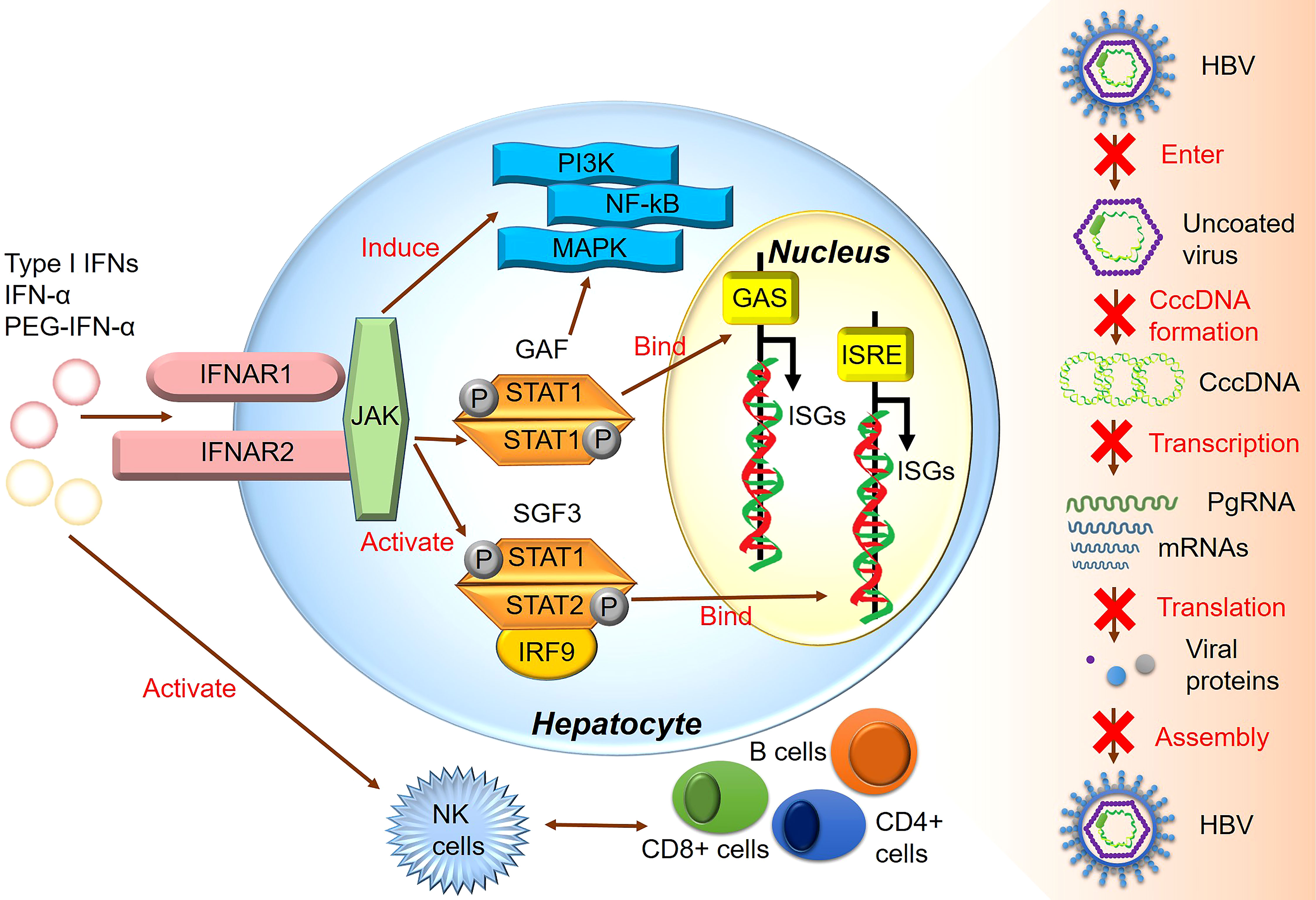
Figure 2 The major signaling pathway through which IFN produces its inhibitory effect on HBV.
The ISG gene pool is complex and large. Next-generation RNA sequencing studies have shown that IFNs regulate ~10% of all human genes. Moreover, studies comprehensively examining ISG expression in transcriptomes of different animals identified 62 core ISGs (96). Furthermore, several antiviral ISGs with critical roles have been discovered by identifying genes that are aberrantly expressed during viral infection inhibition. Among them, anti-myxovirus protein (MX)1 is the first classical effector molecule found to inhibit virus entry, primarily by preventing early-stage viral replication (97). In addition, interferon-induced transmembrane protein 3 (IFITM3) prevented the membrane fusion process of virus entry into cells via the endocytic pathway (98). Protein kinase R (PKR) and zinc antiviral protein are typical ISGs that inhibit viral protein production (99, 100). Therefore, different ISGs can block the HBV life cycle via corresponding pathways and play an essential role in regulating IFN-induced immune response and antiviral processes.
IFN-α is an antiviral drug with a limited treatment course. It acts on important biological processes including HBV replication and transcription by enhancing immune cell function, increasing cytokine levels, inducing ISG expression, and activating multi-antiviral proteins via the IFN signaling pathway, thereby playing a dual role in immune and antiviral regulation (10). Various ISGs exert anti-HBV effects in the host via different mechanisms (Table 2). Host cells infected with viruses can immediately recognize their pathogen-associated molecular pattern, promoting the viability of B cells activated by transcription factors IFN regulator 3 or 7 and NF-κB. This process initiates the expression of the genes of type I IFNs and proinflammatory cytokines, inducing downstream ISGs to establish an antiviral host cell environment with antiviral effects (97).
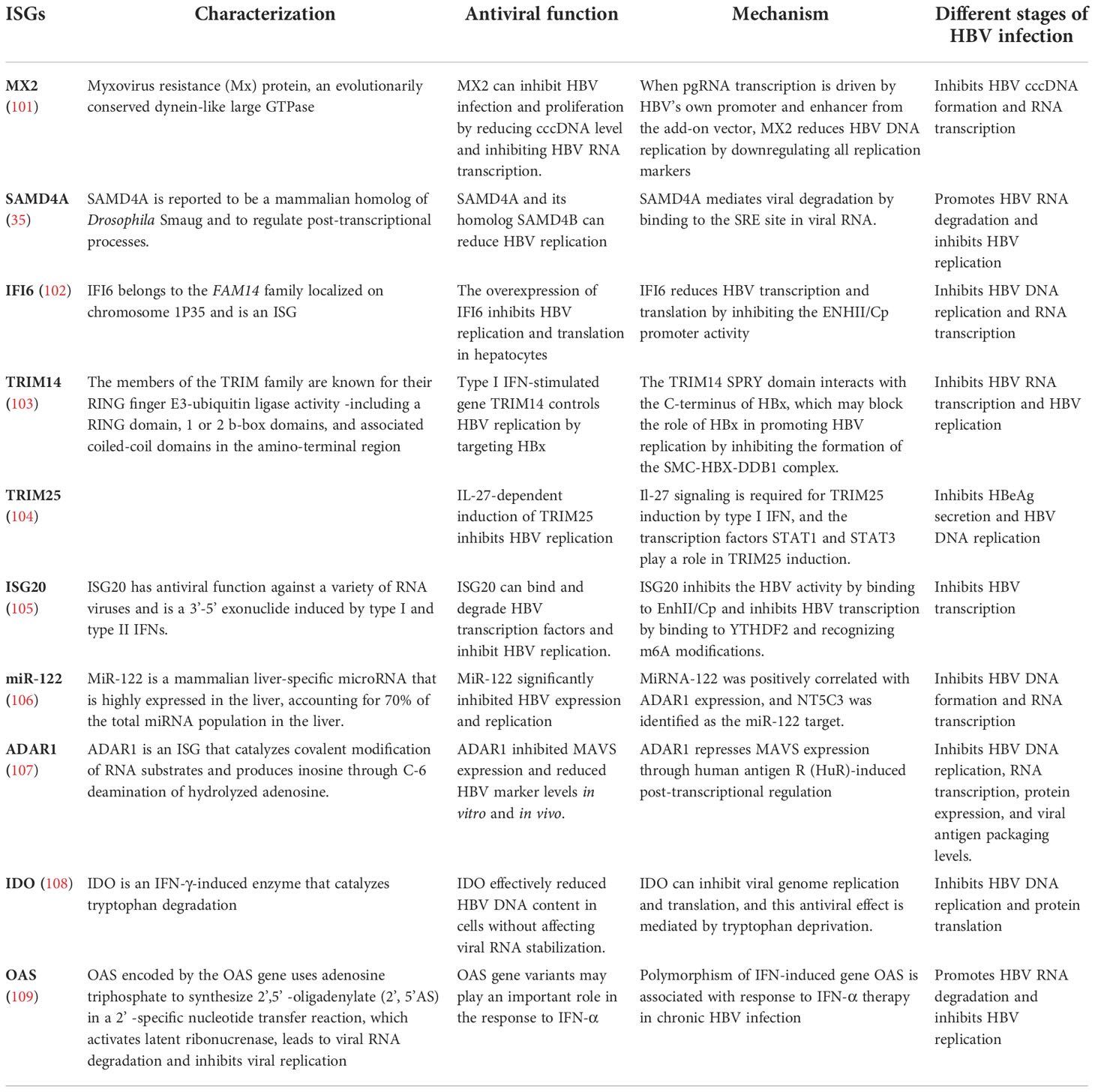
Table 2 Summary of major anti-HBV ISGs.
IFN-α-induced ISG MX2 reduces HBV cccDNA expression by inhibiting viral RNA synthesis, an important anti-HBV function. MX2 represents a novel HBV inhibitor with therapeutic potential (101). APOBEC3G is an IFN-α-induced cytosine deaminase that deaminates cytosine to uracil in single-stranded DNA replication, inhibiting the coding and replication ability of HBV (110). In a study, cell-based assays were performed to screen 285 human ISGs to check their anti-HBV activity, finding SAMD4A to be an important anti-HBV ISG and a strong repressor of HBV replication. It can be used in IFN-HBV treatment. SAMD4A/B expression was associated with human HBV sensitivity (35). In addition, IFN-α-inducible protein 6 (IFI6) inhibited HBV replication in cell and mouse model by reducing the expression of the gene of HBV enhancer II and core promoter (EnhII/Cp); thus, increasing IFI6 expression may be a potential therapeutic approach for inhibiting HBV infection (102).
Another study showed that the SPRY domain of tripartite motif containing 14 (TRIM14) interacted with the C-terminus of the HBV X protein (HBx) and might block HBV replication by inhibiting the formation of the structural maintenance of chromosome protein (SMC)-HBx- DNA damage-binding protein 1 (DDB1) complex (103). Other studies have shown that the IFN-interleukin (IL)-27-TRIM25 signaling pathway is induced by type I IFNs and inhibits HBV replication, identifying the ISG TRIM25 as a potential therapeutic target for HBV infection (104). In addition, TRIM5γ and TRIM31 were identified as key genes interacting with HBx that promote its degradation among the 145 ISGs examined, identifying them as potential therapeutic strategies for IFN-resistant patients with HBV infection (111). ISG20 is a 3′-5′ exonuclease that binds and degrades HBV transcripts (105). ISG20 is primarily induced by IFN-β, reducing HBV gene expression and inhibiting HBV enhancer activity by binding to EnhII/Cp regions (112). Moreover, m6A reader protein YTH domain family 2 (YTHDF2) regulates ISG20 expression by selectively recognizing and processing N6-methyladenosine (m6A)-modified HBV transcripts for degradation (105).
Moreover, studies have shown that IFN-α treatment significantly decreases microRNA-122 (miR-122) expression in hepatocytes, targeting ISG 5′-nucleotidase, cytosolic III (NT5C3), an inhibitor of miR-122 expression, and potentially inhibiting IFN-α function in HBV treatment (106). Other studies have shown that hepatocyte-specific miR-122 expression positively correlates with adenosine deaminase acting on RNA gene (ADAR1) expression. Exogenous miR-122 reduces HBV RNA and DNA, and p53 is also involved in the ADAR1-mediated reduction of HBV RNA (113). In addition, studies have shown that IFN-α attenuates mitochondrial signaling protein (MAVS) by RNA editing, which is mediated by ADAR1 antiviral therapy. These results indicate that combining MAVS with IFN-α has potential clinical applications in the studies on HBV infection (107).
ISG stimulator of interferon response the cyclic guanosine monophosphate-adenosine monophosphate (cGAMP) interactor (STING) is an important DNA-mediated regulator regulating the natural immune response of the body and a potential therapeutic target in HBV infection (114). Studies have shown that activation of STING signaling pathway can effectively reduce the severity of liver injury in chronic HBV mouse models, which may be a promising approach to prevent HBV virus proliferation and HBV-related liver fibrosis (115, 116). Furthermore, IFN-α reduces HBV cccDNA content by regulating the general control non-repressed 5 protein-mediated succinylation of histone H3K79 in HBV-infected human liver-chimeric mice. Therefore, IFN-α can inhibit HBV transcription at the epigenetic level (117). Indoleamine 2, 3-dioxygenase (IDO) is an ISG that can effectively reduce intracellular HBV DNA levels and the main IFN-γ regulatory gene in hepatocytes to produce an anti-HBV response (108). Moreover, the downstream signaling pathway of IFN-λ was identified by a proteomic method. IFITM3, 5′-3′ exoribonuclease 2 (XRN2), and 5’-nucleotidase, cytosolic IIIA (NT5C3A) expression were upregulated, and ISG transcription was activated to inhibit HBV replication (118).
In addition, ISG expression as a predictor of clinical efficacy is also an attractive strategy. Single nucleotide polymorphisms (SNPs) in the 2′,5′-oligoadenylate synthetase gene (OAS) in patients play a major role in predicting the efficacy of IFN treatment against CHB (119). Additionally, SNPs in IL28B and OAS were correlated with the clinical efficacy of IFN therapy in children with CHB, suggesting that they might be a new important consideration in treating CHB with IFNs (120).
According to recent studies, ISGs may also be involved in the mechanism by which HBV antagonizes IFNs and inhibits IFN efficacy. Studies have shown that IFN-α treatment activates STAT1 nuclear translocation and ISG expression. Therefore, HBV inhibits STAT1 nuclear translocation and interferes with ISG transcription in hepatocytes, blocking IFN-α signaling and causing a poor treatment response (71). In addition, HBV has molecular mechanisms that promote resistance to IFN therapy. HBV infection increases HBV polymerase levels and inhibits ISG induction, resulting in the poor antiviral efficacy of IFN-α in HBV mouse model (121). In addition, HBV precore protein P22 can reduce ISG expression and IFN-stimulated response element activity and inhibit IFN-α signaling by blocking the JAK/STAT signaling pathway and STAT nuclear translocation (122). Spliceosome-associated factor 1 can reduce the antiviral activity of IFN-α by attenuating JAK/STAT signaling and reducing the expression of ISGs such as MX, OAS, and PKR in HepG2 cells (123).
Moreover, IL-6 expression impaired the efficiency of IFN-α-mediated HBV suppression in hepatocytes by upregulating the suppressor of the cytokine signaling 3 genes (SOCS3). Therefore, SOCS3 downregulation can improve the antiviral activity of IFNs in HBV-replicating hepatocytes to a certain extent, representing a novel therapeutic strategy that may effectively target HBV infection (124). Other studies have shown that HBV can promote miR-146a transcription, inhibiting STAT1 and leading to IFN resistance. Therefore, this mechanism represents a promising research target for recovering the effects of IFN-α in HBV treatment (125). Moreover, the homologous to the E6-AP carboxyl terminus and RLD domain containing E3 ubiquitin-protein ligase 5-mediated modification of HBx by ISG15 increased HBV replication, resulting in HBV resistance to IFN-α therapy (126). Understanding the interaction between HBV and ISGs and ISG regulation by HBV to produce IFN antagonism will be helpful for further anti-HBV research (Figure 3).
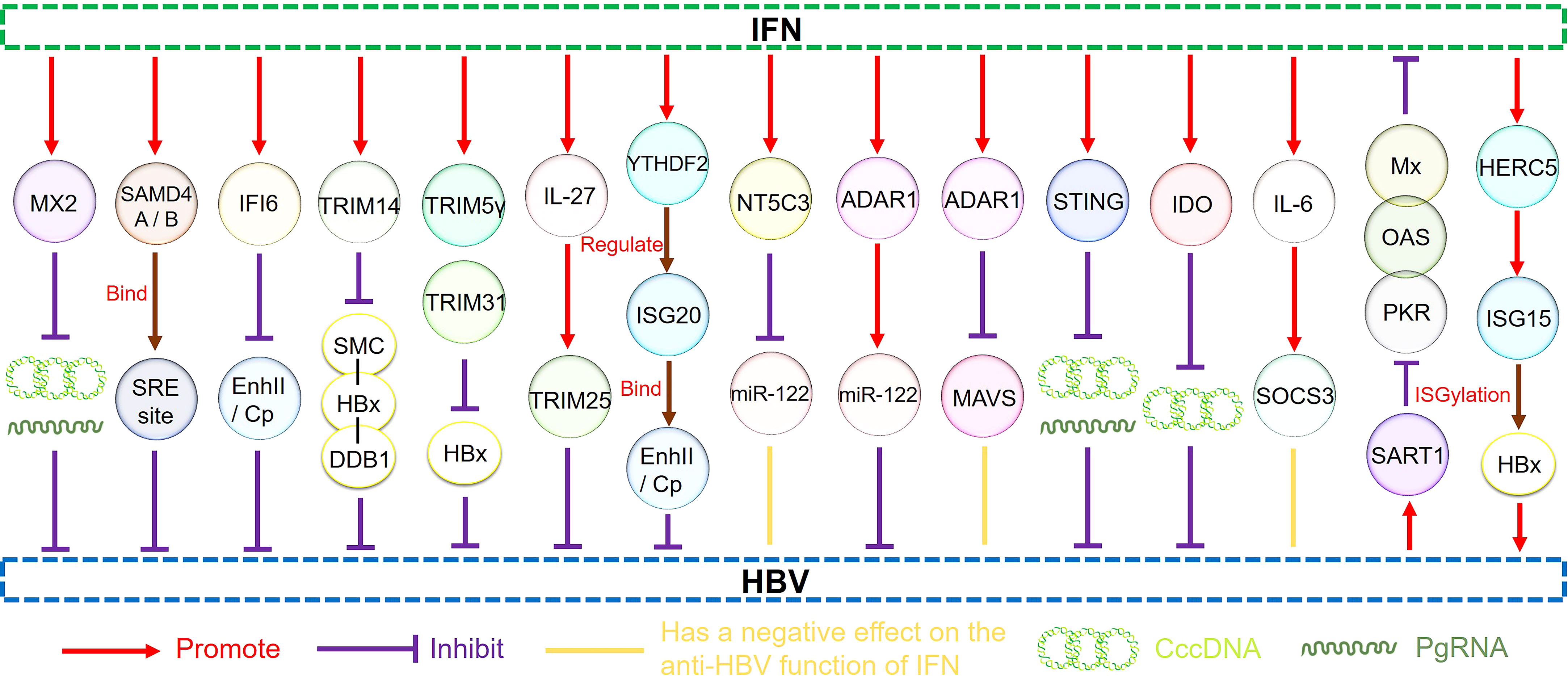
Figure 3 The regulatory pathways of ISGs.
As CHB can lead to immune impairment and tolerance, immunomodulatory IFN therapy offers particular mechanistic advantages in antiviral regulation than NAs, which cannot directly target the viral cccDNA reservoir (10). Studies have shown that IFN-α treatment can promote the degradation of HBV pgRNA in transgenic mice and induce the epigenetic inhibition of cccDNA in human hepatocytes both in vitro and in vitro (127, 128). The PEGylated form of IFNs is an immunomodulator providing the highest functional cure rate over a fixed treatment period (129). However, IFN therapy also has certain disadvantages. Loss of HBsAg associated with HBV DNA suppression is a desirable outcome of antiviral therapy. However, only 3%–11% of patients benefit from IFN therapy, and most need to continue drug therapy indefinitely (130). PEG-IFN-α is effective in only ~20% of patients, and its use is limited by its side effects. Therefore, developing new therapies that can be used in limited therapeutic courses to cure HBV infection is imperative (131). IFN therapy requires new drug combination strategies, IFN optimization, and more reliable biomarkers for clinical diagnosis. New IFN subtypes and delivery methods can be explored to improve the clinical effect of IFN treatment. Besides, with the development of animal models, more and more HBV animal models such as mice, chimpanzees, ducks, woodchucks and monkeys have been used to study the mechanism of IFN regulation of ISG, which helps us to further understand the method of suppressing HBV in vivo. Moreover, IFN-induced ISGs also play an important role in HBV progression. An important research direction might be to improve the efficacy of IFN treatment by ISGs targeting HBV.
Studies have shown that IFNs can achieve its powerful antiviral performance by inducing ISGs, regulating the immune response of the body, and acting directly on the enhancer and promoter sequences of infected viruses (97). Many ISGs are upregulated by IFN signaling and target different phases of the HBV life cycle (132). ISGs can act as effectors produced by IFN stimulation to exert a direct antiviral effect. The overexpression of ISGs that inhibit HBV HBeAg expression, including SAMD4A, MX2, IFI6, TRIM family members, ISG20, miR-122, ADAR1, and IDO, is conducive to the use of IFNs in HBV treatment. Other studies have shown that ISGs such as MAVS, NT5C3, and SOCS3 attenuate the anti-HBV effect of IFNs, and the downregulation of their expression may be an effective treatment strategy.
In addition, the efficacy of IFN treatment against CHB varies greatly among patients. Previous studies have shown that ISGs may be related to the outcome and antiviral efficacy after HBV infection, making them promising biomarkers for predicting the clinical efficacy of IFN treatment (120). However, HBV can also regulate ISGs to inhibit IFN signal transduction and promote viral proliferation. Hence, the mechanism of HBV acting on ISGs can also be used as a breakthrough point for treatment (122). Moreover, some important ISGs may contribute to the development of adjuvants for viral vaccines. IDO expression is increased in hemodialysis patients and affects the immune response to HBV vaccination (133). In addition, the induction of humoral and cellular immune responses to HBV vaccine can be upregulated by the STING ligand cGAMP (134). Studies have shown that ISG15 plays a critical role in MDA5-mediated antiviral response, and this mechanism may facilitate the development of new antiviral drugs and vaccines against COVID-19 (135). Besides, toll-like receptor (TLR) has been shown to control ISG mRNA levels, and a variety of vaccines with TLR as adjuvants have been shown to be effective in preclinical studies (136). It has also been demonstrated that the regulation of constitutive ISGs in tumor cells contributes to the enhancement of the antitumor response to Newcastle disease virus-infected tumor vaccines (137). Therefore, the regulatory mechanism of ISGs is a promising direction for the research of HBV vaccine adjuvants. IFNs can induce many ISGs. At present, only a few ISGs are associated with the antiviral activity of HBV, and the related biology and antiviral action mechanism of most remaining ISGs still need to be explored in depth. In addition, no unified model for predicting the efficacy of IFN treatment on CHB is available, and studies on the predictive efficacy of ISGs are limited. Further basic and clinical studies are needed to identify the target and mechanism of IFNs in HBV treatment by the combined effect of IFNs and ISG regulation, which may be a more promising strategy for clinical research to cure HBV.
HBV treatment remains an important medical problem. IFNs are commonly used immunomodulatory agent that suppresses HBV. The inhibitory mechanism of IFNs on HBV is complex and includes regulating ISG to inhibit HBV, which has received much attention. IFNs induces various ISGs to reduce HBV transcription, replication, and translation. Understanding the mechanism of ISG regulation of HBV will help identify new targets that promote the therapeutic effect of IFNs and develop new clinical strategies for HBV treatment.
Conceptualization, QL, YG and DW. Funding acquisition, DW. Project administration, DW. Supervision, DW. Writing – original draft, QL. Writing – review & editing, QL, BS, YZ, ZJ, RL, CL, YJ, YG and DW. All authors contributed to the article and approved the submitted version.
This work was supported by the Jilin Province Science and Technology Development Program under Grant 20210204013YY, the National Natural Science Foundation of China under Grant 82003985, the Foundation of Jilin Educational Committee under Grant JJKH20210995KJ, the Jilin Scientific and Technological Development Program under Grant 20220505033ZP, and the Grain, Oil and Food Deep Processing Scientific Research Project of Key Laboratories of Colleges and Universities in Jilin Province under Grant [2019] No. 004.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Llovet JM, Zucman-Rossi J, Pikarsky E, Sangro B, Schwartz M, Sherman M, et al. Hepatocellular carcinoma. Nat Rev Dis Primers (2016) 2:16018. doi: 10.1038/nrdp.2016.18
2. Seeger C, Mason WS. Molecular biology of hepatitis b virus infection. Virology (2015) 479-480:672–86. doi: 10.1016/j.virol.2015.02.031
3. Heathcote EJL. Viral hepatitis and liver disease. edited by a. j. zuckerman, 1136 pp. new York: Alan r. liss, Inc., 1988. $350.00. Hepatology (1989) 10(1):120–1. doi: 10.1002/hep.1840100127
4. Newbold JE, Xin H, Tencza M, Sherman G, Dean J, Bowden S, et al. The covalently closed duplex form of the hepadnavirus genome exists in situ as a heterogeneous population of viral minichromosomes. J Virol (1995) 69(6):3350–7. doi: 10.1128/JVI.69.6.3350-3357.1995
5. Xia Y, Guo H. Hepatitis b virus cccdna: Formation, regulation and therapeutic potential. Antiviral Res (2020) 180:104824. doi: 10.1016/j.antiviral.2020.104824
6. Zeisel MB, Lucifora J, Mason WS, Sureau C, Beck J, Levrero M, et al. Towards an hbv cure: State-of-the-Art and unresolved questions–report of the anrs workshop on hbv cure. Gut (2015) 64(8):1314–26. doi: 10.1136/gutjnl-2014-308943
7. Yuen MF, Chen DS, Dusheiko GM, Janssen HLA, Lau DTY, Locarnini SA, et al. Hepatitis b virus infection. Nat Rev Dis Primers (2018) 4:18035. doi: 10.1038/nrdp.2018.35
8. Foster GR. Pegylated interferon with ribavirin therapy for chronic infection with the hepatitis c virus. Expert Opin Pharmacother (2003) 4(5):685–91. doi: 10.1517/14656566.4.5.685
9. Gish RG, Given BD, Lai CL, Locarnini SA, Lau JY, Lewis DL, et al. Chronic hepatitis b: Virology, natural history, current management and a glimpse at future opportunities. Antiviral Res (2015) 121:47–58. doi: 10.1016/j.antiviral.2015.06.008
10. Ye J, Chen J. Interferon and hepatitis b: Current and future perspectives. Front Immunol (2021) 12:733364. doi: 10.3389/fimmu.2021.733364
11. Hoffmann HH, Schneider WM, Rice CM. Interferons and viruses: An evolutionary arms race of molecular interactions. Trends Immunol (2015) 36(3):124–38. doi: 10.1016/j.it.2015.01.004
12. MacMicking JD. Interferon-inducible effector mechanisms in cell-autonomous immunity. Nat Rev Immunol (2012) 12(5):367–82. doi: 10.1038/nri3210
13. Namineni S, O’Connor T, Faure-Dupuy S, Johansen P, Riedl T, Liu K, et al. A dual role for hepatocyte-intrinsic canonical nf-kappab signaling in virus control. J Hepatol (2020) 72(5):960–75. doi: 10.1016/j.jhep.2019.12.019
14. Schoggins JW. Interferon-stimulated genes: What do they all do? Annu Rev Virol (2019) 6(1):567–84. doi: 10.1146/annurev-virology-092818-015756
15. Revill PA, Chisari FV, Block JM, Dandri M, Gehring AJ, Guo H, et al. A global scientific strategy to cure hepatitis b. Lancet Gastroenterol Hepatol (2019) 4(7):545–58. doi: 10.1016/S2468-1253(19)30119-0
16. Iannacone M, Guidotti LG. Immunobiology and pathogenesis of hepatitis b virus infection. Nat Rev Immunol (2022) 22(1):19–32. doi: 10.1038/s41577-021-00549-4
17. Hillis WD. Viral hepatitis associated with Sub-human primates. Transfusion (1963) 3:445–54. doi: 10.1111/j.1537-2995.1963.tb04673.x
18. Levrero M, Zucman-Rossi J. Mechanisms of hbv-induced hepatocellular carcinoma. J Hepatol (2016) 64(1 Suppl):S84–S101. doi: 10.1016/j.jhep.2016.02.021
19. Suslov A, Boldanova T, Wang X, Wieland S, Heim MH. Hepatitis b virus does not interfere with innate immune responses in the human liver. Gastroenterology (2018) 154(6):1778–90. doi: 10.1053/j.gastro.2018.01.034
20. Sunbul M. Hepatitis b virus genotypes: Global distribution and clinical importance. World J Gastroenterol (2014) 20(18):5427–34. doi: 10.3748/wjg.v20.i18.5427
21. Erhardt A, Blondin D, Hauck K, Sagir A, Kohnle T, Heintges T, et al. Response to interferon Alfa is hepatitis b virus genotype dependent: Genotype a is more sensitive to interferon than genotype d. Gut (2005) 54(7):1009–13. doi: 10.1136/gut.2004.060327
22. Liaw YF. Hbeag seroconversion as an important end point in the treatment of chronic hepatitis b. Hepatol Int (2009) 3(3):425–33. doi: 10.1007/s12072-009-9140-3
23. Shi YH, Shi CH. Molecular characteristics and stages of chronic hepatitis b virus infection. World J Gastroenterol (2009) 15(25):3099–105. doi: 10.3748/wjg.15.3099
24. Chu CM, Liaw YF. Hepatitis b surface antigen seroclearance during chronic hbv infection. Antivir Ther (2010) 15(2):133–43. doi: 10.3851/IMP1497
25. Tang LSY, Covert E, Wilson E, Kottilil S. Chronic hepatitis b infection: A review. JAMA (2018) 319(17):1802–13. doi: 10.1001/jama.2018.3795
26. Jiang B, Hildt E. Intracellular trafficking of hbv particles. Cells (2020) 9(9):2023. doi: 10.3390/cells9092023
27. Sureau C, Salisse J. A conformational heparan sulfate binding site essential to infectivity overlaps with the conserved hepatitis b virus a-determinant. Hepatology (2013) 57(3):985–94. doi: 10.1002/hep.26125
28. Le Seyec J, Chouteau P, Cannie I, Guguen-Guillouzo C, Gripon P. Infection process of the hepatitis b virus depends on the presence of a defined sequence in the pre-S1 domain. J Virol (1999) 73(3):2052–7. doi: 10.1128/JVI.73.3.2052-2057.1999
29. Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z, et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis b and d virus. Elife (2012) 1:e00049. doi: 10.7554/eLife.00049
30. Nguyen MH, Wong G, Gane E, Kao JH, Dusheiko G. Hepatitis b virus: Advances in prevention, diagnosis, and therapy. Clin Microbiol Rev (2020) 33(2):e00046–19. doi: 10.1128/CMR.00046-19
31. Chuang YC, Tsai KN, Ou JJ. Pathogenicity and virulence of hepatitis b virus. Virulence (2022) 13(1):258–96. doi: 10.1080/21505594.2022.2028483
32. Zlotnick A, Venkatakrishnan B, Tan Z, Lewellyn E, Turner W, Francis S. Core protein: A pleiotropic keystone in the hbv lifecycle. Antiviral Res (2015) 121:82–93. doi: 10.1016/j.antiviral.2015.06.020
33. Seitz S, Habjanic J, Schutz AK, Bartenschlager R. The hepatitis b virus envelope proteins: Molecular gymnastics throughout the viral life cycle. Annu Rev Virol (2020) 7(1):263–88. doi: 10.1146/annurev-virology-092818-015508
34. Hu J, Lin YY, Chen PJ, Watashi K, Wakita T. Cell and animal models for studying hepatitis b virus infection and drug development. Gastroenterology (2019) 156(2):338–54. doi: 10.1053/j.gastro.2018.06.093
35. Wang Y, Fan X, Song Y, Liu Y, Liu R, Wu J, et al. Samd4 family members suppress human hepatitis b virus by directly binding to the smaug recognition region of viral rna. Cell Mol Immunol (2021) 18(4):1032–44. doi: 10.1038/s41423-020-0431-x
36. Ullah H, Sajid M, Yan K, Feng J, He M, Shereen MA, et al. Antiviral activity of interferon alpha-inducible protein 27 against hepatitis b virus gene expression and replication. Front Microbiol (2021) 12:656353. doi: 10.3389/fmicb.2021.656353
37. Kim JH, Luo JK, Zhang DE. The level of hepatitis b virus replication is not affected by protein Isg15 modification but is reduced by inhibition of Ubp43 (Usp18) expression. J Immunol (2008) 181(9):6467–72. doi: 10.4049/jimmunol.181.9.6467
38. Giersch K, Allweiss L, Volz T, Helbig M, Bierwolf J, Lohse AW, et al. Hepatitis delta Co-infection in humanized mice leads to pronounced induction of innate immune responses in comparison to hbv mono-infection. J Hepatol (2015) 63(2):346–53. doi: 10.1016/j.jhep.2015.03.011
39. Engle RE, De Battista D, Danoff EJ, Nguyen H, Chen Z, Lusso P, et al. Distinct cytokine profiles correlate with disease severity and outcome in longitudinal studies of acute hepatitis b virus and hepatitis d virus infection in chimpanzees. mBio (2020) 11(6):e02580–20. doi: 10.1128/mBio.02580-20
40. Menne S, Cote PJ. The woodchuck as an animal model for pathogenesis and therapy of chronic hepatitis b virus infection. World J Gastroenterol (2007) 13(1):104–24. doi: 10.3748/wjg.v13.i1.104
41. Li Y, Liu Z, Hui L, Liu X, Feng A, Wang W, et al. Transbody against virus core protein potently inhibits hepadnavirus replication in vivo: Evidence from a duck model of hepatitis b virus. Br J Pharmacol (2017) 174(14):2261–72. doi: 10.1111/bph.13811
42. Fletcher SP, Chin DJ, Cheng DT, Ravindran P, Bitter H, Gruenbaum L, et al. Identification of an intrahepatic transcriptional signature associated with self-limiting infection in the woodchuck model of hepatitis b. Hepatology (2013) 57(1):13–22. doi: 10.1002/hep.25954
43. Lanford RE, Chavez D, Brasky KM, Burns RB 3rd, Rico-Hesse R. Isolation of a hepadnavirus from the woolly monkey, a new world primate. Proc Natl Acad Sci U S A (1998) 95(10):5757–61. doi: 10.1073/pnas.95.10.5757
44. Liu Y, Maya S, Ploss A. Animal models of hepatitis b virus infection-success, challenges, and future directions. Viruses (2021) 13(5):777. doi: 10.3390/v13050777
45. Du Y, Broering R, Li X, Zhang X, Liu J, Yang D, et al. In vivo mouse models for hepatitis b virus infection and their application. Front Immunol (2021) 12:766534. doi: 10.3389/fimmu.2021.766534
46. Wieland SF. The chimpanzee model for hepatitis b virus infection. Cold Spring Harb Perspect Med (2015) 5(6):a021469. doi: 10.1101/cshperspect.a021469
47. Burwitz BJ, Zhou Z, Li W. Animal models for the study of human hepatitis b and d virus infection: New insights and progress. Antiviral Res (2020) 182:104898. doi: 10.1016/j.antiviral.2020.104898
48. Locarnini S, Hatzakis A, Chen DS, Lok A. Strategies to control hepatitis b: Public policy, epidemiology, vaccine and drugs. J Hepatol (2015) 62(1 Suppl):S76–86. doi: 10.1016/j.jhep.2015.01.018
49. Lin FC, Young HA. Interferons: Success in anti-viral immunotherapy. Cytokine Growth Factor Rev (2014) 25(4):369–76. doi: 10.1016/j.cytogfr.2014.07.015
50. Tian Y, Chen WL, Kuo CF, Ou JH. Viral-Load-Dependent effects of liver injury and regeneration on hepatitis b virus replication in mice. J Virol (2012) 86(18):9599–605. doi: 10.1128/JVI.01087-12
51. Nguyen DH, Gummuluru S, Hu J. Deamination-independent inhibition of hepatitis b virus reverse transcription by Apobec3g. J Virol (2007) 81(9):4465–72. doi: 10.1128/JVI.02510-06
52. Samuel CE. Antiviral actions of interferons. Clin Microbiol Rev (2001) 14(4):778–809. doi: 10.1128/Cmr.14.4.778-809.2001
53. Zhang M, Zhang Z, Imamura M, Osawa M, Teraoka Y, Piotrowski J, et al. Infection courses, virological features and ifn-alpha responses of hbv genotypes in cell culture and animal models. J Hepatol (2021) 75(6):1335–45. doi: 10.1016/j.jhep.2021.07.030
54. Chen J, Li Y, Lai F, Wang Y, Sutter K, Dittmer U, et al. Functional comparison of interferon-alpha subtypes reveals potent hepatitis b virus suppression by a concerted action of interferon-alpha and interferon-gamma signaling. Hepatology (2021) 73(2):486–502. doi: 10.1002/hep.31282
55. Thimme R, Dandri M. Dissecting the divergent effects of interferon-alpha on immune cells: Time to rethink combination therapy in chronic hepatitis b? J Hepatol (2013) 58(2):205–9. doi: 10.1016/j.jhep.2012.11.007
56. Belloni L, Allweiss L, Guerrieri F, Pediconi N, Volz T, Pollicino T, et al. Ifn-alpha inhibits hbv transcription and replication in cell culture and in humanized mice by targeting the epigenetic regulation of the nuclear cccdna minichromosome. J Clin Invest (2012) 122(2):529–37. doi: 10.1172/JCI58847
57. Zhou L, Ren JH, Cheng ST, Xu HM, Chen WX, Chen DP, et al. A functional variant in ubiquitin conjugating enzyme E2 L3 contributes to hepatitis b virus infection and maintains covalently closed circular DNA stability by inducing degradation of apolipoprotein b mrna editing enzyme catalytic subunit 3a. Hepatology (2019) 69(5):1885–902. doi: 10.1002/hep.30497
58. Li J, Liu K, Liu Y, Xu Y, Zhang F, Yang H, et al. Exosomes mediate the cell-to-Cell transmission of ifn-Alpha-Induced antiviral activity. Nat Immunol (2013) 14(8):793–803. doi: 10.1038/ni.2647
59. Berraondo P, Di Scala M, Korolowicz K, Thampi LM, Otano I, Suarez L, et al. Liver-directed gene therapy of chronic hepadnavirus infection using interferon alpha tethered to apolipoprotein a-I. J Hepatol (2015) 63(2):329–36. doi: 10.1016/j.jhep.2015.02.048
60. Yan R, Zhao X, Cai D, Liu Y, Block TM, Guo JT, et al. The interferon-inducible protein tetherin inhibits hepatitis b virus virion secretion. J Virol (2015) 89(18):9200–12. doi: 10.1128/JVI.00933-15
61. Youngster S, Wang YS, Grace M, Bausch J, Bordens R, Wyss DF. Structure, biology, and therapeutic implications of pegylated interferon alpha-2b. Curr Pharm Des (2002) 8(24):2139–57. doi: 10.2174/1381612023393242
62. Cooksley WG, Piratvisuth T, Lee SD, Mahachai V, Chao YC, Tanwandee T, et al. Peginterferon alpha-2a (40 kda): An advance in the treatment of hepatitis b e antigen-positive chronic hepatitis b. J Viral Hepat (2003) 10(4):298–305. doi: 10.1046/j.1365-2893.2003.00450.x
63. Ning Q, Han M, Sun Y, Jiang J, Tan D, Hou J, et al. Switching from entecavir to pegifn Alfa-2a in patients with hbeag-positive chronic hepatitis b: A randomised open-label trial (Osst trial). J Hepatol (2014) 61(4):777–84. doi: 10.1016/j.jhep.2014.05.044
64. Kozlowski A, Charles SA, Harris JM. Development of pegylated interferons for the treatment of chronic hepatitis c. BioDrugs (2001) 15(7):419–29. doi: 10.2165/00063030-200115070-00001
65. Nishio A, Bolte FJ, Takeda K, Park N, Yu ZX, Park H, et al. Clearance of pegylated interferon by kupffer cells limits nk cell activation and therapy response of patients with hbv infection. Sci Transl Med (2021) 13(587):eaba6322. doi: 10.1126/scitranslmed.aba6322
66. Chen J, Wang Y, Wu XJ, Li J, Hou FQ, Wang GQ. Pegylated interferon alpha-2b up-regulates specific Cd8+ T cells in patients with chronic hepatitis b. World J Gastroenterol (2010) 16(48):6145–50. doi: 10.3748/wjg.v16.i48.6145
67. Wu W, Wu D, Yan W, Wang Y, You J, Wan X, et al. Interferon-induced macrophage-derived exosomes mediate antiviral activity against hepatitis b virus through mir-574-5p. J Infect Dis (2021) 223(4):686–98. doi: 10.1093/infdis/jiaa399
68. Sun X, Qin W, Zhou R, Wang L, Li Y, Zhao L. Effect of conventional interferon-alpha in patients with hbeag-positive chronic hepatitis b: A systematic review and meta-analysis. J Evid Based Med (2010) 3(4):220–5. doi: 10.1111/j.1756-5391.2010.01100.x
69. Hu Y, Ye YZ, Ye LJ, Wang XH, Yu H. Efficacy and safety of interferon alpha-2b versus pegylated interferon alpha-2a monotherapy in children with chronic hepatitis b: A real-life cohort study from shanghai, China. World J Pediatr (2019) 15(6):595–600. doi: 10.1007/s12519-019-00303-w
70. Wong DK, Cheung AM, O’Rourke K, Naylor CD, Detsky AS, Heathcote J. Effect of alpha-interferon treatment in patients with hepatitis b e antigen-positive chronic hepatitis b. A Meta-Analysis Ann Intern Med (1993) 119(4):312–23. doi: 10.7326/0003-4819-119-4-199308150-00011
71. Lutgehetmann M, Bornscheuer T, Volz T, Allweiss L, Bockmann JH, Pollok JM, et al. Hepatitis b virus limits response of human hepatocytes to interferon-alpha in chimeric mice. Gastroenterology (2011) 140(7):2074–83. doi: 10.1053/j.gastro.2011.02.057
72. Zoulim F, Lebosse F, Levrero M. Current treatments for chronic hepatitis b virus infections. Curr Opin Virol (2016) 18:109–16. doi: 10.1016/j.coviro.2016.06.004
73. Janssen HL, van Zonneveld M, Senturk H, Zeuzem S, Akarca US, Cakaloglu Y, et al. Pegylated interferon Alfa-2b alone or in combination with lamivudine for hbeag-positive chronic hepatitis b: A randomised trial. Lancet (2005) 365(9454):123–9. doi: 10.1016/S0140-6736(05)17701-0
74. Wu F, Lu R, Liu Y, Wang Y, Tian Y, Li Y, et al. Efficacy and safety of peginterferon alpha monotherapy in Chinese inactive chronic hepatitis b virus carriers. Liver Int (2021) 41(9):2032–45. doi: 10.1111/liv.14897
75. Janssen HLA, van Zonneveld M, Senturk H, Zeuzem S, Akarca US, Cakaloglu Y, et al. Pegylated interferon Alfa-2b alone or in combination with lamivudine for hbeag-positive chronic hepatitis b: A randomised. Lancet (2005) 365(9454):123–9. doi: 10.1016/S0140-6736(05)17701-0
76. Lau GK. Hbeag-positive chronic hepatitis b: Why do I treat my patients with pegylated interferon. Liver Int (2009) 29 Suppl 1:125–9. doi: 10.1111/j.1478-3231.2008.01946.x
77. Huang D, Wu D, Wang P, Wang Y, Yuan W, Hu D, et al. End-of-Treatment hbcrag and hbsab levels identify durable functional cure after peg-Ifn-Based therapy in patients with chb. J Hepatol (2022) 77(1):42–54. doi: 10.1016/j.jhep.2022.01.021
78. Cotonat T, Quiroga JA, Lopez-Alcorocho JM, Clouet R, Pardo M, Manzarbeitia F, et al. Pilot study of combination therapy with ribavirin and interferon Alfa for the retreatment of chronic hepatitis b e antibody-positive patients. Hepatology (2000) 31(2):502–6. doi: 10.1002/hep.510310234
79. Serfaty L, Thabut D, Zoulim F, Andreani T, Chazouilleres O, Carbonell N, et al. Sequential treatment with lamivudine and interferon monotherapies in patients with chronic hepatitis b not responding to interferon alone: Results of a pilot study. Hepatology (2001) 34(3):573–7. doi: 10.1053/jhep.2001.26819
80. Gane EJ, Schwabe C, Walker K, Flores L, Hartman GD, Klumpp K, et al. Phase 1a safety and pharmacokinetics of nvr 3-778, a potential first-in-Class hbv core inhibitor. Hepatology (2014) 60(6):1279a–a.
81. Yuen MF, Kim DJ, Weilert F, Chan HLY, Lalezari JP, Hwang SG, et al. Nvr 3-778, a first-in-Class hbvcore inhibitor, alone and in combination with peg-interferon (Pegifn), in treatment-naive hbeag-positive patients: Early reductions in hbv DNA and hbeag. J Hepatol (2016) 64:S210–S1. doi: 10.1016/S0168-8278(16)00175-6
82. Klumpp K, Shimada T, Allweiss L, Volz T, Lutgehetmann M, Hartman G, et al. Efficacy of nvr 3-778, alone and in combination with pegylated interferon, vs entecavir in Upa/Scid mice with humanized livers and hbv infection. Gastroenterology (2018) 154(3):652–62.e8. doi: 10.1053/j.gastro.2017.10.017
83. Yoshida K, Enomoto M, Tamori A, Nishiguchi S, Kawada N. Combination of entecavir or tenofovir with pegylated interferon-alpha for long-term reduction in hepatitis b surface antigen levels: Simultaneous, sequential, or add-on combination therapy. Int J Mol Sci (2021) 22(3):1456. doi: 10.3390/ijms22031456
84. Brouwer WP, Xie Q, Sonneveld MJ, Zhang N, Zhang Q, Tabak F, et al. Adding pegylated interferon to entecavir for hepatitis b e antigen-positive chronic hepatitis b: A multicenter randomized trial (Ares study). Hepatology (2015) 61(5):1512–22. doi: 10.1002/hep.27586
85. Liu JP, McIntosh H, Lin H. Chinese Medicinal herbs for chronic hepatitis b. Cochrane Database Syst Rev (2001) 1):CD001940. doi: 10.1002/14651858.CD001940
86. Cui X, Wang Y, Kokudo N, Fang D, Tang W. Traditional Chinese medicine and related active compounds against hepatitis b virus infection. Biosci Trends (2010) 4(2):39–47.
87. Zhang L, Wang G, Hou W, Li P, Dulin A, Bonkovsky HL. Contemporary clinical research of traditional Chinese medicines for chronic hepatitis b in China: An analytical review. Hepatology (2010) 51(2):690–8. doi: 10.1002/hep.23384
88. Wang T, Wang X, Zhuo Y, Si C, Yang L, Meng L, et al. Antiviral activity of a polysaccharide from radix isatidis (Isatis indigotica fortune) against hepatitis b virus (Hbv) in vitro Via activation of Jak/Stat signal pathway. J Ethnopharmacol (2020) 257:112782. doi: 10.1016/j.jep.2020.112782
89. Ooi EL, Chan ST, Cho NE, Wilkins C, Woodward J, Li M, et al. Novel antiviral host factor, Tnk1, regulates ifn signaling through serine phosphorylation of Stat1. Proc Natl Acad Sci U.S.A. (2014) 111(5):1909–14. doi: 10.1073/pnas.1314268111
90. Tan G, Song H, Xu F, Cheng G. When hepatitis b virus meets interferons. Front Microbiol (2018) 9:1611. doi: 10.3389/fmicb.2018.01611
91. Yuliantie E, Dai X, Yang D, Crack PJ, Wang MW. High-throughput screening for small molecule inhibitors of the type-I interferon signaling pathway. Acta Pharm Sin B (2018) 8(6):889–99. doi: 10.1016/j.apsb.2018.07.005
92. Wang BX, Fish EN. The yin and yang of viruses and interferons. Trends Immunol (2012) 33(4):190–7. doi: 10.1016/j.it.2012.01.004
93. Li J, Chen F, Zheng M, Zhu H, Zhao D, Liu W, et al. Inhibition of Stat1 methylation is involved in the resistance of hepatitis b virus to interferon alpha. Antiviral Res (2010) 85(3):463–9. doi: 10.1016/j.antiviral.2009.10.011
94. Chen K, Liu J, Liu S, Xia M, Zhang X, Han D, et al. Methyltransferase Setd2-mediated methylation of Stat1 is critical for interferon antiviral activity. Cell (2017) 170(3):492–506.e14. doi: 10.1016/j.cell.2017.06.042
95. Forster SC, Tate MD, Hertzog PJ. Microrna as type I interferon-regulated transcripts and modulators of the innate immune response. Front Immunol (2015) 6:334. doi: 10.3389/fimmu.2015.00334
96. Shaw AE, Hughes J, Gu Q, Behdenna A, Singer JB, Dennis T, et al. Fundamental properties of the mammalian innate immune system revealed by multispecies comparison of type I interferon responses. PLoS Biol (2017) 15(12):e2004086. doi: 10.1371/journal.pbio.2004086
97. Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: A complex web of host defenses. Annu Rev Immunol (2014) 32:513–45. doi: 10.1146/annurev-immunol-032713-120231
98. Chesarino NM, Compton AA, McMichael TM, Kenney AD, Zhang L, Soewarna V, et al. Ifitm3 requires an amphipathic helix for antiviral activity. EMBO Rep (2017) 18(10):1740–51. doi: 10.15252/embr.201744100
99. Pindel A, Sadler A. The role of protein kinase r in the interferon response. J Interferon Cytokine Res (2011) 31(1):59–70. doi: 10.1089/jir.2010.0099
100. Chemudupati M, Kenney AD, Bonifati S, Zani A, McMichael TM, Wu L, et al. From apobec to zap: Diverse mechanisms used by cellular restriction factors to inhibit virus infections. Biochim Biophys Acta Mol Cell Res (2019) 1866(3):382–94. doi: 10.1016/j.bbamcr.2018.09.012
101. Wang YX, Niklasch M, Liu T, Wang Y, Shi B, Yuan W, et al. Interferon-inducible Mx2 is a host restriction factor of hepatitis b virus replication. J Hepatol (2020) 72(5):865–76. doi: 10.1016/j.jhep.2019.12.009
102. Sajid M, Ullah H, Yan K, He M, Feng J, Shereen MA, et al. The functional and antiviral activity of interferon alpha-inducible Ifi6 against hepatitis b virus replication and gene expression. Front Immunol (2021) 12:634937. doi: 10.3389/fimmu.2021.634937
103. Tan G, Xu F, Song H, Yuan Y, Xiao Q, Ma F, et al. Identification of Trim14 as a type I ifn-stimulated gene controlling hepatitis b virus replication by targeting hbx. Front Immunol (2018) 9:1872. doi: 10.3389/fimmu.2018.01872
104. Tan G, Xiao Q, Song H, Ma F, Xu F, Peng D, et al. Type I ifn augments il-27-Dependent Trim25 expression to inhibit hbv replication. Cell Mol Immunol (2018) 15(3):272–81. doi: 10.1038/cmi.2016.67
105. Imam H, Kim GW, Mir SA, Khan M, Siddiqui A. Interferon-stimulated gene 20 (Isg20) selectively degrades N6-methyladenosine modified hepatitis b virus transcripts. PLoS Pathog (2020) 16(2):e1008338. doi: 10.1371/journal.ppat.1008338
106. Hao J, Jin W, Li X, Wang S, Zhang X, Fan H, et al. Inhibition of alpha interferon (Ifn-Alpha)-Induced microrna-122 negatively affects the anti-hepatitis b virus efficiency of ifn-alpha. J Virol (2013) 87(1):137–47. doi: 10.1128/JVI.01710-12
107. Li T, Yang X, Li W, Song J, Li Z, Zhu X, et al. Adar1 stimulation by ifn-alpha downregulates the expression of mavs Via rna editing to regulate the anti-hbv response. Mol Ther (2021) 29(3):1335–48. doi: 10.1016/j.ymthe.2020.11.031
108. Mao R, Zhang J, Jiang D, Cai D, Levy JM, Cuconati A, et al. Indoleamine 2,3-dioxygenase mediates the antiviral effect of gamma interferon against hepatitis b virus in human hepatocyte-derived cells. J Virol (2011) 85(2):1048–57. doi: 10.1128/JVI.01998-10
109. Hovanessian AG, Justesen J. The human 2’-5’oligoadenylate synthetase family: Unique interferon-inducible enzymes catalyzing 2’-5’ instead of 3’-5’ phosphodiester bond formation. Biochimie (2007) 89(6-7):779–88. doi: 10.1016/j.biochi.2007.02.003
110. Harris RS, Liddament MT. Retroviral restriction by apobec proteins. Nat Rev Immunol (2004) 4(11):868–77. doi: 10.1038/nri1489
111. Tan G, Yi Z, Song H, Xu F, Li F, Aliyari R, et al. Type-I-Ifn-Stimulated gene Trim5gamma inhibits hbv replication by promoting hbx degradation. Cell Rep (2019) 29(11):3551–63.e3. doi: 10.1016/j.celrep.2019.11.041
112. Park YK, Lee SY, Lee AR, Kim KC, Kim K, Kim KH, et al. Antiviral activity of interferon-stimulated gene 20, as a putative repressor binding to hepatitis b virus enhancer ii and core promoter. J Gastroenterol Hepatol (2020) 35(8):1426–36. doi: 10.1111/jgh.14986
113. Liu G, Ma X, Wang Z, Wakae K, Yuan Y, He Z, et al. Adenosine deaminase acting on rna-1 (Adar1) inhibits hepatitis b virus (Hbv) replication by enhancing microrna-122 processing. J Biol Chem (2019) 294(38):14043–54. doi: 10.1074/jbc.RA119.007970
114. Kumada T, Toyoda H, Tada T, Kiriyama S, Tanikawa M, Hisanaga Y, et al. Effect of Nucleos(T)Ide analogue therapy on hepatocarcinogenesis in chronic hepatitis b patients: A propensity score analysis. J Hepatol (2013) 58(3):427–33. doi: 10.1016/j.jhep.2012.10.025
115. Li Y, He M, Wang Z, Duan Z, Guo Z, Wang Z, et al. Sting signaling activation inhibits hbv replication and attenuates the severity of liver injury and hbv-induced fibrosis. Cell Mol Immunol (2022) 19(1):92–107. doi: 10.1038/s41423-021-00801-w
116. Guo F, Tang L, Shu S, Sehgal M, Sheraz M, Liu B, et al. Activation of stimulator of interferon genes in hepatocytes suppresses the replication of hepatitis b virus. Antimicrob Agents Chemother (2017) 61(10):e00771–17. doi: 10.1128/AAC.00771-17
117. Yuan Y, Yuan H, Yang G, Yun H, Zhao M, Liu Z, et al. Ifn-alpha confers epigenetic regulation of hbv cccdna minichromosome by modulating Gcn5-mediated succinylation of histone H3k79 to clear hbv cccdna. Clin Epigenet (2020) 12(1):135. doi: 10.1186/s13148-020-00928-z
118. Makjaroen J, Somparn P, Hodge K, Poomipak W, Hirankarn N, Pisitkun T. Comprehensive proteomics identification of ifn-Lambda3-Regulated antiviral proteins in hbv-transfected cells. Mol Cell Proteomics (2018) 17(11):2197–215. doi: 10.1074/mcp.RA118.000735
119. Ren S, Yu H, Zhang H, Liu Y, Huang Y, Ma L, et al. Polymorphisms of interferon-inducible genes oas associated with interferon-alpha treatment response in chronic hbv infection. Antiviral Res (2011) 89(3):232–7. doi: 10.1016/j.antiviral.2011.01.006
120. Domagalski K, Pawlowska M, Zalesna A, Pilarczyk M, Rajewski P, Halota W, et al. Impact of Il28b and oas gene family polymorphisms on interferon treatment response in Caucasian children chronically infected with hepatitis b virus. World J Gastroenterol (2016) 22(41):9186–95. doi: 10.3748/wjg.v22.i41.9186
121. Chen J, Wu M, Zhang X, Zhang W, Zhang Z, Chen L, et al. Hepatitis b virus polymerase impairs interferon-Alpha-Induced sta T activation through inhibition of importin-Alpha5 and protein kinase c-delta. Hepatology (2013) 57(2):470–82. doi: 10.1002/hep.26064
122. Mitra B, Wang J, Kim ES, Mao R, Dong M, Liu Y, et al. Hepatitis b virus precore protein P22 inhibits alpha interferon signaling by blocking stat nuclear translocation. J Virol (2019) 93(13):e00196–19. doi: 10.1128/JVI.00196-19
123. Li Y, Zhu C, Wang F, Zhu T, Li J, Liu S, et al. Expression of interferon effector gene Sart1 correlates with interferon treatment response against hepatitis b infection. Mediators Inflammation (2016) 2016:3894816. doi: 10.1155/2016/3894816
124. Yang K, Guan S, Zhang H, Chen Z. Induction of interleukin 6 impairs the anti-hbv efficiency of ifn-alpha in human hepatocytes through upregulation of Socs3. J Med Virol (2019) 91(5):803–12. doi: 10.1002/jmv.25382
125. Hou ZH, Han QJ, Zhang C, Tian ZG, Zhang J. Mir146a impairs the ifn-induced anti-hbv immune response by downregulating Stat1 in hepatocytes. Liver Int (2014) 34(1):58–68. doi: 10.1111/liv.12244
126. Bawono RG, Abe T, Qu M, Kuroki D, Deng L, Matsui C, et al. Herc5 E3 ligase mediates isgylation of hepatitis b virus X protein to promote viral replication. J Gen Virol (2021) 102(10). doi: 10.1099/jgv.0.001668
127. Wieland SF, Eustaquio A, Whitten-Bauer C, Boyd B, Chisari FV. Interferon prevents formation of replication-competent hepatitis b virus rna-containing nucleocapsids. Proc Natl Acad Sci U.S.A. (2005) 102(28):9913–7. doi: 10.1073/pnas.0504273102
128. Xu C, Guo H, Pan XB, Mao R, Yu W, Xu X, et al. Interferons accelerate decay of replication-competent nucleocapsids of hepatitis b virus. J Virol (2010) 84(18):9332–40. doi: 10.1128/JVI.00918-10
129. Fung S, Choi HSJ, Gehring A, Janssen HLA. Getting to hbv cure: The promising paths forward. Hepatology (2022) 76(1):233–50. doi: 10.1002/hep.32314
130. Terrault NA, Bzowej NH, Chang KM, Hwang JP, Jonas MM, Murad MH, et al. Aasld guidelines for treatment of chronic hepatitis b. Hepatology (2016) 63(1):261–83. doi: 10.1002/hep.28156
131. Asselah T, Loureiro D, Boyer N, Mansouri A. Targets and future direct-acting antiviral approaches to achieve hepatitis b virus cure. Lancet Gastroenterol Hepatol (2019) 4(11):883–92. doi: 10.1016/S2468-1253(19)30190-6
132. Li MM, MacDonald MR, Rice CM. To translate, or not to translate: Viral and host mrna regulation by interferon-stimulated genes. Trends Cell Biol (2015) 25(6):320–9. doi: 10.1016/j.tcb.2015.02.001
133. Eleftheriadis T, Liakopoulos V, Antoniadi G, Stefanidis I, Galaktidou G. Indoleamine 2,3-dioxygenase is increased in hemodialysis patients and affects immune response to hepatitis b vaccination. Vaccine (2011) 29(12):2242–7. doi: 10.1016/j.vaccine.2011.01.051
134. Ito H, Kanbe A, Hara A, Ishikawa T. Induction of humoral and cellular immune response to hbv vaccine can be up-regulated by sting ligand. Virology (2019) 531:233–9. doi: 10.1016/j.virol.2019.03.013
135. Liu G, Lee JH, Parker ZM, Acharya D, Chiang JJ, van Gent M, et al. Isg15-dependent activation of the rna sensor Mda5 and its antagonism by the sars-Cov-2 papain-like protease. bioRxiv (2020) 6(4):467–478. doi: 10.1101/2020.10.26.356048
136. Albin TJ, Tom JK, Manna S, Gilkes AP, Stetkevich SA, Katz BB, et al. Linked toll-like receptor triagonists stimulate distinct, combination-dependent innate immune responses. ACS Cent Sci (2019) 5(7):1137–45. doi: 10.1021/acscentsci.8b00823
Keywords: HBV, IFN, ISGs, IFN-α, Peg-IFN-α
Citation: Li Q, Sun B, Zhuo Y, Jiang Z, Li R, Lin C, Jin Y, Gao Y and Wang D (2022) Interferon and interferon-stimulated genes in HBV treatment. Front. Immunol. 13:1034968. doi: 10.3389/fimmu.2022.1034968
Received: 02 September 2022; Accepted: 09 November 2022;
Published: 01 December 2022.
Edited by:
Yongye Huang, Northeastern University, ChinaReviewed by:
Yuan Liao, Tsinghua University, ChinaCopyright © 2022 Li, Sun, Zhuo, Jiang, Li, Lin, Jin, Gao and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongjian Gao, Z2FveWoxMjNAamx1LmVkdS5jbg==; Dongxu Wang, d2FuZ19kb25nX3h1QGpsdS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.