 Maha Abdeladhim
Maha Abdeladhim Jodi L. Karnell
Jodi L. Karnell Sadiye Amcaoglu Rieder
Sadiye Amcaoglu Rieder
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 15 December 2022
Sec. Immunological Tolerance and Regulation
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1033705
Regulatory T cells (Tregs) are the master regulators of immunity and they have been implicated in different disease states such as infection, autoimmunity and cancer. Since their discovery, many studies have focused on understanding Treg development, differentiation, and function. While there are many players in the generation and function of truly suppressive Tregs, the role of checkpoint pathways in these processes have been studied extensively. In this paper, we systematically review the role of different checkpoint pathways in Treg homeostasis and function. We describe how co-stimulatory and co-inhibitory pathways modulate Treg homeostasis and function and highlight data from mouse and human studies. Multiple checkpoint pathways are being targeted in cancer and autoimmunity; therefore, we share insights from the clinic and discuss the effect of experimental and approved therapeutics on Treg biology.
The immune system employs multiple players and mechanisms to protect the host from infections, autoimmunity, and cancer. Innate and adaptive arms of the immune system collaborate to maintain homeostasis and to protect the host from foreign antigens while maintaining the distinction between foreign and self (1). One integral lymphoid population in the adaptive immune system is the T cell and broadly there are two major populations—effector T cells (Teffs, including helper, cytotoxic, memory and γδ subsets) and regulatory T cells (Tregs). While Teffs are necessary to mount an effective immune response against foreign antigens, Tregs are essential for maintaining tolerance to self (2). Like all T cells, Tregs are developed and trained in the thymus (3). T cells with high affinity to self-antigens are deleted during negative selection while cells with intermediate affinity to self-antigens develop into Tregs. Due to imperfections of the negative selection process, Teffs with high affinity TCRs for self-antigens are sometimes released into the periphery (4). One of the main functions of Tregs is to control these self-reactive cells and maintain peripheral tolerance (5).
Natural Tregs (nTregs) emerge from the thymus, and they express the master transcription factor Foxp3 (forkhead box P3) (6). The most studied Treg population is CD4+; however there have been reports of CD8+ Tregs in the recent years (7, 8). In addition, there is a suppressive T cell population named Tr1 cells that are CD4+ Foxp3- and these cells express the co-inhibitory marker LAG3, CD49b and the inhibitory cytokine IL-10 (9). Tregs can also be induced from the naïve T effector cell populations (iTregs) and these cells have shown to be important in tissues such as the gut (10). Like Teffs, Tregs are heterogenous and different subpopulations exist—naïve/resting, central memory, effector memory and effector Tregs (11). The subpopulations are defined by differential expression of surface receptors such as CCR7, CD45RA, CD45RO and CD27 expression in humans; and CD62L and CD44 expression in mice (12, 13). Additionally, specific chemokine receptors and adhesion molecules are expressed, especially in tissue-directed and tissue-resident Tregs (14). For example, CCR4 is important for migration to skin and CCR9 is needed for homing to intestine (15, 16). Furthermore, memory Tregs can be divided into Th1, Th17 and Th2 Tregs, and these subpopulations emerge for two main reasons (17–19). Firstly, Tregs may differentiate into T helper-like phenotypes in order to adapt to the microenvironment, and better control Teff immune responses. Secondly, in certain inflammatory settings, Tregs lose their suppressive capabilities, and secrete pro-inflammatory cytokines (17–19). Figure 1 summarizes some of these different subtypes of human Tregs and their associated markers.
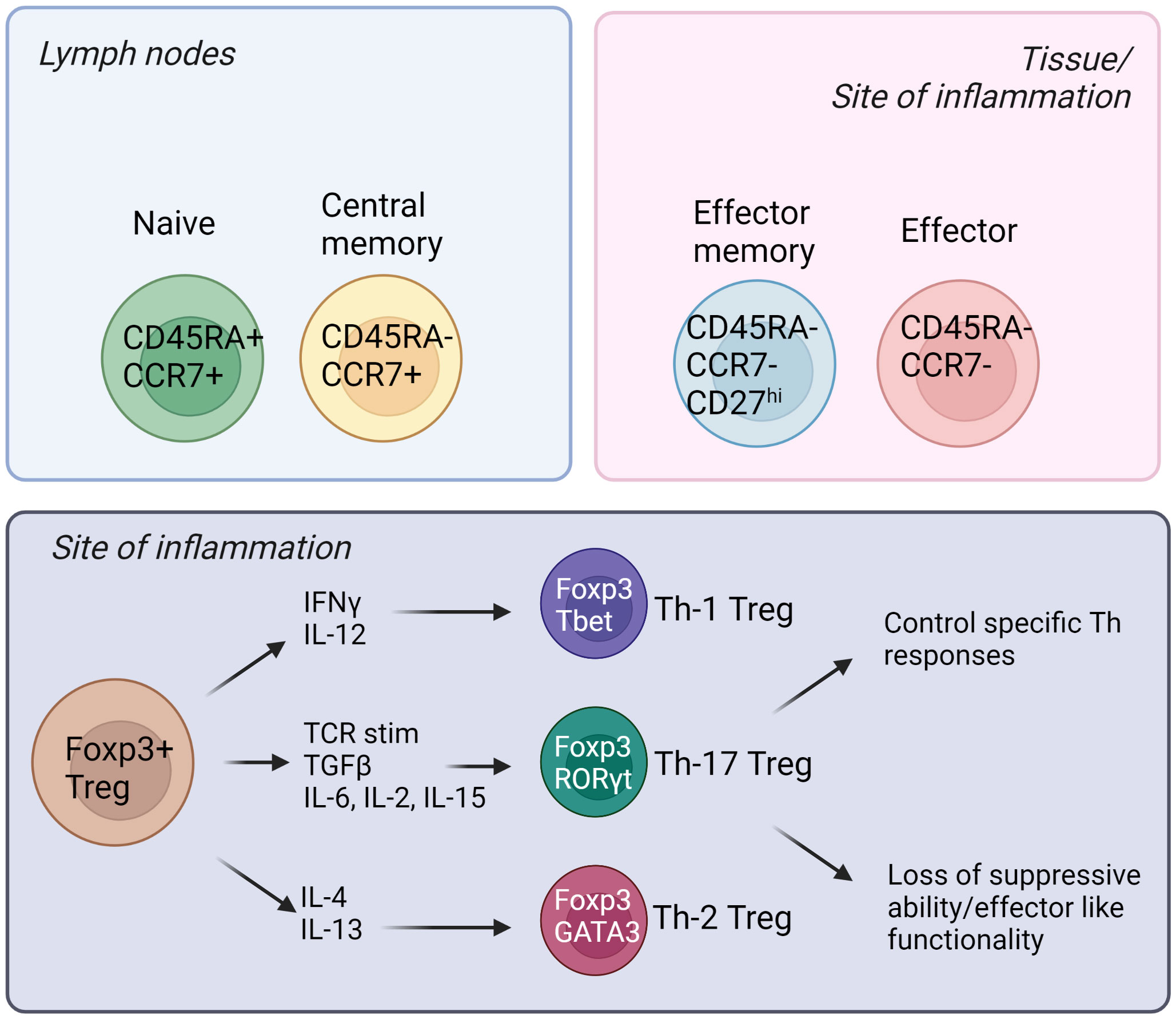
Figure 1 Different subsets of human Tregs in the lymph nodes, tissues, and sites of inflammation. Tregs emerge from the thymus and migrate into secondary lymphoid organs. Naïve and central memory Tregs reside in the lymph nodes while effector memory and effector Tregs can be found in the tissues and sites of inflammation. (Created by BioRender.com).
Tregs exert their suppressive role in different ways including cell-cell contact, secretion of inhibitory cytokines, cytolysis, metabolic disruption, and modulation of DC maturation. Tregs can suppress the immune system in an antigen-specific way but also through infectious tolerance mechanisms (20). A well balanced Treg function is key for a healthy immune system. Defects in Treg homeostasis and function leads to autoimmunity and chronic inflammation while increased number and function of Tregs can aid in the establishment and spread of cancer (21). Tregs can be activated through various axes, including antigen recognition, cytokines and in recent years checkpoint molecules have been widely studied as important pathways which can regulate Treg development and function (22, 23).
Activation and regulation of T cells including Tregs require dual signals. The first signal (referred to as signal 1) is delivered when the T cell receptor (TCR) recognizes antigen in the context of Major Histocompatibility complex (MHC) and antigen on the antigen presenting cell (APC). The second signal (referred to as signal 2) is engaged when CD28—the major co-stimulatory molecule engages CD80 and CD86 on the APC (further discussed in detail below). Without either of the two signals, T cells and Tregs cannot be activated (24). Notably, the initiation and activation of T cells are further regulated by other checkpoint pathways. The immune response is fundamentally shaped and modulated by co-stimulatory and co-inhibitory receptors and their corresponding ligands and the balance between pro and anti-inflammatory signaling is maintained during homeostasis (25). In tumorigenic environments, cancer cells recruit Tregs, induce T cell tolerance and/or anergy, and stimulate inhibitory immune checkpoints (26). Inversely, in autoimmune settings, inflammatory molecules and cytokines activate stimulatory checkpoint pathways. Stimulatory checkpoint molecules are composed of two major families; CD28 superfamily composed of CD28 and ICOS; and Tumor Necrosis Factor receptor superfamily containing CD40, OX40, GITR, CD137 and CD27 (25, 27). Inhibitory checkpoint molecules are composed of a much larger number of described molecules such as CTLA-4, PD-1, TIGIT, LAG3, TIM3, LILRB4, VISTA, KIR, 2B4 and many others (28) (Figure 2). Deeper understanding of Tregs and different immune checkpoint pathways shaped the strong interest in targeting them as potential therapeutics in autoimmunity, transplantation, chronic allergic and inflammatory diseases (29, 30). Moreover, the finding that Tregs are present at tumor sites also raised the importance of targeting them to promote anti-tumor immunity (31). Therefore, all fields converged to the same objective to selectively manipulate Tregs to inhibit or promote their function in disease. While there is rich literature on the role of immune checkpoint pathways in modulating Treg homeostasis and function, there remains some controversies and outstanding questions. Comprehensive understanding of the impact of co-stimulatory and co-inhibitory pathways on Treg modulation will empower next generation therapeutics such as biologics, small molecules and Chimeric Antigen Receptor (CAR) cell therapy approaches. In this review, we discuss the biological impact of different checkpoints pathways, provide perspective from mouse and human studies, and share some insights about the modulation of these pathways in the clinic.
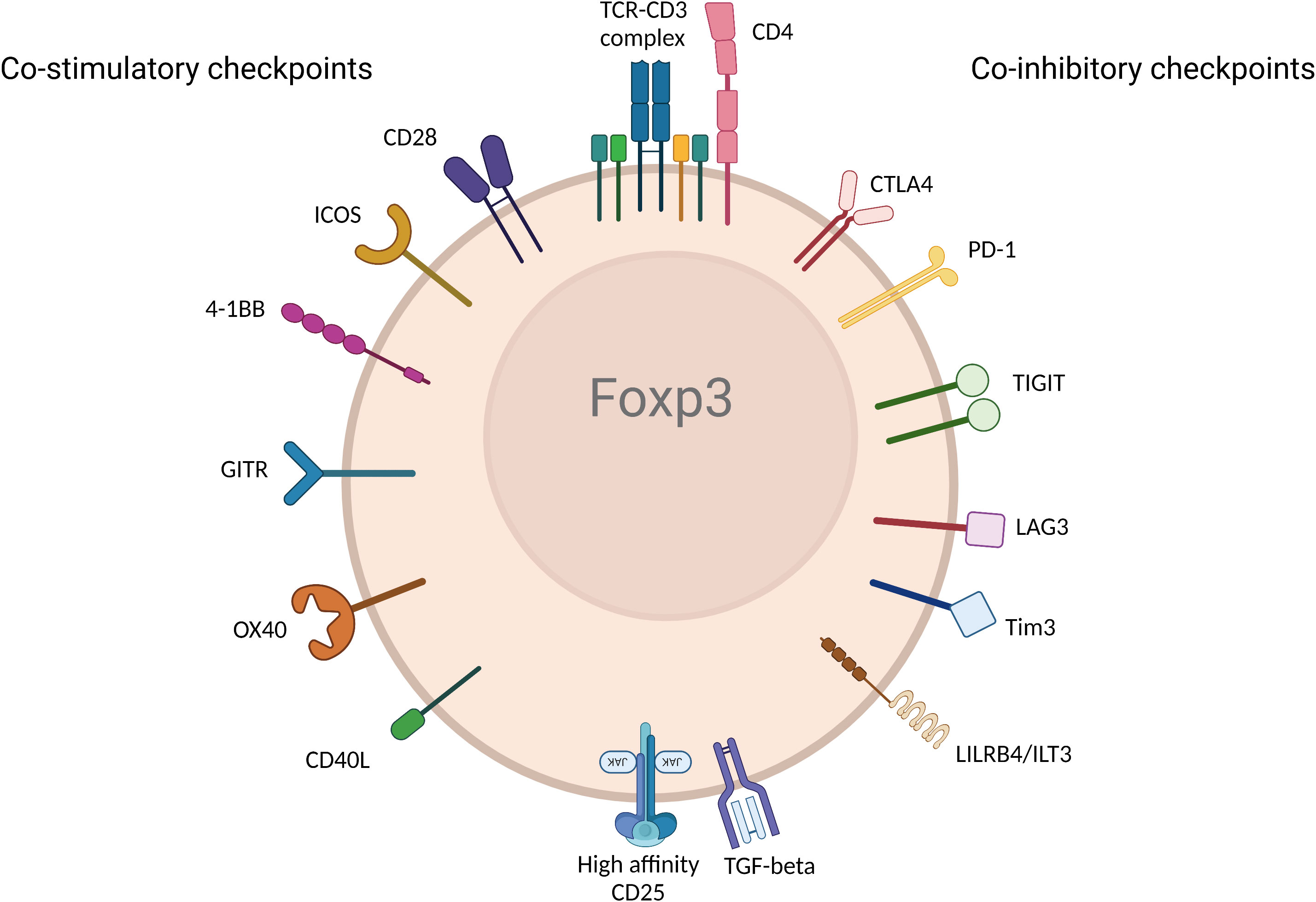
Figure 2 Immune checkpoint receptors expressed by Tregs. Various immune checkpoint receptors deliver unique signals to Tregs following engagement with their ligands expressed on APC and/or cells in tissue or diseased cells in different disease environments. The figure represents the most described co-stimulatory (left) and co-inhibitory checkpoints. (Created with BioRender.com).
CD28 is (44kDa, type I transmembrane protein) expressed on the surface of the majority of naïve CD4 and CD8 T cells as a glycosylated, disulfide-linked homodimer, and consists of a single extracellular Ig-V-like domain. CD28 was first characterized by a monoclonal antibody generated by John Hansen and Paul Martin in 1980 (32). They demonstrated that CD28 is a differentiation antigen expressed on thymocytes and majority of mature T cells. Cloning and expression of CD28 unveiled that each CD28 monomer contains 134 extracellular amino acids with a single transmembrane domain and a short cytoplasmic tail with no enzymatic activity. Each of the four exons contained in the CD28 gene defines a functional domain of the molecule (33, 34). CD28 binds both ligands CD80 (B7 or B7-1) and CD86 (B7-2). Additionally, CD28 can bind ICOS (35). Furthermore, cytotoxic T lymphocyte-associated protein-4 (CTLA-4) can oppose CD28, competes for binding the same ligands on APCs and induces CD28 downregulation via endocytosis (36). CD28 can generate a costimulatory signal (signal 2) once the TCR is engaged but is unable to activate T cells independently. In the absence of TCR stimulation, CD28 binds to CD80 monovalently and this weak interaction doesn’t generate a strong signal. However, in the presence of TCR activation, the homodimer form of CD28 binds to CD80 bivalently. Upon co-ligation, CD28 helps amplify the TCR signal in T cells. CD28 engagement on T cells induce the formation of distinct molecular patterns. It also induces cytoskeletal rearrangements and formation of an immunological synapse (37). CD28 is constitutively expressed in 80% of CD4 and 50% of CD8 T cells in human while all mouse T cells express CD28. Loss of CD28 expression is observed mainly in the CD8 T cells and was reported in human during aging and autoimmune disease (38). However, its expression is never downregulated in mice (39). The co-stimulatory function of CD28 has been utilized as a therapeutic factor in immunotherapy in oncology, especially in Chimeric Antigen Receptor (CAR) cell therapies and biologics (40, 41). However, it has also been an intricate balance to fully unleash the activating potential of CD28, since overstimulation can lead to cytokine storm and toxicity. Some of these CD28 activating therapies are discussed later in the review.
Like all other T cells, co-stimulatory signals are required for Tregs to become fully functional. Early studies have yielded conflicting results on the role of CD28 signaling in Tregs. This first became apparent in mice with the observation that spontaneous diabetes is exacerbated and the number of CD4+CD25+ Tregs is decreased in both CD28-deficient and B7-1/B7-2-deficient NOD mice (42). Tai et al. unveiled the important role of CD28 co-stimulation in thymic Treg differentiation. They reported that CD28 knock out mice had fewer Tregs in the thymus and these cells were unable to suppress the proliferative response of T cells in vitro (43). Likewise, Liang et al., showed that CD4+CD25- cells can be converted into Tregs in vivo through a B7-dependent pathway in a thymic independent manner. Later using CD28 knock out T cells, Guo et al. attributed this thymic independent peripheral Treg generation process to CD28 rather than CTLA-4 ligation of B7 both in vitro and in vivo (44–47). The role of CD28 in Treg function was further confirmed by Zhang et al. They developed CD28-deficient Treg mice in which CD28 is deleted under control of the FOXP3 promoter. While Treg numbers were preserved in these mice, these cells had lower levels of CTLA-4, PD-1 and CCR6 and they developed a systemic autoimmune condition affecting mainly the liver and the skin. This result was consistent with loss of suppressive function in CD28-deficient Tregs and failed to maintain immune surveillance in vivo (48). Other studies confirmed the stipulation of CD28 for in vitro Treg generation, as enriched Treg populations can only maintain their Treg phenotype and suppressive activity when co-stimulated with anti-CD28, even when expanded in the presence of rapamycin (49). To further assess the role of CD28 in Treg maturation and function, Zhang et al. performed a detailed analysis of manifestation of skin disease in CD28 Treg deficient mice. They demonstrated that CD28 activation is essential for optimal maturation of CD44loCD62Lhi central Tregs (cTregs or naïve Tregs in the secondary lymphoid organs) into CD44hiCD62Llo effector Tregs (eTregs) and induction of CCR6 in the latter cells. While CD28-deficient Tregs had normal function when injected directly to the skin, they were unable to home to inflamed skin due to the downregulation of CCR6 which is required for tissue homing (25).
Hombach et al. highlighted the role of CD28 co-stimulation in human Treg cells by comparing the activation requirement of resting human CD4+CD25+ Tregs to CD4+CD25- T cells. They demonstrated that the stimulatory conditions that induce T cell proliferation are not sufficient to induce Treg proliferation since Tregs require a combination of an intense TCR signaling with a very strong CD28 co-stimulation. Despite high levels of CD25 expression in Tregs, high concentrations of IL-2 could not substitute CD28 signaling (50). With the expansion of immunotherapy and the use of Treg as living therapies for autoimmune conditions, there has been great interest in generating high quality Tregs. He et al. demonstrated that a single CD28 stimulation of FACS sorted nTregs induces proliferation, high levels of Foxp3 expression, reduces pro-inflammatory cytokine production potential, preserves the TSDR demethylation and maintains high suppressive capacity (51).
CD28 engagement activates several cytosolic signaling pathways. Using CD28 transgenic mice, several groups uncovered different signaling pathways of CD28 that are distinct in Tregs and T cells. Tai et al. proved the important role of the PYAP motif within the cytoplasmic domain of CD28 that require an intact Lck-binding motif. This CD28 cytosolic tail interaction with Lck is required both for efficient Treg generation and for IL-2 production. Similarly, CD28 activation in CD4+CD25- T cells lead to STAT3 Tyr705 phosphorylation in an Lck-dependent manner to drive Foxp3 expression (52). Vang et al. demonstrated that CD28 drives Treg differentiation in the thymus via the PYAP motif through a subsequent activation of the proto-oncogene c-Rel and Nuclear factor kappa B (NFkB). Engagement of c-Rel induces high-level expression of the IL-2R complex on Treg progenitors which allow Tregs to respond to IL-2 (53). (Figure 3). Other studies pointed the role of Phosphoinositide 3-kinase (PI3K) pathway in Foxp3 transcription induction. Scotta et al. demonstrated that CD28 signals independent from TCR, results in PI3K/AKT pathway activation and this is sufficient to induce the transcription of FOXP3 in a small proportion of primary CD4+CD25- T cells committed to express Foxp3 (54). CD28 ligation was also sufficient to activate PI3K target protein kinase B (PKB; c-AKT) and inactivate phosphorylation of PKB target glycogen synthase kinase-3 (GSK-3) (55). These results were complemented using CD28 superagonist in FACS sorted Tregs in the presence or absence of PI3K-inhibitor or mTOR-inhibitor. The Treg activation in the presence of exogenous rhIL-2 induced an increase in Foxp3 expression and a decrease in inflammatory cytokine production that was dependent on distal mTOR and proximal PI3K signaling (51). Collectively, these results suggest a key role for CD28 co-stimulation in promoting Treg function.
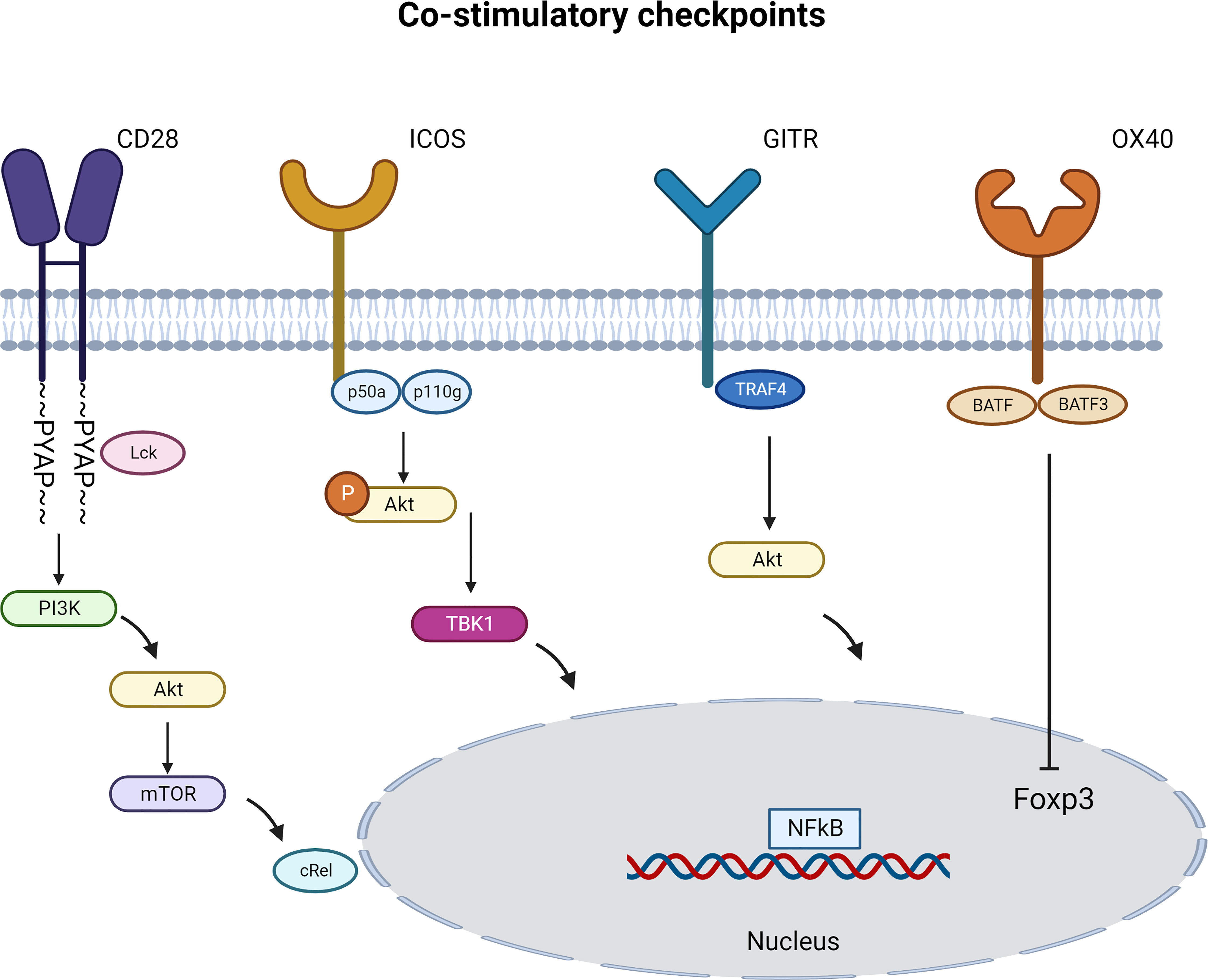
Figure 3 Immune Co-stimulatory signaling pathways specific to Tregs. Immune co-stimulatory checkpoints signaling are described in different cell types but only some are detailed in Tregs. The figure represents the most studied pathways. (Created with BioRender.com).
ICOS (55-60kDa) is a disulfide-linked homodimeric T-cell surface glycoprotein. First identified in humans two decades ago, ICOS is the third member of the CD28 co-stimulatory family that is expressed on different populations of activated T cells comprising CD8+ cytotoxic T lymphocytes (CTL), CD4+ (Th1, Th2, Th17 and Tfh) and CD4+Foxp3+ Tregs (56). TCR engagement and/or CD28 co-stimulatory signaling induce increased expression of ICOS on CD4 and CD8 T cells. ICOS engagement by ICOS ligand (ICOSL) induces a wide range of cytokines and increases Treg suppressive function. Thus, distinct expression pattern of ICOSL can regulate ICOS ligation (57, 58).
During homeostasis, ICOS is constitutively expressed in approximately 20% of Foxp3+ Tregs, with the majority of ICOS+ Treg being CXCR3+ (Th1-like Tregs) or CXCR3-CCR6+CCR10- (Th17-like Tregs) (59). Despite substantial homology with the other family members, co-stimulatory CD28 and co-inhibitory CTLA-4, ICOS cannot bind the same receptor due to the lack of MYPPY motif (60). However, it binds its unique ligand (ICOSL/B7-H2) constitutively expressed on B cells, macrophages, DCs, and somatic cells (61, 62). ICOS can be induced by TNF-alpha on various non-lymphoid cells including endothelial cells (63), lung epithelial cells (64), mesenchymal stem cells (65), fibroblasts (66) and tumor cells (67). Likewise, ICOS is highly expressed on tonsillar T cells. These latter cells are imperative for germinal center formation and B-lymphocyte maturation (56). The crosslinking of ICOS and ICOSL has stimulating activities, promoting an anti-tumor response by Th1, CTL and T follicular helper (Tfh) and a pro-tumor response mediated by Tregs and Th2 cells in the tumor microenvironment. On the other hand, ICOS engagement by its ligand is essential for the generation, function and maintenance of Tfh cells that help germinal center formation and auto-antibody production in autoimmunity (25, 58, 68). In contrast to the CD28 pathway, ICOS co-stimulation results in inefficient IL-2 production by activated T cells. Nevertheless, other cytokines including IFN-γ, IL-4, IL-10 and IL-21 are frequently more efficiently produced (69, 70). Mechanistic studies by Chen et al., proved that ICOS signal plays a key role in regulating suppressive function and survival of Tregs. In vitro activation of CD3 and ICOS favor the interaction between nuclear factor of activated T cells (NFAT) and Foxp3 leading to upregulation of Foxp3 downstream regulatory genes. ICOS promoted anti-apoptotic activity through PI3K-AKT pathway increasing Treg survival and function (71–73).
ICOS engagement leads to the recruitment of both p50α for AKT activity (72, 74) and p85α (75) regulatory subunits of PI3K, in parallel to the recruitment of the p110δ catalytic subunit. Recently, the recruitment of the p85α subunit was linked to the differentiation of Tfh cells (72). Contrarily to CD28, ICOS induces a more robust PI3K signaling and a less robust MAPK signaling with a specific recruitment of TBK1 (58). Using an ICOS YF mice model that are incapable of activating PI3K signaling, O’Brien et al. demonstrated that PI3K-independent signaling downstream of ICOS plays a crucial role in Treg stability in the context of chronic inflammation (76). Activation of PI3K increases calcium mobilization triggered by the TCR. ICOS ligation induces a stronger AKT activation than CD28 engagement. The entire cytoplasmic domain of ICOS is necessary for its co-stimulation and calcium mobilization. This co-stimulatory function depends on the unique transmembrane domain (TMD) of ICOS, responsible for promoting association with the tyrosine kinase Lck. Transmembrane domain-enabled Lck association is essential for p85 recruitment to ICOS and consequent PI3K activation. Lck triggers both the bystander and co-stimulatory signaling activity of ICOS. Replacement of ICOS transmembrane domain, while keeping the cytoplasmic domain intact, fails to support Tfh development of germinal center formation in vivo. When the ICOS transmembrane domain was transplanted onto a CAR, it boosts interactions between T cells and antigen-presenting target cells (77). Therefore, the third generation of CAR T cell therapy comprising the ICOS in a transmembrane signaling demonstrated a superior efficacy and an increased persistence in vivo (78).
Abrogation of the ICOS pathway in NOD mice exacerbated the T1D disease pointing to the important role of this pathway in tolerance. In this model, ICOS expression discriminated Foxp3- T cells from Foxp3+ Tregs and specifically designated a subset of intra-islet Tregs with amplified potential to expand, produce IL-10 and mediate suppressive function both in vitro and in vivo. Blockade or genetic deficiency of ICOS abrogated Treg mediated protection from T1D and exacerbated the disease in BDC2.5-NOD mice (79). There is a strong core of evidence that ICOS+ Treg are more sensitive to IL-2 than their ICOS- counterparts. In a colitis model, ICOS deficiency resulted in an increased induction of IL-10 in CD4 T cells but reduced accumulation of Foxp3+ cells in large intestine. ICOS- Treg displayed increased methylation of Foxp3 conserved non-coding sequence 2 (CNS2) and preferentially downregulated Foxp3, which renders these cells unable of reverting the gut inflammation (80). In a model of hypersensitivity to 2,4-dinitrofluorobenzene, Vocanson et al. demonstrated the Hapten-specific Tregs expanding in response to their cognate antigen in vivo are mainly ICOS+ Treg, and by using reporter mice they demonstrated that these cells were derived from the expansion of natural Treg and were dependent on innate cells such as DCs (81). In Toxoplasma and Mycobacterium tuberculosis models, they described an expansion of Teffs and a loss of Treg frequency in the brain and lung of ICOS deficient mice respectively (76, 82).
In human melanoma patients treated with high dose IL-2 therapy, the ICOS+ Treg population was the most expanded and the most proliferative lymphocyte population in the blood. Melanoma patients with enhanced expansion of ICOS+ Treg in blood following treatment had unfavorable clinical outcomes than patients with fewer ICOS+ Tregs (83). For instance, ICOShi Tregs had superior immunosuppressive capacity compared to ICOSlo Treg isolated from melanoma patients. These ICOShi Tregs were also able to induce suppressive IL-10 producing Tr1 cells from CD4+T cells (61). Similarly, a high infiltration of ICOS+ Treg in hepatocellular carcinoma indicated a worse prognosis (84). Moreover, in the tumor microenvironment, Tregs express increased levels of ICOS and Foxp3 and secrete higher levels of IL-10 and TGF-b (59). There is a clear understanding of the exacerbating role of ICOS+ Tregs in the disease in the immune-oncology field; however on the flip side, data are controversial in autoimmunity. In active SLE patients the ratio of ICOS+ Tregs to ICOS+ T responder CD4+ cells is significantly reduced and ICOS- Treg reduction is observed (85). Whereas in RA patients, ICOS+ Tregs were increased compared to normal controls, and such an increase was accentuated in patients with inactive RA compared to patients with active RA. Additionally for the patients with active RA the expression of suppressive cytokines (IL-10, TGF-β and IL-35) decreased while, expression of IL-17 increased compared with inactive RA, suggesting that ICOS+ Tregs may play an inflammatory and inhibitory function in a context-dependent manner (77).
TNFRSF9 also known as 4-1BB or CD137 is an inducible member of the tumor necrosis factor receptor (TNFR) family that plays the role of a co-stimulatory receptor. It was first discovered in 1989 on activated T lymphocytes (86, 87). It is expressed on activated CD4 and CD8 T cells but also on NK cells, B cells, neutrophils, dendritic cells, eosinophils, mast cells (88–91), endothelial cells, and some tumor cells (92–94). Tregs also expresses CD137 and its expression increases after activation (95–98). Whereas, its only known TNF-related ligand, 4-1BBL, is expressed by activated APCs (99, 100). When 4-1BB engagement by 4-1BBL is coupled with a strong TCR signaling, it provides co-stimulatory signals to T cells even in the absence of CD28 signaling. It enhances IL-2 and IFN-g production by Teffs and CTLs, respectively. Although, CD4 and CD8 T cells are both stimulated with 4-1BB in vitro, 4-1BBL/4-1BB crosslinking preferentially activates CD8 T cells (91, 101). There is also support that 4-1BB signaling in conventional T cells drives an excessive production of IL-2 responsible for escaping the Treg suppressive effect (102). Zheng et al., demonstrated that engagement of 4-1BB on thymic Tregs induces their proliferation both in vitro and in vivo while maintaining their suppressive ability despite the absence of detectable IL-2 indicating this pathway doesn’t involve IL-2 production (103). Similarly, data from Elpek et al. support the use of 4-1BBL as a means of proliferating Tregs in vivo (104). Likewise, 4-1BB is also able to downregulate the functions of Th cells, either by anergizing them or by enhancing the generation of Tregs (105–107). On the contrary, Akhmetzyanova et al. showed that 4-1BB stimulation converted Foxp3+ Tregs into cytotoxic killer cells that were able to contribute to antigen specific tumor rejection in vivo (108).
In a type 1 diabetes model, targeting 4-1BB with a monoclonal antibody suppressed the disease in NOD mice when the treatment is initiated before the development of auto-reactive T cells and is dependent on Treg induction (109). In a colitis model administration of anti-4-1BB agonistic antibody led to the reduction of incidence and severity of colitis. This effect was linked to a reduction of IL-2 expression by Th1 cells and an increase in Tregs (110, 111). Similarly, in an Experimental Autoimmune Encephalomyelitis (EAE) and Imiquimod-induced psoriasis-like dermatitis models, 4-1BB agonist was able to reduce disease by modulating Th17 versus Treg balance (112, 113). The generation of iTregs in the gut was attributed to the expression of retinal dehydrogenase (RALDH) by DC, an enzyme that promotes retinoic acid that aids differentiation of iTregs in the intestinal mucosa (114, 115). Another mechanism by which Treg engagement of CD137 tunes down Teff activation is by transferring and internalizing the CD137-CD137L complex formed between Tregs and APCs to Tregs via trygocytosis and depriving APC from their CD137L stimulating ability (107). As many of the members of the tumor necrosis factor superfamily (TNFRSF), 4-1BB ligation recruits TRAF adapter proteins, particularly that result in increased NFκB and MAP-kinase signaling (67, 116–119).
Despite the controversial function of 4-1BB in Tregs, agonistic anti-CD137 mAbs are being tested as therapies for cancer and autoimmune diseases. While, their mechanisms of action are different in different disease settings, they remain not fully studied and possible collateral effect on immune responses needs to be further investigated (120). Tregs with low 4-1BB expression was associated with enhanced overall survival in non-small cell lung cancer, confirming the role of 4-1BB in promoting suppressive function in tumor microenvironment (121). Also, using a genome-wide RNA-Seq analysis of multiple human cancer types and purified Tregs, Freeman et al. demonstrated that 4-1BB had an increased selectivity for human tumor Tregs (122). In autoimmunity, data support the role of 4-1BB in promoting Tregs (27). Altogether, 4-1BB can affect the biology of Treg cells and drive the crosstalk with other immunomodulatory cells.
Glucocorticoid-induced tumor necrosis factor (TNF) receptor (GITR) is a member of the TNF receptor (TNFR) superfamily. GITR is a 228-amino acids type I transmembrane protein distinguished by three cysteine pseudorepeats in the extracellular domain. It was described for the first time in 1997 by Nocentini in DEX-treated murine T cell lines (123). GITR is constitutively expressed on CD25+CD4+ Tregs (97, 124). After activation, its expression is upregulated on all T cell subsets (125). GITR is also detected on various myeloid cells; which include monocytes, macrophages, DCs and MDSCs (126, 127). GITR is engaged by its unique ligand (GITRL) that is expressed on activated APCs and endothelial cells (128–130). GITR and GITRL expression is reported on numerous cell types and is not limited to hematopoietic cells. A moderate expression is observed on keratinocytes and osteoclast precursors, and elevated expression on endothelial cells stimulated with type I IFN (128). GITR engagement with GITRL rescues T cells from anti-CD3 induced apoptosis, preserves their activation, proliferation and cytokine production (131, 132). GITR has a distinctive role for CD8+ and CD4+ T cells. GITR signaling induces the expression of CD137 on CD8 memory T cells, it could also lower the threshold for CD28 signaling on CD8 T cells (133, 134). While, on Tregs, GITR is a critical receptor in the differentiation of thymic Tregs (tTregs), and expansion of both tTregs and peripheral Tregs (pTregs) (135, 136). GITR signaling is mediated by recruitment of TRAF2 and TRAF5 that induce NF-κB, resulting in the upregulation of Bcl-Xi, an anti-apoptotic protein on activated CD8 T cell (137). GITR engagement by GITRL induces IL-9 production by a subest of Th cells in a TRAF6 and NF-κB dependent manner, yielding enhanced tumor-specific CTLs response (138). However, in the context of Treg, GITR induces a TRAF4 mediated induction of NF-κB (139). To sum it up, Ephrem et al., demonstrated that the effect of GITR signaling is complex and depend on the activation state of the Tregs and Teffs as well as the physiologic environment of the host (140).
Mouse studies showed that GITR signaling abrogates the suppressive function of Tregs both in vitro and in vivo (97, 141). In contrast to data obtained in mice, however, the engagement of GITR by huGITRL did not abrogate Treg suppression and promoted their expansion (142–144). In mouse tumor models, treatment with anti-mouse GITR agonistic Ab, did not affect GITR expression in Tregs among Tumor Infiltrating Lymphocytes (TILs) that remained high. Nonetheless, non-Treg CD4 and CD8 TILs exhibited high GITR expression as well. Interestingly, the GITR expression on CD8 and CD4 TILs corroborated the anti-tumor effects of GITR treatment (145). In a mouse melanoma model GITR signaling destabilized Treg by reducing Foxp3 expression in intratumoral Tregs but not in circulating one which improved anti-tumor immunity (146). Similarly, targeting GITR in a glioblastoma model using an agonistic antibody promoted CD4 Treg differentiation into CD4 Teffs, alleviated Treg-mediated suppression of anti-tumor immune response, and induced potent anti-tumor effector cells (147). Recent evidence from murine tumor models suggests that anti-GITR antibodies selectively reprogrammed Tumor infiltrating-Tregs (TI-Tregs). These antibodies caused downregulation of Foxp3, Helios and IL-10, while increased the levels of IFN-g production from TI-Tregs. GITR targeting antibodies play a crucial role via in regulating Helios expression, as anti-GITR treatment phenocopies Helios genetic deletion in Tregs. Similar observation was made in the context of Myasthenia Gravis model where GITR directly regulated Helios expression in Tregs (148, 149).
On the flip side, blockade of GITR and GITRL interaction in autoimmunity alleviates disease severity. In NOD mice, an agonistic treatment with GITR accelerated disease onset significantly. The activity was not attributed to a decline in Tregs but rather an activation of diabetogenic T cells. The role of Tregs was confirmed when a similar observation was made is CD28-/- NOD mice which lack Tregs. Likewise, agonistic treatment with anti-GITR (DTA-1) during the induction phase of EAE significantly enhanced the level of clinical severity (150, 151). In contrast, blockade of GITR with a neutralizing antibody led to a significant protection from diabetes even at late-stage disease (151). Comparison of cells from GITR-/- to GITR +/+ showed that GITR-/- Tregs are more suppressive in vitro and GITR-/- mice are less susceptible to developing RA in a CIA disease model (152).
OX40 (also called CD134 and TNFRSF4) is another co-stimulatory receptor of the TNFRSF of an approximately 50kD which is a type 1 transmembrane glycoprotein first described in 1987 (153, 154). OX-40 is a well-known Tcell activation marker. It is induced in activated CD4 and CD8 T cells, as well as in neutrophils, NK and NKTs (155, 156). OX40 is a late costimulatory receptor. Artificial engagement by OX40L induces proliferation, cytokine production, and T cell survival. This effect is partly induced by the expression of antiapoptotic molecules of the Bcl-2 family including Bcl-xL, Bcl-2 and Bfl-1 as well as survivin (157). However, it does not interfere with priming naïve T cells, as OX40-deficient T cells show normal differentiation to Teffs via TCR signaling but are not able to remain alive (158, 159). OX40 is also considered as a key marker of Tfh cells that can promote their generation and maintenance. OX40 synergizes with ICOS to maximize Tfh responses and formation and maintenance of germinal centers (160). In mouse Tregs, OX40 is constitutively expressed and the OX40 ligand (OX40L or CD252) is broadly expressed on activated APCs, including DCs, B cells, and macrophages, as well as non-hematopoietic cells, comprising endothelial cells and smooth muscle cells (161). Some studies show that when OX40 is engaged on Tregs, it not only inhibits their function and generation but can additionally, impede the generation, differentiation, and suppressive function of IL-10-producing CD4 type 1 Tregs significantly (162, 163). Peloso et al. demonstrated that engaging OX40 on Tregs by an OX40 agonist does not intrinsically impair their function but rather enhances their inflammatory cytokines production such as IFN-g, TNF-a and Granzyme B (164). In a different study by Piconese et al., they showed that mast cells counteracted the Treg function by ligating the OX40, and together with IL-6 production they induced a differentiation toward Th17 (165). On the other hand, OX40L expression on ILC2s induced tissue-restricted T cell co-stimulation that was crucial for Th2 and Treg responses in the lung and adipose tissue. Additionally, the IL-33 administration resulted in an organ specific expression of OX40L on ILC2 with a concomitant expansion of Tregs (166).
In T cells, OX40 activates both PI3K and NF-κB pathways by forming a signalosome containing TRAF2, IKKa, IKKb, PI3k and AKT (157). However, in Treg precursors, OX40 co-stimulatory activates BATF3 and BATF that induce the production of a closed chromatin to repress Foxp3 expression in Sirt1/7-dependent manner. Additionally, OX40 can also activate PI3K-AKT-mTOR pathway and inhibit Foxp3 induction by phosphorylation and nuclear exclusion of the transcription factor FoxO (46, 167). Much of the signaling work for OX-40 has been done in Teffs and Tfh cells, and further detailed studies for signaling in Tregs would be beneficial.
Numerous tumor mouse models and preclinical studies investigated OX40 agonistic signaling as an anti-tumor therapy through TIL regulation. The anti-tumor effects of OX40 agonist antibodies has been reported in several mouse models and human preclinical studies (168). Considering the constitutive expression of OX40 in Tregs, OX40 signaling dominantly affects Tregs. However, OX40 stimulation has numerous effects that lead to tumor elimination. OX40 engagement not only mediated Treg inhibition that unleashed nearby DCs allowing the induction of an adaptive immune response, but concomitantly delivered a fitness signal to activated T cells (164, 165). In autoimmunity, inhibition of OX40L ameliorated CIA disease scores in mice even when the signal on OX40 activated cells is intact (169). Contrarily, Griseri et al. demonstrated that OX40 is indispensable for Tregs accumulation in the colon and it plays a crucial role in Treg mediated suppression of colitis. OX40 deficient Tregs underwent enhanced activation induced death which corroborated the importance of OX40 in delivering the survival signal after activation. This could be explained by the importance of OX40 in Tregs that allows them to compete with colitogenic Th1 OX40+ cells for interaction with OX40L in DC (170). In a mouse model of pemphigus, Iriki et al. showed that OX40+ Tregs played a essential role in constraining OX40 signaling in autoreactive T cells. Likewise, Tregs signaling through OX40L on DCs suppressed the expression of OX40L itself (171). In a GVHD model, blocking OX40 reduced infiltration of human T cells to target organs. It also decreased IL-21 and TNF producing T cells while promoting Treg responses without compromising the cytolytic activity of CD8 T cells (172).
In active SLE patients, Treg dysfunction is mediated by APCs in an OX40L-dependent manner. In active skin lesions, Tregs and OX40L expression colocalized. Ligation of OX40 by OX40L resulted in downregulation of Foxp3 in Tregs (173). The data is conflicting regarding the effect of OX40 agonist on Tregs with some studies demonstrating that OX40 blocks the suppressive function of Tregs, while others showing enhanced Treg proliferation (46, 164, 165). Many studies in the autoimmunity setting demonstrated consistently that OX40 limits Treg function (169, 172–175). Altogether, these results indicate that OX40 signaling may regulate Tregs in several ways, depending on numerous factors, such as cytokines and other stimulation conditions (70).
CD40L or CD154 is the ligand for CD40. It is a 33-39kD type II membrane glycoprotein (176, 177). CD154 was first reported in 1992 by Lederman and colleagues (178). It was described as an activation-induced surface T cell marker that is involved in mediating contact-dependent Teff function. It engages CD40 on B cells driving B cell activation, maturation, and germinal center formation. But it is also essential for the final differentiation of CD4 T cells and the selection of TCR clonotypes during a T-dependent humoral immune response. Recent data demonstrated that CD154 is critical for the selection of T-cell clones throughout the negative selection exercise in the thymus (179). CD154 expression has been described in a variety of other cells, including platelets, mast cells, macrophages, basophils, NK cells, B lymphocytes, and non-haematopoietic cells (180). Increased expression of CD154 on T cells has been reported in different autoimmune patients (181, 182). Naïve Tregs express basal levels of CD154 that are upregulated upon TCR-triggered activation (183). Several studies demonstrated that expression of CD40 on DCs is essential to induce T cell tolerance and Treg accumulation. This signaling in IL-10-differentiated DC-10 contributes importantly to the expression of IL-10 potent activator and this is a modulator of Treg activation and function (183–185). Other studies pointed the important role of CD40-activated B cells in inducing and expanding Tregs from naïve precursors (186, 187). The abrogation of the CD40-CD40L interaction hinders the homeostasis of thymic resident Tregs by changing the levels of IL-2 but does not affect their precursor development (188). Nicole et al. demonstrated that CD154+ nTreg could be efficiently expanded by specific antigenic activation while preserving their suppressive activity (189, 190). In a HIV chronic infection model, soluble CD154 induced immunosuppression by expanding Tregs (191). Similarly, CD40 upregulation on mature DCs increased the Th17/Treg ratio in vitro during the pathogenesis of periodontitis in young patients (192). The blockade of CD154 was able to modulate this ratio and prolonged the survival of allogeneic corneal grafts in mice (193). In an influenza model, Ballesteros-Tato et al. showed that Teffs and Tregs compete for CD40 ligation on DCs, and CD4+ T cells are only required for robust influenza specific CD8 response when Tregs are present (194). Ferrer et al. extensively studied the effect of CD154 blockade in extending graft survival. They demonstrated that CD154 blockade reduced antigen-specific CD4+ T-cell accumulation and promoted migration of Foxp3+ Tregs to the graft. They also showed that anti-CD154 promotes conversion of donor reactive CD4+ T cells into CD25+ Foxp3+ induced Tregs (195). Using a DEREG mouse model Lee et al. demonstrated that a short-term blockade of CD154 could lead to Treg mediated immune tolerance in the intrahepatic murine allogeneic islet transplantation model (196). Animal studies are supported by clinical data where CD154 blockade in relapsing-remitting multiple sclerosis patient demonstrated an increase of CD25+ T cells and anti-inflammatory cytokines (197).
Cytotoxic T lymphocyte antigen-4 (CTLA-4) or CD152, is a critical regulator of T cell responses. It was first described by Brunet et al. in 1987 in mice. It encodes 223 amino acid protein and was classified as a member of the Ig superfamily (198). Highly homologous to CD28, CTLA-4 binds to their shared ligands, CD80 and CD86, with greater affinity thereby preventing CD28 costimulation of T cells (199). Engagement of CTLA-4 also initiates signaling events, including recruitment of phosphatases and activation of ubiquitin ligases, which inhibit T cell activation (200–202). Unlike CD28, CTLA-4 has rapid and constitutive endocytosis process (203). While its expression is upregulated rapidly on conventional T cells following TCR engagement (204), CTLA-4 is transcriptionally regulated by Foxp3 (205) and therefore constitutively expressed on CD4+ Foxp3+ Tregs (206). Mice deficient in CTLA-4 develop lethal multi-organ autoimmune disease driven by CD4+ T cells (207–209). Disease also develops in mice with a Treg-specific depletion of CTLA-4, suggesting CTLA-4 plays a critical role in Treg biology (210). Evaluation of Tregs from these mice demonstrated that Tregs develop and survive in the absence CTLA-4 but are unable to suppress the activation of conventional CD4+ T cells. The dysregulated T cell response that developed when CTLA-4 was deficient on Tregs was associated with ineffective downregulation of CD80 and CD86 on APCs (210). Biochemical analyses have demonstrated that an interaction between CTLA-4 and the protein kinase C in Tregs is required for reduced CD86 expression by APCs (211).
There are numerous in vivo animal models that support a role for CTLA-4 in Treg function (212–214); however in vitro systems have yielded conflicting findings. Some reports have suggested that the suppressive capacity of human Tregs is largely dependent on CTLA-4, with CTLA-4 blocking antibodies reversing the inhibitory effect of Tregs on CD4+ CD25- T cell proliferation (215). Further, depletion of CD25+ cells also reduced the effect of CTLA-4 blocking antibodies on human T cell proliferation, suggesting that the impact of anti-CTLA-4 antibodies is in part mediated through Tregs (216). However, other in vitro studies revealed no impact of CTLA-4 blockade on the suppressive capacity of human Tregs (217). This could reflect variation in assay conditions as well as discrepancies between in vitro assays and complex in vivo systems. Indeed, there are reports that CTLA-4-deficient Tregs can retain functionality in vitro yet be unable to control inflammation in vivo (214). Additionally, conditional deletion of CTLA-4 in Tregs in adult mice was associated with protection from autoimmunity (218). Expression of the extracellular portion of CTLA-4 only by Tregs was sufficient to suppress the proliferative response of conventional T cells both in vitro and in vivo, while the intracellular portion of the protein was required to maintain TCR hypo-signaling in Tregs (219). Cumulatively, these data suggest that CTLA-4 can contribute to the suppressive capacity of Tregs, but its role may be context-dependent where the nature of the stimulus or the presence of compensatory suppressive mechanisms may influence the overall activity of Treg.
Programmed death 1 (PD-1) or CD279 is also a member of the Ig superfamily and is encoded by PDCD1 gene and composed of 288 amino acid residues (220, 221). PD-1 contains an intracellular immunoreceptor tyrosine-based inhibitory motif (ITIM) and serves as a central negative regulator of immune responses (222). It is expressed on the surfaces of activated T-cells, B-cells, dendritic cells, monocytes and natural killer cells (223–225). In 1989, Smith et al. described the phenomena of apoptosis or programmed cell death that the abnormal thymic T cells undergo in the thymus during cell maturation (226). This observation led to the discovery of the gene responsible for this programmed death by Honjo’s team in 1991 and elucidating its real function as a negative regulator of the immune responses of T cells (227). PD-1 can bind two ligands PD-L1 (228) and PD-L2 (229). PD-L1 is broadly expressed in inflamed tissues (221), while expression of PD-L2 is restricted to APCs (229). PD-L2 has three-fold higher affinity for PD-1 than PD-L1 (230). Mice deficient in PD-1 spontaneously develop autoimmunity, with the specific manifestations linked to the genetic strain on the animal, suggesting that in the absence of PD-1, the overall immune response is not differentially skewed but rather unleashed (231–234).
In addition to its broad expression pattern on a variety of activated immune cell populations, PD-1 as well as its ligand PD-L1, are expressed on the surface of Tregs. Using mice that selectively lack PD-1 in Tregs, Tan et al. demonstrated an improvement of the experimental autoimmune encephalomyelitis (EAE) and protection from diabetes in nonobese diabetic (NOD) mice (235). PD-1 deficient Tregs display an activated phenotype and an enhanced immunosuppressive function linked to a reduced signaling through the PI3K–AKT pathway (235). Likewise, the presence of PD-1 expressing Tregs in the tumor microenvironment is a signature of dysfunctional exhausted Tregs that have an increased IFN-g secretion and are unable to suppress the anti-tumor immune response (236). In the clinic, the use of PD-1 blockade led to an augmented proliferation and suppressive function of tumor infiltrating Tregs that resulted in a rapid cancer progression called hyper-progressive disease in 10% of advanced gastric cancer patients (237). Animal studies demonstrated that PD-1 signaling blocks the AKT/mTOR pathway and promotes of Foxp3 and the development of iTregs from naïve T cells (238). These observations were confirmed in a human system, where PDL-1 expressing cells were able to induce conversion of Th1 cells into Foxp3 expressing Tregs capable of conferring protection in a GvHD and colitis model (239, 240). Chronic stimulation leads to further upregulation of PD-1 levels on Tregs where, in settings of viral infection, its expression is critical for suppressing the anti-viral T cell response (241, 242). Additional exploration of human PD-1+ Tregs, including single-cell approaches, will be needed to better understand the consequence of PD-1 expression on Tregs in settings of infection, cancer and autoimmunity.
Tregs can directly inhibit Teff responses or can act on these cells indirectly by regulating APCs. Human Tregs have been shown to promote immune suppression by inducing upregulation of PD-L1 on DCs, with PD-L1 blockade reversing the Treg-induced suppressive activity of DCs (243). The ability of Tregs to directly inhibit B cell responses also involves the PD-1 pathway. Engagement of PD-1 on B cells by PD-L1/2 expressing Tregs directly inhibited activation, proliferation and antibody production from self-reactive B cells and promoted their apoptosis (244). Interestingly, the levels of expression of PD-1 are important for regulating Treg cell function as well. Performing a series of in vitro and in vivo testing of PD-1 pathway blockade, Wong et al. demonstrated that Tregs are functionally intact when the level of PD-1 expression is neither too high nor too low and that varied degrees of expression or engagement of PD-1 can trigger different immune responses (245).
T cell immunoglobulin with ITIM domain (TIGIT) also designated as VSTM3, VSIG9 or WUCAM is a co-inhibitory receptor that was first described in 2009 expressed by T cells (246). Studies have confirmed its expression by other immune cells including B cells, NK cells, ILCs and pDCs (247–252). Later, studies demonstrated that TIGIT is highly expressed by Tregs (253–256). TIGIT is composed of an extracellular immunoglobulin variable domain, a type 1 transmembrane domain and a cytoplasmic tail with two inhibitory motifs and ITIM (immunoreceptor tyrosine-based inhibitory motif) and an ITT-like motif (257). TIGIT has multiple ligands including CD155, CD112, CD113, PVLR4 (258). However, TIGIT has the highest binding affinity to CD155, preventing the association between CD155 and the T cell costimulatory molecule CD226 (258, 259). Binding to either of the two ligands CD155 or CD112 on APCs prevents their maturation and confers a tolerogenic phenotype (246). Studies in mice have demonstrated that TIGIT expression serves as a marker for activated Tregs and Tregs expressing TIGIT have been demonstrated to be more potent suppressors than TIGIT negative Tregs (260). More specifically, TIGIT+ Tregs express elevated levels of markers including CTLA-4, CD25 and GITR as well as the transcription factor Foxp3. In addition, TIGIT+ Tregs produced higher amounts of effector molecules including IL-10, Fgl2 and granzyme B compared to TIGIT- Tregs. Ligation of TIGIT with an agonistic antibody directly induced production of both IL-10 and Fgl2 by Tregs in vitro and neutralization of Fgl2 in vitro reversed the enhanced suppressive capacity of TIGIT+ Tregs, demonstrating that Fgl2 is a key mechanism by which TIGIT drives enhanced Treg suppression (260).
TIGIT is highly expressed on human thymic-derived Tregs where its expression was associated with robust suppressive activity (254). TIGIT expression in combination with additional phenotypic markers can be used to define subsets of human Tregs. Fuhrman et al, for example, demonstrate that human Tregs which co-express both TIGIT and CD226 can produce cytokines such as IL-10 and effector genes, where TIGIT+ CD226- Tregs do not (254). Additionally, the combination of TIGIT and FCRL5 identifies a population of Helios+ Foxp3+ Tregs which do not produce pro-inflammatory cytokines and is highly enriched for suppressive activity (261). These studies highlight the heterogeneity that exists within the Treg lineage. Further evaluation of these specialized cell populations in different disease states and tissue compartments will be necessary to dissect the contribution of these various Treg populations to overall immune control and aid in our ability to better target these cell types for therapeutic benefit.
In vitro studies using natural ligand have provided insight into the mechanism by which TIGIT engagement impacts Treg signaling and function. TIGIT signaling leads to phosphorylation and nuclear localization of FoxO1 and suppression of AKT activity, ultimately preventing the reprogramming of human Tregs to pro-inflammatory Th1 Tregs by repressing IFNγ and T-bet expression (262). Studies in both mouse and human also suggest that TIGIT signaling may promote Treg stability. Agonism of TIGIT led to reduced expression of TCF7, a factor known to antagonize Foxp3 (254, 263). Further, signaling through TIGIT reduced the phosphorylation of both AKT and S6 and blocked the downregulation of Foxp3 in T cells stimulated through the TCR, supporting the idea that TIGIT plays a role in stabilizing the Treg lineage (264).
Lymphocyte activation gene-3 (LAG-3) or CD223 is a 70kDa transmembrane glycoprotein. It is a member of the immunoglobulin superfamily and contains four extracellular Ig-like domains (265). LAG-3 is a CD4 ancestral homolog (265, 266). Similarly to CD4, LAG-3 binds MHC class II (MHCII) (267), additionally it was described to bind other receptors including fibrinogen-like protein 1 (FGL-1) (268), alpha-synuclein fibrils (alpha-syn) (269), galectin-3 (Gal-3) (270) and lymph node sinusoidal endothelial cell C-type lectin (LSECtin) (271). LAG-3 is another co-inhibitory receptor whose expression is transiently upregulated on effector cells following activation and more persistently expressed by Tregs, including thymic-derived Tregs, iTregs and Tr1 cells (272). While it is the most studied on T cells and Tregs, LAG-3 can also be expressed on unconventional T cells including gamma-delta T cells (273), mucosal-associated invariant T cells (274), invariant NKT cells (275), B cells (276, 277), plasmacytoid DC (278) and neurons (279).
There is evidence that LAG-3 can contribute to the regulatory capacity of Tregs through multiple mechanisms, including both cell-intrinsic and -extrinsic pathways. Antagonistic LAG-3 antibodies blunted the ability of both natural and iTregs to suppress the proliferation of Teffs in vitro and in vivo and ectopic expression of LAG-3 was sufficient to confer regulatory properties to T cells (272). Notably, the ability of ectopic LAG-3 to regulate T cell responses required expression of full-length LAG-3, with intracellular mutants not sufficient to confer suppressive activity. These data suggest that signaling through LAG-3 is required for T cell-intrinsic regulation. The intracellular pathways involved downstream of LAG-3 remain to be elucidated. Identification and characterization of the key signaling pathways regulated by LAG-3 will be critical for the development of therapeutics aimed at targeting LAG-3 activity.
Additional studies demonstrate that LAG-3 can also promote suppressive activity independently of its ability to deliver signals to the Treg. In vitro murine Treg/DC co-culture assays have shown that Tregs can inhibit antigen specific DC maturation in a LAG-3/MHCII-dependent manner, as LAG-3 deficient Tregs lost the ability to suppress upregulation of CD86 on DCs (280). Transwell studies demonstrated that the capacity of LAG-3 to block DC maturation was dependent on cell-cell contact, did not require the intracellular signaling domain of LAG-3 and involved a novel ITAM-mediated signaling event in DCs that involved the recruitment of SHP-1 (280). Cumulatively, these data highlight the potential for LAG-3 to induce inhibition in a bidirectional manner.
LAG-3 also represents a critical marker of Tr1 cells, a population of regulatory cells that produce high levels of IL-10 but do not express Foxp3 (281, 282). Forced expression of the transcription factor Egr-2 induced naive CD4+ T cells to express LAG-3 and IL-10 and to exhibit antigen-specific suppressive activity, suggesting a role for Egr-2 in LAG-3 regulation and Tr1 development (283). Anti-LAG-3 antagonist antibody treatment reversed the immunological tolerance induced by Tr1 cells in a mouse model of pancreatic islet transplantation supporting a role for LAG-3 in the activity of Tr1 cells, however, the precise role of LAG-3 in Tr1 function remains to be determined (284). Similarly, Jha et al., reported that blocking LAG-3 was associated with increased susceptibility to mercury-induced autoimmunity, and this response reduced Treg-mediated inhibition of DC maturation (285). Zhang et al. demonstrated that NOD mice lacking the cell surface expression of LAG-3 on Tregs exhibit delayed onset of Type 1 diabetes, and this was ascribed to the augmented Treg cell proliferation and activity (286). Altogether, these data suggest the important role of LAG-3 in modulating the Treg function and influencing the overall immune response in different disease settings.
T cell immunoglobulin and mucin domain-containing protein-3 (TIM-3) also known as HAVCR2 is a member of the TIM family of immunoregulatory proteins. It was first described in 2002 by Monney and colleagues (287). TIM-3 has multiple ligands binding different epitopes on the TIM-3 extracellular V domain (288). The ligands include Galectin 9 which is a C type lectin (289), phosphatidylserine (PtdSer) (290), the glycoprotein CEACAM1 (291) and the alarmin high mobility group protein B1 (HMGB1) (292). TIM-3 has a unique feature because of the lack of known inhibitory signaling motifs in its cytoplasmic tail (288). It was originally described as a marker for exhausted effector T cells or IFN-γ producing CD4 and CD8 T cells (287, 288) but later described on a variety of immune cell types including myeloid cells (293), NK cells (294), mast cells (295) and Tregs (296). Subsets of Tregs have been described to express TIM-3, including some tissue resident Tregs. TIM-3 expressing Foxp3+ Tregs, in fact, make up minor fraction of the total Treg pool in a naïve mouse, but both the number and frequency of these cells increases significantly in both lymphoid and graft tissue during an allograft response (297). In vitro studies demonstrated that TIM-3 expression is upregulated on Tregs undergoing proliferation and its expression marks Tregs with enhanced suppressive capacity compared to TIM-3 negative Tregs. The enhanced suppressive capacity of TIM-3+ Tregs was associated with increased expression of CTLA-4, CD25 and IL-10 (297). Adoptive transfer studies revealed that while these cells have potent suppressive capacity, they did not prolong allograft survival as well as TIM-3 negative cells in vivo suggesting this represents a short-lived population of Tregs (297).
Oncology studies in both mouse and human support these observations, demonstrating an enrichment of TIM-3 expressing Tregs within the tumor microenvironment, with few of these cells observed in the blood or peripheral tissues (298, 299). In this setting as well, TIM-3 expressing Tregs demonstrate enhanced suppressive capacity and express higher levels of suppressive effector molecules than TIM-3 negative Tregs. Co-blockade of TIM-3 and PD-1 in preclinical tumor models was associated with downregulation of molecules that support the function of Tregs as well as an overall improvement in tumor clearance. Given the co-administration of TIM-3 and PD-1 blockade in these studies and the direct potential impact of these approaches on effector cells, additional studies are required to more specifically tease apart the impact of the TIM-3 pathway on Treg function.
Leukocyte immunoglobulin like receptor B4 (LILRB4, also known as ILT3, LIR5, CD85K or HM18) is an ITIM-containing member of the LILR family of proteins. It was identified in 1997 as the homolog for the mouse gp49B1 inhibitory receptor (300). Several ligands have been reported to bind LILRB4 including CD166 (301), ApoE (302) and CNTFR (303). LILRB4 is broadly expressed across the myeloid lineage (including monocytes, macrophages, and DCs) as well as on some plasma cells (304). LILRB4 is highly expressed by tolerogenic DCs and thought to contribute to Treg induction by these cells (305). Expression of LILRB4 either on the surface of APCs or in soluble form, can engage with ligands expressed by activated T cells to drive T cell anergy and activation of Tregs (306). LILRB4 and LILRB2 expressing DCs support the conversion of alloreactive CD4+CD45RO+CD25+ T cells to Tregs (307). Some studies suggest that subsets of Tregs can themselves express LILRB4 (308). LILRB4+ Treg shows attenuated T cell receptor mediated signaling (308). LILRB4+ Tregs represent a small fraction of Foxp3+ cells in healthy peripheral blood but were found to be expanded in the circulation of allergic patients and in tumor infiltrating immune cells in various tumor models (309). Mouse studies demonstrate that the serine threonine kinase CK2 regulates the expression of LILRB4 in Tregs and highlights a role for this Treg subset in the control of Th2-cell driven inflammation. In the absence of CK2, LILRB4 expression was elevated on Tregs and this was associated with impaired Treg suppressive capacity and expansion of Th2-inducing dendritic cell populations (308). The authors hypothesize that LILRB4 upregulation on Tregs may represent a transient mechanism by which immune suppression is blocked when a productive immune response is required. These studies suggest that LILRB4 may play a complex role in Treg biology with the potential to either promote the induction of Tregs or to suppress Treg responses depending on the context and cell-type in which the pathway is engaged. As differences exist between murine and human LILRB4 both in terms of protein structure and expression, it will be important to demonstrate some of these mechanisms and explore the role of LILRB4+ Tregs in human assay systems or humanized mouse models.
The importance of checkpoint pathways in modulating different diseases have been highlighted with the presence of multiple approved therapeutics and many more in the clinical and pre-clinical development pipelines (85, 303, 310). Blocking and activating approaches with different modalities for different pathways have been pursued, and in 2018, James P. Allison and Tasuku Honjo was awarded the Nobel Prize in Medicine for the development of immune checkpoint blockade in cancer (311). In instances in which conflicting data is existent, this may be due to short-term versus long-term effects of treatment, different dosing strategies, and different patient cohorts. It may be even as simple as how the samples were collected, how the cells were isolated and how the samples were stored. Tregs are specifically sensitive to different storage conditions, and this can affect their viability. Below, we will discuss insights on the effect of these molecules on Treg biology.
Targeting CD28 with a superagonist antibody (TGN1412) in a first in human trial was a tragic failure, since the administered antibody induced a severe systemic inflammatory response and a cytokine storm in healthy volunteers (312). To overcome these adverse effects, this antibody was further engineered into TAB08 and tested clinically in healthy volunteers at very low doses and showed induction of IL-10 release in the circulation which is a signature of Treg activation. The positive results granted a phase Ib trial in RA patients followed by a double blinded phase II study to confirm the therapeutic effect in patients (313).
FR104 is a CD28 antagonist (a pegylated Fab antibody) and is currently being tested in Ph2 trials. In non-human primates, it has been demonstrated that this therapeutic is Treg sparring or Treg stimulating, yet data from further clinical studies are needed to confirm this finding (314). Acazicolcept (ALPN-101) is yet another experimental therapeutic in the clinic being tested by Alpine Immune Sciences in collaboration with Abbvie, and it simultaneously blocks CD28 and ICOS co-stimulation. Phase 1 healthy volunteer study demonstrated the safety, tolerability and PK/PD properties of this therapeutic, and currently being tested in a randomized, double-blind, placebo controlled Ph2 trials for SLE (315). It will be important to track the effect of ALP101 on Tregs among other things in these studies. Recently, Alpine Immune Sciences terminated enrollment in their NEON-2 study with Davoceticept (ALPN-202, CD28 co-stimulator, PD-L1 and CTLA-4 blocker) and pembroluzimab due to cardiogenic shock. It becomes more evident with every additional study that the modulation of immune cells (both effector and Treg) requires a sweet spot for therapeutic efficacy and safety and more precision medicine approaches will need to be utilized for future studies.
As discussed above, CTLA-4 is constitutively expressed on Tregs and some of the suppressive capability of Tregs is mediated by CTLA-4 expression. Abatacept (CTLA-4 Ig fusion protein, Orencia™) was first approved in 2005 approved for Rheumatoid Arthritis, then later for Juvenile Idiopathic Arthritis (2008) and Psoriatic Arthritis (2017), Acute Graft versus Host Disease (2021) and it is being tested in the clinic for many other indications such as psoriasis, dermatomyositis, GvHD and others (316, 317). One study investigated the effects of Abatacept in RA patients and measured Treg numbers in the periphery at week 4 and 12 after treatment. 45 total RA patients with active disease were studied—30 received Abatacept and 15 received placebo treatment. The study showed that while Treg numbers were diminished in the periphery, the ex vivo functionality of Tregs were improved upon Abatacept treatment (318). In another study in juvenile idiopathic arthritis, Abatacept (10mg/kg i.v.) was administered to 10 patients (on day 0, 15 and 30 and monthly thereafter). Similarly in this setting, the number of peripheral Tregs was reduced at day 90 compared to normal controls (319). Bonelli et al., demonstrated that Abatacept treatment in RA patients increased proportions of Tregs but inhibited their suppressive capacity. This study also investigated the activation markers on Tregs such as CD69, CD71, HLA-DR and showed that activation markers were reduced after 2 and 4 weeks of therapy. There was also a reduction in CD95 and apoptosis mediated by this molecule (320). Diamanti et al., investigated the effect of Abatacept in anti-TNF therapy refractory patients (20 moderate to severe patients) and demonstrated that Treg frequency was not changed in the periphery. In this report, Abatacept treatment (10mg/kg) partially recovered the suppressive capability of Tregs after 6 months of treatment. One important point to note is that Tregs were defined as CD4+CD25+ cells in this study which is not the most robust way of identifying human Tregs (321). Szentpetery et al., investigated Treg numbers in the synovium and the psoriatic skin of 15 biologics-naïve psoriatic arthritis patients. Patients were randomized to receive i.v. Abatacept (10mg/kg) or placebo treatment. Treg (CD4+ Foxp3+) numbers in the synovium but not in the psoriatic skin of the patients were diminished with Abatacept treatment significantly improving clinical measures. It was interesting to see that Abatacept treatment was efficacious for joint-related outcomes but not for skin-related outcomes (322). These data points are good examples of how these co-stimulatory and co-inhibitory pathways can have tissue-specific effects. While tumor biopsy tissue has been more readily accessible for analysis of Treg modulation beyond the periphery, tissue data from autoimmune settings has been scarce. With the emergence of new technologies such as high dimensional immunofluorescence and spatial transcriptomics, our scientific understanding of tissue immunology and Treg function will deepen in the upcoming years.
Belatacept (Nulojix™) has 4 times higher affinity for CD86 and 2 times higher affinity for CD80 and is the newer generation of CTLA-4 blocker (323). In 2011, it was approved for prophylaxis of organ rejection in adults (324). In a small cohort study, Grimbert et al., studied the effect of Belatacept on mRNA levels of Foxp3 in biopsies from renal transplants and demonstrated that the Foxp3 levels were reduced only in the graft. This did not correlate with clinical efficacy since Belatacept treated patients had better functionality of graft after 1 year (325).
Anti-CTLA-4 blocking therapies have been approved and used for oncology indications mainly because they are able to remove the inhibitory breaks on effector cells. However, due to their constitutive CTLA-4 expression, Tregs are also a direct target of this therapy. Ipilimumab (Yervoy™), the first anti-CTLA-4 was first approved in 2011 and since then the indications for this medicine has been expanding along with combination therapy testing with other checkpoint inhibitors (326). One study by Sharma et al., demonstrated that the levels of Tregs were transiently increased 6 weeks after Ipilimumab therapy but returned to baseline by 3 months and stayed unchanged for 6 and 9 months in melanoma patients (10mg/kg iv every 3 weeks) (327). To mirror these results, a different study demonstrated the same outcome in melanoma patients that Tregs were transiently increased at week 6 but returned to normal after 3, 6 and 9 months and there was no difference in relapsed and relapse-free groups (328). Zhou et al., studied Ipilimumab treatment in patients with relapsed malignancy following allogeneic hematopoietic stem cell transplantation and demonstrated that the absolute counts of Tregs (CD4+CD25highFoxp3+) did not change during the study period (329). Kavanagh et al., studied a small cohort of progressive metastatic hormone-refractory prostate cancer patients in a Ph1 study. The subjects were dosed with escalating dose of Ipilimumab with a fixed dose of GM-CSF in a separate cohort. 3mg/kg anti-CTLA-4 only treatment resulted in increase of CD4+ Foxp3+ frequency and that Tregs maintained their level of both surface and intracellular reserves of CTLA-4 (330). One proposed mechanism of action is depletion of CTLA-4hi cells, especially Tregs by ADCC in the tumor microenvironment. Specifically, some approaches are trying to generate second generation of anti-CTLA-4 therapies with deeper Treg-depleting capability such as with enhanced Fc binding. One group demonstrated that Fc-enhanced CTLA-4 antibodies had better efficacy in tumor bearing FcγR humanized mice due to their Treg depleting capabilities (331). Another study by Semmrich et al., generated proof of concept in mice that intratumorally administered viral vectors containing anti-CTLA-4 or anti-CTLA-4 in combination with GM-CSF was better than systemic administration of anti-CTLA-4. A follow up Ph1 study is ongoing with viral vector (anti-CTLA-4 and GMCSF) administration in patients with metastatic or advanced solid tumors (NCT04725331 (331)).
Tremelimumab is another anti-CTLA-4 fully human IgG2 antibody currently in development. There are multiple Phase 3 and Phase 2 studies ongoing worldwide (332). Comin-Anduix et al., studied the cell signaling events in PBMCs from melanoma patients that were treated with tremelimumab after TCR and cytokine receptor stimulation. Looking at CD4, CD8 T cells and monocytes, they demonstrated that pp38, pSTAT1 and pSTAT3 were increased, pLck, pERK1/2 and pSTAT5 levels were decreased (333). In a very small Phase 1 study, the effect of local radiation and tremelimumab treatment was studied in inoperable locally recurrent or metastatic breast cancer patients. The data demonstrated that one week post treatment, there was an increase in ki-67+ proliferating Tregs in 5 out of 6 patients (334). In a different study, Khan et al., demonstrated that Tremelimumab treatment increased proliferation of Teffs, secretion of IL-2 and IFN-γ however, did not change the proportion of Tregs while reducing their suppressive capacity (335).
Nivolumab (Opdivo™) is a PD-1 antagonist and has been approved by FDA for the treatment of several different cancer types such as melanoma, non-small cell lung cancer, hepatocellular carcinoma, cervical cancer and head and neck squamous cell carcinoma (FDA: Hematology/oncology (cancer) approvals & safety notifications. 2019). One study studied the effect of Nivolumab in oral cavity squamous cell carcinoma in a Ph2 study with treatment on day 1, 14 and 28 (3mg/kg i.v.). Peripheral blood samples were collected before Nivolumab treatment and at the time of definitive oral resection. Progressive patients on day 28 proceeded with surgery while the patients improving received a fourth dose of Nivolumab on day 48. The findings demonstrated that while anti-PD-1 had opposing effects on CD4 (reduced) and CD8 (increased), it increased the proportion of Foxp3+ expressing cells in the periphery (336). In a different study, it was shown that treatment with Nivolumab pre-transplant of HSCT resulted in increased IFNg+ effector cells and potential rejection of the graft. Interestingly, posttransplant treatment of these patients with cyclophosphamide increased Tregs (337). To further build on this data set, Ikegawa et al., took a closer look into Treg numbers and phenotype in an exploratory study pre- and post-transplant with cyclophosphamide treatment. Their study demonstrated that cyclophosphamide treatment helps with the control of T effector cells and robust recovery of Tregs in the presence of pre-treatment with Nivolumab (338).
Another PD-1 antagonist is Pembrolizumab (Keytruda™, humanized IgG4k antibody) and its effects on different T cell populations have been studied. In vitro, Toor et al., demonstrated that treatment of PBMCs from primary cancer patients did not change Treg related markers such as CTLA-4, CD15s, LAP and ki-67. In this study, Pembrolizumab had the greatest reduction effect on PD-1 expression in CD4+CD25- cells compared to CD4+CD25+ T cells. As a follow up, the same group studied the effect of Pembrolizumab on iTreg generation in vitro and showed that pembrolizumab treatment inhibited iTreg generation and rendered Tregs less suppressive, especially at higher ratios. This was due to reduced IL-10 expression, mTOR and STAT1 activation and subsequent MAPK inhibition (339).
AMG557 (MEDI5872) is an anti-ICOSL antagonist and has been tested in the clinic in SLE, Lupus arthritis, Sjogren’s syndrome and Psoriasis. Single and multiple doses of AMG557 were tested in 112 SLE patients, and it was shown to be safe and tolerable with no change in the Treg frequency in the periphery (340). In a human clinical trial, OX40 agonist expanded non-Treg CD4 and CD8 T cells and upregulated OX40 expression on TIL Tregs (341, 342). GSK has developed an anti-LAG3 depleting mAb (ADCC enhanced, afucosylated) and this molecule has completed Ph1 testing in healthy volunteers and patients with plaque psoriasis. Overall, there was a reduction in LAG3+ cells; however no specific data on Tregs was reported.
In this review, we provided a summary of our understanding of how immune checkpoints regulate Treg homeostasis and function. While our understanding has gotten deeper over the years, there is still much to explore. Some outstanding areas to investigate include interplay between different immune checkpoints and how they modulate Treg function, the role of immune checkpoint pathways in Treg and other immune and non-immune cell interactions, and the role of checkpoint pathways in the non-immunological functions of Tregs such as wound healing and repair. In conclusion, immune checkpoints play fundamental roles in controlling Treg biology and further evidence, especially from the clinic, will be necessary in order to further our understanding and build the next generation of therapeutics.
MA, JK and SR all contributed to the writing and editing of the manuscript. All authors contributed to the article and approved the submitted version.
MA, JK and SR were employed by Horizon Therapeutics.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Chaplin DD. Overview of the immune response. J Allergy Clin Immunol (2010) 125(2 Suppl 2):S3–23. doi: 10.1016/j.jaci.2009.12.980
2. Golubovskaya V, Wu L. Different subsets of T cells, memory, effector functions, and CAR-T immunotherapy. Cancers (Basel) (2016) 8(3):36. doi: 10.3390/cancers8030036
3. Thapa P, Farber DL. The role of the thymus in the immune response. Thorac Surg Clin (2019) 29(2):123–31. doi: 10.1016/j.thorsurg.2018.12.001
4. Klein L, Kyewski B, Allen PM, Hogquist KA. Positive and negative selection of the T cell repertoire: what thymocytes see (and don't see). Nat Rev Immunol (2014) 14(6):377–91. doi: 10.1038/nri3667
5. Shevach EM, Thornton AM. tTregs, pTregs, and iTregs: similarities and differences. Immunol Rev (2014) 259(1):88–102. doi: 10.1111/imr.12160
6. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol (2003) 4(4):330–6. doi: 10.1038/ni904
7. Mishra S, Srinivasan S, Ma C, Zhang N. CD8(+) regulatory T cell - a mystery to be revealed. Front Immunol (2021) 12:708874. doi: 10.3389/fimmu.2021.708874
8. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell (2008) 133(5):775–87. doi: 10.1016/j.cell.2008.05.009
9. Roncarolo MG, Gregori S, Bacchetta R, Battaglia M. Tr1 cells and the counter-regulation of immunity: natural mechanisms and therapeutic applications. Curr Top Microbiol Immunol (2014) 380:39–68. doi: 10.1007/978-3-662-43492-5_3
10. Pabst O, Bernhardt G. On the road to tolerance–generation and migration of gut regulatory T cells. Eur J Immunol (2013) 43(6):1422–5. doi: 10.1002/eji.201243154
11. Shevyrev D, Tereshchenko V. Treg heterogeneity, function, and homeostasis. Front Immunol (2019) 10:3100. doi: 10.3389/fimmu.2019.03100
12. Godoy GJ, Paira DA, Olivera C, Breser ML, Sanchez LR, Motrich RD, et al. Differences in T regulatory cells between mouse strains frequently used in immunological research: Treg cell quantities and subpopulations in NOD, B6 and BALB/c mice. Immunol Lett (2020) 223:17–25. doi: 10.1016/j.imlet.2020.04.006
13. Khantakova JN, Bulygin AS, Sennikov SV. The Regulatory-T-Cell Memory Phenotype: What We Know. Cells (2022) 11(10). doi: 10.3390/cells11101687
14. Shao Q, Gu J, Zhou J, Wang Q, Li X, Deng Z, et al. Tissue Tregs and Maintenance of Tissue Homeostasis. Front Cell Dev Biol (2021) 9:717903. doi: 10.3389/fcell.2021.717903
15. Evans-Marin HL, Cao AT, Yao S, Chen F, He C, Liu H, et al. Unexpected Regulatory Role of CCR9 in Regulatory T Cell Development. PloS One (2015) 10(7):e0134100. doi: 10.1371/journal.pone.0134100
16. Yoshie O, Matsushima K. CCR4 and its ligands: from bench to bedside. Int Immunol (2015) 27(1):11–20. doi: 10.1093/intimm/dxu079
17. Du R, Zhao H, Yan F, Li H. IL-17+Foxp3+ T cells: an intermediate differentiation stage between Th17 cells and regulatory T cells. J Leukoc Biol (2014) 96(1):39–48. doi: 10.1189/jlb.1RU0114-010RR
18. Halim L, Romano M, McGregor R, Correa I, Pavlidis P, Grageda N, et al. An Atlas of Human Regulatory T Helper-like Cells Reveals Features of Th2-like Tregs that Support a Tumorigenic Environment. Cell Rep (2017) 20(3):757–70. doi: 10.1016/j.celrep.2017.06.079
19. Kitz A, Dominguez-Villar M. Molecular mechanisms underlying Th1-like Treg generation and function. Cell Mol Life Sci (2017) 74(22):4059–75. doi: 10.1007/s00018-017-2569-y
20. Kitz A, Singer E, Hafler D. Regulatory T cells: From discovery to autoimmunity. Cold Spring Harb Perspect Med (2018) 8(12). doi: 10.1101/cshperspect.a029041
21. Sakaguchi S, Mikami N, Wing JB, Tanaka A, Ichiyama K, Ohkura N. Regulatory T cells and human disease. Annu Rev Immunol (2020) 38:541–66. doi: 10.1146/annurev-immunol-042718-041717
22. Kolios AGA, Tsokos GC, Klatzmann D. Interleukin-2 and regulatory T cells in rheumatic diseases. Nat Rev Rheumatol (2021) 17(12):749–66. doi: 10.1038/s41584-021-00707-x
23. Lucca LE, Dominguez-Villar M. Modulation of regulatory T cell function and stability by co-inhibitory receptors. Nat Rev Immunol (2020) 20(11):680–93. doi: 10.1038/s41577-020-0296-3
24. Wingren AG, Parra E, Varga M, Kalland T, Sjogren HO, Hedlund G, et al. T Cell activation pathways: B7, LFA-3, and ICAM-1 shape unique T cell profiles. Crit Rev Immunol (2017) 37(2-6):463–81. doi: 10.1615/CritRevImmunol.v37.i2-6.130
25. Zhang Q, Vignali DA. Co-Stimulatory and Co-inhibitory pathways in autoimmunity. Immunity (2016) 44(5):1034–51. doi: 10.1016/j.immuni.2016.04.017
26. Li C, Jiang P, Wei S, Xu X, Wang J. Regulatory T cells in tumor microenvironment: new mechanisms, potential therapeutic strategies and future prospects. Mol Cancer (2020) 19(1):116. doi: 10.1158/1557-3125.HIPPO19-B11
27. Copsel S, Wolf D, Komanduri KV, Levy RB. The promise of CD4(+)FoxP3(+) regulatory T-cell manipulation in vivo: applications for allogeneic hematopoietic stem cell transplantation. Haematologica (2019) 104(7):1309–21. doi: 10.3324/haematol.2018.198838
28. Attanasio J, Wherry EJ. Costimulatory and coinhibitory receptor pathways in infectious disease. Immunity (2016) 44(5):1052–68. doi: 10.1016/j.immuni.2016.04.022
29. Atif M, Conti F, Gorochov G, Oo YH, Miyara M. Regulatory T cells in solid organ transplantation. Clin Transl Immunol (2020) 9(2):e01099. doi: 10.1002/cti2.1099
30. Roth-Walter F, Adcock IM, Benito-Villalvilla C, Bianchini R, Bjermer L, Boyman O, et al. Immune modulation via T regulatory cell enhancement: Disease-modifying therapies for autoimmunity and their potential for chronic allergic and inflammatory diseases-an EAACI position paper of the task force on immunopharmacology (TIPCO). Allergy (2021) 76(1):90–113. doi: 10.1111/all.14478
31. Huppert LA, Green MD, Kim L, Chow C, Leyfman Y, Daud AI, et al. Tissue-specific tregs in cancer metastasis: opportunities for precision immunotherapy. Cell Mol Immunol (2022) 19(1):33–45. doi: 10.1038/s41423-021-00742-4
32. Hansen JA, Martin PJ, Nowinski RC. Monoclonal antibodies identifying a novel T-cell antigen and ia antigens of human lymphocytes. Immunogenetics (1980) 10(1):247–60. doi: 10.1007/BF01561573
33. Brzostek J, Gascoigne NR, Rybakin V. Cell type-specific regulation of immunological synapse dynamics by B7 ligand recognition. Front Immunol (2016) 7:24. doi: 10.3389/fimmu.2016.00024
35. Singh SS, Jois SD. Homo- and heterodimerization of proteins in cell signaling: Inhibition and drug design. Adv Protein Chem Struct Biol (2018) 111:1–59. doi: 10.1016/bs.apcsb.2017.08.003
36. Rudd CE, Taylor A, Schneider H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol Rev (2009) 229(1):12–26. doi: 10.1111/j.1600-065X.2009.00770.x
37. Beyersdorf N, Kerkau T, Hunig T. CD28 co-stimulation in T-cell homeostasis: a recent perspective. Immunotargets Ther (2015) 4:111–22. doi: 10.2147/ITT.S61647
38. Weng NP, Akbar AN, Goronzy J. CD28(-) T cells: their role in the age-associated decline of immune function. Trends Immunol (2009) 30(7):306–12. doi: 10.1016/j.it.2009.03.013
39. Ortiz-Suarez A, Miller RA. A subset of CD8 memory T cells from old mice have high levels of CD28 and produce IFN-gamma. Clin Immunol (2002) 104(3):282–92. doi: 10.1006/clim.2002.5221
40. Lamarthee B, Marchal A, Charbonnier S, Blein T, Leon J, Martin E, et al. Transient mTOR inhibition rescues 4-1BB CAR-tregs from tonic signal-induced dysfunction. Nat Commun (2021) 12(1):6446. doi: 10.1038/s41467-021-26844-1
41. Sadelain M, Brentjens R, Riviere I. The basic principles of chimeric antigen receptor design. Cancer Discovery (2013) 3(4):388–98. doi: 10.1158/2159-8290.CD-12-0548
42. Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, et al. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity (2000) 12(4):431–40. doi: 10.1016/S1074-7613(00)80195-8
43. Tai X, Cowan M, Feigenbaum L, Singer A. CD28 costimulation of developing thymocytes induces Foxp3 expression and regulatory T cell differentiation independently of interleukin 2. Nat Immunol (2005) 6(2):152–62. doi: 10.1038/ni1160
44. Guo F, Iclozan C, Suh WK, Anasetti C, Yu XZ. CD28 controls differentiation of regulatory T cells from naive CD4 T cells. J Immunol (2008) 181(4):2285–91. doi: 10.4049/jimmunol.181.4.2285
45. Holt MP, Punkosdy GA, Glass DD, Shevach EM. TCR signaling and CD28/CTLA-4 signaling cooperatively modulate T regulatory cell homeostasis. J Immunol (2017) 198(4):1503–11. doi: 10.4049/jimmunol.1601670
46. Kumar P, Marinelarena A, Raghunathan D, Ragothaman VK, Saini S, Bhattacharya P, et al. Critical role of OX40 signaling in the TCR-independent phase of human and murine thymic treg generation. Cell Mol Immunol (2019) 16(2):138–53. doi: 10.1038/cmi.2018.8
47. Liang S, Alard P, Zhao Y, Parnell S, Clark SL, Kosiewicz MM. Conversion of CD4+ CD25- cells into CD4+ CD25+ regulatory T cells in vivo requires B7 costimulation, but not the thymus. J Exp Med (2005) 201(1):127–37. doi: 10.1084/jem.20041201
48. Zhang R, Huynh A, Whitcher G, Chang J, Maltzman JS, Turka LA. An obligate cell-intrinsic function for CD28 in tregs. J Clin Invest (2013) 123(2):580–93. doi: 10.1172/JCI65013
49. Golovina TN, Mikheeva T, Suhoski MM, Aqui NA, Tai VC, Shan X, et al. CD28 costimulation is essential for human T regulatory expansion and function. J Immunol (2008) 181(4):2855–68. doi: 10.4049/jimmunol.181.4.2855
50. Hombach AA, Kofler D, Hombach A, Rappl G, Abken H. Effective proliferation of human regulatory T cells requires a strong costimulatory CD28 signal that cannot be substituted by IL-2. J Immunol (2007) 179(11):7924–31. doi: 10.4049/jimmunol.179.11.7924
51. He X, Smeets RL, van Rijssen E, Boots AM, Joosten I, Koenen HJ. Single CD28 stimulation induces stable and polyclonal expansion of human regulatory T cells. Sci Rep (2017) 7:43003. doi: 10.1038/srep43003
52. Pallandre JR, Brillard E, Crehange G, Radlovic A, Remy-Martin JP, Saas P, et al. Role of STAT3 in CD4+CD25+FOXP3+ regulatory lymphocyte generation: implications in graft-versus-host disease and antitumor immunity. J Immunol (2007) 179(11):7593–604. doi: 10.4049/jimmunol.179.11.7593
53. Vang KB, Yang J, Pagan AJ, Li LX, Wang J, Green JM, et al. Cutting edge: CD28 and c-rel-dependent pathways initiate regulatory T cell development. J Immunol (2010) 184(8):4074–7. doi: 10.4049/jimmunol.0903933
54. Scotta C, Soligo M, Camperio C, Piccolella E. FOXP3 induced by CD28/B7 interaction regulates CD25 and anergic phenotype in human CD4+CD25- T lymphocytes. J Immunol (2008) 181(2):1025–33. doi: 10.4049/jimmunol.181.2.1025
55. Appleman LJ, van Puijenbroek AA, Shu KM, Nadler LM, Boussiotis VA. CD28 costimulation mediates down-regulation of p27kip1 and cell cycle progression by activation of the PI3K/PKB signaling pathway in primary human T cells. J Immunol (2002) 168(6):2729–36. doi: 10.4049/jimmunol.168.6.2729
56. Hutloff A, Dittrich AM, Beier KC, Eljaschewitsch B, Kraft R, Anagnostopoulos I, et al. ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature (1999) 397(6716):263–6. doi: 10.1038/16717
57. McAdam AJ, Chang TT, Lumelsky AE, Greenfield EA, Boussiotis VA, Duke-Cohan JS, et al. Mouse inducible costimulatory molecule (ICOS) expression is enhanced by CD28 costimulation and regulates differentiation of CD4+ T cells. J Immunol (2000) 165(9):5035–40. doi: 10.4049/jimmunol.165.9.5035
58. Wikenheiser DJ, Stumhofer JS. ICOS Co-stimulation: Friend or foe? Front Immunol (2016) 7:304. doi: 10.3389/fimmu.2016.00304
59. Li DY, Xiong XZ. ICOS(+) tregs: A functional subset of tregs in immune diseases. Front Immunol (2020) 11:2104. doi: 10.3389/fimmu.2020.02104
60. Peach RJ, Bajorath J, Brady W, Leytze G, Greene J, Naemura J, et al. Complementarity determining region 1 (CDR1)- and CDR3-analogous regions in CTLA-4 and CD28 determine the binding to B7-1. J Exp Med (1994) 180(6):2049–58. doi: 10.1084/jem.180.6.2049
61. Solinas C, Gu-Trantien C, Willard-Gallo K. The rationale behind targeting the ICOS-ICOS ligand costimulatory pathway in cancer immunotherapy. ESMO Open (2020) 5(1). doi: 10.1136/esmoopen-2019-000544
62. Yoshinaga SK, Whoriskey JS, Khare SD, Sarmiento U, Guo J, Horan T, et al. T-Cell co-stimulation through B7RP-1 and ICOS. Nature (1999) 402(6763):827–32. doi: 10.1038/45582
63. Khayyamian S, Hutloff A, Buchner K, Grafe M, Henn V, Kroczek RA, et al. ICOS-ligand, expressed on human endothelial cells, costimulates Th1 and Th2 cytokine secretion by memory CD4+ T cells. Proc Natl Acad Sci USA (2002) 99(9):6198–203. doi: 10.1073/pnas.092576699
64. Qian X, et al. The ICOS-ligand B7-H2, expressed on human type II alveolar epithelial cells, plays a role in the pulmonary host defense system. Eur J Immunol (2006) 36(4):906–18. doi: 10.1002/eji.200535253
65. Lee HJ, Kim SN, Jeon MS, Yi T, Song SU. ICOSL expression in human bone marrow-derived mesenchymal stem cells promotes induction of regulatory T cells. Sci Rep (2017) 7:44486. doi: 10.1038/srep44486
66. Swallow MM, Wallin JJ, Sha WC. B7h, a novel costimulatory homolog of B7.1 and B7.2, is induced by TNFalpha. Immunity (1999) 11(4):423–32. doi: 10.1016/s1074-7613(00)80117-x
67. Martin-Orozco N, Li Y, Wang Y, Liu S, Hwu P, Liu YJ, et al. Melanoma cells express ICOS ligand to promote the activation and expansion of T-regulatory cells. Cancer Res (2010) 70(23):9581–90. doi: 10.1158/0008-5472.CAN-10-1379
68. Odegard JM, Marks BR, DiPlacido LD, Poholek AC, Kono DH, Dong C, et al. ICOS-dependent extrafollicular helper T cells elicit IgG production via IL-21 in systemic autoimmunity. J Exp Med (2008) 205(12):2873–86. doi: 10.1084/jem.20080840
69. Arimura Y, Kato H, Dianzani U, Okamoto T, Kamekura S, Buonfiglio D, et al. A co-stimulatory molecule on activated T cells, H4/ICOS, delivers specific signals in t(h) cells and regulates their responses. Int Immunol (2002) 14(6):555–66. doi: 10.1093/intimm/dxf022
70. Jeong S, Park SH. Co-Stimulatory receptors in cancers and their implications for cancer immunotherapy. Immune Netw (2020) 20(1):e3. doi: 10.4110/in.2020.20.e3
71. Chen Q, Mo L, Cai X, Wei L, Xie Z, Li H, et al. ICOS signal facilitates Foxp3 transcription to favor suppressive function of regulatory T cells. Int J Med Sci (2018) 15(7):666–73. doi: 10.7150/ijms.23940
72. Lownik JC, Conrad DH, Martin RK. T Cell receptor signaling defines the fate and pathway of ICOS internalization. Biochem Biophys Rep (2020) 24:100803. doi: 10.1016/j.bbrep.2020.100803
73. Mittelsteadt KL, Hayes ET, Campbell DJ. ICOS signaling limits regulatory T cell accumulation and function in visceral adipose tissue. J Exp Med (2021) 218(6). doi: 10.1084/jem.20201142
74. Fos C, Salles A, Lang V, Carrette F, Audebert S, Pastor S, et al. ICOS ligation recruits the p50alpha PI3K regulatory subunit to the immunological synapse. J Immunol (2008) 181(3):1969–77. doi: 10.4049/jimmunol.181.3.1969
75. Leavenworth JW, Verbinnen B, Yin J, Huang H, Cantor H. A p85alpha-osteopontin axis couples the receptor ICOS to sustained bcl-6 expression by follicular helper and regulatory T cells. Nat Immunol (2015) 16(1):96–106. doi: 10.1038/ni.3050
76. O'Brien CA, Harris TH. ICOS-deficient and ICOS YF mutant mice fail to control toxoplasma gondii infection of the brain. PloS One (2020) 15(1):e0228251. doi: 10.1371/journal.pone.0228251
77. Wang HX, Kang X, Chu S, Li H, Li X, Yin X, et al. Dysregulated ICOS(+) proinflammatory and suppressive regulatory T cells in patients with rheumatoid arthritis. Exp Ther Med (2018) 16(4):3728–34. doi: 10.3892/etm.2018.6657
78. Guedan S, Posey AD Jr, Shaw C, Wing A, Da T, Patel PR, et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight (2018) 3(1). doi: 10.1172/jci.insight.96976
79. Kornete M, Sgouroudis E, Piccirillo CA. ICOS-dependent homeostasis and function of Foxp3+ regulatory T cells in islets of nonobese diabetic mice. J Immunol (2012) 188(3):1064–74. doi: 10.4049/jimmunol.1101303
80. Landuyt AE, Klocke BJ, Colvin TB, Schoeb TR, Maynard CL. Cutting edge: ICOS-deficient regulatory T cells display normal induction of Il10 but readily downregulate expression of Foxp3. J Immunol (2019) 202(4):1039–44. doi: 10.4049/jimmunol.1801266
81. Vocanson M, Rozieres A, Hennino A, Poyet G, Gaillard V, Renaudineau S, et al. Inducible costimulator (ICOS) is a marker for highly suppressive antigen-specific T cells sharing features of TH17/TH1 and regulatory T cells. J Allergy Clin Immunol (2010) 126(2):280–9, 289.e1-7. doi: 10.1016/j.jaci.2010.05.022
82. Nouailles G, Day TA, Kuhlmann S, Loewe D, Dorhoi A, Gamradt P, et al. Impact of inducible co-stimulatory molecule (ICOS) on T-cell responses and protection against mycobacterium tuberculosis infection. Eur J Immunol (2011) 41(4):981–91. doi: 10.1002/eji.201040608
83. Sim GC, Martin-Orozco N, Jin L, Yang Y, Wu S, Washington E, et al. IL-2 therapy promotes suppressive ICOS+ treg expansion in melanoma patients. J Clin Invest (2014) 124(1):99–110. doi: 10.1172/JCI46266
84. Tu JF, Ding YH, Ying XH, Wu FZ, Zhou XM, Zhang DK, et al. Regulatory T cells, especially ICOS(+) FOXP3(+) regulatory T cells, are increased in the hepatocellular carcinoma microenvironment and predict reduced survival. Sci Rep (2016) 6:35056. doi: 10.1038/srep35056
85. Kalble F, Wu L, Lorenz HM, Zeier M, Schaier M, Steinborn A. Impaired differentiation of highly proliferative ICOS(+)-tregs is involved in the transition from low to high disease activity in systemic lupus erythematosus (SLE) patients. Int J Mol Sci (2021) 22(17). doi: 10.3390/ijms22179501
86. Kwon BS, Weissman SM. cDNA sequences of two inducible T-cell genes. Proc Natl Acad Sci U.S.A. (1989) 86(6):1963–7. doi: 10.1073/pnas.86.6.1963
87. Pollok KE, Kim YJ, Zhou Z, Hurtado J, Kim KK, Pickard RT, et al. Inducible T cell antigen 4-1BB. analysis of expression and function. J Immunol (1993) 150(3):771–81.
88. Futagawa T, Akiba H, Kodama T, Takeda K, Hosoda Y, Yagita H, et al. Expression and function of 4-1BB and 4-1BB ligand on murine dendritic cells. Int Immunol (2002) 14(3):275–86. doi: 10.1093/intimm/14.3.275
89. Melero I, Johnston JV, Shufford WW, Mittler RS, Chen L. NK1.1 cells express 4-1BB (CDw137) costimulatory molecule and are required for tumor immunity elicited by anti-4-1BB monoclonal antibodies. Cell Immunol (1998) 190(2):167–72. doi: 10.1006/cimm.1998.1396
90. Vinay DS, Kwon BS. 4-1BB signaling beyond T cells. Cell Mol Immunol (2011) 8(4):281–4. doi: 10.1038/cmi.2010.82
91. Wong HY, Schwarz H. CD137 / CD137 ligand signalling regulates the immune balance: A potential target for novel immunotherapy of autoimmune diseases. J Autoimmun (2020) 112:102499. doi: 10.1016/j.jaut.2020.102499
92. Broll K, Richter G, Pauly S, Hofstaedter F, Schwarz H. CD137 expression in tumor vessel walls. high correlation with malignant tumors. Am J Clin Pathol (2001) 115(4):543–9. doi: 10.1309/E343-KMYX-W3Y2-10KY
93. Ho WT, Pang WL, Chong SM, Castella A, Al-Salam S, Tan TE, et al. Expression of CD137 on Hodgkin and reed-sternberg cells inhibits T-cell activation by eliminating CD137 ligand expression. Cancer Res (2013) 73(2):652–61. doi: 10.1158/0008-5472.CAN-12-3849
94. Wang Q, Zhang P, Zhang Q, Wang X, Li J, Ma C, et al. Analysis of CD137 and CD137L expression in human primary tumor tissues. Croat Med J (2008) 49(2):192–200. doi: 10.3325/cmj.2008.2.192
95. Bartkowiak T, Curran MA. 4-1BB agonists: Multi-potent potentiators of tumor immunity. Front Oncol (2015) 5:117. doi: 10.3389/fonc.2015.00117
96. Buchan SL, Dou L, Remer M, Booth SG, Dunn SN, Lai C, et al. Antibodies to costimulatory receptor 4-1BB enhance anti-tumor immunity via T regulatory cell depletion and promotion of CD8 T cell effector function. Immunity (2018) 49(5):958–970.e7. doi: 10.1016/j.immuni.2018.09.014
97. McHugh RS, Whitters MJ, Piccirillo CA, Young DA, Shevach EM, Collins M, et al. CD4(+)CD25(+) immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity (2002) 16(2):311–23. doi: 10.1016/S1074-7613(02)00280-7
98. Nowak A, Lock D, Bacher P, Hohnstein T, Vogt K, Gottfreund J, et al. CD137+CD154- expression as a regulatory T cell (Treg)-specific activation signature for identification and sorting of stable human tregs from in vitro expansion cultures. Front Immunol (2018) 9:199. doi: 10.3389/fimmu.2018.00199
99. Summers KL, Hock BD, McKenzie JL, Hart DN. Phenotypic characterization of five dendritic cell subsets in human tonsils. Am J Pathol (2001) 159(1):285–95. doi: 10.1016/S0002-9440(10)61694-X
100. Wilcox RA, Chapoval AI, Gorski KS, Otsuji M, Shin T, Flies DB, et al. Cutting edge: Expression of functional CD137 receptor by dendritic cells. J Immunol (2002) 168(9):4262–7. doi: 10.4049/jimmunol.168.9.4262
101. Choi BK, Lee DY, Lee DG, Kim YH, Kim SH, Oh HS, et al. 4-1BB signaling activates glucose and fatty acid metabolism to enhance CD8(+) T cell proliferation. Cell Mol Immunol (2017) 14(9):748–57. doi: 10.1038/cmi.2016.02
102. Barsoumian HB, Yolcu ES, Shirwan H. 4-1BB signaling in conventional T cells drives IL-2 production that overcomes CD4+CD25+FoxP3+ T regulatory cell suppression. PloS One (2016) 11(4):e0153088. doi: 10.1371/journal.pone.0153088
103. Zheng G, Wang B, Chen A. The 4-1BB costimulation augments the proliferation of CD4+CD25+ regulatory T cells. J Immunol (2004) 173(4):2428–34. doi: 10.4049/jimmunol.173.4.2428
104. Elpek KG, Yolcu ES, Franke DD, Lacelle C, Schabowsky RH, Shirwan H. Ex vivo expansion of CD4+CD25+FoxP3+ T regulatory cells based on synergy between IL-2 and 4-1BB signaling. J Immunol (2007) 179(11):7295–304. doi: 10.4049/jimmunol.179.11.7295
105. Eun SY, Lee SW, Xu Y, Croft M. 4-1BB ligand signaling to T cells limits T cell activation. J Immunol (2015) 194(1):134–41. doi: 10.4049/jimmunol.1401383
106. Foell J, Strahotin S SP, McCausland MM, Suwyn C, Haber M, et al. CD137 costimulatory T cell receptor engagement reverses acute disease in lupus-prone NZB x NZW F1 mice. J Clin Invest (2003) 111(10):1505–18. doi: 10.1172/JCI200317662
107. Luu K, Patwardhan MV, Zeng Q, Wickstrom SL, Lundqvist A, Schwarz H. Regulatory T cells inhibit T cell activity by downregulating CD137 ligand via CD137 trogocytosis. Cells (2021) 10(2). doi: 10.3390/cells10020353
108. Akhmetzyanova I, Zelinskyy G, Littwitz-Salomon E, Malyshkina A, Dietze KK, Streeck H, et al. CD137 agonist therapy can reprogram regulatory T cells into cytotoxic CD4+ T cells with antitumor activity. J Immunol (2016) 196(1):484–92. doi: 10.4049/jimmunol.1403039
109. Irie J, Wu Y, Kachapati K, Mittler RS, Ridgway WM. Modulating protective and pathogenic CD4+ subsets via CD137 in type 1 diabetes. Diabetes (2007) 56(1):186–96. doi: 10.2337/db06-0793
110. Haga T, Suzuki J, Kosuge H, Ogawa M, Saiki H, Haraguchi G, et al. Attenuation of experimental autoimmune myocarditis by blocking T cell activation through 4-1BB pathway. J Mol Cell Cardiol (2009) 46(5):719–27. doi: 10.1016/j.yjmcc.2009.02.003
111. Lee J, Lee EN, Kim EY, Park HJ, Chang CY, Jung DY, et al. Administration of agonistic anti-4-1BB monoclonal antibody leads to the amelioration of inflammatory bowel disease. Immunol Lett (2005) 101(2):210–6. doi: 10.1016/j.imlet.2005.06.001
112. Kim YH, Choi BK, Shin SM, Kim CH, Oh HS, Park SH, et al. 4-1BB triggering ameliorates experimental autoimmune encephalomyelitis by modulating the balance between Th17 and regulatory T cells. J Immunol (2011) 187(3):1120–8. doi: 10.4049/jimmunol.1002681
113. Yoo JK, Choo YK, Kwak DH, Lee JM, Lim CY, Lee JH, et al. Protective effects of agonistic anti-4-1BB antibody on the development of imiquimod-induced psoriasis-like dermatitis in mice. Immunol Lett (2016) 178:131–9. doi: 10.1016/j.imlet.2016.08.012
114. Jin J, Jung IH, Moon SH, Jeon S, Jeong SJ, Sonn SK, et al. CD137 signaling regulates acute colitis via RALDH2-expressing CD11b(-)CD103(+) DCs. Cell Rep (2020) 30(12):4124–4136.e5. doi: 10.1016/j.celrep.2020.02.103
115. Lee SW, Park Y, Eun SY, Madireddi S, Cheroutre H, Croft M. Cutting edge: 4-1BB controls regulatory activity in dendritic cells through promoting optimal expression of retinal dehydrogenase. J Immunol (2012) 189(6):2697–701. doi: 10.4049/jimmunol.1201248
116. Chester C, Sanmamed MF, Wang J, Melero I. Immunotherapy targeting 4-1BB: mechanistic rationale, clinical results, and future strategies. Blood (2018) 131(1):49–57. doi: 10.1182/blood-2017-06-741041
117. Sabbagh L, Pulle G, Liu Y, Tsitsikov EN, Watts TH. ERK-dependent bim modulation downstream of the 4-1BB-TRAF1 signaling axis is a critical mediator of CD8 T cell survival in vivo. J Immunol (2008) 180(12):8093–101. doi: 10.4049/jimmunol.180.12.8093
118. Saoulli K, Lee SY, Cannons JL, Yeh WC, Santana A, Goldstein MD, et al. CD28-independent, TRAF2-dependent costimulation of resting T cells by 4-1BB ligand. J Exp Med (1998) 187(11):1849–62. doi: 10.1084/jem.187.11.1849
119. Soderstrom LA, Tarnawski L, Olofsson PS. CD137: A checkpoint regulator involved in atherosclerosis. Atherosclerosis (2018) 272:66–72. doi: 10.1016/j.atherosclerosis.2018.03.007
120. Hong JP, Reynoso GV, Andhey PS, Swain A, Turner JS, Boon ACM, et al. An agonistic anti-CD137 antibody disrupts lymphoid follicle structure and T-Cell-Dependent antibody responses. Cell Rep Med (2020) 1(3). doi: 10.1016/j.xcrm.2020.100035
121. Cho JW, Son J, Ha SJ, Lee I. Systems biology analysis identifies TNFRSF9 as a functional marker of tumor-infiltrating regulatory T-cell enabling clinical outcome prediction in lung cancer. Comput Struct Biotechnol J (2021) 19:860–8. doi: 10.1016/j.csbj.2021.01.025
122. Freeman ZT, Nirschl TR, Hovelson DH, Johnston RJ, Engelhardt JJ, Selby MJ, et al. A conserved intratumoral regulatory T cell signature identifies 4-1BB as a pan-cancer target. J Clin Invest (2020) 130(3):1405–16. doi: 10.1172/JCI128672
123. Nocentini G, Giunchi L, Ronchetti S, Krausz LT, Bartoli A, Moraca R, et al. A new member of the tumor necrosis factor/nerve growth factor receptor family inhibits T cell receptor-induced apoptosis. Proc Natl Acad Sci U.S.A. (1997) 94(12):6216–21. doi: 10.1073/pnas.94.12.6216
124. Mahmud SA, Manlove LS, Schmitz HM, Xing Y, Wang Y, Owen DL, et al. Costimulation via the tumor-necrosis factor receptor superfamily couples TCR signal strength to the thymic differentiation of regulatory T cells. Nat Immunol (2014) 15(5):473–81. doi: 10.1038/ni.2849
125. Tian J, Zhang B, Rui K, Wang S. The role of GITR/GITRL interaction in autoimmune diseases. Front Immunol (2020) 11:588682. doi: 10.3389/fimmu.2020.588682
126. Shami A, Atzler D, Bosmans LA, Winkels H, Meiler S, Lacy M, et al. Glucocorticoid-induced tumour necrosis factor receptor family-related protein (GITR) drives atherosclerosis in mice and is associated with an unstable plaque phenotype and cerebrovascular events in humans. Eur Heart J (2020) 41(31):2938–48. doi: 10.1093/eurheartj/ehaa484
127. Tian J, Rui K, Hong Y, Wang X, Xiao F, Lin X, et al. Increased GITRL impairs the function of myeloid-derived suppressor cells and exacerbates primary sjogren syndrome. J Immunol (2019) 202(6):1693–703. doi: 10.4049/jimmunol.1801051
128. Nocentini G, Riccardi C. GITR: a modulator of immune response and inflammation. Adv Exp Med Biol (2009) 647:156–73. doi: 10.1007/978-0-387-89520-8_11
129. Shevach EM, Stephens GL. The GITR-GITRL interaction: co-stimulation or contrasuppression of regulatory activity? Nat Rev Immunol (2006) 6(8):613–8. doi: 10.1038/nri1867
130. Wang F, Chau B, West SM, Kimberlin CR, Cao F, Schwarz F, et al. Structures of mouse and human GITR-GITRL complexes reveal unique TNF superfamily interactions. Nat Commun (2021) 12(1):1378. doi: 10.1038/s41467-021-21563-z
131. Kanamaru F, Youngnak P, Hashiguchi M, Nishioka T, Takahashi T, Sakaguchi S, et al. Costimulation via glucocorticoid-induced TNF receptor in both conventional and CD25+ regulatory CD4+ T cells. J Immunol (2004) 172(12):7306–14. doi: 10.4049/jimmunol.172.12.7306
132. Ronchetti S, Nocentini G, Riccardi C, Pandolfi PP. Role of GITR in activation response of T lymphocytes. Blood (2002) 100(1):350–2. doi: 10.1182/blood-2001-12-0276
133. Durham NM, Holoweckyj N, MacGill RS, McGlinchey K, Leow CC, Robbins SH. GITR ligand fusion protein agonist enhances the tumor antigen-specific CD8 T-cell response and leads to long-lasting memory. J Immunother Cancer (2017) 5:47. doi: 10.1186/s40425-017-0247-0
134. Knee DA, Hewes B, Brogdon JL. Rationale for anti-GITR cancer immunotherapy. Eur J Cancer (2016) 67:1–10. doi: 10.1016/j.ejca.2016.06.028
135. Ronchetti S, Ricci E, Petrillo MG, Cari L, Migliorati G, Nocentini G, et al. Glucocorticoid-induced tumour necrosis factor receptor-related protein: a key marker of functional regulatory T cells. J Immunol Res (2015) 2015:171520. doi: 10.1155/2015/171520
136. Uraushihara K, Kanai T, Ko K, Totsuka T, Makita S, Iiyama R, et al. Regulation of murine inflammatory bowel disease by CD25+ and CD25- CD4+ glucocorticoid-induced TNF receptor family-related gene+ regulatory T cells. J Immunol (2003) 171(2):708–16. doi: 10.4049/jimmunol.171.2.708
137. Snell LM, McPherson AJ, Lin GH, Sakaguchi S, Pandolfi PP, Riccardi C, et al. CD8 T cell-intrinsic GITR is required for T cell clonal expansion and mouse survival following severe influenza infection. J Immunol (2010) 185(12):7223–34. doi: 10.4049/jimmunol.1001912
138. Kim IK, Kim BS, Koh CH, Seok JW, Park JS, Shin KS, et al. Glucocorticoid-induced tumor necrosis factor receptor-related protein co-stimulation facilitates tumor regression by inducing IL-9-producing helper T cells. Nat Med (2015) 21(9):1010–7. doi: 10.1038/nm.3922
139. Esparza EM, Arch RH. TRAF4 functions as an intermediate of GITR-induced NF-kappaB activation. Cell Mol Life Sci (2004) 61(24):3087–92. doi: 10.1007/s00018-004-4417-0
140. Ephrem A, Epstein AL, Stephens GL, Thornton AM, Glass D, Shevach EM. Modulation of treg cells/T effector function by GITR signaling is context-dependent. Eur J Immunol (2013) 43(9):2421–9. doi: 10.1002/eji.201343451
141. Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S. Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. Nat Immunol (2002) 3(2):135–42. doi: 10.1038/ni759
142. Liao G, Nayak S, Regueiro JR, Berger SB, Detre C, Romero X, et al. GITR engagement preferentially enhances proliferation of functionally competent CD4+CD25+FoxP3+ regulatory T cells. Int Immunol (2010) 22(4):259–70. doi: 10.1093/intimm/dxq001
143. Petrillo MG, Ronchetti S, Ricci E, Alunno A, Gerli R, Nocentini G, et al. GITR+ regulatory T cells in the treatment of autoimmune diseases. Autoimmun Rev (2015) 14(2):117–26. doi: 10.1016/j.autrev.2014.10.011
144. Tuyaerts S, Meirvenne Van S, Bonehill A, Heirman C, Corthals J, Waldmann H, et al. Expression of human GITRL on myeloid dendritic cells enhances their immunostimulatory function but does not abrogate the suppressive effect of CD4+CD25+ regulatory T cells. J Leukoc Biol (2007) 82(1):93–105. doi: 10.1189/jlb.0906568
145. Vence L, Bucktrout SL, Curbelo Fernandez I, Blando J, Smith BM, Mahne AE, et al. Characterization and comparison of GITR expression in solid tumors. Clin Cancer Res (2019) 25(21):6501–10. doi: 10.1158/1078-0432.CCR-19-0289
146. Cohen AD, Schaer DA, Liu C, Li Y, Hirschhorn-Cymmerman D, Kim SC, et al. Agonist anti-GITR monoclonal antibody induces melanoma tumor immunity in mice by altering regulatory T cell stability and intra-tumor accumulation. PloS One (2010) 5(5):e10436. doi: 10.1371/journal.pone.0010436
147. Amoozgar Z, Kloepper J, Ren J, Tay RE, Kazer SW, Kiner E, et al. Targeting treg cells with GITR activation alleviates resistance to immunotherapy in murine glioblastomas. Nat Commun (2021) 12(1):2582. doi: 10.1038/s41467-021-22885-8
148. Li Y, Yang S, Li Z, Meng H, Jin W, Yang H, et al. Soluble glucocorticoid-induced tumor necrosis factor receptor regulates Helios expression in myasthenia gravis. J Transl Med (2019) 17(1):168. doi: 10.1186/s12967-019-1916-1
149. Moreno Ayala MA, Li Z, DuPage M. Treg programming and therapeutic reprogramming in cancer. Immunology (2019) 157(3):198–209. doi: 10.1111/imm.13058
150. Kohm AP, Williams JS, Miller SD. Cutting edge: ligation of the glucocorticoid-induced TNF receptor enhances autoreactive CD4+ T cell activation and experimental autoimmune encephalomyelitis. J Immunol (2004) 172(8):4686–90. doi: 10.4049/jimmunol.172.8.4686
151. You S, Poulton L, Cobbold S, Liu CP, Rosenzweig M, Ringler D, et al. Key role of the GITR/GITRLigand pathway in the development of murine autoimmune diabetes: a potential therapeutic target. PloS One (2009) 4(11):e7848. doi: 10.1371/journal.pone.0007848
152. Cuzzocrea S, Ayroldi E, Paola Di R, Agostini M, Mazzon E, Bruscoli S, et al. Role of glucocorticoid-induced TNF receptor family gene (GITR) in collagen-induced arthritis. FASEB J (2005) 19(10):1253–65. doi: 10.1096/fj.04-3556com
153. Mallett S, Fossum S, Barclay AN. Characterization of the MRC OX40 antigen of activated CD4 positive T lymphocytes–a molecule related to nerve growth factor receptor. EMBO J (1990) 9(4):1063–8. doi: 10.1002/j.1460-2075.1990.tb08211.x
154. Paterson DJ, Jefferies WA, Green JR, Brandon MR, Corthesy P, Puklavec M, et al. Antigens of activated rat T lymphocytes including a molecule of 50,000 Mr detected only on CD4 positive T blasts. Mol Immunol (1987) 24(12):1281–90. doi: 10.1016/0161-5890(87)90122-2
155. Webb GJ, Hirschfield GM, Lane PJ. OX40, OX40L and autoimmunity: a comprehensive review. Clin Rev Allergy Immunol (2016) 50(3):312–32. doi: 10.1007/s12016-015-8498-3
156. Willoughby J, Griffiths J, Tews I, Cragg MS. OX40: Structure and function - what questions remain? Mol Immunol (2017) 83:13–22. doi: 10.1016/j.molimm.2017.01.006
157. Croft M, So T, Duan W, Soroosh P. The significance of OX40 and OX40L to T-cell biology and immune disease. Immunol Rev (2009) 229(1):173–91. doi: 10.1111/j.1600-065X.2009.00766.x
158. Redmond WL, Ruby CE, Weinberg AD. The role of OX40-mediated co-stimulation in T-cell activation and survival. Crit Rev Immunol (2009) 29(3):187–201. doi: 10.1615/CritRevImmunol.v29.i3.10
159. Song J, Salek-Ardakani S, Rogers PR, Cheng M, Parijs Van L, Croft M. The costimulation-regulated duration of PKB activation controls T cell longevity. Nat Immunol (2004) 5(2):150–8. doi: 10.1038/ni1030
160. Fu N, Xie F, Sun Z, Wang Q. The OX40/OX40L axis regulates T follicular helper cell differentiation: Implications for autoimmune diseases. Front Immunol (2021) 12:670637. doi: 10.3389/fimmu.2021.670637
161. Fu Y, Lin Q, Zhang Z, Zhang L. Therapeutic strategies for the costimulatory molecule OX40 in T-cell-mediated immunity. Acta Pharm Sin B (2020) 10(3):414–33. doi: 10.1016/j.apsb.2019.08.010
162. Ito T, Wang YH, Duramad O, Hanabuchi S, Perng OA, Gilliet M, et al. OX40 ligand shuts down IL-10-producing regulatory T cells. Proc Natl Acad Sci U.S.A. (2006) 103(35):13138–43. doi: 10.1073/pnas.0603107103
163. Vu MD, Xiao X, Gao W, Degauque N, Chen M, Kroemer A, et al. OX40 costimulation turns off Foxp3+ tregs. Blood (2007) 110(7):2501–10. doi: 10.1182/blood-2007-01-070748
164. Polesso F, Sarker M, Weinberg AD, Murray SE, Moran AE. OX40 agonist tumor immunotherapy does not impact regulatory T cell suppressive function. J Immunol (2019) 203(7):2011–9. doi: 10.4049/jimmunol.1900696
165. Piconese S, Gri G, Tripodo C, Musio S, Gorzanelli A, Frossi B, et al. Mast cells counteract regulatory T-cell suppression through interleukin-6 and OX40/OX40L axis toward Th17-cell differentiation. Blood (2009) 114(13):2639–48. doi: 10.1182/blood-2009-05-220004
166. Halim TYF, Rana BMJ, Walker JA, Kerscher B, Knolle MD, Jolin HE, et al. Tissue-restricted adaptive type 2 immunity is orchestrated by expression of the costimulatory molecule OX40L on group 2 innate lymphoid cells. Immunity (2018) 48(6):1195–1207.e6. doi: 10.1016/j.immuni.2018.05.003
167. Zhang X, Xiao X, Lan P, Li J, Dou Y, Chen W, et al. OX40 costimulation inhibits Foxp3 expression and treg induction via BATF3-dependent and independent mechanisms. Cell Rep (2018) 24(3):607–18. doi: 10.1016/j.celrep.2018.06.052
168. Linch SN, McNamara MJ, Redmond WL. OX40 agonists and combination immunotherapy: Putting the pedal to the metal. Front Oncol (2015) 5:34. doi: 10.3389/fonc.2015.00034
169. Gwyer Findlay E, Danks L, Madden J, Cavanagh MM, McNamee K, McCann F, et al. OX40L blockade is therapeutic in arthritis, despite promoting osteoclastogenesis. Proc Natl Acad Sci U.S.A. (2014) 111(6):2289–94. doi: 10.1073/pnas.1321071111
170. Griseri T, Asquith M, Thompson C, Powrie F. OX40 is required for regulatory T cell-mediated control of colitis. J Exp Med (2010) 207(4):699–709. doi: 10.1084/jem.20091618
171. Iriki H, Takahashi H, Wada N, Nomura H, Mukai M, Kamata A, et al. Peripheral tolerance by treg via constraining OX40 signal in autoreactive T cells against desmoglein 3, a target antigen in pemphigus. Proc Natl Acad Sci U.S.A. (2021) 118(49). doi: 10.1073/pnas.2026763118
172. Tripathi T, Yin W, Xue Y, Zurawski S, Fujita H, Hanabuchi S, et al. Central roles of OX40L-OX40 interaction in the induction and progression of human T cell-driven acute graft-versus-Host disease. Immunohorizons (2019) 3(3):110–20. doi: 10.4049/immunohorizons.1900001
173. Jacquemin C, Augusto JF, Scherlinger M, Gensous N, Forcade E, Douchet I, et al. OX40L/OX40 axis impairs follicular and natural treg function in human SLE. JCI Insight (2018) 3(24). doi: 10.1172/jci.insight.122167
174. Artinger K, Kirsch AH, Mooslechner AA, Cooper DJ, Aringer I, Schuller M, et al. Blockade of tumor necrosis factor superfamily members CD30 and OX40 abrogates disease activity in murine immune-mediated glomerulonephritis. Kidney Int (2021) 100(2):336–48. doi: 10.1016/j.kint.2021.02.039
175. Kumari R, Chakraborty S, Jain R, Mitra S, Mohan A, Guleria R, et al. Inhibiting OX40 restores regulatory T-cell function and suppresses inflammation in pulmonary sarcoidosis. Chest (2021) 160(3):969–82. doi: 10.1016/j.chest.2021.04.032
176. Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell (2001) 104(4):487–501. doi: 10.1016/S0092-8674(01)00237-9
177. Schonbeck U, Mach F, Libby P. CD154 (CD40 ligand). Int J Biochem Cell Biol (2000) 32(7):687–93. doi: 10.1016/S1357-2725(00)00016-9
178. Lederman S, Yellin MJ, Krichevsky A, Belko J, Lee JJ, Chess L. Identification of a novel surface protein on activated CD4+ T cells that induces contact-dependent b cell differentiation (help). J Exp Med (1992) 175(4):1091–101. doi: 10.1084/jem.175.4.1091
179. Fahnrich A, Klein S, Serge A, Nyhoegen C, Kombrink S, Moller S, et al. CD154 costimulation shifts the local T-cell receptor repertoire not only during thymic selection but also during peripheral T-dependent humoral immune responses. Front Immunol (2018) 9:1019. doi: 10.3389/fimmu.2018.01019
180. Schonbeck U, Libby P. The CD40/CD154 receptor/ligand dyad. Cell Mol Life Sci (2001) 58(1):4–43. doi: 10.1007/pl00000776
181. Berner B, Wolf G, Hummel KM, Muller GA, Reuss-Borst MA. Increased expression of CD40 ligand (CD154) on CD4+ T cells as a marker of disease activity in rheumatoid arthritis. Ann Rheum Dis (2000) 59(3):190–5. doi: 10.1136/ard.59.3.190
182. Daoussis D, Antonopoulos I, Andonopoulos AP, Liossis SN. Increased expression of CD154 (CD40L) on stimulated T-cells from patients with psoriatic arthritis. Rheumatol (Oxford) (2007) 46(2):227–31. doi: 10.1093/rheumatology/kel229
183. Guiducci C, Valzasina B, Dislich H, Colombo MP. CD40/CD40L interaction regulates CD4+CD25+ T reg homeostasis through dendritic cell-produced IL-2. Eur J Immunol (2005) 35(2):557–67. doi: 10.1002/eji.200425810
184. Dawicki W, Huang H, Ma Y, Town J, Zhang X, Rudulier CD, et al. CD40 signaling augments IL-10 expression and the tolerogenicity of IL-10-induced regulatory dendritic cells. PloS One (2021) 16(4):e0248290. doi: 10.1371/journal.pone.0248290
185. Pan PY, Ma G, Weber KJ, Ozao-Choy J, Wang G, Yin B, et al. Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Res (2010) 70(1):99–108. doi: 10.1158/0008-5472.CAN-09-1882
186. Tu W, Lau YL, Zheng J, Liu Y, Chan PL, Mao H, et al. Efficient generation of human alloantigen-specific CD4+ regulatory T cells from naive precursors by CD40-activated b cells. Blood (2008) 112(6):2554–62. doi: 10.1182/blood-2008-04-152041
187. Zheng J, Liu Y, Lau YL, Tu W. CD40-activated b cells are more potent than immature dendritic cells to induce and expand CD4(+) regulatory T cells. Cell Mol Immunol (2010) 7(1):44–50. doi: 10.1038/cmi.2009.103
188. Cuss SM, Green EA. Abrogation of CD40-CD154 signaling impedes the homeostasis of thymic resident regulatory T cells by altering the levels of IL-2, but does not affect regulatory T cell development. J Immunol (2012) 189(4):1717–25. doi: 10.4049/jimmunol.1200588
189. Litjens NH, Boer K, Betjes MG. Identification of circulating human antigen-reactive CD4+ FOXP3+ natural regulatory T cells. J Immunol (2012) 188(3):1083–90. doi: 10.4049/jimmunol.1101974
190. Taylor PA, Friedman TM, Korngold R, Noelle RJ, Blazar BR. Tolerance induction of alloreactive T cells via ex vivo blockade of the CD40:CD40L costimulatory pathway results in the generation of a potent immune regulatory cell. Blood (2002) 99(12):4601–9. doi: 10.1182/blood.V99.12.4601
191. Jenabian MA, Patel M, Kema I, Vyboh K, Kanagaratham C, Radzioch D, et al. Soluble CD40-ligand (sCD40L, sCD154) plays an immunosuppressive role via regulatory T cell expansion in HIV infection. Clin Exp Immunol (2014) 178(1):102–11. doi: 10.1111/cei.12396
192. Su X, Zhang J, Qin X. CD40 up-regulation on dendritic cells correlates with Th17/Treg imbalance in chronic periodontitis in young population. Innate Immun (2020) 26(6):482–9. doi: 10.1177/1753425920917731
193. Tan X, Zeng H, Jie Y, Zhang Y, Xu Q, Pan Z. CD154 blockade modulates the ratio of treg to Th1 cells and prolongs the survival of allogeneic corneal grafts in mice. Exp Ther Med (2014) 7(4):827–34. doi: 10.3892/etm.2014.1527
194. Ballesteros-Tato A, Leon B, Lund FE, Randall TD. CD4+ T helper cells use CD154-CD40 interactions to counteract T reg cell-mediated suppression of CD8+ T cell responses to influenza. J Exp Med (2013) 210(8):1591–601. doi: 10.1084/jem.20130097
195. Ferrer IR, Wagener ME, Song M, Kirk AD, Larsen CP, Ford ML. Antigen-specific induced Foxp3+ regulatory T cells are generated following CD40/CD154 blockade. Proc Natl Acad Sci U.S.A. (2011) 108(51):20701–6. doi: 10.1073/pnas.1105500108
196. Lee SJ, Kim HJ, Byun NR, Park CG. Donor-specific regulatory T cell-mediated immune tolerance in an intrahepatic murine allogeneic islet transplantation model with short-term anti-CD154 mAb single treatment. Cell Transplant (2020) 29. doi: 10.1177/0963689720913876
197. Fadul CE, Mao-Draayer Y, Ryan KA, Noelle RJ, Wishart HA, Channon JY, et al. Safety and immune effects of blocking CD40 ligand in multiple sclerosis. Neurol Neuroimmunol Neuroinflamm (2021) 8(6). doi: 10.1212/NXI.0000000000001096
198. Brunet JF, Denizot F, Luciani MF, Roux-Dosseto M, Suzan M, Mattei MG, et al. A new member of the immunoglobulin superfamily–CTLA-4. Nature (1987) 328(6127):267–70. doi: 10.1038/328267a0
199. Walunas TL, Bakker CY, Bluestone JA. CTLA-4 ligation blocks CD28-dependent T cell activation. J Exp Med (1996) 183(6):2541–50. doi: 10.1084/jem.183.6.2541
200. Chuang E, Fisher TS, Morgan RW, Robbins MD, Duerr JM, Heiden Vander MG, et al. The CD28 and CTLA-4 receptors associate with the serine/threonine phosphatase PP2A. Immunity (2000) 13(3):313–22. doi: 10.1016/S1074-7613(00)00031-5
201. Hoff H, Kolar P, Ambach A, Radbruch A, Brunner-Weinzierl MC. CTLA-4 (CD152) inhibits T cell function by activating the ubiquitin ligase itch. Mol Immunol (2010) 47(10):1875–81. doi: 10.1016/j.molimm.2010.03.017
202. Marengere LE, Waterhouse P, Duncan GS, Mittrucker HW, Feng GS, Mak TW. Regulation of T cell receptor signaling by tyrosine phosphatase SYP association with CTLA-4. Science (1996) 272(5265):1170–3. doi: 10.1126/science.272.5265.1170
203. Chuang E, Alegre ML, Duckett CS, Noel PJ, Heiden Vander MG, Thompson CB. Interaction of CTLA-4 with the clathrin-associated protein AP50 results in ligand-independent endocytosis that limits cell surface expression. J Immunol (1997) 159(1):144–51.
204. Brunner MC, Chambers CA, Chan FK, Hanke J, Winoto A, Allison JP. CTLA-4-Mediated inhibition of early events of T cell proliferation. J Immunol (1999) 162(10):5813–20.
205. Wu Y, Borde M, Heissmeyer V, Feuerer M, Lapan AD, Stroud JC, et al. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell (2006) 126(2):375–87. doi: 10.1016/j.cell.2006.05.042
206. Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, Sakaguchi N, et al. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med (2000) 192(2):303–10. doi: 10.1084/jem.192.2.303
207. Chambers CA, Sullivan TJ, Allison JP. Lymphoproliferation in CTLA-4-deficient mice is mediated by costimulation-dependent activation of CD4+ T cells. Immunity (1997) 7(6):885–95. doi: 10.1016/S1074-7613(00)80406-9
208. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity (1995) 3(5):541–7. doi: 10.1016/1074-7613(95)90125-6
209. Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative disorders with early lethality in mice deficient in ctla-4. Science (1995) 270(5238):985–8. doi: 10.1126/science.270.5238.985
210. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science (2008) 322(5899):271–5. doi: 10.1126/science.1160062
211. Kong KF, Fu G, Zhang Y, Yokosuka T, Casas J, Canonigo-Balancio AJ, et al. Protein kinase c-eta controls CTLA-4-mediated regulatory T cell function. Nat Immunol (2014) 15(5):465–72. doi: 10.1038/ni.2866
212. Friedline RH, Brown DS, Nguyen H, Kornfeld H, Lee J, Zhang Y, et al. CD4+ regulatory T cells require CTLA-4 for the maintenance of systemic tolerance. J Exp Med (2009) 206(2):421–34. doi: 10.1084/jem.20081811
213. Read S, Greenwald R, Izcue A, Robinson N, Mandelbrot D, Francisco L, et al. Blockade of CTLA-4 on CD4+CD25+ regulatory T cells abrogates their function in vivo. J Immunol (2006) 177(7):4376–83. doi: 10.4049/jimmunol.177.7.4376
214. Schmidt EM, Wang CJ, Ryan GA, Clough LE, Qureshi OS, Goodall M, et al. Ctla-4 controls regulatory T cell peripheral homeostasis and is required for suppression of pancreatic islet autoimmunity. J Immunol (2009) 182(1):274–82. doi: 10.4049/jimmunol.182.1.274
215. Annunziato F, Cosmi L, Liotta F, Lazzeri E, Manetti R, Vanini V, et al. Phenotype, localization, and mechanism of suppression of CD4(+)CD25(+) human thymocytes. J Exp Med (2002) 196(3):379–87. doi: 10.1084/jem.20020110
216. Manzotti CN, Tipping H, Perry LC, Mead KI, Blair PJ, Zheng Y, et al. Inhibition of human T cell proliferation by CTLA-4 utilizes CD80 and requires CD25+ regulatory T cells. Eur J Immunol (2002) 32(10):2888–96. doi: 10.1002/1521-4141(2002010)32:10<2888::AID-IMMU2888>3.0.CO;2-F
217. Jonuleit H, Schmitt E, Stassen M, Tuettenberg A, Knop J, Enk AH. Identification and functional characterization of human CD4(+)CD25(+) T cells with regulatory properties isolated from peripheral blood. J Exp Med (2001) 193(11):1285–94. doi: 10.1084/jem.193.11.1285
218. Paterson AM, Lovitch SB, Sage PT, Juneja VR, Lee Y, Trombley JD, et al. Deletion of CTLA-4 on regulatory T cells during adulthood leads to resistance to autoimmunity. J Exp Med (2015) 212(10):1603–21. doi: 10.1084/jem.20141030
219. Tai X, Laethem Van F, Pobezinsky L, Guinter T, Sharrow SO, Adams A, et al. Basis of CTLA-4 function in regulatory and conventional CD4(+) T cells. Blood (2012) 119(22):5155–63. doi: 10.1182/blood-2011-11-388918
220. Cai J, Wang D, Zhang G, Guo X. The role of PD-1/PD-L1 axis in treg development and function: Implications for cancer immunotherapy. Onco Targets Ther (2019) 12:8437–45. doi: 10.2147/OTT.S221340
221. Liang SC, Latchman YE, Buhlmann JE, Tomczak MF, Horwitz BH, Freeman GJ, et al. Regulation of PD-1, PD-L1, and PD-L2 expression during normal and autoimmune responses. Eur J Immunol (2003) 33(10):2706–16. doi: 10.1002/eji.200324228
222. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J (1992) 11(11):3887–95. doi: 10.1002/j.1460-2075.1992.tb05481.x
223. Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, Yagita H, et al. Expression of the PD-1 antigen on the surface of stimulated mouse T and b lymphocytes. Int Immunol (1996) 8(5):765–72. doi: 10.1093/intimm/8.5.765
224. Gianchecchi E, Delfino DV, Fierabracci A. Recent insights into the role of the PD-1/PD-L1 pathway in immunological tolerance and autoimmunity. Autoimmun Rev (2013) 12(11):1091–100. doi: 10.1016/j.autrev.2013.05.003
225. Qin W, Hu L, Zhang X, Jiang S, Li J, Zhang Z, et al. The diverse function of PD-1/PD-L pathway beyond cancer. Front Immunol (2019) 10:2298. doi: 10.3389/fimmu.2019.02298
226. Smith CA, Williams GT, Kingston R, Jenkinson EJ, Owen JJ. Antibodies to CD3/T-cell receptor complex induce death by apoptosis in immature T cells in thymic cultures. Nature (1989) 337(6203):181–4. doi: 10.1038/337181a0
227. Ishida Y. PD-1: Its discovery, involvement in cancer immunotherapy, and beyond. Cells (2020) 9(6). doi: 10.3390/cells9061376
228. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med (2000) 192(7):1027–34. doi: 10.1084/jem.192.7.1027
229. Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol (2001) 2(3):261–8. doi: 10.1038/85330
230. Philips EA, Garcia-Espana A, Tocheva AS, Ahearn IM, Adam KR, Pan R, et al. The structural features that distinguish PD-L2 from PD-L1 emerged in placental mammals. J Biol Chem (2020) 295(14):4372–80. doi: 10.1074/jbc.AC119.011747
231. Wang J, Okazaki IM, Yoshida T, Chikuma S, Kato Y, Nakaki F, et al. PD-1 deficiency results in the development of fatal myocarditis in MRL mice. Int Immunol (2010) 22(6):443–52. doi: 10.1093/intimm/dxq026
232. Wang J, Yoshida T, Nakaki F, Hiai H, Okazaki T, Honjo T. Establishment of NOD-Pdcd1-/- mice as an efficient animal model of type I diabetes. Proc Natl Acad Sci U.S.A. (2005) 102(33):11823–8. doi: 10.1073/pnas.0505497102
233. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity (1999) 11(2):141–51. doi: 10.1016/S1074-7613(00)80089-8
234. Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science (2001) 291(5502):319–22. doi: 10.1126/science.291.5502.319
235. Tan CL, Kuchroo JR, Sage PT, Liang D, Francisco LM, Buck J, et al. PD-1 restraint of regulatory T cell suppressive activity is critical for immune tolerance. J Exp Med (2021) 218(1). doi: 10.1084/jem.20182232
236. Lowther DE, Goods BA, Lucca LE, Lerner BA, Raddassi K, van Dijk D, et al. PD-1 marks dysfunctional regulatory T cells in malignant gliomas. JCI Insight (2016) 1(5). doi: 10.1172/jci.insight.85935
237. Kamada T, Togashi Y, Tay C, Ha D, Sasaki A, Nakamura Y, et al. PD-1(+) regulatory T cells amplified by PD-1 blockade promote hyperprogression of cancer. Proc Natl Acad Sci U.S.A. (2019) 116(20):9999–10008. doi: 10.1073/pnas.1822001116
238. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med (2009) 206(13):3015–29. doi: 10.1084/jem.20090847
239. Amarnath S, Mangus CW, Wang JC, Wei F, He A, Kapoor V, et al. The PDL1-PD1 axis converts human TH1 cells into regulatory T cells. Sci Transl Med (2011) 3(111):111ra120. doi: 10.1126/scitranslmed.3003130
240. Totsuka T, Kanai T, Makita S, Fujii R, Nemoto Y, Oshima S, et al. Regulation of murine chronic colitis by CD4+CD25- programmed death-1+ T cells. Eur J Immunol (2005) 35(6):1773–85. doi: 10.1002/eji.200425109
241. Park HJ, Park JS, Jeong YH, Son J, Ban YH, Lee BH, et al. PD-1 upregulated on regulatory T cells during chronic virus infection enhances the suppression of CD8+ T cell immune response via the interaction with PD-L1 expressed on CD8+ T cells. J Immunol (2015) 194(12):5801–11. doi: 10.4049/jimmunol.1401936
242. Punkosdy GA, Blain M, Glass DD, Lozano MM, O'Mara L, Dudley JP, et al. Regulatory T-cell expansion during chronic viral infection is dependent on endogenous retroviral superantigens. Proc Natl Acad Sci U.S.A. (2011) 108(9):3677–82. doi: 10.1073/pnas.1100213108
243. Amarnath S, Costanzo CM, Mariotti J, Ullman JL, Telford WG, Kapoor V, et al. Regulatory T cells and human myeloid dendritic cells promote tolerance via programmed death ligand-1. PloS Biol (2010) 8(2):e1000302. doi: 10.1371/journal.pbio.1000302
244. Gotot J, Gottschalk C, Leopold S, Knolle PA, Yagita H, Kurts C, et al. Regulatory T cells use programmed death 1 ligands to directly suppress autoreactive b cells in vivo. Proc Natl Acad Sci U.S.A. (2012) 109(26):10468–73. doi: 10.1073/pnas.1201131109
245. Wong M, La Cava A, Hahn BH. Blockade of programmed death-1 in young (New Zealand black x new Zealand White)F1 mice promotes the suppressive capacity of CD4+ regulatory T cells protecting from lupus-like disease. J Immunol (2013) 190(11):5402–10. doi: 10.4049/jimmunol.1202382
246. Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol (2009) 10(1):48–57. doi: 10.1038/ni.1674
247. Ercolano G, Wyss T, Salome B, Romero P, Trabanelli S, Jandus C. Distinct and shared gene expression for human innate versus adaptive helper lymphoid cells. J Leukoc Biol (2020) 108(2):723–37. doi: 10.1002/JLB.5MA0120-209R
248. Hasan MM, Nair SS, O'Leary JG, Thompson-Snipes L, Nyarige V, Wang J, et al. Implication of TIGIT(+) human memory b cells in immune regulation. Nat Commun (2021) 12(1):1534. doi: 10.1038/s41467-021-21413-y
249. Johnston RJ, Comps-Agrar L, Hackney J, Yu X, Huseni M, Yang Y, et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell (2014) 26(6):923–37. doi: 10.1016/j.ccell.2014.10.018
250. Lee MY, Upadhyay AA, Walum H, Chan CN, Dawoud RA, Grech C, et al. Tissue-specific transcriptional profiling of plasmacytoid dendritic cells reveals a hyperactivated state in chronic SIV infection. PloS Pathog (2021) 17(6):e1009674. doi: 10.1371/journal.ppat.1009674
251. Meng F, Li L, Lu F, Yue J, Liu Z, Zhang W, et al. Overexpression of TIGIT in NK and T cells contributes to tumor immune escape in myelodysplastic syndromes. Front Oncol (2020) 10:1595. doi: 10.3389/fonc.2020.01595
252. Wang FF, Wang Y, Wang L, Wang TS, Bai YP. TIGIT expression levels on CD4+ T cells are correlated with disease severity in patients with psoriasis. Clin Exp Dermatol (2018) 43(6):675–82. doi: 10.1111/ced.13414
253. Boles KS, Vermi W, Facchetti F, Fuchs A, Wilson TJ, Diacovo TG, et al. A novel molecular interaction for the adhesion of follicular CD4 T cells to follicular DC. Eur J Immunol (2009) 39(3):695–703. doi: 10.1002/eji.200839116
254. Fuhrman CA, Yeh WI, Seay HR, Lakshmi Saikumar P, Chopra G, Zhang L, et al. Divergent phenotypes of human regulatory T cells expressing the receptors TIGIT and CD226. J Immunol (2015) 195(1):145–55. doi: 10.4049/jimmunol.1402381
255. Levin SD, Taft DW, Brandt CS, Bucher C, Howard ED, Chadwick EM, et al. Vstm3 is a member of the CD28 family and an important modulator of T-cell function. Eur J Immunol (2011) 41(4):902–15. doi: 10.1002/eji.201041136
256. Stanietsky N, Simic H, Arapovic J, Toporik A, Levy O, Novik A, et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc Natl Acad Sci U.S.A. (2009) 106(42):17858–63. doi: 10.1073/pnas.0903474106
257. Chauvin JM, Zarour HM. TIGIT in cancer immunotherapy. J Immunother Cancer (2020) 8(2). doi: 10.1136/jitc-2020-000957
258. Yeo J, Ko M, Lee DH, Park Y, Jin HS. TIGIT/CD226 axis regulates anti-tumor immunity. Pharm (Basel) (2021) 14(3). doi: 10.3390/ph14030200
259. Yasutomi M, Christiaansen AF, Imai N, Martin-Orozco N, Forst CV, Chen G, et al. CD226 and TIGIT cooperate in the differentiation and maturation of human tfh cells. Front Immunol (2022) 13:840457. doi: 10.3389/fimmu.2022.840457
260. Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C, et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity (2014) 40(4):569–81. doi: 10.1016/j.immuni.2014.02.012
261. Bin Dhuban K, d'Hennezel E, Nashi E, Bar-Or A, Rieder S, Shevach EM, et al. Coexpression of TIGIT and FCRL3 identifies helios+ human memory regulatory T cells. J Immunol (2015) 194(8):3687–96. doi: 10.4049/jimmunol.1401803
262. Lucca LE, Axisa PP, Singer ER, Nolan NM, Dominguez-Villar M, Hafler DA. TIGIT signaling restores suppressor function of Th1 tregs. JCI Insight (2019) 4(3). doi: 10.1172/jci.insight.124427
263. Kurtulus S, Sakuishi K, Ngiow SF, Joller N, Tan DJ, Teng MW, et al. TIGIT predominantly regulates the immune response via regulatory T cells. J Clin Invest (2015) 125(11):4053–62. doi: 10.1172/JCI81187
264. Sato K, Yamashita-Kanemaru Y, Abe F, Murata R, Nakamura-Shinya Y, Kanemaru K, et al. DNAM-1 regulates Foxp3 expression in regulatory T cells by interfering with TIGIT under inflammatory conditions. Proc Natl Acad Sci U.S.A. (2021) 118(21). doi: 10.1073/pnas.2021309118
265. Mastrangeli R, Micangeli E, Donini S. Cloning of murine LAG-3 by magnetic bead bound homologous probes and PCR (gene-capture PCR). Anal Biochem (1996) 241(1):93–102. doi: 10.1006/abio.1996.0382
266. Huard B, Gaulard P, Faure F, Hercend T, Triebel F. Cellular expression and tissue distribution of the human LAG-3-encoded protein, an MHC class II ligand. Immunogenetics (1994) 39(3):213–7. doi: 10.1007/BF00241263
267. Baixeras E, Huard B, Miossec C, Jitsukawa S, Martin M, Hercend T, et al. Characterization of the lymphocyte activation gene 3-encoded protein. a new ligand for human leukocyte antigen class II antigens. J Exp Med (1992) 176(2):327–37. doi: 10.1084/jem.176.2.327
268. Wang J, Sanmamed MF, Datar I, Su TT, Ji L, Sun J, et al. Fibrinogen-like protein 1 is a major immune inhibitory ligand of LAG-3. Cell (2019) 176(1-2):334–347.e12. doi: 10.1016/j.cell.2018.11.010
269. Mao X, Ou MT, Karuppagounder SS, Kam TI, Yin X, Xiong Y, et al. Pathological alpha-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science (2016) 353(6307). doi: 10.1126/science.aah3374
270. Kouo T, Huang L, Pucsek AB, Cao M, Solt S, Armstrong T, et al. Galectin-3 shapes antitumor immune responses by suppressing CD8+ T cells via LAG-3 and inhibiting expansion of plasmacytoid dendritic cells. Cancer Immunol Res (2015) 3(4):412–23. doi: 10.1158/2326-6066.CIR-14-0150
271. Xu F, Liu J, Liu D, Liu B, Wang M, Hu Z, et al. LSECtin expressed on melanoma cells promotes tumor progression by inhibiting antitumor T-cell responses. Cancer Res (2014) 74(13):3418–28. doi: 10.1158/0008-5472.CAN-13-2690
272. Huang CT, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, et al. Role of LAG-3 in regulatory T cells. Immunity (2004) 21(4):503–13. doi: 10.1016/j.immuni.2004.08.010
273. He W, Hu Y, Chen D, Li Y, Ye D, Zhao Q, et al. Hepatocellular carcinoma-infiltrating gammadelta T cells are functionally defected and allogenic Vdelta2(+) gammadelta T cell can be a promising complement. Clin Transl Med (2022) 12(4):e800. doi: 10.1002/ctm2.800
274. Schubert K, Karkossa I, Schor J, Engelmann B, Steinheuer LM, Bruns T, et al. A multi-omics analysis of mucosal-Associated-Invariant T cells reveals key drivers of distinct modes of activation. Front Immunol (2021) 12:616967. doi: 10.3389/fimmu.2021.616967
275. Juno JA, Stalker AT, Waruk JL, Oyugi J, Kimani M, Plummer FA, et al. Elevated expression of LAG-3, but not PD-1, is associated with impaired iNKT cytokine production during chronic HIV-1 infection and treatment. Retrovirology (2015) 12:17. doi: 10.1186/s12977-015-0142-z
276. Keane C, Law SC, Gould C, Birch S, Sabdia MB, Long Merida L, et al. LAG3: a novel immune checkpoint expressed by multiple lymphocyte subsets in diffuse large b-cell lymphoma. Blood Adv (2020) 4(7):1367–77. doi: 10.1182/bloodadvances.2019001390
277. Kisielow M, Kisielow J, Capoferri-Sollami G, Karjalainen K. Expression of lymphocyte activation gene 3 (LAG-3) on b cells is induced by T cells. Eur J Immunol (2005) 35(7):2081–8. doi: 10.1002/eji.200526090
278. Camisaschi C, De Filippo A, Beretta V, Vergani B, Villa A, Vergani E, et al. Alternative activation of human plasmacytoid DCs in vitro and in melanoma lesions: involvement of LAG-3. J Invest Dermatol (2014) 134(7):1893–902. doi: 10.1038/jid.2014.29
279. Liu Y, Sorce S, Nuvolone M, Domange J, Aguzzi A. Lymphocyte activation gene 3 (Lag3) expression is increased in prion infections but does not modify disease progression. Sci Rep (2018) 8(1):14600. doi: 10.1038/s41598-018-32712-8
280. Liang B, Workman C, Lee J, Chew C, Dale BM, Colonna L, et al. Regulatory T cells inhibit dendritic cells by lymphocyte activation gene-3 engagement of MHC class II. J Immunol (2008) 180(9):5916–26. doi: 10.4049/jimmunol.180.9.5916
281. Gagliani N, Magnani CF, Huber S, Gianolini ME, Pala M, Licona-Limon P, et al. Coexpression of CD49b and LAG-3 identifies human and mouse T regulatory type 1 cells. Nat Med (2013) 19(6):739–46. doi: 10.1038/nm.3179
282. Huang W, Solouki S, Carter C, Zheng SG, August A. Beyond type 1 regulatory T cells: Co-expression of LAG3 and CD49b in IL-10-Producing T cell lineages. Front Immunol (2018) 9:2625. doi: 10.3389/fimmu.2018.02625
283. Okamura T, Fujio K, Shibuya M, Sumitomo S, Shoda H, Sakaguchi S, et al. CD4+CD25-LAG3+ regulatory T cells controlled by the transcription factor egr-2. Proc Natl Acad Sci U.S.A. (2009) 106(33):13974–9. doi: 10.1073/pnas.0906872106
284. Jofra T, Di Fonte R, Galvani G, Kuka M, Iannacone M, Battaglia M, et al. Tr1 cell immunotherapy promotes transplant tolerance via de novo Tr1 cell induction in mice and is safe and effective during acute viral infection. Eur J Immunol (2018) 48(8):1389–99. doi: 10.1002/eji.201747316
285. Jha V, Workman CJ, McGaha TL, Li L, Vas J, Vignali DA, et al. Lymphocyte activation gene-3 (LAG-3) negatively regulates environmentally-induced autoimmunity. PloS One (2014) 9(8):e104484. doi: 10.1371/journal.pone.0104484
286. Zhang Q, Chikina M, Szymczak-Workman AL, Horne W, Kolls JK, Vignali KM, et al. LAG3 limits regulatory T cell proliferation and function in autoimmune diabetes. Sci Immunol (2017) 2(9). doi: 10.1126/sciimmunol.aah4569
287. Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature (2002) 415(6871):536–41. doi: 10.1038/415536a
288. Wolf Y, Anderson AC, Kuchroo VK. TIM3 comes of age as an inhibitory receptor. Nat Rev Immunol (2020) 20(3):173–85. doi: 10.1038/s41577-019-0224-6
289. Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol (2005) 6(12):1245–52. doi: 10.1038/ni1271
290. Kobayashi N, Karisola P, Pena-Cruz V, Dorfman DM, Jinushi M, Umetsu SE, et al. TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity (2007) 27(6):927–40. doi: 10.1016/j.immuni.2007.11.011
291. Huang YH, Zhu C, Kondo Y, Anderson AC, Gandhi A, Russell A, et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature (2015) 517(7534):386–90. doi: 10.1038/nature13848
292. Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol (2012) 13(9):832–42. doi: 10.1038/ni.2376
293. Anderson AC, Anderson DE, Bregoli L, Hastings WD, Kassam N, Lei C, et al. Promotion of tissue inflammation by the immune receptor Tim-3 expressed on innate immune cells. Science (2007) 318(5853):1141–3. doi: 10.1126/science.1148536
294. Ndhlovu LC, Lopez-Verges S, Barbour JD, Jones RB, Jha AR, Long BR, et al. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood (2012) 119(16):3734–43. doi: 10.1182/blood-2011-11-392951
295. Phong BL, Avery L, Sumpter TL, Gorman JV, Watkins SC, Colgan JD, et al. Tim-3 enhances FcepsilonRI-proximal signaling to modulate mast cell activation. J Exp Med (2015) 212(13):2289–304. doi: 10.1084/jem.20150388
296. Gao X, Zhu Y, Li G, Huang H, Zhang G, Wang F, et al. TIM-3 expression characterizes regulatory T cells in tumor tissues and is associated with lung cancer progression. PloS One (2012) 7(2):e30676. doi: 10.1371/journal.pone.0030676
297. Gupta S, Thornley TB, Gao W, Larocca R, Turka LA, Kuchroo VK, et al. Allograft rejection is restrained by short-lived TIM-3+PD-1+Foxp3+ tregs. J Clin Invest (2012) 122(7):2395–404. doi: 10.1172/JCI45138
298. Sakuishi K, Ngiow SF, Sullivan JM, Teng MW, Kuchroo VK, Smyth MJ, et al. TIM3(+)FOXP3(+) regulatory T cells are tissue-specific promoters of T-cell dysfunction in cancer. Oncoimmunology (2013) 2(4):e23849. doi: 10.4161/onci.23849
299. Yan J, Zhang Y, Zhang JP, Liang J, Li L, Zheng L. Tim-3 expression defines regulatory T cells in human tumors. PloS One (2013) 8(3):e58006. doi: 10.1371/journal.pone.0058006
300. Arm JP, Nwankwo C, Austen KF. Molecular identification of a novel family of human ig superfamily members that possess immunoreceptor tyrosine-based inhibition motifs and homology to the mouse gp49B1 inhibitory receptor. J Immunol (1997) 159(5):2342–9.
301. Xu Z, Chang CC, Li M, Zhang QY, Vasilescu EM, D'Agati V, et al. ILT3.Fc-CD166 interaction induces inactivation of p70 S6 kinase and inhibits tumor cell growth. J Immunol (2018) 200(3):1207–19. doi: 10.4049/jimmunol.1700553
302. Deng M, Gui X, Kim J, Xie L, Chen W, Li Z, et al. LILRB4 signalling in leukaemia cells mediates T cell suppression and tumour infiltration. Nature (2018) 562(7728):605–9. doi: 10.1038/s41586-018-0615-z
303. Verschueren E, Husain B, Yuen K, Sun Y, Paduchuri S, Senbabaoglu Y, et al. The immunoglobulin superfamily receptome defines cancer-relevant networks associated with clinical outcome. Cell (2020) 182(2):329–344.e19. doi: 10.1016/j.cell.2020.06.007
304. Liu J, Wu Q, Shi J, Guo W, Jiang X, Zhou B, et al. LILRB4, from the immune system to the disease target. Am J Transl Res (2020) 12(7):3149–66.
305. Chang CC, Ciubotariu R, Manavalan JS, Yuan J, Colovai AI, Piazza F, et al. Tolerization of dendritic cells by T(S) cells: the crucial role of inhibitory receptors ILT3 and ILT4. Nat Immunol (2002) 3(3):237–43. doi: 10.1038/ni760
306. Suciu-Foca N, Cortesini R. Central role of ILT3 in the T suppressor cell cascade. Cell Immunol (2007) 248(1):59–67. doi: 10.1016/j.cellimm.2007.01.013
307. Manavalan JS, Rossi PC, Vlad G, Piazza F, Yarilina A, Cortesini R, et al. High expression of ILT3 and ILT4 is a general feature of tolerogenic dendritic cells. Transpl Immunol (2003) 11(3-4):245–58. doi: 10.1016/S0966-3274(03)00058-3
308. Ulges A, Klein M, Reuter S, Gerlitzki B, Hoffmann M, Grebe N, et al. Protein kinase CK2 enables regulatory T cells to suppress excessive TH2 responses in vivo. Nat Immunol (2015) 16(3):267–75. doi: 10.1038/ni.3083
309. Sharma N, Atolagbe OT, Ge Z, Allison JP. LILRB4 suppresses immunity in solid tumors and is a potential target for immunotherapy. J Exp Med (2021) 218(7). doi: 10.1084/jem.20201811
310. Paluch C, Santos AM, Anzilotti C, Cornall RJ, Davis SJ. Immune checkpoints as therapeutic targets in autoimmunity. Front Immunol (2018) 9:2306. doi: 10.3389/fimmu.2018.02306
311. Baglini E, Salerno S, Barresi E, Marzo T, Settimo FD, Taliani S. Cancer immunotherapy: An overview on small molecules as inhibitors of the immune checkpoint PD-1/PD-L1 (2015-2021). Mini Rev Med Chem (2022). doi: 10.2174/1389557522666220217110925
312. Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med (2006) 355(10):1018–28. doi: 10.1056/NEJMoa063842
313. Tyrsin D, Chuvpilo S, Matskevich A, Nemenov D, Romer PS, Tabares P, et al. From TGN1412 to TAB08: the return of CD28 superagonist therapy to clinical development for the treatment of rheumatoid arthritis. Clin Exp Rheumatol (2016) 34(4 Suppl 98):45–8.
314. Poirier N, Azimzadeh AM, Zhang T, Dilek N, Mary C, Nguyen B, et al. Inducing CTLA-4-dependent immune regulation by selective CD28 blockade promotes regulatory T cells in organ transplantation. Sci Transl Med (2010) 2(17):17ra10. doi: 10.1126/scitranslmed.3000116
315. Yang J, Lickliter JD, Hillson JL, Means GD, Sanderson RJ, Carley K, et al. First-in-human study of the safety, tolerability, pharmacokinetics, and pharmacodynamics of ALPN-101, a dual CD28/ICOS antagonist, in healthy adult subjects. Clin Transl Sci (2021) 14(4):1314–26. doi: 10.1111/cts.12983
316. Blair HA, Deeks ED. Abatacept: A review in rheumatoid arthritis. Drugs (2017) 77(11):1221–33. doi: 10.1007/s40265-017-0775-4
317. Hosseini A, Gharibi T, Marofi F, Babaloo Z, Baradaran B. CTLA-4: From mechanism to autoimmune therapy. Int Immunopharmacol (2020) 80:106221. doi: 10.1016/j.intimp.2020.106221
318. Alvarez-Quiroga C, Abud-Mendoza C, Doniz-Padilla L, Juarez-Reyes A, Monsivais-Urenda A, Baranda L, et al. CTLA-4-Ig therapy diminishes the frequency but enhances the function of treg cells in patients with rheumatoid arthritis. J Clin Immunol (2011) 31(4):588–95. doi: 10.1007/s10875-011-9527-5
319. Maggi L, Cimaz R, Capone M, Santarlasci V, Rossi MC, Mazzoni A, et al. Immunosuppressive activity of abatacept on circulating T helper lymphocytes from juvenile idiopathic arthritis patients. Int Arch Allergy Immunol (2016) 171(1):45–53. doi: 10.1159/000450948
320. Bonelli M, Goschl L, Bluml S, Karonitsch T, Hirahara K, Ferner E, et al. Abatacept (CTLA-4Ig) treatment reduces T cell apoptosis and regulatory T cell suppression in patients with rheumatoid arthritis. Rheumatol (Oxford) (2016) 55(4):710–20. doi: 10.1093/rheumatology/kev403
321. Picchianti Diamanti A, Rosado MM, Scarsella M, Germano V, Giorda E, Cascioli S, et al. Abatacept (cytotoxic T lymphocyte antigen 4-immunoglobulin) improves b cell function and regulatory T cell inhibitory capacity in rheumatoid arthritis patients non-responding to anti-tumour necrosis factor-alpha agents. Clin Exp Immunol (2014) 177(3):630–40. doi: 10.1111/cei.12367
322. Szentpetery A, Heffernan E, Gogarty M, Mellerick L, McCormack J, Haroon M, et al. Abatacept reduces synovial regulatory T-cell expression in patients with psoriatic arthritis. Arthritis Res Ther (2017) 19(1):158. doi: 10.1186/s13075-017-1364-3
323. El-Charabaty E, Geara AS, Ting C, El-Sayegh S, Azzi J. Belatacept: a new era of immunosuppression? Expert Rev Clin Immunol (2012) 8(6):527–36. doi: 10.1586/eci.12.42
324. Vincenti F, Dritselis A, Kirkpatrick P. Belatacept. Nat Rev Drug Discovery (2011) 10(9):655–6. doi: 10.1038/nrd3536
325. Grimbert P, Audard V, Diet C, Matignon M, Plonquet A, Mansour H, et al. T-Cell phenotype in protocol renal biopsy from transplant recipients treated with belatacept-mediated co-stimulatory blockade. Nephrol Dial Transplant (2011) 26(3):1087–93. doi: 10.1093/ndt/gfq453
326. Korman AJ, Garrett-Thomson SC, Lonberg N. The foundations of immune checkpoint blockade and the ipilimumab approval decennial. Nat Rev Drug Discovery (2022) 21(7):509–28. doi: 10.1038/s41573-021-00345-8
327. Sharma A, Subudhi SK, Blando J, Scutti J, Vence L, Wargo J, et al. Anti-CTLA-4 immunotherapy does not deplete FOXP3(+) regulatory T cells (Tregs) in human cancers. Clin Cancer Res (2019) 25(4):1233–8. doi: 10.1158/1078-0432.CCR-18-0762
328. Martens A, Wistuba-Hamprecht K, Foppen Geukes M, Yuan J, Postow MA, Wong P, et al. Baseline peripheral blood biomarkers associated with clinical outcome of advanced melanoma patients treated with ipilimumab. Clin Cancer Res (2016) 22(12):2908–18. doi: 10.1158/1078-0432.CCR-15-2412
329. Zhou J, Bashey A, Zhong R, Corringham S, Messer K, Pu M, et al. CTLA-4 blockade following relapse of malignancy after allogeneic stem cell transplantation is associated with T cell activation but not with increased levels of T regulatory cells. Biol Blood Marrow Transplant (2011) 17(5):682–92. doi: 10.1016/j.bbmt.2010.08.005
330. Kavanagh B, O'Brien S, Lee D, Hou Y, Weinberg V, Rini B, et al. CTLA4 blockade expands FoxP3+ regulatory and activated effector CD4+ T cells in a dose-dependent fashion. Blood (2008) 112(4):1175–83. doi: 10.1182/blood-2007-11-125435
331. Arce Vargas F, Furness AJS, Litchfield K, Joshi K, Rosenthal R, Ghorani E, et al. Fc effector function contributes to the activity of human anti-CTLA-4 antibodies. Cancer Cell (2018) 33(4):649–663.e4. doi: 10.1016/j.ccell.2018.02.010
333. Comin-Anduix B, Sazegar H, Chodon T, Matsunaga D, Jalil J, von Euw E, et al. Modulation of cell signaling networks after CTLA4 blockade in patients with metastatic melanoma. PloS One (2010) 5(9):e12711. doi: 10.1371/journal.pone.0012711
334. Jiang DM, Fyles A, Nguyen LT, Neel BG, Sacher A, Rottapel R, et al. Phase I study of local radiation and tremelimumab in patients with inoperable locally recurrent or metastatic breast cancer. Oncotarget (2019) 10(31):2947–58. doi: 10.18632/oncotarget.26893
335. Khan S, Burt DJ, Ralph C, Thistlethwaite FC, Hawkins RE, Elkord E. Tremelimumab (anti-CTLA4) mediates immune responses mainly by direct activation of T effector cells rather than by affecting T regulatory cells. Clin Immunol (2011) 138(1):85–96. doi: 10.1016/j.clim.2010.09.011
336. Xiong Y, Neskey DM, Horton JD, Paulos CM, Knochelmann HM, Armeson KE, et al. Immunological effects of nivolumab immunotherapy in patients with oral cavity squamous cell carcinoma. BMC Cancer (2020) 20(1):229. doi: 10.1186/s12885-020-06726-3
337. Nieto JC, Roldan E, Jimenez I, Fox L, Carabia J, Orti G, et al. Posttransplant cyclophosphamide after allogeneic hematopoietic cell transplantation mitigates the immune activation induced by previous nivolumab therapy. Leukemia (2020) 34(12):3420–5. doi: 10.1038/s41375-020-0851-8
338. Ikegawa S, Meguri Y, Mizuhara K, Fukumi T, Kobayashi H, Sumii Y, et al. Pretransplant nivolumab further enhanced treg expansion after posttransplant cyclophosphamide; another aspect for immune tolerance by PTCy after nivolumab. Leukemia (2021) 35(3):929–31. doi: 10.1038/s41375-021-01167-8
339. Sasidharan Nair V, Toor SM, Taouk G, Pfister G, Ouararhni K, Alajez NM, et al. Pembrolizumab interferes with the differentiation of human FOXP3(+)-induced T regulatory cells, but not with FOXP3 stability, through activation of mTOR. J Immunol (2020) 204(1):199–211. doi: 10.4049/jimmunol.1900575
340. Sullivan BA, Tsuji W, Kivitz A, Peng J, Arnold GE, Boedigheimer MJ, et al. Inducible T-cell co-stimulator ligand (ICOSL) blockade leads to selective inhibition of anti-KLH IgG responses in subjects with systemic lupus erythematosus. Lupus Sci Med (2016) 3(1):e000146. doi: 10.1136/lupus-2016-000146
341. Curti BD, Kovacsovics-Bankowski M, Morris N, Walker E, Chisholm L, Floyd K, et al. OX40 is a potent immune-stimulating target in late-stage cancer patients. Cancer Res (2013) 73(24):7189–98. doi: 10.1158/0008-5472.CAN-12-4174
Keywords: Treg - regulatory T cell, immune checkpoint, costimulatory pathway, coinhibition modulation, cancer, autoimmunity
Citation: Abdeladhim M, Karnell JL and Rieder SA (2022) In or out of control: Modulating regulatory T cell homeostasis and function with immune checkpoint pathways. Front. Immunol. 13:1033705. doi: 10.3389/fimmu.2022.1033705
Received: 31 August 2022; Accepted: 16 November 2022;
Published: 15 December 2022.
Edited by:
Ethan Menahem Shevach, National Institutes of Health (NIH), United StatesReviewed by:
Lesley Ann Smyth, University of East London, United KingdomCopyright © 2022 Abdeladhim, Karnell and Rieder. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sadiye Amcaoglu Rieder, c3JpZWRlckBob3Jpem9udGhlcmFwZXV0aWNzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.