 Aswath P. Chandrasekar
Aswath P. Chandrasekar Andrew D. Badley
Andrew D. Badley
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 20 October 2022
Sec. Viral Immunology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1033609
While modern HIV therapy can effectively suppress viral replication, the persistence of the latent reservoir posits the greatest hurdle to complete cure. The “shock and kill” strategy is under investigation for HIV therapy, aiming to reactivate latent HIV, and subsequently eliminate it through anti-retroviral therapy and host immune function. However, thus far, studies have yielded suboptimal results, stemming from a combination of ineffective latency reversal and poor immune clearance. Concomitantly, studies have now revealed the importance of the BCL-2 anti-apoptotic protein as a critical mediator of infected cell survival, reservoir maintenance and immune evasion in HIV. Furthermore, BCL-2 inhibitors are now recognized for their anti-HIV effects in pre-clinical studies. This minireview aims to examine the intersection of BCL-2 inhibition and current shock and kill efforts, hoping to inform future studies which may ultimately yield a cure for HIV.
Modern combination anti-retroviral therapy (cART) is highly effective at suppressing HIV viremia to levels below the limit of detection. However, infected individuals continue to harbor integrated, transcriptionally silent proviral forms in a pool of infected cells known as the latent reservoir (1). Studies have now highlighted the dynamic nature of the reservoir, which can propagate even under the coverage of cART through low level transcription and clonal expansion (2–4). Strategies to target the latent reservoir aim to achieve either complete elimination of infected cells (a sterilizing cure), or permanently prevent HIV transcription in these cells (a functional cure) (5).
The “Shock and Kill” HIV cure strategy centers around the former principle, aiming to reactivate latent HIV, and subsequently eliminate it through anti-retroviral therapy and host immune function (6). However, even in the setting of efficient viral reactivation, numerous barriers have been identified which prevent host immunity from maximally clearing HIV infected cells in-vivo including poor immune recognition, immune exhaustion, the effects of immune-modulatory cytokines, and upregulation of prosurvival proteins (7–12).
Through a combination of mechanisms, these changes allow for either infected cell survival, or compromised immune effector function, or both. One such factor that has been recognized to facilitate HIV persistence by both priming infected cells for survival, and simultaneously antagonizing the host immune response, is the BCL-2 pro-survival protein (13). This minireview aims to examine the implications of this protein to HIV pathogenesis and to therapeutic targeting for HIV “shock and kill” efforts.
BCL-2 and its homologs are intracellular regulators of multiple cellular processes, the most crucial of which is cell survival and apoptosis. Prototypical members of the BCL-2 family are characterized by the presence of four conserved homology domains: BH1, BH2, BH3 and BH4 (14). At homeostasis, cell apoptosis is controlled by the balance between BCL-2 (and other anti-apoptotic homologs) and the pro-apoptotic homologs, with a subset of proteins containing only the BH3 domain, serving as modulators of this balance. The proposed mechanisms through which these interactions occur have been reviewed extensively elsewhere (15). The BCL-2 protein prevents cell death through the stabilization of the mitochondrial membrane via the sequestration of pro-apoptotic proteins which, canonically, occupy a hydrophobic groove in the BH3 domain of anti-apoptotic BCL-2 family members. The HIV life cycle is dependent on host cell apoptosis to facilitate the release of viral progeny, and various HIV proteins have been shown to modulate the levels of BCL-2 family members to either promote or prevent the death of infected cells. [Reviewed in detail in (13)]
Apoptosis is crucial to the HIV life cycle, and one of the mechanisms through which HIV achieves infected cell apoptosis is through the Casp8p41 pathway. Casp8p41 is a 41 kilodalton, BH3-like protein that is generated due to the cleavage of host procaspase8 by HIV protease (16). Through the direct potentiation of the pro-apoptotic BCL-2 family members BAK and BAX and subsequent mitochondrial membrane depolarization, Casp8p41 results in host cell apoptosis (17, 18). Clinically, it was observed that intracellular Casp8p41 levels in HIV infected individuals correlated inversely with CD4 T-cell counts (19), and that patients on suppressive ART who demonstrated continuous production of Casp8p41 exhibited an increased risk of CD4 losses (20).
Of particular importance was the recognition that latently infected cells produced Casp8p41 upon reactivation, which was observed to be bound by host BCL-2 and degraded by the proteasome, antagonizing its pro-apoptotic phenotype, thereby facilitating HIV survival (21, 22). The ability of BCL-2 to abrogate Casp8p41 induced apoptosis led to the hypothesis that BCL-2 contributed to the establishment of the latent reservoir.
Ex vivo studies have now verified that the cell subsets that are classically described to harbor the latent HIV reservoir, namely central memory T-cells, have been observed to express higher levels of BCL-2 (21), and that BCL-2high cells have been seen to occur more frequently in HIV infected individuals compared to uninfected controls (9). Studies of simian immuno deficiency (SIV) infection in ART suppressed macaques revealed that CTLA+ PD1- Memory CD4 cells that harbored significant quantities of replication competent provirus demonstrated significantly higher BCL-2 expression compared to other subsets (23). Most importantly, it has now been recognized that the inducible HIV reservoir is preferentially enriched in cells with higher expression of BCL-2 in-vivo (11).
HIV latency has been recapitulated through lentiviral transfection of primary CD4 cells with a BCL-2 construct, in an in-vitro model which allowed for sustained HIV replication, and recapitulated changes seen during the natural course of HIV infection, further illustrating the critical role of BCL-2 in HIV latency (24–26).
Overall, the critical role of BCL-2 for HIV persistence and latency is supported by a growing body of evidence (Figure 1).
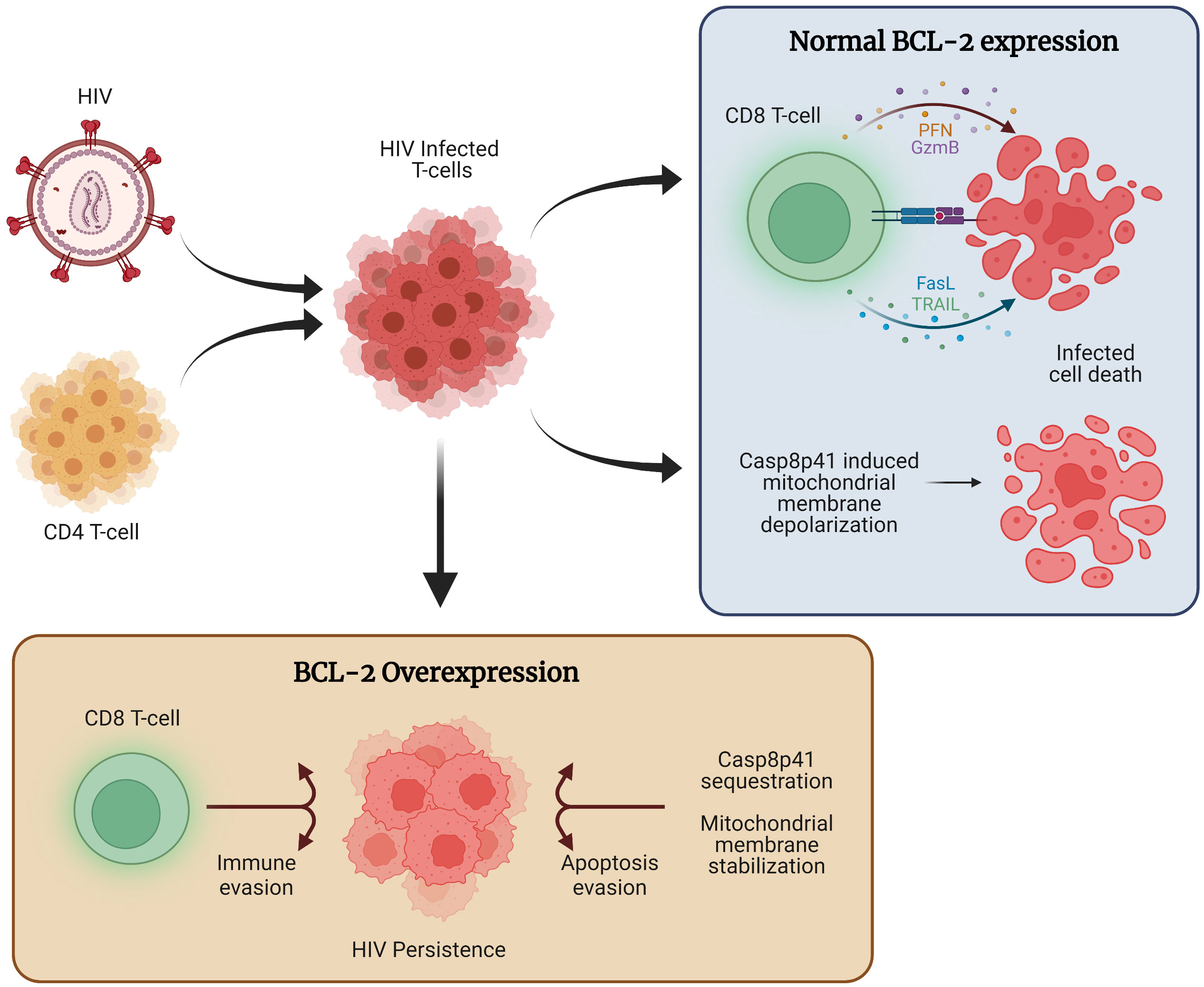
Figure 1 The role of BCL-2 in HIV persistence. Following HIV infection, a combination of host immune function and direct viral cytopathic effects, such as those mediated through Casp8p41 cause the majority of cells to die. However, a subset of infected cells expresses higher levels of BCL-2, allowing for apoptosis evasion and compromised immune function, ultimately allowing for the persistence of HIV infected cells.
In-vivo, cell mediated, cytotoxic immune responses are mediated by cytotoxic CD8 T-cells and Natural killer (NK) cells. These cells achieve target cell apoptosis through either granule mediated, or cytotoxic ligand mediated signalling. BCL-2 serves as a key regulatory factor for both mechanisms (27–30). CD8 and NK cells are crucial HIV control and dysregulated immune response is a key factor leading to the persistence of the HIV reservoir (1). CD8 and NK cells respond to antigenic challenge through overlapping mechanisms which ultimately result in target cell elimination. These mechanisms include granule mediated cytotoxicity and pro-apoptotic ligands such as FasL and TNF-related apoptosis inducting ligand (TRAIL) (27, 31, 32). The intersection of the aforementioned cytotoxic mechanisms utilized by effector cells, and the anti-apoptotic BCL-2 protein, will be examined further below.
Perforin/Granzyme B is a key effector pathway that is utilized by both CD8 T-cells and NK cells (27, 33, 34). Briefly, following the recognition of a cognate antigen, effector cells degranulate, leading to the release of Perforin and Granzyme B. Perforin binds to the target cell wall and causes pore formation in a manner that is postulated to be similar to bacterial cytolysins, allowing for the intracellular delivery of granzyme B (34). Granzyme B initiates the apoptotic singling cascade either through direct activation of caspases, or through the activation of BID. Activated BID subsequently activated the pro-apoptotic BAX protein, which in turn leads to mitochondrial membrane depolarization and efflux of apoptotic mediators, ultimately leading to cell death.
In vitro BCL-2 has been seen to antagonize granzyme mediated cell death through the inhibition of downstream Bax activation (35). Additionally, it was observed that BCL-2 strongly antagonized Perforin mediated Granzyme translocation to the nucleus, leading to blunted cytotoxicity (36). Overexpression of BCL-2 in mouse cell lines was seen to inhibit granzyme mediated apoptosis in-vitro; however it was observed that cytotoxic T-cells retained their apoptotic functionality, likely through granzyme B independent mechanisms (28). Overall, it has now been well established that BCL-2 overexpression antagonizes apoptosis induced by perforin and granzyme B.
Fas ligand (FasL) is a key effector molecule that is produced by both CD8 and NK cells and has been widely studied in the context of HIV infection and CD4 depletion (27, 33, 37–39). FasL binds its cognate receptor, Fas, on the surface of target cells, and recruits the “Fas associated death domain” (FADD) (40) to activate the apoptotic pathway. Fas has been shown to elicit apoptosis through Bid/Bax/Bak dependent and independent pathways in lymphocytes (41). Fas signaling is also a crucial to chimeric antigen receptor T- cell (CAR-T) mediated clearance in acute lymphoblastic leukemia (ALL) (42, 43).
The role of BCL-2 in preventing Fas mediated cell death is controversial and multiple reports have suggested that FasL can act in a BCL-2 independent fashion to induce cell death (44). However, it has also been demonstrated that BCL-2 overexpression can prevent Fas mediated cell death in lymphoid cell lines and in breast cancer (29, 45), and can abrogate cytotoxic T-lymphocyte (CTL) responses in cells overexpressing Fas receptor (46) suggesting that even though Fas signalling can function independently of pro-apoptotic BCL-2 family members, BCL-2 may play a role in blunting the apoptotic effect of Fas (47). BCL-2 has also been shown to downregulate the expression of FasL, through the disruption of the nuclear translocation of nuclear factor of activated T-cells (NFAT), via the inhibition of calcineurin, conferring drug resistance to FasL dependent drugs (48).
TNF-related apoptosis inducting ligand (TRAIL) is another key effector molecule that is essential for CD8 and NK function (27, 32). Following the binding of TRAIL to its cognate receptor, TRAIL Receptors 1 and 2, FADD is recruited to initiate apoptotic signalling (49). Similar to Fas, TRAIL too has been identified as a crucial regulator of CAR-T function in lymphoid malignancy (42, 43). TRAIL mediated apoptosis has been described to be directly inhibited by BCL-2 overexpression in multiple cell lines, though similar to FasL, lymphoid cells exhibited apoptosis through a BCL-2 independent pathway (30).
In terms of cell mediated cytotoxicity, upregulation of the BCL-2 homologs MCL-2 and BCL-2A1 were recently described to inhibit cytotoxic T-cell mediated clearance of melanoma cells (50). BCL-2 upregulation in CD8 effector cells in lymphoid malignancies has been demonstrated to directly impair the ability of immune effectors to degranulate efficiently, blunting the immune response. These cells were seen to also co-express PD-1 and TIM-3, both of which have been implicated in HIV immune escape (51–53). It has now also been demonstrated that cytotoxic T cells exhibit impaired clearance of target cells overexpressing BCL-2 (11).
Therefore, given the ability of BCL-2 to antagonize both direct viral cytopathic effects and cytotoxicity mediated by immune effector mechanisms, targeted therapy against BCL-2 has become a topic of growing importance in HIV shock and kill studies. The intersection of these will be examined further below.
The first pillar of the “shock and kill” strategy is the reactivation of the latent reservoir. Various classes of drugs, known as latency reversal agents (LRAs) are under investigation for the reversal of HIV latency in-vivo. These include Histone deacetylase (HDAC) inhibitors, Protein kinase C agonists, second mitochondrial-derived activator of caspases (SMAC) mimetics, Proteasome inhibitors, histone methyltransferase (HMT) inhibitors, DNA methyltransferase inhibitors, bromodomain inhibitors, Toll-like receptor (TLR) agonists and cytokines such as IL-15 super-agonist (N-803) (54, 55).
The crucial role that BCL-2 plays in the persistence of malignancy lead to the development of now clinically approved BCL-2 inhibitors. Given our growing understanding that BCL-2 is also a critical determinant of the HIV latent reservoir, studies have aimed to assess the impact of BCL-2 inhibition on HIV dynamics. Thus far, it has been established that selective BCL-2 inhibition can decrease the number of virally infected cells, decrease the size of the inducible latent reservoir, and restore the ability of HIV specific cytotoxic lymphocytes to target latently infected cells following reactivation (11, 21, 22, 56).
However, a suitable combination of an LRA and BCL-2 inhibitor that allows for maximal reactivation and selective elimination of HIV is yet to be identified. The following text aims to examine the effects of known latency reversal agents in combination with BCL-2 inhibitors (Table 1).

Table 1 Shock and kill agents and their interactions with BCL-2 and its inhibitors.
PKC agonists such as bryostatin and prostratin are amongst the most studied latency reversal agents. These drugs have demonstrated latency reversal in-vitro, and in in-vivo models, though have been disappointing in clinical settings (70, 71). A recent study identified that treatment of CD4 cells with PKC agonists induced phosphorylation on the serine 70 (ser70) residue of BCL-2, that was seen to inhibit the susceptibility of infected cells to cytotoxic stimuli (9). It has been demonstrated that the ser70 phosphorylation can directly inhibit the binding and efficacy of BCL-2 antagonists such as Navitoclax, ABT-737 and Venetoclax (57). These findings suggest that treatment with PKC agonists would likely not allow for maximal clearance of HIV infected cells, necessitating the development of novel PKC agonists which do not have the phosphorylating effect on BCL2. Additionally, the inhibitory effect of the ser70 phosphorylation on BCL-2 inhibitor activity would also likely preclude its clinical efficacy in this specific combination.
Proteasome inhibitors are another class of drugs that have shown latency reversal effect both in-vitro and in-vivo (22, 58, 72). The combination of the proteasome inhibitor Ixazomib with venetoclax was shown to exhibit strong synergistic activity against lymphoma cells (59). However, in HIV, the combination of proteasome and BCL-2 inhibitors was recently demonstrated to effectively target infected cells in cell line models but was associated with significant toxicity in ex vivo cells (58). A multi-center clinical trial is currently ongoing to determine the safety and efficacy of this combination in patients with multiple myeloma (Clinical trial number NCT03399539), which may inform future efforts at repurposing this combination in setting of HIV.
HDAC inhibitors such as Romidepsin, Vorinostat and Panobinostat have now demonstrated measurable latency reversal in the clinical setting (73–76). In ex vivo studies of cutaneous T cell lymphoma, combination of BCL-2 and HDAC inhibitors was seen to be synergistic at high doses, and paradoxically antagonistic at low doses, suggesting that regulated dosing and maintenance of drug levels may be necessary to achieve good therapeutic efficacy with this combination. Treatment with HDACi also has been shown to lead to the upregulation of the pro-apoptotic BCL-2 family proteins BCL2L11 and BMF (60).
The TLR 9 agonist MGN1703 has demonstrated in-vivo latency reversal, while also resulting in immune activation, but had no effect on the latent reservoir (77). TLR9 signaling has previously been demonstrated to result in upregulated expression of BCL-2 in primary activated CD4 cells (61), and it stands to be determined if the absence of reservoir depletion in trials of TLR9 agonists was as a result of BCL-2 upregulation. Consequently, the combination of TLR9 agonism and BCL-2 inhibition may prove an effective combination to target reactivated cells that are apoptosis resistant. However, a recent study involving ex vivo peripheral blood mononuclear cells (PBMCs) from patients with hairy cell leukemia (HCL) revealed that the cytotoxic effects of venetoclax against HCL cells was significantly reduced following TLR9 stimulation with CpG (62).
SMAC mimetics are another group of drugs that have shown efficacy in in-vivo animal models (63). The combination of SMAC mimetics and BCL-2 inhibitors have shown synergistic effect in hepatocellular carcinomas (64). This combination therefore may represent a possible combination to target the latent reservoir.
The IL-15 super-agonist N-803 was demonstrated to cause latency reversal and reservoir depletion in SIV infected macaques (78), and has recently demonstrated safety in a phase I clinical trial (79). IL-15 has been noted to regulate T-cell survival by altering the expression of Bim and MCL-1, overall favoring a pro-survival phenotype (65). In line with this observation, the combination of IL-15 and Venetoclax did not result in any observable decline in the number of HIV infected cells in an in-vitro model of latency (58). Further studies are required to assess if this combination would prove efficacious in vivo.
Immune function is a critical determinant of HIV reservoir size but varies widely, with the majority of infected individuals succumbing to a combination of viral escape mutations and host immune exhaustion, ultimately allowing for the persistence of HIV infection (80–82). The second pillar of the “shock and kill” strategy centers around the clearance of infected cells by immune effector cells and anti-retroviral therapy.
As monotherapy BCL-2 inhibition has been shown to independently result in the selective elimination of HIV infected cells and decrease the size of the HIV reservoir in in-vitro and ex-vivo studies (7, 11, 21, 22, 56, 58). BCL-2 inhibition has been shown to improve the efficacy of both NK cells and CTLs in human malignancies (66, 67). BCL-2 inhibition has now demonstrated measurably improved clearance of infected cells by CTLs in in-vitro and ex-vivo models of HIV (11), though further studies are required to demonstrate similar results in in-vivo settings.
Immune checkpoint blockade in HIV has demonstrated improved HIV specific CD8 degranulation in vitro (83, 84). Modest improvement in CD8 function has also been demonstrated in vivoin vitro (85). In human malignancy, the combination of immune checkpoint blockade and BCL-2 inhibition was seen to improve anti-tumor CD8 activity (68), suggesting that this combinatorial approach may also help improve anti-HIV immune function, though no studies have examined this combination to date.
HIV specific chimeric antigen receptor (CAR) T cells have demonstrated promise as a viable treatment option for HIV, having demonstrated efficacy at clearing HIV in-vitro and in-vivo, demonstrating the ability to traffic to lymphoid tissues and reduce the viral reservoir (86–89). BCL-2 inhibition has been demonstrated to improve the efficacy of CAR-Ts in the setting of malignancy (69), suggesting that combinations in HIV may allow for superior clearance, however further investigations are required to study this combination.
Successful Shock and kill therapy for HIV will ultimately require a potent and effective combination of latency reversal and immune function. Evidence thus far suggests that BCL-2 is a critical determinant of HIV survival and persistence. BCL-2 inhibition allows for the selective elimination of HIV infected cells, both as a monotherapy, and in combination with host immune cells, suggesting that targeted BCL-2 inhibition may lead to the potentiation of shock and kill treatment regimens, and ultimately HIV cure.
APC organized and wrote the manuscript. APC and ADB were involved in conceptualization, review and final drafting. Both authors contributed to the article and approved the submitted version.
Portions of this work were funded through grants (grants AI110173 and AI120698) from the National Institute of Allergy and Infectious Diseases of the NIH and the Mayo Clinic Foundation. The opinions expressed are solely of the authors and do not necessarily represent the opinions of the funding organization(s).
We thank the innumerable persons who have participated in research studies, without which many of the described observations would not have been possible. Figure 1 was created using Biorender.com
ADB is supported by grants from NIAID grants AI110173 and AI120698 Amfar #109593 and Mayo Clinic HH Sheikh Khalifa Bin Zayed Al-Nahyan Named Professorship of Infectious Diseases. ADB is a paid consultant for Abbvie, Gilead, Freedom Tunnel, Pinetree therapeutics Primmune, Immunome, MarPam, Rion, Symbiosis, NexImmune and Flambeau Diagnostics, is a paid member of the DSMB for Corvus Pharmaceuticals, Equilium, CSL Behring, and Excision Biotherapeutics, has received fees for speaking for Reach MD, Peer Voice, and Medscape, owns equity for scientific advisory work in Tier 1 Bio, Zentalis, Rion, and Nference, and is founder and President of Splissen therapeutics, and Member of the Board of Attivare.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Deeks SG, Overbaugh J, Phillips A, Buchbinder S. HIV Infection. Nat Rev Dis Primers (2015) 1:15035. doi: 10.1038/nrdp.2015.35
2. Bachmann N, von Siebenthal C, Vongrad V, Turk T, Neumann K, Beerenwinkel N, et al. Determinants of HIV-1 reservoir size and long-term dynamics during suppressive ART. Nat Commun (2019) 10(1):3193. doi: 10.1038/s41467-019-10884-9
3. Hosmane NN, Kwon KJ, Bruner KM, Capoferri AA, Beg S, Rosenbloom DI, et al. Proliferation of latently infected CD4(+) T cells carrying replication-competent HIV-1: Potential role in latent reservoir dynamics. J Exp Med (2017) 214(4):959–72. doi: 10.1084/jem.20170193
4. Yeh YJ, Yang K, Razmi A, Ho YC. The clonal expansion dynamics of the HIV-1 reservoir: Mechanisms of integration site-dependent proliferation and HIV-1 persistence. Viruses (2021) 13(9):1858. doi: 10.3390/v13091858
5. Deeks SG, Archin N, Cannon P, Collins S, Jones RB, de Jong M, et al. Research priorities for an HIV cure: International AIDS society global scientific strategy 2021. Nat Med (2021) 27(12):2085–98. doi: 10.1038/s41591-021-01590-5
7. Huang SH, Ren Y, Thomas AS, Chan D, Mueller S, Ward AR, et al. Latent HIV reservoirs exhibit inherent resistance to elimination by CD8+ T cells. J Clin Invest (2018) 128(2):876–89. doi: 10.1172/JCI97555
8. Quigley M, Pereyra F, Nilsson B, Porichis F, Fonseca C, Eichbaum Q, et al. Transcriptional analysis of HIV-specific CD8+ T cells shows that PD-1 inhibits T cell function by upregulating BATF. Nat Med (2010) 16(10):1147–51. doi: 10.1038/nm.2232
9. French AJ, Natesampillai S, Krogman A, Correia C, Peterson KL, Alto A, et al. Reactivating latent HIV with PKC agonists induces resistance to apoptosis and is associated with phosphorylation and activation of BCL2. PloS Pathog (2020) 16(10):e1008906. doi: 10.1371/journal.ppat.1008906
10. Kuo HH, Ahmad R, Lee GQ, Gao C, Chen HR, Ouyang Z, et al. Anti-apoptotic protein BIRC5 maintains survival of HIV-1-Infected CD4(+) T cells. Immunity (2018) 48(6):1183–94.e5. doi: 10.1016/j.immuni.2018.04.004
11. Ren Y, Huang SH, Patel S, Alberto WDC, Magat D, Ahimovic D, et al. BCL-2 antagonism sensitizes cytotoxic T cell-resistant HIV reservoirs to elimination ex vivo. J Clin Invest (2020) 130(5):2542–59. doi: 10.1172/JCI132374
12. Nie Z, Aboulnasr F, Natesampillai S, Burke SP, Krogman A, Bren GD, et al. Both HIV-infected and uninfected cells express TRAILshort, which confers TRAIL resistance upon bystander cells within the microenvironment. J Immunol (2018) 200(3):1110–23. doi: 10.4049/jimmunol.1701113
13. Chandrasekar AP, Cummins NW, Badley AD. The role of the BCL-2 family of proteins in HIV-1 pathogenesis and persistence. Clin Microbiol Rev (2019) 33(1):e00107–19. doi: 10.1128/CMR.00107-19
14. Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer (2002) 2(9):647–56. doi: 10.1038/nrc883
15. Shamas-Din A, Kale J, Leber B, Andrews DW. Mechanisms of action of bcl-2 family proteins. Cold Spring Harb Perspect Biol (2013) 5(4):a008714. doi: 10.1101/cshperspect.a008714
16. Nie Z, Bren GD, Vlahakis SR, Schimnich AA, Brenchley JM, Trushin SA, et al. Human immunodeficiency virus type 1 protease cleaves procaspase 8 in vivo. J Virol (2007) 81(13):6947–56. doi: 10.1128/JVI.02798-06
17. Sainski AM, Dai H, Natesampillai S, Pang YP, Bren GD, Cummins NW, et al. Casp8p41 generated by HIV protease kills CD4 T cells through direct bak activation. J Cell Biol (2014) 206(7):867–76. doi: 10.1083/jcb.201405051
18. Sainski AM, Natesampillai S, Cummins NW, Bren GD, Taylor J, Saenz DT, et al. The HIV-1-specific protein Casp8p41 induces death of infected cells through Bax/Bak. J Virol (2011) 85(16):7965–75. doi: 10.1128/JVI.02515-10
19. Cummins NW, Jiang W, McGinty J, Bren GD, Bosch RJ, Landay A, et al. Intracellular Casp8p41 content is inversely associated with CD4 T cell count. J Infect Dis (2010) 202(3):386–91. doi: 10.1086/653705
20. Cummins NW, Neuhaus J, Sainski AM, Strausbauch MA, Wettstein PJ, Lewin SR, et al. Short communication: CD4 T cell declines occurring during suppressive antiretroviral therapy reflect continued production of Casp8p41. AIDS Res Hum Retroviruses (2014) 30(5):476–9. doi: 10.1089/aid.2013.0243
21. Cummins NW, Sainski AM, Dai H, Natesampillai S, Pang YP, Bren GD, et al. Prime, shock, and kill: Priming CD4 T cells from HIV patients with a BCL-2 antagonist before HIV reactivation reduces HIV reservoir size. J Virol (2016) 90(8):4032–48. doi: 10.1128/JVI.03179-15
22. Natesampillai S, Cummins NW, Nie Z, Sampath R, Baker JV, Henry K, et al. HIV Protease-generated Casp8p41, when bound and inactivated by Bcl2, is degraded by the proteasome. J Virol (2018) 92(13):e00037–18. doi: 10.1128/JVI.00037-18
23. McGary CS, Deleage C, Harper J, Micci L, Ribeiro SP, Paganini S, et al. CTLA-4(+)PD-1(-) memory CD4(+) T cells critically contribute to viral persistence in antiretroviral therapy-suppressed, SIV-infected rhesus macaques. Immunity (2017) 47(4):776–88.e5. doi: 10.1016/j.immuni.2017.09.018
24. Kim M, Hosmane NN, Bullen CK, Capoferri A, Yang HC, Siliciano JD, et al. A primary CD4(+) T cell model of HIV-1 latency established after activation through the T cell receptor and subsequent return to quiescence. Nat Protoc (2014) 9(12):2755–70. doi: 10.1038/nprot.2014.188
25. Yang HC, Xing S, Shan L, O’Connell K, Dinoso J, Shen A, et al. Small-molecule screening using a human primary cell model of HIV latency identifies compounds that reverse latency without cellular activation. J Clin Invest (2009) 119(11):3473–86. doi: 10.1172/JCI39199
26. Sunshine S, Kirchner R, Amr SS, Mansur L, Shakhbatyan R, Kim M, et al. HIV Integration site analysis of cellular models of HIV latency with a probe-enriched next-generation sequencing assay. J Virol (2016) 90(9):4511–9. doi: 10.1128/JVI.01617-15
27. Prager I, Liesche C, van Ooijen H, Urlaub D, Verron Q, Sandström N, et al. NK cells switch from granzyme b to death receptor-mediated cytotoxicity during serial killing. J Exp Med (2019) 216(9):2113–27. doi: 10.1084/jem.20181454
28. Sutton VR, Vaux DL, Trapani JA. Bcl-2 prevents apoptosis induced by perforin and granzyme b, but not that mediated by whole cytotoxic lymphocytes. J Immunol (1997) 158(12):5783–90.
29. Kawahara A, Kobayashi T, Nagata S. Inhibition of fas-induced apoptosis by bcl-2. Oncogene (1998) 17(20):2549–54. doi: 10.1038/sj.onc.1202192
30. Fulda S, Meyer E, Debatin KM. Inhibition of TRAIL-induced apoptosis by bcl-2 overexpression. Oncogene (2002) 21(15):2283–94. doi: 10.1038/sj.onc.1205258
31. Collins DR, Gaiha GD, Walker BD. CD8(+) T cells in HIV control, cure and prevention. Nat Rev Immunol (2020) 20(8):471–82. doi: 10.1038/s41577-020-0274-9
32. Mirandola P, Ponti C, Gobbi G, Sponzilli I, Vaccarezza M, Cocco L, et al. Activated human NK and CD8+ T cells express both TNF-related apoptosis-inducing ligand (TRAIL) and TRAIL receptors but are resistant to TRAIL-mediated cytotoxicity. Blood (2004) 104(8):2418–24. doi: 10.1182/blood-2004-04-1294
33. Hassin D, Garber OG, Meiraz A, Schiffenbauer YS, Berke G. Cytotoxic T lymphocyte perforin and fas ligand working in concert even when fas ligand lytic action is still not detectable. Immunology (2011) 133(2):190–6. doi: 10.1111/j.1365-2567.2011.03426.x
34. Voskoboinik I, Whisstock JC, Trapani JA. Perforin and granzymes: Function, dysfunction and human pathology. Nat Rev Immunol (2015) 15(6):388–400. doi: 10.1038/nri3839
35. Heibein JA, Goping IS, Barry M, Pinkoski MJ, Shore GC, Green DR, et al. Granzyme b-mediated cytochrome c release is regulated by the bcl-2 family members bid and bax. J Exp Med (2000) 192(10):1391–402. doi: 10.1084/jem.192.10.1391
36. Jans DA, Sutton VR, Jans P, Froelich CJ, Trapani JA. BCL-2 blocks perforin-induced nuclear translocation of granzymes concomitant with protection against the nuclear events of apoptosis. J Biol Chem (1999) 274(7):3953–61. doi: 10.1074/jbc.274.7.3953
37. Zamai L, Ahmad M, Bennett IM, Azzoni L, Alnemri ES, Perussia B. Natural killer (NK) cell-mediated cytotoxicity: Differential use of TRAIL and fas ligand by immature and mature primary human NK cells. J Exp Med (1998) 188(12):2375–80. doi: 10.1084/jem.188.12.2375
38. Badley AD, McElhinny JA, Leibson PJ, Lynch DH, Alderson MR, Paya CV. Upregulation of fas ligand expression by human immunodeficiency virus in human macrophages mediates apoptosis of uninfected T lymphocytes. J Virol (1996) 70(1):199–206. doi: 10.1128/jvi.70.1.199-206.1996
39. Dockrell DH, Badley AD, Villacian JS, Heppelmann CJ, Algeciras A, Ziesmer S, et al. The expression of fas ligand by macrophages and its upregulation by human immunodeficiency virus infection. J Clin Invest (1998) 101(11):2394–405. doi: 10.1172/JCI1171
40. Scott FL, Stec B, Pop C, Dobaczewska MK, Lee JJ, Monosov E, et al. The fas-FADD death domain complex structure unravels signalling by receptor clustering. Nature (2009) 457(7232):1019–22. doi: 10.1038/nature07606
41. Jost PJ, Grabow S, Gray D, McKenzie MD, Nachbur U, Huang DC, et al. XIAP discriminates between type I and type II FAS-induced apoptosis. Nature (2009) 460(7258):1035–9. doi: 10.1038/nature08229
42. Singh N, Lee YG, Shestova O, Ravikumar P, Hayer KE, Hong SJ, et al. Impaired death receptor signaling in leukemia causes antigen-independent resistance by inducing CAR T-cell dysfunction. Cancer Discov (2020) 10(4):552–67. doi: 10.1158/2159-8290.CD-19-0813
43. Dufva O, Koski J, Maliniemi P, Ianevski A, Klievink J, Leitner J, et al. Integrated drug profiling and CRISPR screening identify essential pathways for CAR T-cell cytotoxicity. Blood (2020) 135(9):597–609. doi: 10.1182/blood.2019002121
44. Huang DC, Hahne M, Schroeter M, Frei K, Fontana A, Villunger A, et al. Activation of fas by FasL induces apoptosis by a mechanism that cannot be blocked by bcl-2 or bcl-x(L). Proc Natl Acad Sci USA (1999) 96(26):14871–6. doi: 10.1073/pnas.96.26.14871
45. Jäättelä M, Benedict M, Tewari M, Shayman JA, Dixit VM. Bcl-x and bcl-2 inhibit TNF and fas-induced apoptosis and activation of phospholipase A2 in breast carcinoma cells. Oncogene (1995) 10(12):2297–305.
46. Lee RK, Spielman J, Podack ER. Bcl-2 protects against fas-based but not perforin-based T cell-mediated cytolysis. Int Immunol (1996) 8(7):991–1000. doi: 10.1093/intimm/8.7.991
47. Croker BA, O’Donnell JA, Nowell CJ, Metcalf D, Dewson G, Campbell KJ, et al. Fas-mediated neutrophil apoptosis is accelerated by bid, bak, and bax and inhibited by bcl-2 and mcl-1. Proc Natl Acad Sci USA (2011) 108(32):13135–40. doi: 10.1073/pnas.1110358108
48. Srivastava RK, Sasaki CY, Hardwick JM, Longo DL. Bcl-2-mediated drug resistance: inhibition of apoptosis by blocking nuclear factor of activated T lymphocytes (NFAT)-induced fas ligand transcription. J Exp Med (1999) 190(2):253–65. doi: 10.1084/jem.190.2.253
49. Sprick MR, Weigand MA, Rieser E, Rauch CT, Juo P, Blenis J, et al. FADD/MORT1 and caspase-8 are recruited to TRAIL receptors 1 and 2 and are essential for apoptosis mediated by TRAIL receptor 2. Immunity (2000) 12(6): 599–609. doi: 10.1016/s1074-7613(00)80211-3
50. Joung J, Kirchgatterer PC, Singh A, Cho JH, Nety SP, Larson RC, et al. CRISPR activation screen identifies BCL-2 proteins and B3GNT2 as drivers of cancer resistance to T cell-mediated cytotoxicity. Nat Commun (2022) 13(1):1606. doi: 10.1038/s41467-022-29205-8
51. Liu L, Cheng X, Yang H, Lian S, Jiang Y, Liang J, et al. BCL-2 expression promotes immunosuppression in chronic lymphocytic leukemia by enhancing regulatory T cell differentiation and cytotoxic T cell exhaustion. Mol Cancer (2022) 21(1):59. doi: 10.1186/s12943-022-01516-w
52. Jones RB, Ndhlovu LC, Barbour JD, Sheth PM, Jha AR, Long BR, et al. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J Exp Med (2008) 205(12):2763–79. doi: 10.1084/jem.20081398
53. Ghiglione Y, Trifone C, Salido J, Rhodes A, Ruiz MJ, Polo ML, et al. PD-1 expression in HIV-specific CD8+ T cells before antiretroviral therapy is associated with HIV persistence. J Acquir Immune Defic Syndr (2019) 80(1):1–6. doi: 10.1097/QAI.0000000000001887
54. Kim Y, Anderson JL, Lewin SR. Getting the “Kill” into “Shock and kill”: Strategies to eliminate latent HIV. Cell Host Microbe (2018) 23(1):14–26. doi: 10.1016/j.chom.2017.12.004
55. Harwood O, O’Connor S. Therapeutic potential of IL-15 and n-803 in HIV/SIV infection. Viruses (2021) 13(9):1750. doi: 10.3390/v13091750
56. Cummins NW, Sainski-Nguyen AM, Natesampillai S, Aboulnasr F, Kaufmann S, Badley AD. Maintenance of the HIV reservoir is antagonized by selective BCL2 inhibition. J Virol (2017) 91(11):e00012–17. doi: 10.1128/JVI.00012-17
57. Song T, Chai G, Liu Y, Yu X, Wang Z, Zhang Z. Bcl-2 phosphorylation confers resistance on chronic lymphocytic leukaemia cells to the BH3 mimetics ABT-737, ABT-263 and ABT-199 by impeding direct binding. Br J Pharmacol (2016) 173(3):471–83. doi: 10.1111/bph.13370
58. Alto A, Natesampillai S, Chandrasekar AP, Krogman A, Misra A, Shweta F, et al. The combination of venetoclax and ixazomib selectively and efficiently kills HIV-infected cell lines but has unacceptable toxicity in primary cell models. J Virol (2021) 95(12). doi: 10.1128/JVI.00138-21
59. Cosenza M, Sacchi S, Pozzi S. Preclinical efficacy and biological effects of venetoclax and ixazomib in combination in lymphoma cells. Blood (2020) 136:37–8. doi: 10.1182/blood-2020-134882
60. Cyrenne BM, Lewis JM, Weed JG, Carlson KR, Mirza FN, Foss FM, et al. Synergy of BCL2 and histone deacetylase inhibition against leukemic cells from cutaneous T-cell lymphoma patients. Blood (2017) 130(19):2073–83. doi: 10.1182/blood-2017-06-792150
61. Zheng L, Asprodites N, Keene AH, Rodriguez P, Brown KD, Davila E. TLR9 engagement on CD4 T lymphocytes represses gamma-radiation-induced apoptosis through activation of checkpoint kinase response elements. Blood (2008) 111(5):2704–13. doi: 10.1182/blood-2007-07-104141
62. Vereertbrugghen A, Colado A, Gargiulo E, Bezares RF, Fernández Grecco H, Cordini G, et al. In vitro sensitivity to venetoclax and microenvironment protection in hairy cell leukemia. Front Oncol (2021) 11:598319. doi: 10.3389/fonc.2021.598319
63. Nixon CC, Mavigner M, Sampey GC, Brooks AD, Spagnuolo RA, Irlbeck DM, et al. Systemic HIV and SIV latency reversal via non-canonical NF-κB signalling in vivo. Nature (2020) 578(7793):160–5. doi: 10.1038/s41586-020-1951-3
64. Chen KF, Lin JP, Shiau CW, Tai WT, Liu CY, Yu HC, et al. Inhibition of bcl-2 improves effect of LCL161, a SMAC mimetic, in hepatocellular carcinoma cells. Biochem Pharmacol (2012) 84(3):268–77. doi: 10.1016/j.bcp.2012.04.023
65. Shenoy AR, Kirschnek S, Häcker G. IL-15 regulates bcl-2 family members bim and mcl-1 through JAK/STAT and PI3K/AKT pathways in T cells. Eur J Immunol (2014) 44(8):2500–7. doi: 10.1002/eji.201344238
66. Murakami S, Suzuki S, Hanamura I, Yoshikawa K, Ueda R, Seto M, et al. Combining T-cell-based immunotherapy with venetoclax elicits synergistic cytotoxicity to b-cell lines in vitro. Hematol Oncol (2020) 38(5):705–14. doi: 10.1002/hon.2794
67. Wu HY, Li KX, Pan WY, Guo MQ, Qiu DZ, He YJ, et al. Venetoclax enhances NK cell killing sensitivity of AML cells through the NKG2D/NKG2DL activation pathway. Int Immunopharmacol (2022) 104:108497. doi: 10.1016/j.intimp.2021.108497
68. Kohlhapp FJ, Haribhai D, Mathew R, Duggan R, Ellis PA, Wang R, et al. Venetoclax increases intratumoral effector T cells and antitumor efficacy in combination with immune checkpoint blockade. Cancer Discov (2021) 11(1):68–79. doi: 10.1158/2159-8290.CD-19-0759
69. Yang M, Wang L, Ni M, Neuber B, Wang S, Gong W, et al. Pre-sensitization of malignant b cells through venetoclax significantly improves the cytotoxic efficacy of CD19.CAR-T cells. Front Immunol (2020) 11:608167. doi: 10.3389/fimmu.2020.608167
70. Marsden MD, Loy BA, Wu X, Ramirez CM, Schrier AJ, Murray D, et al. In vivo activation of latent HIV with a synthetic bryostatin analog effects both latent cell “kick” and “kill” in strategy for virus eradication. PloS Pathog (2017) 13(9):e1006575. doi: 10.1371/journal.ppat.1006575
71. Gutiérrez C, Serrano-Villar S, Madrid-Elena N, Pérez-Elías MJ, Martín ME, Barbas C, et al. Bryostatin-1 for latent virus reactivation in HIV-infected patients on antiretroviral therapy. Aids (2016) 30(9):1385–92. doi: 10.1097/QAD.0000000000001064
72. Cummins NW, Baker J, Chakraborty R, Dean PG, Garcia-Rivera E, Krogman A, et al. Single center, open label dose escalating trial evaluating once weekly oral ixazomib in ART-suppressed, HIV positive adults and effects on HIV reservoir size in vivo. EClinicalMedicine (2021) 42:101225. doi: 10.1016/j.eclinm.2021.101225
73. Archin NM, Kirchherr JL, Sung JA, Clutton G, Sholtis K, Xu Y, et al. Interval dosing with the HDAC inhibitor vorinostat effectively reverses HIV latency. J Clin Invest (2017) 127(8):3126–35. doi: 10.1172/JCI92684
74. Rasmussen TA, Tolstrup M, Brinkmann CR, Olesen R, Erikstrup C, Solomon A, et al. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: a phase 1/2, single group, clinical trial. Lancet HIV (2014) 1(1):e13–21. doi: 10.1016/S2352-3018(14)70014-1
75. Rosás-Umbert M, Ruiz-Riol M, Fernández MA, Marszalek M, Coll P, Manzardo C, et al. In vivo effects of romidepsin on T-cell activation, apoptosis and function in the BCN02 HIV-1 Kick&Kill clinical trial. Front Immunol (2020) 11:418. doi: 10.3389/fimmu.2020.00418
76. Gruell H, Gunst JD, Cohen YZ, Pahus MH, Malin JJ, Platten M, et al. Effect of 3BNC117 and romidepsin on the HIV-1 reservoir in people taking suppressive antiretroviral therapy (ROADMAP): a randomised, open-label, phase 2A trial. Lancet Microbe (2022) 3(3):e203–e14. doi: 10.1016/S2666-5247(21)00239-1
77. Vibholm L, Schleimann MH, Højen JF, Benfield T, Offersen R, Rasmussen K, et al. Short-course toll-like receptor 9 agonist treatment impacts innate immunity and plasma viremia in individuals with human immunodeficiency virus infection. Clin Infect Dis (2017) 64(12):1686–95. doi: 10.1093/cid/cix201
78. McBrien JB, Mavigner M, Franchitti L, Smith SA, White E, Tharp GK, et al. Robust and persistent reactivation of SIV and HIV by n-803 and depletion of CD8(+) cells. Nature (2020) 578(7793):154–9. doi: 10.1038/s41586-020-1946-0
79. Miller JS, Davis ZB, Helgeson E, Reilly C, Thorkelson A, Anderson J, et al. Safety and virologic impact of the IL-15 superagonist n-803 in people living with HIV: a phase 1 trial. Nat Med (2022) 28(2):392–400. doi: 10.1038/s41591-021-01651-9
80. Betts MR, Nason MC, West SM, De Rosa SC, Migueles SA, Abraham J, et al. HIV Nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood (2006) 107(12):4781–9. doi: 10.1182/blood-2005-12-4818
81. Shasha D, Karel D, Angiuli O, Greenblatt A, Ghebremichael M, Yu X, et al. Elite controller CD8+ T cells exhibit comparable viral inhibition capacity, but better sustained effector properties compared to chronic progressors. J Leukoc Biol (2016) 100(6):1425–33. doi: 10.1189/jlb.4A0915-422R
82. Marras F, Nicco E, Bozzano F, Di Biagio A, Dentone C, Pontali E, et al. Natural killer cells in HIV controller patients express an activated effector phenotype and do not up-regulate NKp44 on IL-2 stimulation. Proc Natl Acad Sci U.S.A. (2013) 110(29):11970–5. doi: 10.1073/pnas.1302090110
83. Chiu CY, Chang JJ, Dantanarayana AI, Solomon A, Evans VA, Pascoe R, et al. Combination immune checkpoint blockade enhances IL-2 and CD107a production from HIV-specific T cells ex vivo in people living with HIV on antiretroviral therapy. J Immunol (2022) 208(1):54–62. doi: 10.4049/jimmunol.2100367
84. Grabmeier-Pfistershammer K, Stecher C, Zettl M, Rosskopf S, Rieger A, Zlabinger GJ, et al. Antibodies targeting BTLA or TIM-3 enhance HIV-1 specific T cell responses in combination with PD-1 blockade. Clin Immunol (2017) 183:167–73. doi: 10.1016/j.clim.2017.09.002
85. Lau JSY, McMahon JH, Gubser C, Solomon A, Chiu CYH, Dantanarayana A, et al. The impact of immune checkpoint therapy on the latent reservoir in HIV-infected individuals with cancer on antiretroviral therapy. Aids (2021) 35(10):1631–6. doi: 10.1097/QAD.0000000000002919
86. Qi J, Ding C, Jiang X, Gao Y. Advances in developing CAR T-cell therapy for HIV cure. Front Immunol (2020) 11:361. doi: 10.3389/fimmu.2020.00361
87. Anthony-Gonda K, Bardhi A, Ray A, Flerin N, Li M, Chen W, et al. Multispecific anti-HIV duoCAR-T cells display broad in vitro antiviral activity and potent in vivo elimination of HIV-infected cells in a humanized mouse model. Sci Transl Med (2019) 11(504). doi: 10.1126/scitranslmed.aav5685
88. Barber-Axthelm IM, Barber-Axthelm V, Sze KY, Zhen A, Suryawanshi GW, Chen IS, et al. Stem cell-derived CAR T cells traffic to HIV reservoirs in macaques. JCI Insight (2021) 6(1):e141502. doi: 10.1172/jci.insight.141502
Keywords: Bcl-2, hiv, HIV cure, shock and kill strategies, apoptosis
Citation: Chandrasekar AP and Badley AD (2022) Prime, shock and kill: BCL-2 inhibition for HIV cure. Front. Immunol. 13:1033609. doi: 10.3389/fimmu.2022.1033609
Received: 31 August 2022; Accepted: 07 October 2022;
Published: 20 October 2022.
Edited by:
Francesca Chiodi, Karolinska Institutet (KI), SwedenReviewed by:
Ana E. Sousa, University of Lisbon, PortugalCopyright © 2022 Chandrasekar and Badley. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrew D. Badley, QmFkbGV5LmFuZHJld0BtYXlvLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.