
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 23 November 2022
Sec. Alloimmunity and Transplantation
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1033490
This article is part of the Research TopicImmunobiology of Allogeneic Stem Cell Transplantation and Acute Graft-Versus-Host-DiseaseView all 11 articles
 Lotus Neidemire-Colley1,2
Lotus Neidemire-Colley1,2 Jérémy Robert3
Jérémy Robert3 Antoine Ackaoui3
Antoine Ackaoui3 Adrienne M. Dorrance2,4
Adrienne M. Dorrance2,4 Martin Guimond3,5,6†
Martin Guimond3,5,6† Parvathi Ranganathan2,4*†
Parvathi Ranganathan2,4*†To date, the only curative treatment for high-risk or refractory hematologic malignancies non-responsive to standard chemotherapy is allogeneic hematopoietic transplantation (allo-HCT). Acute graft-versus-host disease (GVHD) is a donor T cell-mediated immunological disorder that is frequently fatal and the leading cause of non-relapse mortality (NRM) in patients post allo-HCT. The pathogenesis of acute GVHD involves recognition of minor and/or major HLA mismatched host antigens by donor T cells followed by expansion, migration and finally end-organ damage due to combination of inflammatory cytokine secretion and direct cytotoxic effects. The endothelium is a thin layer of endothelial cells (EC) that line the innermost portion of the blood vessels and a key regulator in vascular homeostasis and inflammatory responses. Endothelial cells are activated by a wide range of inflammatory mediators including bacterial products, contents released from dying/apoptotic cells and cytokines and respond by secreting cytokines/chemokines that facilitate the recruitment of innate and adaptive immune cells to the site of inflammation. Endothelial cells can also be damaged prior to transplant as well as by alloreactive donor T cells. Prolonged EC activation results in dysfunction that plays a role in multiple post-transplant complications including but not limited to veno-occlusive disease (VOD), transplant associated thrombotic microangiopathy (TA-TMA), and idiopathic pneumonia syndrome. In this mini review, we summarize the biology of endothelial cells, factors regulating EC activation and the role of ECs in inflammation and GVHD pathogenesis.
Endothelial cells (ECs) form a single cell layer that line the inside of all blood and lymphatic vessels controlling the exchange of nutrients and oxygen between blood and tissues/organs (1, 2). In addition, they transport immune cells across the body to reach tissues and organs regulating immune surveillance under steady-state as well as infectious complications and malignant disorders. Under normal conditions, EC tight junctions regulate paracellular diffusion and homeostasis of tissues and organs. However, under inflammatory conditions, dramatic changes occur to the junction ultrastructures allowing the entry of immune cells (2–4). Acute graft-versus-host disease (GVHD) mediated by alloreactive T cells in the donor graft is a frequently fatal complication and the leading cause of non-relapse mortality (NRM) in patients post allo-HCT. Transplant-associated microangiopathy (TA-TMA), veno-occlusive disease (VOD), idiopathic pneumonia syndrome and accelerated arteriosclerosis are vascular injury syndromes that occur after allo-HCT (5). During allo-HCT, ECs can be directly damaged and/or activated via multiple mechanisms – i) chemotherapy and radiation included in the conditioning regimen; ii) cytokines released by injured tissues; iii) translocation of endotoxins through the damaged gastrointestinal tract as well as iv) immunosuppressive prophylactic regimens used to prevent acute GVHD, Figure 1. Apart from these early factors, ECs are also a target of alloreactive donor T cells that recognize the HLA mismatched antigens on ECs and mediate EC damage (5–7).
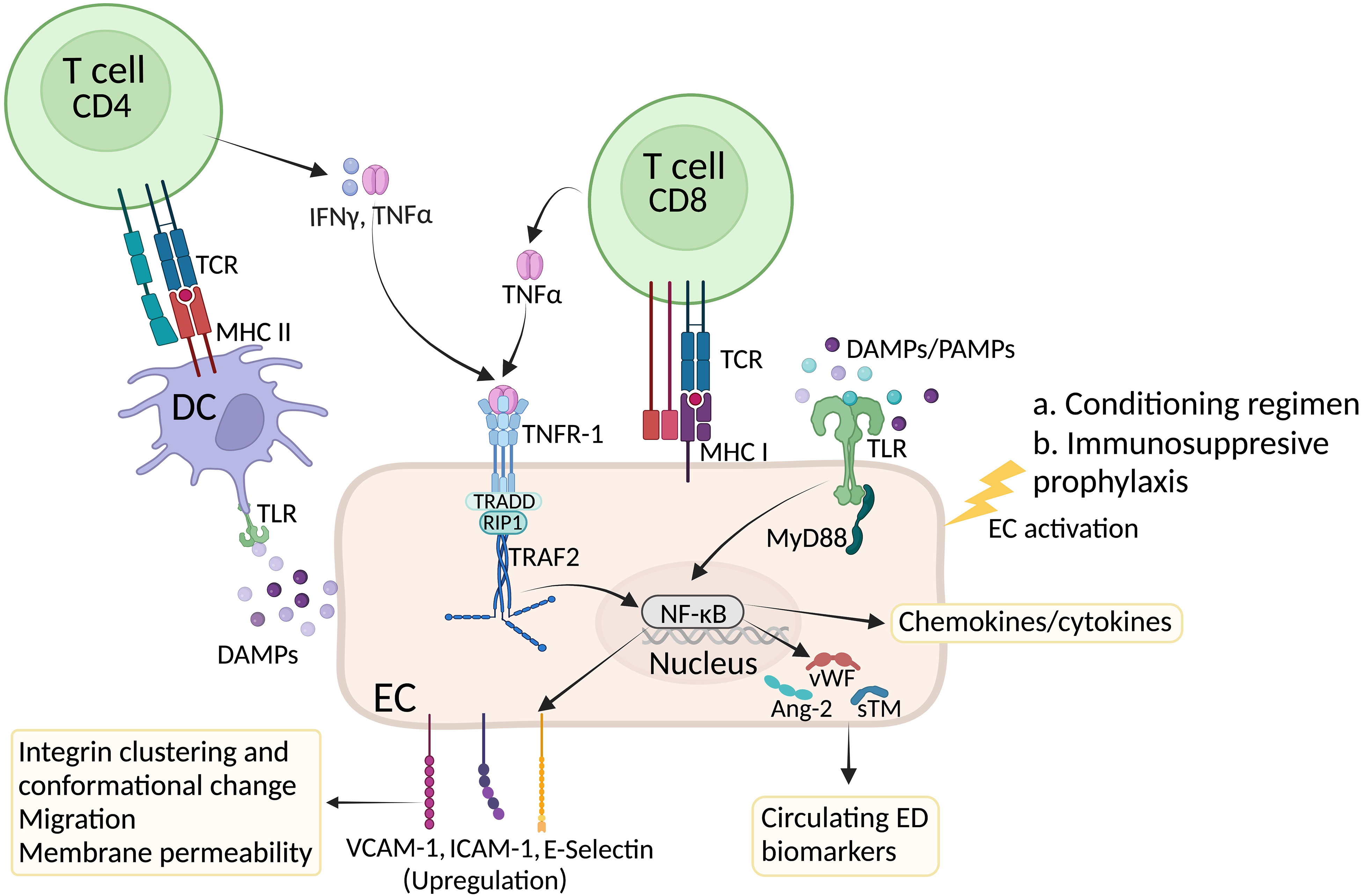
Figure 1 Factors affecting EC activation in GVHD. High dose chemotherapy induces systemic inflammation and endothelial cell damage. In response to inflammatory cytokines and Damage Associated Molecular Patterns (DAMPs), ECs undergo activation and express E and P selectins as well as VCAM-1 and ICAM-1 integrins to recruit innate and later adaptive immune cells at the inflammatory site. Inside the lymph node, presentation of allogeneic peptides by host dendritic will induce activation of CD4+ and CD8+ T cells. While cytotoxic CD8+ T cells further damage ECs, release of inflammatory cytokines such as IFN-γ and TNF-α by CD4+ T cells can bind to their receptors on ECs and further contribute to EC activation. In response to damage and activation, ECs produce and secrete von Willebrand factor (vWf) as well as angiotensin 2 (Ang-2) and soluble thrombomodulin (sTM). Circulating levels of these proteins are considered a biomarker of EC injury. Created with BioRender.com.
The permeability of the vascular system is regulated at the junctions of adjacent ECs separated by adherent or tight junctions (1, 8, 9), Figure 2. Even though ECs are found across the whole circuit, their heterogeneity is quite spectacular. For example, inside the kidney glomerulus, pores are present in tight junction ultrastructures which increase permeability to fluids and allow the filtration of glucose, urea, and sodium (8, 10, 11). At the other end of the spectrum, ECs in the brain present tight junctions which form a blood-brain barrier to prevent the entry of blood born cells/pathogens, suggesting barrier permeability is highly regulated and is tissue/organ specific (1, 2, 9, 12).
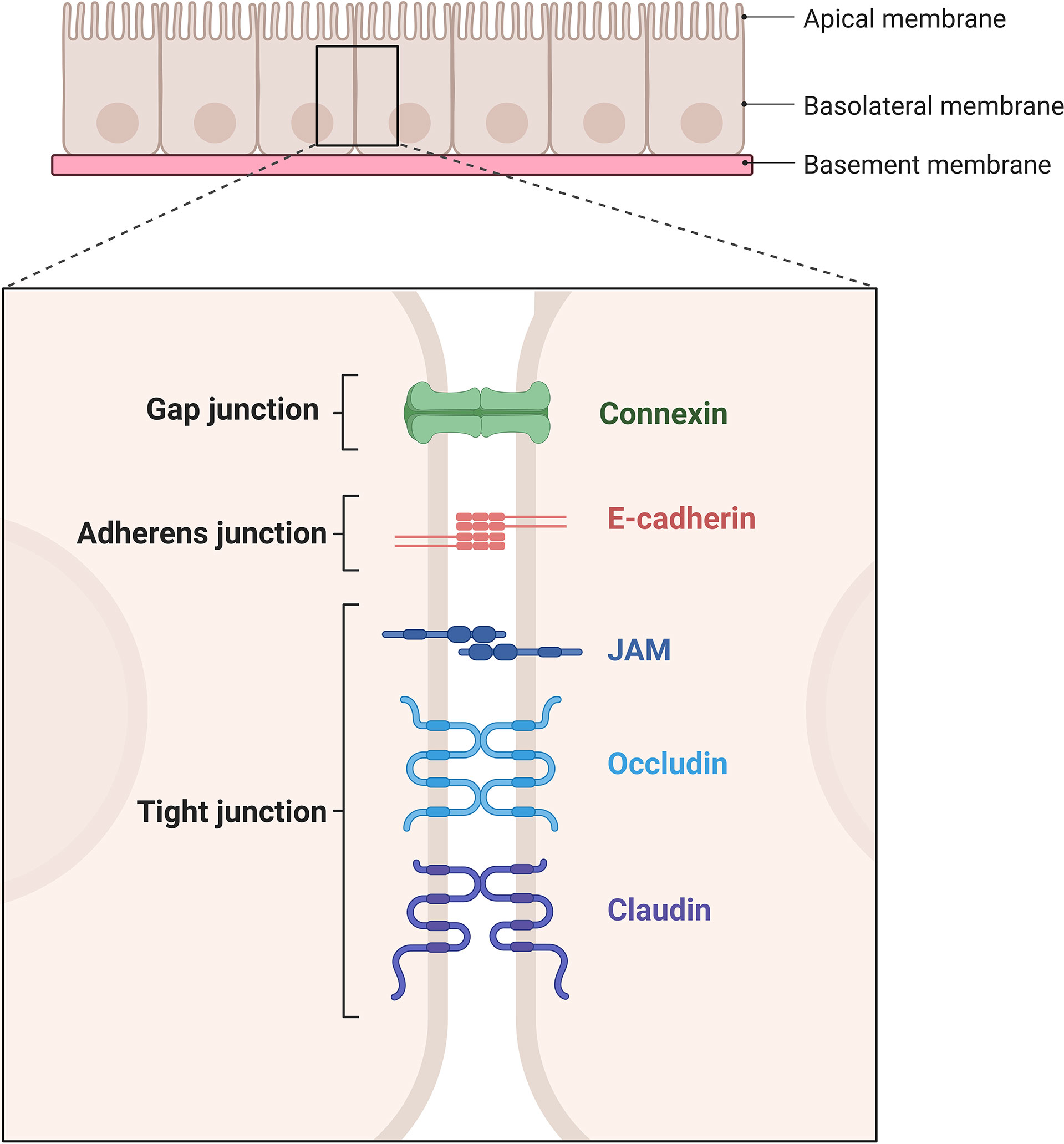
Figure 2 Schematic representation of gap junction, adherent junction, and tight junction. Gap junction is mediated by connexins. Adherent junction is mediated by two E-cadherins. Tight junction is mediated by JAMs, occludins and claudins. Created with BioRender.com.
The vascular endothelial cadherin (VE-cadherin) is found across the entire endothelium, forming adherent junctions and is particularly important for maintaining endothelial permeability (4, 8). Apart from their roles in adhesion, transmembrane proteins forming adherent junctions can prevent growth through contact-inhibition and importantly allow passage of leukocytes through the endothelium (13). The structure of tight junctions is largely dependent on claudin and occludin along with intermediary proteins including catenin (α, β, p120) and zona occluden (ZO) families (1, 12), while junctional adhesion molecules (JAMs) maintain the apicobasal polarity of cells (9). Reduced expression of tight junction (ZO-1) and adherent junction (VE-Cadherin) proteins result in increased endothelial leakage in GVHD target organs liver and colon in experimental models of GVHD (14) showing the importance of barrier function in disease pathogenesis. There was also evidence of increased endothelial cell apoptosis in experimental models of GVHD and patients with GI-GVHD (14).
Cell adhesion molecules such as selectins and integrins play a crucial function in the interaction between ECs and immune cells. Selectins are sub-divided into three groups: P-selectin (platelets and ECs), E-selectin (ECs) and L-selectin (leukocytes) (2, 15). L-selectin allows leukocytes to access lymph nodes and following lymphocyte activation, their surface expression is generally downregulated to prevent lymph node homing (16). P-selectin glycoprotein ligand 1 (PSGL-1) is the main ligand for all three types of selectins and requires posttranslational modifications for its activation (17). Type II leukocyte adhesion deficiency (LAD-II) is a disease caused by deficiency in the posttranslational modification of PSGL-1, resulting in the inability of leukocyte binding to any selectin, leading to bacterial infection of the mucosal membrane and skin (18, 19).
ECs can rapidly initiate the inflammatory response since they store pre-formed molecules in specialized organelles called Weibel-Palade bodies. Weibel-Palade bodies contain a wide range of inflammatory and angiogenic factors including but not limited to von Willebrand factor, P selectin, Angiotensin-2, IL-8, endothelin, and their content can vary based on the microenvironment (20). Pre-formed P-selectin is found in the Weibel-Palade bodies and upon EC activation, P-selectin is rapidly expressed to initiate recruitment of innate immune cells to the inflammatory site. Unlike P-selectin, E-selectin is not pre-formed; the synthesis of E-selectin occurs during EC activation and for this reason, its expression is normally delayed compared with P-selectin. In preclinical GVHD mouse models, recipients deficient for P-selectin displayed reduced GVHD mortality with associated reduction in alloreactive T cell infiltration into GVHD target organs (21).
PSGL-1 and CD44 expressed on leukocytes can bind E-selectin to modulate the rolling of immune cells on the endothelium (15, 19, 22–24). Innate immune cells such as neutrophils constitutively express PSGL-1 whereas T cells require cytokines and antigen presentation to induce posttranslational modifications required for PSGL-1 function (15, 17, 25) and subsequent rolling. As a result, neutrophils are the first cell type to be recruited at inflammatory sites while homing of T lymphocytes to inflammatory sites occurs later because expression of functional PSGL-1 requires activation (22, 25, 26). However, donor T cells deficient for PSGL-1 displayed similar migration patterns and caused GVHD similar to wildtype donor T cells in experimental acute GVHD mouse models suggesting that other P-selectin ligands might be involved in T cell infiltration in GVHD (21). In experimental models of chronic GVHD, it was shown that donor PSGL1hiCD4+ peripheral T cells differentiate into PSGL1loCD4+ tissue-resident memory T cells that in turn support B cell differentiation and autoreactive antibody production (27).
Integrins are a large family of heterodimers containing α- and β- chains forming a receptor at the cell surface (9, 28) that are critical for tethering and rolling of leukocytes on the endothelium. Neutrophils express integrin macrophage-1 (MAC-1) that has a wide range of ligands whereas T cells express lymphocyte function-associated antigen-1 (LFA-1) and α1β1 (VLA-1), α4 and β7 integrins that bind fewer types of ligands, mainly intracellular adhesion molecules-1-5 (ICAM-1-5), JAM-1 and vascular cell adhesion molecule-1 (VCAM-1) present on leukocytes, epithelial and endothelial cells (29–31).
Integrins have three different conformational forms that affect their affinity to the ligand: bent-closed (inactive, basal state), extended-closed (active, low-affinity) and extended-open (active, high-affinity) (4, 32). Conformational changes of integrins can be induced by signaling via chemokine receptors, selectins and Toll-like receptors (TLRs), passing from an intermediate to high affinity state (33–35). Following this change, integrins bind to their specific ligands and are immobilized. The glycocalyx found on ECs is rich in glycosaminoglycans that immobilize important chemokines forming a chemotactic gradient thereby facilitating the homing of leukocytes bearing the cognate chemokine receptors (CCR4/CCR10 in the skin, CCR9 in the gut and CCR7 in secondary lymphoid organs) and contribute to the tissue-tropism involved in GVHD pathogenesis (36–39). Each type of TLR can respond to pathogen-associated molecular patterns (PAMPs) or damage associated molecular pattern molecules (DAMPs) released by tissue injury caused by the conditioning regimen pre allo-HCT. Release of reactive oxygen species and DAMPs such as high mobility group box 1 (HMGB1) further amplify inflammatory cytokine production via TLR signaling thereby positioning ECs as both a target and contributor of the “cytokine storm” that perpetuates GVHD (40–42). TLR stimulation signals through mitogen-activated protein kinases (MAPKs), promoting the release of pro-inflammatory cytokines and increasing expression of E/P-selectin and integrins ICAM-1 and VCAM-1 on ECs (35, 43, 44). In mouse models of acute GVHD, allogeneic recipients showed upregulation of VCAM-1, ICAM-1 in the GI tract compared to syngeneic recipients with concomitant increase in T cell infiltration in GVHD target organs of skin, liver and GI tract (45). Lymphocytes in the intestinal mucosa express β7 integrins that bind to mucosal addressing cell adhesion molecule-1 (MadCAM-1) and E-cadherin, expressed on mucosal endothelium and intestinal epithelial cells resulting in donor T cell infiltration into the intestine (38, 46). Absence of β2 integrins on donor T cells resulted in significant downregulation of T cell infiltration in experimental GVHD (47).
In addition, integrin binding on the endothelium can promote the expression of pro-inflammatory genes, suggesting that integrin signaling can influence inflammatory microenvironment (48). Circulating pro-inflammatory cytokines such as TNFα, IFNγ, IL-1, and IL-6 are elevated during acute GVHD and can activate ECs (49, 50). Binding of TNFα to its receptor on ECs (TNFRI) activates a complex cascade of signaling events resulting in upregulation of adhesion molecules (VCAM-1, E-selectin, and ICAM-1) enabling transmigration of leukocytes (51, 52). Signaling through TNFRI also results in elevation of Angiopoetin-2 (Ang-2) that increases EC vulnerability in part by destabilizing cell junctions resulting in increased permeability (5, 53, 54) creating a more permissive environment for T cell extravasation. Apart from TNFRI, ECs also express TNFRII that can have both pro- and anti- inflammatory effects. Endothelial Progenitor Cells (EPCs) are undifferentiated ECs with stem cell like features, present in circulation. EPCs express TNF receptor II (TNFRII) on their surface that binds to TNFα and exerts an immunosuppressive effect on T cells in part by secretion of anti-inflammatory TGFβ, IL-10 and HLA-G cytokines (55). Additionally, TNF-TNFRII activated endothelial cells produce CCR2 ligands, that can in turn promote the differentiation of CCR2+ monocytes into immature macrophages that can promote inflammation (56).
The final step of the diapedesis/leukocyte extravasation is the transmigration of leukocytes through the pericyte and vascular basement membrane by the receptors ICAM-1, MAC-1, LFA-1 and platelet ECs adhesion molecule-1 (PECAM-1) to reach the site of inflammation (26, 57). ECs can also activate alloreactive T cells by presenting antigens in the context of MHC class I on their surface (58–60), while DAMPs such as HMGB1 released by ECs activate dendritic cells that in turn promote T cell inflammatory responses (42, 61, 62). ECs are also a target of alloreactive T cells, and the subsequent tissue-related EC damage and death is a hallmark of acute GVHD end-organ damage (63, 64).
Prolonged EC activation results in irreversible damage termed endothelial dysfunction. The von Willebrand factor (vWF) is stored in the Weibel-Palade bodies and plays a central role in the recruitment of platelets (65, 66) to adhere to injured endothelial cells/blood vessels, and thus is a key regulator of the coagulation cascade (65, 67, 68). Multimeric vWF is cleaved by ADAMTS13 metalloprotease to prevent excessive platelet aggregation (69). The massive and rapid release vWF in the bloodstream make this protein an ideal clinical marker of inflammation, EC activation and EC damage (65, 67).
Multiple markers of EC damage such as vWF, soluble VCAM-1 (sVCAM-1), ADAMTS-13 activity, and soluble tumor necrosis factor receptor-1 (sTNFRI) are upregulated in the plasma after use of conditioning regimens (5, 70). Use of conditioning agents busulfan and cyclophosphamide in mice resulted in vascular endothelial injury in mice associated with increased mobilization of endothelial progenitor cells, increased circulating ECs and structural changes observed by transmission electron microscopy (71). Paradoxically, the use of broadly immunosuppressive prophylactic regimens such as calcineurin and mechanistic target of rapamycin (mTOR) inhibitors to prevent acute GVHD can mediate endothelial damage (72, 73) and is associated with increased circulating levels of vWF, soluble thrombomodulin (sTM), and ICAM-1, predictive of VOD (72). Sirolimus, an inhibitor of mTOR, has been shown to directly inhibit endothelial cell proliferation and function in vitro (74) and in patients with coronary artery disease who receive sirolimus coated artery stents (75). Calcineurin inhibitors such as cyclosporine and tacrolimus can cause varying degrees of endothelial dysfunction caused primarily by a reduction in the release of endothelial protective nitric oxide (NO), increasing formation of free radicals leading to ED (74, 76).
Studies have shown that levels of circulating Ang-2, ST2, and sTM are increased prior to HCT suggesting endothelium is already damaged by underlying disease. The Endothelial Activation and Stress Index (EASIX) score (that measures creatinine, lactate dehydrogenase and platelets) was developed as surrogate of endothelial dysfunction. A study by Luft et al. used the EASIX score prior to conditioning regimens (EASIX-pre) to predict mortality after alloSCT. EASIX-pre was successful in predicting overall survival and risk of TAM after allo-SCT. However, EASIX-pre only showed an association with higher risk of grade 3-4 acute GVHD and no correlation with Ang-2 and ST2 levels (77). The EASIX score has since been adopted as a prognostic tool for predicting outcomes for a number of diseases including small cell lung cancer, bilirubinemia, and myelodysplastic syndromes (78–81).These studies suggest while ED is not specific to GVHD onset, ED and related systemic inflammation contribute to pathogenesis of GVHD, bolstering the use of EASIX/endothelial dysfunction to predict non-relapse mortality after transplant.
Supporting the role of EC activation or dysfunction in GVHD pathogenesis, histologic analysis of patients with cutaneous GVHD showed evidence of increased adhesion markers VCAM-1, endothelial leukocyte adhesion molecule-1 (ELAM-1) and vWF extravasation (82), while upregulation of vWF and thrombomodulin (TM) levels was observed in patients who developed acute GVHD post-transplant compared to those who did not (83), and serum levels of sICAM-1 and skin biopsies of E-selectin were both increased in acute GVHD patients (84).
Soluble levels of vWF and TNFRI at day 7 post-transplant could positively predict the development of acute GVHD in majority of patients who (90%) expressed higher than cut-off levels of these markers (52). Circulating levels of Ang-2, was reported to be significantly higher by day 21 post HCT in patients who went on to develop acute GVHD compared to the non-GVHD group (85) and has shown to be an effective biomarker for patients who develop endothelial damage post allo-HCT (86, 87). More significant levels of vWF led to more severe acute GVHD. Other damage-associated angiogenic factors that indicate tissue damage and inflammation such as follistatin (FS) and soluble endoglin (sEng) are elevated at day +28 post HCT and predict one-year NRM (88).
Interventions to prevent or restore EC damage, activation, and dysfunction are being explored as potential therapeutic agents in ameliorating GVHD. Treatment with an anticoagulant agent, recombinant TM, significantly reduced levels of sCAMs that predict EC dysfunction level and acute GVHD frequency (89). Another treatment for coagulation and thrombotic disorders, defibrotide, has been shown to protect ECs by preserving EC homeostasis. Defibrotide lowers vWF, VCAM-1, and sICAM-1 levels in GVHD patients by suppressing EC proliferation (90, 91). Epidermal growth factor-like domain 7 (EGFL7) inhibits EC activation by pro-inflammatory cytokines through a negative feedback loop. Using mouse models of disease, we have shown that treatment with recombinant EGFL7 reduced VCAM-1 expression on ECs and led to reduction of T cell infiltration, resulting in significant GVHD improvement (92).
High-dose corticosteroids remain the first-line therapy for GVHD patients, despite poor response rates. Patients with steroid-refractory GVHD do poorly with less than 50% survival at 6-months highlighting the need for novel treatment approaches (93). Steroid-refractory acute GVHD (SR-aGVHD) immunotherapies eradicate alloreactive T cells but fail to stop organ damage suggesting there are additional mechanisms that are relevant independent from initial T cell insult. Endothelial damage in patients has been correlated with pathogenesis of steroid resistant GVHD and increased NRM (94). A seminal study by Luft et al. showed that serum levels of Ang-2 were higher in SR-GVHD patients compared to steroid-sensitive GVHD while T cell activation patterns between groups were similar. In the same study, soluble TM (sTM) levels increased steadily in steroid-refractory patients and remained constant in patients who responded to corticosteroids. Both Ang-2 and sTM levels differentiated GVHD patients and the category of therapy response within those patients (95). Suppression of tumorigenicity 2 (ST2), a marker of endothelial injury, has high-risk association with SR-aGVHD and emerged as an important biomarker for treatment-resistant GVHD, NRM (96) as well as TA-TMA NRM at 6 months (97). A recent study demonstrated the endothelium protective effects of PDE5 inhibitor sildenafil, and showed promising results in steroid-refractory experimental mouse models of GVHD (14). Alpha-1 Antitrypsin (AAT), a serine protease inhibitor modulates inflammatory response of ECs to TNFα (98) and enhances T regulatory cell recovery in experimental mouse models of acute GVHD (86, 99). Multiple clinical trials have shown that AAT is well-tolerated in allo-HCT patients with varying efficacy in the treatment of steroid refractory GVHD (100–102), however, pre-emptive use AAT in of patients at high risk of developing SR-GVHD, did not change the incidence of steroid-resistance (103).
In recent years, there has been an increasing appreciation of the role played by ECs in the pathology of GVHD. While it was believed that EC dysfunction resulted from complications of allo-HCT and GVHD, our current understanding of the biology of ECs suggests that EC dysfunction and associated systemic inflammation also contribute to the onset and pathogenesis of acute GVHD. While chemotherapeutic insults can induce significant damage to ECs, the inflammatory milieu probably add to EC dysfunction by allowing uncontrolled migration of immune cells between the blood and tissues. Biomarkers of endothelial cell dysfunction are typically involved in hemostasis and while they might be reliable to measure EC damage, it remains uncertain as to whether these markers can be used to diagnose or predict acute GVHD onset, severity and/or overall allo-HCT outcomes. Early detection of biomarkers could lead to prevention of irreversible ED, and strategies to improve EC function and restore vascular barrier presents an attractive regenerative-based approach to prevent or treat GVHD. Given the importance of the vascular system in nutrient and gas exchange, reversing, or preventing EC dysfunction after allo-HCT may perhaps surpass the benefit of actual immunosuppressive therapies currently used to treat GVHD.
LN-C, AA, JR, MG and PR wrote and reviewed the manuscript. AD edited the manuscript. MG and AD hold a revisionary patent for the use of EGFL7 in the treatment of GVHD. All authors contributed to the article and approved the submitted version.
This work was supported by grants from NIH R01CA252469 (PR), American Cancer Society RSG RSG-22-053-01-IBCD (PR), NIH R01HL163849 (PR and AD), the American Cancer Society RSG-18-170-01-LIB (AD), the Cancer Research Society of Canada 24380 (MG), and Cancer Research Society and the Leukemia Lymphoma Society of Canada 840026 (MG). AA has received the Andy & Léna Chabot Cancer Research Society Studentship.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Bauer HC, Krizbai IA, Bauer H, Traweger A. "You shall not pass"-tight junctions of the blood brain barrier. Front Neurosci (2014) 8:392. doi: 10.3389/fnins.2014.00392
2. Wettschureck N, Strilic B, Offermanns S. Passing the vascular barrier: Endothelial signaling processes controlling extravasation. Physiol Rev (2019) 99(3):1467–525. doi: 10.1152/physrev.00037.2018
3. Vestweber D. Regulation of endothelial cell contacts during leukocyte extravasation. Curr Opin Cell Biol (2002) 14(5):587–93. doi: 10.1016/S0955-0674(02)00372-1
4. Vestweber D. How leukocytes cross the vascular endothelium. Nat Rev Immunol (2015) 15(11):692–704. doi: 10.1038/nri3908
5. Milone G, Bellofiore C, Leotta S, Milone GA, Cupri A, Duminuco A, et al. Endothelial dysfunction after hematopoietic stem cell transplantation: A review based on physiopathology. J Clin Med (2022) 11(3):1–29. doi: 10.3390/jcm11030623
6. Sumransub N, El Jurdi N, Chiraphapphaiboon W, Maakaron JE. Putting function back in dysfunction: Endothelial diseases and current therapies in hematopoietic stem cell transplantation and cellular therapies. Blood Rev (2022) 51:100883. doi: 10.1016/j.blre.2021.100883
7. Holtan SG, Penack O. Immune biology of allogeneic hematopoietic stem cell Transplantation. Chapter 21: The endothelium during allogeneic stem cell transplantation. 2nd ed: 401–414. (2019). Cambridge, MA: Academic Press.
8. Duong CN, Vestweber D. Mechanisms ensuring endothelial junction integrity beyond VE-cadherin. Front Physiol (2020) 11:519. doi: 10.3389/fphys.2020.00519
9. Zihni C, Mills C, Matter K, Balda MS. Tight junctions: from simple barriers to multifunctional molecular gates. Nat Rev Mol Cell Biol (2016) 17(9):564–80. doi: 10.1038/nrm.2016.80
10. Jambusaria A, Hong Z, Zhang L, Srivastava S, Jana A, Toth PT, et al. Endothelial heterogeneity across distinct vascular beds during homeostasis and inflammation. Elife (2020) 9:1–32. doi: 10.7554/eLife.51413
11. Ribatti D, Nico B, Vacca A, Roncali L, Dammacco F. Endothelial cell heterogeneity and organ specificity. J Hematother Stem Cell Res (2002) 11(1):81–90. doi: 10.1089/152581602753448559
12. Cong X, Kong W. Endothelial tight junctions and their regulatory signaling pathways in vascular homeostasis and disease. Cell Signal (2020) 66:109485. doi: 10.1016/j.cellsig.2019.109485
13. Dejana E. Endothelial cell-cell junctions: Happy together. Nat Rev Mol Cell Biol (2004) 5(4):261–70. doi: 10.1038/nrm1357
14. Cordes S, Mokhtari Z, Bartosova M, Mertlitz S, Riesner K, Shi Y, et al. Endothelial damage and dysfunction in acute graft-versus-host disease. Haematologica (2021) 106(8):2147–60. doi: 10.3324/haematol.2020.253716
15. Moore KL, Patel KD, Bruehl RE, Li F, Johnson DA, Lichenstein HS, et al. P-selectin glycoprotein ligand-1 mediates rolling of human neutrophils on p-selectin. J Cell Biol (1995) 128(4):661–71. doi: 10.1083/jcb.128.4.661
16. Chao CC, Jensen R, Dailey MO. Mechanisms of L-selectin regulation by activated T cells. J Immunol (1997) 159(4):1686–94.
17. McEver RP, Cummings RD. Perspectives series: cell adhesion in vascular biology. role of PSGL-1 binding to selectins in leukocyte recruitment. J Clin Invest (1997) 100(3):485–91. doi: 10.1172/JCI119556
18. Bunting M, Harris ES, McIntyre TM, Prescott SM, Zimmerman GA. Leukocyte adhesion deficiency syndromes: adhesion and tethering defects involving beta 2 integrins and selectin ligands. Curr Opin Hematol (2002) 9(1):30–5. doi: 10.1097/00062752-200201000-00006
19. Kappelmayer J, Nagy B Jr. The interaction of selectins and PSGL-1 as a key component in thrombus formation and cancer progression. BioMed Res Int (2017) 2017:6138145. doi: 10.1155/2017/6138145
20. Schillemans M, Karampini E, Kat M, Bierings R. Exocytosis of weibel-palade bodies: How to unpack a vascular emergency kit. J Thromb Haemost (2019) 17(1):6–18. doi: 10.1111/jth.14322
21. Lu SX, Holland AM, Na IK, Terwey TH, Alpdogan O, Bautista JL, et al. Absence of p-selectin in recipients of allogeneic bone marrow transplantation ameliorates experimental graft-versus-host disease. J Immunol (2010) 185(3):1912–9. doi: 10.4049/jimmunol.0903148
22. Ali AJ, Abuelela AF, Merzaban JS. An analysis of trafficking receptors shows that CD44 and p-selectin glycoprotein ligand-1 collectively control the migration of activated human T-cells. Front Immunol (2017) 8:492. doi: 10.3389/fimmu.2017.00492
23. Baaten BJ, Tinoco R, Chen AT, Bradley LM. Regulation of antigen-experienced T cells: Lessons from the quintessential memory marker CD44. Front Immunol (2012) 3:23. doi: 10.3389/fimmu.2012.00023
24. Hidalgo A, Peired AJ, Wild M, Vestweber D, Frenette PS. Complete identification of e-selectin ligands on neutrophils reveals distinct functions of PSGL-1, ESL-1, and CD44. Immunity (2007) 26(4):477–89. doi: 10.1016/j.immuni.2007.03.011
25. Abadier M, Pramod AB, McArdle S, Marki A, Fan Z, Gutierrez E, et al. Effector and regulatory T cells roll at high shear stress by inducible tether and sling formation. Cell Rep (2017) 21(13):3885–99. doi: 10.1016/j.celrep.2017.11.099
26. Voisin MB, Nourshargh S. Neutrophil transmigration: emergence of an adhesive cascade within venular walls. J Innate Immun (2013) 5(4):336–47. doi: 10.1159/000346659
27. Kong X, Zeng D, Wu X, Wang B, Yang S, Song Q, et al. Tissue-resident PSGL1loCD4+ T cells promote b cell differentiation and chronic graft-versus-host disease-associated autoimmunity. J Clin Invest (2021) 131(1):1–20. doi: 10.1172/JCI135468
29. Harjunpaa H, Llort Asens M, Guenther C, Fagerholm SC. Cell adhesion molecules and their roles and regulation in the immune and tumor microenvironment. Front Immunol (2019) 10:1078. doi: 10.3389/fimmu.2019.01078
30. Muller WA. Mechanisms of transendothelial migration of leukocytes. Circ Res (2009) 105(3):223–30. doi: 10.1161/CIRCRESAHA.109.200717
31. Pinte S, Caetano B, Le Bras A, Havet C, Villain G, Dernayka R, et al. Endothelial cell activation is regulated by epidermal growth factor-like domain 7 (Egfl7) during inflammation. J Biol Chem (2016) 291(46):24017–28. doi: 10.1074/jbc.M116.731331
32. Nishida N, Xie C, Shimaoka M, Cheng Y, Walz T, Springer TA. Activation of leukocyte beta2 integrins by conversion from bent to extended conformations. Immunity (2006) 25(4):583–94. doi: 10.1016/j.immuni.2006.07.016
33. Alon R, Feigelson SW. Chemokine signaling to lymphocyte integrins under shear flow. Microcirculation (2009) 16(1):3–16. doi: 10.1080/10739680802026076
34. Campbell ID, Humphries MJ. Integrin structure, activation, and interactions. Cold Spring Harb Perspect Biol (2011) 3(3):1–14. doi: 10.1101/cshperspect.a004994
35. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol (2007) 7(9):678–89. doi: 10.1038/nri2156
36. Graham GJ, Handel TM, Proudfoot AEI. Leukocyte adhesion: Reconceptualizing chemokine presentation by glycosaminoglycans. Trends Immunol (2019) 40(6):472–81. doi: 10.1016/j.it.2019.03.009
37. Rot A, von Andrian UH. Chemokines in innate and adaptive host defense: basic chemokinese grammar for immune cells. Annu Rev Immunol (2004) 22:891–928. doi: 10.1146/annurev.immunol.22.012703.104543
38. Wysocki CA, Panoskaltsis-Mortari A, Blazar BR, Serody JS. Leukocyte migration and graft-versus-host disease. Blood (2005) 105(11):4191–9. doi: 10.1182/blood-2004-12-4726
39. Cooke KR, Coghill JM, Serody JS. Immune biology of allogeneic hematopoietic stem cell transplantation. Chapter 18: Chemokines and graft-Versus-Host disease. 2nd ed: 323–47. (2019) Cambridge, MA: Academic Press.
40. Heidegger S, van den Brink MR, Haas T, Poeck H. The role of pattern-recognition receptors in graft-versus-host disease and graft-versus-leukemia after allogeneic stem cell transplantation. Front Immunol (2014) 5:337. doi: 10.3389/fimmu.2014.00337
41. Kornblit B, Muller K. Sensing danger: toll-like receptors and outcome in allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant (2017) 52(4):499–505. doi: 10.1038/bmt.2016.263
42. Toubai T, Mathewson ND, Magenau J, Reddy P. Danger signals and graft-versus-host disease: Current understanding and future perspectives. Front Immunol (2016) 7:539. doi: 10.3389/fimmu.2016.00539
43. Khakpour S, Wilhelmsen K, Hellman J. Vascular endothelial cell toll-like receptor pathways in sepsis. Innate Immun (2015) 21(8):827–46. doi: 10.1177/1753425915606525
44. Bhagwani A, Thompson AAR, Farkas L. When innate immunity meets angiogenesis-the role of toll-like receptors in endothelial cells and pulmonary hypertension. Front Med (Lausanne) (2020) 7:352. doi: 10.3389/fmed.2020.00352
45. Eyrich M, Burger G, Marquardt K, Budach W, Schilbach K, Niethammer D, et al. Sequential expression of adhesion and costimulatory molecules in graft-versus-host disease target organs after murine bone marrow transplantation across minor histocompatibility antigen barriers. Biol Blood Marrow Transplant (2005) 11(5):371–82. doi: 10.1016/j.bbmt.2005.02.002
46. Tanaka T, Ohtsuka Y, Yagita H, Shiratori Y, Omata M, Okumura K. Involvement of alpha 1 and alpha 4 integrins in gut mucosal injury of graft-versus-host disease. Int Immunol (1995) 7(8):1183–9. doi: 10.1093/intimm/7.8.1183
47. Liang Y, Liu C, Djeu JY, Zhong B, Peters T, Scharffetter-Kochanek K, et al. Beta2 integrins separate graft-versus-host disease and graft-versus-leukemia effects. Blood (2008) 111(2):954–62. doi: 10.1182/blood-2007-05-089573
48. Ramgolam VS, Markovic-Plese S. Regulation of suppressors of cytokine signaling as a therapeutic approach in autoimmune diseases, with an emphasis on multiple sclerosis. J Signal Transduction (2011) 2011:635721. doi: 10.1155/2011/635721
49. Hill GR, Koyama M. Cytokines and costimulation in acute graft-versus-host disease. Blood (2020) 136(4):418–28. doi: 10.1182/blood.2019000952
50. Kim S, Reddy P. Targeting signal 3 extracellularly and intracellularly in graft-Versus-Host disease. Front Immunol (2020) 11:722. doi: 10.3389/fimmu.2020.00722
51. Millan J, Hewlett L, Glyn M, Toomre D, Clark P, Ridley AJ. Lymphocyte transcellular migration occurs through recruitment of endothelial ICAM-1 to caveola- and f-actin-rich domains. Nat Cell Biol (2006) 8(2):113–23. doi: 10.1038/ncb1356
52. Mir E, Palomo M, Rovira M, Pereira A, Escolar G, Penack O, et al. Endothelial damage is aggravated in acute GvHD and could predict its development. Bone Marrow Transplant (2017) 52(9):1317–25. doi: 10.1038/bmt.2017.121
53. Fiedler U, Reiss Y, Scharpfenecker M, Grunow V, Koidl S, Thurston G, et al. Angiopoietin-2 sensitizes endothelial cells to TNF-alpha and has a crucial role in the induction of inflammation. Nat Med (2006) 12(2):235–9. doi: 10.1038/nm1351
54. Hakanpaa L, Sipila T, Leppanen VM, Gautam P, Nurmi H, Jacquemet G, et al. Endothelial destabilization by angiopoietin-2 via integrin beta1 activation. Nat Commun (2015) 6:5962. doi: 10.1038/ncomms6962
55. Naserian S, Abdelgawad ME, Afshar Bakshloo M, Ha G, Arouche N, Cohen JL, et al. The TNF/TNFR2 signaling pathway is a key regulatory factor in endothelial progenitor cell immunosuppressive effect. Cell Commun Signal (2020) 18(1):94. doi: 10.1186/s12964-020-00564-3
56. Mysore V, Tahir S, Furuhashi K, Arora J, Rosetti F, Cullere X, et al. Monocytes transition to macrophages within the inflamed vasculature via monocyte CCR2 and endothelial TNFR2. J Exp Med (2022) 219(5):1–25. doi: 10.1084/jem.20210562
57. Filippi MD. Mechanism of diapedesis: Importance of the transcellular route. Adv Immunol (2016) 129:25–53. doi: 10.1016/bs.ai.2015.09.001
58. Kreisel D, Krupnick AS, Gelman AE, Engels FH, Popma SH, Krasinskas AM, et al. Non-hematopoietic allograft cells directly activate CD8+ T cells and trigger acute rejection: an alternative mechanism of allorecognition. Nat Med (2002) 8(3):233–9. doi: 10.1038/nm0302-233
59. Limmer A, Ohl J, Kurts C, Ljunggren HG, Reiss Y, Groettrup M, et al. Efficient presentation of exogenous antigen by liver endothelial cells to CD8+ T cells results in antigen-specific T-cell tolerance. Nat Med (2000) 6(12):1348–54. doi: 10.1038/82161
60. Manes TD, Pober JS. Antigen presentation by human microvascular endothelial cells triggers ICAM-1-dependent transendothelial protrusion by, and fractalkine-dependent transendothelial migration of, effector memory CD4+ T cells. J Immunol (2008) 180(12):8386–92. doi: 10.4049/jimmunol.180.12.8386
61. Apostolova P, Zeiser R. The role of danger signals and ectonucleotidases in acute graft-versus-host disease. Hum Immunol (2016) 77(11):1037–47. doi: 10.1016/j.humimm.2016.02.005
62. Toubai T, Hou G, Mathewson N, Liu C, Wang Y, Oravecz-Wilson K, et al. Siglec-G-CD24 axis controls the severity of graft-versus-host disease in mice. Blood (2014) 123(22):3512–23. doi: 10.1182/blood-2013-12-545335
63. Biedermann BC, Sahner S, Gregor M, Tsakiris DA, Jeanneret C, Pober JS, et al. Endothelial injury mediated by cytotoxic T lymphocytes and loss of microvessels in chronic graft versus host disease. Lancet (2002) 359(9323):2078–83. doi: 10.1016/S0140-6736(02)08907-9
64. Tichelli A, Gratwohl A. Vascular endothelium as 'novel' target of graft-versus-host disease. Best Pract Res Clin Haematol (2008) 21(2):139–48. doi: 10.1016/j.beha.2008.02.002
65. Lelas A, Greinix HT, Wolff D, Eissner G, Pavletic SZ, Pulanic D. Von Willebrand factor, factor VIII, and other acute phase reactants as biomarkers of inflammation and endothelial dysfunction in chronic graft-Versus-Host disease. Front Immunol (2021) 12:676756. doi: 10.3389/fimmu.2021.676756
66. Mannucci PM. Von willebrand factor: a marker of endothelial damage? Arterioscler Thromb Vasc Biol (1998) 18(9):1359–62. doi: 10.1161/01.ATV.18.9.1359
67. Federici AB. The factor VIII/von willebrand factor complex: basic and clinical issues. Haematologica (2003) 88(6):EREP02.
68. Lenting PJ, Casari C, Christophe OD, Denis CV. Von willebrand factor: the old, the new and the unknown. J Thromb Haemost (2012) 10(12):2428–37. doi: 10.1111/jth.12008
69. Dong JF, Moake JL, Nolasco L, Bernardo A, Arceneaux W, Shrimpton CN, et al. ADAMTS-13 rapidly cleaves newly secreted ultralarge von willebrand factor multimers on the endothelial surface under flowing conditions. Blood (2002) 100(12):4033–9. doi: 10.1182/blood-2002-05-1401
70. Palomo M, Diaz-Ricart M, Carbo C, Rovira M, Fernandez-Aviles F, Martine C, et al. Endothelial dysfunction after hematopoietic stem cell transplantation: role of the conditioning regimen and the type of transplantation. Biol Blood Marrow Transplant (2010) 16(7):985–93. doi: 10.1016/j.bbmt.2010.02.008
71. Zeng L, Jia L, Xu S, Yan Z, Ding S, Xu K. Vascular endothelium changes after conditioning in hematopoietic stem cell transplantation: role of cyclophosphamide and busulfan. Transplant Proc (2010) 42(7):2720–4. doi: 10.1016/j.transproceed.2010.04.024
72. Cutler C, Kim HT, Ayanian S, Bradwin G, Revta C, Aldridge J, et al. Prediction of veno-occlusive disease using biomarkers of endothelial injury. Biol Blood Marrow Transplant (2010) 16(8):1180–5. doi: 10.1016/j.bbmt.2010.02.016
73. Cutler C, Logan B, Nakamura R, Johnston L, Choi S, Porter D, et al. Tacrolimus/sirolimus vs tacrolimus/methotrexate as GVHD prophylaxis after matched, related donor allogeneic HCT. Blood (2014) 124(8):1372–7. doi: 10.1182/blood-2014-04-567164
74. Jeanmart H, Malo O, Carrier M, Nickner C, Desjardins N, Perrault LP. Comparative study of cyclosporine and tacrolimus vs newer immunosuppressants mycophenolate mofetil and rapamycin on coronary endothelial function. J Heart Lung Transplant (2002) 21(9):990–8. doi: 10.1016/S1053-2498(02)00429-1
75. Fuke S, Maekawa K, Kawamoto K, Saito H, Sato T, Hioka T, et al. Impaired endothelial vasomotor function after sirolimus-eluting stent implantation. Circ J (2007) 71(2):220–5. doi: 10.1253/circj.71.220
76. Hoskova L, Malek I, Kopkan L, Kautzner J. Pathophysiological mechanisms of calcineurin inhibitor-induced nephrotoxicity and arterial hypertension. Physiol Res (2017) 66(2):167–80. doi: 10.33549/physiolres.933332
77. Luft T, Benner A, Terzer T, Jodele S, Dandoy CE, Storb R, et al. EASIX and mortality after allogeneic stem cell transplantation. Bone Marrow Transplant (2020) 55(3):553–61. doi: 10.1038/s41409-019-0703-1
78. Dai H, Penack O, Radujkovic A, Schult D, Majer-Lauterbach J, Blau IW, et al. Early bilirubinemia after allogeneic stem cell transplantation-an endothelial complication. Bone Marrow Transplant (2021) 56(7):1573–83. doi: 10.1038/s41409-020-01186-6
79. Go SI, Park S, Kang MH, Kim HG, Kang JH, Kim JH, et al. Endothelial activation and stress index (EASIX) as a predictive biomarker in small cell lung cancer. Cancer Biomark (2022) 35(2):217–225. doi: 10.3233/CBM-220032
80. Merz A, Germing U, Kobbe G, Kaivers J, Jauch A, Radujkovic A, et al. EASIX for prediction of survival in lower-risk myelodysplastic syndromes. Blood Cancer J (2019) 9(11):85. doi: 10.1038/s41408-019-0247-z
81. Varma A, Rondon G, Srour SA, Chen J, Ledesma C, Champlin RE, et al. Endothelial activation and stress index (EASIX) at admission predicts fluid overload in recipients of allogeneic stem cell transplantation. Biol Blood Marrow Transplant (2020) 26(5):1013–20. doi: 10.1016/j.bbmt.2020.01.028
82. Shen N, Ffrench P, Guyotat D, Ffrench M, Fiere D, Bryon PA, et al. Expression of adhesion molecules in endothelial cells during allogeneic bone marrow transplantation. Eur J Haematol (1994) 52(5):296–301. doi: 10.1111/j.1600-0609.1994.tb00099.x
83. Salat C, Holler E, Kolb HJ, Pihusch R, Reinhardt B, Hiller E. Endothelial cell markers in bone marrow transplant recipients with and without acute graft-versus-host disease. Bone Marrow Transplant (1997) 19(9):909–14. doi: 10.1038/sj.bmt.1700767
84. Matsuda Y, Hara J, Osugi Y, Tokimasa S, Fujisaki H, Takai K, et al. Serum levels of soluble adhesion molecules in stem cell transplantation-related complications. Bone Marrow Transplant (2001) 27(9):977–82. doi: 10.1038/sj.bmt.1703026
85. Nie DM, Wu QL, Zhu XX, Zhang R, Zheng P, Fang J, et al. Angiogenic factors are associated with development of acute graft-versus-host disease after allogeneic hematopoietic stem cell transplantation. J Huazhong Univ Sci Technolog Med Sci (2015) 35(5):694–9. doi: 10.1007/s11596-015-1492-4
86. Tatekawa S, Kohno A, Ozeki K, Watamoto K, Ueda N, Yamaguchi Y, et al. A novel diagnostic and prognostic biomarker panel for endothelial cell damage-related complications in allogeneic transplantation. Biol Blood Marrow Transplant (2016) 22(9):1573–81. doi: 10.1016/j.bbmt.2016.05.018
87. Ueda N, Chihara D, Kohno A, Tatekawa S, Ozeki K, Watamoto K, et al. Predictive value of circulating angiopoietin-2 for endothelial damage-related complications in allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant (2014) 20(9):1335–40. doi: 10.1016/j.bbmt.2014.04.030
88. Newell LF, DeFor TE, Cutler C, Verneris MR, Blazar BR, Miller JS, et al. Follistatin and soluble endoglin predict 1-year nonrelapse mortality after allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant (2020) 26(3):606–11. doi: 10.1016/j.bbmt.2019.11.006
89. Nomura S, Ishii K, Fujita S, Nakaya A, Satake A, Ito T. Associations between acute GVHD-related biomarkers and endothelial cell activation after allogeneic hematopoietic stem cell transplantation. Transpl Immunol (2017) 43-44:27–32. doi: 10.1016/j.trim.2017.06.004
90. Richardson PG, Palomo M, Kernan NA, Hildebrandt GC, Chao N, Carreras E. The importance of endothelial protection: the emerging role of defibrotide in reversing endothelial injury and its sequelae. Bone Marrow Transplant (2021) 56(12):2889–96. doi: 10.1038/s41409-021-01383-x
91. Martinez-Sanchez J, Hamelmann H, Palomo M, Mir E, Moreno-Castano AB, Torramade S, et al. Acute graft-vs.-Host disease-associated endothelial activation in vitro is prevented by defibrotide. Front Immunol (2019) 10:2339. doi: 10.3389/fimmu.2019.02339
92. Dorrance AM, Moutuou MM, Goda C, Sell NE, Kalyan S, Karunasiri M, et al. Modulating endothelial cells with EGFL7 to diminish aGVHD after allogeneic bone marrow transplantation in mice. Blood Adv (2022) 6(7):2403–8. doi: 10.1182/bloodadvances.2021005498
93. Toubai T, Magenau J. Immunopathology and biology-based treatment of steroid-refractory graft-versus-host disease. Blood (2020) 136(4):429–40. doi: 10.1182/blood.2019000953
94. Dietrich S, Falk CS, Benner A, Karamustafa S, Hahn E, Andrulis M, et al. Endothelial vulnerability and endothelial damage are associated with risk of graft-versus-host disease and response to steroid treatment. Biol Blood Marrow Transplant (2013) 19(1):22–7. doi: 10.1016/j.bbmt.2012.09.018
95. Luft T, Dietrich S, Falk C, Conzelmann M, Hess M, Benner A, et al. Steroid-refractory GVHD: T-cell attack within a vulnerable endothelial system. Blood (2011) 118(6):1685–92. doi: 10.1182/blood-2011-02-334821
96. Vander Lugt MT, Braun TM, Hanash S, Ritz J, Ho VT, Antin JH, et al. ST2 as a marker for risk of therapy-resistant graft-versus-host disease and death. N Engl J Med (2013) 369(6):529–39. doi: 10.1056/NEJMoa1213299
97. Rotz SJ, Dandoy CE, Davies SM. ST2 and endothelial injury as a link between GVHD and microangiopathy. N Engl J Med (2017) 376(12):1189–90. doi: 10.1056/NEJMc1700185
98. Lockett AD, Kimani S, Ddungu G, Wrenger S, Tuder RM, Janciauskiene SM, et al. alpha(1)-antitrypsin modulates lung endothelial cell inflammatory responses to TNF-alpha. Am J Respir Cell Mol Biol (2013) 49(1):143–50. doi: 10.1165/rcmb.2012-0515OC
99. Tawara I, Sun Y, Lewis EC, Toubai T, Evers R, Nieves E, et al. Alpha-1-antitrypsin monotherapy reduces graft-versus-host disease after experimental allogeneic bone marrow transplantation. Proc Natl Acad Sci USA (2012) 109(2):564–9. doi: 10.1073/pnas.1117665109
100. Giannoni L, Morin F, Robin M, Peyneau M, Schlageter MH, Desmier D, et al. Human-derived alpha1-antitrypsin is still efficacious in heavily pretreated patients with steroid-resistant gastrointestinal graft-versus-Host disease. Biol Blood Marrow Transplant (2020) 26(9):1620–6. doi: 10.1016/j.bbmt.2020.05.014
101. Magenau JM, Goldstein SC, Peltier D, Soiffer RJ, Braun T, Pawarode A, et al. alpha1-antitrypsin infusion for treatment of steroid-resistant acute graft-versus-host disease. Blood (2018) 131(12):1372–9. doi: 10.1182/blood-2017-11-815746
102. Marcondes AM, Hockenbery D, Lesnikova M, Dinarello CA, Woolfrey A, Gernsheimer T, et al. Response of steroid-refractory acute GVHD to alpha1-antitrypsin. Biol Blood Marrow Transplant (2016) 22(9):1596–601. doi: 10.1016/j.bbmt.2016.05.011
Keywords: endothelial cell, endotheial dysfunction, GvHD, inflammation, integrins, selectins
Citation: Neidemire-Colley L, Robert J, Ackaoui A, Dorrance AM, Guimond M and Ranganathan P (2022) Role of endothelial cells in graft-versus-host disease. Front. Immunol. 13:1033490. doi: 10.3389/fimmu.2022.1033490
Received: 31 August 2022; Accepted: 07 November 2022;
Published: 23 November 2022.
Edited by:
Tomomi Toubai, Yamagata University, JapanReviewed by:
Federico Simonetta, Hôpitaux universitaires de Genève (HUG), SwitzerlandCopyright © 2022 Neidemire-Colley, Robert, Ackaoui, Dorrance, Guimond and Ranganathan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Parvathi Ranganathan, cGFydmF0aGkucmFuZ2FuYXRoYW5Ab3N1bWMuZWR1
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.