 Amelia Kulle
Amelia Kulle Ajitha Thanabalasuriar
Ajitha Thanabalasuriar Taylor S. Cohen
Taylor S. Cohen Marta Szydlowska
Marta Szydlowska- 1Department of Pharmacology and Therapeutics, McGill University, Montreal, QC, Canada
- 2Late Stage Development, Vaccines and Immune Therapies (V&I), BioPharmaceuticals R&D, AstraZeneca, Gaithersburg, MD, United States
- 3Bacteriology and Vaccine Discovery, Research and Early Development, Vaccines and Immune Therapies (V&I), BioPharmaceuticals R&D, AstraZeneca, Gaithersburg, MD, United States
Resident macrophages play a unique role in the maintenance of tissue function. As phagocytes, they are an essential first line defenders against pathogens and much of the initial characterization of these cells was focused on their interaction with viral and bacterial pathogens. However, these cells are increasingly recognized as contributing to more than just host defense. Through cytokine production, receptor engagement and gap junction communication resident macrophages tune tissue inflammatory tone, influence adaptive immune cell phenotype and regulate tissue structure and function. This review highlights resident macrophages in the liver and lung as they hold unique roles in the maintenance of the interface between the circulatory system and the external environment. As such, we detail the developmental origin of these cells, their contribution to host defense and the array of tools these cells use to regulate tissue homeostasis.
Introduction
Tissue resident macrophages are long-lived innate immune cells that persist within various tissues in the body. These cells play many roles in health and disease and are unique when compared to more transient innate immune cells not just in function but also genetic profile. Herein we explore the origins and functions of tissue-resident macrophages, with a focus on those populations found in the lung and liver. Moreover, we define the role tissue resident macrophages play in maintaining tissue homeostasis and the ability of these cells to drive disease when improperly regulated.
Macrophages are a type of innate immune cells known for their ability to phagocytose debris and pathogens and present antigens to direct adaptive immunity. The discovery of macrophages is credited to Ilya Metchnikoff, a French-Russian zoologist who identified the cell population while studying phagocytosis (1). Shortly thereafter, macrophages were designated as tissue phagocytes within the reticuloendothelial system which proposed that endothelial cells and reticulocytes, otherwise known as phagocytes, shared a common origin (2). However, with the identification of morphological and functional differences between phagocytes and endothelial cells, this theory did not accurately depict cell lineage. Until recently, the prevailing belief was that all macrophages are terminally differentiated cells derived from circulating macrophages and monocytes (3). This belief was first proposed by Ralph van Furth in the 1960s and led to the establishment of the mononuclear phagocyte system (4). Although contradictory evidence has been published for many years, recent technological advances have allowed the definitive rejection of the mononuclear phagocyte system and the recognition of tissue-resident macrophages as unique populations independent of monocyte differentiation (5–7).
Herein we review the current understanding of resident macrophage populations in two major organs: the lung which is constantly exposed to the external environment and the liver which acts to filter the blood as it circulates through the body. In addition we cover the origin and maintenance of these cell populations, their role in maintaining tissue homeostasis and defense against invading pathogens. Finally, we discuss the implications of resident macrophage dysfunction in various diseases.
Origins
Evidence of long-term persistence and proliferation in tissues led researchers to investigate the origins of tissue-resident macrophages. Several studies provided this evidence through the analysis of macrophages in organ transplant patients which demonstrated long-term maintenance of the donor macrophage population years after surgery (8–10). From there, evidence of macrophage populations in the yolk sac before the establishment of monocyte precursors suggested an embryonic origin for some macrophages (11). Indeed, further studies elucidated that tissue-resident macrophage populations in most organs of both mice and humans are established during embryogenesis and maintained through self-renewal (12). Classically, mature fully differentiated cells lose the capacity to proliferate as differentiation coincides with cell cycle withdrawal. Therefore, tissue-resident macrophages are unique in their ability to re-enter the cell cycle as fully differentiated cells (13). It must be noted that most findings on tissue-resident macrophage origins were established in mice as the study of tissue-resident macrophage ontogeny in human organs is limited (14).
Most mammalian organs have populations of tissue-resident macrophages. Some of the most well-known include Langerhans cells in the skin, alveolar macrophages in the lung, Kupffer cells in the liver, microglia in the brain, and red pulp macrophages in the spleen. These cells take on a number of different functions, that at first were defined by M1 or M2 polarization (15, 16). M1-macrophages are defined by a pro-inflammatory polarization as they secrete higher levels of pro-inflammatory factors and expression of the surface marker CD86. The second population, M2-macrophages, were thought to be immune-suppressive, support tissue repair, and characterized by the surface marker CD206 (17). More recently, it became clear that this paradigm does not mirror the high level of plasticity in the polarization of these cells, that often show a spectrum of features typical of M1 or M2 cells. For example, four different subtypes of M2 polarization were recognized: M2a-M2d, together with M4, Mox and M(Hb) polarization types based on the activating factors and function of each of mentioned populations (18). Resident cells respond to several different stimuli by modifying both function and surface marker expression, and as such are referred to as plastic.
Based on mouse studies, tissue resident macrophages, are generated in three waves. The first wave referred to as the primitive wave begins on embryonic day 6.5 (E6.5) in the yolk sac (19). This wave establishes a population of macrophage-exclusive progenitors which then differentiate into erythroid and myeloid progenitors (EMPs) between E8.5 and E10.5 during the pro-definitive wave (20). An intermediate pre-macrophage population (p-Macs) are derived from EMPs without first becoming monocytes (21). The population of EMPs and p-Macs expand for several days in the yolk sac and migrate to the fetal liver by E14.5. Between E12.5 and E17.5, p-Macs in the fetal liver migrate and seed other tissues to establish life-long populations of tissue-resident macrophages. The third wave of hematopoiesis, the definitive wave, begins around E17.5 and involves the establishment of hematopoietic stem cells in the bone marrow. Although all tissue-resident macrophages originate from fetal liver p-Macs, bone marrow derived macrophages are found to replace the fetal population in the intestines, spleen, skin, and heart (22). However, it must be noted that these hematopoietic stem cell derived macrophage populations are maintained through self-renewal with minimal contribution from circulating monocytes following their colonization of the tissue (23).
Once seeded, tissue-resident macrophage mobility is restricted to their colonized tissue. Intriguingly, during the process of seeding embryonic tissues they are highly mobile. Therefore, researchers have wondered whether early macrophages are committed to a specific tissue before leaving the fetal liver, or if all tissues are seeded blindly and local cues guide tissue-specification? As mentioned, p-Macs begin to colonize embryonic tissues around E12.5. The migration of p-Macs is dependent on the expression of the chemokine receptor CX3CR1, which is not expressed by EMPs (24). In a study by Mass et al., single cell sequencing found that p-Macs lack tissue-specific signatures, thus suggesting that tissue-resident macrophages gain their specialized phenotype after colonization (21). Macrophage development is known to be regulated by the transcription factor PU.1 and is involved in tissue-resident macrophage specification by acting as a scaffold for histone modifiers that can induce chromatin remodeling (25–27). Once p-Macs seed a tissue, tissue-specific signals initiate the enrichment of transcriptional regulators to generate tissue-resident macrophages with specialized functions (Figure 1) (28). For example, the tissue-specific signals required for Langerhans cell development include IL-34 and TGF-β which induce the expression of the transcription factors RUNX3 and ID2 (29). In the lung, alveolar macrophages develop in response to granulocyte-macrophage colony-stimulating factor (GM-CSF) causing the upregulation of the transcription factor PPARγ. In addition, the transcription factors BACH2 and CEBPβ have been implicated in alveolar macrophage development (24, 30). Kupffer cell development in the liver has been found to be dependent on the transcription factor ID3 which is regulated by TGF-β (21). In the central nervous system, SALL1 is a microglia-specific transcription factor involved in their development (31). Finally, splenic red pulp macrophage development is induced by the presence of heme causing an upregulation in the transcription factor SPIC.
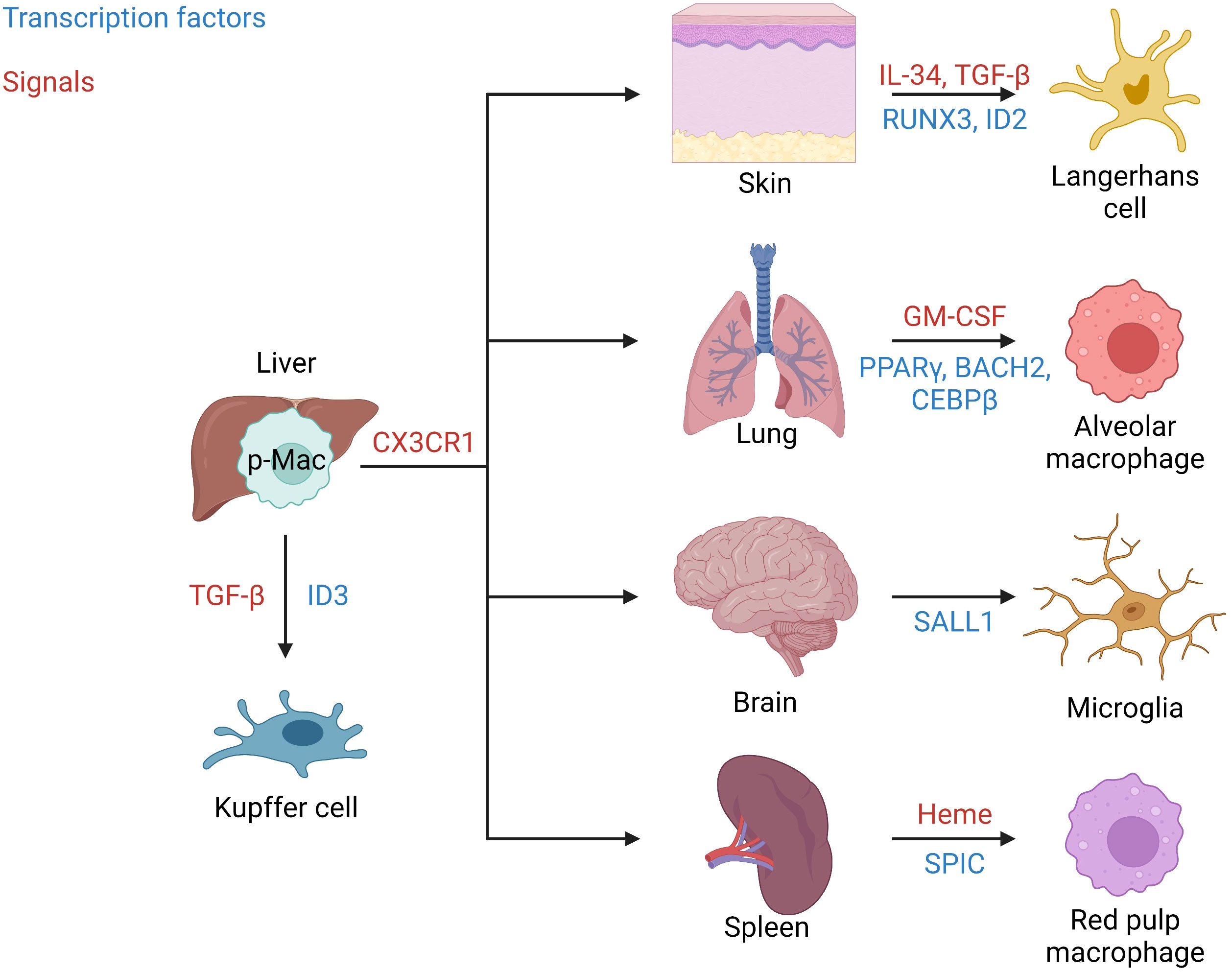
Figure 1 Tissue seeding. Signals and transcription factors involved in seeding of well-known tissue-resident macrophages from fetal liver p-Macs. Migration from the liver occurs in a CX3CR1 dependent manner. In the skin IL-34 and TGF-β induce the transcription factors RUNX3 and ID2 to generate Langerhans cells. In the lung GM-CSF induces the transcription factors PPARγ, BACH2, and CEBPβ to produce alveolar macrophages. In the brain/central nervous system, microglia are generated in response to the induction of the transcription factor SALL1. Red pulp macrophages in the spleen are produced in response to Heme and the transcription factor SPIC. Finally, in the liver Kupffer cells are generated in response to TGF-β and the transcription factor ID3.
The remainder of this review will highlight the roles of tissue-resident macrophages in the liver and lung at steady-state and in disease. However, a comprehensive list of all tissue-resident macrophages, their origin, and functions is detailed in Table 1. The importance of the liver in our understanding of tissue-resident macrophages begins with its role as the reservoir of p-Macs to seed other tissues. In addition, during homeostasis it also maintains its own population of tissue-resident macrophages. In addition to the liver, the lung and brain are the only other tissues in which the population of tissue-resident macrophages is not replaced by hematopoietic stem cell derived macrophages following the initial colonization by fetal liver p-Macs. As such, these tissues have unique embryonic macrophage populations unlike the rest of the body. However, we will not be discussing tissue-resident macrophages in the brain as the blood-brain barrier isolates these macrophages from the external environment. In contrast the liver and lungs act as an interface between our circulatory systems and the external environment. Therefore, the tissue-resident macrophages in these organs play an essential role in host defense at steady-state as pathogens that enter circulation can cause systemic complications.
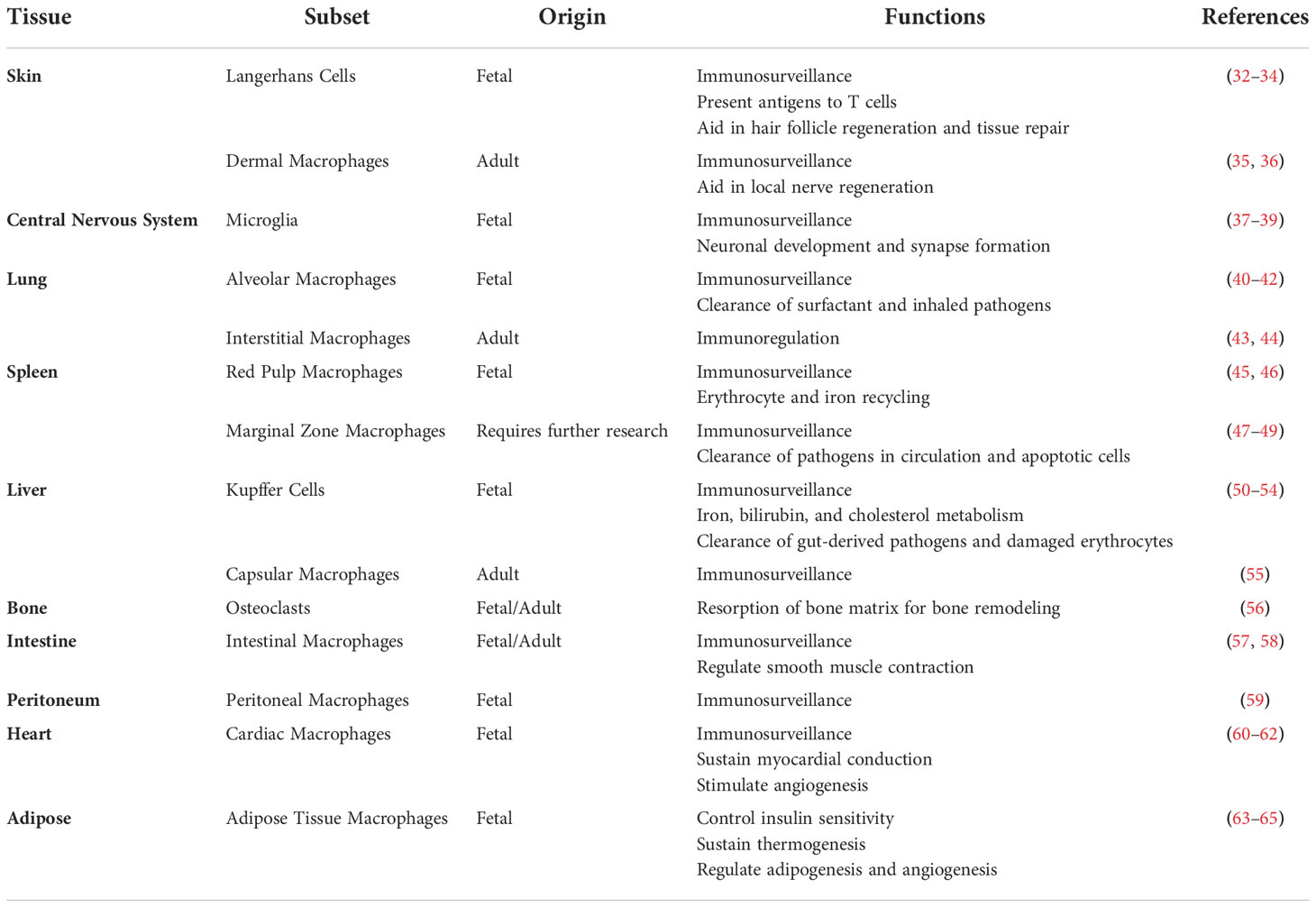
Table 1 Summary of tissue-resident macrophage subsets, origins, and functions.
Tissue-resident macrophages in the liver
The fetal liver acts as a reservoir of tissue-resident macrophages, which then seed the organs during development, including the liver. It was long suspected that the population of macrophages in the liver consists of different subsets. So far, mostly thanks to preclinical studies performed on mice, researchers were able to characterize a few subpopulations based on origin and a high degree of phenotypic and functional specificity (Figure 2) (Table 2). In addition to the heterogeneity of the population, these cells are highly plastic and adapt to the dynamically changing liver microenvironment (66, 73–75). Classically, we would refer to two main subpopulations of macrophages in the liver, Kupffer Cells (KCs) and monocyte-derived macrophages (MoMFs). Kupffer cells (KCs) represent the major fraction of phagocytic cells in the liver at steady state (76, 77). The half-life of KCs in mice is estimated to be 12.4 days, while in humans transplanted donor-derived macrophages can be detected for as long as 1 year after surgery (78–80). Under physiological conditions, this population is replenished by self-renewal and does not depend on bone marrow-derived progenitors. The second subset, MoMFs, are not established embryonically, but serve to repopulate KCs during liver injury and/or chronic inflammation, where increased death of KCs, in most of cases apoptotic, can be observed (81–83). These newly recruited cells differentiate and acquire some of the phenotypic and functional features of KCs to replenish their population (67, 84). An additional population of hepatic macrophages reside in the capsule surrounding the whole organ referred to as liver capsular macrophages (LCMs), in mice expressing F4/80, CD11c and CX3XR1 and particularly enriched in CD207 in humans. This subset originates from adult circulating monocytes and are phenotypically distinct from other two populations, although further studies are needed to fully understand their role in liver homeostasis (55, 76).
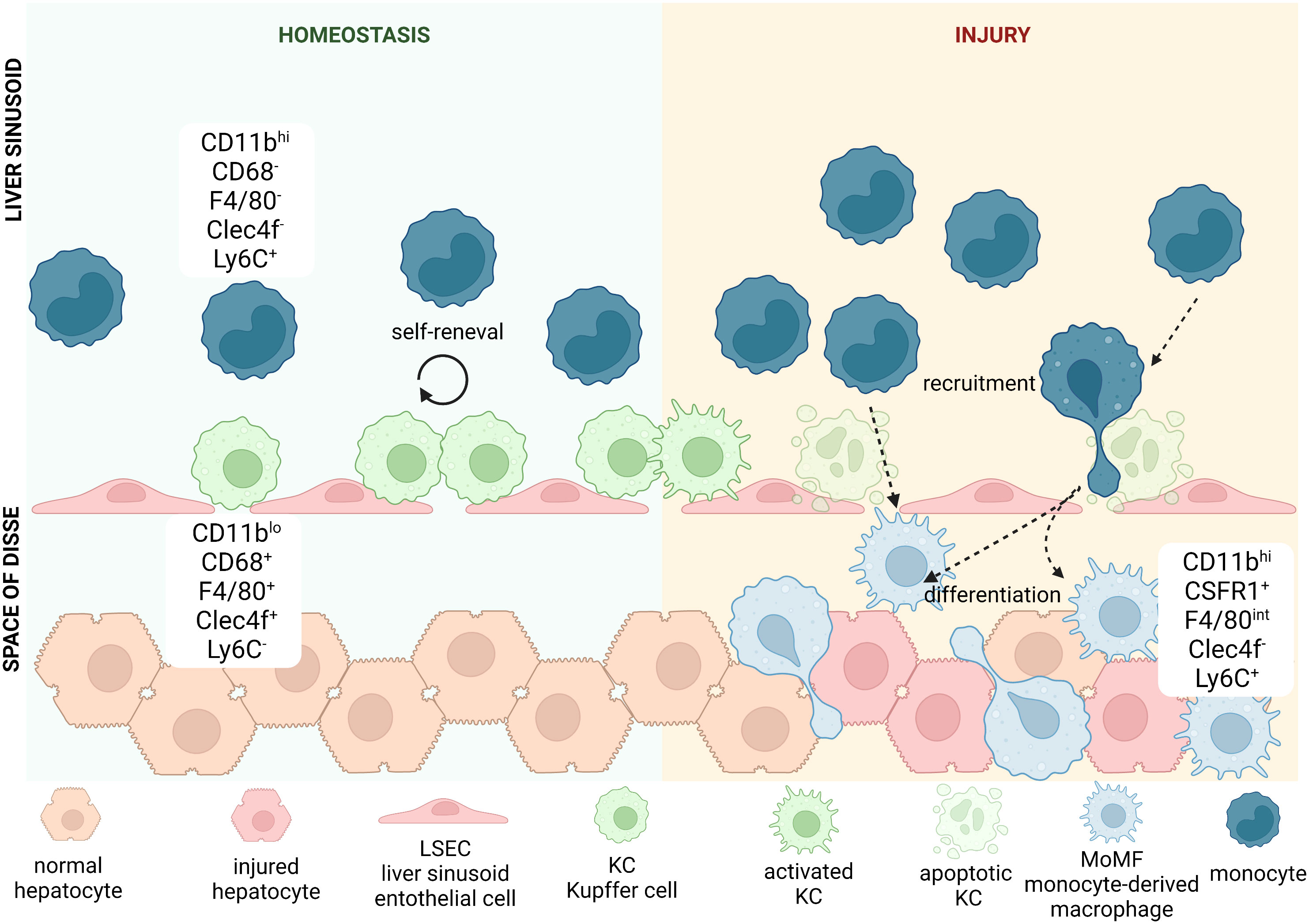
Figure 2 Heterogeneity of hepatic macrophages. In mice, liver-resident macrophages known as Kupffer cells (KCs) are classically defined by positive expression of CD11b, CD68, F4/80, Clec4f and negativity for Ly6C. They are located in liver sinusoids, where they adhere to liver sinusoid endothelial cells (LSECs). KCs thanks to their particular location remain in close contact with blood stream, which allows them to detect a variety of antigens. During homeostasis the pool of KCs is being replenished by cell renewal. During acute or chronic liver injury KCs get activated and secrete cytokines and chemokines that can recruit other immune cells from the circulating blood. Some of the secreted cytokines or chemokines thanks to fenestrae present between LSECs can reach liver parenchyma and directly affect hepatocytes and other immune cells located there. Haptic injury and/or chronic inflammation increases apoptotic rate of KCs and when self-renewal does not suffice to maintain their population, we can observe increased recruitment of monocytes, characterized by positivity for CD11b and Ly6C, with concomitant negativity to CD68, F4/80 and Clec4f. Once they enter parenchyma through endothelial fenestration, they differentiate into KC-like cells called monocyte-derived macrophages (MoMFs), that resemble the phenotype and function of KCs. These highly pro-inflammatory cells can be recognized by positivity for some markers typical for both KCs and monocytes: CD11b, F4/80 and Ly6C, while they remain negative for Clec4f. They also repopulate hepatic macrophages niche after increased death of KCs due to injury.
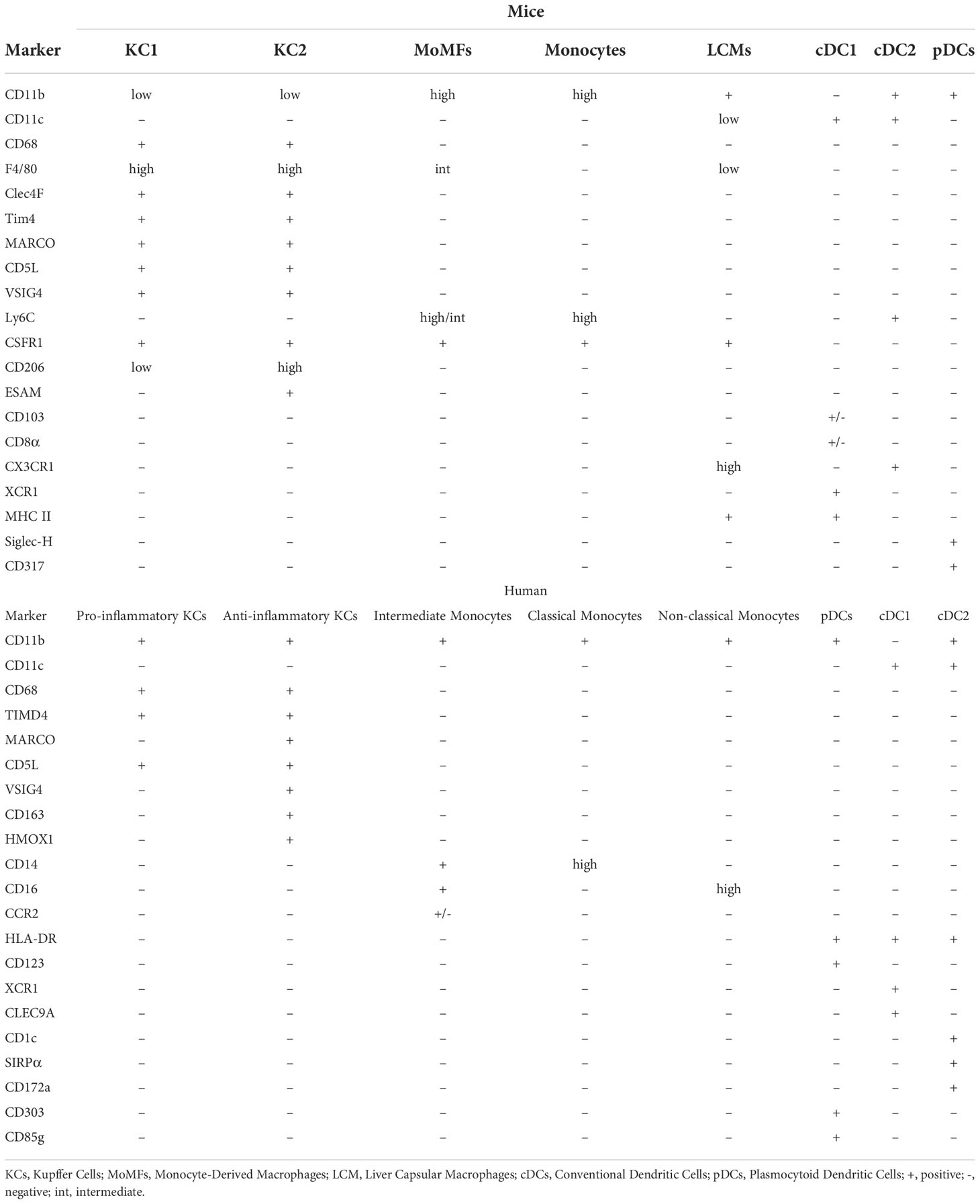
Liver macrophage populations can be distinguished from each other, and from infiltrating monocytes, by the differential expression of protein surface markers. Murine macrophages express CD11b and macrophage marker CD68, while a combination of other markers e.g., F4/80 and Clec4F (KC markers) or Ly6C (monocyte marker) help identify subpopulations (68). KCs can be identified by high expression of F4/80 and Clec4F, negative or low expression of Ly6C (CD11bloCD68+F4/80hiClec4F+Ly6C) and expression of scavenger receptors TLR4 and TLR9 (85). MoMFs are positive for CSF1R and Ly6C and express increasing levels of F4/80 as they differentiate into KC-like cells (CD11bhiCSF1R+ F4/80intLy6Chi/lo). In parallel, levels of Ly6C decrease as they become KC-like (67, 69). LCMs residing in hepatic capsule share some of the surface antigens with both KCs (F4/80) and MoMFs (CSF1R), while also expressing the dendritic cell marker CD11c (55). Non-resident monocytes are also found in the liver and can be separated from resident cells based on negative expression of F4/80 or Clec4F and high expression of Ly6C (86).
In addition to these classical populations, recent data obtained using single-cell RNA sequencing (scRNA seq) and further corroborated by flow cytometry, proteomics and mechanistic studies led to a discovery of two distinct populations of KCs in the liver: KC1 (consisting of 85% of KCs) and KC2 (15%). While both share all markers typical for KCs, they can be distinguished by their differential expression of CD206 and ESAM with KC1 cells identified as CD206lo and ESAM- and KC2 as CD206hi and ESAM+. The ratio between both populations in the hepatic tissue remains stable in the steady state, with similar localization in sinusoids and zonation. On the other hand, in-depth transcriptomic analysis revealed particular enrichment of KC2 cells in genes involved in carbohydrate and lipid metabolism, what suggested their role in liver metabolic disorders (87).
The definition and characteristics of different human hepatic macrophage populations are less clear due to the limited availability of appropriate samples, although recently published scRNAseq data shed more light into that topic and discovered multiple novel markers allowing identification of cell subsets (Table 2). The main subsets of monocytes and macrophages are identified by expression of CD68, MARCO, TIMD4, CCR2, CD14 and CD16 (67, 69–71). KCs are defined as CD68+TIMD4+ cells and some, MARCO+KCs, in addition express VSIG4, CD163 and HMOX1 that suggest an immune-tolerogenic or immunosuppressive role. scRNAseq revealed that apart from mentioned markers, these cells show as well enriched expression of e.g. CD5L, VCAM1 or KLF4 (66). Meanwhile CD68+MARCO- macrophages produce IL-18 and have a transcriptomic profile associated with more pro-inflammatory polarization, as they are enriched in LYZ, CSTA or CD74 genes (66, 67, 69, 88). CD14hiCD16- are defined as classical monocytes, CD14+CD16+ as intermediate and CD14-CD16hi as non-classical monocytes, with CD14+CD16+/-CCR2+ cells most likely corresponding to pro-inflammatory murine MoMFs (70). All of mentioned populations and markers for both murine and human liver macrophages, monocytes and dendritic cells are summarized in the Table 2.
Macrophages in the liver during homeostasis and inflammation
Six main functions of hepatic macrophages have been observed. As immune cells they carry immune surveillance while maintaining immune tolerance (I) but also serve as a first line of anti-microbial defense (II) (89–91). Moreover, they perform the clearance of the cellular debris and metabolites (III), maintain the iron homeostasis through phagocytosis of red blood cells (RBCs) (IV) and regulate cholesterol metabolism (V) (50–54, 92, 93). Lastly, through the interaction with other cell types in the liver they are important players in hepatic tissue repair (VI) (94, 95). The liver is exposed to the nutrients, metabolites and bacteria absorbed in the digestive tract (96). Hepatic macrophages serve an essential role in monitoring the gut-liver axis for invading pathogens and toxins (90). KCs are the most abundant immune cell population in the liver, residing in hepatic sinusoids (97). KCs express classic pattern recognition receptors to identify pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), including LPS, bacterial wall parts, DNA and lipoproteins. Moreover, they express CRig receptor that are able to effectively catch bacteria from the blood stream (55, 98–105).
Although KCs play a crucial role in the activation of an inflammatory response, they are equally important players in immunotolerance. They are capable of not only suppressing activity of effector T cells, but they are able to activate regulatory T cells (106). Liver macrophages can recognize, digest, and dispose of apoptotic and necrotic bodies. Immediate reaction from phagocytes and removal of necrotic bodies is particularly important, as cell contents released during necrosis are potent activators of immune response, therefore could sustain inflammation and delay tissue regeneration (107). Apart from removing the cellular debris from the hepatic parenchyma, liver macrophages also play an important role in iron homeostasis through the removal of damaged or aged RBCs and vesicles containing hemoglobin (Hb). Their involvement in this process depends on their expression of scavenger receptors which recognize polyinosinic acid or phosphatidylserine. In addition, KCs take up Hb-containing vesicles which helps to prevent the undesired loss of iron that could lead to its deficiency, or excess release of iron to extracellular matrix that could cause iron-induced toxicity (52, 108). Lastly, KCs play a crucial role in the modulation of cholesterol metabolism. KCs ingest and transfer LDL-derived cholesterol to hepatocytes and maintain HDL and VLDL levels through the surface receptor cholesteryl ester transfer protein (CETP) (109, 110).
Tissue-resident macrophages in the lung
Tissue resident macrophages in the lung make the biggest journey in their life shortly after birth and are seeded from fetal liver macrophages within the first 3 days following birth (111, 112). The average person inhales more than 10,000 liters of air daily (113). As such, the lungs are constantly exposed to foreign particulates and pathogens from the external environment. Tissue-resident macrophages in the lung play a major role in filtering inhaled air and maintaining tissue homeostasis, to protect the host from airborne pathogens. Our lungs have two main populations of tissue-resident macrophages with specialized functions based on their anatomical compartment (Figure 3) (114). Alveolar macrophages (AMs) are found within the alveolus, while interstitial macrophages (IMs) are found in the surrounding tissue (115). Macrophages located within the upper airways have been proposed as a third population but are generally grouped with AMs (116). Our understanding of AMs is far more advanced than IMs as they are more accessible through bronchoalveolar lavage compared to tissue digestion required to isolate IMs. Recent technological advances have shed light on the importance of both subsets in maintaining the integrity of our lungs. We will explore the roles of AMs and IMs during homeostasis, infection, injury, repair, and disease.
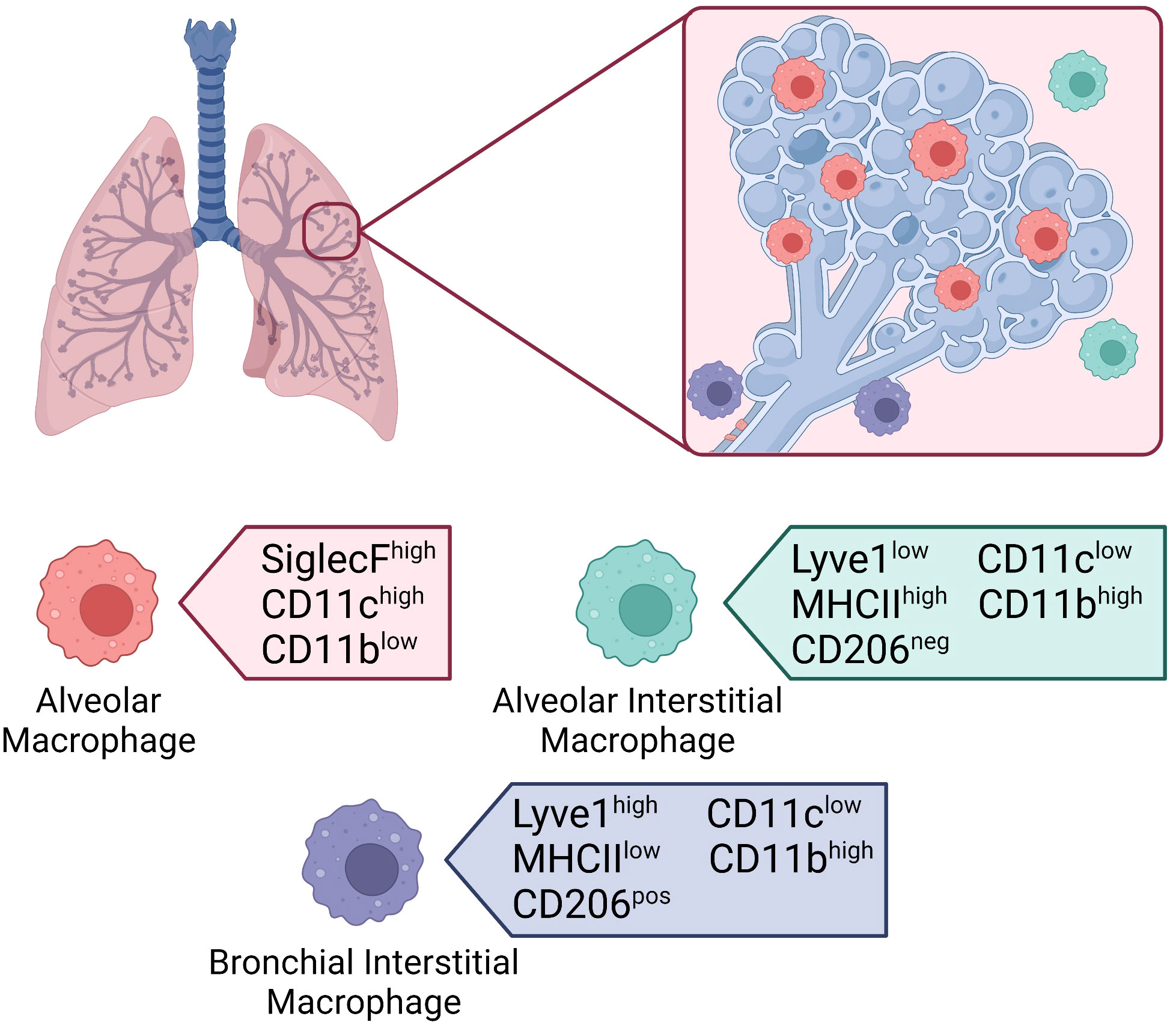
Figure 3 Macrophages in the lung. Location of tissue-resident macrophages in the lung and their distinguishing surface markers at steady-state. Alveolar macrophages located within the alveolar lumen express high levels of SiglecF and CD11c, and low levels of CD11b. Interstitial macrophages located in the alveolar interstitium express low levels of Lyve1, high levels of MHCII, and are CD206 negative. Interstitial macrophages located in the bronchial interstitium express high levels of Lyve1, low levels of MHCII, and are CD206 positive. Both interstitial macrophage subsets express low levels of CD11c and high levels of CD11b.
Lung resident macrophages in homeostasis and inflammation
Alveolar macrophages
AMs are the most abundant tissue-resident macrophages in the respiratory tract (8, 113). Shortly after birth, the AM population is established in response to the production of GM-CSF by alveolar epithelial cells (14). GM-CSF upregulates the transcription factor peroxisome proliferator-activated γ (PPARγ) in embryonic precursors triggering the terminal differentiation into AMs. The establishment of the AM population coincides with alveologenesis – the process by which the alveolar space is created. As such, AMs interact with alveolar epithelial cells to contribute to alveologenesis (115, 117). Maintenance of the population is dependent on GM-CSF from alveolar epithelial cells and transforming growth factor β (TGF-β) which is released by AMs themselves (Figure 4) (116). Although AMs are generally considered to be anti-inflammatory in nature, they are highly adaptable, and their phenotype is dependent on the surrounding microenvironment (115). Under homeostatic conditions, murine AMs can be identified by the high expression of Siglec F and the dendritic cell marker CD11c, and low expression of CD11b (Figure 3) (112). As different subsets of mononuclear phagocytes express similar markers, additional information on their defining markers and the differences between subsets in both mice and humans can be found in Table 3. Until recently, AMs were considered to be sessile like most other tissue-resident macrophages (122). However, in vivo intravital imaging has revealed that populations of AMs are motile and move between alveoli through pores of Kohn (123). The ability of AMs to move through the airways and between alveoli makes them one of the most unique tissue resident macrophages, who are classical static after being seeded.
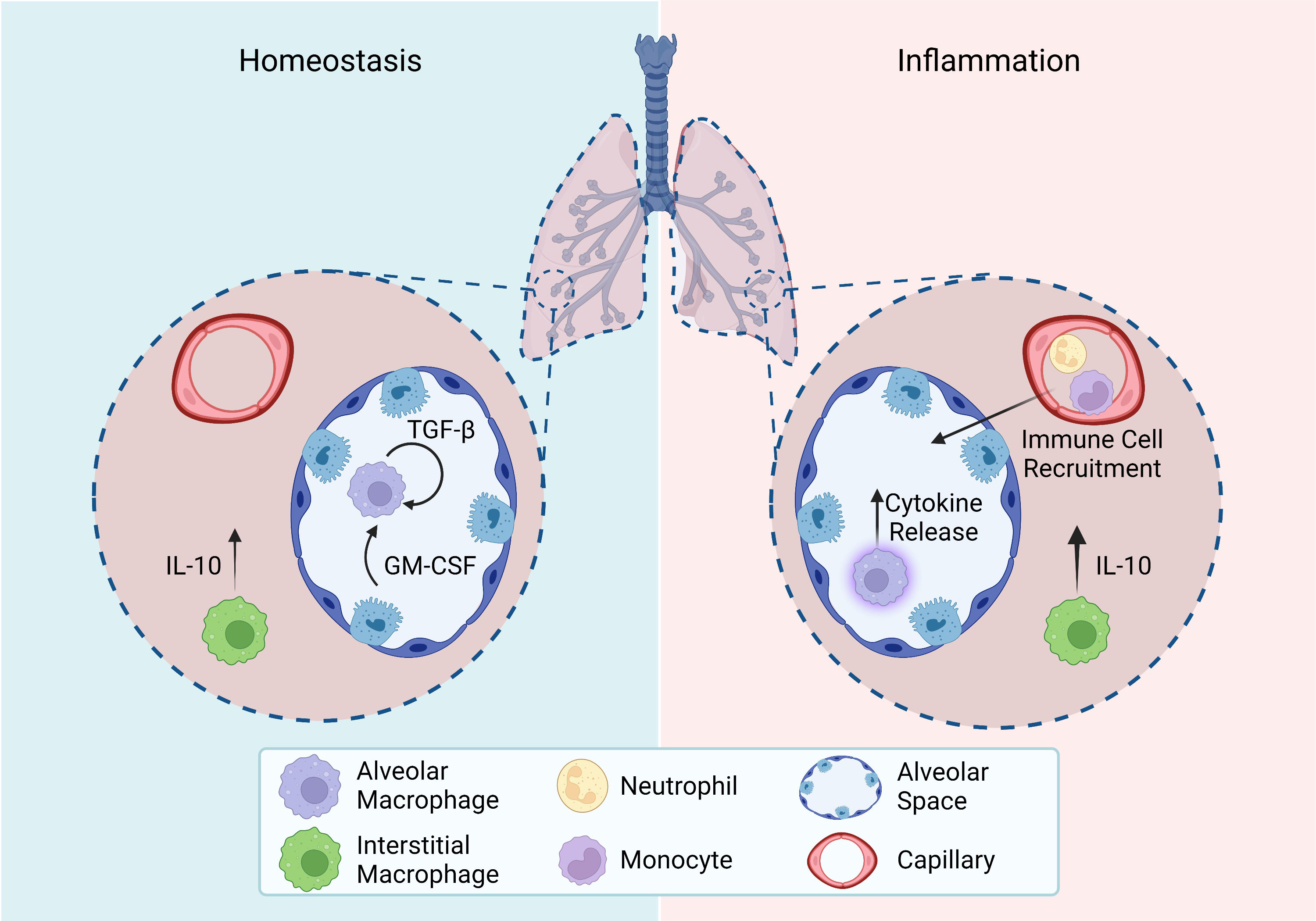
Figure 4 Homeostasis and inflammation in the lung. Roles of tissue-resident macrophage subsets in the lung during homeostasis and inflammation. Homeostasis: Alveolar macrophage maintenance relies on autocrine TGF-β and epithelial cell release of GM-CSF. Interstitial macrophages constitutively release IL-10. Inflammation: Activated alveolar macrophages release cytokines to recruit other immune cells and promote an inflammatory response. Interstitial macrophages upregulate their release of IL-10.All figures were created with BioRender.com.
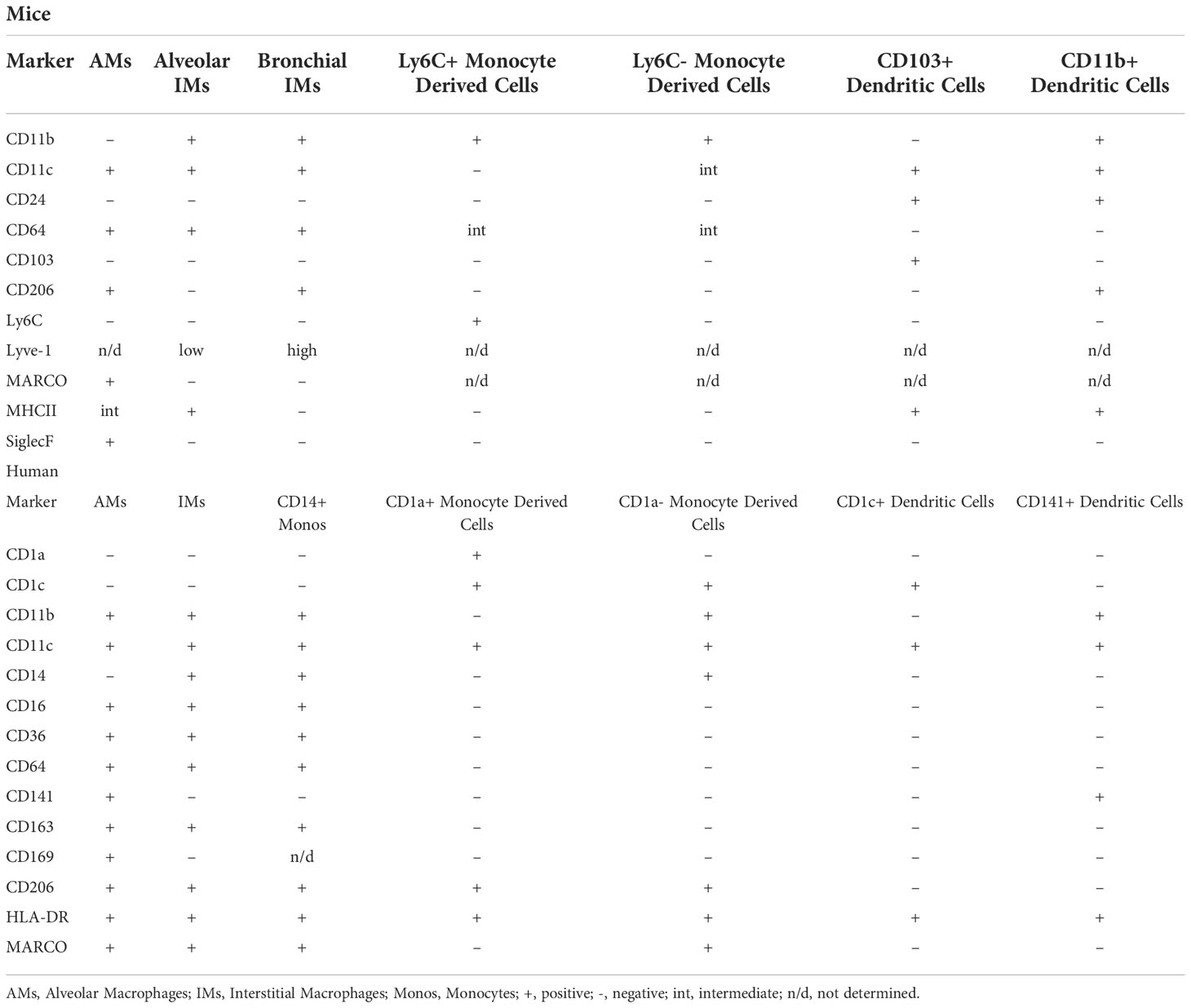
Table 3 Defining markers of pulmonary mononuclear phagocyte subsets in mice and humans at steady state (118–121).
Often referred to as housekeeping cells, AMs patrol the alveolar lumen phagocytosing debris to maintain homeostasis. AMs are also essential for the catabolism of surfactant proteins (113). By cleaning up the airways, AMs prevent unwarranted inflammatory responses to harmless particulates and maintain gas exchange. At steady state, AMs have several immunosuppressive functions. The release of TGF-β by AMs prevent their activation through an autocrine loop and convert naïve or activated T cells into regulatory T cells which are also immunosuppressive. In addition, AMs can release vesicles that suppress cytokine secretion from alveolar epithelial cells. Overall, AMs maintain tissue homeostasis by removing debris and preventing excess inflammation.
As mentioned, AMs are located within the airways making them the first immune cell to encounter inhaled pathogens. This localization enables AMs to control the intensity of the inflammatory response. Under states of infection or injury, AMs detect PAMPs and DAMPs through pattern recognition receptors including toll-like receptors (113). These signals are integrated with other stimuli to determine the strength of the resulting response. AMs then release either pro-inflammatory mediators such as IL-1β, IL-6, and tumor necrosis factor alpha (TNF-α), or anti-inflammatory mediators including IL-10 and TGF-β (Figure 4). Both pro- and anti-inflammatory AMs can exist at the same time and can be differentiated based on the expression of CXCL2 (124). Additionally, pro-inflammatory AMs are essential for recruiting other immune cells (Figure 4) (115). In the case of severe infection, injury, or inflammation tissue-resident AMs are depleted and can be supplemented by circulating monocytes. These recruited monocytes can gradually transition to resemble and act like AMs while the tissue-resident population is replenished by self-renewal. Although AMs play a role in the initiation of inflammation, they also contribute to inflammation resolution. The phagocytic properties of AMs are essential in clearing apoptotic cells to reduce tissue injury. Additionally, AMs release several repair mediators including resolvin, protectin, and amphiregulin which enhance healing.
Interstitial macrophages
Previously, IMs were thought to be an intermediate subtype between recruited and tissue-resident AMs. However, recent advances such as single cell RNA sequencing have allowed researchers to gain a better understanding of their phenotype and function (125). IMs differ from AMs as the embryonically derived IMs present in the lungs before birth are gradually replaced by circulating monocytes and have a shorter life span (126). Two distinct populations of IMs have been identified in mice based on their location and expression of several surface markers at steady state, mainly hyaluronan receptor (Lyve1), major histocompatibility complex II (MHCII), and lung macrophage mannose receptor (CD206) (Figure 3). IMs residing in the bronchial interstitium express high levels of the receptor Lyve1, low levels of MHCII and are CD206 positive. IMs located in the alveolar interstitium express low levels of Lyve1, high levels of MHCII and are CD206 negative. Unlike AMs, both subsets of IMs express high levels of CD11b and low levels of CD11c. In addition, IMs express monocyte-specific markers, further proving that the population is replenished by circulating monocytes (43). Each subset has specific roles based on their location. IMs within the alveolar interstitium are involved in antigen presentation while those within the bronchial interstitium play a role in healing and repair (125). However, both subsets constitutively express the immunosuppressive cytokine IL-10, and thus contribute to immunoregulation. Although our understanding of IMs is not as advanced as AMs, some key functions and characteristics have been uncovered.
As with our knowledge on the function of IMs during steady state, our knowledge of their role during inflammation is not well understood. Upon encountering unmethylated CpG regions of bacterial DNA, the IM population expands and a subsequent increase in IL-10 is seen (Figure 4). These newly differentiated IMs express higher levels of the classic pro-inflammatory macrophage markers CD40, CD80, and CD86, compared to steady state IMs (127). IMs located within the bronchial interstitium can regulate the permeability of surrounding blood vessels to control the influx of immune cells into the lung. In addition, IMs have been found to possess greater antigen-presentation capabilities compared to AMs. Overall, in response to injury, IMs are anti-inflammatory in nature to maintain tissue homeostasis. One way in which this phenotype is mediated is through the release of Rspondin3 by endothelial cells, which promotes inflammation resolution (128). These functions are likely only the tip of the iceberg, and more work is needed to uncover further functions in inflammation.
Tissue resident macrophages and diseases
Tissue resident macrophages make up a small portion of tissues cells but their effect on tissue homeostasis is mighty. If the function of tissue-resident macrophages is impaired, several disease states can develop (129). Diseases such as non-alcoholic steatohepatitis, alcoholic steatohepatitis, autoimmune hepatitis, and toxic liver injury worsen as a result of the overactivation and necroptosis of KCs (Table 4). However, KCs are also found to play protective roles in viral hepatitis and liver cancer such that their depletion results in a worse prognosis. In the lung, defective phagocytosis by AMs results in pulmonary alveolar proteinosis, an inflammatory condition caused improper catabolism of surfactant (14). In addition, dysfunctional phagocytosis is also seen in asthma, chronic obstructive pulmonary disease (COPD), and cystic fibrosis (Table 5). We will now explore how the tissue-resident macrophages in the liver and lungs are implicated in these disease states.
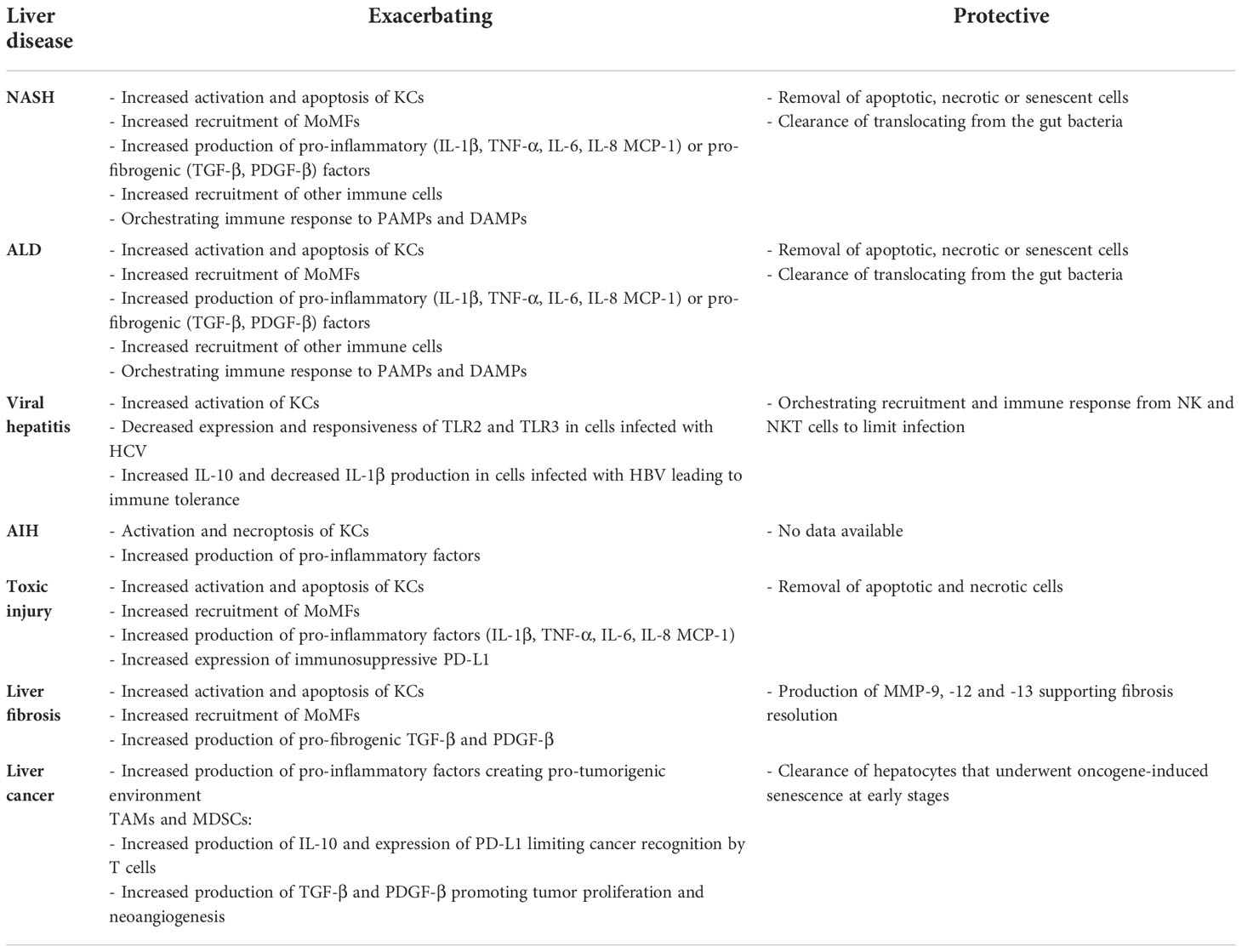
Table 4 Effects of disease on tissue-resident macrophages in the liver.

Table 5 Effects of disease on tissue-resident macrophages in the lung.
Non-alcoholic steatohepatitis (NASH)
NASH is a liver condition characterized by hepatic steatosis (intrahepatic lipid accumulation in >5% hepatocytes), accompanied by inflammation and fibrosis in the absence of excessive alcohol consumption (130). To date, multiple analyses of human, biopsy-derived liver samples have shown reduced numbers of KCs and increased MoMFs correlating with severity of inflammation, level of liver damage, and stage of fibrosis (131–134). NASH development and progression are associated with altered gut microbiome composition (dysbiosis) and increased intestinal permeability leading to increased levels of DAMPs and PAMPs reaching the liver. These molecules are one of the main activators of KCs through TLR4 and TLR9 signaling (135, 136). Moreover, lipid accumulation in liver parenchyma over time causes increased cell death and oxidative stress due to lipid peroxidation, that leads to the activation of an immune response in the liver (137, 138). In NASH, platelets and KCs are the first immune cells affected by lipid peroxidation. Activated KCs orchestrate recruitment of other immune cells to the liver and secrete pro-inflammatory cytokines and chemokines e.g., IL-1β, TNF-α, IL-6, IL-8, or MCP-1. Moreover, activated KCs also produce TGF-β and PDGF-β, both chemokines known for their pro-fibrogenic proprieties (139–143). In NASH, dietary lipids and cholesterol induce pro-inflammatory transcriptomic changes in KCs e.g., increased expression of Macrophage scavenger receptor 1 (MSR1) (144). Reversing some of mentioned changes could potentially provide some therapeutic benefit, as shown by the example of MSR1-blocking Ab, that attenuated the disease progression in experimental animal model (145). The crucial role of KCs in the development and progression of NASH was further demonstrated by cell depletion experiments (143, 146).
In addition to well-established changes in KCs during liver injury, recent discovery of their two new subpopulations: KC1 and KC2 shed a new light into particular roles of these cells in metabolic changes in the liver in NASH. KC2 (described in more detail above) show enrichment in genes involved in lipid and glucose regulation in steady state, which get further upregulated in metabolic disorders, e.g. hepatic steatosis. One of the most upregulated genes was CD36 known for its role in lipid uptake and oxidative stress. Indeed, experimental data showed that mice lacking KC2 macrophages in their hepatic tissue were protected from obesity, showed decreased oxidative stress and liver steatosis. This interesting finding requires further investigation, although the data collected so far suggests KC2 cells might become one of the important potential targets for the therapy of liver steatosis and/or NASH (87).
Constant activation of KCs during NASH results in the exhaustion and apoptotic death of these cells (81). In chronic inflammation self-renewal of the KC population is severely impaired, therefore MoMFs are recruited from the peripheral circulation to repopulate the tissue. MoMFs are able to acquire some of the features of KCs but show a highly pro-inflammatory phenotype that maintains liver inflammation and prevents its resolution (81, 143). Since the recruitment of inflammatory Ly6Chigh MoMFs to liver parenchyma occurs in CCR2-dependent manner, its pharmacological inhibition by cenicriviroc could be a potential therapeutic strategy, since its use in mouse model of NASH significantly improved liver inflammation and fibrosis (88).
Alcohol-associated liver disease
Alcohol-associated liver disease (ALD) is a spectrum of liver pathologies related to excessive alcohol consumption, ranging from simple steatosis, through alcoholic hepatitis (AH) or alcoholic steatohepatitis (ASH), to alcohol-associated cirrhosis (AC) potentially resulting in development of HCC. In addition, we can distinguish a more acute AH showing characteristics of acute-on-chronic liver failure (147). ASH is characterized by the same pathophysiological features as in NASH driven by excessive alcohol consumption (148). As observed in NASH, AH or ASH are both characterized by increased numbers and activation of macrophages in both human liver biopsies and in animal models, which translates to higher levels of produced pro-inflammatory cytokines and pro-fibrogenic factors (149, 150). Similarly, activation of liver macrophages in ASH results from steatosis-related lipotoxicity, increased gut permeability, and endotoxemia (151–153). In both conditions, KC activation by LPS is induced through TLR4, although it was shown that it occurs in a slightly different manner. In NASH, MyD88 is crucial for KC activation, while in ASH this occurs in MyD88-independent manner that involves type I interferon signaling through the IRF3-dependent pathway (154). The crucial role of liver macrophages in the pathogenesis of ASH was ultimately confirmed by results of KC depletion by Clodronate, which reduced liver damage in chronic-binge ethanol-feeding mouse model of ASH (155).
Viral hepatitis
Hepatitis B and hepatitis C are chronic liver infections caused by the hepatitis B virus (HBV) and hepatitis C virus (HCV), respectively (156). Despite vaccine preventing HBV infection and available treatment for hepatitis C, both diseases have high prevalence putting numerous patients in risk for development of fibrosis/cirrhosis and HCC (157). The chronicity of viral hepatic diseases depends on ability of immune system to clear the virus and its persistence (158, 159). Since both HBV and HCV pose high-risk for laboratory personnel and these viruses do not infect rodents, most of the data about hepatic viral infections come from studies performed on mice infected with lymphocytic choriomeningitis virus (LCMV) or in vitro experiments with primary hepatocytes (160). In the early stages of infection, KCs recognize viral particles through TLR2 and present viral antigens to T cells. Additional KC activation occurs in response to IFNγ which is secreted by activated CD8+ cytotoxic T cells (161). Simultaneously, activated KCs secrete pro-inflammatory factors IL-1β, IL-6, TNF-α and CXCL8 that recruits NK and NKT cells to help limit the infection (162). The role of KCs in control of infection was further confirmed by increased viral dissemination and enhanced liver damage following KC depletion (161, 163).
Surprisingly, some data suggest KCs also play an important role in immune tolerance against the virus. It was shown that primary monocytes differentiated to M1- or M2-like phenotypes and treated ex vivo with HBV decreased production of the pro-inflammatory cytokine IL-1β and increased release of anti-inflammatory IL-10 (164). The decreased production of IL-1β resulted from inhibition of NLRP3 inflammasome activation by viral antigen HBeAg (165). Apart from limited cytokine production, monocytes and macrophages from patients with chronic hepatitis C have altered TLR responsiveness corresponding with reduced expression of TLR2 and TLR3 (163, 166). Altogether these data suggest that role of liver macrophages in viral hepatitis is complex and more studies are needed to better understand their role in these pathologies.
Auto-immune hepatitis
Auto-immune hepatitis (AIH) is characterized by liver necro-inflammation with lymphoplasmacytic infiltrates in hepatic tissue and a presence of circulating autoantibodies. Immune cell driven inflammation is a central mechanism in the development and progression of AIH, and targeting this inflammation with immunosuppressive therapies is an effective treatment. Among the few reports discussing the role of KCs in AIH, most focused on the finding of hyaline droplets in the cytoplasm of these cells (167, 168). In adults this histological feature was shared between samples with AIH and primary biliary cholangitis (PBC). In pediatric patients it was a key indicator of diagnosis of AIH versus other pediatric liver diseases, although the exact function of these cells is unknown (167–169). One of the recent studies focusing on the role of KCs in AIH suggest that AIH is associated with dysbiosis and leaky gut, resulting in activation of necroptosis signaling through receptor interacting protein kinase 3 (RIPk3) in hepatic macrophages. Induction of necroptosis in KCs leads to the release of pro-inflammatory cytokines and chemokines resulting in the aggravation of inflammation (170). These first reports suggest liver macrophages might have an important role in pathogenesis of AIH, although further studies are needed to shed some light on mechanism of its development.
Toxic liver injury
Acute liver injury usually results from an overdose of hepatotoxic agents, e.g., acetaminophen (known as APAP) or carbon tetrachloride (CCl4), resulting in extensive and irreversible hepatocyte damage with a high-mortality risk (171, 172). Necrosis occurring in the liver parenchyma leads to high levels of oxidative stress, together with DAMPs released from dying hepatocytes, stimulate KC activation. One of the recent studies showed, using scRNAseq, that APAP-induced liver failure leads to activation of around 51% of KCs. Further transcriptome analysis revealed that activated KCs differed from quiescent ones by upregulation of genes involved in not only immune response, but also in chemotaxis, cell migration, as well as interferon response (173). The latter is not surprising, since it was observed in all major liver pathologies, that activated KCs secrete a variety of pro-inflammatory factors that recruit and activate other cell types leading to acute inflammation (174). One of the key factors involved in the process of KC activation in APAP-induced liver injury is macrophage-inducible C-type lectin (Mincle). Mincle recognizes spliceosome-associated protein 130 (SAP130) released by necrotic hepatocytes. KCs are the main source of Mincle, and Mincle KO mice subjected to APAP overdose were less prone to liver injury. These animals showed fewer necrotic lesions, lower levels of alanine aminotransferase and aspartate aminotransferase in their plasma, and decreased production of IL-1β (175).
Interestingly, recent evidence suggest that KCs can also exhibit an immune-suppressive role during APAP-induced liver injury. In acute injury KCs and intrahepatic T cells express high levels of PD-1 and PD-L1, which suppresses the anti-bacterial function of liver macrophages. These data suggest that check-point inhibitors could restore anti-bacterial function of KCs by targeting PD-1/PD-L1 signaling (176). Additionally, IL-10 secreted by KCs directly affected surrounding hepatocytes, which express higher levels of CXCR2 in response. Mice lacking KCs, treated with high doses of APAP and a CXCR2 inhibitor were more susceptible to liver damage and had impaired liver regeneration, implicating role of hepatic macrophages in organ recovery in CXCR2-dependent manner (177, 178). Acute liver injury and inflammation observed during hepatotoxicity results in the cell death of large numbers of hepatic macrophages, resulting in dramatically increased recruitment of MoMFs and monocytes from the circulation, like the liver repopulation process observed in NASH or ASH (179).
Liver fibrosis
Liver fibrosis is a process of pathological scarring occurring within liver parenchyma, as a result of sustained activation of hepatic stellate cells (HSCs) due to chronic hepatic injury occurring e.g. in NASH, ALD or viral hepatitis (180). Activation of HSCs leads to excessive deposition of extracellular matrix (ECM) consisting mainly of collagen, which affects hepatic architecture and flow of oxygen and nutrients, that can further accelerate the injury causing progression of fibrosis towards cirrhosis (181, 182).
Although HSCs are responsible for ECM deposits, multiple other cell types are involved in fibrogenesis, including damaged hepatocytes and activated immune cells. Among them, activated during injury KCs play an important role through the production of pro-fibrogenic cytokines and chemokines, including PDGF-β and TGF-β. At the same time cytokines and chemokines released from injured hepatocytes, activated HSCs and immune cells (e.g. CCl2, CCl5), lead to increased recruitment of CCR2+Ly6Chi monocytes that further differentiate towards Ly6Chi MoMFs with highly pro-inflammatory and pro-fibrogenic proprieties (179, 183, 184). In turn, these cells can, through PDGF-β, TGF-β and CTGF, further stimulate ECM production by HSCs and promote their survival by IL-1 β and TNF-α (182, 185). On the other hand, after the peak of fibrogenesis process, MoMFs start to differentiate towards more restorative cells that are a source of matrix metalloproteinases MMP-9, -12 and -13 that were associated with fibrosis resolution (186–188). Unfortunately, the latter can be counteracted by tissue inhibitors of matrix metalloproteinases (TIMPs) produced by activated HSCs, preventing resolution of fibrosis during chronic pathologies (186, 189). Indeed, dual role of liver macrophages in liver fibrosis largely depends on the stage of fibrogenesis. In the mouse model of liver fibrosis induced by treatment with CCl4 depletion of hepatic macrophages during the progression of the disease reduced scarring. On the other hands depletion of the same cells during the phase of resolution limited ECM degradation and hepatic recovery, making liver macrophages a complicated target for therapy of liver fibrosis (190).
Liver cancer
Hepatocellular carcinoma (HCC) and intrahepatic cholangiocarcinoma (iCC) are the two most frequent primary liver malignancies, with HCC consisting of 70% of the cases (191). Tumors develop due to sustained injury of hepatocytes or cholangiocytes leading to continuous cell death and compensatory proliferation, increasing the risk of genetic mutations. Some of these mutations lead to cell cycle arrest and apoptosis, while others initiate the process of hepatocarcinogenesis increasing the growth rate of these cells (192, 193). Chronic inflammation due to higher levels of cytokines, chemokines, and growth factors present in liver parenchyma create a microenvironment that promotes liver cancer onset and progression (194, 195). In the context of HCC, KCs play a protective role in the early phases of tumor initiation. Upon liver injury, senescent hepatocytes secrete CCL2, which in turn recruits CCR2+ macrophages. These cells remove hepatocytes that underwent oncogene-induced senescence from liver tissue and prevent initiation of carcinogenesis (76, 196, 197). Conversely, increased levels of CCL2 and recruitment of CCR2+ macrophages correlate with increased tumor burden and poor prognosis, suggesting that these cells might play pro-tumorigenic role. This hypothesis was confirmed by studies in which a CCR2 antagonist, RDC018, suppressed the development of liver cancer (198, 199).
Non-resident macrophages, such as tumor-associated macrophages (TAMs), induce immune tolerance against tumors by producing high levels of IL-10 and increasing the expression of PD-L1, thus limiting the immune response of T cells to cancer cells (200, 201). Another subset of macrophages with similar proprieties are called myeloid-derived suppressor cells (MDSCs) and an increase in both TAM and MDSC numbers was associated with increased tumor burden and higher metastasis rate in both preclinical models and human patients (202). Moreover, multiple factors released by TAMs, TNF-α, IL-1β, and IL-6 support tumorigenesis by maintaining a pro-inflammatory environment (203). Others, like TGF-β or PDGF-β promote fibrogenesis and act as growth factors promoting tumor proliferation or neoangiogenesis in fully formed tumors that increases the supply of nutrients and oxygen within the tumor (204, 205).
Considering the important role of liver macrophages in the maintenance of immune tolerance against tumors, targeting these cells to reverse their M2 phenotype or use of anti-PD-L1 antibodies might be a potential therapeutic strategy for the treatment of liver cancer. Preventing liver macrophages from inducing immune tolerance could activate T and NK cells and provoke anti-tumor immunity leading to inhibition of tumor growth. This approach, together with therapeutic strategies that are already in place, could increase the efficacy of treatment leading to the increased survival and a better quality of life for patients.
Chronic obstructive pulmonary disease (COPD)
Patients with COPD have increased numbers of macrophages within their airways and interstitium. However, the function of these macrophages is highly impaired. To begin, AMs in COPD show impaired expression of toll-like receptors that are essential for detecting DAMPs and PAMPs (206). In addition, these AMs have dysfunctional phagocytic abilities which can lead to increased inflammation (207). Increased levels of IL-8, TNF-α, reactive oxygen species, and matrix metalloproteinase 12 are produced by AMs in COPD patients (129). These factors exacerbate inflammation and cause tissue damage. As for IMs, little is known about their role in COPD. However, murine studies suggest that they contribute to the release of the pro-inflammatory factors TNF-α and IL-6 (208). Current therapeutic options focus on reducing the symptoms of COPD in lieu of reversing disease progression. Therefore, researchers have looked at targeting lung macrophages to restore lung function. Shifting macrophage polarization towards an anti-inflammatory phenotype by reducing oxidative stress and supressing pro-inflammatory mediators release have been found to restore the phagocytic function of lung macrophages thus improving disease pathogenesis (209, 210).
Asthma
Asthma and allergic reactions are attributed to the dysfunctional response of T helper 2 (Th2) cells, and tissue-resident lung macrophages play an important role in the regulation and maintenance of these T cells in the lung (211, 212). Th2 cells secrete the cytokines IL-4, IL-5, and IL-13 and stimulate a type II immune response. As the primary immune cell responsible for responding to allergens and pathogens in the airway, AM dysfunction can by linked to the development of T-cell mediated pathologies such as asthma. As such, pathways more commonly linked to macrophage function, such as the inflammasome, have been demonstrated to influence Th2 and regulatory T-cell development (213). In a mouse model of allergic asthma, loss of NLRP6 prevented development of Th2 mediated lung inflammation (214). In response to inflammation AMs release IL-27, an essential regulator of airway hyperresponsiveness known to be reduced in individuals with asthma (114, 215–217). When AMs are depleted, the lack of IL-27 production delays inflammation resolution exacerbating allergic inflammation (218). The intrinsic expression of TGF-β by AMs and IL-10 by IMs aid in controlling responses to inhaled allergens through influence on regulatory T-cells (219). Resident macrophages also play a central role in the clearance of dead and dying cells, which if left in the lung can be a driver of inflammation. In murine models, the efferocytosis of apoptotic cells by AMs prevented development of asthma (220, 221). Defective efferocytosis in AMs is in part due to altered expression of Axl receptor kinase (222). Blocking this pathway has been shown to prevent pathology during viral asthma exacerbation, suggesting that targeting macrophage function could be a therapeutic opportunity for the treatment or prevention of asthma (223).
Cystic fibrosis
Cystic fibrosis (CF) is a genetic disease caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR), which results in exaggerated airway inflammation, airway edema and impaired host defense (224). Due to the primary function of CFTR as an epithelial ion pump, the role of macrophages in CF is somewhat understudied. In patients, macrophage derived factors including IL-10, IL-8, TNF-α, and IL-1β were increased (225, 226). In addition, macrophages in these patients show impaired phagocytosis and bactericidal killing due to an increased lysosomal pH (227, 228). Murine studies have further demonstrated that loss of CFTR influences macrophage function. AMs and bone marrow-derived macrophages from mice lacking CFTR produced higher amounts of inflammatory cytokines ex vivo following simulation with LPS, in part due to altered trafficking of TLR4 in these cells (229, 230). Interestingly, defects in phagocytosis or phagolysosome maturation have not been confirmed, suggesting that while the inflammatory response of the AM is altered in CF, their ability to eliminate bacteria may not be impacted (231, 232).
Influenza
Influenza is a highly prevalent viral infection affecting an estimated 1 billion people every year. As with bacterial infections, AMs play a critical role in host defense. In response to the detection of PAMPs by TLRs, AMs release cytokines including interferons, IL-6, and IL-12 (233). The importance of AMs in combatting influenza has been shown through depletion experiments resulting in uncontrolled viral infection (234, 235). However, influenza can also have deleterious effects on the AM population. Verma et al., found that influenza activates IFNγR signaling in AMs resulting in reduced antibacterial abilities (236). This deficient antibacterial activity leaves the host vulnerable to secondary infections. In addition, influenza infections induce premature apoptosis and depletion of AMs (237). Impaired AM function can cause surfactant accumulation leading to respiratory distress. Overall, AMs can play a critical role in both the defense and pathogenesis of influenza infections. As such, AMs could be therapeutic target to lessen disease severity. The primary way in which AMs have been targeted has been to downregulate the release of pro-inflammatory cytokines from dysregulated macrophages to prevent lung injury (238, 239).
Conclusion and future perspectives
Tissue resident macrophages are a unique sub-arm of the innate immune system. These cells guard our organs from infection and play pivotal roles in tissue homeostasis. Resident macrophages seed the major organs, such as liver and lung, during embryonic development, and they stay with the organ throughout life. These cells direct organ development, regulate the phenotype of other immune cells and protect the tissue from microbial infection. With such an essential role in directing organ function and protecting from infection, it is understandable that a number of diseases are associated with macrophage dysfunction. Our understanding of tissue resident macrophages is still limited, particularly chronic respiratory disease. Due to the myriad of functions driven by macrophages, they are an attractive target for therapeutic intervention. Additional research is required to more deeply understand the molecular signaling regulating macrophage function and to safely develop therapies directed at this essential cell type.
Author contributions
AK and MS wrote the manuscript, AT, TSC, and MS revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
AK was supported by The Fighting Blindness Canada Funding.
Conflict of interest
TSC and MS are employed by AstraZeneca.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Gordon S. Elie metchnikoff: father of natural immunity. Eur J Immunol (2008) 38:3257–64. doi: 10.1002/eji.200838855
2. Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue-resident macrophages. Nat Immunol (2013) 14:986–95. doi: 10.1038/ni.2705
3. Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity (2014) 41:21–35. doi: 10.1016/j.immuni.2014.06.013
4. van Furth R, Cohn ZA, Hirsch JG, Humphrey JH, Spector WG, Langevoort HL. The mononuclear phagocyte system: a new classification of macrophages, monocytes, and their precursor cells. Bull World Health Organ (1972) 46:845–52.
5. Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, et al. A lineage of myeloid cells independent of myb and hematopoietic stem cells. Science (2012) 336:86–90. doi: 10.1126/science.1219179
6. Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity (2013) 38:792–804. doi: 10.1016/j.immuni.2013.04.004
7. Sieweke MH, Allen JE. Beyond stem cells: self-renewal of differentiated macrophages. Science (2013) 342:1242974. doi: 10.1126/science.1242974
8. Byrne AJ, Powell JE, O'Sullivan BJ, Ogger PP, Hoffland A, Cook J, et al. Dynamics of human monocytes and airway macrophages during healthy aging and after transplant. J Exp Med (2020) 217. doi: 10.1084/jem.20191236
9. Collin MP, Hart DN, Jackson GH, Cook G, Cavet J, Mackinnon S, et al. The fate of human langerhans cells in hematopoietic stem cell transplantation. J Exp Med (2006) 203:27–33. doi: 10.1084/jem.20051787
10. Bittmann I, Bottino A, Baretton GB, Gerbes AL, Zachoval R, Rau HG, et al. The role of graft-resident kupffer cells and lymphocytes of donor type during the time course after liver transplantation–a clinico-pathological study. Virchows Arch (2003) 443:541–8. doi: 10.1007/s00428-003-0861-8
11. Naito M, Umeda S, Yamamoto T, Moriyama H, Umezu H, Hasegawa G, et al. Development, differentiation, and phenotypic heterogeneity of murine tissue macrophages. J Leukoc Biol (1996) 59:133–8. doi: 10.1002/jlb.59.2.133
12. Ginhoux F, Guilliams M. Tissue-resident macrophage ontogeny and homeostasis. Immunity (2016) 44:439–49. doi: 10.1016/j.immuni.2016.02.024
13. Aziz A, Soucie E, Sarrazin S, Sieweke MH. MafB/c-maf deficiency enables self-renewal of differentiated functional macrophages. Science (2009) 326:867–71. doi: 10.1126/science.1176056
14. Nobs SP, Kopf M. Tissue-resident macrophages: guardians of organ homeostasis. Trends Immunol (2021) 42:495–507. doi: 10.1016/j.it.2021.04.007
15. Sica A, Invernizzi P, Mantovani A. Macrophage plasticity and polarization in liver homeostasis and pathology. Hepatology (2014) 59:2034–42. doi: 10.1002/hep.26754
16. Yunna C, Mengru H, Lei W, Weidong C. Macrophage M1/M2 polarization. Eur J Pharmacol (2020) 877:173090. doi: 10.1016/j.ejphar.2020.173090
17. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
18. Wang C, Ma C, Gong L, Guo Y, Fu K, Zhang Y, et al. Macrophage polarization and its role in liver disease. Front Immunol (2021) 12:803037. doi: 10.3389/fimmu.2021.803037
19. Samokhvalov IM. Deconvoluting the ontogeny of hematopoietic stem cells. Cell Mol Life Sci (2014) 71:957–78. doi: 10.1007/s00018-013-1364-7
20. Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature (2015) 518:547–51. doi: 10.1038/nature13989
21. Mass E, Ballesteros I, Farlik M, Halbritter F, Günther P, Crozet L, et al. Specification of tissue-resident macrophages during organogenesis. Sci (2016) 353(6304):aaf4238. doi: 10.1126/science.aaf4238
22. Wu Y, Hirschi KK. Tissue-resident macrophage development and function. Front Cell Dev Biol (2020) 8:617879. doi: 10.3389/fcell.2020.617879
23. Haldar M, Murphy KM. Origin, development, and homeostasis of tissue-resident macrophages. Immunol Rev (2014) 262:25–35. doi: 10.1111/imr.12215
24. Mass E. Delineating the origins, developmental programs and homeostatic functions of tissue-resident macrophages. Int Immunol (2018) 30:493–501. doi: 10.1093/intimm/dxy044
25. T'Jonck W, Guilliams M, Bonnardel J. Niche signals and transcription factors involved in tissue-resident macrophage development. Cell Immunol (2018) 330:43–53. doi: 10.1016/j.cellimm.2018.02.005
26. van Riel B, Rosenbauer F. Epigenetic control of hematopoiesis: the PU.1 chromatin connection. Biol Chem (2014) 395:1265–74. doi: 10.1515/hsz-2014-0195
27. Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell (2014) 159:1312–26. doi: 10.1016/j.cell.2014.11.018
28. Cox N, Pokrovskii M, Vicario R, Geissmann F. Origins, biology, and diseases of tissue macrophages. Annu Rev Immunol (2021) 39:313–44. doi: 10.1146/annurev-immunol-093019-111748
29. Fainaru O, Woolf E, Lotem J, Yarmus M, Brenner O, Goldenberg D, et al. Runx3 regulates mouse TGF-beta-mediated dendritic cell function and its absence results in airway inflammation. EMBO J (2004) 23:969–79. doi: 10.1038/sj.emboj.7600085
30. Nakamura A, Ebina-Shibuya R, Itoh-Nakadai A, Muto A, Shima H, Saigusa D, et al. Transcription repressor Bach2 is required for pulmonary surfactant homeostasis and alveolar macrophage function. J Exp Med (2013) 210:2191–204. doi: 10.1084/jem.20130028
31. Buttgereit A, Lelios I, Yu X, Vrohlings M, Krakoski NR, Gautier EL, et al. Sall1 is a transcriptional regulator defining microglia identity and function. Nat Immunol (2016) 17:1397–406. doi: 10.1038/ni.3585
32. Ohman J, Magnusson B, Telemo E, Jontell M, Hasseus B. Langerhans cells and T cells sense cell dysplasia in oral leukoplakias and oral squamous cell carcinomas–evidence for immunosurveillance. Scand J Immunol (2012) 76:39–48. doi: 10.1111/j.1365-3083.2012.02701.x
33. Xie Y, Li Y, Zhang Q, Stiller MJ, Wang CL, Streilein JW. Haptoglobin is a natural regulator of langerhans cell function in the skin. J Dermatol Sci (2000) 24:25–37. doi: 10.1016/S0923-1811(00)00078-5
34. Paus R, van der Veen C, Eichmuller S, Kopp T, Hagen E, Müller-Röver S, et al. Generation and cyclic remodeling of the hair follicle immune system in mice. J Invest Dermatol (1998) 111:7–18. doi: 10.1046/j.1523-1747.1998.00243.x
35. Tamoutounour S, Guilliams M, Montanana Sanchis F, Liu H, Terhorst D, Malosse C, et al. Origins and functional specialization of macrophages and of conventional and monocyte-derived dendritic cells in mouse skin. Immunity (2013) 39:925–38. doi: 10.1016/j.immuni.2013.10.004
36. Kolter J, Feuerstein R, Zeis P, Hagemeyer N, Paterson N, d'Errico P, et al. A subset of skin macrophages contributes to the surveillance and regeneration of local nerves. Immunity (2019) 50:1482–97 e7. doi: 10.1016/j.immuni.2019.05.009
37. Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic pruning by microglia is necessary for normal brain development. Science (2011) 333:1456–8. doi: 10.1126/science.1202529
38. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science (2005) 308:1314–8. doi: 10.1126/science.1110647
39. Chertoff M, Shrivastava K, Gonzalez B, Acarin L, Gimenez-Llort L. Differential modulation of TREM2 protein during postnatal brain development in mice. PloS One (2013) 8:e72083. doi: 10.1371/journal.pone.0072083
40. Nagre N, Cong X, Pearson AC, Zhao X. Alveolar macrophage phagocytosis and bacteria clearance in mice. J Vis Exp (2019) (145):10.3791/59088. doi: 10.3791/59088
41. Malur A, Kavuru MS, Marshall I, Barna BP, Huizar I, Karnekar R, et al. Rituximab therapy in pulmonary alveolar proteinosis improves alveolar macrophage lipid homeostasis. Respir Res (2012) 13:46. doi: 10.1186/1465-9921-13-46
42. Dong Y, Arif AA, Guo J, Ha Z, Lee-Sayer SSM, Poon GFT, et al. CD44 loss disrupts lung lipid surfactant homeostasis and exacerbates oxidized lipid-induced lung inflammation. Front Immunol (2020) 11:29. doi: 10.3389/fimmu.2020.00029
43. Reddy SP, Mehta D. Lung interstitial macrophages redefined: It is not that simple anymore. Am J Respir Cell Mol Biol (2017) 57:135–6. doi: 10.1165/rcmb.2017-0158ED
44. Bedoret D, Wallemacq H, Marichal T, Desmet C, Quesada Calvo F, Henry E, et al. Lung interstitial macrophages alter dendritic cell functions to prevent airway allergy in mice. J Clin Invest (2009) 119:3723–38. doi: 10.1172/JCI39717
45. Kohyama M, Ise W, Edelson BT, Wilker PR, Hildner K, Mejia C, et al. Role for spi-c in the development of red pulp macrophages and splenic iron homeostasis. Nature (2009) 457:318–21. doi: 10.1038/nature07472
46. Youssef LA, Rebbaa A, Pampou S, Weisberg SP, Stockwell BR, Hod EA, et al. Increased erythrophagocytosis induces ferroptosis in red pulp macrophages in a mouse model of transfusion. Blood (2018) 131:2581–93. doi: 10.1182/blood-2017-12-822619
47. Miyake Y, Asano K, Kaise H, Uemura M, Nakayama M, Tanaka M. Critical role of macrophages in the marginal zone in the suppression of immune responses to apoptotic cell-associated antigens. J Clin Invest (2007) 117:2268–78. doi: 10.1172/JCI31990
48. Tada R, Nagao K, Tanaka R, Yamada S, Watanabe A, Negishi Y. Involvement of splenic marginal zone macrophages in the recognition of systemically administered phosphatidylserine-coated liposomes in mice. Int Immunopharmacol (2022) 112:109209. doi: 10.1016/j.intimp.2022.109209
49. AG N, Guillen JA, Gallardo G, Diaz M, de la Rosa JV, Hernandez IH, et al. The nuclear receptor LXRalpha controls the functional specialization of splenic macrophages. Nat Immunol (2013) 14:831–9. doi: 10.1038/ni.2622
50. Deppermann C, Kratofil RM, Peiseler M, David BA, Zindel J, Castanheira FVES, et al. Macrophage galactose lectin is critical for kupffer cells to clear aged platelets. . J Exp Med (2020) 217(4):e20190723. doi: 10.1084/jem.20190723
51. Davies SP, Reynolds GM, Stamataki Z. Clearance of apoptotic cells by tissue epithelia: A putative role for hepatocytes in liver efferocytosis. Front Immunol (2018) 9:44. doi: 10.3389/fimmu.2018.00044
52. Willekens FL, Werre JM, Kruijt JK, Roerdinkholder-Stoelwinder B, Groenen-Döpp YA, van den Bos AG, et al. Liver kupffer cells rapidly remove red blood cell-derived vesicles from the circulation by scavenger receptors. Blood (2005) 105:2141–5. doi: 10.1182/blood-2004-04-1578
53. Loegering DJ, Commins LM, Minnear FL, Gary LA, Hill LA. Effect of kupffer cell phagocytosis of erythrocytes and erythrocyte ghosts on susceptibility to endotoxemia and bacteremia. Infect Immun (1987) 55:2074–80. doi: 10.1128/iai.55.9.2074-2080.1987
54. Nenseter MS, Gudmundsen O, Roos N, Maelandsmo G, Drevon CA, Berg T. Role of liver endothelial and kupffer cells in clearing low density lipoprotein from blood in hypercholesterolemic rabbits. J Lipid Res (1992) 33:867–77.
55. Sierro F, Evrard M, Rizzetto S, Melino M, Mitchell AJ, Florido M, et al. A liver capsular network of monocyte-derived macrophages restricts hepatic dissemination of intraperitoneal bacteria by neutrophil recruitment. Immunity (2017) 47:374–88 e6. doi: 10.1016/j.immuni.2017.07.018
56. Jacome-Galarza CE, Percin GI, Muller JT, Mass E, Lazarov T, Eitler J, et al. Developmental origin, functional maintenance and genetic rescue of osteoclasts. Nature (2019) 568:541–5. doi: 10.1038/s41586-019-1105-7
57. De Schepper S, Verheijden S, Aguilera-Lizarraga J, Viola MF, Boesmans W, Stakenborg N, et al. Self-maintaining gut macrophages are essential for intestinal homeostasis. Cell (2018) 175:400–15 e13. doi: 10.1016/j.cell.2018.07.048
58. Muller PA, Koscso B, Rajani GM, Stevanovic K, Berres ML, Hashimoto D, et al. Crosstalk between muscularis macrophages and enteric neurons regulates gastrointestinal motility. Cell (2014) 158:300–13. doi: 10.1016/j.cell.2014.04.050
59. Ghosn EE, Cassado AA, Govoni GR, Fukuhara T, Yang Y, Monack DM, et al. Two physically, functionally, and developmentally distinct peritoneal macrophage subsets. Proc Natl Acad Sci U.S.A. (2010) 107:2568–73. doi: 10.1073/pnas.0915000107
60. Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity (2014) 40:91–104. doi: 10.1016/j.immuni.2013.11.019
61. Hulsmans M, Clauss S, Xiao L, Aguirre AD, King KR, Hanley A, et al. Macrophages facilitate electrical conduction in the heart. Cell (2017) 169:510–22 e20. doi: 10.1016/j.cell.2017.03.050
62. Pinto AR, Paolicelli R, Salimova E, Gospocic J, Slonimsky E, Bilbao-Cortes D, et al. An abundant tissue macrophage population in the adult murine heart with a distinct alternatively-activated macrophage profile. PloS One (2012) 7:e36814. doi: 10.1371/journal.pone.0036814
63. Li P, Spann NJ, Kaikkonen MU, Lu M, Oh DY, Fox JN, et al. NCoR repression of LXRs restricts macrophage biosynthesis of insulin-sensitizing omega 3 fatty acids. Cell (2013) 155:200–14. doi: 10.1016/j.cell.2013.08.054
64. Nguyen KD, Qiu Y, Cui X, Goh YP, Mwangi J, David T, et al. Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature (2011) 480:104–8. doi: 10.1038/nature10653
65. Nishimura S, Manabe I, Nagasaki M, Hosoya Y, Yamashita H, Fujita H, et al. Adipogenesis in obesity requires close interplay between differentiating adipocytes, stromal cells, and blood vessels. Diabetes (2007) 56:1517–26. doi: 10.2337/db06-1749
66. MacParland SA, Liu JC, Ma XZ, Innes BT, Bartczak AM, Gage BK, et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat Commun (2018) 9:4383. doi: 10.1038/s41467-018-06318-7
67. Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol (2017) 17:306–21. doi: 10.1038/nri.2017.11
68. van der Heide D, Weiskirchen R, Bansal R. Therapeutic targeting of hepatic macrophages for the treatment of liver diseases. Front Immunol (2019) 10:2852. doi: 10.3389/fimmu.2019.02852
69. Wen Y, Lambrecht J, Ju C, Tacke F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell Mol Immunol (2021) 18:45–56. doi: 10.1038/s41423-020-00558-8
70. Ingersoll MA, Spanbroek R, Lottaz C, Gautier EL, Frankenberger M, Hoffmann R, et al. Comparison of gene expression profiles between human and mouse monocyte subsets. Blood (2010) 115:e10-9. doi: 10.1182/blood-2009-07-235028
71. Guilliams M, Bonnardel J, Haest B, Vanderborght B, Wagner C, Remmerie A, et al. Spatial proteogenomics reveals distinct and evolutionarily conserved hepatic macrophage niches. Cell (2022) 185:379–96 e38. doi: 10.1016/j.cell.2021.12.018
72. Dalod M, Chelbi R, Malissen B, Lawrence T. Dendritic cell maturation: functional specialization through signaling specificity and transcriptional programming. EMBO J (2014) 33:1104–16. doi: 10.1002/embj.201488027
73. Ramachandran P, Dobie R, Wilson-Kanamori JR, Dora EF, Henderson BEP, Luu NT, et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature (2019) 575:512–8. doi: 10.1038/s41586-019-1631-3
74. Ritz T, Krenkel O, Tacke F. Dynamic plasticity of macrophage functions in diseased liver. Cell Immunol (2018) 330:175–82. doi: 10.1016/j.cellimm.2017.12.007
75. Singanayagam A, Triantafyllou E. Macrophages in chronic liver failure: Diversity, plasticity and therapeutic targeting. Front Immunol (2021) 12:661182. doi: 10.3389/fimmu.2021.661182
76. Elchaninov AV, Fatkhudinov TK, Vishnyakova PA, Lokhonina AV, Sukhikh GT. Phenotypical and functional polymorphism of liver resident macrophages. Cells (2019) 8(9):1032. doi: 10.3390/cells8091032
77. Hoeffel G, Chen J, Lavin Y, Low D, Almeida FF, See P, et al. C-myb(+) erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity (2015) 42:665–78. doi: 10.1016/j.immuni.2015.03.011
78. Yamamoto T, Naito M, Moriyama H, Umezu H, Matsuo H, Kiwada H, et al. Repopulation of murine kupffer cells after intravenous administration of liposome-encapsulated dichloromethylene diphosphonate. Am J Pathol (1996) 149:1271–86.
79. Wacker HH, Radzun HJ, Parwaresch MR. Kinetics of kupffer cells as shown by parabiosis and combined autoradiographic/immunohistochemical analysis. Virchows Arch B Cell Pathol Incl Mol Pathol (1986) 51:71–8. doi: 10.1007/BF02899017
80. Steinhoff G BM, Sorg C, Wonigeit K, Pichlmayr R. Sequential analysis of macrophage tissue differentiation and kupffer cell exchange after human liver transplantation. Kupffer Cell Found (1989).
81. Tran S, Baba I, Poupel L, Dussaud S, Moreau M, Gélineau A, et al. Impaired kupffer cell self-renewal alters the liver response to lipid overload during non-alcoholic steatohepatitis. Immunity (2020) 53:627–40 e5. doi: 10.1016/j.immuni.2020.06.003
82. Li Z, Zhao J, Zhang S, Weinman SA. FOXO3-dependent apoptosis limits alcohol-induced liver inflammation by promoting infiltrating macrophage differentiation. Cell Death Discovery (2018) 4:16. doi: 10.1038/s41420-017-0020-7
83. Liu C, Tao Q, Sun M, Wu JZ, Yang W, Jian P, et al. Kupffer cells are associated with apoptosis, inflammation and fibrotic effects in hepatic fibrosis in rats. Lab Invest (2010) 90:1805–16. doi: 10.1038/labinvest.2010.123
84. Baeck C, Wehr A, Karlmark KR, Heymann F, Vucur M, Gassler N, et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut (2012) 61:416–26. doi: 10.1136/gutjnl-2011-300304
85. Sato R, Reuter T, Hiranuma R, Shibata T, Fukui R, Motoi Y, et al. The impact of cell maturation and tissue microenvironments on the expression of endosomal toll-like receptors in monocytes and macrophages. Int Immunol (2020) 32:785–98. doi: 10.1093/intimm/dxaa055
86. Triantafyllou E, Woollard KJ, McPhail MJW, Antoniades CG, Possamai LA. The role of monocytes and macrophages in acute and acute-on-Chronic liver failure. Front Immunol (2018) 9:2948. doi: 10.3389/fimmu.2018.02948
87. Bleriot C, Barreby E, Dunsmore G, Ballaire R, Chakarov S, Ficht X, et al. A subset of kupffer cells regulates metabolism through the expression of CD36. Immunity (2021) 54:2101–16 e6. doi: 10.1016/j.immuni.2021.08.006
88. Krenkel O, Puengel T, Govaere O, Abdallah AT, Mossanen JC, Kohlhepp M, et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology (2018) 67:1270–83. doi: 10.1002/hep.29544
89. Racanelli V, Rehermann B. The liver as an immunological organ. Hepatology (2006) 43:S54–62. doi: 10.1002/hep.21060
90. Zhao D, Yang F, Wang Y, Li S, Li Y, Hou F, et al. ALK1 signaling is required for the homeostasis of kupffer cells and prevention of bacterial infection. J Clin Invest (2022) 132(3):e150489. doi: 10.1172/JCI150489
91. Bleriot C, Dupuis T, Jouvion G, Eberl G, Disson O, Lecuit M. Liver-resident macrophage necroptosis orchestrates type 1 microbicidal inflammation and type-2-mediated tissue repair during bacterial infection. Immunity (2015) 42:145–58. doi: 10.1016/j.immuni.2014.12.020
92. Bradshaw G, Gutierrez A, Miyake JH, Davis KR, Li AC, Glass CK, et al. Facilitated replacement of kupffer cells expressing a paraoxonase-1 transgene is essential for ameliorating atherosclerosis in mice. Proc Natl Acad Sci U.S.A. (2005) 102:11029–34. doi: 10.1073/pnas.0502677102
93. Chan IH, Van Hoof D, Abramova M, Bilardello M, Mar E, Jorgensen B, et al. PEGylated IL-10 activates kupffer cells to control hypercholesterolemia. PloS One (2016) 11:e0156229. doi: 10.1371/journal.pone.0156229
94. Coelho I, Duarte N, Barros A, Macedo MP, Penha-Goncalves C. Trem-2 promotes emergence of restorative macrophages and endothelial cells during recovery from hepatic tissue damage. Front Immunol (2020) 11:616044. doi: 10.3389/fimmu.2020.616044
95. Feng M, Ding J, Wang M, Zhang J, Zhu X, Guan W. Kupffer-derived matrix metalloproteinase-9 contributes to liver fibrosis resolution. Int J Biol Sci (2018) 14:1033–40. doi: 10.7150/ijbs.25589
96. Bozward AG, Ronca V, Osei-Bordom D, Oo YH. Gut-liver immune traffic: Deciphering immune-pathogenesis to underpin translational therapy. Front Immunol (2021) 12:711217. doi: 10.3389/fimmu.2021.711217
97. Trefts E, Gannon M, Wasserman DH. The liver. Curr Biol (2017) 27:R1147–R51. doi: 10.1016/j.cub.2017.09.019
98. Wong CH, Jenne CN, Petri B, Chrobok NL, Kubes P. Nucleation of platelets with blood-borne pathogens on kupffer cells precedes other innate immunity and contributes to bacterial clearance. Nat Immunol (2013) 14:785–92. doi: 10.1038/ni.2631
99. Zeng Z, Surewaard BG, Wong CH, Geoghegan JA, Jenne CN, Kubes P. CRIg functions as a macrophage pattern recognition receptor to directly bind and capture blood-borne gram-positive bacteria. Cell Host Microbe (2016) 20:99–106. doi: 10.1016/j.chom.2016.06.002
100. Armengol C, Bartoli R, Sanjurjo L, Serra I, Amézaga N, Sala M, et al. Role of scavenger receptors in the pathophysiology of chronic liver diseases. Crit Rev Immunol (2013) 33:57–96. doi: 10.1615/CritRevImmunol.2013006794
101. Mihm S. Danger-associated molecular patterns (DAMPs): Molecular triggers for sterile inflammation in the liver. Int J Mol Sci (2018) 19(10):3104. doi: 10.3390/ijms19103104
102. Shan Z, Li L, Atkins CL, Wang M, Wen Y, Jeong J, et al. Chitinase 3-like-1 contributes to acetaminophen-induced liver injury by promoting hepatic platelet recruitment. Elife (2021) 10:e68571. doi: 10.7554/eLife.68571.sa2
103. McDonald B, Jenne CN, Zhuo L, Kimata K, Kubes P. Kupffer cells and activation of endothelial TLR4 coordinate neutrophil adhesion within liver sinusoids during endotoxemia. Am J Physiol Gastrointest Liver Physiol (2013) 305:G797–806. doi: 10.1152/ajpgi.00058.2013
104. Lohse AW, Knolle PA, Bilo K, Uhrig A, Waldmann C, Ibe M, et al. Antigen-presenting function and B7 expression of murine sinusoidal endothelial cells and kupffer cells. Gastroenterology (1996) 110:1175–81. doi: 10.1053/gast.1996.v110.pm8613007
105. Strauss O, Dunbar PR, Bartlett A, Phillips A. The immunophenotype of antigen presenting cells of the mononuclear phagocyte system in normal human liver–a systematic review. J Hepatol (2015) 62:458–68. doi: 10.1016/j.jhep.2014.10.006
106. Huang H, Lu Y, Zhou T, Gu G, Xia Q. Innate immune cells in immune tolerance after liver transplantation. Front Immunol (2018) 9:2401. doi: 10.3389/fimmu.2018.02401
107. Westman J, Grinstein S, Marques PE. Phagocytosis of necrotic debris at sites of injury and inflammation. Front Immunol (2019) 10:3030. doi: 10.3389/fimmu.2019.03030
108. Terpstra V, van Berkel TJ. Scavenger receptors on liver kupffer cells mediate the in vivo uptake of oxidatively damaged red blood cells in mice. Blood (2000) 95:2157–63. doi: 10.1182/blood.V95.6.2157
109. Demetz E, Tymoszuk P, Hilbe R, Volani C, Haschka D, Heim C, et al. The haemochromatosis gene hfe and kupffer cells control LDL cholesterol homeostasis and impact on atherosclerosis development. Eur Heart J (2020) 41:3949–59. doi: 10.1093/eurheartj/ehaa140
110. Wang Y, van der Tuin S, Tjeerdema N, van Dam AD, Rensen SS, Hendrikx T, et al. Plasma cholesteryl ester transfer protein is predominantly derived from kupffer cells. Hepatology (2015) 62:1710–22. doi: 10.1002/hep.27985
111. Evren E, Ringqvist E, Doisne JM, Thaller A, Sleiers N, Flavell RA, et al. CD116+ fetal precursors migrate to the perinatal lung and give rise to human alveolar macrophages. J Exp Med (2022) 219(2):e20210987. doi: 10.1084/jem.20210987
112. Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med (2013) 210:1977–92. doi: 10.1084/jem.20131199
113. Hartl D, Tirouvanziam R, Laval J, Greene CM, Habiel D, Sharma L, et al. Innate immunity of the lung: From basic mechanisms to translational medicine. J Innate Immun (2018) 10:487–501. doi: 10.1159/000487057
114. Hou F, Xiao K, Tang L, Xie L. Diversity of macrophages in lung homeostasis and diseases. Front Immunol (2021) 12:753940. doi: 10.3389/fimmu.2021.753940
115. Ardain A, Marakalala MJ, Leslie A. Tissue-resident innate immunity in the lung. Immunology (2020) 159:245–56. doi: 10.1111/imm.13143
116. Shi T, Denney L, An H, Ho LP, Zheng Y. Alveolar and lung interstitial macrophages: Definitions, functions, and roles in lung fibrosis. J Leukoc Biol (2021) 110:107–14. doi: 10.1002/JLB.3RU0720-418R
117. Cheng P, Li S, Chen H. Macrophages in lung injury, repair, and fibrosis. Cells (2021) 10(2):436. doi: 10.3390/cells10020436
118. Misharin AV, Morales-Nebreda L, Mutlu GM, Budinger GR, Perlman H. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am J Respir Cell Mol Biol (2013) 49:503–10. doi: 10.1165/rcmb.2013-0086MA
119. Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol (2012) 13:1118–28. doi: 10.1038/ni.2419
120. Leach SM, Gibbings SL, Tewari AD, Atif SM, Vestal B, Danhorn T, et al. Human and mouse transcriptome profiling identifies cross-species homology in pulmonary and lymph node mononuclear phagocytes. Cell Rep (2020) 33:108337. doi: 10.1016/j.celrep.2020.108337
121. Sandhu SS, Lower WR. In situ assessment of genotoxic hazards of environmental pollution. Toxicol Ind Health (1989) 5:73–83. doi: 10.1177/074823378900500107
122. Westphalen K, Gusarova GA, Islam MN, Subramanian M, Cohen TS, Prince AS, et al. Sessile alveolar macrophages communicate with alveolar epithelium to modulate immunity. Nature (2014) 506:503–6. doi: 10.1038/nature12902
123. Neupane AS, Willson M, Chojnacki AK, Vargas E Silva Castanheira F, Morehouse C, Carestia A, et al. Patrolling alveolar macrophages conceal bacteria from the immune system to maintain homeostasis. Cell (2020) 183:110–25 e11. doi: 10.1016/j.cell.2020.08.020
124. Xu-Vanpala S, Deerhake ME, Wheaton JD, Parker ME, Juvvadi PR, MacIver N, et al. Functional heterogeneity of alveolar macrophage population based on expression of CXCL2. Sci Immunol (2020) 5(50):eaba7350. doi: 10.1126/sciimmunol.aba7350
125. Schyns J, Bai Q, Ruscitti C, Radermecker C, De Schepper S, Chakarov S, et al. Non-classical tissue monocytes and two functionally distinct populations of interstitial macrophages populate the mouse lung. Nat Commun (2019) 10:3964. doi: 10.1038/s41467-019-11843-0
126. Evren E, Ringqvist E, Willinger T. Origin and ontogeny of lung macrophages: from mice to humans. Immunology (2020) 160:126–38. doi: 10.1111/imm.13154
127. Schyns J, Bureau F, Marichal T. Lung interstitial macrophages: Past, present, and future. J Immunol Res (2018) 2018:5160794. doi: 10.1155/2018/5160794
128. Zhou B, Magana L, Hong Z, Huang LS, Chakraborty S, Tsukasaki Y, et al. The angiocrine Rspondin3 instructs interstitial macrophage transition via metabolic-epigenetic reprogramming and resolves inflammatory injury. Nat Immunol (2020) 21:1430–43. doi: 10.1038/s41590-020-0764-8
129. Byrne AJ, Mathie SA, Gregory LG, Lloyd CM. Pulmonary macrophages: key players in the innate defence of the airways. Thorax (2015) 70:1189–96. doi: 10.1136/thoraxjnl-2015-207020
130. Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med (2018) 24:908–22. doi: 10.1038/s41591-018-0104-9
131. Tosello-Trampont AC, Landes SG, Nguyen V, Novobrantseva TI, Hahn YS. Kuppfer cells trigger nonalcoholic steatohepatitis development in diet-induced mouse model through tumor necrosis factor-alpha production. J Biol Chem (2012) 287:40161–72. doi: 10.1074/jbc.M112.417014
132. Reid DT, Reyes JL, McDonald BA, Vo T, Reimer RA, Eksteen B. Kupffer cells undergo fundamental changes during the development of experimental NASH and are critical in initiating liver damage and inflammation. PloS One (2016) 11:e0159524. doi: 10.1371/journal.pone.0159524
133. Daemen S, Gainullina A, Kalugotla G, He L, Chan MM, Beals JW, et al. Dynamic shifts in the composition of resident and recruited macrophages influence tissue remodeling in NASH. Cell Rep (2021) 34:108626. doi: 10.1016/j.celrep.2020.108626
134. Remmerie A, Martens L, Thone T, Castoldi A, Seurinck R, Pavie B, et al. Osteopontin expression identifies a subset of recruited macrophages distinct from kupffer cells in the fatty liver. Immunity (2020) 53:641–57 e14. doi: 10.1016/j.immuni.2020.08.004
135. Arrese M, Cabrera D, Kalergis AM, Feldstein AE. Innate immunity and inflammation in NAFLD/NASH. Dig Dis Sci (2016) 61:1294–303. doi: 10.1007/s10620-016-4049-x
136. Alisi A, Carsetti R, Nobili V. Pathogen- or damage-associated molecular patterns during nonalcoholic fatty liver disease development. Hepatology (2011) 54:1500–2. doi: 10.1002/hep.24611
137. Tao L, Yi Y, Chen Y, Zhang H, Orning P, Lien E, et al. RIP1 kinase activity promotes steatohepatitis through mediating cell death and inflammation in macrophages. Cell Death Differ (2021) 28:1418–33. doi: 10.1038/s41418-020-00668-w
138. Peters KM, Wilson RB, Borradaile NM. Non-parenchymal hepatic cell lipotoxicity and the coordinated progression of non-alcoholic fatty liver disease and atherosclerosis. Curr Opin Lipidol (2018) 29:417–22. doi: 10.1097/MOL.0000000000000535
139. Leroux A, Ferrere G, Godie V, Cailleux F, Renoud ML, Gaudin F, et al. Toxic lipids stored by kupffer cells correlates with their pro-inflammatory phenotype at an early stage of steatohepatitis. J Hepatol (2012) 57:141–9. doi: 10.1016/j.jhep.2012.02.028
140. Luo W, Xu Q, Wang Q, Wu H, Hua J. Effect of modulation of PPAR-gamma activity on kupffer cells M1/M2 polarization in the development of non-alcoholic fatty liver disease. Sci Rep (2017) 7:44612. doi: 10.1038/srep44612
141. Kanamori Y, Tanaka M, Itoh M, Ochi K, Ito A, Hidaka I, et al. Iron-rich kupffer cells exhibit phenotypic changes during the development of liver fibrosis in NASH. iScience (2021) 24:102032. doi: 10.1016/j.isci.2020.102032
142. Mekala S, Tulimilli SV, Geesala R, Manupati K, Dhoke NR, Das A. Cellular crosstalk mediated by platelet-derived growth factor BB and transforming growth factor beta during hepatic injury activates hepatic stellate cells. Can J Physiol Pharmacol (2018) 96:728–41. doi: 10.1139/cjpp-2017-0768
143. Malehmir M, Pfister D, Gallage S, Szydlowska M, Inverso D, Kotsiliti E, et al. Platelet GPIbalpha is a mediator and potential interventional target for NASH and subsequent liver cancer. Nat Med (2019) 25:641–55. doi: 10.1038/s41591-019-0379-5
144. McGettigan B, McMahan R, Orlicky D, Burchill M, Danhorn T, Francis P, et al. Dietary lipids differentially shape nonalcoholic steatohepatitis progression and the transcriptome of kupffer cells and infiltrating macrophages. Hepatology (2019) 70:67–83. doi: 10.1002/hep.30401
145. Govaere O, Petersen SK, Martinez-Lopez N, Wouters J, Van Haele M, Mancina RM, et al. Macrophage scavenger receptor 1 mediates lipid-induced inflammation in non-alcoholic fatty liver disease. J Hepatol (2022) 76:1001–12. doi: 10.1016/j.jhep.2021.12.012
146. Hasuzawa N, Tatsushima K, Wang L, Kabashima M, Tokubuchi R, Nagayama A, et al. Clodronate, an inhibitor of the vesicular nucleotide transporter, ameliorates steatohepatitis and acute liver injury. Sci Rep (2021) 11:5192. doi: 10.1038/s41598-021-83144-w
147. Crabb DW, Im GY, Szabo G, Mellinger JL, Lucey MR. Diagnosis and treatment of alcohol-associated liver diseases: 2019 practice guidance from the American association for the study of liver diseases. Hepatology (2020) 71:306–33. doi: 10.1002/hep.30866
148. Glaser T, Baiocchi L, Zhou T, Francis H, Lenci I, Grassi G, et al. Pro-inflammatory signalling and gut-liver axis in non-alcoholic and alcoholic steatohepatitis: Differences and similarities along the path. J Cell Mol Med (2020) 24:5955–65. doi: 10.1111/jcmm.15182
149. Ambade A, Lowe P, Kodys K, Catalano D, Gyongyosi B, Cho Y, et al. Pharmacological inhibition of CCR2/5 signaling prevents and reverses alcohol-induced liver damage, steatosis, and inflammation in mice. Hepatology (2019) 69:1105–21. doi: 10.1002/hep.30249
150. Petrasek J, Bala S, Csak T, Lippai D, Kodys K, Menashy V, et al. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J Clin Invest (2012) 122:3476–89. doi: 10.1172/JCI60777
151. Mir H, Meena AS, Chaudhry KK, Shukla PK, Gangwar R, Manda B, et al. Occludin deficiency promotes ethanol-induced disruption of colonic epithelial junctions, gut barrier dysfunction and liver damage in mice. Biochim Biophys Acta (2016) 1860:765–74. doi: 10.1016/j.bbagen.2015.12.013
152. Rao R. Endotoxemia and gut barrier dysfunction in alcoholic liver disease. Hepatology (2009) 50:638–44. doi: 10.1002/hep.23009
153. Suraweera DB, Weeratunga AN, Hu RW, Pandol SJ, Hu R. Alcoholic hepatitis: The pivotal role of kupffer cells. World J Gastrointest Pathophysiol (2015) 6:90–8. doi: 10.4291/wjgp.v6.i4.90
154. Petrasek J, Csak T, Ganz M, Szabo G. Differences in innate immune signaling between alcoholic and non-alcoholic steatohepatitis. J Gastroenterol Hepatol (2013) 28(Suppl 1):93–8. doi: 10.1111/jgh.12020
155. Roh YS, Zhang B, Loomba R, Seki E. TLR2 and TLR9 contribute to alcohol-mediated liver injury through induction of CXCL1 and neutrophil infiltration. Am J Physiol Gastrointest Liver Physiol (2015) 309:G30–41. doi: 10.1152/ajpgi.00031.2015
156. Mysore KR, Leung DH. Hepatitis b and c. Clin Liver Dis (2018) 22:703–22. doi: 10.1016/j.cld.2018.06.002
157. Higuera-de la Tijera F, Servin-Caamano A, Servin-Abad L. Progress and challenges in the comprehensive management of chronic viral hepatitis: Key ways to achieve the elimination. World J Gastroenterol (2021) 27:4004–17. doi: 10.3748/wjg.v27.i26.4004
158. Chiale C, Marchese AM, Robek MD. Innate immunity and HBV persistence. Curr Opin Virol (2021) 49:13–20. doi: 10.1016/j.coviro.2021.04.003
159. Zhou Y, Zhang Y, Moorman JP, Yao ZQ, Jia ZS. Viral (hepatitis c virus, hepatitis b virus, HIV) persistence and immune homeostasis. Immunology (2014) 143:319–30. doi: 10.1111/imm.12349
160. Rehermann B. Pathogenesis of chronic viral hepatitis: differential roles of T cells and NK cells. Nat Med (2013) 19:859–68. doi: 10.1038/nm.3251
161. Lang PA, Recher M, Honke N, Scheu S, Borkens S, Gailus N, et al. Tissue macrophages suppress viral replication and prevent severe immunopathology in an interferon-i-dependent manner in mice. Hepatology (2010) 52:25–32. doi: 10.1002/hep.23640
162. Tu Z, Bozorgzadeh A, Pierce RH, Kurtis J, Crispe IN, Orloff MS. TLR-dependent cross talk between human kupffer cells and NK cells. J Exp Med (2008) 205:233–44. doi: 10.1084/jem.20072195
163. Boltjes A, Movita D, Boonstra A, Woltman AM. The role of kupffer cells in hepatitis b and hepatitis c virus infections. J Hepatol (2014) 61:660–71. doi: 10.1016/j.jhep.2014.04.026
164. Faure-Dupuy S, Delphin M, Aillot L, Dimier L, Lebossé F, Fresquet J, et al. Hepatitis b virus-induced modulation of liver macrophage function promotes hepatocyte infection. J Hepatol (2019) 71:1086–98. doi: 10.1016/j.jhep.2019.06.032
165. Yu X, Lan P, Hou X, Han Q, Lu N, Li T, et al. HBV inhibits LPS-induced NLRP3 inflammasome activation and IL-1beta production via suppressing the NF-kappaB pathway and ROS production. J Hepatol (2017) 66:693–702. doi: 10.1016/j.jhep.2016.12.018
166. Riordan SM, Skinner N, Kurtovic J, Locarnini S, Visvanathan K. Reduced expression of toll-like receptor 2 on peripheral monocytes in patients with chronic hepatitis b. Clin Vaccine Immunol (2006) 13:972–4. doi: 10.1128/CVI.00396-05
167. Tucker SM, Jonas MM, Perez-Atayde AR. Hyaline droplets in kupffer cells: a novel diagnostic clue for autoimmune hepatitis. Am J Surg Pathol (2015) 39:772–8. doi: 10.1097/PAS.0000000000000395
168. Khedr MA, Adawy NM, Salim TA, Salem ME, Ghazy RM, Elharoun AS, et al. Kupffer cell hyaline globules in children with autoimmune hepatitis. J Clin Exp Hepatol (2022) 12:20–8. doi: 10.1016/j.jceh.2021.04.013
169. Himoto T, Kadota K, Fujita K, Nomura T, Morishita A, Yoneyama H, et al. The pathological appearance of hyaline droplets in kupffer cells is not specific to patients with autoimmune hepatitis. Int J Clin Exp Pathol (2017) 10:8703–8.
170. Zhang H, Liu M, Zhong W, Zheng Y, Li Y, Guo L, et al. Leaky gut driven by dysbiosis augments activation and accumulation of liver macrophages via RIP3 signaling pathway in autoimmune hepatitis. Front Immunol (2021) 12:624360. doi: 10.3389/fimmu.2021.624360
171. Weber LW, Boll M, Stampfl A. Hepatotoxicity and mechanism of action of haloalkanes: carbon tetrachloride as a toxicological model. Crit Rev Toxicol (2003) 33:105–36. doi: 10.1080/713611034
172. Mossanen JC, Tacke F. Acetaminophen-induced acute liver injury in mice. Lab Anim (2015) 49:30–6. doi: 10.1177/0023677215570992
173. Kolodziejczyk AA, Federici S, Zmora N, Mohapatra G, Dori-Bachash M, Hornstein S, et al. Acute liver failure is regulated by MYC- and microbiome-dependent programs. Nat Med (2020) 26:1899–911. doi: 10.1038/s41591-020-1102-2
174. Roth K, Strickland J, Copple BL. Regulation of macrophage activation in the liver after acute injury: Role of the fibrinolytic system. World J Gastroenterol (2020) 26:1879–87. doi: 10.3748/wjg.v26.i16.1879
175. Zhao J, Kim JW, Zhou Z, Qi J, Tian W, Lim CW, et al. Macrophage-inducible c-type lectin signaling exacerbates acetaminophen-induced liver injury by promoting kupffer cell activation in mice. Mol Pharmacol (2021) 99:92–103. doi: 10.1124/molpharm.120.000043
176. Triantafyllou E, Gudd CL, Mawhin MA, Husbyn HC, Trovato FM, Siggins MK, et al. PD-1 blockade improves kupffer cell bacterial clearance in acute liver injury. J Clin Invest (2021) 131(4):e140196. doi: 10.1172/JCI140196
177. Ju C, Reilly TP, Bourdi M, Radonovich MF, Brady JN, George JW, et al. Protective role of kupffer cells in acetaminophen-induced hepatic injury in mice. Chem Res Toxicol (2002) 15:1504–13. doi: 10.1021/tx0255976
178. Nguyen NT, Umbaugh DS, Sanchez-Guerrero G, Ramachandran A, Jaeschke H. Kupffer cells regulate liver recovery through induction of chemokine receptor CXCR2 on hepatocytes after acetaminophen overdose in mice. Arch Toxicol (2022) 96:305–20. doi: 10.1007/s00204-021-03183-0
179. Mossanen JC, Krenkel O, Ergen C, Govaere O, Liepelt A, Puengel T, et al. Chemokine (C-c motif) receptor 2-positive monocytes aggravate the early phase of acetaminophen-induced acute liver injury. Hepatology (2016) 64:1667–82. doi: 10.1002/hep.28682
180. Pinzani M, Macias-Barragan J. Update on the pathophysiology of liver fibrosis. Expert Rev Gastroenterol Hepatol (2010) 4:459–72. doi: 10.1586/egh.10.47
182. Roehlen N, Crouchet E, Baumert TF. Liver fibrosis: Mechanistic concepts and therapeutic perspectives. Cells (2020) 9(4):875. doi: 10.3390/cells9040875
183. She S, Wu X, Zheng D, Pei X, Ma J, Sun Y, et al. PSMP/MSMP promotes hepatic fibrosis through CCR2 and represents a novel therapeutic target. J Hepatol (2020) 72:506–18. doi: 10.1016/j.jhep.2019.09.033
184. Sasaki R, Devhare PB, Steele R, Ray R, Ray RB. Hepatitis c virus-induced CCL5 secretion from macrophages activates hepatic stellate cells. Hepatology (2017) 66:746–57. doi: 10.1002/hep.29170
185. Fabregat I, Caballero-Diaz D. Transforming growth factor-beta-Induced cell plasticity in liver fibrosis and hepatocarcinogenesis. Front Oncol (2018) 8:357. doi: 10.3389/fonc.2018.00357
186. Tacke F, Weiskirchen R. Update on hepatic stellate cells: pathogenic role in liver fibrosis and novel isolation techniques. Expert Rev Gastroenterol Hepatol (2012) 6:67–80. doi: 10.1586/egh.11.92
187. Dal-Secco D, Wang J, Zeng Z, Kolaczkowska E, Wong CH, Petri B, et al. A dynamic spectrum of monocytes arising from the in situ reprogramming of CCR2+ monocytes at a site of sterile injury. J Exp Med (2015) 212:447–56. doi: 10.1084/jem.20141539
188. Ramachandran P, Pellicoro A, Vernon MA, Boulter L, Aucott RL, Ali A, et al. Differential ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc Natl Acad Sci U.S.A. (2012) 109:E3186–95. doi: 10.1073/pnas.1119964109
189. Friedman SL, Pinzani M. Hepatic fibrosis 2022: Unmet needs and a blueprint for the future. Hepatology (2022) 75:473–88. doi: 10.1002/hep.32285
190. Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest (2005) 115:56–65. doi: 10.1172/JCI200522675
191. Massarweh NN, El-Serag HB. Epidemiology of hepatocellular carcinoma and intrahepatic cholangiocarcinoma. Cancer Control (2017) 24:1073274817729245. doi: 10.1177/1073274817729245
192. Wang C, Chen WJ, Wu YF, You P, Zheng SY, Liu CC, et al. The extent of liver injury determines hepatocyte fate toward senescence or cancer. Cell Death Dis (2018) 9:575. doi: 10.1038/s41419-018-0622-x
193. Karin M, Dhar D. Liver carcinogenesis: from naughty chemicals to soothing fat and the surprising role of NRF2. Carcinogenesis (2016) 37:541–6. doi: 10.1093/carcin/bgw060
194. Yu LX, Ling Y, Wang HY. Role of nonresolving inflammation in hepatocellular carcinoma development and progression. NPJ Precis Oncol (2018) 2:6. doi: 10.1038/s41698-018-0048-z
195. Yang YM, Kim SY, Seki E. Inflammation and liver cancer: Molecular mechanisms and therapeutic targets. Semin Liver Dis (2019) 39:26–42. doi: 10.1055/s-0038-1676806
196. Eggert T, Wolter K, Ji J, Ma C, Yevsa T, Klotz S, et al. Distinct functions of senescence-associated immune responses in liver tumor surveillance and tumor progression. Cancer Cell (2016) 30:533–47. doi: 10.1016/j.ccell.2016.09.003
197. Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature (2011) 479:547–51. doi: 10.1038/nature10599
198. Ding T, Xu J, Wang F, Shi M, Zhang Y, Li SP, et al. High tumor-infiltrating macrophage density predicts poor prognosis in patients with primary hepatocellular carcinoma after resection. Hum Pathol (2009) 40:381–9. doi: 10.1016/j.humpath.2008.08.011
199. Li X, Yao W, Yuan Y, Chen P, Li B, Li J, et al. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut (2017) 66:157–67. doi: 10.1136/gutjnl-2015-310514
200. Huang Y, Ge W, Zhou J, Gao B, Qian X, Wang W. The role of tumor associated macrophages in hepatocellular carcinoma. J Cancer (2021) 12:1284–94. doi: 10.7150/jca.51346
201. Zhu Y, Yang J, Xu D, Gao XM, Zhang Z, Hsu JL, et al. Disruption of tumour-associated macrophage trafficking by the osteopontin-induced colony-stimulating factor-1 signalling sensitises hepatocellular carcinoma to anti-PD-L1 blockade. Gut (2019) 68:1653–66. doi: 10.1136/gutjnl-2019-318419
202. Medina-Echeverz J, Eggert T, Han M, Greten TF. Hepatic myeloid-derived suppressor cells in cancer. Cancer Immunol Immunother (2015) 64:931–40. doi: 10.1007/s00262-015-1736-y
203. Arvanitakis K, Koletsa T, Mitroulis I, Germanidis G. Tumor-associated macrophages in hepatocellular carcinoma pathogenesis, prognosis and therapy. Cancers (Basel) (2022) 14(1):226. doi: 10.3390/cancers14010226
204. Zhou D, Luan J, Huang C, Li J. Tumor-associated macrophages in hepatocellular carcinoma: Friend or foe? Gut Liver (2021) 15:500–16. doi: 10.5009/gnl20223
205. Bartneck M, Schrammen PL, Mockel D, Govaere O, Liepelt A, Krenkel O, et al. The CCR2(+) macrophage subset promotes pathogenic angiogenesis for tumor vascularization in fibrotic livers. Cell Mol Gastroenterol Hepatol (2019) 7:371–90. doi: 10.1016/j.jcmgh.2018.10.007
206. Berenson CS, Kruzel RL, Eberhardt E, Dolnick R, Minderman H, Wallace PK, et al. Impaired innate immune alveolar macrophage response and the predilection for COPD exacerbations. Thorax (2014) 69:811–8. doi: 10.1136/thoraxjnl-2013-203669
207. Belchamber KBR, Singh R, Batista CM, Whyte MK, Dockrell DH, Kilty I, et al. Defective bacterial phagocytosis is associated with dysfunctional mitochondria in COPD macrophages. Eur Respir J (2019) 54(4):1802244. doi: 10.1183/13993003.02244-2018
208. Xiong Z, Leme AS, Ray P, Shapiro SD, Lee JS. CX3CR1+ lung mononuclear phagocytes spatially confined to the interstitium produce TNF-alpha and IL-6 and promote cigarette smoke-induced emphysema. J Immunol (2011) 186:3206–14. doi: 10.4049/jimmunol.1003221
209. Moon C, Lee YJ, Park HJ, Chong YH, Kang JL. N-acetylcysteine inhibits RhoA and promotes apoptotic cell clearance during intense lung inflammation. Am J Respir Crit Care Med (2010) 181:374–87. doi: 10.1164/rccm.200907-1061OC
210. Akata K, van Eeden SF. Lung macrophage functional properties in chronic obstructive pulmonary disease. Int J Mol Sci (2020) 21(3):853. doi: 10.3390/ijms21030853
211. Pei W, Zhang Y, Li X, Luo M, Chen T, Zhang M, et al. LncRNA AK085865 depletion ameliorates asthmatic airway inflammation by modulating macrophage polarization. Int Immunopharmacol (2020) 83:106450. doi: 10.1016/j.intimp.2020.106450
212. Chung S, Kim JY, Song MA, Park GY, Lee YG, Karpurapu M, et al. FoxO1 is a critical regulator of M2-like macrophage activation in allergic asthma. Allergy (2019) 74:535–48. doi: 10.1111/all.13626
213. Zhu C, Zhang L, Liu Z, Li C, Bai Y, Wang L. Atractylenolide III reduces NLRP3 inflammasome activation and Th1/Th2 imbalances in both in vitro and in vivo models of asthma. Clin Exp Pharmacol Physiol (2020) 47:1360–7. doi: 10.1111/1440-1681.13306
214. Chenuet P, Marquant Q, Fauconnier L, Youness A, Mellier M, Marchiol T, et al. NLRP6 negatively regulates type 2 immune responses in mice. Allergy (2022) 77(11):3320–36. doi: 10.1111/all.15388
215. Pyle CJ, Uwadiae FI, Swieboda DP, Harker JA. Early IL-6 signalling promotes IL-27 dependent maturation of regulatory T cells in the lungs and resolution of viral immunopathology. PloS Pathog (2017) 13:e1006640. doi: 10.1371/journal.ppat.1006640
216. Lu D, Lu J, Ji X, Ji Y, Zhang Z, Peng H, et al. IL27 suppresses airway inflammation, hyperresponsiveness and remodeling via the STAT1 and STAT3 pathways in mice with allergic asthma. Int J Mol Med (2020) 46:641–52. doi: 10.3892/ijmm.2020.4622
217. Qin L, Li Z, Fan Y, Fang X, Zhang C, Yue J, et al. Exploration of plasma interleukin-27 levels in asthma patients and the correlation with lung function. Respir Med (2020) 175:106208. doi: 10.1016/j.rmed.2020.106208
218. Mathie SA, Dixon KL, Walker SA, Tyrrell V, Mondhe M, O'Donnell VB, et al. Alveolar macrophages are sentinels of murine pulmonary homeostasis following inhaled antigen challenge. Allergy (2015) 70:80–9. doi: 10.1111/all.12536
219. Saradna A, Do DC, Kumar S, Fu QL, Gao P. Macrophage polarization and allergic asthma. Transl Res (2018) 191:1–14. doi: 10.1016/j.trsl.2017.09.002
220. Miki H, Pei H, Gracias DT, Linden J, Croft M. Clearance of apoptotic cells by lung alveolar macrophages prevents development of house dust mite-induced asthmatic lung inflammation. J Allergy Clin Immunol (2021) 147:1087–92 e3. doi: 10.1016/j.jaci.2020.10.005
221. Erriah M, Pabreja K, Fricker M, Baines KJ, Donnelly LE, Bylund J, et al. Galectin-3 enhances monocyte-derived macrophage efferocytosis of apoptotic granulocytes in asthma. Respir Res (2019) 20:1. doi: 10.1186/s12931-018-0967-9
222. Grabiec AM, Denny N, Doherty JA, Happonen KE, Hankinson J, Connolly E, et al. Diminished airway macrophage expression of the axl receptor tyrosine kinase is associated with defective efferocytosis in asthma. J Allergy Clin Immunol (2017) 140:1144–6 e4. doi: 10.1016/j.jaci.2017.03.024
223. Shibata T, Habiel DM, Coelho AL, Kunkel SL, Lukacs NW, Hogaboam CM. Axl receptor blockade ameliorates pulmonary pathology resulting from primary viral infection and viral exacerbation of asthma. J Immunol (2014) 192:3569–81. doi: 10.4049/jimmunol.1302766
224. Cohen TS, Prince A. Cystic fibrosis: a mucosal immunodeficiency syndrome. Nat Med (2012) 18:509–19. doi: 10.1038/nm.2715
225. Bonfield TL, Panuska JR, Konstan MW, Hilliard KA, Hilliard JB, Ghnaim H, et al. Inflammatory cytokines in cystic fibrosis lungs. Am J Respir Crit Care Med (1995) 152:2111–8. doi: 10.1164/ajrccm.152.6.8520783
226. Kopp BT, Abdulrahman BA, Khweek AA, Kumar SB, Akhter A, Montione R, et al. Exaggerated inflammatory responses mediated by burkholderia cenocepacia in human macrophages derived from cystic fibrosis patients. Biochem Biophys Res Commun (2012) 424:221–7. doi: 10.1016/j.bbrc.2012.06.066
227. Del Porto P, Cifani N, Guarnieri S, Domenico EG, Mariggiò MA, Spadaro F, et al. Dysfunctional CFTR alters the bactericidal activity of human macrophages against pseudomonas aeruginosa. PloS One (2011) 6:e19970. doi: 10.1371/journal.pone.0019970
228. Pang Z, Xu Y, Zhu Q. Early growth response 1 suppresses macrophage phagocytosis by inhibiting NRF2 activation through upregulation of autophagy during pseudomonas aeruginosa infection. Front Cell Infect Microbiol (2021) 11:773665. doi: 10.3389/fcimb.2021.773665
229. Bruscia EM, Zhang PX, Ferreira E, Caputo C, Emerson JW, Tuck D, et al. Macrophages directly contribute to the exaggerated inflammatory response in cystic fibrosis transmembrane conductance regulator-/- mice. Am J Respir Cell Mol Biol (2009) 40:295–304. doi: 10.1165/rcmb.2008-0170OC
230. Bruscia EM, Zhang PX, Satoh A, Caputo C, Medzhitov R, Shenoy A, et al. Abnormal trafficking and degradation of TLR4 underlie the elevated inflammatory response in cystic fibrosis. J Immunol (2011) 186:6990–8. doi: 10.4049/jimmunol.1100396
231. Law SM, Stanfield SJ, Hardisty GR, Dransfield I, Campbell CJ, Gray RD. Human cystic fibrosis monocyte derived macrophages display no defect in acidification of phagolysosomes when measured by optical nanosensors. J Cyst Fibros (2020) 19:203–10. doi: 10.1016/j.jcf.2019.09.003
232. Li C, Wu Y, Riehle A, Ma J, Kamler M, Gulbins E, et al. Staphylococcus aureus survives in cystic fibrosis macrophages, forming a reservoir for chronic pneumonia. Infect Immun (2017) 85(5):e00883-16. doi: 10.1128/IAI.00883-16
233. Connolly E, Morgan DJ, Franklin M, Simpson A, Shah R, Brand OJ, et al. Neurturin regulates the lung-resident macrophage inflammatory response to viral infection. Life Sci Alliance (2020) 3(12):e202000780. doi: 10.26508/lsa.202000780
234. Kim HM, Kang YM, Ku KB, Park EH, Yum J, Kim JC, et al. The severe pathogenicity of alveolar macrophage-depleted ferrets infected with 2009 pandemic H1N1 influenza virus. Virology (2013) 444:394–403. doi: 10.1016/j.virol.2013.07.006
235. Kim HM, Lee YW, Lee KJ, Kim HS, Cho SW, van Rooijen N, et al. Alveolar macrophages are indispensable for controlling influenza viruses in lungs of pigs. J Virol (2008) 82:4265–74. doi: 10.1128/JVI.02602-07
236. Verma AK, Bansal S, Bauer C, Muralidharan A, Sun K. Influenza infection induces alveolar macrophage dysfunction and thereby enables noninvasive streptococcus pneumoniae to cause deadly pneumonia. J Immunol (2020) 205:1601–7. doi: 10.4049/jimmunol.2000094
237. Halstead ES, Chroneos ZC. Lethal influenza infection: Is a macrophage to blame? Expert Rev Anti Infect Ther (2015) 13:1425–8. doi: 10.1586/14787210.2015.1094375
238. Xu Y, Liu L. Curcumin alleviates macrophage activation and lung inflammation induced by influenza virus infection through inhibiting the NF-kappaB signaling pathway. Influenza Other Respir Viruses (2017) 11:457–63. doi: 10.1111/irv.12459
Keywords: macrophages, liver, lung, homeostasis, inflammation, Kupffer cells, alveolar macrophages, interstitial macrophages
Citation: Kulle A, Thanabalasuriar A, Cohen TS and Szydlowska M (2022) Resident macrophages of the lung and liver: The guardians of our tissues. Front. Immunol. 13:1029085. doi: 10.3389/fimmu.2022.1029085
Received: 26 August 2022; Accepted: 09 November 2022;
Published: 30 November 2022.
Edited by:
Prakash Ramachandran, University of Edinburgh, United KingdomReviewed by:
Frank Tacke, Charité Universitätsmedizin Berlin, GermanyJoanna Kirman, University of Otago, New Zealand
Yan Chun Li, The University of Chicago, United States
Copyright © 2022 Kulle, Thanabalasuriar, Cohen and Szydlowska. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marta Szydlowska, bWFydGEuc3p5ZGxvd3NrYUBhc3RyYXplbmVjYS5jb20=