 Günther Schönrich
Günther Schönrich Mohammed O. Abdelaziz1
Mohammed O. Abdelaziz1 Martin J. Raftery
Martin J. Raftery
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Immunol., 07 October 2022
Sec. Autoimmune and Autoinflammatory Disorders : Autoimmune Disorders
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1028972
This article is part of the Research TopicImmune Evasion Mechanisms and Their Role in the Pathogenesis of Autoimmune DisordersView all 6 articles
Multiple Sclerosis (MS) is an autoimmune disease that is characterized by inflammation and demyelination of nerve cells. There is strong evidence that Epstein-Barr virus (EBV), a human herpesvirus infecting B cells, greatly increases the risk of subsequent MS. Intriguingly, EBV not only induces human interleukin-10 but also encodes a homologue of this molecule, which is a key anti-inflammatory cytokine of the immune system. Although EBV-encoded IL-10 (ebvIL-10) has a high amino acid identity with its cellular counterpart (cIL-10), it shows more restricted and partially weaker functionality. We propose that both EBV-induced cIL-10 and ebvIL-10 act in a temporally and functionally coordinated manner helping the pathogen to establish latency in B cells and, at the same time, to balance the function of antiviral T cells. As a result, the EBV load persisting in the immune system is kept at a constant but individually different level (set point). During this immunological tug of war between virus and host, however, MS can be induced as collateral damage if the set point is too high. Here, we discuss a possible role of ebvIL-10 and EBV-induced cIL-10 in EBV-driven pathogenesis of MS.
Epstein–Barr virus (EBV), a human gammaherpesvirus, persists in 95% of the world population due to sophisticated immune evasion strategies (1). After transmission of EBV to naïve individuals, the first site of replication is in the orophayryngeal epithelium (2). Subsequently, the virus crosses the mucosal barrier, infects naïve B cells, and establishes latency within a few days post infection (3). Latency requires expression of viral proteins that induce proliferation, growth transformation and differentiation of naïve B cells into memory B cells (4, 5). EBV persists lifelong as episomal DNA in resting memory B cells that percolate through lymphoid tissue and are detectable in the circulation (6). After stimulation, latently infected memory B cells terminally differentiate into plasmablasts and plasma cells resulting in reactivation, lytic replication and production of infectious particles, which infect new naïve B cells completing the viral life cycle and replenishing the EBV reservoir (7, 8). EBV reactivation is tightly controlled by memory T cells, which keep the EBV-load for each individual at a constant level (9–11). During EBV reactivation, epithelial cells of the oropharynx can be reinfected and shed viral particles into the saliva (12), which passes EBV on to new individuals (13).
EBV like human cytomegalovirus encodes a viral IL-10 homologue (ebvIL-10) of cellular IL-10 (cIL-10) (14). ebvIL-10 has 82% amino acid identity to cIL-10 (15, 16), a key anti-inflammatory cytokine of the host with pleiotropic biological activity (17–19). EBV exploits cIL-10 and its viral homologue to evade host immunity and establish a balance with antiviral T cells similar to other viruses that persist in the host (20–23). ebvIL-10 is expressed within a few hours of starting the EBV lytic cycle (24, 25). Approximately 20-30 hours later, in the pre-latent phase, EBV-encoded latent membrane protein-1 (LMP-1) and EBV-encoded small non-coding RNAs (EBERs) upregulate the production of cIL-10 (25–28).
EBV infection seems to be a conditio sine qua non for development of multiple sclerosis (MS), the most common chronic inflammatory disease of the central nerve system (CNS) (29–34). The hallmark of MS pathogenesis is an immune attack against axons and their insulating myelin sheath together with disruption of the blood-brain barrier (BBB) (35). As a consequence, inflammation, demyelination, remyelination, neurodegeneration and glial scar formation occur (36). These pathological lesions are either focally or diffusely distributed in the white and grey matter of the brain and spinal cord and result in neurological deficits that differ substantially among patients and over the disease course (37). The global prevalence of MS is rising with nearly 3 million people living with MS worldwide, mostly adults between 20 years and 40 years with females affected twice as likely as males (38, 39). With increasing age, new demyelinated lesions appear less often, but some inflammatory plaques remain. A body of evidence indicates that the immune response is critically involved in MS pathogenesis (40, 41). For development of effective MS therapy and prophylaxis, it is crucial to understand the detailed mechanisms of how EBV drives MS underlying immunopathology.
In this article we discuss how ebvIL-10 and EBV-induced cIL-10 could help the virus evade the immune system and persist in then host while at same time drive inflammatory processes that contribute to MS pathogenesis.
Biologically active IL-10 binds with high-affinity to a private receptor subunit (IL-10R1), and with low-affinity to a public receptor subunit (IL-10R2) shared in common with other members of the cytokine class II family (42). IL-10R1 represents the ligand binding subunit of the receptor complex, whereas IL-10R2 is the signaling subunit. Upon binding to IL-10, IL-10R1 induces a conformational change in IL-10R2, permitting IL-10R2 to also bind IL-10. Subsequently, the intracellular Janus tyrosine kinases Jak1 and Tyk2 are activated resulting in phosphorylation of signal transducer and activator of transcription 3 (STAT3), which induces the cellular responses.
The affinity of IL-10R1 for ebvIL-10 is approximately 1000-fold less than for cIL-10 (43). Thus, ebvIL-10 is a selective agonist with impaired binding to the IL-10R1 (16). This implies that cIL-10 and ebvIL-10 share some but not all biological activities. In accordance, ebvIL-10 is unable to signal thymocytes and mast cells, which both express low levels of IL-10R1, whereas cIL-10 signals normally (44–46). Moreover, ebvIL-10 does not upregulate MHC class II molecules on B cells (47). In comparison to cIL-10, ebvIL-10 impacts only weakly on DC function (48). Furthermore, ebvIL-10 can inhibit the effects of cIL-10 on monocytes (49). In fact, ebvIL-10 reduced cIL-10 induced STAT3 phosphorylation to levels similar to monocytes stimulated with ebvIL-10 alone (49). In contrast, ebvIL-10 has retained the ability to increase B cell growth and differentiation (50).
Thus, ebvIL-10 has evolved to retain some but not all functions of cIL-10 and in some circumstances can even compete with cIL-10. Both molecules act in a coordinated fashion, however, to facilitate EBV persistence in the host by balancing viral load and antiviral immune responses.
Primary EBV infection in developing countries takes place during childhood and is in general asymptomatic (51). In contrast, primary infection in developed countries occurs during adolescence (11 to 19 years of age) and young adulthood (20 to 24 years of age) and is associated with infectious mononucleosis (IM) (52). IM is a self-limiting disease with fever, sore throat, skin rash, tender lymphoadenopathy and hepatosplenomegaly. These IM symptoms are a consequence of an exaggerated antiviral T cell response involving predominantly CD8+ T cells (53–55). In IM patients, cIL-10 levels as well as the EBV load in circulating B cells are increased compared to healthy controls (56–58). Moreover, EBV-infected B cells dramatically enlarge the CD8+ T cell compartment resulting in up to 50% of CD8+ T cells reacting to lytic EBV antigens (59). The disease severity correlates with both blood EBV load and proliferation of CD8+ T cells (53). Although the blood EBV load is high in both asymptomatic primary infection and IM, only IM patients showed exaggerated T cell responses (60). Strikingly, the risk for MS is absent in EBV-seronegative individuals, increases after EBV infection, is high after IM in adolescence and particularly high after IM in early adulthood (56, 61–66). Accordingly, developed countries experience most cases of IM and have considerably higher prevalences and incidences of MS compared to developing countries (38, 67).
Taken together, primary EBV infection in adolescents and young adults is associated with IM, high blood EBV load, high cIL-10 levels, exaggerated T cell responses, and a high risk for MS.
After primary infection, T cells continuously control and interact with EBV-infected B cells (68). The size of the peripheral EBV reservoir is determined to a large part by the frequency and functional activity of CD8+ T cells eliminating EBV-infected B cells. This loss is countered by periodic virus reactivation and fresh infection of naïve B cells, which are then reprogrammed into latently EBV-infected memory B cells within a few days post infection (3). Healthy EBV-seropositive humans vary greatly in the number of latently infected memory B cells, from 1 to 50 per 106 peripheral B cells (69). However, in each person the levels of latent EBV remains stable over time, defining a steady state or “set point” for each individual (69–71). After IM, the EBV loads in the saliva are persistently high and the pool of latently EBV-infected B cells remains elevated for a considerable time period as compared to healthy EBV carriers without a record of IM (53, 72).
An elevated set point of the EBV load may facilitate and drive MS. In accordance, peripheral blood mononuclear cells from MS patients harbor increased numbers of latently EBV-infected B cells compared to healthy controls (73). Moreover, monoclonal antibodies directed against CD20+ cells, which reduce MS pathology, spare plasma cells and do not reduce the amount of immunoglobulin but remove B cells including those with latent EBV thereby decreasing the set point (74, 75). Accordingly, the effectiveness of anti-CD20 treatment in MS patients is probably not based on the reduction of autoantibody levels but rather on depleting B cells that drive MS by presenting auto-antigens (76). Consistent with this view, individuals overexpressing B-cell activating factor (BAFF), which increases B-cell activation, differentiation, and survival, have a higher risk for MS (77, 78). Moreover, adoptive transfer of EBV-specific T cells, a promising strategy in MS treatment, reduces the pool size of latently EBV-infected B cells (79–81). The observation that high serum titres of antibodies against EBV nuclear antigen 1 (EBNA1), an essential viral protein for EBV latency, enhance the risk for MS supports this notion (82). Importantly, anti-EBNA1 antibody titres were already significantly elevated 5 or more years prior to MS onset suggesting that a high EBV set point leads MS and not vice versa (83, 84). In MS, the anti-EBNA1 IgG titre correlate inversely with the frequency of EBV-specific CD8+ T cells supporting the notion that these immune cells control EBV reactivation and the set point of the virus load (85). Intriguingly, the anti-EBNA1 antibody levels after primary EBV infection are to a large part genetically determined, for example by variants of MHC class II molecules (86–89). Moreover, co-infection with Malaria increases the circulating EBV load (90). Thus, genetic and environmental factors such as coinfections with other pathogns or gut microbiota influence the EBV load and MS development, for example through modulating the cytokine network and triggering EBV reactivation (55, 68, 91–96).
Altogether, these data support the idea that an increased load of latently EBV-infected memory B cells after primary infection is due to genetic and environmental factors and represents an important risk factor for MS.
The coordinated action of EBV-induced cIL-10 and ebvIL-10 regulates the set point of the EBV load not only by enhancing differentiation, proliferation and survival of EBV-infected naive B cells but also by regulating the activity of antiviral T cells (Figure 1). Dysregulation of this delicate balance could facilitate the spread of latent EBV in the memory B cell compartment thereby increasing the set point and the risk for MS. In line with this view, MS patients show decreased T cell reactivity against autologous lymphoblastoid cell lines (LCLs) indicating an attenuation of the immune surveillance (97). LCLs are EBV-transformed B-cell lines that continuously proliferate in vitro. They can be easily established by EBV infection or derived spontaneously ex vivo from peripheral blood B lymphocytes in the absence or inhibition of T and NK cells. Emphasizing the regulatory role of IL-10 in this context, in a mouse model gammaherpesvirus-induced IL-10 was required for expansion and differentiation of latently infected B cells, while at the same time interfering with the activity of antiviral CD8+ T cells (98).
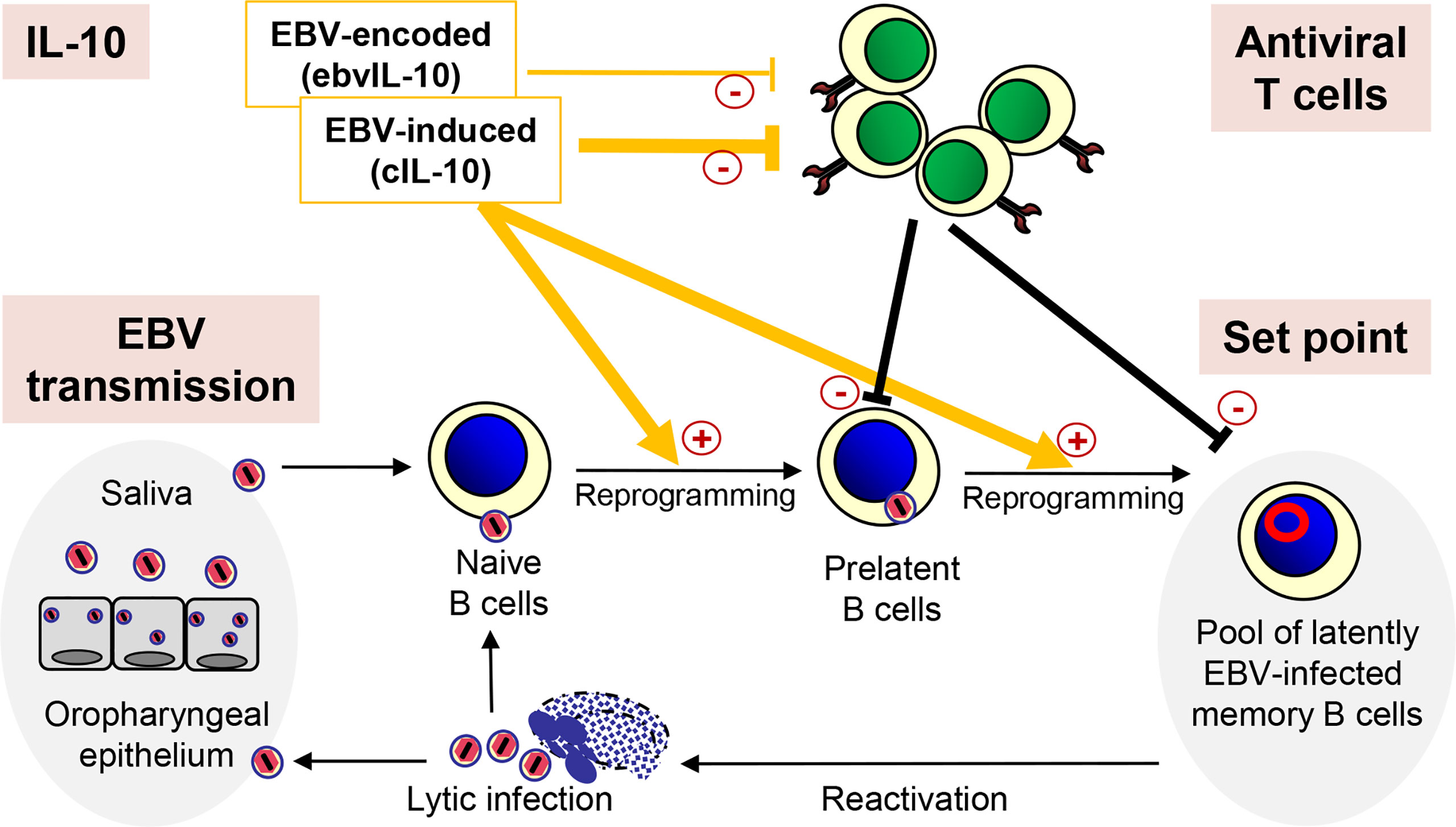
Figure 1 Regulation of the EBV load by IL-10. After transmission, EBV infects naïve B cells, which are then reprogrammed and replenish the pool of latently EBV-infected memory B cells. Occasionally, EBV reactivates and lytically infected plasma cells produce new virus particles, which infect new naïve B cells completing the viral life cycle. After reinfection, epithelial cells of the oropharynx shed viral particles into the saliva, which passes EBV on to new individuals. Importantly, the coordinated action of EBV-induced cIL-10 and ebvIL-10 regulates the individual set point of the EBV load not only by reprogramming EBV-infected naïve B cells but also by regulating the activity of antiviral T cells that eliminate newly infected B cells.
It has been shown that cIL-10 can inhibit CD8+ T cell function directly thereby facilitating persistent virus infection (99). EBV-induced cIL-10 abrogates the capacity of T cells to inhibit the outgrowth of autologous LCLs (100). Similarly, ebvIL-10 has been reported to interfere with elimination of newly infected B cells by CD8+ T cells (100–102). As a potential mechanism, both EBV-induced cIL-10 and ebvIL-10 downregulate the transport of peptides into the endoplasmic reticulum resulting in reduced surface expression of MHC class I molecules and presentation of viral epitopes to CD8+ T cells (103). However, other investigators reported that ebvIL-10 can also enhance the activity of EBV-specific CD8+ T cells (104). Moreover, cIL-10-stimulatory effects on functionality of CD8+ T cells have also been described (105–107). Thus, the coordinated action of ebvIL-10 and EBV-induced cIL-10 may allow minimal activity of EBV-specific CD8+ T cells that is required to push EBV back into latency and prevent outgrowth of B cell lymphomas due to opportunistic expansion of latently infected B cells.
In conclusion, ebvIL-10 and EBV-induced cIL-10, which are regulated by genetic host factors and environmental cues, play a crucial role in defining the set point of the individual EBV load in the periphery.
Individuals with high EBV load display virus-specific CD8+ T cells showing signs of exhaustion such as surface upregulation of programmed cell death protein 1 (PD-1) (108). In a vicious cycle, EBV-specific CD8+ T cells confronted with a high EBV load may be further stimulated to proliferate without being able to re-establish a balance (68, 109). As a consequence, control of EBV reactivation in MS patients becomes defective resulting in further expansion of latently EBV-infected B cells that may also include autoreactive B cells contributing to MS pathogenesis (85). Possibly due to T cell exhaustion the impairment of EBV control by antiviral T cells in MS patients worsens with age (110) whereas strong CD8+ EBV-specific T cell responses are found in patients with early and active MS (111, 112).
A high EBV load is associated with frequent but stochastic reactivation that is randomly distributed in lymphoid tissue of the host. The local release of infectious EBV particles could create an inflammatory microenvironment, periodically flaring up before subsiding again. These oscillating stimulatory events increase the likelihood of breaking self-tolerance and molecular mimicry (113). An increased number of B cells could present EBV-derived peptides that contain molecular mimicry motifs allowing stimulation of auto-reactive T cells (114). In accordance, peripheral memory B cells drive proliferation of CD4+ T cells that recognize peptides expressed in MS brain lesions (115). Indeed, homologies between EBV-encoded proteins on the one hand and myelin and other CNS antigens on the other have been found (34). T cell clones from MS patients recognizing myelin basic protein are activated by peptides derived from EBV-encoded DNA polymerase, which is expressed during lytic infection (116, 117). EBNA-1 is not only expressed in all forms of latent infection but also during the lytic phase of infection (118). A recent study demonstrated that antibodies against EBNA-1 cross-react with glial cell adhesion protein (GlialCAM), a self-antigen expressed in the CNS (119, 120). EBV-specific B cells cross the BBB and form ectopic lymphoid-like structures, which are often found in infection and autoimmunity (121). After undergoing somatic hypermutation, B cells that produce cross-reactive antibodies with high affinity for GlialCAM are selected by ENBA-1 specific CD4+ T follicular helper cells and follicular dendritic cells. These cross-reactive antibodies subsequently damage myelin-producing glial cells (119). Moreover, clonally expanded EBNA1-specific CD4+ T cells cross-reacting with myelin are observed in MS patients (122).
Taken together, a high EBV load in the periphery could facilitate pathogenic B–T cell interactions resulting in stimulation and accumulation of auto-reactive immune cells that cross the BBB and drive demyelination.
B cells crossing the BBB represent an important immune axis between periphery and CNS of MS patients (123–125). Thus, the latently EBV-infected B cell reservoir in the CNS is continuously replenished by the immune axis between periphery and CNS. The presence of EBV-infected B cells in the CNS of MS patients has been reported by numerous studies (126). In ectopic lymphoid-like structures of the CNS, latently EBV-infected B cells differentiate into plasma cells, resulting in EBV reactivation, lytic infection and stimulation of cytotoxic CD8+ T cells (127–129). EBV gene expression patterns found in the brain of MS patients support the idea of EBV reactivation and EBV entry into the lytic cycle (130). Moreover, cytotoxic CD8+ T cells interacting with plasma cells lytically infected with EBV were observed in inflammatory white matter lesions and meninges from post-mortem MS brain samples (111). In these lesions, CD8+ T cells recognizing lytic EBV antigens tended to be more frequent than those recognizing EBV latent proteins (131). Thus, ebvIL-10 and EBV-induced cIL-10 are likely released in the CNS thereby maintaining a reservoir of latently EBV-infected B cells that stimulate pathogenic T cells also at this site. In accordance, cIL-10 drives CNS inflammation by promoting survival of pathogenic T cells in mouse models of CNS autoimmunity (132, 133). Accordingly, periodic reactivation of EBV and the release of infectious virus particles could create an inflammatory environment that facilitates MS immunopathology. In line with this view, lytic EBV was restricted to chronic MS plaques (134).
The coordinated action of both EBV-induced cIL-10 and ebvIL-10 plays an important role in viral immune evasion and virus persistence. By reprogramming EBV-infected naïve B cells and regulating the antiviral T cell responses, these cytokines could define the set point of the latently EBV-infected B cell reservoir, which varies from person to person but remains stable in each person over time. At a low set point, EBV reactivates only rarely and is not efficiently passed on from one person to another due to minimal oral shedding coincident with low MS risk (Figure 2). In striking contrast, if the set point is too high as observed after IM, reactivation and release of infectious EBV particles occurs frequently in lymphoid tissue that harbors latently EBV-infected memory B cells creating an inflammatory microenvironment. This may allow not only persistent oral shedding with high virus transmission but also drastically increases the risk for pathogenic immune responses that initiate and drive MS (Figure 2). In the future, it might be sensible to calculate the risk of young adults with a previous record of IM by quantifying the EBV load and develop prophylactic and therapeutic measures that adjust the set point of a disproportionally high EBV load downwards. Most importantly, an effective vaccine against EBV infection could prevent MS and its deleterious consequences.
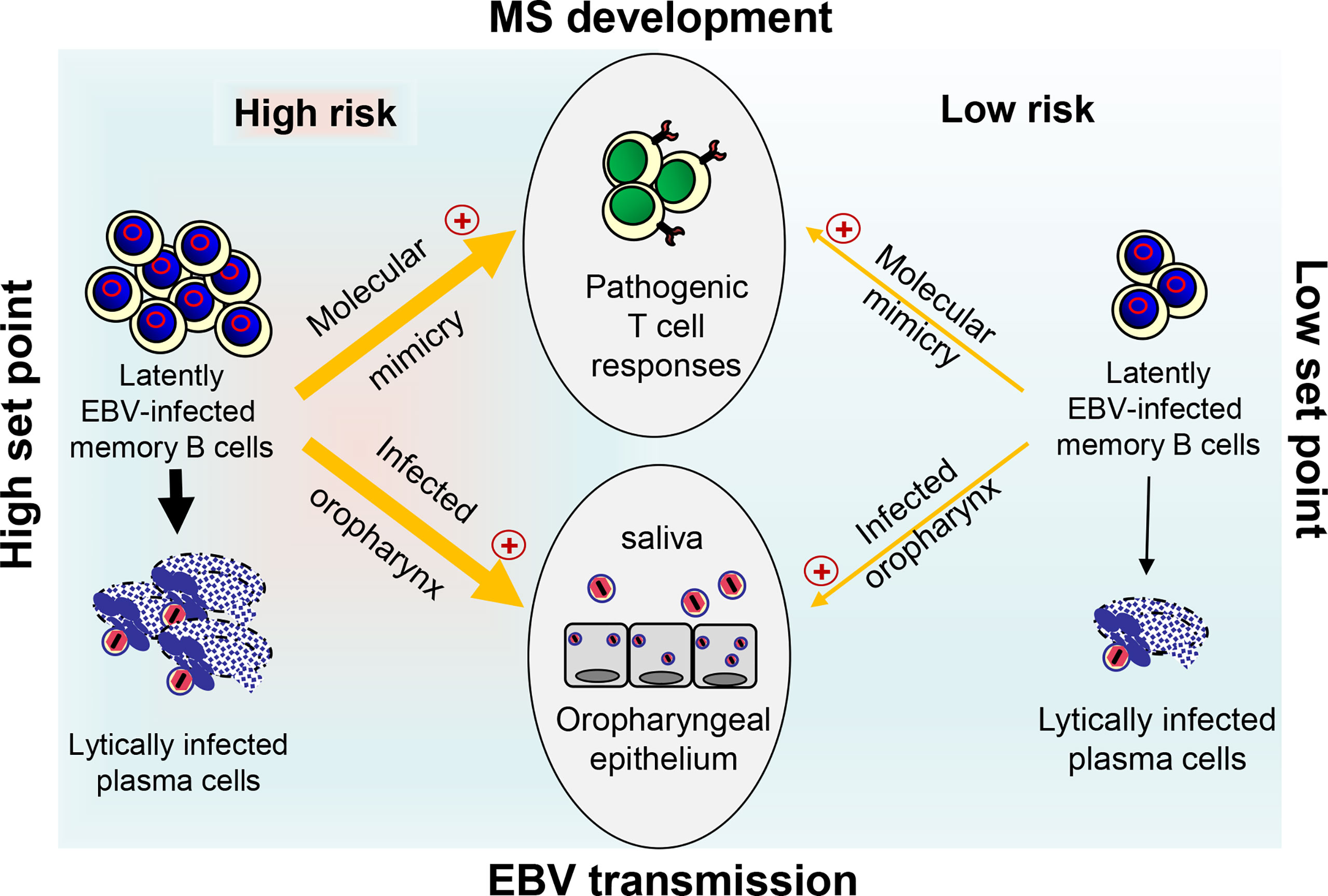
Figure 2 Link between EBV load, risk for MS development, and EBV transmission. At a high set point of the EBV load (left side), reactivation of EBV in latently infected memory B cells and release of infectious EBV particles occurs frequently in lymphoid tissue. This may allow not only persistent oral shedding with high EBV transmission but also facilitate pathogenic B–T cell interactions thereby drastically increasing the risk for pathogenic T ell responses that initiate and drive MS through molecular mimicry. The latter occurs when B cells present EBV-derived peptides that are similar to self-peptides found in CNS antigens. At a low set point (right side), however, EBV reactivates only rarely resulting in inefficient EBV transmission due to reduced oral shedding and only a low risk for MS.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
GS, MOA, and MR drafted and corrected the text and figure. All authors contributed to the article and approved the submitted version.
This work was supported by the Berlin Institute of Health (BIH).
The authors apologize that many research articles with relevance to the field were not included in this review due to space limitations.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Ressing ME, van Gent M, Gram AM, Hooykaas MJ, Piersma SJ, Wiertz EJ. Immune Evasion by Epstein-Barr Virus. Curr Top Microbiol Immunol (2015) 391:355–81. doi: 10.1007/978-3-319-22834-1_12
2. Temple RM, Zhu J, Budgeon L, Christensen ND, Meyers C, Sample CE. Efficient replication of Epstein-Barr virus in stratified epithelium in vitro. Proc Natl Acad Sci USA (2014) 111:16544–9. doi: 10.1073/pnas.1400818111
3. Mrozek-Gorska P, Buschle A, Pich D, Schwarzmayr T, Fechtner R, Scialdone A, et al. Epstein-Barr Virus reprograms human B lymphocytes immediately in the prelatent phase of infection. Proc Natl Acad Sci USA (2019) 116:16046–55. doi: 10.1073/pnas.1901314116
4. Kempkes B, Robertson ES. Epstein-Barr Virus latency: current and future perspectives. Curr Opin Virol (2015) 14:138–44. doi: 10.1016/j.coviro.2015.09.007
5. Price AM, Luftig MA. To be or not IIb: a multi-step process for Epstein-Barr virus latency establishment and consequences for B cell tumorigenesis. PLoS Pathog (2015) 11:e1004656. doi: 10.1371/journal.ppat.1004656
6. Babcock GJ, Decker LL, Volk M, Thorley-Lawson DA. EBV persistence in memory B cells in vivo. Immunity (1998) 9:395–404. doi: 10.1016/S1074-7613(00)80622-6
7. Laichalk LL, Thorley-Lawson DA. Terminal differentiation into plasma cells initiates the replicative cycle of Epstein-Barr virus in vivo. J Virol (2005) 79:1296–307. doi: 10.1128/JVI.79.2.1296-1307.2005
8. Liang X, Collins CM, Mendel JB, Iwakoshi NN, Speck SH. Gammaherpesvirus-driven plasma cell differentiation regulates virus reactivation from latently infected B lymphocytes. PLoS Pathog (2009) 5:e1000677. doi: 10.1371/journal.ppat.1000677
9. Hislop AD, Taylor GS, Sauce D, Rickinson AB. Cellular responses to viral infection in humans: lessons from Epstein-Barr virus. Annu Rev Immunol (2007) 25:587–617. doi: 10.1146/annurev.immunol.25.022106.141553
10. Thorley-Lawson DA, Hawkins JB, Tracy SI, Shapiro M. The pathogenesis of Epstein–Barr virus persistent infection. Curr Opin Virol (2013) 3:227–32. doi: 10.1016/j.coviro.2013.04.005
11. Münz C. Latency and lytic replication in Epstein-Barr virus-associated oncogenesis. Nat Rev Microbiol (2019) 17:691–700. doi: 10.1038/s41579-019-0249-7
12. Johnson KH, Webb C-H, Schmeling DO, Brundage RC, Balfour HH. Epstein–Barr Virus dynamics in asymptomatic immunocompetent adults: an intensive 6-month study. Clin Transl Immunol (2016) 5:e81. doi: 10.1038/cti.2016.28
13. Byrne CM, Johnston C, Orem J, Okuku F, Huang M-L, Rahman H, et al. Examining the dynamics of Epstein-Barr virus shedding in the tonsils and the impact of HIV-1 coinfection on daily saliva viral loads. PLoS Comput Biol (2021) 17:e1009072. doi: 10.1371/journal.pcbi.1009072
14. Schonrich G, Abdelaziz MO, Raftery MJ. Herpesviral capture of immunomodulatory host genes. Virus Genes (2017) 53:762–73. doi: 10.1007/s11262-017-1460-0
15. Hsu DH, de Waal Malefyt R, Fiorentino DF, Dang MN, Vieira P, de Vries J, et al. Expression of interleukin-10 activity by Epstein-Barr virus protein BCRF1. Science (1990) 250:830–2. doi: 10.1126/science.2173142
16. Liu Y, de Waal Malefyt R, Briere F, Parham C, Bridon JM, Banchereau J, et al. The EBV IL-10 homologue is a selective agonist with impaired binding to the IL-10 receptor. J Immunol (1997) 158:604–13.
17. Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol (2001) 19:683–765. doi: 10.1146/annurev.immunol.19.1.683
18. Ouyang W, Rutz S, Crellin NT, Valdez PA, Hymowitz SG. Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annu Rev Immunol (2011) 29:71–109. doi: 10.1146/annurev-immunol-031210-101312
19. Saraiva M, Vieira P, O’Garra A. Biology and therapeutic potential of interleukin-10. J Exp Med (2020) 217:e20190418. doi: 10.1084/jem.20190418
20. Blackburn SD, Wherry EJ. IL-10, T cell exhaustion and viral persistence. Trends Microbiol (2007) 15:143–6. doi: 10.1016/j.tim.2007.02.006
21. Ng CT, Oldstone MB. IL-10: Achieving balance during persistent viral infection. Curr Top Microbiol Immunol (2014) 380:129–44. doi: 10.1007/978-3-662-43492-5_6
22. Rojas JM, Avia M, Martín V, Sevilla N. IL-10: A multifunctional cytokine in viral infections. J Immunol Res (2017) 2017:6104054. doi: 10.1155/2017/6104054
23. Wilson EB, Brooks DG. The role of IL-10 in regulating immunity to persistent viral infections. Curr Top Microbiol Immunol (2011) 350:39–65. doi: 10.1007/82_2010_96
24. McKenzie J, Lopez-Giraldez F, Delecluse HJ, Walsh A, El-Guindy A. The Epstein-Barr virus immunoevasins BCRF1 and BPLF1 are expressed by a mechanism independent of the canonical late pre-initiation complex. PLoS Pathog (2016) 12:e1006008. doi: 10.1371/journal.ppat.1006008
25. Miyazaki I, Cheung RK, Dosch HM. Viral interleukin 10 is critical for the induction of B cell growth transformation by Epstein-Barr virus. J Exp Med (1993) 178:439–47. doi: 10.1084/jem.178.2.439
26. Burdin N, Péronne C, Banchereau J, Rousset F. Epstein-Barr Virus transformation induces B lymphocytes to produce human interleukin 10. J Exp Med (1993) 177:295–304. doi: 10.1084/jem.177.2.295
27. Samanta M, Iwakiri D, Takada K. Epstein–Barr Virus-encoded small RNA induces IL-10 through RIG-i-mediated IRF-3 signaling. Oncogene (2008) 27:4150–60. doi: 10.1038/onc.2008.75
28. Taga H, Taga K, Wang F, Chretien J, Tosato G. Human and viral interleukin-10 in acute Epstein-Barr virus-induced infectious mononucleosis. J Infect Dis (1995) 171:1347–50. doi: 10.1093/infdis/171.5.1347
29. Bjornevik K, Cortese M, Healy Brian C, Kuhle J, Mina Michael J, Leng Y, et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science (2022) 375:296–301. doi: 10.1126/science.abj8222
30. Fernández-Menéndez S, Fernández-Morán M, Fernández-Vega I, Pérez-Álvarez A, Villafani-Echazú J. Epstein–Barr Virus and multiple sclerosis. from evidence to therapeutic strategies. J Neurological Sci (2016) 361:213–9. doi: 10.1016/j.jns.2016.01.013
31. Hacohen Y, Ciccarelli O. New evidence for Epstein-Barr virus infection as a cause of multiple sclerosis. Neurology (2022) 98:605–6. doi: 10.1212/WNL.0000000000200243
32. Levin LI, Munger KL, O’Reilly EJ, Falk KI, Ascherio A. Primary infection with the Epstein-Barr virus and risk of multiple sclerosis. Ann Neurol (2010) 67:824–30. doi: 10.1002/ana.21978
33. Munger KL, Levin LI, O’Reilly EJ, Falk KI, Ascherio A. Anti-Epstein-Barr virus antibodies as serological markers of multiple sclerosis: a prospective study among united states military personnel. Mult Scler (2011) 17:1185–93. doi: 10.1177/1352458511408991
34. Robinson William H, Steinman L. Epstein-Barr Virus and multiple sclerosis. Science (2022) 375:264–5. doi: 10.1126/science.abm7930
35. Compston A, Coles A. Multiple sclerosis. Lancet (2008) 372:1502–17. doi: 10.1016/S0140-6736(08)61620-7
36. Lassmann H, van Horssen J, Mahad D. Progressive multiple sclerosis: pathology and pathogenesis. Nat Rev Neurol (2012) 8:647–56. doi: 10.1038/nrneurol.2012.168
37. Ford H. Clinical presentation and diagnosis of multiple sclerosis. Clin Med (2020) 20:380–3. doi: 10.7861/clinmed.2020-0292
38. Wallin MT, Culpepper WJ, Nichols E, Bhutta ZA, Gebrehiwot TT, Hay SI, et al. Global, regional, and national burden of multiple sclerosis 1990-2016: a systematic analysis for the global burden of disease study 2016. Lancet Neurol (2019) 18:269–85. doi: 10.1016/S1474-4422(18)30443-5
39. Walton C, King R, Rechtman L, Kaye W, Leray E, Marrie RA, et al. Rising prevalence of multiple sclerosis worldwide: Insights from the atlas of MS, third edition. Multiple sclerosis (Houndmills Basingstoke England) (2020) 26:1816–21. doi: 10.1177/1352458520970841
40. Attfield KE, Jensen LT, Kaufmann M, Friese MA, Fugger L. The immunology of multiple sclerosis. Nat Rev Immunol (2022). doi: 10.1038/s41577-022-00718-z
41. Hemmer B, Kerschensteiner M, Korn T. Role of the innate and adaptive immune responses in the course of multiple sclerosis. Lancet Neurol (2015) 14:406–19. doi: 10.1016/S1474-4422(14)70305-9
42. Walter MR. The molecular basis of IL-10 function: from receptor structure to the onset of signaling. Curr Top Microbiol Immunol (2014) 380:191–212. doi: 10.1007/978-3-662-43492-5_9
43. Jones BC, Logsdon NJ, Josephson K, Cook J, Barry PA, Walter MR. Crystal structure of human cytomegalovirus IL-10 bound to soluble human IL-10R1. Proc Natl Acad Sci USA (2002) 99:9404–9. doi: 10.1073/pnas.152147499
44. Ding Y, Qin L, Zamarin D, Kotenko SV, Pestka S, Moore KW, et al. Differential IL-10R1 expression plays a critical role in IL-10-mediated immune regulation. J Immunol (2001) 167:6884–92. doi: 10.4049/jimmunol.167.12.6884
45. MacNeil IA, Suda T, Moore KW, Mosmann TR, Zlotnik A. IL-10, a novel growth cofactor for mature and immature T cells. J Immunol (1990) 145:4167–73.
46. Vieira P, de Waal-Malefyt R, Dang MN, Johnson KE, Kastelein R, Fiorentino DF, et al. Isolation and expression of human cytokine synthesis inhibitory factor cDNA clones: homology to Epstein-Barr virus open reading frame BCRFI. Proc Natl Acad Sci USA (1991) 88:1172–6. doi: 10.1073/pnas.88.4.1172
47. Go NF, Castle BE, Barrett R, Kastelein R, Dang W, Mosmann TR, et al. Interleukin 10, a novel B cell stimulatory factor: unresponsiveness of X chromosome-linked immunodeficiency B cells. J Exp Med (1990) 172:1625–31. doi: 10.1084/jem.172.6.1625
48. Raftery MJ, Wieland D, Gronewald S, Kraus AA, Giese T, Schonrich G. Shaping phenotype, function, and survival of dendritic cells by cytomegalovirus-encoded IL-10. J Immunol (2004) 173:3383–91. doi: 10.4049/jimmunol.173.5.3383
49. Jog NR, Chakravarty EF, Guthridge JM, James JA. Epstein Barr Virus interleukin 10 suppresses anti-inflammatory phenotype in human monocytes. Front Immunol (2018) 9:2198. doi: 10.3389/fimmu.2018.02198
50. Rousset F, Garcia E, Defrance T, Péronne C, Vezzio N, Hsu DH, et al. Interleukin 10 is a potent growth and differentiation factor for activated human B lymphocytes. Proc Natl Acad Sci USA (1992) 89:1890–3. doi: 10.1073/pnas.89.5.1890
51. Dunmire SK, Verghese PS, Balfour HH. Primary Epstein-Barr virus infection. J Clin Virol (2018) 102:84–92. doi: 10.1016/j.jcv.2018.03.001
52. Cohen JI. Epstein–Barr Virus infection. N Engl J Med (2000) 343:481–92. doi: 10.1056/NEJM200008173430707
53. Balfour HHJR, Odumade OA, Schmeling DO, Mullan BD, Ed JA, Knight JA, et al. Behavioral, virologic, and immunologic factors associated with acquisition and severity of primary Epstein–Barr virus infection in university students. J Infect Dis (2012) 207:80–8. doi: 10.1093/infdis/jis646
54. Long HM, Meckiff BJ, Taylor GS. The T-cell response to Epstein-Barr virus-new tricks from an old dog. Front Immunol (2019) 10:2193. doi: 10.3389/fimmu.2019.02193
55. Taylor GS, Long HM, Brooks JM, Rickinson AB, Hislop AD. The immunology of Epstein-Barr virus-induced disease. Annu Rev Immunol (2015) 33:787–821. doi: 10.1146/annurev-immunol-032414-112326
56. Endriz J, Ho PP, Steinman L. Time correlation between mononucleosis and initial symptoms of MS. Neurol Neuroimmunol Neuroinflamm (2017) 4:e308. doi: 10.1212/NXI.0000000000000308
57. Rocchi G, Felici A, Ragona G, Heinz A. Quantitative evaluation of Epstein-barr-virus-infected mononuclear peripheral blood leukocytes in infectious mononucleosis. N Engl J Med (1977) 296:132–4. doi: 10.1056/NEJM197701202960302
58. Tanner JE, Diaz-Mitoma F, Rooney CM, Alfieri C. Anti-interleukin-10 antibodies in patients with chronic active Epstein-Barr virus infection. J Infect Dis (1997) 176:1454–61. doi: 10.1086/514141
59. Callan MF, Steven N, Krausa P, Wilson JD, Moss PA, Gillespie GM, et al. Large Clonal expansions of CD8+ T cells in acute infectious mononucleosis. Nat Med (1996) 2:906–11. doi: 10.1038/nm0896-906
60. Silins SL, Sherritt MA, Silleri JM, Cross SM, Elliott SL, Bharadwaj M, et al. Asymptomatic primary Epstein-Barr virus infection occurs in the absence of blood T-cell repertoire perturbations despite high levels of systemic viral load. Blood (2001) 98:3739–44. doi: 10.1182/blood.V98.13.3739
61. Biström M, Jons D, Engdahl E, Gustafsson R, Huang J, Brenner N, et al. Epstein–Barr Virus infection after adolescence and human herpesvirus 6A as risk factors for multiple sclerosis. Eur J Neurol (2021) 28:579–86. doi: 10.1111/ene.14597
62. Jacobs BM, Giovannoni G, Cuzick J, Dobson R. Systematic review and meta-analysis of the association between Epstein-Barr virus, multiple sclerosis and other risk factors. Mult Scler (2020) 26:1281–97. doi: 10.1177/1352458520907901
63. Handel AE, Williamson AJ, Disanto G, Handunnetthi L, Giovannoni G, Ramagopalan SV. An updated meta-analysis of risk of multiple sclerosis following infectious mononucleosis. PLoS One (2010) 5:e12496. doi: 10.1371/journal.pone.0012496
64. Nielsen TR, Rostgaard K, Nielsen NM, Koch-Henriksen N, Haahr S, Sørensen PS, et al. Multiple sclerosis after infectious mononucleosis. Arch Neurol (2007) 64:72–5. doi: 10.1001/archneur.64.1.72
65. Thacker EL, Mirzaei F, Ascherio A. Infectious mononucleosis and risk for multiple sclerosis: A meta-analysis. Ann Neurol (2006) 59:499–503. doi: 10.1002/ana.20820
66. Xu Y, Hiyoshi A, Smith KA, Piehl F, Olsson T, Fall K, et al. Association of infectious mononucleosis in childhood and adolescence with risk for a subsequent multiple sclerosis diagnosis among siblings. JAMA Network Open (2021) 4:e2124932–e2124932. doi: 10.1001/jamanetworkopen.2021.24932
67. Moghaddam VK, Dickerson AS, Bazrafshan E, Seyedhasani SN, Najafi F, Hadei M, et al. Socioeconomic determinants of global distribution of multiple sclerosis: an ecological investigation based on global burden of disease data. BMC Neurol (2021) 21:145. doi: 10.1186/s12883-021-02170-3
68. Veroni C, Aloisi F. The CD8 T cell-Epstein-Barr virus-B cell trialogue: A central issue in multiple sclerosis pathogenesis. Front Immunol (2021) 12:665718. doi: 10.3389/fimmu.2021.665718
69. Khan G, Miyashita EM, Yang B, Babcock GJ, Thorley-Lawson DA. Is EBV persistence in vivo a model for B cell homeostasis? Immunity (1996) 5:173–9. doi: 10.1016/S1074-7613(00)80493-8
70. Hoshino Y, Katano H, Zou P, Hohman P, Marques A, Tyring SK, et al. Long-term administration of valacyclovir reduces the number of Epstein-Barr virus (EBV)-infected B cells but not the number of EBV DNA copies per B cell in healthy volunteers. J Virol (2009) 83:11857–61. doi: 10.1128/JVI.01005-09
71. Miyashita EM, Yang B, Lam KMC, Crawford DH, Thorley-Lawson DA. A novel form of Epstein-Barr virus latency in normal B cells in vivo. Cell (1995) 80:593–601. doi: 10.1016/0092-8674(95)90513-8
72. Fafi-Kremer S, Morand P, Brion J-P, Pavese P, Baccard M, Germi R, et al. Long-term shedding of infectious Epstein-Barr virus after infectious mononucleosis. J Infect Dis (2005) 191:985–9. doi: 10.1086/428097
73. Fraser KB, Millar JHD, Haire M, McCrea S. Increased tendency to spontaneous in-vitro lymphocyte transformation in clinically active multiple sclerosis. Lancet (1979) 314:715–7. doi: 10.1016/S0140-6736(79)90643-3
74. Cencioni MT, Mattoscio M, Magliozzi R, Bar-Or A, Muraro PA. B cells in multiple sclerosis - from targeted depletion to immune reconstitution therapies. Nat Rev Neurol (2021) 17:399–414. doi: 10.1038/s41582-021-00498-5
75. Li R, Patterson KR, Bar-Or A. Reassessing B cell contributions in multiple sclerosis. Nat Immunol (2018) 19:696–707. doi: 10.1038/s41590-018-0135-x
76. Molnarfi N, Schulze-Topphoff U, Weber MS, Patarroyo JC, Prod’homme T, Varrin-Doyer M, et al. MHC class II-dependent B cell APC function is required for induction of CNS autoimmunity independent of myelin-specific antibodies. J Exp Med (2013) 210:2921–37. doi: 10.1084/jem.20130699
77. Korn T, Oukka M. A BAFFling association between malaria resistance and the risk of multiple sclerosis. N Engl J Med (2017) 376:1680–1. doi: 10.1056/NEJMe1700720
78. Steri M, Orrù V, Idda ML, Pitzalis M, Pala M, Zara I, et al. Overexpression of the cytokine BAFF and autoimmunity risk. N Engl J Med (2017) 376:1615–26. doi: 10.1056/NEJMoa1610528
79. Ioannides ZA, Csurhes PA, Douglas NL, Mackenroth G, Swayne A, Thompson KM, et al. Sustained clinical improvement in a subset of patients with progressive multiple sclerosis treated with Epstein–Barr virus-specific T cell therapy. Front Neurol (2021) 12:652811. doi: 10.3389/fneur.2021.652811
80. Pender MP, Csurhes PA, Smith C, Beagley L, Hooper KD, Raj M, et al. Epstein–Barr Virus-specific adoptive immunotherapy for progressive multiple sclerosis. Mult Scler (2014) 20:1541–4. doi: 10.1177/1352458514521888
81. Pender MP, Csurhes PA, Smith C, Douglas NL, Neller MA, Matthews KK, et al. Epstein-Barr Virus-specific T cell therapy for progressive multiple sclerosis. JCI Insight (2018) 3:e124714. doi: 10.1172/jci.insight.124714
82. Ascherio A, Munger KL, Lennette ET, Spiegelman D, Hernán MA, Olek MJ, et al. Epstein-Barr Virus antibodies and risk of multiple SclerosisA prospective study. JAMA (2001) 286:3083–8. doi: 10.1001/jama.286.24.3083
83. Levin LI, Munger KL, Rubertone MV, Peck CA, Lennette ET, Spiegelman D, et al. Temporal relationship between elevation of Epstein-Barr virus antibody titers and initial onset of neurological symptoms in multiple sclerosis. JAMA (2005) 293:2496–500. doi: 10.1001/jama.293.20.2496
84. Sundström P, Juto P, Wadell G, Hallmans G, Svenningsson A, Nyström L, et al. An altered immune response to Epstein-Barr virus in multiple sclerosis. Neurology (2004) 62:2277–82. doi: 10.1212/01.WNL.0000130496.51156.D7
85. Pender MP, Csurhes PA, Burrows JM, Burrows SR. Defective T-cell control of Epstein–Barr virus infection in multiple sclerosis. Clin Transl Immunol (2017) 6:e126. doi: 10.1038/cti.2016.87
86. Rubicz R, Yolken R, Drigalenko E, Carless MA, Dyer TD, Bauman L, et al. A genome-wide integrative genomic study localizes genetic factors influencing antibodies against Epstein-Barr virus nuclear antigen 1 (EBNA-1). PLoS Genet (2013) 9:e1003147. doi: 10.1371/journal.pgen.1003147
87. Sundström P, Nyström M, Ruuth K, Lundgren E. Antibodies to specific EBNA-1 domains and HLA DRB1*1501 interact as risk factors for multiple sclerosis. J Neuroimmunol (2009) 215:102–7. doi: 10.1016/j.jneuroim.2009.08.004
88. Zdimerova H, Murer A, Engelmann C, Raykova A, Deng Y, Gujer C, et al. Attenuated immune control of Epstein–Barr virus in humanized mice is associated with the multiple sclerosis risk factor HLA-DR15. Eur J Immunol (2021) 51:64–75. doi: 10.1002/eji.202048655
89. Zhou Y, Zhu G, Charlesworth JC, Simpson S, Rubicz R, Göring HH, et al. Genetic loci for Epstein-Barr virus nuclear antigen-1 are associated with risk of multiple sclerosis. Mult Scler J (2016) 22:1655–64. doi: 10.1177/1352458515626598
90. Budiningsih I, Dachlan YP, Hadi U, Middeldorp JM. Quantitative cytokine level of TNF-α, IFN-γ, IL-10, TGF-β and circulating Epstein-Barr virus DNA load in individuals with acute malaria due to p. falciparum or p. vivax or double infection in a malaria endemic region in Indonesia. PLoS One (2022) 16:e0261923.
91. Baranzini SE, Oksenberg JR. The genetics of multiple sclerosis: From 0 to 200 in 50 years. Trends Genet (2017) 33:960–70. doi: 10.1016/j.tig.2017.09.004
92. Correale J, Hohlfeld R, Baranzini SE. The role of the gut microbiota in multiple sclerosis. Nat Rev Neurol (2022) 18:544–58. doi: 10.1038/s41582-022-00697-8
93. Ganeshan K, Chawla A. Metabolic regulation of immune responses. Annu Rev Immunol (2014) 32:609–34. doi: 10.1146/annurev-immunol-032713-120236
94. Olsson T, Barcellos LF, Alfredsson L. Interactions between genetic, lifestyle and environmental risk factors for multiple sclerosis. Nat Rev Neurol (2017) 13:25–36. doi: 10.1038/nrneurol.2016.187
95. Reuss E, Fimmers R, Kruger A, Becker C, Rittner C, Höhler T. Differential regulation of interleukin-10 production by genetic and environmental factors – a twin study. Genes Immun (2002) 3:407–13. doi: 10.1038/sj.gene.6363920
96. International Multiple Sclerosis Genetics Consortium. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science (2019) 365:eaav7188. doi: 10.1126/science.aav7188
97. Pender MP, Csurhes PA, Lenarczyk A, Pfluger CMM, Burrows SR. Decreased T cell reactivity to Epstein–Barr virus infected lymphoblastoid cell lines in multiple sclerosis. J Neurol Neurosurg Psychiatry (2009) 80:498–505. doi: 10.1136/jnnp.2008.161018
98. Siegel AM, Herskowitz JH, Speck SH. The MHV68 M2 protein drives IL-10 dependent B cell proliferation and differentiation. PLoS Pathog (2008) 4:e1000039. doi: 10.1371/journal.ppat.1000039
99. Smith LK, Boukhaled GM, Condotta SA, Mazouz S, Guthmiller JJ, Vijay R, et al. Interleukin-10 directly inhibits CD8+ T cell function by enhancing n-glycan branching to decrease antigen sensitivity. Immunity (2018) 48:299–312.e5. doi: 10.1016/j.immuni.2018.01.006
100. Bejarano MT, Masucci MG. Interleukin-10 abrogates the inhibition of Epstein-Barr virus–induced B-cell transformation by memory T-cell responses. Blood (1998) 92:4256–62. doi: 10.1182/blood.V92.11.4256.423k12_4256_4262
101. Jochum S, Ruiss R, Moosmann A, Hammerschmidt W, Zeidler R. RNAs in Epstein-Barr virions control early steps of infection. Proc Natl Acad Sci USA (2012) 109:E1396–404. doi: 10.1073/pnas.1115906109
102. Jochum S, Moosmann A, Lang S, Hammerschmidt W, Zeidler R. The EBV immunoevasins vIL-10 and BNLF2a protect newly infected B cells from immune recognition and elimination. PLoS Pathog (2012) 8:e1002704. doi: 10.1371/journal.ppat.1002704
103. Zeidler R, Eissner Gn, Meissner P, Uebel S, Tampeí R, Lazis S, et al. Downregulation of TAP1 in B lymphocytes by cellular and Epstein-Barr virus–encoded interleukin-10. Blood (1997) 90:2390–7. doi: 10.1182/blood.V90.6.2390
104. Stewart JP, Rooney CM. The interleukin-10 homolog encoded by Epstein-Barr virus enhances the reactivation of virus-specific cytotoxic T cell and HLA-unrestricted killer cell responses. Virology (1992) 191:773–82. doi: 10.1016/0042-6822(92)90253-L
105. Groux H, Bigler M, de Vries JE, Roncarolo MG. Inhibitory and stimulatory effects of IL-10 on human CD8+ T cells. J Immunol (1998) 160:3188–93.
106. Mumm John B, Emmerich J, Zhang X, Chan I, Wu L, Mauze S, et al. IL-10 elicits IFNγ-dependent tumor immune surveillance. Cancer Cell (2011) 20:781–96. doi: 10.1016/j.ccr.2011.11.003
107. Saxton RA, Tsutsumi N, Su LL, Abhiraman GC, Mohan K, Henneberg LT, et al. Structure-based decoupling of the pro- and anti-inflammatory functions of interleukin-10. Science (2021) 371:eabc8433. doi: 10.1126/science.abc8433
108. Macedo C, Webber SA, Donnenberg AD, Popescu I, Hua Y, Green M, et al. EBV-specific CD8+ T cells from asymptomatic pediatric thoracic transplant patients carrying chronic high EBV loads display contrasting features: Activated phenotype and exhausted function. J Immunol (2011) 186:5854. doi: 10.4049/jimmunol.1001024
109. Guerrera G, Ruggieri S, Picozza M, Piras E, Gargano F, Placido R, et al. EBV-specific CD8 T lymphocytes and B cells during glatiramer acetate therapy in patients with MS. Neurol Neuroimmunol Neuroinflamm (2020) 7:e876. doi: 10.1212/NXI.0000000000000876
110. Pender MP, Csurhes PA, Pfluger CMM, Burrows SR. CD8 T cell deficiency impairs control of Epstein–Barr virus and worsens with age in multiple sclerosis. J Neurol Neurosurg Psychiatry (2012) 83:353–4. doi: 10.1136/jnnp-2011-300213
111. Angelini DF, Serafini B, Piras E, Severa M, Coccia EM, Rosicarelli B, et al. Increased CD8+ T cell response to Epstein-Barr virus lytic antigens in the active phase of multiple sclerosis. PLoS Pathog (2013) 9:e1003220. doi: 10.1371/journal.ppat.1003220
112. Jilek S, Schluep M, Meylan P, Vingerhoets F, Guignard L, Monney A, et al. Strong EBV-specific CD8+ T-cell response in patients with early multiple sclerosis. Brain (2008) 131:1712–21. doi: 10.1093/brain/awn108
113. Oldstone MBA. Molecular mimicry and autoimmune disease. Cell (1987) 50:819–20. doi: 10.1016/0092-8674(87)90507-1
114. Soldan SS, Lieberman PM. Epstein-Barr Virus and multiple sclerosis. Nat Rev Microbiol (2022). doi: 10.1038/s41579-022-00770-5:1-14
115. Jelcic I, Al Nimer F, Wang J, Lentsch V, Planas R, Jelcic I, et al. Memory B cells activate brain-homing, autoreactive CD4+ T cells in multiple sclerosis. Cell (2018) 175:85–100.e23. doi: 10.1016/j.cell.2018.08.011
116. Wucherpfennig KW, Strominger JL. Molecular mimicry in T cell-mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell (1995) 80:695–705. doi: 10.1016/0092-8674(95)90348-8
117. Wucherpfennig KW. Structural basis of molecular mimicry. J Autoimmun (2001) 16:293–302. doi: 10.1006/jaut.2000.0499
118. Kang M-S, Kieff E. Epstein–Barr Virus latent genes. Exp Mol Med (2015) 47:e131–1. doi: 10.1038/emm.2014.84
119. Lanz TV, Brewer RC, Ho PP, Moon J-S, Jude KM, Fernandez D, et al. Clonally expanded B cells in multiple sclerosis bind EBV EBNA1 and GlialCAM. Nature (2022) 603:321–7. doi: 10.1038/s41586-022-04432-7
120. Wekerle H. Epstein-Barr Virus sparks brain autoimmunity in multiple sclerosis. Nature (2022) 603:230–2. doi: 10.1038/d41586-022-00382-2
121. Pitzalis C, Jones GW, Bombardieri M, Jones SA. Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat Rev Immunol (2014) 14:447–62. doi: 10.1038/nri3700
122. Lünemann JD, Jelcić I, Roberts S, Lutterotti A, Tackenberg B, Martin R, et al. EBNA1-specific T cells from patients with multiple sclerosis cross react with myelin antigens and co-produce IFN-gamma and IL-2. J Exp Med (2008) 205:1763–73. doi: 10.1084/jem.20072397
123. Lu DR, Robinson WH. Street-Experienced peripheral B cells traffic to the brain. Sci Transl Med (2014) 6:248fs31. doi: 10.1126/scitranslmed.3009919
124. Palanichamy A, Apeltsin L, Kuo TC, Sirota M, Wang S, Pitts SJ, et al. Immunoglobulin class-switched B cells form an active immune axis between CNS and periphery in multiple sclerosis. Sci Transl Med (2014) 6:248ra106. doi: 10.1126/scitranslmed.3008930
125. Soldan SS, Lieberman PM. Epstein-Barr Virus infection in the development of neurological disorders. Drug Discov Today Dis Models (2020) 32:35–52. doi: 10.1016/j.ddmod.2020.01.001
126. Bar-Or A, Pender MP, Khanna R, Steinman L, Hartung HP, Maniar T, et al. Epstein-Barr Virus in multiple sclerosis: Theory and emerging immunotherapies. Trends Mol Med (2020) 26:296–310. doi: 10.1016/j.molmed.2019.11.003
127. Magliozzi R, Serafini B, Rosicarelli B, Chiappetta G, Veroni C, Reynolds R, et al. B-cell enrichment and Epstein-Barr virus infection in inflammatory cortical lesions in secondary progressive multiple sclerosis. J Neuropathol Exp Neurol (2013) 72:29–41. doi: 10.1097/NEN.0b013e31827bfc62
128. Serafini B, Rosicarelli B, Franciotta D, Magliozzi R, Reynolds R, Cinque P, et al. Dysregulated Epstein-Barr virus infection in the multiple sclerosis brain. J Exp Med (2007) 204:2899–912. doi: 10.1084/jem.20071030
129. Serafini B, Severa M, Columba-Cabezas S, Rosicarelli B, Veroni C, Chiappetta G, et al. Epstein-Barr Virus latent infection and BAFF expression in B cells in the multiple sclerosis brain: implications for viral persistence and intrathecal B-cell activation. J Neuropathol Exp Neurol (2010) 69:677–93. doi: 10.1097/NEN.0b013e3181e332ec
130. Veroni C, Serafini B, Rosicarelli B, Fagnani C, Aloisi F. Transcriptional profile and Epstein-Barr virus infection status of laser-cut immune infiltrates from the brain of patients with progressive multiple sclerosis. J Neuroinflamm (2018) 15:18. doi: 10.1186/s12974-017-1049-5
131. Serafini B, Rosicarelli B, Veroni C, Mazzola Gina A, Aloisi F, Longnecker Richard M. Epstein-Barr Virus-specific CD8 T cells selectively infiltrate the brain in multiple sclerosis and interact locally with virus-infected cells: Clue for a virus-driven immunopathological mechanism. J Virol (2019) 93:e00980–19. doi: 10.1128/JVI.00980-19
132. Liu X, Alli R, Steeves M, Nguyen P, Vogel P, Geiger TL. The T cell response to IL-10 alters cellular dynamics and paradoxically promotes central nervous system autoimmunity. J Immunol (2012) 189:669–78. doi: 10.4049/jimmunol.1200607
133. Yogev N, Bedke T, Kobayashi Y, Brockmann L, Lukas D, Regen T, et al. CD4+ T-cell-derived IL-10 promotes CNS inflammation in mice by sustaining effector T cell survival. Cell Rep (2022) 38:110565. doi: 10.1016/j.celrep.2022.110565
Keywords: viral IL-10, IL-10, antiviral immune responses, viral immune evasion, virus-induced immunopathogenesis, multiple sclerosis, viruses
Citation: Schönrich G, Abdelaziz MO and Raftery MJ (2022) Epstein-Barr virus, interleukin-10 and multiple sclerosis: A ménage à trois. Front. Immunol. 13:1028972. doi: 10.3389/fimmu.2022.1028972
Received: 26 August 2022; Accepted: 23 September 2022;
Published: 07 October 2022.
Edited by:
Fabiana Rizzo, National Institute of Health (ISS), ItalyReviewed by:
Evangelia Kesidou, Aristotle University of Thessaloniki, GreeceCopyright © 2022 Schönrich, Abdelaziz and Raftery. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Günther Schönrich, Z3VlbnRoZXIuc2Nob2VucmljaEBjaGFyaXRlLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.