 Jessica Astorga1
Jessica Astorga1 Naschla Gasaly1,2,3
Naschla Gasaly1,2,3 Karen Dubois-Camacho1,4
Karen Dubois-Camacho1,4 Marjorie De la Fuente5
Marjorie De la Fuente5 Glauben Landskron5
Glauben Landskron5 Klaas Nico Faber3
Klaas Nico Faber3 Félix A. Urra4
Félix A. Urra4 Marcela A. Hermoso1,3*
Marcela A. Hermoso1,3*- 1Laboratory of Innate Immunity, Program of Immunology, Institute of Biomedical Sciences, Faculty of Medicine, Universidad de Chile, Santiago, Chile
- 2Immunoendocrinology, Division of Medical Biology, Department of Pathology and Medical Biology, University Medical Center Groningen, Groningen, Netherlands
- 3Department of Gastroenterology and Hepatology, University Medical Center Groningen, Groningen, Netherlands
- 4Laboratory of Metabolic Plasticity and Bioenergetics, Program of Molecular and Clinical Pharmacology, Institute of Biomedical Sciences, Faculty of Medicine, Universidad de Chile, Santiago, Chile
- 5Laboratory of Biomedicine Research, School of Medicine, Universidad Finis Terrae, Santiago, Chile
Inflammatory Bowel Disease (IBD) is characterized by a loss of intestinal barrier function caused by an aberrant interaction between the immune response and the gut microbiota. In IBD, imbalance in cholesterol homeostasis and mitochondrial bioenergetics have been identified as essential events for activating the inflammasome-mediated response. Mitochondrial alterations, such as reduced respiratory complex activities and reduced production of tricarboxylic acid (TCA) cycle intermediates (e.g., citric acid, fumarate, isocitric acid, malate, pyruvate, and succinate) have been described in in vitro and clinical studies. Under inflammatory conditions, mitochondrial architecture in intestinal epithelial cells is dysmorphic, with cristae destruction and high dynamin-related protein 1 (DRP1)-dependent fission. Likewise, these alterations in mitochondrial morphology and bioenergetics promote metabolic shifts towards glycolysis and down-regulation of antioxidant Nuclear erythroid 2-related factor 2 (Nrf2)/Peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1α) signaling. Although the mechanisms underlying the mitochondrial dysfunction during mucosal inflammation are not fully understood at present, metabolic intermediates and cholesterol may act as signals activating the NLRP3 inflammasome in IBD. Notably, dietary phytochemicals exhibit protective effects against cholesterol imbalance and mitochondrial function alterations to maintain gastrointestinal mucosal renewal in vitro and in vivo conditions. Here, we discuss the role of cholesterol and mitochondrial metabolism in IBD, highlighting the therapeutic potential of dietary phytochemicals, restoring intestinal metabolism and function.
Introduction
Inflammatory bowel disease (IBD), including ulcerative colitis (UC) and Crohn’s disease (CD), is characterized by a loss of intestinal barrier function and chronic inflammation of the gastrointestinal tract caused by an aberrant interaction between immune response and gut microbiota in genetically susceptible subjects (1–3).
IBD is a 21st century global disease (4), with industrialized countries having a stable incidence (12 to 26 per 100,000 people) as well as a rising prevalence (5), based on genetic susceptibility, early diagnosis, and environmental factors, including diet, lifestyle, and comorbidities (6, 7). Systematic reviews of MEDLINE and EMBASE from 1990 until 2015 established the highest prevalence values for IBD in Europe (UC, 505 per 100,000 people in Norway; CD, 322 per 100,000 people in Germany) and North America (UC, 279 per 100,000 people in the USA; CD, 319 per 100,000 people in Canada) (4). In contrast, IBD incidence in newly industrialized countries, has increased with low prevalence, including countries in Asia, the Middle East, South America, and Africa (5).
The early diagnosis allows possibilities to treat short-term complications and reduce their severity to avoid hospitalization and surgery (8). Treatment possibilities for CD include corticosteroids, azathioprine, or immunomodulators such as methotrexate, while for UC, 5-aminosalicylates (5-ASA), corticosteroids, or azathioprine (9). Biologic drugs are currently used for treating severe to moderate IBD, such as anti-TNF blockers (infliximab, adalimumab, certolizumab pegol, and golimumab), enhancing intestinal mucosa healing, symptom relief, reduction in hospitalization rates, and improvement in quality of life (8). However, a third of patients are non-responsive to anti-TNF therapy or lose their response to treatment over time (10–12). Other biological drugs have been developed and used as primary therapy, or for those who failed anti-TNF therapy, such as anti-integrin agents (13). Nevertheless, these treatments as well as the hospitalization remain costly for IBD patients (14). Diet can be considered as a booster in the treatment of IBD patients, since some dietary components have been shown benefits preventing the onset of this disease (15).
Therefore, the innate immune system is essential for the first response of the host against infection or damage in the intestinal mucosa. Pattern Recognition Receptors (PRRs) recognize pathogens-associated molecular patterns (PAMPs) and/or damage-associated molecular patterns (DAMPs), such as Toll-like receptors (TLRs), nucleotide-binding oligomerization-domain protein-like receptors (NLRs), and C-type lectin receptors. There is evidence that inflammasome participates during IBD development, and its activation in the intestinal mucosa enhance the pathogen clearance (16, 17). The inflammasome is a supramolecular assembly recognizing PAMPs and/or DAMPs, serving as a platform for caspase-1 activation and production of mainly IL-1β and IL-18. The most studied inflammasome corresponds to the NLR family pyrin domain containing 3 (NLRP3), triggering intestinal mucosa inflammation. High IL-1β expression is related to lack of primary response to anti-TNF therapy. Therefore, NLRP3 inflammasome and IL-1β are crucial players during IBD development (18, 19).
Inflammasome activation requires two signals: the first is an NF-κB-mediated signal for NLRP3 and pro-IL1B gene transcription. The second signal (required for its assembly) can be of diverse origins, including lipopolysaccharide (LPS), toxins, and viral and bacterial nucleic acid. Interestingly, second signals can also arise from cellular metabolism, such as excess intracellular cholesterol, oxidized low density lipoprotein (oxLDL), mitochondria-derived components (cardiolipin, reactive oxygen species (ROS), mitochondrial DNA), or endoplasmic reticulum (ER) stress, all currently related to IBD pathogenesis (20–23). This review summarizes the relevance of intracellular cholesterol changes and how alterations in mitochondrial bioenergetics influence inflammasome activation in IBD. Finally, the role of dietary compounds, especially phytochemicals, in the restitution of mitochondrial function and cholesterol homeostasis in this disease is considered.
Alterations in gut barrier homeostasis and cell diversity during IBD
The intestinal barrier comprises the intestinal epithelium and mucus, coexisting with the lamina propria, the gut-associated lymphoid tissue (GALT), and the commensal gut microbiota (24). It allows digestion and absorption of dietary components and protects us against pathogens. A proper epithelial barrier function and an eubiotic gut microbiome are crucial for intestinal homeostasis (24, 25). Thus, during some gastrointestinal disorders, such as IBD, an imbalanced gut barrier appears, with increased pro-inflammatory mediators causing epithelial damage and, therefore, epithelial permeability, accompanied by an imbalanced gut microbiome, known as “gut dysbiosis” (24).
Located at the intestinal crypts, tissue-resident stem cells proliferate and differentiate into mature enterocytes, mucus-secreting goblet cells, or enteroendocrine cells, while migrating upwards through the crypt to the villus. Essential for adherens junctions between epithelial cells, tight junctions maintain intestinal barrier integrity, which can be disrupted by inflammation or gut dysbiosis (25). Tight junctions include adherens junctions, desmosomes, and gap junctions and they are formed by transmembrane proteins such as occludins, claudins, junctional adhesion molecules, tricelluin, and intracellular scaffolds proteins (zonula occludens (ZO) proteins). Intestinal stem cells have tight junctions distributed heterogeneously and different composition of transmembrane proteins, avoiding permeability closer to the crypt (26). Notably, UC is characterized by reduced epithelial cell diversity and decreased stem cell differentiation and migration, negatively affecting intestinal healing and mucus production by goblet cells (27).
Coordinating the immune response underneath the epithelial layer, immune cells (T cells, B cells, intraepithelial lymphocytes- IELs, and phagocytes) are organized in GALT, such as Peyer’s patches, mesenteric lymph nodes, and isolated lymphoid follicles (28).
Although, loss of gut homeostasis in IBD involves the participation of lamina propria and infiltrating immune cells, such as granulocytes, antigen-presenting cells (APCs), macrophages (Mø), B and T cells (29–31), as well as inflammatory cytokines, such as inflammasome-regulated IL-1β and IL-18, amongst others (TNF-α, IL-6, IL-12, IL-17, IFN-γ, GSDM (32, 33)). Despite an evident relationship between immune cells, metabolism, and inflammasome activation is expected, the role of metabolic mediators, such as cholesterol in IBD remains not fully understood.
Intracellular cholesterol accumulation in IBD
Cholesterol sources and homeostasis
Cholesterol is a sterol that occurs in two forms: free cholesterol and esterified cholesterol, the latter of which is a result of the conjugation of a cholesterol molecule to a fatty acid (FA, usually palmitic or oleic acid, and less frequently, linoleic acid) via an ester bond. Physiologically, cholesterol is an essential lipid of animal cell plasma membranes and a precursor of a variety of biologically active compounds, such as bile salts, hormones, and vitamin D3 (34). Additionally, cholesterol interacts with phospholipid FAs, decreasing the mobility of their hydrocarbon chains and consequently reducing the plasma membrane fluidity.
Dietary absorption (“exogenous cholesterol”) and endogenous synthesis (“endogenous cholesterol”) are the main sources of cholesterol in the body. Accordingly, principal cholesterol metabolic routes refer to its function in biosynthetic processes, excretion in the digestive tract, and an alternative route of removal, known as trans-intestinal C excretion (TICE) to complete the cholesterol balance (35). To maintain physiological cholesterol levels, also known as “cholesterol homeostasis”, all pathways are subjected to regulatory mechanisms responsible for cholesterol balance in the plasma membrane and intracellular.
Endogenous cholesterol is synthesized by a long and complex route, whose limiting enzyme HMG-CoA reductase (HMGCR) expression is activated by the sterol regulatory element-binding protein 2 (SREBP-2) transcription factor, localized in the ER (36).
Approximately 40-50% of exogenous cholesterol comprising the conversion of cholesterol to bile acids (BAs) and the excreted cholesterol via mixed micelles is reabsorbed through enterocyte pathways and once esterified, is incorporated in intestinal lipoproteins, named the chylomicrons (35).
In the intestinal lumen, the enzyme cholesterol esterase starts cholesterol hydrolysis, resulting in both free cholesterol and esterified FA. Then, interacting with the mixed micelles and their content, dietary cholesterol, monoacylglycerols (MAGs), and free FA, reaches the enterocytes. A selective cholesterol absorption results from regulated mechanisms differentiating from other micellar lipid components. The cholesterol transport from micelles, contacting the intestinal cell microvilli, is carried out by the membrane protein Niemann-Pick C1 Like 1 (NPC1L1) through cholesterol-regulated clathrin-mediated endocytosis (37, 38). Once in the enterocyte, free cholesterol is re-esterified with FA (of endogenous or dietary origin) by the catalytic action of the enzyme acyl-CoA-cholesterol-acyltransferase (ACAT). Also, triacylglycerides (TG) from dietary fats and oils are hydrolyzed in the intestinal lumen and absorbed as free FA and MAGs, being re-esterified by the enterocytes and, in combination with cholesterol esters, phospholipids, and apoprotein B-48 (apo B-48), forming the chylomicrons. The latter are then secreted from the basolateral membrane into the lymphatic vessels and subsequently into circulation (39). Figure 1 briefly summarizes the metabolism of endogenous and exogenous lipids and sterols by enterocytes.
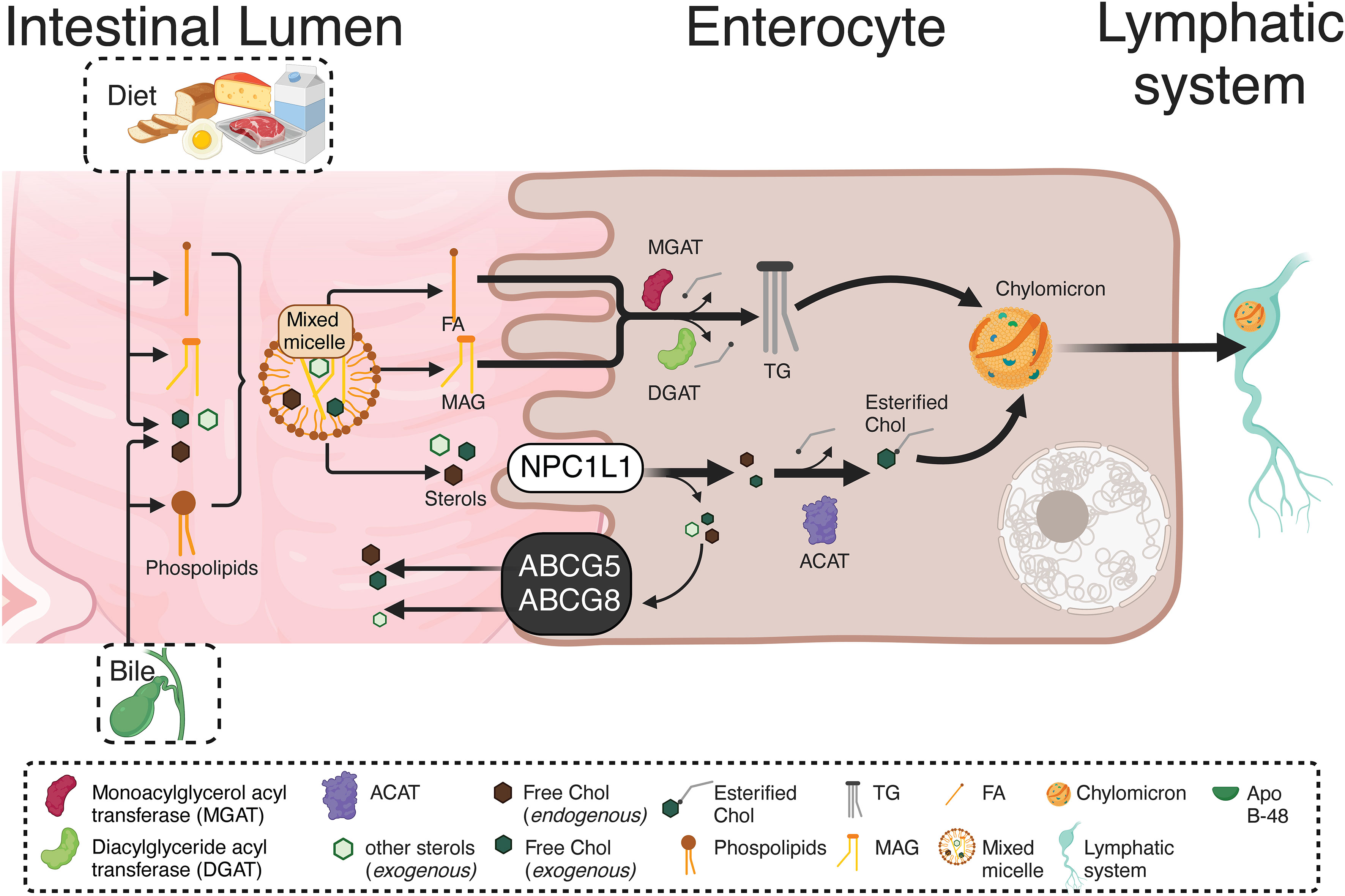
Figure 1 Absorption and re-esterification of exogenous and endogenous lipids and sterols by intestinal epithelial cells. Dietary cholesterol, MAGs, and free FA, inside of mixed micelles reach the enterocytes. A selective cholesterol absorption results from regulated mechanisms differentiating from other micellar lipid components. The cholesterol transport from micelles, contacting the intestinal cell microvilli, is carried out by NPCL1 through cholesterol-regulated clathrin-mediated endocytosis. Once in the enterocyte, the TG re-esterification is carried out by two enzymes: MGAT and DGAT. Both incorporate FA, the first to a MAG, while the second to a DAG, resulting in TG formation. Likewise, free cholesterol is re-esterified with FA by the ACAT. Endogenous and exogenous FA are used in re-esterification, with the TG produced by re-esterification, leaving the enterocytes for their distribution to different tissues. Therefore, in the enterocyte, re-esterified TG and cholesterol, as well as phospholipids and apo B-48, form chylomicrons. These are secreted from the basolateral cell region to the lymphatic vessels and afterward into the circulation. MAGs, Monoacylglycerols; NPCL1, Niemann-Pick C1 Like 1; TG, Triacylglyceride; MGAT, Monoacylglycerol acyltransferase DGAT, Diacylglyceride acyltransferase; FA, Fatty acids; DAG, Diacylglycerol; ACAT, Acyl-CoA-cholesterol-acyltransferase.. This figure was created with BioRender.com.
The endogenous and exogenous cholesterol trafficking in cells is mediated by transporters that allow cholesterol influx or efflux. Some of these transporters are affected in IBD, causing impaired cellular cholesterol trafficking (40, 41) and abnormal accumulation of intracellular cholesterol in ER or plasma membranes. This recruits the NLRP3 and the adaptor molecule apoptosis-associated speck-like protein containing a CARD (ASC), thus promoting inflammasome oligomerization (42–44). Despite there being no information in IBD, this perturbation could contribute to the severity of the pathology.
Cellular cholesterol dynamics in IBD
Cellular lipid fate starts with intestinal absorption of endogenous and exogenous sources to the lymphatic circulation, then transported to the liver, and finally to the bloodstream by LDL lipoproteins. Selective cellular LDL and HDL lipoprotein uptake by LDL receptors comprise a route through classical-clathrin coat endosomal/lysosomal route (45) and scavenger receptor class B type 1 (SR-BI), respectively (46). Intracellular cholesterol excess is delivered to HDL (reverse cholesterol transport) by ATP-Binding Cassette Transporter A1 (ABCA1), ATP-binding cassette transporter G1 (ABCG1), and SR-BI (47). Additionally, other transporters are steroidogenic acute regulatory (StAR) family members, including StARD1 and StARD3, mediating cholesterol movement from endosomes to mitochondria and ER (48, 49). Furthermore, vesicular transporter NPC1 mediates cholesterol efflux from late endosomes/lysosomes to the ER and plasma membranes (50). As expected, NPC1 deficiency in Niemann Pick C disease (NPC) is characterized by cholesterol accumulation in lysosomes and altered cholesterol content in mitochondria, plasma membranes, and ER (51), whereas NPC1 overexpression in CHO cells increases plasma membrane and ER cholesterol content (52). Notably, Npc1-null mice show impaired autophagy, intestinal granuloma, and a CD phenotype (53). Likewise, CD patients with an NPC1 disorder develop a granulomatous aggressive phenotype (54). Interestingly, IBD patients show increased LDL-R, NPC1, and decreased ABCG1 and SR-BI transcript levels (40, 41) suggesting that cellular cholesterol dynamics and homeostasis are affected in the intestinal mucosa (Figure 2).
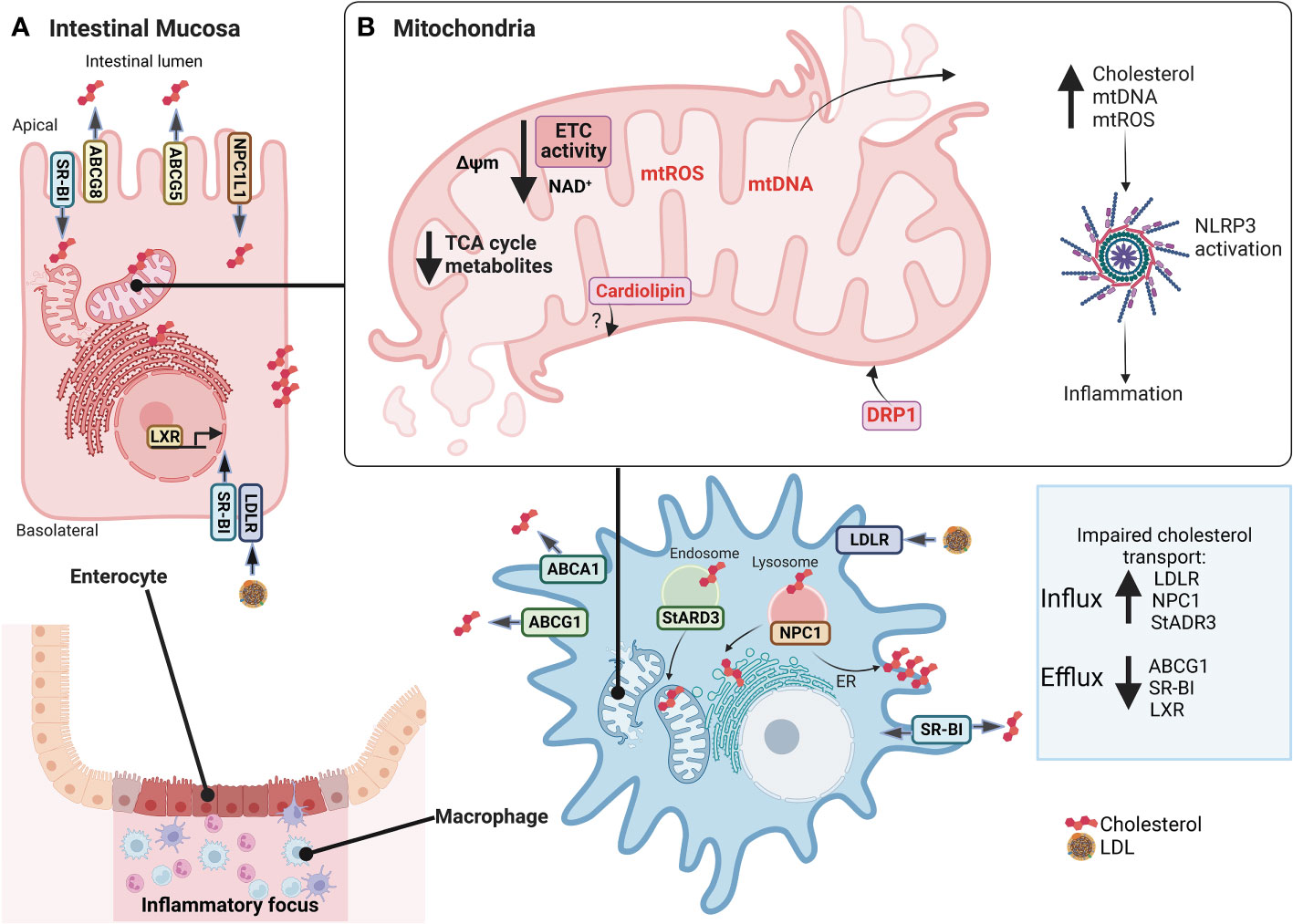
Figure 2 Alterations in cholesterol dynamics and mitochondrial dysfunction in IBD mucosa. In (A), inflamed intestinal mucosa shows enterocytes and macrophages have impaired transport cholesterol in IBD, with increased LDL-R, NPC1, and StADR3 allowing cholesterol influx and decreased ABCG1, SR-BI, and LXR preventing cholesterol efflux. In (B), also, mitochondria lose their function, promoting mitochondrial dysfunction, characterized by decreased expression of mitochondrial and nuclear genes associated with ETC and ATP synthase, NAD+ depletion, reduced levels of TCA cycle intermediates, reduction of Δψm, increased mtROS production, mitochondrial morphological changes with increased of DRP1 expression to regulate mitophagy and mitochondrial fission, and release of mtDNA. An imbalance in cholesterol traffic and mitochondrial function can activate NLRP3 inflammasome and promote inflammation of intestine mucosa, such as cholesterol, mtDNA, mtROS, and cardiolipin. However, there are no studies linking cardiolipin to IBD. IBD, Inflammatory Bowel Disease; LDL-R, low-density lipoprotein receptor; NPC1, Niemann–Pick C1 protein cholesterol transporter; StADR3, Steroidogenic acute regulatory; ABCG1, ATP Binding Cassette Subfamily G Member 1; SR-BI, scavenger receptor class B type I; LXR, Liver X receptor; ETC, Electron transport chain; TCA, Tricarboxylic acid; Δψm, mitochondrial membrane potential; mtROS, mitochondrial reactive oxygen species; DRP1, dynamin-related protein 1; mtDNA, mitochondrial DNA; NLRP3, NLR (Nod-like receptor) family pyrin domain containing 3. This figure was created with BioRender.com.
Also involved in cholesterol homeostasis and TG metabolism in the intestinal mucosa are the nuclear receptors (NR), liver X receptor (LXRs) α (NR1H3) and β (NR1H2) (55), as evidenced by decreased LXR expression in IBD patients, and several colitis mouse models (Il-10-null mice and dextran sodium sulfate, DSS or 2,4,6-trinitrobenzene sulfonic acid, TNBS-induced colitis) (56, 57) accompanied by decreased expression of ABCA1 (an LXR target gene) (57).
Interestingly, IBD etiology is associated with an unbalanced peripheral lipid profile with low total cholesterol, HDL, and LDL (58, 59). Furthermore, disease severity is related to high TG levels (59) and hyperlipidemia in IBD patients, and colitis mouse models (60, 61) suggesting the strong linkage between lipid metabolism and IBD. Moreover, a recent study demonstrates that the visceral adipose tissue is associated with therapy response and lower remission rates in IBD patients (62). In addition, the use of statins (drugs lowering circulating cholesterol levels) in IBD is associated with reduced steroids use (63) and disease relapse risk.
Being a component of plasma membranes and endomembranes, cholesterol is a critical constituent in lipid rafts. Cholesterol is decreased in lipid rafts and increased in non-lipid rafts fraction in an in vitro DSS-induced IBD-like phenotype of intestinal Caco-2 cells, with ER showing altered trafficking of protein, glycoprotein, and cholesterol. All this suggests inflammation promotes both unbalanced cell polarity and membrane integrity (64). In line, macromolecule trafficking is also disturbed by ER stress in IBD, characterized by increased expression of ER stress proteins (immunoglobulin-binding protein (BiP), C/EBP homologous protein (CHOP), transcription factor activation protein 4 (ATF4), and X-box binding protein (XBP1)) (64, 65). Mitochondria-associated ER membranes (MAMs) are microdomains that allow the calcium and lipid trafficking between mitochondria and ER, although disturbed regulation contributes to pathological conditions (66). Under these ER stress conditions, interruption of this ER-mitochondria communication produces oxidative stress and mitochondrial depolarization, impairing mitochondrial bioenergetics. A pro-inflammatory in vitro model of the M1-like Mø phenotype exhibits reduced ER-mitochondria communication and upregulated genes involved in mitochondrial fusion (MFN1, MFN2, and OPA1) and fission (DRP1 and FIS1), compared to M0- and M2-like Mø. Additionally, M1-like Mø converts superoxide anions into hydrogen peroxide (H2O2), generating mitochondrial ROS (mtROS) release (67, 68). Although changes in ER-mitochondria communication in cells from inflamed mucosa, such as Mø in IBD, are so far unknown, in vitro M1-like Mø models suggest alterations in the reciprocity platform between organelles (67).
Concomitantly, mucosal StARD3 expression is also increased in IBD patients (69), suggesting an increased cholesterol content in mitochondria. Accordingly, StARD3 upregulation increases mitochondrial cholesterol content in mice on a cholesterol-rich diet (70). Similarly, hepatocytes overexpressing StARD3 cause mitochondrial dysfunction mediated by oxidative stress, comparable to Npc1-null mice suffering severe mitochondrial oxidative stress (71). Currently, no studies demonstrate cholesterol levels within the mitochondrial membrane in the intestinal cell types in IBD patients or IBD in vivo models. On the other hand, cholesterol sulfate, which is catalyzed by hydroxysteroid sulfotransferase 2B1 (SULT2B1), and intestinal epithelium-specific knock of Sult2b1 has been significantly associated with down-regulation of cholesterol biosynthesis genes. In addition, cholesterol sulfate administration improved colitis features in DSS mice model; however, further studies are necessary to evaluate its anti-inflammatory effect in IBD (72).
Impaired mitochondrial metabolism in IBD
Mitochondrial dysfunction refers to loss of electron transport chain (ETC) efficiency, reduced ATP synthesis, mitochondrial morphological changes, and increased mtROS, and these are present in chronic diseases (58) such as IBD (Figure 2) (73–76). Regarding IBD, decreased expression of some mitochondrial and nuclear genes, associated with ETC (e.g. Complex I, III, and IV), ATP synthase (Complex V), and tricarboxylic acid (TCA) cycle, as well as reduced ETC activity has been observed in inflamed colonic biopsies versus non-inflamed mucosa (73). Despite the above, conflicting evidence on the contribution of ETC to IBD suggests a possibly high intrinsic variability in patients. Whether this is related to the severity of the pathology or the response to therapy currently remains unclear.
Complex I activity is a limiting step in oxidative phosphorylation (OXPHOS) and catalyzes the oxidation of nicotinamide adenine dinucleotide (NADH) (77, 78). Simultaneously, complex II links ETC with TCA cycle, oxidizing succinate to fumarate and reducing flavin adenine dinucleotide (FAD+) to hydroquinone form (FADH) (79). In UC patients, NAD+ is depleted in inflamed tissue (80). Although no FAD+ data is available in IBD, lower levels are expected due to decreased complex II activity; however, further studies are necessary to verify this.
Consistent with this reduced respiratory complex activity, reduced levels of TCA cycle intermediates citric acid, fumarate, isocitric acid, malate, pyruvate, and succinate have been reported in inflamed mucosa of UC patients (81). Seemingly even more significant, citrate, aconitate, α-ketoglutarate, succinate, fumarate, and malate are decreased in CD compared to UC and healthy patients (82), suggesting a differential metabolic phenotype related to bioenergetics dysfunction and inflammation in CD.
Notably, a metabolic shift towards glycolysis to compensate for a deficiency in mitochondrial ATP synthesis is shown in PBMCs from IBD patients when compared to healthy donors (83). In this IBD metabolic context, colonocytes change to glycolytic metabolism (increased intracellular lactate), and bacteria in the intestinal lumen consume epithelium oxygen, causing dysbiosis and promoting mucosal inflammation (84).
Colonic epithelial cells present a reduction of mitochondrial membrane potential (Δψm) in inflamed tissue (73) that could be promoted by the reduction of ETC activity and the presence of uncoupling proteins (UCP), which permit the proton leak from the mitochondrial intermembrane space to the matrix (85). Ucp2 overexpression was reported in colon of DSS-treated mice (86), with a reduced inflammation severity in the Ucp2 knockdown (87) that could promote the loss of Δψm. However, discordant results were obtained in DSS-treated Ucp2 null mice, which developed more severe colitis that was associated with higher ROS and decreased expression of ZO-1 and JAM1 (88) (related to tight junction permeability and epithelial architecture loss). Furthermore, Ucp2 expression in colonocytes in the small intestine from TNBS-treated mice was not significantly increased, despite the reduction of respiration in isolated mitochondria (89) possibly reflecting the participation of other proteins of the mitochondrial membrane. More studies are necessary to evaluate the participation of UCP2 in IBD bioenergetics and physiopathology.
Regarding mitochondrial morphology, CD patients with active disease show severe ultrastructural mitochondrial abnormalities in Paneth cells, goblet cells, and enterocytes (90). Intestinal enterocyte mitochondria appear dysmorphic, with cristae destruction and abnormal architecture in mouse models of intestinal inflammation. Dysfunctional mitochondria undergo BCL2 interacting protein 3-like (BNIP3L)/NIX-mediated mitophagy and mitochondrial fission, evidenced by increased dynamin-related protein 1 (DRP1) expression. As a physiological response to stress and mitochondrial damage, hypoxia-inducible factor alpha (HIF1α) and mtROS are enhanced to regulate mitophagy during intestinal inflammation, eliminating dysfunctional mitochondria from the cell and preventing further ROS accumulation (91).
Mitochondrial dysfunction in IBD has also been related to decreased PPARGC1A expression (encoding Peroxisome proliferator- activated receptor-alpha; PGC1-α), which is considered a master regulator of mitochondrial biogenesis and bioenergetics (69, 73). Under stress conditions, PGC-1α potently stimulates mitochondrial turnover and antioxidant activity (92, 93). Different roles for PGC-1α have been suggested from in vitro and in vivo UC models. PGC-1α is highly expressed in the murine intestinal epithelium; however, during colitis, acetylation inactivates PGC-1α, followed by ubiquitination and proteasomal degradation (92). Moreover, hypermethylation of the PPARGC1A gene in severe UC, when compared to mild UC patients, is related to decreased PGC-1α expression (73, 94). In addition, reduced mitochondrial mass in the intestinal mucosa of UC patients can be a consequence of decreased PGC-1α and/or increased mitophagy (95). Epithelium-specific Ppargc1a null mice are more susceptible to DSS-induced colitis compared to wild-type mice (92), highlighting its participation in colitis pathophysiology. Contradictorily, Paneth cells of CD patients showed upregulated PGC-1α signaling, which may represent an adaptive response to ongoing mitochondrial damage during IBD (90).
Post-translational regulation of PGC-1α involves highly conserved NAD+-dependent deacetylase Sirtuins (SIRTs), including SIRT1. SIRT1 senses metabolic stress and thus promotes upregulation of mitochondrial function and FA oxidation genes (96, 97). Also, SIRT1 deacetylates histones (H1, H3, and H4) and transcription factors, such as NF-κBp65, inhibiting pro-inflammatory gene transcription. Accordingly, decreased SIRT1 in IBD mucosa promotes the inflammatory environment (98).
Conventional therapy in IBD, such as 5-ASA (5-aminosalicylic acid), reverses mitochondrial dysfunction by activating Peroxisome proliferator- activated receptor-gamma (PPAR-γ) (84, 99, 100). PPAR-α and PPAR-γ nuclear receptors (encoded by PPARA and PPARG), expressed in crypt upper epithelial cells and Mø, are associated with cellular processes, such as mitochondrial biogenesis and translation initiation. Interestingly, PPAR-α and PPAR-γ have been associated with ROS detoxification and cholesterol biosynthesis, while specifically PPAR-γ is associated with TCA cycle and NF-κB signaling inhibition, whose gene expression is decreased in IBD, affecting these processes (73, 101). Overexpression of PPAR-γ (in a hepatocyte cell line) decreases HMGCR and SREBP-2 gene and protein levels, increasing CYP7A1, ABCG5, and ABCG8 gene and protein expression, which contribute to a decrease in cholesterol synthesis, an increased conversion of cholesterol to BAs and cholesterol efflux (102).
In mouse Mø treated with soluble (free) cholesterol, it was observed that cholesterol was absorbed and accumulated in the cytoplasm and mitochondria, leading to increased mitochondrial mass, decreased oxygen consumption rate, and OXPHOS (103). This suggests that an accumulation of cholesterol in cellular compartments, such as the mitochondria, could cause changes in mitochondrial metabolism and bioenergetics. Further studies of intracellular cholesterol accumulation and trafficking with its metabolic consequences in IBD are still required.
Inflammasome involvement in IBD
Inflammasome activation
Participating in innate antimicrobial responses against PAMPs, DAMPs, and stress, the inflammasome is a supramolecular complex composed of a sensor protein, a caspase, and an ASC adapter protein (104). Sensor proteins are PRRs of innate immunity, belonging to the NOD-like receptor family (NLR), such as NLRP1, NLRP3/NLRP6, NLRC4, and NAIP, or the PYHIN family (pyrin and HIN200 (hematopoietic interferon-inducible nuclear antigens with 200 amino acid repeat domain-containing protein)), such as AIM2/IFI16 (105, 106). So far, NLRP3 is the type most studied. Inactive NLRP3 resides mainly in the ER but, upon activation, translocates to MAMs in the perinuclear space and recruits ASC and caspase-1, thereby enabling cytoplasmic NLRP3 inflammasome assembly (107). In modulating NLRP3 activation, its filamentous assembly is ASC-dependent, containing a central nucleotide-binding, self-oligomerization domain (NBD) with ATPase activity and an LRR (leucine-rich repeat) domain conserved at the C-terminal. Once activated, NLRP3 oligomerizes, assembling multiple ASC filaments, through interactions between both N-terminal PYD (pyrin domain) of NLRP3 and ASC. Alternatively, ASC contains a caspase activation and recruiting domain (CARD), which interacts with the caspase-1 CARD, linking caspase domains for dimerization, activating self-cleavage, and finally, caspase-1 activation (108).
The canonical inflammasome pathway activation involves caspase-1-mediated cleavage of pro-IL-1β and pro-IL-18 into active IL-1β and IL-18 (104). Moreover, the non-canonical pathway consists of ASC-independent murine caspase 11 or human caspase 4/5, NRLP3 (109). Caspase-1 and caspase-4/5/11 cleave gasdermin D (GSDMD), releasing gasdermin-amino terminal domain and inducing pyroptosis, a variant of programmed cell death characterized by pore formation in the plasma membranes. This allows cytoplasmic content to be released from infected cells, which are then eliminated by neutrophils (110).
As mentioned before, inflammasome activation requires two signals, one being NF-κB-mediated NLRP3 and pro-IL1B gene transcription, and a second signal may arise from PAMPs, such as LPS, toxins, viral and bacterial nucleic acids and leads to oligomerization and activation. In addition, the inflammasome senses stress and cell damage, such as high ATP concentrations, mitochondrial-derived components (cardiolipin, ROS, mitochondrial DNA (mtDNA)), ER stress, intracellular calcium influx, uric acid, serum amyloid A, and cholesterol crystals (20–23).
Inflammasome activation in IBD
Inflammasome activation in the intestinal mucosa promotes pathogen clearance and Mø-mediated epithelial barrier recovery, with NLRP3 expressed both in colonic epithelial cells and Mø (111). The inflammasome is linked to IBD, as higher NLRP3, Caspase-1, NIMA-related kinase 7 (NEK7), and GSDMD expression were observed in inflamed versus non-inflamed tissue of UC patients (18). Also, hypomethylation of genes, such as NLRP3, NLRC4, NLRP12, and IL-10, correlated with higher gene transcription in severe compared to mild UC patients (94).
Furthermore, intestinal barrier dysfunction in DSS-induced colitis in mice allows the translocation of bacteria to stimulate lamina propria Mø via TLR/NF-κB, leading to pro-IL-1β and pro-IL-18 transcription, consequently NLRP3 activation (18, 112). This observation was confirmed by DSS-treated Mø in vitro, producing high IL-1β levels in a caspase-1-, NLRP3- and ASC-dependent manner (29). Nonetheless, conflicting evidence on the role of Nlrp3 and Caspase 1 genes was observed in IBD models, reporting in nlrp3-null mice less severe colitis and lower colonic pro-inflammatory cytokine production (29, 112). Whilst another study showed worse survival and clinical scores than wild-type mice (113). Recently, a GWAS study demonstrated the presence of inflammasome activation components in an IBD risk loci (33).
Additionally, the inflammasome is essential for protection against colitis-associated dysplasia and tumorigenesis towards colorectal cancer (CRC) (31, 113). Both Nlrp3- and Caspase 1-null mice treated with AOM (azoxymethane)/DSS develop large tumors with significant hyperplasia, and inflammation, with reduced production of IL-18 and the presence of proliferating dysplastic cells in dysplasia regions. NLRP3-dependent IL-18 production prevents neoplasia, with IL-18 participating in intestinal epithelial barrier repair via IFN-γ production and STAT1 signaling (31). In data analysis on the Oncomine® platform from patients with CRC, a decreased expression of inflammasome NLRP1, NLRP3, NLRC4, and AIM2 was observed compared to healthy controls (114).
Thus, evidence from in vivo colitis models shows that other molecular factors could interact with the inflammasome.
Cholesterol balance and inflammasome activation
Altered cholesterol transport leads to its accumulation in ER or plasma membranes, recruiting NLRP3 and ASC, facilitating inflammasome oligomerization (42–44), thus, impacting inflammation in atherosclerosis and Alzheimer’s disease (115). Initially, cholesterol uptake in intestinal epithelial cells by NPC1L1 promotes acute intestinal inflammation, recruiting myeloid cells, such as Mø, which capture cholesterol by NPC1 and activate NLRP3 inflammasome by activating caspase-1 and IL-1β secretion (42, 116). Additionally, dietary cholesterol has been shown to exacerbate CRC in murine models by activating NLRP3 inflammasome (20), and its blockade improves metabolism in obese mice (117). Additionally, cholesterol accumulation in plasma membranes (rafts/caveolae domains) affects its integrity, mainly represented by a changed distribution of caveolin-1 (CAV-1), becoming a platform for the inflammasome (118), and TLR4 activation in immune cells (44, 119–122). Importantly, TLR4 monomer is inactive in the plasma membranes and recruited into lipid raft domains once stimulated, allowing the toll-interleukin-1 receptor (TIR) domain to interact with membrane-associated co-receptor MAL/MyD88, initiating signaling for pro-IL-1β production (123). Reinforcing the role of cholesterol transport in inflammasome activation, ABCA1/G1 deficiency promotes cholesterol accumulation in Mø (43, 124, 125). Similarly, treatment with the NPC1 inhibitor U18666A (126) or NPC1 deficiency leads to the cholesterol accumulation in late-endosomes/lysosomes, reducing cholesterol transport to the ER and plasma membranes, thus blocking inflammasome assembly (42). These observations demonstrate a role for NPC1 in cholesterol homeostasis and TLR4 activation impacting inflammasome assembly. However, these mechanisms have not been established yet in IBD.
Inflammasome activation by mitochondrial mediators in IBD
The NLRP3 inflammasome can be activated by mitochondrial DAMPs which are released from intestinal mucosa during mitochondrial dysfunction in IBD (127). Mitochondrial damage causes mtDNA fragmentation and mtROS production, which oxidizes mtDNA into an oxidized form (ox-mtDNA) and acts as a second signal to activate the NLRP3 inflammasome (128). On the other hand, the first signal in LPS-stimulated Mø induces de novo mtDNA synthesis via the MyD88/TRIF-dependent TLR4, which activates IRF1 to induce CMPK2 expression, a mitochondrial nucleotide kinase (129). This de novo mtDNA is susceptible to oxidation and fragmentation, resulting in ox-mtDNA fragments, which can activate the inflammasome (129). Also, NLRP3 inflammasome activation in Mø induces pyroptosis by GSDMD. This process is characterized by plasma membrane permeabilization, causing changes in ion homeostasis that contribute to mitochondrial collapse, thus accumulating cytosolic mtDNA to be released to the extracellular environment when the cell undergoes apoptosis or pyroptosis (130). Both IBD patients with active disease and DSS-induced colitis in mice show increased plasma mtDNA levels, corroborating mitochondrial damage and suggesting inflammasome activation by mtDNA during IBD development (127).
NLRP3 inflammasome can also be activated by direct interaction with mitochondrial cardiolipin, in a mtROS-independent manner (21). Cardiolipin is a phospholipid locates exclusively in the inner mitochondrial membrane (IMM). It can translocate to the outer mitochondrial membrane (OMM) by mtROS production, PAMPs (e.g., LPS), or pro-apoptotic stimuli, thus promoting mitophagy (131). This suggests that cardiolipin could have a role in IBD pathogenesis. However, there are still no studies linking cardiolipin to IBD.
The interplay between mitochondrial dysfunction, cholesterol metabolism, and inflammasome activation upon the gut microbiota in IBD
Important host’s physiological functions are influenced by their gut microbiota, whose closest association became a single unit, called “holobiont”, where mtROS levels have important roles. Because of mtDNA mutations, mtROS levels are increased, reducing the ATP production, with MtDNA variations have an important impact on intestinal health. Mice strains with increased OXPHOS are less susceptible to developing colitis (PMID: 23872498), while mtDNA variations lead to different mtROS levels, influencing gut microbiota (132, 133). Furthermore, mice with mtDNA variants related to higher ROS production had a reduced gut microbiome diversity (133). In IBD, there is reduced microbial diversity, lower SCFAs and ATP production, impaired mucus production and antimicrobial peptides (134, 135), as a consequence of higher pathogenic bacteria and mtROS levels. IBD patients present different microbiota compositions; some having eubiotic (homeostatic) or dysbiotic (imbalanced) microbiota. Increased facultative anaerobes and aerobic bacteria is seen in dysbiotic IBD patients compared to healthy control and eubiotic IBD patients (characterized by high obligate anaerobe abundance) (136). This evidence is consistent with the hemoglobin (oxygen carrier) and ROS release to the intestinal lumen, increasing oxygen levels to overgrown facultative anaerobes and aerobic bacteria (137). This dysbiotic environment promotes increased gut permeability, thus favoring the passage of PAMPs, which can activate PRRs, such as the TLR4/NLRP3 inflammasome signaling pathway, and triggering inflammation.
Disruptions in cholesterol metabolism are described in IBD, where BAs are end products of cholesterol, and their synthesis is regulated by the farnesoid X receptor (FXR). In IBD, there is a decreasing activity of FXR (138), a consequence of exacerbated NF-κB activation (139, 140).
Despite the pathogenic role of BAs in IBD being controversial, an impaired reabsorption of conjugated BAs occurs in enterocytes, and consequently, many side effects such as maldigestion of lipids, steatorrhea, impaired intestinal motility, and dysbiosis can occur (141). Regarding bile acid malabsorption, a parallel stimulation of cholesterol synthesis, cholesterol degradation, and LDL receptor expression, results in reduced LDL cholesterol in IBD patients (142). On the other hand, in humans, conjugated BAs are secreted in the intestine, being substrates of bacterial microbiota, resulting in the production of secondary BAs, such as deoxycholic and lithocholic acids (DCA and LCA, respectively) (143). These can activate TGR5 and induce the TGR5-cAMP-PKA signaling pathway, promoting NLRP3 ubiquitination (144). Accordingly, several studies demonstrate reduced DCA and LCA levels in active IBD patients, associated with gut dysbiosis (143), and consequently, NLRP3 can be permanently activated, exacerbating the inflammatory response.
Dietary phytochemicals to restore mitochondrial function and cholesterol balance in IBD
Since mtROS overproduction and cholesterol unbalance are considered detrimental events in IBD, novel antioxidant strategies targeting the mitochondrial metabolism may be promising alternatives and/or supportive therapies (90, 145, 146). Notably, dietary compounds (mainly phytochemicals, such as polyphenols) may counteract oxidative stress, mitochondrial dysfunction, and intracellular cholesterol trafficking via Nuclear erythroid 2-related factor 2 (Nrf2) (Figure 3), PPAR-γ (Figure 4) and LXR signaling. These effects have been extensively explored and reviewed elsewhere. However, the main findings relevant to this review regarding phytochemicals are summarized in Table 1. In addition, phytochemicals can act as prebiotics, promoting an eubiotic microbiota that produces higher amounts of bacterial metabolites, enhancing the epithelial barrier function, and reducing the inflammatory response, which could be related to the improvement of symptoms in IBD patients (Figure 3) (1, 174).
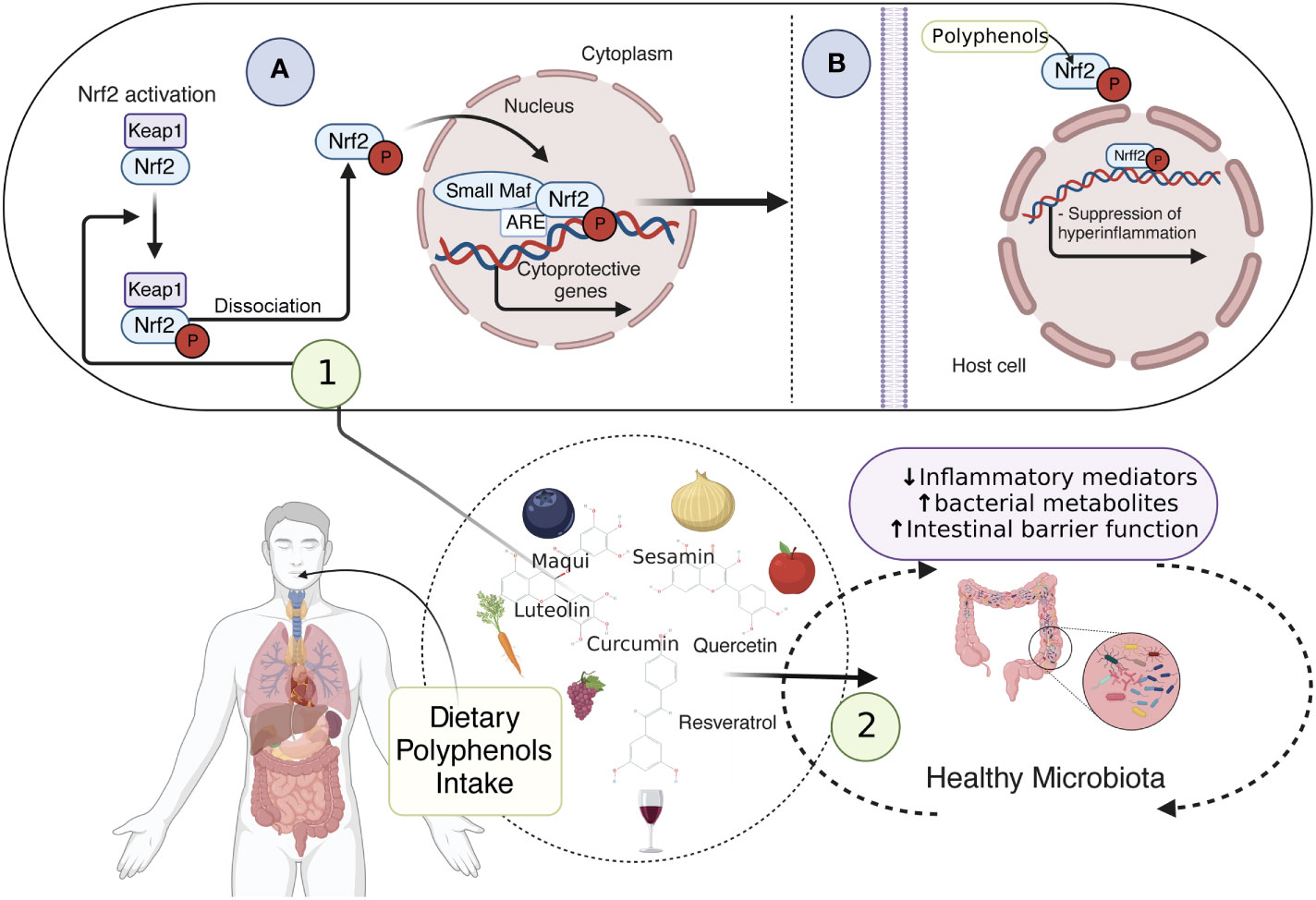
Figure 3 Dietary compounds and activation of Nrf2 pathway preventing inflammation. dietary compounds, such as polyphenols, prevent oxidative stress, mitochondrial dysfunction, and intracellular cholesterol trafficking via Nrf2 signaling. Sequestered in the cytoplasm by Keap1, Nrf2 is inactive, however, when phosphorylated by polyphenols, it can be released from Keap1, translocate to the nucleus, and form a heterodimer with small Maf proteins. This allows cellular protection and adaptive responses through the cytoprotective gene expression (1A), thus suppressing the inflammation (1B). Additionally, phytochemicals acting as prebiotics promote a healthy microbiota, which ensures an accurate amount of bacterial metabolites, enhancing the epithelial barrier function and reducing the inflammatory response (2). Nrf2, Nuclear erythroid 2-related factor 2; Keap1, Kelch-like ECH-associated protein1. This figure was created with BioRender.com.
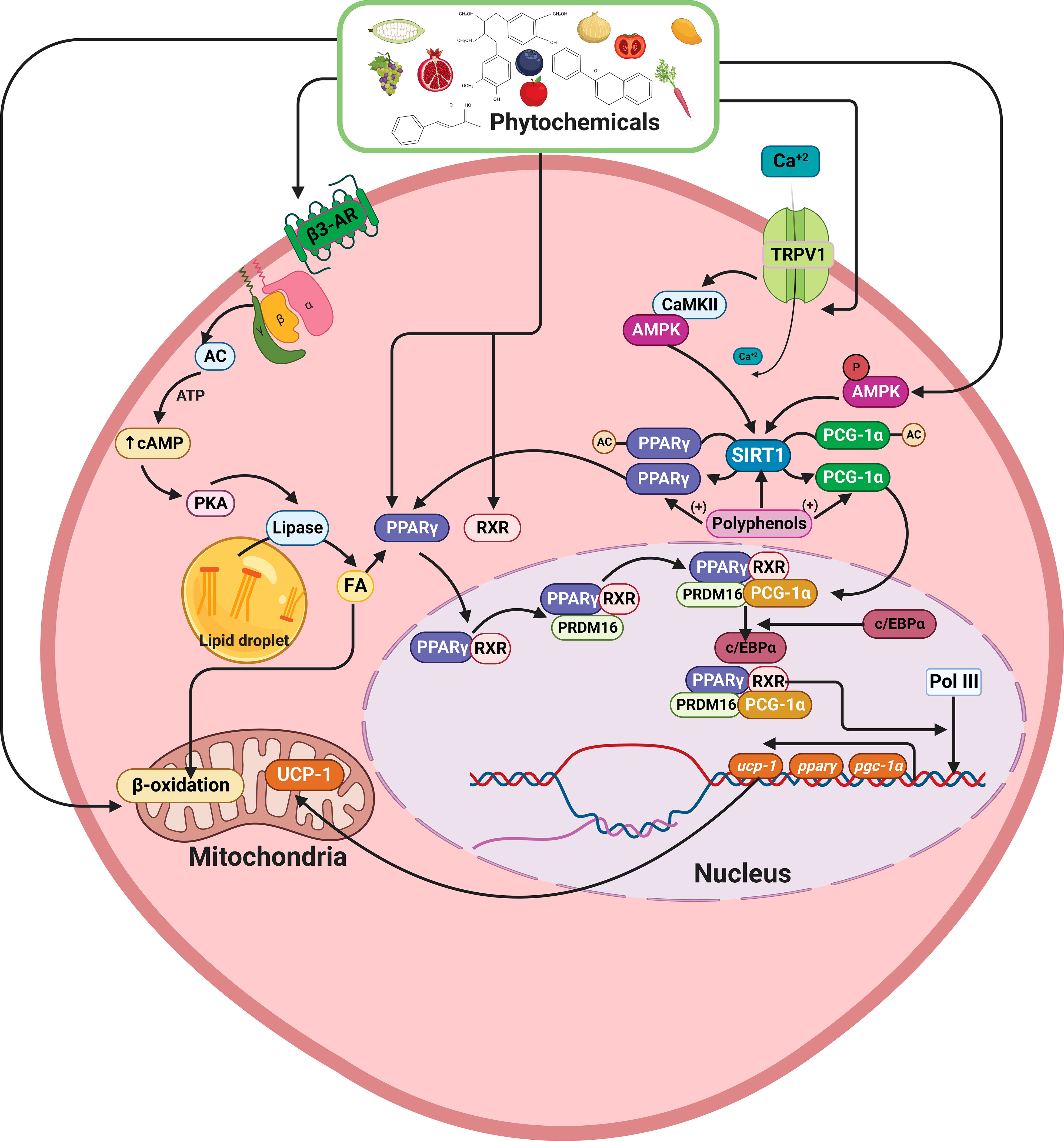
Figure 4 Schematic model showing the effects of phytochemicals on lipid metabolism, increase of PPAR-γ SIRT1, PGC-1α, and UCP1 expression. Polyphenols can deacetylate PPAR-γ and PRDM16, promoting the UCP1, PPAR-γ, SIRT1, and PGC-1α overexpression. On the other hand, via TRPV1, polyphenols can increase SIRT1 expression, and through β3-AR activation, increase cAMP levels, activating MAPKs pathway, resulting in TG hydrolysis, FA oxidation, and increased mitochondrial UCP1 transcription and activity. PPAR-γ, Peroxisome proliferator- activated receptor-gamma; SIRT1, Sirtuin 1; PGC-1α, Peroxisome proliferator- activated receptor-alpha; PRDM16, PR domaing containing 16; UCP1, Uncoupling protein 1; TRPV1, Transient receptor potential vanilloid 1; β3-AR, β3-adrenergic receptor; TG, Triacylglyceride; FA, Fatty acid. This figure was created with BioRender.com.
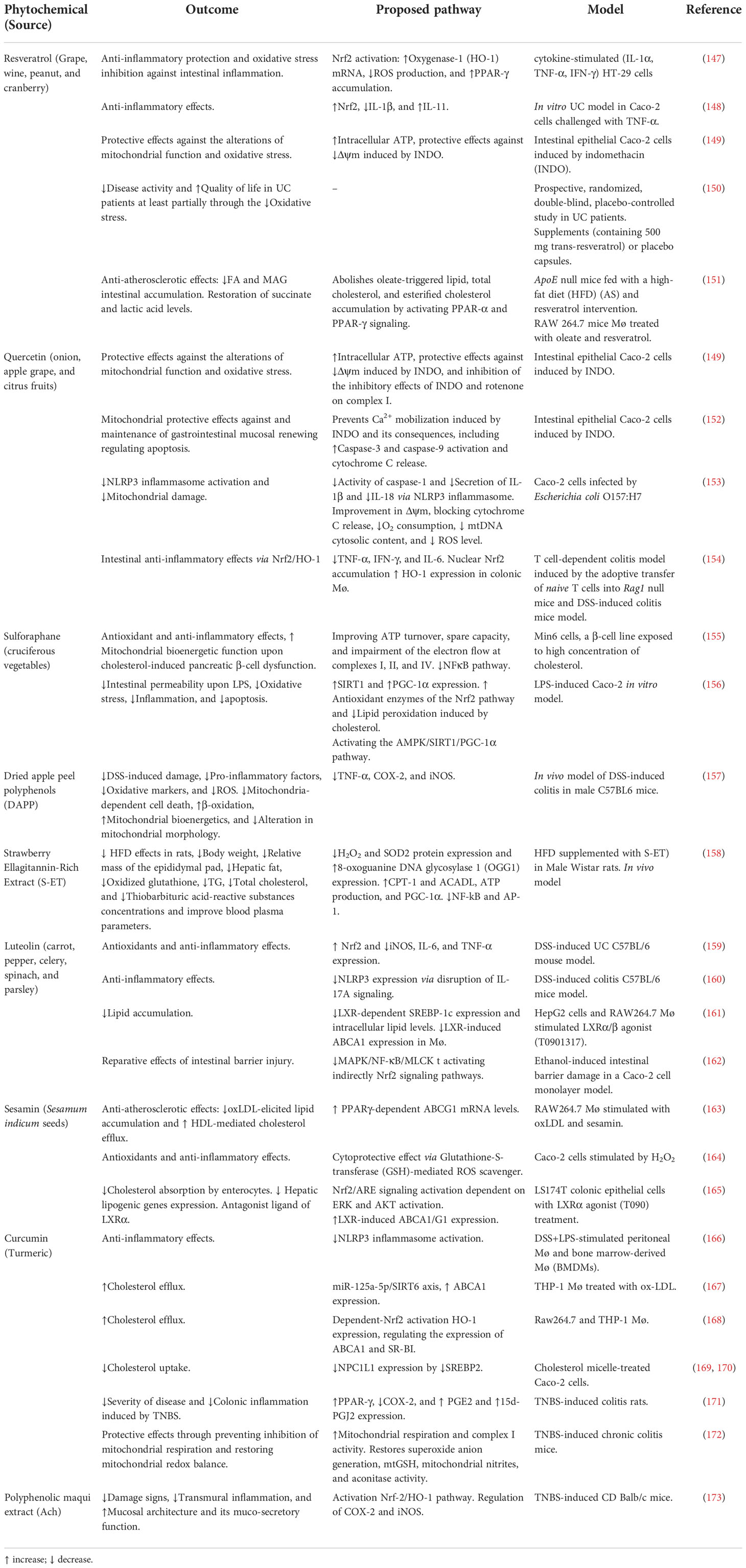
Table 1 Dietary phytochemicals and their restorative effects on mitochondrial function and lipid homeostasis.
Nrf2 is a redox-sensitive transcription factor sequestered in the cytoplasm by Kelch-like ECH-associated protein1 (Keap1), remaining inactive under physiological conditions and being degraded by the proteasome (175). Upon oxidative stress, Nrf2 is released from Keap1, translocates to the nucleus, and forms a heterodimer with small Maf proteins (175). This heterodimer allows cellular protection and adaptive responses through the expression of phase II-detoxifying enzymes (glutathione S-transferase (GST), NAD(P)H: quinone oxidoreductase (NQO1)), stress-responsive proteins (heme oxygenase-1 (HO-1)), and ROS scavenging enzymes (glutathione peroxidase (GPx), superoxide dismutases (SOD) (175, 176) (Figure 3). Additionally, Nrf2 activation regulates mitochondrial biogenesis by activating nuclear respiratory factor-1 (NRF-1), transcribing mitochondrial transcription factor A (TFAM), and mitochondrial transcription factor B2 (TFBM2) and regulating PGC-1α expression (177). Interestingly, evidence suggests that down-regulation of Nrf2/PGC-1α axis contributes to susceptibility to inflammatory bowel disease (92, 178, 179), indicating that activation of Nrf2 signaling by dietary phytochemicals may alleviate oxidative stress and mitochondrial dysfunction in IBD.
In addition, phytochemicals, especially polyphenols, are players in the deacetylation process of PPAR-γ and PRDM16, causing the UCP1 overexpression and increase of PPAR-γ, PPAR-α, SIRT1, and PGC-1α expression (Figure 4). Moreover, via transient receptor potential vanilloid 1 (TRPV1) some phytochemicals can also increase the SIRT1 expression (180). In addition, other phytochemicals, such as quercetin, curcumin, and resveratrol can activate the β3-adrenergic pathway (β3-AR), increasing cAMP levels by activating the MAPKs pathway, resulting in TG hydrolysis, FA oxidation and increasing mitochondrial UCP1 transcription and activity (181). In DSS-treated mice, reduced UCP1 expression in subcutaneous adipose tissues was found in conjunction with increased intestinal permeability (86). These findings could suggest a role in IBD pathogenesis or disease progression. However, no data about its expression in epithelial or lamina propria cells, nor its bioenergetics outcomes, has been addressed so far.
On the other hand, LXR expression is decreased in IBD (56, 57). LXR activation enhances the intestinal ABCA1 and ABCG1 expression while it reduces NPC1L1 expression, resulting in increased intracellular cholesterol efflux and limiting exogenous cholesterol absorption by enterocytes (39, 182). Furthermore, plasma-derived cholesterol from the enterocyte basolateral membrane is excreted to the gut lumen (e.g., transintestinal cholesterol excretion) by ABCG5 and ABCG8, both of which are regulated by LXR (183, 184). Of note, in atherosclerosis, diabetes, and diabetic retinopathy using a SIRT1 agonist, LXR is deacetylated by SIRT1 at a specific conserved lysine (K432 in LXRα and K433 in LXRβ), thereby activating LXR (185–188) suggesting that SIRT1 may be a potential IBD target to promote LXR-mediated cholesterol efflux.
Hence, Nrf2-, PPAR-γ-, and LXR-mediated signaling pathway regulation by dietary phytochemicals enhance the antioxidant defense and reduce inflammation in both in vitro and in vivo IBD models (Table 1). Some phytochemicals such as luteolin, sesamin, and maqui extract have an unknown direct effect on mitochondrial dysfunction in IBD, becoming an interesting topic of study. Additionally, further studies of Nrf2, PPAR-γ, and LXR upstream pathways are required for a better understanding of the antioxidant, anti-arteriosclerotic, and anti-inflammatory effects of these dietary compounds.
Conclusion and future perspectives
IBD is characterized by a loss of intestinal barrier function and inflammation. During IBD pathogenesis, trafficking of intracellular cholesterol is altered, with decreased cholesterol transporter expression, reducing cholesterol efflux and increasing influx. Since cholesterol is an important component of the plasma membranes and endomembranes (ER and mitochondria), abnormal cholesterol accumulation promotes ER stress and mitochondrial dysfunction during IBD. Furthermore, mitochondria in IBD are characterized by decreased ETC expression and activity, Δψm drop, mtROS production, and decreased TCA cycle intermediates, leading to a metabolic shift towards glycolysis as an adaptive response. Chronic inflammation during IBD may be mediated by NLRP3 inflammasome activation by DAMPs, such as cholesterol, mtROS, and mtDNA, generating an IL-1β-mediated pro-inflammatory response. Notably, the recovery of the mitochondrial function with correct cholesterol trafficking could be a potential therapeutic strategy for IBD. In addition, it has been discussed that the high prevalence and incidence in industrialized countries could be related to a Western diet, due to the high intake of saturated fatty acids, cholesterol, refined carbohydrates, and sugar may impact the inflammatory status becoming essential during disease pathogenesis (189). Therefore, changing towards an increase in the intake of whole grains, vegetables, fruit, seeds, and polyunsaturated fatty acids, with are part of a Mediterranean diet characterized by a higher content of phytochemicals and essential fatty acids could bring benefits to IBD patients. In this line, several dietary phytochemicals have been identified as efficient agents reducing inflammasome activation through the recovery of cholesterol efflux and mitochondrial function, which could reduce immunopathogenic effects in IBD patients.
The role of altered cholesterol trafficking and accumulation in IBD inflammation is an emerging area that offers new perspectives to understanding the pathogenic mechanisms of IBD. This may represent new opportunities for the discovery of drugs with mitochondrial functions that inhibit NLRP3 inflammasome activation.
Author contributions
JA wrote most of the review. NG contributed to writing and made the figures. FU, KD, and MAH contributed to the writing and participated reviewing and critically correcting the manuscript, MAH corrected on manuscript structure and supervised the work. KN, MF, and GL participated reviewing and critically correcting the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by the Agencia Nacional de Investigación y Desarrollo (ANID)/PhD fellowship #21220889 (JA), and #21200669 (NG), FONDECYT Grants #1220702 (MAH), #11201322 (FAU), MiBi: interdisciplinary group on mitochondrial targeting and bioenergetics ACT 210097 (FAU); FONDECYT postdoctoral grant #3210367 (KD-C).
Acknowledgments
We wish to thank David Cox for his editing. Figures were created with BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Gasaly N, de Vos P, Hermoso MA. Impact of bacterial metabolites on gut barrier function and host immunity: A focus on bacterial metabolism and its relevance for intestinal inflammation. Front Immunol (2021) 12:658354. doi: 10.3389/fimmu.2021.658354
2. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature (2012) 491(7422):119–24. doi: 10.1038/nature11582
3. Zhang YZ, Li YY. Inflammatory bowel disease: pathogenesis. World J Gastroenterol (2014) 20(1):91–9. doi: 10.3748/wjg.v20.i1.91
4. Ng SC, Shi HY, Hamidi N, Underwood FE, Tang W, Benchimol EI, et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet (2017) 390(10114):2769–78. doi: 10.1016/S0140-6736(17)32448-0
5. Kaplan GG, Windsor JW. The four epidemiological stages in the global evolution of inflammatory bowel disease. Nat Rev Gastroenterol Hepatol (2021) 18(1):56–66. doi: 10.1038/s41575-020-00360-x
6. Kaplan GG. The global burden of IBD: from 2015 to 2025. Nat Rev Gastroenterol Hepatol (2015) 12(12):720–7. doi: 10.1038/nrgastro.2015.150
7. Molodecky NA, Soon IS, Rabi DM, Ghali WA, Ferris M, Chernoff G, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology (2012) 142(1):46–54.e42. doi: 10.1053/j.gastro.2011.10.001
8. Pagnini C, Pizarro TT, Cominelli F. Novel pharmacological therapy in inflammatory bowel diseases: Beyond anti-tumor necrosis factor. Front Pharmacol (2019) 10:671. doi: 10.3389/fphar.2019.00671
9. Neurath MF, Travis SP. Mucosal healing in inflammatory bowel diseases: a systematic review. Gut (2012) 61(11):1619–35. doi: 10.1136/gutjnl-2012-302830
10. Gisbert JP, Panés J. Loss of response and requirement of infliximab dose intensification in crohn's disease: a review. Am J Gastroenterol (2009) 104(3):760–7. doi: 10.1038/ajg.2008.88
11. Guerra I, Bermejo F. Management of inflammatory bowel disease in poor responders to infliximab. Clin Exp Gastroenterol (2014) 7:359–67. doi: 10.2147/CEG.S45297
12. Wong U, Cross RK. Primary and secondary nonresponse to infliximab: mechanisms and countermeasures. Expert Opin Drug Metab Toxicol (2017) 13(10):1039–46. doi: 10.1080/17425255.2017.1377180
13. Abraham BP, Ahmed T, Ali T. Inflammatory bowel disease: Pathophysiology and current therapeutic approaches. Handb Exp Pharmacol (2017) 239:115–46. doi: 10.1007/164_2016_122
14. Berg DR, Colombel JF, Ungaro R. The role of early biologic therapy in inflammatory bowel disease. Inflammation Bowel Dis (2019) 25(12):1896–905. doi: 10.1093/ibd/izz059
15. Kolodziejczyk AA, Zheng D, Elinav E. Diet-microbiota interactions and personalized nutrition. Nat Rev Microbiol (2019) 17(12):742–53. doi: 10.1038/s41579-019-0256-8
16. de Souza HS, Fiocchi C. Immunopathogenesis of IBD: current state of the art. Nat Rev Gastroenterol Hepatol (2016) 13(1):13–27. doi: 10.1038/nrgastro.2015.186
17. Zhen Y, Zhang H. NLRP3 inflammasome and inflammatory bowel disease. Front Immunol (2019) 10:276. doi: 10.3389/fimmu.2019.00276
18. Chen X, Liu G, Yuan Y, Wu G, Wang S, Yuan L. NEK7 interacts with NLRP3 to modulate the pyroptosis in inflammatory bowel disease via NF-κB signaling. Cell Death Dis (2019) 10(12):906. doi: 10.1038/s41419-019-2157-1
19. West NR, Hegazy AN, Owens BMJ, Bullers SJ, Linggi B, Buonocore S, et al. Oncostatin m drives intestinal inflammation and predicts response to tumor necrosis factor-neutralizing therapy in patients with inflammatory bowel disease. Nat Med (2017) 23(5):579–89. doi: 10.1038/nm.4307
20. Du Q, Wang Q, Fan H, Wang J, Liu X, Wang H, et al. Dietary cholesterol promotes AOM-induced colorectal cancer through activating the NLRP3 inflammasome. Biochem Pharmacol (2016) 105:42–54. doi: 10.1016/j.bcp.2016.02.017
21. Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity (2013) 39(2):311–23. doi: 10.1016/j.immuni.2013.08.001
22. Juliana C, Fernandes-Alnemri T, Kang S, Farias A, Qin F, Alnemri ES. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem (2012) 287(43):36617–22. doi: 10.1074/jbc.M112.407130
23. Zmora N, Levy M, Pevsner-Fishcer M, Elinav E. Inflammasomes and intestinal inflammation. Mucosal Immunol (2017) 10(4):865–83. doi: 10.1038/mi.2017.19
24. Chen Y, Cui W, Li X, Yang H. Interaction between commensal bacteria, immune response and the intestinal barrier in inflammatory bowel disease. Front Immunol (2021) 12:761981. doi: 10.3389/fimmu.2021.761981
25. Wells JM, Brummer RJ, Derrien M, MacDonald TT, Troost F, Cani PD, et al. Homeostasis of the gut barrier and potential biomarkers. Am J Physiol Gastrointest Liver Physiol. (2017) 312(3):G171–g93. doi: 10.1152/ajpgi.00048.2015
26. Pearce SC, Al-Jawadi A, Kishida K, Yu S, Hu M, Fritzky LF, et al. Marked differences in tight junction composition and macromolecular permeability among different intestinal cell types. BMC Biol (2018) 16(1):19. doi: 10.1186/s12915-018-0481-z
27. Parikh K, Antanaviciute A, Fawkner-Corbett D, Jagielowicz M, Aulicino A, Lagerholm C, et al. Colonic epithelial cell diversity in health and inflammatory bowel disease. Nature (2019) 567(7746):49–55. doi: 10.1038/s41586-019-0992-y
28. Rathinam VAK, Chan FK. Inflammasome, inflammation, and tissue homeostasis. Trends Mol Med (2018) 24(3):304–18. doi: 10.1016/j.molmed.2018.01.004
29. Bauer C, Duewell P, Mayer C, Lehr HA, Fitzgerald KA, Dauer M, et al. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut (2010) 59(9):1192–9. doi: 10.1136/gut.2009.197822
30. Uzzan M, Martin JC, Mesin L, Livanos AE, Castro-Dopico T, Huang R, et al. Ulcerative colitis is characterized by a plasmablast-skewed humoral response associated with disease activity. Nat Med (2022) 28(4):766–79. doi: 10.1038/s41591-022-01680-y
31. Zaki MH, Vogel P, Body-Malapel M, Lamkanfi M, Kanneganti TD. IL-18 production downstream of the Nlrp3 inflammasome confers protection against colorectal tumor formation. J Immunol (2010) 185(8):4912–20. doi: 10.4049/jimmunol.1002046
32. Leppkes M, Neurath MF. Cytokines in inflammatory bowel diseases - update 2020. Pharmacol Res (2020) 158:104835. doi: 10.1016/j.phrs.2020.104835
33. Momozawa Y, Dmitrieva J, Théâtre E, Deffontaine V, Rahmouni S, Charloteaux B, et al. IBD risk loci are enriched in multigenic regulatory modules encompassing putative causative genes. Nat Commun (2018) 9(1):2427. doi: 10.1038/s41467-018-04365-8
34. Lu K, Lee MH, Patel SB. Dietary cholesterol absorption; more than just bile. Trends Endocrinol Metab (2001) 12(7):314–20. doi: 10.1016/S1043-2760(01)00433-7
35. Stellaard F. From dietary cholesterol to blood cholesterol, physiological lipid fluxes, and cholesterol homeostasis. Nutrients (2022) 14(8):1643. doi: 10.3390/nu14081643
36. Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell (2006) 124(1):35–46. doi: 10.1016/j.cell.2005.12.022
37. Betters JL, Yu L. NPC1L1 and cholesterol transport. FEBS Lett (2010) 584(13):2740–7. doi: 10.1016/j.febslet.2010.03.030
38. Paalvast Y, de Boer JF, Groen AK. Developments in intestinal cholesterol transport and triglyceride absorption. Curr Opin Lipidol (2017) 28(3):248–54. doi: 10.1097/MOL.0000000000000415
39. Chen HC. Molecular mechanisms of sterol absorption. J Nutr (2001) 131(10):2603–5. doi: 10.1093/jn/131.10.2603
40. Masson CJ, Plat J, Mensink RP, Namiot A, Kisielewski W, Namiot Z, et al. Fatty acid- and cholesterol transporter protein expression along the human intestinal tract. PloS One (2010) 5(4):e10380. doi: 10.1371/journal.pone.0010380
41. Verma N, Ahuja V, Paul J. Profiling of ABC transporters during active ulcerative colitis and in vitro effect of inflammatory modulators. Dig Dis Sci (2013) 58(8):2282–92. doi: 10.1007/s10620-013-2636-7
42. de la Roche M, Hamilton C, Mortensen R, Jeyaprakash AA, Ghosh S, Anand PK. Trafficking of cholesterol to the ER is required for NLRP3 inflammasome activation. J Cell Biol (2018) 217(10):3560–76. doi: 10.1083/jcb.201709057
43. Li J, Liu J, Yu Y, Liu Y, Guan X. NF-κB/ABCA1 pathway aggravates ox-LDL-induced cell pyroptosis by activation of NLRP3 inflammasomes in THP-1-derived macrophages. Mol Biol Rep (2022) 49(7):6161–71. doi: 10.1007/s11033-022-07408-y
44. Yvan-Charvet L, Welch C, Pagler TA, Ranalletta M, Lamkanfi M, Han S, et al. Increased inflammatory gene expression in ABC transporter-deficient macrophages: free cholesterol accumulation, increased signaling via toll-like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation (2008) 118(18):1837–47. doi: 10.1161/CIRCULATIONAHA.108.793869
45. Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science (1986) 232(4746):34–47. doi: 10.1126/science.3513311
46. Rhainds D, Bourgeois P, Bourret G, Huard K, Falstrault L, Brissette L. Localization and regulation of SR-BI in membrane rafts of HepG2 cells. J Cell Sci (2004) 117(Pt 15):3095–105. doi: 10.1242/jcs.01182
47. Zannis VI, Fotakis P, Koukos G, Kardassis D, Ehnholm C, Jauhiainen M, et al. HDL biogenesis, remodeling, and catabolism. Handb Exp Pharmacol (2015) 224:53–111. doi: 10.1007/978-3-319-09665-0_2
48. Torres S, Balboa E, Zanlungo S, Enrich C, Garcia-Ruiz C, Fernandez-Checa JC. Lysosomal and mitochondrial liaisons in niemann-pick disease. Front Physiol (2017) 8:982. doi: 10.3389/fphys.2017.00982
49. Wilhelm LP, Wendling C, Védie B, Kobayashi T, Chenard MP, Tomasetto C, et al. STARD3 mediates endoplasmic reticulum-to-endosome cholesterol transport at membrane contact sites. EMBO J (2017) 36(10):1412–33. doi: 10.15252/embj.201695917
50. Yu XH, Jiang N, Yao PB, Zheng XL, Cayabyab FS, Tang CK. NPC1, intracellular cholesterol trafficking and atherosclerosis. Clin Chim Acta (2014) 429:69–75. doi: 10.1016/j.cca.2013.11.026
51. Ikonen E, Hölttä-Vuori M. Cellular pathology of niemann-pick type c disease. Semin Cell Dev Biol (2004) 15(4):445–54. doi: 10.1016/j.semcdb.2004.03.001
52. Millard EE, Srivastava K, Traub LM, Schaffer JE, Ory DS. Niemann-pick type C1 (NPC1) overexpression alters cellular cholesterol homeostasis. J Biol Chem (2000) 275(49):38445–51. doi: 10.1074/jbc.M003180200
53. Cougnoux A, Movassaghi M, Picache JA, Iben JR, Navid F, Salman A, et al. Gastrointestinal tract pathology in a BALB/c niemann-pick disease type C1 null mouse model. Dig Dis Sci (2018) 63(4):870–80. doi: 10.1007/s10620-018-4914-x
54. Schwerd T, Pandey S, Yang HT, Bagola K, Jameson E, Jung J, et al. Impaired antibacterial autophagy links granulomatous intestinal inflammation in niemann-pick disease type C1 and XIAP deficiency with NOD2 variants in crohn's disease. Gut (2017) 66(6):1060–73. doi: 10.1136/gutjnl-2015-310382
55. Korach-André M, Gustafsson J. Liver X receptors as regulators of metabolism. Biomol Concepts (2015) 6(3):177–90. doi: 10.1515/bmc-2015-0007
56. Jakobsson T, Vedin LL, Hassan T, Venteclef N, Greco D, D'Amato M, et al. The oxysterol receptor LXRβ protects against DSS- and TNBS-induced colitis in mice. Mucosal Immunol (2014) 7(6):1416–28. doi: 10.1038/mi.2014.31
57. Miranda-Bautista J, Rodríguez-Feo JA, Puerto M, López-Cauce B, Lara JM, González-Novo R, et al. Liver X receptor exerts anti-inflammatory effects in colonic epithelial cells via ABCA1 and its expression is decreased in human and experimental inflammatory bowel disease. Inflammation Bowel Dis (2021) 27(10):1661–73. doi: 10.1093/ibd/izab034
58. Nicolson GL. Mitochondrial dysfunction and chronic disease: Treatment with natural supplements. Integr Med (Encinitas) (2014) 13(4):35–43. doi: 10.1007/978-3-319-73344-9_22
59. Suzuki S, Tajima T, Sassa S, Kudo H, Okayasu I, Sakamoto S. Preventive effect of fluvastatin on ulcerative colitis-associated carcinogenesis in mice. Anticancer Res (2006) 26(6b):4223–8.
60. Cheng L, Jin H, Qiang Y, Wu S, Yan C, Han M, et al. High fat diet exacerbates dextran sulfate sodium induced colitis through disturbing mucosal dendritic cell homeostasis. Int Immunopharmacol (2016) 40:1–10. doi: 10.1016/j.intimp.2016.08.018
61. Tien N, Wu TY, Lin CL, Wu CJ, Hsu CY, Fang YJ, et al. Impact of inflammatory bowel disease (IBD) and IBD medications on risk of hyperlipidemia and in vitro hepatic lipogenic-related gene expression: A population-based cohort study. Front Med (Lausanne) (2022) 9:910623. doi: 10.3389/fmed.2022.910623
62. Yarur A, Bruss A, Nunez L, Moosreiner A, Stein D, Agrawal D, et al. Higher visceral adipose tissue is associated with lower rates of steroi-free deep and endoscopic remission in patients starting biologic therapy for inflammatory bowel diseases: results from the constellation study. Gastroenterology (2021) 160:S–79. doi: 10.1016/S0016-5085(21)00935-5
63. Crockett SD, Hansen RA, Stürmer T, Schectman R, Darter J, Sandler RS, et al. Statins are associated with reduced use of steroids in inflammatory bowel disease: a retrospective cohort study. Inflammation Bowel Dis (2012) 18(6):1048–56. doi: 10.1002/ibd.21822
64. Toutounji M, Wanes D, El-Harakeh M, El-Sabban M, Rizk S, Naim HY. Dextran sodium sulfate-induced impairment of protein trafficking and alterations in membrane composition in intestinal caco-2 cell line. Int J Mol Sci (2020) 21(8):2726. doi: 10.3390/ijms21082726
65. Kaser A, Lee AH, Franke A, Glickman JN, Zeissig S, Tilg H, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell (2008) 134(5):743–56. doi: 10.1016/j.cell.2008.07.021
66. Missiroli S, Patergnani S, Caroccia N, Pedriali G, Perrone M, Previati M, et al. Mitochondria-associated membranes (MAMs) and inflammation. Cell Death Dis (2018) 9(3):329. doi: 10.1038/s41419-017-0027-2
67. Assis LHP, Dorighello GG, Oliveira HCF. Pro-inflammatory polarization of macrophages is associated with reduced endoplasmic reticulum-mitochondria interaction. Biochem Biophys Res Commun (2022) 606:61–7. doi: 10.1016/j.bbrc.2022.03.086
68. Willems PH, Rossignol R, Dieteren CE, Murphy MP, Koopman WJ. Redox homeostasis and mitochondrial dynamics. Cell Metab (2015) 22(2):207–18. doi: 10.1016/j.cmet.2015.06.006
69. Dubois-Camacho K, Diaz-Jimenez D, de la Fuente M, Quera R, Simian D, Martínez M, et al. Inhibition of miR-378a-3p by inflammation enhances IL-33 levels: A novel mechanism of alarmin modulation in ulcerative colitis. Front Immunol (2019) 10:2449. doi: 10.3389/fimmu.2019.02449
70. Solsona-Vilarrasa E, Fucho R, Torres S, Nuñez S, Nuño-Lámbarri N, Enrich C, et al. Cholesterol enrichment in liver mitochondria impairs oxidative phosphorylation and disrupts the assembly of respiratory supercomplexes. Redox Biol (2019) 24:101214. doi: 10.1016/j.redox.2019.101214
71. Balboa E, Castro J, Pinochet MJ, Cancino GI, Matías N, Sáez PJ, et al. MLN64 induces mitochondrial dysfunction associated with increased mitochondrial cholesterol content. Redox Biol (2017) 12:274–84. doi: 10.1016/j.redox.2017.02.024
72. Xu D, Ma R, Ju Y, Song X, Niu B, Hong W, et al. Cholesterol sulfate alleviates ulcerative colitis by promoting cholesterol biosynthesis in colonic epithelial cells. Nat Commun (2022) 13(1):4428. doi: 10.1038/s41467-022-32158-7
73. Haberman Y, Karns R, Dexheimer PJ, Schirmer M, Somekh J, Jurickova I, et al. Ulcerative colitis mucosal transcriptomes reveal mitochondriopathy and personalized mechanisms underlying disease severity and treatment response. Nat Commun (2019) 10(1):38. doi: 10.1038/s41467-018-07841-3
74. Kłos P, Dabravolski SA. The role of mitochondria dysfunction in inflammatory bowel diseases and colorectal cancer. Int J Mol Sci (2021) 2(21):11673. doi: 10.3390/ijms222111673
75. Novak EA, Mollen KP. Mitochondrial dysfunction in inflammatory bowel disease. Front Cell Dev Biol (2015) 3:62. doi: 10.3389/fcell.2015.00062
76. Rath E, Moschetta A, Haller D. Mitochondrial function - gatekeeper of intestinal epithelial cell homeostasis. Nat Rev Gastroenterol Hepatol (2018) 15(8):497–516. doi: 10.1038/s41575-018-0021-x
77. Urra FA, Muñoz F, Lovy A, Cárdenas C. The mitochondrial Complex(I)ty of cancer. Front Oncol (2017) 7:118. doi: 10.3389/fonc.2017.00118
78. Zickermann V, Wirth C, Nasiri H, Siegmund K, Schwalbe H, Hunte C, et al. Structural biology. mechanistic insight from the crystal structure of mitochondrial complex I. Science (2015) 347(6217):44–9. doi: 10.1126/science.1259859
79. Hroudová J, Fišar Z. Control mechanisms in mitochondrial oxidative phosphorylation. Neural Regener Res (2013) 8(4):363–75. doi: 10.3969/j.issn.1673-5374.2013.04.009
80. Kang YH, Tucker SA, Quevedo SF, Inal A, Korzenik JR, Haigis MC. Metabolic analyses reveal dysregulated NAD+ metabolism and altered mitochondrial state in ulcerative colitis. PloS One (2022) 17(8):e0273080. doi: 10.1371/journal.pone.0273080
81. Ooi M, Nishiumi S, Yoshie T, Shiomi Y, Kohashi M, Fukunaga K, et al. GC/MS-based profiling of amino acids and TCA cycle-related molecules in ulcerative colitis. Inflammation Res (2011) 60(9):831–40. doi: 10.1007/s00011-011-0340-7
82. Scoville EA, Allaman MM, Brown CT, Motley AK, Horst SN, Williams CS, et al. Alterations in lipid, amino acid, and energy metabolism distinguish crohn's disease from ulcerative colitis and control subjects by serum metabolomic profiling. Metabolomics (2018) 14(1):17. doi: 10.1007/s11306-017-1311-y
83. Winkelmann P, Unterweger AL, Khullar D, Beigel F, Koletzko L, Siebeck M, et al. The PI3K pathway as a therapeutic intervention point in inflammatory bowel disease. Immun Inflammation Dis (2021) 9(3):804–18. doi: 10.1002/iid3.435
84. Lee JY, Cevallos SA, Byndloss MX, Tiffany CR, Olsan EE, Butler BP, et al. High-fat diet and antibiotics cooperatively impair mitochondrial bioenergetics to trigger dysbiosis that exacerbates pre-inflammatory bowel disease. Cell Host Microbe (2020) 28(2):273–84.e6. doi: 10.1016/j.chom.2020.06.001
85. Busiello RA, Savarese S, Lombardi A. Mitochondrial uncoupling proteins and energy metabolism. Front Physiol (2015) 6:36. doi: 10.3389/fphys.2015.00036
86. Kwon J, Lee C, Heo S, Kim B, Hyun CK. DSS-induced colitis is associated with adipose tissue dysfunction and disrupted hepatic lipid metabolism leading to hepatosteatosis and dyslipidemia in mice. Sci Rep (2021) 11(1):5283. doi: 10.1038/s41598-021-84761-1
87. Jin X, Chen D, Zheng RH, Zhang H, Chen YP, Xiang Z. miRNA-133a-UCP2 pathway regulates inflammatory bowel disease progress by influencing inflammation, oxidative stress and energy metabolism. World J Gastroenterol (2017) 23(1):76–86. doi: 10.3748/wjg.v23.i1.76
88. Zhang H, Kuai XY, Yu P, Lin L, Shi R. Protective role of uncoupling protein-2 against dextran sodium sulfate-induced colitis. J Gastroenterol Hepatol (2012) 27(3):603–8. doi: 10.1111/j.1440-1746.2011.06879.x
89. Al Ojaimi Y, Khachab M, Bazzi S, Bahr GM, Echtay KS. Mitochondrial bioenergetics, uncoupling protein-2 activity, and reactive oxygen species production in the small intestine of a TNBS-induced colitis rat model. Mol Cell Biochem (2020) 470(1-2):87–98. doi: 10.1007/s11010-020-03749-z
90. Alula KM, Jackson DN, Smith AD, Kim DS, Turner K, Odstrcil E, et al. Targeting mitochondrial damage as a therapeutic for ileal crohn's disease. Cells (2021) 10(6):1349. doi: 10.3390/cells10061349
91. Vincent G, Novak EA, Siow VS, Cunningham KE, Griffith BD, Comerford TE, et al. Nix-mediated mitophagy modulates mitochondrial damage during intestinal inflammation. Antioxid Redox Signal (2020) 33(1):1–19. doi: 10.1089/ars.2018.7702
92. Cunningham KE, Vincent G, Sodhi CP, Novak EA, Ranganathan S, Egan CE, et al. Peroxisome proliferator-activated receptor-γ coactivator 1-α (PGC1α) protects against experimental murine colitis. J Biol Chem (2016) 291(19):10184–200. doi: 10.1074/jbc.M115.688812
93. Rius-Pérez S, Torres-Cuevas I, Millán I, Ortega ÁL, Pérez S. PGC-1α, inflammation, and oxidative stress: An integrative view in metabolism. Oxid Med Cell Longev (2020) 2020:1452696. doi: 10.1155/2020/1452696
94. Taman H, Fenton CG, Anderssen E, Florholmen J, Paulssen RH. DNA Hypo-methylation facilitates anti-inflammatory responses in severe ulcerative colitis. PloS One (2021) 16(4):e0248905. doi: 10.1371/journal.pone.0248905
95. Schneider AM, Özsoy M, Zimmermann FA, Brunner SM, Feichtinger RG, Mayr JA, et al. Expression of oxidative phosphorylation complexes and mitochondrial mass in pediatric and adult inflammatory bowel disease. Oxid Med Cell Longev (2022) 2022:9151169. doi: 10.1155/2022/9151169
96. Gerhart-Hines Z, Rodgers JT, Bare O, Lerin C, Kim SH, Mostoslavsky R, et al. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. EMBO J (2007) 26(7):1913–23. doi: 10.1038/sj.emboj.7601633
97. Wellman AS, Metukuri MR, Kazgan N, Xu X, Xu Q, Ren NSX, et al. Intestinal epithelial sirtuin 1 regulates intestinal inflammation during aging in mice by altering the intestinal microbiota. Gastroenterology (2017) 153(3):772–86. doi: 10.1053/j.gastro.2017.05.022
98. Caruso R, Marafini I, Franzè E, Stolfi C, Zorzi F, Monteleone I, et al. Defective expression of SIRT1 contributes to sustain inflammatory pathways in the gut. Mucosal Immunol (2014) 7(6):1467–79. doi: 10.1038/mi.2014.35
99. Rousseaux C, Lefebvre B, Dubuquoy L, Lefebvre P, Romano O, Auwerx J, et al. Intestinal antiinflammatory effect of 5-aminosalicylic acid is dependent on peroxisome proliferator-activated receptor-gamma. J Exp Med (2005) 201(8):1205–15. doi: 10.1084/jem.20041948
100. Vetuschi A, Pompili S, Gaudio E, Latella G, Sferra R. PPAR-γ with its anti-inflammatory and anti-fibrotic action could be an effective therapeutic target in IBD. Eur Rev Med Pharmacol Sci (2018) 22(24):8839–48. doi: 10.26355/eurrev_201812_16652
101. Katkar GD, Sayed IM, Anandachar MS, Castillo V, Vidales E, Toobian D, et al. Artificial intelligence-rationalized balanced PPARα/γ dual agonism resets dysregulated macrophage processes in inflammatory bowel disease. Commun Biol (2022) 5(1):231. doi: 10.1038/s42003-022-03168-4
102. Han T, Lv Y, Wang S, Hu T, Hong H, Fu Z. PPARγ overexpression regulates cholesterol metabolism in human L02 hepatocytes. J Pharmacol Sci (2019) 139(1):1–8. doi: 10.1016/j.jphs.2018.09.013
103. Chiu YC, Chu PW, Lin HC, Chen SK. Accumulation of cholesterol suppresses oxidative phosphorylation and altered responses to inflammatory stimuli of macrophages. Biochem Biophys Rep (2021) 28:101166. doi: 10.1016/j.bbrep.2021.101166
104. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell (2002) 10(2):417–26. doi: 10.1016/S1097-2765(02)00599-3
105. Schroder K, Tschopp J. The inflammasomes. Cell (2010) 140(6):821–32. doi: 10.1016/j.cell.2010.01.040
106. Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature (2012) 481(7381):278–86. doi: 10.1038/nature10759
107. Zhou Y, Tong Z, Jiang S, Zheng W, Zhao J, Zhou X. The roles of endoplasmic reticulum in NLRP3 inflammasome activation. Cells (2020) 9(5):1219. doi: 10.3390/cells9051219
108. Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell (2014) 156(6):1193–206. doi: 10.1016/j.cell.2014.02.008
109. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature (2011) 479(7371):117–21. doi: 10.1038/nature10558
110. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature (2015) 526(7575):660–5. doi: 10.1038/nature15514
111. Bording-Jorgensen M, Armstrong H, Wickenberg M, LaPointe P, Wine E. Macrophages and epithelial cells mutually interact through NLRP3 to clear infection and enhance the gastrointestinal barrier. Immuno (2022) 2(1):13–25. doi: 10.3390/immuno2010002
112. Hirota SA, Ng J, Lueng A, Khajah M, Parhar K, Li Y, et al. NLRP3 inflammasome plays a key role in the regulation of intestinal homeostasis. Inflammation Bowel Dis (2011) 17(6):1359–72. doi: 10.1002/ibd.21478
113. Allen IC, TeKippe EM, Woodford RM, Uronis JM, Holl EK, Rogers AB, et al. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J Exp Med (2010) 207(5):1045–56. doi: 10.1084/jem.20100050
114. Liu R, Truax AD, Chen L, Hu P, Li Z, Chen J, et al. Expression profile of innate immune receptors, NLRs and AIM2, in human colorectal cancer: correlation with cancer stages and inflammasome components. Oncotarget (2015) 6(32):33456–69. doi: 10.18632/oncotarget.5587
115. Westerterp M, Fotakis P, Ouimet M, Bochem AE, Zhang H, Molusky MM, et al. Cholesterol efflux pathways suppress inflammasome activation, NETosis, and atherogenesis. Circulation (2018) 138(9):898–912. doi: 10.1161/CIRCULATIONAHA.117.032636
116. Progatzky F, Sangha NJ, Yoshida N, McBrien M, Cheung J, Shia A, et al. Dietary cholesterol directly induces acute inflammasome-dependent intestinal inflammation. Nat Commun (2014) 5:5864. doi: 10.1038/ncomms6864
117. Cañadas-Lozano D, Marín-Aguilar F, Castejón-Vega B, Ryffel B, Navarro-Pando JM, Ruiz-Cabello J, et al. Blockade of the NLRP3 inflammasome improves metabolic health and lifespan in obese mice. Geroscience (2020) 42(2):715–25. doi: 10.1007/s11357-019-00151-6
118. Li G, Chen Z, Bhat OM, Zhang Q, Abais-Battad JM, Conley SM, et al. NLRP3 inflammasome as a novel target for docosahexaenoic acid metabolites to abrogate glomerular injury. J Lipid Res (2017) 58(6):1080–90. doi: 10.1194/jlr.M072587
119. Fernandez-Lizarbe S, Montesinos J, Guerri C. Ethanol induces TLR4/TLR2 association, triggering an inflammatory response in microglial cells. J Neurochem (2013) 126(2):261–73. doi: 10.1111/jnc.12276
120. Płóciennikowska A, Hromada-Judycka A, Borzęcka K, Kwiatkowska K. Co-Operation of TLR4 and raft proteins in LPS-induced pro-inflammatory signaling. Cell Mol Life Sci (2015) 72(3):557–81. doi: 10.1007/s00018-014-1762-5
121. Sun Y, Ishibashi M, Seimon T, Lee M, Sharma SM, Fitzgerald KA, et al. Free cholesterol accumulation in macrophage membranes activates toll-like receptors and p38 mitogen-activated protein kinase and induces cathepsin K. Circ Res (2009) 104(4):455–65. doi: 10.1161/CIRCRESAHA.108.182568
122. Triantafilou M, Gamper FG, Lepper PM, Mouratis MA, Schumann C, Harokopakis E, et al. Lipopolysaccharides from atherosclerosis-associated bacteria antagonize TLR4, induce formation of TLR2/1/CD36 complexes in lipid rafts and trigger TLR2-induced inflammatory responses in human vascular endothelial cells. Cell Microbiol (2007) 9(8):2030–9. doi: 10.1111/j.1462-5822.2007.00935.x
123. Ruysschaert JM, Lonez C. Role of lipid microdomains in TLR-mediated signalling. Biochim Biophys Acta (2015) 1848(9):1860–7. doi: 10.1016/j.bbamem.2015.03.014
124. Noti M, Corazza N, Tuffin G, Schoonjans K, Brunner T. Lipopolysaccharide induces intestinal glucocorticoid synthesis in a TNFalpha-dependent manner. FASEB J (2010) 24(5):1340–6. doi: 10.1096/fj.09-140913
125. Tall AR, Westerterp M. Inflammasomes, neutrophil extracellular traps, and cholesterol. J Lipid Res (2019) 60(4):721–7. doi: 10.1194/jlr.S091280
126. Lu F, Liang Q, Abi-Mosleh L, Das A, De Brabander JK, Goldstein JL, et al. Identification of NPC1 as the target of U18666A, an inhibitor of lysosomal cholesterol export and Ebola infection. Elife (2015) 4:e12177. doi: 10.7554/eLife.12177
127. Boyapati RK, Dorward DA, Tamborska A, Kalla R, Ventham NT, Doherty MK, et al. Mitochondrial DNA is a pro-inflammatory damage-associated molecular pattern released during active IBD. Inflammation Bowel Dis (2018) 24(10):2113–22. doi: 10.1093/ibd/izy095
128. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity (2012) 36(3):401–14. doi: 10.1016/j.immuni.2012.01.009
129. Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin XJ, et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature (2018) 560(7717):198–203. doi: 10.1038/s41586-018-0372-z
130. de Torre-Minguela C, Gómez AI, Couillin I, Pelegrín P. Gasdermins mediate cellular release of mitochondrial DNA during pyroptosis and apoptosis. FASEB J (2021) 35(8):e21757. doi: 10.1096/fj.202100085R
131. Pizzuto M, Pelegrin P. Cardiolipin in immune signaling and cell death. Trends Cell Biol (2020) 30(11):892–903. doi: 10.1016/j.tcb.2020.09.004
132. Hirose M, Künstner A, Schilf P, Sünderhauf A, Rupp J, Jöhren O, et al. Mitochondrial gene polymorphism is associated with gut microbial communities in mice. Sci Rep (2017) 7(1):15293. doi: 10.1038/s41598-017-15377-7
133. Yardeni T, Tanes CE, Bittinger K, Mattei LM, Schaefer PM, Singh LN, et al. Host mitochondria influence gut microbiome diversity: A role for ROS. Sci Signal (2019) 12(588):eaaw3159. doi: 10.1126/scisignal.aaw3159
134. Gophna U, Sommerfeld K, Gophna S, Doolittle WF, Veldhuyzen van Zanten SJ. Differences between tissue-associated intestinal microfloras of patients with crohn's disease and ulcerative colitis. J Clin Microbiol (2006) 44(11):4136–41. doi: 10.1128/JCM.01004-06
135. Ott SJ, Musfeldt M, Wenderoth DF, Hampe J, Brant O, Fölsch UR, et al. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut (2004) 53(5):685–93. doi: 10.1136/gut.2003.025403
136. Chamorro N, Montero DA, Gallardo P, Farfán M, Contreras M, de la Fuente M, et al. Landscapes and bacterial signatures of mucosa-associated intestinal microbiota in Chilean and Spanish patients with inflammatory bowel disease. Microb Cell (2021) 8(9):223–38. doi: 10.15698/mic2021.09.760
137. Rigottier-Gois L. Dysbiosis in inflammatory bowel diseases: the oxygen hypothesis. Isme J (2013) 7(7):1256–61. doi: 10.1038/ismej.2013.80
138. Nijmeijer RM, Gadaleta RM, van Mil SW, van Bodegraven AA, Crusius JB, Dijkstra G, et al. Farnesoid X receptor (FXR) activation and FXR genetic variation in inflammatory bowel disease. PloS One (2011) 6(8):e23745. doi: 10.1371/journal.pone.0023745
139. Rogler G, Brand K, Vogl D, Page S, Hofmeister R, Andus T, et al. Nuclear factor kappaB is activated in macrophages and epithelial cells of inflamed intestinal mucosa. Gastroenterology (1998) 115(2):357–69. doi: 10.1016/S0016-5085(98)70202-1
140. Wang YD, Chen WD, Wang M, Yu D, Forman BM, Huang W. Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology (2008) 48(5):1632–43. doi: 10.1002/hep.22519
141. Vítek L. Bile acid malabsorption in inflammatory bowel disease. Inflammation Bowel Dis (2015) 21(2):476–83. doi: 10.1097/MIB.0000000000000193
142. Akerlund JE, Reihnér E, Angelin B, Rudling M, Ewerth S, Björkhem I, et al. Hepatic metabolism of cholesterol in crohn's disease. Effect Partial Resection Ileum Gastroenterol (1991) 100(4):1046–53. doi: 10.1016/0016-5085(91)90281-O
143. Fiorucci S, Carino A, Baldoni M, Santucci L, Costanzi E, Graziosi L, et al. Bile acid signaling in inflammatory bowel diseases. Dig Dis Sci (2021) 66(3):674–93. doi: 10.1007/s10620-020-06715-3
144. Guo C, Xie S, Chi Z, Zhang J, Liu Y, Zhang L, et al. Bile acids control inflammation and metabolic disorder through inhibition of NLRP3 inflammasome. Immunity (2016) 45(4):802–16. doi: 10.1016/j.immuni.2016.09.008
145. Dashdorj A, Jyothi KR, Lim S, Jo A, Nguyen MN, Ha J, et al. Mitochondria-targeted antioxidant MitoQ ameliorates experimental mouse colitis by suppressing NLRP3 inflammasome-mediated inflammatory cytokines. BMC Med (2013) 11:178. doi: 10.1186/1741-7015-11-178
146. Murphy MP. How mitochondria produce reactive oxygen species. Biochem J (2009) 417(1):1–13. doi: 10.1042/BJ20081386
147. Serra D, Almeida LM, Dinis TC. Anti-inflammatory protection afforded by cyanidin-3-glucoside and resveratrol in human intestinal cells via Nrf2 and PPAR-γ: Comparison with 5-aminosalicylic acid. Chem Biol Interact (2016) 260:102–9. doi: 10.1016/j.cbi.2016.11.003
148. Sabzevary-Ghahfarokhi M, Soltani A, Luzza F, Larussa T, Rahimian G, Shirzad H, et al. The protective effects of resveratrol on ulcerative colitis via changing the profile of Nrf2 and IL-1β protein. Mol Biol Rep (2020) 47(9):6941–7. doi: 10.1007/s11033-020-05753-4
149. Carrasco-Pozo C, Mizgier ML, Speisky H, Gotteland M. Differential protective effects of quercetin, resveratrol, rutin and epigallocatechin gallate against mitochondrial dysfunction induced by indomethacin in caco-2 cells. Chem Biol Interact (2012) 195(3):199–205. doi: 10.1016/j.cbi.2011.12.007
150. Samsamikor M, Daryani NE, Asl PR, Hekmatdoost A. Resveratrol supplementation and Oxidative/Anti-oxidative status in patients with ulcerative colitis: A randomized, double-blind, placebo-controlled pilot study. Arch Med Res (2016) 47(4):304–9. doi: 10.1016/j.arcmed.2016.07.003
151. Ye G, Chen G, Gao H, Lin Y, Liao X, Zhang H, et al. Resveratrol inhibits lipid accumulation in the intestine of atherosclerotic mice and macrophages. J Cell Mol Med (2019) 23(6):4313–25. doi: 10.1111/jcmm.14323
152. Carrasco-Pozo C, Pastene E, Vergara C, Zapata M, Sandoval C, Gotteland M. Stimulation of cytosolic and mitochondrial calcium mobilization by indomethacin in caco-2 cells: modulation by the polyphenols quercetin, resveratrol and rutin. Biochim Biophys Acta (2012) 1820(12):2052–61. doi: 10.1016/j.bbagen.2012.09.015
153. Xue Y, Du M, Zhu MJ. Quercetin suppresses NLRP3 inflammasome activation in epithelial cells triggered by escherichia coli O157:H7. Free Radic Biol Med (2017) 108:760–9. doi: 10.1016/j.freeradbiomed.2017.05.003
154. Ju S, Ge Y, Li P, Tian X, Wang H, Zheng X, et al. Dietary quercetin ameliorates experimental colitis in mouse by remodeling the function of colonic macrophages via a heme oxygenase-1-dependent pathway. Cell Cycle (2018) 17(1):53–63. doi: 10.1080/15384101.2017.1387701
155. Carrasco-Pozo C, Tan KN, Gotteland M, Borges K. Sulforaphane protects against high cholesterol-induced mitochondrial bioenergetics impairments, inflammation, and oxidative stress and preserves pancreatic β-cells function. Oxid Med Cell Longev (2017) 2017:3839756. doi: 10.1155/2017/3839756
156. Zhang YJ, Wu Q. Sulforaphane protects intestinal epithelial cells against lipopolysaccharide-induced injury by activating the AMPK/SIRT1/PGC-1α pathway. Bioengineered (2021) 12(1):4349–60. doi: 10.1080/21655979.2021.1952368
157. Yeganeh PR, Leahy J, Spahis S, Patey N, Desjardins Y, Roy D, et al. Apple peel polyphenols reduce mitochondrial dysfunction in mice with DSS-induced ulcerative colitis. J Nutr Biochem (2018) 57:56–66. doi: 10.1016/j.jnutbio.2018.03.008
158. Żary-Sikorska E, Fotschki B, Jurgoński A, Kosmala M, Milala J, Kołodziejczyk K, et al. Protective effects of a strawberry ellagitannin-rich extract against pro-oxidative and pro-inflammatory dysfunctions induced by a high-fat diet in a rat model. Molecules (2020) 25(24):5874. doi: 10.3390/molecules25245874
159. Li Y, Shen L, Luo H. Luteolin ameliorates dextran sulfate sodium-induced colitis in mice possibly through activation of the Nrf2 signaling pathway. Int Immunopharmacol (2016) 40:24–31. doi: 10.1016/j.intimp.2016.08.020
160. Lin TJ, Yin SY, Hsiao PW, Yang NS, Wang IJ. Transcriptomic analysis reveals a controlling mechanism for NLRP3 and IL-17A in dextran sulfate sodium (DSS)-induced colitis. Sci Rep (2018) 8(1):14927. doi: 10.1038/s41598-018-33204-5
161. Francisco V, Figueirinha A, Costa G, Liberal J, Ferreira I, Lopes MC, et al. The flavone luteolin inhibits liver X receptor activation. J Nat Prod (2016) 79(5):1423–8. doi: 10.1021/acs.jnatprod.6b00146
162. Yuan J, Che S, Zhang L, Ruan Z. Reparative effects of ethanol-induced intestinal barrier injury by flavonoid luteolin via MAPK/NF-κB/MLCK and Nrf2 signaling pathways. J Agric Food Chem (2021) 69(14):4101–10. doi: 10.1021/acs.jafc.1c00199
163. Liu N, Wu C, Sun L, Zheng J, Guo P. Sesamin enhances cholesterol efflux in RAW264.7 macrophages. Molecules (2014) 19(6):7516–27. doi: 10.3390/molecules19067516
164. Bai X, Gou X, Cai P, Xu C, Cao L, Zhao Z, et al. Sesamin enhances Nrf2-mediated protective defense against oxidative stress and inflammation in colitis via AKT and ERK activation. Oxid Med Cell Longev (2019) 2019:2432416. doi: 10.1155/2019/2432416
165. Tai TS, Tien N, Shen HY, Chu FY, Wang CCN, Lu CH, et al. Sesamin, a naturally occurring lignan, inhibits ligand-induced lipogenesis through interaction with liver X receptor alpha (LXRα) and pregnane X receptor (PXR). Evid Based Complement Alternat Med (2019) 2019:9401648. doi: 10.1155/2019/9401648
166. Gong Z, Zhao S, Zhou J, Yan J, Wang L, Du X, et al. Curcumin alleviates DSS-induced colitis via inhibiting NLRP3 inflammsome activation and IL-1β production. Mol Immunol (2018) 104:11–9. doi: 10.1016/j.molimm.2018.09.004
167. Tan C, Zhou L, Wen W, Xiao N. Curcumin promotes cholesterol efflux by regulating ABCA1 expression through miR-125a-5p/SIRT6 axis in THP-1 macrophage to prevent atherosclerosis. J Toxicol Sci (2021) 46(5):209–22. doi: 10.2131/jts.46.209
168. Zhong Y, Feng J, Fan Z, Li J. Curcumin increases cholesterol efflux via heme oxygenase−1−mediated ABCA1 and SR−BI expression in macrophages. Mol Med Rep (2018) 17(4):6138–43. doi: 10.3892/mmr.2018.8577
169. Feng D, Ohlsson L, Duan RD. Curcumin inhibits cholesterol uptake in caco-2 cells by down-regulation of NPC1L1 expression. Lipids Health Dis (2010) 9:40. doi: 10.1186/1476-511X-9-40
170. Kumar P, Malhotra P, Ma K, Singla A, Hedroug O, Saksena S, et al. SREBP2 mediates the modulation of intestinal NPC1L1 expression by curcumin. Am J Physiol Gastrointest Liver Physiol (2011) 301(1):G148–55. doi: 10.1152/ajpgi.00119.2011
171. Zhang M, Deng C, Zheng J, Xia J, Sheng D. Curcumin inhibits trinitrobenzene sulphonic acid-induced colitis in rats by activation of peroxisome proliferator-activated receptor gamma. Int Immunopharmacol (2006) 6(8):1233–42. doi: 10.1016/j.intimp.2006.02.013
172. Mouzaoui S, Banerjee S, Djerdjouri B. Low-dose curcumin reduced TNBS-associated mucin depleted foci in mice by scavenging superoxide anion and lipid peroxides, rebalancing matrix NO synthase and aconitase activities, and recoupling mitochondria. Inflammopharmacology (2020) 28(4):949–65. doi: 10.1007/s10787-019-00684-4
173. Ortiz T, Argüelles-Arias F, Illanes M, García-Montes JM, Talero E, Macías-García L, et al. Polyphenolic maqui extract as a potential nutraceutical to treat TNBS-induced crohn's disease by the regulation of antioxidant and anti-inflammatory pathways. Nutrients (2020) 12(6):1752. doi: 10.3390/nu12061752
174. Gasaly N, Riveros K, Gotteland M. Fitoquímicos: una nueva clase de prebióticos. Rev Chil Nutrición (2020) 47:317–27. doi: 10.4067/S0717-75182020000200317
175. Bellezza I, Giambanco I, Minelli A, Donato R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim Biophys Acta Mol Cell Res (2018) 1865(5):721–33. doi: 10.1016/j.bbamcr.2018.02.010
176. Piotrowska M, Swierczynski M, Fichna J, Piechota-Polanczyk A. The Nrf2 in the pathophysiology of the intestine: Molecular mechanisms and therapeutic implications for inflammatory bowel diseases. Pharmacol Res (2021) 163:105243. doi: 10.1016/j.phrs.2020.105243
177. He F, Ru X, Wen T. NRF2, a transcription factor for stress response and beyond. Int J Mol Sci (2020) 21(13):4777. doi: 10.3390/ijms21134777
178. Khor TO, Huang MT, Kwon KH, Chan JY, Reddy BS, Kong AN. Nrf2-deficient mice have an increased susceptibility to dextran sulfate sodium-induced colitis. Cancer Res (2006) 66(24):11580–4. doi: 10.1158/0008-5472.CAN-06-3562
179. Li J, Wang H, Zheng Z, Luo L, Wang P, Liu K, et al. Mkp-1 cross-talks with Nrf2/Ho-1 pathway protecting against intestinal inflammation. Free Radic Biol Med (2018) 124:541–9. doi: 10.1016/j.freeradbiomed.2018.07.002
180. Zhu SL, Wang ML, He YT, Guo SW, Li TT, Peng WJ, et al. Capsaicin ameliorates intermittent high glucose-mediated endothelial senescence via the TRPV1/SIRT1 pathway. Phytomedicine (2022) 100:154081. doi: 10.1016/j.phymed.2022.154081
181. Sacks D, Baxter B, Campbell BCV, Carpenter JS, Cognard C, Dippel D, et al. Multisociety consensus quality improvement revised consensus statement for endovascular therapy of acute ischemic stroke. Int J Stroke (2018) 13(6):612–32. doi: 10.1016/j.jvir.2017.11.026
182. Duval C, Touche V, Tailleux A, Fruchart JC, Fievet C, Clavey V, et al. Niemann-pick C1 like 1 gene expression is down-regulated by LXR activators in the intestine. Biochem Biophys Res Commun (2006) 340(4):1259–63. doi: 10.1016/j.bbrc.2005.12.137
183. Ko CW, Qu J, Black DD, Tso P. Regulation of intestinal lipid metabolism: current concepts and relevance to disease. Nat Rev Gastroenterol Hepatol (2020) 17(3):169–83. doi: 10.1038/s41575-019-0250-7
184. van der Veen JN, van Dijk TH, Vrins CL, van Meer H, Havinga R, Bijsterveld K, et al. Activation of the liver X receptor stimulates trans-intestinal excretion of plasma cholesterol. J Biol Chem (2009) 284(29):19211–9. doi: 10.1074/jbc.M109.014860
185. Fan W, Zhang R, Han D, Jiang Z, Li S, Zhang J, et al. Reduced Sirtuin1 signalling exacerbates diabetic mice hindlimb ischaemia injury and inhibits the protective effect of a liver X receptor agonist. J Cell Mol Med (2020) 24(10):5476–90. doi: 10.1111/jcmm.15201
186. Hammer SS, Beli E, Kady N, Wang Q, Wood K, Lydic TA, et al. The mechanism of diabetic retinopathy pathogenesis unifying key lipid regulators, sirtuin 1 and liver X receptor. EBioMedicine (2017) 22:181–90. doi: 10.1016/j.ebiom.2017.07.008
187. Li X, Zhang S, Blander G, Tse JG, Krieger M, Guarente L. SIRT1 deacetylates and positively regulates the nuclear receptor LXR. Mol Cell (2007) 28(1):91–106. doi: 10.1016/j.molcel.2007.07.032
188. Zeng HT, Fu YC, Yu W, Lin JM, Zhou L, Liu L, et al. SIRT1 prevents atherosclerosis via liver−X−receptor and NF−κB signaling in a U937 cell model. Mol Med Rep (2013) 8(1):23–8. doi: 10.3892/mmr.2013.1460
Keywords: IBD - inflammatory bowel disease, intracellular cholesterol accumulation, mitochondrial dysfunction, inflammasome, NLRP3 inflammasome, diet phytochemicals
Citation: Astorga J, Gasaly N, Dubois-Camacho K, De la Fuente M, Landskron G, Faber KN, Urra FA and Hermoso MA (2022) The role of cholesterol and mitochondrial bioenergetics in activation of the inflammasome in IBD. Front. Immunol. 13:1028953. doi: 10.3389/fimmu.2022.1028953
Received: 26 August 2022; Accepted: 26 October 2022;
Published: 18 November 2022.
Edited by:
Silvia Melgar, University College Cork, IrelandReviewed by:
Chunying Li, Georgia State University, United StatesElizabeth A. Novak, University of Pittsburgh, United States
Copyright © 2022 Astorga, Gasaly, Dubois-Camacho, De la Fuente, Landskron, Faber, Urra and Hermoso. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marcela A. Hermoso, bWhlcm1vc29AbWVkLnVjaGlsZS5jbA==; bS5hLmhlcm1vc29AdW1jZy5ubA==