 Jodie Deborah Ouahed
Jodie Deborah Ouahed
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 29 September 2022
Sec. Primary Immunodeficiencies
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1026511
This article is part of the Research TopicInborn Errors of Immunity and Mucosal ImmunityView all 6 articles
Inflammatory bowel diseases (IBD) are chronic inflammatory conditions of the gastrointestinal tract, including Crohn’s disease, ulcerative colitis and inflammatory bowel disease-undefined (IBD-U). IBD are understood to be multifactorial, involving genetic, immune, microbial and environmental factors. Advances in next generation sequencing facilitated the growing identification of over 80 monogenic causes of IBD, many of which overlap with Inborn errors of immunity (IEI); Approximately a third of currently identified IEI result in gastrointestinal manifestations, many of which are inflammatory in nature, such as IBD. Indeed, the gastrointestinal tract represents an opportune system to study IEI as it consists of the largest mass of lymphoid tissue in the body and employs a thin layer of intestinal epithelial cells as the critical barrier between the intestinal lumen and the host. In this mini-review, a selection of pertinent IEI resulting in monogenic IBD is described involving disorders in the intestinal epithelial barrier, phagocytosis, T and B cell defects, as well as those impairing central and peripheral tolerance. The contribution of disrupted gut-microbiota-host interactions in disturbing intestinal homeostasis among patients with intestinal disease is also discussed. The molecular mechanisms driving pathogenesis are reviewed along with the personalized therapeutic interventions and investigational avenues this growing knowledge has enabled.
The gastrointestinal tract juggles many roles including absorption of nutrients, transport of electrolytes and fluids, all while maintaining mucosal homeostasis. It is perhaps not surprising that it harbors the largest collection of immune cells in the body, sophistically organized to orchestrate an appropriate immune response against harmful pathogens, while permitting tolerance toward commensal organisms. The immune compartments of the gastrointestinal tract have been categorized into: inductive sites and effector sites (1). Inductive sites include the mesenteric lymph nodes and the gut associated lymphoid tissue armored with T cells, B cells and innate immune cells such as mast cells, granulocytes, dendritic cells and group 3 innate lymphoid cells. It is in these inductive sites where adaptive immune cells undergo priming and differentiation. This works alongside effector sites, such as the lamina propria and the epithelium, where primed adaptive immune cells reside to assure appropriate immunity against harmful pathogens (1).
Inborn errors of immunity (IEI) are monogenic germline mutations that result in primary immunodeficies (2). Well over 400 IEI have been identified (3), with roughly a third involving a gastrointestinal manifestation, most being inflammatory in nature (4, 5). Inflammatory bowel diseases (IBD) are chronic inflammatory diseases of the gastrointestinal tract. They include Crohn’s disease (CD), a transmural disease, that can affect the mouth to the anus; ulcerative colitis (UC) that results in mucosal inflammation of the colon; and IBD-undefined comprising a mixed picture. The pathophysiology of IBD is believed to involve one’s immune system, genetic predisposition, microbiome and environmental factors.
There are over 80 monogenic causes of IBD (6–8), most of which are IEI (Table 1). Consequences of the breakdown of the immune system are robustly reflected in the gastrointestinal tract and often result in disease at an especially young age. Monogenic IBD is predominantly identified in patients diagnosed prior to 6 years old, known as very early onset IBD (VEOIBD) (9). Two large North American centers identified the prevalence of monogenic VEOIBD to be roughly 8% (10, 11), but this varies depending on the population studied (12, 13).

Table 1 Monogenic causes of inflammatory bowel disease revealing strong overlap among inborn errors of immunity.
Interleukin 10 (IL10) signaling defects exemplify IEI where life-threatening infantile-onset IBD is the most striking feature (14). However, in most cases of IEI resulting in IBD, intestinal inflammation represents one of many disorders. IBD from underlying IEI can be categorized as: epithelial barrier defects that lead to mucosal inflammation; phagocytic defects that lead to alterations in pathogen clearance; T and B cell defects that present with intestinal inflammation; and defects in central and peripheral tolerance that lead to disruption of intestinal homeostasis. Inclusion in one group does not preclude function in another. In this non-exhaustive mini-review, a selection of IEI from each of these categories is described, along with mechanisms driving disease, and associated therapeutic implications (Table 2). Importance of the gut microbiota in intestinal inflammation is highlighted.
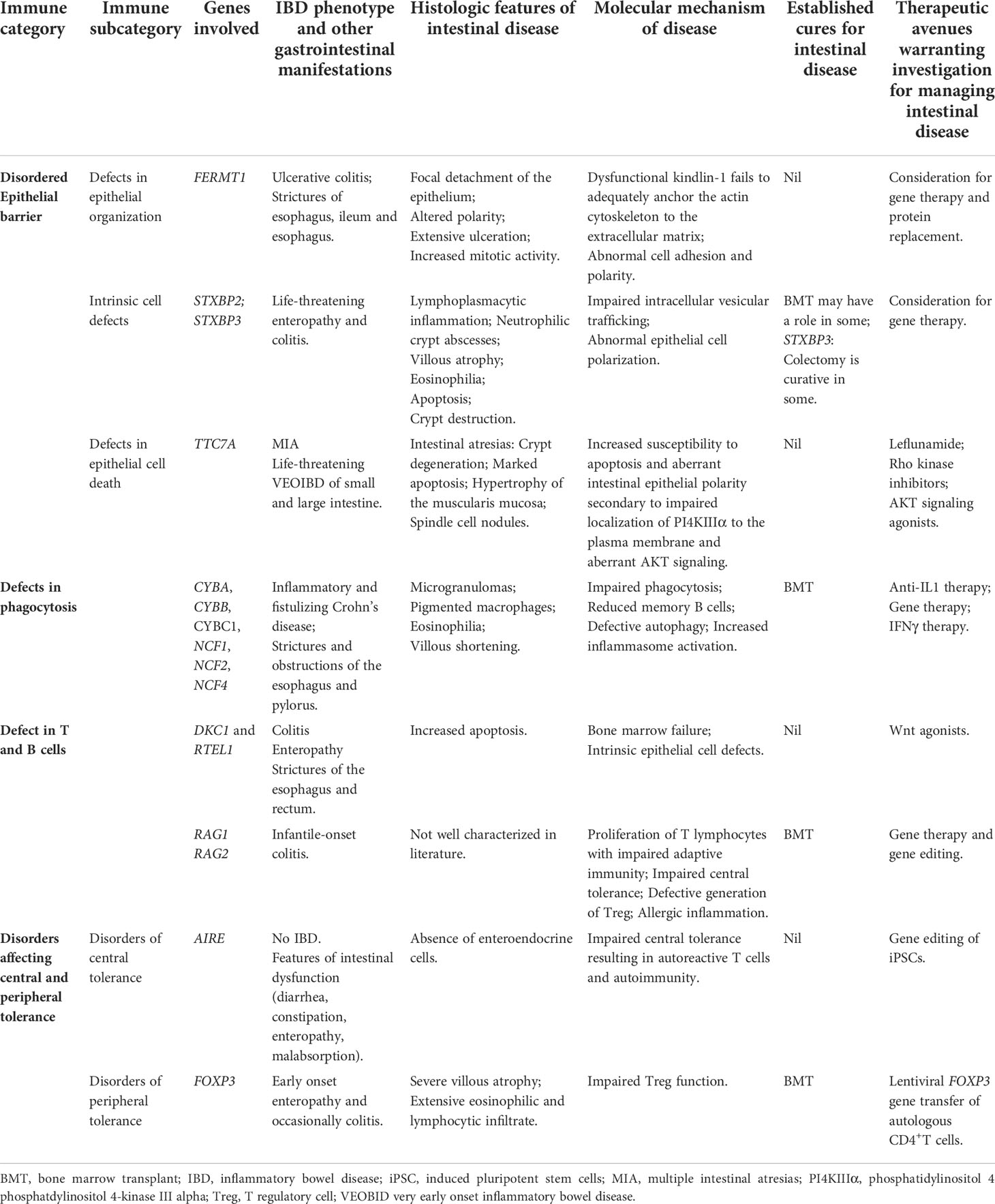
Table 2 Selection of IEI resulting in IBD, histologic features and molecular mechanisms of intestinal disease and summary of available cures and therapeutics warranting further investigation.
The intestinal epithelial barrier consists of a single layer of epithelial cells, a mucous layer, and an organization of immune cells. A delicate balance of cell turnover is imperative for appropriate barrier function. Disruption of the epithelial barrier can be subdivided into: defects in epithelial organization; intrinsic cell defects; and defects in epithelial cell death (8).
Mutations in KINDLIN-1, also known as FERMT1, result in kindler syndrome, an IEI that manifests primarily as a skin disorder, characterized by acral blistering as neonates and poikiloderma with age (15). Gastrointestinal manifestations include anal, esophageal and ileal strictures (16, 17). Approximately 15% of patients develop UC (18). Kindlin-1 knock out mice exhibit severe colitis with extensive epithelial detachment, ensuing perinatal lethality (19). In humans, the severity of UC is milder and does not correlate with specific genotypes (20).
The molecular basis of kindler syndrome results from dysfunctional kindlin-1, a cytoplasmic adaptor protein that normally anchors the actin cytoskeleton to the extracellular matrix (15, 21, 22). It is important in cell adhesion and polarity (23). In the colon and rectum, kindlin-1 is localized in the periphery of epithelial cells, while there is only minimal expression in the terminal ileum (23), consistent with patients’ colitis phenotype. Microscopically, the colon displays extensive ulceration with focal detachment and loss of epithelium from the underlying tissue, along with altered cell polarity (23), increased mitotic activity and infiltration of plasma cells and eosinophils (23, 24). The breach of intestinal barrier is thought to result in penetration of antigens, inflammation, and mucosal changes (23).
While the contribution of barrier dysfunction to mechanisms generating IBD remains incompletely understood, mutations in FERMT1 illustrate how breakdown of this barrier can result in colitis (23). At present there is no cure for kindler syndrome. In some, colitis is managed with anti-inflammatory agents (16), others respond to immune modulators (25), while others require colectomy (23). Potential therapeutic avenues include gene therapy or protein replacement (26).
Mutations in STXBP2 result in an IEI responsible for familial hemophagocytic lymphohistiocytosis, often associated with hypogammaglobulinemia, hearing loss, and bleeding tendencies (27, 28). About 38% of patients develop IBD, characterized by enteropathy and colitis, often within months of life (28, 29). There is no identifiable genotype-phenotype correlation (30). STXBP2 is expressed in intestinal epithelial cells (IECs) and is important for regulating intracellular granule trafficking and docking at the plasma membrane (29, 31, 32). Damaging mutations in STXBP2 result in impaired degranulation of NK cells and cytotoxic T cells, ensuing compromised cytotoxicity (33, 34) and diminished granule fusion in neutrophils, resulting in impaired bacterial killing (35). Additionally, mutations in STXBP2 result in altered epithelial cell polarity (29, 30).
Damaging mutations in STXBP3, a related gene also important in regulating intracellular vesicular trafficking, results in life-threatening infantile-onset IBD of both large and small intestine, usually associated with hearing loss (36). STXBP3 is expressed in epithelial and immune cells and is required for IEC polarization (36). Intestinal disease is histologically characterized by lymphoplasmacytic inflammation, neutrophilic crypt abscesses, villous atrophy, eosinophilia, apoptosis, and crypt destruction (36). Colitis is refractory to immune suppression (36). One patient described exhibited remission of colitis following bone marrow transplant (BMT), while colectomies were curative in four (36).
The unifying molecular functions of STXBP2 and STXBP3 point to their importance in membrane trafficking and maintaining a polarized IEC layer. Disruption in polarity seems, at least partly, responsible for the IBD that persists in a subset of patients with mutations in STXBP2 following successful BMT (29). Additional studies are necessary to delineate mechanisms and cellular components whereby STXBP2 and STXBP3 maintain mucosal homeostasis.
Patients with damaging mutations in TTC7A can present with multiple intestinal atresias (MIA) of the large and small intestine (37), VEOIBD, and a combined immune deficiency (CID) (38–41). VEOIBD from TTC7A deficiency results in profuse bloody diarrhea shortly after birth, usually necessitating parenteral nutrition (39–41). Intestinal disease is microscopically characterized by atresias, architectural distortion, crypt degeneration, pronounced apoptosis, hypertrophy of the muscularis mucosa and spindle cell nodules (42). While all patients present with intestinal manifestations (MIA or VEOIBD, with or without CID), it remains unclear what drives the phenotypic variability.
TTC7A is a scaffolding protein normally expressed in the plasma membrane of intestinal cells (40). It localizes phosphatidylinositol 4 phosphatdylinositol 4-kinase III alpha (PI4KIIIα) to the plasma membrane to facilitate synthesis of PI4-phosphate (40). PI4-phosphate is an upstream precursor of the AKT signaling pathway, critical in preventing apoptosis and promoting proliferation (43). TTC7A deficiency thereby results in increased susceptibility to apoptosis, consistent with patients’ histologic features (40), and aberrant IEC polarity, in line with intestinal atresias (38). Intestinal organoids derived from patients demonstrate disrupted apico-basal polarity, poor epithelial cell integrity, reduced proliferation and absence of luminal space (38). This illustrates the damaging consequences of deficient TTC7A on the intestinal epithelial barrier, in absence of effects from the microbiome and immune system. TTC7A is also expressed in the thymus, but its contribution to CID remains undefined (44).
TTC7A deficiency is fatal in over 50% of patients (45). While BMT can correct the CID, it does not rectify intestinal disease (39, 41, 45), which remains life-threatening (38, 40, 41, 46). VEOIBD is resistant to immune suppression (41, 45) and MIA recur following surgical resection (47). Administration of a RhoA kinase inhibitor to patient-derived organoids reverses defects in polarity and ameliorates proliferation via mechanisms that remain to be elucidated (38). Jardine et al. identified that leflunomide reduces apoptosis, optimizes structure and ion transport in patient-derived colonoids and also restores gut function in a ttc7a-/- zebrafish model (48). While the mechanism remains unclear, leflunomide rescues defective AKT signaling, critical in preventing apoptosis and promoting proliferation (43, 48). TTC7A deficiency highlights the importance of these functions in maintaining mucosal homeostasis.
The nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex, normally generates hydrogen peroxide and reactive oxygen species in phagocytes. Damaging variants in genes encoding components of the NADPH oxidase complex (CYBA, CYBB, CYBC1 (49), NCF1, NCF2, and NCF4) result in impaired anti-microbial activities in phagocytes (50), and thereby chronic granulomatous disease (CGD) (51). Patients have increased susceptibility to infections, especially catalase positive bacteria and fungi (52). Intestinal manifestations include inflammatory and fistulizing CD, which can be mistaken for non-monogenic IBD (51, 53–55). The colon is usually affected, histologically characterized by microgranulomas, pigmented macrophages, eosinophilia and occasionally the small intestine displays villous shortening (56). Esophageal, pyloric and intestinal strictures and obstructions are additional gastrointestinal manifestations (56–59).
The specific mechanisms driving intestinal inflammation in patients with CGD remains to be elucidated. Patient survival is related to the amount of residual NADPH oxidase function (60). In addition to impaired phagocytosis, evidence has pointed to reduced memory B cells (61), defective autophagy and increased inflammasome activation with increased IL1β activity (62, 63).
BMT is curative for both susceptibility to infections and IBD (64–66). Yet, prior to transplant, management of CD in CGD patients is challenging. One needs to balance the inherent high susceptibility to infection with need for immune suppression to control intestinal inflammation. Anti-TNF agents, conventionally used in CD, are avoided given risk of life-threatening infection (67). To minimize extra-intestinal immune suppression, the gut-specific agent, Vedolizumab (an anti-α4β7 integrin blocker), has been tried, but does not result in endoscopic improvement (68). Anti-IL1 agents have been used in small numbers, and while it restores autophagy, reports on whether it improves colitis are mixed (62, 63, 69). This is being further studied (70). IFNγ is used in CGD patients for infection prophylaxis by stimulating superoxide release, but its effects on IBD remains unclear (71, 72). Gene therapy is being investigated (73).
Mutations in DKC1 and RTEL1, result in dyskeratosis congenita (DC), telomeropathies with accelerated shortening or damage of telomeres and bone marrow failure (74–76). The classic clinical triad is dysplastic nails, abnormal skin pigmentation and oral leukoplakia (74). Other features include pulmonary fibrosis, liver disease, and cancer predisposition (74, 77, 78). Hoyeraal-Hreidarsson syndrome is a severe form of DC with intrauterine growth retardation, microcephaly, cerebellar hypoplasia and often VEOIBD (78–81), in addition to esophageal and rectal strictures (79–81). Patients with DC have progressive T, B and NK cell lymphopenia (77–81). Mutations in DKC1 result in medically refractory enterocolitis (76, 80, 81). Similarly, mutated RTEL1 leads to refractory colitis or enteropathy (74, 75, 77). Histologically, intestinal disease is characterized by increased apoptosis, whether attributed to mutations in DKC1 (76, 80) or RTEL1 (74, 77).
The molecular basis of IBD in DC is incompletely understood. DKC1 encodes dyskerin, which is ubiquitously expressed, including the colon, small intestine and peripheral blood cells (82). Dyskerin is important in ribosome biogenesis, preserving telomere integrity (83), responding to DNA damage (84), and vesicular trafficking (85). RTEL1 is expressed in murine intestinal crypts, where intestinal stem cells reside, which are imperative in intestinal homeostasis given their self-renewal capacity (86). RTEL1 is essential for DNA replication and telomere maintenance (87). RTEL1-deficient cells exhibit spontaneous apoptosis and senescence (77).
The course of IBD following BMT in DC patients is poorly described: some report resolution in patients with RTEL1 mutation (77), while IBD persisted in at least one patient with DKC1 mutation (81). This variable response suggests that the underlying mechanism of disease may extend beyond effects of bone marrow failure. This notion is supported by identifying that intestinal organoids generated from a patient with mutated DKC1 exhibit poor growth and differentiation with decreased epithelial markers and reduced Wnt signaling, resulting in attenuated intestinal stem cell renewal (88). These features, observable in absence of input of the immune system, reversed following treatment with a Wnt agonist (88). Similar reversible features were recapitulated in mice lacking telomerase when treated with a Wnt agonist (89). This suggests that telomeropathies have intrinsic epithelial defects in addition to immune deficiencies and bone marrow failure that contribute to IBD. These findings can be harnessed by considering Wnt agonists in the management of this medically refractory IBD (90, 91).
Omenn syndrome (OS), is an IEI that typically presents within weeks of life with immunodeficiency, lymphadenopathy, hepatosplenomegaly, erythroderma and fever, often with infantile-onset colitis (92–95). Patients have a dysplastic thymus, impaired central tolerance resulting in oligoclonality of T cells (2, 95) and absent mature T and B cells (96, 97). OS can result from various IEI, including mutations in RAG1 and RAG2 responsible for somatic V(D)J recombination which defines B and T cell repertoire (95). RAG1 and RAG2 are primarily expressed in T and B lymphocytes (98–100). Genotype-phenotype correlations are variable (101).
The mechanisms driving IBD in OS are unclear. It is thought that impaired central tolerance, proliferation of T lymphocytes with impaired adaptive immunity, defective generation of immune suppressing T regulatory cells (Treg), absent mature T and B cells, and allergic inflammation are all involved (95). Murine models of OS exhibit prominent gastrointestinal inflammation driven by T cells, as their adoptive transfer in immunodeficient hosts suffices to result in colitis, whereas depletion of CD4+ T cells improves their intestinal inflammation (102).
BMT reverses intestinal inflammation and cures OS, which is otherwise fatal (103). The striking life-threatening disease in OS illustrates the critical role of mature T and B cells in the gastrointestinal tract and beyond. Gene therapy and gene editing are potential therapeutic approaches (104).
While OS holds features of disordered central tolerance, this is better illustrated by mutations in AIRE resulting in autoimmune polyendocrinopathy, candidiasis ecto-dermal dystrophy (APECED). AIRE is normally expressed in the medulla of the thymus and drives negative selection of autoreactive T cells (105). Patients manifest autoimmunity of endocrine and non-endocrine tissues and chronic mucocutaneous candidiasis (106). While patients do not develop IBD, they experience various gastrointestinal disorders: autoimmune hepatitis, atrophic gastritis, malabsorption, and poorly described chronic diarrhea and constipation (106–108). Some exhibit enteropathy with antibodies against tryptophan hydroxylase (109, 110). A unique histologic feature among some APECED patients is loss of enteroendocrine cells (110–112). Studies are warranted to better characterize these gastrointestinal disorders. Symptomatic management is employed as there is no cure. A potential future curative intervention is to restore AIRE expression in patient-derived induced pluripotent stem cells (113).
Aside from central tolerance, peripheral tolerance is also critical for maintaining intestinal homeostasis. Tregs normally maintain peripheral immune tolerance and suppress excessive immune responses (114–118). FOXP3 is critical for the development and function of Tregs (116, 119, 120). FOXP3+ cells are densely expressed in lymphoid follicles and scattered in the lamina propria (121). They expand following intestinal inflammation from IBD (121, 122). Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome results from damaging mutations in FOXP3 (123–125). The typical clinical triad is endocrinopathy, enteropathy, and dermatitis, with enteropathy often being the first and most severe phenotype. Intestinal disease most commonly affects the small bowel, but patients can also have colitis (126). The histologic hallmark is severe villous atrophy with extensive eosinophil and lymphocytic infiltrates (126, 127). Other features of IPEX syndrome include: allergies, autoimmune hematologic disorders, hepatitis, nephropathy, and susceptibility to infection (126, 128). There is extensive phenotypic variability, even among patients with identical genotype (126). IPEX syndrome and other IPEX-like diseases (129) underscore the imperative role of peripheral tolerance in maintaining homeostasis in various systems, especially the intestine.
IPEX syndrome can be managed with immune suppression, ideally using sirolimus to spare Tregs (130). However, disease-free survival is poor following immune suppression (126). BMT is currently the only effective cure, with the most significant predictor of survival being the lowest organ involvement (126). A clinical trial is ongoing with lentiviral FOXP3 gene transfer of autologous CD4+T cells (131, 132), hypothesized to reverse the life-threatening multisystemic effects of IPEX syndrome.
The gut microbiota and host immune system are closely intertwined such that each affects development and function of the other (133–135). The bidirectional crosstalk between the gut microbiota and host immune system is disrupted in IBD. Patients with non-monogenic IBD have dysbiosis (136–138) and reduced microbial diversity (139, 140) which may affect intestinal permeability and promote inflammation (141). Those with an IEI driving IBD also have distinct variations of their microbiome as described in CGD and TTC7A deficiency (142) among others (141). Moreover, microbiota changes have been identified following therapeutic interventions, such as increased bacterial diversity in an IPEX patient after fecal microbial transplantation (FMT) (143) and changes in the microbiome of patients with severe combined immune deficiency after BMT (144). Despite limited efficacy to date, eagerness remains in therapeutically targeting dysbiosis via antibiotics, prebiotics, probiotics and FMT in IBD (145–147).
Herein, a selection of IEI is presented, highlighting how interruption of various components of immunity contribute to IBD development. Detrimental consequences ensue when IEI disrupt the necessary mechanisms that maintain intestinal homeostasis.
Among patients with monogenic IBD, employing conventional therapeutics is oftentimes inadequate. Indeed, biologics are only effective in 25.5% of patients with monogenic IBD (9). Moreover, among VEOIBD patients, monogenic disease is a driver of disease severity, including death (10). The growing number of IEI manifesting with IBD, and interaction with the microbiome permits a deeper understanding of the molecular mechanisms driving intestinal inflammation. This knowledge holds important prospects to facilitate personalized therapeutic options and optimized prognostics (148, 149). In some scenarios, understanding the mechanisms driving intestinal inflammation reveals pathways of importance that merit evaluation for therapeutic targeting. Examples include consideration of leflunomide to rescue defective AKT signaling in patients with TTC7A deficiency and refurbishing IL1 antagonists after learning that CGD involves defective autophagy with increased IL1β activity. In other situations, gained insight serves to avoid potentially life-threatening interventions, such as use of anti-TNF agents in CGD patients, or BMT for patients with epithelial barrier defects. Finally, in other circumstances, curative interventions are enabled in otherwise life-threatening disease, such as BMT for IL10 signaling defects, CGD, IPEX syndrome and OS, and sets the groundwork for investigating gene therapy and manipulation of the microbiome.
Clinicians managing patients with IBD from underlying IEI are challenged to think outside the box, delve into the pathologic mechanism at play, and consider innovative personalized approaches. While understanding the molecular mechanisms of IEI driving IBD inspired critical advancements in personalized therapeutics, there remains an urgent need to further advance this field. Developments have been achieved by studying murine models, intestinal organoids, and transcriptomics. More is attainable by harnessing multi-omic efforts in a collaborative and interdisciplinary fashion.
The author confirms being the sole contributor of this work and has approved it for publication.
Research reported in this publication was supported by the National Institute Of Diabetes And Digestive And Kidney Diseases of the National Institutes of Health under Award Number K08DK122133. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Morbe UM, Jorgensen PB, Fenton TM, von Burg N, Riis LB, Spencer J, et al. Human gut-associated lymphoid tissues (GALT); diversity, structure, and function. Mucosal Immunol (2021) 14:793–802. doi: 10.1038/s41385-021-00389-4
2. Sogkas G, Atschekzei F, Adriawan IR, Dubrowinskaja N, Witte T, Schmidt RE. Cellular and molecular mechanisms breaking immune tolerance in inborn errors of immunity. Cell Mol Immunol (2021) 18:1122–40. doi: 10.1038/s41423-020-00626-z
3. Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. Human inborn errors of immunity: 2022 update on the classification from the international union of immunological societies expert committee. J Clin Immunol (2022) 24:1–35. doi: 10.1007/s10875-022-01289-3
4. Hartono S, Ippoliti MR, Mastroianni M, Torres R, Rider NL. Gastrointestinal disorders associated with primary immunodeficiency diseases. Clin Rev Allergy Immunol (2019) 57:145–65. doi: 10.1007/s12016-018-8689-9
5. Picard C, Bobby Gaspar H, Al-Herz W, Bousfiha A, Casanova JL, Chatila T, et al. International union of immunological societies: 2017 primary immunodeficiency diseases committee report on inborn errors of immunity. J Clin Immunol (2018) 38:96–128. doi: 10.1007/s10875-017-0464-9
6. Uhlig HH, Schwerd T, Koletzko S, Shah N, Kammermeier J, Elkadri A, et al. The diagnostic approach to monogenic very early onset inflammatory bowel disease. Gastroenterology (2014) 147:990–1007.e1003. doi: 10.1053/j.gastro.2014.07.023
7. Uhlig HH, Charbit-Henrion F, Kotlarz D, Shouval DS, Schwerd T, Strisciuglio C, et al. Clinical genomics for the diagnosis of monogenic forms of inflammatory bowel disease: A position paper from the paediatric IBD Porto group of European society of paediatric gastroenterology, hepatology and nutrition. J Pediatr Gastroenterol Nutr (2021) 72:456–73. doi: 10.1097/MPG.0000000000003017
8. Pazmandi J, Kalinichenko A, Ardy RC, Boztug K. Early-onset inflammatory bowel disease as a model disease to identify key regulators of immune homeostasis mechanisms. Immunol Rev (2019) 287:162–85. doi: 10.1111/imr.12726
9. Nambu R, Warner N, Mulder DJ, Kotlarz D, McGovern DPB, Cho J, et al. A systematic review of monogenic inflammatory bowel disease. Clin Gastroenterol Hepatol (2022) 20:e653–63. doi: 10.1016/j.cgh.2021.03.021
10. Collen LV, Kim DY, Field M, Okoroafor I, Saccocia G, Whitcomb SD, et al. Clinical phenotypes and outcomes in monogenic versus non-monogenic very early onset inflammatory bowel disease. J Crohns Colitis (2022) 16(9):1380–96. doi: 10.1093/ecco-jcc/jjac045
11. Crowley E, Warner N, Pan J, Khalouei S, Elkadri A, Fiedler K, et al. Prevalence and clinical features of inflammatory bowel diseases associated with monogenic variants, identified by whole-exome sequencing in 1000 children at a single center. Gastroenterology (2020) 158(8):2208–20. doi: 10.1053/j.gastro.2020.02.023
12. Kammermeier J, Dziubak R, Pescarin M, Drury S, Godwin H, Reeve K, et al. Phenotypic and genotypic characterisation of inflammatory bowel disease presenting before the age of 2 years. J Crohns Colitis (2016) 11(1):60–9. doi: 10.1093/ecco-jcc/jjw118
13. Charbit-Henrion F, Parlato M, Hanein S, Duclaux-Loras R, Nowak J, Begue B, et al. Diagnostic yield of next-generation sequencing in very early-onset inflammatory bowel diseases: A multicenter study. J Crohns Colitis (2018) 12(9):1104–12. doi: 10.1093/ecco-jcc/jjy068
14. Glocker EO, Kotlarz D, Boztug K, Gertz EM, Schaffer AA, Noyan F, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med (2009) 361:2033–45. doi: 10.1056/NEJMoa0907206
15. White SJ, McLean WH. Kindler surprise: mutations in a novel actin-associated protein cause kindler syndrome. J Dermatol Sci (2005) 38:169–75. doi: 10.1016/j.jdermsci.2004.12.026
16. Freeman EB, Koglmeier J, Martinez AE, Mellerio JE, Haynes L, Sebire NJ, et al. Gastrointestinal complications of epidermolysis bullosa in children. Br J Dermatol (2008) 158:1308–14. doi: 10.1111/j.1365-2133.2008.08507.x
17. Shimizu S, Nishie W, Tsuchiya K, Inokuma D, Koguchi-Yoshioka H, Kikuchi K, et al. Kindler syndrome with severe intestinal involvement: A 31-year follow-up. Acta Derm Venereol (2014) 94:351–2. doi: 10.2340/00015555-1696
18. Has C, Castiglia D, del Rio M, Diez MG, Piccinni E, Kiritsi D, et al. Kindler syndrome: extension of FERMT1 mutational spectrum and natural history. Hum Mutat (2011) 32:1204–12. doi: 10.1002/humu.21576
19. Ussar S, Moser M, Widmaier M, Rognoni E, Harrer C, Genzel-Boroviczeny O, et al. Loss of kindlin-1 causes skin atrophy and lethal neonatal intestinal epithelial dysfunction. PloS Genet (2008) 4:e1000289. doi: 10.1371/journal.pgen.1000289
20. Rognoni E, Ruppert R, Fassler R. The kindlin family: Functions, signaling properties and implications for human disease. J Cell Sci (2016) 129:17–27. doi: 10.1242/jcs.161190
21. Jobard F, Bouadjar B, Caux F, Hadj-Rabia S, Has C, Matsuda F, et al. Identification of mutations in a new gene encoding a FERM family protein with a pleckstrin homology domain in kindler syndrome. Hum Mol Genet (2003) 12:925–35. doi: 10.1093/hmg/ddg097
22. Siegel DH, Ashton GH, Penagos HG, Lee JV, Feiler HS, Wilhelmsen KC, et al. Loss of kindlin-1, a human homolog of the caenorhabditis elegans actin-extracellular-matrix linker protein UNC-112, causes kindler syndrome. Am J Hum Genet (2003) 73:174–87. doi: 10.1086/376609
23. Kern JS, Herz C, Haan E, Moore D, Nottelmann S, von Lilien T, et al. Chronic colitis due to an epithelial barrier defect: the role of kindlin-1 isoforms. J Pathol (2007) 213:462–70. doi: 10.1002/path.2253
24. Sadler E, Klausegger A, Muss W, Deinsberger U, Pohla-Gubo G, Laimer M, et al. Novel KIND1 gene mutation in kindler syndrome with severe gastrointestinal tract involvement. Arch Dermatol (2006) 142:1619–24. doi: 10.1001/archderm.142.12.1619
25. Ashton JJ, Andreoletti G, Coelho T, Haggarty R, Batra A, Afzal NA, et al. Identification of variants in genes associated with single-gene inflammatory bowel disease by whole-exome sequencing. Inflamm Bowel Dis (2016) 22:2317–27. doi: 10.1097/MIB.0000000000000890
26. Ren A, Yin W, Miller H, Westerberg LS, Candotti F, Park CS, et al. Novel discoveries in immune dysregulation in inborn errors of immunity. Front Immunol (2021) 12:725587. doi: 10.3389/fimmu.2021.725587
27. Meeths M, Entesarian M, Al-Herz W, Chiang SC, Wood SM, Al-Ateeqi W, et al. Spectrum of clinical presentations in familial hemophagocytic lymphohistiocytosis type 5 patients with mutations in STXBP2. Blood (2010) 116:2635–43. doi: 10.1182/blood-2010-05-282541
28. Pagel J, Beutel K, Lehmberg K, Koch F, Maul-Pavicic A, Rohlfs AK, et al. Distinct mutations in STXBP2 are associated with variable clinical presentations in patients with familial hemophagocytic lymphohistiocytosis type 5 (FHL5). Blood (2012) 119:6016–24. doi: 10.1182/blood-2011-12-398958
29. Stepensky P, Bartram J, Barth TF, Lehmberg K, Walther P, Amann K, et al. Persistent defective membrane trafficking in epithelial cells of patients with familial hemophagocytic lymphohistiocytosis type 5 due to STXBP2/MUNC18-2 mutations. Pediatr Blood Cancer (2013) 60:1215–22. doi: 10.1002/pbc.24475
30. Dhekne HS, Pylypenko O, Overeem AW, Zibouche M, Ferreira RJ, van der Velde KJ, et al. MYO5B, STX3, and STXBP2 mutations reveal a common disease mechanism that unifies a subset of congenital diarrheal disorders: A mutation update. Hum Mutat (2018) 39:333–44. doi: 10.1002/humu.23386
31. Vogel GF, van Rijn JM, Krainer IM, Janecke AR, Posovszky C, Cohen M, et al. Disrupted apical exocytosis of cargo vesicles causes enteropathy in FHL5 patients with Munc18-2 mutations. JCI Insight (2017) 2. doi: 10.1172/jci.insight.94564
32. Riento K, Jantti J, Jansson S, Hielm S, Lehtonen E, Ehnholm C, et al. A sec1-related vesicle-transport protein that is expressed predominantly in epithelial cells. Eur J Biochem (1996) 239:638–46. doi: 10.1111/j.1432-1033.1996.0638u.x
33. zur Stadt U, Rohr J, Seifert W, Koch F, Grieve S, Pagel J, et al. Familial hemophagocytic lymphohistiocytosis type 5 (FHL-5) is caused by mutations in Munc18-2 and impaired binding to syntaxin 11. Am J Hum Genet (2009) 85:482–92. doi: 10.1016/j.ajhg.2009.09.005
34. Cote M, Menager MM, Burgess A, Mahlaoui N, Picard C, Schaffner C, et al. Munc18-2 deficiency causes familial hemophagocytic lymphohistiocytosis type 5 and impairs cytotoxic granule exocytosis in patient NK cells. J Clin Invest (2009) 119:3765–73. doi: 10.1172/JCI40732
35. Zhao XW, Gazendam RP, Drewniak A, van Houdt M, Tool AT, van Hamme JL, et al. Defects in neutrophil granule mobilization and bactericidal activity in familial hemophagocytic lymphohistiocytosis type 5 (FHL-5) syndrome caused by STXBP2/Munc18-2 mutations. Blood (2013) 122:109–11. doi: 10.1182/blood-2013-03-494039
36. Ouahed J, Kelsen JR, Spessott WA, Kooshesh K, Sanmillan ML, Dawany N, et al. Variants in STXBP3 are associated with very early onset inflammatory bowel disease, bilateral sensorineural hearing loss and immune dysregulation. J Crohns Colitis (2021) 15:1908–19. doi: 10.1093/ecco-jcc/jjab077
37. Samuels ME, Majewski J, Alirezaie N, Fernandez I, Casals F, Patey N, et al. Exome sequencing identifies mutations in the gene TTC7A in French-Canadian cases with hereditary multiple intestinal atresia. J Med Genet (2013) 50:324–9. doi: 10.1136/jmedgenet-2012-101483
38. Bigorgne AE, Farin HF, Lemoine R, Mahlaoui N, Lambert N, Gil M, et al. TTC7A mutations disrupt intestinal epithelial apicobasal polarity. J Clin Invest (2014) 124:328–37. doi: 10.1172/JCI71471
39. Lemoine R, Pachlopnik-Schmid J, Farin HF, Bigorgne A, Debre M, Sepulveda F, et al. Immune deficiency-related enteropathy-lymphocytopenia-alopecia syndrome results from tetratricopeptide repeat domain 7A deficiency. J Allergy Clin Immunol (2014) 134:1354–64.e1356. doi: 10.1016/j.jaci.2014.07.019
40. Avitzur Y, Guo C, Mastropaolo LA, Bahrami E, Chen H, Zhao Z, et al. Mutations in tetratricopeptide repeat domain 7A result in a severe form of very early onset inflammatory bowel disease. Gastroenterology (2014) 146:1028–39. doi: 10.1053/j.gastro.2014.01.015
41. Lien R, Lin YF, Lai MW, Weng HY, Wu RC, Jaing TH, et al. Novel mutations of the tetratricopeptide repeat domain 7A gene and Phenotype/Genotype comparison. Front Immunol (2017) 8:1066. doi: 10.3389/fimmu.2017.01066
42. Dannheim K, Ouahed J, Field M, Snapper S, Raphael BP, Glover SC, et al. Pediatric gastrointestinal histopathology in patients with tetratricopeptide repeat domain 7A (TTC7A) germline mutations: A rare condition leading to multiple intestinal atresias, severe combined immunodeficiency, and congenital enteropathy. Am J Surg Pathol (2022) 46:846–53. doi: 10.1097/PAS.0000000000001856
43. Tan J, Brill JA. Cinderella Story: PI4P goes from precursor to key signaling molecule. Crit Rev Biochem Mol Biol (2014) 49:33–58. doi: 10.3109/10409238.2013.853024
44. Chen R, Giliani S, Lanzi G, Mias GI, Lonardi S, Dobbs K, et al. Whole-exome sequencing identifies tetratricopeptide repeat domain 7A (TTC7A) mutations for combined immunodeficiency with intestinal atresias. J Allergy Clin Immunol (2013) 132:656–64.e617. doi: 10.1016/j.jaci.2013.06.013
45. Kammermeier J, Lucchini G, Pai SY, Worth A, Rampling D, Amrolia P, et al. Stem cell transplantation for tetratricopeptide repeat domain 7A deficiency: long-term follow-up. Blood (2016) 128:1306–8. doi: 10.1182/blood-2016-01-696385
46. Fullerton BS, Velazco CS, Hong CR, Carey AN, Jaksic T. High rates of positive severe combined immunodeficiency screening among newborns with severe intestinal failure. JPEN J Parenter Enteral Nutr (2018) 42:239–46. doi: 10.1002/jpen.1013
47. Ali YA, Rahman S, Bhat V, Al Thani S, Ismail A, Bassiouny I. Hereditary multiple intestinal atresia (HMIA) with severe combined immunodeficiency (SCID): a case report of two siblings and review of the literature on MIA, HMIA and HMIA with immunodeficiency over the last 50 years. BMJ Case Rep (2011) 2011. doi: 10.1136/bcr.05.2010.3031
48. Jardine S, Anderson S, Babcock S, Leung G, Pan J, Dhingani N, et al. Drug screen identifies leflunomide for treatment of inflammatory bowel disease caused by TTC7A deficiency. Gastroenterology (2020) 158:1000–15. doi: 10.1053/j.gastro.2019.11.019
49. Arnadottir GA, Norddahl GL, Gudmundsdottir S, Agustsdottir AB, Sigurdsson S, Jensson BO, et al. A homozygous loss-of-function mutation leading to CYBC1 deficiency causes chronic granulomatous disease. Nat Commun (2018) 9:4447. doi: 10.1038/s41467-018-06964-x
50. Segal AW, Peters TJ. Characterisation of the enzyme defect in chronic granulomatous disease. Lancet (1976) 1:1363–5. doi: 10.1016/s0140-6736(76)93021-x
51. Schappi MG, Smith VV, Goldblatt D, Lindley KJ, Milla PJ. Colitis in chronic granulomatous disease. Arch Dis Child (2001) 84:147–51. doi: 10.1136/adc.84.2.147
52. Winkelstein JA, Marino MC, Johnston RB, Jr. , Boyle J, Curnutte J, Gallin JI, et al. Chronic granulomatous disease. report on a national registry of 368 patients. Med (Baltimore) (2000) 79:155–69. doi: 10.1097/00005792-200005000-00003
53. Matute JD, Arias AA, Wright NA, Wrobel I, Waterhouse CC, Li XJ, et al. A new genetic subgroup of chronic granulomatous disease with autosomal recessive mutations in p40 phox and selective defects in neutrophil NADPH oxidase activity. Blood (2009) 114:3309–15. doi: 10.1182/blood-2009-07-231498
54. Al-Bousafy A, Al-Tubuly A, Dawi E, Zaroog S, Schulze I. Libyan Boy with autosomal recessive trait (P22-phox defect) of chronic granulomatous disease. Libyan J Med (2006) 1:162–71. doi: 10.4176/060905
55. Marciano BE, Rosenzweig SD, Kleiner DE, Anderson VL, Darnell DN, Anaya-O'Brien S, et al. Gastrointestinal involvement in chronic granulomatous disease. Pediatrics (2004) 114:462–8. doi: 10.1542/peds.114.2.462
56. Alimchandani M, Lai JP, Aung PP, Khangura S, Kamal N, Gallin JI, et al. Gastrointestinal histopathology in chronic granulomatous disease: a study of 87 patients. Am J Surg Pathol (2013) 37:1365–72. doi: 10.1097/PAS.0b013e318297427d
57. Huang A, Abbasakoor F, Vaizey CJ. Gastrointestinal manifestations of chronic granulomatous disease. Colorectal Dis (2006) 8:637–44. doi: 10.1111/j.1463-1318.2006.01030.x
58. Varma VA, Sessions JT, Kahn LB, Lipper S. Chronic granulomatous disease of childhood presenting as gastric outlet obstruction. Am J Surg Pathol (1982) 6:673–6. doi: 10.1097/00000478-198210000-00009
59. Laskey HL, Gopal L, Gallin JI, Holland SM, Heller T. Twenty-year follow-up of esophageal involvement in chronic granulomatous disease. Am J Gastroenterol (2009) 104:2368–70. doi: 10.1038/ajg.2009.274
60. Kuhns DB, Alvord WG, Heller T, Feld JJ, Pike KM, Marciano BE, et al. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med (2010) 363:2600–10. doi: 10.1056/NEJMoa1007097
61. Mohsenzadegan M, Fattahi F, Fattahi F, Mirshafiey A, Fazlollahi MR, Naderi Beni F, et al. Altered pattern of naive and memory b cells and B1 cells in patients with chronic granulomatous disease. Iran J Allergy Asthma Immunol (2014) 13:157–65.
62. de Luca A, Smeekens SP, Casagrande A, Iannitti R, Conway KL, Gresnigt MS, et al. IL-1 receptor blockade restores autophagy and reduces inflammation in chronic granulomatous disease in mice and in humans. Proc Natl Acad Sci USA (2014) 111:3526–31. doi: 10.1073/pnas.1322831111
63. Meissner F, Seger RA, Moshous D, Fischer A, Reichenbach J, Zychlinsky A. Inflammasome activation in NADPH oxidase defective mononuclear phagocytes from patients with chronic granulomatous disease. Blood (2010) 116:1570–3. doi: 10.1182/blood-2010-01-264218
64. Kato K, Kojima Y, Kobayashi C, Mitsui K, Nakajima-Yamaguchi R, Kudo K, et al. Successful allogeneic hematopoietic stem cell transplantation for chronic granulomatous disease with inflammatory complications and severe infection. Int J Hematol (2011) 94:479–82. doi: 10.1007/s12185-011-0932-6
65. Gungor T, Teira P, Slatter M, Stussi G, Stepensky P, Moshous D, et al. Reduced-intensity conditioning and HLA-matched haemopoietic stem-cell transplantation in patients with chronic granulomatous disease: a prospective multicentre study. Lancet (2014) 383:436–48. doi: 10.1016/S0140-6736(13)62069-3
66. Marsh RA, Leiding JW, Logan BR, Griffith LM, Arnold DE, Haddad E, et al. Chronic granulomatous disease-associated IBD resolves and does not adversely impact survival following allogeneic HCT. J Clin Immunol (2019) 39:653–67. doi: 10.1007/s10875-019-00659-8
67. Uzel G, Orange JS, Poliak N, Marciano BE, Heller T, Holland SM. Complications of tumor necrosis factor-alpha blockade in chronic granulomatous disease-related colitis. Clin Infect Dis (2010) 51:1429–34. doi: 10.1086/657308
68. Kamal N, Marciano B, Curtin B, Strongin A, DeRavin SS, Bousvaros A, et al. The response to vedolizumab in chronic granulomatous disease-related inflammatory bowel disease. Gastroenterol Rep (Oxf) (2020) 8:404–6. doi: 10.1093/gastro/goaa005
69. Hahn KJ, Ho N, Yockey L, Kreuzberg S, Daub J, Rump A, et al. Treatment with anakinra, a recombinant IL-1 receptor antagonist, unlikely to induce lasting remission in patients with CGD colitis. Am J Gastroenterol (2015) 110:938–9. doi: 10.1038/ajg.2015.135
71. LaBere B, Gutierrez MJ, Wright H, Garabedian E, Ochs HD, Fuleihan RL, et al. Chronic granulomatous disease with inflammatory bowel disease: Clinical presentation, treatment, and outcomes from the USIDNET registry. J Allergy Clin Immunol Pract (2022) 10:1325–33.e1325. doi: 10.1016/j.jaip.2021.12.035
74. Ballew BJ, Joseph V, De S, Sarek G, Vannier JB, Stracker T, et al. A recessive founder mutation in regulator of telomere elongation helicase 1, RTEL1, underlies severe immunodeficiency and features of hoyeraal hreidarsson syndrome. PloS Genet (2013) 9:e1003695. doi: 10.1371/journal.pgen.1003695
75. Le Guen T, Jullien L, Touzot F, Schertzer M, Gaillard L, Perderiset M, et al. Human RTEL1 deficiency causes hoyeraal-hreidarsson syndrome with short telomeres and genome instability. Hum Mol Genet (2013) 22:3239–49. doi: 10.1093/hmg/ddt178
76. Sznajer Y, Baumann C, David A, Journel H, Lacombe D, Perel Y, et al. Further delineation of the congenital form of X-linked dyskeratosis congenita (Hoyeraal-hreidarsson syndrome). Eur J Pediatr (2003) 162:863–7. doi: 10.1007/s00431-003-1317-5
77. Speckmann C, Sahoo SS, Rizzi M, Hirabayashi S, Karow A, Serwas NK, et al. Clinical and molecular heterogeneity of RTEL1 deficiency. Front Immunol (2017) 8:449. doi: 10.3389/fimmu.2017.00449
78. Hreidarsson S, Kristjansson K, Johannesson G. & johannsson, j. h. a syndrome of progressive pancytopenia with microcephaly, cerebellar hypoplasia and growth failure. Acta Paediatr Scand (1988) 77:773–5. doi: 10.1111/j.1651-2227.1988.tb10751.x
79. Ziv A, Werner L, Konnikova L, Awad A, Jeske T, Hastreiter M, et al. An RTEL1 mutation links to infantile-onset ulcerative colitis and severe immunodeficiency. J Clin Immunol (2020) 40:1010–9. doi: 10.1007/s10875-020-00829-z
80. Borggraefe I, Koletzko S, Arenz T, Fuehrer M, Hoffmann F, Dokal I, et al. Severe variant of x-linked dyskeratosis congenita (Hoyeraal-hreidarsson syndrome) causes significant enterocolitis in early infancy. J Pediatr Gastroenterol Nutr (2009) 49:359–63. doi: 10.1097/MPG.0b013e3181a15b94
81. Knight SW, Heiss NS, Vulliamy TJ, Aalfs CM, McMahon C, Richmond P, et al. Unexplained aplastic anaemia, immunodeficiency, and cerebellar hypoplasia (Hoyeraal-hreidarsson syndrome) due to mutations in the dyskeratosis congenita gene, DKC1. Br J Haematol (1999) 107:335–9. doi: 10.1046/j.1365-2141.1999.01690.x
82. Heiss NS, Knight SW, Vulliamy TJ, Klauck SM, Wiemann S, Mason PJ, et al. X-Linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nat Genet (1998) 19:32–8. doi: 10.1038/ng0598-32
83. Angrisani A, Vicidomini R, Turano M, Furia M. Human dyskerin: beyond telomeres. Biol Chem (2014) 395:593–610. doi: 10.1515/hsz-2013-0287
84. Gu BW, Bessler M, Mason PJ. A pathogenic dyskerin mutation impairs proliferation and activates a DNA damage response independent of telomere length in mice. Proc Natl Acad Sci USA (2008) 105:10173–8. doi: 10.1073/pnas.0803559105
85. Di Maio N, Vicidomini R, Angrisani A, Belli V, Furia M, Turano M. A new role for human dyskerin in vesicular trafficking. FEBS Open Bio (2017) 7:1453–68. doi: 10.1002/2211-5463.12307
86. Seshadri N, Sandhu S, Wu X, Liu W, Ding H. Generation of an Rtel1-CreERT2 knock-in mouse model for lineage tracing RTEL1+ stem cells during development. Transgenic Res (2018) 27:571–8. doi: 10.1007/s11248-018-0093-y
87. Walne AJ, Vulliamy T, Kirwan M, Plagnol V, Dokal I. Constitutional mutations in RTEL1 cause severe dyskeratosis congenita. Am J Hum Genet (2013) 92:448–53. doi: 10.1016/j.ajhg.2013.02.001
88. Woo DH, Chen Q, Yang TL, Glineburg MR, Hoge C, Leu NA, et al. Enhancing a wnt-telomere feedback loop restores intestinal stem cell function in a human organotypic model of dyskeratosis congenita. Cell Stem Cell (2016) 19:397–405. doi: 10.1016/j.stem.2016.05.024
89. Yang TB, Chen Q, Deng JT, Jagannathan G, Tobias JW, Schultz DC, et al. Mutual reinforcement between telomere capping and canonical wnt signalling in the intestinal stem cell niche. Nat Commun (2017) 8:14766. doi: 10.1038/ncomms14766
90. Fernandez RJ 3rd, Johnson FB. A regulatory loop connecting WNT signaling and telomere capping: possible therapeutic implications for dyskeratosis congenita. Ann N Y Acad Sci (2018) 1418:56–68. doi: 10.1111/nyas.13692
91. Fernandez RJ 3rd, Gardner ZJG, Slovik KJ, Liberti DC, Estep KN, Yang W, et al. GSK3 inhibition rescues growth and telomere dysfunction in dyskeratosis congenita iPSC-derived type II alveolar epithelial cells. Elife (2022) 11. doi: 10.7554/eLife.64430
92. Omenn GS. Familial reticuloendotheliosis with eosinophilia. N Engl J Med (1965) 273:427–32. doi: 10.1056/NEJM196508192730806
93. Chou J, Hanna-Wakim R, Tirosh I, Kane J, Fraulino D, Lee YN, et al. A novel homozygous mutation in recombination activating gene 2 in 2 relatives with different clinical phenotypes: Omenn syndrome and hyper-IgM syndrome. J Allergy Clin Immunol (2012) 130:1414–6. doi: 10.1016/j.jaci.2012.06.012
94. Felgentreff K, Perez-Becker R, Speckmann C, Schwarz K, Kalwak K, Markelj G, et al. Clinical and immunological manifestations of patients with atypical severe combined immunodeficiency. Clin Immunol (2011) 141:73–82. doi: 10.1016/j.clim.2011.05.007
95. Villa A, Notarangelo LD, Roifman CM. Omenn syndrome: inflammation in leaky severe combined immunodeficiency. J Allergy Clin Immunol (2008) 122:1082–6. doi: 10.1016/j.jaci.2008.09.037
96. Schwarz K, Gauss GH, Ludwig L, Pannicke U, Li Z, Lindner D, et al. RAG mutations in human b cell-negative SCID. Science (1996) 274:97–9. doi: 10.1126/science.274.5284.97
97. Villa A, Sobacchi C, Vezzoni P. Omenn syndrome in the context of other b cell-negative severe combined immunodeficiencies. Isr Med Assoc J (2002) 4:218–21.
98. Girschick HJ, Grammer AC, Nanki T, Mayo M, Lipsky PE. RAG1 and RAG2 expression by b cell subsets from human tonsil and peripheral blood. J Immunol (2001) 166:377–86. doi: 10.4049/jimmunol.166.1.377
99. Mansson R, Hultquist A, Luc S, Yang L, Anderson K, Kharazi S, et al. Molecular evidence for hierarchical transcriptional lineage priming in fetal and adult stem cells and multipotent progenitors. Immunity (2007) 26:407–19. doi: 10.1016/j.immuni.2007.02.013
100. Kuo TC, Schlissel MS. Mechanisms controlling expression of the RAG locus during lymphocyte development. Curr Opin Immunol (2009) 21:173–8. doi: 10.1016/j.coi.2009.03.008
101. Cifaldi C, Rivalta B, Amodio D, Mattia A, Pacillo L, Di Cesare S, et al. Clinical, immunological, and molecular variability of RAG deficiency: A retrospective analysis of 22 RAG patients. J Clin Immunol (2022) 42:130–45. doi: 10.1007/s10875-021-01130-3
102. Rigoni R, Fontana E, Guglielmetti S, Fosso B, D'Erchia AM, Maina V, et al. Intestinal microbiota sustains inflammation and autoimmunity induced by hypomorphic RAG defects. J Exp Med (2016) 213:355–75. doi: 10.1084/jem.20151116
103. Mazzolari E, Moshous D, Forino C, De Martiis D, Offer C, Lanfranchi A, et al. Hematopoietic stem cell transplantation in omenn syndrome: a single-center experience. Bone Marrow Transplant (2005) 36:107–14. doi: 10.1038/sj.bmt.1705017
104. Bosticardo M, Pala F, Notarangelo LD. RAG deficiencies: Recent advances in disease pathogenesis and novel therapeutic approaches. Eur J Immunol (2021) 51:1028–38. doi: 10.1002/eji.202048880
105. Anderson MS, Su MA. Aire and T cell development. Curr Opin Immunol (2011) 23:198–206. doi: 10.1016/j.coi.2010.11.007
106. Ferre EM, Rose SR, Rosenzweig SD, Burbelo PD, Romito KR, Niemela JE, et al. Redefined clinical features and diagnostic criteria in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. JCI Insight (2016) 1. doi: 10.1172/jci.insight.88782
107. Skrabic V, Skrabic I, Skrabic R, Roje B, Simunovic M. Clinical characteristics in the longitudinal follow-up of APECED syndrome in southern Croatia-case series. Genes (Basel) (2022) 13. doi: 10.3390/genes13040558
108. Kluger N, Jokinen M, Krohn K, Ranki A. Gastrointestinal manifestations in APECED syndrome. J Clin Gastroenterol (2013) 47:112–20. doi: 10.1097/MCG.0b013e31827356e1
109. Ekwall O, et al. Identification of tryptophan hydroxylase as an intestinal autoantigen. Lancet (1998) 352:279–83. doi: 10.1016/S0140-6736(97)11050-9
110. Posovszky C, Lahr G, von Schnurbein J, Buderus S, Findeisen A, Schroder C, et al. Loss of enteroendocrine cells in autoimmune-polyendocrine-candidiasis-ectodermal-dystrophy (APECED) syndrome with gastrointestinal dysfunction. J Clin Endocrinol Metab (2012) 97:E292–300. doi: 10.1210/jc.2011-2044
111. Oliva-Hemker M, Berkenblit GV, Anhalt GJ, Yardley JH. Pernicious anemia and widespread absence of gastrointestinal endocrine cells in a patient with autoimmune polyglandular syndrome type I and malabsorption. J Clin Endocrinol Metab (2006) 91:2833–8. doi: 10.1210/jc.2005-2506
112. Ohsie S, Gerney G, Gui D, Kahana D, Martin MG, Cortina G. A paucity of colonic enteroendocrine and/or enterochromaffin cells characterizes a subset of patients with chronic unexplained diarrhea/malabsorption. Hum Pathol (2009) 40:1006–14. doi: 10.1016/j.humpath.2008.12.016
113. Karvonen E, Krohn KJE, Ranki A, Hau A. Generation and characterization of iPS cells derived from APECED patients for gene correction. Front Endocrinol (Lausanne) (2022) 13:794327. doi: 10.3389/fendo.2022.794327
114. Sakaguchi S, Fukuma K, Kuribayashi K, Masuda T. Organ-specific autoimmune diseases induced in mice by elimination of T cell subset. i. evidence for the active participation of T cells in natural self-tolerance; deficit of a T cell subset as a possible cause of autoimmune disease. J Exp Med (1985) 161:72–87. doi: 10.1084/jem.161.1.72
115. Sakaguchi S. Animal models of autoimmunity and their relevance to human diseases. Curr Opin Immunol (2000) 12:684–90. doi: 10.1016/s0952-7915(00)00163-1
116. Yagi H, Nomura T, Nakamura K, Yamazaki S, Kitawaki T, Hori S, et al. Crucial role of FOXP3 in the development and function of human CD25+CD4+ regulatory T cells. Int Immunol (2004) 16:1643–56. doi: 10.1093/intimm/dxh165
117. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell (2008) 133:775–87. doi: 10.1016/j.cell.2008.05.009
118. Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol (2010) 10:490–500. doi: 10.1038/nri2785
119. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science (2003) 299:1057–61. doi: 10.1126/science.1079490
120. Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for scurfin in CD4+CD25+ T regulatory cells. Nat Immunol (2003) 4:337–42. doi: 10.1038/ni909
121. Uhlig HH, Coombes J, Mottet C, Izcue A, Thompson C, Fanger A, et al. Characterization of Foxp3+CD4+CD25+ and IL-10-secreting CD4+CD25+ T cells during cure of colitis. J Immunol (2006) 177:5852–60. doi: 10.4049/jimmunol.177.9.5852
122. Mitsialis V, Wall S, Liu P, Ordovas-Montanes J, Parmet T, Vukovic M, et al. Single-cell analyses of colon and blood reveal distinct immune cell signatures of ulcerative colitis and crohn's disease. Gastroenterology (2020) 159:591–608.e510. doi: 10.1053/j.gastro.2020.04.074
123. Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, et al. X-Linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet (2001) 27:18–20. doi: 10.1038/83707
124. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet (2001) 27:20–1. doi: 10.1038/83713
125. Chatila TA, Blaeser F, Ho N, Lederman HM, Voulgaropoulos C, Helms C, et al. JM2, encoding a fork head-related protein, is mutated in X-linked autoimmunity-allergic disregulation syndrome. J Clin Invest (2000) 106:R75–81. doi: 10.1172/JCI11679
126. Barzaghi F, Amaya Hernandez LC, Neven B, Ricci S, Kucuk ZY, Bleesing JJ, et al. Long-term follow-up of IPEX syndrome patients after different therapeutic strategies: An international multicenter retrospective study. J Allergy Clin Immunol (2018) 141:1036–49.e1035. doi: 10.1016/j.jaci.2017.10.041
127. Torgerson TR, Ochs HD. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked: forkhead box protein 3 mutations and lack of regulatory T cells. J Allergy Clin Immunol (2007) 120:744–50; quiz 751-742. doi: 10.1016/j.jaci.2007.08.044
128. Gambineri E, Torgerson TR, Ochs HD. Immune dysregulation, polyendocrinopathy, enteropathy, and X-linked inheritance (IPEX), a syndrome of systemic autoimmunity caused by mutations of FOXP3, a critical regulator of T-cell homeostasis. Curr Opin Rheumatol (2003) 15:430–5. doi: 10.1097/00002281-200307000-00010
129. Baxter SK, Walsh T, Casadei S, Eckert MM, Allenspach EJ, Hagin D, et al. Molecular diagnosis of childhood immune dysregulation, polyendocrinopathy, and enteropathy, and implications for clinical management. J Allergy Clin Immunol (2022) 149:327–39. doi: 10.1016/j.jaci.2021.04.005
130. Passerini L, Barzaghi F, Curto R, Sartirana C, Barera G, Tucci F, et al. Treatment with rapamycin can restore regulatory T-cell function in IPEX patients. J Allergy Clin Immunol (2020) 145:1262–1271.e1213. doi: 10.1016/j.jaci.2019.11.043
131. Sato Y, Passerini L, Piening BD, Uyeda MJ, Goodwin M, Gregori S, et al. Human-engineered treg-like cells suppress FOXP3-deficient T cells but preserve adaptive immune responses in vivo. Clin Transl Immunol (2020) 9:e1214. doi: 10.1002/cti2.1214
133. Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol (2009) 9:313–23. doi: 10.1038/nri2515
134. Kamada N, Nunez G. Regulation of the immune system by the resident intestinal bacteria. Gastroenterology (2014) 146:1477–88. doi: 10.1053/j.gastro.2014.01.060
135. Zheng D, Liwinski T, Elinav E. Interaction between microbiota and immunity in health and disease. Cell Res (2020) 30:492–506. doi: 10.1038/s41422-020-0332-7
136. Andoh A, Sakata S, Koizumi Y, Mitsuyama K, Fujiyama Y, Benno Y. Terminal restriction fragment length polymorphism analysis of the diversity of fecal microbiota in patients with ulcerative colitis. Inflammation Bowel Dis (2007) 13:955–62. doi: 10.1002/ibd.20151
137. Fujimoto T, Imaeda H, Takahashi K, Kasumi E, Bamba S, Fujiyama Y, et al. Decreased abundance of faecalibacterium prausnitzii in the gut microbiota of crohn's disease. J Gastroenterol Hepatol (2013) 28:613–9. doi: 10.1111/jgh.12073
138. Nishino K, Nishida A, Inoue R, Kawada Y, Ohno M, Sakai S, et al. Analysis of endoscopic brush samples identified mucosa-associated dysbiosis in inflammatory bowel disease. J Gastroenterol (2018) 53:95–106. doi: 10.1007/s00535-017-1384-4
139. Walker AW, Sanderson JD, Churcher C, Parkes GC, Hudspith BN, Rayment N, et al. High-throughput clone library analysis of the mucosa-associated microbiota reveals dysbiosis and differences between inflamed and non-inflamed regions of the intestine in inflammatory bowel disease. BMC Microbiol (2011) 11:7. doi: 10.1186/1471-2180-11-7
140. Manichanh C, Rigottier-Gois L, Bonnaud E, Gloux K, Pelletier E, Frangeul L, et al. Reduced diversity of faecal microbiota in crohn's disease revealed by a metagenomic approach. Gut (2006) 55:205–11. doi: 10.1136/gut.2005.073817
141. Castagnoli R, Pala F, Bosticardo M, Licari A, Delmonte OM, Villa A, et al. Gut microbiota-host interactions in inborn errors of immunity. Int J Mol Sci (2021) 22. doi: 10.3390/ijms22031416
142. Sokol H, Mahlaoui N, Aguilar C, Bach P, Join-Lambert O, Garraffo A, et al. Intestinal dysbiosis in inflammatory bowel disease associated with primary immunodeficiency. J Allergy Clin Immunol (2019) 143:775–778.e776. doi: 10.1016/j.jaci.2018.09.021
143. Wu W, Shen N, Luo L, Deng Z, Chen J, Tao Y, et al. Fecal microbiota transplantation before hematopoietic stem cell transplantation in a pediatric case of chronic diarrhea with a FOXP3 mutation. Pediatr Neonatol (2021) 62:172–80. doi: 10.1016/j.pedneo.2020.11.003
144. Lane JP, Stewart CJ, Cummings SP, Gennery AR. Gut microbiome variations during hematopoietic stem cell transplant in severe combined immunodeficiency. J Allergy Clin Immunol (2015) 135:1654–6. doi: 10.1016/j.jaci.2015.01.024
145. Sartor RB, Wu GD. Roles for intestinal bacteria, viruses, and fungi in pathogenesis of inflammatory bowel diseases and therapeutic approaches. Gastroenterology (2017) 152:327–39.e324. doi: 10.1053/j.gastro.2016.10.012
146. Nishida A, Inoue R, Inatomi O, Bamba S, Naito Y, Andoh A. Gut microbiota in the pathogenesis of inflammatory bowel disease. Clin J Gastroenterol (2018) 11:1–10. doi: 10.1007/s12328-017-0813-5
147. Paramsothy S, Paramsothy R, Rubin DT, Kamm MA, Kaakoush NO, Mitchell HM, et al. Faecal microbiota transplantation for inflammatory bowel disease: A systematic review and meta-analysis. J Crohns Colitis (2017) 11:1180–99. doi: 10.1093/ecco-jcc/jjx063
148. Kelsen JR, Sullivan KE, Rabizadeh S, Singh N, Snapper S, Elkadri A, et al. North American society for pediatric gastroenterology, hepatology, and nutrition position paper on the evaluation and management for patients with very early-onset inflammatory bowel disease. J Pediatr Gastroenterol Nutr (2020) 70:389–403. doi: 10.1097/MPG.0000000000002567
Keywords: inborn errors of immunity (IEI), monogenic inflammatory bowel disease, mechanisms of disease, very early onset IBD (VEOIBD), genetics
Citation: Ouahed JD (2022) Understanding inborn errors of immunity: A lens into the pathophysiology of monogenic inflammatory bowel disease. Front. Immunol. 13:1026511. doi: 10.3389/fimmu.2022.1026511
Received: 24 August 2022; Accepted: 13 September 2022;
Published: 29 September 2022.
Edited by:
Elena Wen-Yuan Hsieh, University of Colorado Anschutz Medical Campus, United StatesReviewed by:
Riccardo Castagnoli, Laboratory of Clinical Immunology and Microbiology (NIH), United StatesCopyright © 2022 Ouahed. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jodie Deborah Ouahed, am9kaWUub3VhaGVkQGNoaWxkcmVucy5oYXJ2YXJkLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.