
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 15 December 2022
Sec. Vaccines and Molecular Therapeutics
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1026052
This article is part of the Research Topic Women in Vaccines and Molecular Therapeutics View all 7 articles
 Ishita N. Waghela1,2Katherine L. Mallory1,2Justin A. Taylor1,3
Ishita N. Waghela1,2Katherine L. Mallory1,2Justin A. Taylor1,3 Cosette G. Schneider1,4Tatyana Savransky5,6
Cosette G. Schneider1,4Tatyana Savransky5,6 Chris J. Janse7Paulo J. C. Lin8
Chris J. Janse7Paulo J. C. Lin8 Ying K. Tam8Drew Weissman9
Ying K. Tam8Drew Weissman9 Evelina Angov1*
Evelina Angov1*The secreted malarial protein, Cell-Traversal protein for Ookinetes and Sporozoites (CelTOS), is highly conserved among Plasmodium species, and plays a role in the invasion of mosquito midgut cells and hepatocytes in the vertebrate host. CelTOS was identified as a potential protective antigen based on a proteomic analysis, which showed that CelTOS stimulated significant effector T cells producing IFN-γ in peripheral blood mononuclear cells (PBMCs) from radiation attenuated sporozoite-immunized, malaria-naïve human subjects. In a rodent malaria model, recombinant full-length CelTOS protein/adjuvant combinations induced sterile protection, and in several studies, functional antibodies were produced that had hepatocyte invasion inhibition and transmission-blocking activities. Despite some encouraging results, vaccine approaches using CelTOS will require improvement before it can be considered as an effective vaccine candidate. Here, we report on the use of mRNA vaccine technology to induce humoral and cell-mediated immune responses using this antigen. Several pfceltos encoding mRNA transcripts were assessed for the impact on protein translation levels in vitro. Protein coding sequences included those to evaluate the effects of signal sequence, N-glycosylation on translation, and of nucleoside substitutions. Using in vitro transfection experiments as a pre-screen, we assessed the quality of the expressed CelTOS target relative to the homogeneity, cellular localization, and durability of expression levels. Optimized mRNA transcripts, which demonstrated highest protein expression levels in vitro were selected for encapsulation in lipid nanoparticles (LNP) and used to immunize mice to assess for both humoral and cellular cytokine responses. Our findings indicate that mRNA transcripts encoding pfceltos while potent for inducing antigen-specific cellular cytokine responses in mice, were less able to mount PfCelTOS-specific antibody responses using a two-dose regimen. An additional booster dose was needed to overcome low seroconversion rates in mice. With respect to antibody fine specificities, N-glycosylation site mutated immunogens yielded lower immune responses, particularly to the N-terminus of the molecule. While it remains unclear the impact on CelTOS antigen as immunogen, this study highlights the need to optimize antigen design for vaccine development.
Malaria caused by Plasmodium parasites remains a major life-threatening disease, resulting in a high burden on public health and economic disruption in afflicted regions. The World Health Organization estimated that 241 million malaria cases occurred in 2020 alone, worldwide (1). A striking increase of 14 million more cases in 2020 compared to 2019 are likely linked to disruptions in efforts to prevent and diagnosis of malaria during the COVID-19 pandemic. Importantly, in 2020, mortality caused by Plasmodium falciparum, compared to P. vivax and to a lesser extent P. malariae, accounted for 96% of reported cases in the WHO African region, a disproportionately high share of the global malaria burden (1). Despite progress in the prevention and control of malaria, emergence of drug and pesticide resistance still poses the greatest challenge to malaria control efforts.
The life cycle of Plasmodium parasites is complex with significant antigenic diversity in the mammalian host. This complexity makes it difficult to develop a vaccine against parasite infection, since the immune response targeting one stage may not be effective against a later stage in the life cycle (2). The mode of transmission of malaria occurs during a blood meal, when an infected female Anopheles mosquito inoculates sporozoites into the skin of the human host. Once sporozoites enter the host, they traverse through cells and ultimately invade hepatocytes, where they undergo maturation and replication followed by cell rupture and release of merozoites into the bloodstream. There, merozoites rapidly invade erythrocytes, transforming from rings, trophozoites to schizonts and on red blood cell rupture releasing merozoites back into the bloodstream thus initiating the cyclic phase of erythrocytic infection (3, 4). An effective pre-erythrocytic vaccine would avert transmission to blood stages and progression to clinical disease (5, 6). Vaccine candidates targeting the pre-erythrocytic stage have demonstrated encouraging vaccine efficacy (7–9). The most advanced of these is based on the P. falciparum circumsporozoite protein (PfCSP), RTS,S/AS01, albeit exhibiting partial vaccine efficacy in phase III clinical trials (10). Despite the limited efficacy, in 2021, the WHO recommended RTS,S/AS01 for prevention of P. falciparum malaria in children residing in regions of moderate to high malaria transmission (1). While the protection induced by this vaccine is encouraging, the road to a durable and highly effective vaccine against malaria remains to be determined.
The Cell-Traversal protein for Ookinetes and Sporozoites (CelTOS) is a micronemal, secreted protein, which plays a role in cell traversal of ookinetes in the mosquito midgut and sporozoites in the human host (11, 12). Jimah et al., demonstrated that CelTOS has enhanced specificity for the cytosolic face of host cell membranes by directly binding to phosphatidic acid, a lipid found in the inner leaflet of plasma membranes (11). In clinical studies of radiation-attenuated sporozoite (RAS) vaccine (13) or from natural exposure (14), CelTOS-specific peptides stimulated peripheral blood mononuclear cells (PBMC’s) to recall significant levels of antigen-specific CD8+ IFN-γ responses. In preclinical studies, adjuvanted, E. coli expressed PfCelTOS induced cellular and humoral immune responses in mice and conferred sterile protection against a heterologous rodent malaria challenge (14–16). While replication-deficient pfceltos encoding, recombinant chimpanzee adenoviral vector 63 and modified vaccinia virus Ankara (ChAd63-MVA) delivered as a heterologous prime-boost regimen, induced higher frequency of antigen-specific cellular responses than was previously reported with a recombinant PfCelTOS/Montanide ISA 720 (16, 17). While a recombinant PfCelTOS produced in Pseudomonas fluorescens induced antibodies that inhibited P. falciparum 3D7 parasites infection of hepatocytes and impaired parasites development within mosquito cells in vitro (18). Immunization with plasmid DNA encoding pfceltos, which was optimized for mRNA stability and human codon usage, yielded modest humoral responses but significant levels of cellular immunity, in both mice and non-human primates (19).
Similar to DNA vaccines, messenger RNA (mRNA) enables the encoded antigen to be directly expressed within cells, however, without altering the host cell genome or requiring access to the nucleus (20, 21). Lipid nanoparticles (LNP) serve to protect mRNA against degradation, efficiently delivering the cargo into cells, having the advantage of providing immunostimulatory signals (22). Nominally, compared to traditional approaches, an advantage of mRNA vaccine technology is the facile design of constructs and the simplicity of manufacture. This phenomenon was particularly evident in the responsiveness to the coronavirus disease 2019 (COVID-19) pandemic, where the mRNA platform was rapidly fielded against SARS-CoV-2 virus clinical endpoints (23). Hence, we sought to investigate factors which may influence translation and translocation of expressed protein, such as signal sequence, an essential feature of mRNA transcripts for protein functioning (24), N-glycosylation of pfceltos mRNA from transfected Chinese Hamster Ovary cells (CHO) and on in vivo immune responses in mice, and finally non-modified (native) and nucleoside-modified (base substitutions of pseudouridine and 5’-methylcytidine) mRNA transcripts on immunogenicity in mice (25, 26).
mRNA transcripts were based on the pfceltos coding sequence of the P. falciparum 3D7 strain parasites and transcripts were synthesized by TriLink Biotechnologies including proprietary 5’ and 3’ UTRs, poly-A tail length. To assess for effect of signal sequence on translation and translocation, pfceltos mRNA transcripts included the P. falciparum CelTOS wild type (Wt-SS) signal sequence, or the mouse Ig light chain signal sequence (IgLC-SS), or the human IgE signal sequence (IgE-SS). A pfceltos transcript lacking a signal sequence (No-SS) was included for comparison. P. falciparum CelTOS contains three putative N-glycosylation motifs (http://www.cbs.dtu.dk/services/NetNGlyc /; NetNGlyc, Glycosylation site prediction tool) (27). The three predicted N-glycosylation sites (NxS/T motif) located in the N-terminus of the protein were mutated to code for glutamine (N>Q) at the N position and designated glycosylation site modified (GM). Table 1 summarizes the TriLink pfceltos encoded mRNA transcripts used to assess for signal sequence and the N-glycosylation status of each transcript. These sequences were codon harmonized for optimal expression in mice (28). In addition, in some experiments, uridine and cytosine residues were modified by ψ-pseudouridine and 5-methylcytosine replacements, respectively, detailed in Figure Legends. Two additional pfceltos mRNAs were obtained and assessed for immunogenicity and efficacy in the mouse model (UPenn). These were in vitro transcribed (IVT) from different plasmid templates (Cap1-TEV), were codon optimized, and translated identical coding regions that were either N-link glycosylated or nonglycosylated, and were nucleoside modified by uridine triphosphate replacement with one-methylpseudouridine (m1Ψ)-5′-triphosphate, and cellulose affinity purified (29–31). The UPenn mRNAs were transcribed to contain 101 nucleotide-long poly (A) tails. Capping was performed co-transcriptionally using the trinucleotide Cap1 analog, CleanCap (TriLink). All IVT mRNAs were analyzed by agarose gel electrophoresis and were stored frozen at either -20° C (UPenn) or -80°C (TriLink).

Table 1 pfceltos mRNAs translation products.
Lipid Nanoparticles (LNP) used in this study were similar to those previously described (30, 32, 33) and contained ionizable lipids and have compositions that are proprietary to Acuitas Therapeutics (pKa in range of 6.0-6.5/DSPC/Cholesterol/PEG-Lipid). The proprietary lipids and LNP composition are described in US patent applications WO 2017/075531 and WO 2017/0041443. All LNP used for the current studies were characterized post-production for their size and polydispersity (PDI) using a Malvern Zetasizer (Zetasizer, Nano DS, Malvern, UK) and for measuring the encapsulation efficiency using ribogreen assay (RG). Characterization results were measured and calculated using Malvern Panalytical Software (Malvern, UK) and are listed here for LNP1 – size range: 68-75nm, PDI: <0.054, Ribogreen: 95-97%; and for LNP3 – size range: 70 nm, PDI: <0.095, Ribogreen: 86%. All mRNA-LNPs were stored at -80°C.
Chinese hamster ovary (CHO) E77.4 cells (34) purchased from ATCC were cultured in media supplemented with RPMI 1640 (Quality Biological 112-025-101), 10% heat inactivated fetal bovine serum (FBS) (Gibco 10082147), 2mM L-Glutamine (Quality Biological 118-084-721) and 100 U/mL Penicillin with 100 µg/mL Streptomycin (Quality Biological 120-095-721). Cells were passaged using 0.05% Trypsin-0.1% EDTA (Quality Biological 118-087-721). CHO cells at passages 9-12 were used for all experiments.
Female, inbred BALB/cJ mice aged five-six weeks were used in all animal studies (The Jackson Laboratories, Sacramento, CA). Mice were immunized intramuscularly two or three times at three-week intervals. The intramuscular injection site was at the posterior thigh muscle with an immunization volume of 50µL. For evaluation of humoral responses, blood samples were collected by lateral tail vein bleeds the day before each immunization and two weeks following the final immunization, at which time terminal cardiocentesis was performed and splenocytes were harvested to measure cellular cytokine responses.
To determine protection, BALB/cJ mice (n = 10 for each group) were challenged two weeks following the final immunization by mosquito bite challenge. Mice were anesthetized using a cocktail of Ketamine, Xylazine (100 µg/mL) and Phosphate buffered saline (PBS; pH 7.4) based on weight and drug response. Four female An. stephensi mosquitoes were placed in a plastic cup container with a mesh covering per challenge. Sedated mice were placed on the mesh with their bellies facing down. Mosquitoes were allowed to take a blood meal during a timed 8-minute period. Evidence of blood in the abdomen was rated as successful injection. The sporozoite ratings of infected mosquitoes for this experiment was 2.4 on a scale of 1-4, in 9-out-of-10 infected mosquitoes. The challenge study used Plasmodium berghei ANKA transgenic strain expressing NF54/3D7 strain PfCelTOS under the Pbuis4 promoter control (Gene model: PF3D7_1216600) (Leiden University Medical Center, Leiden, The Netherlands) (17). Parasitemia detection was assessed as previously described (30). Protection in mice was defined as the lack of blood stage parasites up to 14 days post challenge. Briefly, parasitemia was observed using microscopy with thin blood smears fixed with methanol and stained with 10% Giemsa stain for 15 minutes at room temperature (Sigma Aldrich GS500). Mice were monitored starting on day 7 following the challenge through day 14, the final day in the study. Any mouse that was not parasitemic by day 14 was considered sterile protected.
To determine in vitro protein expression, transient mRNA transfections were performed using the TransIT-mRNA kit (Mirus Bio MIR 2225) according to the manufacturer’s protocol. Briefly, CHO cells were plated at 300,000 cells/mL in a 24-well tissue culture plate and incubated at 37°C with 5% carbon dioxide for 24 hours. Following the incubation, when cells achieved ~80% confluence, cells were transfected with 0.5 µg/well mRNA and returned to 37°C in a 5% carbon dioxide incubator. Transfected cell culture supernatants and pellets were harvested at 8-, 24-, and 48-hour time-points. A negative control with TransIT reagents without mRNA (no mRNA) was included for each time point to provide background detection levels. For gel loading, at the time of cell harvest, cells were counted and normalized for equal loading. Translated proteins from the culture supernatant and cell lysates (cell pellet) were analyzed using Tris-Glycine SDS-PAGE (Novex XP04205BOX) with SeeBlue Pre-stained protein standard marker and transferred onto nitrocellulose membranes for protein detection by Western blot.
Following transfer, membranes were blocked with 5% non-fat dried milk (w/v) in phosphate buffered saline, pH 7.4 with 0.1% Tween-20 (PBS-T) (w/v) for 1 hour at room temperature with gentle rocking. Membranes were washed three times with PBS-T for 5 minutes at room temperature with gentle rocking between each step. The primary antibody; SVP-09-011, a rabbit polyclonal anti-PfCelTOS antibody (generated at Spring Valley Laboratories, Sykesville MD) was diluted in PBS-T at a ratio of 1:10,000 (antibody:diluent). The primary antibody was incubated for 1 hour at room temperature (RT) with gentle rocking. Following the wash, the membrane was probed with alkaline phosphatase-conjugated anti-rabbit IgG (Southern Biotech 4030-04) diluted in PBS at a ratio of 1:10,000 (antibody:diluent). Following the wash, membranes were developed for 5 minutes using 5-bromo-4-chloro-3-indolyl-phosphate (BCIP) with nitro blue tetrazolium (NBT) (Sigma Aldrich Lot# 16853420, 14799531, respectively) in alkaline phosphatase buffer. Reactions were stopped with deionized water and membranes were air dried.
Densitometric detection of proteins in Western blots were quantified with ImageJ software (35). The 10ng recombinant PfCelTOS (rPfCelTOS) protein served as a relative control for quantification. Bands in the experimental lanes were individually selected and circumscribed with the rectangular selection option, followed by quantification of peak areas of the obtained histograms. The data acquired were reported as arbitrary area units.
To determine antibodies specific to PfCelTOS, mice were bled via lateral tail veins and blood samples were collected prior to each immunization and the day of challenge. ELISA was performed as previously described (16, 36). Briefly, 96-well 2HB Immunolon plates (Thermo Scientific, Waltham, MA) were coated with 100µL/well of 25ng/well of rPfCelTOS in 1X PBS, pH 7.4 (Quality Biological, Gaithersburg, MD) and incubated overnight at 4°C in a humidified chamber. Plates were washed with 1X PBS/0.1% Tween 20 and then blocked with 1X PBS/1% BSA at 22°C for 1 hour. Diluted sera samples were incubated for 2 hours at 37°C. Plates were washed and then incubated with alkaline phosphatase-conjugated goat anti-mouse IgG (Southern Biotechnology, Birmingham, AL) diluted in PBS at 1:1000 (antibody:diluent) for 1 hour at room temperature (RT). Anti-PfCelTOS IgG was detected using BluePhos substrate (Kirkegaard Perry, MD) for 15 minutes at RT. The reaction was arrested with stop solution and read at 630nm using M2 spectrophotometer (Molecular Devices, Downington, PA). The antibody concentration was determined against a purified mouse standard IgG curve (run in parallel with each assay) (Invitrogen, Waltham, MA). For each serum tested, we determined a concentration that was within the linear portion of the reaction curve and used this dilution to extrapolate the actual antibody concentration in the assay wells (16).
Fine specificity of antibody responses was assessed by using rPfCelTOS, recombinant protein fragments PfCelTOS N-term and PfCelTOS C-term and PfCelTOS Peptides: Peptide 1-2, Peptide 2-3, and Peptide 4 (Supplementary Table 1). Briefly, microtiter plates, 4HBX (ThermoFisher Scientific, Waltham, MA), were coated with 100µL/well of 25ng/well of rPfCelTOS, each protein fragment, and Peptides 1-2, 2-3, and 4, and incubated overnight at 4°C in a humidified chamber. Plates were washed with 1X PBS/0.1% Tween 20 and then blocked with 1X PBS/1% BSA at 22°C for 1 hour. Sera were diluted serially two-fold and incubated at 22°C for 2 hours. Plates were washed and then incubated with goat anti-mouse IgG (H+L) conjugated with horseradish peroxidase and diluted 1:4000 (KPL Gaithersburg, MD, USA) for 1 hour at 22°C. Antibody titers were detected using ABTS substrate (KPL Gaithersburg, MD, USA), measured at OD405 after 1 hour with an M2 spectrophotometer (Molecular Devices, Downington, PA). Antibody titers were reported as the serum dilution required to achieve an optical density equal to 1.0.
To determine cellular responses against PfCelTOS, mouse IFN-γ and IL-4 ELISpot assay (R&D systems SEL485 SEL404, respectively) were performed according to the manufacturer’s instructions. Briefly, spleens were harvested following terminal cardiocentesis and processed under sterile conditions. Hydrophobic multiscreen plates (Millipore) were coated with IFN-γ and IL-4 capture antibodies in sterile 1X PBS and incubated overnight at 4°C in humidified chamber. The plates were washed with D-MEM (Quality Biological 112-013-101) followed by blocking with complete media. Wells were plated with splenocytes at 200,000 cells/well (16). The cells were stimulated with recombinant PfCelTOS (10 μg/mL) for 42 hours at 37°C with 5% carbon dioxide incubator. Following wash, IFN-γ and IL-4 detection antibodies in sterile PBS/1%BSA were added to the plates and incubated overnight at 4°C in a humidified chamber. Plates were then incubated for 2 hours at RT with streptavidin AP conjugate followed by development with BCIP/NBT substrate (R&D systems P201257). Spot counting was performed with appropriate settings using an ELISpot reader (Autoimmune Diagnostika, Strassberg, Germany).
Cytokine detection was determined with MSD assay and as previously described (16, 36). Briefly, following splenocytes harvest from mice, splenocytes were plated at 400,000 cells/well in 96-well flat bottom plates (Costar 3595). The stimulating antigen was either 10µg/mL rPfCelTOS 3D7 protein or 15mer overlapping CelTOS peptide pools (1 µg/mL). Cells were stimulated for 48 hours by incubation at 37°C with 5% carbon dioxide. Cell culture supernatant was harvested, and pro-inflammatory cytokines measured using the V-PLEX Proinflammatory Panel 1 Mouse Kit (Meso Scale Discovery, K15048G-2) according to the manufacturer’s instructions. Cytokine levels detected with cells incubated in culture media alone served to normalize for background cytokine secretion.
Statistical analysis of mouse serological and cellular immune responses where p<0.05 is considered significant, were evaluated using parametric two-tailed, unpaired T-tests and Mann-Whitney tests (GraphPad Prism, v 8.4.1, San Diego, CA).
Signal sequences play a crucial role in cellular localization of target proteins and are essential for their proper function (24). In eukaryotes, signal sequences or signal peptides, direct secretory and membrane proteins to the Sec61 translocon in the endoplasmic reticulum (ER) membrane. To determine an optimal signal sequence for in vitro expression in CHO cells and targeting to the cellular secretion pathway, we explored pfceltos translation and localization using the homologous wild-type parasitic pfceltos signal sequence (Wt-SS), and two heterologous signal sequences, one derived from human IgE (IgE-SS) and second derived from mouse Ig light chain (IgLC-SS) (Table 1). While these signal sequences have no homology in their primary structures, several common features are inherent, such as the positively charged N-terminal end, a hydrophobic core and a polar C-terminal end that serves as the recognition sequence for processing by signal peptidases (37). A pfceltos mRNA transcript lacking a signal sequence (No-SS) was also assessed for protein expression and retention in the cell lysate of transfected cells by Western blot (Figure 1A). Other general features of pfceltos mRNA transcripts used in this experiment were inclusion of Cap1 analog modification at the 5’-end and codon harmonization of coding regions using WRAIR’s proprietary codon harmonization (CH) algorithm for optimal expression in mice (28).
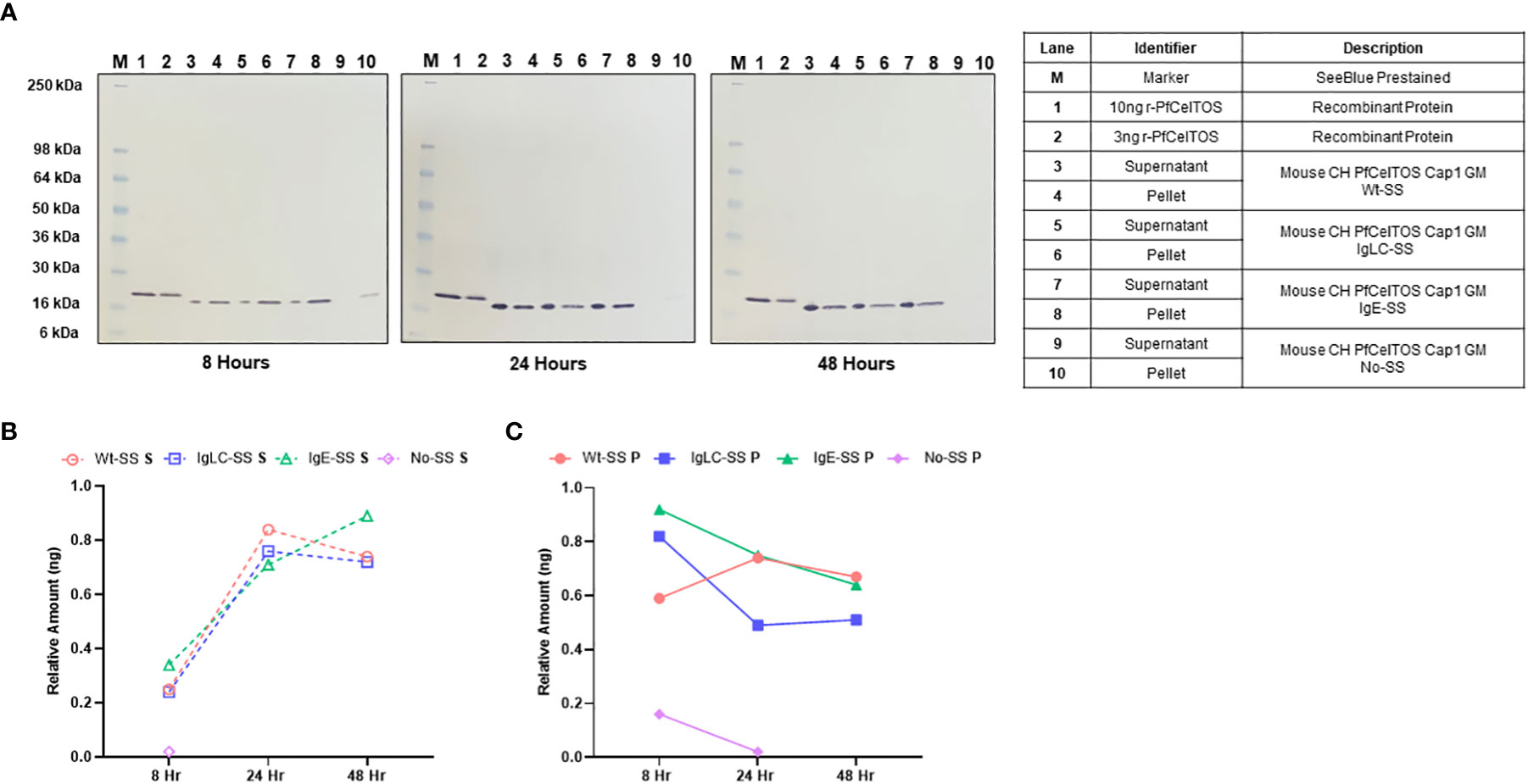
Figure 1 In vitro translation of pfceltos mRNAs with different signal sequences. (A) CHO cells were transfected with pfceltos mRNAs, having identical non-coding and coding sequences except for the signal sequences, and harvested at 8-, 24-, and 48-hours post-transfection. Western blot analysis was performed to assess for in vitro translation and translocation of PfCelTOS. The right panel outlines each lane and provides a detailed description of the test samples. Semi-quantitative analysis of the PfCelTOS protein levels at the different time points was performed by using ImageJ for the (B) supernatant and (C) cell lysate relative to the 10ng recombinant PfCelTOS protein (Lane 1, for each panel).
To assess for protein translation in vitro, CHO cell culture supernatant and cell lysate fractions were harvested at 8-, 24-, and 48-hour time-points post transient transfection. High levels of translated protein were detected from pfceltos mRNAs incorporating the Wt-SS, IgLC-SS and IgE-SS signal sequences, with expression detected both in the cell culture supernatant and the cell lysate until 48 hours post transfection by Western blot (Figure 1A). In contrast, pfceltos mRNA without a signal sequence (No-SS) had significantly reduced protein levels at 8 hours with partitioning exclusively to the cell lysate, and a barely visible band detected at 24 hours (Figure 1A, 24 hours, Ln 10). A functional signal sequence on secreted proteins has a role in protecting mRNA in the cytosol from degradation and the lack of such here, may account for the reduced protein translation levels for the pfceltos No-SS mRNA transcript. To estimate protein expression profiles of pfceltos mRNA’s incorporating different signal sequences, relative density plots based on the 10ng rPfCelTOS reference standard, were analyzed using ImageJ, a Java-based image processing program, on Western blots (35). Differences in protein migration observed between the E. coli expressed reference protein and the in vitro translated protein is approximately 2KDa and can be accounted for by the inclusion of a 6-His+linker sequence on the rPfCelTOS. The semi-quantitative analysis reinforced that pfceltos mRNAs encoding with each, Wt-SS, mouse IgLC-SS and human IgE-SS transcripts, were translated at comparable protein kinetics (Figures 1B, C). Maximal protein expression was observed between 24 and 48 hours, providing evidence of peak translation. Additional time points beyond 48 hours are needed to define the limits of in vitro translation in transfected CHO cells. Density plots indicated that at earlier time points, 8 hours, PfCelTOS protein was predominantly found in cell lysates (Figure 1C), while between 24-48 hours, the PfCelTOS protein was predominantly found in the culture supernatant fractions (Figures 1B, C). Since no significant differences in protein levels or translocation kinetics were detected for the mRNA encoding WT-SS, IgLC-SS and IgE-SS, then the pfceltos encoding mRNAs with Wt-SS and human IgE-SS were selected for subsequent experiments to assess for in vivo responses in mice.
Post-translational modifications by N-glycosylation of proteins are essential for cells to diversify their function, enabling recognition by endocytic receptors of antigen-presenting cells and preventing proteolysis in the endolysosome by steric obstruction (38, 39). Common across all eukaryotes, N-linked glycosylation imparts a wide range of properties to proteins including their localization, cellular growth, and host immune regulation (40). The relevance of N-glycans, in Plasmodium has been much in debate for several decades. A recent study of the Plasmodium genome demonstrated that Plasmodium expresses the essential oligosaccharyl transferase complex needed for N-glycan modification of asparagines on proteins (41). Notwithstanding these findings, little is known on the extent and biological function of N-glycosylation in Plasmodium and thus the impact on vaccine induced immune responses of Plasmodium proteins. A scan of PfCelTOS amino acid sequence revealed three putative asparagine N-linked glycosylation sites all located at the N-proximal end (Table 1). In practice, post-translation modifications (PTM) of protein N-linked glycosylation can be assessed by SDS-PAGE, the extent of which is seen as shifts in protein molecular weight and retardation of migration. PTM by N-link glycosylation can impact protein quality attributes including protein folding, stability, and solubility. We evaluated the influence of N-linked glycosylation on CelTOS in vitro translation and translocation using ARCA-capped pfceltos mRNA transcripts with and without N-glycosylation recognition sites (pfceltos ARCA GM and pfceltos ARCA, respectively) and compared these to a similar coding Cap1 pfceltos mRNA transcript with native N-glycosylation sites as a reference. Analysis of CHO cell supernatants and cell lysates, harvested at 24 hours post-transfection, showed a qualitative difference between the two transcripts, i.e. pfceltos ARCA and pfceltos ARCA GM (Figure 2A; Lanes 5, 6, and 9, 10), respectively, with the latter having a single homogenous band that was distributed between the cell supernatant and the cell lysate at 24 hours, similar to what was observed in Figure 1A. Both mRNA’s coding for native sequence N-glycosylation sites, whether having ARCA or Cap1 analogs at the 5’-end (Figure 2A; Lanes 5, 6 and 7, 8, respectively), exhibited a more complex, heterogeneous protein banding from ~25-36kDa. We speculate that the increased molecular weight of translated proteins are isoforms having varying extent of glycosylation. More in depth analysis is required to directly characterize these glycans. No protein bands are detected in the negative control lanes, 3 and 4, which are CHO cells transfected with Mirus reagent but without mRNAs. While glycans added to secreted proteins play an essential role in their function; including immunogenicity, potency and stability, little is known of the effects of introduction of ‘non-native’ N-glycans on vaccine efficacy, thus addressing this knowledge gap can aid in vaccine design strategies (38).
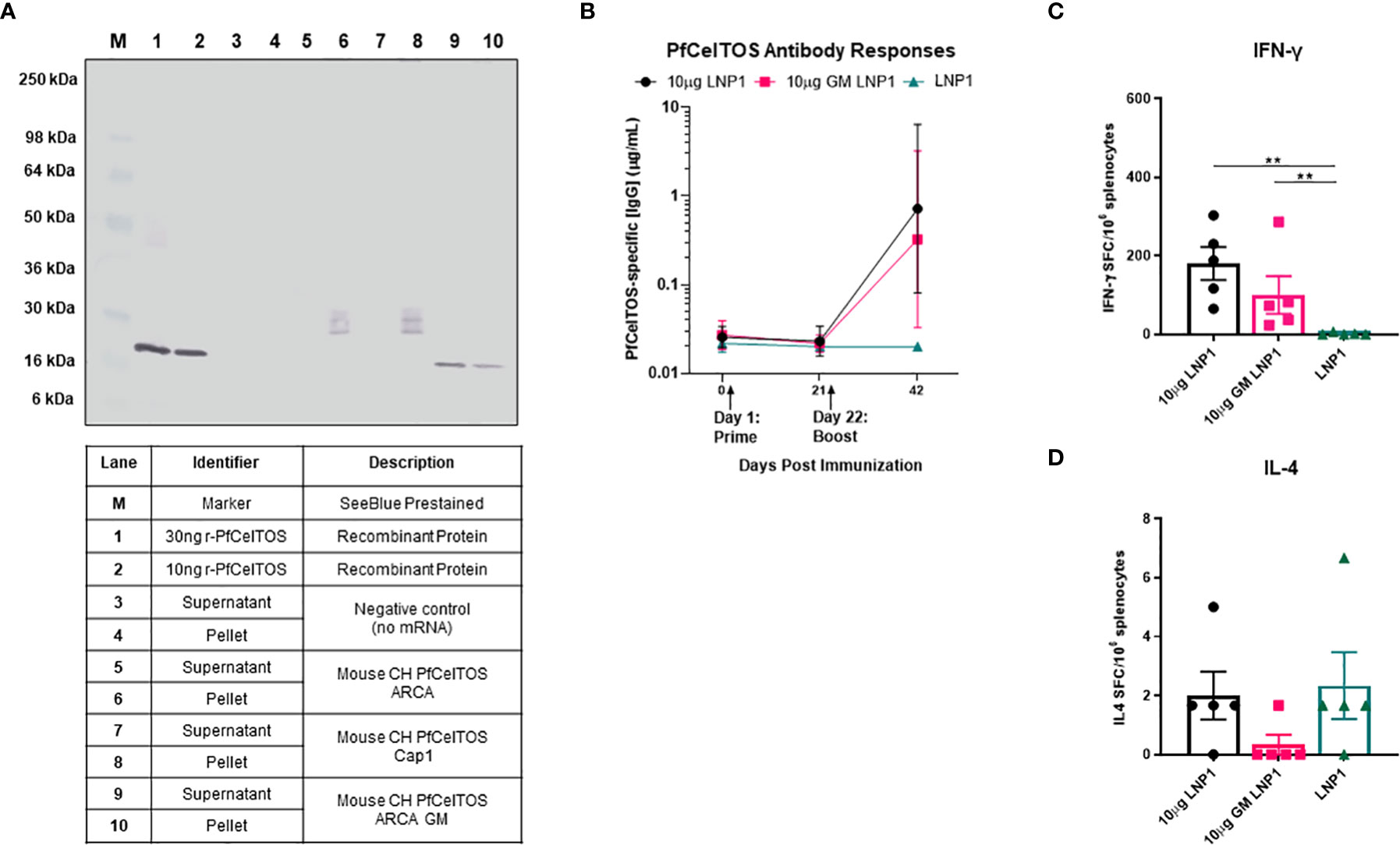
Figure 2 Effect of N-glycosylation on in vitro and in vivo responses. (A) Western blot analysis of CHO cells transfected with mRNAs pfceltos ARCA, pfceltos Cap1 and pfceltos ARCA GM. pfceltos mRNA transcripts were codon harmonized (CH) for optimal expression in mice and encoded with the native falciparum celtos signal sequence (Wt-SS). Cell culture supernatants and cell lysates were harvested at 24 hours post-transfection to assess for the effect of N-glycosylation on protein translation. (B) BALB/cJ mice were immunized intramuscularly (IM) two times at a three-week interval with 10µg of pfceltos mRNA encapsulated into LNP1 (10µg LNP1), 10µg pfceltos mRNA with glycosylation site modified (GM) in LNP1 (10µg GM LNP1) or LNP1 alone (n=5 per group). PfCelTOS-specific IgG antibody concentrations (µg/mL) were quantified in sera at pre-immunization, three weeks after the primary dose and two weeks after the final dose by ELISA. Antibody concentrations are reported as the geometric mean and 95% confidence intervals. (C, D) Splenocytes were harvested and IFN-γ and IL-4 cytokines were detected by ELISpot assay. The mean number of spot-forming cells (SFC) per splenocytes were reported with standard errors of the mean (SEM). Statistical analysis was performed using Mann-Whitney test (**p<0.01).
To evaluate the impact of N-glycosylation on in vivo immune responses in mice, we evaluated pfceltos mRNA transcripts without (pfceltos LNP1) and with N-glycosylation site modifications (pfceltos GM LNP1), that were 5’-capped with ARCA analog, included the WT-SS and were codon harmonized for expression in mice. mRNAs were packaged in biodegradable ionizable lipid nanoparticles (LNPs) consisting of phospholipids, cholesterol and polyethylene glycol (PEG) containing lipids for in vivo delivery (42). Female BALB/cJ mice were immunized intramuscularly (IM) with 10µg of each mRNA, encapsulated in LNP1, twice, at three-week intervals. Mouse sera were collected on day -1, day 21, and day 42 and PfCelTOS-specific antibody responses were measured by enzyme-linked immunosorbent assay (ELISA) in which, the coating antigen was the rPfCelTOS produced in Escherichia coli and, therefore, not N-glycosylated, and lacked the native signal sequence. Whether the N-glycosylation sites on pfceltos mRNA transcripts were modified or not, had little effect on the antigen-specific antibody responses (Figure 2B). Using a two-dose regimen, neither the mice vaccinated with pfceltos mRNA-LNP1 nor those vaccinated with the pfceltos GM mRNA-LNP1, induced significant PfCelTOS-specific antibody concentrations. ELISpot assays, on the other hand, demonstrated significant numbers of splenocytes producing antigen-specific IFN-γ, while not statistically different between the two pfceltos mRNA groups (Figure 2C). In contrast, no significant IL-4 production was detected above background (Figure 2D), suggesting that the responses were biased toward a T helper 1 (Th1) response using this regimen. To assess in more depth cytokine responses, Meso Scale Discovery (MSD) was performed. Responses were recalled using the rPfCelTOS protein, as with ELISpot. The results showed that the concentrations of CelTOS-specific IFN-γ, IL-2, TNF-α, IL-10, and IL-6 for pfceltos mRNA-LNP1 and pfceltos GM mRNA-LNP1 were not significantly different (Supplementary Figure 1). Like the ELISpot responses, MSD revealed a biased response toward Th1 cytokines, that was not restricted by potential non-natural N-glycans on the translated protein.
To date, vaccines comprised of nucleoside-modified mRNA-lipid nanoparticle (LNP) have shown promise for inducing potent neutralizing antibodies against an array of global and emerging infectious disease threats (43, 44). We investigated pfceltos mRNA dose and nucleoside substitutions on immunogenicity in mice. pfceltos mRNA transcripts comprised of identical pfceltos coding sequences mutated at the three predicted N-glycosylation sites (N>Q) (glycosylation site modified, GM), encoded for human IgE signal sequence (IgE-SS), were codon harmonized for optimal expression in mice, were 5’-CleanCap1 (Cap1), and were encapsulated in one of two LNPs (LNP1 or LNP3). Nucleoside modifications of 5-methylcytidine (m5C) and pseudouridine (ψ) (PU5MC), for cytidine and uridine, respectively, were introduced to dampen innate immune responses while purportedly increasing translation efficiency (45). As above, female, BALB/cJ mice were immunized by the IM route, twice at three-week intervals with either 10µg or 30µg of pfceltos encoding mRNAs; GM-IgE-SS or GM-PU5MC-IgE-SS. PfCelTOS-specific antibody concentrations were measured by ELISA and splenocytes were harvested to measure cytokine profiles by either ELISpot or MSD. The findings revealed the following: first, antibody concentrations were relatively low, and increasing the mRNA dose from 10 to 30µg, was not a factor for improving antibody responses (Figure 3A). Humoral responses across groups were by-and-large “all-or-none”, as was also seen by Huysmans et al. (46). Although several mice had higher concentrations of antibodies, particularly in groups where mRNA was encapsulated in LNP1, overall, the antibody responses were low. Second, cytokine responses detected by ELISpot suggested that lower doses of mRNA were superior for both PfCelTOS-specific IFN-γ and IL-4 responses, particularly when encapsulated in LNP3 (Figures 3B, C). With regards to nucleoside modification, a similar trend was observed that lower doses, i.e., 10 µg pfceltos GM PU5MC IgE-SS mRNA-LNP3 yielded significant numbers of antigen specific IFN-γ secreting splenocytes. MSD revealed a similar bias toward Th1 and pro-inflammatory cytokines (IFN-γ, IL-4, TNF-α, IL-12p70, IL-2, IL-1β, IL-10, IL-6, KC/GRO) (Supplementary Figure 2). IFN-γ responses affirmed those of ELISpot that 10µg of pfceltos GM PU5MC IgE-SS mRNA-LNP3 induced significant pro-inflammatory cytokines compared to the same mRNA encapsulated in LNP1. Notably, the presence of IFN-γ and IL-10 cytokines are implicated in immune response regulation. With respect to Th2 cytokines, 10µg of pfceltos GM PU5MC IgE-SS mRNA-LNP3 induced significantly higher levels of IL-4 compared to the mRNA encapsulated in LNP1 which was also observed for mRNAs in Figure 2D using an mRNA-LNP1 coding for ARCA capped pfceltos with P. falciparum signal sequence, suggesting that at least for IL-4 encapsulation with LNP3 was superior for inducing significant numbers of IL-4 secreting splenocytes. Characterization of LNPs by dynamic light scattering (DLS) suggest similar particle sizes, while there was greater efficiency of encapsulation seen with LNP1 compared to LNP3. These findings emphasize the need for optimizing composition and encapsulation of LNPs.
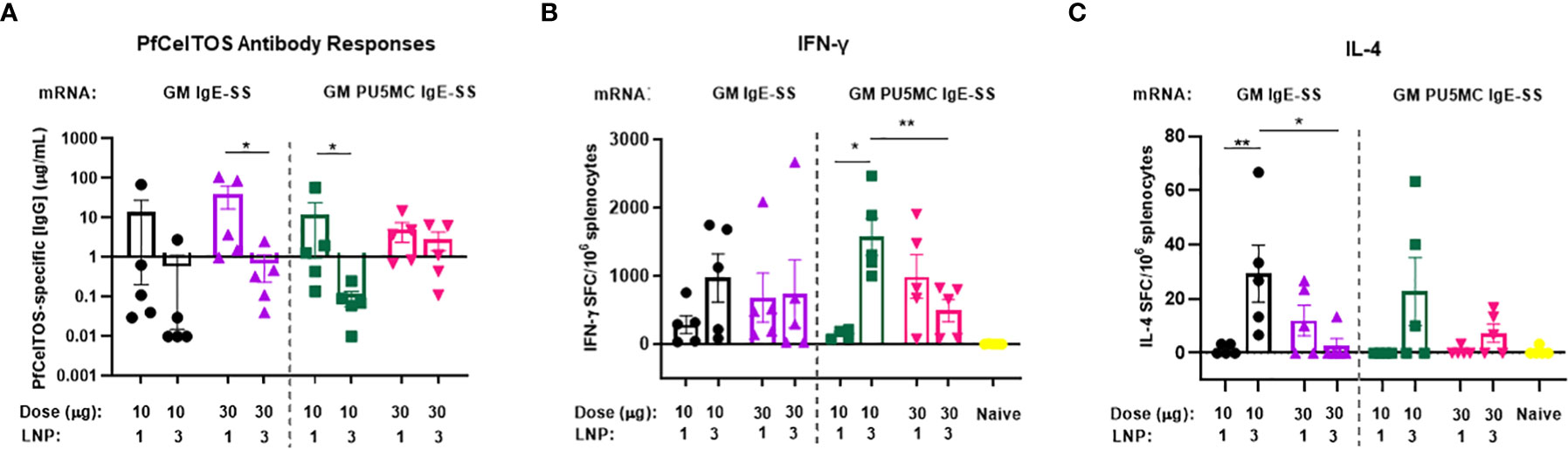
Figure 3 Effect of mRNA dose and nucleoside modification on immune responses. BALB/cJ mice were immunized intramuscularly (IM), two times at three-week intervals, with a low dose (10µg) or a high dose (30µg) of pfceltos mRNA that was N-glycosylation site modified (GM), containing human IgE signal sequence (IgE-SS) (GM IgE-SS), and with nucleoside ψ-pseudouridine and 5’-methylcytosine substitutions (PU5MC), (GM PU5MC IgE-SS), in LNP (LNP1 or LNP3), (n=5 per group). (A) Antigen-specific IgG antibody concentrations against PfCelTOS were quantified in sera two weeks after the final dose by ELISA. Antibody concentrations are represented as the mean and standard deviation (SD). Statistical analysis was performed using an unpaired t-test (*p<0.05). (B, C) IFN-γ and IL-4 cytokines were detected by ELISpot. The mean number of spot-forming cells (SFC) per splenocytes were reported with standard errors of the mean (SEM). Statistical analysis was performed using Mann-Whitney test, (*p<0.05, **p<0.01).
The magnitude of antibody responses depends on several factors including the nature of the antigen, administration route and the dosing schedule (30, 47). An experiment was conducted to examine if a second boost of pfceltos encoding mRNA-LNP improved immunogenicity to CelTOS. Female BALB/cJ mice were vaccinated by IM route, thrice, at three-week intervals with either 10µg pfceltos TriLink GM encapsulated in LNP1 or the same mRNA, 10µg pfceltos TriLink GM, encapsulated in LNP3. These mRNAs had glycosylation site modified (GM), human IgE signal sequence, codon harmonized for expression in mice and incorporated PU5MC nucleoside modifications. To address the role of innate immunity on adaptive immune responses, and to dampen innate immune sensor activation and improve on translational efficiency, these pfceltos mRNAs underwent and additional HPLC purification step (48). Contrary to the preceding experiments, two booster doses of pfceltos mRNA-LNP, either TriLink GM LNP1 or TriLink GM LNP3, yielded overall higher PfCelTOS-specific antibody concentrations (Figure 4A). This suggests that a second booster dose may be necessary for pfceltos mRNAs to overcome the “all-or-none” pattern of humoral immune responses, also corroborating similar findings observed for malaria antigen, pfcsp encoding mRNA-LNP1 (30). In addition to the mRNA transcripts (TriLink), we tested two additional pfceltos encoding mRNAs that were essentially identical in their coding regions but differing in their upstream and downstream untranslated regions (UTRs) and poly A tail lengths (UPenn GM LNP1 and UPenn LNP1). mRNA in vitro transcription (IVT) and LNP encapsulation for the UPenn mRNAs was as previously described (29, 30). The UPenn pfceltos mRNAs (Cap1-TEV) were codon optimized and like the TriLink mRNAs modified to either retain or remove N-linked glycosylation sites. Female, BALB/cJ were vaccinated as above, by the IM route, with 10µg of each mRNA, three times at a three-week interval. Two weeks after the final dose, these mice were challenged by infectious mosquito bite with P. berghei PfCelTOS NF54/3D7 transgenic parasites. Serum was analyzed for antigen-specific antibody concentrations and splenocytes were assessed for production of cytokines. The kinetics of the antibody responses revealed that the pfceltos mRNA-LNP1 (UPenn) boosted higher responses after two primary doses compared to either TriLink encoding pfceltos mRNA-LNP1 or pfceltos mRNA-LNP3, as seen by nonoverlapping 95% confidence intervals (Figure 4A). Similarly, both pfceltos mRNA-LNP1 (UPenn GM LNP1 and UPenn LNP1) yielded significantly higher antibodies to PfCelTOS two weeks after the final booster dose. IFN-γ cytokine responses (Figure 4B) were not different for the two mRNAs that were glycosylation site modified (GM), i.e., the TriLink GM LNP1 versus UPenn GM LNP1). MSD revealed a higher pro-inflammatory immune response for the TriLink mRNAs with significantly higher concentrations of IL-1β, IL-10, KC/GRO, and IL-6 cytokines for both TriLink GM LNP1 and LNP3 compared to the two UPenn transcripts, pfceltos UPenn LNP1 (glycosylated) and UPenn GM LNP1 (glycosylation site modified) (Supplementary Figure 3). Relative to the responses measured here, UPenn mRNA encoding pfceltos either glycosylated or nonglycosylated were superior for inducing antibody responses compared to the TriLink mRNA (TriLink GM LNP1 versus TriLink GMP LNP3). Interestingly, the mRNA TriLink GM LNP3 induced the highest IFN-γ responses compared to all groups. These findings suggest a bias toward cellular responses using the TriLink mRNA while the UPenn mRNA preferentially induced higher antibody responses.
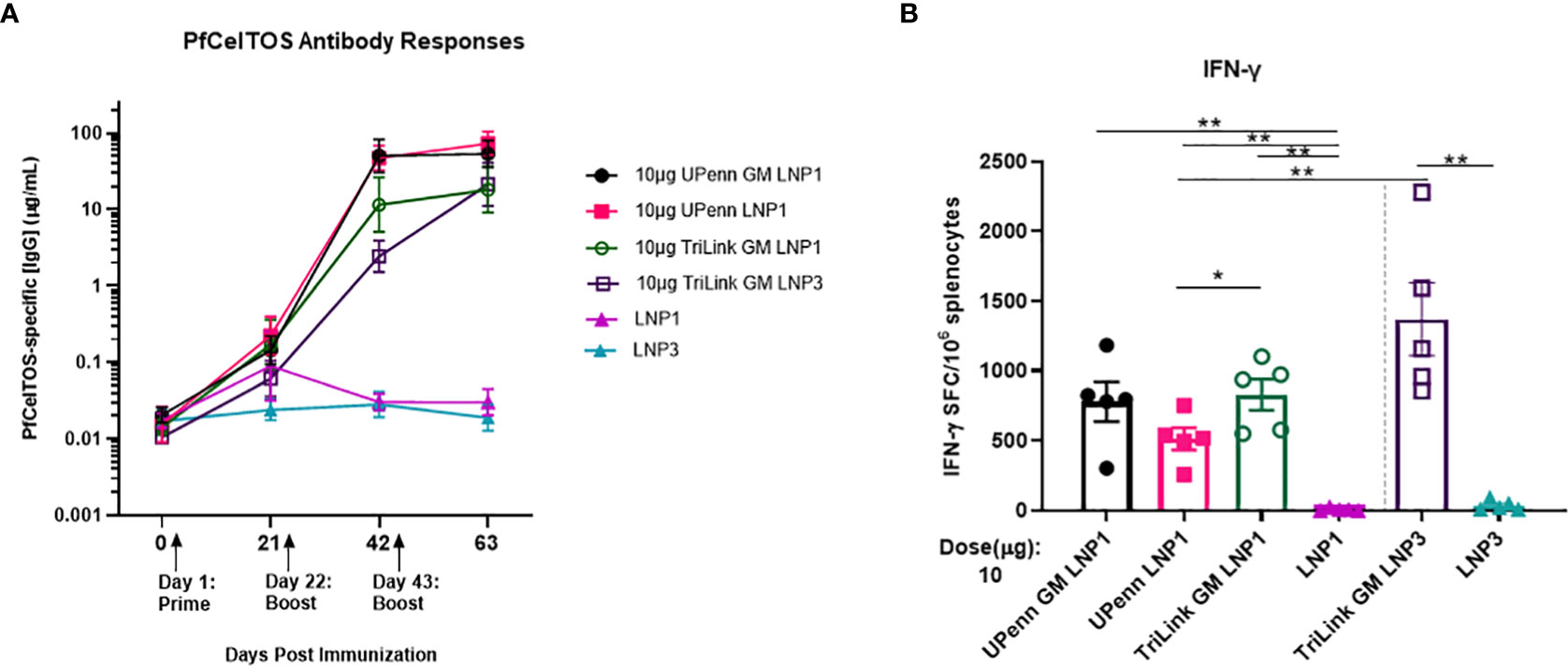
Figure 4 A three-dose regime overcomes an “all-or-none” pattern for humoral immune responses. Mice were immunized intramuscularly (IM) thrice at a three-week interval with 10µg of University of Pennsylvania (UPenn) N-glycosylation site modified (GM) and encapsulated in LNP1 (UPenn GM LNP1), or without N-glycosylation modification (UPenn LNP1) that were one-methylpseudouridine (m1Ψ)-5′-triphosphate modified, and cellulose affinity purified or with TriLink pfceltos mRNA with N-glycosylation site modified (GM) in LNP1 (TriLink GM LNP1) or LNP3 (TriLink GM LNP3) that were ψ-pseudouridine and 5-methylcytosine modified (n = 15 per group; n = 10 in LNP alone groups). (A) Kinetics of PfCelTOS antibody concentrations measured by ELISA. Antibody concentrations are reported as the geometric mean and 95% confidence intervals. (B) IFN-γ cytokine responses were detected by ELISpot, (n=5 per group). The mean number of spot-forming cells (SFC) per splenocytes were reported with standard errors of the mean (SEM). Statistical analysis was performed using Mann-Whitney test, (*p<0.05, **p<0.01).
To address fine-specificity of antibody responses, post third dose sera from UPenn GM LNP1, UPenn LNP1 and TriLink GM LNP1 mRNA vaccinated mice were analyzed through ELISAs PfCelTOS, PfCelTOS N-term, PfCelTOS C-term, full length PfCelTOS proteins and three PfCelTOS peptides (14), Peptide 1-2, 2-3, and 4 (amino acid sequence information, Supplementary Table 1). Antibody titers to subunit regions of PfCelTOS revealed significant differences between the UPenn GM LNP1 (nonglycosylated) and UPenn LNP1 (glycosylated) coding mRNAs specifically targeted to the N-terminus, i.e., to N-term protein (Figure 5A), CelTOS Peptide 1-2 (Figure 5B), and CelTOS Peptide 2-3 (Figure 5C). In each case, responses to the ‘native’ protein sequences were higher (UPenn LNP1). Both CelTOS Peptides 1-2 and 2-3 comprise a region of the N-terminus, with Peptide 1-2 fully encompassing the three predicted N-glycosylation motifs (amino acid 25-60 of the native sequence). These findings suggest that mutating the N-glycosylation sites altered induction of antibodies to this region. Interestingly, for TriLink GM LNP1 vaccinated mice, fewer mice had positive seroconversion rates with antibody levels below the assay limits of detection, i.e., PfCelTOS N-term subunit (n=5 out of 15) (Figure 5A), CelTOS Peptide 1-2 (N= 6 out of 15) (Figure 5B), and CelTOS Peptide 2-3 (N=3 out of 15) (Figure 5C). All three mRNA encoding immunogens responded equally to the PfCelTOS C-term subunit protein (Figure 5D) and the PfCelTOS C-term Peptide 4 (Figure 5E). Interestingly, antibody titers to the full length PfCelTOS after the final immunization revealed no difference between the two UPenn mRNAs, UPenn LNP1 (glycosylated) and UPenn GM LNP1 (nonglycosylated); and similarly, no significant differences for the UPenn GM LNP1 (nonglycosylated) and TriLink GM LNP1 (nonglycosylated) groups (Figure 5F). Notwithstanding the possibility of technical variations, and despite comparing inbred mice of the same sex and age, other factors may contribute to the variability in responses observed, include animal epigenetic differences, housing, interactions, and the local microbiome. These effects have previously been well documented (49, 50).
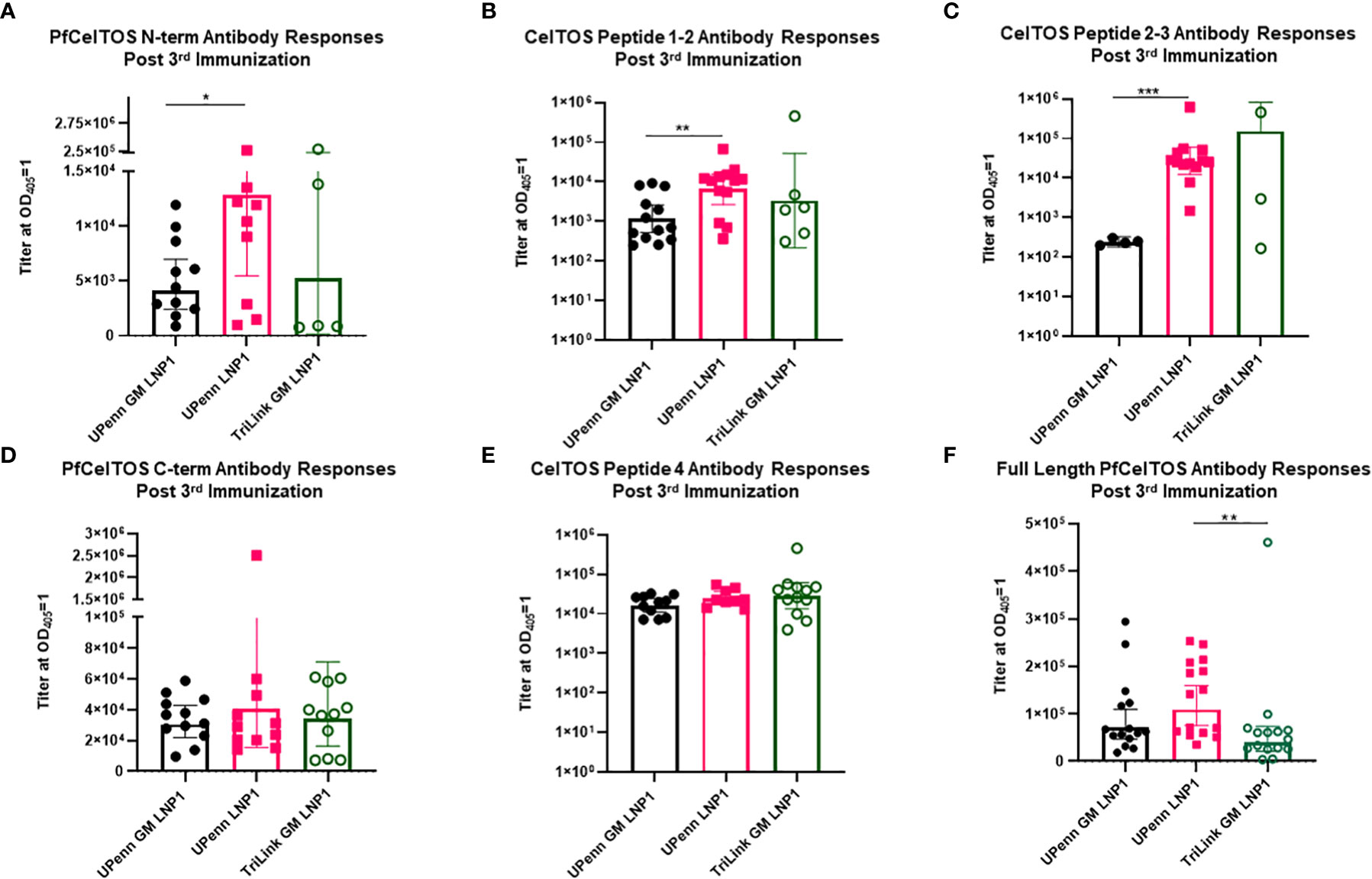
Figure 5 Fine specificity of antibody responses. Sera from mice that were immunized IM thrice at a three-week interval with 10µg mRNA pfceltos UPenn N-glycosylation site modified (GM) in LNP1 (UPenn GM LNP1), or without N-glycosylation modification (UPenn LNP1) that were one-methylpseudouridine (m1Ψ)-5′-triphosphate, and cellulose affinity purified or with TriLink pfceltos mRNA that were N-glycosylation site modified (GM) in LNP1 (TriLink GM LNP1) and were ψ-pseudouridine and 5-methylcytosine modified were characterized for antibody specificities by ELISA against (A) PfCelTOS N-term, (B) CelTOS Peptide 1-2, (C) CelTOS Peptide 2-3, (D) PfCelTOS C-term, (E) CelTOS Peptide 4 and (F) Full length PfCelTOS protein (n = 15 per group). Antibody titers are reported as the geometric mean and 95% confidence interval of the dilution required to achieve an OD405 = 1. Statistical analysis was performed using a Mann-Whitney test (*p<0.05, **p<0.01, ***p<0.001).
Given that the three-dose regimen yielded superior antibody and cellular responses compared to the two-dose regimen, we next sought to evaluate whether the mRNAs encoding pfceltos were protective against rodent malaria challenge. The challenge was performed by a four-mosquito bite inoculum of the P. berghei ANKA expressing P. falciparum NF54/3D7 CelTOS (PbANKA PfCelTOS) parasites two weeks following the third immunizations, a challenge model that is more sensitive to the role of antibodies than the intravenous sporozoite challenge route. All mice (n =10 per group) including challenge controls were parasitemic on day 7, thus neither the UPenn nor TriLink pfceltos mRNA-LNP (neither LNP1 nor LNP3) elicited sterile protection (data not shown). A possible explanation for the lack of protection the transgenic P. berghei line used in this study expresses an additional copy of the pfceltos gene under control of the UIS4 promoter (17). The additional copy of pfceltos may not correctly expressed PfCelTOS with regards to timing of expression which may affect its localization. Thus, the observations made in immunization/challenge studies using transgenic/chimeric mouse models while informative can differ from outcomes from immunization studies in humans using P. falciparum. Unlike the extensive characterization of the protective potential of P. falciparum CSP antigen in preclinical and clinical studies, the CelTOS antigen has yet to be robustly validated as a protective antigen.
In response to the COVID-19 pandemic caused by the SARS-CoV-2 virus, mRNA-LNP vaccines have demonstrated their versatility to overcome manufacturing obstacles associated with traditional vaccine platforms (51, 52). mRNA have unique advantages over traditional vaccine technologies, such as efficient delivery and in vivo translation obviating the need for long development processes for difficult purification/isolation steps. Unlike nucleic acid or vectored approaches, mRNA transcripts are delivered directly to the cell cytosol, thus avoiding the safety concerns of host genome integration which can have detrimental effects due to insertional mutagenesis (48, 53). An essential feature of mRNA-LNP vaccines is the efficient delivery and uptake of transcripts encoding immunogens to target cells. Moreover, targeting directly to the cell cytosol eliminates the rate-limiting step of nuclear translocation (54). Conversely, mRNA’s utility for in vivo protein expression is potentially limited by translational capacity and rapid mRNA turnover rates, compromising induced immune responses. LNPs, the preferred delivery platform for mRNA, enable efficient encapsulation, protection from nucleases, and in vivo transport into cells. LNPs are comprised of phospholipids, cholesterol, PEGylated lipids, and cationic or ionizable lipids. The phospholipids and cholesterol serve structural and stabilizing roles, while PEGylated lipids support persistence in circulation. Cationic and ionizable lipids form complexes with the negatively charged mRNA molecules and allow for exiting from the endosome to the cytosol for translation. Proprietary ionizable cationic lipids decrease cytotoxicity and inflammation, while retaining some adjuvanting activities (55). In vaccine applications, mRNA-LNPs induce strong immune responses, antigen-specific antibody responses and T-cell responses. In fact, LNP formulations promote T follicular helper (Tfh) cell adjuvant activities, and the induction of robust germinal center B cell responses and proinflammatory IL-6 cytokine responses (56). Administration of mRNA-LNPs by the intramuscular route results in a local inflammation that drives recruitment of neutrophils and antigen presenting cells at the site of injection (56). In this study, we addressed factors such as the effect of signal peptides and N-linked glycosylation on PfCelTOS protein quality, translation levels, and the influence of nucleoside modifications of pfceltos encoding mRNA transcripts on immune responses in BALB/cJ mice. While the goal was to select a signal peptide that facilitated higher extracellular secretion of PfCelTOS protein compared to the native Plasmodium CelTOS signal peptide, we observed overall similar translational profiles in transfected CHO cells; with no significant differences seen in the kinetics of translation nor in the rate of localization of target antigen to the extracellular space, nor total protein translation levels. The selected signal peptides had similar hydrophobicity indices (Table 1) and were either derived from the native or from antibody sequences naturally targeted to the cell membrane, i.e., falciparum Wt-SS, and mouse IgLC-SS, human IgE-SS, respectively, thus achieving the desired evidence of adequate translational levels and extracellular translocation. While eukaryotic signal sequences may lack sequence homology, they generally share common structural characteristics; a charged N-terminal region, a hydrophobic core and a less hydrophobic region which includes the cleavage site information. Notionally, cells control translation levels of proteins in a sequence-dependent manner by controlling the efficiency by which ribosomes recognize the initiation complex and the adjacent nucleotides to the initiation codon (57, 58). Therefore, selecting an optimal signal peptide is a critical feature in antigen design for eukaryotic cell expression. All things being considered, the results showed that the introduction of the heterologous signal peptides at the 5’-end of the coding region had little overall influence on PfCelTOS protein translation and the protein output, confirming the ubiquity of primary signal sequences for protein translation, at least in vitro.
Glycosylation is a critical post translational modification to consider when developing vaccine strategies. An ideal vaccine target must share the same antigenic determinants as the native antigen of the pathogen. Thus, removal of N-linked glycans is a reasonable strategy toward avoiding altering immunity. The nature of N-glycosylation on Plasmodium parasite antigens, and effect on induction of functional immune responses is a critical decision point in vaccine antigen design (59–62). N-glycosylation on parasite proteins may play a role in survival, infectivity and antigenicity, of which the exact nature is still not well understood (60). Metabolic labeling reveals that Plasmodium parasites produce shortened N-glycans of N-acetylglucosamine, (i.e., GlcNac and GlcNac2), that recognize the Griffonia simplicifolia Lectin-II (GSL-II) (63), and localize to Plasmodium rhoptry organelle, the endoplasmic reticulum (ER), and cell surface. Previously, the effect of N-link glycosylation on vaccine induced responses raised for two lead blood-stage subunit vaccine candidates, i.e. - P. falciparum apical membrane antigen 1 (AMA1) (55), and merozoite surface protein 1 (MSP1) (64, 65), and the transmission blocking vaccines, Pfs25 (61) and Pfs48/45 was investigated (66). For PfAMA1 and the transmission blocking vaccine Pfs25, post-translational modifications had little effect on functional immunity. While, a milk secreted, N-glycosylated version of MSP142 yielded poor efficacy in vaccinated Aotus nancymai monkeys, a similar non-glycosylated form protected monkeys against virulent challenge with P. falciparum FVO parasites. In the same study, a baculovirus expressed MSP142, which was glycosylated, was able to induce protective responses in monkeys, revealing that the extent and/or complexity of glycosylation contributed to efficacy outcomes. These findings highlight that the effect of N-glycosylation on malarial antigens may vary depending on the antigen, the site of modification, localization, and the type of glycans presented (63). Expression of complex mammalian glycans on plasmodial proteins through nucleic acid vaccines can either adversely influence or redirect the immune responses. Thus non-’natural’ glycans can mask epitopes and impede antigenicity and immunogenicity (67, 68). Translocating misfolded proteins to the endoplasmic signals cell apoptosis and induction of MHC class I-specific CD8+ T cell responses. Cytotoxic, antigen-specific CD8+ T cells can then function against malaria-infected hepatocytes (69). To evaluate the influence of N-linked glycosylation on CelTOS protein translation, mutated transcripts of pfceltos were constructed introducing N>Q amino acids substitutions at the three predicted N-glycosylation sites (NxS/T), all located on the N-terminal end of the protein. For PfCelTOS, we observed that mutating NxS/T motifs altered antibody responses to the N-terminus, while responses to B-cell dominant epitopes localized to the C-terminal end were relatively similar across mRNA transcripts. These findings suggest that the mutated PfCelTOS had altered structure as a more likely explanation of the reduced recognition since these responses were measured against either recombinant protein derived from E. coli expression or synthetic linear peptides.
In the present study, mRNA-LNP encoding pfceltos with varying signal sequences, N-glycosylation, nucleoside substitutions along with LNP (LNP1 versus LNP3) and coding/noncoding regions of mRNA (TriLink versus UPenn) were explored to identify an optimal configuration for in vivo delivery. Importantly, the results reveal that a third dose of pfceltos mRNA-LNPs can overcome an “all-or-none” immune response pattern seen for the two-dose regimen. Currently, nucleoside modified of mRNA transcripts are commonly applied to evade innate immune activation and improve on antigen stability and expression (48, 70), we observed differences that were associated with mRNA dose and/or LNP composition, over N-glycosylation or nucleoside modifications. Interestingly, unlike the pfceltos mRNA (TriLink), pfceltos mRNA (UPenn) induced higher levels of antibody after three doses whether modified for N-link glycosylation (GM) or as glycosylated protein. One distinction between the two mRNAs was that the UPenn mRNAs were N1-methylpseudouridine-modified while TriLink mRNAs were modified with ψ-pseudouridine and 5-methylcytosine. While not confirmed, this could explain for the differences in immune responses and the improved antibody responses with the UPenn mRNAs. N1-methylpseudouridine modified mRNA have been previously reported as superior to pseudouridine, demonstrating reduced sensing by TLR3 and improved in vivo expression levels (25), by prolonged antigen availability, increased expression of antigen in antigen presenting cells and/or a favorable cytokine environment (55, 71).
CelTOS is a soluble, micronemal secreted protein, previously identified as a promising vaccine candidate against malaria. We and others demonstrated that CelTOS vaccines induce potent humoral and cellular immune responses that are capable of functional immunity (15, 16, 18, 19). Structural conserved elements of the N- and C-terminus are prerequisite to oligomerization and function of CelTOS in pore formation (72), therefore preservation of the natural protein assembly and folding is essential (73) for inducing relevant immune responses (11, 72). Given the complexity of the parasite life cycle and infection, presentation of a structurally and functionally conserved CelTOS could augment immunity and yield improved breadth of immune responses. The potential role of antibodies and T cells against this function are still not fully elucidated. Factors such as coding and noncoding content on mRNA, immunization schedule and delivery modalities such as LNP composition are likely critical to optimal immunogenicity. Novel antigens and delivery platforms which target both humoral and/or cellular immunity are needed to ensure a robust pipeline of next-generation malaria vaccine candidates. To date, relatively limited success has been achieved in translating novel antigens such as the P. falciparum CelTOS from preclinical to clinical studies. A more rigorous effort to optimize antigens with unique and/or essential biological roles would yield novel high-priority targets for translation to clinically relevant vaccines.
The original data presented in the article included in the article/Supplementary Materials. Further inquiries can be directed to the corresponding author.
All animal procedures were conducted in compliance with the Institutional Animal Care and Use Committee (IACUC) at Walter Reed Army Institute of Research, Silver Spring, MD. This material has been reviewed and approved by the Walter Reed Army Institute of Research. There is no objection to its publication. Research was conducted in an AAALACi accredited facility in compliance with the Animal Welfare Act and other federal statutes and regulations relating to animals and experiments involving animals and adheres to principles stated in the Guide for the Care and Use of Laboratory Animals, NRC Publication, 2011 edition. This work was conducted under WRAIR Protocol Number: 20-MVD-25.
Study conceptualization and design EA; IW and EA drafted the manuscript; all authors reviewed and edited manuscript versions; KM, JT, IW, and CS, performed immune assays and acquired the data; DW designed and produced pfceltos mRNA transcripts (UPenn); CJ provided P. berghei PfCelTOS NF54/3D7 transgenic parasites; TS performed rodent transgenic mosquito bite challenge; PL and YT designed, prepared and performed mRNA-LNP-encapsulations. All authors contributed to the article and approved the submitted version.
This research was funded by the U.S. Agency for International Development (USAID) and U.S. Military Infectious Diseases Research Program (MIDRP).
We acknowledge Dr. Shahid Khan, Leiden University Medical Center (LUMC), The Netherlands, for his significant contributions to the field of biology of the malaria parasite and his contributions to the development of transgenic parasites for use in malaria vaccine research. We also thank Neeraja Punde and Michael Sibilo for their technical assistance and data acquisition.
Authors IW and KM were employed by Parsons Corporation. PL and YT are employees of Acuitas Therapeutics Inc., a company focused on the development of lipid nanoparticulate nucleic acid delivery systems for therapeutic applications. YT is named on patents describing the use of modified mRNA lipid nanoparticles. In accordance with the University of Pennsylvania policies and procedures and our ethical obligations as researchers, we report that DW is named on patents that describe the use of nucleoside-modified mRNA as a platform to deliver therapeutic proteins and vaccines. We have disclosed those interests fully to the University of Pennsylvania, and we have in place an approved plan for managing any potential conflicts arising from licensing of our patents.
The remaining authors declare that the research was conducted in the absences of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Material has been reviewed by the Walter Reed Army Institute of Research. There is no objection to its presentation and/or publication. The opinions or assertions contained herein are the private views of the author, and are not to be construed as official, or as reflecting true views of the Department of the Army or the Department of Defense. Research was conducted under an approved animal use protocol in an AAALAC International-accredited facility in compliance with the Animal Welfare Act and all other federal statutes and regulations relating to animals and experiments involving animals and adheres to principles stated in the Guide for Care and Use of Laboratory Animals, NRC Publication, 2011 edition. The submitted manuscript has been reviewed and is cleared for PMI policy. The findings and conclusions in this report are those of the author(s) and do not necessarily represent the official position of the US Agency for International Development. EA is an employee of the U.S. Government. This work was prepared as part of her official duties. Title 17, U.S.C., §105 provides that copyright protection under this title is not available for any work of the U.S. Government. Title 17, U.S.C., §101 defines a U.S. Government work as work prepared by a military Service member or employee of the U.S. Government as part of that person’s official duties.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.1026052/full#supplementary-material
2. Hoffman SL, Vekemans J, Richie TL, Duffy PE. The march toward malaria vaccines. Am J Prev Med (2015) 49(6 Suppl 4):S319–33. doi: 10.1016/j.amepre.2015.09.011
3. Atre T, Robinson TM, Savransky T, Dutta S, Epstein JE, Bergmann-Leitner ES. Novel sporozoite-based ELISpot assay to assess frequency of parasite-specific b cells after vaccination with irradiated sporozoites. Malar J (2019) 18(1):186. doi: 10.1186/s12936-019-2819-6
4. Tuteja R. Malaria - an overview. FEBS J (2007) 274(18):4670–9. doi: 10.1111/j.1742-4658.2007.05997.x
5. Marques-da-Silva C, Peissig K, Kurup SP. Pre-erythrocytic vaccines against malaria. Vaccines (Basel). (2020) 8(3):1–15. doi: 10.3390/vaccines8030400
6. Muller K, Gibbins MP, Roberts M, Reyes-Sandoval A, Hill AVS, Draper SJ, et al. Low immunogenicity of malaria pre-erythrocytic stages can be overcome by vaccination. EMBO Mol Med (2021) 13(4):e13390. doi: 10.15252/emmm.202013390
7. Duffy PE, Sahu T, Akue A, Milman N, Anderson C. Pre-erythrocytic malaria vaccines: Identifying the targets. Expert Rev Vaccines (2012) 11(10):1261–80. doi: 10.1586/erv.12.92
8. Hill AV. Pre-erythrocytic malaria vaccines: Towards greater efficacy. Nat Rev Immunol (2006) 6(1):21–32. doi: 10.1038/nri1746
9. Zheng J, Pan H, Gu Y, Zuo X, Ran N, Yuan Y, et al. Prospects for malaria vaccines: Pre-erythrocytic stages, blood stages, and transmission-blocking stages. BioMed Res Int (2019) 2019:9751471. doi: 10.1155/2019/9751471
10. Sanchez L, Vidal M, Jairoce C, Aguilar R, Ubillos I, Cuamba I, et al. Antibody responses to the RTS,S/AS01E vaccine and plasmodium falciparum antigens after a booster dose within the phase 3 trial in Mozambique. NPJ Vaccines (2020) 5:46. doi: 10.1038/s41541-020-0192-7
11. Jimah JR, Salinas ND, Sala-Rabanal M, Jones NG, Sibley LD, Nichols CG, et al. Malaria parasite CelTOS targets the inner leaflet of cell membranes for pore-dependent disruption. Elife (2016) 5:1–11. doi: 10.7554/eLife.20621
12. Rodrigues-da-Silva RN, Soares IF, Lopez-Camacho C, Martins da Silva JH, Perce-da-Silva DS, Teva A, et al. Plasmodium vivax cell-traversal protein for ookinetes and sporozoites: Naturally acquired humoral immune response and b-cell epitope mapping in Brazilian Amazon inhabitants. Front Immunol (2017) 8:77. doi: 10.3389/fimmu.2017.00077
13. Sedegah M, Hollingdale MR, Ganeshan H, Belmonte M, Huang J, Belmonte A, et al. IMRAS-immunization with radiation-attenuated plasmodium falciparum sporozoites by mosquito bite: Cellular immunity to sporozoites, CSP, AMA1, TRAP and CelTOS. PloS One (2021) 16(8):e0256396. doi: 10.1371/journal.pone.0256396
14. Bergmann-Leitner ES, Chaudhury S, Steers NJ, Sabato M, Delvecchio V, Wallqvist AS, et al. Computational and experimental validation of b and T-cell epitopes of the in vivo immune response to a novel malarial antigen. PloS One (2013) 8(8):e71610. doi: 10.1371/journal.pone.0071610
15. Bergmann-Leitner ES, Legler PM, Savranskaya T, Ockenhouse CF, Angov E. Cellular and humoral immune effector mechanisms required for sterile protection against sporozoite challenge induced with the novel malaria vaccine candidate CelTOS. Vaccine. (2011) 29(35):5940–9. doi: 10.1016/j.vaccine.2011.06.053
16. Bergmann-Leitner ES, Mease RM, de la Vega P, Savranskaya T, Polhemus M, Ockenhouse C, et al. Immunization with pre-erythrocytic antigen CelTOS from plasmodium falciparum elicits cross-species protection against heterologous challenge with plasmodium berghei. PloS One (2010) 5(8):e12294. doi: 10.1371/journal.pone.0012294
17. Longley RJ, Salman AM, Cottingham MG, Ewer K, Janse CJ, Khan SM, et al. Comparative assessment of vaccine vectors encoding ten malaria antigens identifies two protective liver-stage candidates. Sci Rep (2015) 5:11820. doi: 10.1038/srep11820
18. Espinosa DA, Vega-Rodriguez J, Flores-Garcia Y, Noe AR, Munoz C, Coleman R, et al. The plasmodium falciparum cell-traversal protein for ookinetes and sporozoites as a candidate for preerythrocytic and transmission-blocking vaccines. Infect Immun (2017) 85(2):1–10. doi: 10.1128/IAI.00498-16
19. Ferraro B, Talbott KT, Balakrishnan A, Cisper N, Morrow MP, Hutnick NA, et al. Inducing humoral and cellular responses to multiple sporozoite and liver-stage malaria antigens using exogenous plasmid DNA. Infect Immun (2013) 81(10):3709–20. doi: 10.1128/IAI.00180-13
20. Thess A, Grund S, Mui BL, Hope MJ, Baumhof P, Fotin-Mleczek M, et al. Sequence-engineered mRNA without chemical nucleoside modifications enables an effective protein therapy in Large animals. Mol Ther (2015) 23(9):1456–64. doi: 10.1038/mt.2015.103
21. Anderson BR, Muramatsu H, Jha BK, Silverman RH, Weissman D, Kariko K. Nucleoside modifications in RNA limit activation of 2'-5'-oligoadenylate synthetase and increase resistance to cleavage by RNase l. Nucleic Acids Res (2011) 39(21):9329–38. doi: 10.1093/nar/gkr586
22. Midoux P, Pichon C. Lipid-based mRNA vaccine delivery systems. Expert Rev Vaccines (2015) 14(2):221–34. doi: 10.1586/14760584.2015.986104
23. Sadeghi S, Kalantari Y, Shokri S, Fallahpour M, Nafissi N, Goodarzi A, et al. Immunologic response, efficacy, and safety of vaccines against COVID-19 infection in healthy and immunosuppressed children and adolescents aged 2 - 21 years old: A systematic review and meta-analysis. J Clin Virol (2022) 153:105196. doi: 10.1016/j.jcv.2022.105196
24. Burdukiewicz M, Sobczyk P, Chilimoniuk J, Gagat P, Mackiewicz P. Prediction of signal peptides in proteins from malaria parasites. Int J Mol Sci (2018) 19(12):1–16. doi: 10.3390/ijms19123709
25. Andries O, Mc Cafferty S, De Smedt SC, Weiss R, Sanders NN, Kitada T. N(1)-methylpseudouridine-incorporated mRNA outperforms pseudouridine-incorporated mRNA by providing enhanced protein expression and reduced immunogenicity in mammalian cell lines and mice. J Control Release. (2015) 217:337–44. doi: 10.1016/j.jconrel.2015.08.051
26. Kauffman KJ, Mir FF, Jhunjhunwala S, Kaczmarek JC, Hurtado JE, Yang JH, et al. Efficacy and immunogenicity of unmodified and pseudouridine-modified mRNA delivered systemically with lipid nanoparticles in vivo. Biomaterials (2016) 109:78–87. doi: 10.1016/j.biomaterials.2016.09.006
27. Gupta R, Brunak S. Prediction of glycosylation across the human proteome and the correlation to protein function pac symp biocomput. (2002). Available from: https://services.healthtech.dtu.dk/service.php?NetNGlyc-1.0.
28. Angov E, Hillier CJ, Kincaid RL, Lyon JA. Heterologous protein expression is enhanced by harmonizing the codon usage frequencies of the target gene with those of the expression host. PloS One (2008) 3(5):e2189. doi: 10.1371/journal.pone.0002189
29. Baiersdorfer M, Boros G, Muramatsu H, Mahiny A, Vlatkovic I, Sahin U, et al. A facile method for the removal of dsRNA contaminant from In vitro-transcribed mRNA. Mol Ther Nucleic Acids (2019) 15:26–35. doi: 10.1016/j.omtn.2019.02.018
30. Mallory KL, Taylor JA, Zou X, Waghela IN, Schneider CG, Sibilo MQ, et al. Messenger RNA expressing PfCSP induces functional, protective immune responses against malaria in mice. NPJ Vaccines (2021) 6(1):84. doi: 10.1038/s41541-021-00345-0
31. Freyn AW, Ramos da Silva J, Rosado VC, Bliss CM, Pine M, Mui BL, et al. A multi-targeting, nucleoside-modified mRNA influenza virus vaccine provides broad protection in mice. Mol Ther (2020) 28(7):1569–84. doi: 10.1016/j.ymthe.2020.04.018
32. Jayaraman M, Ansell SM, Mui BL, Tam YK, Chen J, Du X, et al. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew Chem Int Ed Engl (2012) 51(34):8529–33. doi: 10.1002/anie.201203263
33. Maier MA, Jayaraman M, Matsuda S, Liu J, Barros S, Querbes W, et al. Biodegradable lipids enabling rapidly eliminated lipid nanoparticles for systemic delivery of RNAi therapeutics. Mol Ther (2013) 21(8):1570–8. doi: 10.1038/mt.2013.124
34. Kang SY, Kim YG, Kang S, Lee HW, Lee EG. A novel regulatory element (E77) isolated from CHO-K1 genomic DNA enhances stable gene expression in Chinese hamster ovary cells. Biotechnol J (2016) 11(5):633–41. doi: 10.1002/biot.201500464
35. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods (2012) 9(7):671–5. doi: 10.1038/nmeth.2089
36. Schneider CG, Taylor JA, Sibilo MQ, Miura K, Mallory KL, Mann C, et al. Orientation of antigen display on self-assembling protein nanoparticles influences immunogenicity. Vaccines (Basel) (2021) 9(2):1–22. doi: 10.3390/vaccines9020103
37. Bird P, Gething MJ, Sambrook J. The functional efficiency of a mammalian signal peptide is directly related to its hydrophobicity. J Biol Chem (1990) 265(15):8420–5. doi: 10.1016/S0021-9258(19)38904-5
38. Goddard-Borger ED, Boddey JA. Implications of plasmodium glycosylation on vaccine efficacy and design. Future Microbiol (2018) 13:609–12. doi: 10.2217/fmb-2017-0284
39. Lisowska E. The role of glycosylation in protein antigenic properties. Cell Mol Life Sci (2002) 59(3):445–55. doi: 10.1007/s00018-002-8437-3
40. Kimura EA, Couto AS, Peres VJ, Casal OL, Katzin AM. N-linked glycoproteins are related to schizogony of the intraerythrocytic stage in plasmodium falciparum. J Biol Chem (1996) 271(24):14452–61. doi: 10.1074/jbc.271.24.14452
41. Tamana S, Promponas VJ. An updated view of the oligosaccharyltransferase complex in plasmodium. Glycobiology. (2019) 29(5):385–96. doi: 10.1093/glycob/cwz011
42. Reichmuth AM, Oberli MA, Jaklenec A, Langer R, Blankschtein D. mRNA vaccine delivery using lipid nanoparticles. Ther Deliv. (2016) 7(5):319–34. doi: 10.4155/tde-2016-0006
43. Alameh MG, Weissman D, Pardi N. Messenger RNA-based vaccines against infectious diseases. Curr Top Microbiol Immunol (2020) 1–35. doi: 10.1007/82_2020_202
44. Pardi N, Hogan MJ, Porter FW, Weissman D. mRNA vaccines - a new era in vaccinology. Nat Rev Drug Discovery (2018) 17(4):261–79. doi: 10.1039/D2SC00670G
45. Bornewasser L, Domnick C, Kath-Schorr S. Stronger together for in-cell translation: natural and unnatural base modified mRNA. Chem Sci (2022) 13(17):4753–61.
46. Huysmans H, Zhong Z, De Temmerman J, Mui BL, Tam YK, Mc Cafferty S, et al. Expression kinetics and innate immune response after electroporation and LNP-mediated delivery of a self-amplifying mRNA in the skin. Mol Ther Nucleic Acids (2019) 17:867–78. doi: 10.1016/j.omtn.2019.08.001
47. Stanley MA, Sudenga SL, Giuliano AR. Alternative dosage schedules with HPV virus-like particle vaccines. Expert Rev Vaccines (2014) 13(8):1027–38. doi: 10.1586/14760584.2014.935767
48. Kariko K, Muramatsu H, Ludwig J, Weissman D. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res (2011) 39(21):e142. doi: 10.1093/nar/gkr695
49. Spor A, Koren O, Ley R. Unravelling the effects of the environment and host genotype on the gut microbiome. Nat Rev Microbiol (2011) 9(4):279–90. doi: 10.1038/nrmicro2540
50. Casellas J. Inbred mouse strains and genetic stability: A review. Animal. (2011) 5(1):1–7. doi: 10.1017/S1751731110001667
51. Maruggi G, Zhang C, Li J, Ulmer JB, Yu D. mRNA as a transformative technology for vaccine development to control infectious diseases. Mol Ther (2019) 27(4):757–72. doi: 10.1016/j.ymthe.2019.01.020
52. Versteeg L, Almutairi MM, Hotez PJ, Pollet J. Enlisting the mRNA vaccine platform to combat parasitic infections. Vaccines (Basel). (2019) 7(4):1–19. doi: 10.3390/vaccines7040122
53. Pardi N, Tuyishime S, Muramatsu H, Kariko K, Mui BL, Tam YK, et al. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J Control Release. (2015) 217:345–51. doi: 10.1016/j.jconrel.2015.08.007
54. Meyer M, Huang E, Yuzhakov O, Ramanathan P, Ciaramella G, Bukreyev A. Modified mRNA-based vaccines elicit robust immune responses and protect Guinea pigs from Ebola virus disease. J Infect Dis (2018) 217(3):451–5. doi: 10.1093/infdis/jix592
55. Pardi N, Hogan MJ, Naradikian MS, Parkhouse K, Cain DW, Jones L, et al. Nucleoside-modified mRNA vaccines induce potent T follicular helper and germinal center b cell responses. J Exp Med (2018) 215(6):1571–88. doi: 10.1084/jem.20171450
56. Alameh MG, Tombacz I, Bettini E, Lederer K, Sittplangkoon C, Wilmore JR, et al. Lipid nanoparticles enhance the efficacy of mRNA and protein subunit vaccines by inducing robust T follicular helper cell and humoral responses. Immunity. (2021) 54(12):2877–92 e7. doi: 10.1016/j.immuni.2021.11.001
57. Gaspar P, Moura G, Santos MA, Oliveira JL. mRNA secondary structure optimization using a correlated stem-loop prediction. Nucleic Acids Res (2013) 41(6):e73. doi: 10.1093/nar/gks1473
58. Mauger DM, Cabral BJ, Presnyak V, Su SV, Reid DW, Goodman B, et al. mRNA structure regulates protein expression through changes in functional half-life. Proc Natl Acad Sci USA (2019) 116(48):24075–83. doi: 10.1073/pnas.1908052116
59. Cole KS, Steckbeck JD, Rowles JL, Desrosiers RC, Montelaro RC. Removal of n-linked glycosylation sites in the V1 region of simian immunodeficiency virus gp120 results in redirection of b-cell responses to V3. J Virol (2004) 78(3):1525–39. doi: 10.1128/JVI.78.3.1525-1539.2004
60. Cova M, Rodrigues JA, Smith TK, Izquierdo L. Sugar activation and glycosylation in plasmodium. Malar J (2015) 14:427. doi: 10.1186/s12936-015-0949-z
61. Datta D, Bansal GP, Kumar R, Ellefsen B, Hannaman D, Kumar N. Evaluation of the impact of codon optimization and n-linked glycosylation on functional immunogenicity of Pfs25 DNA vaccines delivered by In vivo electroporation in preclinical studies in mice. Clin Vaccine Immunol (2015) 22(9):1013–9. doi: 10.1128/CVI.00185-15
62. Rudd PM, Elliott T, Cresswell P, Wilson IA, Dwek RA. Glycosylation and the immune system. Science. (2001) 291(5512):2370–6. doi: 10.1126/science.291.5512.2370
63. Bushkin GG, Ratner DM, Cui J, Banerjee S, Duraisingh MT, Jennings CV, et al. Suggestive evidence for Darwinian selection against asparagine-linked glycans of plasmodium falciparum and toxoplasma gondii. Eukaryot Cell (2010) 9(2):228–41. doi: 10.1128/EC.00197-09
64. Stowers AW, Chen Lh LH, Zhang Y, Kennedy MC, Zou L, Lambert L, et al. A recombinant vaccine expressed in the milk of transgenic mice protects aotus monkeys from a lethal challenge with plasmodium falciparum. Proc Natl Acad Sci USA (2002) 99(1):339–44. doi: 10.1073/pnas.012590199
65. Giersing B, Miura K, Shimp R, Wang J, Zhou H, Orcutt A, et al. Posttranslational modification of recombinant plasmodium falciparum apical membrane antigen 1: Impact on functional immune responses to a malaria vaccine candidate. Infect Immun (2005) 73(7):3963–70. doi: 10.1128/IAI.73.7.3963-3970.2005
66. Lee SM, Hickey JM, Miura K, Joshi SB, Volkin DB, King CR, et al. A c-terminal Pfs48/45 malaria transmission-blocking vaccine candidate produced in the baculovirus expression system. Sci Rep (2020) 10(1):395. doi: 10.1038/s41598-019-56642-1
67. Dubrovskaya V, Guenaga J, de Val N, Wilson R, Feng Y, Movsesyan A, et al. Targeted n-glycan deletion at the receptor-binding site retains HIV env NFL trimer integrity and accelerates the elicited antibody response. PloS Pathog (2017) 13(9):e1006614. doi: 10.1371/journal.ppat.1006614
68. Zhou T, Doria-Rose NA, Cheng C, Stewart-Jones GBE, Chuang GY, Chambers M, et al. Quantification of the impact of the HIV-1-Glycan shield on antibody elicitation. Cell Rep (2017) 19(4):719–32. doi: 10.1016/j.celrep.2017.04.013
69. Seder RA, Chang LJ, Enama ME, Zephir KL, Sarwar UN, Gordon IJ, et al. Protection against malaria by intravenous immunization with a nonreplicating sporozoite vaccine. Science. (2013) 341(6152):1359–65. doi: 10.1126/science.1241800
70. Zhang C, Maruggi G, Shan H, Li J. Advances in mRNA vaccines for infectious diseases. Front Immunol (2019) 10:594. doi: 10.3389/fimmu.2019.00594
71. Tam HH, Melo MB, Kang M, Pelet JM, Ruda VM, Foley MH, et al. Sustained antigen availability during germinal center initiation enhances antibody responses to vaccination. Proc Natl Acad Sci USA (2016) 113(43):E6639–E48. doi: 10.1073/pnas.1606050113
72. Kumar H, Jimah JR, Misal SA, Salinas ND, Fried M, Schlesinger PH, et al. Implications of conformational flexibility, lipid binding, and regulatory domains in cell-traversal protein CelTOS for apicomplexan migration. J Biol Chem (2022) 298(9):102241. doi: 10.1016/j.jbc.2022.102241
Keywords: malaria, Plasmodium falciparum, malaria vaccine, mRNA, CelTOS, lipid nanoparticles, N-glycosylation, nucleoside modifications
Citation: Waghela IN, Mallory KL, Taylor JA, Schneider CG, Savransky T, Janse CJ, Lin PJC, Tam YK, Weissman D and Angov E (2022) Exploring in vitro expression and immune potency in mice using mRNA encoding the Plasmodium falciparum malaria antigen, CelTOS. Front. Immunol. 13:1026052. doi: 10.3389/fimmu.2022.1026052
Received: 23 August 2022; Accepted: 18 November 2022;
Published: 15 December 2022.
Edited by:
Alexandra Jane Spencer, The University of Newcastle, AustraliaReviewed by:
Aneesh Thakur, Vaccine and Infectious Disease Organization, International Vaccine Centre (VIDO-InterVac), CanadaCopyright © 2022 Waghela, Mallory, Taylor, Schneider, Savransky, Janse, Lin, Tam, Weissman and Angov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Evelina Angov, RXZlbGluYS5hbmdvdi5jaXZAaGVhbHRoLm1pbA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.