 Eirini Nikolouli
Eirini Nikolouli Janne Reichstein1
Janne Reichstein1 Gesine Hansen
Gesine Hansen Nico Lachmann
Nico Lachmann
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 17 November 2022
Sec. Primary Immunodeficiencies
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1024935
This article is part of the Research Topic Advances in primary Immunodeficiencies (Inborn Errors of Immunity) in Central-Eastern Europe: Volume II View all 20 articles
In the last two decades, the exponential progress in the field of genetics could reveal the genetic impact on the onset and progression of several diseases affecting the immune system. This knowledge has led to the discovery of more than 400 monogenic germline mutations, also known as “inborn errors of immunity (IEI)”. Given the rarity of various IEI and the clinical diversity as well as the limited available patients’ material, the continuous development of novel cell-based in vitro models to elucidate the cellular and molecular mechanisms involved in the pathogenesis of these diseases is imperative. Focusing on stem cell technologies, this review aims to provide an overview of the current available in vitro models used to study IEI and which could lay the foundation for new therapeutic approaches. We elaborate in particular on the use of induced pluripotent stem cell-based systems and their broad application in studying IEI by establishing also novel infection culture models. The review will critically discuss the current limitations or gaps in the field of stem cell technology as well as the future perspectives from the use of these cell culture systems.
In the last decades, the major progress in the field of genetics and the availability of high-throughput DNA sequencing techniques contributed to the discovery of more than 400 monogenic germline mutations affecting our immune system. These mutations are referred to as inborn errors of immunity (IEI) and can lead either to the loss of expression or loss/gain of function of the respective protein (1–3). The prevalence of IEI in the overall population is in the range of 1/10,000-1/50,000 (4). In most cases, IEI are identified early in life upon recurring infections such as bronchitis or sinusitis and can be life-threatening if the patients do not receive proper treatment. The clinical phenotype of IEI shows a variety of disorders, including autoimmune or inflammatory diseases, allergies, cancer, and increased susceptibility to several pathogens.
According to the International Union of Immunological Societies, the IEI are classified into the following ten categories of conditions: Combined immunodeficiencies; Combined immunodeficiencies with syndromic features; Predominantly antibody deficiencies; Diseases of immune dysregulation; Congenital defects of phagocytes; Defects in intrinsic and innate immunity; Autoinflammatory diseases; Complement deficiencies, Bone marrow failure, and Phenocopies of IEI (2–5).
Due to the variable clinical features of IEI-related disorders, the medical care and treatment of these young patients is extremely challenging and requires a careful fine-tuning of the immune system. Children with IEI are usually treated with immunosuppressants, such as rapamycin or corticosteroids to decrease inflammation, however, this leads to a broad range of side effects. In addition, these types of treatment can only alleviate the symptoms but do not offer a curative solution for the patient. Other therapeutic strategies include the long-term usage of anti-fungal, anti-viral, or anti-bacterial agents, increasing the risk for the development of drug-resistant pathogens, which can cause life-threatening infections. In some cases, like in the severe combined immunodeficiency (SCID) syndrome, allogenic hematopoietic stem cell transplantation (HSCT) (or autologous HSC-gene therapy) is the only curative therapy (6, 7). However, HSCT always lurks the risk of immunological rejection or development of graft versus host disease with devastating consequences for the patient, pointing towards the need of suitable alternatives.
For these reasons, more targeted therapeutic approaches, which can directly modulate specific cell types or intracellular pathways, are preferred. These approaches include the use of specific inhibitors or biologics (antibodies or recombinant proteins). For the safe use of these emerging therapeutic agents, a detailed study of the pathophysiological mechanisms of the diseases is necessary. Given the rarity of IEI and the technical difficulties (obtaining sufficient samples from children or the low number of affected cells), the study of IEI-related diseases remains challenging. Thus, the development of novel systems to unravel the cellular and molecular mechanisms involved in the pathophysiology of the various IEI is of great importance.
In the last years, the establishment of novel IEI in vitro systems has contributed enormously to the current understanding of the immunopathology involved in the various clinical features of different IEI-related diseases. As a consequence, these insights allowed for the development of new therapeutic approaches. The most appropriate in vitro models developed for these purposes are stem-cell based since stem cells have the capacity for self-renewal and differentiation into specialized cell types. The two main approaches used are based either on adult hematopoietic stem cells (HSCs) or induced pluripotent stem cells (iPSCs).
Adult HSCs are primary cells isolated from different sources such as peripheral blood, bone marrow, or umbilical cord. Their low number, inefficient long-term expansion, and heterogeneity however, impact their use in disease modeling and clinical applications.
As an alternative, iPSC-based in vitro models have proven to be one of the most successful options to adequately study IEI (8). Reprogramming of a few somatic cells, isolated from a patient, leads to the generation of stable, pluripotent, and patient-specific iPSC lines, which can give rise indefinitely to various cell types (Figure 1). Thus, iPSC technology becomes a promising tool to investigate the possible mechanisms involved in the pathophysiology of IEI using various cell types generated through specialized differentiation protocols from a single iPSC line. In addition, the fact that the generation of iPSCs is based on less-invasive methods for the patient renders the iPSC-derived cells preferable in comparison to primary specialized cells isolated consecutively from patients with more laborious and invasive procedures. Of note, this plays a particular role when children are affected.
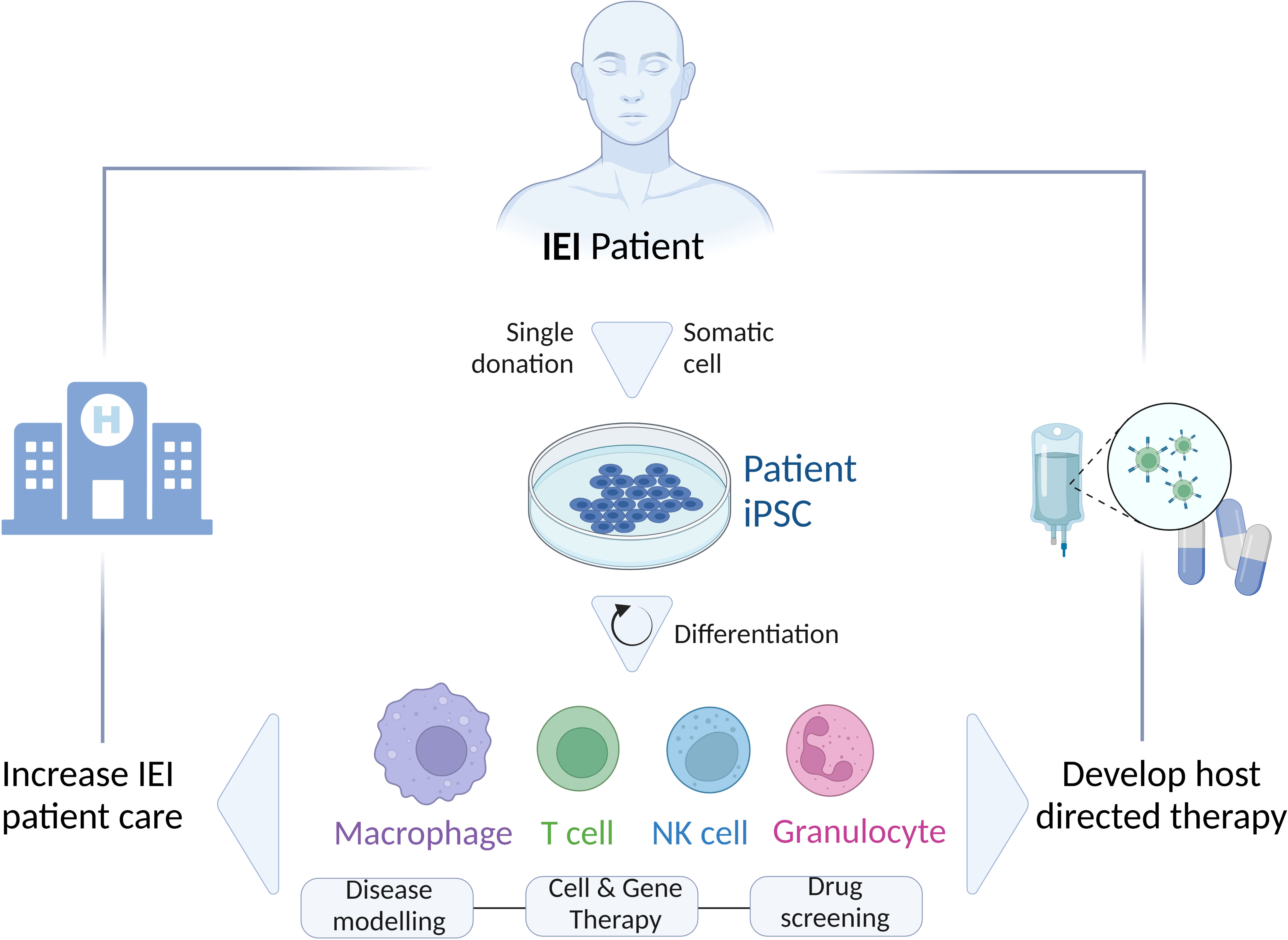
Figure 1 Schematic representation of the generation and differentiation of patient’s-specific iPSCs to different cell types (Macrophages, T cells, NK cells, Granulocytes) for various applications (Disease modeling; Cell and Gene therapy; Drug screening) to study IEI and improve the patient’s care by developing novel host directed therapies. IEI, inborn errors of immunity; iPSC, induced-pluripotent stem cells.
Studying the immune system using animal models has given us insights into its function. However, in cases of IEI, which affect hematopoiesis and immune development, the inter-species differences within hematopoietic development are a considerable limitation for the use of animal models to adequately study IEI (9). In those cases, human iPSC-derived cells are better suited to clarify the role of specific IEI in the hematopoietic system.
Furthermore, the use of iPSC-derived cells is not only advantageous for studying IEI to unravel the pathomechanism of various diseases but also introduces alternative therapeutic strategies. The phenotypical and functional similarities of iPSC-derived immune cells such as granulocytes, macrophages, and dendritic cells with their respective primary counterparts (10) further support the use of iPSC-derived cells to study the onset of diseases and to develop novel cell therapy concepts. As a consequence, in the last years continuous optimization of the iPSC-based differentiation protocols improved both the quality and quantity of the derived cell types, aiming to fulfill the requirements for clinical application.
The fact that various tissues and cell types may be affected in different IEI underlines the complexity of IEI-related diseases and the growing need for specialized and highly standardized immune cells to study the onset and progression of these diseases. The high demand for adequate numbers of the affected patient-specific immune cells led, upon the discovery of iPSCs in 2006, to the establishment of numerous hematopoietic differentiation protocols able to generate different lineages of the lympho-hematopoietic system. In recent years, several iPSC-based differentiation protocols have been established for the generation of for instance macrophages, granulocytes, dendritic cells (DCs), natural killer cells (NK), NKT cells, and T lymphocytes. In general, the iPSC differentiation protocols include as first step the differentiation of iPSCs to hematopoietic progenitors either by the support of stromal cells and the use of cytokines or by the formation of so-called embryonic bodies (EBs), which are aggregates containing cells of the three germ layers. As an example, in the context of macrophages, recent differentiation techniques result in the generation of cells using EBs. The differentiation to hematopoietic or myeloid progenitors within the cell aggregates happens either autonomously from factors produced by the cell aggregates (11–13) or by the addition of exogenous factors such as BMP4, VEGF, SCF, Flt3-ligand, and TPO (13–15). Of note, using modern differentiation media authentic macrophages can be generated from human iPSCs, which share phenotypical and functional hallmarks with their in vivo counterparts (16). Similarly, simplified two-step protocols have also been established for the generation of iPSC-derived NK cells. In these differentiation platforms, the cytokines IL-3, IL-7, SCF, IL-15 and Flt3-ligand are often used for the differentiation of iPSC-derived progenitor cells towards NK cells (17, 18). Although still quite challenging to generate from human iPSC, some progress has been made to produce iPSC-derived T lymphocytes (19–28). Some of the strategies that have been developed include the reprogramming of antigen-specific T cells to iPSCs and the subsequent differentiation to T cells with the respective antigen-specificity (20, 23, 27) or the generation of custom-made antigen-specific T cells using T-cell receptor (TCR)-transduced iPSCs (22, 26). Of note, the functional resemblance of the iPSC-derived lymphocytes to the in vivo lymphocytes raises hopes for the use of these cells for the treatment of several diseases.
Moving from the use of iPSC-derived immune cells for disease modelling towards cell-based therapies targeting IEI, required the development of differentiation protocols which allow a continuous and scalable production of iPSC-derived immune cells such as macrophages (12, 29), NK cells (30), and T lymphocytes (22). The successful use of iPSC-derived immune cells as a cell-based therapy requires authentic immune cells, which are functionally indistinguishable from their in vivo counterparts. For instance, iPSC-derived macrophages show typical morphological and phenotypical characteristics. When tested for their ability to secrete cytokines and to perform phagocytosis, iPSC-macrophages showed a similar cytokine secretion profile to the monocyte-derived macrophages and high phagocytic capacity, respectively (11, 12, 31). In addition, iPSC-macrophages have been shown to react highly similar to monocyte-derived macrophages to a variety of pathogens (12, 32–34). Similarly, iPSC-derived NK cells show typical NK characteristics and full functionality, as proven in several studies to be able to eradicate HIV-infected CD4+ T cells (17), myeloma or pancreatic tumor cells (18) as well as ovarian cancer cells (35). Likewise, iPSC-derived antigen-specific cytotoxic T cells (CTLs) directed against the melanoma epitope MART1 (25) or the WT1 antigen (20), showed antigen-specific reactivity upon stimulation with the respective antigen, proving their functional similarity to in vivo CTLs. These protocols are constantly adapted and pave the way for the generation of highly standardized, well-characterized cells from iPSCs, which are derived from healthy or diseased individuals and which can now be used to model IEI in vitro. Furthermore, the existence of such differentiation protocols makes these iPSC-derived cell products promising therapeutic agents for “bench to bedside” applications.
The role and importance of iPSC-derived immune cells for the field of IEI is constantly growing and opens new possibilities to study novel forms of treatment. The establishment of numerous disease models for the discovery of the responsible molecular and cellular factors for the clinical phenotype and, at the same time, establishment of promising alternative therapeutic strategies either through the discovery of potent drugs by drug screening approaches or through genetic manipulation of the cells for cell-based therapies, are of great importance. In the chapter below, we cite representative studies for most of the categories of IEI and for which iPSC-derived immune cells have been used to study IEI. A broader overview of the different studies published in this field in the last decade can also be seen in Table 1.
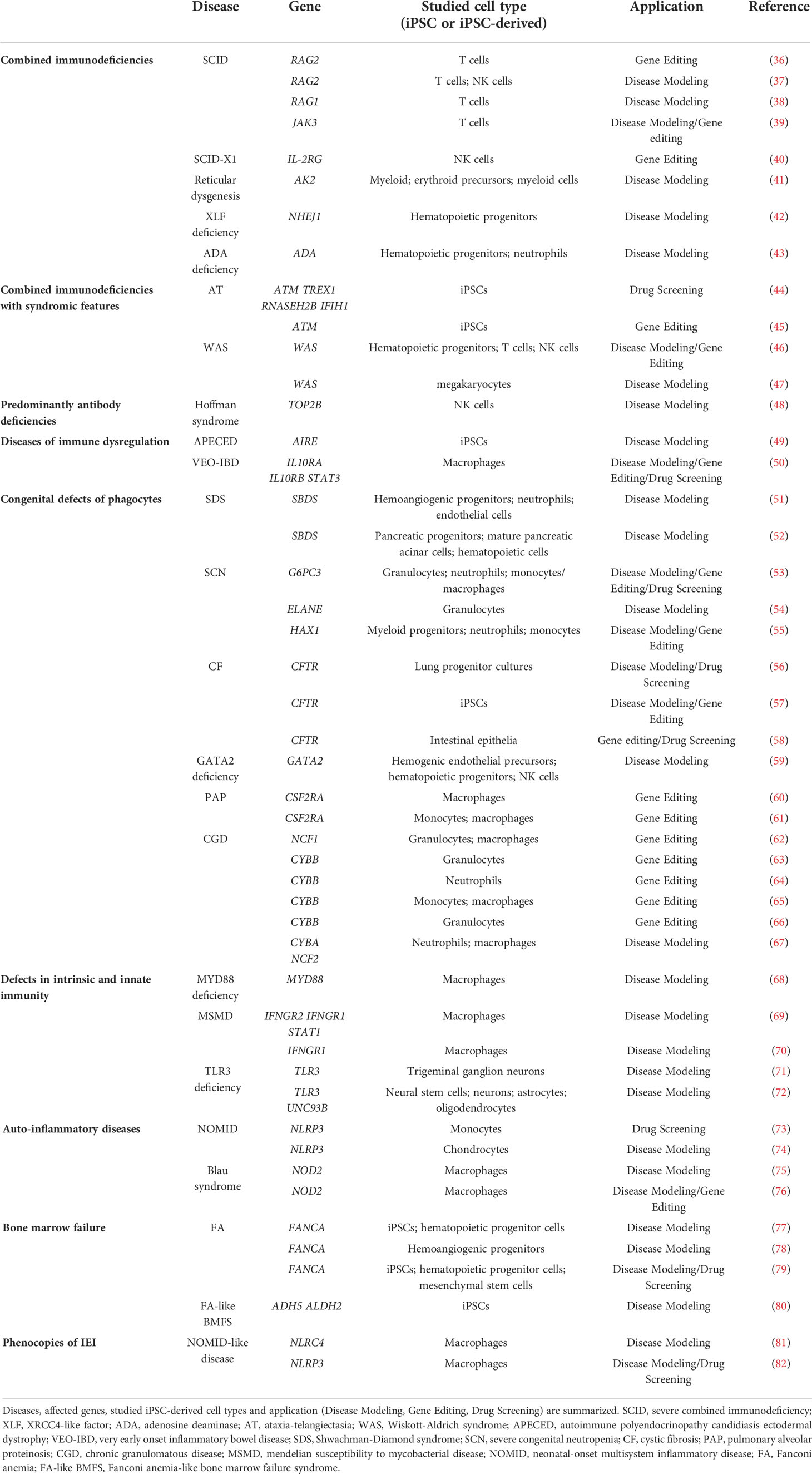
Table 1 An overview of the latest studies using iPSC-derived cells to study IEI.
One of the most common diseases of this category is severe combined immunodeficiency (SCID), which is characterized by a lack of CD3+ T cells. SCID is a life-threatening syndrome with a prevalence of 1/50.000-100.000 worldwide. IL2RG, IL7R, JAK3, ADA, RAG1/2, and DCLRE1C are the most common genes identified to be impaired in SCID patients, resulting in various clinical phenotypes. The current therapeutic approach for SCID patients, apart from antimicrobial drugs, is HSCT partially in combination with gene therapy. The first trial to generate iPSCs from a SCID-patient (adenosine deaminase; ADA deficient-SCID) was conducted in 2008 by Park et al. (83). Later, in 2015 Chang et al. used a patient-specific iPSC line with a mutation in the JAK3 gene to generate T cells using a two-step OP9 and OP9-DL4 system (39). Studying these iPSC-derived JAK-deficient-T cells showed that JAK deficiency negatively impacts the differentiation of the cells into an early T cell progenitor stage, unraveling the mechanism of immunodeficiency in these patients (39). Correction of the JAK3 mutation in iPSCs using CRISP/Cas9 technology restored normal T cell development (39). This highlights the importance of iPSC-based in vitro systems for studying human lymphopoiesis while developing novel gene correction strategies for human immunodeficiencies at the same time.
ADA deficiency causes abnormal differentiation and function of T cells leading to a severe combined immunodeficiency (84, 85). Recent data from ADA-deficient patients indicated that ADA deficiency impacts myeloid cells, such as neutrophils (43, 86). Given the difficulty in isolating neutrophils from ADA-deficient patients for follow-up studies, using patient-specific iPSCs for generating ADA-deficient neutrophils is very beneficial. Here Tsui et al. could show that ADA-deficient iPSCs generate lower numbers of neutrophils with increased frequency of hyper lobular neutrophils, characterized by decreased phagocytic capacity (43). Thus, the iPSCs technology was able to further associate the contributing mechanisms to the phenotype of ADA-deficient patients (43).
Ataxia telangiectasia (AT) is an inherited disease characterized by a severe neurological phenotype with a poor prognosis and a lack of efficient accessible treatment. AT is caused by a mutation in the ataxia-telangiectasia mutated gene (ATM), leading to a combined immunodeficiency in patients and an increased risk for the development of autoimmunity (87). Not differentiated towards immune cells, iPSCs generated from an AT patient were used as an in vitro model to study the cytotoxic effects of the potentially effective immunomodulators thioguanine, mercaptopurine, dexamethasone, mepacrine, thalidomide, and lenalidomide (44). In detail, AT iPSCs were more resistant to thioguanine compared to wild-type iPSCs and at the highest tested concentration of thalidomide and lenalidomide slightly higher cytotoxic effect was observed in AT iPSCs (44). Both AT and wild-type iPSCs were resistant to dexamethasone (44).
As another example, Wiskott-Aldrich syndrome (WAS) is an X-linked inherited immunodeficiency characterized by micro thrombocytopenia, autoimmunity, and hematological malignancies (28). The disease is caused by various mutations in the WAS protein gene. Generation of WAS-specific iPSCs and subsequent differentiation to megakaryocytes and platelets contributed to understanding the disease and identifying the responsible molecular and cellular players. More specifically, WAS-iPSC-derived megakaryocytes showed an abnormal pattern of F-actin distribution with abnormal pro-platelet processes, indicating dysregulated cytoskeletal protein rearrangement during pro-platelet formation (47). In this case, the use of patient-derived iPSCs could highlight the importance of the WAS protein for normal platelet production. Similar to the SCID studies, overexpression of the healthy WAS protein in patient- derived iPSCs could rescue the phenotype, paving the way for new therapeutic options. Similarly, Laskowski et al. restored the WAS protein function in patient-derived iPSCs using zinc finger nucleases technology and the differentiated hematopoietic lineages were restored (46). Of note, while differentiation of both healthy and WAS-iPSCs towards non-lymphoid cells was sufficient, a clear reduction in the generation of CD4/CD8 double positive T cells and NK cells was observed, which could be restored upon targeted correction of the WAS gene locus.
Genetic forms of inflammatory bowel disease (IBD) are caused by mutations in genes that are involved in the IL-10 signaling pathway (88). IBD is characterized by severe bowel inflammation and is developed within the first 6 years of life (89). Many of the patients do not respond to anti-inflammatory and immunosuppressive treatments. To study the pathophysiology of IBD and contribute to novel therapeutic strategies, KO iPSC models for the genes IL10R, IL10RB, STAT1, and STAT3 were generated using sgRNA-directed CRISPR-Cas9 lentiviral vectors (50). Using macrophages derived from these KO-iPSC lines these studies could show that defects in any of the IL10R chains or in STAT3 result in absence of BCL3 expression and reduced secretion of defined IL-10R-/STAT3-dependent cytokines (50). Of note, the phenotype of the KO-iPSC-derived macrophages could however be restored (reduced pro-inflammatory cytokines) using lentiviral vectors overexpressing the IL-10R gene. Using the same iPSC-macrophage system, small anti-inflammatory agents (SB202190 and Filgotinib) were tested and could confirm their anti-inflammatory effect by the reduction of TNF-α, IL-6, and CCL5, while no negative impact could be observed on iPSC-derived macrophages with respect to cell viability (50).
Chronic granulomatous disease (CGD) is characterized by severe, recurrent, and life-threatening bacterial and fungal infections due to defects in the oxidative burst in phagocytes. Its prevalence is 1/250,000 individuals. CGD can be caused by mutations in any of the four components of the NADPH oxidase complex. The most common mutation is found in the CYBB gene, which encodes for the gp91phox subunit and is X-linked. To date, the only available treatment is allogeneic or autologous (genetic corrected) HSCT. To elaborate on new treatments for X-CGD patients, several studies have used X-CGD-patient-specific iPSCs to genetically modify the cells using zing finger nuclease-mediated gene targeting (90), transcription activator-like effector nucleases (TALENs) (66) or bacterial artificial chromosomes (BAC) transgenesis (64), respectively. In all iPSC-based studies, the CYBB function was successfully restored, leading to sufficient oxidative activity and ROS production in iPSC-derived granulocytes, proven the suitability of gene therapy to restore the anti-microbial function in immune cells. In a completely different approach, the NADPH oxidase activity of X-CGD iPSC-derived macrophages was restored using NOX2/p22phox proteoliposomes, which were transported into the macrophages (91). The combination of patient-specific iPSC-derived cells with recombinant therapeutic proteoliposomes could in the future lead to the development of alternative antibacterial or antifungal therapies for patients with IEI. Similar to the aforementioned approaches, the iPSC system has also been used to establish and test gene correction of p47phox deficiency. Introducing a functional NCF1 minigene into the intron 1 of the NCF1 gene using CRISPR/Cas9 (62) or targeted correction of the mutation (GT deletion in NCF1 pseudogenes) using zinc-finger nucleases (92) into p47-CGD iPSCs could restore oxidase function in iPSC-derived immune cells, highlighting the suitability of the iPSC system to test novel gene therapy concepts.
Mendelian susceptibility to mycobacterial disease (MSMD) is characterized by increased susceptibility to weakly virulent mycobacteria (e.g. Mycobacterium bovis Bacillus Calmette-Guerin; BCG). The genetic etiology of MSMD is complex, with a variety of genes and mutations involved, which all affect the sufficient breakdown of mycobacteria. Mutations can affect either T cells (e.g. IL12RB1, IL12RB2, TYK2) or macrophages (e.g. IFNGR1, INFGR2, IRF8, CYBB, NEMO), which lead to an impaired crosstalk of these two cell types. As an example, the clinical phenotype can be severe, as seen in patients with complete IFN-gamma receptor 1 or 2 deficiency (IFNGR1/2). In contrast, the clinical phenotype can also be mild to moderate, as seen in patients suffering from STAT1, IL-12/IL-23 receptor, or tyrosine kinase 2 deficiency. Of note, clinical symptoms and the impaired function of e.g. macrophages can be improved by treating patients with high dose IFNγ therapy. However, this kind of treatment is unsuitable for patients who suffer from complete IFNGR1 or IFNGR2 deficiency. The generation of iPSCs from patients harboring mutations in genes involved in the IFNγ signaling, such as IFNGR1, IFNGR2, and also STAT1 were able to demonstrate in detail the impact of IFNγ on macrophages and the importance of this cell type in the onset and progression of mycobacterial susceptibility (69, 70). These studies revealed iPSC-derived macrophages with an impaired type II IFN system showing normal macrophage differentiation and phenotype but severely impaired intracellular killing activity for BCG (69, 70).
Neonatal-onset multisystem inflammatory disease (NOMID), also known as chronic infantile neurologic cutaneous articular syndrome (CINCA), is a rare genetic disease present from birth and caused by mutations mainly in the NLRP3 locus. It is inherited in an autosomal dominant way, and the patients suffer from uncontrolled inflammation in several systems of the body, such as skin, joints, and central nervous system. The clinical phenotype varies and includes urticarial-like skin rash, arthritis, and chronic meningitis, which increases the risk of neurological problems. So far, anti-IL-1β treatment (e.g. Anakinra) using specific inhibitors is the preferable therapeutic option. However, its effectiveness is highly dependent on the severity of the disease phenotype. In addition, the complete IL-1β blockade involves the risk of uncontrolled immunosuppression. For this reason, selective NLRP3 inhibitors would be more beneficial as therapeutic option and several NLRP3 inhibitors have already entered clinical trials (93). However, given the different mutations observed in NOMID patients, it is possible that some NLRP3 mutants escape an efficient inhibition from already known inhibitors (94). Therefore, discovery of novel NLRP3 inhibitors is necessary. To test and screen for new therapeutic compounds, patient-specific NOMID-iPSCs could be used as a screening platform. Seki et al. generated iPSC-derived immortalized myeloid cell lines from wild-type and NLRP3-mutated iPSC clones and subsequently differentiated these into macrophages (73). Generated macrophages were further used for developing a high throughput system to identify compounds that show inhibitory effects specifically against the secretion of IL-1β and the activation of mutant NLRP3 (73). Out of almost 5,000 tested compounds, seven candidates were sufficiently blocking the IL-1β secretion. Interestingly those were already introduced in previous studies as NLRP3 inhibitors, indicating the effectiveness of the system (73).
In addition to increased susceptibility and recurrent bacterial infections, deficiencies in the complement pathway have been linked to age-related macular degeneration (AMD). AMD is an ideal example underlining the interconnection of IEI with various tissues and organs of the body. Thus, using iPSCs generated from patients with AMD or from healthy individuals shed light on the mechanisms involved in the disease and showed the impact of an IEI in a complement protein on the progression of AMD. In this case, retinal pigment epithelium derived from AMD-derived iPSCs was used to show impaired mitochondrial function under stress conditions and its link to the presence of the high-risk allele for the complement factor H (CFH locus) (95). More generated iPSC lines from three patients carrying the rare variants in the CFH locus and suffering from AMD are available tools for further cellular studies and the development of novel treatments (96).
Fanconi anemia (FA) is an inherited condition diagnosed usually in children between the age of 3 and 14, and it is characterized, among other symptoms, by failure of bone marrow function. It is caused by mutations in at least 22 different genes, which are involved in the FA pathway, responsible for the DNA repair process, with the genes FANCA, FANCC, and FANCG to be utmost affected. The only curative treatment so far is HSCT, with gene therapy on the horizon (97). Given the difficulty that exists in recapitulating FA pathophysiology using mouse models, further understanding of the disease pathogenesis was accomplished using FA patient-specific iPSCs for disease modelling (77, 79). Marion et al. revealed that activation of the p53-p21 axis leads to accelerated erythroid differentiation in FANCA-deficient HPCs. Use of exogenous recombinant human GAS6 resulted in restored hematopoiesis, providing alternative options for improving therapy of FA in the future (77). In contrast, another study utilized the iPSC technology to screen and evaluate novel compounds, discovering that Tremulacin was able to rescue the hematopoietic defects of FA patient by suppressing the transcription of the inflammatory cytokine TNFα (79), which impressively shows the potential of the iPSC technology.
The numerous studies that have used iPSC-derived cells for disease modeling and drug screening for several IEI-related conditions could lay the foundation for developing novel therapies. The reason for this success is the unique potential of iPSCs to differentiate into almost all cells of the hematopoietic system and beyond. Although established protocols for the differentiation of a plethora of iPSC-derived immune cells exist, still the lack of protocols for a robust generation of T or B lymphocytes that resemble their in vivo counterparts as much as possible, is currently a limitation of the technology and must be confronted in the coming years.
While most of the aforementioned reports have used iPSCs as a disease modeling platform, the use of iPSC-based platforms for establishing drug screening, drug toxicology, and drug-drug interactions in the context of IEI is highly warranted and will accelerate the progress of personalized medicine further. Along this line, several studies used iPSC-derived cells for targeted drug testing or screening (44, 50, 53, 56, 58, 79). However, these attempts did not result in developing a widely accepted drug so far. Consequently, researchers have not fully exploited the full potential of iPSCs until now, and novel approaches are currently underway to solve this issue. Besides drug discovery in the context of IEI, the further improvement of existing differentiation protocols from feeder-based GMP-incompatible systems to xeno-free and GMP-compatible protocols, drove iPSC-derived cells to the first clinical trials. In 2021, 19 therapeutic clinical studies were globally ongoing (98). Interestingly, none of them was related to IEI, highlighting the effort that should be invested in the next years to bring the undoubtable benefits that we can gain from the iPSC technology closer to the clinic. One of the main challenges in using iPSC-derived cells as a cell-based therapeutic intervention are the immunohistocompatibility issues arising from the use of allogeneic cells. Most clinical trials currently use allogeneic cells since the generation of autologous iPSCs is time-consuming, which becomes a particular issue when the recipient urgently needs cell therapy. Therefore several studies have tried to elaborate alternative options, either by developing cell banks with homozygous iPSC lines or by generating immunocompatible iPSCs through genetic manipulation (99–101).
Given the very young age of most patients with IEI, the elimination of the possible tumorigenicity of iPSCs and iPSC-derived cells, should be highly warranted, before the use of iPSC-derived cells for the treatment of children suffering from IEI. In order to diminish the impact of the integrating vectors on the iPSCs genetic stability and to provide a reliable tool to generate novel therapies against IEI, different reprogramming strategies have been developed, such as the use of non-integrating vectors, synthetic mRNAs, or integrating vectors that can be excised. Establishing a universal and highly standardized procedure for confirming the genetic stability and purity of iPSCs to get approved for clinical use, could be very beneficial. Usage of Next Generation Sequencing analysis should also be considered to guarantee the detection of all possible genetic anomalies and to ensure the production of high-quality iPSCs with limited risk for tumorigenicity (102). To further minimize the tumorigenic risk, in the last two decades, several systems have been developed for the elimination of aberrant cells using suicide gene technology (103–108). Most recently, immunodepletion has also been used to selectively deplete contaminating iPSCs with the help of monoclonal antibodies (109, 110) or chimerized monoclonal antibodies (111). Further optimization of these tools will significantly assist in facilitating the safe use of iPSC-derived cells in the clinical setting in the future.
While iPSC and thereof derived cells are used frequently for modeling IEI the clinical translation of cells to treat IEI is more in the future.
EN, JR, GH, and NL designed, wrote and approved the manuscript. All authors contributed to the article and approved the submitted version.
This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement No. 852178). The work is also funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy - EXC 2155 - project number 390874280 and REBIRTH Research Center for Translational Regenerative Medicine “Förderung aus Mitteln des Niedersächsischen Vorab” (grant: ZN3340).
We thank Shifaa Abdin and Mania Ackermann for critical review of the manuscript. The figure has been created with BioRender.com.
NL filed and licensed patents in the field of iPSC-derived macrophages outside of the MS. NL is a consultant for CATALENT outside of the MS.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Picard C, Bobby Gaspar H, Al-Herz W, Bousfiha A, Casanova JL, Chatila T, et al. International union of immunological societies: 2017 primary immunodeficiency diseases committee report on inborn errors of immunity. J Clin Immunol (2018) 38(1):96–128. doi: 10.1007/s10875-017-0464-9
2. Bousfiha A, Moundir A, Tangye SG, Picard C, Jeddane L, Al-Herz W, et al. The 2022 update of IUIS phenotypical classification for human inborn errors of immunity. J Clin Immunol (2022). doi: 10.1007/s10875-022-01352-z
3. Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. Human inborn errors of immunity: 2022 update on the classification from the international union of immunological societies expert committee. J Clin Immunol Published online June 2022, 1–35. doi: 10.1007/s10875-022-01289-3
4. Bousfiha A, Jeddane L, Picard C, Al-Herz W, Ailal F, Chatila T, et al. Human inborn errors of immunity: 2019 update of the IUIS phenotypical classification. J Clin Immunol (2020) 40(1):66–81. doi: 10.1007/s10875-020-00758-x
5. Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human inborn errors of immunity: 2019 update on the classification from the international union of immunological societies expert committee. J Clin Immunol (2020) 40(1):24–64. doi: 10.1007/s10875-019-00737-x
6. Pai S-Y, Logan BR, Griffith LM, Buckley RH, Parrott RE, Dvorak CC, et al. Transplantation outcomes for severe combined immunodeficiency, 2000-2009. N Engl J Med (2014) 371(5):434–46. doi: 10.1056/NEJMoa1401177
7. Speckmann C, Doerken S, Aiuti A, Albert MH, Al-Herz W, Allende LM, et al. A prospective study on the natural history of patients with profound combined immunodeficiency: An interim analysis. J Allergy Clin Immunol (2017) 139(4):1302–10.e4. doi: 10.1016/j.jaci.2016.07.040
8. Ye Z, Chou B-K, Cheng L. Promise and challenges of human iPSC-based hematologic disease modeling and treatment. Int J Hematol (2012) 95(6):601–9. doi: 10.1007/s12185-012-1095-9
9. Parekh C, Crooks GM. Critical differences in hematopoiesis and lymphoid development between humans and mice. J Clin Immunol (2013) 33(4):711–5. doi: 10.1007/s10875-012-9844-3
10. Monkley S, Krishnaswamy JK, Göransson M, Clausen M, Meuller J, Thörn K, et al. Optimised generation of iPSC-derived macrophages and dendritic cells that are functionally and transcriptionally similar to their primary counterparts. PloS One (2020) 15(12):e0243807. doi: 10.1371/journal.pone.0243807
11. van Wilgenburg B, Browne C, Vowles J, Cowley SA. Efficient, long term production of monocyte-derived macrophages from human pluripotent stem cells under partly-defined and fully-defined conditions. PloS One (2013) 8(8):e71098. doi: 10.1371/journal.pone.0071098
12. Ackermann M, Kempf H, Hetzel M, Hesse C, Hashtchin AR, Brinkert K, et al. Bioreactor-based mass production of human iPSC-derived macrophages enables immunotherapies against bacterial airway infections. Nat Commun (2018) 9(1):5088. doi: 10.1038/s41467-018-07570-7
13. Lyadova I, Gerasimova T, Nenasheva T. Macrophages derived from human induced pluripotent stem cells: The diversity of protocols, future prospects, and outstanding questions. Front Cell Dev Biol (2021) 9:640703. doi: 10.3389/fcell.2021.640703
14. Zhang H, Xue C, Shah R, Bermingham K, Hinkle CC, Li W, et al. Functional analysis and transcriptomic profiling of iPSC-derived macrophages and their application in modeling mendelian disease. Circ Res (2015) 117(1):17–28. doi: 10.1161/CIRCRESAHA.117.305860
15. Buchrieser J, James W, Moore MD. Human induced pluripotent stem cell-derived macrophages share ontogeny with MYB-independent tissue-resident macrophages. Stem Cell Rep (2017) 8(2):334–45. doi: 10.1016/j.stemcr.2016.12.020
16. Vaughan-Jackson A, Stodolak S, Ebrahimi KH, Browne C, Reardon PK, Pires E, et al. Differentiation of human induced pluripotent stem cells to authentic macrophages using a defined, serum-free, open-source medium. Stem Cell Rep (2021) 16(7):1735–48. doi: 10.1016/j.stemcr.2021.05.018
17. Ni Z, Knorr DA, Clouser CL, Hexum MK, Southern P, Mansky LM, et al. Human pluripotent stem cells produce natural killer cells that mediate anti-HIV-1 activity by utilizing diverse cellular mechanisms. J Virol (2011) 85(1):43–50. doi: 10.1128/JVI.01774-10
18. Knorr DA, Ni Z, Hermanson D, Hexum MK, Bendzick L, Cooper LJN, et al. Clinical-scale derivation of natural killer cells from human pluripotent stem cells for cancer therapy. Stem Cells Transl Med (2013) 2(4):274–83. doi: 10.5966/sctm.2012-0084
19. Flippe L, Gaignerie A, Sérazin C, Baron O, Saulquin X, Themeli M, et al. Rapid and reproducible differentiation of hematopoietic and T cell progenitors from pluripotent stem cells. Front Cell Dev Biol (2020) 8:577464. doi: 10.3389/fcell.2020.577464
20. Kawamoto H, Masuda K, Nagano S, Maeda T. Cloning and expansion of antigen-specific T cells using iPS cell technology: development of “off-the-shelf” T cells for the use in allogeneic transfusion settings. Int J Hematol (2018) 107(3):271–7. doi: 10.1007/s12185-018-2399-1
21. Iriguchi S, Yasui Y, Kawai Y, Arima S, Kunitomo M, Sato T, et al. A clinically applicable and scalable method to regenerate T-cells from iPSCs for off-the-shelf T-cell immunotherapy. Nat Commun (2021) 12(1):430. doi: 10.1038/s41467-020-20658-3
22. Nishimura T, Kaneko S, Kawana-Tachikawa A, Tajima Y, Goto H, Zhu D, et al. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell (2013) 12(1):114–26. doi: 10.1016/j.stem.2012.11.002
23. Wakao H, Yoshikiyo K, Koshimizu U, Furukawa T, Enomoto K, Matsunaga T, et al. Expansion of functional human mucosal-associated invariant T cells via reprogramming to pluripotency and redifferentiation. Cell Stem Cell (2013) 12(5):546–58. doi: 10.1016/j.stem.2013.03.001
24. Vizcardo R, Masuda K, Yamada D, Ikawa T, Shimizu K, Fujii SI, et al. Regeneration of human tumor antigen-specific T cells from iPSCs derived from mature CD8(+) T cells. Cell Stem Cell (2013) 12(1):31–6. doi: 10.1016/j.stem.2012.12.006
25. Themeli M, Kloss CC, Ciriello G, Fedorov VD, Perna F, Gonen M, et al. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat Biotechnol (2013) 31(10):928–33. doi: 10.1038/nbt.2678
26. Maeda T, Nagano S, Ichise H, Kataoka K, Yamada D, Ogawa S, et al. Regeneration of CD8αβ T cells from T-cell-Derived iPSC imparts potent tumor antigen-specific cytotoxicity. Cancer Res (2016) 76(23):6839–50. doi: 10.1158/0008-5472.CAN-16-1149
27. Minagawa A, Yoshikawa T, Yasukawa M, Hotta A, Kunitomo M, Iriguchi S, et al. Enhancing T cell receptor stability in rejuvenated iPSC-derived T cells improves their use in cancer immunotherapy. Cell Stem Cell (2018) 23(6):850–858.e4. doi: 10.1016/j.stem.2018.10.005
28. Orange JS, Stone KD, Turvey SE, Krzewski K. The wiskott-Aldrich syndrome. Cell Mol Life Sci (2004) 61(18):2361–85. doi: 10.1007/s00018-004-4086-z
29. Ackermann M, Rafiei Hashtchin A, Manstein F, Carvalho Oliveira M, Kempf H, Zweigerdt R, et al. Continuous human iPSC-macrophage mass production by suspension culture in stirred tank bioreactors. Nat Protoc (2022) 17(2):513–39. doi: 10.1038/s41596-021-00654-7
30. Zhu H, Kaufman DS. An improved method to produce clinical-scale natural killer cells from human pluripotent stem cells. Methods Mol Biol (2019) 2048:107–19. doi: 10.1007/978-1-4939-9728-2_12
31. Panicker LM, Miller D, Park TS, Patel B, Azevedo JL, Awad O, et al. Induced pluripotent stem cell model recapitulates pathologic hallmarks of gaucher disease. Proc Natl Acad Sci U S A. (2012) 109(44):18054–9. doi: 10.1073/pnas.1207889109
32. Rafiei Hashtchin A, Fehlhaber B, Hetzel M, Manstein F, Stalp JL, Glage S, et al. Human iPSC-derived macrophages for efficient staphylococcus aureus clearance in a murine pulmonary infection model. Blood Adv (2021) 5(23):5190–201. doi: 10.1182/bloodadvances.2021004853
33. Hale C, Yeung A, Goulding D, Pickard D, Alasoo K, Powrie F, et al. Induced pluripotent stem cell derived macrophages as a cellular system to study salmonella and other pathogens. PloS One (2015) 10(5):e0124307. doi: 10.1371/journal.pone.0124307
34. Nenasheva T, Gerasimova T, Serdyuk Y, Grigor’eva E, Kosmiadi G, Nikolaev A, et al. Macrophages derived from human induced pluripotent stem cells are low-activated “Naïve-like” cells capable of restricting mycobacteria growth. Front Immunol (2020) 11:1016. doi: 10.3389/fimmu.2020.01016
35. Hermanson DL, Bendzick L, Pribyl L, McCullar V, Vogel RI, Miller JS, et al. Induced pluripotent stem cell-derived natural killer cells for treatment of ovarian cancer. Stem Cells (2016) 34(1):93–101. doi: 10.1002/stem.2230
36. Gardner CL, Pavel-Dinu M, Dobbs K, Bosticardo M, Reardon PK, Lack J, et al. Gene editing rescues In vitro T cell development of RAG2-deficient induced pluripotent stem cells in an artificial thymic organoid system. J Clin Immunol (2021) 41(5):852–62. doi: 10.1007/s10875-021-00989-6
37. Themeli M, Chhatta A, Boersma H, Prins HJ, Cordes M, de Wilt E, et al. iPSC-based modeling of RAG2 severe combined immunodeficiency reveals multiple T cell developmental arrests. Stem Cell Rep (2020) 14(2):300–11. doi: 10.1016/j.stemcr.2019.12.010
38. Brauer PM, Pessach IM, Clarke E, Rowe JH, Ott de Bruin L, Lee YN, et al. Modeling altered T-cell development with induced pluripotent stem cells from patients with RAG1-dependent immune deficiencies. Blood (2016) 128(6):783–93. doi: 10.1182/blood-2015-10-676304
39. Chang C-W, Lai Y-S, Westin E, Khodadadi-Jamayran A, Pawlik KM, Lamb LSJ, et al. Modeling human severe combined immunodeficiency and correction by CRISPR/Cas9-enhanced gene targeting. Cell Rep (2015) 12(10):1668–77. doi: 10.1016/j.celrep.2015.08.013
40. Menon T, Firth AL, Scripture-Adams DD, Galic Z, Qualls SJ, Gilmore WB, et al. Lymphoid regeneration from gene-corrected SCID-X1 subject-derived iPSCs. Cell Stem Cell (2015) 16(4):367–72. doi: 10.1016/j.stem.2015.02.005
41. Rissone A, Weinacht KG, la Marca G, Bishop K, Giocaliere E, Jagadeesh J, et al. Reticular dysgenesis-associated AK2 protects hematopoietic stem and progenitor cell development from oxidative stress. J Exp Med (2015) 212(8):1185–202. doi: 10.1084/jem.20141286
42. Tilgner K, Neganova I, Singhapol C, Saretzki G, Al-Aama JY, Evans J, et al. Brief report: a human induced pluripotent stem cell model of cernunnos deficiency reveals an important role for XLF in the survival of the primitive hematopoietic progenitors. Stem Cells (2013) 31(9):2015–23. doi: 10.1002/stem.1456
43. Tsui M, Min W, Ng S, Dobbs K, Notarangelo LD, Dror Y, et al. The use of induced pluripotent stem cells to study the effects of adenosine deaminase deficiency on human neutrophil development. Front Immunol (2021) 12:748519. doi: 10.3389/fimmu.2021.748519
44. Genova E, Cavion F, Lucafò M, Pelin M, Lanzi G, Masneri S, et al. Biomarkers and precision therapy for primary immunodeficiencies: An In vitro study based on induced pluripotent stem cells from patients. Clin Pharmacol Ther (2020) 108(2):358–67. doi: 10.1002/cpt.1837
45. Ovchinnikov DA, Withey SL, Leeson HC, Lei UW, Sundarrajan A, Junday K, et al. Correction of ATM mutations in iPS cells from two ataxia-telangiectasia patients restores DNA damage and oxidative stress responses. Hum Mol Genet (2020) 29(6):990–1001. doi: 10.1093/hmg/ddaa023
46. Laskowski TJ, Van Caeneghem Y, Pourebrahim R, Ma C, Ni Z, Garate Z, et al. Gene correction of iPSCs from a wiskott-Aldrich syndrome patient normalizes the lymphoid developmental and functional defects. Stem Cell Rep (2016) 7(2):139–48. doi: 10.1016/j.stemcr.2016.06.003
47. Ingrungruanglert P, Amarinthnukrowh P, Rungsiwiwut R, Maneesri-le Grand S, Sosothikul D, Suphapeetiporn K, et al. Wiskott-Aldrich syndrome iPS cells produce megakaryocytes with defects in cytoskeletal rearrangement and proplatelet formation. Thromb Haemost (2015) 113(4):792–805. doi: 10.1160/TH14-06-0503
48. Broderick L, Clay GM, Blum RH, Liu Y, McVicar R, Papes F, et al. Disease-associated mutations in topoisomerase IIβ result in defective NK cells. J Allergy Clin Immunol (2022) 149(6):2171–6.e3. doi: 10.1016/j.jaci.2021.12.792
49. Karvonen E, Krohn KJE, Ranki A, Hau A. Generation and characterization of iPS cells derived from APECED patients for gene correction. Front Endocrinol (Lausanne) (2022) 13:794327. doi: 10.3389/fendo.2022.794327
50. Sens J, Hoffmann D, Lange L, Vollmer Barbosa P, Morgan M, Falk CS, et al. Knockout-induced pluripotent stem cells for disease and therapy modeling of IL-10-Associated primary immunodeficiencies. Hum Gene Ther (2021) 32(1–2):77–95. doi: 10.1089/hum.2020.235
51. Hamabata T, Umeda K, Kouzuki K, Tanaka T, Daifu T, Nodomi S, et al. Pluripotent stem cell model of shwachman-diamond syndrome reveals apoptotic predisposition of hemoangiogenic progenitors. Sci Rep (2020) 10(1):14859. doi: 10.1038/s41598-020-71844-8
52. Tulpule A, Kelley JM, Lensch MW, McPherson J, Park IH, Hartung O, et al. Pluripotent stem cell models of shwachman-diamond syndrome reveal a common mechanism for pancreatic and hematopoietic dysfunction. Cell Stem Cell (2013) 12(6):727–36. doi: 10.1016/j.stem.2013.04.002
53. Hoffmann D, Kuehle J, Lenz D, Philipp F, Zychlinski D, Lachmann N, et al. Lentiviral gene therapy and vitamin B3 treatment enable granulocytic differentiation of G6PC3-deficient induced pluripotent stem cells. Gene Ther (2020) 27(6):297–306. doi: 10.1038/s41434-020-0127-y
54. Nayak RC, Trump LR, Aronow BJ, Myers K, Mehta P, Kalfa T, et al. Pathogenesis of ELANE-mutant severe neutropenia revealed by induced pluripotent stem cells. J Clin Invest (2015) 125(8):3103–16. doi: 10.1172/JCI80924
55. Pittermann E, Lachmann N, MacLean G, Emmrich S, Ackermann M, G’hring G, et al. Gene correction of HAX1 reversed kostmann disease phenotype in patient-specific induced pluripotent stem cells. Blood Adv (2017) 1(14):903–14. doi: 10.1182/bloodadvances.2016003798
56. Jiang JX, Wellhauser L, Laselva O, Utkina I, Bozoky Z, Gunawardena T, et al. A new platform for high-throughput therapy testing on iPSC-derived lung progenitor cells from cystic fibrosis patients. Stem Cell Rep (2021) 16(11):2825–37. doi: 10.1016/j.stemcr.2021.09.020
57. Smirnikhina SA, Kondrateva EV, Adilgereeva EP, Anuchina AA, Zaynitdinova MI, Slesarenko YS, et al. P.F508del editing in cells from cystic fibrosis patients. PloS One (2020) 15(11):e0242094. doi: 10.1371/journal.pone.0242094
58. Merkert S, Schubert M, Olmer R, Engels L, Radetzki S, Veltman M, et al. High-throughput screening for modulators of CFTR activity based on genetically engineered cystic fibrosis disease-specific iPSCs. Stem Cell Rep (2019) 12(6):1389–403. doi: 10.1016/j.stemcr.2019.04.014
59. Jung M, Cordes S, Zou J, Yu SJ, Guitart X, Hong SG, et al. GATA2 deficiency and human hematopoietic development modeled using induced pluripotent stem cells. Blood Adv (2018) 2(23):3553–65. doi: 10.1182/bloodadvances.2018017137
60. Kuhn A, Ackermann M, Mussolino C, Cathomen T, Lachmann N, Moritz T. TALEN-mediated functional correction of human iPSC-derived macrophages in context of hereditary pulmonary alveolar proteinosis. Sci Rep (2017) 7(1):15195. doi: 10.1038/s41598-017-14566-8
61. Lachmann N, Happle C, Ackermann M, Lüttge D, Wetzke M, Merkert S, et al. Gene correction of human induced pluripotent stem cells repairs the cellular phenotype in pulmonary alveolar proteinosis. Am J Respir Crit Care Med (2013) 189(2):131126070906004. doi: 10.1164/rccm.201306-1012OC
62. Klatt D, Cheng E, Philipp F, Selich A, Dahlke J, Schmidt RE, et al. Targeted repair of p47-CGD in iPSCs by CRISPR/Cas9: Functional correction without cleavage in the highly homologous pseudogenes. Stem Cell Rep (2019) 13(4):590–8. doi: 10.1016/j.stemcr.2019.08.008
63. Sweeney CL, Zou J, Choi U, Merling RK, Liu A, Bodansky A, et al. Targeted repair of CYBB in X-CGD iPSCs requires retention of intronic sequences for expression and functional correction. Mol Ther (2017) 25(2):321–30. doi: 10.1016/j.ymthe.2016.11.012
64. Laugsch M, Rostovskaya M, Velychko S, Richter C, Zimmer A, Klink B, et al. Functional restoration of gp91phox-oxidase activity by BAC transgenesis and gene targeting in X-linked chronic granulomatous disease iPSCs. Mol Ther (2016) 24(4):812–22. doi: 10.1038/mt.2015.154
65. Flynn R, Grundmann A, Renz P, Hänseler W, James WS, Cowley SA, et al. CRISPR-mediated genotypic and phenotypic correction of a chronic granulomatous disease mutation in human iPS cells. Exp Hematol (2015) 43(10):838–848.e3. doi: 10.1016/j.exphem.2015.06.002
66. Dreyer A-K, Hoffmann D, Lachmann N, Ackermann M, Steinemann D, Timm B, et al. TALEN-mediated functional correction of X-linked chronic granulomatous disease in patient-derived induced pluripotent stem cells. Biomaterials (2015) 69:191–200. doi: 10.1016/j.biomaterials.2015.07.057
67. Brault J, Goutagny E, Telugu N, Shao K, Baquié M, Satre V, et al. Optimized generation of functional neutrophils and macrophages from patient-specific induced pluripotent stem cells: Ex vivo models of X(0)-linked, AR22(0)- and AR47(0)- chronic granulomatous diseases. Biores Open Access (2014) 3(6):311–26. doi: 10.1089/biores.2014.0045
68. Craig-Mueller N, Hammad R, Elling R, Alzubi J, Timm B, Kolter J, et al. Modeling MyD88 deficiency In vitro provides new insights in its function. Front Immunol (2020) 11:608802. doi: 10.3389/fimmu.2020.608802
69. Haake K, Neehus A-L, Buchegger T, Kühnel MP, Blank P, Philipp F, et al. Patient iPSC-Derived Macrophages to Study Inborn Errors IFN-γ Responsive Pathway. Cells (2020) 9(2):483. doi: 10.3390/cells9020483
70. Neehus A-L, Lam J, Haake K, Merkert S, Schmidt N, Mucci A, et al. Impaired IFNγ-signaling and mycobacterial clearance in IFNγR1-deficient human iPSC-derived macrophages. Stem Cell Rep (2018) 10(1):7–16. doi: 10.1016/j.stemcr.2017.11.011
71. Zimmer B, Ewaleifoh O, Harschnitz O, Lee YS, Peneau C, McAlpine JL, et al. Human iPSC-derived trigeminal neurons lack constitutive TLR3-dependent immunity that protects cortical neurons from HSV-1 infection. Proc Natl Acad Sci USA (2018) 115(37):E8775–82. doi: 10.1073/pnas.1809853115
72. Lafaille FG, Pessach IM, Zhang S-Y, Ciancanelli MJ, Herman M, Abhyankar A, et al. Impaired intrinsic immunity to HSV-1 in human iPSC-derived TLR3-deficient CNS cells. Nature (2012) 491(7426):769–73. doi: 10.1038/nature11583
73. Seki R, Ohta A, Niwa A, Sugimine Y, Naito H, Nakahata T, et al. Induced pluripotent stem cell-derived monocytic cell lines from a NOMID patient serve as a screening platform for modulating NLRP3 inflammasome activity. PloS One (2020) 15(8):e0237030. doi: 10.1371/journal.pone.0237030
74. Yokoyama K, Ikeya M, Umeda K, Oda H, Nodomi S, Nasu A, et al. Enhanced chondrogenesis of induced pluripotent stem cells from patients with neonatal-onset multisystem inflammatory disease occurs via the caspase 1-independent cAMP/protein kinase A/CREB pathway. Arthritis Rheumatol (Hoboken NJ). (2015) 67(1):302–14. doi: 10.1002/art.38912
75. Kitagawa Y, Kawasaki Y, Yamasaki Y, Kambe N, Takei S, Saito MK. Anti-TNF treatment corrects IFN-γ-dependent proinflammatory signatures in blau syndrome patient-derived macrophages. J Allergy Clin Immunol (2022) 149(1):176–188.e7. doi: 10.1016/j.jaci.2021.05.030
76. Takada S, Kambe N, Kawasaki Y, Niwa A, Honda-Ozaki F, Kobayashi K, et al. Pluripotent stem cell models of blau syndrome reveal an IFN-γ-dependent inflammatory response in macrophages. J Allergy Clin Immunol (2018) 141(1):339–49.e11. doi: 10.1016/j.jaci.2017.04.013
77. Marion W, Boettcher S, Ruiz-Torres S, Lummertz da Rocha E, Lundin V, Morris V, et al. An induced pluripotent stem cell model of fanconi anemia reveals mechanisms of p53-driven progenitor cell differentiation. Blood Adv (2020) 4(19):4679–92. doi: 10.1182/bloodadvances.2020001593
78. Suzuki NM, Niwa A, Yabe M, Hira A, Okada C, Amano N, et al. Pluripotent cell models of fanconi anemia identify the early pathological defect in human hemoangiogenic progenitors. Stem Cells Transl Med (2015) 4(4):333–8. doi: 10.5966/sctm.2013-0172
79. Liu G-H, Suzuki K, Li M, Qu J, Montserrat N, Tarantino C, et al. Modelling fanconi anemia pathogenesis and therapeutics using integration-free patient-derived iPSCs. Nat Commun (2014) 5:4330. doi: 10.1038/ncomms5330
80. Mu A, Hira A, Niwa A, Osawa M, Yoshida K, Mori M, et al. Analysis of disease model iPSCs derived from patients with a novel fanconi anemia-like IBMFS ADH5/ALDH2 deficiency. Blood (2021) 137(15):2021–32. doi: 10.1182/blood.2020009111
81. Kawasaki Y, Oda H, Ito J, Niwa A, Tanaka T, Hijikata A, et al. Identification of a high-frequency somatic NLRC4 mutation as a cause of autoinflammation by pluripotent cell-based phenotype dissection. Arthritis Rheumatol (Hoboken NJ). (2017) 69(2):447–59. doi: 10.1002/art.39960
82. Tanaka T, Takahashi K, Yamane M, Tomida S, Nakamura S, Oshima K, et al. Induced pluripotent stem cells from CINCA syndrome patients as a model for dissecting somatic mosaicism and drug discovery. Blood (2012) 120(6):1299–308. doi: 10.1182/blood-2012-03-417881
83. Park I-H, Arora N, Huo H, Maherali N, Ahfeldt T, Shimamura A, et al. Disease-specific induced pluripotent stem cells. Cell (2008) 134(5):877–86. doi: 10.1016/j.cell.2008.07.041
84. Apasov SG, Blackburn MR, Kellems RE, Smith PT, Sitkovsky MV. Adenosine deaminase deficiency increases thymic apoptosis and causes defective T cell receptor signaling. J Clin Invest (2001) 108(1):131–41. doi: 10.1172/JCI10360
85. Carson DA, Kaye J, Seegmiller JE. Lymphospecific toxicity in adenosine deaminase deficiency and purine nucleoside phosphorylase deficiency: possible role of nucleoside kinase(s). Proc Natl Acad Sci USA (1977) 74(12):5677–81. doi: 10.1073/pnas.74.12.5677
86. Kim VHD, Roifman CR, Grunebaum E. Neutropenia in patients with adenosine deaminase deficiency. Allergy Asthma Clin Immunol Off J Can Soc Allergy Clin Immunol (2014) 10(Suppl 1):A41. doi: 10.1186/1710-1492-10-S1-A41
87. Zaki-Dizaji M, Akrami SM, Azizi G, Abolhassani H, Aghamohammadi A. Inflammation, a significant player of ataxia-telangiectasia pathogenesis? Inflamm Res off J Eur Histamine Res Soc (2018) 67(7):559–70. doi: 10.1007/s00011-018-1142-y
88. Glocker EO, Kotlarz D, Boztug K, Gertz EM, Schäffer AA, Noyan F, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med (2009) 361(21):2033–45. doi: 10.1056/NEJMoa0907206
89. Ruemmele FM, El Khoury MG, Talbotec C, Maurage C, Mougenot JF, Schmitz J, et al. Characteristics of inflammatory bowel disease with onset during the first year of life. J Pediatr Gastroenterol Nutr (2006) 43(5):603–9. doi: 10.1097/01.mpg.0000237938.12674.e3
90. Zou J, Sweeney CL, Chou BK, Choi U, Pan J, Wang H, et al. Oxidase-deficient neutrophils from X-linked chronic granulomatous disease iPS cells: functional correction by zinc finger nuclease-mediated safe harbor targeting. Blood (2011) 117(21):5561–72. doi: 10.1182/blood-2010-12-328161
91. Brault J, Vaganay G, Le Roy A, Lenormand J-L, Cortes S, Stasia MJ. Therapeutic effects of proteoliposomes on X-linked chronic granulomatous disease: proof of concept using macrophages differentiated from patient-specific induced pluripotent stem cells. Int J Nanomedicine (2017) 12:2161–77. doi: 10.2147/IJN.S128611
92. Merling RK, Kuhns DB, Sweeney CL, Wu X, Burkett S, Chu J, et al. Gene-edited pseudogene resurrection corrects p47(phox)-deficient chronic granulomatous disease. Blood Adv (2017) 1(4):270–8. doi: 10.1182/bloodadvances.2016001214
93. Schwaid AG, Spencer KB. Strategies for targeting the NLRP3 inflammasome in the clinical and preclinical space. J Med Chem (2021) 64(1):101–22. doi: 10.1021/acs.jmedchem.0c01307
94. Vande Walle L, Stowe IB, Šácha P, Lee Bl, Demon D, Fossoul A, et al. MCC950/CRID3 potently targets the NACHT domain of wild-type NLRP3 but not disease-associated mutants for inflammasome inhibition. PloS Biol (2019) 17(9):e3000354. doi: 10.1371/journal.pbio.3000354
95. Ebeling MC, Geng Z, Kapphahn RJ, Roehrich H, Montezuma SR, Dutton JR, et al. Impaired Mitochondrial Funct iPSC-Retinal Pigment Epithelium Complement Factor H Polymorphism Age-Related Macular Degeneration. Cells (2021) 10(4):789. doi: 10.3390/cells10040789
96. Koolen L, Gagliardi G, Ten Brink SCA, de Breuk A, Heesterbeek TJ, Hoyng CB, et al. Generation and characterization of human induced pluripotent stem cells (iPSCs) from three patients with age-related macular degeneration carrying rare variants in the CFH gene. Stem Cell Res (2022) 60:102669. doi: 10.1016/j.scr.2022.102669
97. Río P, Navarro S, Wang W, Sánchez-Domínguez R, Pujol RM, Segovia JC, et al. Successful engraftment of gene-corrected hematopoietic stem cells in non-conditioned patients with fanconi anemia. Nat Med (2019) 25(9):1396–401. doi: 10.1038/s41591-019-0550-z
98. Kim JY, Nam Y, Rim YA, Ju JH. Review of the current trends in clinical trials involving induced pluripotent stem cells. Stem Cell Rev Rep (2022) 18(1):142–54. doi: 10.1007/s12015-021-10262-3
99. Jang Y, Choi J, Park N, Kang J, Kim M, Kim Y, et al. Development of immunocompatible pluripotent stem cells via CRISPR-based human leukocyte antigen engineering. Exp Mol Med (2019) 51(1):1–11. doi: 10.1038/s12276-018-0190-2
100. Rim YA, Park N, Nam Y, Ham DS, Kim JW, Ha HY, et al. Recent progress of national banking project on homozygous HLA-typed induced pluripotent stem cells in south Korea. J Tissue Eng Regener Med (2018) 12(3):e1531–6. doi: 10.1002/term.2578
101. Lanza R, Russell DW, Nagy A. Engineering universal cells that evade immune detection. Nat Rev Immunol (2019) 19(12):723–33. doi: 10.1038/s41577-019-0200-1
102. Zhong C, Liu M, Pan X, Zhu H, et al. Tumorigenicity risk of iPSCs in vivo: nip it in the bud. Precis Clin Med (2022) 5(1):pbac004. doi: 10.1093/pcmedi/pbac004
103. Sułkowski M, Konieczny P, Chlebanowska P, Majka M. Introduction of exogenous HSV-TK suicide gene increases safety of keratinocyte-derived induced pluripotent stem cells by providing genetic “Emergency exit” switch. Int J Mol Sci (2018) 19(1):197. doi: 10.3390/ijms19010197
104. Liang Q, Monetti C, Shutova MV, Neely EJ, Hacibekiroglu S, Yang H. Linking a cell-division gene and a suicide gene to define and improve cell therapy safety. Nature (2018) 563(7733):701–4. doi: 10.1038/s41586-018-0733-7
105. Shi Z-D, Tchao J, Wu L, Carman AJ. Precision installation of a highly efficient suicide gene safety switch in human induced pluripotent stem cells. Stem Cells Transl Med (2020) 9(11):1378–88. doi: 10.1002/sctm.20-0007
106. Dahlke J, Schott JW, Vollmer Barbosa P, Klatt D, Selich A, Lachmann N, et al. Efficient genetic safety switches for future application of iPSC-derived cell transplants. J Pers Med (2021) 11(6):565. doi: 10.3390/jpm11060565
107. Lee M, Ha J, Son YS, Ahn H, Jung KB, Son MY, et al. Efficient exogenous DNA-free reprogramming with suicide gene vectors. Exp Mol Med (2019) 51(7):1–12. doi: 10.1038/s12276-019-0282-7
108. Zhou X, Naik S, Dakhova O, Dotti G, Heslop HE, Brenner MK. Serial activation of the inducible caspase 9 safety switch after human stem cell transplantation. Mol Ther (2016) 24(4):823–31. doi: 10.1038/mt.2015.234
109. Park J, Lee DG, Lee NG, Kwon MG, Son YS, Son MY, et al. Monoclonal antibody K312-based depletion of pluripotent cells from differentiated stem cell progeny prevents teratoma formation. BMB Rep (2022) 55(3):142–7. doi: 10.5483/BMBRep.2022.55.3.090
110. Tang C, Lee AS, Volkmer J-P, Sahoo D, Nag D, Mosley AR, et al. An antibody against SSEA-5 glycan on human pluripotent stem cells enables removal of teratoma-forming cells. Nat Biotechnol (2011) 29(9):829–34. doi: 10.1038/nbt.1947
Keywords: inborn errors of immunity, iPSCs, disease modeling, cell therapies, immune cells, macrophages
Citation: Nikolouli E, Reichstein J, Hansen G and Lachmann N (2022) In vitro systems to study inborn errors of immunity using human induced pluripotent stem cells. Front. Immunol. 13:1024935. doi: 10.3389/fimmu.2022.1024935
Received: 22 August 2022; Accepted: 28 October 2022;
Published: 17 November 2022.
Edited by:
Irina A. Tuzankina, Institute of Immunology and Physiology (RAS), RussiaReviewed by:
Ute Modlich, Paul-Ehrlich-Institut (PEI), GermanyCopyright © 2022 Nikolouli, Reichstein, Hansen and Lachmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nico Lachmann, TGFjaG1hbm4ubmljb0BtaC1oYW5ub3Zlci5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.