 Jiawen Huang
Jiawen Huang Xiaobing Huang
Xiaobing Huang Juan Huang
Juan Huang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 28 September 2022
Sec. Cancer Immunity and Immunotherapy
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1019115
This article is part of the Research Topic Community Series in Manipulating the Immunological Tumor Microenvironment - Volume II View all 20 articles
In the past decade, the emergence of chimeric antigen receptor (CAR) T-cell therapy has led to a cellular immunotherapy revolution against various cancers. Although CAR-T cell therapies have demonstrated remarkable efficacy for patients with certain B cell driven hematological malignancies, further studies are required to broaden the use of CAR-T cell therapy against other hematological malignancies. Moreover, treatment failure still occurs for a significant proportion of patients. CAR antigen loss on cancer cells is one of the most common reasons for cancer relapse. Additionally, immune evasion can arise due to the hostile immunosuppressive tumor microenvironment and the impaired CAR-T cells in vivo persistence. Other than direct antitumor activity, the adverse effects associated with CAR-T cell therapy are another major concern during treatment. As a newly emerged treatment approach, numerous novel preclinical studies have proposed different strategies to enhance the efficacy and attenuate CAR-T cell associated toxicity in recent years. The major obstacles that impede promising outcomes for patients with hematological malignancies during CAR-T cell therapy have been reviewed herein, along with recent advancements being made to surmount them.
In recent years, cellular immunotherapy has emerged as one of the leading areas of on-going researches and clinical therapies. Allogeneic hematopoietic stem cell transplantation (HSCT) is one of the oldest forms of cellular immunotherapy, and it forms the theoretical basis for cellular immunotherapy and provides us the ability to harness immune system to eradicate malignancies. The underlying curative mechanism associated with HSCT against hematological malignancies is the graft-versus-leukemia (GVL) or graft-versus-tumor (GVT) effect (i.e., donor immune cells against abnormal leukemia/tumor cells in patient’s body) (1, 2). The ability of immune cells to recognize target cells to exert their function forms the basis of GVL or GVT. Therefore, a new era in cellular immunotherapy was initiated with the advent of genetically engineering T cell receptor to enable the targeting of a specific tumor antigen, which became the rational basis for chimeric antigen receptor (CAR) T-cell therapy (3, 4).
Since its first Food and Drug Administration (FDA) approval in 2017, CAR-T cell therapy has shown promising efficacy against certain hematological malignancies and even provides a curative treatment strategy for patients with advanced diseases, such as B cell acute lymphoblastic leukemia (B-ALL) or lymphoma. Currently, the major approved CAR-T cell therapy targets are B cell maturation antigen (BCMA) for multiple myeloma (MM) (5) and CD19 for various lymphoid malignancies including B-ALL and diffuse large B-cell lymphoma (DLBCL) (6–9).
Despite great success, barriers still remain that prevent the extension of CAR-T cell therapy beyond B cell/plasma cell driven hematological malignancies. Moreover, even for B cell driven cancers, the efficacy of CAR-T cell therapy is far from being satisfactory and great efforts should be done to improve clinical outcomes. As a newly emerged therapeutic strategy, newly developed direct modifications or combination therapies for CAR-T cell therapy are booming in recent years and they have deeply reshaped CAR-T cell therapy in hematological malignancies. These approaches can be categorized into four major aspects, aiming to address the four major hurdles for CAR-T cell therapy in hematological malignancies: tumor heterogeneity/antigen loss, immunosuppressive tumor microenvironment, CAR-T cell exhaustion (or poor persistence) and severe adverse-effects. This review will focus on recent novel approaches that have provided refinements and potential solutions to these limitations.
Despite the robust efficacy of the currently approved CAR-T cell products, follow-up studies still demonstrated a high rate of posttherapy relapse (10). The most common cause of resistance is via antigen loss/downregulation, which is for the selective pressure of CAR-T cells (11, 12). Such target antigen-negative relapses after CAR-T cell therapy usually arise from pre-existing antigen-negative cancer cells, or cells with altered/mutated antigen expression (12). Therefore, expanding the potential CAR-T cell target repertoires or overcoming antigen loss are key to ensure outcomes of CAR-T cell therapy. Besides the commonly adopted multi-specific CAR or dual CAR strategies, here we will discuss several novel aspects of antigen selections and methods to overcome antigen loss.
Currently, CAR-T cell targets have mostly been restricted to extracellular tumor-associated antigens (TAA), which greatly limits the wider application of this promising therapy. This is a more pressing concern for hematological malignancies, as their mutation burden is lower, leading to a lower abundance of usable TAAs (13). Whereas, a rich range of intracellular oncoproteins have not been explored as targets. Therefore, T-cell receptor (TCR)-like CARs have been proposed as a means to utilize these targets. For example, intracellular protein Wilms tumor 1 (WT1) is an oncogene that promotes cell proliferation and differentiation. It is overexpressed in leukemia and lymphoma with limited expression in normal tissues (14, 15). The use of TCR-like WT1-CAR-T cell therapy has led to enhanced survival of leukemia-bearing mice (16, 17). Its principle is to combine an extracellular single chain variable fragment (scFv) that recognize the WT1 peptide-human leukocyte antigens (HLA) complex (as a full TCR do) with other CAR elements to generate TCR-like CAR-T cells (16, 17). Besides WT1, other intracellular antigen peptide-HLA complexes for CAR-T cell therapy have been investigated, including NY-ESO-1/HLA-A2 for potentially MM (18), HA-1H (derived from minor histocompatibility antigen 1, HMHA1)/HLA-A2 for chronic myelogenous leukemia (19), and SSX2/HLA∗0201 for acute myeloid leukemia (AML) (20). However, one of the difficulties of TCR-like CAR is that the target peptide and its associated HLA allele vary in different patients and cancers (e.g., WT1235-243/HLA-A*2402 (16) or WT1126-134/HLA-A*02:01 (17)). Hence, the applicability of TCR-like CAR-T cell needs to be evaluated on a case-by-case basis.
The increasing potential target repertoires enable the development of more comprehensive strategies against tumor heterogeneity and antigen loss. However, we should not limit our sights in simply identifying an excessive number of antigens and generating numerous CAR-T cell products to combat antigen loss. Previous studies suggest that the combination of multiple types of CAR-T cells may lead to growth competition, which severely impairs CAR-T cell proliferation (21). With proper design, a monospecific CAR-T product can be directed to multiple targets. For instance, the B-cell activating factor (BAFF) ligand binds to three different receptors BAFF receptor, BCMA, as well as transmembrane activator and CAML interactor (TACI) on mature B cells (22). Almost all B cell driven cancers express at least one of these receptors (23–27). Therefore, a single ligand-based BAFF-CAR-T cell product is capable of binding to all three of these receptors, which avoids the possibility of growth competition, enables broad antitumor activity and greatly minimizes the possibility of antigen escape (28).
Natural killer (NK) cells are ideal candidate for the elimination of antigen-negative cancer cells. Therefore, CAR-T cells can be converted into NK-like cells by pharmacologic agents to obtain the ability to overcome antigen loss and tumor heterogeneity. Bryostatin is an oxygenated macrolide that increases CD22 expression in ALL and chronic lymphocytic leukemia cells (29, 30). Combination therapy of bryostatin and CD22-CAR-T cells increases the CAR-T cell activity by increasing CD22 expression and sensitizing leukemia cells to CAR-T cell antigen-non-specific killing (31). However, this effect is limited to B-ALL cells, and does not affect Burkitt lymphoma cells.
Perhaps the simplest way to prevent an antigen-negative relapse is to actively avoid antigen loss. Moreover, CAR-T cell activity is closely related to the expression level of its antigen on the cell surface. Therefore, treatments that maintain high antigen concentration on the cell surface may be able to prevent loss-of-antigen relapse. Combination of bryostatin and CD22-CAR-T cells discussed in previous section is an example of this strategy (31). In addition, gamma (γ)-secretase cleaves BCMA on cell surface (32) and therefore leads to loss of targets for BCMA-CAR-T cells. Inhibition of γ-secretase in murine models can reduce BCMA antigen loss and improve BCMA-CAR-T cell anti-tumor effect (33). Clinical trials combining γ-secretase inhibitors and BCMA-CAR-T cells are currently ongoing (NCT03502577). Similarly, all-trans retinoic acid (ATRA) can enhance BCMA expression on MM cells. It acts synergistically with the γ-secretase inhibitor to boost the function of BCMA-CAR-T cells (34).
Another direct way to prevent loss-of-antigen relapse is to artificially display antigen on cancer cells. Su et al. (35), designed a novel CAR-T cell engager which binds CD20 and displays CD19 on CD20+ lymphoma cells. In mice transplanted with JeKo mantle cell lymphoma cells, co-administration of CAR-T cell engager and CD19-CAR-T cells successfully eliminates both CD19+ and CD19- lymphoma cells and prevent antigen-negative relapse. By changing the specificity of the engager, single type of CAR-T cells can be directed to different cancer subclones to overcome tumor heterogeneity. Moreover, by using single type of CAR-T cells, growth competition of multiple types of CAR-T cells can be avoided, further enhancing the anti-tumor activity.
The tumor microenvironment (TME) can greatly impact the efficacy of CAR-T cell therapies. Numerous signals from cancer cells and surrounding cells have significant impacts on anti-tumor immunity (36–39). As the TME of hematological malignancy is mainly immunosuppressive, therefore it becomes another major cause of treatment failure. Indeed, the inhibitory immune checkpoint ligands Programmed death-1 ligand-1/2 (PD-L1/L2) are expressed in B cell driven malignancies (40). They bind to Programmed death-1 (PD-1) expressed on activated CAR-T cells, inhibiting their function. Recent study also showed that the TME of large B cell lymphoma is the key determinant of CD19-CAR-T cell activity (41). Several strategies have been proposed to overcome the immunosuppressive effects of the TME, including the blockade of immune checkpoints, promoting CAR-T cell activity and remodeling the TME (Figure 1).
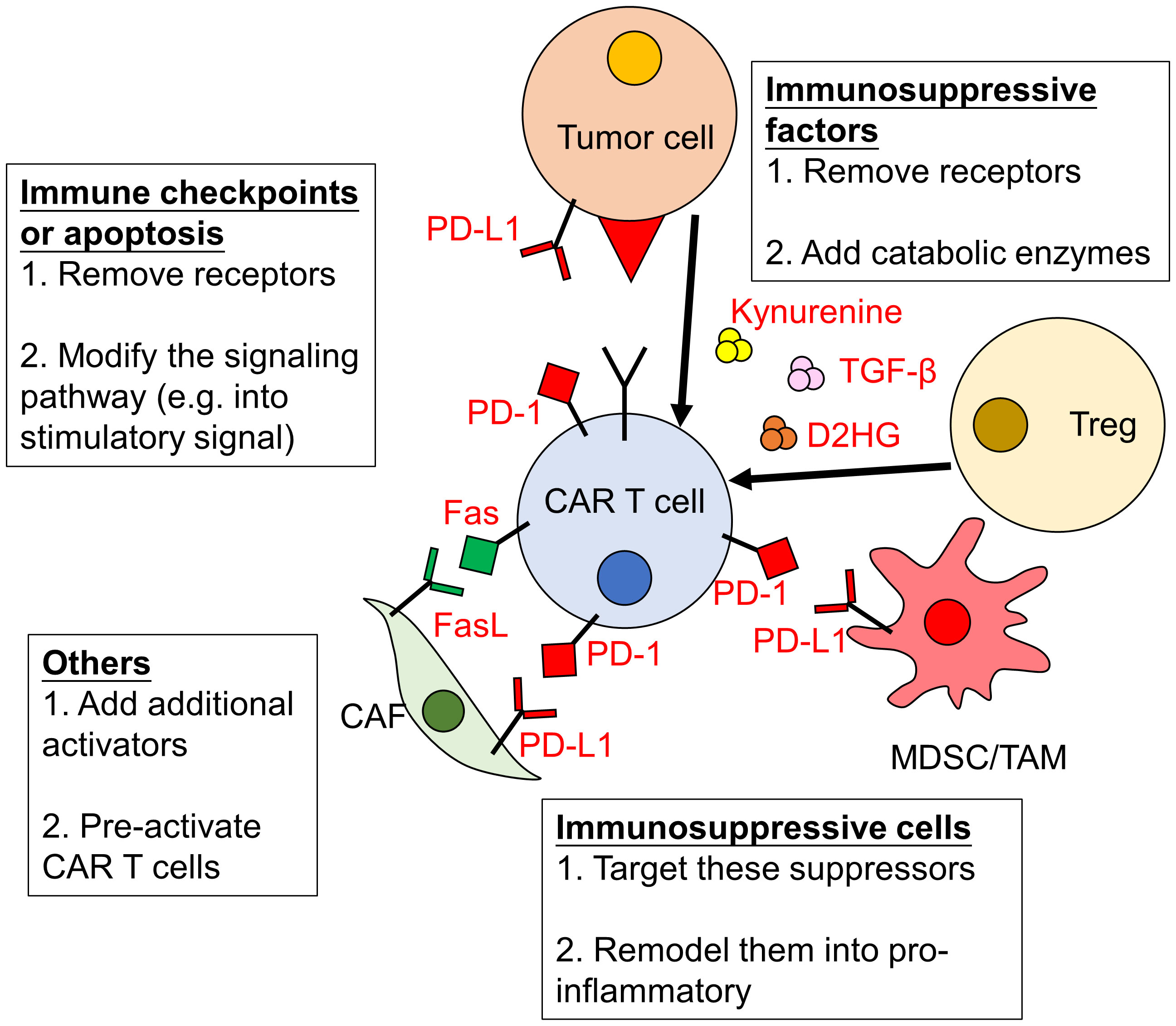
Figure 1 Immunosuppressive effects of the TME on CAR-T cells and strategies to overcome them. The activity of CAR-T cells can be severely impaired by the tumor microenvironment (TME). The immunosuppressive effects of the TME are due to increased level of immune checkpoints/inhibitors and immunosuppressive factors/cells. Strategies to overcome them include: (1) removing or modifying related signaling pathways; (2) eliminating or remodeling immunosuppressive cells and (3) enhancing the activation of CAR-T cells. PD-1, Programmed death-1; PD-L1, Programmed death-1 ligand-1; CAF, cancer-associated fibroblasts; MDSC, myeloid-derived suppressor cells; TAM, tumor associated macrophages; Treg, regulatory T cells; TGF-β, transforming growth factor -β; D2HG, D-2-hydroxyglutarate.
By directly removing or blocking the immune checkpoints on CAR-T cells, their inhibitory effects might be avoided and therefore the anti-tumor effects of CAR-T cells are promoted. For example, the production of PD-1 resistant CD19-, CD133- and C-type lectin-like molecule-1 (CLL-1)-CAR-T cells achieved by the knockout of PD-1/PD-1 homolog genes can enhance CAR-T cell therapy (42–44). Recently, two relapsed/refractory AML patients who received HSCT and CD38-CAR-T cell therapy before showed continuous remission after receiving PD-1-silenced CLL-1-CAR-T cell therapy (45). Resistance to PD-1 signaling could also be acquired by CAR-T cells secreting soluble PD-1-blocking scFv (46). This strategy demonstrated matching efficacies against hematological malignancy models when compared to a combination therapy that incorporated CAR-T cells and an anti-PD1 antibody.
Besides direct blockade of PD-1, high levels of PD-L1 in the TME can also be leveraged by generating CD19-directed CAR-T cells that secrete soluble PD-1. These cells exhibited enhanced eradication efficiency against CD19+PD-L1+ cancer cells. It is thought that the increased efficacy associated with PD-1 secretion is due to a protective effect from apoptosis that enhances CAR-T cell persistence (47). Similar approaches are used for another immune checkpoint T-cell immunoglobulin and mucin-domain containing-3 (TIM-3) by generating TIM-3-CD28 fusion proteins in CD19- or CD138-CAR-T cells. It converts the immunosuppressive TIM-3 signaling to CAR-T cells immunostimulation (48, 49).
Other than PD-1 and TIM-3, Carnevale et al. (50), identified a novel immune checkpoint, RAS P21 Protein Activator 2 (RASA2), in T cells. RASA2 is a RAS GTPase-activating protein and is upregulated upon chronic stimulation. As demonstrated in multiple murine models including leukemia, CD19-CAR-T cells with RASA2-ablation experience increased RAS/MAPK signaling and activation, leading to enhanced and persistent anti-tumor activity (50).
However, it is notable that the effect of PD-1 removal in CAR-T cells should be carefully evaluated. Studies showed that using protein trap to remove PD-1 in CD19-CAR-T cells is associated with increased CAR-T cell activation, but also with diminished T cell survival and therefore reduced T cell cytotoxicity (51). Furthermore, these CAR-T cells also mature earlier and are quickly exhausted by T cell immunoreceptor with Ig and ITIM domains (TIGIT) upregulation (51).
Other than immune checkpoints like PD-1 signaling, numerous pathways have been linked to the modulation of CAR-T cell activity within an immunosuppressive TME. Although transforming growth factor beta (TGF-β) is a tumor suppressor in the premalignant stage, it promotes tumor transformation and growth at later disease stages (52). TGF-β also suppresses T cell function and therefore T cells with a dominant-negative form of the TGF-β receptor are resistant to TGF-β-mediated inhibition (53). As TGF-β is commonly expressed in NY-ESO-1+ cancers (53), the combination of TCR-like CAR-T cells targeting NY-ESO-1/HLA (18) with TGF-β blockade could be applicable for NY-ESO-1+ hematological malignancies as well.
Additionally, expression of Fas ligand in the TME can induce T cell apoptosis (37). Therefore, the expression of a dominant negative form of Fas receptor protects T cells in adoptive cell therapy (ACT) from Fas-induced apoptosis (54). Furthermore, Fas signal can be converted into T cell stimulatory signal via the transduction of T cells with Fas-4-1BB, which linked the Fas intracellular tail with costimulatory 4-1BB (CD137, Tumor necrosis factor receptor superfamily 9). This construct leads to increased T cell survival and enhanced T cell therapy against murine leukemia models (55). Similar strategies could be applied to CAR-T cells as well.
The TME modulates CAR-T cell metabolism as well. In the TME, kynurenine is often present, which inhibits glucose uptake and T cell function (56). Therefore, overexpression of kynureninase in CD19-CAR-T cells can protect them from immunosuppression by catabolizing kynurenine, and they exhibit excellent anti-tumor activity in mice bearing ALL cells (56). Similarly, the production of D-2-hydroxyglutarate (D2HG) by IDH1/2-mutated cancer cells can have critical implications for tumorigenesis and immunosuppression in the TME (38, 39). The overexpression of D2HG dehydrogenase (D2HGDH) in CD19-CAR-T cells decreases serum D2HG in mice bearing NALM6 leukemia cells with mutation IDH1 leading to enhanced T cell function and persistence (57).
The site of injection is also important for CAR-T cells activity. The intracerebroventricular (ICV) injection, rather than intravenous (IV) injection, of CD19-CAR-T cells is most effective (in terms of complete and durable eradication) for the treatment of murine model of central nervous system and systemic lymphoma (58). The superior CAR-T cell activity in this case is due to cerebrospinal fluid exposure of CAR-T cells in the ICV environment, which may alter cellular metabolism (58). Hence, the infusion of CAR-T cells to an immunostimulatory environment can pre-activate CAR-T cells and prevent immunosuppression.
Rather than targeting immunosuppressive signaling, the activation of CAR-T cells can increase cytotoxicity within the TME. CD58 is a ligand for T cell costimulatory receptor CD2 (59, 60), which is frequently mutated in B cell driven malignancies. Notably, these mutations are associated with poor CD19-CAR-T cell therapy outcomes (61). The linkage of a second CAR construct (the CD2-signaling domain with a CAR intracellular domain) allows CAR-T cells to be activated in the absence of CD58 expression and increases their function (61). However, further investigations are needed to examine whether this results in T cell exhaustion.
In addition to CD58, other methods to promote CAR-T cell activity focus on improvements to their persistence, which are discussed in a separate section (“Approaches to prolong the persistence of CAR-T cells”).
Besides promoting CAR-T cell resistance to immunosuppression, directly targeting or even remodeling the TME provides an alternative approach to overcome its immunosuppressive effect.
In MM, cancer-associated fibroblasts (CAFs) in the immunosuppressive TME play important roles in promoting tumor growth and inhibiting CAR-T cell function (62, 63). Therefore, Sakemura et al. (63), developed a dual-targeting CAR-T cell strategy, which targets both bone marrow CAFs (via SLAMF7 or Fibroblast activation protein/FAP) and MM cells (via BCMA). Under this condition, the anti-tumor activity of CAR-T cells is significantly improved. Besides CAFs, tumor associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs) are also major contributors to the immunosuppressive effects in the TME via producing immunosuppressive cytokines and expressing immune checkpoints, which lead to T cell inactivation (64). Ultimately, these factors can cause CAR-T cell treatment failure. For example, it has been demonstrated that increased TAM infiltration is associated with a poorer CD19-CAR-T cell therapy outcome against B-cell non-Hodgkin’s lymphoma (65). Therefore, targeting MDSCs/TAMs by Gemtuzumab ozogamicin (66, 67), or Phosphoinositide 3-kinase (PI3K) blockade (68) are able to boost CAR-T cell activity against multiple cancer types, such as non-Hodgkin’s lymphoma and AML.
TME remodulation is an alternative strategy for the prevention of immunosuppressive effects on CAR-T cells, which can even further increase CAR-T cell functions and contribute to sustained cancer remission. The use of CAR-T cells expressing immunostimulants or cytokines has been proposed for this purpose. IL-12 can enhance T cell activity, interferon γ (IFNγ) production and Th1 polarization, as well as serve as chemoattractant for various immune cells (69). Thus, CD19-CAR-T cells with IL-12-secreting ability could reshape the TME and induce robust T cell activity against lymphoma (70). However, constitutive IL-12 secretion is associated with toxic adverse effects, which necessitates the development of an inducible IL-12 secretion system (71). Recently, IL-12 nanostimulant-engineered CAR-T cells (INS-CAR T) provides a potential solution for this problem. In the presence of tumor antigen, IL-12-loaded human serum albumin (HSA) nanoparticles are released from INS-CAR T, allowing for local and controlled IL-12 release (and recruitment of other CAR-T cells) (72). Besides IL-12, IL-18 also enhances Th1 function, and IL-18-expressing CD19-CAR-T cells exhibit augmented activity in mice bearing ALL tumor (73).
Furthermore, whilst TAMs and MDSCs are known to be immunosuppressor, they have the plasticity to be transformed into M1-like pro-inflammatory phenotypes (74), which can enhance CAR-T cell activity. This transformation can be achieved by the co-administration of folate-targeted Toll-like receptor 7 agonist (FA-TLR7-1A) (75) or attenuated bacterial strain (Brucella melitensis) with TME homing ability (76). Engineered CAR-T cells secreting immunostimulatory RNA RN7SL1 are also able to inhibit MDSC development and activate immune cells in the TME (77). Although these approaches are initially designed to improve CAR-T cell activity against solid tumors, CD19-CAR-T cells are still used in these studies (75, 77). Therefore, it is worthy to investigate if similar strategies can be applied to certain hematological malignancies like lymphomas.
Recently, an immunostimulatory gene therapy using oncolytic viruses has been developed. Oncolytic viruses can specifically infect and kill tumor cells, promote TME remodeling and induce an enhanced immune response (78, 79). In murine models of human B-cell lymphoma, oncolytic adenovirus LOAd703-infected lymphoma cells exhibited enhanced immunogenic profiles with upregulated co-stimulatory molecules and chemokines, leading to enhanced CD19-CAR-T cell recruitment and anti-tumor activity (80). However, type I IFN expressed upon oncolytic virus infection may dampen the efficacy of CAR-T cells (81). Therefore, further investigations are required to optimize the combination therapy of oncolytic viruses and CAR-T cells.
The poor persistence of CAR-T cells is another major obstacle that stands in the way of long-term cancer remission of CAR-T cell therapy (82). This is often due to a combination of inter-related factors, including T cell exhaustion, transcriptional changes, metabolic changes and influences from the TME (83, 84). The persistence of CAR-T cells can be prolonged by enhancing CAR-T cell activity and expansion, improving fitness, reducing exhaustion, engineering the cells to have a less-differentiated state, altering their metabolism, and improving CAR design (Figure 2). These factors are inter-related. For example, cytokines that regulate T cell activity will alter the state of T cell differentiation and metabolism (85). The following sub-sections mostly focus on the major direct effects associated with each approach.
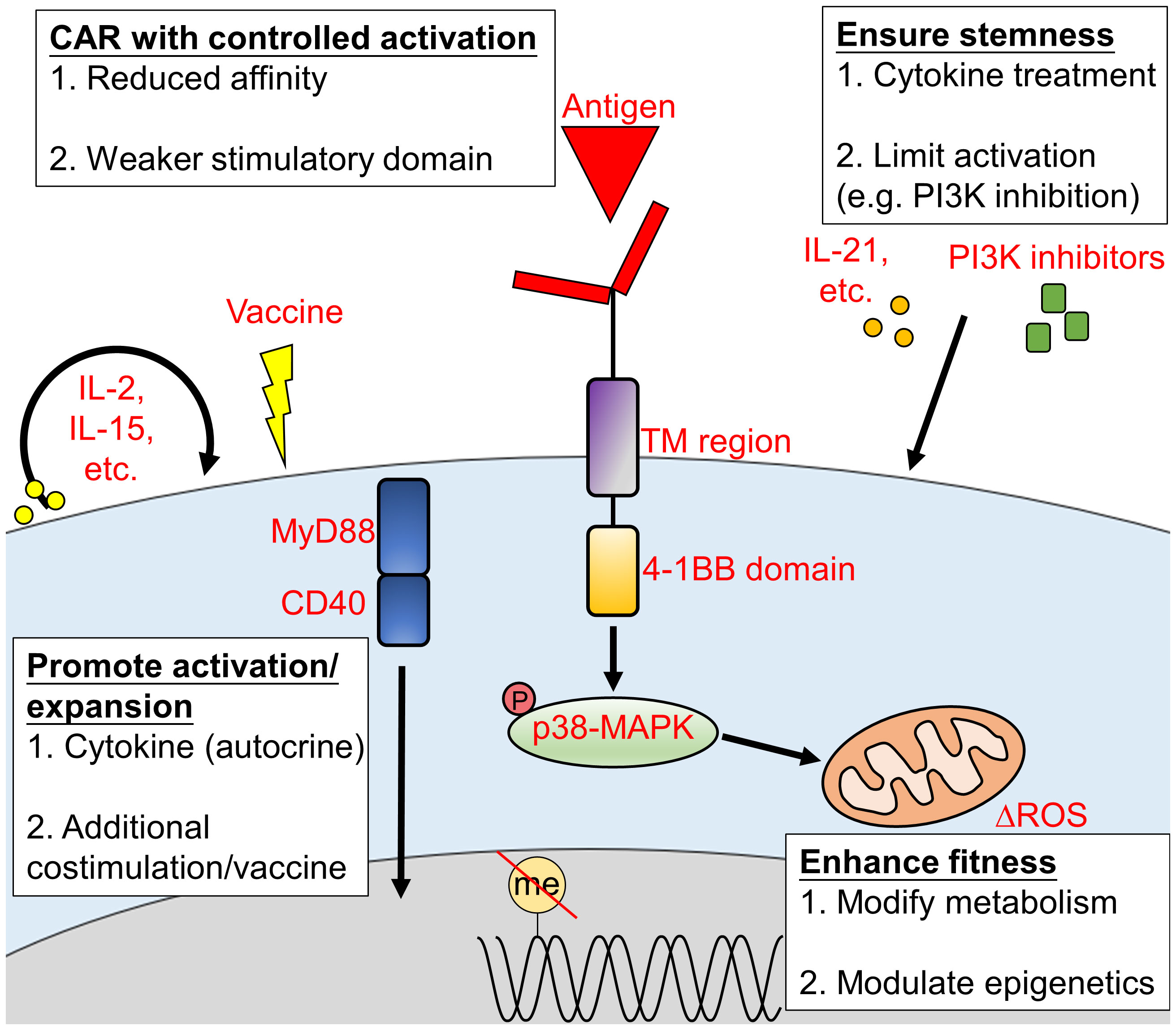
Figure 2 Approaches to promote CAR-T cell persistence in vivo. The persistence of CAR-T cells during treatment is crucial to achieve a durable and long-lasting outcome. CAR-T cell persistence can be extended via (1) the promotion of CAR-T cell activation or expansion; (2) enhanced CAR-T cell fitness; (3) proper CAR construct design and (4) increased CAR-T cells stemness. TM, transmembrane; ROS, reactive oxygen species.
Various cytokines are closely related to a diverse array of T cell behaviors. Thus, efforts have been made to engineer CAR-T cells with the ability to deliver cytokines in situ, or with special cytokine-induced signaling pathways to promote their efficacy. Cytokines can function in paracrine fashion that remodeling the surrounding TME (as introduced in the “Remodeling the immunosuppressive TME” section). They can also directly enhance CAR-T cells in an autocrine manner to boost their activity and expansion. IL-2 is one of the most important cytokines for T cell proliferation and activation. Therefore, the expression of orthogonal human IL-2Rβ on CD19-CAR-T cells allows for controllable expansion upon administration of orthogonal human IL-2, which leads to enhanced antitumor activity against leukemia and lymphoma (86, 87).
Additionally, IL-15 has been shown to promote CD8+ T cell activation and proliferation, which can enhance antitumor activity (88). A case report has described the use of CD19-CAR-T cells with membrane-bound IL-15 for B-ALL (following CD19- and CD22-CAR-T cell therapy failure) (89), demonstrating such IL-15-expressing CAR-T cells can be applied to hematological malignancies. These CD19-CAR-T cells with membrane-bound IL-15 are phenotypically similar to stem-cell memory T cells with prolonged proliferating persistence (90). Additionally, CD123- or CLL-1-CAR-T cells that express IL-15 also have a superior capacity of expansion in AML animal models (91, 92). However, this strategy is also associated with tumor necrosis factor α (TNFα)-induced acute toxicity, which requires an adjuvant anti-TNFα treatment (91).
IL-7 is another valuable cytokine, which is required for T cell development, survival, homeostasis and expansion (93). Studies have demonstrated that co-expression of IL-7 and CCL19 prolongs the survival and improves the activity of CD20-CAR-T cells (94). Furthermore, CD19-CAR-T cells expressing IL-7 and CCL19 are currently being explored in a clinical trial for relapsed/refractory B cell lymphoma (NCT04833504). A similar clinical trial for relapsed/refractory DLBCL is to use CD19-CAR-T cells with IL-7 and CCL19 co-expression in combination with an anti-PD1 monoclonal antibody (NCT04381741). Similar strategy has also been proven for T cell adoptive cellular therapy (ACT) (95), which further strengthened the case for IL-7 and CCL19 overexpression in CAR-T cells.
Recently, IL-36γ-secreting CD19-CAR-T cells have been generated to promote CAR-T cell expansion and persistence (in an autocrine manner), resulting in superior anti-tumor activity in mice bearing EL4-CD19+ T lymphoblast tumor. Moreover, IL-36γ also activates surrounding antigen presenting cells (APCs) and endogenous T cells, which might even facilitate the formation of an immune memory against antigen-negative tumors (96).
The provision of a constitutive signal is an option that is being explored to ensure durable CAR-T cell persistence. Toll-like receptor (TLR) adaptor MyD88 and tumor necrosis factor family member CD40 are nonconventional costimulatory molecules for the activation of T cells. Unlike the conventional stimulatory domains like CD28 or 4-1BB commonly used for CAR-T cells, MyD88/CD40 are unable to trigger T cell expansion upon transduction. However, they provide a costimulatory signal to the conventional CAR-T cell activation pathway, which greatly enhances CAR-T cell proliferation and expansion. This effect has been tested in CD123- or CD19-CAR-T cells against hematological malignancies (97, 98). However, MyD88/CD40 co-stimulated CAR-T cell therapy (like IL-15-expressing CAR-T cells) also requires anti-TNFα treatment to prevent severe toxicity (98).
Besides MyD88/CD40, in a murine model of B cell malignancies, miR155 overexpression together with Lysine-specific demethylase 1 (LSD1) knocking down can induce rapid expansion of CD19-CAR-T cells and improve their antitumor function (although the detailed mechanism remains unclear) (99).
Several combination therapies have been reported to promote in vivo CAR-T cell expansion. The well-known immunomodulatory drug, lenalidomide, induces WT1235-243/HLA-A*2402-directed CAR-T cell expansion, leading to enhanced anti-tumor activity in mice inoculated with chronic myelogenous leukemia K526 cells (100).
Additionally, CAR-T cell expansion can be induced by vaccination. Vaccination with dendritic cells containing a modified WT1 peptide (WT1236Y) has successfully achieved in vivo WT1235-243/HLA-A*2402-CAR-T cell expansion, which enhanced CAR-T cell efficacy against murine K526 cell tumor model (16). Moreover, cytomegalovirus (CMV)-specific T cells from patients are isolated, enriched and transduced with a CD19-CAR. Therefore, CMV vaccination is able to promote the expansion of such CAR-T cells. These CMV-CD19-CAR-T cells are studied in CD19+ tumor bearing murine models, and have potential utility for B cell non-Hodgkin’s lymphoma treatment (101, 102). Similarly, amphiphile-bound ligand of CAR can bind to endogenous albumin upon injection, which traffics to lymph nodes as well as inserts into cell membranes of APCs (103). APCs can present these ligands to CAR-T cells together with endogenous cytokines and costimulation signaling, leading to significant CAR-T cell expansion (including CD19-CAR-T cells). Hence, the amphiphile-bound ligands serve as booster vaccine (104).
CAR-T cell exhaustion and the subsequent reduction of CAR-T cell fitness are among the factors that diminish long-lasting beneficial therapeutic outcomes. T cell fitness and exhaustion are determined by metabolic alterations, transcriptional/epigenetic regulations, treatment strategies, and signaling dynamics. Newly developed approaches that enhance CAR-T cell fitness from the listed aspects are assessed herein.
Proper CAR design plays an essential role in CAR-T cell fitness, as it determines the fate and manner of T cell activation. By limiting the activation level of CAR-T cells, T cell exhaustion is prevented and thereby T cell persistence is maintained (105). While the co-stimulation of CAR-T cells via the CD28 pathway can lead to PI3K-AKT activation and rapid T cell exhaustion; 4-1BB-depedent co-stimulation via the p38-MAPK pathway can improve mitochondrial biogenesis (106), which enhances CD19- and CD33-CAR-T cell activity and persistence (106–108). However, contrasting results were obtained for CD28-based CD33-CAR-T cells, which are more potent than 4-1BB-based CD33-CAR-T cells against patient-derived xenograft (PDX) models of childhood AML (109). Therefore, the use of CD28-based or 4-1BB-based CARs needs to be evaluated with caution. It is notable that 4-1BB-based CAR-T cells appear to be less hematological toxic than CD28-based CAR-T cells (110), which is perhaps due to their lower activation capacity. Other than 4-1BB, integration of costimulatory domains, ICOS and CD27, can promote CAR-T cell fitness as well (111, 112), although further investigations are required to validate these constructs. Additionally, inclusion of the aforementioned MyD88/CD40 in CAR has also been proposed. However, study has reported that such integration severely affects the stability of CAR (98).
Regions other than the costimulatory domain also play important roles in the regulation of CAR-T cell activation. Increasing TCR affinity above a certain threshold is known to reduce T cell activity (113, 114), which might also be similar for CAR-T cells. Indeed, Ghorashian et al. (115), generated a CD19-CAR with low affinity to CD19, namely CAT. They demonstrated that this CAT T-cells exhibits improved proliferation and antitumor activity, without a significant increase in toxicity (115). This strategy is proven effective in clinical study as well (NCT02443831), as 12/14 relapsed/refractory pediatric B-ALL patients achieved molecular remission and CAR-T cell persistence was found in 11/14 patients (115). Furthermore, the reduction of hinge domain flexibility also decreases the affinity of CD19-CAR-T cells to CD19, which leads to reduced proinflammatory cytokine secretion without changing their specific cytotoxicity. When being tested in murine model of leukemia, these CAR-T cells with reduced affinity are able to prolong the survival of mice (116). Alternatively, mutation of the immunoreceptor tyrosine-based activation motif (ITAM) region of CD3ζ of CAR also results in lower affinity of CD19-CAR. Therefore, it can modulate CAR-T cell activation and prolong CAR-T cell persistence (117).
Similar efforts to limit CD19-CAR-T cell activation have been made by increasing the hinge and transmembrane region of CAR (such construct is called CD19-BBz (86) CAR) (105). These CAR-T cells are activated at lower levels, proliferate slowly and retain their cytotoxicity. A clinical trial using CD19-BBz (86) CAR-T cells enabled 6/11 patients with B cell lymphoma to achieve complete remission (NCT02842138) (105).
During chronic activation, mitochondrial dysfunction accumulates in CD8+ T cells. This is accompanied by a significant increase in reactive oxygen species (ROS), which impairs mitochondrial function and contributes to T cell exhaustion (118). Therefore, methods that optimize metabolism can provide a straightforward mechanism for the improvement of CAR-T cell fitness. The expression of H2O2 catalase in CAR-T cells can protect CAR-T cells from H2O2-induced oxidative stress and limit ROS accumulation, leading to maintained fitness (119). Although it was initially validated in solid tumors, similar strategies can be applied in hematological malignancies. It is noteworthy that ROS is not always harmful. Peroxisome proliferator-activated receptor-gamma coactivator (PGC)-1α activation can lead to increased mitochondrial metabolism and ROS accumulation in T cells, improving their antitumor effects (120, 121). Therefore, CD19-CAR-T cells with PGC1α-overexpression are able to improve mitochondrial function and T cells activity (122). The double-edged role of ROS in CAR-T cells probably results from complex influence of ROS on T cells (123). Therefore, further knowledge about the regulation of ROS and T cells is required. Besides ROS metabolism, the inhibition of cholesterol acyltransferase with avasimibe can alter T cell metabolism and increase CD19-CAR-T cells fitness and activity (124).
T cell exhaustion is closely regulated by transcriptional/epigenetic alterations. Recently, a study reported that CD8+ CD19-CAR-T cells in ALL patients undergo exhaustion-related DNA methylation (125), thus demethylation therapy might provide means to ensure CAR-T cell fitness. Indeed, Khawanky et al. (126). employed a 5′-Azacitidine (AZA) demethylation therapy in combination with CD123-CAR-T cell therapy against AML. AZA increases the number of Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4)-negative CD123-CAR-T cells, preventing T cell exhaustion, although anti-TNFα treatment may be required to reduce the accompanied toxicity (126). Similarly, DNA methyltransferase 3α (DNMT3A) deletion in CD19-CAR-T cells also prevents T cell exhaustion and improves CAR-T cell efficacy (127).
Anti-cancer drugs can impact CAR-T cell fitness too, although the precise mechanisms remain unclear. An in vitro study demonstrated that JQ1, an epigenetic inhibitor, reverses the exhaustion of CAR-T cells obtained from nonresponding chronic lymphocytic leukemia patients (128). Ibrutinib, a bruton tyrosine kinase inhibitor for mantle cell lymphoma and chronic lymphocytic leukemia, was also shown to increase CD19-CAR-T cell fitness against leukemia (129, 130).
The possibility of rejuvenating exhausted CAR-T cells has also been explored. Luo et al. (131) constructed a fluorescein-targeted-CAR-T cells with both antigen recognition and drug internalization ability. These CAR-T cells were directed toward the folate receptor+ cancer cells via a fluorescein-folate bispecific adaptor. Upon CAR-T cell exhaustion, TLR7-1A linked fluorescein is administered and then internalized by fluorescein-CAR-T cells, leading to their reactivation. This type of strategy could also be applied to hematological malignancies via the use of a bispecific adaptor targeting CD19 or CD22.
Clinical studies have indicated that CAR-T cells with a less-differentiated phenotype (such as memory stem cell-like T cells, TSCM-like) can exhibit longer proliferation and superior antitumor function (132–134). Therefore, generating CAR-T cells in a less differentiated state can guarantee their persistence and enormous achievements have been made in recent years. Although these protocols vary in different platforms, the key step is to inhibit terminal differentiation of CAR-T cells. While IL-21 plays a key role in the maintenance of memory T cells (including TSCM and central memory T cells, TCM) by inducing naïve T cells into an early differentiation phenotype (135), IL-15 prevents T cell differentiation by inhibiting mTORC1 (85). Therefore IL-21 or IL-15 were commonly administrated during manufacturing. Indeed, several studies reported that the addition of IL-21 or IL-15 results in increased proportion of CAR-TSCM and TCM (including WT1- and CD19-CAR-T cells) associated with enhanced persistence and anti-tumor effects (85, 136–140). Furthermore, induced secretion of IL-21 by CD19-CAR-T cells can even maintain CAR-T cells in early memory phenotype and enhance anti-tumor activity in vivo, which was already demonstrated in mice transplanted with Ramos B cells (139).
Besides cytokine administration, other methods could be applied to move CAR transduction toward a less differentiated phenotype. Upon costimulatory molecule CD81-mediated ex vivo T cell activation during manufacturing, CD19-CAR-T cells are enriched with naïve T cell-derived. These T cells are less differentiated with increased CAR expression and antitumor activity (141). Moreover, CAR-T cell stemness can also be maintained by modulating signaling pathways. PI3K or AKT inhibition can preserve CAR-T cell persistence by preventing T cell differentiation. Therefore, CAR-T cells are maintained in a less differentiated state, as demonstrated in CD33- (142), CD5- (143), CD19- (144, 145) and Minor histocompatibility antigen (MiHA)- (146) CAR-T cells against various hematological malignancies. Besides the PI3K-AKT pathways, the modulation of other pathways could maintain CAR-T cells memory stem cell-like features and even revive exhausted CAR-T cells, including Notch activation (147) and tyrosine kinase inhibition (148, 149) for CD19-CAR-T cells against humanized murine leukemia model. Recently, the role of MEK inhibition in CD8+ TSCM formation has been discovered (150), making it another strategy for CAR-TSCM generation.
Besides exhaustion, CAR-T cells also undergo activation-induced cell death (AICD) upon repeated activation, which is a manner of cell apoptosis through Fas-dependent pathways (151). Methods of Fas blockade or limiting CAR activation were introduced in previous sections (“Targeting immunosuppressive effects (other than immune checkpoints)” and “Regulation of signaling dynamics by CAR design”). ACID can also be prevented by Bcl-2 activation. CD20-CAR-T cells with Bcl-2 overexpression exhibit enhanced viability and antitumor activity in mouse xenograft lymphoma model (152).
As discussed above, many strategies that enhance CAR-T cell activity and expansion also increase the risk of adverse effect, some even need adjuvant treatment. However, as lessons learnt from HSCT, the onset of a mild graft-versus-host disease (GVHD, the major complication) may be associated with a lower risk of disease relapse after HSCT (153). Indeed, enhanced function of T cells probably leads to improved immune response toward both cancer cells and health cells. Therefore, adverse effects might not be solely a bad thing and researchers should treat them accordingly. Nevertheless, some adverse effects of CAR-T cells are life-threatening and require special attentions. These will be covered in the next sections.
Despite the superior therapeutic success and efficacy that have been achieved by CAR-T cell treatment, several concerning adverse effects require special attention, including: cytokine release syndrome (CRS)/neurotoxicity, on-target-off-tumor effects and off-target effects. These adverse effects greatly limit patient prognosis and restrict the outcomes associated with CAR-T cell therapy. Extensive efforts have been made to minimize these adverse effects. In the following sub-sections, we will focus on the strategies that directly modify CAR-T cells to minimize unintended toxicity and related preclinical studies.
Upon CAR-T cell activation, massive cytokines are produced by CAR-T cells and bystander cells. High concentrations of circulating pro-inflammatory cytokines can lead to the hyperactivation of the cytokine signaling network, and potentially lead to cytokine toxicity or CRS (154). It has been suggested that the presence and severity of CRS may be related to the treatment outcomes (155), but this notion remains controversial. Nevertheless, CRS is a life-threatening symptom and requires urgent therapeutic interventions (156). Immune effector cell-associated neurotoxicity syndrome (ICANS) is the second most frequent adverse effect associated with CAR-T cell therapy. Its underlying mechanisms appear to be similar to that of CRS, i.e., Both of them are due to the action of CAR-T cells and bystander cells (157). Therefore, numerous improved CAR-T cell designs and treatment strategies have been proposed to mitigate CRS/neurotoxicity by avoiding excessive cytokine release. IL-6 and IL-1 are closely related to CRS (158–160). Therefore, CD19- or BCMA-CAR-T cells with the capacity of anti-IL-6 scFv and IL-1 receptor antagonist (IL-1RA) secretion can help self-neutralize IL-6 and IL-1β. CRS and neurotoxicity are minimized in patients treated with these anti-IL-6 scFv and IL-1RA-secreting CAR-T cells (ChiCTR2000032124; ChiCTR2000031868) (161). Besides IL-6 and IL-1, cytokine-profiling assay identified granulocyte-macrophage colony-stimulating factor (GMCSF) as a key CRS-promoting protein released from CAR-T cells (162). Therefore, the reduction of GMCSF from CAR-T cells via antibody-mediated neutralization or GMCSF knockout significantly decreases the secretion of key CRS-associated cytokines from CAR-T cells without affecting their antitumor function, as shown in CD19-CAR-T cells in murine leukemia model (163) and CD22-CAR-T cells (162).
IFNγ inhibition is considered to be a possible salvage option for refractory CRS when IL-6 inhibition and glucocorticoids treatment fail. Whilst IFNγ production is an indicator of CAR-T cell activity, IFNγ blockade or deletion do not significantly impair the antitumor effect of CD19-CAR-T cells in murine models. Moreover, IFNγ removal results in attenuated cytokine toxicities due to reduced bystander macrophage activation. It also reduces the immune checkpoints expression on CAR-T cells and ensures their persistence (164). Moreover, it should be noted that IFNγ blockade is only applicable in CAR-T cells against hematological malignancies, as IFNγ receptor pathway is required for CAR-T cell killing activity in solid tumors (165).
For most forms of cancers, it is rare to find tumor-specific antigens that are exclusively expressed in cancer cells and shared by most cancer patients. Therefore, on-target off-tumor toxicity is a major concern of CAR-T cell therapy as the target antigens are also expressed on normal tissues. The development of single cell sequencing might improve the target antigen analysis, and provide valuable guidance on on-target, off-tumor toxicity evaluation (166). Nevertheless, several strategies have been proposed to attenuate or mitigate on-target off-tumor effect of CAR-T cells, including refined T cell regulation and the administration of preventive measures.
To avoid the cross-reactivity on normal tissues, a variety of gating strategies of AND, OR and NOT have been implemented for CAR-T cells for a long time. In recent years, several studies have proposed further improvements to this gated system for a more refined regulation of CAR-T cell activity.
AND-gated CAR-T cells have two (or more) CAR-like receptors and are only activated upon two (or more) antigens binding. Further increased modulation of this AND-gated CAR-T cell system can be achieved by the incorporation of a synthetic Notch receptor. The synthetic Notch receptor recognizes the first antigen and triggers the expression of a second CAR that recognizes another antigen (167). Roybal et al. (167) introduced the use of CAR-T cells with a CD19-synNotch receptor and an inducible α-mesothelin CAR gene in vitro. These CAR-T cells are not activated in the presence of mesothelin alone, they need to encounter CD19 first to express the α-mesothelin CAR. The author further demonstrated that synNotch receptors are able to induce tumor-localized CAR expression in vivo, by using CAR-T cells with GFP synNotch receptor and inducible CD19-CAR gene against GFP+ CD19+ tumor cells (167).
NOT-gated CAR-T cells, or inhibitory CAR (iCAR) T-cells are CAR-T cells with an activating receptor and an inhibitory receptor. The inclusion of an inhibitory receptor that targets antigens expressed on normal tissues minimizes the potential for cross reactivity. For example, CAR-T cells with CD19-CAR and HLA-A*02-iCAR have been constructed (168). Such CAR-T cells exploit the loss of heterozygosity (LOH) commonly observed in tumors. The presence of HLA-A*02-iCAR allows CAR-T cells to selectively eliminate hemizygous lymphoma cells which lost HLA-A*02 via LOH among heterozygous normal cells. By switching different types of HLA-iCAR, this approach can be extended to patients beyond HLA-A*02 heterozygotes.
And-, NOT- and OR-gated (the OR-gated strategy, i.e., the bispecific CAR) can be combined to produce more complex/refined regulation networks. Williams et al. (169) developed a system with multiple input circuits, which enables the production of CAR-T cells with complex activation mode like “A and B and C”, “A and B not C” or “[A or B] and C”. Moreover, multiple synNotch receptors have also been included in this system. Several combinations have been investigated, including an HER2-NOT-gate with an AND-gate of CD19 and GFP against K562 cells. However, most of the gated designs in this study were only tested in vitro and in vivo for K562 cells engineered to express different combinations of the antigens. Whether such multiple input circuits system can be applied to unmodified or spontaneous tumor models requires further investigation.
Preventive measures provide an alternative approach that can be applied to relieve damage incurred by on-target off-tumor toxicity. For example, whilst CD7-CAR-T cells are able to target T-ALL, normal CD7+ T cells and NK cells are eliminated as well, leading to immunodeficiency. To protect normal cells, Kim et al. genetically deleted CD7 in hematopoietic stem cells (HSCs) which were then engrafted into mice prior to the CD7-CAR-T cell treatment. These CD7-KO HSCs generated functional CD7- T cells and NK cells, although they are different from CD7+ T cells/NK cells. As the results, the mice are protected from CD7-CAR-T cell-induced immunodeficiency (170). Similarly, infusion of CD33-KO HSCs is able to restore immunity upon CD33-CAR-T cell treatment against AML (171, 172). Although the infusion of these antigen-removed HSCs provides an ideal strategy to maintain hematopoiesis during CAR-T cell therapy, concerns about the safety of genetic modification and phenotype of the antigen-removed HSCs exist. Moreover, in case of antigen-negative relapse and a second type of CAR-T cells is need, these antigen-removed HSCs become useless. It will only be applicable to clinics after all related techniques are mature.
The development of an On-switch for CAR-T cells enables the tight regulation of CAR-T cell activity and toxicity. Recently, On-switch system which takes the advantage of the tetracycline-on (Tet-On) system (173) has been developed. In the presence of doxycycline (Dox), reverse Tet transactivator (rtTA) fusion protein drives the expression of protein of interest. This Tet-On system has been applied to CD19-CAR-T cells against leukemia cells (174), and CD38-CAR-T cells against MM cells (175). In these studies, peak CAR expression is observed within 24 to 48 hours of Dox administration; and upon Dox withdrawal, CAR expression decay occurs within 24 to 48 hours. As Dox is generally well tolerated and allergic reactions are uncommon (176), this system may have a wide application.
Hypoxia is closely related to gene regulation and is a common feature shared by various TME. Therefore, a hypoxia-inducible transcription amplification system (HiTA-system) has been developed to control the expression of CAR in T cells (HiTA-CAR-T) (177). He et al. (177) have demonstrated that Her2-directed HiTA-CAR expression and HiTA-CAR-T activity are tightly restricted to hypoxic environments, with nearly no CAR expression in normoxic tissues in a mouse xenograft liver cancer model (177). As bone marrow niches are hypoxic, this technique has great potential for the treatment of hematological malignancies as well.
The light-switchable CAR (LiCAR) T-cells is a light-controlled system of On-switch. The intracellular domains of CAR are split into half, each half with a photoresponsive module. The two halves of CAR can only reassemble in the presence of near-infrared (NIR)-to-blue light, thereby allowing the transduction of T cell activation signals. This spatial-temporal regulation system can achieve attenuated toxicity and has been tested for CD19-CAR-T cells against lymphoma both in vitro and in vivo (178). Moreover, Kobayashi et al. (179) introduced a light-controlled binary switch system for CAR-T cell activation. They used the aforementioned fluorescein-directed CAR-T cells and an adaptor in which fluorescein is conjugated to small molecules targeting CD38. Therefore, the CAR-T cell activity is directed to CD38+ Ramos B cells in vitro. Such adaptors are initially trapped in an ultraviolet (UV)-light-sensitive cage. They are released upon UV exposure to induce CAR-T cell function in vitro (179).
Besides light, light-induced heat serves as another means of spatial-temporal CAR expression modulation, which is developed by Miller et al. (180). In this system, CD19-CAR expression is controlled by synthetic gene switches. When temperature reaches 40-42°C by photothermal heating, CAR expression is induced. In murine models with B cell driven cancer (i.e., Raji cells), plasmonic gold nanorods (AuNRs) were injected intravenously to convert NIR light into heat, together with heat-controlled CD19-CAR-T cells. When heating with NIR laser light directly to induce local CAR expressions, CAR was expressed in T cells and the tumors were irradiated with enhanced safety and efficacy (180). The major concern of these light/heat-based systems is that whether light/heat can penetrate into body. It is unlikely for a UV-based system (179) to reach site of hematological malignancies. By contrary, deep-tissue-penetrable NIR light is able to directly target tumor sites for precise control of CAR-T cells, as demonstrated in murine models of lymphoma (178, 180). However, further investigations are required to explore whether NIR light-controlled CAR-T cells are applicable in large and complex human body.
Besides switches that activate CAR-T cells, CAR-T cell activity can be switched off by the incorporation of a suicide genes. By overexpressing an inducible caspase9 (iCasp9 by AP1903 induction), majority of CD20- or CD19-CAR-T cells are eliminated within 72 hours upon AP1903 administration (181, 182). Other studies have further demonstrated that it improves CAR-T cell therapy safety through the use of iCasp9 in CD33-CAR-T cell against AML (183), interleukin-1 receptor accessory protein (IL-1RAP)-CAR-T cell against chronic myeloid leukemia (184) and CD19-CAR-T cell against various hematological malignancies (98).
The binding of monoclonal antibodies to cells induces antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity, leading to cell elimination. This phenomenon has been exploited for CAR-T cell elimination and the overexpressed CD20 or CD34 in lymphocytes are good candidates for elimination targets (185–187). For example, Sommer et al. (188) developed a rituximab-base (targeting CD20) off-switch system to eliminate FLT3-CAR-T cells in AML models. The application of this strategy enables rapid elimination of CAR-T cells to enhance bone marrow recovery without impairing antitumor activity (188). However, as CAR-T cells for hematological malignancies are likely to impair immunity, whether patients can survive the side effects of another lymphocyte-targeting treatment requires careful evaluation.
The small molecule–assisted shutoff (SMASh) system was firstly developed by Chung et al. (189), which offers another strategy to eliminate CAR-T cells. In this system, proteins of interest are fused to a SMAsh tag (consisting of an HCV protease and a degron) at the C-terminal via HCV NS3 protease recognition site. After protein folding, the internal protease cleaves at the fusion site to release the protein of interest, while the SMAsh tag is degraded due to degron activity. HCV protease inhibitors can block tag removal and the degron then leads to rapid degradation of the protein-tag fusion (189). This SMAsh strategy was implemented into CAR construct (SWIFF-CAR) by Juillerat et al. (190). In the absence of asunaprevir (a protease inhibitor), CAR is normally expressed on the cell surface in vitro. Upon administration of asunaprevir to the medium, the protease activity is inhibited within 48 hours, leading to rapid degradation of the newly synthesized CAR (190). Cao et al. (191) further applied this SMAsh technique in vivo: CD19-CAR-T cells with the SMAsh tag can efficiently kill human CD19+ tumor cells in mice. They also demonstrated that asunaprevir administration/withdrawal can control repeated surface CAR expression in a dose-dependent manner (191). Such repeated control of CAR expression might be helpful to avoid chronic antigen stimulation and prevent CAR-T cell exhaustion.
As discussed above, the current limitations of CAR-T cells include antigen-negative relapse, immunosuppressive TME, poor persistence and severe toxicities. To address these limitations, Gardner et al. (192) developed a novel class of CAR-T cells, namely synthetic enzyme-armed killer (SEAKER) cells. By taking advantages of the high specificity of CAR-T cells, engineered SEAKER cells which express prodrug-activating enzymes can activate systemically administered small molecule prodrugs specifically at tumor site. In murine lymphoma and leukemia models, CD19-directed SEAKER cells exhibited enhanced anti-tumor activity which is due to both direct CAR-T cell intrinsic function and activated prodrugs. Moreover, activation of prodrug allows CD19-directed SEAKER cells to kill CD19-negative cancer cells as well, making this strategy possible to eradicate heterogeneous tumor and prevent antigen-negative relapse. In addition, prodrug-activating capability is maintained in exhausted CD19-SNEAKER cells after multiple courses of prodrug treatment, indicating the superior persistence of this therapy. Finally, as prodrugs activation is independent of the immune activity of SEAKER cells, it is not affected by the immunosuppressive TME and the dose of infused SEAKER cell can be reduced to minimize immune toxicity (192). Therefore, SEAKER cell is an excellent approach to address multiple major limitations of CAR-T cells in hematological malignancies.
Currently, majority of CAR-T cell therapies for hematological malignancies are against B cell driven cancers. A major difficulty of the development of CAR-T cells against T cell driven cancers is that the attractive antigens (such as CD7) are expressed on normal T cell surfaces as well, leading to potential fratricide of CAR-T cells (i.e., CAR-T cells attack each other due to the presence of CD7). Such fratricide can be avoided through genetic modification of CD7 in CAR-T cells (193, 194). However, Freiwan et al. (195), developed a modification-free approach by isolating the naturally occurring CD7-negative T cells to generate CD7-negative CD7-CAR-T cells. These CD7-negative CD7-CAR-T cells closely resemble CD4+ memory T cells and exhibit promising persistence. Moreover, CD7-negative CD19-CAR-T cells also show enhanced anti-tumor activity compared to normal CD19-CAR-T cells. Therefore, CD7-negative T cells may be an ideal source of CAR-T cells for T cell driven cancers and other hematological malignancies.
Besides CD7-negative CD7-CAR-T cells, naturally selected CD7-CAR (NS7CAR)-T cells may be another promising modification-free therapy against T cell driven cancers. These CD7-negative NS7CAR-T cells were generated from fratricidal “natural selection” after the transduction of CD7-CAR constructs into bulk T cells. When compared to CD7-negative CD7-CAR-T cells, NS7CAR-T cells exhibited similar anti-tumor properties and have already passed phase 1 clinical trial recruiting patients with T-ALL or T-cell lymphoblastic lymphoma (NCT04572308) (196). However, extensive immune response occurs during “natural selection”. Whether this leads to pre-mature exhaustion of NS7CAR-T cells requires further study.
CAR-T cell therapy has revolutionized both the field of immunotherapy as well as the treatment of hematological malignancies. Although limitations still exist, great endeavor has been done to address them. Continuous development and improvement of antigen discovery, together with strategies to avoid antigen loss, impart us an arsenal to tackle tumor heterogeneity and antigen-negative relapse. Barriers of the immunosuppressive TME can be overcame via circumventing, targeting or remodeling TME. In addition, CAR-T cell persistence inside patient’s body is also crucial for a durable response of the treatment and it can be prolonged by proper CAR construct design, as well as ensuring CAR-T cell expansion, fitness and stemness. Moreover, strategies to enhance, refine and control CAR-T cell activity through various switch or gated systems are being established to mitigate the adverse effects during CAR-T cell therapy (Figure 3).
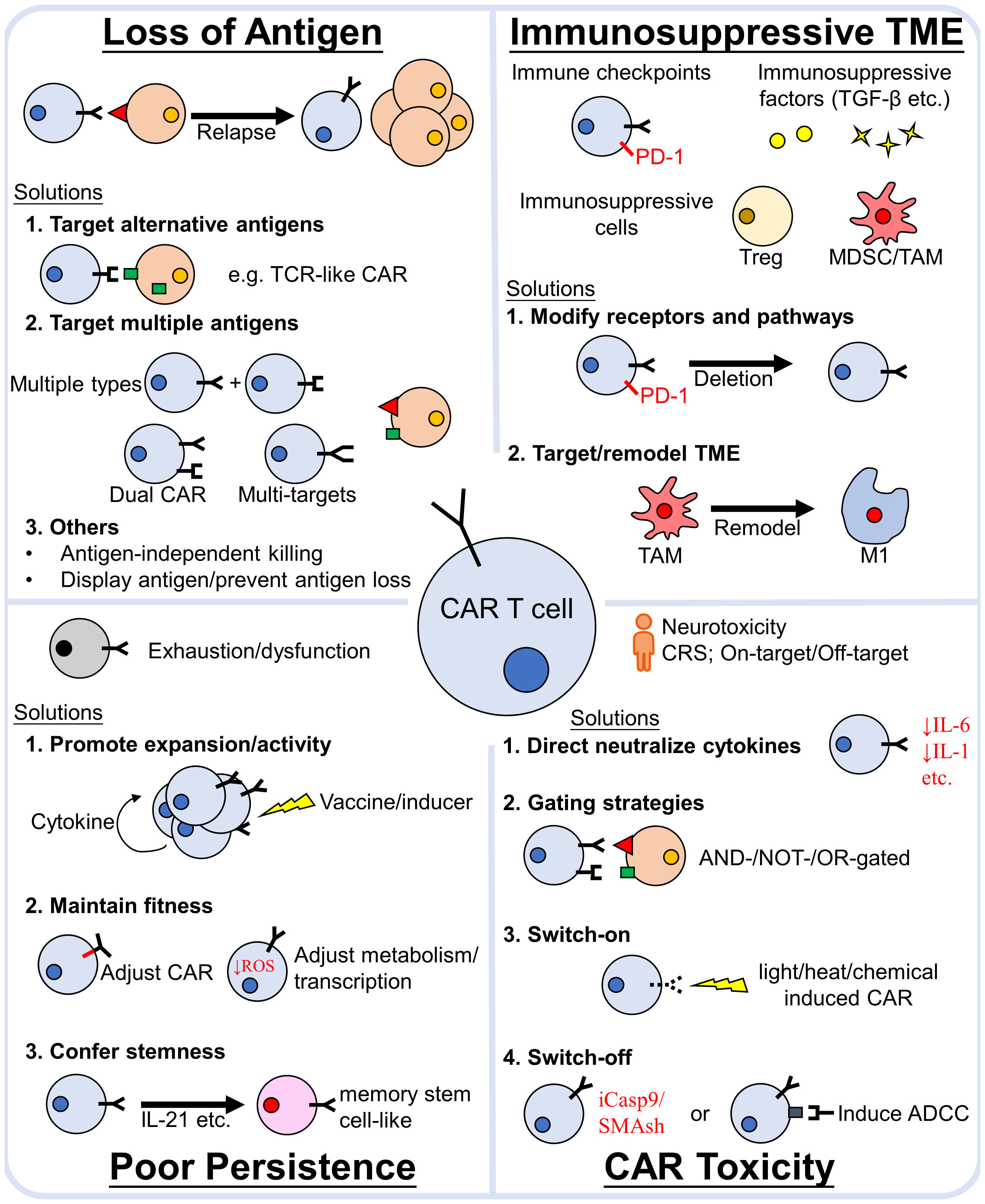
Figure 3 Major causes of CAR-T cell treatment failure and strategies to avoid them. The main reasons for CAR-T cell treatment failure are (1) loss of antigen relapse, (2) the immunosuppressive TME, (3) poor CAR-T cell poor persistence in vivo and (4) severe toxicity. Numerous efforts have been made to propose strategies to overcome these obstacles. The efficacy of CAR-T cell therapy can be enhanced by addressing these factors. TCR, T-cell receptor; TME, tumor microenvironment; PD-1, Programmed death-1; TGF-β, transforming growth factor -β; MDSC, myeloid-derived suppressor cells; TAM, tumor associated macrophages; Treg, regulatory T cells; CRS, cytokine release syndrome; iCasp9, inducible caspase9; SMAsh, Small molecule–assisted shutoff; ADCC, antibody-dependent cellular cytotoxicity.
CAR-T cell therapy is a booming field that incorporates both fundamental and clinical researches. Its great success in certain hematological malignancies has completely reshaped the approach to cancer and cancer therapies. The sophisticated immune evasion strategies of cancer cells and the complex immune microenvironment still limit CAR-T cell activity, but we are on the right track to unveil them. Fortunately, numerous solutions are already on the horizon. We envision that there will be a giant leap for CAR-T cell therapy once these recently developed preclinical improvements are translated into clinics. Besides the aspects related to the direct modifications on CAR-T cells discussed in this review, other factors that need to be further addressed for CAR-T cell therapy include: improving manufacturing practices with a shorter processing time, allogeneic or off-the-shelf CAR-T cells, improving CAR-T cell infiltration for solid tumors, pre-conditioning strategies for patients and the use of other immune cells for CAR transduction (e.g. NK cells, macrophages etc.). With advances in the understanding of cancer biology, immune systems and bioengineering/biomaterials, ways have been paved for the rapid developments of CAR-based therapies from different disciplines. We hope this review may provide new viewpoints for multidisciplinary approaches and the obstacles of effective CAR-T cell therapy will be overcome in the near future.
Conceptualization, JWH and JH; Investigation, JWH; Writing - Original Draft, JWH; Writing -Review and Editing, JWH, XH, and JH; Supervision, JH. All authors contributed to the article and approved the submitted version.
This work was financially supported by National Natural Science Foundation of China (NSFC 81500173).
We acknowledge Dr. Yangbao Miao for his ongoing advices and Dr. Yiguo Hu for language editing. We apologize to colleagues whose work we could not cite due to space limitations.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.

1. Weiden PL, Flournoy N, Thomas ED, Prentice R, Fefer A, Buckner CD, et al. Antileukemic effect of graft-versus-host disease in human recipients of allogeneic-marrow grafts. N Engl J Med (1979) 300(19):1068–73. doi: 10.1056/NEJM197905103001902
2. Horowitz MM, Gale RP, Sondel PM, Goldman JM, Kersey J, Kolb HJ, et al. Graft-versus-leukemia reactions after bone marrow transplantation. Blood (1990) 75(3):555–62. doi: 10.1182/blood.V75.3.555.555
3. Rafiq S, Hackett CS, Brentjens RJ. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat Rev Clin Oncol (2020) 17(3):147–67. doi: 10.1038/s41571-019-0297-y
4. Cheadle EJ, Gornall H, Baldan V, Hanson V, Hawkins RE, Gilham DE. CAR T cells: driving the road from the laboratory to the clinic. Immunol Rev (2014) 257(1):91–106. doi: 10.1111/imr.12126
5. Munshi NC, Anderson LD Jr., Shah N, Madduri D, Berdeja J, Lonial S, et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N Engl J Med (2021) 384(8):705–16. doi: 10.1056/NEJMoa2024850
6. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory Large b-cell lymphoma. N Engl J Med (2017) 377(26):2531–44. doi: 10.1056/NEJMoa1707447
7. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with b-cell lymphoblastic leukemia. N Engl J Med (2018) 378(5):439–48. doi: 10.1056/NEJMoa1709866
8. Shah BD, Ghobadi A, Oluwole OO, Logan AC, Boissel N, Cassaday RD, et al. KTE-X19 for relapsed or refractory adult b-cell acute lymphoblastic leukaemia: Phase 2 results of the single-arm, open-label, multicentre ZUMA-3 study. Lancet (2021) 398(10299):491–502. doi: 10.1016/S0140-6736(21)01222-8
9. Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang M, Arnason J, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large b-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet (2020) 396(10254):839–52. doi: 10.1016/S0140-6736(20)31366-0
10. Strati P, Neelapu SS. CAR-T failure: Beyond antigen loss and T cells. Blood (2021) 137(19):2567–8. doi: 10.1182/blood.2020010462
11. Majzner RG, Mackall CL. Tumor antigen escape from CAR T-cell therapy. Cancer Discovery (2018) 8(10):1219–26. doi: 10.1158/2159-8290.CD-18-0442
12. Lemoine J, Ruella M, Houot R. Born to survive: how cancer cells resist CAR T cell therapy. J Hematol Oncol (2021) 14(1):199. doi: 10.1186/s13045-021-01209-9
13. Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med (2017) 9(1):34. doi: 10.1186/s13073-017-0424-2
14. Yang L, Han Y, Suarez Saiz F, Minden MD. And oncogene: the WT1 story. Leukemia (2007) 21(5):868–76. doi: 10.1038/sj.leu.2404624
15. Hohenstein P, Hastie ND. The many facets of the wilms' tumour gene, WT1. Hum Mol Genet (2006) 15. Spec No 2:R196–201. doi: 10.1093/hmg/ddl196
16. Akahori Y, Wang L, Yoneyama M, Seo N, Okumura S, Miyahara Y, et al. Antitumor activity of CAR-T cells targeting the intracellular oncoprotein WT1 can be enhanced by vaccination. Blood (2018) 132(11):1134–45. doi: 10.1182/blood-2017-08-802926
17. Rafiq S, Purdon TJ, Daniyan AF, Koneru M, Dao T, Liu C, et al. Optimized T-cell receptor-mimic chimeric antigen receptor T cells directed toward the intracellular wilms tumor 1 antigen. Leukemia (2017) 31(8):1788–97. doi: 10.1038/leu.2016.373
18. Liu X, Xu Y, Xiong W, Yin B, Huang Y, Chu J, et al. Development of a TCR-like antibody and chimeric antigen receptor against NY-ESO-1/HLA-A2 for cancer immunotherapy. J Immunother Cancer (2022) 10(3):e004035. doi: 10.1136/jitc-2021-004035
19. Inaguma Y, Akahori Y, Murayama Y, Shiraishi K, Tsuzuki-Iba S, Endoh A, et al. Construction and molecular characterization of a T-cell receptor-like antibody and CAR-T cells specific for minor histocompatibility antigen HA-1H. Gene Ther (2014) 21(6):575–84. doi: 10.1038/gt.2014.30
20. Raskin S, Van Pelt S, Toner K, Balakrishnan PB, Dave H, Bollard CM, et al. Novel TCR-like CAR-T cells targeting an HLA *0201-restricted SSX2 epitope display strong activity against acute myeloid leukemia. Mol Ther Methods Clin Dev (2021) 23:296–306. doi: 10.1016/j.omtm.2021.09.008
21. Zah E, Lin MY, Silva-Benedict A, Jensen MC, Chen YY. T Cells expressing CD19/CD20 bispecific chimeric antigen receptors prevent antigen escape by malignant b cells. Cancer Immunol Res (2016) 4(6):498–508. doi: 10.1158/2326-6066.CIR-15-0231
22. Mackay F, Schneider P, Rennert P, Browning J. BAFF AND APRIL: A tutorial on b cell survival. Annu Rev Immunol (2003) 21:231–64. doi: 10.1146/annurev.immunol.21.120601.141152
23. Yarchoan M, Ho WJ, Mohan A, Shah Y, Vithayathil T, Leatherman J, et al. Effects of b cell-activating factor on tumor immunity. JCI Insight (2020) 5(10):e136417. doi: 10.1172/jci.insight.136417
24. Tussiwand R, Rauch M, Fluck LA, Rolink AG. BAFF-r expression correlates with positive selection of immature b cells. Eur J Immunol (2012) 42(1):206–16. doi: 10.1002/eji.201141957
25. Parameswaran R, Muschen M, Kim YM, Groffen J, Heisterkamp N. A functional receptor for b-cell-activating factor is expressed on human acute lymphoblastic leukemias. Cancer Res (2010) 70(11):4346–56. doi: 10.1158/0008-5472.CAN-10-0300
26. Parameswaran R, Lim M, Fei F, Abdel-Azim H, Arutyunyan A, Schiffer I, et al. Effector-mediated eradication of precursor b acute lymphoblastic leukemia with a novel fc-engineered monoclonal antibody targeting the BAFF-r. Mol Cancer Ther (2014) 13(6):1567–77. doi: 10.1158/1535-7163.MCT-13-1023
27. Novak AJ, Grote DM, Stenson M, Ziesmer SC, Witzig TE, Habermann TM, et al. Expression of BLyS and its receptors in b-cell non-Hodgkin lymphoma: correlation with disease activity and patient outcome. Blood (2004) 104(8):2247–53. doi: 10.1182/blood-2004-02-0762
28. Wong DP, Roy NK, Zhang K, Anukanth A, Asthana A, Shirkey-Son NJ, et al. A BAFF ligand-based CAR-T cell targeting three receptors and multiple b cell cancers. Nat Commun (2022) 13(1):217. doi: 10.1038/s41467-021-27853-w
29. Ramakrishna S, Highfill SL, Walsh Z, Nguyen SM, Lei H, Shern JF, et al. Modulation of target antigen density improves CAR T-cell functionality and persistence. Clin Cancer Res (2019) 25(17):5329–41. doi: 10.1158/1078-0432.CCR-18-3784
30. Biberacher V, Decker T, Oelsner M, Wagner M, Bogner C, Schmidt B, et al. The cytotoxicity of anti-CD22 immunotoxin is enhanced by bryostatin 1 in b-cell lymphomas through CD22 upregulation and PKC-betaII depletion. Haematologica (2012) 97(5):771–9. doi: 10.3324/haematol.2011.049155
31. Wang L, Zhang Y, Anderson E, Lamble A, Orentas RJ. Bryostatin activates CAR T-cell antigen-Non-Specific killing (CTAK), and CAR-T NK-like killing for pre-b ALL, while blocking cytolysis of a burkitt lymphoma cell line. Front Immunol (2022) 13:825364. doi: 10.3389/fimmu.2022.825364
32. Laurent SA, Hoffmann FS, Kuhn PH, Cheng Q, Chu Y, Schmidt-Supprian M, et al. Gamma-secretase directly sheds the survival receptor BCMA from plasma cells. Nat Commun (2015) 6:7333. doi: 10.1038/ncomms8333
33. Pont MJ, Hill T, Cole GO, Abbott JJ, Kelliher J, Salter AI, et al. Gamma-secretase inhibition increases efficacy of BCMA-specific chimeric antigen receptor T cells in multiple myeloma. Blood (2019) 134(19):1585–97. doi: 10.1182/blood.2019000050
34. Garcia-Guerrero E, Rodriguez-Lobato LG, Sierro-Martinez B, Danhof S, Bates S, Frenz S, et al. ATRA works synergistically with the gamma-secretase inhibitor crenigacestat to augment BCMA on multiple myeloma and the efficacy of BCMA-CAR T-cells. Haematologica (2022). doi: 10.3324/haematol.2022.281339
35. Su L, Wu L, Lobb RR, Rennert PD, Ambrose C. CAR-T engager proteins optimize anti-CD19 CAR-T cell therapies for lymphoma. Oncoimmunology (2022) 11(1):2111904. doi: 10.1080/2162402X.2022.2111904
36. Wang G, Xu J, Zhao J, Yin W, Liu D, Chen W, et al. Arf1-mediated lipid metabolism sustains cancer cells and its ablation induces anti-tumor immune responses in mice. Nat Commun (2020) 11(1):220. doi: 10.1038/s41467-019-14046-9
37. Contini P, Zocchi MR, Pierri I, Albarello A, Poggi A. In vivo apoptosis of CD8(+) lymphocytes in acute myeloid leukemia patients: involvement of soluble HLA-I and fas ligand. Leukemia (2007) 21(2):253–60. doi: 10.1038/sj.leu.2404494
38. Bunse L, Pusch S, Bunse T, Sahm F, Sanghvi K, Friedrich M, et al. Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat Med (2018) 24(8):1192–203. doi: 10.1038/s41591-018-0095-6
39. Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature (2009) 462(7274):739–44. doi: 10.1038/nature08617
40. Goodman A, Patel SP, Kurzrock R. PD-1-PD-L1 immune-checkpoint blockade in b-cell lymphomas. Nat Rev Clin Oncol (2017) 14(4):203–20. doi: 10.1038/nrclinonc.2016.168
41. Scholler N, Perbost R, Locke FL, Jain MD, Turcan S, Danan C, et al. Tumor immune contexture is a determinant of anti-CD19 CAR T cell efficacy in large b cell lymphoma. Nat Med (2022). doi: 10.1038/s41591-022-01916-x
42. Hu L, Chen L, Xiao Z, Zheng X, Chen Y, Xian N, et al. Ablation of T cell-associated PD-1H enhances functionality and promotes adoptive immunotherapy. JCI Insight (2022) 7(2):e148247. doi: 10.1172/jci.insight.148247.
43. Lin G, Zhang Y, Yu L, Wu D. Cytotoxic effect of CLL1 CART cell immunotherapy with PD1 silencing on relapsed/refractory acute myeloid leukemia. Mol Med Rep (2021) 23(3):208. doi: 10.3892/mmr.2021.11847
44. Hu B, Zou Y, Zhang L, Tang J, Niedermann G, Firat E, et al. Nucleofection with plasmid DNA for CRISPR/Cas9-mediated inactivation of programmed cell death protein 1 in CD133-specific CAR T cells. Hum Gene Ther (2019) 30(4):446–58. doi: 10.1089/hum.2017.234
45. Ma YJ, Dai HP, Cui QY, Cui W, Zhu WJ, Qu CJ, et al. Successful application of PD-1 knockdown CLL-1 CAR-T therapy in two AML patients with post-transplant relapse and failure of anti-CD38 CAR-T cell treatment. Am J Cancer Res (2022) 12(2):615–21.
46. Rafiq S, Yeku OO, Jackson HJ, Purdon TJ, van Leeuwen DG, Drakes DJ, et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat Biotechnol (2018) 36(9):847–56. doi: 10.1038/nbt.4195
47. Zhang A, Sun Y, Wang S, Du J, Gao X, Yuan Y, et al. Secretion of human soluble programmed cell death protein 1 by chimeric antigen receptor-modified T cells enhances anti-tumor efficacy. Cytotherapy (2020) 22(12):734–43. doi: 10.1016/j.jcyt.2020.05.007
48. Zhao S, Wang C, Lu P, Lou Y, Liu H, Wang T, et al. Switch receptor T3/28 improves long-term persistence and antitumor efficacy of CAR-T cells. J Immunother Cancer (2021) 9(12):e003176. doi: 10.1136/jitc-2021-003176
49. Blaeschke F, Ortner E, Stenger D, Mahdawi J, Apfelbeck A, Habjan N, et al. Design and evaluation of TIM-3-CD28 checkpoint fusion proteins to improve anti-CD19 CAR T-cell function. Front Immunol (2022) 13:845499. doi: 10.3389/fimmu.2022.845499
50. Carnevale J, Shifrut E, Kale N, Nyberg WA, Blaeschke F, Chen YY, et al. RASA2 ablation in T cells boosts antigen sensitivity and long-term function. Nature (2022) 609(7925):174–82. doi: 10.1038/s41586-022-05126-w
51. Kalinin RS, Ukrainskaya VM, Chumakov SP, Moysenovich AM, Tereshchuk VM, Volkov DV, et al. Engineered removal of PD-1 from the surface of CD19 CAR-T cells results in increased activation and diminished survival. Front Mol Biosci (2021) 8:745286. doi: 10.3389/fmolb.2021.745286
52. David CJ, Massague J. Contextual determinants of TGFbeta action in development, immunity and cancer. Nat Rev Mol Cell Biol (2018) 19(7):419–35. doi: 10.1038/s41580-018-0007-0
53. Silk JD, Abbott RJM, Adams KJ, Bennett AD, Brett S, Cornforth TV, et al. Engineering cancer antigen-specific T cells to overcome the immunosuppressive effects of TGF-beta. J Immunol (2022) 208(1):169–80. doi: 10.4049/jimmunol.2001357
54. Yamamoto TN, Lee PH, Vodnala SK, Gurusamy D, Kishton RJ, Yu Z, et al. T Cells genetically engineered to overcome death signaling enhance adoptive cancer immunotherapy. J Clin Invest (2019) 129(4):1551–65. doi: 10.1172/JCI121491
55. Oda SK, Anderson KG, Ravikumar P, Bonson P, Garcia NM, Jenkins CM, et al. A fas-4-1BB fusion protein converts a death to a pro-survival signal and enhances T cell therapy. J Exp Med (2020) 217(12):e20191166. doi: 10.1084/jem.20191166
56. Yang Q, Hao J, Chi M, Wang Y, Xin B, Huang J, et al. Superior antitumor immunotherapy efficacy of kynureninase modified CAR-T cells through targeting kynurenine metabolism. Oncoimmunology (2022) 11(1):2055703. doi: 10.1080/2162402X.2022.2055703
57. Yang Q, Hao J, Chi M, Wang Y, Li J, Huang J, et al. D2HGDH-mediated D2HG catabolism enhances the anti-tumor activities of CAR-T cells in an immunosuppressive microenvironment. Mol Ther (2022) 30(3):1188–200. doi: 10.1016/j.ymthe.2022.01.007
58. Wang X, Huynh C, Urak R, Weng L, Walter M, Lim L, et al. The cerebroventricular environment modifies CAR T cells for potent activity against both central nervous system and systemic lymphoma. Cancer Immunol Res (2021) 9(1):75–88. doi: 10.1158/2326-6066.CIR-20-0236
59. Leitner J, Herndler-Brandstetter D, Zlabinger GJ, Grubeck-Loebenstein B, Steinberger P. CD58/CD2 is the primary costimulatory pathway in human CD28-CD8+ T cells. J Immunol (2015) 195(2):477–87. doi: 10.4049/jimmunol.1401917
60. Bullens DM, Rafiq K, Charitidou L, Peng X, Kasran A, Warmerdam PA, et al. Effects of co-stimulation by CD58 on human T cell cytokine production: a selective cytokine pattern with induction of high IL-10 production. Int Immunol (2001) 13(2):181–91. doi: 10.1093/intimm/13.2.181
61. Majzner RG, Frank MJ, Mount C, Tousley A, Kurtz DM, Sworder B, et al. CD58 aberrations limit durable responses to CD19 CAR in Large b cell lymphoma patients treated with axicabtagene ciloleucel but can be overcome through novel CAR engineering. Blood (2020) 136:53–4. doi: 10.1182/blood-2020-139605
62. De Veirman K, Rao L, De Bruyne E, Menu E, Van Valckenborgh E, Van Riet I, et al. Cancer associated fibroblasts and tumor growth: focus on multiple myeloma. Cancers (Basel) (2014) 6(3):1363–81. doi: 10.3390/cancers6031363
63. Sakemura R, Hefazi M, Siegler EL, Cox MJ, Larson DP, Hansen MJ, et al. Targeting cancer-associated fibroblasts in the bone marrow prevents resistance to CART-cell therapy in multiple myeloma. Blood (2022) 139(26):3708–21. doi: 10.1182/blood.2021012811
64. Ugel S, De Sanctis F, Mandruzzato S, Bronte V. Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages. J Clin Invest (2015) 125(9):3365–76. doi: 10.1172/JCI80006
65. Yan ZX, Li L, Wang W, OuYang BS, Cheng S, Wang L, et al. Clinical efficacy and tumor microenvironment influence in a dose-escalation study of anti-CD19 chimeric antigen receptor T cells in refractory b-cell non-hodgkin's lymphoma. Clin Cancer Res (2019) 25(23):6995–7003. doi: 10.1158/1078-0432.CCR-19-0101
66. Fultang L, Panetti S, Ng M, Collins P, Graef S, Rizkalla N, et al. MDSC targeting with gemtuzumab ozogamicin restores T cell immunity and immunotherapy against cancers. EBioMedicine (2019) 47:235–46. doi: 10.1016/j.ebiom.2019.08.025
67. Liu Y, Wang S, Schubert ML, Lauk A, Yao H, Blank MF, et al. CD33-directed immunotherapy with third-generation chimeric antigen receptor T cells and gemtuzumab ozogamicin in intact and CD33-edited acute myeloid leukemia and hematopoietic stem and progenitor cells. Int J Cancer (2022) 150(7):1141–55. doi: 10.1002/ijc.33865
68. Chandrasekaran S, Funk CR, Kleber T, Paulos CM, Shanmugam M, Waller EK. Strategies to overcome failures in T-cell immunotherapies by targeting PI3K-delta and -gamma. Front Immunol (2021) 12:718621. doi: 10.3389/fimmu.2021.718621
69. Lasek W, Zagozdzon R, Jakobisiak M. Interleukin 12: still a promising candidate for tumor immunotherapy? Cancer Immunol Immunother (2014) 63(5):419–35. doi: 10.1007/s00262-014-1523-1
70. Kueberuwa G, Kalaitsidou M, Cheadle E, Hawkins RE, Gilham DE. CD19 CAR T cells expressing IL-12 eradicate lymphoma in fully lymphoreplete mice through induction of host immunity. Mol Ther Oncolytics (2018) 8:41–51. doi: 10.1016/j.omto.2017.12.003
71. Nguyen KG, Vrabel MR, Mantooth SM, Hopkins JJ, Wagner ES, Gabaldon TA, et al. Localized interleukin-12 for cancer immunotherapy. Front Immunol (2020) 11:575597. doi: 10.3389/fimmu.2020.575597
72. Luo Y, Chen Z, Sun M, Li B, Pan F, Ma A, et al. IL-12 nanochaperone-engineered CAR T cell for robust tumor-immunotherapy. Biomaterials (2022) 281:121341. doi: 10.1016/j.biomaterials.2021.121341
73. Hu B, Ren J, Luo Y, Keith B, Young RM, Scholler J, et al. Augmentation of antitumor immunity by human and mouse CAR T cells secreting IL-18. Cell Rep (2017) 20(13):3025–33. doi: 10.1016/j.celrep.2017.09.002
74. Murray PJ. Macrophage polarization. Annu Rev Physiol (2017) 79:541–66. doi: 10.1146/annurev-physiol-022516-034339
75. Luo W, Napoleon JV, Zhang F, Lee YG, Wang B, Putt KS, et al. Repolarization of tumor-infiltrating myeloid cells for augmentation of CAR T cell therapies. Front Immunol (2022) 13:816761. doi: 10.3389/fimmu.2022.816761
76. Guo F, Das JK, Kobayashi KS, Qin QM, AF T, Alaniz RC, et al. Live attenuated bacterium limits cancer resistance to CAR-T therapy by remodeling the tumor microenvironment. J Immunother Cancer (2022) 10(1):e003760. doi: 10.1136/jitc-2021-003760
77. Johnson LR, Lee DY, Eacret JS, Ye D, June CH, Minn AJ. The immunostimulatory RNA RN7SL1 enables CAR-T cells to enhance autonomous and endogenous immune function. Cell (2021) 184(19):4981–95.e14. doi: 10.1016/j.cell.2021.08.004
78. Pol J, Bloy N, Obrist F, Eggermont A, Galon J, Cremer I, et al. Trial watch:: Oncolytic viruses for cancer therapy. Oncoimmunology (2014) 3:e28694. doi: 10.4161/onci.28694
79. Bridle BW, Stephenson KB, Boudreau JE, Koshy S, Kazdhan N, Pullenayegum E, et al. Potentiating cancer immunotherapy using an oncolytic virus. Mol Ther (2010) 18(8):1430–9. doi: 10.1038/mt.2010.98
80. Wenthe J, Naseri S, Labani-Motlagh A, Enblad G, Wikstrom KI, Eriksson E, et al. Boosting CAR T-cell responses in lymphoma by simultaneous targeting of CD40/4-1BB using oncolytic viral gene therapy. Cancer Immunol Immunother (2021) 70(10):2851–65. doi: 10.1007/s00262-021-02895-7
81. Evgin L, Huff AL, Wongthida P, Thompson J, Kottke T, Tonne J, et al. Oncolytic virus-derived type I interferon restricts CAR T cell therapy. Nat Commun (2020) 11(1):3187. doi: 10.1038/s41467-020-17011-z
82. Byrne M, Oluwole OO, Savani B, Majhail NS, Hill BT, Locke FL. Understanding and managing Large b cell lymphoma relapses after chimeric antigen receptor T cell therapy. Biol Blood Marrow Transplant (2019) 25(11):e344–e51. doi: 10.1016/j.bbmt.2019.06.036
83. Shen C, Zhang Z, Zhang Y. Chimeric antigen receptor T cell exhaustion during treatment for hematological malignancies. BioMed Res Int (2020) 2020:8765028. doi: 10.1155/2020/8765028
84. Tang L, Zhang Y, Hu Y, Mei H. T Cell exhaustion and CAR-T immunotherapy in hematological malignancies. BioMed Res Int (2021) 2021:6616391. doi: 10.1155/2021/6616391
85. Alizadeh D, Wong RA, Yang X, Wang D, Pecoraro JR, Kuo CF, et al. IL15 enhances CAR-T cell antitumor activity by reducing mTORC1 activity and preserving their stem cell memory phenotype. Cancer Immunol Res (2019) 7(5):759–72. doi: 10.1158/2326-6066.CIR-18-0466
86. Aspuria PJ, Vivona S, Bauer M, Semana M, Ratti N, McCauley S, et al. An orthogonal IL-2 and IL-2Rbeta system drives persistence and activation of CAR T cells and clearance of bulky lymphoma. Sci Transl Med (2021) 13(625):eabg7565. doi: 10.1126/scitranslmed.abg7565
87. Zhang Q, Hresko ME, Picton LK, Su L, Hollander MJ, Nunez-Cruz S, et al. A human orthogonal IL-2 and IL-2Rbeta system enhances CAR T cell expansion and antitumor activity in a murine model of leukemia. Sci Transl Med (2021) 13(625):eabg6986. doi: 10.1126/scitranslmed.abg6986
88. Berraondo P, Sanmamed MF, Ochoa MC, Etxeberria I, Aznar MA, Perez-Gracia JL, et al. Cytokines in clinical cancer immunotherapy. Br J Cancer (2019) 120(1):6–15. doi: 10.1038/s41416-018-0328-y
89. Sun Y, Su Y, Wang Y, Liu N, Li Y, Chen J, et al. CD19 CAR-T cells with membrane-bound IL-15 for b-cell acute lymphoblastic leukemia after failure of CD19 and CD22 CAR-T cells: Case report. Front Immunol (2021) 12:728962. doi: 10.3389/fimmu.2021.728962
90. Hurton LV, Singh H, Najjar AM, Switzer KC, Mi T, Maiti S, et al. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proc Natl Acad Sci U S A (2016) 113(48):E7788–E97. doi: 10.1073/pnas.1610544113
91. Ataca Atilla P, McKenna MK, Tashiro H, Srinivasan M, Mo F, Watanabe N, et al. Modulating TNFalpha activity allows transgenic IL15-expressing CLL-1 CAR T cells to safely eliminate acute myeloid leukemia. J Immunother Cancer (2020) 8(2):e001229. doi: 10.1136/jitc-2020-001229
92. Mu-Mosley H, Ostermann L, Muftuoglu M, Vaidya A, Bonifant CL, Velasquez MP, et al. Transgenic expression of IL15 retains CD123-redirected T cells in a less differentiated state resulting in improved anti-AML activity in autologous AML PDX models. Front Immunol (2022) 13:880108. doi: 10.3389/fimmu.2022.880108
93. Lin J, Zhu Z, Xiao H, Wakefield MR, Ding VA, Bai Q, et al. The role of IL-7 in immunity and cancer. Anticancer Res (2017) 37(3):963–7. doi: 10.21873/anticanres.11405
94. Adachi K, Kano Y, Nagai T, Okuyama N, Sakoda Y, Tamada K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat Biotechnol (2018) 36(4):346–51. doi: 10.1038/nbt.4086
95. Tokunaga Y, Sasaki T, Goto S, Adachi K, Sakoda Y, Tamada K. Enhanced antitumor responses of tumor antigen-specific TCR T cells genetically engineered to produce IL7 and CCL19. Mol Cancer Ther (2022) 21(1):138–48. doi: 10.1158/1535-7163.MCT-21-0400
96. Li X, Daniyan AF, Lopez AV, Purdon TJ, Brentjens RJ. Cytokine IL-36gamma improves CAR T-cell functionality and induces endogenous antitumor response. Leukemia (2021) 35(2):506–21. doi: 10.1038/s41375-020-0874-1
97. Foster AE, Mahendravada A, Shinners NP, Chang WC, Crisostomo J, Lu A, et al. Regulated expansion and survival of chimeric antigen receptor-modified T cells using small molecule-dependent inducible MyD88/CD40. Mol Ther (2017) 25(9):2176–88. doi: 10.1016/j.ymthe.2017.06.014
98. Collinson-Pautz MR, Chang WC, Lu A, Khalil M, Crisostomo JW, Lin PY, et al. Constitutively active MyD88/CD40 costimulation enhances expansion and efficacy of chimeric antigen receptor T cells targeting hematological malignancies. Leukemia (2019) 33(9):2195–207. doi: 10.1038/s41375-019-0417-9
99. Zhang J, Zhu J, Zheng G, Wang Q, Li X, Feng Y, et al. Co-Expression of miR155 or LSD1 shRNA increases the anti-tumor functions of CD19 CAR-T cells. Front Immunol (2021) 12:811364. doi: 10.3389/fimmu.2021.811364
100. Zhang L, Jin G, Chen Z, Yu C, Li Y, Li Y, et al. Lenalidomide improves the antitumor activity of CAR-T cells directed toward the intracellular wilms tumor 1 antigen. Hematology (2021) 26(1):818–26. doi: 10.1080/16078454.2021.1981534
101. Wang X, Urak R, Walter M, Guan M, Han T, Vyas V, et al. Large-Scale manufacturing and characterization of CMV-CD19CAR T cells. J Immunother Cancer (2022) 10(1):e003461. doi: 10.1136/jitc-2021-003461
102. Wang X, Wong CW, Urak R, Mardiros A, Budde LE, Chang WC, et al. CMVpp65 vaccine enhances the antitumor efficacy of adoptively transferred CD19-redirected CMV-specific T cells. Clin Cancer Res (2015) 21(13):2993–3002. doi: 10.1158/1078-0432.CCR-14-2920
103. Liu H, Kwong B, Irvine DJ. Membrane anchored immunostimulatory oligonucleotides for in vivo cell modification and localized immunotherapy. Angew Chem Int Ed Engl (2011) 50(31):7052–5. doi: 10.1002/anie.201101266
104. Ma L, Dichwalkar T, Chang JYH, Cossette B, Garafola D, Zhang AQ, et al. Enhanced CAR-T cell activity against solid tumors by vaccine boosting through the chimeric receptor. Science (2019) 365(6449):162–8. doi: 10.1126/science.aav8692
105. Ying Z, Huang XF, Xiang X, Liu Y, Kang X, Song Y, et al. A safe and potent anti-CD19 CAR T cell therapy. Nat Med (2019) 25(6):947–53. doi: 10.1038/s41591-019-0421-7
106. Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med (2015) 21(6):581–90. doi: 10.1038/nm.3838
107. Li G, Boucher JC, Kotani H, Park K, Zhang Y, Shrestha B, et al. 4-1BB enhancement of CAR T function requires NF-kappaB and TRAFs. JCI Insight (2018) 3(18):e121322. doi: 10.1172/jci.insight.121322
108. Li S, Tao Z, Xu Y, Liu J, An N, Wang Y, et al. CD33-specific chimeric antigen receptor T cells with different Co-stimulators showed potent anti-leukemia efficacy and different phenotype. Hum Gene Ther (2018) 29(5):626–39. doi: 10.1089/hum.2017.241
109. Qin H, Yang L, Chukinas JA, Shah N, Tarun S, Pouzolles M, et al. Systematic preclinical evaluation of CD33-directed chimeric antigen receptor T cell immunotherapy for acute myeloid leukemia defines optimized construct design. J Immunother Cancer (2021) 9(9):e003149. doi: 10.1136/jitc-2021-003149
110. Luo W, Li C, Zhang Y, Du M, Kou H, Lu C, et al. Adverse effects in hematologic malignancies treated with chimeric antigen receptor (CAR) T cell therapy: A systematic review and meta-analysis. BMC Cancer (2022) 22(1):98. doi: 10.1186/s12885-021-09102-x
111. Song DG, Ye Q, Poussin M, Harms GM, Figini M, Powell DJ Jr. CD27 costimulation augments the survival and antitumor activity of redirected human T cells in vivo. Blood (2012) 119(3):696–706. doi: 10.1182/blood-2011-03-344275
112. Guedan S, Chen X, Madar A, Carpenito C, McGettigan SE, Frigault MJ, et al. ICOS-based chimeric antigen receptors program bipolar TH17/TH1 cells. Blood (2014) 124(7):1070–80. doi: 10.1182/blood-2013-10-535245
113. Thomas S, Xue SA, Bangham CR, Jakobsen BK, Morris EC, Stauss HJ. Human T cells expressing affinity-matured TCR display accelerated responses but fail to recognize low density of MHC-peptide antigen. Blood (2011) 118(2):319–29. doi: 10.1182/blood-2010-12-326736
114. Schmid DA, Irving MB, Posevitz V, Hebeisen M, Posevitz-Fejfar A, Sarria JC, et al. Evidence for a TCR affinity threshold delimiting maximal CD8 T cell function. J Immunol (2010) 184(9):4936–46. doi: 10.4049/jimmunol.1000173
115. Ghorashian S, Kramer AM, Onuoha S, Wright G, Bartram J, Richardson R, et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat Med (2019) 25(9):1408–14. doi: 10.1038/s41591-019-0549-5
116. Zhang A, Sun Y, Du J, Dong Y, Pang H, Ma L, et al. Reducing hinge flexibility of CAR-T cells prolongs survival In vivo with low cytokines release. Front Immunol (2021) 12:724211. doi: 10.3389/fimmu.2021.724211
117. Feucht J, Sun J, Eyquem J, Ho YJ, Zhao Z, Leibold J, et al. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat Med (2019) 25(1):82–8. doi: 10.1038/s41591-018-0290-5
118. Vardhana SA, Hwee MA, Berisa M, Wells DK, Yost KE, King B, et al. Impaired mitochondrial oxidative phosphorylation limits the self-renewal of T cells exposed to persistent antigen. Nat Immunol (2020) 21(9):1022–33. doi: 10.1038/s41590-020-0725-2
119. Ligtenberg MA, Mougiakakos D, Mukhopadhyay M, Witt K, Lladser A, Chmielewski M, et al. Coexpressed catalase protects chimeric antigen receptor-redirected T cells as well as bystander cells from oxidative stress-induced loss of antitumor activity. J Immunol (2016) 196(2):759–66. doi: 10.4049/jimmunol.1401710
120. Wan H, Xu B, Zhu N, Ren B. PGC-1alpha activator-induced fatty acid oxidation in tumor-infiltrating CTLs enhances effects of PD-1 blockade therapy in lung cancer. Tumori (2020) 106(1):55–63. doi: 10.1177/0300891619868287
121. Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, et al. The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity (2016) 45(2):374–88. doi: 10.1016/j.immuni.2016.07.009
122. Atkins RM, Menges MA, Bauer A, Turner JG, Locke FL. Metabolically flexible CAR T cells (mfCAR-T), with constitutive expression of PGC-1α resistant to post translational modifications, exhibit superior survival and function in vitro. Blood (2020) 136:30. doi: 10.1182/blood-2020-143217
123. Belikov AV, Schraven B, Simeoni L. T Cells and reactive oxygen species. J BioMed Sci (2015) 22:85. doi: 10.1186/s12929-015-0194-3
124. Zhao L, Li J, Liu Y, Kang L, Chen H, Jin Y, et al. Cholesterol esterification enzyme inhibition enhances antitumor effects of human chimeric antigen receptors modified T cells. J Immunother (2018) 41(2):45–52. doi: 10.1097/CJI.0000000000000207
125. Zebley CC, Brown C, Mi T, Fan Y, Alli S, Boi S, et al. CD19-CAR T cells undergo exhaustion DNA methylation programming in patients with acute lymphoblastic leukemia. Cell Rep (2021) 37(9):110079. doi: 10.1016/j.celrep.2021.110079
126. El Khawanky N, Hughes A, Yu W, Myburgh R, Matschulla T, Taromi S, et al. Demethylating therapy increases anti-CD123 CAR T cell cytotoxicity against acute myeloid leukemia. Nat Commun (2021) 12(1):6436. doi: 10.1038/s41467-021-26683-0
127. Prinzing B, Zebley CC, Petersen CT, Fan Y, Anido AA, Yi Z, et al. Deleting DNMT3A in CAR T cells prevents exhaustion and enhances antitumor activity. Sci Transl Med (2021) 13(620):eabh0272. doi: 10.1126/scitranslmed.abh0272
128. Kong W, Dimitri A, Wang W, Jung IY, Ott CJ, Fasolino M, et al. BET bromodomain protein inhibition reverses chimeric antigen receptor extinction and reinvigorates exhausted T cells in chronic lymphocytic leukemia. J Clin Invest (2021) 131(16):e145459. doi: 10.1172/JCI145459
129. Fraietta JA, Beckwith KA, Patel PR, Ruella M, Zheng Z, Barrett DM, et al. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood (2016) 127(9):1117–27. doi: 10.1182/blood-2015-11-679134
130. Gauthier J, Hirayama AV, Purushe J, Hay KA, Lymp J, Li DH, et al. Feasibility and efficacy of CD19-targeted CAR T cells with concurrent ibrutinib for CLL after ibrutinib failure. Blood (2020) 135(19):1650–60. doi: 10.1182/blood.2019002936
131. Luo Q, Napoleon JV, Liu X, Zhang B, Zheng S, Low PS. Targeted rejuvenation of exhausted chimeric antigen receptor T-cells regresses refractory solid tumors. Mol Cancer Res (2022) 20(5):823–33. doi: 10.1158/1541-7786.MCR-21-0711
132. Xu Y, Zhang M, Ramos CA, Durett A, Liu E, Dakhova O, et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood (2014) 123(24):3750–9. doi: 10.1182/blood-2014-01-552174
133. Sabatino M, Hu J, Sommariva M, Gautam S, Fellowes V, Hocker JD, et al. Generation of clinical-grade CD19-specific CAR-modified CD8+ memory stem cells for the treatment of human b-cell malignancies. Blood (2016) 128(4):519–28. doi: 10.1182/blood-2015-11-683847
134. Sommermeyer D, Hudecek M, Kosasih PL, Gogishvili T, Maloney DG, Turtle CJ, et al. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia (2016) 30(2):492–500. doi: 10.1038/leu.2015.247
135. Hinrichs CS, Spolski R, Paulos CM, Gattinoni L, Kerstann KW, Palmer DC, et al. IL-2 and IL-21 confer opposing differentiation programs to CD8+ T cells for adoptive immunotherapy. Blood (2008) 111(11):5326–33. doi: 10.1182/blood-2007-09-113050
136. Bonte S, de Munter S, Billiet L, Goetgeluk G, Ingels J, Jansen H, et al. In vitro OP9-DL1 co-culture and subsequent maturation in the presence of IL-21 generates tumor antigen-specific T cells with a favorable less-differentiated phenotype and enhanced functionality. Oncoimmunology (2021) 10(1):1954800. doi: 10.1080/2162402X.2021.1954800
137. Štach M, Musil J, Cetkovsky P, Otahal P. Interleukin 21 enhances survival and expansion of CAR T cells Via inhibition of their terminal differentiation during interaction with tumor target cells. Blood (2018) 132:4545. doi: 10.1182/blood-2018-99-116294
138. Ptackova P, Musil J, Stach M, Lesny P, Nemeckova S, Kral V, et al. A new approach to CAR T-cell gene engineering and cultivation using piggyBac transposon in the presence of IL-4, IL-7 and IL-21. Cytotherapy (2018) 20(4):507–20. doi: 10.1016/j.jcyt.2017.10.001
139. Stach M, Ptackova P, Mucha M, Musil J, Klener P, Otahal P. Inducible secretion of IL-21 augments anti-tumor activity of piggyBac-manufactured chimeric antigen receptor T cells. Cytotherapy (2020) 22(12):744–54. doi: 10.1016/j.jcyt.2020.08.005
140. Kranz E, Kuhlmann CJ, Chan J, Kim PY, Chen ISY, Kamata M. Efficient derivation of chimeric-antigen receptor-modified TSCM cells. Front Immunol (2022) 13:877682. doi: 10.3389/fimmu.2022.877682
141. Schultz LM, Czerwinski DK, Levy R, Levy S. CD81 costimulation skews CAR transduction toward naive T cells. Proc Natl Acad Sci U S A (2022) 119(5):e1910844119. doi: 10.1073/pnas.1910844119
142. Zheng W, O'Hear CE, Alli R, Basham JH, Abdelsamed HA, Palmer LE, et al. PI3K orchestration of the in vivo persistence of chimeric antigen receptor-modified T cells. Leukemia (2018) 32(5):1157–67. doi: 10.1038/s41375-017-0008-6
143. Petersen CT, Hassan M, Morris AB, Jeffery J, Lee K, Jagirdar N, et al. Improving T-cell expansion and function for adoptive T-cell therapy using ex vivo treatment with PI3Kdelta inhibitors and VIP antagonists. Blood Adv (2018) 2(3):210–23. doi: 10.1182/bloodadvances.2017011254
144. Urak R, Walter M, Lim L, Wong CW, Budde LE, Thomas S, et al. Ex vivo akt inhibition promotes the generation of potent CD19CAR T cells for adoptive immunotherapy. J Immunother Cancer (2017) 5:26. doi: 10.1186/s40425-017-0227-4
145. Klebanoff CA, Crompton JG, Leonardi AJ, Yamamoto TN, Chandran SS, Eil RL, et al. Inhibition of AKT signaling uncouples T cell differentiation from expansion for receptor-engineered adoptive immunotherapy. JCI Insight (2017) 2(23):e95103. doi: 10.1172/jci.insight.95103
146. Mousset CM, Hobo W, Ji Y, Fredrix H, De Giorgi V, Allison RD, et al. Ex vivo AKT-inhibition facilitates generation of polyfunctional stem cell memory-like CD8(+) T cells for adoptive immunotherapy. Oncoimmunology (2018) 7(10):e1488565. doi: 10.1080/2162402X.2018.1488565
147. Kondo T, Ando M, Nagai N, Tomisato W, Srirat T, Liu B, et al. The NOTCH-FOXM1 axis plays a key role in mitochondrial biogenesis in the induction of human stem cell memory-like CAR-T cells. Cancer Res (2020) 80(3):471–83. doi: 10.1158/0008-5472.CAN-19-1196
148. Zhang H, Hu Y, Shao M, Teng X, Jiang P, Wang X, et al. Dasatinib enhances anti-leukemia efficacy of chimeric antigen receptor T cells by inhibiting cell differentiation and exhaustion. J Hematol Oncol (2021) 14(1):113. doi: 10.1186/s13045-021-01117-y
149. Weber EW, Parker KR, Sotillo E, Lynn RC, Anbunathan H, Lattin J, et al. Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Science (2021) 372(6537):eaba1786. doi: 10.1126/science.aba1786
150. Verma V, Jafarzadeh N, Boi S, Kundu S, Jiang Z, Fan Y, et al. MEK inhibition reprograms CD8(+) T lymphocytes into memory stem cells with potent antitumor effects. Nat Immunol (2021) 22(1):53–66. doi: 10.1038/s41590-020-00818-9
151. Arakaki R, Yamada A, Kudo Y, Hayashi Y, Ishimaru N. Mechanism of activation-induced cell death of T cells and regulation of FasL expression. Crit Rev Immunol (2014) 34(4):301–14. doi: 10.1615/CritRevImmunol.2014009988
152. Wang H, Han P, Qi X, Li F, Li M, Fan L, et al. Bcl-2 enhances chimeric antigen receptor T cell persistence by reducing activation-induced apoptosis. Cancers (Basel) (2021) 13(2):197. doi: 10.3390/cancers13020197
153. Ringden O, Hermans J, Labopin M, Apperley J, Gorin NC, Gratwohl A. The highest leukaemia-free survival after allogeneic bone marrow transplantation is seen in patients with grade I acute graft-versus-host disease. acute and chronic leukaemia working parties of the European group for blood and marrow transplantation (EBMT). Leuk Lymphoma (1996) 24(1-2):71–9. doi: 10.3109/10428199609045715
154. Cosenza M, Sacchi S, Pozzi S. Cytokine release syndrome associated with T-Cell-Based therapies for hematological malignancies: Pathophysiology, clinical presentation, and treatment. Int J Mol Sci (2021) 22(14):7652. doi: 10.3390/ijms22147652
155. Dong R, Jiang S, Chen Y, Ma Y, Sun L, Xing C, et al. Prognostic significance of cytokine release syndrome in b cell hematological malignancies patients after chimeric antigen receptor T cell therapy. J Interferon Cytokine Res (2021) 41(12):469–76. doi: 10.1089/jir.2021.0057
156. Kalaitsidou M, Kueberuwa G, Schutt A, Gilham DE. CAR T-cell therapy: toxicity and the relevance of preclinical models. Immunotherapy (2015) 7(5):487–97. doi: 10.2217/imt.14.123
157. Gust J, Ponce R, Liles WC, Garden GA, Turtle CJ. Cytokines in CAR T cell-associated neurotoxicity. Front Immunol (2020) 11:577027. doi: 10.3389/fimmu.2020.577027
158. Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med (2018) 24(6):739–48. doi: 10.1038/s41591-018-0036-4
159. Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, Sadelain M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med (2018) 24(6):731–8. doi: 10.1038/s41591-018-0041-7
160. Teachey DT, Lacey SF, Shaw PA, Melenhorst JJ, Maude SL, Frey N, et al. Identification of predictive biomarkers for cytokine release syndrome after chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Cancer Discovery (2016) 6(6):664–79. doi: 10.1158/2159-8290.CD-16-0040
161. Xue L, Yi Y, Xu Q, Wang L, Yang X, Zhang Y, et al. Chimeric antigen receptor T cells self-neutralizing IL6 storm in patients with hematologic malignancy. Cell Discovery (2021) 7(1):84. doi: 10.1038/s41421-021-00299-6
162. Sachdeva M, Duchateau P, Depil S, Poirot L, Valton J. Granulocyte-macrophage colony-stimulating factor inactivation in CAR T-cells prevents monocyte-dependent release of key cytokine release syndrome mediators. J Biol Chem (2019) 294(14):5430–7. doi: 10.1074/jbc.AC119.007558
163. Sterner RM, Sakemura R, Cox MJ, Yang N, Khadka RH, Forsman CL, et al. GM-CSF inhibition reduces cytokine release syndrome and neuroinflammation but enhances CAR-T cell function in xenografts. Blood (2019) 133(7):697–709. doi: 10.1182/blood-2018-10-881722
164. Bailey SR, Vatsa S, Larson RC, Bouffard AA, Scarfo I, Kann MC, et al. Blockade or deletion of IFNgamma reduces macrophage activation without compromising CAR T-cell function in hematologic malignancies. Blood Cancer Discovery (2022) 3(2):136–53. doi: 10.1158/2643-3230.BCD-21-0181
165. Larson RC, Kann MC, Bailey SR, Haradhvala NJ, Llopis PM, Bouffard AA, et al. CAR T cell killing requires the IFNgammaR pathway in solid but not liquid tumours. Nature (2022) 604(7906):563–70. doi: 10.1038/s41586-022-04585-5
166. Zhang Y, Li Y, Cao W, Wang F, Xie X, Li Y, et al. Single-cell analysis of target antigens of CAR-T reveals a potential landscape of "On-target, off-tumor toxicity". Front Immunol (2021) 12:799206. doi: 10.3389/fimmu.2021.799206
167. Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS, et al. Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell (2016) 164(4):770–9. doi: 10.1016/j.cell.2016.01.011
168. Hamburger AE, DiAndreth B, Cui J, Daris ME, Munguia ML, Deshmukh K, et al. Engineered T cells directed at tumors with defined allelic loss. Mol Immunol (2020) 128:298–310. doi: 10.1016/j.molimm.2020.09.012
169. Williams JZ, Allen GM, Shah D, Sterin IS, Kim KH, Garcia VP, et al. Precise T cell recognition programs designed by transcriptionally linking multiple receptors. Science (2020) 370(6520):1099–104. doi: 10.1126/science.abc6270
170. Kim MY, Cooper ML, Jacobs MT, Ritchey JK, Hollaway J, Fehniger TA, et al. CD7-deleted hematopoietic stem cells can restore immunity after CAR T cell therapy. JCI Insight (2021) 6(16):e149819. doi: 10.1172/jci.insight.149819
171. Kim MY, Yu KR, Kenderian SS, Ruella M, Chen S, Shin TH, et al. Genetic inactivation of CD33 in hematopoietic stem cells to enable CAR T cell immunotherapy for acute myeloid leukemia. Cell (2018) 173(6):1439–53.e19. doi: 10.1016/j.cell.2018.05.013
172. Borot F, Wang H, Ma Y, Jafarov T, Raza A, Ali AM, et al. Gene-edited stem cells enable CD33-directed immune therapy for myeloid malignancies. Proc Natl Acad Sci U S A (2019) 116(24):11978–87. doi: 10.1073/pnas.1819992116
173. Loew R, Heinz N, Hampf M, Bujard H, Gossen M. Improved tet-responsive promoters with minimized background expression. BMC Biotechnol (2010) 10:81. doi: 10.1186/1472-6750-10-81
174. Gu X, He D, Li C, Wang H, Yang G. Development of inducible CD19-CAR T cells with a tet-on system for controlled activity and enhanced clinical safety. Int J Mol Sci (2018) 19(11):3455. doi: 10.3390/ijms19113455
175. Drent E, Poels R, Mulders MJ, van de Donk N, Themeli M, Lokhorst HM, et al. Feasibility of controlling CD38-CAR T cell activity with a tet-on inducible CAR design. PloS One (2018) 13(5):e0197349. doi: 10.1371/journal.pone.0197349
176. Scholar E. Doxycycline. In: Enna SJ, Bylund DB, editors. xPharm: The comprehensive pharmacology reference. New York: Elsevier (2007). p. 1–8.
177. He H, Liao Q, Zhao C, Zhu C, Feng M, Liu Z, et al. Conditioned CAR-T cells by hypoxia-inducible transcription amplification (HiTA) system significantly enhances systemic safety and retains antitumor efficacy. J Immunother Cancer (2021) 9(10):e002755. doi: 10.1136/jitc-2021-002755
178. Nguyen NT, Huang K, Zeng H, Jing J, Wang R, Fang S, et al. Nano-optogenetic engineering of CAR T cells for precision immunotherapy with enhanced safety. Nat Nanotechnol (2021) 16(12):1424–34. doi: 10.1038/s41565-021-00982-5
179. Kobayashi A, Nobili A, Neier SC, Sadiki A, Distel R, Zhou ZS, et al. Light-controllable binary switch activation of CAR T cells. ChemMedChem (2022) 17(12):e202100722. doi: 10.1002/cmdc.202100722
180. Miller IC, Zamat A, Sun LK, Phuengkham H, Harris AM, Gamboa L, et al. Enhanced intratumoural activity of CAR T cells engineered to produce immunomodulators under photothermal control. Nat BioMed Eng (2021) 5(11):1348–59. doi: 10.1038/s41551-021-00781-2
181. Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, et al. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia (2010) 24(6):1160–70. doi: 10.1038/leu.2010.75
182. Budde LE, Berger C, Lin Y, Wang J, Lin X, Frayo SE, et al. Combining a CD20 chimeric antigen receptor and an inducible caspase 9 suicide switch to improve the efficacy and safety of T cell adoptive immunotherapy for lymphoma. PloS One (2013) 8(12):e82742. doi: 10.1371/journal.pone.0082742
183. Minagawa K, Jamil MO, Al-Obaidi M, Pereboeva L, Salzman D, Erba HP, et al. In vitro pre-clinical validation of suicide gene modified anti-CD33 redirected chimeric antigen receptor T-cells for acute myeloid leukemia. PloS One (2016) 11(12):e0166891. doi: 10.1371/journal.pone.0166891
184. Warda W, Larosa F, Neto Da Rocha M, Trad R, Deconinck E, Fajloun Z, et al. CML hematopoietic stem cells expressing IL1RAP can be targeted by chimeric antigen receptor-engineered T cells. Cancer Res (2019) 79(3):663–75. doi: 10.1158/0008-5472.CAN-18-1078
185. Philip B, Kokalaki E, Mekkaoui L, Thomas S, Straathof K, Flutter B, et al. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood (2014) 124(8):1277–87. doi: 10.1182/blood-2014-01-545020
186. Vogler I, Newrzela S, Hartmann S, Schneider N, von Laer D, Koehl U, et al. An improved bicistronic CD20/tCD34 vector for efficient purification and in vivo depletion of gene-modified T cells for adoptive immunotherapy. Mol Ther (2010) 18(7):1330–8. doi: 10.1038/mt.2010.83
187. Griffioen M, van Egmond EH, Kester MG, Willemze R, Falkenburg JH, Heemskerk MH. Retroviral transfer of human CD20 as a suicide gene for adoptive T-cell therapy. Haematologica (2009) 94(9):1316–20. doi: 10.3324/haematol.2008.001677
188. Sommer C, Cheng HY, Nguyen D, Dettling D, Yeung YA, Sutton J, et al. Allogeneic FLT3 CAR T cells with an off-switch exhibit potent activity against AML and can be depleted to expedite bone marrow recovery. Mol Ther (2020) 28(10):2237–51. doi: 10.1016/j.ymthe.2020.06.022
189. Chung HK, Jacobs CL, Huo Y, Yang J, Krumm SA, Plemper RK, et al. Tunable and reversible drug control of protein production via a self-excising degron. Nat Chem Biol (2015) 11(9):713–20. doi: 10.1038/nchembio.1869
190. Juillerat A, Tkach D, Busser BW, Temburni S, Valton J, Duclert A, et al. Modulation of chimeric antigen receptor surface expression by a small molecule switch. BMC Biotechnol (2019) 19(1):44. doi: 10.1186/s12896-019-0537-3
191. Cao W, Geng ZZ, Wang N, Pan Q, Guo S, Xu S, et al. A reversible chemogenetic switch for chimeric antigen receptor T cells. Angew Chem Int Ed Engl (2022) 61(10):e202109550. doi: 10.1002/anie.202109550
192. Gardner TJ, Lee JP, Bourne CM, Wijewarnasuriya D, Kinarivala N, Kurtz KG, et al. Engineering CAR-T cells to activate small-molecule drugs in situ. Nat Chem Biol (2022) 18(2):216–25. doi: 10.1038/s41589-021-00932-1
193. Gomes-Silva D, Srinivasan M, Sharma S, Lee CM, Wagner DL, Davis TH, et al. CD7-edited T cells expressing a CD7-specific CAR for the therapy of T-cell malignancies. Blood (2017) 130(3):285–96. doi: 10.1182/blood-2017-01-761320
194. Georgiadis C, Rasaiyaah J, Gkazi SA, Preece R, Etuk A, Christi A, et al. Base-edited CAR T cells for combinational therapy against T cell malignancies. Leukemia (2021) 35(12):3466–81. doi: 10.1038/s41375-021-01282-6
195. Freiwan A, Zoine J, Crawford JC, Vaidya A, Schattgen SA, Myers J, et al. Engineering naturally occurring CD7 negative T cells for the immunotherapy of hematological malignancies. Blood (2022). doi: 10.1182/blood.2021015020
Keywords: CAR-T cell therapy, hematological malignancies, immune evasion, tumor microenvironment, toxicity
Citation: Huang J, Huang X and Huang J (2022) CAR-T cell therapy for hematological malignancies: Limitations and optimization strategies. Front. Immunol. 13:1019115. doi: 10.3389/fimmu.2022.1019115
Received: 14 August 2022; Accepted: 13 September 2022;
Published: 28 September 2022.
Edited by:
Peng Qu, National Institutes of Health (NIH), United StatesReviewed by:
Guohao Wang, National Institutes of Health (NIH), United StatesCopyright © 2022 Huang, Huang and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juan Huang, aHVhbmdqdWFueHlAbWVkLnVlc3RjLmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.