
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 17 October 2022
Sec. Viral Immunology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1018053
This article is part of the Research Topic Interferon and its Antiviral Effect in Response to HBV Infection View all 5 articles
 Zhijing Yang1,3†
Zhijing Yang1,3† Baozhen Sun2†Jingcheng Xiang1,3Han Wu1,3Shaoning Kan1,3
Baozhen Sun2†Jingcheng Xiang1,3Han Wu1,3Shaoning Kan1,3 Ming Hao1,3
Ming Hao1,3 Lu Chang1,3
Lu Chang1,3 Huimin Liu1,3
Huimin Liu1,3 Dongxu Wang4*
Dongxu Wang4* Weiwei Liu1,3*
Weiwei Liu1,3*Human hepatitis B virus (HBV) is a small, enveloped DNA virus that causes acute and chronic hepatitis. Chronic hepatitis B (CHB) is associated with hepatocellular carcinoma pathogenesis. Interferons (IFNs) have been used for the treatment of CHB for a long time, with advantages including less treatment duration and sustained virological response. Presently, various evidence suggests that epigenetic modification of the viral covalently closed circular DNA (cccDNA) and the host genome is crucial for the regulation of viral activity. This modification includes histone acetylation, DNA methylation, N6-methyladenosine, and non-coding RNA modification. IFN treatment for CHB can stimulate multiple IFN-stimulated genes for inhibiting virus replication. IFNs can also affect the HBV life cycle through epigenetic modulation. In this review, we summarized the different mechanisms through which IFN-α inhibits HBV replication, including epigenetic regulation. Moreover, the mechanisms underlying IFN activity are discussed, which indicated its potential as a novel treatment for CHB. It is proposed that epigenetic changes such as histone acetylation, DNA methylation, m6A methylation could be the targets of IFN, which may offer a novel approach to HBV treatment.
Hepatitis B is a global epidemic that remains a great challenge (1, 2). After acute infection with the hepatitis B virus (HBV), 90%–95% of patients can be cured completely; however, the remaining 5%–10% can act as carriers, allowing chronic HBV infections to spread within populations (3). Moreover, HBV infection leads to liver cirrhosis and hepatocellular carcinoma (HCC), which can be fatal (4, 5). China is the country with the heaviest burden of HBV infection and liver cancer is the second most common cancer in China (6, 7).
HBV is a small DNA virus belonging to the family Hepadnaviridae. Its genetic material is 3.2 kb long and consists of relaxed, circular, partially double-stranded DNA, which is enclosed within the nucleocapsid core of the virus (8). HBV enters hepatocytes through a receptor-mediated pathway and binds to the receptor sodium taurocholate cotransporter polypeptide (NTCP) on the surface of hepatocytes via its envelope proteins PreS1 and PreS2 (9). After entering the hepatocytes, the relaxed, circular, partially double-stranded DNA (rcDNA) of HBV is released from the viral nucleocapsid and transported into the nucleus (10). Redundant sequences at the pol-linked terminal on minus strand DNA and RNA oligonucleotides at the 5’ ends of plus strand DNA are removed from rcDNA and gaps on both strands are filled in and connected to generate cccDNA (11–13). CccDNA is used as a template for the transcription of pregenomic RNA (pgRNA) and subgenomic mRNA (Figure 1) (14, 15). PgRNA was catalyzed by HBV polymerase to synthesize viral genomic DNA. Meanwhile, subgenomic mRNA is translated into various viral proteins as a part of the HBV life cycle. Among the proteins, the packaging proteins are packaged with the nascent viral DNA to form progeny virus and released from the cells (16). HBV cccDNA is important for chronic infection (17) and exists in the nucleus as a minichromosome, bound to histones and non-histone proteins. The epigenetic modification of cccDNA contributes to the viral replication and affects the prognosis of chronic HBV infection (18). Therefore, epigenetic modifications, such as DNA methylation, RNA methylation, histone acetylation, miRNA regulation, and chromatin remodeling, can regulate the activity of cccDNA and offer new therapeutic targets for HBV infection.
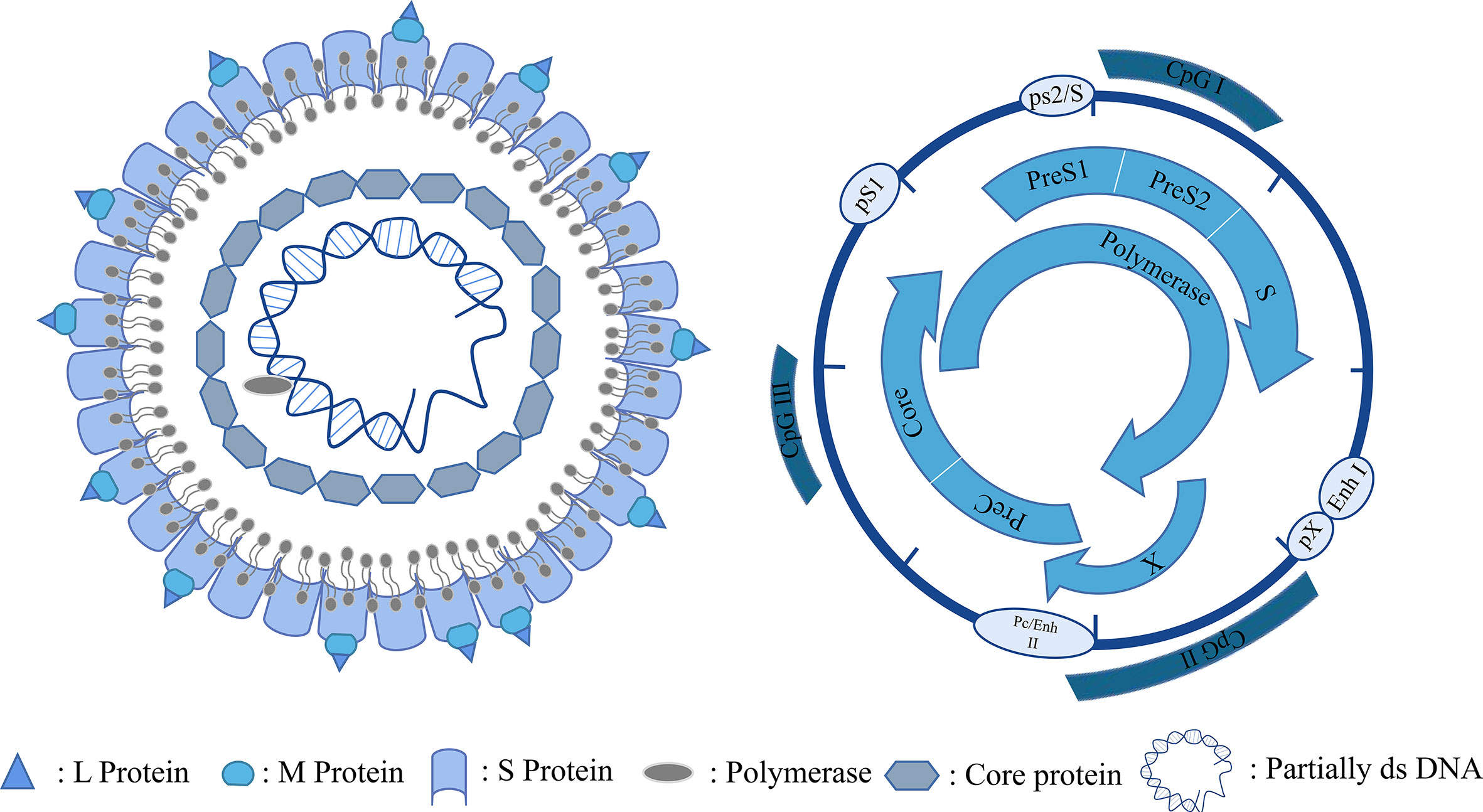
Figure 1 HBV particles and genome.
Current Food and Drug Administration (FDA)-approved treatments for chronic HBV infection include interferon-α (IFN-α) and nucleoside analogs (such as lamivudine, adefovir dipivoxil, entecavir, telbivudine, and tenofovir fumarate dipivoxil) (19). IFN-α has various therapeutic advantages, which include less treatment time and higher clearance of hepatitis B antigen and surface antigen (20). Moreover, IFN-α has both immunomodulatory and antiviral effects (21) and can inhibit cccDNA activity through epigenetic repression (22). In this review, we have discussed the mechanism underlying the IFN treatment for HBV infection and described the epigenetic modification of HBV through IFN treatment.
IFN is a natural immune substance with broad-spectrum antiviral activity (23). It is released by virus-infected cells. The following three types of IFNs have been identified: type I IFN (including IFN-α, -β, -ϵ, -κ, -ω, and others), type II IFN (IFN-γ), and type III IFN (IFN-λ) (24). IFN inhibits virus replication by promoting the expression of downstream IFN-stimulated genes (ISGs) via multiple signaling pathways (25, 26). Different types of IFNs bind to distinct receptors on the surfaces of cognate cells. IFN-I binds to IFN-α receptor 1 (IFNAR1) and 2 (IFNAR2) heterodimers, whereas IFN-III binds to interleukin-10 receptor 2 (IL-10R2) and IFN-λ receptor 1 (IFNLR1) heterodimers, and IFN-II binds to heterodimers consisting of IFN-γ receptor 1 (IFNGR1) and 2 (IFNGR2) (27). The binding of IFNs to their receptors initiates the signaling cascades via the Janus kinase (JAK)- signal transducer and activator of transcription (STAT) pathway, leading to the transcriptional regulation of several genes (28). The binding of IFN-I and IFN-III to their respective receptors leads to the phosphorylation of TYK-2 near JAK1, which is followed by further activation of STAT1 and STAT2. Phosphorylated STAT1 and STAT2 bind to IRF-9, which forms the heterotrimeric complex ISGF3 (29). Subsequently, ISGF3 is transported into the nucleus where it binds to ISREs and activates ISG transcription. The intracellular domains of the IFN-II receptors, IFNGR1 and IFNGR2, bind and activate JAK1 and JAK2 kinases, respectively. They induce phosphorylation of STAT1 and STAT2, which subsequently form homodimeric GAF. This is transported into the nucleus where it binds to GAS and induces ISG transcription (30–32) (Figure 2).
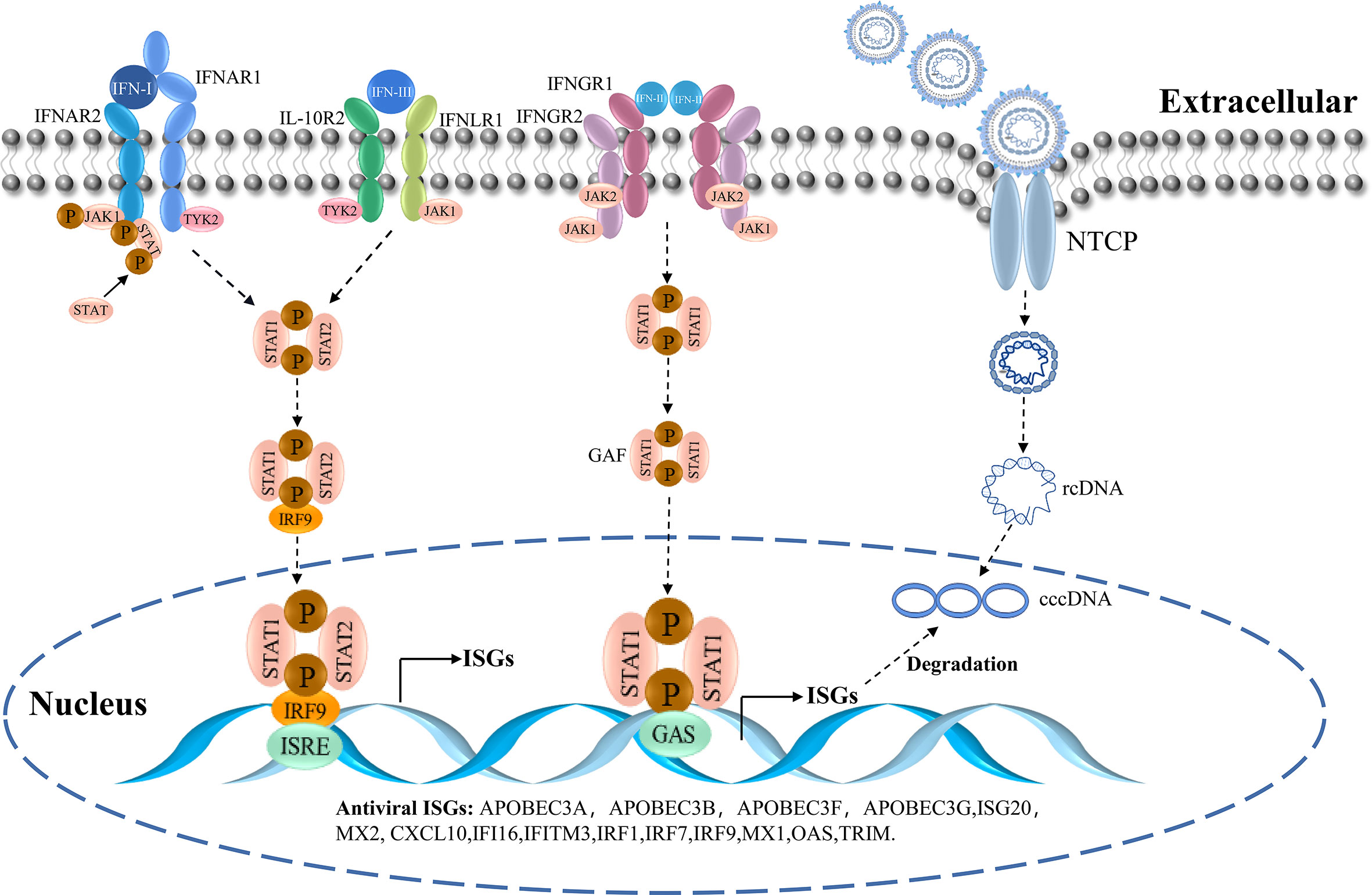
Figure 2 The IFN-signaling cascade and the entry of HBV.
ISGs play an important role in host resistance to HBV infection. It inhibits viral entry and exit as well as viral replication, transcription, translation, and post-translational modification (33, 34). For the invasive phase of the virus, ISGs include the myxovirus resistance gene (Mx) and the IFN -induced transmembrane proteins CH25H, viperin, and tetherin. IFN-induced proteins are capable of inhibiting viral replication and transcription. They include 2′-5′-oligoadenylate synthetase (OAS), protein kinase R (PKR), ZAP, IFN-induced protein with tetratricopeptide repeats, ISG15, and members of the TRIM family. Furthermore, the release phase of the virus is inhibited by proteins, including viperin and tetherin (35, 36). The SMAD4A can bind to the Smaug recognition region (SRE) sequence in the HBV virus sequence to trigger the degradation of the virus (37). TRIM25 is downregulated in HBV patients, and TRIM25 overexpression results in increased IFN production and decreased HBV replication (38). The proteomic data analysis showed that after IFN treatment, the levels of the proteins RIG-I and RIG-G (known to be suppressed by HBV) are restored. Moreover, RNA metabolism, translation, and endoplasmic reticulum targeting are differentially regulated in the biological process (39). Therefore, as a class of effector molecules mediating antiviral effects, ISGs can target virtually all the processes of viral invasion, uncoating, genome replication, and virion assembly and release, thereby inhibiting viral proliferation in vivo (Figure 2).
Presently, seven therapies have been approved by the FDA for their use in HBV clinical treatment, including standard IFN and pegylated interferon (peg-IFN) (19). Peg-IFN has been recommended by the guidelines of the major liver associations (40, 41). The course of peg-IFN therapy is finite and can lead to long-term benefits, such as continuous and cumulative responses. Moreover, the progression of hepatitis to fibrosclerosis and hepatocellular carcinoma can be reduced (42). The clinical applications of interferon and its mechanisms underlying the HBV treatment are discussed below.
Even though the mechanisms underlying IFN treatment for hepatitis B are still being elucidated, IFN has been used in the clinical treatment of HBV for decades and remains an effective therapy. For patients with chronic hepatitis B (CHB), standard (peg-IFN) monotherapy is administered subcutaneously once weekly for 48 weeks, which has decreased treatment time and sustained virological response (43). After peg-IFN-α treatment, some patients with CHB maintained a functional cure, related to lower HBcrAg and higher HBsAb levels (44). Clinical trial results have shown that peg-IFN-α2b treatment can lead to a greater decrease in HBV DNA in patients with HBeAg positive compared with patients who received an only placebo (45). In addition, peg-IFN-α2b was effective in approximately one-third of patients who were refractory to standard IFN or lamivudine therapy (46). Compared with HBeAg-negative patients who received Entecavir (ETV) monotherapy, HBeAg-negative patients who received peg-IFN monotherapy showed a significantly greater decline in HBsAg levels (47).
IFN can also be used as an adjuvant with other types of drugs to achieve better HBV treatment effects. The peg-IFN-nucleoside analog (NA) sequential optimization therapy (SOT) can lower HBsAg, undetectable HBV DNA, and ALT normalization compared with peg-IFN monotherapy (48). One study showed that after 48 weeks of treatment, HBsAg in the early combined treatment group (ETV plus Peg-IFN-α-2a) decreased by more than 1500 IU/mL, and the average HBsAg level was significantly lower than that in the late combined treatment group and the NA monotherapy group (P <0.05) (49). In another study, after peg-IFN treatment, responses were doubled in patients that were not treated with peg-IFN with HBsAg below 4000 IU/mL and HBV DNA below 50 IU/mL. These patients are the candidates for peg-IFN add-on therapy (50). In addition, the combination therapy after hepatitis B vaccination significantly increased the seroclearance of HBsAg (51).
Presently, HBV cannot be completely cured by current approved clinical drugs. Reducing the loss of serum HBV DNA and HBsAg is presently a method that can reduce the probability of transformation to hepatitis and hepatic cell carcinoma. IFN therapy has the advantages of a shorter treatment cycle and fewer treatment times that can improve the patient’s compliance and treatment effectiveness. Moreover, IFN therapy has advantages over NAs in immunology mechanisms and can alter the state of immune tolerance. However, despite the side effects of IFN therapy, the relatively low response rates are a problem. NAs do not target the cccDNA; therefore, the HBV can reactivate. Therefore, combination therapy can exert a better effect on the treatment of CHB.
IFN can develop direct anti-viral effects and develop a complex immune response in the treatment of HBV. IFN-α14 can suppress the transcription of cccDNA and the production of HBsAg and HBeAg (52). IFN has been shown to increase T cell survival, the expression of T cell antigens, and IL 12 (53). It can induce antiviral activity in hepatocytes through the regulation of gene expression and protein translation, thereby playing a non-cytolytic antiviral role in many stages of the HBV life cycle (54). For example, the MX2 induced by IFN-α can decrease the number of cccDNA and the levels of HBV RNA (55). Furthermore, TRIM14 is an important molecule in the IFN signaling pathway, playing a crucial role in HBV suppression mediated by IFN-I. The TRIM14 SPRY domain interacts with the C-terminus of HBx, which can block the role of HBx in promoting HBV replication by inhibiting the formation of the Smc-HBx-DDB1 complex (56). To elucidate the mechanism underlying IFN treatment in HBV, high-throughput bimolecular fluorescence complementation screening was performed to determine the interactions between HBx protein and 145 ISGs. The results showed that seven HBx-interacting ISGs (GBP2, PVRL4, CBFβ, TRIM38, TRIM5γ, TRIM25, and Gadd45γ) exerted strong inhibitory effects on HBV replication. Among them, TRIM5γ and TRIM31 promoted HBx degradation, which can offer a novel therapeutic method in cases of IFN drug resistance (57). Under the action of a key nuclease for IFN-triggered cccDNA clearance, IFN-α induced the non-cytolytic removal of HBV cccDNA from the nucleus of infected hepatocytes in an A3A deaminase-dependent manner (22, 58). Further cell-based assays were performed to detect the anti-viral activity of ISGs. These results showed that SMAD4A was the strongest suppressor of HBV replication, functioning through an SRE-like sequence located in viral RNA. Moreover, the SMAD family inhibited HBV in mouse models (37). Elucidating the mechanism of action of IFN in the treatment of HBV can contribute to the development of IFN-related drugs in clinical practice and provide new targets for HBV treatment.
The cccDNA is a major hindrance in the treatment of HBV infection (12, 59). After HBV treatment, cccDNA molecules persist in low numbers, which causes CHB. Epigenetic modification regulates the transcriptional activity of cccDNA chromosomes (60). Therefore, studying the epigenetic modification of cccDNA can aid the development of new therapeutic drugs to eliminate cccDNA (61).
HBV has a tightly condensed DNA genome of only 3.2 kb in length. The four functional viral proteins are produced in overlapping open reading frames (ORFs), which are orientated in the same direction and encoded by negative chains (62), which are as follows: (i) preS/S, encoding the three viral surface proteins (HBs) (termed the small [S], medium [M], and large [L] surface antigens) that bind to the viral envelope and mediate viral entry; (ii) Precore/core, encoding the HBV core protein (HBc) and the secreted e-antigen (HBeAg); (iii) viral polymerase POL, which participates in viral replication and packaging; and (iv) the X region, encoding the X protein (HBx), which has been proved to have multiple functions, such as viral replication, and is the main cause of liver cancer caused by HBV (63, 64). In addition, two enhancers (enhancers I and II) and three predicted CpG islands constitute the structure of HBV cccDNA. HBV cccDNA exists as a minichromosome and undergoes post-translational modification (PTM) of the bound histones (65). Furthermore, the transcriptional activity of cccDNA is highly affected by epigenetic modification (15). IFN and NA treatment have limited treatment effects because they cannot eliminate HBV cccDNA. Therefore, the understanding of epigenetic modifications in the replication and disease development of HBV is important for degrading cccDNA and developing new therapeutic approaches. The epigenetic changes involved in the process of HBV infection are mentioned below.
DNA methylation typically occurs on CpG dinucleotides through the activity of DNA methyltransferases (DNMTs) and is often associated with gene transcriptional silencing (66). The HBc maintains its binding to cccDNA by binding to CpG island 2 in the HBV genome, and its relative abundance is highly associated with the serum HBV DNA levels in patients (67, 68). One study showed that patients with occult hepatitis B infection had a high methylation density at CpG island 2 (69). CpG island 2 methylation was significantly higher in patients with HBeAg-negative. In addition, HBV DNA methylation was higher in CpG islands 2 and 3 in HCC tissues compared with infected and cirrhotic tissues (70–72). HBx expression increased total DNMT activity by upregulating DNMT1, DNMT3A1, and DNMT3A2 and selectively promoted the regional hypermethylation of specific tumor suppressor genes, which caused hepatocarcinogenesis (73). HBV infection cause hypermethylation of the promoter region of the tumor suppressor E-cadherin, the expression of which is frequently absent in HCC (74). A study showed that the HBV DNA demethylation and increased abundance of 5hmc residues in viral CpG sequences is associated with the HBV replication (75). In general, these reports indicated that the DNA modification plays an important role in the HBV replication and HCC development, which may provide a new target for HBV treatment.
Histone acetylation also occurs during the process of HBV infection. Many histone modifying enzymes, such as acetyltransferases (HATs), deacetylases (HDACs), lysine methyltransferases, and protein arginine methyltransferases, can modify histones associated with cccDNA (76). Transcription of HBV minichromosomes is regulated by epigenetic changes in cccDNA-bound histones, whereas the acetylation status of H3/H4 histones that regulate cccDNA binding affects HBV replication (77). Chromatin immunoprecipitation (ChIP)-sequencing assays of HBV cccDNA showed non-randomly distributed PTMs, which strongly suggested that PTMs in the stained cccDNA are specifically introduced after histone assembly. High levels of H3K4me3, H3K27ac, and H3K122ac have been detected in infected cells, which indicates their importance for HBV transcription (78). Therefore, understanding the role of histone acetylation in cccDNA transcription is important for HBV treatment.
While DNA methylation and histone acetylation are targeted to DNA and proteins, RNA transcriptional regulation is also involved in epigenetic modifications in HBV. N6-methyladenosine (m6A) modification is the most common form of modification in eukaryotic cells and viruses, which includes HBV, regulating RNA transcription, splicing, degradation, and translation without changing the base sequence (79, 80). m6A regulates HBV RNA stability and reverse transcription during the HBV life cycle (81). HBV can affect m6A levels in host cells to achieve HBV-directed immune evasion. A study has shown that HBV significantly increased m6A modification of PTEN RNA, which resulted in decreased levels of PTEN protein. In the absence of PTEN, IRF-3 dephosphorylation at Ser97 was diminished and IFN synthesis was impaired (82). Moreover, ChIP analysis of wild-type HBV and HBx-null virus-infected primary human hepatocytes and HepG2-NTCP cells revealed recruitment of METTL3/14 proteins by HBx at transcriptional initiation sites, which led to internal RNA m6A modification (83). Overall, m6A modification occurring during HBV replication directly affected co-transcriptional synthesis and modification of viral RNA.
MiRNA is an important factor in HBV. Cellular miRNAs are specific noncoding RNA molecules and 21–25 nucleotides in length. They regulate gene expression by preventing translation or accelerating RNA degradation by targeting specific mRNAs (84). Viruses can also encode miRNAs that regulate viral replication by altering host gene expression (85). miRNAs can affect HBV replication directly by binding to HBV transcripts or indirectly by targeting cytokines involved in HBV replication (86). HBV-miR-3, an HBV-encoded miRNA whose expression is highly related to HBV activity, promotes the anti-HBV effects of IFN and also induces the production of IL-6 in M1 macrophages (87). Liver tissue samples from 52 of 87 patients with CHB showed expression of HBV-miR-6. The levels of HBV-mir-6 correlate with hepatic HBV DNA and plasma HBsAg levels, suggesting that this molecule can participate in viral excretion or particle formation (88). Therefore, miRNA encoded by HBV can be a new target for HBV treatment.
To summarize, epigenetic modifications involved in the HBV life cycle include DNA methylation, histone acetylation, m6A modification, and miRNA expression. Therefore, a deeper investigation into epigenetic regulatory processes can provide new avenues for controlling CHB.
Epigenetic repression of HBV by IFN-α is one of the most important mechanisms of modulating cccDNA modulation (89). IFN-α can regulate the HBV cccDNA minichromosome by modulation of GCN5-mediated succinylation of histone H3K79, facilitating clearance of HBV cccDNA (90). IFN-α2b reportedly increased the HDAC3-mediated de-2-hydroxyisobutyrylation of histone H4 lysine 8 (H4K8) on the HBV cccDNA minichromosome, restricting transcription of the cccDNA in liver (91). Another study showed that the proteins YY1 and Ezh2 (components of the transcriptional repressor complex PRC2) and the histone deacetylases HDAC1 and hSirt1 were actively recruited to cccDNA after IFN-α treatment, which led to the hypoacetylation of cccDNA-bound histones (89) (Figure 3). Furthermore, the high-dose IFN-α-mediated upregulation of cytidine deaminase or NFκB pathway induction by antibody-mediated activation of the lymphotoxin-β receptor (LTβR) can promote partial cccDNA degradation (92).
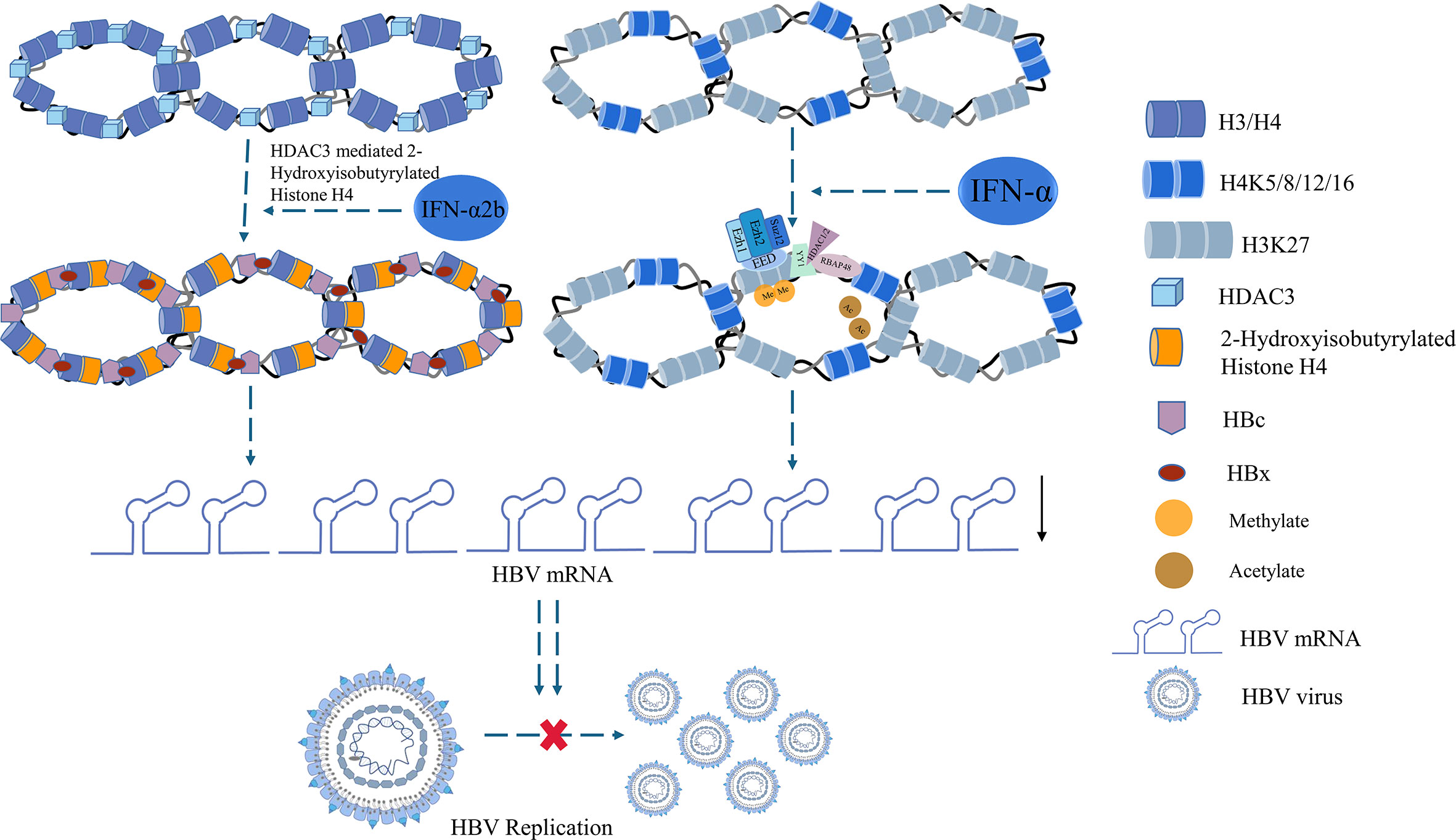
Figure 3 Histone acetylation involved in IFN treatment.
Further anti−HBV effects of IFN−α involve m6A modification (Figure 4). For example, increased m6A modification of pgRNA was observed after IFN-α2a treatment. The expression of METTL3 and METTL14 was significantly upregulated, whereas that of FTO was downregulated in IFN-α2a-treated HepG2.2.15 cells. These results suggested that IFN-α2a can regulate m6A RNA modification (93). IFN-α can induce ISG20 to selectively degrade m6A-containing HBV RNA. ISG20 can form a complex with YTHDF2 to recruit ISG20, which leads to the degradation of HBV transcripts (94). Overall, IFNs can epigenetically regulate RNA transcription, which leads to transcriptional repression of cccDNA.
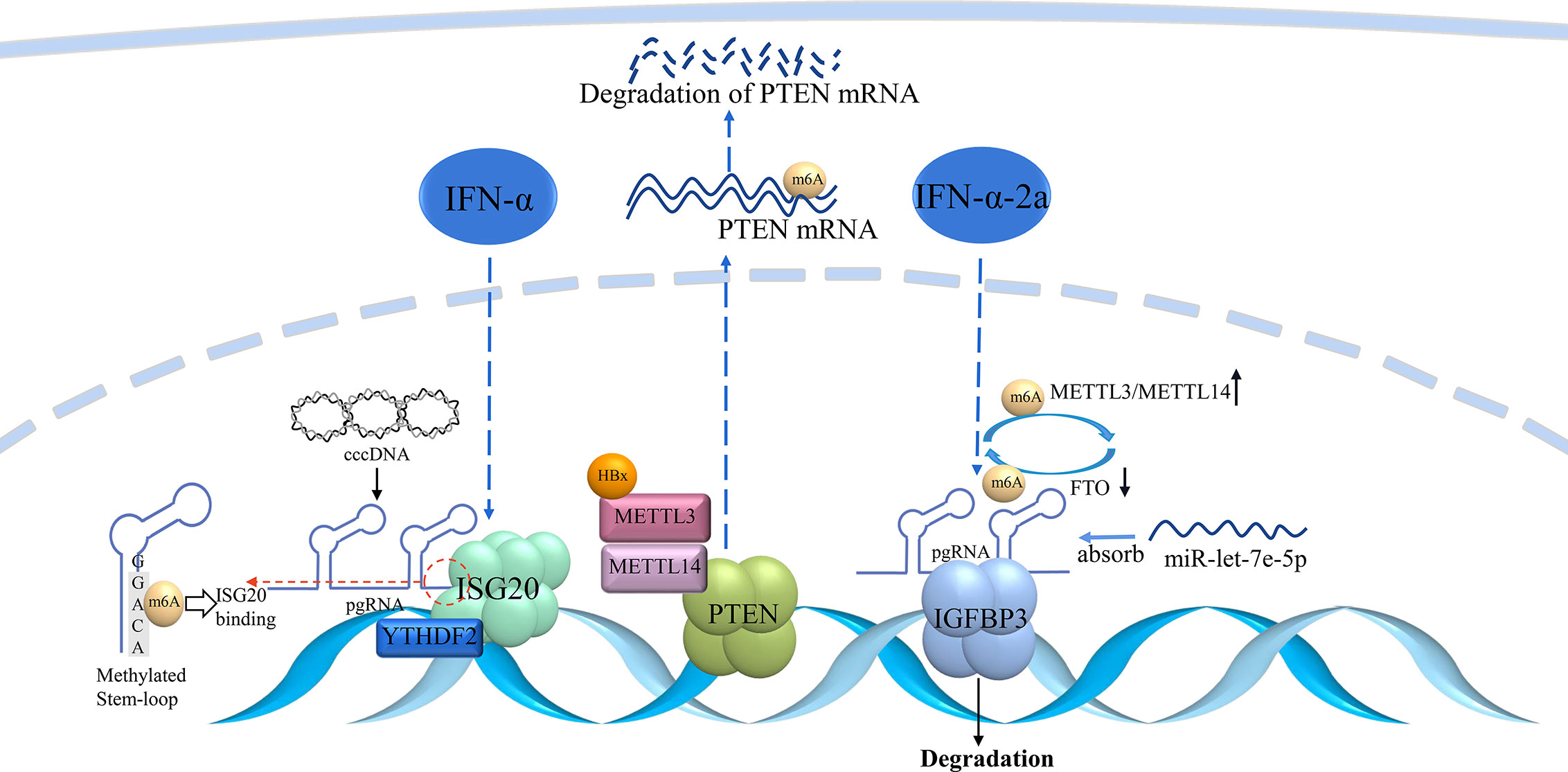
Figure 4 m6A modulation involved in IFN treatment.
Moreover, a study showed that during the peg-IFN treatment, the serum mir-6126 is high, which predicted HBsAg 1-log drop, and the mir-6126 can inhibit HBsAg production and HBV replication in vitro (95). A lentivirus-mediated RNAi high-throughput screen for epigenetic modifiers was performed, and it was found that the methyltransferase SETD2 was a key factor involved in the IFN-α mediated antiviral response. It can directly mediate STAT1 methylation on lysine 525 through its methyltransferase activity to increase IFN-α activated STAT1 phosphorylation and antiviral cellular responses (96). IFN can exert the anti-viral effect which inhibits the cccDNA transcription and improve its anti-viral activity through epigenetic modification. The study of epigenetic modification in IFN treatment in HBV infection can offer new insight into the combination of epigenetic drugs and IFN for CHB treatment.
Disease caused by HBV is a global health burden, with 256 million people chronically afected (97, 98). HBV is an enveloped DNA virus, belonging to the Hepadnaviridae family (99, 100). After the entry of the virus into the nucleus, cccDNA is formed through the action of DNA polymerase (101), acting as a stable template for viral transcription (102). Even when antigen levels fulfill the treatment criteria, cccDNA remains in the infected cells and tissues, which can reactivate again (103). Owing to the inherent stability of cccDNA, it is now recognized as the main marker of virus persistence and the main obstacle in curing CHB. IFN therapy is a common therapy used for the treatment of CHB based on its immune response; however, drug resistance is a problem. IFN-α significantly induces IL-6 expression in HBV replicated hepatocytes, which upregulates the expression of cytokine signal transduction inhibitor-3 (SOCS3) and downregulates the expression of downstream effectors of IFN-α. This impairs IFN-α anti-HBV efficiency and leads to IFN resistance (104). Moreover, IFN therapy leads to side effects such as fatigue, thyroid dysfunction, depression, and cognitive slowing because of off-target effects (105). Therefore, IFN in combination with epigenetic modification inhibitors may decrease the off-target effect to achieve a better therapeutic effect.
Because the function of cccDNA is dependent on its epigenetic modification, silencing the activity of cccDNA through epigenetic mechanisms can be an efficient and practical approach to control CHB (18, 106). Epigenetic drugs have been used for the treatment of HBV to silence the transcription of cccDNA. The HAT inhibitor C646 can downregulate H3K27ac and H3K122ac, thereby silencing cccDNA and decreasing HBV transcription (78). Moreover, Gs5801 (Gilead), a prodrug that specifically inhibits lysine demethylase 5, can inhibit cccDNA transcription by removing H3K4me3 (107). SAM can decrease the methylation level of STAT1, which can increase the antiviral effect of IFN-α (108). Because IFN can modulate the epigenetic status of cccDNA, a combination of epigenetic drugs and IFN therapy can be more effective for patients with CHB in the future. To develop such therapies, further elucidation of the molecular mechanisms underlying cccDNA epigenetic regulation in IFN treatment is required.
IFN is crucial to HBV treatment, and it can also inhibit HBV replication through ISGs and some epigenetic modification occurred during its treatment. HBV is a DNA virus whose infection is tightly linked to its cccDNA. Epigenetic changes such as histone acetylation, DNA methylation, m6A modification, and miRNA expression is involved in its infection process. Therefore, it is important to understand the epigenetic changes involved in the pathogenesis and treatment of CHB which may provide a new therapeutic target to develop new treatment methods.
ZY and DW wrote the manuscript; ZY, BS, JX, HW, HL, LC, MH, SK, and WL collected the references and drew the figures. All authors contributed to the article and approved the submitted version.
This work was supported by the Fundamental Research Funds of the Jilin Province, Department of Finance (Grant No. jcsz202189313), the Jilin Scientific and Technological Development Program (Grant Nos. 81602377), the Changchun Scientific and Technological Development Program (Grant Nos. 21ZY26), the Jilin Province Scientific and Technological Development Program (Grant Nos. 20200801077GH) and the Jilin Province Scientific and Technological Development Program (Grant Nos. 20220505033ZP).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a shared parent affiliation with the authors at the time of review.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Nelson NP, Easterbrook PJ, McMahon BJ. Epidemiology of hepatitis b virus infection and impact of vaccination on disease. Clin Liver Dis (2016) 20:607–28. doi: 10.1016/j.cld.2016.06.006
2. Wang FS, Fan JG, Zhang Z, Gao B, Wang HY. The global burden of liver disease: The major impact of China. Hepatology (2014) 60:2099–108. doi: 10.1002/hep.27406
3. Mani SKK, Andrisani O. Interferon signaling during hepatitis b virus (HBV) infection and HBV-associated hepatocellular carcinoma. Cytokine (2019) 124:154518. doi: 10.1016/j.cyto.2018.08.012
4. Su S, Wong WC, Zou Z, Cheng DD, Ong JJ, Chan P, et al. Cost-effectiveness of universal screening for chronic hepatitis b virus infection in China: an economic evaluation. Lancet Glob Health (2022) 10:e278–87. doi: 10.1016/S2214-109X(21)00517-9
5. Chen Y, Tian Z. HBV-induced immune imbalance in the development of HCC. Front Immunol (2019) 10:2048. doi: 10.3389/fimmu.2019.02048
6. Ou G, Liu X, Xu H, Ji X, Liu X, Wang J, et al. Variation and expression of HLA-DPB1 gene in HBV infection. Immunogenetics (2021) 73:253–61. doi: 10.1007/s00251-021-01213-w
7. Liu J, Liang W, Jing W, Liu M. Countdown to 2030: eliminating hepatitis b disease, China. Bull World Health Organ (2019) 97:230–8. doi: 10.2471/BLT.18.219469
8. Seeger C, Mason WS. Molecular biology of hepatitis b virus infection. Virology (2015) 479-480:672–86. doi: 10.1016/j.virol.2015.02.031
9. Herrscher C, Roingeard P, Blanchard E. Hepatitis b virus entry into cells. Cells (2020) 9:1486. doi: 10.3390/cells9061486
10. Tuttleman JS, Pourcel C, Summers J. Formation of the pool of covalently closed circular viral DNA in hepadnavirus-infected cells. Cell (1986) 47:451–60. doi: 10.1016/0092-8674(86)90602-1
11. Qiu LP, Chen KP. Anti-HBV agents derived from botanical origin. Fitoterapia (2013) 84:140–57. doi: 10.1016/j.fitote.2012.11.003
12. Zhu A, Liao X, Li S, Zhao H, Chen L, Xu M, et al. HBV cccDNA and its potential as a therapeutic target. J Clin Transl Hepatol (2019) 7:258–62. doi: 10.14218/JCTH.2018.00054
13. Tsukuda S, Watashi K. Hepatitis b virus biology and life cycle. Antiviral Res (2020) 182:104925. doi: 10.1016/j.antiviral.2020.104925
14. Bock CT, Schwinn S, Locarnini S, Fyfe J, Manns MP, Trautwein C, et al. Structural organization of the hepatitis b virus minichromosome. J Mol Biol (2001) 307:183–96. doi: 10.1006/jmbi.2000.4481
15. Levrero M, Pollicino T, Petersen J, Belloni L, Raimondo G, Dandri M. Control of cccDNA function in hepatitis b virus infection. J Hepatol (2009) 51:581–92. doi: 10.1016/j.jhep.2009.05.022
16. Li H, Zhuang Q, Wang Y, Zhang T, Zhao J, Zhang Y, et al. HBV life cycle is restricted in mouse hepatocytes expressing human NTCP. Cell Mol Immunol (2014) 11:175–83. doi: 10.1038/cmi.2013.66
17. Alter H, Block T, Brown N, Brownstein A, Brosgart C, Chang KM, et al. A research agenda for curing chronic hepatitis b virus infection. Hepatology (2018) 67:1127–31. doi: 10.1002/hep.29509
18. Hong X, Kim ES, Guo H. Epigenetic regulation of hepatitis b virus covalently closed circular DNA: Implications for epigenetic therapy against chronic hepatitis b. Hepatology (2017) 66:2066–77. doi: 10.1002/hep.29479
19. Cornberg M, Lok AS, Terrault NA, Zoulim F, Faculty E-AHTEC. Guidance for design and endpoints of clinical trials in chronic hepatitis b - report from the 2019 EASL-AASLD HBV treatment endpoints conference(double dagger). J Hepatol (2020) 72:539–57. doi: 10.1016/j.jhep.2019.11.003
20. Konerman MA, Lok AS. Interferon treatment for hepatitis b. Clin Liver Dis (2016) 20:645–65. doi: 10.1016/j.cld.2016.06.002
21. Maher SG, Sheikh F, Scarzello AJ, Romero-Weaver AL, Baker DP, Donnelly RP, et al. IFNalpha and IFNlambda differ in their antiproliferative effects and duration of JAK/STAT signaling activity. Cancer Biol Ther (2008) 7:1109–15. doi: 10.4161/cbt.7.7.6192
22. Lucifora J, Xia Y, Reisinger F, Zhang K, Stadler D, Cheng X, et al. Specific and nonhepatotoxic degradation of nuclear hepatitis b virus cccDNA. Science (2014) 343:1221–8. doi: 10.1126/science.1243462
23. Lazear HM, Schoggins JW, Diamond MS. Shared and distinct functions of type I and type III interferons. Immunity (2019) 50:907–23. doi: 10.1016/j.immuni.2019.03.025
24. Zhang SY, Boisson-Dupuis S, Chapgier A, Yang K, Bustamante J, Puel A, et al. Inborn errors of interferon (IFN)-mediated immunity in humans: insights into the respective roles of IFN-alpha/beta, IFN-gamma, and IFN-lambda in host defense. Immunol Rev (2008) 226:29–40. doi: 10.1111/j.1600-065X.2008.00698.x
25. Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol (2014) 32:513–45. doi: 10.1146/annurev-immunol-032713-120231
26. Crosse KM, Monson EA, Beard MR, Helbig KJ. Interferon-stimulated genes as enhancers of antiviral innate immune signaling. J Innate Immun (2018) 10:85–93. doi: 10.1159/000484258
27. Schoggins JW. Interferon-stimulated genes: What do they all do? Annu Rev Virol (2019) 6:567–84. doi: 10.1146/annurev-virology-092818-015756
28. Mazewski C, Perez RE, Fish EN, Platanias LC. Type I interferon (IFN)-regulated activation of canonical and non-canonical signaling pathways. Front Immunol (2020) 11:606456. doi: 10.3389/fimmu.2020.606456
29. Stark GR, Darnell JE Jr. The JAK-STAT pathway at twenty. Immunity (2012) 36:503–14. doi: 10.1016/j.immuni.2012.03.013
30. Au-Yeung N, Horvath CM. Transcriptional and chromatin regulation in interferon and innate antiviral gene expression. Cytokine Growth Factor Rev (2018) 44:11–7. doi: 10.1016/j.cytogfr.2018.10.003
31. Schreiber G. The molecular basis for differential type I interferon signaling. J Biol Chem (2017) 292:7285–94. doi: 10.1074/jbc.R116.774562
32. Domanski P, Colamonici OR. The type-I interferon receptor. the long and short of it. Cytokine Growth Factor Rev (1996) 7:143–51. doi: 10.1016/1359-6101(96)00017-2
33. Michalska A, Blaszczyk K, Wesoly J. & bluyssen, h. a. r. a positive feedback amplifier circuit that regulates interferon (IFN)-stimulated gene expression and controls type I and type II IFN responses. Front Immunol (2018) 9:1135. doi: 10.3389/fimmu.2018.01135
34. Hayes CN, Chayama K. Interferon stimulated genes and innate immune activation following infection with hepatitis b and c viruses. J Med Virol (2017) 89:388–96. doi: 10.1002/jmv.24659
35. Rojas M, Luz-Crawford P, Soto-Rifo R, Reyes-Cerpa S, Toro-Ascuy D. The landscape of IFN/ISG signaling in HIV-1-Infected macrophages and its possible role in the HIV-1 latency. Cells (2021) 10:2378. doi: 10.3390/cells10092378
36. Wang W, Xu L, Su J, Peppelenbosch MP, Pan Q. Transcriptional regulation of antiviral interferon-stimulated genes. Trends Microbiol (2017) 25:573–84. doi: 10.1016/j.tim.2017.01.001
37. Wang Y, Fan X, Song Y, Liu Y, Liu R, Wu J, et al. SAMD4 family members suppress human hepatitis b virus by directly binding to the smaug recognition region of viral RNA. Cell Mol Immunol (2021) 18:1032–44. doi: 10.1038/s41423-020-0431-x
38. Tan G, Xiao Q, Song H, Ma F, Xu F, Peng D, et al. Type I IFN augments IL-27-dependent TRIM25 expression to inhibit HBV replication. Cell Mol Immunol (2018) 15:272–81. doi: 10.1038/cmi.2016.67
39. Makjaroen J, Somparn P, Hodge K, Poomipak W, Hirankarn N, Pisitkun T, et al. Comprehensive proteomics identification of IFN-lambda3-regulated antiviral proteins in HBV-transfected cells. Mol Cell Proteomics (2018) 17:2197–215. doi: 10.1074/mcp.RA118.000735
40. European Association for the study of the, l. EASL clinical practice guidelines: Management of chronic hepatitis b virus infection. J Hepatol (2012) 57:167–85. doi: 10.1016/j.jhep.2012.02.010
41. Tan G, Song H, Xu F, Cheng G. When hepatitis b virus meets interferons. Front Microbiol (2018) 9:1611. doi: 10.3389/fmicb.2018.01611
42. Battistella S, Lynch EN, Gambato M, Zanetto A, Pellone M, Shalaby S, et al. Hepatocellular carcinoma risk in patients with HBV-related liver disease receiving antiviral therapy. Minerva Gastroenterol (Torino) (2021) 67:38–49. doi: 10.23736/S2724-5985.20.02791-9
43. Hu C, Song Y, Tang C, Li M, Liu J, Liu J, et al. Effect of pegylated interferon plus tenofovir combination on higher hepatitis b surface antigen loss in treatment-naive patients with hepatitis b e antigen -positive chronic hepatitis b: A real-world experience. Clin Ther (2021) 43:572–581 e573. doi: 10.1016/j.clinthera.2020.12.022
44. Huang D, Wu D, Wang P, Wang Y, Yuan W, Hu D, et al. End-of-treatment HBcrAg and HBsAb levels identify durable functional cure after peg-IFN-based therapy in patients with CHB. J Hepatol (2022) 77:42–54. doi: 10.1016/j.jhep.2022.01.021
45. Hansen BE, Rijckborst V, Ter Borg MJ, Janssen HL. HBV DNA suppression in HBeAg-positive chronic hepatitis b patients treated with peginterferon or placebo. J Med Virol (2011) 83:1917–23. doi: 10.1002/jmv.22208
46. Flink HJ, Hansen BE, Heathcote EJ, Feinman SV, Simsek H, Karayalcin S, et al. Successful treatment with peginterferon alfa-2b of HBeAg-positive HBV non-responders to standard interferon or lamivudine. Am J Gastroenterol (2006) 101:2523–9. doi: 10.1111/j.1572-0241.2006.00812.x
47. Reijnders JG, Rijckborst V, Sonneveld MJ, Scherbeijn SM, Boucher CA, Hansen BE, et al. Kinetics of hepatitis b surface antigen differ between treatment with peginterferon and entecavir. J Hepatol (2011) 54:449–54. doi: 10.1016/j.jhep.2010.07.046
48. Xu W, Li Q, Huang C, Hu Q, Qi X, Huang Y, et al. Efficacy of peg-interferon-nucleoside analog sequential optimization therapy in HBeAg-positive patients with CHB. Hepatol Int (2021) 15:51–9. doi: 10.1007/s12072-020-10095-1
49. Qi W, Zhang Q, Xu Y, Wang X, Yu F, Zhang Y, et al. Peg-interferon and nucleos(t)ide analogue combination at inception of antiviral therapy improves both anti-HBV efficacy and long-term survival among HBV DNA-positive hepatocellular carcinoma patients after hepatectomy/ablation. J Viral Hepat (2020) 27:387–96. doi: 10.1111/jvh.13236
50. Liem KS, van Campenhout MJH, Xie Q, Brouwer WP, Chi H, Qi X, et al. Low hepatitis b surface antigen and HBV DNA levels predict response to the addition of pegylated interferon to entecavir in hepatitis b e antigen positive chronic hepatitis b. Aliment Pharmacol Ther (2019) 49:448–56. doi: 10.1111/apt.15098
51. Lee JH, Lee YB, Cho EJ, Yu SJ, Yoon JH, Kim YJ.. Entecavir plus pegylated interferon and sequential hepatitis b virus vaccination increases hepatitis b surface antigen seroclearance: A randomized controlled proof-of-Concept study. Clin Infect Dis (2021) 73:e3308–16. doi: 10.1093/cid/ciaa807
52. Chen J, Li Y, Lai F, Wang Y, Sutter K, Dittmer U, et al. Functional comparison of interferon-alpha subtypes reveals potent hepatitis b virus suppression by a concerted action of interferon-alpha and interferon-gamma signaling. Hepatology (2021) 73:486–502. doi: 10.1002/hep.31282
53. Rehermann B, Bertoletti A. Immunological aspects of antiviral therapy of chronic hepatitis b virus and hepatitis c virus infections. Hepatology (2015) 61:712–21. doi: 10.1002/hep.27323
54. Ye J, Chen J. Interferon and hepatitis b: Current and future perspectives. Front Immunol (2021) 12:733364. doi: 10.3389/fimmu.2021.733364
55. Wang YX, Niklasch M, Liu T, Wang Y, Shi B, Yuan W, et al. Interferon-inducible MX2 is a host restriction factor of hepatitis b virus replication. J Hepatol (2020) 72:865–76. doi: 10.1016/j.jhep.2019.12.009
56. Tan G, Xu F, Song H, Yuan Y, Xiao Q, Ma F, et al. Identification of TRIM14 as a type I IFN-stimulated gene controlling hepatitis b virus replication by targeting HBx. Front Immunol (2018) 9:1872. doi: 10.3389/fimmu.2018.01872
57. Tan G, Yi Z, Song H, Xu F, Li F, Aliyari R, et al. Type-I-IFN-Stimulated gene TRIM5gamma inhibits HBV replication by promoting HBx degradation. Cell Rep (2019) 29:3551–3563.e3553. doi: 10.1016/j.celrep.2019.11.041
58. Stadler D, Kachele M, Jones AN, Hess J, Urban C, Schneider J, et al. Interferon-induced degradation of the persistent hepatitis b virus cccDNA form depends on ISG20. EMBO Rep (2021) 22:e49568. doi: 10.15252/embr.201949568
59. Nassal M. HBV cccDNA: viral persistence reservoir and key obstacle for a cure of chronic hepatitis b. Gut (2015) 64:1972–84. doi: 10.1136/gutjnl-2015-309809
60. Dandri M. Epigenetic modulation in chronic hepatitis b virus infection. Semin Immunopathol (2020) 42:173–85. doi: 10.1007/s00281-020-00780-6
61. Wang Y, Li Y, Zai W, Hu K, Zhu Y, Deng Q, et al. HBV covalently closed circular DNA minichromosomes in distinct epigenetic transcriptional states differ in their vulnerability to damage. Hepatology (2022) 75:1275–88. doi: 10.1002/hep.32245
62. Jiang B, Hildt E. Intracellular trafficking of HBV particles. Cells (2020) 9:2023. doi: 10.3390/cells9092023
63. Wei L, Ploss A. Mechanism of hepatitis b virus cccDNA formation. Viruses (2021) 13:1463. doi: 10.3390/v13081463
64. Tong S, Revill P. Overview of hepatitis b viral replication and genetic variability. J Hepatol (2016) 64:S4–S16. doi: 10.1016/j.jhep.2016.01.027
65. Pollicino T, Belloni L, Raffa G, Pediconi N, Squadrito G Raimondo G, et al. Hepatitis b virus replication is regulated by the acetylation status of hepatitis b virus cccDNA-bound H3 and H4 histones. Gastroenterology (2006) 130:823–37. doi: 10.1053/j.gastro.2006.01.001
66. Kulis M, Esteller M. DNA Methylation and cancer. Adv Genet (2010) 70:27–56. doi: 10.1016/B978-0-12-380866-0.60002-2
67. Chong CK, Cheng CYS, Tsoi SYJ, Huang FY, Liu F, Seto WK , et al. Role of hepatitis b core protein in HBV transcription and recruitment of histone acetyltransferases to cccDNA minichromosome. Antiviral Res (2017) 144:1–7. doi: 10.1016/j.antiviral.2017.05.003
68. Guo YH, Li YN, Zhao JR, Zhang J, Yan Z. HBc binds to the CpG islands of HBV cccDNA and promotes an epigenetic permissive state. Epigenetics (2011) 6:720–6. doi: 10.4161/epi.6.6.15815
69. Mak LY, Wong DK, Pollicino T, Raimondo G, Hollinger FB, Yuen MF . Occult hepatitis b infection and hepatocellular carcinoma: Epidemiology, virology, hepatocarcinogenesis and clinical significance. J Hepatol (2020) 73:952–64. doi: 10.1016/j.jhep.2020.05.042
70. Guo Y, Li Y, Mu S, Zhang J, Yan Z. Evidence that methylation of hepatitis b virus covalently closed circular DNA in liver tissues of patients with chronic hepatitis b modulates HBV replication. J Med Virol (2009) 81:1177–83. doi: 10.1002/jmv.21525
71. Jain S, Chang TT, Chen S, Boldbaatar B , Clemens A, Lin SY , et al. Comprehensive DNA methylation analysis of hepatitis b virus genome in infected liver tissues. Sci Rep (2015) 5:10478. doi: 10.1038/srep10478
72. Vivekanandan P, Thomas D, Torbenson M. Hepatitis b viral DNA is methylated in liver tissues. J Viral Hepat (2008) 15:103–7. doi: 10.1111/j.1365-2893.2007.00905.x
73. Park IY, Sohn BH, Yu E, Suh DJ, Chung YH, Lee JH, et al. Aberrant epigenetic modifications in hepatocarcinogenesis induced by hepatitis b virus X protein. Gastroenterology (2007) 132:1476–94. doi: 10.1053/j.gastro.2007.01.034
74. Wei Y, Van Nhieu JT, Prigent S, Srivatanakul P Tiollais P, Buendia MA. Altered expression of e-cadherin in hepatocellular carcinoma: correlations with genetic alterations, beta-catenin expression, and clinical features. Hepatology (2002) 36:692–701. doi: 10.1053/jhep.2002.35342
75. Oropeza CE, Tarnow G, Taha TY, Shalaby RE, Hyde MV, Maienschein-Cline M, et al. Relative DNA methylation and demethylation efficiencies during postnatal liver development regulate hepatitis b virus biosynthesis. J Virol (2021) 95:e02148-20. doi: 10.1128/JVI.02148-20
76. Chong CK, Cheng CYS, Tsoi SYJ, Huang FY, Liu F, Fung J, et al. HBV X protein mutations affect HBV transcription and association of histone-modifying enzymes with covalently closed circular DNA. Sci Rep (2020) 10:802. doi: 10.1038/s41598-020-57637-z
77. Belloni L, Pollicino T, De Nicola F, Guerrieri F, Raffa G, Fanciulli M, et al. Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc Natl Acad Sci U.S.A. (2009) 106:19975–9. doi: 10.1073/pnas.0908365106
78. Tropberger P, Mercier A, Robinson M, Zhong W, Ganem DE, Holdorf M. Mapping of histone modifications in episomal HBV cccDNA uncovers an unusual chromatin organization amenable to epigenetic manipulation. Proc Natl Acad Sci U.S.A. (2015) 112:E5715–5724. doi: 10.1073/pnas.1518090112
79. Sun T, Wu R, Ming L. The role of m6A RNA methylation in cancer. BioMed Pharmacother (2019) 112:108613. doi: 10.1016/j.biopha.2019.108613
80. Kostyusheva A, Brezgin S, Glebe D, Kostyushev D, Chulanov V. Host-cell interactions in HBV infection and pathogenesis: the emerging role of m6A modification. Emerg Microbes Infect (2021) 10:2264–75. doi: 10.1080/22221751.2021.2006580
81. Imam H, Khan M, Gokhale NS, McIntyre ABR, Kim GW, Jang JY, et al. N6-methyladenosine modification of hepatitis b virus RNA differentially regulates the viral life cycle. Proc Natl Acad Sci U.S.A. (2018) 115:8829–34. doi: 10.1073/pnas.1808319115
82. Kim GW, Imam H, Khan M, Mir SA, Kim SJ, Yoon SK, et al. HBV-induced increased N6 methyladenosine modification of PTEN RNA affects innate immunity and contributes to HCC. Hepatology (2021) 73:533–47. doi: 10.1002/hep.31313
83. Kim GW, Siddiqui A. Hepatitis b virus X protein recruits methyltransferases to affect cotranscriptional N6-methyladenosine modification of viral/host RNAs. Proc Natl Acad Sci U.S.A. (2021) 118:e2019455118. doi: 10.1073/pnas.2019455118
84. Hill M, Tran N. miRNA interplay: mechanisms and consequences in cancer. Dis Model Mech (2021) 14:dmm047662. doi: 10.1242/dmm.047662
85. Gallo A, Miceli V, Bulati M, Iannolo G, Contino F, Conaldi PG. Viral miRNAs as active players and participants in tumorigenesis. Cancers (Basel) (2020) 12:358. doi: 10.3390/cancers12020358
86. Petrini E, Caviglia GP, Abate ML, Fagoonee S, Smedile A, Pellicano R.. MicroRNAs in HBV-related hepatocellular carcinoma: functions and potential clinical applications. Panminerva Med (2015) 57:201–9.
87. Zhao X, Sun L, Mu T, Yi J, Ma C, Xie H, et al. An HBV-encoded miRNA activates innate immunity to restrict HBV replication. J Mol Cell Biol (2020) 12:263–76. doi: 10.1093/jmcb/mjz104
88. Loukachov V, van Dort KA, Jansen L, Reesink HW, Kootstra NA. Identification of a novel HBV encoded miRNA using next generation sequencing. Viruses (2022) 14:1223. doi: 10.3390/v14061223
89. Belloni L, Allweiss L, Guerrieri F, Pediconi N, Volz T, Pollicino T, et al. IFN-alpha inhibits HBV transcription and replication in cell culture and in humanized mice by targeting the epigenetic regulation of the nuclear cccDNA minichromosome. J Clin Invest (2012) 122:529–37. doi: 10.1172/JCI58847
90. Yuan Y, Yuan H, Yang G, Yun H, Zhao M, Liu Z, et al. IFN-alpha confers epigenetic regulation of HBV cccDNA minichromosome by modulating GCN5-mediated succinylation of histone H3K79 to clear HBV cccDNA. Clin Epigenet (2020) 12:135. doi: 10.1186/s13148-020-00928-z
91. Zhao LN, Yuan HF, Wang YF, Yun HL, Zheng W, Yuan Y, et al. IFN-alpha inhibits HBV transcription and replication by promoting HDAC3-mediated de-2-hydroxyisobutyrylation of histone H4K8 on HBV cccDNA minichromosome in liver. Acta Pharmacol Sin (2022) 43:1484–94. doi: 10.1038/s41401-021-00765-7
92. Ramos JC, Lossos IS. Newly emerging therapies targeting viral-related lymphomas. Curr Oncol Rep (2011) 13:416–26. doi: 10.1007/s11912-011-0186-8
93. Ding WB, Wang MC, Yu J, Huang G, Sun DP, Liu L, et al. HBV/Pregenomic RNA increases the stemness and promotes the development of HBV-related HCC through reciprocal regulation with insulin-like growth factor 2 mRNA-binding protein 3. Hepatology (2021) 74:1480–95. doi: 10.1002/hep.31850
94. Imam H, Kim GW, Mir SA, Khan M, Siddiqui A. Interferon-stimulated gene 20 (ISG20) selectively degrades N6-methyladenosine modified hepatitis b virus transcripts. PloS Pathog (2020) 16:e1008338. doi: 10.1371/journal.ppat.1008338
95. Fujita K, Mimura S, Iwama H, Nakahara M, Oura K, Tadokoro T , et al. Serum miRNAs predicting sustained HBs antigen reduction 48 weeks after pegylated interferon therapy in HBe antigen-negative patients. Int J Mol Sci (2018) 19. doi: 10.3390/ijms19071940
96. Chen K, Liu J, Liu S, Xia M, Zhang X, Han D, et al. Methyltransferase SETD2-mediated methylation of STAT1 is critical for interferon antiviral activity. Cell (2017) 170:492–506.e414. doi: 10.1016/j.cell.2017.06.042
97. Revill PA, Chisari FV Block JM, Dandri M Gehring AJ, Guo H, et al. A global scientific strategy to cure hepatitis b. Lancet Gastroenterol Hepatol (2019) 4:545–58. doi: 10.1016/S2468-1253(19)30119-0
98. Xia Y, Guo H. Hepatitis b virus cccDNA: Formation, regulation and therapeutic potential. Antiviral Res (2020) 180:104824. doi: 10.1016/j.antiviral.2020.104824
99. Rajoriya N, Combet C, Zoulim F, Janssen HLA. How viral genetic variants and genotypes influence disease and treatment outcome of chronic hepatitis b. time for an individualised approach? J Hepatol (2017) 67:1281–97. doi: 10.1016/j.jhep.2017.07.011
100. Roca Suarez AA, Testoni B, Zoulim F. HBV 2021: New therapeutic strategies against an old foe. Liver Int (2021) 41 Suppl 1:15–23. doi: 10.1111/liv.14851
101. Dong J, Ying J, Qiu X, Lu Y, Zhang M. Advanced strategies for eliminating the cccDNA of HBV. Dig Dis Sci (2018) 63:7–15. doi: 10.1007/s10620-017-4842-1
102. Mohd-Ismail NK, Lim Z, Gunaratne J, Tan YJ. Mapping the interactions of HBV cccDNA with host factors. Int J Mol Sci (2019) 20:4276. doi: 10.3390/ijms20174276
103. Goh ZY, Ren EC, Ko HL. Intracellular interferon signalling pathways as potential regulators of covalently closed circular DNA in the treatment of chronic hepatitis b. World J Gastroenterol (2021) 27:1369–91. doi: 10.3748/wjg.v27.i14.1369
104. Yang K, Guan S, Zhang H, Chen Z. Induction of interleukin 6 impairs the anti-HBV efficiency of IFN-alpha in human hepatocytes through upregulation of SOCS3. J Med Virol (2019) 91:803–12. doi: 10.1002/jmv.25382
105. Malik UR, Makower DF, Wadler S. Interferon-mediated fatigue. Cancer (2001) 92:1664–8. doi: 10.1002/1097-0142(20010915)92:6+<1664::aid-cncr1494>3.0.co;2-9
106. Yang Y, Zhao X, Wang Z, Shu W, Li L, Li Y, et al. Nuclear sensor interferon-inducible protein 16 inhibits the function of hepatitis b virus covalently closed circular DNA by integrating innate immune activation and epigenetic suppression. Hepatology (2020) 71:1154–69. doi: 10.1002/hep.30897
107. Gilmore S, Tam D, Dick R, Appleby T, Birkus G, Willkom M, et al. Antiviral activity of GS-5801, a liver-targeted prodrug of a lysine demethylase 5 inhibitor, in a hepatitis b virus primary human hepatocyte infection model. J Hepatol (2017) 66:S690–1. doi: 10.1016/S0168-8278(17)31855-X
Keywords: HBV, IFN therapy, ISGs, epigenetic regulation, CccDNA
Citation: Yang Z, Sun B, Xiang J, Wu H, Kan S, Hao M, Chang L, Liu H, Wang D and Liu W (2022) Role of epigenetic modification in interferon treatment of hepatitis B virus infection. Front. Immunol. 13:1018053. doi: 10.3389/fimmu.2022.1018053
Received: 12 August 2022; Accepted: 27 September 2022;
Published: 17 October 2022.
Edited by:
Guangyun Tan, Institute of Translational Medicine, The First Hospital of Jilin University, ChinaReviewed by:
Mingmin Zhao, Inner Mongolia Agricultural University, ChinaCopyright © 2022 Yang, Sun, Xiang, Wu, Kan, Hao, Chang, Liu, Wang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dongxu Wang, d2FuZ19kb25nX3h1QGpsdS5lZHUuY24=; Weiwei Liu, bGl1d2Vpd0BqbHUuZWR1LmNu
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.