 Zhenya Guo1,2,3†
Zhenya Guo1,2,3† Xiude Fan
Xiude Fan Stephen Tomlinson
Stephen Tomlinson Guandou Yuan
Guandou Yuan Songqing He
Songqing He- 1Division of Hepatobiliary Surgery, The First Affiliated Hospital of Guangxi Medical University, Nanning, China
- 2Key Laboratory of Early Prevention and Treatment for Regional High Frequency Tumor (Guangxi Medical University), Ministry of Education, Nanning, China
- 3Guangxi Key Laboratory of Immunology and Metabolism for Liver Diseases, Guangxi Medical University, Nanning, China
- 4Department of Endocrinology, Shandong Provincial Hospital Affiliated to Shandong First Medical University, Jinan, China
- 5Department of Microbiology and Immunology, Medical University of South Carolina, Charleston, SC, United States
Nonalcoholic fatty liver disease (NAFLD) has become a leading cause of chronic liver diseases globally. NAFLD includes a range of hepatic manifestations, starting with liver steatosis and potentially evolving towards nonalcoholic steatohepatitis, cirrhosis or even hepatocellular carcinoma. Although the pathogenesis of NAFLD is incompletely understood, insulin resistance and lipid metabolism disorder are implicated. The complement system is an essential part of the immune system, but it is also involved in lipid metabolism. In particular, activation of the alternative complement pathway and the production of complement activation products such as C3a, C3adesArg (acylation stimulating protein or ASP) and C5a, are strongly associated with insulin resistance, lipid metabolism disorder, and hepatic inflammation. In this review, we briefly summarize research on the role of the complement system in NAFLD, aiming to provide a basis for the development of novel therapeutic strategies for NAFLD.
Introduction
Nonalcoholic fatty liver disease (NAFLD) is defined as a condition of excess fatty deposition in the liver in the absence of excessive alcohol consumption. NAFLD is a common liver disorder, with a wide spectrum range from simple steatosis to nonalcoholic steatohepatitis (NASH), cirrhosis, or even hepatocellular carcinoma. Given the worldwide prevalence of around 25%, NAFLD is a significant global health burden (1, 2). The early stage of NAFLD, nonalcoholic fatty liver (NAFL), is characterized by simple hepatocyte steatosis without significant inflammation that leads to liver injury. NAFL is considered benign and reversible. However, the progressive stage, NASH, accompanied by inflammation and liver damage, may progress to cirrhosis and even liver cancer (3). The pathogenesis of NAFLD has often been explained in terms of a “double-hit’’ hypothesis. The “first hit” is fat deposition in hepatocytes, which originates from an imbalance between lipid in (i.e., fatty acid uptake and lipogenesis) and lipid out (i.e., fatty acid oxidation and secretion in the form of very low-density lipoprotein) (4). The “second hit” refers to oxidative stress resulting from lipid peroxidation, leading to liver inflammation and later fibrosis (5, 6). As research has progressed, a “multiple hit” hypothesis was proposed to better describe the multiple pathways that contribute to NASH (7). According to the “multiple hit” hypothesis, multiple insults act together to induce NAFLD. However, no matter which hypothesis is applied, insulin resistance and lipid metabolism disorder are considered key mechanisms contributing to NAFLD. In this review, we discuss the role of the complement system in the progression of NAFLD, aiming to elucidate the pathogenesis of NAFLD and to provide a premise for novel potential therapeutic strategies.
Overview of the complement system
The complement system, an important component of immunity, consists of blood proteins, receptors, and soluble and membrane-bound regulatory factors, with the soluble components being mainly synthesized by the liver. The complement system can be activated through three pathways, namely the classical pathway (CP), the alternative pathway (AP), and the lectin pathway (LP) (refer to Figure 1). The CP is commonly activated by C1q recognition of immune complexes, leading to the activation of the serine proteases C1r and C1s. These proteases then activate C4 and C2, leading to the formation of the CP C3 convertase (C4bC2b). The LP is initiated by pattern recognition molecules (mannose binding lectin, ficolins) binding to certain carbohydrates and injured cells (pathogen-associated molecular patterns), leading to activation of LP-associated serine proteases and the formation of the CP C3 convertase. The AP, on the other hand, can be initiated spontaneously through a process termed “C3 tick-over”. The “C3 tick-over“ theory was proposed to explain the presence of the initial C3b molecules in human plasma under normal conditions. According to this theory, C3 is hydrolyzed by nucleophilic attack of H2O to form C3(H2O). Then C3(H2O) binds factor B, which is in turn cleaved by factor D, to generate the initial C3 convertase C3(H2O)Bb. The spontaneous “C3 tick-over” reaction is at a slow but constant rate in the plasm (8). The resulting C3(H2O)Bb cleaves C3 into C3b to generate the AP C3 convertase C3bBb in an amplification loop. The molecule properdin binds to and stabilizes C3bBb, which can bind an additional C3b molecule to form the C5 convertase, (C3b)2BbP. The AP also functions as an amplification loop for the CP and LP. Factor H is a critical negative regulator of the AP, which inhibits formation of, accelerates the decay of, and inactivates C3(H2O)Bb and C3bBb. The absence of factor H results in uncontrolled amplification of the AP (9). Interestingly, though the convertase activity of C3(H2O)Bb is much lower than that of C3bBb, C3(H2O)Bb is more resistant to the inactivation of factor H (8).
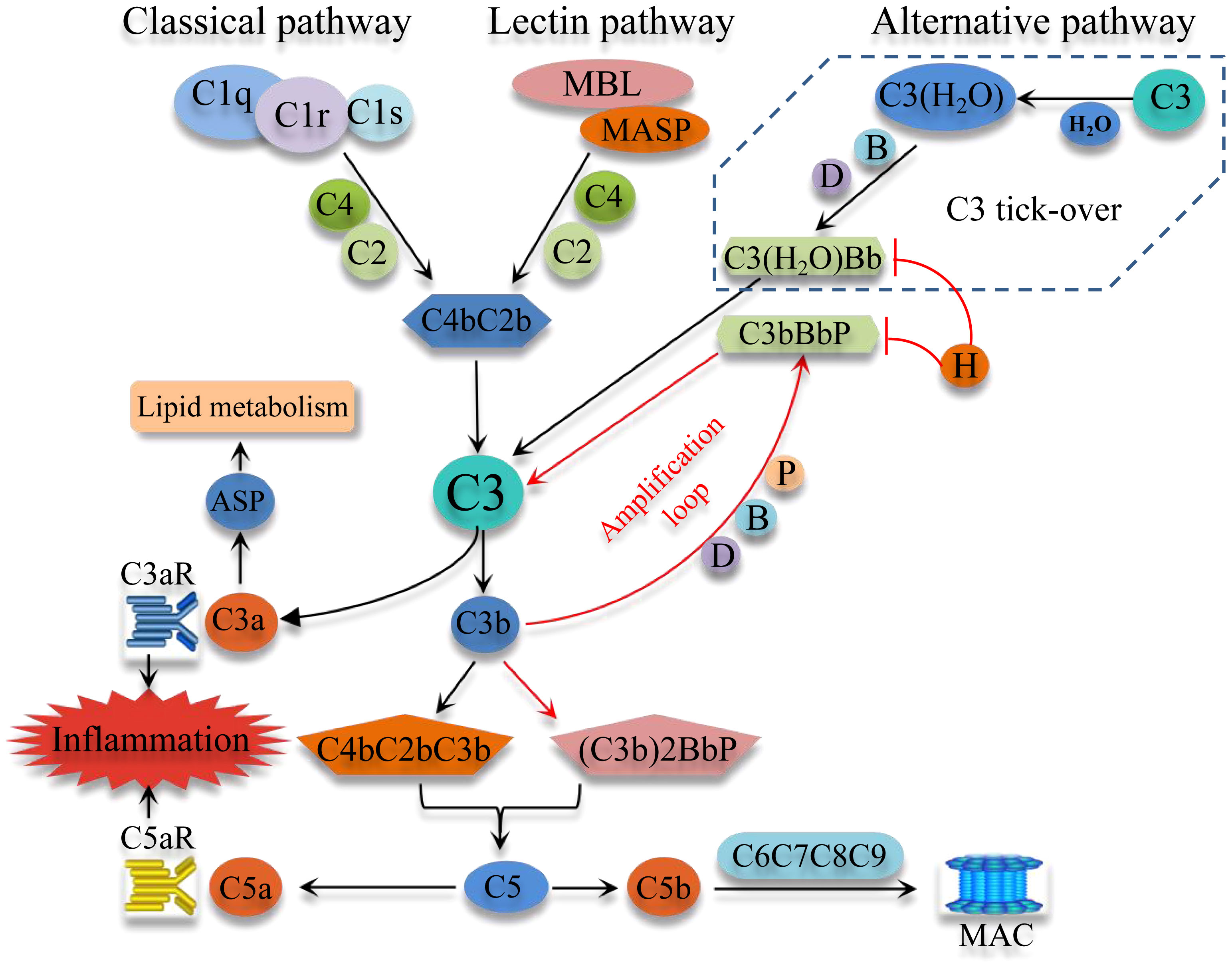
Figure 1 Schematic overview of the complement system activation. Complement system can be commonly activated through three pathways, the classical pathway, the Lectin pathway, and the alternative pathway (AP). Importantly, the AP can be initiated spontaneously by the “C3 tick-over”, which keeps AP idling and has the ability of fast-response through an amplification loop. These three pathways converge at C3 and produce C3 convertase and C5 convertase, which cleave C3 and C5, thereby generating C3a and C5a, important potent pro-inflammatory molecules. The activation of C5 eventually activates the downstream C6, C7, C8 and C9 to form the MAC. MBL, mannose-binding lectin; MASP, mannose-binding lectin-associated serine protease; MAC, membrane attack complex; ASP, acylation stimulating protein; B, factor B; D, factor D; H, factor H; P, properdin.
All three pathways converge at C3 and produce C3 and C5 convertases, which respectively cleave the central components C3 and C5 to generate the anaphylatoxins C3a and C5a. These bioactive peptides have diverse effects and play important roles in inflammation and immunity. The cleavage of C5 and generation of C5b results in the recruitment and self-assembly of C6, C7, C8 and C9 to form the cytolytic membrane attack complex (MAC).
The complement is involved in NAFLD
The complement system was traditionally considered a component of innate immunity and inflammation, although it is now recognized as a central component of both innate and adaptive immunity. In recent years, an increasing number of studies have shown that the complement system also plays key roles in NAFLD. A Chinese cross-sectional study indicated that serum C3 levels positively correlated with the prevalence and severity of NAFLD (10); the data showed that people with higher serum C3 levels were more prone to have NAFLD. Moreover, NAFLD patients with higher serum C3 levels were more likely to have increased liver enzyme levels, which is considered a more serious form of NAFLD (11). A Turkish study came to a similar conclusion that patients with NAFLD correlate with higher serum levels of C3 and acylation stimulating protein (ASP), the desarginated form of C3a (C3adesArg). In addition, a Dutch cross-sectional study suggested that plasma C3a levels were associated with liver fat percentage (12). Histological studies provided further evidence of a link between the complement system and NAFLD. It was found that in 74% of NAFLD patients, there was deposition of activated forms of C3 and C4 in the liver tissue; in most C3-positive livers, C1q and MBL deposition was also observed, and 50% of activated C3-positive patients presented MAC formation in the liver. Interestingly, NASH is more prevalent in MAC or activated C3 positive patients (13). Moreover, in another clinical trial, it was found that AP-related C3 activation contributes to hepatic inflammation in NASH; deposition of properdin, a positive regulator of the AP, and of the C3 activation product C3c, positively correlated with liver inflammation level in NASH (14). These findings imply that the complement system can be activated through all three pathways in NASH, and that the level of complement activation is positively related to the severity of NASH. Activators of the complement system are diverse, but in NAFLD apoptotic cells may initiate complement activation. Lipid accumulation is considered the “first hit” initiating extrinsic apoptosis in NAFLD (15, 16). Both C1q and MBL can recognize apoptotic cells, thus resulting in activation of the CP and LP (17, 18). Together, the above data suggest that the complement is closely involved in the development of NAFLD.
The complement system in insulin resistance and lipid metabolism disorder
Although the pathogenesis of NAFLD is incompletely understood, obesity, insulin resistance, and lipid metabolism disorders are implicated. Accumulating data indicate that NAFLD is a hepatic manifestation of a systemic metabolic disease rather than a confined liver disease (19). While components of the complement system are mainly synthesized by the liver, adipose tissues can also produce some complement components, such as C3, factor B and factor D (20, 21). In particular, adipose tissue is the main source of the AP protein, factor D (22). A clinical trial showed that a positive correlation between serum C3, C4, and ASP levels with obesity. Specifically, the level of C3 increased proportionally to the total amount of adipose tissue (23, 24). Moreover, increases in factor B and factor D levels were observed in obesity, while weight loss resulted in reduction of these factors. In addition, there is a positive correlation of circulating factor H concentration with obesity and insulin resistance (25). Importantly, serum C3 level is considered as a pro-inflammatory biomarker contributing to low-grade inflammation and insulin resistance in obesity (26). Under physiological conditions, the AP is activated spontaneously through the “C3 tick-over”, which requires the participation of factors B and D (27). This implies that AP activation is involved in obesity and insulin resistance. As expected, it has been shown that inhibiting the AP by factor D deficiency downregulated the expression of the genes contributing to fatty acid uptake and de novo lipogenesis in the liver, which eventually led to the alleviation of NAFLD (28). On the other hand, acceleration of the AP by factor H deficiency resulted in lipid accumulation and a pro-inflammatory environment in the liver (29). “C3 tick-over” also leads to the generation of low-level serum C3a, which is further degraded to ASP by serum carboxypeptidase. ASP has insulin-like effects, and stimulates the synthesis of triacylglycerols by transporting free fatty acids (FFAs) into adipocytes (24, 30). Previous studies have demonstrated that plasma ASP levels in obese individuals are significantly higher than those in nonobese people, and when obese individuals lose weight, the circulating ASP level decreases (31, 32). A study of hyperlipidemia patients showed that fasting plasma C3 and ASP levels were higher in hyperlipidemia patients than that of healthy people; oral fat loading test in healthy subjects led to a significant increase in plasma C3, while in hyperlipidemia patients, a delayed C3 response was observed (33). It was also observed that postprandial C3 increase negatively correlated with postprandial FFAs (33). Chylomicron, the form in which dietary fat is carried, can accelerate the “C3 tick-over” and activate the AP, thereby inducing the overproduction of ASP by regulating factor H (34). In addition, hydrolysis of C3 to C3(H2O) in the “C3 tick-over” could also be significantly accelerated by the interaction between C3 and a number of lipid surfaces and complexes (35). This implies that the “C3 tick-over” may be involved with lipid metabolism. Since C3 is the precursor for ASP and acts on FFA uptake by adipocytes, it is reasonable to assume that in hyperlipidemia patients, an impaired C3/ASP response may be associated with impaired FFA uptake, leading to increased plasma FFA concentration and subsequently enhanced FFA flux to the liver (Figure 2).
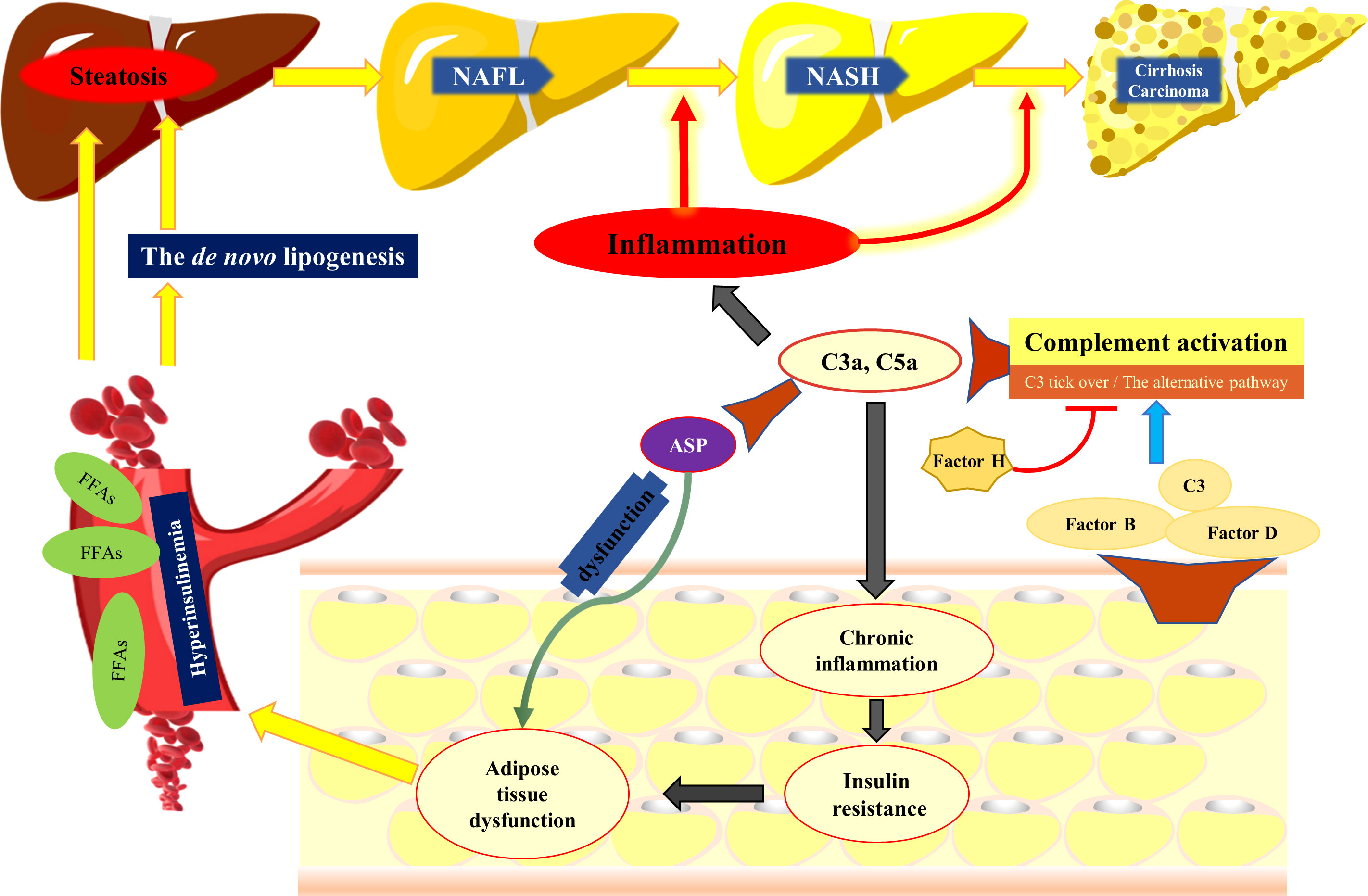
Figure 2 The complement system participates in the development of NAFLD. Adipose tissue can produce complement components, such as C3, factor B and factor D, which are critical for the activation of the alternative pathway. Activation of the complement system results in generation of C3a and C5a, both of which have potent pro-inflammatory activity and contribute to insulin resistance. ASP, the desarginated form of C3a, has insulin like effects, which stimulates the synthesis of triacylglycerol by transporting FFASs into adipocytes. However, when the dysfunction of ASP and insulin resistance compromises the ability of adipocytes to store fat, FFAs are released into the circulation, available for uptake by the liver. In addition, hyperinsulinemia promotes the de novo lipogenesis and suppresses the b-oxidation of FFAs, contributing to lipid accumulation in the liver. The complement system is also involved in NASH by regulating hepatic inflammatory response. NAFL, nonalcoholic fatty liver; NASH, nonalcoholic steatohepatitis, FFAs, free fatty acids; ASP, acylation stimulating protein.
Accumulation of triglycerides (TGs) in hepatocytes is a hallmark of NAFLD (4). The liver can synthesize TGs by uptake of peripheral FFA or by producing lipids from carbohydrates (the de novo lipogenesis). When insulin resistance compromises the ability of adipocytes to store fat, FFAs are released into the circulation, and become available for uptake by the liver (36, 37). Accumulation of FFAs in hepatocytes enhances the synthesis of TGs, during which time the liver is also exposed to high levels of insulin (hyperinsulinemia) as well as to high levels of FFAs. The hyperinsulinemia leads to the upregulation of sterol regulatory element binding protein-1c (38), a critical transcriptional regulator of the genes involved in de novo lipogenesis, contributing up to 25% of total hepatic lipid stores (39). Hyperinsulinemia can suppress the β-oxidation of FFAs, contributing to lipid accumulation in the liver (40). A previous study indicated that increased C3 levels lead to higher levels of ASP in obese people, and it is conceivable that “C3 tick-over” also results in the activation of C5 and generation of C3a and C5a (23). In a high-fat diet mouse model it was shown that C5 deficiency resulted in less steatosis and lower liver inflammation, and additionally inhibited the expression of several pro-inflammatory cytokines, including IL-17 and TGF-β (41). IL-17 aggravates liver steatosis and inflammation (42), and TGF-β is related with insulin resistance (43, 44). Additionally, C3a receptor (C3aR) deficiency was protective against murine NAFLD. In this regard, C3aR is highly expressed in adipocytes and macrophages in adipose tissues (45), and C3 expression is significantly increased in adipose tissue of mice fed a high-fat diet. Interestingly, C3aR expression in the liver does not vary significantly (45). This implies that C3aR deficiency may attenuate NAFLD indirectly through affecting the adipose tissues. Importantly, the data support that the C3a/C3aR and the C5a/C5aR axes contribute to macrophage accumulation and M1 polarization in the adipose tissues of obese individuals, thereby leading to low-grade inflammation and insulin resistance (45, 46). In addition, data from rat model showed that C3aR and C5aR antagonists dramatically attenuated high-fat diet-induced obesity, insulin resistance, and adipose inflammation (47).
Taken together, the above data indicate that under physiological conditions, complement activation participates in lipid metabolism, while aberrant complement activation is involved in insulin resistance and lipid metabolism disorder which contributes to hepatocyte steatosis.
The complement system in NAFLD progression
The involvement of complement in metabolic disorder could be regarded as its contribution to the “first hit”. Nevertheless, complement also participates in the “second hit”, that is, the progression of NAFLD. Entirely distinct from NAFL, NASH is characterized by inflammation and hepatocyte injury. However, the mechanisms driving simple steatosis to NASH are unclear. The progression of NAFLD occurs together with an aberrant activation of the immune system promoted by multiple genetic and environmental factors (48). Apart from parenchymal cells, the liver also contains a large number of complex immune cells, such as macrophages, stellate cells, and lymphoid cells. Therefore, the liver is considered not only as a metabolic organ, but also as an immune organ, which maintains homeostasis under physiological conditions. Disruption of homeostasis could be triggered by aberrant innate immune response, resulting in liver injury, fibrosis, and even carcinogenesis (49, 50). Complement, a key player in an innate immune response, can thus participate in the progression of NAFLD by modulating a variety of immune cells in the liver.
The complement and macrophages in NAFLD
The pathogenesis of NAFLD is multifactorial, although it has been shown that an innate immune response is a key contributing factor, with macrophages playing a critical role (51). The liver harbors the largest proportion of macrophages in the body, which mainly comprise liver-resident macrophages, or Kuppfer cells (KCs), and monocyte-derived macrophages (MoMF) (52). There is abundant evidence that macrophages are involved in the development of liver steatosis, inflammation, and fibrosis in NAFLD (51, 53). Upon stimulation, KCs and MoMFs can differentiate into either pro-inflammatory so called M1-like macrophages or into anti-inflammatory M2-like macrophages. Although there is a wide spectrum of macrophage differentiation that is no longer categorized into M1 and M2, it is recognized that macrophage polarization plays a key role in the regulation of an inflammatory response. Importantly, it has been reported that KCs and MoMFs present a remarkable shift toward an M1-like phenotype in the liver of NASH mice (54), which is closely associated with the progression of NAFLD (51, 55). Activated Kupffer cells/macrophages in the liver secrete inflammatory chemokines that recruit peripheral monocytes into the liver, which further expands the inflammatory response in NAFLD (48). In addition to a pro-inflammatory effect, activated macrophages can promote liver fibrosis by regulating hepatic stellate cells (HSCs). By producing cytokines, such as TGF-β, TNF-α, and IL-1β, activated KCs promote HSC differentiation into a pro-fibrogenic phenotype, which synthesizes abundant collagen and α-smooth muscle actin, leading to liver fibrosis (56). It is well documented that complement activation products, particularly C3a and C5a, interact with a variety of complement receptors on macrophages, thereby modulating an inflammatory response. Both C3a and C5a have potent chemoattractant and pro-inflammatory properties, and their receptors are widely expressed, including on macrophages (45, 57). It has been shown that by inducing NLRP3 inflammasome activation through regulating the efflux of adenosine triphosphate, the C3a/C3aR axis contributes to IL-1β secretion in macrophages (58). C5a can also trigger NLRP3 inflammasome activation, including the release of IL-1β and TNFα release (59). In a high-fat diet mouse model, Mamane et al. (45) found that both macrophages in white adipose tissue and Kupffer cells in the liver express significant amounts of C3aR. Furthermore, C3aR deficiency improved insulin sensitivity, decreased macrophage infiltration, and inhibited the pro-inflammatory effect of M1-like macrophages. Importantly, C3aR deficiency significantly alleviated liver injury in mice fed a high-fat diet (45). Interestingly, in another study, the C5a/C5aR axis was demonstrated to also contribute to macrophage accumulation and M1-like polarization in obese white adipose tissues and thereby to insulin resistance (46). Thus, it can reasonably be speculated that the C3a/C3aR and C5a/C5aR axis may play an important role in the pathogenesis of NAFLD via activating Kupffer/macrophages.
The complement and neutrophils in NAFLD
In addition to macrophages, neutrophils have also been shown to be involved in the pathogenesis of NAFLD. Several studies in humans and mice have suggested that neutrophil infiltration in the liver contributes to the progression of NAFLD. The level of circulating neutrophils and their infiltration are closely related to the severity of NAFLD (60–63). Consistent with these findings, reactive oxygen species (ROS) produced by infiltrating neutrophils contribute to the progression to NASH (64). In NASH patients, plasma myeloperoxidase (MPO) levels are higher (65), and MPO induces liver cell death and activation of HSCs, which contribute to liver fibrosis. In contrast, inhibiting neutrophil infiltration in the mouse liver can significantly reduce NAFLD progression (66). MPO deficiency alleviates liver injury by suppressing HSC activation, leading to decreased TGF-β levels and fibrosis in mice (67). C3a and C5a also have potent biological activities for neutrophils. The effect of C3a on neutrophils is somewhat unclear. While C3a is often considered pro-inflammatory and its receptor C3aR is highly expressed on neutrophils (68), emerging data show that the C3a/C3aR axis also has anti-inflammatory effects (69, 70). Nevertheless, it is clear that C5a has a pro-inflammatory effect on macrophages. The C5a/C5aR axis is closely associated with neutrophil functions such as chemotaxis and respiratory burst (71). In addition, C5a can inhibit apoptosis of neutrophils (72), lead to increased vascular permeability, and promote the expression of P-selectin, thereby contributing to the infiltration of neutrophils (73). The terminal activation product of complement, the MAC, is not only cytolytic, but at sublytic concentrations can also promote the release of chemokines for neutrophil infiltration. Thus, hepatic neutrophil infiltration is also associated with MAC deposition (13).
The complement and T cells in NAFLD
There is emerging evidence that T cells play an important role in the pathogenesis of NASH. In basic functional terms, T cells can be divided into cytotoxic CD8 T cells, CD4 T helper (Th) cells, and CD4 regulatory T cells (Tregs) (74). CD8 T cells are increased in the livers of patients with NASH (75, 76). In a mouse model, depletion of CD8 T cells alleviated NASH (77). It has been implied that CD8 T cells trigger NASH progression through generation of pro-inflammatory cytokines and non-specific killing of hepatocytes (75, 78). Moreover, an imbalance between Th1 (which secrete pro-inflammatory cytokines) and Th2 (secrete anti-inflammatory cytokines) cells appears to contribute to the pathogenesis of NAFLD (79). Inflammatory Th1 and Th17 cells have also been reported to be increased in NASH (80, 81). In addition, the progression from NAFL to NASH is marked by an increased Th17/resting Tregs ratio in the liver (80). Complement has widely been recognized to participate in the differentiation of T cells and regulate their biological function. For example, the C3a/C3aR axis is involved in modulating Th2 cell differentiation through mediating the secretion of IL-12 by antigen-presenting cells (82). It has also been suggested that C3aR and C5aR signaling contributes to the differentiation of Th1 and Th17 cells, and C5aR signaling in dendritic cells is critical for inducing T-cell differentiation into Th17 (83).
Conclusions and future perspective
In summary, as a participant in lipid metabolism as well as immune responses, the complement system plays an important role in multiple stages of NAFLD. Components of the AP, C3, C5, and ASP have all been shown to be involved in insulin resistance, lipid metabolism disorder, and an inflammatory response (Figure 2). Together, the reviewed studies suggest that the complement system may be a promising therapeutic target for treating human NAFLD. Indeed, in several pre-clinical models, intervention in the complement system showed therapeutic benefit. Nevertheless, note should be taken that the complement system is an essential component of immunity and provides an important defense against pathogens. It also has multiple important physiological and homeostatic functions such as tissue repair, immune complex catabolism, and the removal of dead and dying cells (84). Therefore, improper or nonspecific intervention in the complement system carries a risk of serious side effects, including increased risk of infection. And while these are important considerations, including for most of the greater than 80 clinical trials currently ongoing with complement inhibitors, there is also a risk-benefit consideration. A strategy that could also be considered is site-targeted complement inhibition, in which complement is inhibited only locally at sites of pathology while leaving systemic activity largely intact (83). To conclude inhibition of complement may provide a promising approach to limit the progression of NAFLD and may open new avenues of treatment.
Author contributions
ZG and GY conceived this study. SH and GY directed the study. ZG, GY, XF, and JY performed literature search. ZG drafted the manuscript. SH, ST, XF, and GY provided critical intellectual revision. SH and GY provided financial support. All authors contributed to the article and approved the final version.
Funding
This study was supported in part by the National Natural Science Foundation of China (No. 91949122 and No. 82160500); the 111 Project (D17011); Guangxi science and technology base and talent project (GuikeAA21220002); Guangxi Key Research and Development Plan (2018AD03001); Special project of central government guiding local science and technology development (ZY20198011); Key Laboratory Base of Liver Injury and Repair of the First Affiliated Hospital of Guangxi Medical University (YYZS2020001).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Cotter TG, Rinella M. Nonalcoholic fatty liver disease 2020: The state of the disease. Gastroenterology (2020) 158(7):1851–64. doi: 10.1053/j.gastro.2020.01.052
2. Powell EE, Wong VW, Rinella M. Non-alcoholic fatty liver disease. Lancet (London England) (2021) 397(10290):2212–24. doi: 10.1016/s0140-6736(20)32511-3
3. Machado MV, Diehl AM. Pathogenesis of nonalcoholic steatohepatitis. Gastroenterology (2016) 150(8):1769–77. doi: 10.1053/j.gastro.2016.02.066
4. Manne V, Handa P, Kowdley KV. Pathophysiology of nonalcoholic fatty liver Disease/Nonalcoholic steatohepatitis. Clinics liver Dis (2018) 22(1):23–37. doi: 10.1016/j.cld.2017.08.007
5. Sheka AC, Adeyi O, Thompson J, Hameed B, Crawford PA, Ikramuddin S. Nonalcoholic steatohepatitis: A review. Jama (2020) 323(12):1175–83. doi: 10.1001/jama.2020.2298
6. Polyzos SA, Kountouras J, Mantzoros CS. Obesity and nonalcoholic fatty liver disease: From pathophysiology to therapeutics. Metabolism: Clin Exp (2019) 92:82–97. doi: 10.1016/j.metabol.2018.11.014
7. Buzzetti E, Pinzani M, Tsochatzis EA. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism: Clin Exp (2016) 65(8):1038–48. doi: 10.1016/j.metabol.2015.12.012
8. Bexborn F, Andersson PO, Chen H, Nilsson B, Ekdahl KN. The tick-over theory revisited: Formation and regulation of the soluble alternative complement C3 convertase (C3(H2O)Bb). Mol Immunol (2008) 45(8):2370–9. doi: 10.1016/j.molimm.2007.11.003
9. Moreno-Navarrete JM, Fernández-Real JM. The complement system is dysfunctional in metabolic disease: Evidences in plasma and adipose tissue from obese and insulin resistant subjects. Semin Cell Dev Biol (2019) 85:164–72. doi: 10.1016/j.semcdb.2017.10.025
10. Xu C, Chen Y, Xu L, Miao M, Li Y, Yu C. Serum complement C3 levels are associated with nonalcoholic fatty liver disease independently of metabolic features in Chinese population. Sci Rep (2016) 6:23279. doi: 10.1038/srep23279
11. Lazo M, Hernaez R, Bonekamp S, Kamel IR, Brancati FL, Guallar E, et al. Non-alcoholic fatty liver disease and mortality among US adults: Prospective cohort study. BMJ (Clinical Res ed (2011) 343:d6891. doi: 10.1136/bmj.d6891
12. Wlazlo N, van Greevenbroek MM, Ferreira I, Jansen EH, Feskens EJ, van der Kallen CJ, et al. Activated complement factor 3 is associated with liver fat and liver enzymes: The CODAM study. Eur J Clin Invest (2013) 43(7):679–88. doi: 10.1111/eci.12093
13. Rensen SS, Slaats Y, Driessen A, Peutz-Kootstra CJ, Nijhuis J, Steffensen R, et al. Activation of the complement system in human nonalcoholic fatty liver disease. Hepatology (2009) 50(6):1809–17. doi: 10.1002/hep.23228
14. Segers FM, Verdam FJ, de Jonge C, Boonen B, Driessen A, Shiri-Sverdlov R, et al. Complement alternative pathway activation in human nonalcoholic steatohepatitis. PloS One (2014) 9(10):e110053. doi: 10.1371/journal.pone.0110053
15. Bessone F, Razori MV, Roma MG. Molecular pathways of nonalcoholic fatty liver disease development and progression. Cell Mol Life sciences: CMLS (2019) 76(1):99–128. doi: 10.1007/s00018-018-2947-0
16. Ipsen DH, Lykkesfeldt J, Tveden-Nyborg P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell Mol Life sciences: CMLS (2018) 75(18):3313–27. doi: 10.1007/s00018-018-2860-6
17. Ogden CA, deCathelineau A, Hoffmann PR, Bratton D, Ghebrehiwet B, Fadok VA, et al. C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J Exp Med (2001) 194(6):781–95. doi: 10.1084/jem.194.6.781
18. Trouw LA, Blom AM, Gasque P. Role of complement and complement regulators in the removal of apoptotic cells. Mol Immunol (2008) 45(5):1199–207. doi: 10.1016/j.molimm.2007.09.008
19. Kaya E, Yilmaz Y. Metabolic-associated fatty liver disease (MAFLD): A multi-systemic disease beyond the liver. J Clin Trans Hepatol (2022) 10(2):329–38. doi: 10.14218/jcth.2021.00178
20. Gabrielsson BG, Johansson JM, Lönn M, Jernås M, Olbers T, Peltonen M, et al. High expression of complement components in omental adipose tissue in obese men. Obes Res (2003) 11(6):699–708. doi: 10.1038/oby.2003.100
21. Matsunaga H, Iwashita M, Shinjo T, Yamashita A, Tsuruta M, Nagasaka S, et al. Adipose tissue complement factor b promotes adipocyte maturation. Biochem Biophys Res Commun (2018) 495(1):740–8. doi: 10.1016/j.bbrc.2017.11.069
22. Cook KS, Min HY, Johnson D, Chaplinsky RJ, Flier JS, Hunt CR, et al. Adipsin: A circulating serine protease homolog secreted by adipose tissue and sciatic nerve. Sci (New York NY) (1987) 237(4813):402–5. doi: 10.1126/science.3299705
23. Nilsson B, Hamad OA, Ahlström H, Kullberg J, Johansson L, Lindhagen L, et al. C3 and C4 are strongly related to adipose tissue variables and cardiovascular risk factors. Eur J Clin Invest (2014) 44(6):587–96. doi: 10.1111/eci.12275
24. Barbu A, Hamad OA, Lind L, Ekdahl KN, Nilsson B. The role of complement factor C3 in lipid metabolism. Mol Immunol (2015) 67(1):101–7. doi: 10.1016/j.molimm.2015.02.027
25. Moreno-Navarrete JM, Martínez-Barricarte R, Catalán V, Sabater M, Gómez-Ambrosi J, Ortega FJ, et al. Complement factor h is expressed in adipose tissue in association with insulin resistance. Diabetes (2010) 59(1):200–9. doi: 10.2337/db09-0700
26. Ursini F, Abenavoli L. The emerging role of complement C3 as a biomarker of insulin resistance and cardiometabolic diseases: Preclinical and clinical evidence. Rev Recent Clin trials (2018) 13(1):61–8. doi: 10.2174/1574887112666171128134552
27. Bajic G, Degn SE, Thiel S, Andersen GR. Complement activation, regulation, and molecular basis for complement-related diseases. EMBO J (2015) 34(22):2735–57. doi: 10.15252/embj.201591881
28. Tsuru H, Osaka M, Hiraoka Y, Yoshida M. HFD-induced hepatic lipid accumulation and inflammation are decreased in factor d deficient mouse. Sci Rep (2020) 10(1):17593. doi: 10.1038/s41598-020-74617-5
29. Laskowski J, Renner B, Pickering MC, Serkova NJ, Smith-Jones PM, Clambey ET, et al. Complement factor h-deficient mice develop spontaneous hepatic tumors. J Clin Invest (2020) 130(8):4039–54. doi: 10.1172/jci135105
30. Maslowska M, Wang HW, Cianflone K. Novel roles for acylation stimulating protein/C3adesArg: A review of recent in vitro and in vivo evidence. Vitamins hormones (2005) 70:309–32. doi: 10.1016/s0083-6729(05)70010-8
31. Sniderman AD, Cianflone KM, Eckel RH. Levels of acylation stimulating protein in obese women before and after moderate weight loss. Int J Obes (1991) 15(5):333–6.
32. Cianflone K, Kalant D, Marliss EB, Gougeon R, Sniderman AD. Response of plasma ASP to a prolonged fast. Int J Obes related Metab disorders: J Int Assoc Study Obes (1995) 19(9):604–9.
33. Meijssen S. Delayed and exaggerated postprandial complement component 3 response in familial combined hyperlipidemia. Arteriosclerosis Thrombosis Vasc Biol (2002) 22(5):811–6. doi: 10.1161/01.atv.0000014079.98335.72
34. Fujita T, Fujioka T, Murakami T, Satomura A, Fuke Y, Matsumoto K. Chylomicron accelerates C3 tick-over by regulating the role of factor h, leading to overproduction of acylation stimulating protein. J Clin Lab Anal (2007) 21(1):14–23. doi: 10.1002/jcla.20158
35. Nilsson B, Nilsson Ekdahl K. The tick-over theory revisited: Is C3 a contact-activated protein? Immunobiology (2012) 217(11):1106–10. doi: 10.1016/j.imbio.2012.07.008
36. Watt MJ, Miotto PM, De Nardo W, Montgomery MK. The liver as an endocrine organ-linking NAFLD and insulin resistance. Endocrine Rev (2019) 40(5):1367–93. doi: 10.1210/er.2019-00034
37. Tanase DM, Gosav EM, Costea CF, Ciocoiu M, Lacatusu CM, Maranduca MA, et al. The intricate relationship between type 2 diabetes mellitus (T2DM), insulin resistance (IR), and nonalcoholic fatty liver disease (NAFLD). J Diabetes Res (2020) 2020:3920196. doi: 10.1155/2020/3920196
38. Stefan N, Kantartzis K, Häring HU. Causes and metabolic consequences of fatty liver. Endocrine Rev (2008) 29(7):939–60. doi: 10.1210/er.2008-0009
39. Brunt EM, Wong VW, Nobili V, Day CP, Sookoian S, Maher JJ, et al. Nonalcoholic fatty liver disease. Nat Rev Dis Primers (2015) 1:15080. doi: 10.1038/nrdp.2015.80
40. Postic C, Girard J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: Lessons from genetically engineered mice. J Clin Invest (2008) 118(3):829–38. doi: 10.1172/jci34275
41. Bavia L, Cogliati B, Dettoni JB, Ferreira Alves VA, Isaac L. The complement component C5 promotes liver steatosis and inflammation in murine non-alcoholic liver disease model. Immunol Lett (2016) 177:53–61. doi: 10.1016/j.imlet.2016.07.014
42. Giles DA, Moreno-Fernandez ME, Divanovic S. IL-17 axis driven inflammation in non-alcoholic fatty liver disease progression. Curr Drug Targets (2015) 16(12):1315–23. doi: 10.2174/1389450116666150531153627
43. Lee JH, Lee JH, Rane SG. TGF-β signaling in pancreatic islet β cell development and function. Endocrinology (2021) 162(3):bqaa233. doi: 10.1210/endocr/bqaa233
44. Tang XW, Qin QX. miR-335-5p induces insulin resistance and pancreatic islet β-cell secretion in gestational diabetes mellitus mice through VASH1-mediated TGF-β signaling pathway. J Cell Physiol (2019) 234(5):6654–66. doi: 10.1002/jcp.27406
45. Mamane Y, Chung Chan C, Lavallee G, Morin N, Xu LJ, Huang J, et al. The C3a anaphylatoxin receptor is a key mediator of insulin resistance and functions by modulating adipose tissue macrophage infiltration and activation. Diabetes (2009) 58(9):2006–17. doi: 10.2337/db09-0323
46. Phieler J, Chung KJ, Chatzigeorgiou A, Klotzsche-von Ameln A, Garcia-Martin R, Sprott D, et al. The complement anaphylatoxin C5a receptor contributes to obese adipose tissue inflammation and insulin resistance. J Immunol (2013) 191(8):4367–74. doi: 10.4049/jimmunol.1300038
47. Lim J, Iyer A, Suen JY, Seow V, Reid RC, Brown L, et al. C5aR and C3aR antagonists each inhibit diet-induced obesity, metabolic dysfunction, and adipocyte and macrophage signaling. FASEB J (2013) 27(2):822–31. doi: 10.1096/fj.12-220582
48. Meli R, Mattace Raso G, Calignano A. Role of innate immune response in non-alcoholic fatty liver disease: Metabolic complications and therapeutic tools. Front Immunol (2014) 5:177. doi: 10.3389/fimmu.2014.00177
49. Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol (2017) 17(5):306–21. doi: 10.1038/nri.2017.11
50. Robinson MW, Harmon C, O’Farrelly C. Liver immunology and its role in inflammation and homeostasis. Cell Mol Immunol (2016) 13(3):267–76. doi: 10.1038/cmi.2016.3
51. Kazankov K, Jørgensen SMD, Thomsen KL, Møller HJ, Vilstrup H, George J, et al. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat Rev Gastroenterol Hepatol (2019) 16(3):145–59. doi: 10.1038/s41575-018-0082-x
52. Tacke F, Zimmermann HW. Macrophage heterogeneity in liver injury and fibrosis. J Hepatol (2014) 60(5):1090–6. doi: 10.1016/j.jhep.2013.12.025
53. Li H, Zhou Y, Wang H, Zhang M, Qiu P, Zhang M, et al. Crosstalk between liver macrophages and surrounding cells in nonalcoholic steatohepatitis. Front Immunol (2020) 11:1169. doi: 10.3389/fimmu.2020.01169
54. Xiong X, Kuang H, Ansari S, Liu T, Gong J, Wang S, et al. Landscape of intercellular crosstalk in healthy and NASH liver revealed by single-cell secretome gene analysis. Mol Cell (2019) 75(3):644–60.e5. doi: 10.1016/j.molcel.2019.07.028
55. Yang Y, Lu Y, Han F, Chang Y, Li X, Han Z, et al. Saxagliptin regulates M1/M2 macrophage polarization via CaMKKβ/AMPK pathway to attenuate NAFLD. Biochem Biophys Res Commun (2018) 503(3):1618–24. doi: 10.1016/j.bbrc.2018.07.090
56. Fujii H, Kawada N. Inflammation and fibrogenesis in steatohepatitis. J Gastroenterol (2012) 47(3):215–25. doi: 10.1007/s00535-012-0527-x
57. Monk PN, Scola AM, Madala P, Fairlie DP. Function, structure and therapeutic potential of complement C5a receptors. Br J Pharmacol (2007) 152(4):429–48. doi: 10.1038/sj.bjp.0707332
58. Asgari E, Le Friec G, Yamamoto H, Perucha E, Sacks SS, Köhl J, et al. C3a modulates IL-1β secretion in human monocytes by regulating ATP efflux and subsequent NLRP3 inflammasome activation. Blood (2013) 122(20):3473–81. doi: 10.1182/blood-2013-05-502229
59. Samstad EO, Niyonzima N, Nymo S, Aune MH, Ryan L, Bakke SS, et al. Cholesterol crystals induce complement-dependent inflammasome activation and cytokine release. J Immunol (Baltimore Md: 1950) (2014) 192(6):2837–45. doi: 10.4049/jimmunol.1302484
60. Luci C, Bourinet M, Leclère PS, Anty R, Gual P. Chronic inflammation in non-alcoholic steatohepatitis: Molecular mechanisms and therapeutic strategies. Front Endocrinol (2020) 11:597648. doi: 10.3389/fendo.2020.597648
61. Antonucci L, Porcu C, Timperi E, Santini SJ, Iannucci G, Balsano C. Circulating neutrophils of nonalcoholic steatohepatitis patients show an activated phenotype and suppress T lymphocytes activity. J Immunol Res (2020) 2020:4570219. doi: 10.1155/2020/4570219
62. Cai J, Zhang XJ, Li H. The role of innate immune cells in nonalcoholic steatohepatitis. Hepatol (Baltimore Md) (2019) 70(3):1026–37. doi: 10.1002/hep.30506
63. Nati M, Haddad D, Birkenfeld AL, Koch CA, Chavakis T, Chatzigeorgiou A. The role of immune cells in metabolism-related liver inflammation and development of non-alcoholic steatohepatitis (NASH). Rev endocrine Metab Disord (2016) 17(1):29–39. doi: 10.1007/s11154-016-9339-2
64. Herrero-Cervera A, Soehnlein O, Kenne EJC. Immunology m. Neutrophils chronic inflammatory diseases. (2022) 19(2):177–91. doi: 10.1038/s41423-021-00832-3
65. Rensen SS, Slaats Y, Nijhuis J, Jans A, Bieghs V, Driessen A, et al. Increased hepatic myeloperoxidase activity in obese subjects with nonalcoholic steatohepatitis. Am J Pathol (2009) 175(4):1473–82. doi: 10.2353/ajpath.2009.080999
66. Zhang Y, Zhou X, Liu P, Chen X, Zhang J, Zhang H, et al. GCSF deficiency attenuates nonalcoholic fatty liver disease through regulating GCSFR-SOCS3-JAK-STAT3 pathway and immune cells infiltration. Am J Physiol Gastrointestinal liver Physiol (2021) 320(4):G531–g42. doi: 10.1152/ajpgi.00342.2020
67. Pulli B, Ali M, Iwamoto Y, Zeller MW, Schob S, Linnoila JJ, et al. Myeloperoxidase-Hepatocyte-Stellate cell cross talk promotes hepatocyte injury and fibrosis in experimental nonalcoholic steatohepatitis. Antioxidants Redox Signaling (2015) 23(16):1255–69. doi: 10.1089/ars.2014.6108
68. Martin U, Bock D, Arseniev L, Tornetta MA, Ames RS, Bautsch W, et al. The human C3a receptor is expressed on neutrophils and monocytes, but not on b or T lymphocytes. J Exp Med (1997) 186(2):199–207. doi: 10.1084/jem.186.2.199
69. Wu KY, Zhang T, Zhao GX, Ma N, Zhao SJ, Wang N, et al. The C3a/C3aR axis mediates anti-inflammatory activity and protects against uropathogenic e coli-induced kidney injury in mice. Kidney Int (2019) 96(3):612–27. doi: 10.1016/j.kint.2019.03.005
70. Coulthard LG, Woodruff TM. Is the complement activation product C3a a proinflammatory molecule? re-evaluating the evidence and the myth. J Immunol (Baltimore Md: 1950) (2015) 194(8):3542–8. doi: 10.4049/jimmunol.1403068
71. Guo RF, Riedemann NC, Ward PA. Role of C5a-C5aR interaction in sepsis. Shock (Augusta Ga) (2004) 21(1):1–7. doi: 10.1097/01.shk.0000105502.75189.5e
72. Ehrnthaller C, Braumüller S, Kellermann S, Gebhard F, Perl M, Huber-Lang M. Complement factor C5a inhibits apoptosis of neutrophils-a mechanism in polytrauma? J Clin Med (2021) 10(14):3157. doi: 10.3390/jcm10143157
73. Markiewski MM, Lambris JD. The role of complement in inflammatory diseases from behind the scenes into the spotlight. Am J Pathol (2007) 171(3):715–27. doi: 10.2353/ajpath.2007.070166
74. Hosokawa H, Rothenberg EV. How transcription factors drive choice of the T cell fate. Nat Rev Immunol (2021) 21(3):162–76. doi: 10.1038/s41577-020-00426-6
75. Dudek M, Pfister D, Donakonda S, Filpe P, Schneider A, Laschinger M, et al. Auto-aggressive CXCR6(+) CD8 T cells cause liver immune pathology in NASH. Nature (2021) 592(7854):444–9. doi: 10.1038/s41586-021-03233-8
76. Wolf MJ, Adili A, Piotrowitz K, Abdullah Z, Boege Y, Stemmer K, et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell (2014) 26(4):549–64. doi: 10.1016/j.ccell.2014.09.003
77. Van Herck MA, Vonghia L, Kwanten WJ, Julé Y, Vanwolleghem T, Ebo DG, et al. Diet reversal and immune modulation show key role for liver and adipose tissue T cells in murine nonalcoholic steatohepatitis. Cell Mol Gastroenterol Hepatol (2020) 10(3):467–90. doi: 10.1016/j.jcmgh.2020.04.010
78. Hirsova P, Bamidele A, Wang H, Povero D, Revelo X. Emerging roles of T cells in the pathogenesis of nonalcoholic steatohepatitis and hepatocellular carcinoma. Front Endocrinol (2021) 12:760860. doi: 10.3389/fendo.2021.760860
79. Maher JJ, Leon P, Ryan JC. Beyond insulin resistance: Innate immunity in nonalcoholic steatohepatitis. Hepatol (Baltimore Md) (2008) 48(2):670–8. doi: 10.1002/hep.22399
80. Rau M, Schilling AK, Meertens J, Hering I, Weiss J, Jurowich C, et al. Progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis is marked by a higher frequency of Th17 cells in the liver and an increased Th17/Resting regulatory T cell ratio in peripheral blood and in the liver. J Immunol (Baltimore Md: 1950) (2016) 196(1):97–105. doi: 10.4049/jimmunol.1501175
81. Her Z, Tan JHL, Lim YS, Tan SY, Chan XY, Tan WWS, et al. CD4(+) T cells mediate the development of liver fibrosis in high fat diet-induced NAFLD in humanized mice. Front Immunol (2020) 11:580968. doi: 10.3389/fimmu.2020.580968
82. Kawamoto S, Yalcindag A, Laouini D, Brodeur S, Bryce P, Lu B, et al. The anaphylatoxin C3a downregulates the Th2 response to epicutaneously introduced antigen. J Clin Invest (2004) 114(3):399–407. doi: 10.1172/jci19082
83. Strainic MG, Shevach EM, An F, Lin F, Medof ME. Absence of signaling into CD4⁺ cells via C3aR and C5aR enables autoinductive TGF-β1 signaling and induction of Foxp3⁺ regulatory T cells. Nat Immunol (2013) 14(2):162–71. doi: 10.1038/ni.2499
Keywords: complement system, nonalcoholic fatty liver disease, site-targeted inhibitor, inflammation, liver
Citation: Guo Z, Fan X, Yao J, Tomlinson S, Yuan G and He S (2022) The role of complement in nonalcoholic fatty liver disease. Front. Immunol. 13:1017467. doi: 10.3389/fimmu.2022.1017467
Received: 12 August 2022; Accepted: 13 September 2022;
Published: 29 September 2022.
Edited by:
Ruchi Bansal, University of Twente, NetherlandsReviewed by:
Yoshihiro Ogawa, Kyushu University, JapanCopyright © 2022 Guo, Fan, Yao, Tomlinson, Yuan and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Songqing He, ZHJfaGVzb25ncWluZ0AxNjMuY29t; Guandou Yuan, ZHJfeXVhbmdkQGd4bXUuZWR1LmNu
†These authors have contributed equally to this work