
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 17 October 2022
Sec. Molecular Innate Immunity
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1008072
This article is part of the Research Topic Identification, Function and Mechanisms of Interferon Induced Genes Associated with Viruses View all 12 articles
 Rui Lang1,2†
Rui Lang1,2† Huiting Li1†
Huiting Li1† Xiaoqin Luo3
Xiaoqin Luo3 Cencen Liu4Yiwen Zhang2
Cencen Liu4Yiwen Zhang2 ShunYu Guo2
ShunYu Guo2 Jingyi Xu1Changshun Bao2,5,6,7*
Jingyi Xu1Changshun Bao2,5,6,7* Wei Dong1*
Wei Dong1* Yang Yu1,3*
Yang Yu1,3*Interferons (IFNs) bind to cell surface receptors and activate the expression of interferon-stimulated genes (ISGs) through intracellular signaling cascades. ISGs and their expression products have various biological functions, such as antiviral and immunomodulatory effects, and are essential effector molecules for IFN function. ISGs limit the invasion and replication of the virus in a cell-specific and region-specific manner in the central nervous system (CNS). In addition to participating in natural immunity against viral infections, studies have shown that ISGs are essential in the pathogenesis of CNS disorders such as neuroinflammation and neurodegenerative diseases. The aim of this review is to present a macroscopic overview of the characteristics of ISGs that restrict viral neural invasion and the expression of the ISGs underlying viral infection of CNS cells. Furthermore, we elucidate the characteristics of ISGs expression in neurological inflammation, neuropsychiatric disorders such as depression as well as neurodegenerative disorders, including Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS). Finally, we summarize several ISGs (ISG15, IFIT2, IFITM3) that have been studied more in recent years for their antiviral infection in the CNS and their research progress in neurological diseases.
Interferons (IFNs) are a class of antiviral cytokines that are stimulated in response to the challenge of host defenses and are crucial for mobilizing the immune response to pathogens in vertebrates. Most virus-infected cells can produce IFNs, which are secreted to bind to their receptors on autologous or other cells and initiate a signaling cascade that leads to the induction of hundreds of interferon-stimulated genes (ISGs) to promote antiviral effects (1, 2). IFNs are divided into three classes (type I, type II, and type III) based on their sequence and cellular receptors. The type I IFN family comprises members encoded by multiple genes, including 13 highly homologous subtypes of IFN-α, and the other isoforms, such as IFN-β, IFN-ϵ, IFN-κ, IFN-ω, IFN-τ, IFN-δ, and IFN-ζ (3–6). The type II IFN class only contains IFN-γ, while the type III IFN family consists of four IFN-λ molecules, including IFN-λ1(IL-29), IFN-λ2 (IL-28A), and IFN-λ3 (IL-28B), as well as IFN-λ4 (7). Type I and III IFNs are considered the classical antiviral IFNs, while type II IFN has multiple roles in the innate and adaptive immune systems (1, 2, 7–10). Additionally, type III IFNs impact antiviral activity at anatomical barriers, such as the blood-brain barrier (BBB) and epithelial cell surfaces (11). Although type I and type III IFNs induce a similar subset of ISGs, differences in cell-type specificity and signaling kinetics result in distinct responses. In general, type I IFNs activate a more robust and rapid ISG response, whereas type III IFNs induce a slower response with lower levels of ISG expression (reviewed in 2). Traditionally, IFN signaling is involved in the induction of host defense-associated ISGs through the Janus tyrosine kinase (JAK)/signal transducer and activator of transcription (STAT) signal pathway (12–14). Type I and III IFNs activate JAK1 and tyrosine kinase 2 (TYK2), resulting in cytoplasmic STAT1 and STAT2 phosphorylation (15). After phosphorylation, STAT1 and STAT2 dimerize and translocate from the cytoplasm to the nucleus, forming the IFN-stimulated gene factor 3 (ISGF3) complex with interferon regulatory factor 9 (IRF9). ISGF3 further binds to interferon stimulatory response elements (ISREs) and stimulates the transcription of ISGs (16). Type II IFN activates JAK1 and JAK2, resulting in the formation of phosphorylated STAT1 (pSTAT1) homodimers known as γ-activated factors (GAF), which translocate to the nucleus and bind to γ-activated sequences (GAS) to induce transcription of ISGs (17). Some interferon regulatory factors (IRFs) such as IRF3 can induce ISGs directly in the absence of IFN production or collaboratively with other transcription factors such as IFN regulatory factor 7 (IRF7), IRF1, and nuclear factor kappa B (NF-κB) to induce type I IFN production (17–26). In addition, some ISGs encode factors that are involved in the IFN production or response pathway through positive or negative feedback loops (27, 28). For instance, the core retinoic acid-inducible gene I (RIG-I) and melanoma differentiation-associated gene 5 (MDA5), members of the mammalian RIG-I-like receptors (RLRs), are found in the cytosol of most cell types and are powerfully activated by IFNs in a positive feedback loop of RNA virus infection (27, 29). Interferon-responsive activation of interferon-induced protein with tetrapeptide repeats 1 (IFIT1, also known as ISG56) positively regulates the expression of RIG-I, MDA5, and IFIT2 (also known as ISG54) (30). Furthermore, IFN-stimulated gene 15 (ISG15) was found to negatively regulate the IFN signaling pathway by coupling to RIG-I (31, 32).
The IFN pathway provides essential protection to the central nervous system (CNS) against viral infections. It is instrumental in immune-related diseases such as allergic reactions, chronic inflammatory diseases, autoimmune diseases, transplant rejection, viral infections, and many more (33). For example, mice with a deficiency in the IFN-I receptor subunit 1 (Ifnar1-/-) are highly susceptible to various viral infections in multiple organs, including the CNS (34–36). Ifnar1-/- mice showed increased viral load after infection with Sindbis virus (SINV), and increased viral load in the CNS was associated with high susceptibility compared with wild-type (WT) mice (37). Several fatal cases of Herpes simplex encephalitis (HSE) in newborns were associated with defects in genes encoding signal transduction factors of the IFN pathway, such as Toll/interleukin-1 receptor domain-containing adaptor-inducing interferon-β (TRIF), TANK-binding kinase 1 (TBK-1), Toll-like receptor 3 (TLR3) or tumor necrosis factor receptor-associated factor 3 (TRAF3). These findings demonstrated the importance of the human IFN response to neurotropic viral infections (38–41). In addition to participating in natural immunity against viral infections, IFNs have been shown to constitute key factors in the neuroinflammatory network and make an essential contribution to the pathogenesis of neurodegenerative diseases such as Parkinson’s disease (PD), Alzheimer’s disease (AD), and amyotrophic lateral sclerosis (ALS) (42–45). The IFN signaling pathway was recently reported to be severely upregulated in AD patients and significantly correlated with disease severity (46–48). Activation of the IFN signaling pathway can induce the expression of hundreds of ISGs. Although ISGs are major antiviral effectors of the IFN response, the antiviral mechanisms of most ISGs have not been described until recently. The aim of this review is to provide a macroscopic overview of the characteristics of ISGs that restrict viral neural invasion and cellular expression of ISGs after viral infection of the CNS, as well as the expression characteristics of ISGs in neurological diseases (Table 1). In addition, substantial progress has been made in our understanding of individual ISGs (ISG15, IFIT2, IFITM3) in CNS viral infection and diseases in recent years (Table 1), providing an essential target for the development of novel antivirals and anti-neurological disease drugs.

Table 1 ISGs expression in antiviral infections, neuronal localization and neurological diseases.
The CNS requires a complex and coordinated immune response to prevent neurological disorders, avoid excessive immune activation and inappropriate inflammatory response, and protect against invading pathogens such as viruses. Although the CNS is immune privileged and protected from toxic substances and pathogens carried in the blood by the BBB and the blood-cerebrospinal fluid barrier (BCSFB) (78), neurotropic viruses are capable of infecting the CNS and staying there for a long time, including herpes simplex virus type 1 (HSV-1), varicella zoster virus (VZV), Japanese encephalitis virus (JEV), West Nile virus (WNV), measles, rabies virus (RABV), poliovirus, and so on (79–81). Neurotropic viruses can spread through multiple pathways to penetrate the CNS. Some viruses can enter the CNS by infecting host immune cells in the periphery and using these cells as “Trojan horses” to carry them across the BBB, or the virus can directly infect endothelial cells, disrupt the BBB, or infect peripheral neuron axons and retrograde upward through the mechanism of axonal transport of cellular cargo, such as through the olfactory pathway (33, 79, 80) (Figure 1).
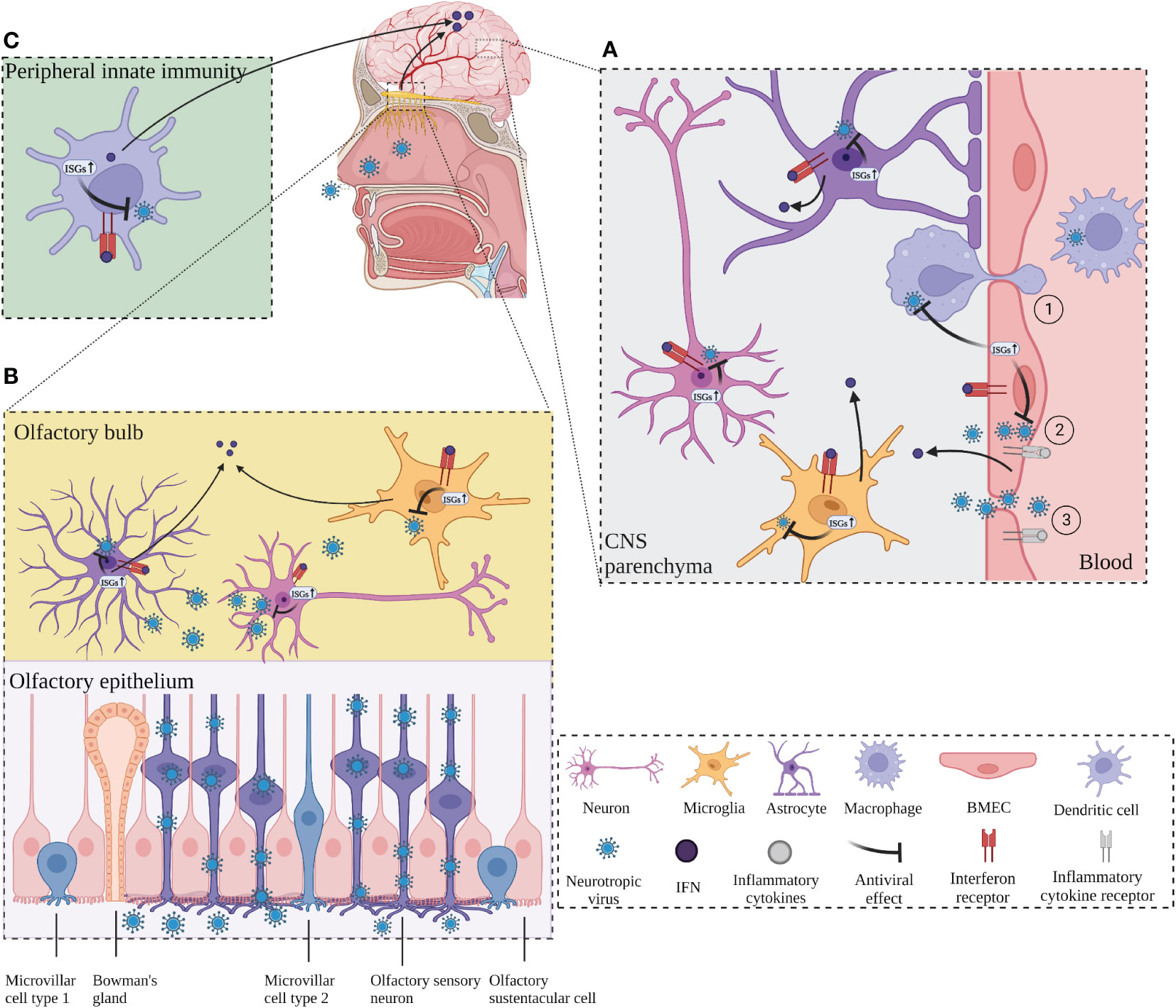
Figure 1 ISGs restrict viral neuroinvasion. (A) ISGs in BMECs, which constitute the blood-brain barrier (BBB), are activated by virus and inflammation, leading to an IFN response and activation of transcription of antiviral ISGs, followed by activation of microglia and astrocytes to release ISGs. BMECs deliver the released ISGs to macrophages to enhance the antiviral response and restrict viral invasion. ① Neurotropic virus crosses the BBB through “Trojan horse” strategy. ② Virus infects endothelial cells across the BBB. ③ Virus enters the CNS by destroying the BBB. (B) The virus invades the CNS retrogradely through the olfactory pathway. The virus invades the olfactory sensory neurons through the olfactory epithelium and retrogrades upward into the olfactory bulb (OB). Virus at OB activates neurons, microglia, and macrophages, which produces IFN response and induces transcription of ISGs, leading to antiviral response. Long-distance signaling of IFN at the OB activates the transcription of ISGs in the brain. (C) IFNs produced by peripheral antiviral response enter the CNS and activate the transcription of ISGs in advance and induces transcription of ISGs, leading to antiviral response. Long-distance signaling of IFN at the OB activates the transcription of ISGs in the brain. (C) IFNs produced by peripheral antiviral response enter the CNS and activate the transcription of ISGs in advance. Created with BioRender.com.
The structural integrity of the BBB is critical to enable the normal neurological function of the CNS and protection from damage from inflammation, virus, and other diseases. Brain microvascular endothelial cells (BMECs) are the structural and functional basis of the BBB and play a critical role in maintaining its normal integrity. One study reported that infection of BMECs by JEV did not affect cell viability but resulted in increased permeability of the endothelial monolayer due to inflammation caused by JEV infection that inhibited the expression of tight junction (TJ) proteins in BMECs, leading to enhanced BBB permeability (81). In vitro experiments with cultured mouse BMECs showed that IFN-λ signaling increased transendothelial resistance, reduced viral movement across the barrier, and modulated TJ protein localization (82). In addition, IFN-λ limits the opening of the BBB by reducing the production of inflammatory cytokines in primary astrocytes and microglia and inducing activation of the JAK/STAT pathway, leading to the production of ISGs (83). Inflammation can play an indirect antiviral role while disrupting the BBB, and in an in vitro model of the BBB with co-culture of astrocytes and BMECs, lipopolysaccharide (LPS) promoted the expression of type I IFN signaling-related proteins such as STAT1, STAT2, ISG15, and SAMHD1 in astrocytes, TNF-α and LPS also induce the production of ISGs (EIF2AK2, ADAR, TRIM25 and ISG15) in astrocyte cultures in vitro (84). BMECs have a critical function in the innate immunity of the BBB to human immunodeficiency virus (HIV) infection by causing activation of TLR3 in BMECs to induce phosphorylation of IRF3 and IRF7 (key regulators of the IFN signaling pathway) and trigger the production of endogenous IFN-β and IFN-λ, thereby significantly inhibiting viral replication (85). In addition, TLR3-activated human BMECs secrete exosomes that inhibit HIV replication by transferring antiviral factors, including several critical IFN-stimulated genes (ISG15, IFIT1, MX2) to macrophages (50). This suggests that human BMECs may help restore the antiviral status of HIV-infected macrophages, which may be a defense mechanism against neural invasion by HIV “Trojan horses” (Figure 1A).
Numerous neurotropic viruses enter the CNS through infection of peripheral nerves, including olfactory neurons and sensory or motor neurons (86). WNV can enter the CNS by infecting sensory nerve endings or olfactory neurons or through the bloodstream (87). RABV and poliovirus transmit from muscle to somatic motor neurons in the spinal cord via the neuromuscular junctions (NMJs) (88). Olfactory pathways play an important role in the invasion of viruses into the CNS via peripheral nerves (Figure 1B). Detje et al. found that blocking the type I IFN pathway promoted the spread of vesicular stomatitis virus (VSV) from the olfactory bulb (OB) to the entire CNS, while local IFN response in the OB effectively controlled viral invasion of the CNS (89). IFN-induced viperin limits the replication of Langat virus (LGTV) in the OB in a region-specific manner (61). Long-range signaling of IFN-β released from infected neurons at the OB after VSV and cytomegalovirus (CMV) infection of the nasal mucosa upregulated the expression of ISGs in uninfected brain regions (90). Similar results were obtained from another investigation, which shows that after intranasal VSV infection, IFN expressed at the OB enters the brain to activate IFIT2 transcription in advance to act as an antiviral agent (67). The accumulation of microglia around the OB and their expression of ISGs form a natural immune barrier that is instrumental in limiting the spread of VSV in the CNS and preventing fatal encephalitis (91) (Figure 1B).
Viral infections rapidly induce IFN in the periphery, which serves to protect most tissues from viral pathogenicity (92). Peripherally induced IFN response can induce ISGs in the brain. Peripheral IFN-α that crosses the BBB directly activates IFN-α/β receptor (IFNAR) signaling in microglia, leading to the upregulation of multiple ISGs (93, 94). As mentioned above, long-range signaling of IFN released from infected neurons at the OB after viral infection of the nasal mucosa upregulated the expression of ISGs (IFIT2, IFIT3, OAS, and MX1) in uninfected brain regions (67, 90). Lukasz et al. reported that adolescent mice injected with the viral mimic poly I:C significantly increased the expression of ISGs (IFIT2, PRKR, MX2, and IRF7) in the hippocampal dentate gyrus (58). Collectively, these findings suggest that early activation of ISGs in the CNS plays a crucial role in limiting viral infection of the CNS (Figure 1C).
The vast majority of cell types in the CNS, comprising neurons, astrocytes, oligodendrocytes, CNS-associated macrophages (CAMs), ventricular epithelial cells, and vascular endothelial cells, are responsive to IFN (95). Activation of astrocytes and microglia in the brain has an essential role in the innate immune response of the CNS to viral infection (96, 97). Different immune cells in the CNS respond to various viral stimuli. Microglia monitor the local environment and rapidly respond to widespread inflammatory stimuli, while astrocytes function as immune response cells and produce large amounts of inflammatory mediators (98). For example, during infection with Taylor mouse encephalomyelitis virus (TMEV), protein levels of ISG15 were elevated mainly in astrocytes and endothelial cells, whereas the protein levels of protein kinase R (PKR) were predominantly increased in microglia/macrophages, oligodendrocytes and neurons (49). Murine hepatitis virus (MHV) is a neurotropic coronavirus, and astrocytes and microglia produce type I IFNs (IFN-α and IFN-β), as well as interleukin (IL-6), TNF-α, IL-12, IL-1α, and IL-1β during experimental MHV infection in mice (99). HSV encephalitis (HSE) is a severe CNS infection caused primarily by HSV-1 and occasionally by HSV-2. Following infection with HSV-1, TLR2 and TLR4 are induced to activate simultaneously, and in turn, TLR2 forms a dimer with TLR1, or TLR6, which then induces IFN-β in neurons and IFN-α in astrocytes, and these IFNs subsequently induce the expression of ISGs such as viperin, CH25H (cholesterol-25-hydroxylase), oligoadenylate synthase2 (OAS2), latent RNase (RNase L), PKR, and IFIT1 (59, 60).
Microglia, a major source of type I IFNs, exert direct antiviral effects by producing type I IFNs to stimulate the expression of ISGs or act indirectly on other cells through type I IFNs to activate the corresponding signaling pathway (100). Microglia express various pattern recognition receptors (PRRs), including TLRs, RNA-sensing RLRs, and cytosolic DNA sensors that are important for virus defense. PRRs recognize viral-associated molecular patterns and induce type I IFN expression in microglia (27, 101). Cyclic GMP-AMP synthase (cGAS) binds to cytoplasmic dsDNA in microglia to produce cyclic GMP-AMP (cGAMP), which activates downstream stimulator of interferon genes (STING) and ultimately activates the transcription factor IRF3. In turn, IRF3 stimulates type I IFN production, and the resulting type I IFNs bind to the heterodimeric receptor IFNAR to initiate a signaling cascade, promote nuclear translocation of heterodimeric STAT1/2, and facilitate transcriptional activation of multiple ISGs (59, 60, 67, 90, 99). Depletion of CNS microglia via CSF1R inactivation has higher viral loads in mice infected with WNV (TX02 strain) or VSV, with increased mortality and viral tissue loads, indicating that microglia are critical for restricting virus transmission (102, 103). The phagocytic activity of microglia recruited via purinergic receptor P2Y12 signaling around infected neurons play an important role in CNS antiviral immunity. Analysis of temporal lobe specimens from patients with HSV-1 encephalitis reveals that there are approximately 1–3 activated microglia around each HSV-1-positive neuron and that P2Y12-positive microglia processes extend to HSV-1-positive cells (104). In addition, the number of microglia recruited to infected neurons was significantly reduced in a P2Y12-deficient mouse model (104). Studies have shown that microglia are the main producers of type I IFNs in viral infections of the CNS. Further studies found that in the mouse model of VSV encephalitis, the infected microglia were found to produce type I IFNs, which caused both infected and uninfected microglia to upregulate the expression of IRF7 and activate innate immunity, thus limiting the trans-synaptic transmission of VSV (105). Microglia also induce IFN and ISG expression by regulating the HSPA8/DNA-PK pathway independently of STING (106). Although phagocytosis of foreign pathogens by microglia is an essential component of neuroprotective immune defense to ensure the function of healthy neurons, excessive microglia activation leads to uncontrolled inflammation that exacerbates neuronal death, causing damage to brain tissue and cells (68, 107). In mice infected with JEV, microglia activation led to uncontrolled inflammation and neuronal death (108). JEV can promote viral replication by infecting microglia and upregulating miR-146a gene expression, inhibiting NF-κB activity, blocking the antiviral JAK/STAT signaling pathway, and downregulating antiviral ISGs (IFIT1 and IFIT2) (68). Phosphorylation of IFN regulators (IRF3 and IRF7) and STAT1/3 are inhibited in HIV-infected microglia, which suppresses the expression of several key anti-HIV ISGs (ISG15, IFIT1, GBP5, MX2, and viperin) (51). Taken together, these findings suggest microglia play an essential role in antiviral defense of the CNS and contribute to explaining how the virus invades microglia and results in a persistent infection.
Astrocytes have essential and significant functions in synaptic plasticity, regulation of the BBB, and maintenance of CNS homeostasis (109, 110). In viral infections, astrocytes play a key role in host defense by supporting a functional BBB, regulating glutamate homeostasis, and engaging in innate and adaptive immune response to viral infections (111). Recent studies have shown that microglia induce astrocytes proliferation through the expression of pro-inflammatory cytokines, including IL-1β, TNF, and IFN-γ (112, 113). During viral infection, astrocytes detect molecular changes in their extracellular environment and neighboring cells. Compared with microglia, astrocytes have low basal mRNA levels of PRRs and ISGs, and poorly induced Ifn-β mRNA following infection, but the upregulated various mRNAs in the IFN-α/β pathway of astrocytes to a higher extent than microglia, suggesting that the response of astrocytes to infection is delayed but stronger compared with that of microglia (114). Genetic astrocyte-specific deletion of the type I IFN receptor IFNAR in a mouse model of viral infection led to an increase of BBB permeability (115). Further studies revealed that abolition of astrocytic IFN-α/β signaling was followed by uncontrolled virus transmission and fatal encephalomyelitis, demonstrating the importance of the inducible IFN signaling pathway within astrocytes in limiting viral infection of the CNS (114). Imaizumi et al. reported that poly I:C upregulated the expression of IFIT2 and IFIT1 in astrocytes via the TLR3/IFN-β pathway and that the expression product IFIT1 positively regulated the expression of IFIT2, RIG-I, and MDA5 to enhance antiviral response (30). In addition, RNA interference (RNAi) knockdown of interferon-induced protein 35 (IFI35) resulted in a decrease in expression of poly I:C-induced IFN-β, pStat1, Rig-1, Cxcl10, and Ccl5, indicating that IFI35 may negatively regulate the astrocyte TLR3/IFN-β/pSTAT1/RIG-I/CXCL10/CCL5 axis and may partially regulate the innate immune response of astrocytes (116). Recent studies of CNS complications due to enterovirus 71 (EV71) infection have shown that in infected astrocytes, phosphorylated and non-phosphorylated STAT3 competes with STAT1 for binding to KPNA1, inhibits nuclear import of pSTAT1 and hinders the formation of the ISGF3 complex, leading to suppression of downstream ISG expression (117). Knockdown of STAT3 attenuated the suppressed IFN-mediated antiviral response to EV71 infection and led to a reduction in viral replication, demonstrating the role of STAT3 in maintaining the balance of inflammatory response in astrocytes and antiviral response in the CNS during infection (117). Borna disease virus (BDV) is a non-hemolytic RNA neurotropic virus, and replication of this virus is effectively blocked in transgenic mice expressing mouse IFN-α in astrocytes (118). Another study showed that rapid type I IFN response protected astrocytes from virus-induced cytopathic effects upon infection with flavivirus, tick-borne encephalitis virus (TBEV), JEV, WNV, and Zika virus (ZIKV), thus limiting the spread of these viruses (119). In addition, type I and type III IFN-independent antiviral pathways were found to be involved in the control of astrocytes during ZIKV infection (120). In summary, the intrinsic structural antiviral response of astrocytes combined with rapid induction of type I IFNs is instrumental in protecting astrocytes and inhibiting viral replication in the CNS (121).
Like microglia and astrocytes, neurons express multiple PRRs, produce innate immune cytokines such as type I IFNs following viral infection, and respond to cytokine stimulation to inhibit viral replication and increase cell survival (122–125). Previous studies have shown neuronal upregulation of key antiviral effector molecules and other ISGs in response to neurotropic virus infection, but the neurons produce a limited amount of IFN and express fewer ISGs compared with microglia (126). Delhaye et al. identified approximately 16% of IFN-producing cells corresponding to neurons, but only 3% of infected neurons produced IFN after infecting mice with two neurotropic viruses that primarily infect neurons (La Crosse virus and Theiler virus) (122). ISGs suppress viral replication by directly interrupting the viral life cycle or by stimulating the production of antiviral factors in infected and adjacent cells, but the effects vary considerably in different regions of the brain (127). Further studies have shown that the induction and response to ISGs vary considerably in diverse neuronal populations. In transgenic mice expressing IFN, the expression of typical IFN response marker MX1 was higher in CA1 and CA2 neurons in comparison with CA3 neurons in the hippocampal region (118). Lucas et al. discovered that the IFN-α-inducible protein 27 like 2A (Ifi27l2a), which is upregulated in the cerebellum, brainstem, and spinal cord after WNV infection, limits viral infection in these regions but not in other neurons and cells, implying that Ifi27l2a contributes to WNV innate immune restriction in certain cell types and tissue-specific manner (76). Furthermore, following LGTV infection in the CNS, the activity of viperin, an interferon-inducible protein that inhibits replication, effectively limits LGTV replication in the OB and brain but does not inhibit virus replication in the cerebellum (128). Viperin also reduced TBEV replication in primary cortical neurons and astrocytes in vitro, but not in cerebellar granule cell neurons (61).
Mutations in human TLR3 are essential in the development of human HSV-1 encephalitis (129–131). TLR3 deficiency impairs the cell-autonomous defense of IPSC-derived cortical neurons and oligodendrocytes against HSV-1 infection, but not that of trigeminal ganglion (TG) neurons, owing to TLR3 control of ISG mRNA expression levels induced in human pluripotent stem cell-derived cortical neurons but not TG neurons (132–134). It has also been reported that neurons at different developmental stages express different levels of ISGs in response to viral infection. During LACV-infected encephalitis, both LACV-infected neural precursor cells and mature neurons undergo apoptosis, but neuronal maturation increases the susceptibility of neurons to LACV-induced apoptosis because mature neurons express less ISGs compared to neural precursor cells (135). Unlike the CNS, the antiviral response of peripheral neurons, such as the dorsal root ganglion (DRG), is more dependent on the dual action of antiviral ISGs and autophagy activation. DRG produces only a small number of type I IFNs and does not effectively induce the production of ISGs (136).
Oligodendrocytes have been shown to be less responsive to IFN in comparison with microglia (137). Mouse oligodendrocytes have lower basal expression levels of PRRs, IFN-α/β, ISGs, and kinases and transcription factors essential for IFN-α/β signaling and displayed a later expression of ISGs by comparison to microglia. Despite the fact that infection increases the expression of ISGs in both cell types, oligodendrocytes have a more limited expression profile and absolute mRNA levels compared with microglia. This limited antiviral response is associated with the inability to upregulate IκB kinase (IkappaB kinase or IKK) and IRF7 transcripts, both of which are required for amplification of the IFN-α/β response (138).
Upregulation of ISGs is associated with neuroinflammation-related diseases, including neuroinflammatory diseases such as multiple sclerosis (MS) (62) and neurodegenerative diseases such as AD (46), PD (139), and ALS (54). MS patients have elevated concentrations of the bile acid precursor 25-hydroxycholesterol (25-HC) in the cerebrospinal fluid (CSF), possibly as a result of the upregulation of the ISG CH25H in macrophages (62). 25-HC is mainly synthesized from cholesterol by CH25H, and has been shown to modulate inflammatory response and oxidative stress in normal or pathological nervous systems (140, 141). Wang et al. reported that ISG15 positively correlated with the degree of neuronal damage in an animal model of ALS with no obvious signs of inflammation, a model of cerebral ischemia, a model of brain injury induced by cortical shocks, and a mouse model of chronic neuronal damage induced by the viral protein HIV gp120, with high and significantly elevated ISG15 levels in areas of neuronal damage (54). This suggests that ISG15 may be a reliable biomarker of pathological changes in the CNS (54). Mutations in the ATM gene contribute to ataxia capillaries (A-T), a rare neurodegenerative and immunodeficiency disorder characterized by cerebellar ataxia capillaris, immunodeficiency, radiosensitivity, and cancer susceptibility. Studies have shown that in A-T, the level of ISG15 is significantly higher in the cerebellum than in the brain (55). In glucosylceramidase1 (GBA1) deficient mice (which causes Parkinson’s disease alpha-synuclein pathology), IFN-β levels are elevated in neurons, and ISGs are elevated in microglia (139). IFP35 is significantly upregulated in patients with untreated MS, demonstrating that IFP35 expression levels predict disease outcome and treatment response in MS (77). TLR3, which is primarily activated in innate immunity due to viral infection and induces the production of downstream ISGs (142), has also been found to be activated in alcohol-induced brain injury (143, 144). McDonough et al. reported TLR4-dependent upregulation of ISGs in ischemia/reperfusion-induced microglia (145). Meanwhile, in an AD model, activated microglia express ISGs, and the microglia are centered around amyloid-β (Aβ) plaques and accumulate in an age-dependent manner (46). Intracerebral injection of recombinant IFN-β activated microglia and eliminated complement C3-dependent synapses. Conversely, selective IFN receptor blockade effectively reduced ongoing microglia proliferation and synapse loss in AD models, demonstrating that ISGs are associated with a reduction in synapses (46). Aicardi-Gtières syndrome (AGS) is a severe inflammatory disease mimicking congenital infection with significant IFN production, characterized by chronic CSF lymphocytosis and elevated IFN-α levels, which can lead to severe neurodevelopmental disorders, spastic dystonia, and abnormal tetraplegia (146). ISGs, such as ISG15, viperin, and IFI27, are consistently elevated in patients with AGS, and these elevated ISGs are highly correlated with disease onset and progression (57). Mutations in adenosine deaminase (ADAR1) are crucial mechanisms for the development of AGS. In ADAR1-mutant mice, the expression of ISGs in neurons and microglia is selectively activated in a patchy manner, and the expression of Isg15 in brain neurons with ADAR1 mutation is upregulated (147). It has also been reported that conditional deletion of ADAR1, specifically in mouse neural spinal cells, leads to overall peripheral nerve depigmentation and myelin loss, and that upregulation of ISGs precedes these defects, suggesting that ISGs may be involved in the production of such defects (148). Collectively, these studies have, in part, revealed a complex relationship between the IFN signaling pathway and neuroinflammation-related diseases.
In recent years, evidence has accumulated to show that ISGs play an important role in psychiatric symptoms caused by CNS disorders. Studies have shown an intrinsic link between type I IFN therapy and severe neuropsychiatric disorders, mainly major depression (149). Considerable evidence suggests that type I IFN is associated with psychiatric disorders, and that the production of type I IFNs as a result of TLR4 induced IRF3 activation and TLR7 induced IRF7 activation may be closely associated with IFN-mediated psychiatric disorders (93). Infant onset of RNaseT2-deficient leukoencephalopathy leads to cystic brain injury, multifocal white matter changes, brain atrophy, and severe psychomotor impairment. Rnaset2-/- mice exhibit upregulation of ISGs and IFNAR-I-dependent neuroinflammation (150). HIV-associated neurocognitive disorders (HAND) also show an upregulation of ISG15 (151). In addition, ISGs may be involved in neuronal and synaptic regulation. The upregulation of inflammatory cytokines induced by maternal immune activation (MIA) promotes ISG15 expression in the offspring’s brain, leading to neuronal dendritic lesions and depression-like behavior (56). In the hippocampus, ISG15 and Ubiquitin-specific peptidase 18 (USP18) mediate IFN-α-induced reduction in neurogenesis through upregulation of ISGylation-associated proteins UBA7, UBE2L6, and HERC5 (152). Adolescent mice injected with the viral mimic poly I:C had significantly increased expression of ISGs (IFIT2, PRKR, MX2, and IRF7) in the hippocampal dentate gyrus and exhibited behavioral deficits of impulse inhibition and impaired recognition of novel objects (58). In summary, upregulation of ISGs in the CNS may regulate various cellular functions and processes, such as neuronal survival and synaptic pruning, in a brain region-dependent manner (94).
To date, type I IFNs remain the most potent, broad-spectrum antiviral agents. The treatment of IFN to cells induces a large set of ISGs that can prevent infection with many viral pathogens. There are currently more than 300 recognized ISGs, but the exact mechanisms of inhibiting virus replication have been identified only in a small subset of ISGs. Detailed mechanistic investigation of the functions of individual ISGs is complicated by the difficulty in dissecting particular processes in virus replication independently of one another. The responses of individual ISGs to different viral infections in different organs may also vary, and there are limited in-depth studies on the effects of individual ISGs on the nervous system. With continuous research in this field, the role of ISGs and their mechanisms are being elucidated. The following subsections summarize several representative ISGs found in viral infections and neurological diseases in recent years.
As mentioned above, Wang et al. found that ISG15 positively correlated with the degree of neuronal injury in animal models of ALS with no obvious signs of inflammation, a model cerebral ischemia, a model of cortical shock-induced brain injury, and a mouse model of chronic neuronal injury caused by the viral protein HIV gp120, with low levels of ISG15 in unaffected areas and high levels of ISG15 in neuronal injury areas. In patients with ALS, elevated levels of ISG15 and ISGylation in the CSF were significantly higher in post-traumatic brain injury ALS compared with those in non-traumatic brain injury ALS (53). It is suggested that ISG15 may be a reliable biomarker of pathological changes in the CNS (54). In cells with mutations in ATM kinase, conjugated ISG15, but not the free form, antagonizes targeted degradation of the ubiquitin pathway, which may lead to progressive neurodegeneration in A-T patients (153). C-Type Lectin Domain Containing 16A (CLEC16A) has been shown to function in autophagy/mitochondrial autophagy and Clec16 knockdown leads to an inflammatory neurodegenerative phenotype similar to spinal cerebellar ataxia in mice. In the whole-body inducible knockout of Clec16a mice model, Clec16a expression was negatively correlated with IGS15 expression, and the expression of ISG15 in neuronal tissues was upregulated, suggesting that ISG15 may be a link between Clec16a and downstream autoimmune inflammatory processes (154). ISG15 binds to a number of key proteins and affects various pathophysiological processes in the CNS. After traumatic brain injury, ISG15 is rapidly elevated and binds covalently to myosin light chain kinase (MLCK), which may promote phosphorylation of the myosin light chain by MLCK and conversion of F-actin to stress actin, which is involved in BBB destruction by disrupting TJs, thus aggravating brain edema (155). IFN-β inhibits the MAPK signaling pathway and attenuates mechanical nociceptive hyperalgesia by elevating both free and conjugated ISG15, an effect that is increased in ubp43-/- mice lacking the key de-binding enzyme (156). Upregulation of MIA-induced inflammatory cytokines promotes ISG15 expression in the offspring brain, leading to neuronal dendritic lesions and depressive-like behavior through a mechanism of ISG15 inhibiting the ubiquitination of Rap2A by NEDD4 (an E3 ubiquitin ligase that ubiquitously inhibits Rap2A activity, leading to dendritic growth and depoliticization), thus inducing Rap2A accumulation (56). In contrast, upregulation of NEDD4 abolishes ISG15-induced dendritic damage (56). In a model of acute inflammation established by LPS-stimulated microglia, increased ISGylation maintained the stability of STAT1 and promoted a sustained immune response during inflammation (157).
Interferon-induced proteins with tetratricopeptide repeats (IFIT) are prominent ISGs, induced following type I IFN- or IRF3-dependent signaling, contribute to the antiviral defense of cells by binding directly to viral RNA or by binding to eukaryotic initiation factor 3 (eIF3) and preventing eIF3 from initiating the viral translational process (158). The human IFIT gene family generally consists of four members: IFIT1, IFIT2 (ISG54, p54), IFIT3 (ISG60, p60), and IFIT5 (ISG58, p58), whereas the mouse IFIT gene family encodes for three relevant genes: IFIT1, IFIT2 and IFIT3 (ISG49, p49), which are induced during IFN signaling pathway, viral infection or other PAMP recognition and have critical roles in host antiviral defense (159, 160). IFIT1 had an antiviral effect in human cytomegalovirus (HCMV)-infected human astrocytes isolated from the fetal brain, but not in HELFs (human embryonic lung fibroblast cells) (69). Recent studies have shown that IFIT2 primarily limits viral infection and protects mice from severe morbidity and mortality following infection with RABV (75), lethal VSV (67, 70), WNV (71), and Sendai virus (SeV) (72). IFIT2 acts as an antiviral in the CNS in several ways. Both VSV and EMCV infections cause neuroinvasive disease and induce IFN-β, IFIT1, and IFIT2 in the brain. However, IFIT2 only prevents VSV invasion of the brain and not EMCV invasion of the brain, suggesting that the antiviral response of IFIT2 in the CNS is virus-specific (67). In Ifit2-/- mice, effective VSV viral replication was restricted to the brain, and the absence of IFIT2 did not affect viral titers in other organs such as the liver or lungs, suggesting that IFIT2 can limit VSV invasion of the nervous system (67). In WNV-infected CNS, viral titers were higher in Ifit2-/- mice compared with those in WT mice only in the OB, cerebral cortex, brainstem, cerebellum, and spinal cord, and in cells with knockdown of IFIT2, increased WNV infection was observed only in cerebellar granule cells and dendritic cells, but not in macrophages, fibroblasts, or cortical neurons (71). Overall, these data suggest that IFIT2 has a crucial role in limiting viral infection in specific regions of the brain and in specific cell types. In experiments with RABV infection of the CNS, IFIT2 exerted antiviral effects predominantly at the level of viral replication and not as a mechanism to restrict viral entry/exit or transport of RABV particles via axons (75). Furthermore, IFIT2 can be involved in antiviral response by inducing and enhancing innate immunity. In neurotropic coronavirus MHV-RSA59 infection, IFIT2 promoted viral clearance by facilitating microglia activation and recruitment of NK1.1 and CD4 T cells to the brain (73). Further studies have shown that IFIT2 and IFIT3 function in a complementary and synergistic manner to restrict RABV in mouse-derived neuroblastoma cells (74). In MHV-induced encephalitis, IFIT2 is a positive regulator of IFNα/β expression rather than a direct antiviral mediator, with Ifit2-/- mice showing significantly reduced expression of IFN-α/β and the downstream ISG mRNAs (Ifit1, Isg15, and Pkr) (52).
The IFN-inducible transmembrane proteins (IFITMs) form a small family of IFN-inducible proteins and have two transmembrane structural domains. The IFITMs were shown to inhibit the cellular entry step of many enveloped viruses such as influenza A, dengue, Ebola, and SARS coronavirus (161). The human IFITM family consists of four proteins, IFITM1, IFITM2, IFITM3 and IFITM5, located on chromosome 11, among them IFITM1, IFITM2 and IFITM3 are well-known ISG proteins (162). IFITMs disrupt the entry of multiple enveloped viruses, and play a role in the transport of viral particles to lysosomes for degradation (163). SARS-CoV-2 infection was recently shown to increase IFITM3 protein expression (63), and in severe SARS-CoV-2 cases, IFITM3 levels are elevated in the frontal cortex and choroid plexus (65). IFITM3 may prevent pathogenesis by limiting early replication and transmission of α-virus in the brain and spinal cord (164). In patients with Rasmussen encephalitis (RE) caused by infection with HCMV viruses, IFITM3 was detected in the neurons of brain tissue, and there was colocalization of HCMV and IFITM3, suggesting that HCMV infection may induce IFITM3 expression in neurons and that IFITM3 can effectively inhibit HCMV infection and participate in the immune response to HCMV infection in RE brain tissue (64). Further studies found that the IFITM3 single nucleotide polymorphism (SNP) rs12252 correlated with the severity of disease caused by viral infection (165–167). The rs12252-C mutant protein IFITM3 ND21 was not flexible enough to effectively prevent the fusion of the virus with the endocytic membrane, which in turn reduced the ability of the immune system to defend against viral infection. Wang et al. found that subjects carrying IFITM3 rs12252 CC genotype were at increased risk of developing RE and were associated with rapid progression of RE disease (64). In addition, the rs12252-C allele was recently reported to be associated with disease severity in patients with SARS-CoV-2 (168). In conclusion, the IFITM3 rs12252-C allele is strongly associated with the severity of some viral infectious diseases (169).
IFITM3 mRNA expression in the cortex and hippocampus is significantly positively correlated with age (ranging from 20 years to 70 years) in humans, according to genotype-tissue expression cohorts (66). IFITM3 protein levels, Aβ production (Aβ42 and Aβ40), and the amount of active IFITM3-γ-secretase were increased in the aging WT mouse brains (66). IFITM3 expression is upregulated in astrocytes and microglia in the brains of the 5xFAD Alzheimer’s disease mouse model, and IFITM3 mRNA and IFITM3 protein are expressed in neurons (66). Pro-inflammatory cytokines (IFN-α or IFN-γ, IL-6, and IL-1β) increase Aβ production in neurons and astrocytes by increasing the formation of IFITM3 protein and active IFITM3-γ-secretase complexes (66). Recently, SARS-CoV-2 has been reported to increase IFITM3 protein (63), and IFITM3 levels are elevated in the frontal cortex and choroid plexus in severe SARS-CoV-2 cases (65). Hur et al. concluded that different inflammatory conditions, such as viral infection and aging, can induce the release of pro-inflammatory cytokines from astrocytes and microglia, which in turn elevate the expression of IFITM3 in neurons and astrocytes, and IFITM3 binds to active γ-secretase complexes, increasing Aβ production and increasing the risk of AD (66).
This review highlights the ISGs involved in resisting the neurotropic viral invasion of the CNS and the mode of activation of these ISGs in viral-infected CNS cells. Furthermore, the expression characteristics of ISGs in the development of CNS disorders are discussed. At last, we summarize in detail several mechanisms of action of individual ISGs in the CNS that have been more studied in recent years. The IFN signal pathways induce hundreds of ISGs to exert antiviral and other physiopathological effects. ISGs are a large family, and many more are still waiting to be identified. Most studies have focused on ISG as a marker of activation of the innate immune response to IFN, whereas the mechanisms of ISG in the pathophysiological response of the CNS remain unclear and need to be investigated in depth. Thus this review summarizes the current research on ISGs in CNS and indicates possible directions for future research. With the development of technologies such as CRISPR-Cas9 gene editing as well as genome-wide RNA-seq and deep proteomics (170), research on the antiviral effects of individual ISG, as well as its other functions in CNS diseases, is expected to evolve rapidly. An improved understanding of the functions of individual ISGs will facilitate the development of ISG-based therapies. Consequently, ISGs may exhibit promise as potential clinical biomarkers as well as therapeutic targets.
RL, YY, CB, and WD conceived the perspective of the work. RL, HL, YY, CB, and WD drafted the manuscript. HL, XL, and CL designed the figure. YZ, SG and JX assisted in collecting and organizing the literature. All authors revised and approved the final version of the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (31871031, 32170968, WD), the Fund of Key Laboratory of Medical Electrophysiology in 2021 (KeyME-2021-01, YY), and China Postdoctoral Science Foundation (2021M692700, YY).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Zhou J-H, Wang Y-N, Chang Q-Y, Ma P, Hu Y, Cao X. Type III interferons in viral infection and antiviral immunity. Cell Physiol Biochem Int J Exp Cell Physiol Biochem Pharmacol (2018) 51:173–85. doi: 10.1159/000495172
2. Lazear HM, Schoggins JW, Diamond MS. Shared and distinct functions of type I and type III interferons. Immunity (2019) 50:907–23. doi: 10.1016/j.immuni.2019.03.025
3. Parker BS, Rautela J, Hertzog PJ. Antitumour actions of interferons: implications for cancer therapy. Nat Rev Cancer (2016) 16:131–44. doi: 10.1038/nrc.2016.14
4. Sprooten J, Agostinis P, Garg AD. Type I interferons and dendritic cells in cancer immunotherapy. Int Rev Cell Mol Biol (2019) 348:217–62. doi: 10.1016/bs.ircmb.2019.06.001
5. Pestka S, Krause CD, Walter MR. Interferons, interferon-like cytokines, and their receptors. Immunol Rev (2004) 202:8–32. doi: 10.1111/j.0105-2896.2004.00204.x
6. Oritani K, Kanakura Y. IFN-? limitin: a member of type I IFN with mild lympho-myelosuppression. J Cell Mol Med (2005) 9:244–54. doi: 10.1111/j.1582-4934.2005.tb00353.x
7. Schoggins JW. Recent advances in antiviral interferon-stimulated gene biology. F1000Research (2018) 7:309. doi: 10.12688/f1000research.12450.1
8. Wells AI, Coyne CB. Type III interferons in antiviral defenses at barrier surfaces. Trends Immunol (2018) 39:848–58. doi: 10.1016/j.it.2018.08.008
9. Kang S, Brown HM, Hwang S. Direct antiviral mechanisms of interferon-gamma. Immune Netw (2018) 18:e33. doi: 10.4110/in.2018.18.e33
10. Billiau A, Matthys P. Interferon-γ: A historical perspective. Cytokine Amp Growth Factor Rev (2009) 20:97–113. doi: 10.1016/j.cytogfr.2009.02.004
11. Lazear HM, Nice TJ, Diamond MS. Interferon-λ: Immune functions at barrier surfaces and beyond. Immunity (2015) 43:15–28. doi: 10.1016/j.immuni.2015.07.001
12. Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol (2014) 32:513–45. doi: 10.1146/annurev-immunol-032713-120231
13. Schoggins JW, Rice CM. Interferon-stimulated genes and their antiviral effector functions. Curr Opin Virol (2011) 1:519–25. doi: 10.1016/j.coviro.2011.10.008
14. Odendall C, Kagan JC. The unique regulation and functions of type III interferons in antiviral immunity. Curr Opin Virol (2015) 12:47–52. doi: 10.1016/j.coviro.2015.02.003
15. Stark GR, Darnell JEJ. The JAK-STAT pathway at twenty. Immunity (2012) 36:503–14. doi: 10.1016/j.immuni.2012.03.013
16. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol (2014) 14:36–49. doi: 10.1038/nri3581
17. Haque SJ, Williams BR. Identification and characterization of an interferon (IFN)-stimulated response element-IFN-stimulated gene factor 3-independent signaling pathway for IFN-alpha. J Biol Chem (1994) 269:19523–9. doi: 10.1016/S0021-9258(17)32200-7
18. Au-Yeung N, Horvath CM. Transcriptional and chromatin regulation in interferon and innate antiviral gene expression. Cytokine Growth Factor Rev (2018) 44:11–7. doi: 10.1016/j.cytogfr.2018.10.003
19. Platanitis E, Decker T. Regulatory networks involving STATs, IRFs, and NFκB in inflammation. Front Immunol (2018) 9:2542. doi: 10.3389/fimmu.2018.02542
20. Orzalli MH, Smith A, Jurado KA, Iwasaki A, Garlick JA, Kagan JC. An antiviral branch of the IL-1 signaling pathway restricts immune-evasive virus replication. Mol Cell (2018) 71:825–40.e6. doi: 10.1016/j.molcel.2018.07.009
21. Rubio D, Xu R-H, Remakus S, Krouse TE, Truckenmiller ME, Thapa RJ, et al. Crosstalk between the type 1 interferon and nuclear factor kappa b pathways confers resistance to a lethal virus infection. Cell Host Microbe (2013) 13:701–10. doi: 10.1016/j.chom.2013.04.015
22. Green R, Ireton RC, Gale MJ. Interferon-stimulated genes: new platforms and computational approaches. Mamm Genome Off J Int Mamm Genome Soc (2018) 29:593–602. doi: 10.1007/s00335-018-9755-6
23. Wang W, Xu L, Su J, Peppelenbosch MP, Pan Q. Transcriptional regulation of antiviral interferon-stimulated genes. Trends Microbiol (2017) 25:573–84. doi: 10.1016/j.tim.2017.01.001
24. Klotz D, Gerhauser I. Interferon-stimulated genes-mediators of the innate immune response during canine distemper virus infection. Int J Mol Sci (2019) 20:1620. doi: 10.3390/ijms20071620
25. Marié I, Durbin JE, Levy DE. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO J (1998) 17:6660–9. doi: 10.1093/emboj/17.22.6660
26. Fujita T, Reis LF, Watanabe N, Kimura Y, Taniguchi T, Vilcek J. Induction of the transcription factor IRF-1 and interferon-beta mRNAs by cytokines and activators of second-messenger pathways. Proc Natl Acad Sci USA (1989) 86:9936–40. doi: 10.1073/pnas.86.24.9936
27. Goubau D, Deddouche S, Reis e Sousa C. Cytosolic sensing of viruses. Immunity (2013) 38:855–69. doi: 10.1016/j.immuni.2013.05.007
28. Sadler AJ, Williams BRG. Interferon-inducible antiviral effectors. Nat Rev Immunol (2008) 8:559–68. doi: 10.1038/nri2314
29. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140:805–20. doi: 10.1016/j.cell.2010.01.022
30. Imaizumi T, Numata A, Yano C, Yoshida H, Meng P, Hayakari R, et al. ISG54 and ISG56 are induced by TLR3 signaling in U373MG human astrocytoma cells: possible involvement in CXCL10 expression. Neurosci Res (2014) 84:34–42. doi: 10.1016/j.neures.2014.03.001
31. Kim M-J, Hwang S-Y, Imaizumi T, Yoo J-Y. Negative feedback regulation of RIG-I-Mediated antiviral signaling by interferon-induced ISG15 conjugation. J Virol (2008) 82:1474–83. doi: 10.1128/JVI.01650-07
32. Du Y, Duan T, Feng Y, Liu Q, Lin M, Cui J, et al. LRRC25 inhibits type I IFN signaling by targeting ISG15-associated RIG-I for autophagic degradation. EMBO J (2018) 37:351–66. doi: 10.15252/embj.201796781
33. McGavern DB, Kang SS. Illuminating viral infections in the nervous system. Nat Rev Immunol (2011) 11:318–29. doi: 10.1038/nri2971
34. Kochs G, Bauer S, Vogt C, Frenz T, Tschopp J, Kalinke U, et al. Thogoto virus infection induces sustained type I interferon responses that depend on RIG-I-Like helicase signaling of conventional dendritic cells. J Virol (2010) 84:12344–50. doi: 10.1128/JVI.00931-10
35. Mrkic B, Pavlovic J, Rülicke T, Volpe P, Buchholz CJ, Hourcade D, et al. Measles virus spread and pathogenesis in genetically modified mice. J Virol (1998) 72:7420–7. doi: 10.1128/JVI.72.9.7420-7427.1998
36. Dhondt KP, Mathieu C, Chalons M, Reynaud JM, Vallve A, Raoul H, et al. Type I interferon signaling protects mice from lethal henipavirus infection. J Infect Dis (2013) 207:142–51. doi: 10.1093/infdis/jis653
37. Ryman KD, Klimstra WB, Nguyen KB, Biron CA, Johnston RE. Alpha/Beta interferon protects adult mice from fatal sindbis virus infection and is an important determinant of cell and tissue tropism. J Virol (2000) 74:3366–78. doi: 10.1128/JVI.74.7.3366-3378.2000
38. Casrouge A, Zhang S-Y, Eidenschenk C, Jouanguy E, Puel A, Yang K, et al. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science (2006) 314:308–12. doi: 10.1126/science.1128346
39. Herman M, Ciancanelli M, Ou Y-H, Lorenzo L, Klaudel-Dreszler M, Pauwels E, et al. Heterozygous TBK1 mutations impair TLR3 immunity and underlie herpes simplex encephalitis of childhood. J Exp Med (2012) 209:1567–82. doi: 10.1084/jem.20111316
40. Pérez de Diego R, Sancho-Shimizu V, Lorenzo L, Puel A, Plancoulaine S, Picard C, et al. Human TRAF3 adaptor molecule deficiency leads to impaired toll-like receptor 3 response and susceptibility to herpes simplex encephalitis. Immunity (2010) 33:400–11. doi: 10.1016/j.immuni.2010.08.014
41. Sancho-Shimizu V, Pérez de Diego R, Lorenzo L, Halwani R, Alangari A, Israelsson E, et al. Herpes simplex encephalitis in children with autosomal recessive and dominant TRIF deficiency. J Clin Invest (2011) 121:4889–902. doi: 10.1172/JCI59259
42. Ma C, Li S, Hu Y, Ma Y, Wu Y, Wu C, et al. AIM2 controls microglial inflammation to prevent experimental autoimmune encephalomyelitis. J Exp Med (2021) 218:e20201796. doi: 10.1084/jem.20201796
43. Ejlerskov P, Hultberg JG, Wang J, Carlsson R, Ambjørn M, Kuss M, et al. Lack of neuronal IFN-β-IFNAR causes lewy body- and parkinson’s disease-like dementia. Cell (2015) 163:324–39. doi: 10.1016/j.cell.2015.08.069
44. Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature (2018) 561:258–62. doi: 10.1038/s41586-018-0448-9
45. McCauley ME, O’Rourke JG, Yáñez A, Markman JL, Ho R, Wang X, et al. C9orf72 in myeloid cells suppresses STING-induced inflammation. Nature (2020) 585:96–101. doi: 10.1038/s41586-020-2625-x
46. Roy ER, Wang B, Wan Y-W, Chiu G, Cole A, Yin Z, et al. Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J Clin Invest (2020) 130:1912–30. doi: 10.1172/JCI133737
47. Baik SH, Kang S, Lee W, Choi H, Chung S, Kim J-I, et al. A breakdown in metabolic reprogramming causes microglia dysfunction in alzheimer’s disease. Cell Metab (2019) 30:493–507.e6. doi: 10.1016/j.cmet.2019.06.005
48. Magusali N, Graham AC, Piers TM, Panichnantakul P, Yaman U, Shoai M, et al. A genetic link between risk for alzheimer’s disease and severe COVID-19 outcomes via the OAS1 gene. Brain (2021) 144:3727–41. doi: 10.1093/brain/awab337
49. Li L, Ulrich R, Baumgärtner W, Gerhauser I. Interferon-stimulated genes-essential antiviral effectors implicated in resistance to theiler’s virus-induced demyelinating disease. J Neuroinflamm (2015) 12:242. doi: 10.1186/s12974-015-0462-x
50. Sun L, Wang X, Zhou Y, Zhou R-H, Ho W-Z, Li J-L. Exosomes contribute to the transmission of anti-HIV activity from TLR3-activated brain microvascular endothelial cells to macrophages. Antiviral Res (2016) 134:167–71. doi: 10.1016/j.antiviral.2016.07.013
51. Liu H, Zhou R-H, Liu Y, Guo L, Wang X, Hu W-H, et al. HIV Infection suppresses TLR3 activation-mediated antiviral immunity in microglia and macrophages. Immunology (2020) 160:269–79. doi: 10.1111/imm.13181
52. Butchi NB, Hinton DR, Stohlman SA, Kapil P, Fensterl V, Sen GC, et al. Ifit2 deficiency results in uncontrolled neurotropic coronavirus replication and enhanced encephalitis via impaired alpha/beta interferon induction in macrophages. J Virol (2014) 88:1051–64. doi: 10.1128/JVI.02272-13
53. Schwartzenburg J, Juncker M, Reed R, Desai S. Increased ISGylation in cases of TBI-exposed ALS veterans. J Neuropathol Exp Neurol (2019) 78:209–18. doi: 10.1093/jnen/nly129
54. Wang R-G, Kaul M, Zhang D-X. Interferon-stimulated gene 15 as a general marker for acute and chronic neuronal injuries. Sheng Li Xue Bao (2012) 64:577–83.
55. Kim CD, Reed RE, Juncker MA, Fang Z, Desai SD. Evidence for the deregulation of protein turnover pathways in atm-deficient mouse cerebellum: An organotypic study. J Neuropathol Exp Neurol (2017) 76:578–84. doi: 10.1093/jnen/nlx038
56. Hu Y, Hong X-Y, Yang X-F, Ma R-H, Wang X, Zhang J-F, et al. Inflammation-dependent ISG15 upregulation mediates MIA-induced dendrite damages and depression by disrupting NEDD4/Rap2A signaling. Biochim Biophys Acta Mol Basis Dis (2019) 1865:1477–89. doi: 10.1016/j.bbadis.2019.02.020
57. Wang BX, Grover SA, Kannu P, Yoon G, Laxer RM, Yeh EA, et al. Interferon-stimulated gene expression as a preferred biomarker for disease activity in aicardi-goutières syndrome. J Interferon Cytokine Res Off J Int Soc Interferon Cytokine Res (2017) 37:147–52. doi: 10.1089/jir.2016.0117
58. Lukasz B, O’Sullivan NC, Loscher JS, Pickering M, Regan CM, Murphy KJ. Peripubertal viral-like challenge and social isolation mediate overlapping but distinct effects on behaviour and brain interferon regulatory factor 7 expression in the adult wistar rat. Brain Behav Immun (2013) 27:71–9. doi: 10.1016/j.bbi.2012.09.011
59. Bansode YD, Chattopadhyay D, Saha B. Transcriptomic analysis of interferon response in toll-like receptor 2 ligand-treated and herpes simplex virus 1-infected neurons and astrocytes. Viral Immunol (2021) 34:256–66. doi: 10.1089/vim.2020.0238
60. Marshall C, Clark ZT, Minckler MR. Aseptic viral meningitis secondary to herpes simplex virus 2 genital infection. Cureus (2021) 13:14535. doi: 10.7759/cureus.14535
61. Lindqvist R, Kurhade C, Gilthorpe JD, Överby AK. Cell-type- and region-specific restriction of neurotropic flavivirus infection by viperin. J Neuroinflamm (2018) 15:80. doi: 10.1186/s12974-018-1119-3
62. Crick PJ, Griffiths WJ, Zhang J, Beibel M, Abdel-Khalik J, Kuhle J, et al. Reduced plasma levels of 25-hydroxycholesterol and increased cerebrospinal fluid levels of bile acid precursors in multiple sclerosis patients. Mol Neurobiol (2017) 54:8009–20. doi: 10.1007/s12035-016-0281-9
63. Hachim MY, Al Heialy S, Hachim IY, Halwani R, Senok AC, Maghazachi AA, et al. Interferon-induced transmembrane protein (IFITM3) is upregulated explicitly in SARS-CoV-2 infected lung epithelial cells. Front Immunol (2020) 11:1372. doi: 10.3389/fimmu.2020.01372
64. Wang Y-S, Luo Q-L, Guan Y-G, Fan D-Y, Luan G-M, Jing A. HCMV infection and IFITM3 rs12252 are associated with rasmussen’s encephalitis disease progression. Ann Clin Transl Neurol (2021) 8:558–70. doi: 10.1002/acn3.51289
65. Yang AC, Kern F, Losada PM, Agam MR, Maat CA, Schmartz GP, et al. Dysregulation of brain and choroid plexus cell types in severe COVID-19. Nature (2021) 595:565–71. doi: 10.1038/s41586-021-03710-0
66. Hur J-Y, Frost GR, Wu X, Crump C, Pan SJ, Wong E, et al. The innate immunity protein IFITM3 modulates γ-secretase in alzheimer’s disease. Nature (2020) 586:735–40. doi: 10.1038/s41586-020-2681-2
67. Fensterl V, Wetzel JL, Ramachandran S, Ogino T, Stohlman SA, Bergmann CC, et al. Interferon-induced Ifit2/ISG54 protects mice from lethal VSV neuropathogenesis. PloS Pathog (2012) 8:13. doi: 10.1371/journal.ppat.1002712
68. Sharma N, Verma R, Kumawat KL, Basu A, Singh SK. miR-146a suppresses cellular immune response during Japanese encephalitis virus JaOArS982 strain infection in human microglial cells. J Neuroinflamm (2015) 12:30. doi: 10.1186/s12974-015-0249-0
69. Zhang L, Wang B, Li L, Qian D-M, Yu H, Xue M-L, et al. Antiviral effects of IFIT1 in human cytomegalovirus-infected fetal astrocytes. J Med Virol (2017) 89:672–84. doi: 10.1002/jmv.24674
70. Fensterl V, Wetzel JL, Sen GC. Interferon-induced protein Ifit2 protects mice from infection of the peripheral nervous system by vesicular stomatitis virus. J Virol (2014) 88:10303–11. doi: 10.1128/JVI.01341-14
71. Cho H, Shrestha B, Sen GC, Diamond MS. A role for Ifit2 in restricting West Nile virus infection in the brain. J Virol (2013) 87:8363–71. doi: 10.1128/JVI.01097-13
72. Wetzel JL, Fensterl V, Sen GC. Sendai Virus pathogenesis in mice is prevented by Ifit2 and exacerbated by interferon. J Virol (2014) 88:13593–601. doi: 10.1128/JVI.02201-14
73. Das Sarma J, Burrows A, Rayman P, Hwang M-H, Kundu S, Sharma N, et al. Ifit2 deficiency restricts microglial activation and leukocyte migration following murine coronavirus (m-CoV) CNS infection. PloS Pathog (2020) 16:e1009034. doi: 10.1371/journal.ppat.1009034
74. Chai B, Tian D, Zhou M, Tian B, Yuan Y, Sui B, et al. Murine Ifit3 restricts the replication of rabies virus both in vitro and in vivo. J Gen Virol (2021) 102:001619. doi: 10.1099/jgv.0.001619
75. Davis BM, Fensterl V, Lawrence TM, Hudacek AW, Sen GC, Schnell MJ. Ifit2 is a restriction factor in rabies virus pathogenicity. J Virol (2017) 91:00889-17. doi: 10.1128/JVI.00889-17
76. Lucas TM, Richner JM, Diamond MS. The interferon-stimulated gene Ifi27l2a restricts West Nile virus infection and pathogenesis in a cell-type- and region-specific manner. J Virol (2015) 90:2600–15. doi: 10.1128/JVI.02463-15
77. De Masi R, Orlando S, Bagordo F, Grassi T. IFP35 is a relevant factor in innate immunity, multiple sclerosis, and other chronic inflammatory diseases: A review. Biology (2021) 10:1325. doi: 10.3390/biology10121325
78. Ransohoff RM, Brown MA. Innate immunity in the central nervous system. J Clin Invest (2012) 122:1164–71. doi: 10.1172/JCI58644
79. Li F, Wang Y, Yu L, Cao S, Wang K, Yuan J, et al. Viral infection of the central nervous system and neuroinflammation precede blood-brain barrier disruption during Japanese encephalitis virus infection. J Virol (2015) 89:5602–14. doi: 10.1128/JVI.00143-15
80. Chen Z, Li G. Immune response and blood-brain barrier dysfunction during viral neuroinvasion. Innate Immun (2021) 27:109–17. doi: 10.1177/1753425920954281
81. Mustafá YM, Meuren LM, Coelho SVA, de Arruda LB. Pathways exploited by flaviviruses to counteract the blood-brain barrier and invade the central nervous system. Front Microbiol (2019) 10:525. doi: 10.3389/fmicb.2019.00525
82. Lazear HM, Daniels BP, Pinto AK, Huang AC, Vick SC, Doyle SE, et al. Interferon-λ restricts West Nile virus neuroinvasion by tightening the blood-brain barrier. Sci Transl Med (2015) 7:284ra59. doi: 10.1126/scitranslmed.aaa4304
83. Li Y, Zhao L, Luo Z, Zhang Y, Lv L, Zhao J, et al. Interferon-λ attenuates rabies virus infection by inducing interferon-stimulated genes and alleviating neurological inflammation. Viruses (2020) 12:405. doi: 10.3390/v12040405
84. Dozio V, Sanchez J-C. Profiling the proteomic inflammatory state of human astrocytes using DIA mass spectrometry. J Neuroinflamm (2018) 15:331. doi: 10.1186/s12974-018-1371-6
85. Li J, Wang Y, Wang X, Ye L, Zhou Y, Persidsky Y, et al. Immune activation of human brain microvascular endothelial cells inhibits HIV replication in macrophages. Blood (2013) 121:2934–42. doi: 10.1182/blood-2012-08-450353
86. Kalinke U, Bechmann I, Detje CN. Host strategies against virus entry via the olfactory system. Virulence (2011) 2:367–70. doi: 10.4161/viru.2.4.16138
87. Singh H, Koury J, Kaul M. Innate immune sensing of viruses and its consequences for the central nervous system. Viruses (2021) 13:170. doi: 10.3390/v13020170
88. Iwasaki A. Immune regulation of antibody access to neuronal tissues. Trends Mol Med (2017) 23:227–45. doi: 10.1016/j.molmed.2017.01.004
89. Detje CN, Lienenklaus S, Chhatbar C, Spanier J, Prajeeth CK, Soldner C, et al. Upon intranasal vesicular stomatitis virus infection, astrocytes in the olfactory bulb are important interferon beta producers that protect from lethal encephalitis. J Virol (2015) 89:2731–8. doi: 10.1128/JVI.02044-14
90. van den Pol AN, Ding S, Robek MD. Long-distance interferon signaling within the brain blocks virus spread. J Virol (2014) 88:3695–704. doi: 10.1128/JVI.03509-13
91. Wheeler DL, Sariol A, Meyerholz DK, Perlman S. Microglia are required for protection against lethal coronavirus encephalitis in mice. J Clin Invest (2018) 128:931–43. doi: 10.1172/JCI97229
92. Trottier MD, Lyles DS, Reiss CS. Peripheral, but not central nervous system, type I interferon expression in mice in response to intranasal vesicular stomatitis virus infection. J Neurovirol (2007) 13:433–45. doi: 10.1080/13550280701460565
93. Thomson CA, McColl A, Cavanagh J, Graham GJ. Peripheral inflammation is associated with remote global gene expression changes in the brain. J Neuroinflamm (2014) 11:73. doi: 10.1186/1742-2094-11-73
94. Aw E, Zhang Y, Carroll M. Microglial responses to peripheral type 1 interferon. J Neuroinflamm (2020) 17:340. doi: 10.1186/s12974-020-02003-z
95. Sorgeloos F, Kreit M, Hermant P, Lardinois C, Michiels T. Antiviral type I and type III interferon responses in the central nervous system. Viruses (2013) 5:834–57. doi: 10.3390/v5030834
96. Butchi NB, Du M, Peterson KE. Interactions between TLR7 and TLR9 agonists and receptors regulate innate immune responses by astrocytes and microglia. Glia (2009) 58:650–64. doi: 10.1002/glia.20952
97. Carroll JA, Race B, Williams K, Striebel JF, Chesebro B. Innate immune responses after stimulation with toll-like receptor agonists in ex vivo microglial cultures and an in vivo model using mice with reduced microglia. J Neuroinflamm (2021) 18:194. doi: 10.1186/s12974-021-02240-w
98. He N, Qu Y-J, Li D-Y, Yue S-W. RIP3 inhibition ameliorates chronic constriction injury-induced neuropathic pain by suppressing JNK signaling. Aging (2021) 13:24417–31. doi: 10.18632/aging.203691
99. Huber AK, Duncker PC, Irani DN. Immune responses to non-tumor antigens in the central nervous system. Front Oncol (2014) 4:328. doi: 10.3389/fonc.2014.00328
100. Chen Z, Zhong D, Li G. The role of microglia in viral encephalitis: a review. J Neuroinflamm (2019) 16:76. doi: 10.1186/s12974-019-1443-2
101. Paludan SR. Activation and regulation of DNA-driven immune responses. Microbiol Mol Biol Rev MMBR (2015) 79:225–41. doi: 10.1128/MMBR.00061-14
102. Rodríguez AM, Rodríguez J, Giambartolomei GH. Microglia at the crossroads of pathogen-induced neuroinflammation. ASN Neuro (2022) 14:175909142211045. doi: 10.1177/17590914221104566
103. Stonedahl S, Leser JS, Clarke P, Tyler KL. Depletion of microglia in an Ex vivo brain slice culture model of West Nile virus infection leads to increased viral titers and cell death. Microbiol Spectr (2022) 10:e00685–22. doi: 10.1128/spectrum.00685-22
104. Fekete R, Cserép C, Lénárt N, Tóth K, Orsolits B, Martinecz B, et al. Microglia control the spread of neurotropic virus infection via P2Y12 signalling and recruit monocytes through P2Y12-independent mechanisms. Acta Neuropathol (Berl) (2018) 136:461–82. doi: 10.1007/s00401-018-1885-0
105. Drokhlyansky E, Göz Aytürk D, Soh TK, Chrenek R, O’Loughlin E, Madore C, et al. The brain parenchyma has a type I interferon response that can limit virus spread. Proc Natl Acad Sci (2017) 114:E95–104. doi: 10.1073/pnas.1618157114
106. Malikov V, Meade N, Simons LM, Hultquist JF, Naghavi MH. FEZ1 phosphorylation regulates HSPA8 localization and interferon-stimulated gene expression. Cell Rep (2022) 38:110396. doi: 10.1016/j.celrep.2022.110396
107. Simmons LJ, Surles-Zeigler MC, Li Y, Ford GD, Newman GD, Ford BD. Regulation of inflammatory responses by neuregulin-1 in brain ischemia and microglial cells in vitro involves the NF-kappa b pathway. J Neuroinflamm (2016) 13:237. doi: 10.1186/s12974-016-0703-7
108. Manangeeswaran M, Ireland DDC, Verthelyi D. Zika (PRVABC59) infection is associated with T cell infiltration and neurodegeneration in CNS of immunocompetent neonatal C57Bl/6 mice. PloS Pathog (2016) 12:e1006004. doi: 10.1371/journal.ppat.1006004
109. Bindocci E, Savtchouk I, Liaudet N, Becker D, Carriero G, Volterra A. Three-dimensional Ca 2+ imaging advances understanding of astrocyte biology. Science (2017) 356:eaai8185. doi: 10.1126/science.aai8185
110. Yoon H, Walters G, Paulsen AR, Scarisbrick IA. Astrocyte heterogeneity across the brain and spinal cord occurs developmentally, in adulthood and in response to demyelination. PloS One (2017) 12:e0180697. doi: 10.1371/journal.pone.0180697
111. DePaula-Silva AB, Bell LA, Wallis GJ, Wilcox KS. Inflammation unleashed in viral-induced epileptogenesis. Epilepsy Curr (2021) 21:433–40. doi: 10.1177/15357597211040939
112. Garber C, Vasek MJ, Vollmer LL, Sun T, Jiang X, Klein RS. Astrocytes decrease adult neurogenesis during virus-induced memory dysfunction via IL-1. Nat Immunol (2018) 19:151–61. doi: 10.1038/s41590-017-0021-y
113. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature (2017) 541:481–7. doi: 10.1038/nature21029
114. Hwang M, Bergmann CC. Alpha/beta interferon (IFN-α/β) signaling in astrocytes mediates protection against viral encephalomyelitis and regulates IFN-γ-dependent responses. J Virol (2018) 92:01901-17. doi: 10.1128/JVI.01901-17
115. Daniels BP, Jujjavarapu H, Durrant DM, Williams JL, Green RR, White JP, et al. Regional astrocyte IFN signaling restricts pathogenesis during neurotropic viral infection. J Clin Invest (2017) 127:843–56. doi: 10.1172/JCI88720
116. Shirai K, Shimada T, Yoshida H, Hayakari R, Matsumiya T, Tanji K, et al. Interferon (IFN)-induced protein 35 (IFI35) negatively regulates IFN-β-phosphorylated STAT1-RIG-I-CXCL10/CCL5 axis in U373MG astrocytoma cells treated with polyinosinic-polycytidylic acid. Brain Res (2017) 1658:60–7. doi: 10.1016/j.brainres.2017.01.018
117. Wang H, Yuan M, Wang S, Zhang L, Zhang R, Zou X, et al. STAT3 regulates the type I IFN-mediated antiviral response by interfering with the nuclear entry of STAT1. Int J Mol Sci (2019) 20:4870. doi: 10.3390/ijms20194870
118. Staeheli P, Sentandreu M, Pagenstecher A, Hausmann JR. Alpha/Beta interferon promotes transcription and inhibits replication of borna disease virus in persistently infected cells. J Virol (2001) 17:8. doi: 10.1128/JVI.75.17.8216-8223.2001
119. Watson Z, Tang S-J. Aberrant synaptic pruning in CNS diseases: A critical player in HIV-associated neurological dysfunction? Cells (2022) 11:1943. doi: 10.3390/cells11121943
120. Das M, Smith ML, Furihata T, Sarker S, O’Shea R, Helbig KJ. Astrocyte control of zika infection is independent of interferon type I and type III expression. Biology (2022) 11:143. doi: 10.3390/biology11010143
121. Lindqvist R, Mundt F, Gilthorpe JD, Wölfel S, Gekara NO, Kröger A, et al. Fast type I interferon response protects astrocytes from flavivirus infection and virus-induced cytopathic effects. J Neuroinflamm (2016) 13:277. doi: 10.1186/s12974-016-0748-7
122. Delhaye S, Paul S, Blakqori G, Minet M, Weber F, Staeheli P, et al. Neurons produce type I interferon during viral encephalitis. Proc Natl Acad Sci U.S.A. (2006) 103:7835–40. doi: 10.1073/pnas.0602460103
123. Préhaud C, Mégret F, Lafage M, Lafon M. Virus infection switches TLR-3-Positive human neurons to become strong producers of beta interferon. J Virol (2005) 79:12893–904. doi: 10.1128/JVI.79.20.12893-12904.2005
124. Wang J, Campbell IL. Innate STAT1-dependent genomic response of neurons to the antiviral cytokine alpha interferon. J Virol (2005) 79:8295–302. doi: 10.1128/JVI.79.13.8295-8302.2005
125. Samuel MA, Whitby K, Keller BC, Marri A, Barchet W, Williams BRG, et al. PKR and RNase l contribute to protection against lethal West Nile virus infection by controlling early viral spread in the periphery and replication in neurons. J Virol (2006) 80:7009–19. doi: 10.1128/JVI.00489-06
126. Ida-Hosonuma M, Iwasaki T, Yoshikawa T, Nagata N, Sato Y, Sata T, et al. The Alpha/Beta interferon response controls tissue tropism and pathogenicity of poliovirus. J Virol (2005) 79:4460–9. doi: 10.1128/JVI.79.7.4460-4469.2005
127. Oo A, Zandi K, Shepard C, Bassit LC, Musall K, Goh SL, et al. Elimination of aicardi–goutières syndrome protein SAMHD1 activates cellular innate immunity and suppresses SARS-CoV-2 replication. J Biol Chem (2022) 298:101635. doi: 10.1016/j.jbc.2022.101635
128. Lindqvist R, Upadhyay A, Överby A. Tick-borne flaviviruses and the type I interferon response. Viruses (2018) 10:340. doi: 10.3390/v10070340
129. De Tiège X, Rozenberg F, Héron B. The spectrum of herpes simplex encephalitis in children. Eur J Paediatr Neurol EJPN Off J Eur Paediatr Neurol Soc (2008) 12:72–81. doi: 10.1016/j.ejpn.2007.07.007
130. Sancho-Shimizu V, Zhang S-Y, Abel L, Tardieu M, Rozenberg F, Jouanguy E, et al. Genetic susceptibility to herpes simplex virus 1 encephalitis in mice and humans. Curr Opin Allergy Clin Immunol (2007) 7:495–505. doi: 10.1097/ACI.0b013e3282f151d2
131. Zhang S-Y, Herman M, Ciancanelli MJ, Pérez de Diego R, Sancho-Shimizu V, Abel L, et al. TLR3 immunity to infection in mice and humans. Curr Opin Immunol (2013) 25:19–33. doi: 10.1016/j.coi.2012.11.001
132. Lafaille FG, Pessach IM, Zhang S-Y, Ciancanelli MJ, Herman M, Abhyankar A, et al. Impaired intrinsic immunity to HSV-1 in human iPSC-derived TLR3-deficient CNS cells. Nature (2012) 491:769–73. doi: 10.1038/nature11583
133. Zimmer B, Ewaleifoh O, Harschnitz O, Lee Y-S, Peneau C, McAlpine JL, et al. Human iPSC-derived trigeminal neurons lack constitutive TLR3-dependent immunity that protects cortical neurons from HSV-1 infection. Proc Natl Acad Sci U.S.A. (2018) 115:E8775–82. doi: 10.1073/pnas.1809853115
134. Gao D, Ciancanelli MJ, Zhang P, Harschnitz O, Bondet V, Hasek M, et al. TLR3 controls constitutive IFN-β antiviral immunity in human fibroblasts and cortical neurons. J Clin Invest (2021) 131:134529. doi: 10.1172/JCI134529
135. Winkler CW, Woods TA, Groveman BR, Carmody AB, Speranza EE, Martens CA, et al. Neuronal maturation reduces the type I IFN response to orthobunyavirus infection and leads to increased apoptosis of human neurons. J Neuroinflamm (2019) 16:229. doi: 10.1186/s12974-019-1614-1
136. Yordy B, Iijima N, Huttner A, Leib D, Iwasaki A. A neuron-specific role for autophagy in antiviral defense against herpes simplex virus. Cell Host Microbe (2012) 12:334–45. doi: 10.1016/j.chom.2012.07.013
137. Li J, Liu Y, Zhang X. Murine coronavirus induces type I interferon in oligodendrocytes through recognition by RIG-I and MDA5. J Virol (2010) 84:6472–82. doi: 10.1128/JVI.00016-10
138. Kapil P, Butchi NB, Stohlman SA, Bergmann CC. Oligodendroglia are limited in type I interferon induction and responsiveness in vivo. Glia (2012) 60:1555–66. doi: 10.1002/glia.22375
139. Vitner EB, Farfel-Becker T, Ferreira NS, Leshkowitz D, Sharma P, Lang KS, et al. Induction of the type I interferon response in neurological forms of gaucher disease. J Neuroinflamm (2016) 13:104. doi: 10.1186/s12974-016-0570-2
140. Dugas B, Charbonnier S, Baarine M, Ragot K, Delmas D, Ménétrier F, et al. Effects of oxysterols on cell viability, inflammatory cytokines, VEGF, and reactive oxygen species production on human retinal cells: cytoprotective effects and prevention of VEGF secretion by resveratrol. Eur J Nutr (2010) 49:435–46. doi: 10.1007/s00394-010-0102-2
141. Jang J, Park S, Jin Hur H, Cho H-J, Hwang I, Pyo Kang Y, et al. 25-hydroxycholesterol contributes to cerebral inflammation of X-linked adrenoleukodystrophy through activation of the NLRP3 inflammasome. Nat Commun (2016) 7:13129. doi: 10.1038/ncomms13129
142. Imaizumi T, Yoshida H, Hayakari R, Xing F, Wang L, Matsumiya T, et al. Interferon-stimulated gene (ISG) 60, as well as ISG56 and ISG54, positively regulates TLR3/IFN-β/STAT1 axis in U373MG human astrocytoma cells. Neurosci Res (2016) 105:35–41. doi: 10.1016/j.neures.2015.09.002
143. Vetreno RP, Crews FT. Adolescent binge drinking increases expression of the danger signal receptor agonist HMGB1 and toll-like receptors in the adult prefrontal cortex. Neuroscience (2012) 226:475–88. doi: 10.1016/j.neuroscience.2012.08.046
144. Coleman LGJ, Zou J, Crews FT. Microglial-derived miRNA let-7 and HMGB1 contribute to ethanol-induced neurotoxicity via TLR7. J Neuroinflamm (2017) 14:22. doi: 10.1186/s12974-017-0799-4
145. McDonough A, Lee RV, Noor S, Lee C, Le T, Iorga M, et al. Ischemia/Reperfusion induces interferon-stimulated gene expression in microglia. J Neurosci Off J Soc Neurosci (2017) 37:8292–308. doi: 10.1523/JNEUROSCI.0725-17.2017
146. Tonduti D, Fazzi E, Badolato R, Orcesi S. Novel and emerging treatments for aicardi-goutières syndrome. Expert Rev Clin Immunol (2020) 16:189–98. doi: 10.1080/1744666X.2019.1707663
147. Guo X, Wiley CA, Steinman RA, Sheng Y, Ji B, Wang J, et al. Aicardi-goutières syndrome-associated mutation at ADAR1 gene locus activates innate immune response in mouse brain. J Neuroinflamm (2021) 18:169. doi: 10.1186/s12974-021-02217-9
148. Gacem N, Kavo A, Zerad L, Richard L, Mathis S, Kapur RP, et al. ADAR1 mediated regulation of neural crest derived melanocytes and schwann cell development. Nat Commun (2020) 11:198. doi: 10.1038/s41467-019-14090-5
149. Neilley LK, Goodin DS, Goodkin DE, Hauser SL. Side effect profile of interferon beta-lb in MS: Results of an open label trial. Neurology (1996) 46:552–3. doi: 10.1212/WNL.46.2.552
150. Kettwig M, Ternka K, Wendland K, Krüger DM, Zampar S, Schob C, et al. Interferon-driven brain phenotype in a mouse model of RNaseT2 deficient leukoencephalopathy. Nat Commun (2021) 12:6530. doi: 10.1038/s41467-021-26880-x
151. Fields J, Dumaop W, Adame A, Ellis RJ, Letendre S, Grant I, et al. Alterations in the levels of vesicular trafficking proteins involved in HIV replication in the brains and CSF of patients with HIV-associated neurocognitive disorders. J Neuroimmune Pharmacol Off J Soc NeuroImmune Pharmacol (2013) 8:1197–209. doi: 10.1007/s11481-013-9511-3
152. Borsini A, Cattaneo A, Malpighi C, Thuret S, Harrison NA, Zunszain PA, et al. Interferon-alpha reduces human hippocampal neurogenesis and increases apoptosis via activation of distinct STAT1-dependent mechanisms. Int J Neuropsychopharmacol (2018) 21:187–200. doi: 10.1093/ijnp/pyx083
153. Wood LM, Sankar S, Reed RE, Haas AL, Liu LF, McKinnon P, et al. A novel role for ATM in regulating proteasome-mediated protein degradation through suppression of the ISG15 conjugation pathway. PloS One (2011) 6:16422. doi: 10.1371/journal.pone.0016422
154. Hain HS, Pandey R, Bakay M, Strenkowski BP, Harrington D, Romer M, et al. Inducible knockout of Clec16a in mice results in sensory neurodegeneration. Sci Rep (2021) 11:9319. doi: 10.1038/s41598-021-88895-0
155. Rossi JL, Todd T, Daniels Z, Bazan NG, Belayev L. Interferon-stimulated gene 15 upregulation precedes the development of blood-brain barrier disruption and cerebral edema after traumatic brain injury in young mice. J Neurotrauma (2015) 32:1101–8. doi: 10.1089/neu.2014.3611
156. Liu S, Karaganis S, Mo R-F, Li X-X, Wen R-X, Song X-J. IFNβ treatment inhibits nerve injury-induced mechanical allodynia and MAPK signaling by activating ISG15 in mouse spinal cord. J Pain (2020) 21:836–47. doi: 10.1016/j.jpain.2019.11.010
157. Przanowski P, Loska S, Cysewski D, Dabrowski M, Kaminska B. ISG’ylation increases stability of numerous proteins including Stat1, which prevents premature termination of immune response in LPS-stimulated microglia. Neurochem Int (2018) 112:227–33. doi: 10.1016/j.neuint.2017.07.013
158. Singh DK, Aladyeva E, Das S, Singh B, Esaulova E, Swain A, et al. Myeloid cell interferon responses correlate with clearance of SARS-CoV-2. Nat Commun (2022) 13:679. doi: 10.1038/s41467-022-28315-7
159. Sen GC, Fensterl V. Crystal structure of IFIT2 (ISG54) predicts functional properties of IFITs. Cell Res (2012) 22:1407–9. doi: 10.1038/cr.2012.130
160. Schoggins JW. Interferon-stimulated genes: roles in viral pathogenesis. Curr Opin Virol (2014) 6:40–6. doi: 10.1016/j.coviro.2014.03.006
161. Diamond MS, Farzan M. The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat Rev Immunol (2013) 13:46–57. doi: 10.1038/nri3344
162. Brass AL, Huang I-C, Benita Y, John SP, Krishnan MN, Feeley EM, et al. The IFITM proteins mediate cellular resistance to influenza a H1N1 virus, West Nile virus, and dengue virus. Cell (2009) 139:1243–54. doi: 10.1016/j.cell.2009.12.017
163. Huang I-C, Bailey CC, Weyer JL, Radoshitzky SR, Becker MM, Chiang JJ, et al. Distinct patterns of IFITM-mediated restriction of filoviruses, SARS coronavirus, and influenza a virus. PloS Pathog (2011) 7:e1001258. doi: 10.1371/journal.ppat.1001258
164. Poddar S, Hyde JL, Gorman MJ, Farzan M, Diamond MS. The interferon-stimulated gene IFITM3 restricts infection and pathogenesis of arthritogenic and encephalitic alphaviruses. J Virol (2016) 90:8780–94. doi: 10.1128/JVI.00655-16
165. Everitt AR, The MOSAIC Investigators, Everitt AR, Clare S, Pertel T, John SP, et al. IFITM3 restricts the morbidity and mortality associated with influenza. Nature (2012) 484:519–23. doi: 10.1038/nature10921
166. Zhao X, Li J, Winkler CA, An P, Guo J-T. IFITM genes, variants, and their roles in the control and pathogenesis of viral infections. Front Microbiol (2019) 9:3228. doi: 10.3389/fmicb.2018.03228
167. Allen EK, Randolph AG, Bhangale T, Dogra P, Ohlson M, Oshansky CM, et al. SNP-mediated disruption of CTCF binding at the IFITM3 promoter is associated with risk of severe influenza in humans. Nat Med (2017) 23:975–83. doi: 10.1038/nm.4370
168. Zhang Y, Qin L, Zhao Y, Zhang P, Xu B, Li K, et al. Interferon-induced transmembrane protein 3 genetic variant rs12252-c associated with disease severity in coronavirus disease 2019. J Infect Dis (2020) 222:34–7. doi: 10.1093/infdis/jiaa224
169. Zhang Y-H, Zhao Y, Li N, Peng Y-C, Giannoulatou E, Jin R-H, et al. Interferon-induced transmembrane protein-3 genetic variant rs12252-c is associated with severe influenza in Chinese individuals. Nat Commun (2013) 4:1418. doi: 10.1038/ncomms2433
170. Schoggins JW. Interferon-stimulated genes: What do they all do? Annu Rev Virol (2019) 6:567–84. doi: 10.1146/annurev-virology-092818-015756

Keywords: interferons, interferon-stimulated genes, central nervous system, interferon-stimulated gene 15, interferon-inducible tetrapeptide repeat protein 2, interferon-inducible transmembrane protein 3, viral infection, neurological diseases
Citation: Lang R, Li H, Luo X, Liu C, Zhang Y, Guo S, Xu J, Bao C, Dong W and Yu Y (2022) Expression and mechanisms of interferon-stimulated genes in viral infection of the central nervous system (CNS) and neurological diseases. Front. Immunol. 13:1008072. doi: 10.3389/fimmu.2022.1008072
Received: 31 July 2022; Accepted: 28 September 2022;
Published: 17 October 2022.
Edited by:
Linzhu Ren, Jilin University, ChinaReviewed by:
Song Wang, Fujian Agriculture and Forestry University, ChinaCopyright © 2022 Lang, Li, Luo, Liu, Zhang, Guo, Xu, Bao, Dong and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yang Yu, eXV5YW5nODBAc3dtdS5lZHUuY24=; Wei Dong, ZG9uZ3dlaUBzd211LmVkdS5jbg==; Changshun Bao, YmNzNzU2QDEyNi5jb20=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.