
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Immunol., 07 November 2022
Sec. Systems Immunology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1006944
 Nicolas Chapelle1†
Nicolas Chapelle1† Aurelie Fantou1,2†
Aurelie Fantou1,2† Thomas Marron3,4,5
Thomas Marron3,4,5 Ephraim Kenigsberg3,4,5
Ephraim Kenigsberg3,4,5 Miriam Merad3,4,5,6
Miriam Merad3,4,5,6 Jerome C. Martin1,2*
Jerome C. Martin1,2*Immunotherapy drugs are transforming the clinical care landscape of major human diseases from cancer, to inflammatory diseases, cardiovascular diseases, neurodegenerative diseases and even aging. In polygenic immune-mediated inflammatory diseases (IMIDs), the clinical benefits of immunotherapy have nevertheless remained limited to a subset of patients. Yet the identification of new actionable molecular candidates has remained challenging, and the use of standard of care imaging and/or histological diagnostic assays has failed to stratify potential responders from non-responders to biotherapies already available. We argue that these limitations partly stem from a poor understanding of disease pathophysiology and insufficient characterization of the roles assumed by candidate targets during disease initiation, progression and treatment. By transforming the resolution and scale of tissue cell mapping, high-resolution profiling strategies offer unprecedented opportunities to the understanding of immunopathogenic events in human IMID lesions. Here we discuss the potential for single-cell technologies to reveal relevant pathogenic cellular programs in IMIDs and to enhance patient stratification to guide biotherapy eligibility and clinical trial design.
Pioneered by anti–tumor necrosis factor (TNF) antibodies (Abs), the introduction of targeted therapies against inflammatory cytokines transformed the clinical outcome of patients with major chronic immune-mediated inflammatory diseases (IMIDs), such as rheumatoid arthritis (RA), psoriasis and inflammatory bowel disease (IBD) (1–3). More recently, the blockade of immune checkpoints resulted in a major paradigm shift in cancer care, which was acknowledged by the 2018 Nobel prize awards in physiology or medicine (4). These spectacular achievements emphasize the extraordinary potential for the discovery of new immunotherapy drug targets that can be leveraged from the vast immunology knowledge generated over the past century.
In monogenic forms of IMIDs where disease pathophysiology is predominantly driven by a unique molecular pathway, substantial clinical success has been achieved. This is for instance the case in autoinflammatory diseases driven by sustained activation of the inflammasome, such as familial Mediterranean fever (FMF) or cryopyrin-associated periodic syndrome (CAPS) (5, 6). Substantial clinical success was also achieved in cancer lesions expressing mismatch repair insufficiency, which drives the expression of high number of neoantigens and increases the immunogenicity of tumors and their responsiveness to PD-1 blockade. This led for the first time in the history of cancer to the FDA approval of PD-1 blockade in all microsatellite instability-high (MSI-H) cancers, regardless of tumor types (7–9).
In contrast, the clinical benefits of immunotherapy in polygenic IMIDs have remained limited to a subset of patients. About 30-40% of IBD patients do not respond or end up not responding to anti-TNF Abs (10, 11). Only two targeted therapies with overall moderate benefits, anifrolumab and belimumab, have been approved for the management of systemic lupus erythematosus (SLE) in more than 50 years of research (12, 13). The limited benefits of immunotherapy in IMIDs partly account for the immunopathological heterogeneity present across similar tissue lesions in IMIDs, as long-time supported by the pleiomorphic auto-antibody patterns for instance. However, while it is now clear that clinical and histological diagnoses are not sufficient to predict efficacy, none of the targeted immunotherapies approved to treat polygenic IMIDs are provided with decision algorithms to maximize chances of therapeutic response.
The sequencing revolution that has occurred over the last decades has propelled enormous enthusiasm and surge to identify the underlying genetic drivers of diseases. Genetic profiling has led to significant advances in the identification, understanding and treatment of monogenic immune diseases such as primary immunodeficiencies (severe combined immunodeficiency (SCID), common variable immunodeficiency (CVID) etc…), auto-inflammatory diseases (FMF, tumor necrosis factor receptor associated periodic syndrome (TRAPS), CAPS, Blau syndrome etc…) and monogenic autoimmune diseases (autoimmune polyendocrinopathy candidiasis ectodermal dystrophy (APECED), immunodysregulation polyendocrinopathy enteropathy X-linked (IPEX), (cytotoxic T-lymphocyte-associated protein-4) CTLA4 deficiency etc…) (5, 14–16). In rare cases of exceptionally strong gene linkage, genetic testing can even contribute to strengthening the diagnosis of multifactorial polygenic diseases such as HLA-B27 in ankylosing spondylitis or HLA-DQ2/DQ8 in celiac disease.
While the clinical benefits of genetic testing are limited to individuals at risk, genome-wide association studies (GWAS) have been extensively used to identify potential mechanisms of diseases in larger patient populations. In most IMIDs, however, only a minor fraction of disease trait variability has been linked to specific genomic loci, and risk gene variants confer small and unpredictable risk of disease occurrence. The role for extragenetic factors has been supported by epidemiologic observations showing discordance of disease expression between homozygous twins, onset frequently occurring during adulthood (17), and the “outbreak” of IMIDs upon change of lifestyle and dietary habits. Accordingly, environmental exposure, infectious history, microbiome composition and epigenetic factors are considered as significant contributors to disease onset, evolution and response to treatment (18, 19). It is thus unlikely that genetic profiling alone will ever be sufficient to help diagnose or predict response to immunotherapy drugs in polygenic IMIDs (17, 20).
Genetic studies should by no means be reduced but it is urgent that we enhance our efforts to understand the cellular contribution to disease pathophysiology. Functional genetics can map risk loci or genes to a cellular phenotype but cellular- and tissue-specific effects of gene mutations often lead to distinct disease pathophysiology. This has been well demonstrated in cancer lesions, where loss-of-function or gain-of-function alterations in specific genes can promote tumorigenesis in some tissues while ineffectual in others (21). For example, inherited BRCA1- and BRCA2-inactivating mutations predispose mainly to breast and ovarian cancer; adenomatous polyposis coli (APC) to colorectal cancer (CRC); and KRAS mutations to pancreas, colon and lung cancer, emphasizing the contribution of the tissue environment to the gene effect (21). IMIDs sharing genetic susceptibility within the IL-23/IL-17 pathway respond differently to IL-17 blockade, and while significant clinical benefit was obtained in axial spondyloarthritis (22) and psoriasis (23, 24), IL-17 blockade led to disease worsening in ileal Crohn’s disease (25). Thus, the pathogenicity of a specific pathway and the outcome of its blockade in polygenic IMIDs may vary across cell types, tissue types but also biological context (26).
Immunologists have excelled in cellular taxonomy (i.e., the definition of cellular phenotypic identity and functional specialization) but the nature of the cellular and molecular pathways that lead to human immune diseases remain sparse. Improving the prediction on gene effects and their therapeutic potential will necessarily depend on the understanding of the cellular phenotypes that populate the diseased organs. In that regard, recent data have emerged to support the potential of population-scale single-cell RNA sequencing (scRNA-seq) analyses to explore multiple expression quantitative trait locus (eQTL) in a cell type-specific manner (26, 27). Combined with the need to gain a better understanding of the cellular programs shaping normal and lesional tissues, such approaches could be instrumental for our understanding of causal mechanisms of complex diseases.
Before that, however, efforts to improve the access to biological samples and the even rarer access to longitudinal sampling of human lesions during disease progression or during treatment must be initiated, as discussed below.
Most drug targets fail to succeed when evaluated in phase 3 trials, hence accounting for both an important economic burden and a distraction for physicians and scientists, which adversely impacts clinical care. The reasons leading to failure are numerous and include factors such as safety issues, insufficient enrollment, suboptimal study design and chiefly, the inability to demonstrate drug efficacy (28). In fact, the majority of FDA-approved drugs every year target known pathophysiological pathways, reflecting the struggle to identify new actionable molecular candidates. We argue that these limitations partly stem from a poor understanding of disease pathophysiology and insufficient characterization of the roles assumed by candidate targets during disease initiation, progression or response to treatment. Animal models led to many key discoveries in immunology, including in the biomedical field (29) but discrepancies between animal and human pathophysiology have been raised as possible obstacles. Still, experiments in mice enabled the development of anti-TNF therapy for RA (30), and of Abs targeting immune checkpoints in cancer (31). While we should discard models that have consistently proved unable to represent human disease, we believe that the limited cellular and molecular information about human lesions actually precluded the development of appropriate models for preclinical studies.
High dimensional mapping strategies now provide with unique opportunities to thoroughly describe the sequence of immunopathogenic events developing along disease course and therapeutic intervention in human lesions (32). Ambitious programs aiming to build data-derived hypotheses as main drivers of mechanistic explorations nevertheless come with heavy loads of administrative and organizational work for both clinical and scientific teams. For immunologists, there is an ever urgent need to access freshly collected tissues as the functionality of immune cells can only be fully revealed on perturbed live cells. It appears critical that such programs be fully embraced at the institutional level to succeed. For instance, by investing more efforts into the creation of professional teams dedicated to patient selection, consent and inclusion, as well as clinical data and tissue collection, distribution, storage and retrieval, the collaboration between physicians and scientists could be largely facilitated. It will thus be important that clinical and scientific teams work in close collaboration together with institutional but also ethical committees to the design of studies offering guarantees of sufficient sustainability (33).
Longitudinal profiling studies, in particular, require that ethically-acceptable conditions be defined and properly framed by ethical review boards. While repeated sampling in organs that would put the patient at risk with no direct clinical needs is simply not ethically acceptable, it remains possible in tissues such as the skin or the digestive tract. In addition, because histology-based decisions are gaining prominence in the therapeutic management of patients with IBD or chronic inflammatory lesions of the stomach for instance (34, 35), this makes it even possible to align molecular profiling with clinical care. IMIDs share several targetable molecular drivers (36, 37), and one can anticipate diseases involving organs not easily accessible to biopsy collection will still benefit from knowledge gains achieved through such longitudinal studies.
Empowered by the progress of next generation sequencing (NGS) technologies, genomic assays are currently being used to search for molecular patterns that can stratify patients’ lesions beyond standard histological assays. Genomic assays that include transcriptomic information contributed to the identification of several molecular signatures of IMIDs, some of which have been successfully correlated to distinct clinical outcomes (38–48). Strategies profiling tissues in bulk only inform on the average expression profile of a mixture of different cell types and cannot resolve cell-specific gene expression programs (49). The development of high-resolution profiling approaches including single-cell technologies have significantly enhanced information obtained from limited human biological specimens. The use of massively parallel single-cell genomics assays can now routinely profile the transcriptome of tens of thousands of cells. Beyond RNA, it is possible to measure DNA, protein and epigenomic profiles with single cell resolution (32). Spatial mapping technologies, which inform on cellular organization and interactions in tissues, provide additional layers of information. Significant progress is being made in the development of robust computational methods to determine cell types, states and spatial location, including through the possibility to integrate multiple datasets made publicly available (50–52). Together, these advances have transformed the resolution and scale of tissue cell mapping of IMIDs lesions.
In particular, single-cell RNA-sequencing (scRNA-seq) provides an unbiased method to identify cellular subtypes and/or states defined by distinct molecular programs based on the analysis of thousands of genes, which has the potential to reveal scarce cellular processes relevant to IMIDs pathogenicity. Using scRNA-seq, the profiling of peripheral blood mononuclear cells (PBMCs) from SLE patients allowed to identify the expansion of populations of IFN-stimulated genes (ISG)hi monocytes, plasmacytoid dendritic cells (pDCs), plasma cells (PCs), and cytotoxic natural killer (NK) cells in patients with the highest disease activity (53). An elevated IFN signature was also observed in kidney tubular cells of non-responder patients (42, 43). In seropositive RA, scRNA-seq studies revealed the expansion of fibroblast activation protein-α (FAPα)+ fibroblasts that comprised subsets of inflammatory FAPα+THY1+, and destructive FAPα+THY1− fibroblasts, respectively localized in the synovial sub-lining and lining layers (54, 55). Building on these findings, further scRNA-seq studies identified central roles of NOTCH3 signaling in mediating the expansion of THY1+ inflammatory fibroblasts (56). Using cytometry by time-of-flight (CyTOF), the same group also described a population of CD4+ T peripheral helper (TPH) cells organizing B-cell responses and antibody production within pathologically inflamed synovial tissues (57). Through scRNA-seq profiling studies, we and others later identified the contribution of similar TPH cells to inflammatory processes involved in ulcerative colitis and type 1 autoimmune hepatitis respectively (58, 59). In atopic dermatitis (AD) and psoriasis, scRNA-seq analyses of skin immune cells suggested pro-inflammatory macrophage subsets could reactivate programs at play during fetal development to promote angiogenesis and leukocyte infiltration (60). In turn, the study of synovial tissue macrophages identified unique molecular states involved in inflammation resolution and tissue repair, which, when present in low proportions during remission phases, predicted increased risk of RA flares (61).
Multimodal measurements provide ways to associate in single cells analyses, several biological features that are essential to the description of cellular states and functions. Combined transcriptome and T-cell receptor (TCR) sequence analyses allow to generate valuable information about T cell clonal fates, dynamics and effector functions. Such approaches revealed the expansion of T cell clones with cytokine signatures relevant to type 2- and type 3-mediated pathogenicity in lesions of AD and psoriasis respectively (60). In Crohn’s disease (CD), Graham and colleagues recently showed that T cell responses to an immunodominant epitope conserved across gut commensal Bacteroidales shifted their cytokine responses from the production of immunoregulatory IL-10 toward IL-17 during disease flares (62). The combined profiling of gene expression and genome-wide chromatin accessibility in single stromal cells suggested the involvement of AP-1 family members in regulating the transition of a discrete subset of LGR5+-scleroderma-associated fibroblasts (ScAFs) from healthy to pathological states in the skin of patients with autoimmune systemic sclerosis (SSc) (44). Interestingly, contrary to immune alterations, which were limited to patients with diffuse SSc, ScAFs transitions were universal to both limited and diffuse phenotypes. By unraveling a previously unappreciated profibrotic cellular state, this study offers new opportunities to target fibrosis not only in all forms of SSc but also in other IMIDs with fibrotic complications such as RA and CD.
Disease-associated cell subtypes do not act in isolation but involve cellular interactions between distinct immune and non-immune cell populations, which together participate in shaping the organization of pathophysiological responses in lesions. Profiling cellular interactions is only possible using technologies that provide single cell resolution at scale. We and others recently identified cellular interactions that correlated with disease course (58, 63–69). Using single cell profiling of advanced ileal CD (iCD) lesions, we described a cellular module consisting of 5 different cell populations that accumulated in inflamed lesions of a subset of patients. We also identified the potential drivers of this pathogenic response through the characterization of ligand-receptor pairs preferentially enriched in these patients and confirmed inferred cellular interactions using spatial mapping by multiplex imaging (63, 70). Finally, we developed an analytical strategy allowing to test in large cohorts of patients, the association between the presence of this cellular response in inflamed tissues and relevant clinical parameters. Using this approach, we characterized the cellular and molecular framework in which lies anti-TNF resistance in iCD. As discussed further below, strategies as ours demonstrate the possibility to use scRNA-seq as a way to agnostically identify clinically meaningful molecular layers of patient stratification in polygenic IMIDs, which can serve as a rational basis to implement the design of clinical trials.
The unique potential for single-cell profiling strategies to identify distinct cellular and molecular organization among clinically and histologically homogenous patients provides enormous opportunities to transform clinical care (Figure 1). The single-cell profiling of human lesions should help reveal distinct cellular and molecular patterns of disease lesions and stratify patients beyond the clinical and histological diagnosis. It is important, however, that such profiling be done first on lesions naïve of treatment to reduce the confounding variable induced upon exposure to drug and maximize the chances to identify relevant molecular drivers of diseases. In clinical trial design, the identification of disease molecular patterns will enable a balanced distribution of patients with distinct immunopathogenic signatures and help reveal the efficacy of drug treatment in specific subset of patients. Such studies will also help the identification of biomarkers of response to treatment and help maximize therapeutic management. In addition, longitudinal mapping of cellular distributions and their molecular dynamics in lesional tissues during treatment should help identify molecular criteria of therapeutic response that are critically needed, as pathogenic processes in tissues can remain active despite apparent clinical recovery. Quantification of cellular and molecular responses could then serve as primary endpoints to guide therapeutic strategies. Dynamic mapping could also help unravel cellular and molecular processes enriched in responder and non-responder patients. Such results should guide ongoing R&D efforts to identify novel potential targets and combination therapies that associate synergistic drugs targeting distinct molecular pathways. Finally, systematic single-cell profiling of human lesions may reveal pathogenic pathways that are shared among distinct clinical and histological entities, and ultimately lead to a molecular classification of diseases and the design of novel therapies that target molecular drivers of diseases irrespective of the clinical diagnosis (71).
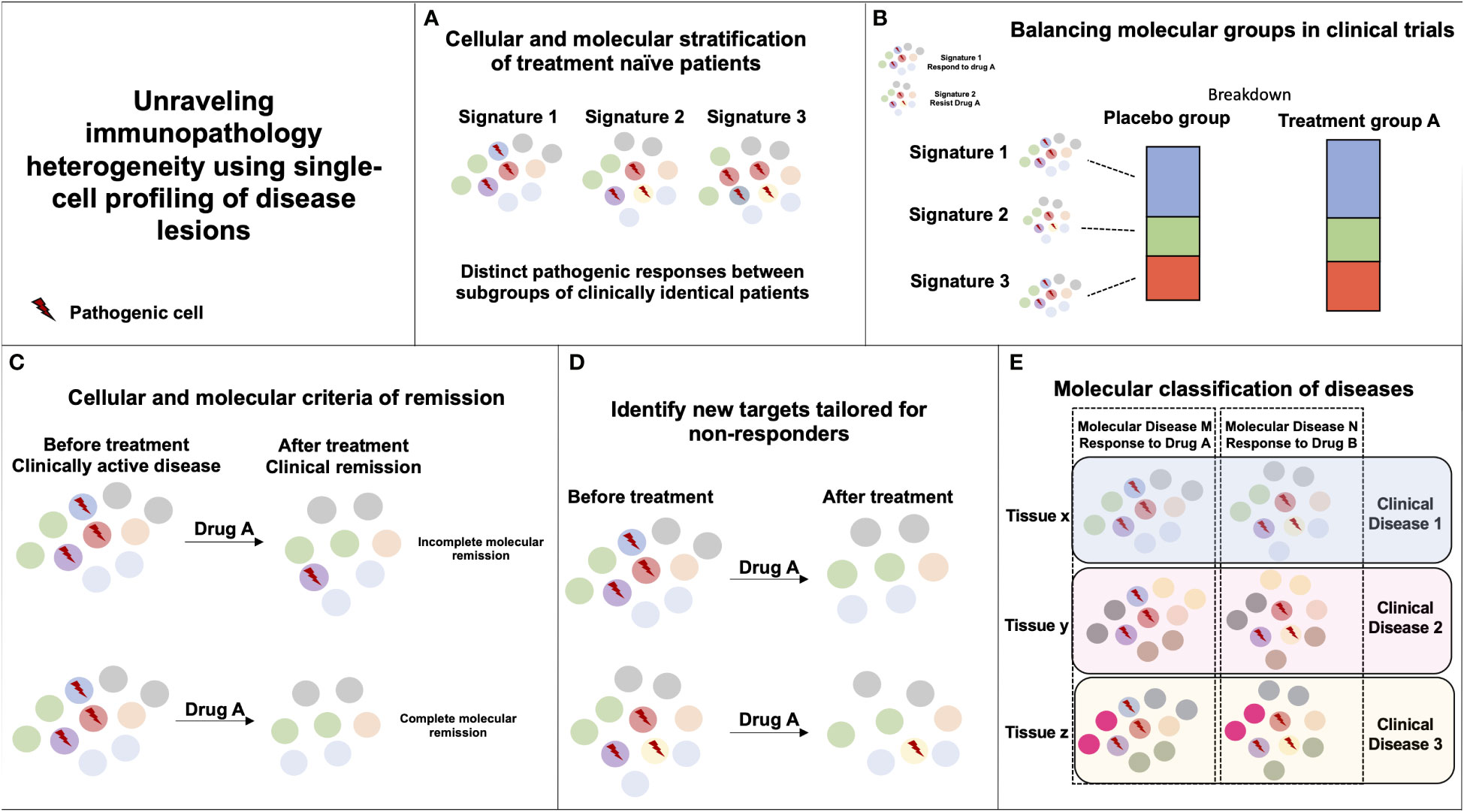
Figure 1 Unraveling immunopathology heterogeneity using single-cell profiling of disease lesions. (A) The characterization of distinct immunopathogenic responses in lesional tissues between subgroups of clinically similar patients can help derive stratifying cellular and molecular signatures. (B) If two pathogenic responses are causing a similar clinical disease but only signature 1 is responding to Drug A, random or biased enrichment of signature 1 in Placebo vs. Drug A group could mislead to Drug A inefficacy in the disease. (C) Monitoring cellular and molecular responses in tissues during the course of treatment can help define molecular endpoints of remission in patients with clinically inactive disease. (D) Monitoring cellular and molecular responses in tissues during the course of treatment can help identify immune pathways unaffected by the drug that could be targeted by another therapy. (E) Similar immunopathogenic responses occurring in distinct tissues and diseases can be targeted by the same immunotherapy.
A major goal of single-cell profiling and multiplex imaging technologies is to associate clinically relevant outcomes with cellular signatures. scRNA-seq enables to agnostically resolve the organization of complex cellular signatures in tissue lesions. Cost and labor constraints, however, restrain its application to small cohorts of patients. Because the distributions of clinical and/or demographic parameters can be highly intercorrelated, it is necessary to disentangle confounding factors in order to extract clinically meaningful molecular signatures, which is only possible with large cohorts. We suggest to overcome these limitations using a 2-steps strategy. In a “molecular discovery” phase, relevant cellular signatures are first identified by applying high-resolution single-cell profiling to a small cohort of patients thoroughly selected by experienced clinicians (Figure 2A). In a “clinical association” phase, large numbers of patients are profiled using approaches with mitigated cellular or molecular resolutions. The clinical value of high-resolution-based molecular signatures is then tested by applying a computational method tailored to project the data from samples profiled at lower-resolution onto the model devised during the “molecular discovery” phase (Figures 2B, C). This 2-steps strategy hence allows to link at minimal cost relevant metadata (clinical records, genetic information etc.) from a large number of patients with the cellular signatures detected by scRNA-seq.
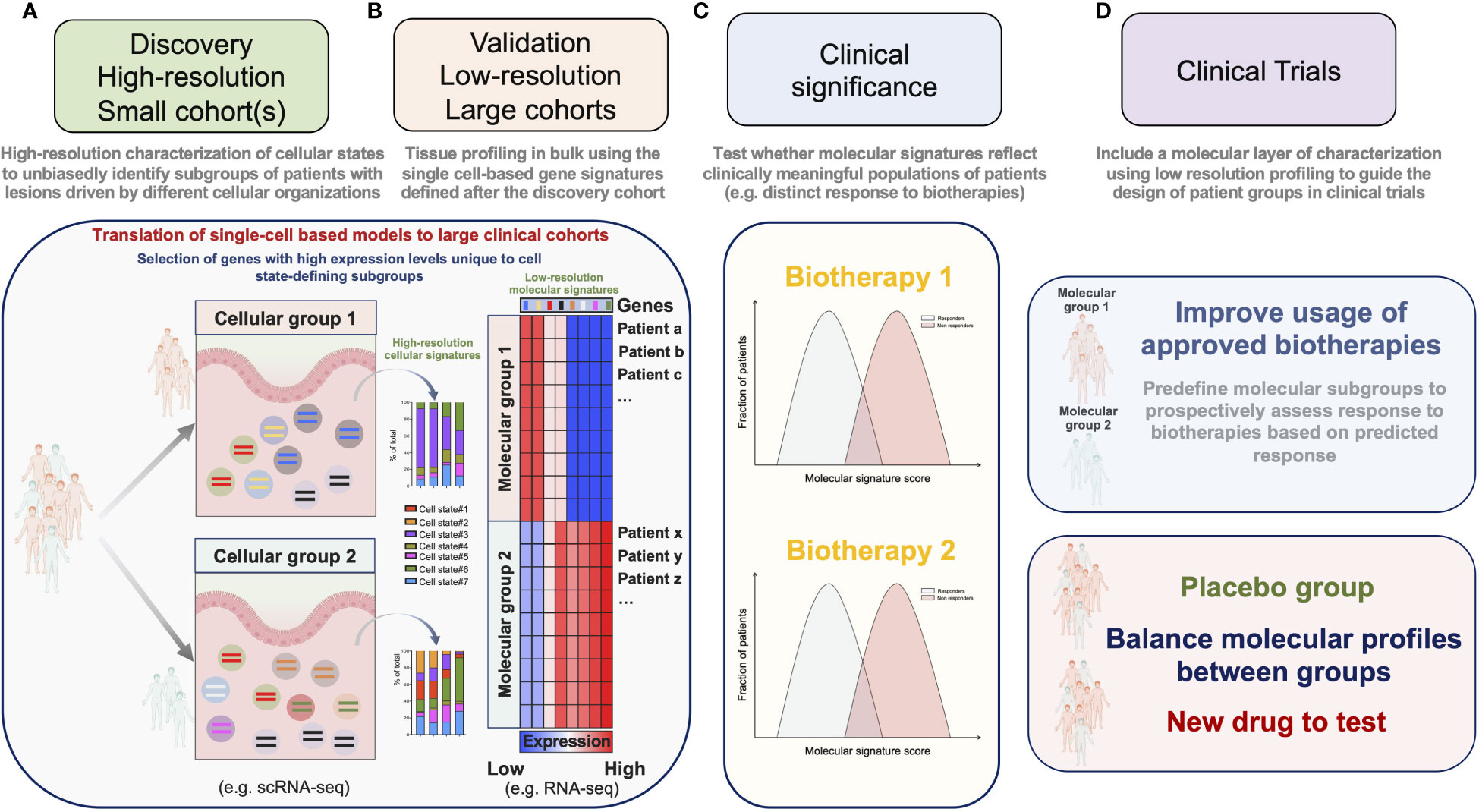
Figure 2 A 2-steps strategy for the implementation of molecular stratification criteria into the design of clinical trials. (A) In a “molecular discovery” phase, relevant cellular signatures are first identified by applying high-resolution single-cell profiling to a small cohort of patients. (B) In a “clinical association” phase high-resolution-based signatures are confirmed on a large number of patients by projecting the data obtained after low-resolution profiling onto a model devised during the “molecular discovery” phase. (C) Molecular scores derived from the agnostically identified signatures are then tested for their association with relevant clinical parameters. (D) Molecular criteria are implemented to guide the design of clinical trials.
It is possible to decrease the molecular resolution of scRNA-seq by reducing the number of readouts per cell and/or by reducing the number of single-cells per sample. Besides, the cost of scRNA-seq has largely decreased over the past decade, and is expected to drop even more in the near future (72). These approaches nevertheless require cost and labor-intensive steps of tissue dissociation for single-cell processing and library-preparation, which are expected to remain significant and challenging for implementation in clinical trials. An alternative approach that we and others privileged (63, 64, 68) was to use data from tissues sequenced in bulk for the “clinical association” phase (Figures 2B, C). By projecting the data obtained after profiling ileal biopsies with low resolution RNA-seq onto a model derived by the high-resolution scRNA-seq analysis of a small cohort of CD patients, we confirmed our findings on a large number of patients and were able to associate the enrichment of this cellular signature with a poor response to anti-TNF Abs (63). The strength and applicability of RNA seq-based stratification to guide group design in clinical trials were recently demonstrated by Pitzalis and colleagues in RA. In this study, the authors showed that the molecular stratification of RA synovial tissue from patients with inadequate response to anti-TNF Abs outcompeted histopathological classification to define subgroups matching clinical responses. Patients with a low or absent B-cell lineage expression signature in synovial tissue better responded to the IL-6 receptor antagonist tocilizumab, than to B-cell depleting rituximab (73). Ancillary analyses in a follow-up study further revealed that humoral immune response gene signatures associated with response to rituximab and tocilizumab, while patients with a stromal/fibroblast signature were refractory to all medications (41). Overall, these studies thus emphasize the importance of including molecular signatures into clinical algorithms to improve the usage of available biotherapies and guide drug development priorities for refractory patients. We believe that the power of such molecular signatures could be significantly enhanced by applying our 2-steps strategy combining profiling approaches and should be translatable to the design of next-generation clinical trials (Figure 2D).
In this perspective we highlight the enormous potential of single-cell profiling strategies to identify cellular and molecular patterns of diseases within clinically and histologically homogenous cohorts of patients. We reflect on our poor understanding of human disease pathophysiology which stems from limited accessibility of human tissues. We argue that single-cell strategies are uniquely able to provide the granularity required to mine complex disease processes and identify cellular and molecular patterns of diseases that can dramatically enhance knowledge of human diseases pathophysiology and potential therapeutic strategies.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
The authors warmfully thank Pr Regis Josien (CR2TI, Nantes Université) for his critical reading of the manuscript and suggestions. JM is supported by NExT “Junior Talent”, ANR JCJC (ANR-20-CE17-0009) and ARC Programmes labellisés (PGA). NC is supported by a Fondation pour la recherche médicale (FRM) PhD-fellowship (FDM202106013475).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Targan SR, Hanauer SB, van Deventer SJ, Mayer L, Present DH, Braakman T, et al. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for crohn’s disease. crohn’s disease cA2 study group. N Engl J Med (1997) 337:1029–35. doi: 10.1056/NEJM199710093371502
2. Elliott MJ, Maini RN, Feldmann M, Kalden JR, Antoni C, Smolen JS, et al. Randomised double-blind comparison of chimeric monoclonal antibody to tumour necrosis factor alpha (cA2) versus placebo in rheumatoid arthritis. Lancet Lond Engl (1994) 344:1105–10. doi: 10.1016/s0140-6736(94)90628-9
3. Reich K, Nestle FO, Papp K, Ortonne J-P, Evans R, Guzzo C, et al. EXPRESS study investigators. infliximab induction and maintenance therapy for moderate-to-severe psoriasis: a phase III, multicentre, double-blind trial. Lancet Lond Engl (2005) 366:1367–74. doi: 10.1016/S0140-6736(05)67566-6
4. Ledford H, Else H, Warren M. Cancer immunologists scoop medicine Nobel prize. Nature (2018) 562:20–1. doi: 10.1038/d41586-018-06751-0
5. Ozen S, Bilginer Y. A clinical guide to autoinflammatory diseases: familial Mediterranean fever and next-of-kin. Nat Rev Rheumatol (2014) 10:135–47. doi: 10.1038/nrrheum.2013.174
6. Lachmann HJ, Kone-Paut I, Kuemmerle-Deschner JB, Leslie KS, Hachulla E, Quartier P, et al. Use of canakinumab in the cryopyrin-associated periodic syndrome. N Engl J Med (2009) 360:2416–25. doi: 10.1056/NEJMoa0810787
7. Lemery S, Keegan P, Pazdur R. First FDA approval agnostic of cancer site - when a biomarker defines the indication. N Engl J Med (2017) 377:1409–12. doi: 10.1056/NEJMp1709968
8. Prasad V, Kaestner V, Mailankody S. Cancer drugs approved based on biomarkers and not tumor type-FDA approval of pembrolizumab for mismatch repair-deficient solid cancers. JAMA Oncol (2018) 4:157–8. doi: 10.1001/jamaoncol.2017.4182
9. First tissue-agnostic drug approval issued. Cancer Discovery (2017) 7:656. doi: 10.1158/2159-8290.CD-NB2017-078
10. Torres J, Mehandru S, Colombel J-F, Peyrin-Biroulet L. Crohn’s disease. Lancet Lond Engl (2017) 389:1741–55. doi: 10.1016/S0140-6736(16)31711-1
11. Ungaro R, Mehandru S, Allen PB, Peyrin-Biroulet L, Colombel J-F. Ulcerative colitis. Lancet Lond Engl (2016) 389(10080):1756–1770. doi: 10.1016/S0140-6736(16)32126-2
12. Morand EF, Furie R, Tanaka Y, Bruce IN, Askanase AD, Richez C, et al. Trial of anifrolumab in active systemic lupus erythematosus. N Engl J Med (2020) 382:211–21. doi: 10.1056/NEJMoa1912196
13. Liu Z, Davidson A. Taming lupus-a new understanding of pathogenesis is leading to clinical advances. Nat Med (2012) 18:871–82. doi: 10.1038/nm.2752
14. Notarangelo LD, Fischer A, Geha RS, Casanova J-L, Chapel H, Conley ME, et al. Primary immunodeficiencies: 2009 update. J Allergy Clin Immunol (2009) 124:1161–78. doi: 10.1016/j.jaci.2009.10.013
15. Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med (2014) 20:1410–6. doi: 10.1038/nm.3746
16. Durandy A, Kracker S, Fischer A. Primary antibody deficiencies. Nat Rev Immunol (2013) 13:519–33. doi: 10.1038/nri3466
17. Cho JH, Feldman M. Heterogeneity of autoimmune diseases: pathophysiologic insights from genetics and implications for new therapies. Nat Med (2015) 21:730–8. doi: 10.1038/nm.3897
18. Javierre BM, Hernando H, Ballestar E. Environmental triggers and epigenetic deregulation in autoimmune disease. Discovery Med (2011) 12:535–45.
19. Bach J-F. The hygiene hypothesis in autoimmunity: the role of pathogens and commensals. Nat Rev Immunol (2018) 18:105–20. doi: 10.1038/nri.2017.111
20. Uffelmann E, Huang QQ, Munung NS, de Vries J, Okada Y, Martin AR, et al. Genome-wide association studies. Nat Rev Methods Primer (2021) 1:1–21. doi: 10.1038/s43586-021-00056-9
21. Salmon H, Remark R, Gnjatic S, Merad M. Host tissue determinants of tumour immunity. Nat Rev Cancer (2019) 19:215–27. doi: 10.1038/s41568-019-0125-9
22. Baeten D, Sieper J, Braun J, Baraliakos X, Dougados M, Emery P, et al. Secukinumab, an interleukin-17A inhibitor, in ankylosing spondylitis. N Engl J Med (2015) 373:2534–48. doi: 10.1056/NEJMoa1505066
23. Langley RG, Elewski BE, Lebwohl M, Reich K, Griffiths CEM, Papp K, et al. Secukinumab in plaque psoriasis–results of two phase 3 trials. N Engl J Med (2014) 371:326–38. doi: 10.1056/NEJMoa1314258
24. Leonardi C, Matheson R, Zachariae C, Cameron G, Li L, Edson-Heredia E, et al. Anti-interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N Engl J Med (2012) 366:1190–9. doi: 10.1056/NEJMoa1109997
25. Hueber W, Sands BE, Lewitzky S, Vandemeulebroecke M, Reinisch W, Higgins PDR, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut (2012) 61:1693–700. doi: 10.1136/gutjnl-2011-301668
26. Perez RK, Gordon MG, Subramaniam M, Kim MC, Hartoularos GC, Targ S, et al. Single-cell RNA-seq reveals cell type-specific molecular and genetic associations to lupus. Science (2022) 376:eabf1970. doi: 10.1126/science.abf1970
27. Yazar S, Alquicira-Hernandez J, Wing K, Senabouth A, Gordon MG, Andersen S, et al. Single-cell eQTL mapping identifies cell type-specific genetic control of autoimmune disease. Science (2022) 376:eabf3041. doi: 10.1126/science.abf3041
28. Fogel DB. Factors associated with clinical trials that fail and opportunities for improving the likelihood of success: A review. Contemp Clin Trials Commun (2018) 11:156–64. doi: 10.1016/j.conctc.2018.08.001
29. Pulendran B, Davis MM. The science and medicine of human immunology. Science (2020) 369:eaay4014. doi: 10.1126/science.aay4014
30. Feldmann M, Maini SRN. Role of cytokines in rheumatoid arthritis: an education in pathophysiology and therapeutics. Immunol Rev (2008) 223:7–19. doi: 10.1111/j.1600-065X.2008.00626.x
31. Sharma P, Allison JP. The future of immune checkpoint therapy. Science (2015) 348:56–61. doi: 10.1126/science.aaa8172
32. Camp JG, Platt R, Treutlein B. Mapping human cell phenotypes to genotypes with single-cell genomics. Science (2019) 365:1401–5. doi: 10.1126/science.aax6648
33. Dickson D, Johnson J, Bergan R, Owens R, Subbiah V, Kurzrock R. The master observational trial: A new class of master protocol to advance precision medicine. Cell (2020) 180:9–14. doi: 10.1016/j.cell.2019.12.009
34. Dinis-Ribeiro M, Areia M, de Vries AC, Marcos-Pinto R, Monteiro-Soares M, O’Connor A, et al. Management of precancerous conditions and lesions in the stomach (MAPS): guideline from the European society of gastrointestinal endoscopy (ESGE), European helicobacter study group (EHSG), European society of pathology (ESP), and the sociedade portuguesa de endoscopia digestiva (SPED). Endoscopy (2012) 44:74–94. doi: 10.1055/s-0031-1291491
35. Turner D, Ricciuto A, Lewis A, D’Amico F, Dhaliwal J, Griffiths AM, et al. STRIDE-II: An update on the selecting therapeutic targets in inflammatory bowel disease (STRIDE) initiative of the international organization for the study of IBD (IOIBD): Determining therapeutic goals for treat-to-Target strategies in IBD. Gastroenterology (2021) 160:1570–83. doi: 10.1053/j.gastro.2020.12.031
36. Schett G, McInnes IB, Neurath MF. Reframing immune-mediated inflammatory diseases through signature cytokine hubs. N Engl J Med (2021) 385:628–39. doi: 10.1056/NEJMra1909094
37. McInnes IB, Gravallese EM. Immune-mediated inflammatory disease therapeutics: past, present and future. Nat Rev Immunol (2021) 21:680–6. doi: 10.1038/s41577-021-00603-1
38. West NR, Hegazy AN, Owens BMJ, Bullers SJ, Linggi B, Buonocore S, et al. Oncostatin m drives intestinal inflammation and predicts response to tumor necrosis factor-neutralizing therapy in patients with inflammatory bowel disease. Nat Med (2017) 23:579–89. doi: 10.1038/nm.4307
39. Banchereau R, Hong S, Cantarel B, Baldwin N, Baisch J, Edens M, et al. Personalized immunomonitoring uncovers molecular networks that stratify lupus patients. Cell (2016) 165:551–65. doi: 10.1016/j.cell.2016.03.008
40. Choy DF, Hart KM, Borthwick LA, Shikotra A, Nagarkar DR, Siddiqui S, et al. TH2 and TH17 inflammatory pathways are reciprocally regulated in asthma. Sci Transl Med (2015) 7:301ra129. doi: 10.1126/scitranslmed.aab3142
41. Rivellese F, Surace AEA, Goldmann K, Sciacca E, Çubuk C, Giorli G, et al. Rituximab versus tocilizumab in rheumatoid arthritis: synovial biopsy-based biomarker analysis of the phase 4 R4RA randomized trial. Nat Med (2022) 28:1256–68. doi: 10.1038/s41591-022-01789-0
42. Arazi A, Rao DA, Berthier CC, Davidson A, Liu Y, Hoover PJ, et al. The immune cell landscape in kidneys of patients with lupus nephritis. Nat Immunol (2019) 20:902–14. doi: 10.1038/s41590-019-0398-x
43. Der E, Suryawanshi H, Morozov P, Kustagi M, Goilav B, Ranabothu S, et al. Tubular cell and keratinocyte single-cell transcriptomics applied to lupus nephritis reveal type I IFN and fibrosis relevant pathways. Nat Immunol (2019) 20:915–27. doi: 10.1038/s41590-019-0386-1
44. Gur C, Wang S-Y, Sheban F, Zada M, Li B, Kharouf F, et al. LGR5 expressing skin fibroblasts define a major cellular hub perturbed in scleroderma. Cell (2022) 185:1373–1388.e20. doi: 10.1016/j.cell.2022.03.011
45. McKinney EF, Lyons PA, Carr EJ, Hollis JL, Jayne DRW, Willcocks LC, et al. A CD8+ T cell transcription signature predicts prognosis in autoimmune disease. Nat Med (2010) 16:586–91. doi: 10.1038/nm.2130
46. McKinney EF, Lee JC, Jayne DRW, Lyons PA, Smith KGC. T-Cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature (2015) 523:612–6. doi: 10.1038/nature14468
47. Friedrich M, Pohin M, Jackson MA, Korsunsky I, Bullers SJ, Rue-Albrecht K, et al. IL-1-driven stromal-neutrophil interactions define a subset of patients with inflammatory bowel disease that does not respond to therapies. Nat Med (2021) 27:1970–81. doi: 10.1038/s41591-021-01520-5
48. Soret P, Le Dantec C, Desvaux E, Foulquier N, Chassagnol B, Hubert S, et al. A new molecular classification to drive precision treatment strategies in primary sjögren’s syndrome. Nat Commun (2021) 12:3523. doi: 10.1038/s41467-021-23472-7
49. Papalexi E, Satija R. Single-cell RNA sequencing to explore immune cell heterogeneity. Nat Rev Immunol (2018) 18:35–45. doi: 10.1038/nri.2017.76
50. Rao A, Barkley D, França GS, Yanai I. Exploring tissue architecture using spatial transcriptomics. Nature (2021) 596:211–20. doi: 10.1038/s41586-021-03634-9
51. Palla G, Fischer DS, Regev A, Theis FJ. Spatial components of molecular tissue biology. Nat Biotechnol (2022) 40:308–18. doi: 10.1038/s41587-021-01182-1
52. Colomé-Tatché M, Theis FJ. Statistical single cell multi-omics integration. Curr Opin Syst Biol (2018) 7:54–9. doi: 10.1016/j.coisb.2018.01.003
53. Nehar-Belaid D, Hong S, Marches R, Chen G, Bolisetty M, Baisch J, et al. Mapping systemic lupus erythematosus heterogeneity at the single-cell level. Nat Immunol (2020) 21:1094–106. doi: 10.1038/s41590-020-0743-0
54. Mizoguchi F, Slowikowski K, Wei K, Marshall JL, Rao DA, Chang SK, et al. Functionally distinct disease-associated fibroblast subsets in rheumatoid arthritis. Nat Commun (2018) 9:789. doi: 10.1038/s41467-018-02892-y
55. Croft AP, Campos J, Jansen K, Turner JD, Marshall J, Attar M, et al. Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature (2019) 570:246–51. doi: 10.1038/s41586-019-1263-7
56. Wei K, Korsunsky I, Marshall JL, Gao A, Watts GFM, Major T, et al. Notch signalling drives synovial fibroblast identity and arthritis pathology. Nature (2020) 582:259–64. doi: 10.1038/s41586-020-2222-z
57. Rao DA, Gurish MF, Marshall JL, Slowikowski K, Fonseka CY, Liu Y, et al. Pathologically expanded peripheral T helper cell subset drives b cells in rheumatoid arthritis. Nature (2017) 542:110–4. doi: 10.1038/nature20810
58. Uzzan M, Martin JC, Mesin L, Livanos AE, Castro-Dopico T, Huang R, et al. Ulcerative colitis is characterized by a plasmablast-skewed humoral response associated with disease activity. Nat Med (2022) 28:766–79. doi: 10.1038/s41591-022-01680-y
59. Renand A, Cervera-Marzal I, Gil L, Dong C, Garcia A, Kervagoret E, et al. Integrative molecular profiling of autoreactive CD4 T cells in autoimmune hepatitis. J Hepatol (2020) 73:1379–90. doi: 10.1016/j.jhep.2020.05.053
60. Reynolds G, Vegh P, Fletcher J, Poyner EFM, Stephenson E, Goh I, et al. Developmental cell programs are co-opted in inflammatory skin disease. Science (2021) 371:eaba6500. doi: 10.1126/science.aba6500
61. Alivernini S, MacDonald L, Elmesmari A, Finlay S, Tolusso B, Gigante MR, et al. Distinct synovial tissue macrophage subsets regulate inflammation and remission in rheumatoid arthritis. Nat Med (2020) 26:1295–306. doi: 10.1038/s41591-020-0939-8
62. Pedersen TK, Brown EM, Plichta DR, Johansen J, Twardus SW, Delorey TM, et al. The CD4+ T cell response to a commensal-derived epitope transitions from a tolerant to an inflammatory state in crohn’s disease. Immunity (2022) 55(10):1909–1923.e6. doi: 10.1016/j.immuni.2022.08.016
63. Martin JC, Chang C, Boschetti G, Ungaro R, Giri M, Grout JA, et al. Single-cell analysis of crohn’s disease lesions identifies a pathogenic cellular module associated with resistance to anti-TNF therapy. Cell (2019) 178:1493–1508.e20. doi: 10.1016/j.cell.2019.08.008
64. Puram SV, Tirosh I, Parikh AS, Patel AP, Yizhak K, Gillespie S, et al. Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell (2017) 171:1611–1624.e24. doi: 10.1016/j.cell.2017.10.044
65. Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su M-J, Melms JC, et al. A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell (2018) 175:984–997.e24. doi: 10.1016/j.cell.2018.09.006
66. Tirosh I, Izar B, Prakadan SM, Wadsworth MH, Treacy D, Trombetta JJ, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science (2016) 352:189–96. doi: 10.1126/science.aad0501
67. Smillie CS, Biton M, Ordovas-Montanes J, Sullivan KM, Burgin G, Graham DB, et al. Intra- and inter-cellular rewiring of the human colon during ulcerative colitis. Cell (2019) 178:714–730.e22. doi: 10.1016/j.cell.2019.06.029
68. Leader AM, Grout JA, Maier BB, Nabet BY, Park MD, Tabachnikova A, et al. Single-cell analysis of human non-small cell lung cancer lesions refines tumor classification and patient stratification. Cancer Cell (2021) 39:1594–1609.e12. doi: 10.1016/j.ccell.2021.10.009
69. Yofe I, Dahan R, Amit I. Single-cell genomic approaches for developing the next generation of immunotherapies. Nat Med (2020) 26:171–7. doi: 10.1038/s41591-019-0736-4
70. Remark R, Merghoub T, Grabe N, Litjens G, Damotte D, Wolchok JD, et al. In-depth tissue profiling using multiplexed immunohistochemical consecutive staining on single slide. Sci Immunol (2016) 1:aaf6925. doi: 10.1126/sciimmunol.aaf6925
71. Buckley CD, Chernajovsky L, Chernajovsky Y, Modis LK, O’Neill LA, Brown D, et al. Immune-mediated inflammation across disease boundaries: breaking down research silos. Nat Immunol (2021) 22:1344–8. doi: 10.1038/s41590-021-01044-7
72. Shalek AK, Benson M. Single-cell analyses to tailor treatments. Sci Transl Med (2017) 9:eaan4730. doi: 10.1126/scitranslmed.aan4730
73. Humby F, Durez P, Buch MH, Lewis MJ, Rizvi H, Rivellese F, et al. Rituximab versus tocilizumab in anti-TNF inadequate responder patients with rheumatoid arthritis (R4RA): 16-week outcomes of a stratified, biopsy-driven, multicentre, open-label, phase 4 randomised controlled trial. Lancet Lond Engl (2021) 397:305–17. doi: 10.1016/S0140-6736(20)32341-2
Keywords: human disease pathophysiology, single-cell technologies, drug discovery, immunotherapy, clinical translation, immune-mediated inflammatory diseases
Citation: Chapelle N, Fantou A, Marron T, Kenigsberg E, Merad M and Martin JC (2022) Single-cell profiling to transform immunotherapy usage and target discovery in immune-mediated inflammatory diseases. Front. Immunol. 13:1006944. doi: 10.3389/fimmu.2022.1006944
Received: 29 July 2022; Accepted: 24 October 2022;
Published: 07 November 2022.
Edited by:
Paul Kaye, University of York, United KingdomReviewed by:
Wim Adriaensen, Institute of Tropical Medicine Antwerp, BelgiumCopyright © 2022 Chapelle, Fantou, Marron, Kenigsberg, Merad and Martin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jerome C. Martin, SmVyb21lLm1hcnRpbkB1bml2LW5hbnRlcy5mcg==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.