 Tasha M. Santiago-Rodriguez
Tasha M. Santiago-Rodriguez Emily B. Hollister
Emily B. Hollister
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 16 September 2022
Sec. Viral Immunology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1005107
This article is part of the Research Topic Host-Virus Interaction at the Omics and Ecology Levels View all 7 articles
Viruses are part of the microbiome and have essential roles in immunology, evolution, biogeochemical cycles, health, and disease progression. Viruses influence a wide variety of systems and processes, and the continued discovery of novel viruses is anticipated to reveal new mechanisms influencing the biology of diverse environments. While the identity and roles of viruses continue to be discovered and understood through viral metagenomics, most of the sequences in virome datasets cannot be attributed to known viruses or may be only distantly related to species already described in public sequence databases, at best. Such viruses are known as the viral dark matter. Ongoing discoveries from the viral dark matter have provided insights into novel viruses from a variety of environments, as well as their potential in immunological processes, virus evolution, health, disease, therapeutics, and surveillance. Increased understanding of the viral dark matter will continue with a combination of cultivation, microscopy, sequencing, and bioinformatic efforts, which are discussed in the present review.
Viruses comprise the most abundant entities on Earth with an estimated 1031 particles (1). Viruses can be found wherever a potential host cell is present, and their numbers vary depending on the environment. For instance, an average of 107 virus-like particles (VLPs) have been identified in a milliliter (mL) of sea water and urine (1, 2), while an average of 107, 108, and 108 to 109 VLPs have been identified in one mL of saliva (3), one gram of stool (4), and one gram of soil (5)(with new viruses increasingly being identified in soil) (6), respectively. Viruses are obligate intracellular parasites, with single-stranded (ss) or double-stranded (ds) DNA or RNA genomes (7), that replicate through a series of steps generally involving attachment to host surface receptors followed by replication and host cell lysis. Persistence of viruses may also depend on host cell availability and physiology (8), and certain viruses can remain dormant until conditions are favorable for replication and host cell lysis (9). Viruses are extremely diverse and include endogenous retroviruses, those infecting human, plant, or other animal and small eukaryotic cells, as well as bacterial viruses (i.e., phage) (10). While viruses are considered part of the microbiome, the term virome refers specifically to the collection of viruses present in a sample or community (11).
Viruses are important and have been implicated in diverse processes ranging from evolution and immunity to biogeochemical cycles. Eukaryotic viruses may be directly or indirectly involved in the evolution of eukaryotic hosts. For instance, human and animal species contain considerable genetic diversity in their resistance against viral diseases, as in the case against certain retroviruses (12). While we often associate viruses with disease, most viruses are not directly pathogenic (13). Indeed, most of the virome is composed of phage, which may be neutral in their effects on the bacterial host, and be directly or indirectly involved in various processes. For instance, phage may impact bacterial evolution by altering genome composition through transduction (Figure 1A) (14). Transduction may also result in the acquisition of genes that may offer evolutionary advantages to the bacterial host (15), such as virulence factors and antibiotic-resistance genes, which in turn can turn out to be detrimental for human and animal health (16). Phage-encoded virulence factors, particularly those that facilitate adhesion in Escherichia coli, Pseudomonas aeruginosa, Streptococcus mitis and Vibrio cholerae, as well as invasion in Salmonella enterica and Staphylococcus aureus, may be associated with disease (17, 18). Similarly, certain phage are known to mediate the transfer of antibiotic resistance genes in E. coli strains including those conferring resistance to beta-lactams, tetracycline, ampicillin and kanamycin (19). Phage may also contribute to health or disease indirectly (discussed in this review) by altering the composition of specific bacterial communities (Figure 1B), and/or acting as a “second immune system” by lysing invading bacterial pathogens (20). In the environment, phage influence biogeochemical cycles by lysing bacteria and archaea, which then become dissolved organic matter used by heterotrophic bacteria. This, in turn, increases available nutrients, respiration, and CO2 production (Figure 1C) (21). Clearly, viruses influence a wide variety of systems and processes, and the continued discovery of novel viruses is anticipated to reveal new mechanisms influencing the biology and ecology of diverse environments.
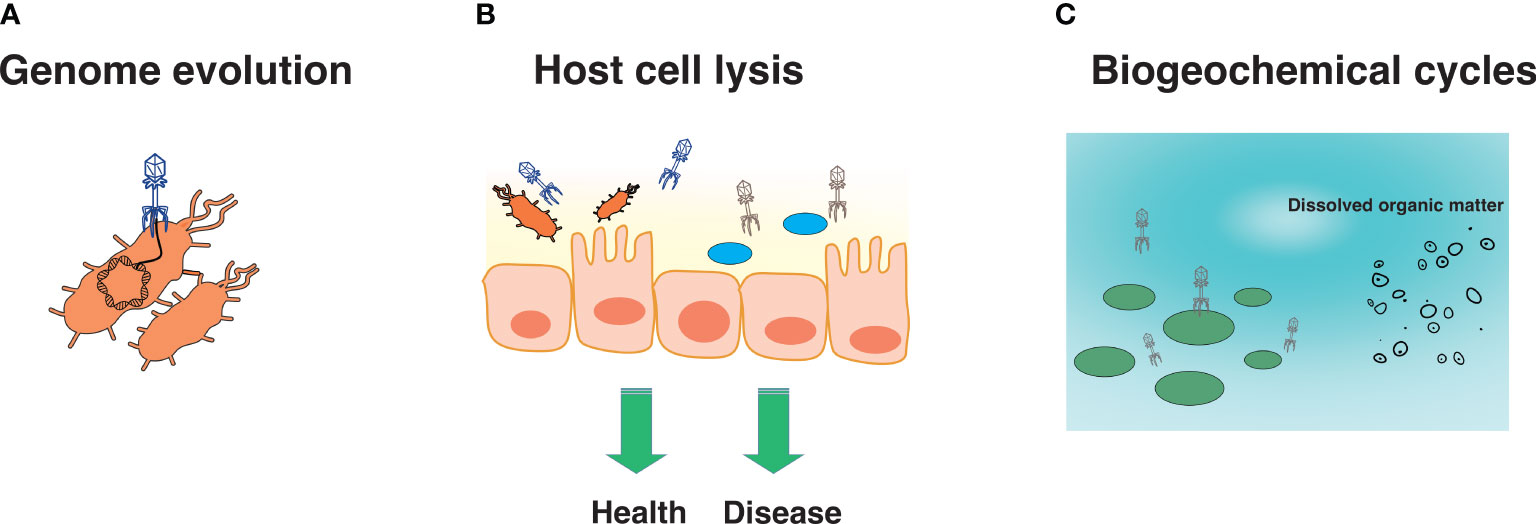
Figure 1 Several roles of viruses. Viruses are known to be involved in genome evolution (A), host cell lysis, which may promote health or disease (B), and biogeochemical cycles (C).
Virus discovery has been essential to understanding the emergence and re-emergence of viral pathogens, discovery, and characterization of non-pathogenic viruses, establishing model systems to study replication and infection mechanisms (e.g., T4 phage), and defining mechanisms that underpin immunological, evolutionary, and environmental processes. Prior the metagenomics era, viruses were typically discovered and understood using a variety of techniques aimed at deciphering replication and infection mechanisms, morphology, and genetic composition. In the following section, culture techniques, electron microscopy (EM), and molecular methods used for virus discovery are described and summarized (Table 1). Sequencing techniques, described later in this review, are also presented in Table 1.
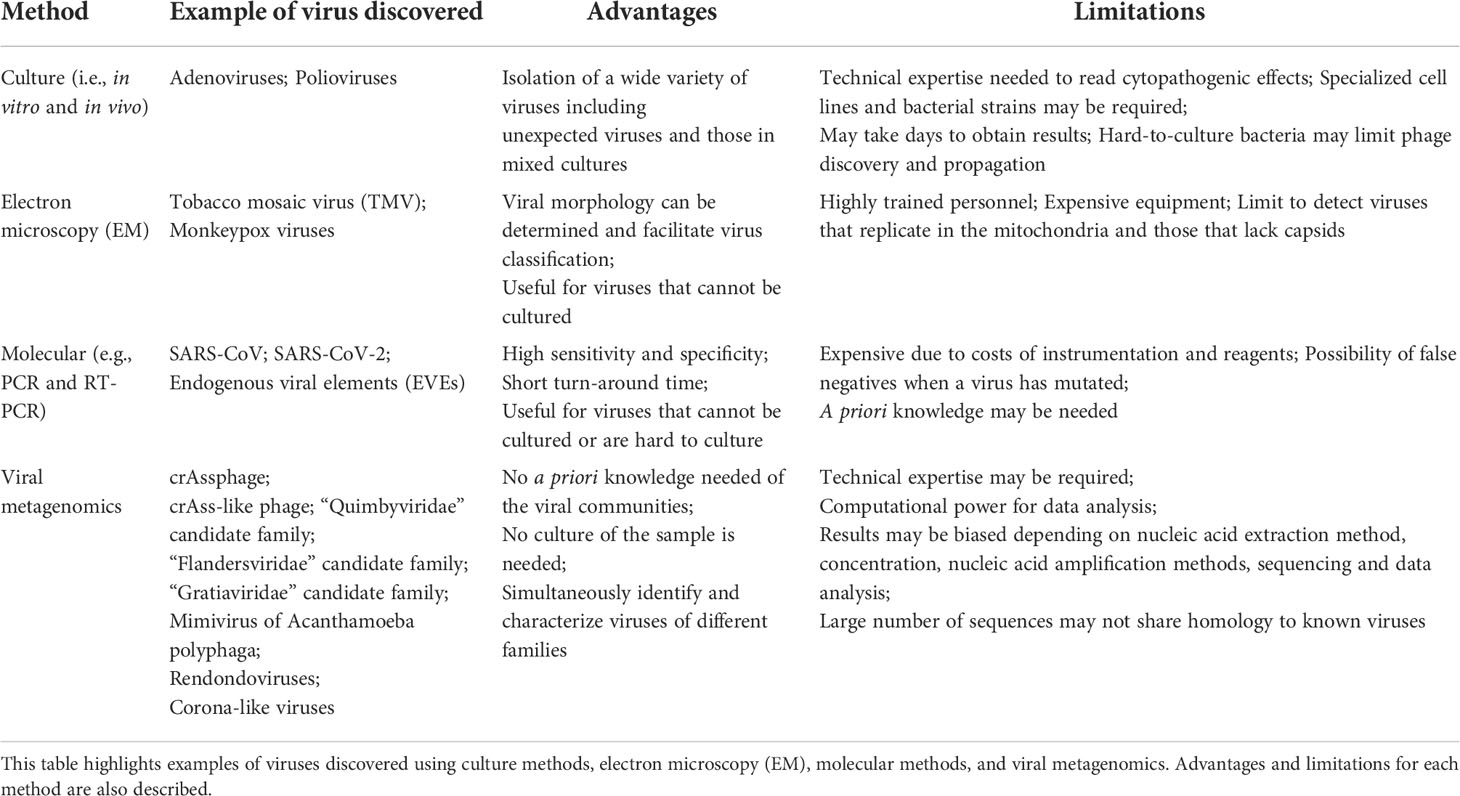
Table 1 Methods used for virus discovery.
Culture methods involving cell lines (eukaryotic viruses) and bacterial strains (phage) have long been the gold standard for the discovery and characterization of viruses. Both in vitro and in vivo culture conditions have been used to identify and isolate a wide variety of viruses, including those that are in mixed culture (Table 1). Human and non-human cell lines have been used to identify viruses stemming from specific disease phenotypes (22). Interestingly, the initial discovery of some viruses did not involve the use of well-characterized cell lines; rather they were discovered by observing cytopathogenic effects in specific cells and tissues. This was the case with adenoviruses, first discovered in the 1950s, when it was noticed that an unknown agent was responsible for the cytopathogenicity of tissues originating from tonsils and adenoids (23). Similarly, poliovirus was originally discovered by the in vivo culture of the virus (which was unknown to be a virus at the time) in several different animals, including monkeys. It was then noticed that the virus caused the same effects to the monkey as those originally observed in humans (24).
Notably, potential drawbacks of in vitro or in vivo culture of viruses are the time and specific conditions required (Table 1). Viruses are not always straightforward to culture and may require technique optimization or the application of additional techniques for their identification and characterization. Hepatitis C (also known as non-A non-B hepatitis) is a prime example of this among eukaryotic viruses (25). Similarly, certain bacteriophage may be challenging to propagate. As with eukaryotic viruses, the identification and propagation of phage often relies on the use of relatively well-characterized cells (i.e., bacterial strains), which must be susceptible to infection and replication. Phage propagation also requires optimal media, temperature, and pH conditions to enable successful bacterial host growth, phage attachment, entry, replication, and bacterial lysis (26) (Table 1). Difficulty culturing phage and/or their bacterial hosts may limit their propagation, hampering our understanding of their morphological structure, and genome composition, as well as infection and replication mechanisms.
Culture techniques are not always ideal for virus discovery and characterization. For this reason, other techniques, particularly EM, have been used in conjunction with culture for the discovery and characterization of viruses. EM, specifically, possesses the advantage over culture-based methods in that organism-specific reagents are not required (Table 1). The first virus visualized using EM was the Tobacco mosaic virus (TMV) in the 1930s. EM has also enabled the confirmation of certain Monkeypox and other poxviruses (27). Despite its benefits, EM can be expensive, requires highly-trained personnel, and may limit viral identification to the family-level as only morphology information can be obtained (Table 1).
Culture methods and EM may be accompanied by molecular techniques for virus discovery and characterization. Virus discovery is also possible through molecular techniques alone (28). Molecular methods have shown to be highly sensitive and specific, provide results with relatively short turn-around times, and be very useful for the identification and characterization of difficult-to-culture viruses (Table 1). Molecular methods using consensus primers, specifically, have also been applied for virus discovery. For example, highly divergent clades of human immunodeficiency virus (HIV) (29), and the Severe Acute Respiratory Syndrome associated coronavirus (SARS-CoV) (30), have been identified using consensus primers. SARS-CoV, in particular, was discovered after a patient’s sample tested negative for influenza, parainfluenza, respiratory syncytial virus, adenovirus, and a variety of bacterial pathogens. Since an unknown agent was suspected, the patient’s nasopharyngeal aspirate was subjected to Reverse Transcription (RT)-PCR using consensus primers targeting the coronavirus pol gene, which revealed a compatible gene product consistent with a novel coronavirus (31). Similarly, a combination of primers targeting coronaviruses aided in the discovery of the agent responsible for the most recent pandemic caused by SARS-CoV-2 (32). Molecular methods in combination with single genome bioinformatics have also enabled the discovery of endogenous viral elements (EVEs), which include retroviruses, DNA viruses, or RNA viruses (33). EVEs are known to be part of eukaryotic genomes, and their divergent hosts shows that some EVEs date back to approximately 100 million years (33). Some of these EVEs include bornaviruses (ss(-)RNA) (34), flaviviruses (ss(+)RNA) (35), circoviruses (ssDNA) (36), and hepadnaviruses (dsDNA) (37), which have been identified in the genomes of mammals, insects, and birds. While molecular techniques are indeed valuable for the discovery and further characterization of viral agents sharing homology to known viruses, a degree of a priori knowledge is typically needed, and false negatives may result in cases when the virus has mutated (Table 1).
For the last 20 years, virus discovery has been facilitated by the application of metagenomic sequencing, and this continues to be an important tool for the discovery of viruses across a variety of sample types, environments and conditions (Figure 2) (2, 3, 28, 32, 38–62). Although metagenomic sequencing typically refers to DNA sequencing of mixed community of organisms, we refer to metagenomics here as the sequencing of DNA, RNA, or both. Viral metagenomics was applied for the first time in 2002 with the characterization of the virome of marine samples. The study found that over 65% of the sequences generated did not match available reference databases and that the identifiable fraction of the marine virome was mostly composed of dsDNA phage and algal viruses (63). Similarly, for the last decade, numerous discoveries have been made regarding the virome. For instance, saliva and other sample types, previously thought to be sterile (e.g., urine, blood, and cerebrospinal fluid), are now known to be home to robust communities of viruses that are not necessarily implicated with disease (2, 3, 51, 56). Another example includes crAssphage, which was discovered using a toolbox of bioinformatic methods aiming to characterize a DNA sequence shared across human stool samples (43). Finally, similar approaches have been applied to discover viral families from unexpected sources, as in the case of the discovery of the Redondoviridae family (55), and more recently, corona-like viruses from petabases (1015) of information (64).
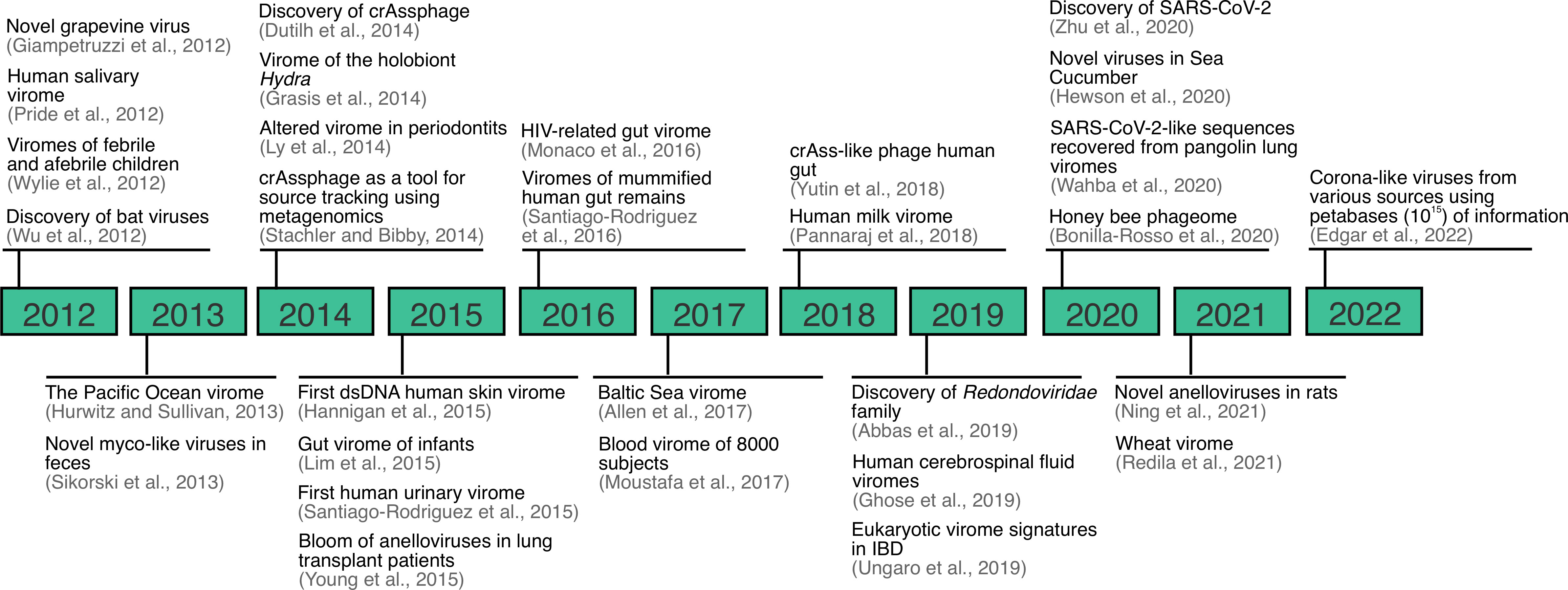
Figure 2 Overview of important viral metagenomic or virome studies landmarks between 2012-2022. Expanded from (28).
Viral metagenomics provides advantages compared to culture, EM, and molecular-based methods in that no culture is required, no a priori knowledge of the viral communities in a sample type is necessarily needed, and diverse viral families can be characterized simultaneously (Table 1). Virus discovery from metagenomic data presents its own challenges, however, including nucleic acid extraction, amplification, library preparation, data analysis, and their associated biases, as described previously (65). An additional challenge associated with viral metagenomic data analysis comes from the lack of similarity of most of the viral sequences to known viruses in reference databases. Viruses are constantly evolving, as are viral databases, highlighting the importance of maintaining updated and curated viral databases to support viral metagenomic classification. For example, in some cases up to 99% of the putative viral sequences in a sample cannot be classified taxonomically due to the high degree of sequence divergence with known viruses (66). While this represents an opportunity for virus discovery, there are several criteria that should ideally be met for a putative virus to be considered novel and which may also be dependent on the bioinformatic methods used. For instance, the criteria for crAssphage to be considered a novel virus included, but were not limited to sharing a low sequence identity to known viruses in databases, and a high identity to putative or uncharacterized sequences originating from the same genome, an average sequence length (bp) of known viruses, ability to predict a putative bacterial host from the assembled genome (in case a phage is being suspected), and the ability to predict Open Reading Frames (ORFs) sharing homology to known viral proteins (43). Similar criteria have been used for the discovery of RNA viruses. In the case of SARS-CoV-2, for instance, the low homology (< 90%) to known viruses within betacoronaviruses was one of the main criteria to support this as a novel virus (67). Given that many constituents of viral dark matter are distantly related (at best) to available reference genomes, additional computational and cultivation efforts may be required to fully characterize a novel virus (68). In the following sections, examples of discoveries made from viral dark matter, and the techniques applied in these efforts, are discussed.
Virus discovery through metagenomic sequencing is not trivial and typically requires the use of a variety of bioinformatic techniques including, but not limited to, in silico host sequence removal, read assembly, binning, alignment, co-occurrence assessment, phylogenetic characterization, and CRISPR analysis (Table 2). A bioinformatics framework incorporating each of these steps led to the discovery of crAssphage from human gut metagenomes and our understanding of Prevotella and Bacteroides spp. as its putative hosts (43, 77). As mentioned, this pioneering work identified a novel phage that we now know to be significantly distributed across the human gut (70), and predominant in the gut of industrialized cultures (78). The discovery of crAssphage also resulted in subsequent studies that enabled the discovery of crAss-like bacteriophage in human gut metagenomes by utilizing a similar suite of bioinformatic techniques, including searching for reads that did not align to nucleotide or protein reference databases, comparison with crAssphage proteins, and the prediction of open reading frames (Table 2) (69).
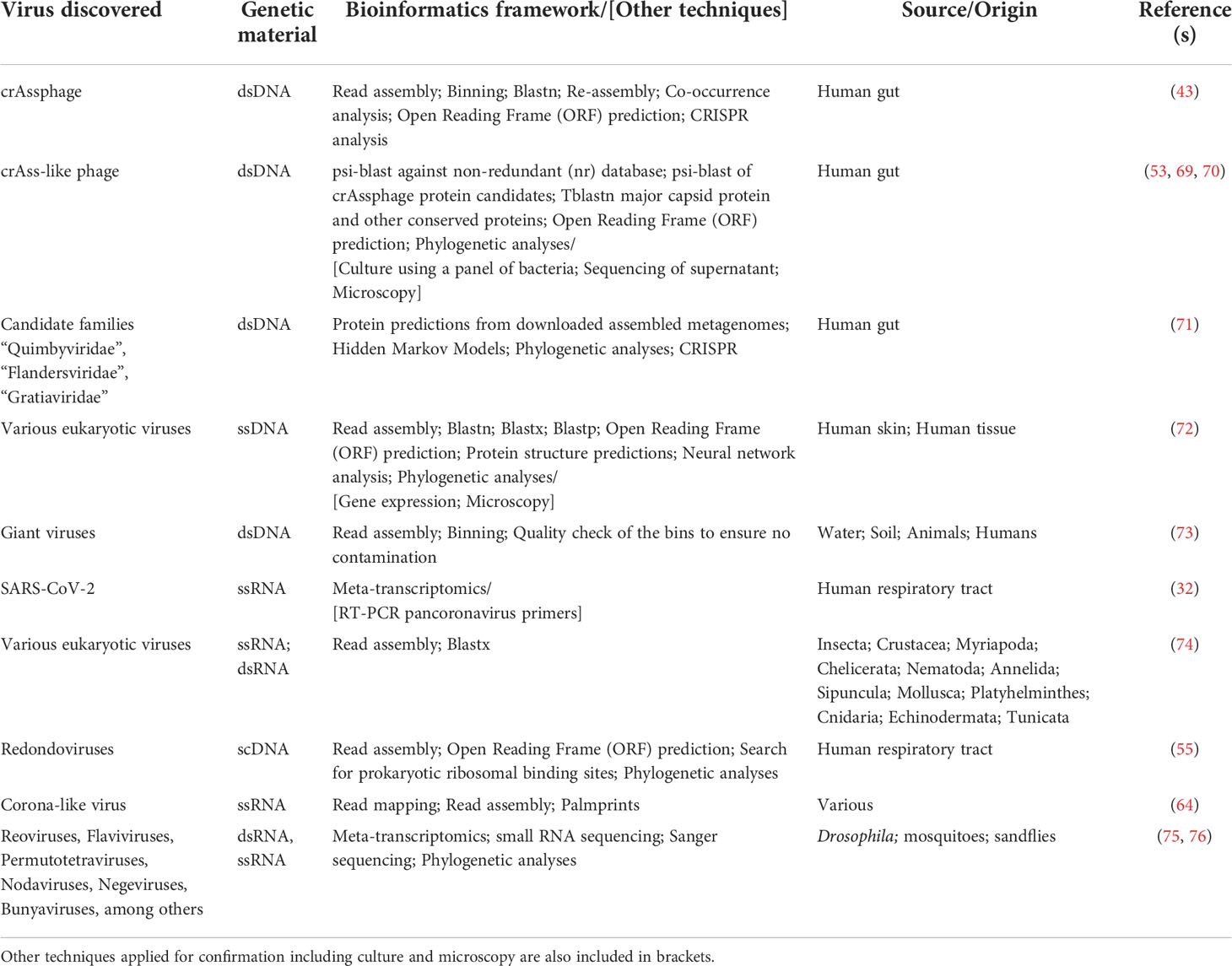
Table 2 Summary of bioinformatic tools used for virus discovery.
Subsequent studies have since classified crAss-like phage sequences into several proposed subfamilies depending on their genome composition and predicted putative host, which involved the use of microscopy and culture-based methods. Microscopy analysis showed that most crAss-like phage possess morphologies (i.e., short, non-contractile tails) typical of the Podoviridae family (53). Additional studies identified a variety of gut bacteria, including Agathobacter, Anaerostipes, several Bacteroides (including Bacteroides intestinalis), Blautia, Clostridium, Collinsella, Enterococcus, and Faecalibacterium spp., as potential putative hosts for crAssphage and crAss-like phage; however, B. intestinalis was eventually confirmed to be crAssphage bacterial host based on sequencing gut filtrates and each bacterial host (79). Discovery, isolation, and characterization of crAssphage and crAss-like phage demonstrate the challenges of expanding currently available viral genomic databases with reliable taxonomic classification. For this reason, it is anticipated that frameworks combining culture, EM, viral metagenomic sequencing, and/or other molecular techniques will continue to be needed to facilitate bacteriophage discovery and characterization.
The approaches used in the discovery of other viruses vary widely but share many similarities with those used in the discovery of crAssphage and crAss-like phage. For instance, a multilevel bioinformatic framework for virus taxonomic classification enabled the discovery of other phage infecting Bacteroidetes, as well as the discovery of potentially novel phage families (80). Briefly, this involved the search of predicted proteins with phage-specific Hidden Markov Models, phylogenetic analysis, phage genome assembly, and CRISPR analyses to identify potential bacterial hosts. This resulted in the discovery of > 3,700 phage genomes covering > 450 genera, and the characterization of the candidate families “Quimbyviridae”, “Flandersviridae” and “Gratiaviridae” (Table 2) (71). Each of the candidate phage families were unique in terms of their genome structure and composition. For instance, phage from the candidate “Gratiaviridae” family encode a HipA-family protein kinase and glycosyltransferase, which suggest a role in host cell wall modification to prevent superinfection by other phage (80). This study is another example of a multi-level bioinformatic framework applied for phage discovery and characterization.
Eukaryotic viruses include viruses that infect humans, non-human primates, plants, fungi, insects, and other eukaryotes. Eukaryotic viruses are diverse in genome structure and composition, morphology, and replication and infection mechanisms; yet, phage are more frequently described in DNA viral dark matter studies than eukaryotic viruses. While eukaryotic viruses are often studied as pathological agents, not all eukaryotic viruses result in disease progression. Indeed, eukaryotic DNA viruses have been identified in samples collected from subjects with no apparent pathologies or history of related comorbidities. This was the case in the characterization of the human urine virome in subjects with and without urinary tract infections, where most of the identifiable reads corresponded to phage and samples from 19 of the 20 study subjects carried reads matching human papillomaviruses (HPVs) (2). These were not HPVs typically associated with urogenital samples, but rather were HPVs that have previously been identified in other sample types (2). This study is intriguing as it demonstrates that, while some eukaryotic viruses may not strictly fit the definition of viral dark matter as they have been previously identified, they can occur in unexpected places.
Similarly, numerous eukaryotic viruses have been identified as part of the DNA virome of humans and other animals but are endogenous to other sample types. For instance, pathogenic eukaryotic DNA viruses known to infect plants and insects have been identified as part of the bat gut virome since plants and insects are part of their diets (81). Specifically, ssDNA viruses, including animal viruses from the Parvoviridae and Circoviridae families, as well as plant viruses from the Geminiviridae family often dominate the DNA viral fraction of bat guano, and the proteins encoded by these viruses often exhibit less than 60% amino acid identity to known viral sequence proteins, suggesting the presence of numerous novel viral species in bats (81). These data highlight the importance of characterizing plant and other animal viruses as these may also provide insights into the virome composition of other samples and environments.
As with phage, there are instances when eukaryotic DNA viruses do not share homology with known sequences, highlighting the importance of searching both the DNA and protein space to enable virus discovery. For instance, ssDNA viral genes from human skin swabs and tissue samples were identified by focusing on sequences that lacked protein structural predictions (72). Leveraging assembly of these unannotated reads in combination with nucleotide, translated, and protein BLAST searches and artificial neural networks, led to the identification of ten novel genome groups containing at least one protein cluster predicted to encode virion structural proteins. A subset of the predicted capsid proteins were then expressed in human-derived 293TT cells and/or in E. coli, and EM results showed that several of the predicted capsid proteins formed rough spherical particles (Table 2) (72). These results are intriguing as they suggest that a suite of bioinformatic tools can positively predict viral sequences that can then be expressed, aiding in virus discovery.
Another intriguing example of DNA viruses infecting eukaryotic cells is that of giant viruses. Giant viruses are defined as viruses with genomes harboring over 500 protein-encoding genes, average genome sizes of over 1.02Mb, and capsids of 370 to 600 nm in diameter. Giant viruses were officially described in 2003 with the discovery of a virus infecting Acanthamoeba polyphaga, an amoeba, using microscopy-based methods (82). This A. polyphaga mimivirus was originally thought to be an intracellular bacterium due to its large structural size (83, 84). Since then, giant viruses have been discovered from various sample types including water, soil, and animals using culture-based methods (83), and more recently, they have been identified in viral dark matter using metagenomics (73). Using a combination of metagenomic assembly, genome binning, and quality assessment to ensure no contamination, it has been shown that giant viruses possess a complex machinery that is not usually found in viruses. Specifically, certain giant viruses have been shown to encode the components necessary for glycolysis and the TCA cycle (73). Other techniques, including a combination of sorting DNA-stained particles, coupled with whole genome amplification and sequencing have been applied for the discovery of giant viruses from soil samples (85). These results demonstrate the range and combination of techniques that can be applied for the discovery of giant viruses in various sample types.
RNA viruses comprise most of the diversity of viruses infecting eukaryotic cells and have been historically discovered and characterized using culture and/or molecular-based methods (86, 87). RNA viruses, however, are not as well characterized as eukaryotic DNA viruses for several reasons (88). For instance, their small genome sizes make RNA viruses harder to detect in metagenomic data compared to DNA viruses, metagenomic sequencing methods targeting DNA tend to be better developed than those for RNA, and reference databases are typically biased towards DNA viruses and pathogens of economic importance. Each of these factors, and all of them in combination, makes RNA virus discovery more complex. High-throughput sequencing of RNA molecules, or meta-transcriptomics requires additional steps beyond a typical DNA-based metagenomic workflow in order to obtain reliable results. These include the synthesis of complementary DNA (cDNA) from messenger RNA (mRNA) and ribosomal RNA (rRNA) depletion.
RNA viruses have historically been of interest due to their potential to drive disease, epidemics, and pandemics, with SARS-CoV-2 being a recent example. Meta-transcriptomics, in combination with PCR techniques using pancoronavirus primers, enabled the discovery of SARS-CoV-2 (67). Meta-transcriptomics, however, is also a useful tool for the detection of non-pathogenic viruses. An example of this was the analysis of sequence data from 220 host species representing nine invertebrate phyla that led to the identification of >1,400 novel RNA viruses (74). In order to detect highly divergent viruses among these samples, the authors performed a combination of read assembly, domain-based blast against the Conserved Domain Database (CDD) for hits to RNA dependent RNA polymerases (RNA_dep_RNAP), and blastx searches of the putative viral assembled reads against the non-redundant (nr) protein database (Table 2), all while trying to balance discovery versus limiting false positive results (74). Several ssRNA, dsRNA, and dsDNA viruses were identified and discovered from the mentioned meta-transcriptomics datasets. As with the discovery of other viruses, this study is another example of the diversity of bioinformatic methods that can be used in conjunction with meta-transcriptomics to enable RNA virus discovery.
RNA virus discovery has also been enabled, in part, through small RNA sequencing. Particularly, small RNA sequencing has unraveled the role of RNA interference (RNAi) as an antiviral protection in insects, such as Drosophila spp. (75). Viruses in Drosophila spp. have been discovered historically using classical methods, which have enabled the discovery of viruses such Drosophila C Virus (DCV) (89), and Drosophila X Virus (DXV) (90). Further, transcriptomic analyses in Drosophila melanogaster revealed the presence of Nora virus (91). However, as with many environments, viral diversity in insects, including Drosophila, remains largely unexplored, but studies thus far have suggested an ongoing antiviral immune response in insects. Specifically, it has been suggested that the presence of RNAi in Drosophila spp. is characteristic of an antiviral response, and that these sequences may represent active viral infections. By using meta-transcriptomics and small RNA sequencing, sequence assembly, and targeted Sanger sequencing to improve completeness, over 20 partial viral genomes that comprised > 3.0% of all the sequences have been identified. The putative viruses identified included, but were not limited to Reoviruses, Flaviviruses, Permutotetraviruses, Nodaviruses, Negeviruses, and Bunyaviruses. Further analyses were not able to place the newly identified viruses within a phylogeny of known viruses, confirming their novelty (75). Similar approaches have been employed for sandflies and mosquitoes, showing six novel viruses belonging to viral families known to be pathogenic to mammals (e.g. Bunyaviridae and Reoviridae families) (76).
As described in the previous sections, virus discovery from the dark matter often relies upon using sequence alignment methods against reference viral genes and genomes, as well as k-mer-based methods, which are used to predict a putative virus based on genomic and sequence signatures. Examples of several bioinformatic tools, as well as advantages and disadvantages for several of these sequence- and k-mer-based methods have previously been described (10). Other approaches, particularly Artificial Intelligence (AI), have also been applied in virus discovery from the viral dark matter. Specifically, deep learning, which uses deep artificial neural networks to ‘learn’ features from a given input and predict the output, have shown to be valuable in virus discovery from metagenomic datasets. Recent tools such as DeepVirFinder (92), and VIBRANT (93), utilize deep learning to predict viruses with success and higher accuracy compared to other methods.
Viruses can be directly or indirectly associated with health and disease; yet, there is much to learn still about the contribution of the viral dark matter in health, disease, therapeutics, and surveillance efforts (Figure 3). The following section discusses known and unknown viruses in association with disease, including inflammatory bowel disease (IBD) and periodontitis, as well as the potential of the viral dark matter in therapeutics, particularly phage therapy, and surveillance.
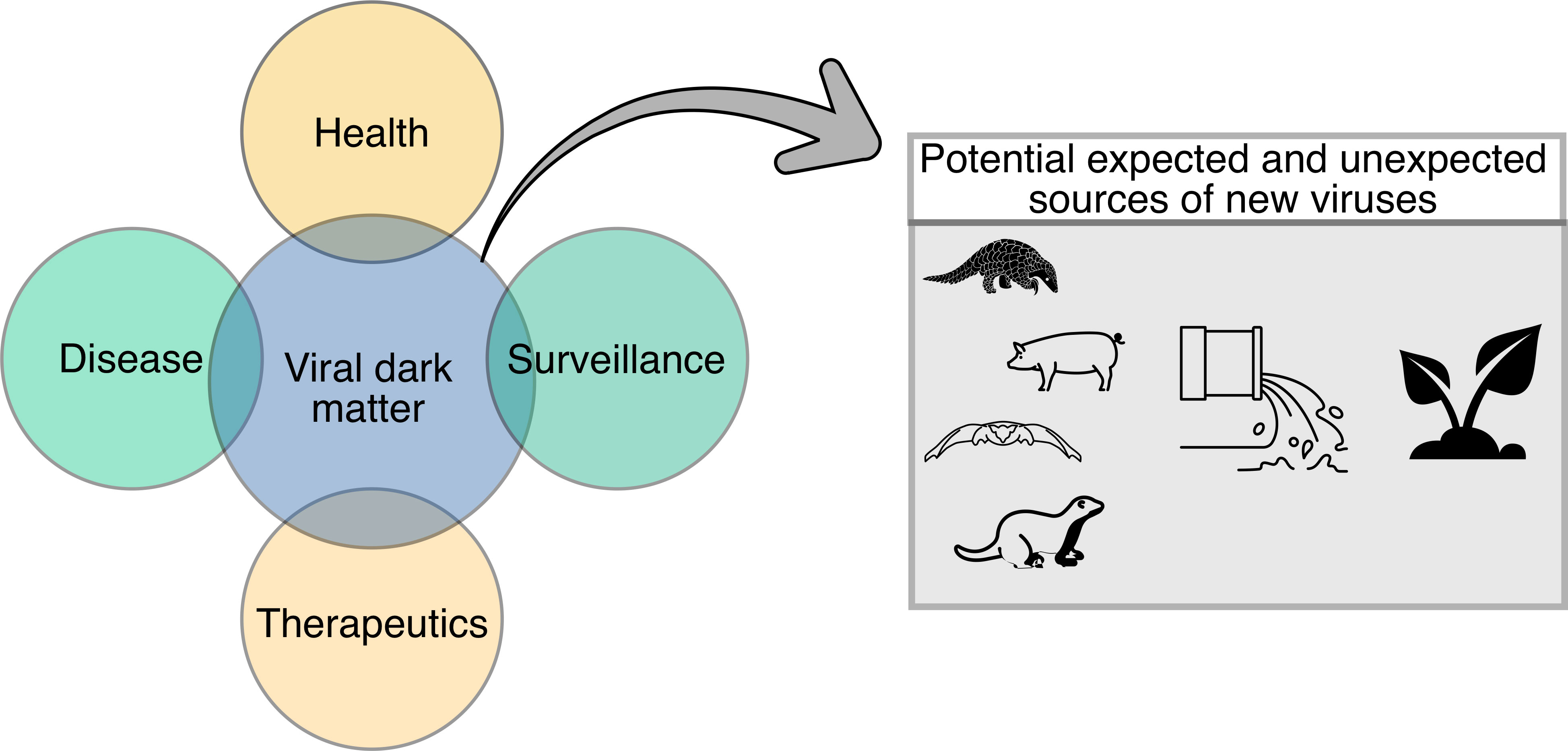
Figure 3 Diagram demonstrating potential applications of the viral dark matter in health, disease, therapeutics, and surveillance. Potential expected and unexpected sources of new viruses (i.e., animal, and environmental) that could contribute to the viral dark matter are also shown.
IBD is a chronic disorder of the intestinal tract that can result in periods of flare and remission. IBD, which includes Crohn’s disease (CD) and ulcerative colitis (UC), has an unknown etiology; yet, IBD appears to be multifactorial in nature and has been repeatedly associated with alterations of the human gut microbiome and more recently, the virome (94–96). Pioneer virome analyses of patients with IBD have found an increased viral richness and a decrease in bacterial diversity relative to control subjects, with the Caudovirales being the predominant viral order (94). Subsequent virome analyses of the same dataset focused on replication cycles (lytic vs lysogenic), as well as CRISPR analyses to determine potential bacterial host. The study identified an increased abundance of viruses belonging to the Siphoviridae and Myoviridae families, specifically those with lysogenic lifestyles (97). This study also found that a healthy gut virome is dominated by lytic phage, suggesting that these may be involved in lysing bacteria associated with disease progression (97). Notably, only 14% of the sequences were associated with known viruses in the original study, suggesting that that viral dark matter may be relevant to IBD onset and progression. These results also illustrate the need to expand viral DNA and RNA databases, which in turn can help elucidate the identity of uncharacterized viruses that are associated with disease.
The human oral cavity is the second most characterized body site after the human gut and is known to possess various biogeographical and ecological niches such as saliva, and subgingival and supragingival plaque. The human oral cavity is home to over 1,000 bacterial species including Streptococcus spp., Lactobacillus spp. and Veillonella spp. (98, 99). Dysbiosis in the oral cavity can result in inflammation and the development of periodontal diseases, which can affect over 30% of the adult population. Historically, periodontitis has been associated with the ‘red complex’ which includes Porphyromonas gingivalis, Treponema denticola and Tannerella forsythia (100). However, periodontitis is likely multifactorial, having both immunological and microbial components (100).
The oral cavity also harbors viruses, many of which are associated with maintaining health or promoting disease (101). Interestingly, the oral cavity is characterized by having robust phage communities from the Siphoviridae, Myoviridae and Podoviridae families, each of which have been associated with health and periodontal disease (3). Specifically, myoviruses in supragingival plaque are predominant in subjects with periodontitis, whereas siphoviruses are predominant in the supragingival plaque of subjects without periodontal disease (45). Interestingly, the opposite has been noted in saliva samples, with myoviruses being predominant in subjects without periodontitis. Viral host taxonomy varies by sample type in subjects with periodontitis. For example, viruses infecting Firmicutes tend to be more abundant in saliva, while viruses infecting Actinobacteria tend to be more abundant in supragingival plaque. Phage infecting Bacteroidetes are also more predominant in individuals without periodontal disease (45). The proportion of identifiable viral reads in the oral cavity also tends to be low, but it has been reported to be as great as 40% in some cases. Given that well over half of the reads cannot be attributed to a specific viral source suggests that viral dark matter comprises most of the oral virome (45) and that the identity of most viruses in the oral cavity and their potential role(s) in health and disease remain to be elucidated.
Specific members of the viral dark matter have been associated with periodontal disease. A recent study showed the association of novel respiratory eukaryotic viruses with periodontal disease (55). Metagenomic sequencing of lung viromes, subsequent de novo read assembly, ORF prediction, prokaryotic ribosomal binding site searches, and phylogenetic analysis resulted in the discovery of a small circular DNA (scDNA) virus from a family named Redondoviridae (Table 2) (55). Further studies demonstrated that these were neither laboratory contaminants, nor phage. Further analysis of metagenomes from subjects with periodontal disease found that redondoviruses were predominant in these datasets (55). These data are intriguing as it shows an association between inflammation and the respiratory viral dark matter.
As described, phage can make up a large fraction of the viral dark matter in some environments. This universality of phage, their host-specificity, and their ability to lyse bacterial hosts make them ideal candidates to treat infections caused by antibiotic-resistant bacteria (ARB). The application of phage in this way, also known as phage therapy, has shown renewed interest in recent years as a way to treat ARB infections (102). Initial disinterest in phage therapy arose from mistakes made during early trials, along with the discovery of antibiotics, which made treatment relatively straightforward. However, phage therapy has again proven to be effective against infection caused by various bacteria including, but not limited to Pseudomonas aeruginosa (102, 103), Clostridium difficile (104), and Enterococcus faecalis (105). Single bacteriophage and phage cocktails are readily available and can be used to target certain ARB.
Phage therapy can also become personalized when available phage and phage cocktails do not efficiently lyse target ARB. Screening for ideal phage candidates to be used in phage therapy can be time and labor intensive. The search for phage that could efficiently lyse target ARB can involve screening samples, such as sewage (as it is an ideal sample type to find human-associated viruses) (106). Once a candidate phage is found, it should be further characterized to understand host range, as ideal phage should be specific to the bacterial strain of interest (106). In some cases, the candidate phage may be known, but in other cases, suitable phage can be unknown and be part of the viral dark matter; therefore, genome sequencing and characterization should be performed to understand genome composition and confirm potential bacterial host. Genome characterization will also ensure that the phage is strictly lytic and that complete or near complete lysis of the bacteria causing infection will be accomplished. In the case of the candidate phage being lysogenic or temperate, genome engineering may be considered as an approach to produce suitable phage candidates for phage therapy against specific pathogens (107).
Understanding the source and evolution of potential emerging and re-emerging pathogens is essential for surveillance efforts. This is when viral metagenomics becomes an important surveillance tool as it allows numerous viruses to be identified simultaneously from various sources and facilitates using one or several of the above-mentioned tools. Virus discovery from viral dark matter is increasingly recognized as an important aspect of surveillance efforts. For instance, while bats are recognized as a resource of novel coronaviruses and an important host for coronavirus evolution (108, 109), viral metagenomic approaches are identifying a variety of novel and unexpected hosts of corona-like viruses (110). Specifically, a recent study leveraging high-throughput compute infrastructure, RNA-dependent RNA polymerase sequences (RdRP), which are characteristic of RNA viruses lacking a DNA stage, and >3 million metagenomics, metatranscriptomics, and virome datasets identified over 130,000 novel RNA viruses, nine of which were novel coronaviruses (64). These novel RNA viruses represented approximately 0.1% of the total virome analyzed. The study also concentrated on corona-like viruses including Microhyla alphaletovirus 1 (MLeV) in the frog Microhyla fissipes, and Pacific salmon nidovirus (PsNV), identifying samples containing corona-like virus-aligned reads and/or k-mers and performing de novo assembly using coronaSPAdes (111). This resulted in 70 species-like operational taxonomic units (sOTUs), 44 of which were found to be described in public databases, and 17 corona-like virus sOTUs contained partial RdRP. The remaining nine sOTUs were identified as novel corona-like viruses, as they exhibit protein domains consistent with a corona-like viral genome (Table 2). This study has revolutionized virome and viral dark matter research in that it has introduced an approach for petabase (1015)-scale genomics, which can aid in reducing the viral dark matter and improve surveillance efforts of potential emergent viruses from unexpected sources.
For the last 20 years, viral metagenomics has shown to be a powerful tool for the discovery of viruses. Viral metagenomics continues to be essential in the characterization of known viruses in diverse sample types, and in association with health, disease, immune system, biogeochemical cycles, therapeutics, and surveillance. Moreover, identification of novel viruses from viral dark matter continues to be possible with viral metagenomics. Virus discovery through viral metagenomics, however, does not come without challenges. Many such challenges that are intrinsic to the technique including viral purification, nucleic acid extraction and amplification, library preparation, sequencing, and bioinformatics. Examples of the bioinformatic frameworks that could be applied for virus discovery from viral dark matter are highlighted in the present review and demonstrate that no universal framework applies to the discovery of all viruses, but are rather diverse and should be suited for the virus(es) of interest. Virus discovery from viral metagenomics data may also require the application of culture, molecular and/or EM techniques.
TS-R original draft preparation. TS-R and EH: review and editing of manuscript draft. All authors contributed to the article and approved the submitted version.
TS-R and EH are current employees of Diversigen, a subsidiary of OraSure Technologies and a microbiome services company.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Breitbart M, Rohwer F. Here a virus, there a virus, everywhere the same virus? Trends Microbiol (2005) 13:278–84. doi: 10.1016/j.tim.2005.04.003
2. Santiago-Rodriguez TM, Ly M, Bonilla N, Pride DT. The human urine virome in association with urinary tract infections. Front Microbiol (2015) 6. doi: 10.3389/fmicb.2015.00014
3. Pride DT, Salzman J, Haynes M, Rohwer F, Davis-Long C, White RA, et al. Evidence of a robust resident bacteriophage population revealed through analysis of the human salivary virome. ISME J (2012) 6:915–26. doi: 10.1038/ismej.2011.169
4. Haynes M, Rohwer F. The human virome. In: Metagenomics of the human body. New York, NY: Springer (2011) p. 63–77.
5. Anand U, Bianco F, Suresh S, Tripathi V, Núñez-Delgado A, Race M. SARS-CoV-2 and other viruses in soil: An environmental outlook. Environ Res (2021) 5:1–13. doi: 10.1016/j.envres.2021.111297
6. Chen Y-M, Sadiq S, Tian J-H, Chen X, Lin X-D, Shen J-J, et al. RNA Viromes from terrestrial sites across China expand environmental viral diversity. Nat Microbiol (2022) 7:1–12. doi: 10.1038/s41564-022-01180-2
7. Lodish H, Berk A, Zipursky SL, Matsudaira P, Baltimore D, Darnell J. Viruses: structure, function, and uses. In: Molecular cell biology, 4th edition. New York, NY: WH Freeman (2000).
8. Hadas H, Einav M, Fishov I, Zaritsky A. Bacteriophage T4 development depends on the physiology of its host escherichia coli. Microbiology (1997) 143:179–85. doi: 10.1099/00221287-143-1-179
9. Traylen CM, Patel HR, Fondaw W, Mahatme S, Williams JF, Walker LR, et al. Virus reactivation: A panoramic view in human infections. Future Virol (2011) 6:451–63. doi: 10.2217/fvl.11.21
10. Santiago-Rodriguez TM, Hollister EB. Human virome and disease: High-throughput sequencing for virus discovery, identification of phage-bacteria dysbiosis and development of therapeutic approaches with emphasis on the human gut. Viruses (2019) 11:1–26. doi: 10.3390/v11070656
11. Wylie KM, Weinstock GM, Storch GA. Emerging view of the human virome. Transl Res (2012) 160:283–90. doi: 10.1016/j.trsl.2012.03.006
12. Van Blerkom LM. Role of viruses in human evolution. Am J Phys Anthropol (2003) 122:14–46. doi: 10.1002/ajpa.10384
13. Gutiérrez D, Fernández L, Martínez B, Rodríguez A, García P. Bacteriophages: The enemies of bad bacteria are our friends! Front Young Minds (2016) 4. doi: 10.3389/frym.2016.00030
14. Harrison E, Brockhurst MA. Ecological and evolutionary benefits of temperate phage: What does or doesn’t kill you makes you stronger. BioEssays (2017) 39:1–6. doi: 10.1002/bies.201700112
15. Canchaya C, Fournous G, Chibani-Chennoufi S, Dillmann ML, Brüssow H. Phage as agents of lateral gene transfer. Curr Opin Microbiol (2003) 6:417–24. doi: 10.1016/S1369-5274(03)00086-9
16. Muniesa M, Colomer-Lluch M, Jofre J. Potential impact of environmental bacteriophages in spreading antibiotic resistance genes. Future Microbiol (2013) 8:739–51. doi: 10.2217/fmb.13.32
17. Wagner PL, Waldor MK. Bacteriophage control of bacterial virulence. Infect Immun (2002) 70:3985–93. doi: 10.1128/IAI.70.8.3985-3993.2002
18. Sharma AK, Dhasmana N, Dubey N, Kumar N, Gangwal A, Gupta M, et al. Bacterial virulence factors: Secreted for survival. Indian J Microbiol (2017) 57:1–10. doi: 10.1007/s12088-016-0625-1
19. Colavecchio A, Cadieux B, Lo A, Goodridge LD. Bacteriophages contribute to the spread of antibiotic resistance genes among foodborne pathogens of the enterobacteriaceae family - a review. Front Microbiol (2017) 8. doi: 10.3389/fmicb.2017.01108
20. Barr JJ, Auro R, Furlan M, Whiteson KL, Erb ML, Pogliano J, et al. Bacteriophage adhering to mucus provide a non-host-derived immunity. Proc Natl Acad Sci U S A (2013) 110:10771–6. doi: 10.1073/pnas.1305923110
21. Rohwer F, Prangishvili D, Lindell D. Roles of viruses in the environment. Environ Microbiol (2009) 11:2771–4. doi: 10.1111/j.1462-2920.2009.02101.x
22. Shioda S, Kasai F, Watanabe K, Kawakami K, Ohtani A, Iemura M, et al. Screening for 15 pathogenic viruses in human cell lines registered at the JCRB cell bank: Characterization of in vitro human cells by viral infection. R Soc Open Sci (2018) 5:1–10. doi: 10.1098/rsos.172472
23. Desheva Y. Introductory chapter: Human adenoviruses. In: Adenoviruses London, UK: IntechOpen (2019). doi: 10.5772/intechopen.82554
24. Skern T. 100 years poliovirus: From discovery to eradication. a meeting report. Arch Virol (2010) 155:1371–81. doi: 10.1007/s00705-010-0778-x
25. Houghton M. Discovery of the hepatitis c virus. Liver Int (2009) 29:82–8. doi: 10.1111/j.1478-3231.2008.01925.x
26. Hyman P. Phages for phage therapy: Isolation, characterization, and host range breadth. Pharmaceuticals (2019) 12:1–23. doi: 10.3390/ph12010035
27. Goldsmith CS, Miller SE. Modern uses of electron microscopy for detection of viruses. Clin Microbiol Rev (2009) 22:552–63. doi: 10.1128/CMR.00027-09
28. Mokili JL, Rohwer F, Dutilh BE. Metagenomics and future perspectives in virus discovery. Curr Opin Virol (2012) 2:63–77. doi: 10.1016/j.coviro.2011.12.004
29. Mokili JLK, Rogers M, Carr JK, Simmonds P, Bopopi JM, Foley BT, et al. Identification of a novel clade of human immunodeficiency virus type 1 in democratic republic of Congo. AIDS Res Hum Retroviruses (2002) 18:817–23. doi: 10.1089/08892220260139567
30. Sridhar S, To KKW, Chan JFW, Lau SKP, Woo PCY, Yuen KY. A systematic approach to novel virus discovery in emerging infectious disease outbreaks. J Mol Diagn (2015) 17:230–41. doi: 10.1016/j.jmoldx.2014.12.002
31. Woo PCY, Lau SKP, Chu C, Chan K, Tsoi H, Huang Y, et al. Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J Virol (2005) 5:1–10. doi: 10.1128/jvi.79.2.884-895.2005
32. Zhou P, Yang X, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature (2020) 579:270–3. doi: 10.1038/s41586-020-2012-7
33. Holmes EC. The evolution of endogenous viral elements. Cell Host Microbe (2011) 10:368–77. doi: 10.1016/j.chom.2011.09.002
34. Belyi VA, Levine AJ, Skalka AM. Unexpected inheritance: Multiple integrations of ancient bornavirus and ebolavirus/marburgvirus sequences in vertebrate genomes. PloS Pathog (2010) 6:1–13. doi: 10.1371/journal.ppat.1001030
35. Crochu S, Cook S, Attoui H, Charrel RN, De Chesse R, Belhouchet M, et al. Sequences of flavivirus-related RNA viruses persist in DNA form integrated in the genome of aedes spp. mosquitoes. J Gen Virol (2004) 85:1971–80. doi: 10.1099/vir.0.79850-0
36. Belyi VA, Levine AJ, Skalka AM. Sequences from ancestral single-stranded DNA viruses in vertebrate genomes: the parvoviridae and circoviridae are more than 40 to 50 million years old. J Virol (2010) 12458–62. doi: 10.1128/jvi.01789-10
37. Gilbert C, Feschotte C. Genomic fossils calibrate the long-term evolution of hepadnaviruses. PloS Biol (2010) 8:1–12. doi: 10.1371/journal.pbio.1000495
38. Giampetruzzi A, Roumi V, Roberto R, Malossini U, Yoshikawa N, La Notte P, et al. A new grapevine virus discovered by deep sequencing of virus- and viroid-derived small RNAs in cv pinot gris. Virus Res (2012) 163:262–8. doi: 10.1016/j.virusres.2011.10.010
39. Wu Z, Ren X, Yang L, Hu Y, Yang J, He G, et al. Virome analysis for identification of novel mammalian viruses in bat species from Chinese provinces. J Virol (2012) 86:10999–1012. doi: 10.1128/jvi.01394-12
40. Wylie KM, Mihindukulasuriya KA, Sodergren E, Weinstock GM, Storch GA. Sequence analysis of the human virome in febrile and afebrile children. PloS One (2012) 7:1–10. doi: 10.1371/journal.pone.0027735
41. Hurwitz BL, Sullivan MB. The pacific ocean virome (POV): A marine viral metagenomic dataset and associated protein clusters for quantitative viral ecology. PloS One (2013) 8:1–12. doi: 10.1371/journal.pone.0057355
42. Sikorski A, Massaro M, Kraberger S, Young LM, Smalley D, Martin DP, et al. Novel myco-like DNA viruses discovered in the faecal matter of various animals. Virus Res (2013) 177:209–16. doi: 10.1016/j.virusres.2013.08.008
43. Dutilh BE, Cassman N, McNair K, Sanchez SE, Silva GGZ, Boling L, et al. A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat Commun (2014) 5:1–11. doi: 10.1038/ncomms5498
44. Grasis JA, Lachnit T, Anton-Erxleben F, Lim YW, Schmieder R, Fraune S, et al. Species-specific viromes in the ancestral holobiont hydra. PloS One (2014) 9:1–13. doi: 10.1371/journal.pone.0109952
45. Ly M, Abeles SR, Boehm TK, Robles-Sikisaka R, Naidu M, Santiago-Rodriguez T, et al. Altered oral viral ecology in association with periodontal disease. MBio (2014) 5:1–13. doi: 10.1128/mBio.01133-14
46. Hannigan GD, Meisel JS, Tyldsley AS, Zheng Q, Hodkinson BP, Sanmiguel AJ, et al. The human skin double-stranded DNA virome: Topographical and temporal diversity, genetic enrichment, and dynamic associations with the host microbiome. MBio (2015) 6:1–13. doi: 10.1128/mBio.01578-15
47. Lim ES, Zhou Y, Zhao G, Bauer IK, Droit L, Ndao IM, et al. Early life dynamics of the human gut virome and bacterial microbiome in infants. Nat Med (2015) 21:1228–34. doi: 10.1038/nm.3950
48. Santiago-Rodriguez TM, Fornaciari G, Luciani S, Dowd SE, Toranzos GA, Marota I, et al. Natural mummification of the human gut preserves bacteriophage DNA. FEMS Microbiol Lett (2015) 363:1–8. doi: 10.1093/femsle/fnv219
49. Young JC, Chehoud C, Bittinger K, Bailey A, Diamond JM, Cantu E, et al. Viral metagenomics reveal blooms of anelloviruses in the respiratory tract of lung transplant recipients. Am J Transplant (2015) 15:200–9. doi: 10.1111/ajt.13031
50. Monaco CL, Gootenberg DB, Zhao G, Handley SA, Ghebremichael MS, Lim ES, et al. Altered virome and bacterial microbiome in human immunodeficiency virus-associated acquired immunodeficiency syndrome. Cell Host Microbe (2016) 19:311–22. doi: 10.1016/j.chom.2016.02.011
51. Moustafa A, Xie C, Kirkness E, Biggs W, Wong E, Turpaz Y, et al. The blood DNA virome in 8,000 humans. PloS Pathog (2017) 13:1–20. doi: 10.1371/journal.ppat.1006292
52. Zeigler Allen L, McCrow JP, Ininbergs K, Dupont CL, Badger JH, Hoffman JM, et al. The Baltic Sea virome: Diversity and transcriptional activity of DNA and RNA viruses. mSystems (2017) 2:1–16. doi: 10.1128/msystems.00125-16
53. Guerin E, Shkoporov A, Stockdale SR, Clooney AG, Ryan FJ, Sutton TDS, et al. Biology and taxonomy of crAss-like bacteriophages, the most abundant virus in the human gut. Cell Host Microbe (2018) 24:653–64. doi: 10.1016/j.chom.2018.10.002
54. Pannaraj PS, Ly M, Cerini C, Saavedra M, Aldrovandi GM, Saboory AA, et al. Shared and distinct features of human milk and infant stool viromes. Front Microbiol (2018) 9:1162. doi: 10.3389/fmicb.2018.01162
55. Abbas AA, Taylor LJ, Dothard MI, Leiby JS, Fitzgerald AS, Khatib LA, et al. Redondoviridae, a family of small, circular DNA viruses of the human oro-respiratory tract associated with periodontitis and critical illness. Cell Host Microbe (2019) 25:719–29. doi: 10.1016/j.chom.2019.04.001
56. Ghose C, Ly M, Schwanemann LK, Shin JH, Atab K, Barr JJ, et al. The virome of cerebrospinal fluid: Viruses where we once thought there were none. Front Microbiol (2019) 10. doi: 10.3389/fmicb.2019.02061
57. Ungaro F, Massimino L, Furfaro F, Rimoldi V, Peyrin-Biroulet L, D’Alessio S, et al. Metagenomic analysis of intestinal mucosa revealed a specific eukaryotic gut virome signature in early-diagnosed inflammatory bowel disease. Gut Microbes (2019) 10:149–58. doi: 10.1080/19490976.2018.1511664
58. Bonilla-Rosso G, Steiner T, Wichmann F, Bexkens E, Engel P. Honey bees harbor a diverse gut virome engaging in nested strain-level interactions with the microbiota. Proc Natl Acad Sci USA (2020) 117:7355–62. doi: 10.1073/pnas.2000228117
59. Hewson I, Johnson MR, Tibbetts IR. An unconventional flavivirus and other RNA viruses in the sea cucumber (Holothuroidea; Echinodermata) virome. Viruses (2020) 12:1–18. doi: 10.3390/v12091057
60. Wahba L, Jain N, Fire AZ, Shoura MJ, Artiles KL, McCoy MJ, et al. An extensive meta-metagenomic search identifies SARS-CoV-2-Homologous sequences in pangolin lung viromes. mSphere (2020) 5:1–8. doi: 10.1128/msphere.00160-20
61. Ning S-Y, Zhou M-M, Yang J, Zeng J, Wang J-P. Viral metagenomics reveals two novel anelloviruses in feces of experimental rats. Virol J (2021) 18:1–7. doi: 10.1186/s12985-021-01723-9
62. Redila CD, Prakash V, Nouri S. Metagenomics analysis of the wheat virome identifies novel plant and fungal-associated viral sequences. Viruses (2021) 13:2457. doi: 10.3390/v13122457
63. Breitbart M, Salamon P, Andresen B, Mahaffy JM, Segall AM, Mead D, et al. Genomic analysis of uncultured marine viral communities. Proc Natl Acad Sci USA (2002) 99:14250–5. doi: 10.1073/pnas.202488399
64. Edgar RC, Taylor J, Lin V, Altman T, Barbera P, Meleshko D, et al. Petabase-scale sequence alignment catalyses viral discovery. Nature (2022) 602:142–7. doi: 10.1038/s41586-021-04332-2
65. Santiago-Rodriguez TM, Hollister EB. Potential applications of human viral metagenomics and reference materials: Considerations for current and future viruses. Appl Environ Microbiol (2020) 86:1–12. doi: 10.1128/AEM.01794-20
66. Shkoporov AN, Hill C. Bacteriophages of the human gut: The “Known unknown” of the microbiome. Cell Host Microbe (2019) 25:195–209. doi: 10.1016/j.chom.2019.01.017
67. Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al. A novel coronavirus from patients with pneumonia in China 2019. N Engl J Med (2020) 382:727–33. doi: 10.1056/nejmoa2001017
68. Krishnamurthy SR, Wang D. Origins and challenges of viral dark matter. Virus Res (2017) 239:136–42. doi: 10.1016/j.virusres.2017.02.002
69. Yutin N, Makarova KS, Gussow AB, Krupovic M, Segall A, Edwards RA, et al. Discovery of an expansive bacteriophage family that includes the most abundant viruses from the human gut. Nat Microbiol (2018) 3:38–46. doi: 10.1038/s41564-017-0053-y
70. Honap TP, Sankaranarayanan K, Schnorr SL, Ozga AT, Warinner C, Lewis CM. Biogeographic study of human gut-associated crAssphage suggests impacts from industrialization and recent expansion. PloS One (2020) 15:1–14. doi: 10.1371/journal.pone.0226930
71. Fujimoto K, Kimura Y, Shimohigoshi M, Satoh T, Sato S, Tremmel G, et al. Metagenome data on intestinal phage-bacteria associations aids the development of phage therapy against pathobionts. Cell Host Microbe (2020) 28:380–9. doi: 10.1016/j.chom.2020.06.005
72. Tisza MJ, Pastrana DV, Welch NL, Stewart B, Peretti A, Starrett GJ, et al. Discovery of several thousand highly diverse circular DNA viruses. Elife (2020) 5:1–10. doi: 10.7554/eLife.51971
73. Moniruzzaman M, Martinez-Gutierrez CA, Weinheimer AR, Aylward FO. Dynamic genome evolution and complex virocell metabolism of globally-distributed giant viruses. Nat Commun (2020) 11:1–11. doi: 10.1038/s41467-020-15507-2
74. Shi M, Lin XD, Tian JH, Chen LJ, Chen X, Li CX, et al. Redefining the invertebrate RNA virosphere. Nature (2016) 540:539–43. doi: 10.1038/nature20167
75. Webster CL, Waldron FM, Robertson S, Crowson D, Ferrari G, Quintana JF, et al. ). the discovery, distribution, and evolution of viruses associated with drosophila melanogaster. PloS Biol (2015) 13:1–33. doi: 10.1371/journal.pbio.1002210
76. Aguiar ERGR, Olmo RP, Paro S, Ferreira FV, De Faria IJDS, Todjro YMH, et al. Sequence-independent characterization of viruses based on the pattern of viral small RNAs produced by the host. Nucleic Acids Res (2015) 43:6191–206. doi: 10.1093/nar/gkv587
77. Dutilh BE, Schmieder R, Nulton J, Felts B, Salamon P, Edwards RA, et al. Reference-independent comparative metagenomics using cross-assembly: CrAss. Bioinformatics (2012) 28:3225–31. doi: 10.1093/bioinformatics/bts613
78. Edwards RA, Vega AA, Norman HM, Ohaeri M, Levi K, Dinsdale EA, et al. Global phylogeography and ancient evolution of the widespread human gut virus crAssphage. Nat Microbiol (2019) 4:1727–36. doi: 10.1038/s41564-019-0494-6
79. Shkoporov AN, Khokhlova EV, Fitzgerald CB, Stockdale SR, Draper LA, Ross RP, et al. ΦCrAss001 represents the most abundant bacteriophage family in the human gut and infects bacteroides intestinalis. Nat Commun (2018) 9:1–8. doi: 10.1038/s41467-018-07225-7
80. Benler S, Yutin N, Antipov D, Rayko M, Shmakov S, Gussow AB, et al. Thousands of previously unknown phages discovered in whole-community human gut metagenomes. Microbiome (2021) 9:1–17. doi: 10.1186/s40168-021-01017-w
81. Li L, Victoria JG, Wang C, Jones M, Fellers GM, Kunz TH, et al. Bat guano virome: Predominance of dietary viruses from insects and plants plus novel mammalian viruses. J Virol (2010) 84:6955–65. doi: 10.1128/jvi.00501-10
82. La Scola B, Audic S, Robert C, Jungang L, de Lamballerie X, Drancourt M, et al. A giant virus in amoebae. Science (2003) 299:2033. doi: 10.1126/science.1081867
83. Colson P, La Scola B, Levasseur A, Caetano-Anollés G, Raoult D. Mimivirus: Leading the way in the discovery of giant viruses of amoebae. Nat Rev Microbiol (2017) 15:243–54. doi: 10.1038/nrmicro.2016.197
84. Brandes N, Linial M. Giant viruses-big surprises. Viruses (2019) 11:1–12. doi: 10.3390/v11050404
85. Schulz F, Alteio L, Goudeau D, Ryan EM, Yu FB, Malmstrom RR, et al. Hidden diversity of soil giant viruses. Nat Commun (2018) 9:1–9. doi: 10.1038/s41467-018-07335-2
86. Marston DA, McElhinney LM, Ellis RJ, Horton DL, Wise EL, Leech SL, et al. Next generation sequencing of viral RNA genomes. BMC Genomics (2013) 14:1–12. doi: 10.1186/1471-2164-14-444
87. Kolakofsky D. A short biased history of RNA viruses. RNA (2015) 21:667–9. doi: 10.1261/rna.049916.115
88. Wolf YI, Kazlauskas D, Iranzo J, Lucía-Sanz A, Kuhn JH, Krupovic M, et al. Origins and evolution of the global RNA virome. MBio (2018) 9:1–31. doi: 10.1128/mBio.02329-18
89. Jousset FX, Bergoin M, Revet B. Characterization of the drosophila c virus. J Gen Virol (1977) 34:269–83. doi: 10.1099/0022-1317-34-2-269
90. Teninges D, Ohanessian A, Richard-Molard C, Contamine D. Isolation and biological properties of drosophila X virus. J Gen Virol (1979) 42:241–54. doi: 10.1099/0022-1317-42-2-241
91. Habayeb MS, Ekengren SK, Hultmark D. Nora Virus, a persistent vitus in drosophila, defines a new picorna-like virus family. J Gen Virol (2006) 87:3045–51. doi: 10.1099/vir.0.81997-0
92. Ren J, Song K, Deng C, Ahlgren NA, Fuhrman JA, Li Y, et al. Identifying viruses from metagenomic data using deep learning. Quant Biol (2020) 8:64–77. doi: 10.1007/s40484-019-0187-4
93. Kieft K, Zhou Z, Anantharaman K. VIBRANT: automated recovery, annotation and curation of microbial viruses, and evaluation of viral community function from genomic sequences. Microbiome (2020) 8:1–23. doi: 10.1186/s40168-020-00867-0
94. Norman JM, Handley SA, Baldridge MT, Droit L, Liu CY, Keller BC, et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell (2015) 160:447–60. doi: 10.1016/j.cell.2015.01.002
95. Lloyd-Price J, Arze C, Ananthakrishnan AN, Schirmer M, Avila-Pacheco J, Poon TW, et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature (2019) 569:655–62. doi: 10.1038/s41586-019-1237-9
96. Liu S, Zhao W, Lan P, Mou X. The microbiome in inflammatory bowel diseases: from pathogenesis to therapy. Protein Cell (2021) 12:331–45. doi: 10.1007/s13238-020-00745-3
97. Clooney AG, Sutton TDS, Shkoporov AN, Holohan RK, Daly KM, O’Regan O, et al. Whole-virome analysis sheds light on viral dark matter in inflammatory bowel disease. Cell Host Microbe (2019) 26:764–78. doi: 10.1016/j.chom.2019.10.009
98. Dewhirst FE, Chen T, Izard J, Paster BJ, Tanner ACR, Yu WH, et al. The human oral microbiome. J Bacteriol (2010) 192:5002–17. doi: 10.1128/JB.00542-10
99. Baker JL, Edlund A. Exploiting the oral microbiome to prevent tooth decay: Has evolution already provided the best tools? Front Microbiol (2019) 9. doi: 10.3389/fmicb.2018.03323
100. Mohanty R, Asopa S, Joseph Md, Singh B, Rajguru J, Saidath K, et al. Red complex: Polymicrobial conglomerate in oral flora: A review. J Fam Med Prim Care (2019) 8:3480–6. doi: 10.4103/jfmpc.jfmpc_759_19
101. Ho SX, Min N, Wong EPY, Chong CY, Chu JJH. Characterization of oral virome and microbiome revealed distinctive microbiome disruptions in paediatric patients with hand, foot and mouth disease. NPJ Biofilms Microbiomes (2021) 7:1–8. doi: 10.1038/s41522-021-00190-y
102. Lin DM, Koskella B, Lin HC. Phage therapy: An alternative to antibiotics in the age of multi-drug resistance. World J Gastrointest Pharmacol Ther (2017) 8:162–73. doi: 10.4292/wjgpt.v8.i3.162
103. Khawaldeh A, Morales S, Dillon B, Alavidze Z, Ginn AN, Thomas L, et al. Bacteriophage therapy for refractory pseudomonas aeruginosa urinary tract infection. J Med Microbiol (2011) 60:1697–700. doi: 10.1099/jmm.0.029744-0
104. Nale JY, Redgwell TA, Millard A, Clokie MRJ. Efficacy of an optimised bacteriophage cocktail to clear clostridium difficile in a batch fermentation model. Antibiotics (2018) 7:1–15. doi: 10.3390/antibiotics7010013
105. Bolocan AS, Upadrasta A, De Almeida Bettio PH, Clooney AG, Draper LA, Ross RP, et al. Evaluation of phage therapy in the context of enterococcus faecalis and its associated diseases. Viruses (2019) 11:1–18. doi: 10.3390/v11040366
106. Gibson SB, Green SI, Liu CG, Salazar KC, Clark JR, Terwilliger AL, et al. Constructing and characterizing bacteriophage libraries for phage therapy of human infections. Front Microbiol (2019) 10. doi: 10.3389/fmicb.2019.02537
107. Chen Y, Batra H, Dong J, Chen C, Rao VB, Tao P. Genetic engineering of bacteriophages against infectious diseases. Front Microbiol (2019) 10. doi: 10.3389/fmicb.2019.00954
108. Tang XC, Zhang JX, Zhang SY, Wang P, Fan XH, Li LF, et al. Prevalence and genetic diversity of coronaviruses in bats from China. J Virol (2006) 80:7481–90. doi: 10.1128/jvi.00697-06
109. Lau SKP, Woo PCY, Li KSM, Huang Y, Wang M, Lam CSF, et al. Complete genome sequence of bat coronavirus HKU2 from Chinese horseshoe bats revealed a much smaller spike gene with a different evolutionary lineage from the rest of the genome. Virology (2007) 367:428–39. doi: 10.1016/j.virol.2007.06.009
110. Hale VL, Dennis PM, McBride DS, Nolting JM, Madden C, Huey D, et al. SARS-CoV-2 infection in free-ranging white-tailed deer. Nature (2022) 602:481–6. doi: 10.1038/s41586-021-04353-x
Keywords: crAssphage, dark matter, microbiome, phage, virome, virus discovery
Citation: Santiago-Rodriguez TM and Hollister EB (2022) Unraveling the viral dark matter through viral metagenomics. Front. Immunol. 13:1005107. doi: 10.3389/fimmu.2022.1005107
Received: 27 July 2022; Accepted: 31 August 2022;
Published: 16 September 2022.
Edited by:
Joel Henrique Ellwanger, Federal University of Rio Grande do Sul, BrazilReviewed by:
Vicente Pérez-Brocal, Fundación para el Fomento de la Investigación Sanitaria y Biomédica de la Comunitat Valenciana (FISABIO), SpainCopyright © 2022 Santiago-Rodriguez and Hollister. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tasha M. Santiago-Rodriguez, dHJvZHJpZ3VlekBkaXZlcnNpZ2VuLmNvbQ==; Emily B. Hollister, ZWhvbGxpc3RlckBkaXZlcnNpZ2VuLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.