
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 19 October 2022
Sec. Alloimmunity and Transplantation
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1000172
This article is part of the Research Topic Targeting Complements in Organ transplantation View all 3 articles
 Isabel F. Delaura1†
Isabel F. Delaura1† Qimeng Gao1†
Qimeng Gao1† Imran J. Anwar1
Imran J. Anwar1 Nader Abraham1
Nader Abraham1 Riley Kahan1
Riley Kahan1 Matthew G. Hartwig2
Matthew G. Hartwig2 Andrew S. Barbas1*
Andrew S. Barbas1*Organ shortages and an expanding waitlist have led to increased utilization of marginal organs. All donor organs are subject to varying degrees of IRI during the transplant process. Extended criteria organs, including those from older donors and organs donated after circulatory death are especially vulnerable to ischemia-reperfusion injury (IRI). Involvement of the complement cascade in mediating IRI has been studied extensively. Complement plays a vital role in the propagation of IRI and subsequent recruitment of the adaptive immune elements. Complement inhibition at various points of the pathway has been shown to mitigate IRI and minimize future immune-mediated injury in preclinical models. The recent introduction of ex vivo machine perfusion platforms provides an ideal window for therapeutic interventions. Here we review the role of complement in IRI by organ system and highlight potential therapeutic targets for intervention during ex vivo machine preservation of donor organs.
Transplantation provides a curative treatment for end-organ pathologies; however, its utility is limited by critical organ shortages. Over the last decade, organ waitlists have continued to grow faster than transplants performed, with 161,758 patients awaiting transplant in the United States in 2020 (1). Accordingly, strategies to combat the increasing organ shortage have focused on expanding the donor pool to include organs from extended criteria donors, the definition of which varies by organ type. In kidney transplantation, extended criteria donors includes donors with acute kidney injury (AKI) (2, 3) and those with a high Kidney Donor Profile Index (KDPI) (4, 5). KDPI considers donor demographics (height, weight, age, ethnicity), comorbidities (diabetes, hypertension), laboratory parameters (creatinine, hepatitis C serology), and other characteristics such as cause of death and donation after circulatory death (DCD) status. Marginal livers—those donor organs with characteristics associated with early graft failure and worse overall outcomes—include allografts with significant steatosis, DCD, and those from elderly and Hepatitis C positive donors (6, 7). Extended criteria heart donors are those older than 65, or with significant comorbidities such as poorly controlled diabetes, renal insufficiency, and peripheral arterial disease (8). In lung transplantation, extended criteria donor fulfills two or more of the following criteria: age ≥ 55, smoking history ≥ 20 pack-year, pO2 ≤ 300, diabetes, purulence found on bronchoscopy, bloodstream infection, and abnormal chest x-ray (9). Among extended criteria organs, transplant using DCD organs has substantially broadened the donor pool. DCD currently accounts for 25% of kidney deceased organ donation (1), 10% of liver donation (10) and 7.4% of all lung donations (11). Since the first DCD heart transplant in the US in 2019, 3.3% of all heart transplants in 2020 used DCD organs (12).
However, increased utilization comes at a cost of potentially increased incidence of early graft dysfunction and compromised long-term outcomes. Both DCD (13) and extended criteria donor (14) kidney transplants result in a higher incidence of graft loss and delayed graft function (DGF). DGF is further associated with increased graft loss and acute rejection in kidney transplant (15). In both liver (16, 17) and heart transplants (8), marginal donors are associated with higher rates of graft loss and recipient mortality. While inclusion of marginal organs in transplantation may provide benefit over remaining on the waitlist, it is associated with compromised short- and long-term outcomes compared to standard criteria donor organs.
Marginal organs are more vulnerable to insults such as ischemia-reperfusion injury (IRI); thus, particular attention is paid to minimizing these insults in DCD and extended criteria organ transplantation. One approach is to minimize cold ischemic time (CIT), which is typically shorter in DCD compared to DBD transplants (18). Recent improvements in liver transplantation outcomes using elderly donors organs have been associated with reduction in cold ischemia time as well (19). Another strategy to minimize IRI is the delivery of therapeutic agents targeting various pathways implicated in IRI pathogenesis, such as the complement system, a multi-functional component of the innate immune system. Various complement-targeting therapeutics have recently become clinically available, offering an opportunity for application to organ transplantation (20). Concurrently, ex vivo machine perfusion technology has been developed for organ preservation and evaluation. This platform may be utilized for the delivery of various therapeutics. In this review, we first outline the role of complement in IRI, and highlight the current evidence evaluating complement inhibition as preventative therapy for IRI. We also review the advantages of and available literature on ex vivo delivery of complement inhibition during donor organ preservation. The review focuses predominantly on kidney transplant, given that most studies have been performed in kidney transplant; however, we will highlight studies performed in other organs as well as important differences in complement inhibition across organ systems.
Complement is a key driver of IRI pathogenesis (21, 22). In brief, the complement system may be activated through three initial pathways: classical via binding of C1q to the Fc region of antibody, lectin via recognition of mannose-binding lectins (MBL) and activation of MASP proteases, or alternative via C3 hydrolysis (Figure 1). All three initial pathways converge on the formation of a C3 convertase and the common pathway. Anaphylatoxins and split products C3a and C5a serve several purposes, including inflammatory mediation and neutrophil chemotaxis. Split product C5b begins the terminal pathway by complexing with proteins C6-9, forming the membrane attack complex (MAC), which facilitates cell lysis. Under normal homeostasis, the complement system is strictly regulated to prevent unwanted tissue damage. Soluble proteins, such as C1 esterase inhibitor (C1 INH), Factor I and H, Clusterin and vitronectin, and membrane-bound proteins, such as membrane cofactor protein (CD46), Decay-accelerating factor (CD55), CD59 and complement receptor I, regulate various steps of the three complement pathways, inhibiting further propagation of the cascade when activated (23–25).
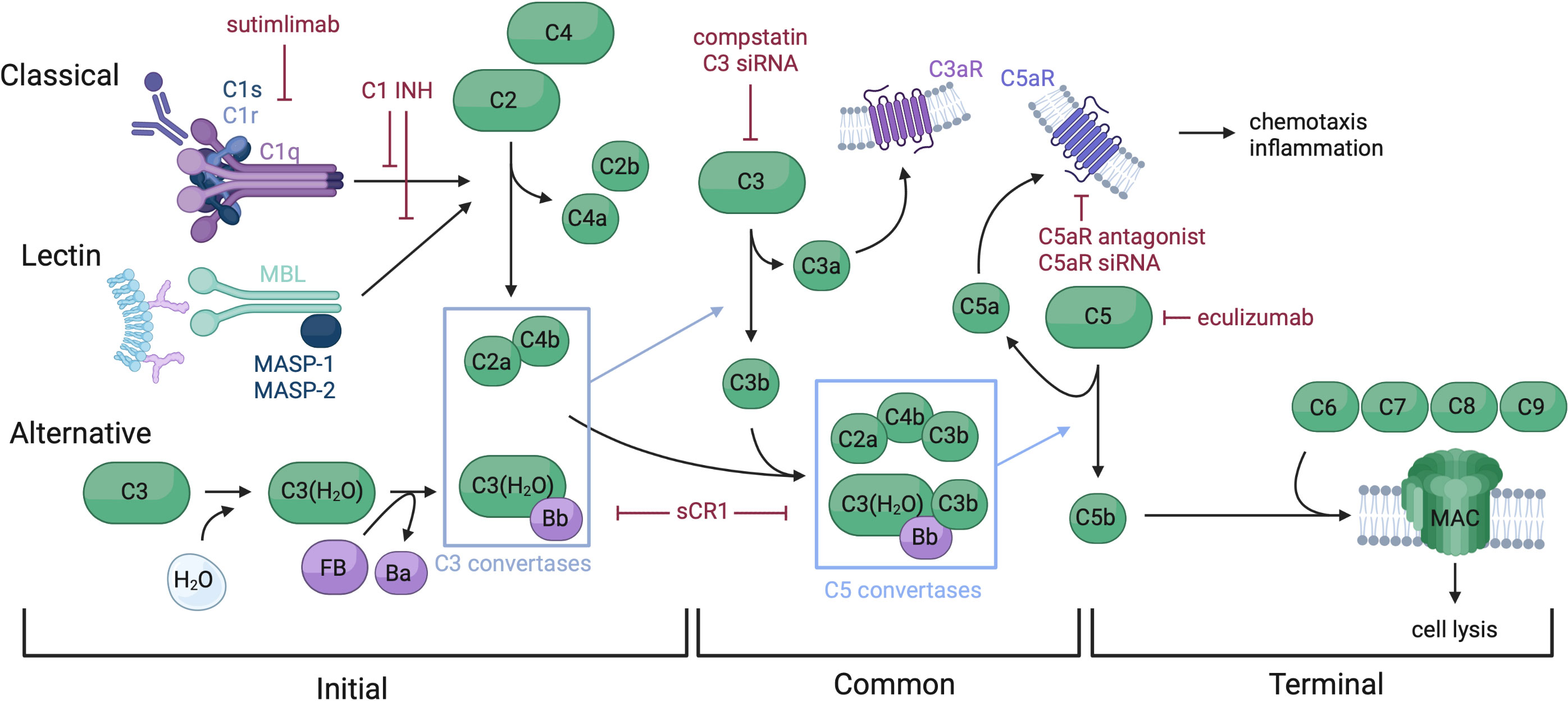
Figure 1 Overview of the complement system and therapeutic targets. The complement system is comprised of soluble, membrane bound, and regulatory proteins, and may be activated via three distinct pathways: classical, lectin, and alternative. These pathways converge on the common pathway with the formation of a C3 convertase which splits C3 into C3a and C3b. C3b complexes to the C3 convertases to form a C5 convertase, which splits C5 and triggers assembly of the MAC, making up the terminal pathway. Complement functions include cell lysis via MAC insertion into cell membranes and chemotaxis and inflammation regulated by split proteins C3a and C5a. Therapeutics targeting several complement components have been developed, including C1 INH which inactivates C1s, C1r and MASP-1 and -2, thus preventing spontaneous activation of the classical and lectin pathways. Sutimlimab is an anti-C1s mAb approved for cold agglutinin disease. sCR1, and versions of this molecule such as mirococept inhibit C3 and C5 convertases, among other functions. Multiple C3 targeting therapeutics have been developed, such as compstatin and C3 siRNA. C5 and C5aR have been targeted by drugs such as eculizumab (anti-C5 mAb), C5aR antagonist, and C5aR siRNA.
From an organ transplantation perspective, the disturbance of such a delicate balance occurs immediately following brain death in organ donors. Systemically, increased levels of C5a (26–28) and C5b-9 were found in the serum of deceased donors, compared to living donors. Upon reperfusion, there appears to be a transient release of C5b-9 detected in recipient serum (29). At the organ level, most complement proteins are synthesized in the liver, while kidney and lung are two of the few organs capable of synthesizing complement locally. Exploring the differences in gene expression profiles between living and deceased donors, Naesens et al. found significant over-expression of many complement components, including C1q, C1s, C1r, C2, C4 and Factor B, a finding later confirmed using microarray technique by Damman et al. (30). In a syngeneic rat kidney transplant model, local C3 deposition on endothelium and glomeruli was found one hour post reperfusion and persisted until posttransplant day 5 in DBD transplant but not living donor kidneys (31). Additionally, higher renal C3 gene expression was observed in DBD rats compared to living donors (32).
The etiology of complement activation in deceased donors is thought to be related to the release of endogenous damage-associated molecular pattern molecules (DAMPs) and sterile inflammation (33). Although most studies have examined complement activation in standard DBD donors, intuitively, the degree of inflammation and thus complement activation may be more profound in DCD and extended criteria donors. Accordingly, comparisons of transcriptional profiles and histology have shown increased activation of inflammatory chemokines, complement, and coagulation in DCD and extended criteria kidneys, compared to standard criteria donors (34, 35).
IRI is inevitable to a degree in the process of organ transplantation, which may be further exacerbated depending on procurement method and storage. The underlying pathways driving IRI are numerous and affect many cellular components: initial ischemia leads to hypoxia, ATP depletion, and subsequent tissue necrosis. Injury is worsened during reperfusion through the production of ROS (36, 37). IRI injury results in the release of DAMPs, which in turn activate complement, perpetuating an ongoing inflammatory cycle. The particulars of complement modulation and pathogenesis of IRI vary widely depending on organ system (21, 38–40).
It is well established that complement mediates renal IRI. Mice lacking common pathway components such as C3 or C5, or C6 knockout mice unable to assemble membrane attack complex are protected from IRI (41). While the involvement of the classical pathway in IRI pathogenesis is well described (42), recent studies indicated an essential role for both lectin and alternative pathways. In murine models, mice deficient in Factor B (alternative pathway) (43), collectin-11 or Mannan-binding lectin (lectin pathway) (44, 45) were protected from renal IRI. Conversely, RAG-1 knockout animals deficient in Ig remained susceptible to IRI (46). Lastly, although the liver synthesizes the majority of circulating complement, complement proteins locally synthesized in the kidney plays an important role in injury. In a mouse kidney transplant model, Pratt et al. showed that donor allografts unable to synthesize C3 experienced reduced allograft rejection when transplanted into recipients with intact complement production; however, transplant of a wild-type kidney into C3 knockout mice did not see the same benefit (47). In human studies, donor C3 allotype was found to influence clinical kidney transplant outcomes (48).
In cardiac IRI models, injury is correlated with C3d deposition, and mouse studies have shown decreased tissue injury, troponin I levels, and immune cell infiltration with C3 knockout (49). In contrast to renal IRI, the classical pathway appears to be the prominent driver of myocardial IRI, with higher rates of IgM and C4d deposition in DBD hearts (49, 50).
The liver is unique as it is the primary location of complement synthesis and contains the largest collection of phagocytic cells in body. Liver tissue also expresses multiple complement receptors such as CR1, CR3, CR4, and C5aR, as well as CR immunoglobulin (51). Complement activation plays a key role in mediating hepatic IRI, as C6-knockout mice produce lower levels of pro-inflammatory cytokines, resulting in improved liver function and less IRI (52). Interestingly, complement activation products C3a and C5a are also key in liver regeneration. In a mouse 70% partial hepatectomy model, C3- and C5-deficient mice succumb to liver failure, a phenotype rescued by the reconstitution of effector molecules C3a and C5a (53, 54). As a result, the use of complement inhibition for prevention and treatment of hepatic IRI must be carried out thoughtfully so as not to compromise liver regeneration.
Complement activation in lung IRI has been recognized and linked to primary graft dysfunction (PGD). Various complement inhibitors have been tested in this setting in both animal models and human studies (55). Shah et al. observed an increase in plasma C5a and C4a in patients with PGD (56) and Westall et al. described significant C3d and C4d staining associated with PGD (57). It is likely all three complement pathways are involved in pulmonary IRI, as increased complement products from all three pathways were found in BAL from patients with PGD compared to those without (58). Like the kidney, the lung is capable of synthesizing complement locally: type II epithelial cells synthesize and secrete complement proteins C2, C3, C4, C5 and factor B (59).
Overall, complement pathways behave differentially across different organ systems in the pathogenesis of IRI, posing unique challenges for complement blockade-based therapeutics.
The complexity of the complement system offers many therapeutic targets, which fall into three broad categories: complement cascade proteins, complement receptors, and complement regulatory proteins that naturally counter complement activation. Many have targeted the common pathway of the complement cascade. In two earlier studies, Zheng et al. used small interfering RNA (siRNA) to silence complement proteins—namely C3 and C5aR—in a murine renal IRI model. Their initial study demonstrated successful C3 and caspase 3 silencing when siRNA was delivered 48 hours prior to the induction of ischemia. Upregulation of C3 was seen only in controls, while the treatment group had lower BUN and creatinine and prolonged mouse survival. Histological exam showed decreased tubular infarction, immune cell infiltration, and necrosis in mice treated with a combination of C3 and caspase 3 siRNA, as well as an 87% reduction in total injury area (60). They went on to test C5aR inhibition using the same RNA interference approach, demonstrating less injury with C5aR inhibition compared to controls (61). Another approach to C5aR antagonism—the use of an acetate salt compound—resulted in improved clinical renal function in a rat model of renal IRI. TNF-alpha was decreased in treated animals, and biopsies showed decreased severity of tubular injury. In comparison to C5 blockade, C5aR antagonism did not result in inhibition of MAC assembly, as demonstrated by comparable hemolytic assay results in the treatment and control groups (62).
Additionally, studies have utilized complement receptor type 1 (CR1), also known as Cb/C4b receptor, which accelerates the degradation of C3 and C5 convertases, as well as activates factor I, leading to degradation of C3b and C4b (63). In a murine renal IRI model, Hameed et al. demonstrated improved serum creatinine, reduced neutrophil influx and superoxide production in the sCR1 treatment group (64). Bongoni et al. evaluated CSL040, a truncated version of sCR1 with greater inhibitory activity and an extended half-life (65). When compared to soluble human CR1 (TP10), CSL040 treatment prior to and after ischemic insult resulted in improved renal function, decreased incidence of tubular injury, and suppression of circulating C3b and C5a. Interestingly, biopsies from the CSL040 group showed decreased deposition of complement proteins and regulators from all pathways—C3d, C4d, C9, C1q, MBL, and Factor Bb—indicating robust complement suppression (66).
An alternative approach is to selectively inhibit upstream pathways. This approach is attractive as IRI in different organs seems to activate the complement system through distinct pathways. C1 INH blocks the classical and lectin pathways and is approved for clinical use in hereditary angioedema. In a brain-dead rat model, C1INH was found to reduce complement activation induced by brain death (67). In a murine renal IRI model, animals pre-treated with C1 INH have significantly better renal function and better survival. Less C5a release, C3b deposition and neutrophil/macrophage infiltration were observed. Interestingly, reduced tissue fibrosis and TGF-β1 levels were noted 30 and 90 days following the initial ischemic insult (68). In a nonhuman primate kidney transplant model, C1 INH has been shown to reduce complement deposition on biopsy and prevent DGF when delivered systemically to the brain-dead donor only (69) or recipient only (70). Lastly, in a porcine kidney autotransplant model, C1 INH treatment promoted more rapid glomerular function recovery and significantly prevented graft fibrosis in the long term (71).
Complement-targeting agents have also been studied in different organs to minimize IRI. Multiple groups have demonstrated the utility of C1 INH in liver IRI. Lehmann et al. showed, in a rat model, improved microvascular perfusion and decreased leukocyte-endothelial cell interaction on in vivo microscopy (72, 73). Heijnen et al. redemonstrated amelioration of hepatic IRI, reporting improved liver function and decreased circulating C4 with C1 INH treatment in a partial hepatic IRI rat model (74). Saidi et al. again used C1 INH in a partial IRI and 70% partial hepatectomy model, demonstrating a decrease in levels of IL-6, a key cytokine implicated in hepatic IRI pathogenesis. Interestingly, they also reported decreased IRI with C1 INH treatment of both wild type and C3 knockout mice, indicating that C1 INH does not act exclusively via blockade of classical activation (75). Clearly, upstream regulation of complement via C1 inhibition results in robust and reproducible amelioration of hepatic IRI.
sCR1 has also been utilized in other organs. In a rat liver transplant model, Chávez-Cartaya et al. reported improved hepatic function, decreased complement activity, and reduced C3 deposition in sCR1-treated rats (76). A similar study also demonstrated increased perfusion, decreased leukocyte adherence to endothelial cells, and decreased phagocytic activity of Kupffer cells, indicating an attenuation of several immunologic processes via complement modulation (77). In a rat lung transplant model, sCR1 improved oxygenation and isograft survival, and decreased circulating and deposited complement levels (78). Additionally, the sCR1-treated rats showed reduced neutrophil migration and lipid peroxidation (79).
Downstream proteins have also proved effective targets in the treatment of hepatic IRI. Kusakabe et al. targeted C5 with anti-C5 monoclonal antibody or C5aR1 antagonist in a murine model. Both approaches resulted in decreased platelet aggregation in hepatic microcirculation, expression of markers of apoptosis and necrosis, and ROS generation compared to controls (80). Another C5aR antagonist molecule trialed by Arumugam et al.—the same antagonist used in this group’s renal IRI study—ameliorated hepatic IRI in both a total and partial hepatic ischemia rat model (81). Both studies of C5aR blockade demonstrated reduction in inflammatory cytokine and chemokine secretion, as well as a decrease in the number of infiltrating immune cells such as macrophages and neutrophils. Zhang et al. took yet another approach to targeting C5 and its split products and succeeded using anti-C5a aptamers conjugated to an antioxidant to treat hepatic IRI in mice (82). As in the treatment of renal IRI, the targeting of C5, and particularly blockade of the C5a/C5aR interaction, decreases the severity of hepatic IRI.
While limited by the feasibility of clinical translation, cobra venom factor (CVF) is a C3 analogue that forms a stable C3/C5 convertase resistant to complement regulatory proteins and depletes components of the complement pathway effectively through unopposed complement activation in vivo. Rats pretreated with CVF had increased survival, decreased CH50 and MAC levels, circulating C5a, and reduced MAC deposition on biopsy (83). CVF has also been shown to reduce myocardial IRI, resulting in preservation of cardiac function and reduced complement deposition on histology (84).
The application of complement blockade for hepatic IRI requires consideration of the balance between tissue injury and regenerative potential, as complement plays an essential role in both processes. Marshall et al. explored this balance using a rat total IRI and 70% partial hepatectomy model. They found that inhibition of both MAC with CR2-CD59 and C3 with CR2-Crry ameliorate hepatic IRI; however, CR2-CD59 treatment leaves liver regeneration intact whereas CR2-Crry does not, indicating the importance of upstream complement activation in hepatic regeneration (85). While blockade of upstream targets such as C1 and C5 may successfully ameliorate hepatic IRI, the impact of these treatments on regenerative potential must also be taken into consideration. Taken together, available data show that IRI can be reduced by targeting various aspects of the complement pathways in a variety of organ systems.
Several complement inhibitors have advanced to the clinic since the introduction of Eculizumab in 2007. Many have been tested off-label in the field of organ transplantation—mostly for antibody mediated rejection (AMR) (86–88). However, the importance of IRI is increasingly recognized as more marginal organs are utilized. In a phase I trial evaluating C1INH as an adjunct for highly sensitized kidney transplant patients following desensitization, Vo et al. noted lower rates of DGF among those that received C1 INH (10% vs 40%, n=10 in each arm) (86). The subsequent phase I/II RCT was performed in patients receiving high risk deceased-donor kidney allografts for the prevention of DGF. The treatment group received C1 INH intraoperatively and at 24 hours postoperatively. There was no significant difference in primary endpoint, defined as the need for hemodialysis within the first week following transplantation, patient or graft survival at one year. However, the treatment group required fewer dialysis sessions at 2 and 4 weeks posttransplant and had better renal function at 1-year compared to the placebo group (89). A three-year post hoc analysis of the trial revealed higher cumulative graft failure and gradually declining eGFR in the control group, with comparable patient survival compared to the C1 INH group (90).
In contrast, the results of Eculizumab trials for the reduction of IRI have been disappointing. Eculizumab has been extensively trialed for AMR prevention and treatment—including in sensitized recipients and HLA-incompatible transplants—with heterogenous results (91–95). A multi-center double-blinded RCT of peri-transplant eculizumab infusion in patients receiving extended criteria kidney allografts showed no difference in rate of DGF, short-term, or long-term graft function between the eculizumab and control groups (96). Additionally, the Alexion-sponsored PROTECT study (NCT02145182), a phase II/III multicenter RCT involving 288 patients from 77 centers that tested the efficacy of two doses of Eculizumab, one prior to and one after reperfusion, for DGF prevention, found no difference in DGF rate or graft survival (96). Interestingly, in a small single-center, non-blinded RCT (29 in the Eculizumab group and 28 in the control group) to assess eculizumab pretreatment in pediatric kidney transplantation, the authors demonstrated better eGFR on post-transplant day 1 and 2, and lower rates of arteriolar hyalinosis and chronic glomerulopathy on biopsy (97). However, several early graft losses were observed among eculizumab-treated children, including 4 within the first two months. Of note, 68% patients included in this study received living donor kidney transplant.
A few studies examined complement inhibitors in other solid organ transplant settings. TP-10, a sCR1, was evaluated in a multicenter double-blinded RTC among lung transplant recipients for the reduction of IRI. Although the incidence of operative deaths, infection and rejection, and length of hospital stay was not significantly different between the treatment and control groups, more patients that received pre-reperfusion TP-10 were extubated by 24-hour posttransplant, compared to controls (50% vs 19%). One study evaluated the use of Nafamostat, a synthetic broad spectrum protease inhibitor that acts on all three complement pathways, in liver transplantation and found a lower incidence of postreperfusion syndrome (98), although it is not clear whether this salutary effect was due to complement inhibition.
Taken together, these studies demonstrate the potential utility of complement blockade in the treatment of IRI, though the success of complement inhibitors in pre-clinical models has not directly translated to the clinical setting. Several issues have emerged. First, systemic administration to the recipient immediately prior to reperfusion may not allow sufficient time for the drug to take effect. Second, systemic administration poorly targets the end organ and may lead to undesired off-target effects. Third, complement activation precedes donor organ procurement and thus complement inhibition administered to recipient may not completely reverse complement activation that began in the donor. The question remains, at which level of the complement cascade should therapeutic agents target to best minimize IRI?
The delivery of complement therapeutics for IRI can be achieved through multiple strategies. Donor preconditioning is commonly used in pre-clinical studies and is not without precedent in humans, as it generally takes a range of pharmacologic support to maintain a brain-dead donor (99); however, various ethical concerns have limited its application in clinical practice. Alternatively, complement therapeutics can be administered ex vivo or systemically during the peri-transplant period. Ex vivo delivery may limit off-target effects seen in systemic delivery.
Ex vivo machine perfusion has recently garnered interest as a method for marginal organ preservation and evaluation prior to transplantation. Several ex vivo machine perfusion devices are currently FDA approved for heart, lung, liver and kidney. The conditions used in ex vivo perfusion devices vary with respect to preservation temperature and perfusate composition, and are extensively reviewed elsewhere (100–102). One potential benefit of ex vivo machine perfusion is that therapeutics may be delivered to the target organ while on the circuit, a particularly attractive strategy for marginal organs. Major interventions, including stem cell therapy, anti-inflammatory and thrombolytic agents, and vasodilators in animal models were reviewed in great detail by Xu et al. (103). Using discarded human organs, several groups have evaluated therapeutic administration during machine perfusion, including high-dose antimicrobials to clear bacterial and fungal infections (104), ultraviolet light to reduce donor organ hepatitis C viral load (105, 106), Rituximab and novel chemokine-based immunotoxin to treat latent CMV-infected monocytes (107, 108), and thrombolytic therapy to preserve liver microvasculature (109).
As applied to complement therapeutics, ex vivo delivery via machine perfusion offers an opportunity to avoid potential deleterious effects of systemic complement blockade, while allowing for localized delivery of therapeutics that may have a greater potential to prevent or treat pathology compared to systemic blockade, especially in organs such as the kidney, lung, and liver that serve as sites of complement production. Local delivery and dosing can also be titrated to further avoid off-target effects and prolonged immunocompromise. Additionally, complement blockade in the donor or administered ex vivo also allows for termination of complement’s downstream effects at an earlier timepoint in the complement cascade. Ex vivo administration also provides an opportunity to monitor organ function and therapeutic efficacy simultaneously.
Several studies to date have evaluated ex vivo delivery of complement therapeutics for IRI prevention in animal models. C5 is a commonly used target, as inhibition results in terminal blockade without compromising the utility of upstream proteins such as C3a, which is involved in opsonization. The administration of an anti-C5 mAb following prolonged cold ischemia time results in a decreased complement deposition in renal tissue and decreased creatinine in rat models (110, 111). Furthermore, addition of a localization sequence allows for complement blockade in the kidneys without decreasing circulating C5, highlighting the utility of tissue-specific localization to not compromise systemic complement function (111). Ex vivo administration of C5aR blockade has also successfully increased graft survival, as well as reduced tissue damage, apoptosis, and cytokine production (112). Antagonism of both C5 and the less central target C5aR improves graft function and decreases tissue injury.
Many have targeted C3 as it is involved in all three pathways and its split product C3a is highly inflammatory. In a murine renal transplant model, Zheng et al. administered an siRNA cocktail targeting C3, RelB, and Fas to the donor prior to graft procurement. Grafts then underwent static cold storage in the same siRNA cocktail. Renal function 48 hours post-transplant, as well as long-term survival was significantly better in the siRNA cocktail group compared to controls. Furthermore, siRNA solution reduced apoptosis of tubular cells and expression of inflammatory cytokines (113). siRNA remains an interesting approach to complement-targeting therapeutics that may be delivered in vivo or ex vivo with improvement in graft survival.
Mirococept (APT070) is a membrane-localizing complement inhibitor that consists of the minimal functional unit of human CR1 with a membrane binding tail designed to bind damaged cell membranes, which also blocks complement activation at the C3 level. Patel et al. perfused ex vivo rat kidney allografts with mirococept prior to subjecting the organs to prolonged cold ischemia time (114). This group first assessed tubular damage and complement deposition on histology and graft function with different lengths of cold ischemia time, finding increased C3 and C9 deposition and a dose-dependent rise in creatinine with increased cold ischemia time. Explanted rat donor kidneys were perfused with mirococept or control agent prior to cold storage and subsequent transplantation. This study reported better renal function at 24 and 72 hours in the treatment group, along with a 35% decrease in tubular damage and reduced complement deposition on histology (114). Indeed, C3 blockade results in robust suppression of inflammation and graft damage corresponding with improved graft function and prolonged survival. Given significant preclinical results, mirococept has since progressed to human trials, discussed below.
Complement-targeting therapeutics for IRI have been administered ex vivo in multiple other organ systems. Bergamachini et al. trialed C1 INH in a porcine liver reperfusion model. They demonstrated reduced complement activity, including generation of C3 and C3 deposition in the graft when C1 INH was added to the preservation solution following liver procurement. Biopsies of control organs had higher inflammatory cell infiltration, microabscess formation, and centrilobular necrosis compared to the treatment group, further supporting the potential of C1 INH to reduce inflammation in multiple organ systems and using multiple routes of administration (115).
Zheng et al. applied the aforementioned complement-silencing siRNA approach in a murine heart transplant model. siRNA solution containing TNF-α-, C3-, and Fas-silencing siRNA was administered to the donor prior to organ harvest. The organ was then preserved in and then flushed with the same siRNA cocktail, and then used for heterotopic transplantation. siRNA treatment resulted in successfully silenced C3 transcription. Additionally, 87.5% of siRNA-treated grafts preserved function at 100 days, compared to 0% in control groups (116). Wei et al. carried out a similar experiment in a porcine model, perfusing and storing cardiac allografts with siRNA targeting C3, caspases-8 and -3, and NF κB-p65, also finding improved graft survival and reduced tissue inflammation and apoptosis with treatment. Upon reperfusion, the cardiac allografts that received siRNA cocktail did not see a decline in hemodynamic parameters observed in controls (117). These experiments showcase the utility of ex vivo administration of complement—specifically C3—targeting siRNA for treatment of IRI in multiple organ systems.
Cheng et al. targeted C3aR in a murine DBD lung transplant model. C3aR is upregulated in lung parenchyma following brain death, thus presenting a particularly relevant target for lung IRI therapy. The authors administered a nebulized C3aR antagonist following brain death and prior to procurement, cold storage, and subsequent transplantation. Mice that received a C3aRA-treated graft had reduced IRI and acute rejection compared to untreated DBD. C3aRA treatment also decreased C3 and C3aR expression, injury on histology, and neutrophil and macrophage infiltration (118). Nebulizers offer a novel localized therapeutic delivery to the lung without the potentially deleterious effects of systemic treatment, that may also be delivered using an ex vivo machine perfusion circuit.
To date, only one complement-targeting drug has been trialed using ex vivo delivery in humans. The EMPIRIKAL trial, a multi-center RCT, assessed the utility of mirococept when delivered ex vivo via machine perfusion, in prevention of IRI (119). 10mg mirococept was added to Soltran solution following initial flushing of the organ, and then administered via a single pass flush through the renal artery. The authors observed comparable levels of DGF between the mirococept and control groups, when stratified by site, pump, and donor type. Secondary outcomes, such as eGFR at one-year, acute rejection episodes, and duration of DGF were also comparable between the two groups (120). Of note, the authors failed to detect any mirococept or mirococept-specific IgG in the recipient serum. No CR1-specific staining above background levels of native CR1 was noted, and no significant difference in serum complement activity was observed between the treatment and control groups. The authors concluded that mirococept was likely not delivered at a clinically significant dose, and thus the study was terminated following the first cohort given the need for dose optimization. Kassimatis et al. conducted a follow-up dose-defining experiment using porcine kidneys, defining the optimal dose of mirococept (APT070) at 80 mg for ex vivo administration. Overall, the impact of complement inhibition in clinical kidney transplant when delivered ex vivo remains unclear; future studies using an optimized dose are needed to better evaluate the utility of this approach.
New solutions to the growing organ shortage, such as DCD and extended criteria organ transplants, have presented new challenges—namely increased vulnerability to IRI and increased rates of DGF, leading to worse outcomes compared to living and DBD organs. Complement activation is implicated in the pathogenesis of IRI across various organ systems and provides a promising target for IRI prevention. Ex vivo organ perfusion has emerged as a strategy not only for maintaining organs prior to transplantation, but also as a route of administration for pre-transplant therapies. In this review, we have highlighted the utility of complement blockade administered both systemically to the recipient and ex vivo to the target organ, with animal models exploring blockade at multiple levels in the complement pathway, including C1, C3, and C5. However, clinical studies so far have seen mixed results. C1INH administered to the recipient during the peri-transplant period may minimize IRI, especially among those receiving high KDPI kidneys. In contrast, human experience with eculizumab for IRI has been disappointing so far. The first human trial of ex vivo delivery of Mirococept also did not show a difference in DGF or graft survival, although mirococept drug dosing clearly requires optimization prior to continued evaluation. Ex vivo pre-treatment of complement blockade is a promising, yet imperfect strategy which requires further investigation prior to large-scale implementation in humans.
QG and AB conceived this review. ID and QG preformed literature review and drafted the manuscript. IA, NA, and RK performed critical review and revision of the manuscript. MH and AB performed critical review and revision of the manuscript and oversaw the drafting process. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Lentine KL, Smith JM, Hart A, Miller J, Skeans MA, Larkin L, et al. OPTN/SRTR 2020 annual data report: Kidney. Am J Transplant (2022) 22(Suppl 2):21–136. doi: 10.1111/ajt.16982
2. Hall IE, Akalin E, Bromberg JS, Doshi MD, Greene T, Harhay MN, et al. Deceased-donor acute kidney injury is not associated with kidney allograft failure. Kidney Int (2019) 95:199–209. doi: 10.1016/j.kint.2018.08.047
3. Heilman RL, Smith ML, Smith BH, Kumar A, Srinivasan A, Huskey JL, et al. Long-term outcomes following kidney transplantation from donors with acute kidney injury. Transplantation (2019) 103:e263–72. doi: 10.1097/TP.0000000000002792
4. Merion RM, Ashby VB, Wolfe RA, Distant DA, Hulbert-Shearon TE, Metzger RA, et al. Deceased-donor characteristics and the survival benefit of kidney transplantation. JAMA (2005) 294:2726–33. doi: 10.1001/jama.294.21.2726
5. Kizilbash SJ, Evans MD, Chinnakotla S, Chavers BM. Use of expanded-criteria donors and > 85 KDPI kidneys for pediatric kidney transplantation in the united states. Am J Transplant (2021) 21:1160–70. doi: 10.1111/ajt.16162
6. Feng S, Goodrich NP, Bragg-Gresham JL, Dykstra DM, Punch JD, Debroy MA, et al. Characteristics associated with liver graft failure: the concept of a donor risk index. Am J Transplant (2006) 6:783–90. doi: 10.1111/j.1600-6143.2006.01242.x
7. Halazun KJ, Quillin RC, Rosenblatt R, Bongu A, Griesemer AD, Kato T, et al. Expanding the margins: High volume utilization of marginal liver grafts among >2000 liver transplants at a single institution. Ann Surg (2017) 266:441–9. doi: 10.1097/SLA.0000000000002383
8. Samsky MD, Patel CB, Owen A, Schulte PJ, Jentzer J, Rosenberg PB, et al. Ten-year experience with extended criteria cardiac transplantation. Circ Heart Fail (2013) 6:1230–8. doi: 10.1161/CIRCHEARTFAILURE.113.000296
9. Christie IG, Chan EG, Ryan JP, Harano T, Morrell M, Luketich JD, et al. National trends in extended criteria donor utilization and outcomes for lung transplantation. Ann Thorac Surg (2021) 111:421–6. doi: 10.1016/j.athoracsur.2020.05.087
10. Kwong AJ, Ebel NH, Kim WR, Lake JR, Smith JM, Schladt DP, et al. OPTN/SRTR 2020 annual data report: Liver. Am J Transplant (2022) 22(Suppl 2):204–309. doi: 10.1111/ajt.16978
11. Valapour M, Lehr CJ, Skeans MA, Smith JM, Miller E, Goff R, et al. OPTN/SRTR 2020 annual data report: Lung. Am J Transplant (2022) 22 (Suppl 2):438–518. doi: 10.1111/ajt.16991
12. Jawitz OK, Bryner BS, Schroder JN, Devore AD. Donation after circulatory death heart transplantation in the united states: An early report of donor characteristics. JTCVS Tech (2022) 12:104–7. doi: 10.1016/j.xjtc.2021.12.014
13. Rijkse E, Ceuppens S, Qi H, Jnm IJ, Hesselink DA, Minnee RC. Implementation of donation after circulatory death kidney transplantation can safely enlarge the donor pool: A systematic review and meta-analysis. Int J Surg (2021) 92:106021. doi: 10.1016/j.ijsu.2021.106021
14. Kayler LK, Magliocca J, Zendejas I, Srinivas TR, Schold JD. Impact of cold ischemia time on graft survival among ECD transplant recipients: A paired kidney analysis. Am J Transplant (2011) 11:2647–56. doi: 10.1111/j.1600-6143.2011.03741.x
15. Yarlagadda SG, Coca SG, Formica RN Jr., Poggio ED, Parikh CR. Association between delayed graft function and allograft and patient survival: A systematic review and meta-analysis. Nephrol Dial Transplant (2009) 24:1039–47. doi: 10.1093/ndt/gfn667
16. Callaghan CJ, Charman SC, Muiesan P, Powell JJ, Gimson AE, van der Meulen JHP, et al. Outcomes of transplantation of livers from donation after circulatory death donors in the UK: A cohort study. BMJ Open (2013) 3:e003287. doi: 10.1136/bmjopen-2013-003287
17. Ruck JM, Jackson KR, Motter JD, Massie AB, Philosophe B, Cameron AM, et al. Temporal trends in utilization and outcomes of DCD livers in the united states. Transplantation (2022) 106:543–51. doi: 10.1097/TP.0000000000003878
18. Gavriilidis P, Inston NG. Recipient and allograft survival following donation after circulatory death versus donation after brain death for renal transplantation: A systematic review and meta-analysis. Transplant Rev (Orlando) (2020) 34:100563. doi: 10.1016/j.trre.2020.100563
19. Gao Q, Mulvihill MS, Scheuermann U, Davis RP, Yerxa J, Yerokun BA, et al. Improvement in liver transplant outcomes from older donors: A US national analysis. Ann Surg (2018) 270(2):333–9. doi: 10.1097/SLA.0000000000002876
20. Ricklin D, Mastellos DC, Reis ES, Lambris JD. The renaissance of complement therapeutics. Nat Rev Nephrol (2018) 14:26–47. doi: 10.1038/nrneph.2017.156
21. Danobeitia JS, Djamali A, Fernandez LA. The role of complement in the pathogenesis of renal ischemia-reperfusion injury and fibrosis. Fibrogenesis Tissue Repair (2014) 7:16. doi: 10.1186/1755-1536-7-16
22. Howard MC, Nauser CL, Farrar CA, Sacks SH. Complement in ischaemia-reperfusion injury and transplantation. Semin Immunopathol (2021) 43:789–97. doi: 10.1007/s00281-021-00896-3
23. Zipfel PF, Skerka C. Complement regulators and inhibitory proteins. Nat Rev Immunol (2009) 9:729–40. doi: 10.1038/nri2620
24. Bajic G, Degn SE, Thiel S, Andersen GR. Complement activation, regulation, and molecular basis for complement-related diseases. EMBO J (2015) 34:2735–57. doi: 10.15252/embj.201591881
25. Ghosh P, Sahoo R, Vaidya A, Chorev M, Halperin JA. Role of complement and complement regulatory proteins in the complications of diabetes. Endocrine Rev (2015) 36:272–88. doi: 10.1210/er.2014-1099
26. Damman J, Seelen MA, Moers C, Daha MR, Rahmel A, Leuvenink HG, et al. Systemic complement activation in deceased donors is associated with acute rejection after renal transplantation in the recipient. Transplantation (2011) 92:163–9. doi: 10.1097/TP.0b013e318222c9a0
27. Van Werkhoven MB, Damman J, Van Dijk M, Daha MR, De Jong IJ, Leliveld A, et al. Complement mediated renal inflammation induced by donor brain death: role of renal C5a-C5aR interaction. Am J Transplant (2013) 13:875–82. doi: 10.1111/ajt.12130
28. Halpern SE, Rush CK, Edwards RW, Brennan TV, Barbas AS, Pollara J. Systemic complement activation in donation after brain death versus donation after circulatory death organ donors. Exp Clin Transplant (2021) 19:635–44. doi: 10.6002/ect.2020.0425
29. De Vries DK, van der Pol P, Van Anken GE, Van Gijlswijk DJ, Damman J, Lindeman JH, et al. Acute but transient release of terminal complement complex after reperfusion in clinical kidney transplantation. Transplantation (2013) 95:816–20. doi: 10.1097/TP.0b013e31827e31c9
30. Damman J, Bloks VW, Daha MR, van der Most PJ, Sanjabi B, van der Vlies P, et al. Hypoxia and complement-and-Coagulation pathways in the deceased organ donor as the major target for intervention to improve renal allograft outcome. Transplantation (2015) 99:1293–300. doi: 10.1097/TP.0000000000000500
31. Kusaka M, Pratschke J, Wilhelm MJ, Ziai F, Zandi-Nejad K, Mackenzie HS, et al. Activation of inflammatory mediators in rat renal isografts by donor brain death. Transplantation (2000) 69:405–10. doi: 10.1097/00007890-200002150-00017
32. Damman J, Nijboer WN, Schuurs TA, Leuvenink HG, Morariu AM, Tullius SG, et al. Local renal complement C3 induction by donor brain death is associated with reduced renal allograft function after transplantation. Nephrol Dial Transplant (2011) 26:2345–54. doi: 10.1093/ndt/gfq717
33. Van Zanden JE, Jager NM, Daha MR, Erasmus ME, Leuvenink HGD, Seelen MA. Complement therapeutics in the multi-organ donor: Do or don't? Front Immunol (2019) 10:329. doi: 10.3389/fimmu.2019.00329
34. Mueller TF, Reeve J, Jhangri GS, Mengel M, Jacaj Z, Cairo L, et al. The transcriptome of the implant biopsy identifies donor kidneys at increased risk of delayed graft function. Am J Transplant (2008) 8:78–85. doi: 10.1111/j.1600-6143.2007.02032.x
35. Hruba P, Krejcik Z, Dostalova Merkerova M, Klema J, Stranecky V, Slatinska J, et al. Molecular fingerprints of borderline changes in kidney allografts are influenced by donor category. Front Immunol (2020) 11:423. doi: 10.3389/fimmu.2020.00423
36. Eltzschig HK, Eckle T. Ischemia and reperfusion–from mechanism to translation. Nat Med (2011) 17:1391–401. doi: 10.1038/nm.2507
37. Franzin R, Stasi A, Fiorentino M, Simone S, Oberbauer R, Castellano G, et al. Renal delivery of pharmacologic agents during machine perfusion to prevent ischaemia-reperfusion injury: From murine model to clinical trials. Front Immunol (2021) 12:673562. doi: 10.3389/fimmu.2021.673562
38. Zwaini Z, Dai H, Stover C, Yang B. Role of complement properdin in renal ischemia-reperfusion injury. Curr Gene Ther (2017) 17:411–23. doi: 10.2174/1566523218666180214093043
39. Hu ZG, Zhou Y, Lin CJ, Yuan GD, He SQ. Emerging recognition of the complement system in hepatic ischemia/reperfusion injury, liver regeneration and recovery (Review). Exp Ther Med (2021) 21:223. doi: 10.3892/etm.2021.9654
40. Talaie T, Dichiacchio L, Prasad NK, Pasrija C, Julliard W, Kaczorowski DJ, et al. Ischemia-reperfusion injury in the transplanted lung: A literature review. Transplant Direct (2021) 7:e652. doi: 10.1097/TXD.0000000000001104
41. Zhou W, Farrar CA, Abe K, Pratt JR, Marsh JE, Wang Y, et al. Predominant role for C5b-9 in renal ischemia/reperfusion injury. J Clin Invest (2000) 105:1363–71. doi: 10.1172/JCI8621
42. Jiang HX, Siegel JN, Gewurz H. Binding and complement activation by c-reactive protein via the collagen-like region of C1q and inhibition of these reactions by monoclonal antibodies to c-reactive protein and C1q. J Immunol (1991) 146:2324–30.
43. Thurman JM, Ljubanovic D, Edelstein CL, Gilkeson GS, Holers VM. Lack of a functional alternative complement pathway ameliorates ischemic acute renal failure in mice. J Immunol (2003) 170:1517–23. doi: 10.4049/jimmunol.170.3.1517
44. Møller-Kristensen M, Wang W, Ruseva M, Thiel S, Nielsen S, Takahashi K, et al. Mannan-binding lectin recognizes structures on ischaemic reperfused mouse kidneys and is implicated in tissue injury. Scand J Immunol (2005) 61:426–34. doi: 10.1111/j.1365-3083.2005.01591.x
45. Farrar CA, Tran D, Li K, Wu W, Peng Q, Schwaeble W, et al. Collectin-11 detects stress-induced l-fucose pattern to trigger renal epithelial injury. J Clin Invest (2016) 126:1911–25. doi: 10.1172/JCI83000
46. Park P, Haas M, Cunningham PN, Bao L, Alexander JJ, Quigg RJ. Injury in renal ischemia-reperfusion is independent from immunoglobulins and T lymphocytes. Am J Physiol Renal Physiol (2002) 282:F352–357. doi: 10.1152/ajprenal.00160.2001
47. Pratt JR, Basheer SA, Sacks SH. Local synthesis of complement component C3 regulates acute renal transplant rejection. Nat Med (2002) 8:582–7. doi: 10.1038/nm0602-582
48. Brown KM, Kondeatis E, Vaughan RW, Kon SP, Farmer CK, Taylor JD, et al. Influence of donor C3 allotype on late renal-transplantation outcome. N Engl J Med (2006) 354:2014–23. doi: 10.1056/NEJMoa052825
49. Atkinson C, Varela JC, Tomlinson S. Complement-dependent inflammation and injury in a murine model of brain dead donor hearts. Circ Res (2009) 105:1094–101. doi: 10.1161/CIRCRESAHA.109.194977
50. Atkinson C, Floerchinger B, Qiao F, Casey S, Williamson T, Moseley E, et al. Donor brain death exacerbates complement-dependent ischemia/reperfusion injury in transplanted hearts. Circulation (2013) 127:1290–9. doi: 10.1161/CIRCULATIONAHA.112.000784
51. Robinson MW, Harmon C, O'farrelly C. Liver immunology and its role in inflammation and homeostasis. Cell Mol Immunol (2016) 13:267–76. doi: 10.1038/cmi.2016.3
52. Fondevila C, Shen XD, Tsuchihashi S, Uchida Y, Freitas MC, Ke B, et al. The membrane attack complex (C5b-9) in liver cold ischemia and reperfusion injury. Liver Transpl (2008) 14:1133–41. doi: 10.1002/lt.21496
53. Mastellos D, Papadimitriou JC, Franchini S, Tsonis PA, Lambris JD. A novel role of complement: mice deficient in the fifth component of complement (C5) exhibit impaired liver regeneration. J Immunol (2001) 166:2479–86. doi: 10.4049/jimmunol.166.4.2479
54. Strey CW, Markiewski M, Mastellos D, Tudoran R, Spruce LA, Greenbaum LE, et al. The proinflammatory mediators C3a and C5a are essential for liver regeneration. J Exp Med (2003) 198:913–23. doi: 10.1084/jem.20030374
55. Ali HA, Pavlisko EN, Snyder LD, Frank M, Palmer SM. Complement system in lung transplantation. Clin Transplant (2018) 32:e13208. doi: 10.1111/ctr.13208
56. Shah RJ, Emtiazjoo AM, Diamond JM, Smith PA, Roe DW, Wille KM, et al. Plasma complement levels are associated with primary graft dysfunction and mortality after lung transplantation. Am J Respir Crit Care Med (2014) 189:1564–7. doi: 10.1164/rccm.201312-2121LE
57. Westall GP, Snell GI, Mclean C, Kotsimbos T, Williams T, Magro C. C3d and C4d deposition early after lung transplantation. J Heart Lung Transplant (2008) 27:722–8. doi: 10.1016/j.healun.2008.03.018
58. Kulkarni HS, Ramphal K, Ma L, Brown M, Oyster M, Speckhart KN, et al. Local complement activation is associated with primary graft dysfunction after lung transplantation. JCI Insight (2020) 5(17):e138358. doi: 10.1172/jci.insight.138358
59. Strunk RC, Eidlen DM, Mason RJ. Pulmonary alveolar type II epithelial cells synthesize and secrete proteins of the classical and alternative complement pathways. J Clin Invest (1988) 81:1419–26. doi: 10.1172/JCI113472
60. Zheng X, Zhang X, Sun H, Feng B, Li M, Chen G, et al. Protection of renal ischemia injury using combination gene silencing of complement 3 and caspase 3 genes. Transplantation (2006) 82:1781–6. doi: 10.1097/01.tp.0000250769.86623.a3
61. Zheng X, Zhang X, Feng B, Sun H, Suzuki M, Ichim T, et al. Gene silencing of complement C5a receptor using siRNA for preventing ischemia/reperfusion injury. Am J Pathol (2008) 173:973–80. doi: 10.2353/ajpath.2008.080103
62. Arumugam TV, Shiels IA, Strachan AJ, Abbenante G, Fairlie DP, Taylor SM. A small molecule C5a receptor antagonist protects kidneys from ischemia/reperfusion injury in rats. Kidney Int (2003) 63:134–42. doi: 10.1046/j.1523-1755.2003.00737.x
63. Iida K, Nussenzweig V. Complement receptor is an inhibitor of the complement cascade. J Exp Med (1981) 153:1138–50. doi: 10.1084/jem.153.5.1138
64. Hameed AM, Lu DB, Burns H, Byrne N, Chew YV, Julovi S, et al. Pharmacologic targeting of renal ischemia-reperfusion injury using a normothermic machine perfusion platform. Sci Rep (2020) 10:6930. doi: 10.1038/s41598-020-63687-0
65. Wymann S, Dai Y, Nair AG, Cao H, Powers GA, Schnell A, et al. A novel soluble complement receptor 1 fragment with enhanced therapeutic potential. J Biol Chem (2021) 296:100200. doi: 10.1074/jbc.RA120.016127
66. Bongoni AK, Vikstrom IB, Mcrae JL, Salvaris EJ, Fisicaro N, Pearse MJ, et al. A potent truncated form of human soluble CR1 is protective in a mouse model of renal ischemia-reperfusion injury. Sci Rep (2021) 11:21873. doi: 10.1038/s41598-021-01423-y
67. Poppelaars F, Jager NM, Kotimaa J, Leuvenink HGD, Daha MR, Van Kooten C, et al. C1-inhibitor treatment decreases renal injury in an established brain-dead rat model. Transplantation (2018) 102:79–87. doi: 10.1097/TP.0000000000001895
68. Danobeitia JS, Ziemelis M, Ma X, Zitur LJ, Zens T, Chlebeck PJ, et al. Complement inhibition attenuates acute kidney injury after ischemia-reperfusion and limits progression to renal fibrosis in mice. PloS One (2017) 12:e0183701. doi: 10.1371/journal.pone.0183701
69. Danobeitia JS, Zens TJ, Chlebeck PJ, Zitur LJ, Reyes JA, Eerhart MJ, et al. Targeted donor complement blockade after brain death prevents delayed graft function in a nonhuman primate model of kidney transplantation. Am J Transplant (2020) 20:1513–26. doi: 10.1111/ajt.15777
70. Eerhart MJ, Reyes JA, Blanton CL, Danobeitia JS, Chlebeck PJ, Zitur LJ, et al. Complement blockade in recipients prevents delayed graft function and delays antibody-mediated rejection in a nonhuman primate model of kidney transplantation. Transplantation (2022) 106:60–71. doi: 10.1097/TP.0000000000003754
71. Delpech PO, Thuillier R, Saintyves T, Danion J, Le Pape S, Van Amersfoort ES, et al. Inhibition of complement improves graft outcome in a pig model of kidney autotransplantation. J Transl Med (2016) 14:277. doi: 10.1186/s12967-016-1013-7
72. Lehmann TG, Heger M, Münch S, Kirschfink M, Klar E. In vivo microscopy reveals that complement inhibition by C1-esterase inhibitor reduces ischemia/reperfusion injury in the liver. Transpl Int (2000) 13(Suppl 1):S547–550. doi: 10.1007/s001470050399
73. Lehmann TG, Koeppel TA, Münch S, Heger M, Kirschfink M, Klar E, et al. Impact of inhibition of complement by sCR1 on hepatic microcirculation after warm ischemia. Microvasc Res (2001) 62:284–92. doi: 10.1006/mvre.2001.2342
74. Heijnen BH, Straatsburg IH, Padilla ND, Van Mierlo GJ, Hack CE, Van Gulik TM. Inhibition of classical complement activation attenuates liver ischaemia and reperfusion injury in a rat model. Clin Exp Immunol (2006) 143:15–23. doi: 10.1111/j.1365-2249.2005.02958.x
75. Saidi RF, Rajeshkumar B, Shariftabrizi A, Dresser K, Walter O. Human C1 inhibitor attenuates liver ischemia-reperfusion injury and promotes liver regeneration. J Surg Res (2014) 187:660–6. doi: 10.1016/j.jss.2013.09.009
76. Chávez-Cartaya RE, Desola GP, Wright L, Jamieson NV, White DJ. Regulation of the complement cascade by soluble complement receptor type 1. protective effect in experimental liver ischemia and reperfusion. Transplantation (1995) 59:1047–52. doi: 10.1097/00007890-199504150-00023
77. Lehmann TG, Koeppel TA, Kirschfink M, Gebhard MM, Herfarth C, Otto G, et al. Complement inhibition by soluble complement receptor type 1 improves microcirculation after rat liver transplantation. Transplantation (1998) 66:717–22. doi: 10.1097/00007890-199809270-00005
78. Naka Y, Marsh HC, Scesney SM, Oz MC, Pinsky DJ. Complement activation as a cause for primary graft failure in an isogeneic rat model of hypothermic lung preservation and transplantation. Transplantation (1997) 64:1248–55. doi: 10.1097/00007890-199711150-00004
79. Stammberger U, Hamacher J, Hillinger S, Schmid RA. sCR1sLe ameliorates ischemia/reperfusion injury in experimental lung transplantation. J Thorac Cardiovasc Surg (2000) 120:1078–84. doi: 10.1067/mtc.2000.111175
80. Kusakabe J, Hata K, Tamaki I, Tajima T, Miyauchi H, Wang Y, et al. Complement 5 inhibition ameliorates hepatic ischemia/reperfusion injury in mice, dominantly via the C5a-mediated cascade. Transplantation (2020) 104:2065–77. doi: 10.1097/TP.0000000000003302
81. Arumugam TV, Woodruff TM, Stocks SZ, Proctor LM, Pollitt S, Shiels IA, et al. Protective effect of a human C5a receptor antagonist against hepatic ischaemia-reperfusion injury in rats. J Hepatol (2004) 40:934–41. doi: 10.1016/j.jhep.2004.02.017
82. Zhang X, Hu J, Becker KV, Engle JW, Ni D, Cai W, et al. Antioxidant and C5a-blocking strategy for hepatic ischemia-reperfusion injury repair. J Nanobiotechnology (2021) 19:107. doi: 10.1186/s12951-021-00858-9
83. Wang B, Xu H, Li J, Gao HM, Xing YH, Lin Z, et al. Complement depletion with cobra venom factor alleviates acute hepatic injury induced by ischemiareperfusion. Mol Med Rep (2018) 18:4523–9. doi: 10.3892/mmr.2018.9484
84. Gorsuch WB, Guikema BJ, Fritzinger DC, Vogel CW, Stahl GL. Humanized cobra venom factor decreases myocardial ischemia-reperfusion injury. Mol Immunol (2009) 47:506–10. doi: 10.1016/j.molimm.2009.08.017
85. Marshall KM, He S, Zhong Z, Atkinson C, Tomlinson S. Dissecting the complement pathway in hepatic injury and regeneration with a novel protective strategy. J Exp Med (2014) 211:1793–805. doi: 10.1084/jem.20131902
86. Vo AA, Zeevi A, Choi J, Cisneros K, Toyoda M, Kahwaji J, et al. A phase I/II placebo-controlled trial of C1-inhibitor for prevention of antibody-mediated rejection in HLA sensitized patients. Transplantation (2015) 99:299–308. doi: 10.1097/TP.0000000000000592
87. Montgomery RA, Orandi BJ, Racusen L, Jackson AM, Garonzik-Wang JM, Shah T, et al. Plasma-derived C1 esterase inhibitor for acute antibody-mediated rejection following kidney transplantation: Results of a randomized double-blind placebo-controlled pilot study. Am J Transplant (2016) 16:3468–78. doi: 10.1111/ajt.13871
88. Viglietti D, Gosset C, Loupy A, Deville L, Verine J, Zeevi A, et al. C1 inhibitor in acute antibody-mediated rejection nonresponsive to conventional therapy in kidney transplant recipients: A pilot study. Am J Transplant (2016) 16:1596–603. doi: 10.1111/ajt.13663
89. Jordan SC, Choi J, Aubert O, Haas M, Loupy A, Huang E, et al. A phase I/II, double-blind, placebo-controlled study assessing safety and efficacy of C1 esterase inhibitor for prevention of delayed graft function in deceased donor kidney transplant recipients. Am J Transplant (2018) 18:2955–64. doi: 10.1111/ajt.14767
90. Huang E, Vo A, Choi J, Ammerman N, Lim K, Sethi S, et al. Three-year outcomes of a randomized, double-blind, placebo-controlled study assessing safety and efficacy of C1 esterase inhibitor for prevention of delayed graft function in deceased donor kidney transplant recipients. Clin J Am Soc Nephrol (2020) 15:109–16. doi: 10.2215/CJN.04840419
91. Stegall MD, Diwan T, Raghavaiah S, Cornell LD, Burns J, Dean PG, et al. Terminal complement inhibition decreases antibody-mediated rejection in sensitized renal transplant recipients. Am J Transplant (2011) 11:2405–13. doi: 10.1111/j.1600-6143.2011.03757.x
92. Orandi BJ, Zachary AA, Dagher NN, Bagnasco SM, Garonzik-Wang JM, Van Arendonk KJ, et al. Eculizumab and splenectomy as salvage therapy for severe antibody-mediated rejection after HLA-incompatible kidney transplantation. Transplantation (2014) 98:857–63. doi: 10.1097/TP.0000000000000298
93. Cornell LD, Schinstock CA, Gandhi MJ, Kremers WK, Stegall MD. Positive crossmatch kidney transplant recipients treated with eculizumab: outcomes beyond 1 year. Am J Transplant (2015) 15:1293–302. doi: 10.1111/ajt.13168
94. Kulkarni S, Kirkiles-Smith NC, Deng YH, Formica RN, Moeckel G, Broecker V, et al. Eculizumab therapy for chronic antibody-mediated injury in kidney transplant recipients: A pilot randomized controlled trial. Am J Transplant (2017) 17:682–91. doi: 10.1111/ajt.14001
95. Marks WH, Mamode N, Montgomery RA, Stegall MD, Ratner LE, Cornell LD, et al. Safety and efficacy of eculizumab in the prevention of antibody-mediated rejection in living-donor kidney transplant recipients requiring desensitization therapy: A randomized trial. Am J Transplant (2019) 19:2876–88. doi: 10.1111/ajt.15364
96. Schroppel B, Akalin E, Baweja M, Bloom RD, Florman S, Goldstein M, et al. Peritransplant eculizumab does not prevent delayed graft function in deceased donor kidney transplant recipients: Results of two randomized controlled pilot trials. Am J Transplant (2020) 20:564–72. doi: 10.1111/ajt.15580
97. Kaabak M, Babenko N, Shapiro R, Zokoyev A, Dymova O, Kim E. A prospective randomized, controlled trial of eculizumab to prevent ischemia-reperfusion injury in pediatric kidney transplantation. Pediatr Transplant (2018) 22(2):e13129. doi: 10.1111/petr.13129
98. Ryu HG, Jung CW, Lee CS, Lee J. Nafamostat mesilate attenuates postreperfusion syndrome during liver transplantation. Am J Transplant (2011) 11:977–83. doi: 10.1111/j.1600-6143.2011.03514.x
99. Mcnally SJ, Harrison EM, Wigmore SJ. Ethical considerations in the application of preconditioning to solid organ transplantation. J Med Ethics (2005) 31:631. doi: 10.1136/jme.2004.011486
100. Hosgood SA, Brown RJ, Nicholson ML. Advances in kidney preservation techniques and their application in clinical practice. Transplantation (2021) 105:e202–14. doi: 10.1097/TP.0000000000003679
101. Serifis N, Matheson R, Cloonan D, Rickert CG, Markmann JF, Coe TM. Machine perfusion of the liver: A review of clinical trials. Front Surg (2021) 8:625394. doi: 10.3389/fsurg.2021.625394
102. Roesel MJ, Wiegmann B, Ius F, Knosalla C, Iske J. The role of ex-situ perfusion for thoracic organs. Curr Opin Organ Transplant (2022) 27(5):466–73. doi: 10.1097/MOT.0000000000001008
103. Xu J, Buchwald JE, Martins PN. Review of current machine perfusion therapeutics for organ preservation. Transplantation (2020) 104:1792–803. doi: 10.1097/TP.0000000000003295
104. Nakajima D, Cypel M, Bonato R, Machuca TN, Iskender I, Hashimoto K, et al. Ex vivo perfusion treatment of infection in human donor lungs. Am J Transplant (2016) 16:1229–37. doi: 10.1111/ajt.13562
105. Galasso M, Feld JJ, Watanabe Y, Pipkin M, Summers C, Ali A, et al. Inactivating hepatitis c virus in donor lungs using light therapies during normothermic ex vivo lung perfusion. Nat Commun (2019) 10:481. doi: 10.1038/s41467-018-08261-z
106. Cypel M, Feld JJ, Galasso M, Pinto Ribeiro RV, Marks N, Kuczynski M, et al. Prevention of viral transmission during lung transplantation with hepatitis c-viraemic donors: An open-label, single-centre, pilot trial. Lancet Respir Med (2020) 8:192–201. doi: 10.1016/S2213-2600(19)30268-1
107. Ku TJY, Ribeiro RVP, Ferreira VH, Galasso M, Keshavjee S, Kumar D, et al. Ex-vivo delivery of monoclonal antibody (Rituximab) to treat human donor lungs prior to transplantation. EBioMedicine (2020) 60:102994. doi: 10.1016/j.ebiom.2020.102994
108. Ribeiro RVP, Ku T, Wang A, Pires L, Ferreira VH, Michaelsen V, et al. Ex vivo treatment of cytomegalovirus in human donor lungs using a novel chemokine-based immunotoxin. J Heart Lung Transplant (2022) 41:287–97. doi: 10.1016/j.healun.2021.10.010
109. Haque O, Raigani S, Rosales I, Carroll C, Coe TM, Baptista S, et al. Thrombolytic therapy during ex-vivo normothermic machine perfusion of human livers reduces peribiliary vascular plexus injury. Front Surg (2021) 8:644859. doi: 10.3389/fsurg.2021.644859
110. Yu ZX, Qi S, Lasaro MA, Bouchard K, Dow C, Moore K, et al. Targeting complement pathways during cold ischemia and reperfusion prevents delayed graft function. Am J Transplant (2016) 16:2589–97. doi: 10.1111/ajt.13797
111. Durigutto P, Sblattero D, Biffi S, De Maso L, Garrovo C, Baj G, et al. Targeted delivery of neutralizing anti-C5 antibody to renal endothelium prevents complement-dependent tissue damage. Front Immunol (2017) 8:1093. doi: 10.3389/fimmu.2017.01093
112. Lewis AG, Kohl G, Ma Q, Devarajan P, Kohl J. Pharmacological targeting of C5a receptors during organ preservation improves kidney graft survival. Clin Exp Immunol (2008) 153:117–26. doi: 10.1111/j.1365-2249.2008.03678.x
113. Zheng X, Zang G, Jiang J, He W, Johnston NJ, Ling H, et al. Attenuating ischemia-reperfusion injury in kidney transplantation by perfusing donor organs with siRNA cocktail solution. Transplantation (2016) 100:743–52. doi: 10.1097/TP.0000000000000960
114. Patel H, Smith RA, Sacks SH, Zhou W. Therapeutic strategy with a membrane-localizing complement regulator to increase the number of usable donor organs after prolonged cold storage. J Am Soc Nephrol (2006) 17:1102–11. doi: 10.1681/ASN.2005101116
115. Bergamaschini L, Gobbo G, Gatti S, Caccamo L, Prato P, Maggioni M, et al. Endothelial targeting with C1-inhibitor reduces complement activation in vitro and during ex vivo reperfusion of pig liver. Clin Exp Immunol (2001) 126:412–20. doi: 10.1046/j.1365-2249.2001.01695.x
116. Zheng X, Lian D, Wong A, Bygrave M, Ichim TE, Khoshniat M, et al. Novel small interfering RNA-containing solution protecting donor organs in heart transplantation. Circulation (2009) 120:1099–107. doi: 10.1161/CIRCULATIONAHA.108.787390
117. Wei J, Chen S, Xue S, Zhu Q, Liu S, Cui L, et al. Blockade of inflammation and apoptosis pathways by siRNA prolongs cold preservation time and protects donor hearts in a porcine model. Mol Ther Nucleic Acids (2017) 9:428–39. doi: 10.1016/j.omtn.2017.10.020
118. Cheng Q, Patel K, Lei B, Rucker L, Allen DP, Zhu P, et al. Donor pretreatment with nebulized complement C3a receptor antagonist mitigates brain-death induced immunological injury post-lung transplant. Am J Transplant (2018) 18:2417–28. doi: 10.1111/ajt.14717
119. Kassimatis T, Qasem A, Douiri A, Ryan EG, Rebollo-Mesa I, Nichols LL, et al. A double-blind randomised controlled investigation into the efficacy of mirococept (APT070) for preventing ischaemia reperfusion injury in the kidney allograft (EMPIRIKAL): study protocol for a randomised controlled trial. Trials (2017) 18:255. doi: 10.1186/s13063-017-1972-x
Keywords: complement inhibitor, ischemia-reperfusion injury, ex vivo delivery, organ transplantation, classic pathway
Citation: Delaura IF, Gao Q, Anwar IJ, Abraham N, Kahan R, Hartwig MG and Barbas AS (2022) Complement-targeting therapeutics for ischemia-reperfusion injury in transplantation and the potential for ex vivo delivery. Front. Immunol. 13:1000172. doi: 10.3389/fimmu.2022.1000172
Received: 21 July 2022; Accepted: 05 October 2022;
Published: 19 October 2022.
Edited by:
Miriam Manook, University of Cambridge, United KingdomReviewed by:
Kang He, Shanghai Jiao Tong University, ChinaCopyright © 2022 Delaura, Gao, Anwar, Abraham, Kahan, Hartwig and Barbas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrew S. Barbas, YW5kcmV3LmJhcmJhc0BkdWtlLmVkdQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.