 Qian Li
Qian Li Yuanjun Deng
Yuanjun Deng Lele Liu
Lele Liu- Division of Nephrology, Department of Internal Medicine, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
Objective: Continuous overactivation of the renal sympathetic nerve is considered to be an important cause of renal fibrosis. Accumulated senescent cells in the damaged kidney have metabolic activities and secrete amounts of proinflammatory factors as part of the SASP (the senescence-associated secretory phenotype), which induce chronic inflammation and fibrosis. It is still unclear whether renal sympathetic nerves affect renal inflammation and fibrosis by regulating cellular senescence. Therefore, we hypothesize that sympathetic activation in the injured kidney induces cellular senescence, which contributes to progressive renal inflammation and fibrosis.
Methods: Renal denervation was performed 2 days before the UUO (unilateral ureteral obstruction) and UIRI (unilateral ischemia-reperfusion injury) models. The effects of renal denervation on renal fibrosis and cellular senescence were observed. In vitro, cellular senescence was induced in renal proximal tubular epithelial cell lines (TKPTS cells) by treatment with norepinephrine (NE). The selective α2A-adrenergic receptor (α2A-AR) antagonists BRL44408 and β-arrestin2 siRNA, were administered to inhibit NE-induced cellular senescence. A significantly altered pathway was identified through immunoblotting, immunofluorescence, immunocytochemistry, and functional assays involved in mitochondrial function.
Results: Renal fibrosis and cellular senescence were significantly increased in UUO and UIRI models, which were partially reversed by renal denervation. In vitro, NE induced epithelial cells secreting proinflammatory cytokines and promoted cell senescence by activating α2A-AR. Importantly, the effects of NE during cellular senescence were blocked by α2A-AR selective antagonist and β-arrestin2 (downstream of α2A-AR) siRNA.
Conclusion: Renal sympathetic activation and cellular senescence are important neurometabolic and neuroimmune mechanisms in the development of renal fibrosis. Renal sympathetic neurotransmitter NE acting on the α2A-AR of epithelial cells promotes cellular senescence through the downstream β-arrestin2 signaling, which is a potential preventive target for renal fibrosis.
Introduction
The incidence and prevalence of chronic kidney disease (CKD) have been rapidly increasing in recent years, especially in the elderly (1, 2). The epidemic surveys showed that the prevalence of CKD in the elderly was 3- to 13-fold higher than that in younger individuals (2). In the elderly, the presence of other diseases, such as hypertension, atherosclerosis, and diabetes, could accelerate the age-related decline of renal function (3). Thus, the elderly was more sensitive to renal injury and more likely developed to CKD (4). Similar to aging kidneys (5, 6), pathological changes of CKD included glomerular sclerosis, tubular atrophy, and interstitial fibrosis (7). These findings suggested that CKD was an “accelerated aging” phenotype (8).
Chronic senescent cell accumulation was found in the renal tissues of various CKDs (9). Premature aging deteriorates the health, life quality, and survival time of patients with CKD (10). Uremic toxins accelerated the progress of cellular senescence in CKD (8), while chronic senescent cells accumulating in aging and damaged kidneys ulteriorly drove renal fibrogenesis (9). These senescent cells underwent metabolic reprogramming and produced important proinflammatory and profibrogenic mediators as part of the SASP, which contributed to chronic inflammation and organ fibrosis (11).
The nerves innervating the kidney are composed of efferent sympathetic nerves and afferent sensory nerves (12). Similar to the cardiovascular system, the renal physiological and pathological functions are also regulated by sympathetic nerves (13, 14). It has been proved that efferent sympathetic nerve fibers are distributed in juxtaglomerular granular cells, renal tubular epithelial cells, and renal vasculature (13). In CKD patients, sympathetic excitability was improved, which contributes to CKD progression. Accumulating studies revealed that sympathetic nerve activities and tissue contents of neurotransmitter norepinephrine are increased in CKD patients and experimental animals (15, 16). Renal denervation has been proved to be an effective intervention of renal interstitial fibrogenesis in obstructive nephropathy and ischemia-reperfusion injury animal models (17, 18). Although the potential mechanisms, including disordered inflammatory response, cell cycle arrest, apoptosis-related signaling pathways, and reduced nitric oxide (NO) availability, were indicated to be related to CKD progression induced by sympathetic excitation (16, 19), the exact role and mechanism of the sympathetic pathway are still unclear.
In this study, we sought to clarify the possible mechanism of sympathetic nerves promoting renal fibrosis by inducing cellular senescence. The findings of our study have not only provided more comprehensive neurometabolic/neuroimmune mechanisms for renal fibrosis but also clarified a detailed signaling pathway involved in sympathetic nerve-induced senescence.
Materials and Methods
Mice and Surgical Preparation
Mice aged 8 weeks were grouped as sham-operated, UUO, UIRI, and renal denervation (DNx). UUO and UIRI surgery were performed as described previously (20, 21). Briefly, the UUO-operated group was conducted by ligating completely the left ureter with 5-0 silk sutures. The UIRI group was established in the mice subjected to clamping of the left renal pedicle for 30 min at 37°C. DNx was performed by stripping the sheath and adventitia of the left renal pedicle through the abdominal incision, as previously reported (17, 22). Briefly, after the renal artery and vein were dissected carefully from the surrounding connective tissue, the visible nerve fibers were cut off. Then the renal vessels were painted for 2 min with sterile gauzes soaked in 10% phenol anhydrous ethanol solution to exterminate the remaining nerves. Notably, the body temperature of the mice was kept between 36.6° and 37.2°C during the whole process. Mice were euthanized 10 days after UUO or 18 days after UIRI surgery. All animal procedures were performed following the Huazhong University of Science and Technology of Health guidelines.
Cell Culture and Treatment
Mouse renal proximal tubular epithelial cell lines (TKPTS cells, American Type Culture Collection, Manassas, VA, USA) were maintained using a DMEM-F12 medium (Gibco, Carlsbad, CA, USA), which was added with 10% fetal bovine serum (Gibco). When 40% confluent, cells were serum-free for 8 hours before norepinephrine (10 nM, HY-13715B, MedChemExpress, USA) or H2O2 (100 μM, Promoter Biotechnology, China) treatment for 48 hours. In some experiments, cells were pretreated with Atipamezole (α2-adrenergic antagonist, HY-12380A, MedChemExpress), Butoxamine (β2-adrenergic antagonist, B1385, Sigma), ICI118551 (β2-adrenergic antagonist, HY-13951, MedChemExpress), and BRL44408 (α2A-adrenergic antagonist, ab120806, Abcam, Cambridge, MA, USA) an hour before norepinephrine stimulation.
Transfection of siRNA
β-arrestin2 small interfering RNA (siRNA) was designed and synthesized by Ribobio Biotechnology (Guangzhou, China). The TKPTS cells were transfected transiently with β-arrestin2 or scrambled siRNA diluted in Lipofectamine iMAX (Thermo Fisher Scientific, USA) 24 hours before norepinephrine treatment.
Western Blot Analysis
Protein homogenates were made from the left kidney or the cultured TKPTS cells. Western blot analyses were conducted following the manufacturer’s instructions with specific primary antibodies: fibronectin (15613-1-AP, Proteintech, Rosemont, IL, USA), College 1 (14695-1-AP, Proteintech), α-SMA (ab5694, Abcam), tyrosine hydroxylase (TH, ab112, Abcam), p53 (A3185, ABclonal Technology, China), p21 (14-6715-81, Invitrogen), γ-H2AX (#9718, Cell Signaling Technology, Danvers, MA, USA), α2A-AR (DF3076, Affinity Biosciences), β-arrestin2 (10171-1-AP, Proteintech), VDAC1 (55259-1-AP, Proteintech), COX IV (11242-1-AP, Proteintech), and NF-κB p65 (sc-8008, Santa Cruz Biotechnology). Anti-GAPDH (AC001, ABclonal Technology) or β-actin antibody (AC026, ABclonal Technology) was adopted for loading controls on membranes. Subsequently, the horseradish peroxidase (HRP)-conjugated secondary antibodies (Servicebio, China) were applied to combine the primary ones for 90 min at room temperature. Finally, the ECL system was used to visualize proteins, and the Image J software (NIH, USA) was applied to analyze bands.
Quantitative Real-Time PCR (qRT-PCR)
RNA in kidney tissues or cells was isolated following the RNA extraction process of the manufacturer, which was assisted by the TRIzol (9109, TaKaRa, Japan). The cDNA was synthesized using 1 μg of RNA through a HiScript II Q RT SuperMix for qPCR kit (R222-01, Vazyme) at 50°C for 15 min and 85°C for 5 s. After using a ChamQTM Universal SYBR® qPCR Master Mix kit (Q711-02, Vazyme), qRT-PCR was performed on a Roche Light 480 II system. Mouse GAPDH or β-actin was used as the housekeeping gene, and the 2-ΔΔCt method was adopted to calculate the relative quantitative evaluation of the target genes. Table 1 shows the primer sequences.

Table 1 The primer sequences used for qRT-PCR.
Histology, Immunohistochemistry, Immunofluorescence
Mouse paraffin-embedded kidney sections (4-µm thickness) fixed in 4% paraformaldehyde were subjected to the Periodic Acid-Schiff (PAS) Stain, Sirius Red Stain, and Masson Stain according to the manufacturer’s procedures. Immunohistochemistry and immunofluorescence were performed following the manufacturer’s protocols with specific antibodies against α2A-AR (A2809, ABclonal Technology) and β-arrestin2 (10171-1-AP, Proteintech). Samples were exposed to peroxidase-conjugated secondary antibodies followed by incubation with hematoxylin to visualize the cell nuclei for immunohistochemistry. As for immunofluorescence, samples were incubated with fluorescence-labeled secondary antibodies (Servicebio), and nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). Finally, representative images were acquired by using a fluorescence microscope.
SA-β-gal, MitoTracker, and TMRE Staining
TKPTS cells cultured on plates were performed SA‐β‐gal,MitoTracker, and TMRE staining according to the guides of the manufacturer. SA‐β‐gal staining was used to test the β‐galactosidase activity (C0602, Beyotime Biotechnology, Shanghai, China) for the detection of senescence. MitoTracker deep red (9082P, Cell Signaling Technology) and Tetramethylrhodamine, Ethyl Ester, Perchlorate (TMRE, T669, Thermo Fisher Scientific) staining were adopted to measure mitochondria and mitochondrial membrane potential. TKPTS cells were incubated in MitoTracker (100 nM) or TMRE (200 nM) at 37°C against the light for 30 min before performing the fluorescence activation.
Transmission Electron Microscopy
TKPTS cells cultured on plates were collected and prepared for assessing mitochondrial morphology using a transmission electron microscope provided by the Electron Microscope Center of Renmin Hospital of Wuhan University.
Immunoprecipitation
Lysates containing 4 mg of total protein were made using NP40 lysis buffer (Promotor, Wuhan, China) in TKPTS cells cultured on plates and were incubated with 4 μg of an anti-β-arrestin2 antibody at 4°C overnight. Normal IgG (2729S, Cell Signaling Technology) was used as a negative control. Then Protein A/G Plus Agarose (sc-2003, Santa Cruz Biotechnology) was put into the compound for 8 hours at 4°C. After the beads were washed 3 times, the lysates were resuspended and boiled. Immunoblotting was operated as a method of Western blot.
Statistical Analyses
Data among multiple groups were assessed by one-way ANOVA followed by Tukey’s multiple comparisons test, and data between two groups were analyzed via nonparametric two-tailed unpaired t-tests through the GraphPad Prism software for Windows Version 8.0 (La Jolla, CA, USA). All data were shown as means ± SEMs. P values < 0.05 mean statistical significance. *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001.
Results
Renal Denervation Inhibits Renal Fibrosis During UUO and UIRI
To assess whether sympathetic nerves are involved in renal fibrosis, we constructed renal denervation (DNx) 2 days before UUO and UIRI. Light microscopy indicated interstitial fibrosis in the UUO-10d and UIRI-18d mice, but not in the sham-operated group, which was dramatically reduced after DNx treatment (Figures 1A, B). The effectiveness of DNx was confirmed by a significantly diminished expression of tyrosine hydroxylase (TH), a marker of sympathetic nerve fibers (17, 18), suggesting that the model of renal denervation was successfully performed (Figures 1C, D). By analyzing the effects of renal denervation on protein and mRNA expression, we showed that renal denervation attenuated profibrotic protein production in UUO or UIRI mice, which was measured by fibronectin (FN), collage 1 (Col 1), and α-SMA (Figures 1C–H). Taken together, these results indicate that sympathetic activation is closely related to renal fibrosis, and renal denervation has an obvious effect on reducing interstitial fibrosis.
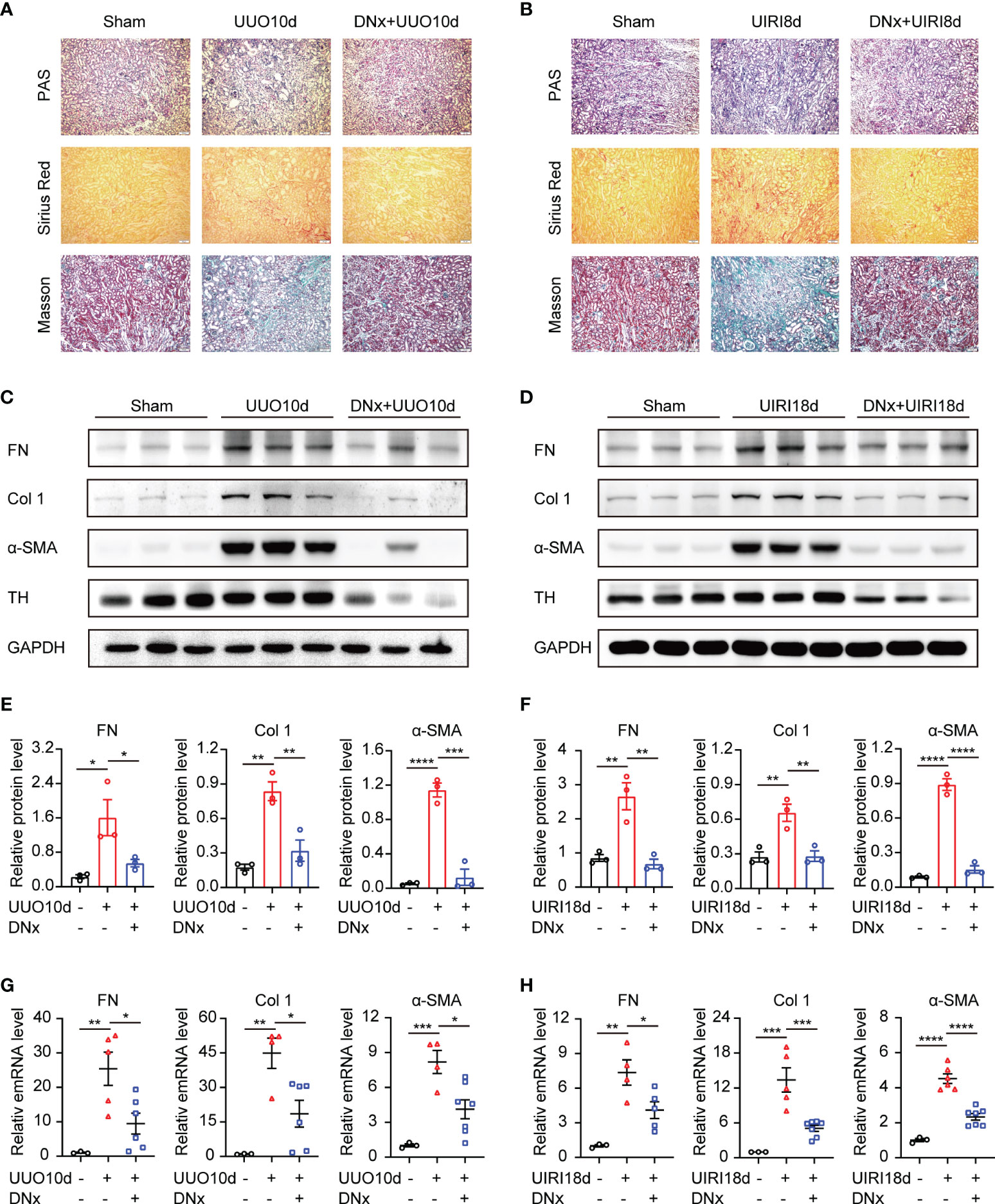
Figure 1 Renal denervation inhibits the fibrotic phenotype during UUO and UIRI. C57BL/6 mice were subjected to UUO or UIRI manipulation 2 days after renal denervation (DNx) or sham operation in the left kidneys. (A, B) Representative histology of kidney sections from sham-operated, UUO/UIRI mice, and DNx+UUO/UIRI mice stained with periodic acid–Schiff, Sirius Red, and Masson’s trichrome stain. Scale bar, 50μm. (C, D) Representative Western blot of fibronectin (FN), Collagen I (Col 1), α-SMA, and Tyrosine Hydroxylase (TH) in the kidney tissue of sham-operated, UUO/UIRI mice, and DNx+UUO/UIRI mice with densitometry analysis (E, F). (G, H) Representative mRNA expressions of FN, Col 1, and α-SMA in the kidney tissue of sham-operated, UUO/UIRI mice, and DNx+UUO/UIRI mice using qRT-PCR analysis. n=3–7 in each group. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Bars represented means ± SEM.
Renal Denervation Reduces Renal Senescence and Inflammation After Kidney Injury
Recent work has indicated that increasing cellular senescence acts as a key contributor to renal inflammation and fibrosis (23, 24). To analyze the effect of renal sympathetic nerves on renal senescence, we detected expressions of senescence-related proteins and senescence-associated secretory phenotype (SASP) in intact or denervated kidneys. Protein expressions of p53, p21, and γ-H2AX (senescent markers) were increased in intact kidneys induced by UUO or UIRI, whereas renal denervation significantly attenuated their expression (Figures 2A–D). Proinflammatory genes, as part of the SASP, are expressed by senescent cells (25). Our results demonstrated that senescence-related symbols (p53, p21, p16, HMGA2, H2AX) and inflammatory components of the SASP (TGF-β1, IL-6, IL-8, and IL-1β) were elevated in mice of UUO or UIRI, which were decreased in denervated kidneys (Figures 2E, F). Taken together, these data demonstrate that cellular senescence is closely related to renal inflammation and fibrosis, and renal denervation inhibits cellular senescence.
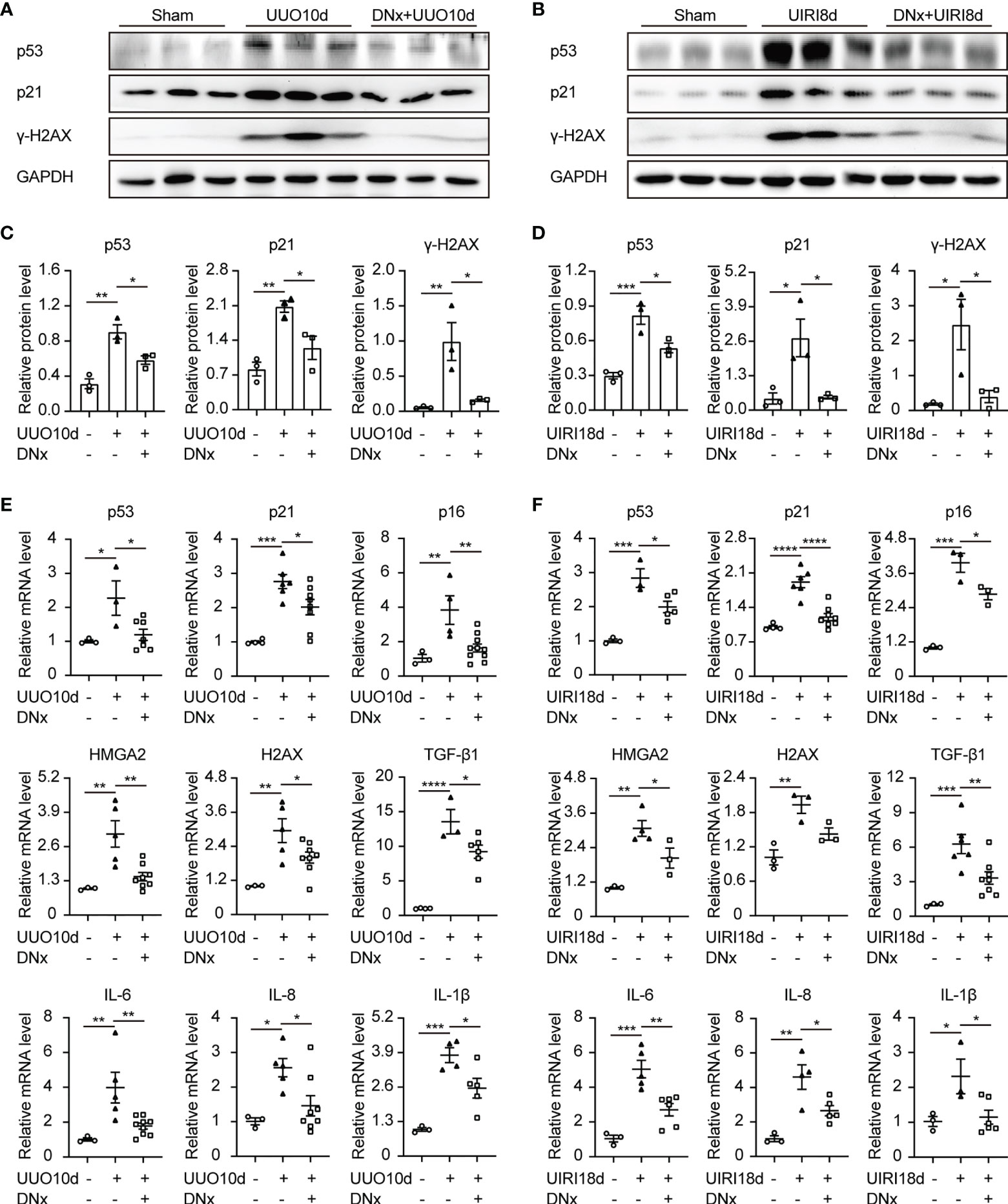
Figure 2 Renal denervation reduces renal senescence and the expression of proinflammatory cytokines after kidney injury. Renal denervation (DNx) or sham operation was performed 2 days before the UUO or UIRI model in the left kidneys of mice. (A, B) Representative Western blot of senescence-related proteins p53, p21, and γ-H2AX in sham-operated, UUO/UIRI, and DNx+UUO/UIRI kidneys with densitometry analysis (C, D). (E, F) mRNA expressions of senescence-related markers p53, p21, p16, HMGA2, H2AX, and senescence-associated secretory phenotype (SASP) TGF-β1, IL-6, IL-8, and IL-1β in the kidney tissue of sham-operated, UUO/UIRI mice, and DNx+UUO/UIRI mice as shown by qRT-PCR analysis. n=3–10 in each group. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Bars represented means ± SEM.
NE Triggers Cellular Senescence and Upregulates Proinflammatory Cytokines and Profibrogenic Factors in Renal Tubular Cells
As a primary neurotransmitter released by the sympathetic nerve fibers, NE participates in kinds of renal physiological functions, including renal blood flow, renal tubular reabsorption of sodium and water, as well as the neural control of renal functions (26). To identify the effect of sympathetic activity on renal fibrosis and cellular senescence, TKPTS cells were utilized and treated with exogenous NE to imitate the state of sympathetic activation. As shown in Figures 3A–D, Western blot analysis revealed that NE markedly increased the levels of senescence-related proteins (p21, H2AX) and profibrotic proteins (FN, Col1) in a time- and concentration-dependent manner. The optimal exposure concentration and time of NE were 10 nM and 48 hours, respectively. Thus, TKPTS cells were harvested with NE (10 nM) for 48 hours in the following experiments. Notably, in the NE-treated group, the expression levels of p53, p21, and γ-H2AX were significantly increased, which was similar to the results from TKPTS cells for the indicated periods after hydrogen peroxide (H2O2) treatment [a classic stimulator of cellular senescence (27)] (Figure 3E). Compared with basal conditions, NE-/H2O2 treatment induced a significant increase in the enzymatic activity of SA-β-Gal (senescence-associated β-galactosidase), a hallmark of senescence (Figure 3F). Furthermore, RT-PCR results showed that the mRNA levels of senescence-related proteins (p53, p21, p16, HMGA2, H2AX), SASP components (IL-6, IL-8, IL-1β, TGF-β1), and profibrogenic growth factor CTGF were increased by NE stimulation, which was consistent with the results in the H2O2 group (Figure 3G). These data suggest that NE contributes to renal tubular cell senescence and upregulates proinflammatory and profibrogenic components.
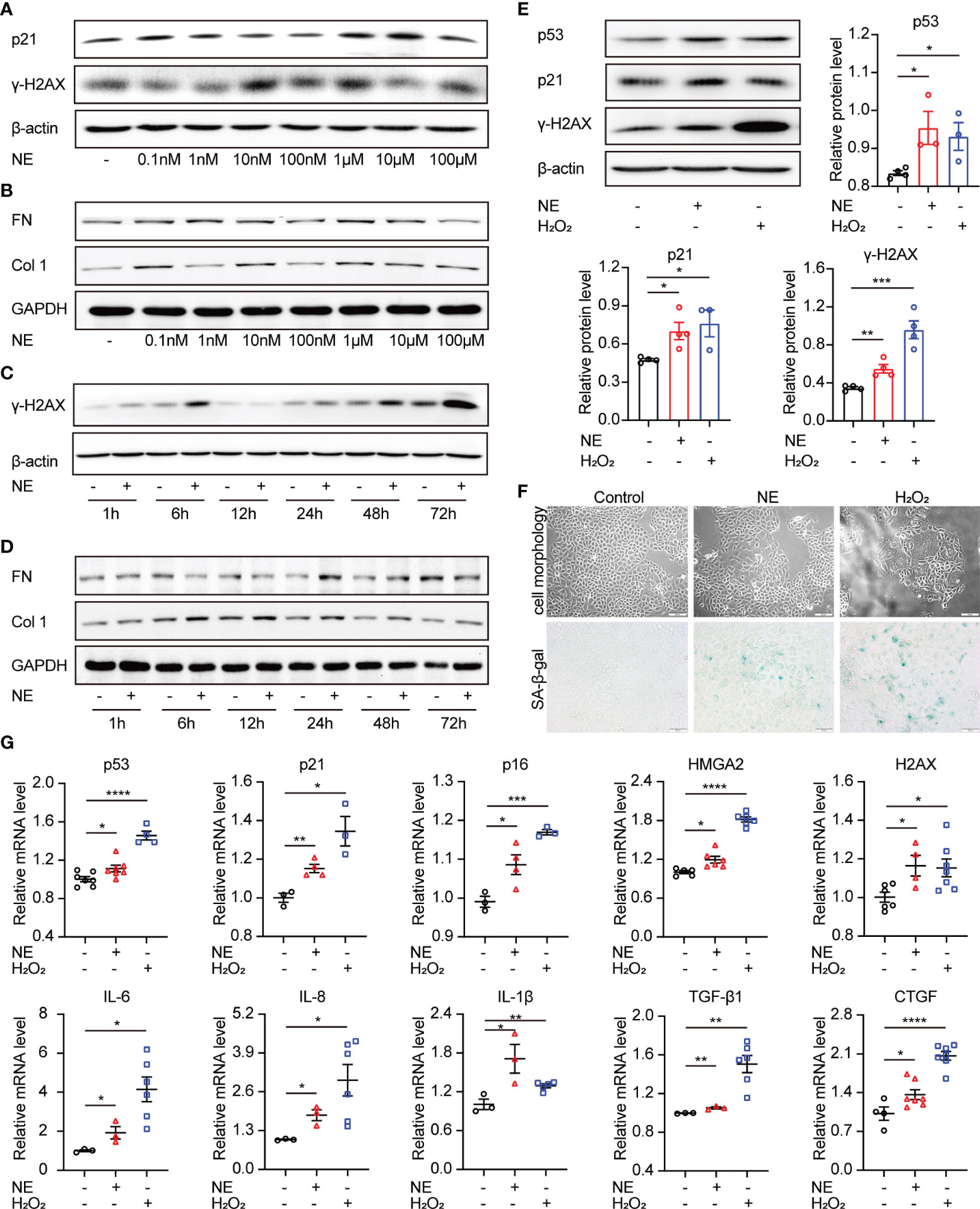
Figure 3 Norepinephrine contributes to cellular senescence and upregulates profibrogenic and proinflammatory cytokines in renal tubular cells. (A, B) Representative Western blot of p21, γ-H2AX or FN, Col 1 in TKPTS cells cultured with PBS (control) or different concentrations of NE (0.1 nM, 1 nM, 10 nM, 100 nM, 1 μM, 10 μM, and 100 μM) (n=3 in each group). (C, D) Representative Western blot of γ-H2AX or FN, Col 1 in TKPTS cells incubated with PBS (control) or NE at a final concentration of 10 nM for the indicated periods (1, 6, 12, 24, 48, and 72 hours) (n=3 in each group). (E–G) TKPTS cells were incubated with NE (10 nM) or H2O2 (100 μM) for 48 hours and were then harvested. (E) Representative Western blot of p53, p21, and γ-H2AX in TKPTS cells treated with PBS, NE, or H2O2 with densitometry analysis (n=3–5 in each group). (F) Representative images of cell morphology and SA-β-gal staining of TKPTS cells treated with PBS, NE, or H2O2 using a microscope (n=3 in each group). Scale bar=50 μm. (G) mRNA levels of senescence-related proteins (p53, p21, p16, HMGA2, H2AX), SASP components (IL-6, IL-8, IL-1β), and profibrotic cytokines (TGF-β1, CTGF) in each group were detected by qRT-PCR analysis (n=3–7 in each group). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Bars represented means ± SEM.
The α2A-Adrenergic Receptor Is Responsible for NE-Induced Cellular Senescence
NE participates in signal transductions through the adrenergic receptors (ARs), a member of the GPCR families. ARs consist of two α-AR (α1-, α2-AR) and three β-AR (β1-, β2-, β3-AR) (28). Thus, we investigated the roles of ARs in cellular senescence using their antagonists. Firstly, cDNA was used to determine the mRNA expression levels of five AR coding genes (ADRA1B, ADRA2A, ADRB1, ADRB2, and ADRB3) (29). Results showed that ADRB2 (coding β2-AR) and ADRA2A (coding α2A-AR) were enriched in TKPTS cells (Figure 4A). Furthermore, the expression of ADRB2 and ADRA2A were upregulated after NE treatment (Figure 4B). To identify which adrenergic receptor mediates renal tubular epithelial cell senescence induced by NE, we pretreated TKPTS cells with β2-AR and α2-AR adrenergic antagonists, respectively. Forty-eight hours after NE stimulation, cells pretreated with α2-AR antagonist Atipamezole (ATI) at the concentrations of 10–100 μM showed significant reductions in p21 and γ-H2AX protein levels (Figure 4C), whereas treatment with the two β2-AR antagonists (Butoxamine and ICI118551) showed no reductions in p21 and γ-H2AX protein expressions (Figures 4D, E). Consistent with the results from Western blot, RT-PCR analysis also showed that TKPTS cells pretreated with α2-AR antagonist ATI significantly reduced the mRNA levels of senescence-related proteins (p53, p21, p16, HMGA2, H2AX) and proinflammatory cytokines (IL-6, IL-8, IL-1β) (Figure 4F). These data suggest that NE induces cellular senescence through α2-AR, and pretreatment with the α2-adrenergic antagonist suppresses the senescence phenotype and inflammatory response induced by NE.
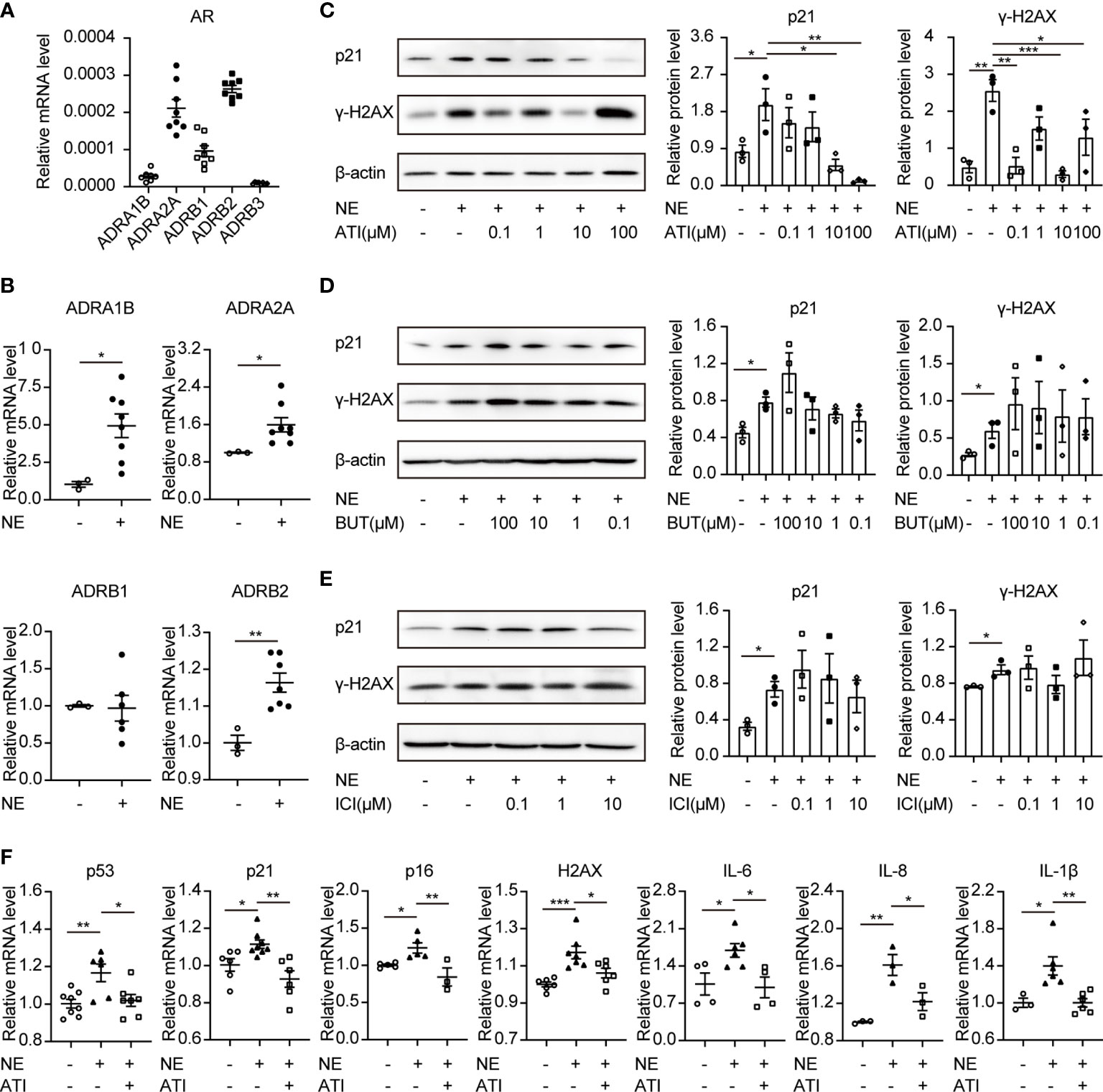
Figure 4 Atipamezole (α2-adrenergic antagonist) reverses cellular senescence induced by NE in renal tubular cells. TKPTS cells were incubated with NE (10 nM) for 48 hours after pretreatment with adrenergic antagonists or PBS for an hour. (A, B) mRNA expressions of ADRA1B, ADRA2A, ADRB1, ADRB2, and ADRB3 in PBS (control) or NE-treated TKPTS cells as shown by qRT-PCR analysis (n=3–8 in each group). (C) Representative Western blotting of p21 and γ-H2AX in TKPTS cells treated with PBS, NE, and NE+ATI (Atipamezole, 100 nM, 1 μM, 10 μM, and 100 μM) with densitometry analysis (n=3 in each group). (D, E) Representative Western blotting of p21 and γ-H2AX in TKPTS cells incubated with PBS, NE, and NE+BUT (Butoxamine)/ICI (ICI118551) (two β2-adrenergic antagonists, 100 μM, 10 μM, 1 μM, and 100 nM) with densitometry analysis, respectively (n=3 in each group). (F) mRNA expressions of p53, p21, p16, H2AX, IL-6, IL-8, and IL-1β in TKPTS cells cultured with PBS, NE, and NE+ATI (10 μM) as displayed by qRT-PCR analysis (n=3–8 in each group). *P < 0.05, **P < 0.01, ***P < 0.001. Bars represented means ± SEM.
α2-AR consists of α2A-AR, α2B-AR, and α2C-AR subtypes. Kable JW et al. demonstrated that the classical pharmacological actions of α2-AR were mainly mediated by the α2A-AR subtype, including antihypertensive, sedative, analgesic, and so on (30). Thus, in the present study, α2A-AR expression in renal tissue and TKPTS cells was further examined. Results showed that α2A-AR was significantly increased in kidney sections from UUO or UIRI mice, whereas renal denervation markedly lessened α2A-AR expression (Figures 5A, B). Moreover, compared with the control group, the expression of α2A-AR was significantly increased in cells treated with NE (Figures 5C, D). To test whether α2A-AR contributes to tubular epithelial cell senescence induced by NE, selective α2A-AR antagonist BRL44408 (BRL, 10 μM) was applied to TKPTS before NE stimulation. Western blot analysis demonstrated that BRL pretreatment significantly decreased the expression of p53, p21, and γ-H2AX compared with that in the NE group (Figure 5E). What is more, BRL pretreatment abolished NE-induced increase in the mRNA expressions of senescence-related proteins (p53, p21, p16, HMGA2, H2AX) and SASP components (TGF-β1, IL-6, IL-8, and IL-1β) (Figure 5F). Taken together, these data suggest that α2A-AR plays a crucial role in tubular epithelial cell senescence and is closely associated with inflammatory cytokine expression induced by NE.
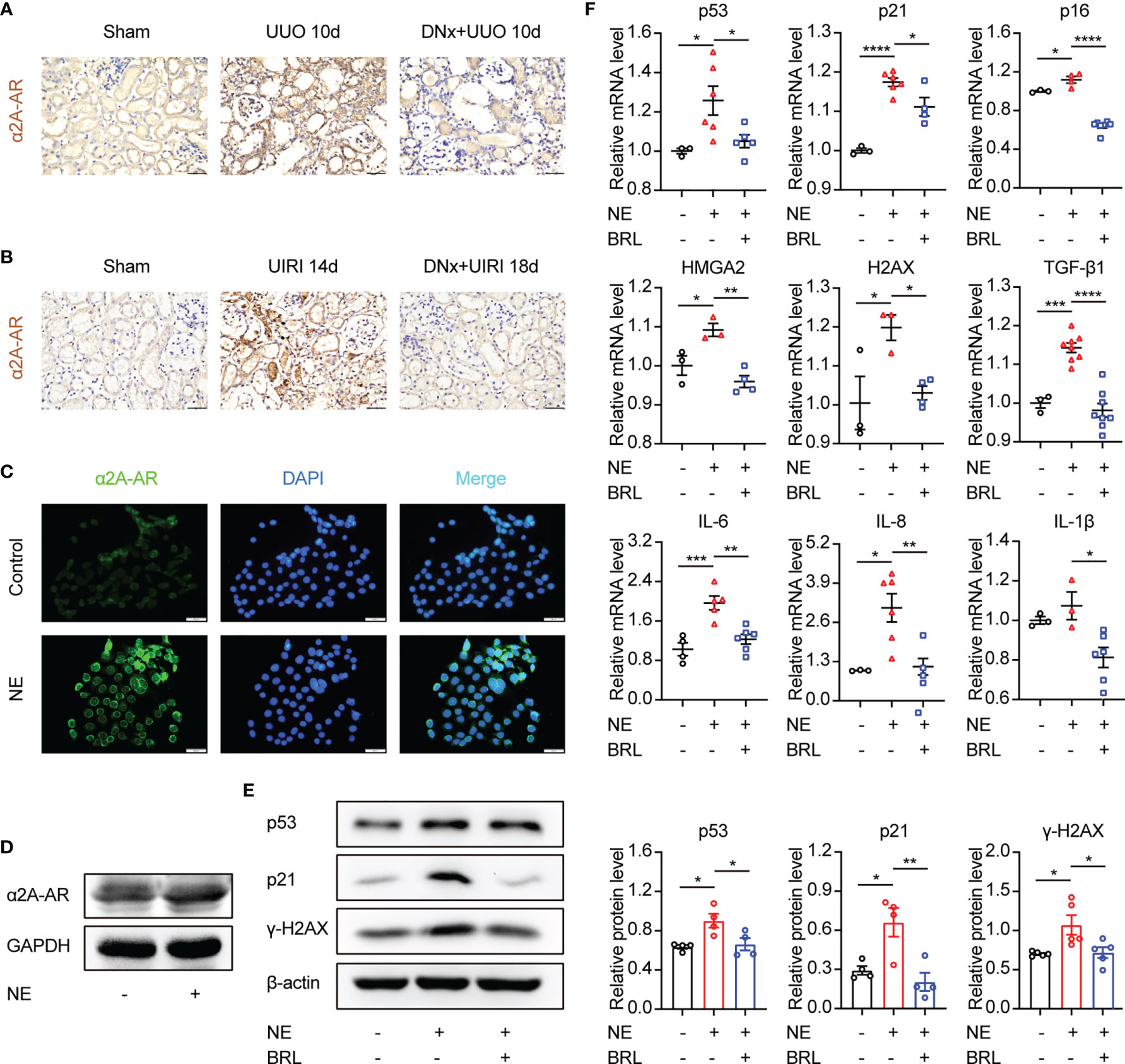
Figure 5 The α2A-adrenergic receptor is responsible for NE-induced tubular epithelial cell senescence. (A, B) Renal denervation (DNx) or sham operation was performed 2 days before UUO or UIRI in the left kidneys of mice. Representative images of immunohistochemistry for α2A-adrenergic receptors (α2A-AR) in sham-operated, UUO/UIRI, mice and DNx+UUO/UIRI mice. Scale bar, 20μm. (C, D) After starving for 8 hours, TKPTS cells were cultured with PBS or NE (10 nM) for 48 hours. α2A-AR expressions were tested by immunofluorescence (C) (Scale bar, 20 μm) and Western Blot (D) (n=3 in each group). (E, F) After pretreatment with PBS or α2A-adrenergic antagonist (BRL4408, BRL, 10 μM) for 1 hour, TKPTS cells were incubated with NE (10 nM) for 48 hours. (E) Representative Western blotting of p53, p21, and γ-H2AX in each group with densitometry analysis (n=4-5). (F) mRNA expressions of p53, p21, p16, HMGA2, H2AX, TGF-β1, IL-6, IL-8, and IL-1β were analyzed using qRT-PCR analysis (n=3–8 in each group). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Bars represented means ± SEM.
α2A-AR/β-Arrestin2 Signaling Contributes to Cellular Senescence Induced by NE
We next explored the signaling pathway downstream of the α2A-AR that promotes cellular senescence. α2A-AR belongs to G protein-coupled receptors (GPCRs). Besides the canonical Gs-cAMP pathway, the GPCR/β-arrestin2 pathway was closely related to organ fibrosis, including liver fibrosis, pulmonary fibrosis, myocardial fibrosis, and renal fibrosis (31–33). In the current study, results showed that the expression of β-arrestin2 was substantially increased on the kidney sections of mice with UUO or UIRI models compared with the sham-operated group as measured by immunohistochemistry and Western blotting (Figures 6A–D). On the other hand, renal denervation lessened the expression of β-arrestin2 compared with the UUO or UIRI group. To further demonstrate the direct effects of the α2A-AR/β-arrestin2 pathway on epithelial cells, we analyzed TKPTS cells in vitro. Immunofluorescence staining showed that β-arrestin2 was substantially increased in the presence of NE compared with control (Figure 6E). Furthermore, we observed the direct colocalization of α2A-AR and β-arrestin2, which was confirmed by analyzing protein–protein interaction using co-immunoprecipitation (Figures 6F, G). Besides, TKPTS pretreated with selective α2A-AR antagonist BRL44408 (BRL) relieved β-arrestin2 expression using Western blot and qRT-PCR (Figures 6H, I), illustrating the direct relationship between β-arrestin2 and α2A-AR. To further illustrate the role of β-arrestin2 on NE-mediated cellular senescence in TKPTS cells, siRNAs (small interfering RNAs) were used to specifically knock down β-arrestin2. As shown in Figures 6J, K, si-Arrb2-1 exhibited the most inhibitory effect on β-arrestin2. Thus, si-Arrb2-1 was used for subsequent experiments. Compared with the NE group, si-Arrb2 restrained the expression of p53 and p21 (Figure 6L). Consistently, the mRNA levels of p21, H2AX, TGF-β1, IL-6, and IL-8 were also prevented by si-Arrb2 pretreatment (Figure 6M). These results indicate that NE activates α2A-AR/β-arrestin2 signaling to induce cellular senescence and proinflammatory responses, and blockade of β-arrestin2 inhibits this process.
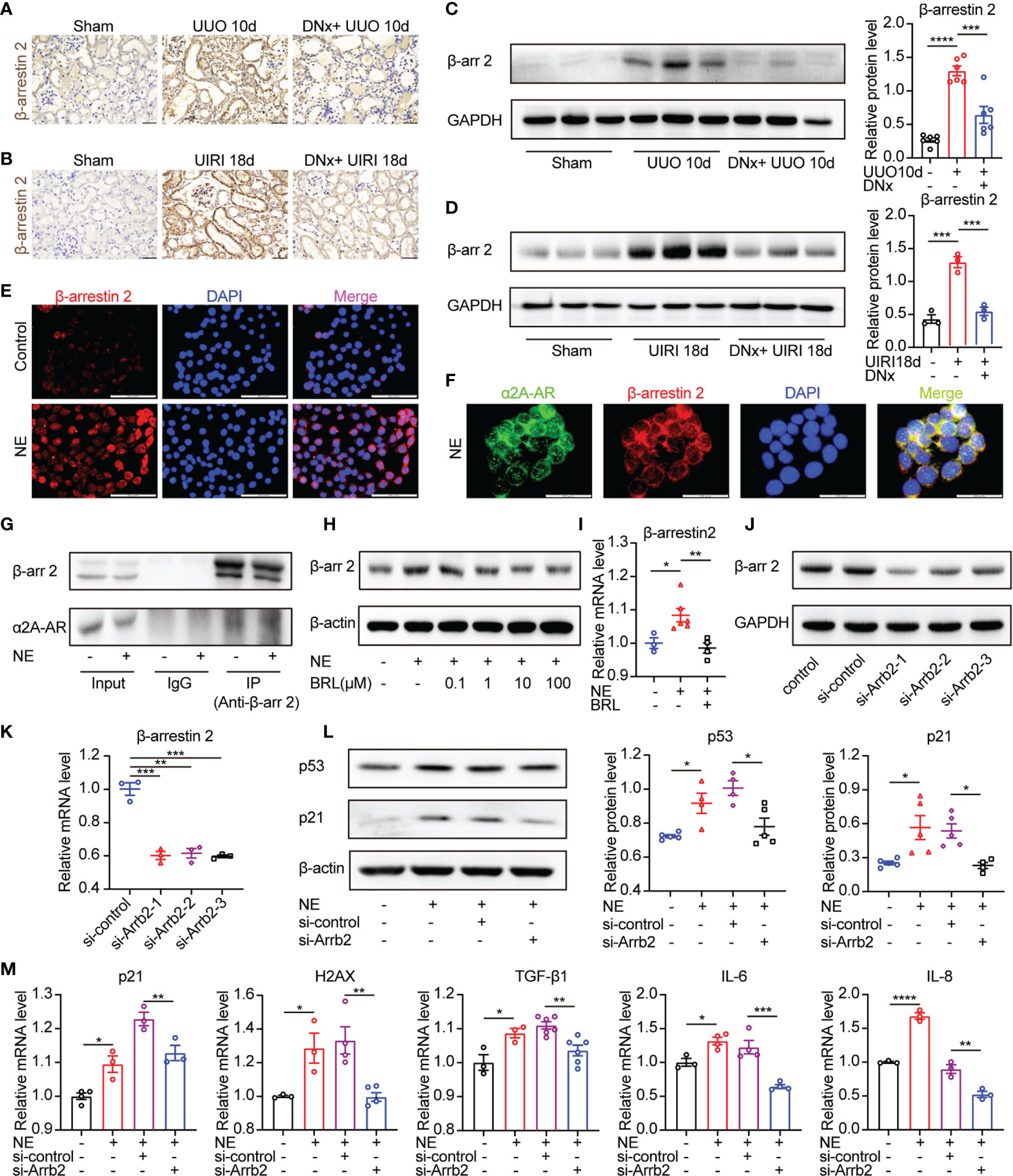
Figure 6 β-arrestin2 is a target of α2A-AR and knockdown of β-arrestin2 ameliorates NE-induced tubular cellular senescence. (A, B) Renal denervation (DNx) or sham operation was performed 2 days before UUO or UIRI in the left kidneys of mice. Representative images of immunohistochemistry for β-arrestin2 in sham-operated, UUO/UIRI mice, and DNx+UUO/UIRI mice. Scale bar, 20 μm. (C, D) Representative Western blotting of β-arrestin2 (β-arr2) in sham-operated, UUO/UIRI, and DNx+UUO/UIRI kidneys with densitometry analysis (n=3–6 in each group). (E–G) TKPTS cells were cultured with PBS or NE (10 nM) for 48 hours. (E) Representative images of immunofluorescence for β-arrestin2 in each group. Scale bar, 20μm. (F) Cells were double-labeled with α2A-AR (green) and β-arrestin2 (red), and cell nuclei were enhanced by staining with DAPI (blue). Colocalization of α2A-AR (green), and β-arrestin2 (red) was visualized as yellow in merged images. Scale bar, 20μm. (G) Interactions between β-arrestin2 and α2A-AR were examined by immunoprecipitation (IP) with an anti-β-arrestin2 antibody, followed by immunoblotting (IB) with anti-α2A-AR antibodies. (H, I) After an hour of pretreatment with PBS or BRL4408, TKPTS cells were stimulated with NE (10 nM) for 48 hours. (H) Representative Western blotting of β-arrestin2 (β-arr2) in TKPTS cells treated with PBS, NE, and NE+BRL (100 nM, 1 μM, 10 μM, and 100 μM) (n=3 in each group). (I) mRNA expression of β-arrestin2 in PBS, NE, and NE+BRL (10 μM)-treated TKPTS cells as measured by qRT-PCR analysis (n=3–6 in each group). (J, K) TKPTS cells were transfected with scrambled siRNA or three β-arrestin2 siRNAs (si-Arrb2-1, si-Arrb2-2, or si-Arrb2-3) for 48 hours. β-arrestin2 (β-arr2) expression was examined by Western blot analysis (J) and qRT-PCR analysis (K) (n=3 in each group). (L, M) TKPTS cells were transfected with scrambled siRNA (si-control) or β-arrestin2 siRNA (si-Arrb2) 24 hours before NE (10 nM) treatment. Representative Western blotting of p53 and p21 in each group with densitometry analysis (L) (n=4–5 in each group). mRNA expressions of p21, H2AX, TGF-β1, IL-6, and IL-8 were tested by qRT-PCR analysis (M) (n=3–6 in each group). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Bars represented means ± SEM.
α2A-AR/β-Arrestin2/NF-κB Signaling Is Involved in Mitochondrial Dysfunction and Cellular Senescence
Considering that the mitochondria status has been closely associated with senescence and aging phenotypes (34), we aimed to determine the response of mitochondria under NE stimulation. We used the voltage-dependent anion channel 1 (VDAC1) and cytochrome c oxidase subunit IV (COX IV), markers representing the mitochondrial outer membrane and the mitochondrial inner membrane, respectively, to represent the mitochondria content (35). VDAC1 and COX IV protein levels were reduced in UUO or UIRI kidneys, whereas renal denervation could rescue their expressions (Figures 7A, B). Besides, NE-treated TKPTS cells showed decreased expression of VDAC1 and COX IV (Figure 7C). These data suggest that sympathetic activation leads to mitochondrial dysfunction.
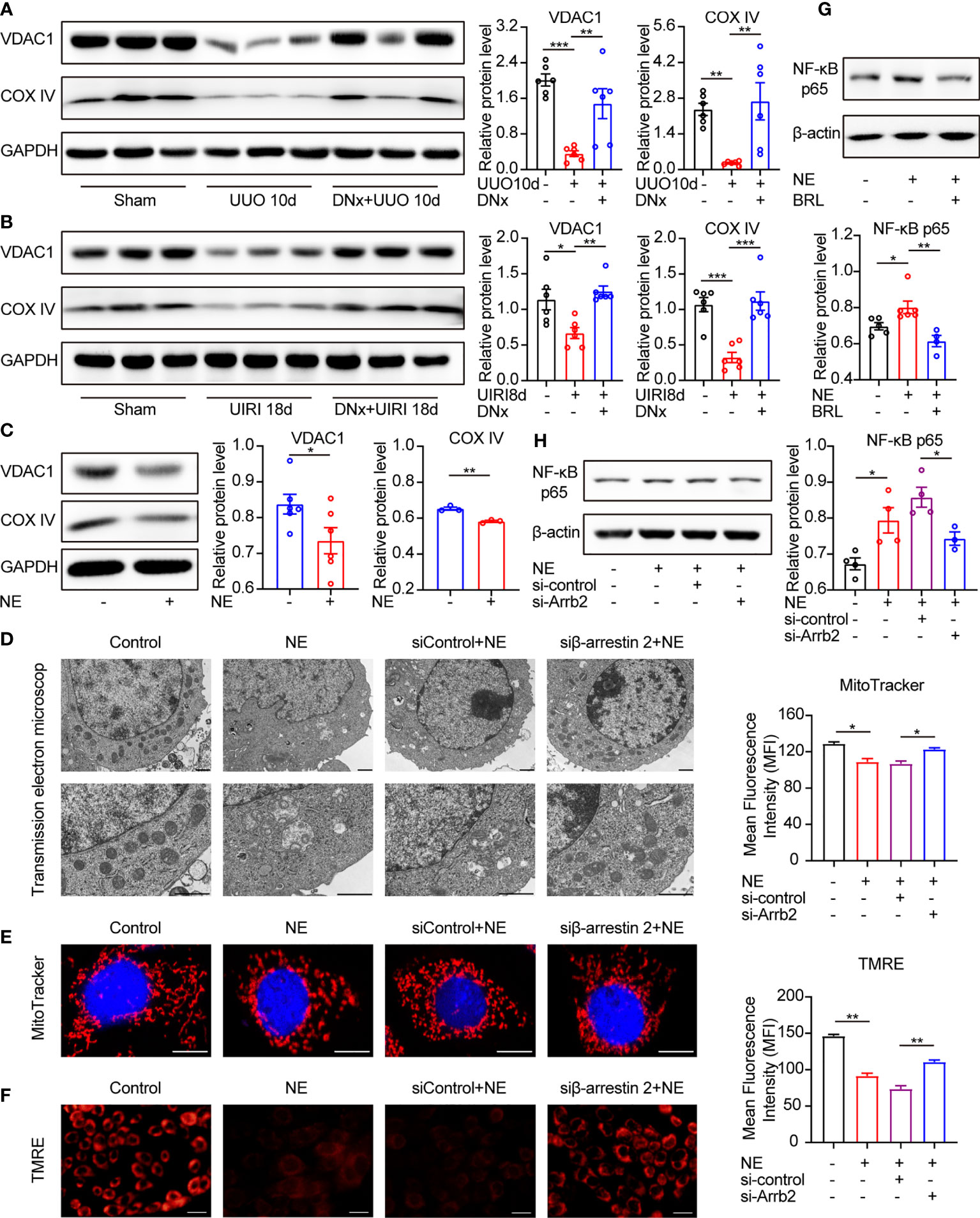
Figure 7 α2A-AR/β-arrestin2/NF-κB signaling contributes to mitochondrial dysfunction and cellular senescence. (A, B) Renal denervation (DNx) or sham operation was performed 2 days before UUO or UIRI in the left kidneys of mice. Representative Western blotting of VDAC1 and COX IV in each group with densitometry analysis (n=6). (C) Representative Western blotting of VDAC1 and COX IV in TKPTS cells treated with PBS or NE (10 nM) for 48 hours with densitometry analysis (n=3–6 in each group). (D–F) TKPTS cells were transfected with scrambled siRNA (si-control) or β-arrestin2 siRNA (si-Arrb2) for 24 hours and then incubated with NE (10 nM) for 48 hours. Mitochondrial ultrastructure was observed by transmission electron microscope (D). Scale bar, 1 μm. Mitochondrial mass was tested by MitoTracker deep red probe (100nM) staining (E). Scale bar, 50 μm. Mitochondrial membrane potential was examined by TMRE fluorescent dye (200 nM) (F). Scale bar, 20 μm. (G) Representative Western blotting of NF-κB p65 in TKPTS cells incubated with PBS, NE, and NE+BRL (10 μM) with densitometry analysis (n=4–6 in each group). (H) Representative Western blotting of NF-κB p65 in TKPTS cells of PBS, NE, and NE+si-control, NE+si-Arrb2 with densitometry analysis (n=3-4 in each group). *P < 0.05, **P < 0.01, ***P < 0.001. Bars represented means ± SEM.
Next, we tested whether β-arrestin2 participated in NE-induced mitochondrial dysfunction. As shown in Figure 7D, in the presence of NE, abnormal mitochondrial morphology occurred, including swelled mitochondria and irregular and fragmented mitochondrial crest. However, pretreatment with β-arrestin2 siRNA markedly ameliorated the abnormal morphology of mitochondria using transmission electron microscopy. This result was further confirmed by MitoTracker deep red staining and TMRE, which showed that the application of β-arrestin2 siRNA meliorated mitochondrial mass (Figure 7E) and mitochondrial membrane potential (Figure 7F) in TKPTS cells. Together, these results indicate that β-arrestin2 is exactly involved in NE-induced mitochondrial dysfunction.
NF-κB signaling was reported to play important roles in mitochondrial function, and using NF-κB inhibitors improved the mitochondrial function (36). Moreover, there has been intensive research using models convincingly demonstrating that NF-κB signaling promotes SASP expression in cellular senescence (37, 38). Thus, we hypothesized that NF-κB is related to NE/α2A-AR/β-arrestin2 mediated senescence. As shown by our results, pretreatment with selective α2A-AR antagonist BRL44408 (BRL, 10μM) declined the protein expression of NF-κB p65 compared with the NE group (Figure 7G). Similarly, NF-κB p65 was higher in NE-treated cells as compared with the control group, which was significantly blocked by β-arrestin2 siRNA (Figure 7H). These results illustrate that NF-κB is downstream of α2A-AR/β-arrestin2 signaling in the NE-induced mitochondrial function, which contributes to cellular senescence and inflammatory responses.
Discussion
In the present study, we have demonstrated that renal denervation significantly improved renal fibrosis and cellular senescence in UUO and UIRI mice. Likewise, in our experiments in vitro, NE stimulation could upregulate profibrotic proteins and proinflammatory cytokines in TKPTS cells accompanied by the activation of α2A-AR/β-arrestin2 signaling.
In previous studies, evidence proved that renal denervation improved physiological changes. Renal sympathetic denervation significantly lessened the damage on podocyte and albuminuria in the rat cardiorenal syndrome model and the diabetic nephropathy model (39, 40). In patients with resistant hypertension, sympathetic renal denervation reduced the incidence of albuminuria and renal resistive index (41, 42). The glomerular filtration rate (GFR) and kidney weight were also improved by renal denervation in the IRI-induced renal fibrosis (18).
Furthermore, neurogenic norepinephrine signaling has been shown to be responsible for kidney inflammation and fibrosis. In UUO mice, norepinephrine supplement after renal denervation increased the levels of inflammatory molecules and infiltration of leukocytes (17). The senescent cell is one of the important resources of these inflammatory molecules. They remain metabolically active and secrete cytokines, chemokines, and growth factors (9). Senescent cells produce SASP consisting of abundant proinflammatory cytokines such as IL-6, IL-8, and IL-1β, which are considered to be important contributors to the proinflammatory phenotype (43). IL-6 disrupted the balance of Th17/Treg differentiated from CD4+ T cells, which pathologically exacerbated the development of inflammatory events (44). IL-8 (or CXCL8), a chemokine, initiated the inflammation process via recruiting and activating neutrophils (45). IL-1β mediated NLRP3 inflammasome-related inflammatory cascade, causing the persistent inflammatory response (46). In the present study, we showed that NE-induced tubular senescence was involved in renal inflammation and fibrosis via the production of inflammatory cytokines IL-6, IL-8, and IL-1β. Therefore, our data suggested that cellular senescence is accurately responsible for renal fibrosis under the control of sympathetic nerves.
The main characteristic of cellular senescence consists of cell cycle arrest, SASP generation, and metabolic dysfunction. The p53/p21CIP1 and p16INK4a/Rb tumor suppressor networks are involved in the development of cell cycle arrest (11, 47). In this study, we found that the increased expressions of p53 and p21 in TKPTS cells treated with NE could be partially reversed by either α2A-AR blockade or β-arrestin2 siRNA, indicating that NE/α2A-AR/β-arrestin2 signaling was an upstream regulator of p53/p21 pathways in this process. A previous report has indicated that renal norepinephrine signals contributed to renal interstitial fibrosis through α2A-AR and α2C-AR in mice with obstructive nephropathy (17). Our data further showed that α2A-AR was rich in renal proximal tubule cells, and blockade of α2A-AR prevented NE-induced cellular senescence, suggesting that NE promoted cellular senescence mainly through the α2A-AR subtype (Figure 5). Moreover, our data showed that β-arrestin2 was a target of α2A-AR in NE-induced cellular senescence. Knockdown of β-arrestin2 reduced senescent markers, suggesting that β-arrestin2 was critically involved in NE-induced cellular senescence (Figure 6).
It was reported that p38/NF-κB signaling, STING/TBK1/NF-κB signaling, JAK/STAT/NF-κB pathway, and other signaling participated in SASP production (48). We proved that the NE/α2A-AR/β-arrestin2 pathway promoted the expression of SASP via NF-κB signaling, and it was supported by data that NF-κB p65 was increased by treatment with NE but attenuated after pretreatment with the selective α2A-AR antagonist and β-arrestin2 siRNA. In addition, we found that mitochondrial dysfunction was also involved in tubular cell senescence induced by NE/α2A-AR/β-arrestin2 signaling. Proximal tubular epithelial cells are rich in mitochondria (49). Damaged mitochondria generated reactive oxygen species (ROS) and led to a cell growth arrest via inducing the DNA damage response (DDR), thus contributing to cellular senescence (50). In this study, sympathetic activation induces mitochondrial disorders, including the reduction of the mitochondrial membrane proteins (VADC1 and COX IV), the increase in abnormal mitochondrial morphology (swollen mitochondria and fractured mitochondrial crest), and the vanishment of the mitochondrial membrane potential (Figure 7). Closed or lost VDAC1 destroyed the integrity of the mitochondrial outer membranes (51), inhibited mitochondrial respiration, and exacerbated mitochondrial division (52). Besides, the lack of electron transport chain proteins, such as the cytochrome c oxidase family (like COX IV) (53), led to electron leakage, which produced ROS-like superoxide free radicals to trigger cellular senescence (54). Partial ablation of β-arrestin2 by siRNA ameliorated mitochondrial disorders, illustrating that β-arrestin2 contributes to mitochondrial dysfunction (Figure 7D-F). In a previous study, β-arrestin2 knockout reduced ROS production through the downregulation of NADPH oxidase 4 (NOX4) in the mice model of hepatic fibrosis (55). Besides, both β-arrestin1 and β-arrestin2 enhanced the production of mitochondrial ROS and superoxide in cardiac fibroblasts isolated from failing adult human left ventricles (56). Based on these studies, we speculate that NE/α2A-AR/β-arrestin2 is one of the upstream regulators of mitochondrial ROS, which then triggers cellular senescence. Furthermore, NF-κB signaling can aggravate mitochondrial disorders (36), suggesting that NF-κB/mitochondrial dysfunction may be the downstream signaling in NE/α2A-AR/β-arrestin2-induced cellular senescence.
It is noteworthy that mitochondrial dysfunction can also contribute to cell apoptosis. The mechanism of mitochondria-induced apoptosis is through increased mitochondrial outer membrane permeabilization, which led to the release of apoptogenic proteins (such as cytochrome c, apoptosis-inducing factor, etc.) to the cytosol and triggered cell apoptosis (57). VDAC1 is the most abundant protein of the mitochondrial outer membrane and is closely associated with cell apoptosis (58). Some studies reported that upregulation of VDAC1 promoted cells apoptosis (59–61). However, in the present study, VDAC1 decreased both in our in vivo and in vitro experiments (Figure 7A–C), suggesting that sympathetic nerves-induced mitochondrial dysfunction contributed to cellular senescence, instead of apoptosis.
In conclusion, this study provides important neurometabolic and neuroimmune mechanisms in the development of renal fibrosis. Renal sympathetic activation triggers tubular senescence, which secretes SASP and promotes renal inflammation and fibrosis. Meanwhile, our work demonstrates that NE-induced tubular cellular senescence is at least partially regulated by α2A-AR/β-arrestin2/NF-κB signaling modules. Inhibiting the actions of α2A-AR or β-arrestin2, alone or in combination, might provide a potential therapeutic strategy to prevent the progression of renal fibrosis.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary files. Further inquiries can be directed to the corresponding authors.
Ethics Statement
The animal study was reviewed and approved by the Animal Ethics Committee at the Huazhong University of Science and Technology.
Author Contributions
MH and GX designed the study. QL and YD prepared the main manuscript text. QL, YD, and LL performed the experiments. CZ, YC, and TZ contributed to experimental data analysis. MH and GX gave crucial guidance to the study and manuscript revision. All authors participated in reviewing the manuscript and approved the final manuscript.
Funding
Grant supports: the National Natural Science Foundation of China (81770686, 81970591, 82000656, and 82000658).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank the Electron Microscope Center of Renmin Hospital of Wuhan University for the transmission electron microscope experiment.
References
1. Jha V, Garcia-Garcia G, Iseki K, Li Z, Naicker S, Plattner B, et al. Chronic Kidney Disease: Global Dimension and Perspectives. Lancet (2013) 382:260–72. doi: 10.1016/s0140-6736(13)60687-x
2. Minutolo R, Borrelli S, De Nicola L. CKD in the Elderly: Kidney Senescence or Blood Pressure-Related Nephropathy? Am J Kidney Dis (2015) 66:184–6. doi: 10.1053/j.ajkd.2015.05.004
3. Hill NR, Fatoba ST, Oke JL, Hirst JA, O’Callaghan CA, Lasserson DS, et al. Global Prevalence of Chronic Kidney Disease - A Systematic Review and Meta-Analysis. PloS One (2016) 11:e0158765. doi: 10.1371/journal.pone.0158765
4. O’Sullivan ED, Hughes J, Ferenbach DA. Renal Aging: Causes and Consequences. J Am Soc Nephrol (2017) 28:407–20. doi: 10.1681/ASN.2015121308
5. Hommos MS, Glassock RJ, Rule AD. Structural and Functional Changes in Human Kidneys With Healthy Aging. J Am Soc Nephrol (2017) 28:2838–44. doi: 10.1681/ASN.2017040421
6. Rule AD, Amer H, Cornell LD, Taler SJ, Cosio FG, Kremers WK, et al. The Association Between Age and Nephrosclerosis on Renal Biopsy Among Healthy Adults. Ann Intern Med (2010) 152:561–7. doi: 10.7326/0003-4819-152-9-201005040-00006
7. Djudjaj S, Boor P. Cellular and Molecular Mechanisms of Kidney Fibrosis. Mol Aspects Med (2019) 65:16–36. doi: 10.1016/j.mam.2018.06.002
8. Stenvinkel P, Larsson TE. Chronic Kidney Disease: A Clinical Model of Premature Aging. Am J Kidney Dis (2013) 62:339–51. doi: 10.1053/j.ajkd.2012.11.051
9. Docherty MH, O’Sullivan ED, Bonventre JV, Ferenbach DA. Cellular Senescence in the Kidney. J Am Soc Nephrol (2019) 30:726–36. doi: 10.1681/ASN.2018121251
10. Ebert T, Pawelzik SC, Witasp A, Arefin S, Hobson S, Kublickiene K, et al. Inflammation and Premature Ageing in Chronic Kidney Disease. Toxins (Basel) (2020) 12:227–48. doi: 10.3390/toxins12040227
11. Herranz N, Gil J. Mechanisms and Functions of Cellular Senescence. J Clin Invest (2018) 128:1238–46. doi: 10.1172/JCI95148
12. Ferguson M, Ryan GB, Bell C. Localization of Sympathetic and Sensory Neurons Innervating the Rat Kidney. J Auton Nerv Syst (1986) 16:279–88. doi: 10.1016/0165-1838(86)90034-2
13. DiBona GF. Physiology in Perspective: The Wisdom of the Body. Neural Control of the Kidney. Am J Physiol Regul Integr Comp Physiol (2005) 289:R633–41. doi: 10.1152/ajpregu.00258.2005
14. Okusa MD, Rosin DL, Tracey KJ. Targeting Neural Reflex Circuits in Immunity to Treat Kidney Disease. Nat Rev Nephrol (2017) 13:669–80. doi: 10.1038/nrneph.2017.132
15. Grassi G, Biffi A, Seravalle G, Bertoli S, Airoldi F, Corrao G, et al. Sympathetic Nerve Traffic Overactivity in Chronic Kidney Disease: A Systematic Review and Meta-Analysis. J Hypertens (2021) 39:408–16. doi: 10.1097/HJH.0000000000002661
16. Noh MR, Jang HS, Kim J, Padanilam BJ. Renal Sympathetic Nerve-Derived Signaling in Acute and Chronic Kidney Diseases. Int J Mol Sci (2020) 21:1647–64. doi: 10.3390/ijms21051647
17. Kim J, Padanilam BJ. Renal Nerves Drive Interstitial Fibrogenesis in Obstructive Nephropathy. J Am Soc Nephrol (2013) 24:229–42. doi: 10.1681/ASN.2012070678
18. Kim J, Padanilam BJ. Renal Denervation Prevents Long-Term Sequelae of Ischemic Renal Injury. Kidney Int (2015) 87:350–8. doi: 10.1038/ki.2014.300
19. Jang HS, Kim J, Padanilam BJ. Renal Sympathetic Nerve Activation via Alpha2-Adrenergic Receptors in Chronic Kidney Disease Progression. Kidney Res Clin Pract (2019) 38:6–14. doi: 10.23876/j.krcp.18.0143
20. Deng Y, Cai Y, Liu L, Lin X, Lu P, Guo Y, et al. Blocking Tyr265 Nitration of Protein Phosphatase 2A Attenuates Nitrosative Stress-Induced Endothelial Dysfunction in Renal Microvessels. FASEB J (2019) 33:3718–30. doi: 10.1096/fj.201800885RR
21. Liu L, Deng Y, Cai Y, Lu P, Guo Y, Zhang C, et al. Ablation of Gsa Impairs Renal Tubule Proliferation After Injury via CDK2/cyclin E. Am J Physiol Renal Physiol (2020) 318:F793–803. doi: 10.1152/ajprenal.00367.2019
22. Zheng H, Liu X, Rao US, Patel KP. Increased Renal ENaC Subunits and Sodium Retention in Rats With Chronic Heart Failure. Am J Physiol Renal Physiol (2011) 300:F641–9. doi: 10.1152/ajprenal.00254.2010
23. Dai L, Qureshi AR, Witasp A, Lindholm B, Stenvinkel P. Early Vascular Ageing and Cellular Senescence in Chronic Kidney Disease. Comput Struct Biotechnol J (2019) 17:721–9. doi: 10.1016/j.csbj.2019.06.015
24. Clements ME, Chaber CJ, Ledbetter SR, Zuk A. Increased Cellular Senescence and Vascular Rarefaction Exacerbate the Progression of Kidney Fibrosis in Aged Mice Following Transient Ischemic Injury. PloS One (2013) 8:e70464. doi: 10.1371/journal.pone.0070464
25. Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular Senescence and the Senescent Secretory Phenotype: Therapeutic Opportunities. J Clin Invest (2013) 123:966–72. doi: 10.1172/JCI64098
26. Johns EJ, Kopp UC, DiBona GF. Neural Control of Renal Function. Compr Physiol (2011) 1:731–67. doi: 10.1002/cphy.c100043
27. Ortiz-Espin A, Morel E, Juarranz A, Guerrero A, Gonzalez S, Jimenez A, et al. An Extract From the Plant Deschampsia Antarctica Protects Fibroblasts From Senescence Induced by Hydrogen Peroxide. Oxid Med Cell Longev (2017) 2017:2694945. doi: 10.1155/2017/2694945
28. Kanagy NL. Alpha(2)-Adrenergic Receptor Signalling in Hypertension. Clin Sci (Lond) (2005) 109:431–7. doi: 10.1042/CS20050101
29. Vasamsetti SB, Florentin J, Coppin E, Stiekema LCA, Zheng KH, Nisar MU, et al. Sympathetic Neuronal Activation Triggers Myeloid Progenitor Proliferation and Differentiation. Immunity (2018) 49:93–106.e7. doi: 10.1016/j.immuni.2018.05.004
30. Kable JW, Murrin LC, Bylund DB. In Vivo Gene Modification Elucidates Subtype-Specific Functions of Alpha(2)-Adrenergic Receptors. J Pharmacol Exp Ther (2000) 293:1–7.
31. Gu YJ, Sun WY, Zhang S, Wu JJ, Wei W. The Emerging Roles of Beta-Arrestins in Fibrotic Diseases. Acta Pharmacol Sin (2015) 36:1277–87. doi: 10.1038/aps.2015.74
32. Nakaya M, Chikura S, Watari K, Mizuno N, Mochinaga K, Mangmool S, et al. Induction of Cardiac Fibrosis by Beta-Blocker in G Protein-Independent and G Protein-Coupled Receptor Kinase 5/Beta-Arrestin2-Dependent Signaling Pathways. J Biol Chem (2012) 287:35669–77. doi: 10.1074/jbc.M112.357871
33. Wang Y, Huang J, Liu X, Niu Y, Zhao L, Yu Y, et al. Beta-Arrestin-Biased AT1R Stimulation Promotes Extracellular Matrix Synthesis in Renal Fibrosis. Am J Physiol Renal Physiol (2017) 313:F1–8. doi: 10.1152/ajprenal.00588.2016
34. Tai H, Wang Z, Gong H, Han X, Zhou J, Wang X, et al. Autophagy Impairment With Lysosomal and Mitochondrial Dysfunction is an Important Characteristic of Oxidative Stress-Induced Senescence. Autophagy (2017) 13:99–113. doi: 10.1080/15548627.2016.1247143
35. Ma L, Chou JW, Snipes JA, Bharadwaj MS, Craddock AL, Cheng D, et al. APOL1 Renal-Risk Variants Induce Mitochondrial Dysfunction. J Am Soc Nephrol (2017) 28:1093–105. doi: 10.1681/ASN.2016050567
36. Yu X, Meng X, Xu M, Zhang X, Zhang Y, Ding G, et al. Celastrol Ameliorates Cisplatin Nephrotoxicity by Inhibiting NF-kappaB and Improving Mitochondrial Function. EBioMedicine (2018) 36:266–80. doi: 10.1016/j.ebiom.2018.09.031
37. Crescenzi E, Pacifico F, Lavorgna A, De Palma R, D’Aiuto E, Palumbo G, et al. NF-kappaB-Dependent Cytokine Secretion Controls Fas Expression on Chemotherapy-Induced Premature Senescent Tumor Cells. Oncogene (2011) 30:2707–17. doi: 10.1038/onc.2011.1
38. Rovillain E, Mansfield L, Caetano C, Alvarez-Fernandez M, Caballero OL, Medema RH, et al. Activation of Nuclear Factor-Kappa B Signalling Promotes Cellular Senescence. Oncogene (2011) 30:2356–66. doi: 10.1038/onc.2010.611
39. Rafiq K, Noma T, Fujisawa Y, Ishihara Y, Arai Y, Nabi AH, et al. Renal Sympathetic Denervation Suppresses De Novo Podocyte Injury and Albuminuria in Rats With Aortic Regurgitation. Circulation (2012) 125:1402–13. doi: 10.1161/CIRCULATIONAHA.111.064097
40. Yao Y, Fomison-Nurse IC, Harrison JC, Walker RJ, Davis G, Sammut IA. Chronic Bilateral Renal Denervation Attenuates Renal Injury in a Transgenic Rat Model of Diabetic Nephropathy. Am J Physiol Renal Physiol (2014) 307:F251–62. doi: 10.1152/ajprenal.00578.2013
41. Mahfoud F, Cremers B, Janker J, Link B, Vonend O, Ukena C, et al. Renal Hemodynamics and Renal Function After Catheter-Based Renal Sympathetic Denervation in Patients With Resistant Hypertension. Hypertension (2012) 60:419–24. doi: 10.1161/HYPERTENSIONAHA.112.193870
42. Ott C, Mahfoud F, Schmid A, Ditting T, Veelken R, Ewen S, et al. Improvement of Albuminuria After Renal Denervation. Int J Cardiol (2014) 173:311–5. doi: 10.1016/j.ijcard.2014.03.017
43. Campisi J. Aging, Cellular Senescence, and Cancer. Annu Rev Physiol (2013) 75:685–705. doi: 10.1146/annurev-physiol-030212-183653
44. Tanaka T, Narazaki M, Kishimoto T. IL-6 in Inflammation, Immunity, and Disease. Cold Spring Harb Perspect Biol (2014) 6:a016295. doi: 10.1101/cshperspect.a016295
45. Rea IM, Gibson DS, McGilligan V, McNerlan SE, Alexander HD, Ross OA. Age and Age-Related Diseases: Role of Inflammation Triggers and Cytokines. Front Immunol (2018) 9:586. doi: 10.3389/fimmu.2018.00586
46. Sims JE, Smith DE. The IL-1 Family: Regulators of Immunity. Nat Rev Immunol (2010) 10:89–102. doi: 10.1038/nri2691
47. Campisi J, d’Adda di Fagagna F. Cellular Senescence: When Bad Things Happen to Good Cells. Nat Rev Mol Cell Biol (2007) 8:729–40. doi: 10.1038/nrm2233
48. Birch J, Gil J. Senescence and the SASP: Many Therapeutic Avenues. Genes Dev (2020) 34:1565–76. doi: 10.1101/gad.343129
49. Gewin LS. Renal Fibrosis: Primacy of the Proximal Tubule. Matrix Biol (2018) 68-69:248–62. doi: 10.1016/j.matbio.2018.02.006
50. Herranz N, Gil J. Mitochondria and Senescence: New Actors for an Old Play. EMBO J (2016) 35:701–2. doi: 10.15252/embj.201694025
51. Vander Heiden MG, Chandel NS, Li XX, Schumacker PT, Colombini M, Thompson CB. Outer Mitochondrial Membrane Permeability can Regulate Coupled Respiration and Cell Survival. Proc Natl Acad Sci USA (2000) 97:4666–71. doi: 10.1073/pnas.090082297
52. Nowak G, Megyesi J, Craigen WJ. Deletion of VDAC1 Hinders Recovery of Mitochondrial and Renal Functions After Acute Kidney Injury. Biomolecules (2020) 10:585–607. doi: 10.3390/biom10040585
53. Kadenbach B. Regulation of Mammalian 13-Subunit Cytochrome C Oxidase and Binding of Other Proteins: Role of NDUFA4. Trends Endocrinol Metab (2017) 28:761–70. doi: 10.1016/j.tem.2017.09.003
54. Barja G. Updating the Mitochondrial Free Radical Theory of Aging: An Integrated View, Key Aspects, and Confounding Concepts. Antioxid Redox Signal (2013) 19:1420–45. doi: 10.1089/ars.2012.5148
55. Du JJ, Sun JC, Li N, Li XQ, Sun WY, Wei W. Beta-Arrestin2 Deficiency Attenuates Oxidative Stress in Mouse Hepatic Fibrosis Through Modulation of NOX4. Acta Pharmacol Sin (2021) 42:1090–100. doi: 10.1038/s41401-020-00545-9
56. Philip JL, Razzaque MA, Han M, Li J, Theccanat T, Xu X, et al. Regulation of Mitochondrial Oxidative Stress by Beta-Arrestins in Cultured Human Cardiac Fibroblasts. Dis Model Mech (2015) 8:1579–89. doi: 10.1242/dmm.019968
57. Abate M, Festa A, Falco M, Lombardi A, Luce A, Grimaldi A, et al. Mitochondria as Playmakers of Apoptosis, Autophagy and Senescence. Semin Cell Dev Biol (2020) 98:139–53. doi: 10.1016/j.semcdb.2019.05.022
58. Shoshan-Barmatz V, Shteinfer-Kuzmine A, Verma A. VDAC1 at the Intersection of Cell Metabolism, Apoptosis, and Diseases. Biomolecules (2020) 10:1485–525. doi: 10.3390/biom10111485
59. Abu-Hamad S, Zaid H, Israelson A, Nahon E, Shoshan-Barmatz V. Hexokinase-I Protection Against Apoptotic Cell Death is Mediated via Interaction With the Voltage-Dependent Anion Channel-1: Mapping the Site of Binding. J Biol Chem (2008) 283:13482–90. doi: 10.1074/jbc.M708216200
60. Zaid H, Abu-Hamad S, Israelson A, Nathan I, Shoshan-Barmatz V. The Voltage-Dependent Anion Channel-1 Modulates Apoptotic Cell Death. Cell Death Differ (2005) 12:751–60. doi: 10.1038/sj.cdd.4401599
Keywords: sympathetic denervation, cellular senescence, neurometabolic, neuroimmune, renal fibrosis.
Citation: Li Q, Deng Y, Liu L, Zhang C, Cai Y, Zhang T, Han M and Xu G (2022) Sympathetic Denervation Ameliorates Renal Fibrosis via Inhibition of Cellular Senescence. Front. Immunol. 12:823935. doi: 10.3389/fimmu.2021.823935
Received: 28 November 2021; Accepted: 27 December 2021;
Published: 24 January 2022.
Edited by:
Liang Ma, Sichuan University, ChinaReviewed by:
Yanggang Yuan, Nanjing Medical University, ChinaXiao-ming Meng, Anhui Medical University, China
Na Liu, Tongji University, China
Copyright © 2022 Li, Deng, Liu, Zhang, Cai, Zhang, Han and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gang Xu, eHVnYW5nQHRqaC50am11LmVkdS5jbg==; Min Han, bWluaGFuQHRqaC50am11LmVkdS5jbg==
†These authors have contributed equally to this work