 Philip B. Busbee1
Philip B. Busbee1 Marpe Bam1
Marpe Bam1 Xiaoming Yang1
Xiaoming Yang1 Osama A. Abdulla1
Osama A. Abdulla1 Juhua Zhou1
Juhua Zhou1 Jay Paul (Jack) Ginsberg1,2Allison E. Aiello3
Jay Paul (Jack) Ginsberg1,2Allison E. Aiello3 Monica Uddin4
Monica Uddin4 Mitzi Nagarkatti1
Mitzi Nagarkatti1 Prakash S. Nagarkatti1*
Prakash S. Nagarkatti1*- 1Department of Pathology, Microbiology, and Immunology, School of Medicine, University of South Carolina, Columbia, SC, United States
- 2Departments of Psychophysiology, Clinical Psychology, and Research Office, Saybrook University, Pasadena, CA, United States
- 3Department of Epidemiology, University of North Carolina (UNC) Gillings School of Global Public Health, University of North Carolina, Mcgavran-Greenberg Hall, Chapel Hill, NC, United States
- 4Genomics Program, University of South Florida College of Public Health, Tampa, FL, United States
Post-traumatic stress disorder (PTSD) is a psychiatric disorder and patients diagnosed with PTSD often express other comorbid health issues, particularly autoimmune and inflammatory disorders. Our previous reports investigating peripheral blood mononuclear cells (PBMCs) from PTSD patients showed that these patients exhibit an increased inflammatory T helper (Th) cell phenotype and widespread downregulation of microRNAs (miRNAs), key molecules involved in post-transcriptional gene regulation. A combination of analyzing prior datasets on gene and miRNA expression of PBMCs from PTSD and Control samples, as well as experiments using primary PBMCs collected from human PTSD and Controls blood, was used to evaluate TP53 expression, DNA methylation, and miRNA modulation on Th17 development. In the current report, we note several downregulated miRNAs were linked to tumor protein 53 (TP53), also known as p53. Expression data from PBMCs revealed that compared to Controls, PTSD patients exhibited decreased TP53 which correlated with an increased inflammatory Th17 phenotype. Decreased expression of TP53 in the PTSD population was shown to be associated with an increase in DNA methylation in the TP53 promotor region. Lastly, the most significantly downregulated TP53-associated miRNA, let-7a, was shown to negatively regulate Th17 T cells. Let-7a modulation in activated CD4+ T cells was shown to influence Th17 development and function, via alterations in IL-6 and IL-17 production, respectively. Collectively, these studies reveal that PTSD patients could be susceptible to inflammation by epigenetic dysregulation of TP53, which alters the miRNA profile to favor a proinflammatory Th17 phenotype.
Introduction
Post-traumatic stress disorder (PTSD) is a psychiatric disorder caused by exposure to a severe traumatic event, such as combat, domestic violence, sexual assault, and natural disasters (1–3). In addition to classic psychiatric and psychologic symptoms of this disorder, such as emotional numbing, repetitive recollections of the traumatic event, and states of hyperarousal (4), other medical disorders have been linked to PTSD, including arthritis, diabetes, cardiovascular-associated conditions, and various inflammatory disorders (5–8). Increased risk of these comorbid medical disorders and medical complications in PTSD patients reduces the effective treatment of these patients, and studies have shown that PTSD patients have increased annual healthcare costs (4.2-9.3%) when compared to patients with other mental disorders (9). While the prevalence of PTSD was relatively low (8%) in the general population for a time (10), PTSD reported in combat veterans was found to be as high as 30% (11, 12), and reports are already showing increased prevalence of PTSD in numerous populations around the world due to the COVID-19 pandemic (13–15). Therefore, it is becoming increasingly critical to better understand why individuals diagnosed with PTSD have increased risk of other comorbid disorders. A better understanding of these mechanisms could provide both better treatment options and significantly improve the overall quality of life of the affected patient population.
Several reports link PTSD with increased inflammation and occurrence of inflammatory disorders (16–25). In an early report by Gola et al, researchers found that peripheral blood mononuclear cells (PBMCs) from PTSD patients exhibited a more pre-activated phenotype, having higher levels of secreting inflammatory cytokines, such as interleukin-6 (IL-6), tumor necrosis factor-alpha (TNF-α), and IL-1β when compared to control samples (26). We have since published extensively on the correlation between PTSD and inflammation, showing how epigenetic modifications (e.g. histone and DNA methylation) and modulation of microRNA (miRNA or miR), small noncoding RNA nucleotides involved in post-transcriptional regulation of genes, could potentially make PTSD patients more prone to inflammatory immune responses (27–31). Among these reports, published results revealed PBMCs from PTSD patients showed increased plasma IL-17 levels and CD4+ T helper-17 (Th17) phenotypes, which correlated with PTSD severity and global downregulation of miRNAs (22). Despite these observations, key regulators or promoters of this Th17 inflammatory phenotype in PTSD patients are yet to be fully understood.
In the current report, we noted several significantly dysregulated miRNA in PTSD PBMCs were associated with TP53, the human gene encoding for tumor protein 53 (TP53 or p53). Analysis of gene expression profile data in the public domain and our own evaluation of human PBMCs revealed TP53 is downregulated in PTSD subjects when compared to Controls. Whole genome methylated DNA immunoprecipitation sequencing (MeDIP-seq) data of PBMCs also showed that there was increased DNA methylation in the TP53 promoter region of PTSD subjects compared to controls, which would suggest decreased transcription of this gene. Elsewhere in this report, we highlight that among the dysregulated miRNAs associated with TP53, let-7a was not only found to be the most significantly altered miRNA, but pathway analysis also revealed let-7a had direct and indirect links to Th17 cells. Lastly, transfection of activated CD4+ T cells purified from human PBMCs to modulate let-7a levels showed that this miRNA is directly involved in both Th17 development and function. The current report highlights an important role for the p53/miRNA(let-7a) axis in promoting a potential inflammatory Th17-skewed phenotype in the PTSD population.
Materials and Methods
Human Patient Samples
Procedures to collect human samples presented in this report were approved and reviewed by the Institutional Review Boards (IRBs) from the University of South Carolina, the University of Michigan, and the University of North Carolina-Chapel Hill. Blood sample collection from participants was performed after obtaining proper informed and written consent. In the current report, human samples were collected from two main sources: 1) veterans from the William Jennings Bryan Dorn Veterans Medical Center (VMC) (27), or 2) study participants from the Detroit Neighborhood Health Study (DNHS) (32). PTSD samples obtained from veterans at the William Jennings Bryan Dorn (VMC) were clinically assessed by medical VMC professionals using psychometric properties of the PTSD Checklist (PCL), Clinician Administered PTSD Scale (CAPS), and formal criteria from the Diagnostic and Statistical Manual of Mental Disorder (DSM-V), as previously described (29). For DNHS samples, PTSD diagnosis was performed using structured telephone interviews and the PTSD Checklist-Civilian Version (PCL-C), which was subsequently validated with in-person clinical interviews, as previously reported (32). The source and number of PTSD and Control samples were used for the following analyses present in the current report: A.) miRNA microarray evaluation of PBMCs: Control (n=4), PTSD (n=8); source: PBMCs from William Jennings Bryan Dorn VA participants; demographics data published in previous report (29). B.) TP53 and IL-17A expression from PBMCs from public database set: Control (n=16), PTSD (n=17); source: Gene expression profiles of PBMCs from combat veterans in public database Gene Expression Omnibus (GEO) database (ID GSE860); demographics data published in previous report (33). C.) TP53/Mdm2 PCR expression and correlation data from collected PBMCs: Control (n=6), PTSD (n=19); source: PBMCs from William Jennings Bryan Dorn VA participants; demographics data available in Table 1. D.) DNA methylation of the TP53 promoter region: Control (n=5); PTSD (n=5); source: PBMCs from DNHS project; demographic data available in Table 1. For Control samples, non-PTSD volunteers were found to be comparable to PTSD samples based on age, race, and sex, as detailed in Table 1. In addition, exclusion criteria for both the Control and PTSD samples included non-adult participants (< 18 years of age), participants with active infection(s) during PBMC collection, or other potentially immunocompromising conditions such as HIV, cancer, active pregnancy among female participants, or recorded history of chronic steroid use.
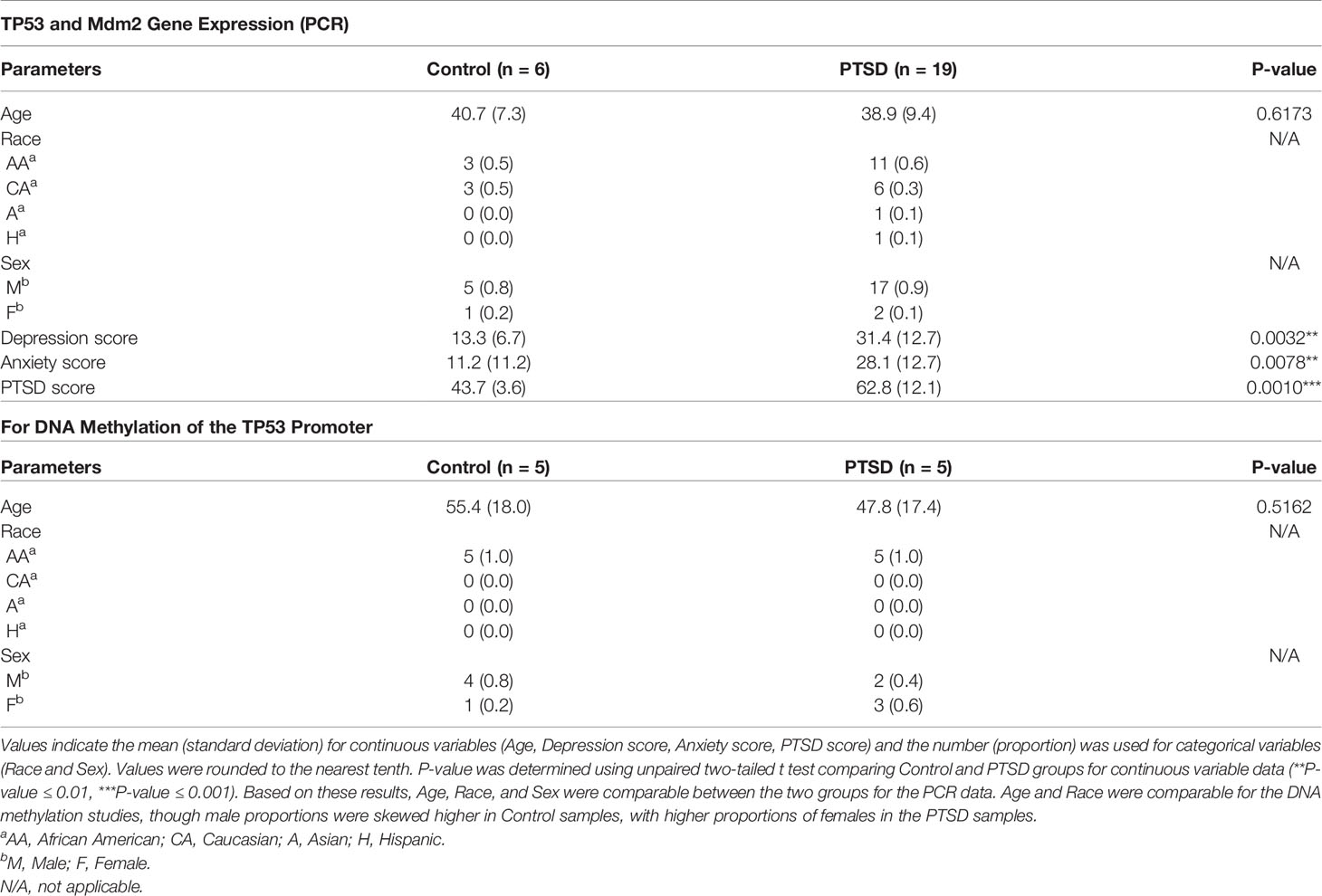
Table 1 Demographics and clinical history of Control and PTSD samples for PCR gene expression and DNA methylation analysis.
PBMC Isolation From Blood Samples
For PBMC isolation, researchers obtained deidentified blood samples (10-20 ml) collected in EDTA-coated tubes and began the isolation procedure within 1-2 hours of the sample collection. Blood samples were diluted in twice the volume of 1X PBS (Sigma-Aldrich, St. Louis, MO) before being layered on top of 20 ml of Ficoll-Paque (GE Healthcare, Uppsala, Sweden) and centrifuged at room temperature for 30 minutes at 1300 rpm. The upper plasma layer was carefully removed before collecting the PBMC interphase layer. PBMCs were diluted in twice the volume of 1X PBS before being centrifuged at 4°C for 5 minutes at 1300 rpm. Isolated PBMCs were washed twice in 1X PBS before further downstream processing of the cells (e.g. DNA/RNA isolation).
miRNA Microarray and Analysis of PTSD and Control Samples
PTSD (n=8) and Control (n=4) samples were processed for miRNA microarray as previously reported (27). In the current report, significantly dysregulated miRNAs associated with direct or indirect links to TP53 were the major focus, thus preventing redundant or overlapping results from the previous report using this dataset. Briefly, total RNA was isolated from PBMC samples using the miRNeasy Mini kit (Qiagen, Valencia, CA), and RNA integrity was analyzed using an Agilent 2100 BioAnalyzer (Agilent Tech, Palo Alto, CA). Microarray was performed by the Deep Sequencing and Microarray Core Facility at John Hopkins Memorial Institute in Baltimore, MD. miRNA hybridization to the Affymetrix miRNA-v1 gene chip (ThermoFisher Scientific, Waltham, MA), microarray data normalization, microarray quality control, and calculation of linear fold change of miRNA between PTSD and Control samples were performed as previously reported (27). Ingenuity Pathway Analysis (Qiagen Valencia, CA), also known as IPA, was used to analyze significantly dysregulated miRNA in PTSD samples compared to Controls (defined as ± 1.5-fold change with p value of < 0.05) in relation to known and predicted gene targets. IPA was also used to identify and visualize dysregulated miRNA, target genes, and the potential interacting networks involved in reported or highly predicted functions (e.g. Diseases and Functions, Molecular and Cellular Functions). Raw miRNA microarray data is available at ArrayExpress (Accession# E-MTAB-4880).
Evaluation of PTSD and Control Gene Expression Data From the Public Database
Initial analysis involving expression of genes of interest based on miRNA microarray data (e.g. TP53, Th17-related genes) was first evaluated in the public database, specifically from the NCBI (www.ncbi.nlm.nih.gov/) GEO database. The dataset used was ID GSE860 (33), which included normalized gene expression array data using the Affymetrix Human Genome U95A Array platform on PBMCs collected from PTSD (n=17) and Control (n=16) samples at two different time points (initial ER visit and 4 months after). Controls were defined as ER patients that experienced psychological trauma but did not develop PTSD over time. Normalized expression data was expressed as fold change relative to the average of gene expression of either Control or PTSD samples during the initial ER visit, prior to evaluation after 4 months. In the current report, results included evaluation of TP53 (31618_at) and IL-17A (1359_at) gene expression in the total of 33 PBMC samples included in the publicly available dataset, thereby reducing overlapping and repetitive results from the previous manuscript for which this data was associated with.
Evaluation of Gene Expression in PTSD and Control Samples Using Quantitative Real-Time PCR
In addition to the aforementioned publicly available dataset, we evaluated gene expression in PBMCs from William Jennings Bryan Dorn VA participants between Control (n=6) and PTSD (n=19) groups for TP53 and the TP53 regulatory protein, Mdm2, using qRT-PCR. PBMC and RNA isolation procedures were performed from collected blood samples as described above. Complementary DNA (cDNA) was synthesized using miScript II RT kit (Qiagen, Valencia, CA), followed by gene transcript detection using iQ universal SYBR Green supermix (Bio-Rad Laboratories, Hercules, CA) on a Bio-Rad CFX384 PCR platform. A total of 10ng of cDNA was used per sample for the PCR reactions, and primers for TP53 (forward: CCTCAGCATCTTATCCGAGTGG; reverse: TGGATGGTGGTACAGTCAGAGC) and Mdm2 (forward: TGTTTGGCGTGCCAAGCTTCTC; reverse: CACAGATGTACCTGAGTCCGATG) were designed and created using Integrated DNA Technologies (IDT, Coralville, IA). Expression of genes were first normalized to 18S and then expressed as fold change using the delta-delta CT method (2–ΔΔCt method). PCR reactions were performed using the following 2-step cycling protocol: initial denaturing and enzyme activation (95°C, 2:00 mins,1 cycle), denaturing (95°C, 15 secs, 40 cycles), annealing and extension (55°C, 30 secs, 40 cycles), melt curve (55-95°C with 0.5°C increments, 30 secs, 1 cycle). Control and PTSD participants used in these PCR assays and subsequent correlation studies were found to be comparable in terms of age, race, and sex.
Methylated DNA Immunoprecipitation Sequencing of PTSD and Control PBMC Aamples
To investigate whole genome DNA methylation patterns, PBMCs were isolated from Control (n=5) and PTSD samples (n=5) provided from the DNHS project as described above. Genomic DNA was purified using a Zymo DNA purification kit (Zymo Research, Irvine, CA) and fragmented by Bioruptor sonicator (Diagenode, Denville, NJ). DNA fragments with sizes from 200 bp to 400 bp were purified and sequencing adaptors were added using Illumina Chip-seq library preparation kit. dsDNA was then denatured and immunoprecipitated with anti 5-methylcytosine antibody using a MeDIP kit from Diagenode. Precipitated DNA was purified and amplified. MeDIP-seq libraries were then sequenced by Nextseq550 with single-end reads. Raw sequencing reads were mapped to human genome build hg19 using Bowtie software by allowing two mismatches in the read (34). The mapped reads were then analyzed with MEDIPS software (35). Generated WIG files were visualized in the IGB genome browser (www.bioviz.org). Control and PTSD participants used in these in the DNA methylation studies were found to be comparable in terms of age and race. However, there were uneven distributions based on sex, with males being more proportionally represented in the Control group compared to females.
MicroRNA let-7a Transfections in Activated and Purified CD4+ T Cell Cultures
PBMCs were isolated from blood of Control, non-PTSD volunteer samples as described above. PBMCs were purified for CD4 using an EasySep kit (StemCell Technologies, Cambridge, MA) and PE-conjugated monoclonal antibody (mAb) (Cat #555347, BD Biosciences, San Jose, CA). Purified CD4+ cells were assessed using FC500 flow cytometer (Beckman Coulter, Indianapolis, IN) and plated at 130,000 cell density per well in 24-well plates in complete RPMI media (Sigma-Aldrich, St. Louis, MO). Cells were left unactivated or activated with Dynabeads Human T-Activator CD3/CD28 (1:1 bead-to-cell ratio) (Cat. #111.61D, ThermoFisher Scientific, Waltham, MA) in 5% CO2 at 37 °C for 24 hours. In addition to activation, some cultures were transfected with either mock, Anti-hsa-let-7a-5p (inhibitor), or syn-has-let-7a-5 (mimic) from Qiagen (Valencia, CA). Transfections were performed using HiPerFect Transfection Reagent (Qiagen, Valencia, CA), following manufacturer’s instructions, with a final concentration of inhibitors and mimics at 20 µM. Mock cultures only contained transfection reagent without addition of either the mimics or inhibitors. After 24 hours, RNA was isolated from cultured cells to test let-7a transfection by PCR as described above. PCR was performed using miScript SYBR Green PCR kit (Qiagen, Valencia, CA) and human let-7a-specific primers using Qiagen-based miScript Primer Assay as per instructions from the manufacturer. In addition, Th17 cell phenotyping by flow cytometry using fluorescently-labeled mABs for CD4 (Cat #555347, BD Biosciences, San Jose, CA) and IL-17A (Cat# 555347, Biolegend, San Diego, CA) was performed. IL-17A intracellular staining was achieved using a BD Cytofix/Cytoperm solution kit as per manufacturer’s instructions (BD Biosciences, San Jose, CA). Cell culture supernatants were also assessed with human IL-17A and IL-6 ELISA kits from Biolegend (San Diego, CA).
Statistical Analysis
Statistical analysis was performed using Graphpad Prism version 9 software. For matching Control and PTSD samples to be comparable with one another, continuous variables (age, PTSD scores, anxiety score, depression score) were expressed as means with standard deviations, and p-values calculated using an unpaired, two-tailed standard t test. For categorical variables (sex and race), these were expressed as numbers with overall proportions represented within each respective group (e.g. PTSD or Control) for qualitative comparisons. For other comparison data (e.g. miRNA fold change, PCR gene expression) between two groups, an unpaired, two-tailed standard t test was performed. When comparing 3 or more groups (e.g. in vitro cell culture studies with flow cytometry, miRNA fold change PCR, and ELISAs), one way ANOVA and post-hoc multiple comparisons Tukey’s test was performed. In vitro assays were performed at least three times and are shown as representative replicate data. For correlation studies, Pearson correlation tests were used to determine significance. Significance was defined as having a p value ≤ 0.05. For DNA methylation studies using Me-DIPseq, qualitative observations with intensity of signal (peak height and width) in the focused gene promoter regions (e.g. TP53) were evaluated as previously reported (28).
Results
Dysregulated miRNA in PTSD PBMCs Is Associated With TP53 and Inflammatory Th17 Response
In our previous study, we noted that compared to healthy controls, PTSD patients had significant downregulation of a number of miRNA (27). Using IPA analysis software, we noted that of the top 6 networks affected by significantly dysregulated miRNA in PTSD samples, 4 of these included TP53 (Table 2). Further analysis revealed that of the 184 miRNAs significantly downregulated in PTSD samples, 21 of these altered miRNAS were found to be associated with TP53 (Figures 1A, B). The most significantly downregulated TP53-associated miRNA with the lowest p value was let-7a (Figure 1C), which was followed by miR-145, miR-16, miR-221, and miR-320b (Figures 1D–G). To better understand what processes and functions were affected by these particular miRNAs, only the 21 TP53-associated miRNAs were input back into IPA to determine how their alteration might affect disease and function. As shown in Figure 1H, Inflammatory Response and Inflammatory Disease were ranked within the top 5 Disease and Functions affected by these specific miRNA. In addition to Disease and Functions, the top Molecular and Cellular processes linked to these altered TP53-associated miRNAs included Cellular Development, as well as Cellular Growth and Proliferation (Figure 1I). Lastly, among the 21 TP53-associated miRNA, let-7a and TP53 were found to have direct and indirect interactions linked to Th17 development and function by way of their association with IL-6 and IL-17A, respectively (Figure 2). Prediction analysis based on the miRNA profiles also suggested that TP53 would be inhibited in PTSD samples, while IL-6 and IL-17A were predicted to be activated. These data suggested that through a TP53/let-7a axis, PTSD patients could be more prone to developing an abnormal Th17 phenotype.
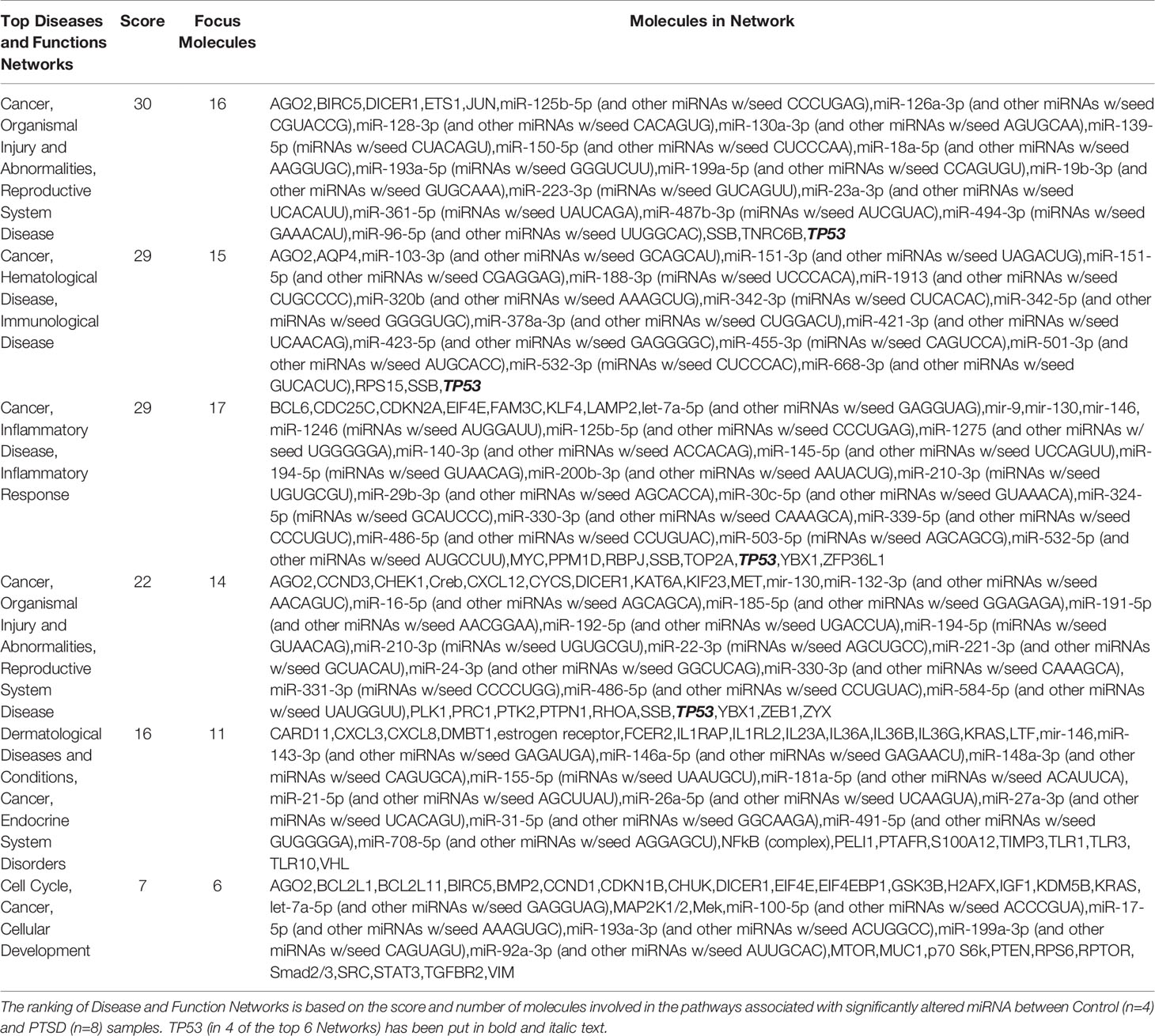
Table 2 IPA-generated networks highlighting dysregulated miRNA and target genes between Control and PTSD samples.
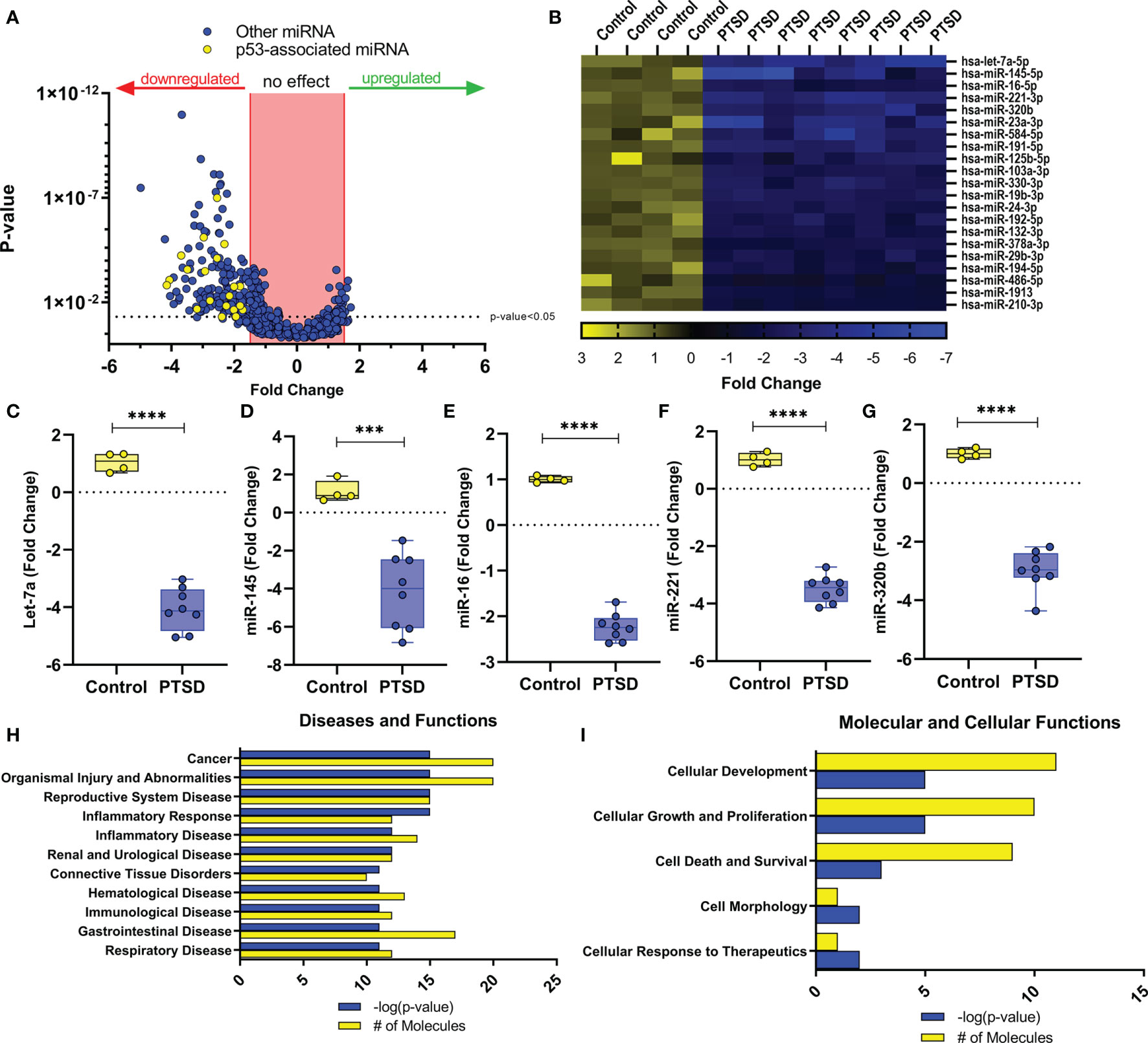
Figure 1 Dysregulated miRNA in PBMCs from PTSD patients are associated with TP53. miRNA array data from PBMCs from Control (n=4) and PTSD subjects (n=8) was analyzed using IPA analysis software. (A) Volcano plot depicting miRNA fold change values (x-axis) of PTSD vs. Control plotted against p-values (y-axis). Yellow dots represent TP53-associated miRNAs. (B) Heat Map depicting 21 TP53-associated miRNA that were significantly downregulated in PTSD samples compared to control. (C–G) Box and whisker plots depicting top 5 downregulated TP53-associated miRNA between Control and PTSD samples to include (C) let-7a, (D) miR-145, (E) miR-16, (F) miR-122, (G) miR-321b. Box and whisker bars depict maximum and minimum values, with middle bar depicted at the median. (H, I) Top ranked networks (as determined by IPA-derived –log p-values and # of molecules) for 21 total dysregulated miRNA associated with TP53 to include (H) Disease and Functions, as well as I) Molecular and Cellular Functions. Significance in linear fold change was determined as any value ≥1.5 or ≤ -1.5 fold, and p-value was determined significant if this value ≤ 0.05 using an unpaired standard t test. P-values: ≤ 0.005 (***), ≤ 0.001 (****).
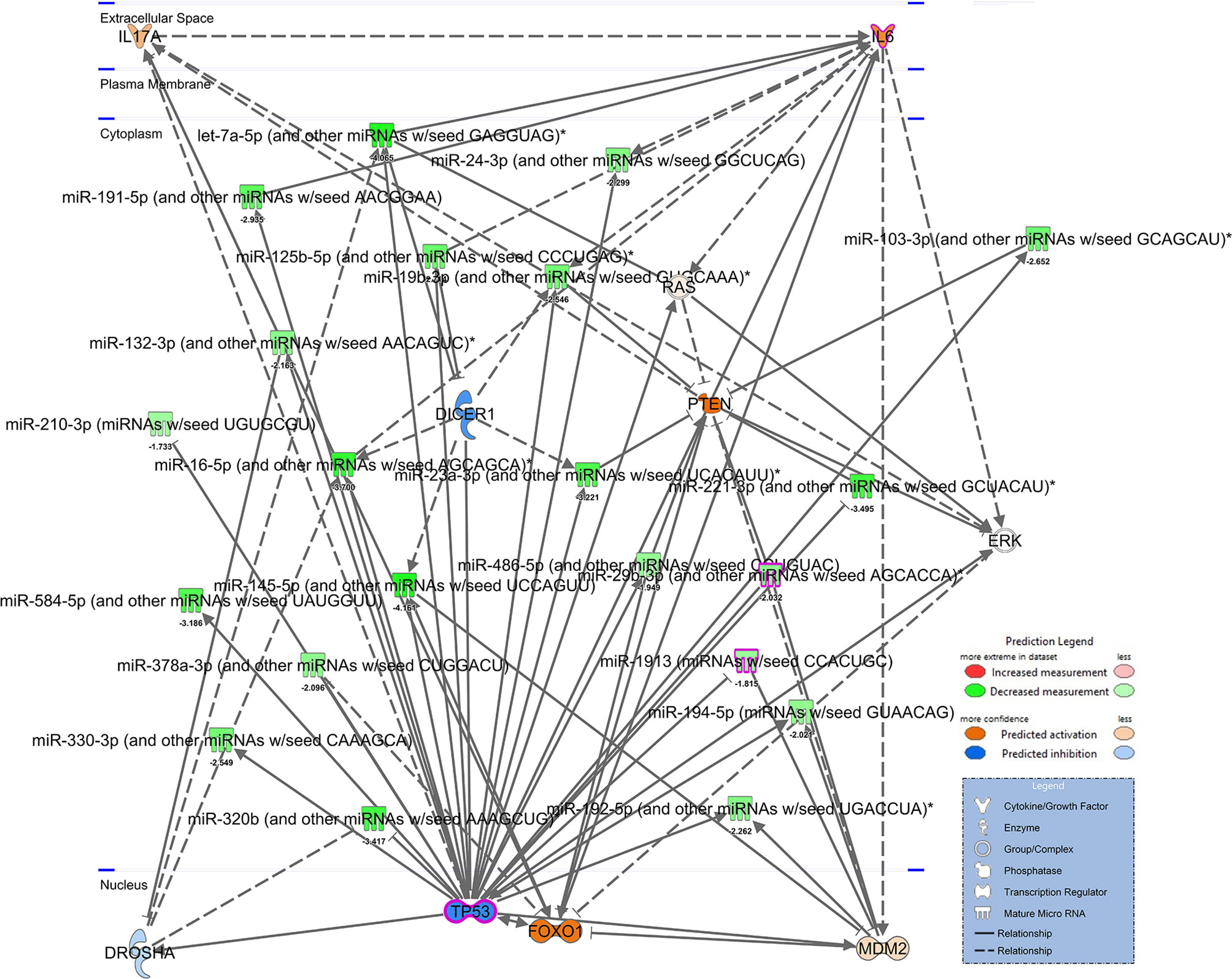
Figure 2 Network of TP53 interactions with dysregulated miRNA from PTSD patients. IPA-derived network depicting dysregulated miRNA associated with TP53 from mean fold changes of Control (n=4) vs. PTSD (n=8) PMBCs. miRNA are depicted in green to denote downregulation (darker green denotes more downregulation) or red to denote decreased expression. Genes depicted in the network are predicted to be activated (orange) or inhibited (blue) based on interactions present, as noted in the Prediction Legend. A separate legend (lower right corner) is provided to identify components depicted in the network. Solid lines indicate direct interactions, and doted lines indicate either indirect or highly-predicted interactions.
TP53 Downregulation in PTSD PBMCs Correlates With Increased Inflammatory IL-17A
Since so many downregulated miRNA in PTSD samples were linked to TP53, initial evaluation of TP53 expression was undertaken in the public database. In database GSE860, normalized expression of gene data was available between PTSD and Control samples during two major time points: 1) a visit to the ER caused by trauma, and then 2) 4 months after their initial ER visits. Control subjects were deemed to not develop PTSD after the 4-month period. Control and PTSD subjects were found to have no significant differences in age, race, or sex, as detailed in the previous published report (33). As the data shows, when compared to Control samples, PBMCs collected from PTSD subjects exhibited a decrease in TP53 4 months after trauma was experienced, which trended towards significance (Figure 3A). In addition, it was interesting to note that in this same cohort of PTSD subjects, IL-17A expression had a noticeable trending increase after 4 months (Figure 3B). While both these observations trended towards significance, data showed there was in fact a significant negative correlation between TP53 and inflammatory cytokine IL-17A levels (Figure 3C). In addition to this publicly available data, we evaluated TP53 expression in PBMCs collected from local veterans at the William’s Jennings Bryan VMC. In this study, we collected PBMCs from both PTSD (n=19) and Control volunteers (n=6), with demographic data supplied in Table 1. Control and PTSD participants matched comparatively in terms of age, race, and sex, with no noted significant differences among these criteria between the two groups. There was however a larger overall proportion of male to female participants in this particular study, due to limited numbers of both Control and PTSD female volunteers. However, we found that veterans diagnosed with PTSD had significantly lower expression of TP53 compared to Controls (Figure 3D). Interestingly, there was no significant difference in the expression of TP53-inhibiting protein Mdm2 (Figure 3E), suggesting that TP53 expression was being specifically altered in the PTSD group, independent of other confounding factors or regulatory mechanisms related to TP53. Indeed, based on these studies, there was a clear significant negative correlation between TP53 expression and PTSD severity (Figure 3F), inasmuch as when the TP53 expression decreased, PTSD scores increased among the sampled population. Additionally, even though PTSD samples in this particular cohort had significantly higher anxiety and depression scores compared to Controls (Table 1), TP53 expression appeared to only be correlated with PTSD score, as there was no significant correlation between TP53 expression and the two commonly comorbid psychological conditions associated with PTSD (Figures 3G–I). Taken together, these data revealed that when compared to Controls, PBMCs from persons diagnosed with PTSD had lower expression of TP53.
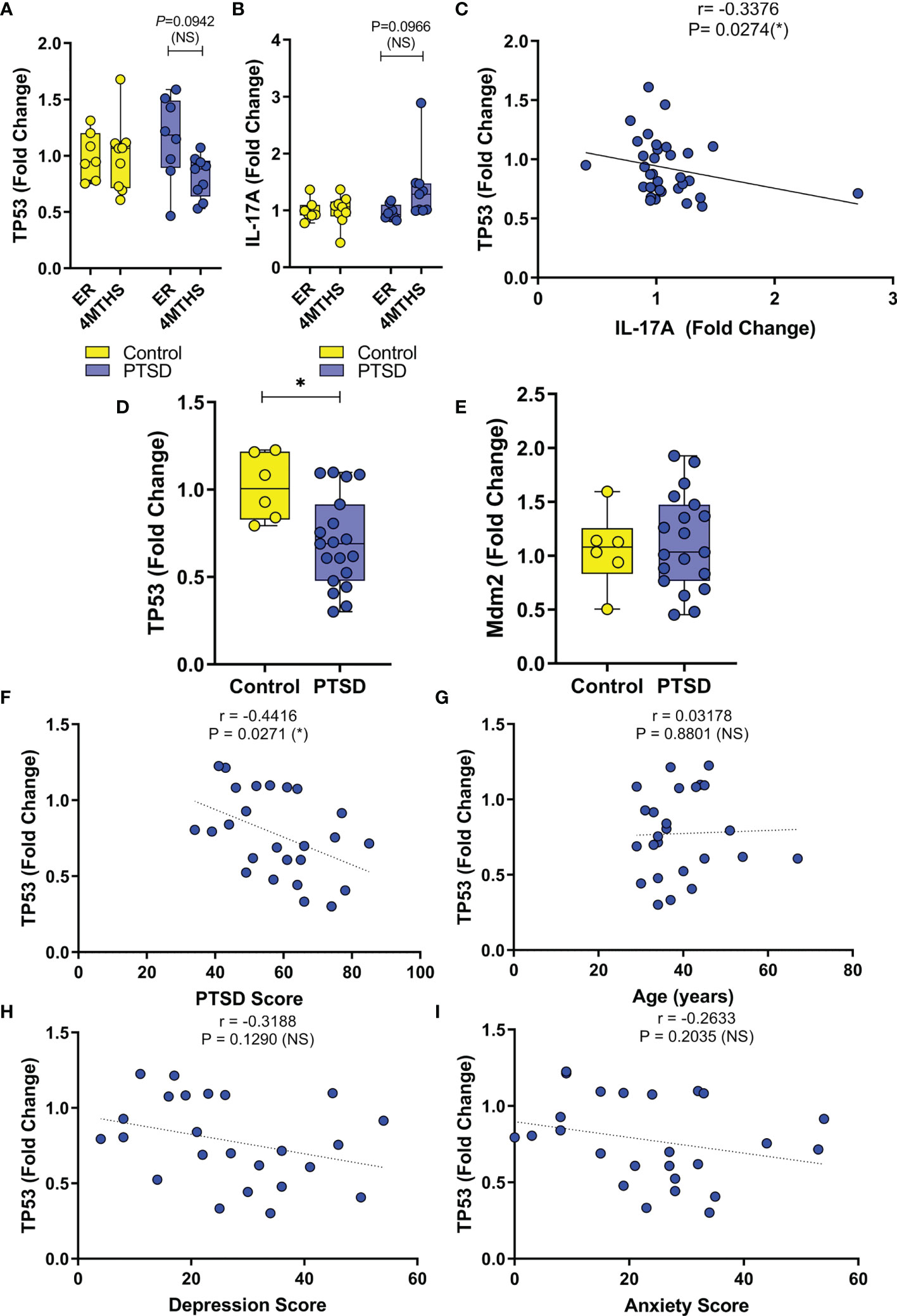
Figure 3 Expression of TP53 in PBMCs is downregulated in PTSD samples compared to Control. (A–C) Normalized expression data was analyzed from public database (ID. GSE860) between PTSD (n=17) and Controls (n=15), with two time points during initial emergency room visit for trauma (ER) and 4 months later (4MTHS). (A) Box and whisker plot depicting fold change expression of TP53. (B) Box and whisker plot depicting fold change expression of IL-17A. (C) Correlation plot between TP53 and IL-17A fold change expression. (D–I) PBMC gene expression data was collected from VMC PTSD (n=19) and Control volunteers (n=6). (D) Box and whisker plot depicting TP53 expression by PCR. (E) Box and whisker plot depicting Mdm2 expression by PCR. Correlation plots depicting (F) TP53 vs. PTSD score, (G) TP53 vs age of cohort, (H) TP53 vs. Depression score, and (I) TP53 vs. Anxiety score. Box and whisker bars depict maximum and minimum values, with middle bar depicted at the median. For box and whisker plots, significance was determined using an unpaired standard t test. For correlation plots, Pearson correlation test was used to determine significance. Significance was defined as having p-value ≤ 0.05 (*).
PBMCs From PTSD Subjects Exhibited Increased Methylation in One of the Known Promoter Regions of TP53
As we already noted that DNA methylation in genes involved in inflammation were altered in PTSD subjects (29), we evaluated the methylation pattern of the TP53 promoter regions in Control (n=5) and PTSD (n=5) PBMC samples obtained from the DHNS project using MeDIP-seq. Patient demographics showed Control and PTSD samples used were comparable to each other in terms of age, race, and sex, though there was a slight disproportionate number of males in the Control samples (Table 1). This more male sex bias proportion was also present when evaluating Control and PTSD TP53 expression in the current report, as noted earlier. The samples collected in the DNHS project were also all African American (AA), which correlated with a larger proportion of this ethnicity in the aforementioned studies evaluating TP53 expression among PTSD samples, at least making the DNA methylation results more comparable to the aforementioned PCR results. In assessing one of the two identified promoter regions of the human TP53 gene on the Ensembl genome browser (ensembl.org), it was found that in general, PTSD PBMC samples had increased methylation intensity in the larger ~5kb promoter region identified in Figure 4A. This increased DNA methylation intensity was even more noticeable when combining the 5 samples into their respective groups (Figure 4B). With increased DNA methylation in the promotor region, it would stand to reason that this would lead to an impediment in the active transcription of TP53, or the gene encoding the p53 protein. This epigenetic modification could explain why PBMCs from PTSD subjects have lower expression of TP53 when compared to healthy controls.
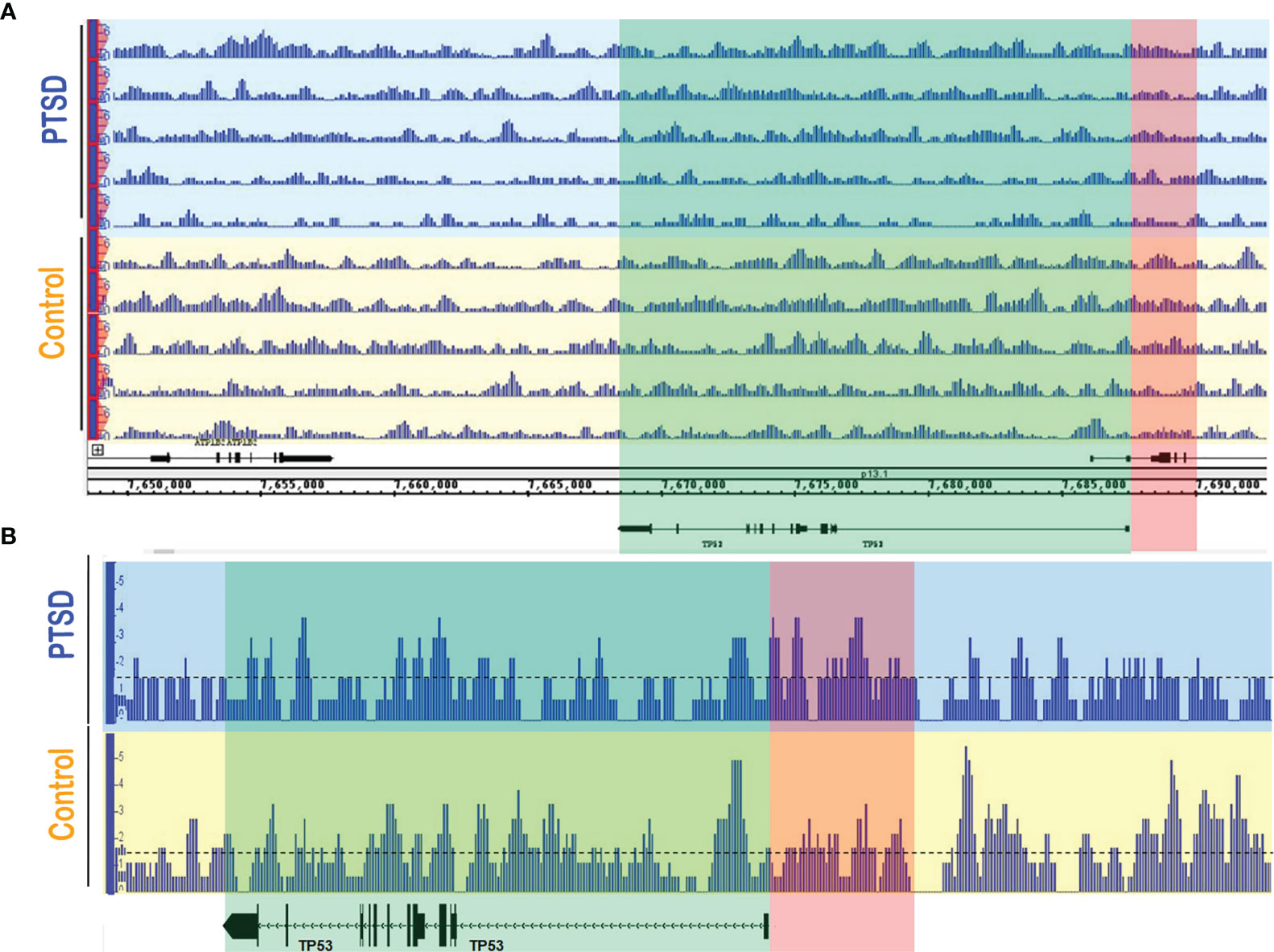
Figure 4 PTSD PBMCs had increased DNA methylation in TP53 promoter region. MeDIP-seq was performed on randomly selected PTSD (n=5) and Control (n=5) PBMC samples. Visualization of the methylated genome was acquired in the IGB genome browser (www.bioviz.org) for (A) individual samples, and (B) combined samples within respective groups. Blue sections indicate PTSD samples, and yellow sections represent Controls. Green section indicates the TP53 gene, while the red section represents one of the known TP53 promoter regions (~5kb) provided in the Ensembl genome browser (www.ensembl.org). For combined data, a line at 1.5 value was drawn to better visualize methylation intensity between the two groups. Differences observed are qualitative measures in which increased signal intensity (e.g. peak height and width) signify more DNA methylation. Signal intensity in the PTSD group was greater overall in the TP53 promoter compared to Control samples.
Modulation of Let-7a in Activated CD4+ T Cells Alters Th17 Development and Function
As it has already been experimentally shown that TP53 modulation affects let-7a expression (36, 37), experiments were carried out focusing on how let-7a alterations affected CD4+ T cell phenotypes during activation. To achieve this, CD4+ cells were purified (~97% consistently) from PBMCs from a Control samples (Figure 5A). Next, these CD4+ cells were either left unactivated, or activated with CD3/CD28 antibodies in the presence or absence of let-7a mimic or inhibitor. Because IPA miRNA analysis pathway linked let-7a to Th17 development and function (Figure 2), Th17 cells (CD4+IL-17A+) were assessed under these conditions using flow cytometry (Figure 5B). As expected, Th17 populations expand under activating conditions when compared to cells left unactivated, but what was interesting was that the Th17 populations were altered by let-7a modulation (Figure 5C). Specifically, when enhancing let-7a levels with the mimic, Th17 percentages significantly decreased, while the opposite was true as inhibiting let-7a led to expansion of the Th17 population in the cultures. It is noteworthy that the activation of these CD4+ cultures resulted in significant decreased expression of let-7a in general when compared to unactivated cells. Let-7a modulation was validated by PCR to show that the mimics significantly enhanced let-7a expression, whereas inhibitors significantly decreased the expression in these cultured conditions (Figure 5D). Once confirmed, Th17 function and development were evaluated by way of IL-17A and Th17-inducing IL-6, respectively. It was shown that under these culturing conditions, increases in let-7a led to a significant decrease in IL-17A production (Figure 5E). Inhibiting let-7a however promoted significant increased secretion of IL-17A. The same trend was observed in IL-6 production levels, where inhibition of let-7a significantly enhanced IL-6 secretion, but a decrease in this miRNA resulted in significantly increased IL-6 production (Figure 5F). Taken altogether, these data demonstrated that let-7a negatively regulates Th17 development and function. Combined with the previous results, the data suggested that decreased TP53 correlates with decreased biogenesis of let-7a in PTSD patients, promoting an increased Th17 phenotype, which would normally be regulated by miRNA let-7a. The overall conclusions suggested that patients diagnosed with PTSD may be more prone to an inflammatory Th17 phenotype due to dysregulation of the P53/let-7a axis, which under normal conditions would aid in regulating this T cell-mediated inflammatory response.
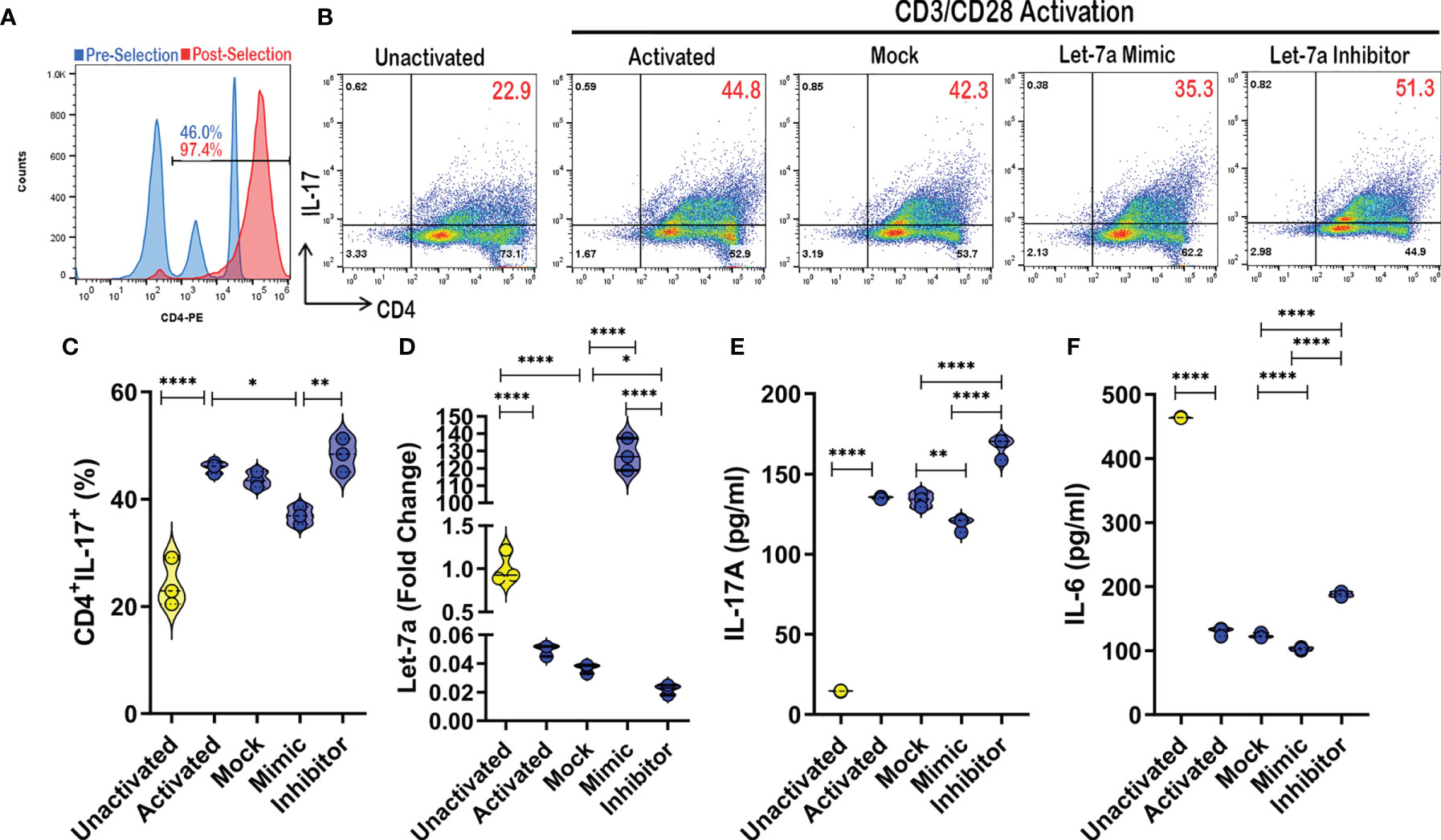
Figure 5 Let-7a modulation affects Th17 development and function. (A) CD4+ cells were purified using magnetic bead separation from PBMCs provided by healthy control donor, which was validated using flow cytometry to ensure purity (>95%). (B) Representative flow cytometry plots depicting Th17 cells (CD4+IL-17A+) in unactivated or CD3/CD28 activation cultures (24 hours) in the absence or presence of mock, let-7a mimic, or let-7a inhibitor. Red numbers indicating double positive TH17 percentage. (C) Violin plot depicting Th17 percentages as assessed by flow cytometry. (D) Violin plot depicting expression of let-7a as assessed by PCR. (E) Violin plot depicting IL-17 concentration as assessed by ELISA from cell culture supernatants. (F) Violin plot depicting IL-6 concentration as assessed by ELISA from cell culture supernatants. In vitro data are representative of at least 3 independent experiments, with 3 samples per culture condition. One way ANOVA was used to determine significance between groups. Significance was determined as p-value ≤ 0.05(*), ≤ 0.01(**), ≤ 0.001(****).
Discussion
PTSD is a psychiatric disorder characterized by exposure to profound traumatic events that result in acute (less than 3 months), chronic (3 months or more), or delayed (occurring months or years after event) onset. Core symptoms are reminders that trigger memories of traumatic events, suppression of memories of the traumatic events, recurrence of disturbing flashbacks, avoidance or emotional numbing, and hyperarousal (4). In addition to these negative impacts on mental health, research has revealed a profound alteration of the immune system in patients with PTSD, which results in a higher risk for developing autoimmune and inflammatory disorders (16). During stressful events, the hypothalamic-pituitary-adrenal (HPA) axis is mobilized, which can activate the sympathetic nervous system to release adrenergic hormones (38). All lymphocytes express adrenergic receptors in varying degrees, including T cells (39, 40). In conjuncture with this notion that stress can have an impact on T cells, research has shown that people suffering from PTSD have increased T cell number and enhanced T cell function (6, 41–43). However, it is also important to note conflicting reports have also shown reduced T cell number and function in some cases of PTSD (44–46). Interestingly, in the cases reporting a reduction, these studies examined non-combat PTSD subjects, while cases showing an increase were from combat-associated PTSD. In this report and our previous report, we noted an increased inflammatory T cell response, but once again, this was observed more in veteran patients (27). Therefore, the type of trauma-inducing PTSD may play a significant role in T cell modulation outcome, and thus should be taken into consideration. More importantly, studies examining PTSD should also take into account certain cell types and not just general PBMCs, particularly in the case of CD4+ T cells, which have pro- and anti-inflammatory roles.
Generally, CD4+ T helper (Th) cells can be divided into pro- (Th1 and Th17) and anti- (Th2 and Treg) inflammatory subsets (47). In our previous report examining CD4+ T helper subsets in PTSD patients, we found that PTSD subjects had increased proportions of pro-inflammatory (Th1 and Th17) and reduced (Treg) or unchanged (Th2) anti-inflammatory CD4+ T cell subsets (27). In support of our findings, Gola and colleagues also showed PBMCs from PTSD patients spontaneously produced higher levels of IL-6, one of the key cytokines involved in Th17 differentiation (26). In our previous reports, we also provided possible mechanisms leading to the development of an increased inflammatory phenotype, which include miRNA modulation and epigenetic modifications to inflammatory-related genes and pathways, such as the Wnt signaling pathway (31). Building upon these results, we also noted an interesting association of PTSD with TP53 in the current study.
Our highlighted mechanism involving TP53 dysregulation in PTSD subjects is both supported and contradicted by other reports analyzing gene expression profiles of PBMCS from PTSD patients (48, 49). In the report from Dong et al., researchers noted TP53 was the most dysregulated gene in PTSD patients when compared to control samples, but interestingly the expression was elevated (48). On the other hand, in support of our current findings, Breen et al. noted that TP53 was downregulated in PTSD-positive samples (49). The discrepancy once again might be better explained with more careful analysis in the future, one that takes into account the type of trauma and cell type, admittedly a limitation even in the current report. In particular, it will be crucial in future studies to validate many of these findings (e.g. TP53 expression and DNA promoter methylation, let-7a levels) in purified CD4+ T cell populations from PTSD and control samples, as opposed to whole circulating PBMC populations, which consists of a variety of different immune cell types. However, it is interesting to take in consideration what we currently know about TP53 and how it regulates the Th17 inflammatory response. TP53 has not only been shown to negatively regulate the function and differentiation of Th17 effector cells (50), but has also been shown to play a role in the biogenesis of several miRNA (51), small non-coding RNA molecules known to regulate several cellular functions including the development of Th17 cells (52). In response to DNA damage or some abnormal oncogene activation, TP53 will act as a tumor suppressor by preventing tumor growth and development through several pathways including apoptosis, senescence, and cell cycle arrest (53). There are an abundant number of resources and research articles dedicated to the role TP53 plays in cancer and tumor development, but more studies are focusing on the role this protein plays in normal cellular processes, particularly in terms of T cell function. Watanabe and colleagues showed that TP53 plays an important role in antigen-specific CD4+ T cell responses, and proliferation of antigen-activated CD4+ T cells was dependent on the downregulation of TP53 (54). TP53 was also found to induce SLAM-associated protein (SAP), a pro-apoptotic factor in lymphocytes, in activated primary human T cells as a means to maintain T cell homeostasis (55). More importantly, in a murine model investigating the effects of autoimmunity in p53null CD45.1 mice, these mice were shown to spontaneously develop autoimmunity, characterized by increased presence of Th17 effectors, as well as elevated serum levels of IL-17 and Th17-producing IL-6 (50). Collectively, these data suggested that TP53 is not only a major player in T cell development and function, but it may also support our current findings suggesting that the TP53/let-7a axis may also negatively regulate Th17 CD4+ subsets, warranting further investigation into the role this protein might be play in the Th17-driven immune response in the PTSD population.
It is important to note that other cell types can produce IL-17A, such as γδ T cells, innate lymphoid type cells (ILCs), mast cells, CD8 T cells, natural killer (NK) cells, and neutrophils (56, 57). However, the source of increased IL-17A observed in PTSD patient PBMCs relative to controls in our previous report (27) and highlighted in the current report is more than likely produced by Th17 cells specifically. IL-17A was co-expressed with either CD4 or a combination of CD3 and CD4. This would exclude cytotoxic CD8 T cells, neutrophils, and mast cells. While there has been reports of some mast cells expressing CD4, current literature suggests that normal mast cells do not express this particular cell surface marker (58). In addition, the source of IL-17A is most likely not attributed to γδ T cells or ILCs since even though they can express the CD4, these cell types represent a very minor population in the human PBMCs. In the case of γδ T cells, they are a minor population in circulating PBMCs (0.5-5%, compared to 50% of αβ T cells), are more often expressed in epithelial cell-rich compartments like skin, the digestive tract, and reproductive organ mucosa, and mainly exhibit a CD3+CD4-CD8- phenotype (59). ILCs (ILC1, ILC2, and ILC3) do not normally express CD4 either, and even though a subset of CD4+ILC1 cells have been identified, these cells shared more characteristics with Th1 phenotypes in their cytokine production, as opposed to Th17 (60). In addition, like γδ T cells, ILCs are mainly tissue-associated and have low frequency in circulation, such as the PBMC population (61). NK/NKT cells are also lowly expressed in PBMCs compared to T helper cell subsets (62), and in addition, NK cells do not normally express T cell-associated CD3. Collectively, these observations strongly suggest the source of increased IL-17A in PTSD patients is most likely derived from CD4+ Th17 cells, which is supported by a report from Hefele et al. which showed trauma can increase IL-17A specifically in Th17 subsets (63).
Regarding the link between TP53 and miRNA let-7a in our current report, TP53 is known to enhance the maturation of several miRNA already, such as the let-7 family, miR-200c, miR-143, miR-16, miR-145, and miR-21 (64–66). miRNA play a crucial role in the regulation of many biological cellular processes, including CD4+ T cell development, proliferation, differentiation, and function (67). Of particular interest is the fact that let-7a, a member of the let-7 family of miRNAs, was shown to decrease IL-6-dependent Th17 differentiation and production of Th17-specific cytokines in a murine model of Con A-induced hepatitis (68). Let-7 members are known to be direct negative regulators of IL-6 and small guanosine triphosphatases (GTPases) Ras proteins, which play important roles in inducing Th17 cells and IL-17, respectively (69, 70). In cancer cells, TP53 plays an important role in the induction of let-7a (37), and let-7a along with TP53 were shown to regulate Ras in a colorectal cancer cell line (36). This would suggest that let-7a-mediated effects on Th17 function involves a possible indirect relationship between this microRNA and IL-17A, since examination of publicly-available miRNA-target gene prediction websites such as miRdatabase (http://mirdb.org/) and TargetScan (http://www.targetscan.org/) revealed human let-7a did not bind directly to IL-17A transcripts. However, as noted in Figure 2 of the current report, let-7a can inhibit Ras proteins, which have been implicated in the development of Th17 and production of IL-17 (71). In studies by Zayoud et al., Ras protein inhibitors were found to suppress in vivo induction of splenic Th17 cells. In the same report, in vitro polarization of Th17 was also perturbed using Ras inhibition. Therefore, it would stand to reason that TP53 downregulation leading to decreased let-7a would prevent directed inhibition of Ras proteins, thereby leading to increased Th17 polarization and IL-17A production in T cells.
Collectively, the current report offers support for the hypothesis that dysregulation of the TP53/let-7a axis could lead to an increased inflammatory Th17 phenotype. This provides evidence in the underreported role of TP53 in PTSD, which is becoming increasingly more prevalent as cases of PTSD are currently on the rise due to the COVID-19 pandemic (72, 73). Despite these exciting findings, the current report does have limitations that must be addressed in future studies. In addition to a relatively overall low sample size, new data generated in the current report contain samples with results more prevalent to both males (sex bias) and African Americans (race/ethnicity bias). Future studies should and must focus now on evaluating these key findings presented in larger and more diverse patient cohorts, with special emphasis on how types of trauma (e.g. combat vs non-combat) and other factors (e.g. sex or race/ethnicity) might affect the TP53/let-7a axis and the subsequent susceptibility to an inflammatory Th17 phenotype in PTSD patients. In addition, and as noted earlier in the discussion, key findings from the current report must be validated specifically in the CD4+ T cell population of patient cohorts as it relates to epigenetic modifications, such as DNA methylation in the TP53 promoter region and let-7a levels. For example, while published reports have shown that TP53 activation can induce let-7a in various human cell lines (74), these findings will need to be further corroborated in human CD4+ T cells by manipulating TP53 (activation and silencing) in this particular cell subset and evaluating how these changes effect let-7a levels. Lastly, based on the more qualitative nature of the MeDIPseq results showing hypermethylation of the TP53 promoter in PTSD patients compared to controls in the current report, ample justification is now provided to examine this observation in a more quantitative assay (e.g. methylation-specific PCR, or MSP) in a larger human subject cohort.
Data Availability Statement
Publicly available datasets were analyzed in this study. This data can be found here: The datasets generated for this study can be found in the following online repositories: ArrayExpress (E-MTAB-4880) (https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-4880/) or Gene Expression Omnibus (GEO; GSE860) (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE860).
Ethics Statement
Procedures to collect human samples presented in this report were approved and reviewed by the Institutional Review Boards (IRBs) from the University of South Carolina, the University of Michigan, and the University of North Carolina-Chapel Hill. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
The following contributions in the current report acknowledge the roles of the listed authors as follows: PB, MN, and PN contributed to conception and design of the study. PB, MB, XY, JZ, OA, and JG conducted the experiments. JG, MU and AA provided samples and assisted with statistical analysis. PB wrote the first draft of the manuscript. PB, MN, PN, MB, XY, JZ, OA, JG, MU, and AA contributed to manuscript revision and read and approved the submitted version.
Funding
The work was supported by funding from the National Institutes of Health (NIH) [R01ES019313, R01MH094755, R01AI123947, R01AI129788, P01AT003961, P20GM103641, R01AT006888].
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Yehuda R. Post-Traumatic Stress Disorder. N Engl J Med (2002) 346(2):108–14. doi: 10.1056/NEJMra012941
2. Kolassa IT, Kolassa S, Ertl V, Papassotiropoulos A, De Quervain DJ. The Risk of Posttraumatic Stress Disorder After Trauma Depends on Traumatic Load and the Catechol-O-Methyltransferase Val(158)Met Polymorphism. Biol Psychiatry (2010) 67(4):304–8. doi: 10.1016/j.biopsych.2009.10.009
3. Gola H, Engler H, Schauer M, Adenauer H, Riether C, Kolassa S, et al. Victims of Rape Show Increased Cortisol Responses to Trauma Reminders: A Study in Individuals With War- and Torture-Related PTSD. Psychoneuroendocrinology (2012) 37(2):213–20. doi: 10.1016/j.psyneuen.2011.06.005
4. American Psychiatric Association, American Psychiatric Association. DSM-5 Task Force. Diagnostic and Statistical Manual of Mental Disorders : DSM-5. 5th ed Vol. xliv, 947. Washington, D.C: American Psychiatric Association (2013). p. ISBN: 9780890425541 (hardcover alk. paper) 9780890425558 (pbk. alk. paper).
5. Schnurr PP, Jankowski MK. Physical Health and Post-Traumatic Stress Disorder: Review and Synthesis. Semin Clin Neuropsychiatry (1999) 4(4):295–304. doi: 10.153/SCNP00400295
6. Boscarino JA. Posttraumatic Stress Disorder and Physical Illness: Results From Clinical and Epidemiologic Studies. Ann N Y Acad Sci (2004) 1032:141–53. doi: 10.1196/annals.1314.011
7. Mikuls TR, Padala PR, Sayles HR, Yu F, Michaud K, Caplan L, et al. Prospective Study of Posttraumatic Stress Disorder and Disease Activity Outcomes in US Veterans With Rheumatoid Arthritis. Arthritis Care Res (Hoboken) (2013) 65(2):227–34. doi: 10.1002/acr.21778
8. Talbot LS, Maguen S, Epel ES, Metzler TJ, Neylan TC. Posttraumatic Stress Disorder Is Associated With Emotional Eating. J Trauma Stress (2013) 26(4):521–5. doi: 10.1002/jts.21824
9. Ivanova JI, Birnbaum HG, Chen L, Duhig AM, Dayoub EJ, Kantor ES, et al. Cost of Post-Traumatic Stress Disorder vs Major Depressive Disorder Among Patients Covered by Medicaid or Private Insurance. Am J Manag Care (2011) 17(8):e314–23.
10. Keane TM, Marshall AD, Taft CT. Posttraumatic Stress Disorder: Etiology, Epidemiology, and Treatment Outcome. Annu Rev Clin Psychol (2006) 2:161–97. doi: 10.1146/annurev.clinpsy.2.022305.095305
11. Schlenger WE, Corry NH, Kulka RA, Williams CS, Henn-Haase C, Marmar CR. Design and Methods of the National Vietnam Veterans Longitudinal Study. Int J Methods Psychiatr Res (2015) 24(3):186–203. doi: 10.1002/mpr.1469
12. Cohen BE, Gima K, Bertenthal D, Kim S, Marmar CR, Seal KH. Mental Health Diagnoses and Utilization of VA non-Mental Health Medical Services Among Returning Iraq and Afghanistan Veterans. J Gen Intern Med (2010) 25(1):18–24. doi: 10.1007/s11606-009-1117-3
13. Holmes MR, Rentrope CR, Korsch-Williams A, King JA. Impact of COVID-19 Pandemic on Posttraumatic Stress, Grief, Burnout, and Secondary Trauma of Social Workers in the United States. Clin Soc Work J (2021) 2:1–10. doi: 10.1007/s10615-021-00795-y
14. Einvik G, Dammen T, Ghanima W, Heir T, Stavem K. Prevalence and Risk Factors for Post-Traumatic Stress in Hospitalized and Non-Hospitalized COVID-19 Patients. Int J Environ Res Public Health (2021) 18(4):2079–91. doi: 10.3390/ijerph18042079
15. Chamberlain SR, Grant JE, Trender W, Hellyer P, Hampshire A. Post-Traumatic Stress Disorder Symptoms in COVID-19 Survivors: Online Population Survey. BJPsych Open (2021) 7(2):e47. doi: 10.1192/bjo.2021.3
16. O'Donovan A, Chao LL, Paulson J, Samuelson KW, Shigenaga JK, Grunfeld C, et al. Altered Inflammatory Activity Associated With Reduced Hippocampal Volume and More Severe Posttraumatic Stress Symptoms in Gulf War Veterans. Psychoneuroendocrinology (2015) 51:557–66. doi: 10.1016/j.psyneuen.2014.11.010
17. Groer MW, Kane B, Williams SN, Duffy A. Relationship of PTSD Symptoms With Combat Exposure, Stress, and Inflammation in American Soldiers. Biol Res Nurs (2015) 17(3):303–10. doi: 10.1177/1099800414544949
18. Lindqvist D, Wolkowitz OM, Mellon S, Yehuda R, Flory JD, Henn-Haase C, et al. Proinflammatory Milieu in Combat-Related PTSD Is Independent of Depression and Early Life Stress. Brain Behav Immun (2014) 42:81–8. doi: 10.1016/j.bbi.2014.06.003
19. Wilson CB, McLaughlin LD, Nair A, Ebenezer PJ, Dange R, Francis J. Inflammation and Oxidative Stress Are Elevated in the Brain, Blood, and Adrenal Glands During the Progression of Post-Traumatic Stress Disorder in a Predator Exposure Animal Model. PloS One (2013) 8(10):e76146. doi: 10.1371/journal.pone.0076146
20. Heath NM, Chesney SA, Gerhart JI, Goldsmith RE, Luborsky JL, Stevens NR, et al. Interpersonal Violence, PTSD, and Inflammation: Potential Psychogenic Pathways to Higher C-Reactive Protein Levels. Cytokine (2013) 63(2):172–8. doi: 10.1016/j.cyto.2013.04.030
21. Pace TW, Wingenfeld K, Schmidt I, Meinlschmidt G, Hellhammer DH, Heim CM. Increased Peripheral NF-kappaB Pathway Activity in Women With Childhood Abuse-Related Posttraumatic Stress Disorder. Brain Behav Immun (2012) 26(1):13–7. doi: 10.1016/j.bbi.2011.07.232
22. Smith AK, Conneely KN, Kilaru V, Mercer KB, Weiss TE, Bradley B, et al. Differential Immune System DNA Methylation and Cytokine Regulation in Post-Traumatic Stress Disorder. Am J Med Genet B Neuropsychiatr Genet (2011) 156B(6):700–8. doi: 10.1002/ajmg.b.31212
23. Gill JM, Saligan L, Woods S, Page G. PTSD Is Associated With an Excess of Inflammatory Immune Activities. Perspect Psychiatr Care (2009) 45(4):262–77. doi: 10.1111/j.1744-6163.2009.00229.x
24. Fesharaki-Zadeh A, Miyauchi JT, St Laurent-Arriot K, Tsirka SE, Bergold PJ. Increased Behavioral Deficits and Inflammation in a Mouse Model of Co-Morbid Traumatic Brain Injury and Post-Traumatic Stress Disorder. ASN Neuro (2020) 12:1759091420979567. doi: 10.1177/1759091420979567
25. Fonkoue IT, Michopoulos V, Park J. Sex Differences in Post-Traumatic Stress Disorder Risk: Autonomic Control and Inflammation. Clin Auton Res (2020) 30(5):409–21. doi: 10.1007/s10286-020-00729-7
26. Gola H, Engler H, Sommershof A, Adenauer H, Kolassa S, Schedlowski M, et al. Posttraumatic Stress Disorder Is Associated With an Enhanced Spontaneous Production of Pro-Inflammatory Cytokines by Peripheral Blood Mononuclear Cells. BMC Psychiatry (2013) 13:40. doi: 10.1186/1471-244X-13-40
27. Zhou J, Nagarkatti P, Zhong Y, Ginsberg JP, Singh NP, Zhang J, et al. Dysregulation in microRNA Expression Is Associated With Alterations in Immune Functions in Combat Veterans With Post-Traumatic Stress Disorder. PloS One (2014) 9(4):e94075. doi: 10.1371/journal.pone.0094075
28. Bam M, Yang X, Zhou J, Ginsberg JP, Leyden Q, Nagarkatti PS, et al. Evidence for Epigenetic Regulation of Pro-Inflammatory Cytokines, Interleukin-12 and Interferon Gamma, in Peripheral Blood Mononuclear Cells From PTSD Patients. J Neuroimmune Pharmacol (2016) 11(1):168–81. doi: 10.1007/s11481-015-9643-8
29. Bam M, Yang X, Zumbrun EE, Zhong Y, Zhou J, Ginsberg JP, et al. Dysregulated Immune System Networks in War Veterans With PTSD Is an Outcome of Altered miRNA Expression and DNA Methylation. Sci Rep (2016) 6:31209. doi: 10.1038/srep31209
30. Bam M, Yang X, Zumbrun EE, Ginsberg JP, Leyden Q, Zhang J, et al. Decreased AGO2 and DCR1 in PBMCs From War Veterans With PTSD Leads to Diminished miRNA Resulting in Elevated Inflammation. Transl Psychiatry (2017) 7(8):e1222. doi: 10.1038/tp.2017.185
31. Bam M, Yang X, Busbee BP, Aiello AE, Uddin M, Ginsberg JP, et al. Increased H3K4me3 Methylation and Decreased miR-7113-5p Expression Lead to Enhanced Wnt/beta-Catenin Signaling in Immune Cells From PTSD Patients Leading to Inflammatory Phenotype. Mol Med (2020) 26(1):110. doi: 10.1186/s10020-020-00238-3
32. Uddin M, Aiello AE, Wildman DE, Koenen KC, Pawelec G, de Los Santos R, et al. Epigenetic and Immune Function Profiles Associated With Posttraumatic Stress Disorder. Proc Natl Acad Sci USA (2010) 107(20):9470–5. doi: 10.1073/pnas.0910794107
33. Segman RH, Shefi N, Goltser-Dubner T, Friedman N, Kaminski N, Shalev AY. Peripheral Blood Mononuclear Cell Gene Expression Profiles Identify Emergent Post-Traumatic Stress Disorder Among Trauma Survivors. Mol Psychiatry (2005) 10(5):500–13, 425. doi: 10.1038/sj.mp.4001636
34. Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and Memory-Efficient Alignment of Short DNA Sequences to the Human Genome. Genome Biol (2009) 10(3):R25. doi: 10.1186/gb-2009-10-3-r25
35. Lienhard M, Grimm C, Morkel M, Herwig R, Chavez L. MEDIPS: Genome-Wide Differential Coverage Analysis of Sequencing Data Derived From DNA Enrichment Experiments. Bioinf (2014) 30(2):284–6. doi: 10.1093/bioinformatics/btt650
36. Luu C, Heinrich EL, Duldulao M, Arrington AK, Fakih M, Garcia-Aguilar J, et al. TP53 and Let-7a Micro-RNA Regulate K-Ras Activity in HCT116 Colorectal Cancer Cells. PloS One (2013) 8(8):e70604. doi: 10.1371/journal.pone.0070604
37. Lee JY, Kim HJ, Yoon NA, Lee WH, Min YJ, Ko BK, et al. Tumor Suppressor P53 Plays a Key Role in Induction of Both Tristetraprolin and Let-7 in Human Cancer Cells. Nucleic Acids Res (2013) 41(11):5614–25. doi: 10.1093/nar/gkt222
38. Griffin GD, Charron D, Al-Daccak R. Post-Traumatic Stress Disorder: Revisiting Adrenergics, Glucocorticoids, Immune System Effects and Homeostasis. Clin Transl Immunol (2014) 3(11):e27. doi: 10.1038/cti.2014.26
39. Anstead MI, Hunt TA, Carlson SL, Burki NK. Variability of Peripheral Blood Lymphocyte Beta-2-Adrenergic Receptor Density in Humans. Am J Respir Crit Care Med (1998) 157(3 Pt 1):990–2. doi: 10.1164/ajrccm.157.3.9704071
40. Landmann R. Beta-Adrenergic Receptors in Human Leukocyte Subpopulations. Eur J Clin Invest (1992) 22(Suppl 1):30–6.
41. Boscarino JA, Chang J. Higher Abnormal Leukocyte and Lymphocyte Counts 20 Years After Exposure to Severe Stress: Research and Clinical Implications. Psychosom Med (1999) 61(3):378–86. doi: 10.1097/00006842-199905000-00019
42. Sommershof A, Aichinger H, Engler H, Adenauer H, Catani C, Boneberg EM, et al. Substantial Reduction of Naive and Regulatory T Cells Following Traumatic Stress. Brain Behav Immun (2009) 23(8):1117–24. doi: 10.1016/j.bbi.2009.07.003
43. Altemus M, Dhabhar FS, Yang R. Immune Function in PTSD. Ann N Y Acad Sci (2006) 1071:167–83. doi: 10.1196/annals.1364.013
44. Kawamura N, Kim Y, Asukai N. Suppression of Cellular Immunity in Men With a Past History of Posttraumatic Stress Disorder. Am J Psychiatry (2001) 158(3):484–6. doi: 10.1176/appi.ajp.158.3.484
45. Ironson G, Wynings C, Schneiderman N, Baum A, Rodriguez M, Greenwood D, et al. Posttraumatic Stress Symptoms, Intrusive Thoughts, Loss, and Immune Function After Hurricane Andrew. Psychosom Med (1997) 59(2):128–41. doi: 10.1097/00006842-199703000-00003
46. Sabioncello A, Kocijan-Hercigonja D, Rabatic S, Tomasic J, Jeren T, Matijevic L, et al. Immune, Endocrine, and Psychological Responses in Civilians Displaced by War. Psychosom Med (2000) 62(4):502–8. doi: 10.1097/00006842-200007000-00008
47. Weinmann AS. Roles for Helper T Cell Lineage-Specifying Transcription Factors in Cellular Specialization. Adv Immunol (2014) 124:171–206. doi: 10.1016/B978-0-12-800147-9.00006-6
48. Dong K, Zhang F, Zhu W, Wang Z. Wang G. Partial Least Squares Based Gene Expression Analysis in Posttraumatic Stress Disorder. Eur Rev Med Pharmacol Sci (2014) 18(16):2306–10.
49. Breen MS, Bierer LM, Daskalakis NP, Bader HN, Makotkine I, Chattopadhyay M, et al. Correction: Differential Transcriptional Response Following Glucocorticoid Activation in Cultured Blood Immune Cells: A Novel Approach to PTSD Biomarker Development. Transl Psychiatry (2020) 10(1):1. doi: 10.1038/s41398-019-0665-5
50. Zhang S, Zheng M, Kibe R, Huang Y, Marrero L, Warren S, et al. Trp53 Negatively Regulates Autoimmunity via the STAT3-Th17 Axis. FASEB J (2011) 25(7):2387–98. doi: 10.1096/fj.10-175299
51. Suzuki HI, Yamagata K, Sugimoto K, Iwamoto T, Kato S, Miyazono K. Modulation of microRNA Processing by P53. Nat (2009) 460(7254):529–33. doi: 10.1038/nature08199
52. Wei B, Pei G. microRNAs: Critical Regulators in Th17 Cells and Players in Diseases. Cell Mol Immunol (2010) 7(3):175–81. doi: 10.1038/cmi.2010.19
53. Kruse JP, Gu W. Modes of P53 Regulation. Cell (2009) 137(4):609–22. doi: 10.1016/j.cell.2009.04.050
54. Watanabe M, Moon KD, Vacchio MS, Hathcock KS, Hodes RJ. Downmodulation of Tumor Suppressor P53 by T Cell Receptor Signaling Is Critical for Antigen-Specific CD4(+) T Cell Responses. Immun (2014) 40(5):681–91. doi: 10.1016/j.immuni.2014.04.006
55. Madapura HS, Salamon D, Wiman KG, Lain S, Klein G, Klein E, et al. P53 Contributes to T Cell Homeostasis Through the Induction of Pro-Apoptotic SAP. Cell Cycle (2012) 11(24):4563–9. doi: 10.4161/cc.22810
56. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol (2009) 27:485–517. doi: 10.1146/annurev.immunol.021908.132710
57. Geha M, Tsokos MG, Bosse RE, Sannikova T, Iwakura Y, Dalle Lucca JJ, et al. IL-17a Produced by Innate Lymphoid Cells Is Essential for Intestinal Ischemia-Reperfusion Injury. J Immunol (2017) 199(8):2921–9. doi: 10.4049/jimmunol.1700655
58. Soliman DS, Al-Sabbagh A, Ibrahim F, Gameil A, Yassin M, El-Omri H. Highly Aggressive CD4-Positive Mast Cell Leukaemia (Leukaemic Variant) Associated With Isolated Trisomy 19 and Hemophagocytosis by Neoplastic Mast Cells: A Case Report With Challenging Experience and Review. Case Rep Hematol (2019) 2019:1805270. doi: 10.1155/2019/1805270
59. Mao Y, Yin S, Zhang J, Hu Y, Huang B, Cui L, et al. A New Effect of IL-4 on Human Gammadelta T Cells: Promoting Regulatory Vdelta1 T Cells via IL-10 Production and Inhibiting Function of Vdelta2 T Cells. Cell Mol Immunol (2016) 13(2):217–28. doi: 10.1038/cmi.2015.07
60. Roan F, Stoklasek TA, Whalen E, Molitor JA, Bluestone JA, Buckner JH, et al. CD4+ Group 1 Innate Lymphoid Cells (ILC) Form a Functionally Distinct ILC Subset That Is Increased in Systemic Sclerosis. J Immunol (2016) 196(5):2051–62. doi: 10.4049/jimmunol.1501491
61. Bonne-Annee S, Bush MC, Nutman TB. Differential Modulation of Human Innate Lymphoid Cell (ILC) Subsets by IL-10 and TGF-Beta. Sci Rep (2019) 9(1):14305. doi: 10.1038/s41598-019-50308-8
62. Angelo LS, Banerjee PP, Monaco-Shawver L, Rosen JB, Makedonas G, Forbes LR, et al. Practical NK Cell Phenotyping and Variability in Healthy Adults. Immunol Res (2015) 62(3):341–56. doi: 10.1007/s12026-015-8664-y
63. Hefele F, Ditsch A, Krysiak N, Caldwell CC, Biberthaler P, van Griensven M, et al. Trauma Induces Interleukin-17a Expression on Th17 Cells and CD4+ Regulatory T Cells as Well as Platelet Dysfunction. Front Immunol (2019) 10:2389. doi: 10.3389/fimmu.2019.02389
64. Boominathan L. The Tumor Suppressors P53, P63, and P73 are Regulators of microRNA Processing Complex. PloS One (2010) 5(5):e10615. doi: 10.1371/journal.pone.0010615
65. Garibaldi F, Falcone E, Trisciuoglio D, Colombo T, Lisek K, Walerych D, et al. Mutant P53 Inhibits miRNA Biogenesis by Interfering With the Microprocessor Complex. Oncogene (2016) 35(29):3760–70. doi: 10.1038/onc.2016.51
66. Gurtner A, Falcone E, Garibaldi F, Piaggio G. Dysregulation of microRNA Biogenesis in Cancer: The Impact of Mutant P53 on Drosha Complex Activity. J Exp Clin Cancer Res (2016) 35:45. doi: 10.1186/s13046-016-0319-x
67. Liu J, Wu CP, Lu BF, Jiang JT. Mechanism of T Cell Regulation by microRNAs. Cancer Biol Med (2013) 10(3):131–7. doi: 10.7497/j.issn.2095-3941.2013.03.002
68. Zhang Y, Wang X, Zhong M, Zhang M, Suo Q, Lv K. MicroRNA Let-7a Ameliorates Con A-Induced Hepatitis by Inhibiting IL-6-Dependent Th17 Cell Differentiation. J Clin Immunol (2013) 33(3):630–9. doi: 10.1007/s10875-012-9840-7
69. Choudhury SN, Li Y. miR-21 and Let-7 in the Ras and NF-kappaB Pathways. Microrna (2012) 1(1):65–9. doi: 10.2174/2211536611201010065
70. Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, et al. RAS is Regulated by the Let-7 microRNA Family. Cell (2005) 120(5):635–47. doi: 10.1016/j.cell.2005.01.014
71. Zayoud M, Vax E, Elad-Sfadia G, Barshack I, Pinkas-Kramarski R, Goldstein I. Inhibition of Ras GTPases Prevents Collagen-Induced Arthritis by Reducing the Generation of Pathogenic CD4(+) T Cells and the Hyposialylation of Autoantibodies. ACR Open Rheumatol (2020) 2(9):512–24. doi: 10.1002/acr2.11169
72. Liu N, Zhang F, Wei C, Jia Y, Shang Z, Sun L, et al. Prevalence and Predictors of PTSS During COVID-19 Outbreak in China Hardest-Hit Areas: Gender Differences Matter. Psychiatry Res (2020) 287:112921. doi: 10.1016/j.psychres.2020.112921
73. Dutheil F, Mondillon L, Navel V. PTSD as the Second Tsunami of the SARS-Cov-2 Pandemic. Psychol Med (2020) 24:1–2. doi: 10.1017/S0033291720001336
Keywords: post-traumatic stress disorder, tumor protein 53, microRNA, inflammation, T helper 17 cells
Citation: Busbee PB, Bam M, Yang X, Abdulla OA, Zhou J, Ginsberg JP (J), Aiello AE, Uddin M, Nagarkatti M and Nagarkatti PS (2022) Dysregulated TP53 Among PTSD Patients Leads to Downregulation of miRNA let-7a and Promotes an Inflammatory Th17 Phenotype. Front. Immunol. 12:815840. doi: 10.3389/fimmu.2021.815840
Received: 15 November 2021; Accepted: 15 December 2021;
Published: 04 January 2022.
Edited by:
Juehua Yu, The First Affiliated Hospital of Kunming Medical University, ChinaReviewed by:
Murugaiyan Gopal, Harvard Medical School, United StatesHong Zan, The University of Texas Health Science Center at San Antonio, United States
Copyright © 2022 Busbee, Bam, Yang, Abdulla, Zhou, Ginsberg, Aiello, Uddin, Nagarkatti and Nagarkatti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Prakash S. Nagarkatti, cHJha2FzaEBtYWlsYm94LnNjLmVkdQ==