 Yuwei Hao
Yuwei Hao Matthew C. Cook
Matthew C. Cook- 1Centre for Personalised Immunology and Department of Immunity and Infectious Diseases, John Curtin School of Medical Research, Australian National University, Acton, ACT, Australia
- 2Department of Immunology, Canberra Hospital, Woden, ACT, Australia
Elucidating links between genotype and phenotype in patients with rare inborn errors of immunity (IEIs) provides insights into mechanisms of immune regulation. In many autosomal dominant IEIs, however, variation in expressivity and penetrance result in complex genotype-phenotype relations, while some autosomal recessive IEIs are so rare that it is difficult to draw firm conclusions. Phenocopies arise when an environmental or non-genetic factor replicates a phenotype conferred by a specific genotype. Phenocopies can result from therapeutic antibodies or autoantibodies that target a protein to replicate aspects of the phenotype conferred by mutations in the gene encoding the same protein. Here, we consider IEIs arising from rare genetic variants in CTLA4 and PDCD1 and compare clinical and laboratory manifestations arising as drug-induced phenocopies (immune related adverse events, IRAEs) in cancer patients treated with immune checkpoint inhibitors (ICI) and identify outstanding questions regarding mechanism of disease.
Introduction
Autosomal dominant loss of function mutations in CTLA4 result in a complex syndrome of immune dysregulation and deficiency (1–3), although the syndrome is characterized by variable expressivity and incomplete penetrance. Recently, human PDCD1 deficiency was described. So far, this syndrome appears to be exceedingly rare, whereas we have extensive experience with ICI that target PD-1 or its ligand., as well as anti-CTLA4 antibodies. Comparing and contrasting these phenocopies with IEIs of CTLA4 and PDCD1 might advance our understanding of the actions of CTLA4 and PD-1, and how defective expression of these molecules cause immune deficiency and dysregulation (4).
CTLA4
CTLA4 is a transmembrane receptor that is structurally similar to CD28 but acts as an inhibitor of T cell activation (5–10). CTLA4 is expressed constitutively by regulatory T cells (Tregs) and is indispensable for immunological self-tolerance and immune homeostasis (11). Conventional T cells upregulate CTLA4 expression upon stimulation, mediated at least in part by nuclear factor of activated T-cells (NFAT) (12). CTLA4 expression has also been reported on B cells, fibroblasts, CD34+ stem cells and granulocytes (13–15), but the significance and action of CTLA4 expression on these cells remains to be determined. CTLA4 is expressed in immune cell malignancies including leukemic B cells and also by breast cancer cells, melanoma, and various carcinomas (16–18).
There is evidence that CTLA4 acts in several ways to modify T cell activation. CTLA4 inhibits co-stimulation by outcompeting CD28 for CD80/86 (19), and real-time competition between CTLA4 and CD28 for translocation into the central-supramolecular activation clusters (cSMAC) of immune synapses has been demonstrated (20). CTLA4 has also been reported to recruit protein phosphatase 2A (PP2A) and tyrosine phosphatase SHP-2 via its cytoplasmic tail, which then dephosphorylates many kinases including AKT, ERK and MEK to inhibit T cell activation (21–23). This cell-intrinsic action has been challenged, however, by reports of a mouse model expressing mutant CTLA4 lacking a cytoplasmic tail, which has no lymphocytic infiltrates or autoimmune disease (24).
Other evidence suggests that CTLA4 acts cell-extrinsically to modify immunity by reducing availability of CD80 and CD86. CTLA4 is a highly endocytic molecule and has been shown to capture and remove CD80/86 from antigen presenting cells, directing these ligands to lysosomes for degradation (25). Other possible actions include regulation of cell adhesion and motility. Ligation of CTLA4 has been postulated to increase T cell motility and reduce contact periods between T cells and antigen-presenting cells, which could prevent inappropriate activation of T cells with low-affinity for peptide-MHC complexes (26). Ligation of CTLA4 recruits PKC-eta, which forms a complex with GIT-2 and PAK-2 to modulate Treg cell-APC interactions. Consistent with this, PKC-eta deficient Tregs fail to dissociate from APCs and exhibit a defect in CD86 capture and transendocytosis (27).
CTLA4 Haploinsufficiency
CTLA4 haploinsufficiency (abbreviated here as CTLA4+/-) leads to a syndrome of immune dysregulation with a broad spectrum of clinical manifestations, and in approximately 30% of carriers, no clinical manifestations at all (1–3). In the largest cohort described to date, the median age of disease onset was 11 years, with a range of 1-59 years. Thus, in many cases, onset of clinical manifestations is not observed until adulthood. Of the clinical phenotypes, lymphoproliferation occurs frequently (73%). Autoimmune and inflammatory manifestations are also common, although there is considerable variability in the end-organs affected. Lymphocytic infiltration of lung, gastrointestinal tract, brain, bone marrow, kidney and retroperitoneal tissue have all been reported. Hematological cytopenia (immune thrombocytopenic purpura, ITP; and autoimmune hemolytic anemia, AIHA) are frequent, while atrophic gastritis, coeliac disease and pancreatitis are uncommon (1–3) (Table 1).
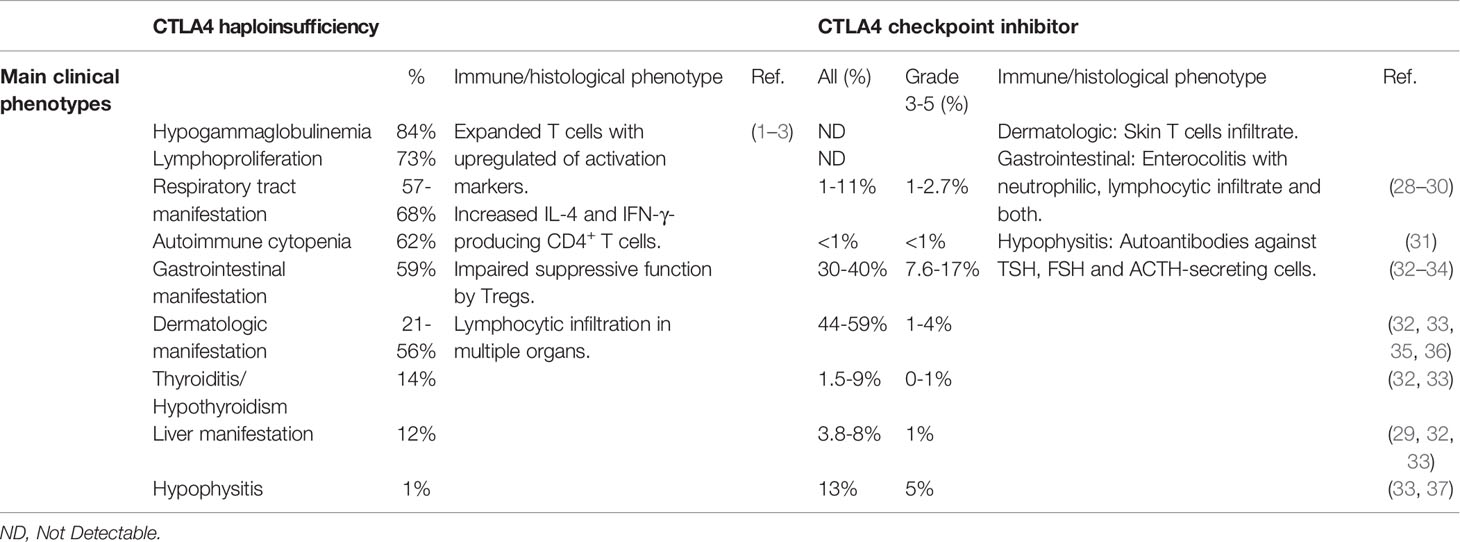
Table 1 Inborn errors of CTLA4 and their phenocopies.
Respiratory manifestations are common in CTLA4+/- patients. In addition to lymphocytic pneumonitis, many CTLA4+/- patients have recurrent respiratory tract infections, including pneumonia, sinusitis and otitis media. Infective complications are accompanied by hypogammaglobulinemia (84%), including reduced IgA (40%), IgG (32%) and IgM (30%) (1–3) (Table 1).
CTLA4+/- patients have hyperactivated effector T cells with increased expression of PD-1 and HLA-DR. In some studies, CD4+ FoxP3+ Treg cells have been reported to be increased (1, 3) but this has not been a consistent finding (2). B cell abnormalities in CTLA4+/- patients include reductions in switched memory B cells, progressive loss of all peripheral B cells, and increased CD21low B cells (1–3). Interestingly, despite the hyperproliferation, T and B cells undergo increased apoptosis in vitro (2).
In the mouse model, Ctla4 haploinsufficiency has not been reported to cause a phenotype but Ctla4-/- mice develop a lethal lymphoproliferative disorder by 3-4 weeks of age. Mice exhibit progressive skewing of the T cell compartment towards CD4+ T cells, lymphocytic infiltrates occur in multiple organs, and pathology is prevented by CD4+ T cell depletion (38). When Ctla4 deficiency is confined to Tregs, mice exhibit delayed lymphoproliferation and fatal T cell-mediated autoimmune disease (including pulmonary lymphocytic infiltrates) by 7 weeks of age (11). T cells are activated with upregulation of activation markers CD44, CD69 and CD25 (9, 10). Ctla4-/- mice also exhibit macrophage and neutrophil infiltration of end-organs, including heart, lung, salivary glands, liver, bone marrow and pancreas. Interestingly, immunodeficiency observed in humans with CTLA4 haploinsufficiency is not observed in Ctla4-/- mice.
Phenocopies of CTLA4 Deficiency
In the 1990s, Allison and colleagues identified the therapeutic potential of CTLA4 inhibition, which culminated in the development and use of ICIs targeting CTLA4 for cancer therapy (39, 40). Ipilimumab and tremelimumab bind to the same region of CTLA4 and interfere with CD80/86 recognition (41–43). Early clinical trials reported that anti-CTLA4 provided durable clinical responses and improved overall survival in a fraction of cancer patients (32, 44). ICIs are now standard of care for many forms of cancer (45). Autoimmunity and inflammatory side effects, however, emerged as significant complications in a proportion of treated patients. Severe IRAEs (Common terminology criteria for adverse events (CTCAE) severity grade of 3-5) have been reported in 19.9-24% of melanoma patients treated with ipilimumab and 52% of melanoma patients treated with tremelimumab (28, 29, 32, 35). The most common severe IRAEs are colitis, dermatitis, and endocrinopathies of hypophysitis and hypothyroidism. Hepatotoxicity, hematological cytopenia and neurologic complications are also observed but are less frequent (31–33) (Table 1).
Colitis of any grade, which most commonly presents with diarrhea, has been reported in 30-40% of patients treated with ipilimumab, while severe colitis/diarrhea is seen in up to 7.6-17% patients on anti-CTLA4 treatment (32, 33) (Table 1). Three histological types of enterocolitis are described: neutrophilic (46%), lymphocytic (15%), and combined neutrophilic and lymphocytic (38%). Neutrophilic inflammation is mainly associated with cryptitis, while lymphocytic inflammation is characterized by increased CD8+ T cells in the crypt epithelium and CD4+ T cells in the lamina propria (34). Gastrointestinal involvement is also common with CTLA4 haploinsufficiency and histology usually reveals extensive T cells infiltration (1–3).
Severe hypophysitis is observed in 5% of melanoma patients treated with ipilimumab but is rare in CTLA4+/- patients (1 of 133) (3, 33) (Table 1). Repeated injection of anti-CTLA4 antibody results in pituitary infiltration of lymphocytes, macrophages and monocytes. In addition, pituitary autoantibodies are detected in mice and melanoma patients after injection of anti-CTLA4 antibody (37). Severe hypothyroidism has been reported in melanoma patients with anti-CTLA4 treatment but is uncommon (~1%) (33). Autoimmune thyroiditis appears to be more frequent in CTLA4 haploinsufficiency (18 of 133) (3). Furthermore, common variants affecting the CTLA4 promoter (49A/G or 60C/T) segregate with autoimmune hypothyroidism (46, 47).
Severe pneumonitis is observed in about 1-2% of patients treated with ipilimumab (28, 29) and bronchospasm has been reported after tremelimumab treatment (2.7%) (30). Respiratory symptoms have been reported in 57-68% patients in different studies. As noted above, many CTLA4+/- patients also suffer from recurrent respiratory tract infections but has been reported infrequently as a complication of anti-CTLA4 treatment (48) (Table 1).
Skin-related IRAEs are common and generally mild in patients receiving anti-CTLA4 treatment. Severe dermatological IRAEs, including pruritus, rash and vitiligo, are observed in up to 4% of patients treated with either ipilimumab or tremelimumab (32, 33, 35, 36). Histological analysis has revealed perivascular immune cell infiltrates in the dermis and epidermis. Although both CD4+ and CD8+ T cells are identified on biopsy of macules, CD4+ T cells dominate the infiltrates reported in melanoma patients receiving ipilimumab (49–51). CTLA4 blockade has also been reported to increase epidermal thickness and infiltrating T cell counts in mice with psoriasis (52). Psoriasis, atopic dermatitis and vitiligo have been observed in in 21-56% of CTLA4+/- patients (1, 3) (Table 1).
Hepatitis has been reported in 3.8-8% of patients receiving ipilimumab but severe hepatic toxicity occurs in less than 1% of patients (29, 32, 33). Liver involvement with lymphocytic infiltrate and liver failure has been observed in 12% of CTLA4+/- patients (1, 3).
Comparison of CTLA4 Deficiency and Its Phenocopy
While the nature and spectrum of IRAEs after anti-CTLA4 treatment is similar to the autoimmune and inflammatory complications of CTLA4 haploinsufficiency, these manifestations appear to be more common in CTLA4 haploinsufficiency. This may reflect the extent of CTLA4 blockade. Consistent with this proposition, IRAEs related to ipilimumab are dose-dependent. Serious IRAEs are more common with higher doses of ipilimumab, mainly due to increased adverse events in gastrointestinal tract, skin and endocrine organs (53). Similarly, low-dose anti-CTLA4 antibody treatment in mice induces anti-parietal autoantibodies, high-dose anti-CTLA4 antibody infection leads to histologically evidence of autoimmunity (54).
Remarkably, hypophysitis is considerably more prevalent in patients treated with anti-CTLA4 antibodies than in patients with CTLA4 haploinsufficiency. CTLA4 is expressed by both human and mice non-hematopoietic cells in the pituitary gland, and anti-CTLA4 antibodies bind to these cells. CTLA4 is also expressed by pituitary cells, particularly those responsible for secreting prolactin and TSH (37).
Differences in Fc receptor binding by ICIs have been investigated for their contributions to therapeutic actions but might also contribute to differences in IRAEs when compared with CTLA4 haploinsufficiency (55). For example, ipilimumab and tremelimumab are IgG1 and IgG2 antibodies, respectively. IgG1 binds to multiple FcRs whereas IgG2 is thought to bind to FcγRIIB and the H131 isoform of FcγRIIA. Different subclasses might also account for the longer half of tremelimumab (22 days) relative to ipilimumab (14 days) (41, 43). Different Fc components might explain other differences as well. Mouse studies have shown FcR binding results in Treg depletion by interaction with tumour-infiltrating myeloid cells, which is crucial for their anti-tumour effects (56, 57). By contrast, CTLA4 haploinsufficiency results in deficiency of ligand binding independently of FcR ligation, which might result in differences in Treg depletion and other effects outside of the tumour environment. Differences in FcR-mediated actions might also identify pathology that is predominantly antibody-mediated. For example, hypophysitis is thought to result from complement activation by C1q binding to the Fc fragment of anti-CTLA4 antibody (37).
One major discrepancy between immune disorders in patients with CTLA4 haploinsufficiency and those receiving anti-CTLA4 treatment is the increased susceptibility to infection in CTLA4+/- patients. Most CTLA4+/- patients present with recurrent respiratory tract infections, which is thought to result from deficiency of B cells and immunoglobulin (1, 3). Serious infections appear to be less frequent in patients receiving immune checkpoint therapy. In one study, they were observed in 54/740 patients (7.3%). Furthermore, the contribution of ICIs to infection is confounded by concurrent immunosuppression to manage IRAEs. In one study, serious infections were observed in 13.5% of melanoma patients treated with either corticosteroids or infliximab but in only 2% in those who did not require immunosuppression (48).
Since LRBA competes with AP-1 for the YVKM motif on the cytoplasmic tail of CTLA4 to protect CTLA4 from lysosomal degradation (58), LRBA deficiency results in reduced CTLA4 expression and could therefore be informative for understanding the CTLA4+/- phenotype. Similar to CTLA4+/- patients, LRBA deficiency also confers increased risk of recurrent respiratory tract infections, and most patients with homozygous or biallelic mutations in LRBA are also diagnosed in early childhood with hypogammaglobulinemia (57%), B cell lymphopenia, particularly affecting memory B cells and plasmablasts (58–60), while heterozygous carriers are healthy. Most LRBA-/- patients have immune dysregulation encompassing enteropathy and hematological cytopenia (AIHA and ITP). Organomegaly, including splenomegaly and lymphadenopathy, is also prevalent, while T1D and hepatitis are less common (58–60). Two groups have reported that Lrba-/- mice do not have any sign of immunological disorders, either at steady state or after challenge with virus or bacterial infection. Another group reported that Lrba-/- mice are susceptible to DSS-induced colitis, although this phenotype was suggested to arise from dysregulation of TLR signaling rather than impaired CTLA4 expression (61–63). In mice, conventional B and T cell development does not appear to be affected by LRBA deletion, although peritoneal B1-a cells are reduced.
PD-1
In the 1990s, Honjo and colleagues discovered and characterised PD-1 as a negative T cell regulator (64–67). The therapeutic potential of PD-1 blockade in cancer therapy was illustrated in Pdcd1-/- mice and then confirmed in cancer patients after anti-PD-L1 treatment (68). PD-1 is expressed by T cells, NK cells, B cells and activated monocytes (69). PD-1 expression is considered to be a marker of cell exhaustion, and PD-1+ cells exhibit reduced cytokine production and reduced proliferative capacity (70–72). Nuclear factor of activated T cells, cytoplasmic 1 (NFATc1) and interferon-stimulated responsive element (ISRE) bind directly to the PD-1 promoter to upregulate PD-1 expression in response to TCR and IFN-α stimulation, respectively (73, 74), whereas T-bet and Blimp-1 suppress PD-1 expression (75, 76). Posttranslational modifications also regulate PD-1 expression, which provides potential novel avenues for PD-1-related therapy. Fucosylation of PD-1 at positions N49 and N74 by Fut8, a core fucosyltransferase, is required for cell-surface expression of PD-1 (77). Moreover, PD-1 is degraded in proteasome after Lys48-linked poly-ubiquitination by the E3 ubiquitin ligase, FBXO38. Deletion of FBXO38 leads to faster tumor progression with increased PD-1 expression in tumor-infiltrating T cells (78).
There are two PD-1 ligands. PD-L1 is expressed on T cells, B cells, dendritic cells, macrophages and non-hematopoietic cells, while PD-L2 is restricted to dendritic cells and macrophages (69). PD-L1 expression is upregulated in many tumors, including melanomas, non-small cell lung cancer and ovarian cancer (79–82). Upon IFN-γ stimulation, PD-L1 upregulation is mediated by various transcription factors, including IRF-1, MyD88, TRAF6 and MEK (83, 84). In addition, chimeric nucleophosmin (NPM) and anaplastic lymphoma kinase (ALK) induce the expression of STAT3, which upregulates PD-L1 expression (85). PD-L1 expression is also regulated post-transcriptionally. Glycogen synthase kinase 3β (GSK 3β) phosphorylates non-glycosylated PD-L1, leading to the proteasomal degradation by β-transducin repeat-containing protein (β-TrCP) (86), while COP9 signalosome 5 (CSN5) induced by NF-κB p65 (RelA) upon TNF-α stimulation inhibit ubiquitination and degradation of PD-L1 (87).
Ligation of PD-1 by PD-L1 results in transduction of a negative signal that suppresses T cell activation, cytokine production, survival and proliferation. LCK phosphorylates the immunoreceptor tyrosine-based switch motif of PD-1 cytoplasmic tail (88), recruiting Src homology region 2 domain containing phosphatase-1 (SHP-1) and SHP-2, which dephosphorylate many signaling molecules, including ZAP70/CD3zeta and ERK in TCR signaling pathway (88, 89), as well as PI3K/AKT/mTOR in CD28 signaling pathway (90–92). There is evidence that PD-1 binds preferentially to SHP-2 and dephosphorylates the CD28 cluster in the immune synapse (93). Additionally, ICOS co-stimulation of T cells for proliferation and cytokine production is also inhibited by PD-1 ligation (94). PD-1 also suppresses TCR-driven signal that stops cellular migration to increase the contact time of antigen-specific T cells with dendritic cells (70). The inhibitory function mediated by PD-1-PD-L1 ligation could also be indirect. In the absence of PD-1 ligation, TCR stimulation upregulates the expression of Ser/Thr protein kinase CK2, which stabilizes PTEN protein as a negative regulator of PI3K/Akt signaling pathway (95).
PDCD1 Deficiency
Recently, a rare, homozygous frameshift mutation (c.105dupC, p.T36Hfs*70) in PDCD1 was identified in a patient with tuberculosis and autoimmunity (96). The mutation was shown to abrogate PD-1 expression. The patient was diagnosed with type 1 diabetes (T1D), hypothyroidism and juvenile idiopathic arthritis (JIA) by the age of 3 years, developed large, multifocal intraperitoneal abscesses and abdominal tuberculosis by the age of 10 years, and died of pneumonitis one year later. Stimulated leukocytes from the patient exhibited reduced IFN-γ production. The numbers of Vδ2+ γδ T cells, mucosal-associated invariant T cells, and CD56bright natural killer cells were decreased, but helper T cell subsets were within normal range. Interestingly, the patient exhibited hepatosplenomegaly with expanded CD38+ activated and RORγT+ CD4- CD8- double-negative αβ T cells, similar to the phenotype displayed by STAT3 GOF patients, and STAT3-dependent cytokines IL-6 and IL-23 were increased (96) (Table 2).
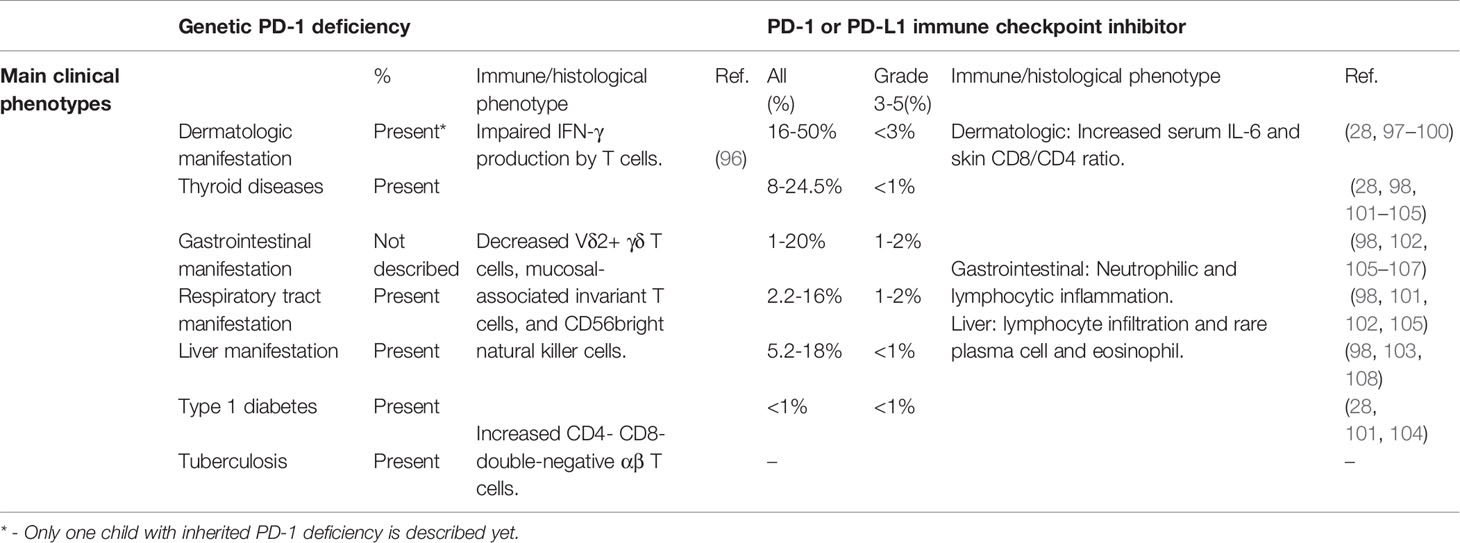
Table 2 Inborn errors of PDCD1 and their phenocopies.
About one third of Pdcd1-/- C57BL/6 mice develop arthritis and mild proliferative glomerulonephritis by 6 months of age, with extensive renal IgG3 and C3 deposition, and these manifestations become more frequent and severe over time (65). On a BALB/c background, only 30% of Pdcd1-/- mice survive to 40 weeks due to autoimmune myocarditis (66).
Phenocopies of PDCD1 Deficiency
IRAEs are less frequent in patients receiving ICIs directed against PD-1 and PD-L1 than with CTLA4 ICIs. Severe IRAEs are have been reported in up to 16.3% of melanoma patients and 10% of non-small-cell lung cancer patients treated with nivolumab (anti-PD-1) (97, 106, 107, 109, 110), 10.1-14.7% of melanoma patients and 9.5-26.6% of non-small-cell lung cancer patients treated with pembrolizumab (anti-PD-1) (28, 101–104), and 11-15% of non-small-cell lung cancer or metastatic urothelial cancer patients with atezolizumab (anti-PD-L1) (105, 111, 112). By contrast with IRAEs after ipilimumab, which appear to be dose-dependent, IRAEs with anti-PD-1/PD-L1 are independent of dose (53, 102, 113). The IRAEs related to anti-PD-1/PD-L1 treatment are mostly mild (28, 97, 105, 107, 110). Pembrolizumab and nivolumab bind to partially overlapping epitopes on PD-1, and both outcompete PD-L1 for binding to PD-1 due to their high affinity. Interestingly, both agents are human IgG4 antibodies, in which Fc regions exhibit low affinity for complement protein C1q and FcRs (114, 115).
Severe colitis has only been reported in 1-2% patients after anti-PD-1/PD-L1 treatment, which is much less frequent than after anti-CTLA4 therapy (98, 102, 105–107) (Table 2). Histology reveals both neutrophilic and lymphocytic inflammation (116). Respiratory IRAEs have been reported in 2.2-16% of patients after anti-PD-1/PD-L1 treatment. Severe pneumonitis was reported in 0.8-2% of patients treated with pembrolizumab (98, 101, 102, 105) (Table 2). Anti-PD-1/PD-L1 therapy related pneumonitis is more likely to occur in non-small cell lung cancer than melanoma and renal cell carcinoma (117). Cryptogenic organizing pneumonia is the main pattern in PD-1 inhibitor-related pneumonia, followed by nonspecific interstitial pneumonia (118).
Thyroiditis can present with either hypothyroidism or hyperthyroidism, and is observed in 8-24.5% patients after anti-PD-1/PD-L1 treatment. Severe hypothyroidism or hyperthyroidism occurs in less than 1% (28, 98, 101–105). Interestingly, individuals with pre-existing anti-thyroid autoantibodies are significantly more susceptible to thyroid IRAEs induced by PD-1 inhibitor (119, 120). T1D and adrenal insufficiency have been observed in about 1% of patients with pembrolizumab treatment (28, 101, 104) (Table 2).
Rash, pruritus and vitiligo occur in 16-50% of patients, while severe skin disorders occur in less than 3% (28, 97, 98) (Table 2). Patients with pre-existing autoantibodies and rheumatoid factor are more susceptible to skin IRAEs (99). In patients with pre-existing psoriasis, anti-PD-1 treatment has been shown to increase CD8+/CD4+ T cells ratio of infiltrating skin lymphocytes. The level of IL-6 but not IL-17A, IFN-γ and IL-8 in serum is significantly increased in cancer patients developing psoriasis-like dermatitis after anti-PD-1 treatment (100). Severe transaminitis occurs in 1% of patients after anti-PD-1 treatment (98, 103). Histology analysis indicates most patients exhibit the lobular inflammation with lymphocytic infiltrate and rare infiltration of plasma cells and eosinophils (108).
Comparison of PDCD1 Deficiency and Its Phenocopies
The spectrum and manifestations of IEIs of PDCD1 and phenocopies arising as IRAEs after PD-1 inhibitors overlap. PDCD1 deficiency (c.105dupC, T36Hfs*70) resulted in T1D and hypothyroidism at the age of 3 years, rash and stomatitis at the age of 11 years (96). A proteome-wide serum autoantibody profile revealed antibodies related to autoimmune thyroiditis and T1D (96).
Experimental models provide insight into these complications. First, PD-1-PD-L1 is critical to maintain intestinal tolerance and prevent experimental autoimmune enteritis. In a transgenic mouse model in which ovalbumin (OVA) was expressed as a neo-self-antigen by intestinal epithelial cells, either PD-L1 deletion and blockade resulted in significant weight loss and intestinal inflammation in mice transferred with OVA-specific CD8+ T cells (121). Similarly, in a model of intestinal injury, PD-L1-/- mice exhibited increased mortality and weight loss, diarrhea and rectal bleeding. PD-L1 expression on non-hematopoietic intestinal parenchyma prevented TNF-α production and conferred protection from intestinal inflammation. Interestingly, PD-L1-/- Rag-/- mice have a significantly higher death rate and morbidity than Rag-/- mice, indicating a contribution by innate immunity (122). Interestingly, however, gastrointestinal abnormalities were not reported in the patient with PDCD1 deficiency (96).
By contrast, destructive thyroiditis was observed in both the PDCD1-deficient patient and in cancer patients treated with anti-PD-1 antibodies. Furthermore, thyroid infiltration of PD-1+ T cells is observed in sporadic Graves’ disease (123), and a mouse thyroiditis model induced by thyroglobulin immunization is exaggerated by anti-PD-1 treatment, which is prevented bydeletion of CD4+ T cells. In this model, thyroid infiltrating CD4+ T cells acquire cytotoxic features and potentially kill thyrocytes via specific recognition of thyroglobulin antigen (120).
T1D is observed in cancer patients after anti-PD-1 treatment and occurred in the PDCD1- deficient patient. A 7146G/A polymorphism in PDCD1 gene has been reported to confer significantly increased susceptibility to T1D (124). T1D is accelerated and completely penetrant after PD-1 deletion in NOD mice, which appears to result in enhanced T cells infiltration of β-islets, with increased IFN-γ production (125) although autoantibodies against insulin were not increased compared with WT NOD mice (125). Deficiencies of PD-L1, PD-L- and PD-L1 have all been shown to accelerate development of diabetes in NOD mice. Pancreatic lymph nodes from Pdl1-/- Pdl2-/- NOD mice also have more IFN-γ and TNF-α producing T cells. Interestingly, PD-L1/PD-L2 expression on nonlymphoid cells is sufficient to control the progression of autoimmune diabetes (126).
Finally, the PDCD1 patient had dermatitis (96). Consistently, mice with Pdcd1 deletion on CD8+ T cells are more susceptible to psoriasis-like dermatitis induced by imiquimod (R848, a toll-like receptor 7/8 agonist), which is ameliorated by anti-IL-6 receptor blockade (100). In another contact hypersensitivity mouse model induced by hapten, PD-1 deletion and anti-PD-L1 treatment also lead to enhanced skin infiltration by CD8+ T cells (127).
Conclusions
IRAEs observed in patients treated with antibodies targeted at CTLA4 and PD-1 and the receptors of their ligands phenocopy the autoimmune manifestations of patients with IEIs of CTLA4 and PDCD1. There are differences in the spectrum of autoimmune manifestations between IEIs and IRAEs, such as hypophysitis. ICIs phenocopy defects in ligand binding, but also result in FcR ligation. This difference merits further investigation as a possible cause of phenotypic discrepancies between IEIs and their phenocopies. Another important observation, however, is that analysis of IEIs and their therapeutic phenocopies suggest that humans are more dependent on checkpoint inhibition than mice for protection against autoimmunity and inflammation.
Clinical manifestations arise in the majority of patients with CTLA4 haploinsufficiency. By contrast, lymphocytic pathology arises only in mice homozygous for Ctla4 deletion. This suggests that in addition to the genetic defect in CTLA4 in its IEI, or after CTLA4 blockade, additional factors may promote inflammatory pathology in humans that do not act in the mouse model, at least not in the specific pathogen free environments in which experimental mice are maintained. This conclusion is supported by observations with IEIs arising from LRBA deficiency, which results in a reduction in CTLA4 expression. LRBA deficiency often results limited autoimmunity in humans, whereas Lrba-deficient mice are either healthy, or exhibit inflammation only after substantial environmental stress (e.g. DSS administration), which might provide further evidence that humans are more sensitive to changes in CTLA4 expression than mice.
The other significant phenotypic discrepancy is immune deficiency. These are common in IEI affecting CTLA4, but are not observed as IRAEs, nor do they feature in mouse models of Ctla4 deficiency. LRBA mutations also result in hypogammaglobulinaemia and B cell deficiencies. Based on the distribution of CTLA4, LRBA and PD-1 expression, the effect is unlikely to be cell-intrinsic to the B cell compartment, but this will need to be resolved empirically. In some patients, CD21low B cells, which are said to be exhausted, have been observed to be expanded in patients with CTLA4 deficiency, and this population has been simultaneously used to explain the hypogammaglobulinemia, and the increased incidence of autoantibodies (128, 129). While antibody deficiency has been observed in humans with LRBA deficiency, and this might reinforce the evidence that CTLA4 deficiency causes antibody deficiency, other potential mechanisms, most notably B cell intrinsic defects in autophagy could explain this phenotype.
Further work will be required to resolve this fascinating discrepancy. Possible contributions include the age of onset of the defect in immune regulation, which is of course congenital with IEIs abut is often only encountered much later with ICIs. The magnitude of inhibition may also be important, since, as described above, there is a dose-response effect of observed with CTLA4 inhibition. Finally, it is plausible that microbiological challenge from infection may exacerbate the phenotype in humans, and that this could be less apparent in mice maintained under specific pathogen-free conditions.
Author Contributions
Both authors contributed to the conceptualization and drafting of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
National Health and Medical Research Council, Australia, GNT1113577 Cancer Australia, GNT1130330.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, et al. Autosomal Dominant Immune Dysregulation Syndrome in Humans With CTLA4 Mutations. Nat Med (2014) 20:1410–6. doi: 10.1038/nm.3746
2. Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT, et al. Immune Dysregulation in Human Subjects With Heterozygous Germline Mutations in CTLA4. Science (2014) 345:1623–7. doi: 10.1126/science.1255904
3. Schwab C, Gabrysch A, Olbrich P, Patiño V, Warnatz K, Wolff D, et al. Phenotype, Penetrance, and Treatment of 133 Cytotoxic T-Lymphocyte Antigen 4 Insufficient Subjects. J Allergy Clin Immunol (2018) 142:1932–46. doi: 10.1016/j.jaci.2018.02.055
4. Boutros C, Tarhini A, Routier E, Lambotte O, Ladurie FL, Carbonnel F, et al. Safety Profiles of Anti-CTLA-4 and Anti-PD-1 Antibodies Alone and in Combination. Nat Rev Clin Oncol (2016) 13:473–86. doi: 10.1038/nrclinonc.2016.58
5. Brunet J-F, Denizot F, Luciani M-F, Roux-Dosseto M, Suzan M, Mattei M-G, et al. A New Member of the Immunoglobulin Superfamily—CTLA4-4. Nature (1987) 328:267–70. doi: 10.1038/328267a0
6. Linsley PS, Greene JL, Tan P, Bradshaw J, Ledbetter JA, Anasetti C, et al. Coexpression and Functional Cooperation of CTLA-4 and CD28 on Activated T Lymphocytes. J Exp Med (1992) 176:1595–604. doi: 10.1084/jem.176.6.1595
7. Green JM, Noel PJ, Sperling AI, Walunas TL, Gray GS, Bluestone JA, et al. Absence of B7-Dependent Responses in CD28-Deficient Mice. Immunity (1994) 1:501–8. doi: 10.1016/1074-7613(94)90092-2
8. Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, et al. CTLA-4 Can Function as a Negative Regulator of T Cell Activation. Immunity (1994) 1:405–13. doi: 10.1016/1074-7613(94)90071-x
9. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 Leads to Massive Lymphoproliferation and Fatal Multiorgan Tissue Destruction, Revealing a Critical Negative Regulatory Role of CTLA-4. Immunity (1995) 3:541–7. doi: 10.1016/1074-7613(95)90125-6
10. Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative Disorders With Early Lethality in Mice Deficient in CTLA-4. Science (1995) 270:985–8. doi: 10.1126/science.270.5238.985
11. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 Control Over Foxp3+ Regulatory T Cell Function. Science (2008) 322:271–5. doi: 10.1126/science.1160062
12. Gibson HM, Hedgcock CJ, Aufiero BM, Wilson AJ, Hafner MS, Tsokos GC, et al. Induction of the CTLA-4 Gene in Human Lymphocytes Is Dependent on NFAT Binding the Proximal Promoter. J Immunol (2007) 179:3831–40. doi: 10.4049/jimmunol.179.6.3831
13. Quandt D, Hoff H, Rudolph M, Fillatreau S, Brunner-Weinzierl MC. A New Role of CTLA-4 on B Cells in Thymus-Dependent Immune Responses In Vivo. J Immunol (2007) 179:7316. doi: 10.4049/jimmunol.179.11.7316
14. Kaufman KA, Bowen JA, Tsai AF, Bluestone JA, Hunt JS, Ober C. The CTLA-4 Gene Is Expressed in Placental Fibroblasts. Mol Hum Reprod (1999) 5:84–7. doi: 10.1093/molehr/5.1.84
15. Pistillo MP, Tazzari PL, Palmisano GL, Pierri I, Bolognesi A, Ferlito F, et al. CTLA-4 Is Not Restricted to the Lymphoid Cell Lineage and Can Function as a Target Molecule for Apoptosis Induction of Leukemic Cells. Blood (2003) 101:202–9. doi: 10.1182/blood-2002-06-1668
16. Oyewole-Said D, Konduri V, Vazquez-Perez J, Weldon SA, Levitt JM, Decker WK. Beyond T-Cells: Functional Characterization of CTLA-4 Expression in Immune and Non-Immune Cell Types. Front Immunol (2020) 11:608024. doi: 10.3389/fimmu.2020.608024
17. Do P, Beckwith KA, Cheney C, Tran M, Beaver L, Griffin BG, et al. Leukemic B Cell CTLA-4 Suppresses Costimulation of T Cells. J Immunol (2019) 202:2806. doi: 10.4049/jimmunol.1801359
18. Mo X, Zhang H, Preston S, Martin K, Zhou B, Vadalia N, et al. Interferon-γ Signaling in Melanocytes and Melanoma Cells Regulates Expression of CTLA-4. Cancer Res (2017) 78:canres.1615.2017. doi: 10.1158/0008-5472.CAN-17-1615
19. Collins AV, Brodie DW, Gilbert RJC, Iaboni A, Manso-Sancho R, Walse B, et al. The Interaction Properties of Costimulatory Molecules Revisited. Immunity (2002) 17:201–10. doi: 10.1016/S1074-7613(02)00362-X
20. Yokosuka T, Kobayashi W, Takamatsu M, Sakata-Sogawa K, Zeng H, Hashimoto-Tane A, et al. Spatiotemporal Basis of CTLA-4 Costimulatory Molecule-Mediated Negative Regulation of T Cell Activation. Immunity (2010) 33:326–39. doi: 10.1016/j.immuni.2010.09.006
21. Chuang E, Fisher TS, Morgan RW, Robbins MD, Duerr JM, Vander Heiden MG, et al. The CD28 and CTLA-4 Receptors Associate With the Serine/Threonine Phosphatase Pp2a. Immunity (2000) 13:313–22. doi: 10.1016/S1074-7613(00)00031-5
22. Schneider H, Mandelbrot DA, Greenwald RJ, Ng F, Lechler R, Sharpe AH, et al. Cutting Edge: CTLA-4 (CD152) Differentially Regulates Mitogen-Activated Protein Kinases (Extracellular Signal-Regulated Kinase and C-Jun N-Terminal Kinase) in CD4+ T Cells From Receptor/Ligand-Deficient Mice. J Immunol (2002) 169:3475. doi: 10.4049/jimmunol.169.7.3475
23. Lee K-M, Chuang E, Griffin M, Khattri R, Hong DK, Zhang W, et al. Molecular Basis of T Cell Inactivation by CTLA-4. Science (1998) 282:2263. doi: 10.1126/science.282.5397.226
24. Masteller EL, Chuang E, Mullen AC, Reiner SL, Thompson CB. Structural Analysis of CTLA-4 Function. J Immunol (2000) 164:5319. doi: 10.4049/jimmunol.164.10.5319
25. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-Endocytosis of CD80 and CD86: A Molecular Basis for the Cell-Extrinsic Function of CTLA-4. Science (2011) 332:600–3. doi: 10.1126/science.1202947
26. Schneider H, Downey J, Smith A, Zinselmeyer BH, Rush C, Brewer JM, et al. Reversal of the TCR Stop Signal by CTLA-4. Science (2006) 313:1972. doi: 10.1126/science.1131078
27. Kong KF, Fu G, Zhang Y, Yokosuka T, Casas J, Canonigo-Balancio AJ, et al. Protein Kinase C-η Controls CTLA-4-Mediated Regulatory T Cell Function. Nat Immunol (2014) 15:465–72. doi: 10.1038/ni.2866
28. Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab Versus Ipilimumab in Advanced Melanoma. N Engl J Med (2015) 372:2521–32. doi: 10.1056/NEJMoa1503093
29. Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, et al. Nivolumab and Ipilimumab Versus Ipilimumab in Untreated Melanoma. N Engl J Med (2015) 372:2006–17. doi: 10.1056/NEJMoa1414428
30. Tarhini AA, Cherian J, Moschos SJ, Tawbi HA, Shuai Y, Gooding WE, et al. Safety and Efficacy of Combination Immunotherapy With Interferon Alfa-2b and Tremelimumab in Patients With Stage IV Melanoma. J Clin Oncol (2011) 30:322–8. doi: 10.1200/JCO.2011.37.5394
31. Michot JM, Lazarovici J, Tieu A, Champiat S, Voisin AL, Ebbo M, et al. Haematological Immune-Related Adverse Events With Immune Checkpoint Inhibitors, How to Manage? Eur J Cancer (2019) 122:72–90. doi: 10.1016/j.ejca.2019.07.014
32. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved Survival With Ipilimumab in Patients With Metastatic Melanoma. N Engl J Med (2010) 363:711–23. doi: 10.1056/NEJMoa1003466
33. Eggermont AMM, Chiarion-Sileni V, Grob J-J, Dummer R, Wolchok JD, Schmidt H, et al. Adjuvant Ipilimumab Versus Placebo After Complete Resection of High-Risk Stage III Melanoma (Eortc 18071): A Randomised, Double-Blind, Phase 3 Trial. Lancet Oncol (2015) 16:522–30. doi: 10.1016/S1470-2045(15)70122-1
34. Beck KE, Blansfield JA, Tran KQ, Feldman AL, Hughes MS, Royal RE, et al. Enterocolitis in Patients With Cancer After Antibody Blockade of Cytotoxic T-Lymphocyte-Associated Antigen 4. J Clin Oncol (2006) 24:2283–9. doi: 10.1200/JCO.2005.04.5716
35. Ribas A, Kefford R, Marshall MA, Punt CJA, Haanen JB, Marmol M, et al. Phase III Randomized Clinical Trial Comparing Tremelimumab With Standard-of-Care Chemotherapy in Patients With Advanced Melanoma. J Clin Oncol (2013) 31:616–22. doi: 10.1200/JCO.2012.44.6112
36. Maio M, Scherpereel A, Calabrò L, Aerts J, Perez SC, Bearz A, et al. Tremelimumab as Second-Line or Third-Line Treatment in Relapsed Malignant Mesothelioma (Determine): A Multicentre, International, Randomised, Double-Blind, Placebo-Controlled Phase 2b Trial. Lancet Oncol (2017) 18:1261–73. doi: 10.1016/s1470-2045(17)30446-1
37. Iwama S, De Remigis A, Callahan MK, Slovin SF, Wolchok JD, Caturegli P. Pituitary Expression of CTLA-4 Mediates Hypophysitis Secondary to Administration of CTLA-4 Blocking Antibody. Sci Transl Med (2014) 6:230ra245. doi: 10.1126/scitranslmed.3008002
38. Chambers CA, Sullivan TJ, Allison JP. Lymphoproliferation in CTLA-4–Deficient Mice Is Mediated by Costimulation-Dependent Activation of CD4+ T Cells. Immunity (1997) 7:885–95. doi: 10.1016/S1074-7613(00)80406-9
39. Leach DR, Krummel MF, Allison JP. Enhancement of Antitumor Immunity by CTLA-4 Blockade. Science (1996) 271:1734. doi: 10.1126/science.271.5256.1734
40. Krummel MF, Allison JP. CD28 and CTLA-4 Have Opposing Effects on the Response of T Cells to Stimulation. J Exp Med (1995) 182:459–65. doi: 10.1084/jem.182.2.459
41. He M, Chai Y, Qi J, Zhang CWH, Tong Z, Shi Y, et al. Remarkably Similar CTLA-4 Binding Properties of Therapeutic Ipilimumab and Tremelimumab Antibodies. Oncotarget (2017) 8:67129–39. doi: 10.18632/oncotarget.18004
42. Ramagopal UA, Liu W, Garrett-Thomson SC, Bonanno JB, Yan Q, Srinivasan M, et al. Structural Basis for Cancer Immunotherapy by the First-in-Class Checkpoint Inhibitor Ipilimumab. Proc Natl Acad Sci USA (2017) 114:E4223. doi: 10.1073/pnas.1617941114
43. Callahan MK, Wolchok JD. At the Bedside: CTLA-4- and PD-1-Blocking Antibodies in Cancer Immunotherapy. J Leukoc Biol (2013) 94:41–53. doi: 10.1189/jlb.1212631
44. Robert C, Thomas L, Bondarenko I, O'Day S, Weber J, Garbe C, et al. Ipilimumab Plus Dacarbazine for Previously Untreated Metastatic Melanoma. N Engl J Med (2011) 364:2517–26. doi: 10.1056/NEJMoa1104621
45. Ribas A, Wolchok JD. Cancer Immunotherapy Using Checkpoint Blockade. Science (2018) 359:1350–5. doi: 10.1126/science.aar4060
46. Ueda H, Howson JM, Esposito L, Heward J, Snook H, Chamberlain G, et al. Association of the T-Cell Regulatory Gene CTLA4 With Susceptibility to Autoimmune Disease. Nature (2003) 423:506–11. doi: 10.1038/nature01621
47. Patel H, Mansuri MS, Singh M, Begum R, Shastri M, Misra A. Association of Cytotoxic T-Lymphocyte Antigen 4 (CTLA4) and Thyroglobulin (Tg) Genetic Variants With Autoimmune Hypothyroidism. PLoS One (2016) 11:e0149441. doi: 10.1371/journal.pone.0149441
48. Del Castillo M, Romero FA, Argüello E, Kyi C, Postow MA, Redelman-Sidi G. The Spectrum of Serious Infections Among Patients Receiving Immune Checkpoint Blockade for the Treatment of Melanoma. Clin Infect Dis (2016) 63:1490–3. doi: 10.1093/cid/ciw539
49. Klein O, Ebert LM, Nicholaou T, Browning J, Russell SE, Zuber M, et al. Melan-A–Specific Cytotoxic T Cells Are Associated With Tumor Regression and Autoimmunity Following Treatment With Anti-CTLA-4. Clin Cancer Res (2009) 15:2507. doi: 10.1158/1078-0432.CCR-08-2424
50. Attia P, Phan GQ, Maker AV, Robinson MR, Quezado MM, Yang JC, et al. Autoimmunity Correlates With Tumor Regression in Patients With Metastatic Melanoma Treated With Anti-Cytotoxic T-Lymphocyte Antigen-4. J Clin Oncol (2005) 23:6043–53. doi: 10.1200/jco.2005.06.205
51. Sanderson K, Scotland R, Lee P, Liu D, Groshen S, Snively J, et al. Autoimmunity in a Phase I Trial of a Fully Human Anti-Cytotoxic T-Lymphocyte Antigen-4 Monoclonal Antibody With Multiple Melanoma Peptides and Montanide Isa 51 for Patients With Resected Stages III and Iv Melanoma. J Clin Oncol (2005) 23:741–50. doi: 10.1200/JCO.2005.01.128
52. Liu P, He Y, Wang H, Kuang Y, Chen W, Li J, et al. The Expression of mCTLA-4 in Skin Lesion Inversely Correlates With the Severity of Psoriasis. J Dermatol Sci (2018) 89:233–40. doi: 10.1016/j.jdermsci.2017.11.007
53. Wolchok JD, Neyns B, Linette G, Negrier S, Lutzky J, Thomas L, et al. Ipilimumab Monotherapy in Patients With Pretreated Advanced Melanoma: A Randomised, Double-Blind, Multicentre, Phase 2, Dose-Ranging Study. Lancet Oncol (2010) 11:155–64. doi: 10.1016/S1470-2045(09)70334-1
54. Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, Sakaguchi N, et al. Immunologic Self-Tolerance Maintained by CD25+ CD4+ Regulatory T Cells Constitutively Expressing Cytotoxic T Lymphocyte-Associated Antigen 4. J Exp Med (2000) 192:303–10. doi: 10.1084/jem.192.2.303
55. Furness AJ, Vargas FA, Peggs KS, Quezada SA. Impact of Tumour Microenvironment and Fc Receptors on the Activity of Immunomodulatory Antibodies. Trends Immunol (2014) 35:290–8. doi: 10.1016/j.it.2014.05.002
56. Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, et al. Fc-Dependent Depletion of Tumor-Infiltrating Regulatory T Cells Co-Defines the Efficacy of Anti-CTLA-4 Therapy Against Melanoma. J Exp Med (2013) 210:1695–710. doi: 10.1084/jem.20130579
57. Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, et al. Anti-CTLA-4 Antibodies of IgG2a Isotype Enhance Antitumor Activity Through Reduction of Intratumoral Regulatory T Cells. Cancer Immunol Res (2013) 1:32–42. doi: 10.1158/2326-6066.Cir-13-0013
58. Lo B, Zhang K, Lu W, Zheng L, Zhang Q, Kanellopoulou C, et al. Patients With LRBA Deficiency Show CTLA4 Loss and Immune Dysregulation Responsive to Abatacept Therapy. Science (2015) 349:436–40. doi: 10.1126/science.aaa1663
59. Gámez-Díaz L, August D, Stepensky P, Revel-Vilk S, Seidel MG, Noriko M, et al. The Extended Phenotype of Lps-Responsive Beige-Like Anchor Protein (LRBA) Deficiency. J Allergy Clin Immunol (2016) 137:223–30. doi: 10.1016/j.jaci.2015.09.025
60. Lopez-Herrera G, Tampella G, Pan-Hammarström Q, Herholz P, Trujillo-Vargas CM, Phadwal K, et al. Deleterious Mutations in LRBA Are Associated With a Syndrome of Immune Deficiency and Autoimmunity. Am J Hum Genet (2012) 90:986–1001. doi: 10.1016/j.ajhg.2012.04.015
61. Gámez-Díaz L, Neumann J, Jäger F, Proietti M, Felber F, Soulas-Sprauel P, et al. Immunological Phenotype of the Murine LRBA Knockout. Immunol Cell Biol (2017) 95:789. doi: 10.1038/icb.2017.52
62. Burnett DL, Parish IA, Masle-Farquhar E, Brink R, Goodnow CC. Murine LRBA Deficiency Causes CTLA-4 Deficiency in Tregs Without Progression to Immune Dysregulation. Immunol Cell Biol (2017) 95:775–88. doi: 10.1038/icb.2017.50
63. Wang K-w, Zhan X, McAlpine W, Zhang Z, Choi JH, Shi H, et al. Enhanced Susceptibility to Chemically Induced Colitis Caused by Excessive Endosomal TLR Signaling in LRBA-Deficient Mice. Proc Natl Acad Sci USA (2019) 116:11380. doi: 10.1073/pnas.1901407116
64. Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubat T, Yagita H, et al. Expression of the PD-1 Antigen on the Surface of Stimulated Mouse T and B Lymphocytes. Int Immunol (1996) 8:765–72. doi: 10.1093/intimm/8.5.765
65. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of Lupus-Like Autoimmune Diseases by Disruption of the PD-1 Gene Encoding an ITIM Motif-Carrying Immunoreceptor. Immunity (1999) 11:141–51. doi: 10.1016/s1074-7613(00)80089-8
66. Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, et al. Autoimmune Dilated Cardiomyopathy in PD-1 Receptor-Deficient Mice. Science (2001) 291:319. doi: 10.1126/science.291.5502.319
67. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced Expression of PD-1, a Novel Member of the Immunoglobulin Gene Superfamily, Upon Programmed Cell Death. EMBO J (1992) 11:3887–95. doi: 10.1002/j.1460-2075.1992.tb05481.x
68. Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on Tumor Cells in the Escape From Host Immune System and Tumor Immunotherapy by PD-L1 Blockade. Proc Natl Acad Sci USA (2002) 99:12293–7. doi: 10.1073/pnas.192461099
69. Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The Function of Programmed Cell Death 1 and Its Ligands in Regulating Autoimmunity and Infection. Nat Immunol (2007) 8:239–45. doi: 10.1038/ni1443
70. Fife BT, Pauken KE, Eagar TN, Obu T, Wu J, Tang Q, et al. Interactions Between PD-1 and PD-L1 Promote Tolerance by Blocking the TCR-Induced Stop Signal. Nat Immunol (2009) 10:1185–92. doi: 10.1038/ni.1790
71. Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, et al. PD-1 Expression on HIV-Specific T Cells Is Associated With T-Cell Exhaustion and Disease Progression. Nature (2006) 443:350–4. doi: 10.1038/nature05115
72. Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, et al. Upregulation of Tim-3 and PD-1 Expression Is Associated With Tumor Antigen-Specific CD8+ T Cell Dysfunction in Melanoma Patients. J Exp Med (2010) 207:2175–86. doi: 10.1084/jem.20100637
73. Oestreich KJ, Yoon H, Ahmed R, Boss JM. NFATc1 Regulates PD-1 Expression Upon T Cell Activation. J Immunol (2008) 181:4832. doi: 10.4049/jimmunol.181.7.4832
74. Terawaki S, Chikuma S, Shibayama S, Hayashi T, Yoshida T, Okazaki T, et al. IFN-α Directly Promotes Programmed Cell Death-1 Transcription and Limits the Duration of T Cell-Mediated Immunity. J Immunol (2011) 186:2772. doi: 10.4049/jimmunol.1003208
75. Kao C, Oestreich KJ, Paley MA, Crawford A, Angelosanto JM, Ali M-AA, et al. Transcription Factor T-Bet Represses Expression of the Inhibitory Receptor PD-1 and Sustains Virus-Specific CD8+ T Cell Responses During Chronic Infection. Nat Immunol (2011) 12:663–71. doi: 10.1038/ni.2046
76. Lu P, Youngblood BA, Austin JW, Rasheed Mohammed AU, Butler R, Ahmed R, et al. Blimp-1 Represses CD8 T Cell Expression of PD-1 Using a Feed-Forward Transcriptional Circuit During Acute Viral Infection. J Exp Med (2014) 211:515–27. doi: 10.1084/jem.20130208
77. Okada M, Chikuma S, Kondo T, Hibino S, Machiyama H, Yokosuka T, et al. Blockage of Core Fucosylation Reduces Cell-Surface Expression of PD-1 and Promotes Anti-Tumor Immune Responses of T Cells. Cell Rep (2017) 20:1017–28. doi: 10.1016/j.celrep.2017.07.027
78. Meng X, Liu X, Guo X, Jiang S, Chen T, Hu Z, et al. FBXO38 Mediates PD-1 Ubiquitination and Regulates Anti-Tumour Immunity of T Cells. Nature (2018) 564:130–5. doi: 10.1038/s41586-018-0756-0
79. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-Associated B7-H1 Promotes T-Cell Apoptosis: A Potential Mechanism of Immune Evasion. Nat Med (2002) 8:793–800. doi: 10.1038/nm730
80. Konishi J, Yamazaki K, Azuma M, Kinoshita I, Dosaka-Akita H, Nishimura M. B7-H1 Expression on Non-Small Cell Lung Cancer Cells and Its Relationship With Tumor-Infiltrating Lymphocytes and Their PD-1 Expression. Clin Cancer Res (2004) 10:5094. doi: 10.1158/1078-0432.CCR-04-0428
81. Hamanishi J, Mandai M, Iwasaki M, Okazaki T, Tanaka Y, Yamaguchi K, et al. Programmed Cell Death 1 Ligand 1 and Tumor-Infiltrating CD8+ T Lymphocytes are Prognostic Factors of Human Ovarian Cancer. Proc Natl Acad Sci USA (2007) 104:3360–5. doi: 10.1073/pnas.0611533104
82. Ohigashi Y, Sho M, Yamada Y, Tsurui Y, Hamada K, Ikeda N, et al. Clinical Significance of Programmed Death-1 Ligand-1 and Programmed Death-1 Ligand-2 Expression in Human Esophageal Cancer. Clin Cancer Res (2005) 11:2947. doi: 10.1158/1078-0432.CCR-04-1469
83. Lee SJ, Jang BC, Lee SW, Yang YI, Suh SI, Park YM, et al. Interferon Regulatory Factor-1 Is Prerequisite to the Constitutive Expression and IFN-Gamma-Induced Upregulation of B7-H1 (Cd274). FEBS Lett (2006) 580:755–62. doi: 10.1016/j.febslet.2005.12.093
84. Liu J, Hamrouni A, Wolowiec D, Coiteux V, Kuliczkowski K, Hetuin D, et al. Plasma Cells From Multiple Myeloma Patients Express B7-H1 (PD-L1) and Increase Expression After Stimulation With IFN-{Gamma} and TLR Ligands via a MyD88-, TRAF6-, and MEK-Dependent Pathway. Blood (2007) 110:296–304. doi: 10.1182/blood-2006-10-051482
85. Marzec M, Zhang Q, Goradia A, Raghunath PN, Liu X, Paessler M, et al. Oncogenic Kinase NPM/ALK Induces Through STAT3 Expression of Immunosuppressive Protein CD274 (PD-L1, B7-H1). Proc Natl Acad Sci USA (2008) 105:20852–7. doi: 10.1073/pnas.0810958105
86. Li C-W, Lim S-O, Xia W, Lee H-H, Chan L-C, Kuo C-W, et al. Glycosylation and Stabilization of Programmed Death Ligand-1 Suppresses T-Cell Activity. Nat Commun (2016) 7:12632. doi: 10.1038/ncomms12632
87. Lim S-O, Li C-W, Xia W, Cha J-H, Chan L-C, Wu Y, et al. Deubiquitination and Stabilization of PD-L1 by CSN5. Cancer Cell (2016) 30:925–39. doi: 10.1016/j.ccell.2016.10.010
88. Sheppard KA, Fitz LJ, Lee JM, Benander C, George JA, Wooters J, et al. PD-1 Inhibits T-Cell Receptor Induced Phosphorylation of the ZAP70/CD3zeta Signalosome and Downstream Signaling to PKCtheta. FEBS Lett (2004) 574:37–41. doi: 10.1016/j.febslet.2004.07.083
89. Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed Cell Death 1 Forms Negative Costimulatory Microclusters That Directly Inhibit T Cell Receptor Signaling by Recruiting Phosphatase SHP2. J Exp Med (2012) 209:1201–17. doi: 10.1084/jem.20112741
90. Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 Associate With Immunoreceptor Tyrosine-Based Switch Motif of Programmed Death 1 Upon Primary Human T Cell Stimulation, But Only Receptor Ligation Prevents T Cell Activation. J Immunol (2004) 173:945. doi: 10.4049/jimmunol.173.2.945
91. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 Receptors Inhibit T-Cell Activation by Distinct Mechanisms. Mol Cell Biol (2005) 25:9543–53. doi: 10.1128/mcb.25.21.9543-9553.2005
92. Kleffel S, Posch C, Barthel SR, Mueller H, Schlapbach C, Guenova E, et al. Melanoma Cell-Intrinsic PD-1 Receptor Functions Promote Tumor Growth. Cell (2015) 162:1242–56. doi: 10.1016/j.cell.2015.08.052
93. Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T Cell Costimulatory Receptor CD28 Is a Primary Target for PD-1-Mediated Inhibition. Science (2017) 355:1428–33. doi: 10.1126/science.aaf1292
94. Bennett F, Luxenberg D, Ling V, Wang IM, Marquette K, Lowe D, et al. Program Death-1 Engagement Upon TCR Activation Has Distinct Effects on Costimulation and Cytokine-Driven Proliferation: Attenuation of ICOS, IL-4, and IL-21, But Not CD28, IL-7, and IL-15 Responses. J Immunol (2003) 170:711. doi: 10.4049/jimmunol.170.2.711
95. Patsoukis N, Li L, Sari D, Petkova V, Boussiotis VA. PD-1 Increases PTEN Phosphatase Activity While Decreasing PTEN Protein Stability by Inhibiting Casein Kinase 2. Mol Cell Biol (2013) 33:3091–8. doi: 10.1128/mcb.00319-13
96. Ogishi M, Yang R, Aytekin C, Langlais D, Bourgey M, Khan T, et al. Inherited PD-1 Deficiency Underlies Tuberculosis and Autoimmunity in a Child. Nat Med (2021) 27:1646–54. doi: 10.1038/s41591-021-01388-5
97. Weber JS, D'Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, et al. Nivolumab Versus Chemotherapy in Patients With Advanced Melanoma Who Progressed After Anti-CTLA-4 Treatment (Checkmate 037): A Randomised, Controlled, Open-Label, Phase 3 Trial. Lancet Oncol (2015) 16:375–84. doi: 10.1016/S1470-2045(15)70076-8
98. Hamid O, Robert C, Daud A, Hodi FS, Hwu W-J, Kefford R, et al. Safety and Tumor Responses With Lambrolizumab (Anti–PD-1) in Melanoma. N Engl J Med (2013) 369:134–44. doi: 10.1056/NEJMoa1305133
99. Toi Y, Sugawara S, Sugisaka J, Ono H, Kawashima Y, Aiba T, et al. Profiling Preexisting Antibodies in Patients Treated With Anti-PD-1 Therapy for Advanced Non-Small Cell Lung Cancer. JAMA Oncol (2019) 5:376–83. doi: 10.1001/jamaoncol.2018.5860
100. Tanaka R, Ichimura Y, Kubota N, Saito A, Nakamura Y, Ishitsuka Y, et al. Activation of CD8 T Cells Accelerates Anti-PD-1 Antibody-Induced Psoriasis-Like Dermatitis Through IL-6. Commun Biol (2020) 3:571. doi: 10.1038/s42003-020-01308-2
101. Eggermont AMM, Blank CU, Mandala M, Long GV, Atkinson V, Dalle S, et al. Adjuvant Pembrolizumab Versus Placebo in Resected Stage Iii Melanoma. N Engl J Med (2018) 378:1789–801. doi: 10.1056/NEJMoa1802357
102. Herbst RS, Baas P, Kim D-W, Felip E, Pérez-Gracia JL, Han J-Y, et al. Pembrolizumab Versus Docetaxel for Previously Treated, PD-L1-Positive, Advanced Non-Small-Cell Lung Cancer (Keynote-010): A Randomised Controlled Trial. Lancet (2016) 387:1540–50. doi: 10.1016/S0140-6736(15)01281-7
103. Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the Treatment of Non–Small-Cell Lung Cancer. N Engl J Med (2015) 372:2018–28. doi: 10.1056/NEJMoa1501824
104. Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csőszi T, Fülöp A, et al. Pembrolizumab Versus Chemotherapy for PD-L1–positive Non–Small-Cell Lung Cancer. N Engl J Med (2016) 375:1823–33. doi: 10.1056/NEJMoa1606774
105. Fehrenbacher L, Spira A, Ballinger M, Kowanetz M, Vansteenkiste J, Mazieres J, et al. Atezolizumab Versus Docetaxel for Patients With Previously Treated Non-Small-Cell Lung Cancer (Poplar): A Multicentre, Open-Label, Phase 2 Randomised Controlled Trial. Lancet (2016) 387:1837–46. doi: 10.1016/S0140-6736(16)00587-0
106. Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in Previously Untreated Melanoma Without Braf Mutation. N Engl J Med (2014) 372:320–30. doi: 10.1056/NEJMoa1412082
107. Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WE, Poddubskaya E, et al. Nivolumab Versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N Engl J Med (2015) 373:123–35. doi: 10.1056/NEJMoa1504627
108. Zhang D, Hart J, Ding X, Zhang X, Feely M, Yassan L, et al. Histologic Patterns of Liver Injury Induced by Anti-PD-1 Therapy. Gastroenterol Rep (2020) 8:50–5. doi: 10.1093/gastro/goz044
109. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med (2015) 373:23–34. doi: 10.1056/NEJMoa1504030
110. Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab Versus Docetaxel in Advanced Nonsquamous Non–Small-Cell Lung Cancer. N Engl J Med (2015) 373:1627–39. doi: 10.1056/NEJMoa1507643
111. Galsky MD, Arija JÁA, Bamias A, Davis ID, De Santis M, Kikuchi E, et al. Atezolizumab With or Without Chemotherapy in Metastatic Urothelial Cancer (Imvigor130): A Multicentre, Randomised, Placebo-Controlled Phase 3 Trial. Lancet (2020) 395:1547–57. doi: 10.1016/S0140-6736(20)30230-0
112. Rittmeyer A, Barlesi F, Waterkamp D, Park K, Ciardiello F, von Pawel J, et al. Atezolizumab Versus Docetaxel in Patients With Previously Treated Non-Small-Cell Lung Cancer (Oak): A Phase 3, Open-Label, Multicentre Randomised Controlled Trial. Lancet (2017) 389:255–65. doi: 10.1016/S0140-6736(16)32517-X
113. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer. N Engl J Med (2012) 366:2443–54. doi: 10.1056/NEJMoa1200690
114. Picardo SL, Doi J, Hansen AR. Structure and Optimization of Checkpoint Inhibitors. Cancers (Basel) (2020) 12:38. doi: 10.3390/cancers12010038
115. Lee JY, Lee HT, Shin W, Chae J, Choi J, Kim SH, et al. Structural Basis of Checkpoint Blockade by Monoclonal Antibodies in Cancer Immunotherapy. Nat Commun (2016) 7:13354. doi: 10.1038/ncomms13354
116. Chen JH, Pezhouh MK, Lauwers GY, Masia R. Histopathologic Features of Colitis Due to Immunotherapy With Anti-PD-1 Antibodies. Am J Surg Pathol (2017) 41:643–54. doi: 10.1097/pas.0000000000000829
117. Khoja L, Day D, Wei-Wu Chen T, Siu LL, Hansen AR. Tumour- and Class-Specific Patterns of Immune-Related Adverse Events of Immune Checkpoint Inhibitors: A Systematic Review. Ann Oncol (2017) 28:2377–85. doi: 10.1093/annonc/mdx286
118. Nishino M, Ramaiya NH, Awad MM, Sholl LM, Maattala JA, Taibi M, et al. PD-1 Inhibitor–Related Pneumonitis in Advanced Cancer Patients: Radiographic Patterns and Clinical Course. Clin Cancer Res (2016) 22:6051. doi: 10.1158/1078-0432.CCR-16-1320
119. Okada N, Iwama S, Okuji T, Kobayashi T, Yasuda Y, Wada E, et al. Anti-Thyroid Antibodies and Thyroid Echo Pattern at Baseline as Risk Factors for Thyroid Dysfunction Induced by Anti-Programmed Cell Death-1 Antibodies: A Prospective Study. Br J Cancer (2020) 122:771–7. doi: 10.1038/s41416-020-0736-7
120. Yasuda Y, Iwama S, Sugiyama D, Okuji T, Kobayashi T, Ito M, et al. CD4+ T Cells Are Essential for the Development of Destructive Thyroiditis Induced by Anti-PD-1 Antibody in Thyroglobulin-Immunized Mice. Sci Transl Med (2021) 13:eabb7495. doi: 10.1126/scitranslmed.abb7495
121. Reynoso ED, Elpek KG, Francisco L, Bronson R, Bellemare-Pelletier A, Sharpe AH, et al. Intestinal Tolerance Is Converted to Autoimmune Enteritis Upon PD-1 Ligand Blockade. J Immunol (2009) 182:2102–12. doi: 10.4049/jimmunol.0802769
122. Scandiuzzi L, Ghosh K, Hofmeyer KA, Abadi YM, Lázár-Molnár E, Lin EY, et al. Tissue-Expressed B7-H1 Critically Controls Intestinal Inflammation. Cell Rep (2014) 6:625–32. doi: 10.1016/j.celrep.2014.01.020
123. Álvarez-Sierra D, Marín-Sánchez A, Ruiz-Blázquez P, de Jesús Gil C, Iglesias-Felip C, González Ó, et al. Analysis of the PD-1/PD-L1 Axis in Human Autoimmune Thyroid Disease: Insights Into Pathogenesis and Clues to Immunotherapy Associated Thyroid Autoimmunity. J Autoimmun (2019) 103:102285. doi: 10.1016/j.jaut.2019.05.013
124. Nielsen C, Hansen D, Husby S, Jacobsen BB, Lillevang ST. Association of a Putative Regulatory Polymorphism in the PD-1 Gene With Susceptibility to Type 1 Diabetes. Tissue Antigens (2003) 62:492–7. doi: 10.1046/j.1399-0039.2003.00136.x
125. Wang J, Yoshida T, Nakaki F, Hiai H, Okazaki T, Honjo T. Establishment of Nod-Pdcd1-/- Mice as an Efficient Animal Model of Type I Diabetes. Proc Natl Acad Sci USA (2005) 102:11823. doi: 10.1073/pnas.0505497102
126. Keir ME, Liang SC, Guleria I, Latchman YE, Qipo A, Albacker LA, et al. Tissue Expression of PD-L1 Mediates Peripheral T Cell Tolerance. J Exp Med (2006) 203:883–95. doi: 10.1084/jem.20051776
127. Ashoori MD, Suzuki K, Tokumaru Y, Ikuta N, Tajima M, Honjo T, et al. Inactivation of the PD-1-Dependent Immunoregulation in Mice Exacerbates Contact Hypersensitivity Resembling Immune-Related Adverse Events. Front Immunol (2021) 11:618711. doi: 10.3389/fimmu.2020.618711
128. Lo B, Abdel-Motal UM. Lessons From CTLA-4 Deficiency and Checkpoint Inhibition. Curr Opin Immunol (2017) 49:14–9. doi: 10.1016/j.coi.2017.07.014
Keywords: CTLA4, PD-1, immune checkpoint inhibitor, phenocopy, immune deficiency
Citation: Hao Y and Cook MC (2022) Inborn Errors of Immunity and Their Phenocopies: CTLA4 and PD-1. Front. Immunol. 12:806043. doi: 10.3389/fimmu.2021.806043
Received: 31 October 2021; Accepted: 29 December 2021;
Published: 28 January 2022.
Edited by:
Paul J. Maglione, Boston University, United StatesReviewed by:
Kelli Wong Williams, Medical University of South Carolina, United StatesYesim Yilmaz Demirdag, University of California, Irvine, United States
Copyright © 2022 Hao and Cook. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthew C. Cook, bWF0dGhldy5jb29rQGFudS5lZHUuYXU=