 Zu Ye
Zu Ye Yin Shi
Yin Shi Susan P. Lees-Miller
Susan P. Lees-Miller John A. Tainer
John A. Tainer
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 14 December 2021
Sec. Cancer Immunity and Immunotherapy
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.797880
This article is part of the Research Topic DNA Repair and Immune Response View all 12 articles
The DNA damage response (DDR) is an organized network of multiple interwoven components evolved to repair damaged DNA and maintain genome fidelity. Conceptually the DDR includes damage sensors, transducer kinases, and effectors to maintain genomic stability and accurate transmission of genetic information. We have recently gained a substantially improved molecular and mechanistic understanding of how DDR components are interconnected to inflammatory and immune responses to stress. DDR shapes both innate and adaptive immune pathways: (i) in the context of innate immunity, DDR components mainly enhance cytosolic DNA sensing and its downstream STimulator of INterferon Genes (STING)-dependent signaling; (ii) in the context of adaptive immunity, the DDR is needed for the assembly and diversification of antigen receptor genes that is requisite for T and B lymphocyte development. Imbalances between DNA damage and repair impair tissue homeostasis and lead to replication and transcription stress, mutation accumulation, and even cell death. These impacts from DDR defects can then drive tumorigenesis, secretion of inflammatory cytokines, and aberrant immune responses. Yet, DDR deficiency or inhibition can also directly enhance innate immune responses. Furthermore, DDR defects plus the higher mutation load in tumor cells synergistically produce primarily tumor-specific neoantigens, which are powerfully targeted in cancer immunotherapy by employing immune checkpoint inhibitors to amplify immune responses. Thus, elucidating DDR-immune response interplay may provide critical connections for harnessing immunomodulatory effects plus targeted inhibition to improve efficacy of radiation and chemotherapies, of immune checkpoint blockade, and of combined therapeutic strategies.
Key cancer hallmarks critically include genomic instability, immune modulation, and altered DNA damage and other stress responses to favor overall cell survival (1, 2). Every day, tens of thousands of damaged DNA lesions occur in each human cell that could impact cell survival and genomic integrity (3). Importantly, the outcome of this DNA damage depends directly upon the nature and actions of the DNA damage response (DDR). Lesions become accurately or inaccurately repaired or left as unrepaired mutations depending upon the DDR. As a result, evolutionary selection ensures that the DDR is a carefully orchestrated response system consisting of multiple signaling pathways that largely maintain genomic stability and fidelity despite high levels of DNA damage (4, 5). Yet, comprehensive analyses of cancer genome databases reveal non-B DNA, mitochondrial dysfunction, and the activation of DNA repair/cell cycle pathways as major factors driving somatic mutation loads in cancer cells (2, 6). From a mechanistic standpoint, the positive correlations of these factors with mutations in cancer cells likely arise from increased reactive oxygen species (ROS), oncogenic replication and transcription stress, and the combination of resulting excessive DNA damage plus its escape from accurate repair.
In particular, DDR are activated by replication obstacles in proliferating cells that lead to replication stress: replication fork stalling, collapse or breakage, such as lesions from oxidation, deamination and alkylation, DNA breaks, protein-DNA cross-links, and non-B DNA structures including R-loops (RNA-DNA hybrids formed by replication-transcription conflicts) (7–9). DNA damage and activation of the DDR from endogenous replication stress are seen at pre- or early stages of oncogenesis, and adaptation to replication stress acts in tumor development (10). In breast-cancer susceptibility gene 2 (BRCA2)-deficient cancer cells, the inactivation of replicative stress response factors (e.g. poly (ADP-ribose) polymerase [PARP1] or ATM and Rad3-related [ATR] inhibition) triggers cyclic GMP-AMP synthase (cGAS)-STING-mediated innate immune responses (11, 12). Furthermore, inherent DNA repair defects in tumors may develop mutation-driven neoantigens that can cause the immune system to recognize the tumor cells as foreign while also increasing the amount of cytosolic DNA to trigger a cGAS-STING response. Thus, the DDR that largely protects against DNA damage in normal cells can often be defective or defeated in proliferating cancer cells with consequent impacts on immune responses. This finding implies a fundamental importance of DDR for cancer biology, for the elucidation of cancer vulnerabilities, and for optimal applications of immunotherapy.
The DDR machinery can conceptually be divided into at least six distinct DNA repair pathways responding to different types of DNA damage: (i) homologous recombination (HR), which repairs double-strand breaks (DSBs) using a homologous DNA template; (ii) non-homologous end joining (NHEJ), which repairs DSBs without a corresponding template; (iii) alternative end-joining (A-EJ), which repairs DSBs with insertion and deletion errors by employing micro-homology; (iv) nucleotide excision repair (NER), which repairs bulky DNA lesions globally or coupled to transcription; (v) mismatch repair (MMR), which repairs DNA single-strand breaks (SSBs) predominantly generated during DNA replication and recombination processes plus mismatches that escaped replication fidelity; and (vi) base excision repair (BER), which removes bases damaged by oxidation, alkylation, deamination, and methylation to avoid replication and transcription blocks and errors (4, 13–15).
The various DDR pathways share similarities in how they respond to the stress of damaged DNA, whereby a damage sensor that can also be a repair effector [e.g., RPA, MUTY, PARP1, Ku70/80, MRE11-RAD50-NBS1 (MRN) complex] recognizes specific DNA damage types (single-stranded DNA, base mismatches, SSBs, and DSBs) before recruiting and activating downstream transducer kinases (such as ATM, ATR, DNA-PKcs), which in turn transduce the signal to effector proteins (such as MRN, CHK1, EXO5, p53, RAD51, and BRCA1/2). The ensuing complexes ultimately orchestrate repair by employing damage removal and sequence replacement by handoffs or dynamic machinery that have evolved to avoid the release of toxic and mutagenic DNA intermediates (15, 16). Thus, the DDR is an ancient and evolutionarily conserved mechanism that is essential for genome stability and cell survival (17, 18).
As the major stress response system essential for surviving infection, the immune response is an evolved network of proteins and complexes that respond to invading pathogens and their associated toxins. Importantly, DDR defects can lead to imbalance between DNA damage and repair, impairing tissue homeostasis and leading to replication and transcription stress, mutation accumulation or outright cell death: this imbalance can drive tumorigenesis as well as secretion of inflammatory cytokines, and aberrant immune responses (19–23). All organisms possess mechanisms to detect and eliminate foreign pathogens via the innate immune system. Additionally, higher vertebrates employ a sophisticated adaptive immune system that includes antibodies as well as B and T lymphocytes with virtually limitless repertoires of receptors that mediate neutralization of foreign pathogens and removal malignant cells (24–26). To stimulate strong anti-tumor immune responses, cancer immunotherapy typically employs immune checkpoint inhibitors for the PD-1/PD-L1 and CTLA-4 pathways to amplify immune system responses and also to harnesses responses to neoantigens that are primarily tumor-specific antigens resulting from the higher mutation load in tumor cells (27–29). The validity of the PD-1/PD-L1 approach requires the functional MHC class I complex, which itself is often deleted during tumor evolution to escape immune regulation (30).
For clarity this review is divided into five overall sections: 1) Introduction, 2) DDR in innate immunity, 3) DDR in adaptive immunity, 4) DDR inhibition in antitumor immunity, and 5) Summary and prospects. Within these sections and their subsections, we furthermore consider how these critical and seemingly distinct DDR and immune stress responses are intertwined and where defining their interconnections may enable novel insights into etiology and advanced molecular-based treatment of cancer and other human diseases.
Innate immunity is the first immunological defense system against pathogens. Activation of the innate immune response relies on Pattern Recognition Receptors (PRRs). These PRRs detect Damage-Associated Molecular Patterns (DAMPs) or Pathogen-Associated Molecular Patterns (PAMPs) to initiate a signaling cascade resulting in production of interferons (IFNs), cytokines and chemokines (24, 26, 31). Importantly, non-self nucleic acids are the most well-characterized stimuli for the innate immune response (32, 33); furthermore, endogenous cytosolic DNA released from the nucleus or mitochondria stimulates the innate immune system.
DNA damage caused by genotoxic stresses or DNA damage stimulus (e.g., cytotoxic chemotherapy and radiation) can create cytosolic chromosomal fragments that may be recognized by cGAS, a cytosolic DNA sensor. Cytosolic exposure of chromosomal DNA by micronuclei rupture, breakage of chromatin bridges, or disintegration of micronuclei-like cytosolic chromatin fragments activates cGAS (34). Once bound to cytosolic DNA, activated cGAS can form dimers and multimer assemblies that undergo liquid–liquid phase separation to form biomolecular condensates that amplify cGAS activation (35). Activated cGAS produces 2´-3´cGAMP (cGAMP) as a second messenger to function in both the host cell and adjacent cells via secretion or by passage through gap junctions, which contributes to the bystander response to radiotherapy in non-irradiated neighboring cells (36–40). In the presence of cGAMP, STING is relocated from the ER to Golgi, where it recruits and activates TANK-binding kinase (TBK1), that activates interferon regulatory factor 3 (IRF3) and NF-κB signaling (41). Activated IRF3 and NF-κB then induce transcription of innate immune response genes, including IFNs and cytokines (36, 37, 42).
Interestingly, cGAS is also found in the nucleus. Nuclear cGAS is inactivated by its acidic patch binding to nucleosome core particles, which prevents DNA binding, thus preventing autoreactivity (34). Moreover, nuclear cGAS is recruited to DNA damage sites by γH2AX, which promotes its interaction with Poly (ADP-ribose) polymerase 1 (PARP1) and impedes formation of PARP1-Timeless complex to thereby suppress HR but not NHEJ (43, 44).
Another important cytosolic DNA sensor is γ-interferon-inducible protein-16 (IFI16). Like cGAS, IFI16 can detect both self and non-self dsDNA to promote IRF3 and NF-κB -dependent interferon production via STING (26, 45).
Emerging data reveal that DNA repair pathways and cytosolic pathological DNA sensing pathways have overlapping effectors that recognize and respond to damaged nuclear DNA, cytosolic endogenous DNA, or foreign DNA (46). These observations provide compelling evidence for inextricable links between the DDR and innate immune responses (Figure 1).
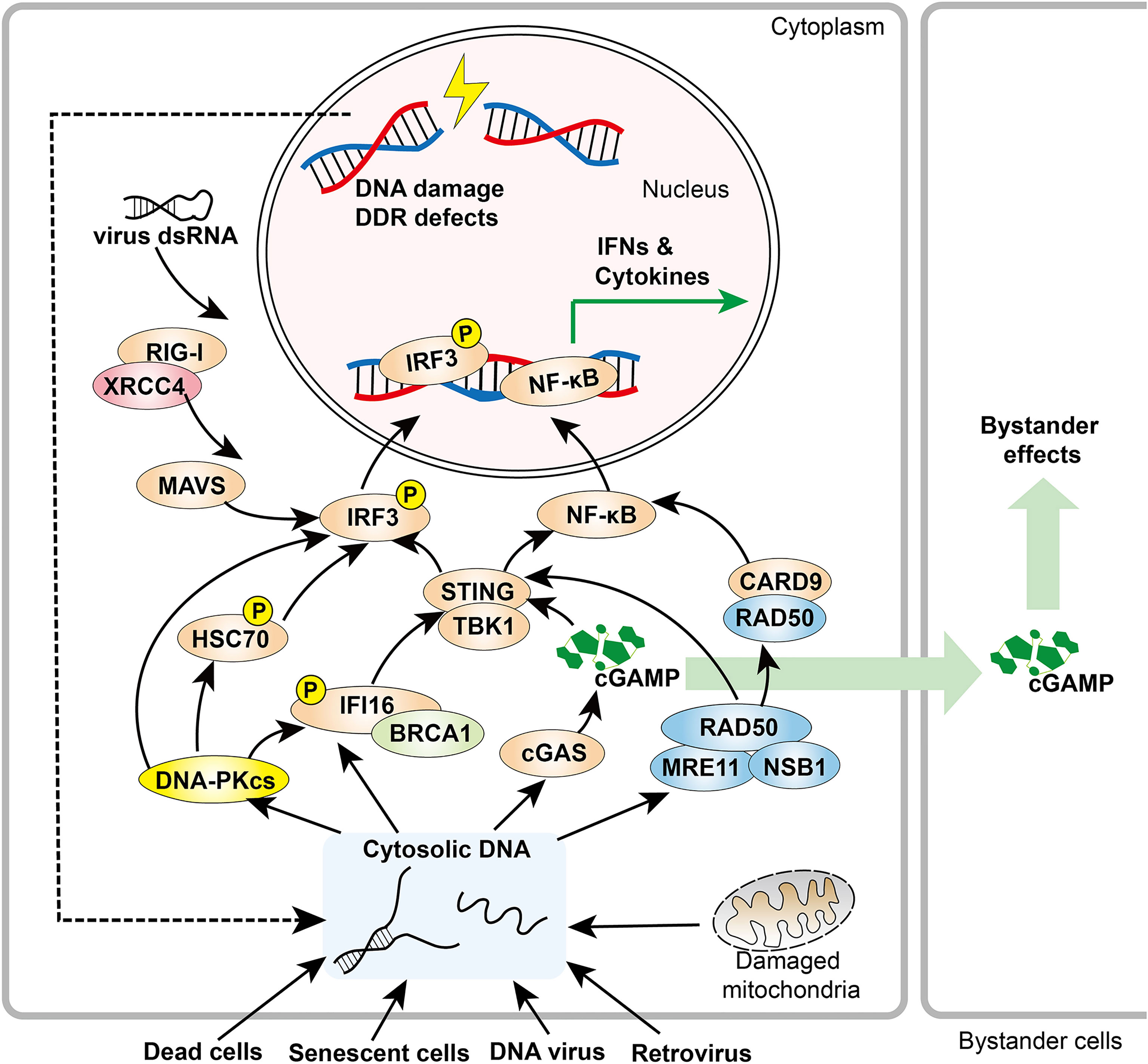
Figure 1 Overview of DDR components in innate immune responses. DDR factors, including DNA-PK and MRE11, promote cytosolic DNA sensing signaling pathways. When activated by cytosolic DNA, cGAS produces cGAMP, a soluble second messenger that initiates STING-IRF3 signaling both within the host cell and adjacent cells. In addition, RAD50 associates with CARD9, leading to NF-κB activation and downstream cytokine production. XRCC4 interacts with RIG-I, which promotes the RIG-I-MAVS-IRF3 pathway.
Interference in DDR signaling elicits innate immune responses. One of the most well-studied examples is PARP inhibition. PARP inhibition generates cytosolic chromatin fragments and significantly potentiates cGAS-STING-dependent immune responses (11, 47–54). Similarly, DNA damage as a result of cytotoxic chemotherapy, ionizing radiation (IR), metabolism, and deficiency of other DDR elements (including BRCA2, ATM, CHK1, RPA, RAD51, TREX1 and FANCD2), also leads to increased IFN signaling–mediated immune responses (11, 19, 55–61).
The RecQ–like BLM helicase partners with EXO5 and EEPD1 nucleases for stalled DNA replication restart and maintenance of genome integrity (62, 63). BLM deficiency in Bloom syndrome (BS) causes increased expression of inflammatory genes through the cGAS–STING–IRF3 pathway, suggesting it prevents unchecked inflammatory gene responses (64). ROS from radiation therapy or cell stress lead to cGAS-STING-mediated immune responses to cancer from DSBs as well as oxidative adducts that must be removed by DNA glycosylases, such as endonuclease VIII (Nei)-like proteins (NEIL) and oxoguanine DNA glycosylase (OGG1) (65–67). Furthermore, high levels of ROS that are not efficiently reduced by superoxide dismutases and catalase can leave unrepaired 8-hydroxyguanosine (8-OHG) (68, 69). 8-OHG stabilizes DNA against degradation by the cytosolic DNA exonuclease TREX1, leading to accumulated cytosolic DNA and increased cGAS activation (70). This ROS effect can be amplified by vicious cycles of oxidative damage and iron release from ROS-sensitive 4Fe-4S co-factors in multiple replication and repair proteins (62, 71–73).
Metabolism and innate immunity converge at the mitochondria, which can orchestrate innate immune signaling pathways in different cancer-relevant metabolic scenarios including a link to PARylation and cell death (74, 75). Metabolic cues including nucleotide imbalance can stimulate the release of mtDNA from mitochondria that drives an interferon response with MRE11 playing a leading role (76). The fundamental importance of DNA breaks in promoting such immune responses is evidenced by the observation that mtDNA breaks synergize with nuclear DNA damage to mount a robust cellular immune response (77).
In general, unresolved DNA damage can act as a mediator linking the DDR and immune recognition, and this can involve the formation of micronuclei as an initiating event in a cascade promoting genomic instability and innate immune responses (78, 79). Moreover, genome instability and imperfect cell cycle checkpoints in tumor cells enhance formation of micronuclei, making them more susceptible to targeting of the innate immune response (5, 22, 79). DNA damage responses occur in minutes to hours. Yet, there is a delayed onset of days for inflammatory cytokines that modify tumor microenvironment by immune cell recruitment as critical for local and systemic (abscopal) tumor responses to radiotherapy.
DNA-dependent protein kinase (DNA-PK) is a trimeric nuclear complex that functions as a central integrator of the DSB repair system. The protein complex consists of a large catalytic subunit, DNA-PKcs, and the Ku70/80 heterodimer (Ku70/80) which recognizes DSB ends (80). DNA-PKcs is a Ser/Thr protein kinase and the largest member of the phosphatidylinositol 3-kinase (PI3K)-related kinase (PIKK) family (81). Once activated by Ku70/80, DNA-PK undergoes autophosphorylation and is then positioned to phosphorylate other repair effectors and promote a synaptic complex for ligation of two dsDNA ends (80, 82–86). In recent decades, emerging evidence revealed that DNA-PK is a critical component of innate immunity against multiple viruses, including human immunodeficiency virus (HIV), Herpes Simplex Virus 1 (HSV-1), alphavirus M1, and vaccinia virus (87–92). As such, DNA-PK is a key DNA sensor that modulates innate immunity through several critical components of innate immune pathways.
In STING-dependent DNA sensing pathways, cGAS, IFI16, and IRF3 are substrates for DNA-PK (89, 93, 94). However, the role of DNA-PK within the cGAS-STING pathway remains controversial. One recent study reported that DNA-PK directly phosphorylates cGAS to suppress its enzymatic activity and thus attenuate innate immune responses (93). To this end, DNA-PKcs deficiency caused by missense mutations in its coding gene, PRKDC, leads to an increased inflammatory response in both human and mouse cells (93). In contrast, a pioneering study showed that DNA-PK interacts with and phosphorylates IRF-3, thus promoting its nuclear translocation (94). In a systematic profiling study, DNA-PKcs directly phosphorylated the DNA sensor IFI16 and promoted IFI16-driven cytokine responses (89). Furthermore, regardless of its partner cGAS, STING can localize to the inner nuclear membrane in breast cancer tumor samples and promote cancer cell survival by resistance to DNA-damaging agents through interacting with DNA-PK (95). Therefore, further studies are warranted to better understand mechanisms governing DNA-PK substrate selection within the context of the innate immune response.
As described above, although a potentially suppressive role of DNA-PK on cGAS was reported which may be context dependent, most studies suggest that DNA-PK promotes a STING-dependent innate immune response (96–100). Mechanistically, the HEXIM1-DNA-PK-paraspeckle components-ribonucleoprotein complex (HDP-RNP), containing DNA-PK subunits and paraspeckle proteins, is required for foreign DNA sensing through the cGAS-STING pathway. The HDP-RNP interacts with cGAS, and when stimulated by cytosolic DNA, the paraspeckle proteins from the complex are released to recruit STING and activate DNA-PK and IRF-3. Knockdown of HDP-RNP subunits including Ku70, the DNA binding subunit in DNA-PK, resulted in loss of IFN stimulatory DNA–mediated immune response (97). In addition, Ku70 was identified as a cytosolic DNA sensor that translocates to the cytoplasm to form a complex with STING and induce production of IFN-λ1 (98, 99).
Besides STING-dependent DNA sensing mechanisms, DNA-PK also acts as a DNA sensor to trigger a robust and broad antiviral response in a STING-independent DNA sensing pathway (SIDSP) in human cells, but not in laboratory mice (101), perhaps a reflection that DNA-PK levels in human cells are much higher than in mouse cells (102–104). A recently characterized DNA-PK partner is LINP1, a lncRNA that can recruit multiple DNA-PK assemblies and promote formation of phase condensates (105). As LINP1 is present in both cytoplasm and nucleus, it will be important to test its potential role in cytosolic immune activation.
Overall, DNA-PK is considered a cytosolic DNA sensor for both STING-dependent and -independent DNA sensing pathways. The extent to which the role of DNA-PKcs in the innate immune response is distinct from its well-characterized nuclear functions in NHEJ is under active investigation.
MRN, a core orchestrator that senses DSB damage and activates DNA repair cascades, is required to maintain genome integrity (13). In recent years, the MRN complex, which acts in DSB sensing, stabilization, signaling, and effector scaffolding (106), has furthermore been found to localize to viral replication sites and trigger innate immune responses (107–109).
An exemplary MRN role in regulating innate immunity comes from the meiotic recombination 11 homolog A (MRE11) nuclease subunit, which recognizes and processes DSB DNA ends as a part of HR repair, replication fork processing, and telomere length maintenance (110, 111). MRE11 has both endonuclease and exonuclease activities that, together, initiate HR repair (112). Furthermore, MRE11 functions as a key cytosolic DNA sensor in recognition of a broad spectrum of dsDNA and activates STING trafficking and type I IFN production in various cell types (108).
An intriguing observation is that nuclease activity is not required for the cytosolic DNA-sensing function of MRE11, which reinforces the notion that besides their nucleotide processing activity DDR nucleases also function to recognize and sculpt specific DNA structures (113–116). In fact, the nuclease-inactive mutant form of MRE11 triggers an even higher immune response than the wild-type form. Therefore, MRE11 may act as a regulatory switch within the STING-dependent immune response, initially functioning as a DNA sensor to activate STING-mediated signaling, then subsequently working as a nuclease to suppress excessive immune responses (108). Obviously, further studies are required to better elucidate the pro- and anti-immune–modulating mechanisms of MRE11 in STING-dependent signaling. Nevertheless, these data suggest that STING-mediated signaling may be activated by one of the existing MRE11 inhibitors (112, 117). It will also be interesting to see if the adaptor regulator GRB2 complex with MRE11, which promotes HR and suppresses A-EJ in the nucleus, plays a role in STING-mediated signaling (118). Intriguingly, multiple GRB2 molecules can also bind to Linker of Activation of T cells (LAT) to mediate its oligomerization, which is important for T-cell signaling under limiting stimulating conditions. Furthermore, GRB2 promotes metabolic reprogramming to support T cell activation (119–121). These and other data support the notion that tight protein and DNA binding plus conformational sculpting can regulate activities and switch DNA repair pathways (122).
MRE11 mutations that result in loss of binding ability to Nijmegen breakage syndrome protein 1 (NBS1) induce type I IFN comparable to wild-type MRE11 (108). This finding suggests that NBS1 is not instrumental for sensing cytosolic DNA and provoking an immune response. This concept is consistent with the mechanistic role implied by the NBS1 structure and its MRE11 interface, to flexibly restrict DNA end processing and homologous recombination activities to the vicinity of DSBs (123). On the basis of prior data showing that NBS1 loss promotes cytosolic MRE11 distribution (124), we propose that a deficiency of NBS1 may enhance cytosolic DNA sensing by MRE11.
The third component of the MRN complex is the ATP-binding cassette-ATPase (RAD50). MRE11 nuclease activity is regulated by ATP-dependent RAD50 helical coiled-coil conformations that switch the MRE11-RAD50 complex between DNA tethering, ATM signaling, and strand resection (125, 126). RAD50 plays an important role in innate immunity via a STING-independent signaling pathway (109). RAD50 binds a proinflammatory signaling adaptor amino-terminal caspase-recruitment domain (CARD9) through its structurally defined zinc-hook region (127). Together with MRE11, RAD50 recognizes cytosolic DNA and interacts with CARD9, which leads to the recruitment of Bcl-10 to induce NF-κB activation and pro-inflammatory cytokine IL-1β generation (109).
BRCA1, which together with the MRN complex plays a central role in HR DNA repair, interacts with IFI16 (128, 129). In herpesvirus-infected cells, BRCA1 is required for IFI16-mediated recognition of foreign DNAs, association with STING, and subsequent IFN-β production (128). Aside from DNA virus sensing, X-ray repair cross-complementing group 4 (XRCC4), a DNA ligase IV (LIG4)-associated protein essential for NHEJ (130–132), acts in an RNA-sensing pathway through interaction with retinoic acid-inducible gene I (RIG-I) (133). XRCC4 promotes oligomerization and ubiquitination of RIG-I, which results in enhancement of the RIG-I-MAVS-IRF3-type I IFN signaling cascade and subsequent suppression of RNA virus replication in host cells. Reciprocally, RIG-I competes with LIG4 to interact with XRCC4, and therefore it impedes XRCC4-dependent NHEJ cascades and hinders retrovirus integration into the host genome by suppressing the NHEJ pathway (133). This finding highlights the critical role of XRCC4 in defense against RNA viruses and in potentiating innate immune response.
Unlike the innate immune system, characterized by rapid sensing and elimination of pathogens as first line of defense, the adaptive immune system provides broader and more accurate discrimination between self and non-self immunogens based on the process of positive and negative selection during lymphocyte development (25). A robust adaptive immune response to any pathogen or biological macromolecule seen for the first time takes weeks to mount. However, subsequent exposure to the same pathogen promotes a rapid “memory” response that is often magnitudes stronger than the response following the first exposure. In adaptive immunity, the DDR is essential for lymphocyte development by facilitating the assembly and diversification of antigen receptor genes (134, 135). Thus, DDR deficiencies are linked with immunological disorders, including autoimmune diseases, such as systemic sclerosis, pediatric systemic lupus erythematosus, and severe sepsis (136–139).
Ataxia-telangiectasia (A-T), a disorder arising from ATM germline mutations, was one of the first-identified disorders whereby immunodeficiency was associated with an aberrant DDR (140–142). Missense mutations of PRKDC, which encodes the catalytic subunit in DNA-PK, were also found in patients with the organ-specific autoimmunity phenotype (93, 143). In addition, autoantibodies directed against Ku 70/80 were detected in autoimmune patient sera (144, 145). Indeed, Ku was first identified via autoantibodies in sera from patients with the autoimmune disease polymyositis-scleroderma overlap syndrome (146).
Adaptive maturation of T and B lymphocytes is guided by the “blueprint” of different cell surface receptors. During the process of lymphocyte maturation, three highly regulated processes, including variable, diversity, and joining [V(D)J] recombination, class-switch recombination (CSR), and somatic hypermutation (SHM) together with negative and positive selection (147), lead to generation of a functional, genetically diverse, and non-autoreactive antigen receptor repertoire. Interestingly, these processes naturally generate DSBs and/or trigger a DDR in adaptive immunity (Figure 2) (136, 148). In this section, we review these pathways highlighting roles of important DNA repair factors.
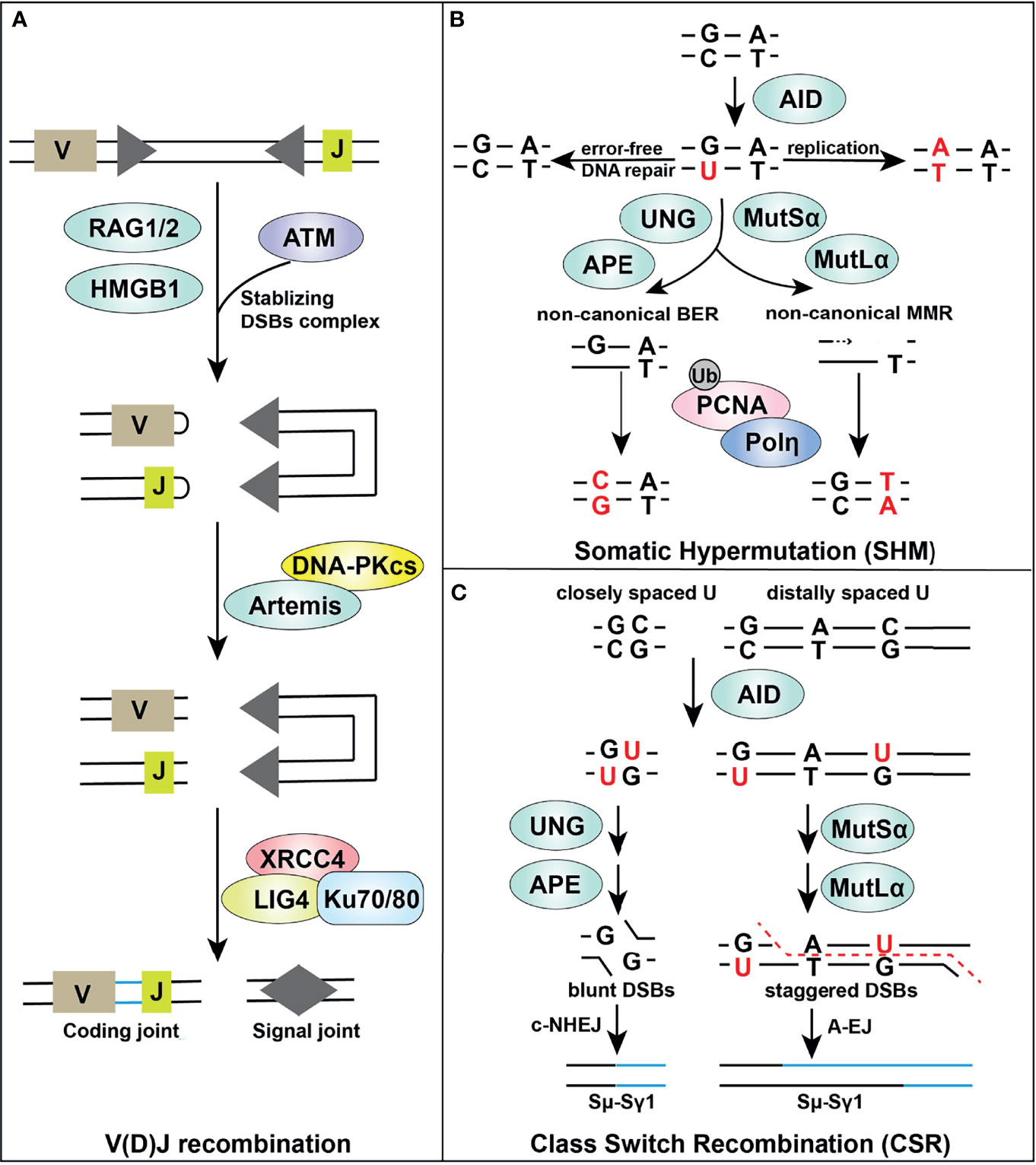
Figure 2 Overview of DDR components in adaptive immune responses. Certain DDR signaling pathways, such as MMR, BER, NHEJ, and A-EJ, are required in V(D)J recombination (A), SHM (B) and CSR (C) processes, supporting successful lymphocyte development.
V(D)J recombination occurs in G1 phase of naive, progenitor T and B lymphocytes, and enables rearrangement of gene segments at both immunoglobulin and T-cell receptor loci in a lineage specific and developmental stage specific manner (148, 149). V(D)J recombination is initiated by the recombinase activating gene (RAG) endonucleases RAG1 and RAG2, which is directed by RAG recognition sequences (recombination signal sequences [RSS]) (Figure 2A). The RAG complex creates a nick between the coding segment and the flanking RSS which leads to a DNA hairpin at the ends of the gene segment containing the coding regions (coding-ends) and a blunt-ended DSB at the end of the RSS, so called signal-ends. Alignment of coding regions, excision, and formation of hairpin-ended coding-ends and blunt-ended signal-ends takes place within the RAG1/2 complex, aided by HMGB1 (148, 149). RAG-mediated DSBs are processed by the NHEJ machinery to assemble genes encoding immunoglobulin, and heterodimeric B- and T-cell receptors (150–152). The rapid repair of RAG-mediated DSBs by NHEJ is essential for normal lymphocyte development. Failure to repair RAG-mediated DSBs in immature B cells leads to a DDR including ATM-mediated upregulation of NF-κB signaling (134, 153–157).
DNA-PKcs in complex with Artemis, a member of the metallo-β-lactamase protein family, is required for successful V(D)J recombination and lymphocyte development. DNA-PKcs interaction is required for Artemis endonuclease and exonuclease activities for the RAG-mediated hairpin-opening step in V(D)J recombination and for 5’ and 3’ overhang processing in NHEJ (158). The two coding-ends, each terminating with a DNA hairpin, are released from the RAG1/2 complex first. Prior to rejoining, the DNA hairpins are opened by the Artemis-DNA-PKcs complex, which cleaves 3’ to the apex of the DNA hairpin. Artemis requires DNA-PKcs for its hairpin opening activity but how this occurs is still an open question (159, 160). Nevertheless, both the interaction of DNA-PKcs with Artemis, and DNA-PKcs phosphorylation are important for Artemis activation (161, 162).
Irrespective of the mechanism, DNA-PKcs protein and Artemis are both required for opening the coding-end hairpins, as the unopened hairpins accumulate in cells lacking either Artemis or DNA-PKcs (158). Indeed, mice, dogs and horses with mutations that compromise DNA-PKcs protein levels are characterized by radiation sensitivity (due to defects in NHEJ and DSB repair) as well as severe loss of T and B cells resulting in severe combined immunodeficiency (SCID) (163–165). Kinase-dead (KD) point mutation in the catalytic domain of DNA-PKcs blocks end-ligation without abolishing hairpin opening in knock-in mouse models (166). However, hairpin opening in the DNA-PKcs-KD mice requires ATM kinase activity (166). While pathogenic PRKDC mutation in humans is rare, six patients with SCID and DNA-PKcs mutation have been identified, five of whom share mutation of L3062R in the C-terminal FAT domain (85). Interestingly, DNA-PKcs with the L3062R mutation maintains full catalytic activity, but the mutation appears to hinder activation of the Artemis nuclease (167). In addition, one patient with two DNA-PKcs mutations that severely impair (but do not completely ablate) catalytic activity presented both with SCID and a severe neurologic deficit incompatible with life (168). Description of this patient has led to speculation that complete loss of DNA-PK in humans is not compatible with life, and may have a unique function in neuronal development. Deficiencies in Artemis are also associated with SCID with radiation sensitivity (RS-SCID) (169–171).
Once the coding end hairpins are opened, they can be acted upon by nucleases, and extended by error prone polymerases such as V(D)J specific terminal deoxynucleotidyl transferase (TdT) and/or the more general NHEJ polymerases mu and lambda (148, 149, 172, 173). This processing of the coding-end creates additional diversity for antigen selectivity. Finally, the processed coding-ends are ligated by the XLF-XRCC4-LIG4 complex in conjunction with Ku (174). The RSS signal ends are released after the coding-ends and directly ligated by the Ku-XLF-XRCC4-LIG4 complex (175–177). DNA-PKcs, but not Artemis, also plays a role in rejoining of signal ends (166, 178, 179).
Although NHEJ is required for both repairing DSBs produced by IR and those produced by the RAG endonuclease in V(D)J recombination, there are both similarities and differences between the two processes. IR introduces complex forms of DNA damage resulting in DSB ends with diverse sequences and overhanging ends, some of which will contain non-ligatable ends (180). Thus, after IR, NHEJ must be able to 1) respond to DSBs wherever they occur in the genome and 2) hold and tether the ends while they are processed before ligating them. The recently determined structures of NHEJ synaptic complexes reveal how NHEJ proteins can both tether and secure DSB ends while DNA-PKcs autophosphorylation provides a mechanism for handover to end processing enzymes and subsequent ligation by the XLF-XRCC4-LIG4 complex (82, 130, 181–184). In V(D)J recombination, defined DSBs with discrete coding-ends and signal-ends are generated and held within the RAG1/2 heterotetrameric complex (185, 186) before being released and opened by DNA-PKcs-Artemis (coding-ends) and ligated by Ku-XRCC4-LIG4 (coding-ends and signal-ends) (177, 185–187). After hairpin opening, coding ends are processed to include both additional antibody diversity (e.g. TdT) and generate ligatable ends. It will be interesting to determine how the NHEJ machinery interfaces with the RAG1/2 complex and the DNA-PK/Artemis hairpin opening complex.
While the role of Artemis in V(D)J recombination is clear, its role in NHEJ after IR is enigmatic (160). It may act to remove overhanging DNA ends, acting at ds-to-ssDNA transitions as a flap-endonuclease or by direct exonuclease activity and/or it may be required to open secondary structure elements formed by looping of ssDNA at the ends of DSBs. It is likely that Artemis is required for repairing only a subset of DSBs after IR, as Artemis-null cells are not as radiation sensitive as those lacking Ku, XRCC4, LIG4 or DNA-PKcs (148, 188–191).
Animals lacking DNA-PKcs, Artemis, Ku70 or Ku80 are viable but radiosensitive due to defects in NHEJ and immune-deficient due to defects in V(D)J recombination (192–195). For V(D)J recombination in mice lacking functional DNA-PKcs or Artemis, unopened coding-end DNA hairpins accumulate, producing a profound defect in coding joint formation (192, 193, 196). Signal joints are unaffected by loss of Artemis whereas mutation of DNA-PKcs has variable effects on signal joints (157, 175). In SCID horses signal ends are profoundly affected by DNA-PKcs mutation, while SCID dogs and mice have intermediate signal end rejoining, indicating species differences in V(D)J recombination at signal ends, possibly due to relative levels of DNA-PKcs and ATM (165). In contrast, in animals lacking Ku70 or Ku80, both coding and signal joins are affected (194, 195). Mice lacking XRCC4 or LIG4 are non-viable, with embryos undergoing neuronal apoptosis, while cells lacking XRCC4 or LIG4 are radiation sensitive and defective in coding and signal joints, consistent with a more severe V(D)J recombination defect (148, 188–190). Notably, deletion of Ku rescued the embryonic lethality, but not the V(D)J recombination defects in LIG4-null mice, likely through aberrant end-resection and the repair by the Alt-EJ pathway (197, 198).
Besides the DNA-PKcs-Artemis/Ku-XRCC4-LIG4 axis, the MRN-associated kinase ATM plays a critical role in lymphocyte development via direct or indirect involvement at various stages of development. Although many details are still unclear, ATM is required for stabilization of the RAG post-cleavage complex that releases the DNA ends to the NHEJ pathway (157, 199, 200). Inactivating somatic ATM mutations are associated with T- and B-cell lymphoma (201, 202); dysregulated V(D)J recombination results in translocations in ATM-deficient lymphocytes, potentially promoting tumorigenesis (203, 204). While XLF-deficient cells have significant V(D)J recombination, ATM kinase activity and its chromatin bound DDR factors (e.g., 53BP1 and H2AX), while dispensable for V(D)J recombination in otherwise wild type cells, become essential for chromosomal NHEJ during V(D)J recombination in XLF-deficient cells (205–207). Indirectly, ATM-related repression of GSK3β and cyclin D3 also plays an important role in thymocytes and pre-B cells (208, 209). DSBs generated by both V(D)J recombination and CSR induce ATM-dependent phosphorylation of GSK3β, which is a constitutively active kinase known to promote cell death (209, 210). The inactivation of GSK3β by DSB-initiated Ser389 phosphorylation protects B cells during V(D)J recombination and CSR that are required for antigen-specific IgG antibody responses following immunization. During T cell development, GSK3β phosphorylation created by V(D)J recombination also promotes survival of DN3 thymocytes undergoing TCRβ rearrangements, mimicking the results described in mice harboring deficiency in several key DDR factors, including ATM, NBS1 and H2AX (209, 211, 212).
The DDR is also essential for additional adaptive immune responses that occur after antigen exposure in germinal center B cells. V(D)J recombination-rearranged immunoglobulin (Ig) variable regions are further modified by the process of SHM, after which antibodies with highest affinity are selected. While in CSR, the constant regions of immunoglobulin genes are excised and rearranged to produce other isotypes (e.g. IgA and IgG) from the initially expressed IgM or IgD isotypes (213, 214) (Figures 2B, C). Both CSR and SHM are initiated by B cell-specific, activation-induced cytidine deaminase (AID), a member of the apolipoprotein B mRNA editing enzyme catalytic polypeptide like (APOBEC) family of deaminases, which converts cytosine to uracil on single-stranded DNA or RNA (215, 216). Various DDR pathways are then involved in both the generation of strand breaks and their repair.
During SHM, AID deaminates a particular trinucleotide sequence in ssDNA of transcriptionally active genes, leaving behind numerous uracil residues and producing predominantly nucleotide substitutions in rearranged V genes on the heavy- and light-chain loci, and switch (S) regions, which precede most C genes on the heavy chain locus (217, 218). The mutagenic outcome of uracil lesions can then be determined by one of the following DDR responses: (i) Uracil can act as a template for replication, resulting in a fixed C-T transition mutation; (ii) U-G mismatches can be recognized by the error‐prone MMR machinery, in which the MutSα complex (MSH2-MSH6) detects the mismatch and recruits MutLα complex (MLH1-PMS2) to nick the DNA, followed by the recruitment of Polη (DNA polymerase η) to generate mutations (219, 220); (iii) Non‐canonical BER initiated by uracil DNA glycosylase (UNG) can be used to recruit proliferating cell nuclear antigen (PCNA) at the lesions, and low‐fidelity polymerases such as Polη, which can increase mutations during replication of common DNA fragile sites (221), then can be recruited by PCNA ubiquitination and utilized by both MMR and BER resulting in mutagenic repair (149, 222–224). The nick generated by the UNG-dependent BER pathway is particularly important for CSR, as UNG1 knock out largely abolishes CSR (225).
During CSR, DSBs are generated in the switch regions that are subsequently ligated by either the canonical NHEJ pathway or A-EJ pathway which involves XRCC1, MRE11, plus FEN1 (which threads and removes DNA flaps) and Pol theta for which there are inhibitors (114, 149, 226–230). Although many details of this important pathway remain to be determined, it has been suggested that UNG removes AID-incorporated uracil to create an abasic site which is then cleaved by apurinic/apyrimidic endonuclease (APE) to create an SSB. Two closely spaced SSBs on opposite DNA strands can create a DSB (213, 231, 232). Indeed, UNG inhibition sensitizes cells to high APOBEC3B deaminase and to floxuridine (5-FdU), which are toxic to tumor cells through incorporation of 5-FU into DNA (233, 234).
Much of what we have learned about CSR has come from disruption of DNA repair genes in mice leading to immunodeficiency characterized by the production of IgM (the first spliced constant region) but not IgA or IgG (products of CSR) (136). As reviewed recently by Zha and colleagues (191), the most dramatic defects (>90% reduction) in CSR have been observed in mice lacking the tumor suppressor p53-binding protein (53BP1). In contrast, more modest defects (50-70% reduction) occur in animals lacking MDC1, H2AX, Ku, XRCC4, LIG4 and mice lacking DNA-PKcs, Artemis or ATM have only a minor defect (<10%) in CSR (191, 235–239). Yet, a recent report revealed that defects of 3’-flap endonuclease XPF-ERCC1 in B cells impairs A-EJ-mediated CSR by impeding joining of resected 3′ flap DSB ends (240). Since 53BP1 and Shieldin both block resection and promote NHEJ, loss of either would be expected to promote resection over NHEJ (241–245). However, even loss of LIG4, which abolishes NHEJ, decreases CSR by only 70% (236, 238). A major feature of CSR is removal of large regions of chromatin between the switch regions to be joined. 53BP1 plays a role in looping DNA at telomeres and is required for rejoining of distal joins in V(D)J recombination (246–248), suggesting that long-range conformational changes in DNA may be disrupted in 53BP1-deficient cells, possibly explaining the importance of 53BP1 in CSR (249, 250). Indeed, loss of components of the shieldin complex, which protects DSB ends to mediate 53BP1-dependent repair, also yield defects in CSR (242, 245, 250–252). Alternatively, 53BP1 also recruits PTIP, an evolutionarily conserved chromatin regulator that binds γH2AX, acts as a major effector of ATM and ATR signaling mechanisms, and is also implicated in CSR (253, 254).
Besides the direct usage of DDR pathways, there are several indirect links between DDR elements in CSR and SHM. For instance, targeting HR by RAD51 inhibitor reduces AID expression, hampering the repair of AID-initiated lesions (255). Interestingly, indirect links reach into RNA-binding proteins such as the autism-associated protein vigilin, which interacts with RAD51 and BRCA1, so its depletion impairs their recruitment to DSB sites (256).
Emerging evidence supports the idea that DDR inhibition in tumor cells remodels the inflammatory microenvironment (10, 257). Impaired DDR typically enhances the tumor foreignness by increasing the number of tumor cell mutations/neoantigens (10, 258). When examined by CIBERSORT analysis through the TIMER2.0 web server (259, 260), the mRNA levels of many DDR factors, such as RPA1, Ku70, Ku80, MRE11A, RAD50, NBS1, PRKDC, RAD51, PARG and XRCC4, were negatively associated with cytotoxic CD8+ T cells infiltration levels across various cancer types (Figure 3A). Indeed, as exemplified in prostate adenocarcinoma, significant negative correlations between gene expression and cytotoxic T cell infiltration levels were found in 19 genes of the 22 DDR related genes we tested (Figure 3A, B). These findings suggest enhanced anticancer immunity in tumors with lower DDR factor expression and imply substantial potential benefits from DNA repair inhibitors. Thus, inhibitors of these DDR factors, such as poly(ADP-ribose) glycohydrolase (PARG) inhibitors that impact DNA break repair and replication fork restart, may be employed to activate the innate immune response (261).
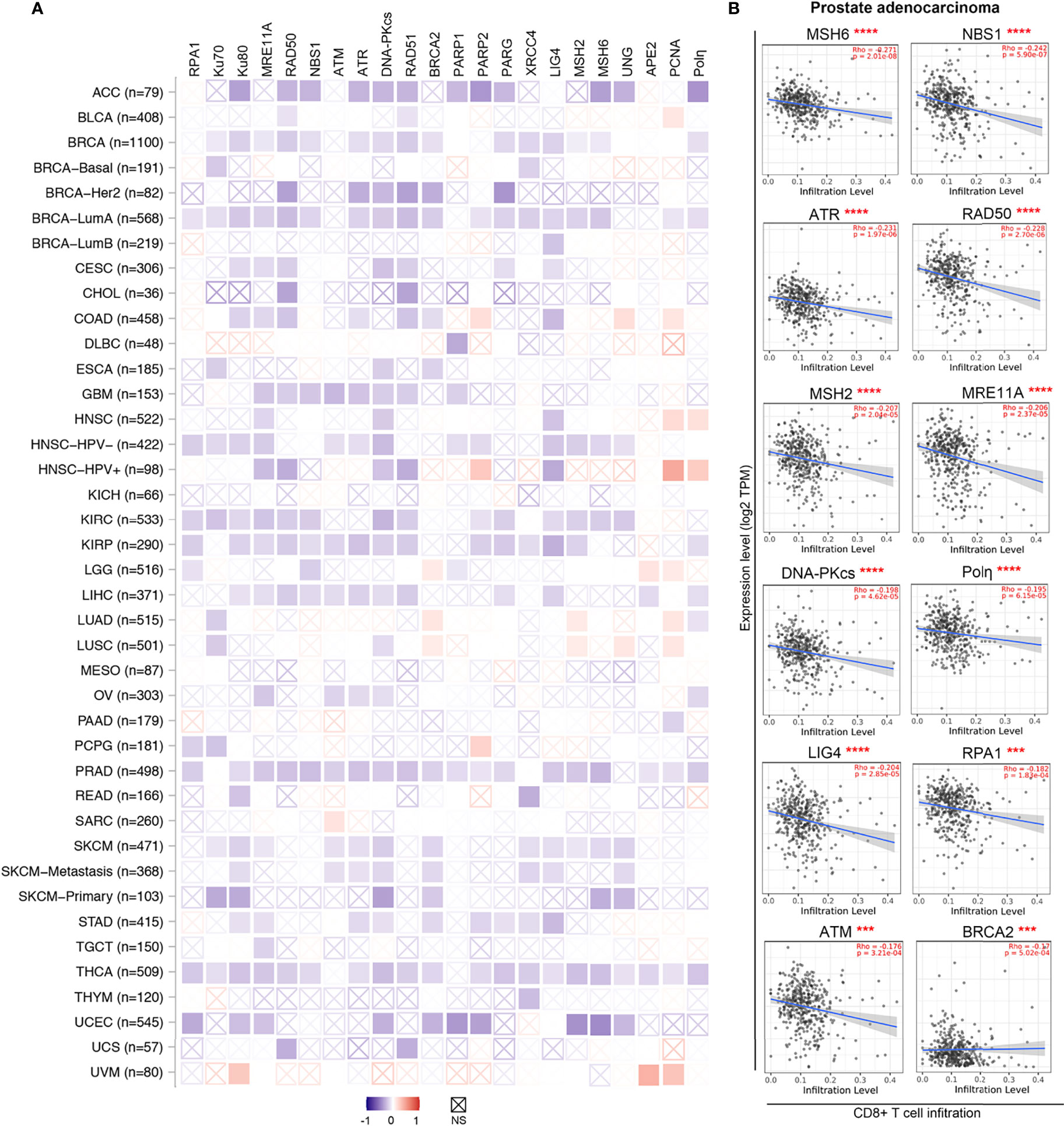
Figure 3 DDR factors negatively associate with CD8+ T cells infiltration levels in diverse cancer types. (A) A heatmap based on the CIBRSORT method shows the purity-adjusted Spearman’s rho of DDR factors with CD8+ T cells across various cancer types. The boxes with indicate non-significant p values (p>0.05). The figures was made using the TIMER2.0 web server based on CIBRSORT analysis (http://timer.cistrome.org/). (B) Detailed correlation between DDR factors and CD8+ T cells in prostate adenocarcinoma (PRAD) from panel (A) The purity-adjusted Spearman’s rho and p value are labeled in red. ***p < 0.001; ****p < 0.00001.
Antitumor immune responses can be promoted and utilized to treat cancer via immune checkpoint blockade with use of agents such as PD-1/PD-L1 and CTLA-4 inhibitors (29, 262, 263). The DDR also offers attractive targets for inhibition (264, 265). Preclinical and clinical efficacy of DDR inhibition in cells with a defective DDR genetic background, are exemplified by the success of PARP inhibitors in BRCA1/2-mutated advanced cancers and of inhibitors to the PARG in cancer cells (261, 266, 267). Emerging evidence has progressively unveiled the involvement of the DDR in antitumor immunity by enhancing STING-dependent immune responses, further supporting the immune-modulatory role of DDR inhibition in anticancer immunity (Figure 4) (134, 135, 268, 269).
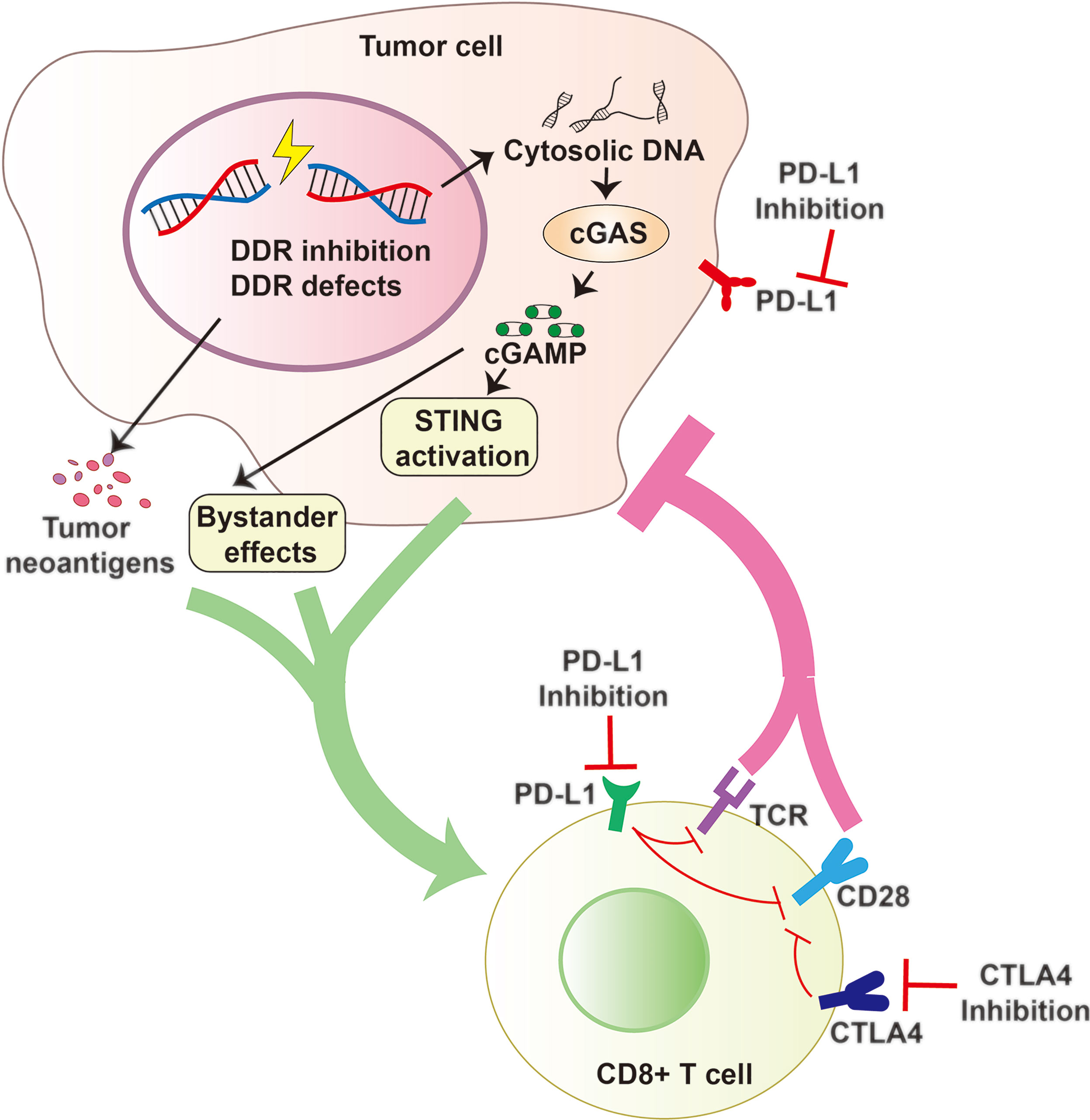
Figure 4 DDR Inhibition and Antitumor Immunity. DDR Inhibition and DDR defects can increase cytosolic DNA that activates the cGAS to generate cGAMP and promote tumor neoantigen production. cGAMP can activate cell intrinsic STING pathway and spread the immunity to bystander cells. All these factors contribute to an inflammatory tumor microenvironment and promote the recruitment of cytotoxic CD8+ T cells and constrict cancer growth effectively. Combining DDR inhibition (such as PARP or PARG inhibition) with Immune checkpoint blockade (including PD-1/PD-L1 or CTLA4 blockade) may be a promising strategy with the potential to improve survival outcomes.
The most studied DDR inhibitors in anticancer immunotherapies are those directed against PARP (PARPi). In line with the usage of PARPi in DDR-deficient tumors (266), PARPi combined with immune checkpoint blockade, including PD-1/PD-L1 and CTLA-4, exerts remarkable efficacy in tumors with BRCA1/2 or ERCC1 mutations via STING-dependent immune responses and infiltration of cytotoxic T cells into tumor (50, 51, 54, 270). There are also findings suggesting that PARPi, with anti-PD-1 inhibitors, have strong therapeutic potential regardless of BRCA1/2 status (49, 271, 272), although the mechanisms involved remain unclear. Besides the STING-dependent pathways, PARPi also increased PD-L1 expression in breast cancer cell lines through inhibition of GSK3β (273), which provided the rationale for combining PARPi with PD-L1 or PD-1 immune checkpoint blockade, a strategy that has been tested in clinical trials (49, 271, 274).
Recently, many other inhibitors targeting DDR components have been developed and are in preclinical study. Recently, several of them, including inhibitors of DNA-PKcs, ATM, ATR, CHK1 and WEE1, have entered into clinical trials (275). Inhibitors of DNA-PKcs promote radiation sensitization through inhibition of NHEJ (276). Their importance in modulating the innate immune response have also been demonstrated. ATR inhibition can further increase cGAS-positive micronuclei and cytokine production in PARPi-treated cancer cells (12). Significantly, inhibition of DNA-PK with AZD7648 resulted in IFN-dependent inhibition of tumor growth following IR in immune competent mouse models, indicating that inhibition of DNA-PK in combination with radiotherapy could lead to durable immune-mediated tumor control in cancer patients (277).
Another important application of DDR in antitumor immunotherapy is the usage of the DDR status as biomarkers to select the patients who are targetable to immune checkpoint blockade. Currently, only a subset of patients respond to immune checkpoint blockade. Predictive biomarkers for reliable response could better guide therapeutic choices (104). As DNA repair deficiencies that promote genome instability are relatively common among tumors, mutational signatures and DDR biomarkers may identify features associated with response to immune-directed therapies. For instance, MMR status was reported to predict response to the PD-1 inhibitor pembrolizumab in a phase 2 study of 41 patients with progressive metastatic carcinoma (278, 279). Also, loss of BRCA1 and defects of MMR in tumors resulted in many somatic mutations, leading to continuous renewal of neoantigens, increased immune response gene expression, and enhancement of immune surveillance (20, 270, 278, 280). In non–small cell lung cancer, deleterious mutations in several DDR-related genes correlated with pembrolizumab clinical efficacy (281). A high mutation level causing a high load of tumor neoantigens suppresses immune evasion. Whereas aneuploidy of large chromosomal regions (arm and whole-chromosome), which cause somatic copy number alterations (SCNAs) and consequent protein imbalances, can weaken cytotoxic immune cell infiltration (282). Importantly, blockade of the immune system PD-1/PD-L1 inhibitory pathway can restore exhausted immune responses as an effective immunological strategy to overcome immune evasion by chronic imbalances and infections (283). For monoclonal antibodies used to block checkpoint molecules, such as PD-1 and PD-L1, to activate immune cells to kill tumor cells more effectively, it may be worth adding designed features such as metal ion binding sites to add to their capabilities or removing free cysteines to improve their stability (284–286).
The DDR shapes how the innate immune system responds to tumors, as well as how the adaptive immune system is recruited to sites of malignancy. Consequently, the interconnections of the DDR and the immune system, which maintain genomic fitness and pathogen protection, can be utilized to improve cancer therapeutic strategies (5, 135, 287–291). Yet, defining how the DDR impacts immune responses has remained challenging as immune activation can evidently be triggered by different types of DDR components including DNA damage sensors, transducers, and effectors (292).
Here, we assessed current molecular and mechanistic data showing how the DDR induces and impacts immune responses. At present, cancer immunotherapy is less widely used than surgery, chemotherapy, or radiation therapy. As only a subset of patients respond to immune checkpoint blockade, enhancements from defining and modulating the DDR along with reliable predictive biomarkers of response are needed to guide and improve therapeutic strategies. DNA repair deficiency is common among tumors, and emerging experimental and clinical evidence suggests that features of genomic instability are associated with response to immune-directed therapies. We propose that advancing all successful cancer therapies will benefit from elucidating key molecular and mechanistic relationships linking DDR, DNA damage outcomes, and immune responses. In fact, the efficacy of conventional chemotherapy and radiotherapy can depend in part upon induction of innate and adaptive immunity.
In innate immunity (Figure 1), MRN (along with its associated ATM and ATR kinases) and DNA-PK complex, which co-regulates DNA DSB repair, can serve as master cytosolic DNA sensors to initiate innate immune response. DNA-PKcs expression with validated immune biomarkers can guide patient selection for DNA-PKcs targeting strategies, DNA-damaging agents, and their combination with an immune-checkpoint blockade (293). Analogously, ATM inhibition induces tumor growth delay and overcomes tumor resistance to anti–PD-1 therapy (294). In addition, other DDR components interact with and promote cytosolic DNA sensing pathways or RIG-I–mediated RNA sensing signaling to trigger innate immune response. Whereas mice and other model systems have proven to be of great value for testing these molecular mechanisms, it is critical to consider possible impacts from the far higher DNA-PKcs levels in human cells compared to laboratory mice (104).
Most immune-related DDR components and immune responses converge upon the STING-IFN signaling pathway, which plays a crucial role in cancer cell immune-surveillance. In adaptive immunity (Figure 2), DDR pathways (including MMR, BER, NHEJ, and A-EJ) are required for V(D)J recombination, CSR and SHM processes, which are critical to lymphocyte development. From a pathology standpoint, DDR modulates anticancer immunity via both innate and adaptive immunity, with the underlying molecular mechanisms being increasingly defined. Such knowledge is likely broadly applicable to human disease, including cancer, infectious disease and atherosclerotic disorders. For instance, SARS CoV-2 proteins, can hijack the human immune response to pathogens and the DNA damage repair system, thereby damaging both innate and adaptive immunity (295, 296). Furthermore, the results of targeting endonuclease V, a ROS response and structure-specific nuclease that cleaves DNA and RNA at inosines as a regulator of innate immune responses, suggests blocking such DDR-related epitranscriptomic modifications to ameliorate carotid atherosclerosis and ischemic stroke (297–299).
For advanced immunotherapeutic strategies, DDR defects plus the increased mutation load in tumor cells produce tumor-specific neoantigens. So chemical tools to alter the DDR in predetermined ways can leverage the full power of cancer immunotherapy. Importantly, advances in structural biology for combining atomic resolution structures with X-ray scattering and computation for solution conformations and assemblies are providing critical enabling methods to define and target dynamic complexes that can generally control mutation rates (66, 300–302). We propose here that the dynamic DNA-PK and MRN-activated ATM and ATR are potential master keys to unlock DDR and their immune system roles. As DNA-PKcs, ATM, and ATR inhibitors are already being evaluated in clinical trials as sensitizers of chemotherapy and radiotherapy, we suggest that these kinases may be both a predictive biomarkers and therapeutic targets for immunotherapy in future clinical trials.
To effectively use such master keys, it will be important to better define the molecular mechanisms orchestrating their activities in DDR and immune system outcomes and their potential as biomarkers for prognosis. We know that with molecular mechanistic knowledge, examination of DDR status can provide informed predictive biomarkers for patient selection and therapeutic approaches (135). Moreover, like immune checkpoint inhibitors, DDR inhibition strategies show great potential to improve cancer treatment efficacy by harnessing their immunomodulatory effects for radiation and chemotherapies, immune checkpoint blockade, and combined therapeutic strategies.
ZY and YS contributed equally to write the original draft. JT and SL-M contributed equally to the conceptualization and revising. All authors listed have made direct and substantial contribution to this work, and approved the final manuscript.
Our research was supported by National Institutes of Health (NIH) grants (R01 CA200231; P01 CA092584; R35 CA220430; 1S10OD012304-01) and by Cancer Prevention Research Institute of Texas (CPRIT) grant (RP180813). JT’s efforts are also supported by a Robert A. Welch Chemistry Chair. YS’ efforts are supported by research grants from National Natural Science Foundation of China (Grant No. 31801161).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We acknowledge useful discussions with members of our laboratories and with members of our program on PARPi and targeting the BRCA interactome (Junjie Chen, Katharina Schlacher, Constance Albarracin, and Banu Arun). We apologize for the many excellent papers not cited due to space limitations and for our focus on results with which we are most familiar for the exemplary work cited. We thank our colleagues Katheryn Meek, Patricia J. Gearhart, James J. Mancuso, Albino Bacolla, Chris Brosey, Natalie Y.L. Ngoi, Shengfeng Xu, and Shan Zha for valuable suggestions. We acknowledge Tamara Locke at Research Medical Library at the University of Texas MD Anderson Cancer Center for editorial assistance.
5-FdU: floxuridine
53BP1: p53-binding protein
8-OHG: 8-hydroxyguanosine
A-EJ: alternative end joining
AgR: antigen receptor
AID: activation-induced cytidine deaminase
APE2: Apurinic/apyrimidinic endodeoxyribonuclease 2
APOBEC: apolipoprotein B mRNA editing enzyme catalytic polypeptide like
ATM: ataxia telangiectasia mutated
A-T: Ataxia-telangiectasia
BCL10: B cell CLL/lymphoma 10
BER: base excision repair
BLM: bloom syndrome RecQ like helicase
BRCA1/2: breast-cancer susceptibility gene 1/2
BS: bloom syndrome
CARD9: caspase-recruitment domain
CHK1: checkpoint kinase 1
CTLA-4: cytotoxic T-lymphocyte-associated antigen 4
cGAMP: 2’-3’cGAMP
cGAS: cyclic GMP-AMP synthase
CSR: class-switch recombination
DAMPs: damage-associated molecular patterns
DDR: DNA damage response
DNA-PK: DNA-dependent protein kinase
DSBs: double-strand breaks
EEPD1: endonuclease/exonuclease/phosphatase family domain containing 1
dsDNA: double-stranded DNA
ERCC1: excision repair cross complementary gene 1
EXO1: exonuclease 1
EXO5: exonuclease 5
FANCD2: Fanconi anemia complementation group D2
FEN1: flap structure-specific endonuclease 1
GSK3β: glycogen synthase kinase 3 beta
GRB2: growth factor receptor bound protein 2
H2AX: H2A histone family member X
HDP-RNP: HEXIM1-DNA-PK-paraspeckle components-ribonucleoprotein complex
HIV: human immunodeficiency virus
HMGB1: high mobility group box 1
HR: homologous recombination
HSPA8: heat shock protein family A (Hsp70) member 8
HSV-1: herpes simplex virus 1
Ig: immunoglobulin
IRF3: interferon regulatory factor 3
IFI16: IFN-inducible protein 16
IFN: interferon
LAT: Linker of Activation of T cells
LIG4: DNA ligase IV
LINP1: lncRNA in nonhomologous end joining (NHEJ) pathway 1
IL7: interleukin 7
IR: ionizing radiation
MAVS: mitochondrial antiviral signaling protein
MDC1: Mediator of DNA damage checkpoint 1
MMR: mismatch repair
MLH1: MutL homolog 1
MRE11: meiotic recombination 11 homolog 1
MRN: MRE11-RAD50-NBS1
MSH2: MutS homolog 2
MSH6: MutS homolog 6
MUTY: MutY DNA glycosylase
NBS1: Nijmegen breakage syndrome protein 1
NEIL: endonuclease VIII (Nei)-like proteins
NER: nucleotide excision repair
NF-κB: nuclear factor kappa B subunit 1
NHEJ: non-homologous end joining
OGG1: oxoguanine DNA glycosylase
p53: tumor protein p53
PARG: poly(ADP-ribose) glycohydrolase
PARP: poly (ADP-ribose) polymerase
PARPi: PARP inhibitors
PAMPs: pathogen associated molecular patterns
PCNA: proliferating cell nuclear antigen
PD-1/PD-L1: programmed cell death protein 1/programmed cell death ligand 1
PIKK: phosphatidylinositol 3-kinase (PI3K)-related kinase
PMS2: PMS1 homolog 2
Polη: DNA polymerase η
pre-BCR: pre-B cell receptor
PRRs: pattern recognition receptors
PRKDC: Protein kinase, DNA-activated, catalytic polypeptide
RAD50: ATP-binding cassette (ABC)-ATPase 50
RAD51: ATP-binding cassette (ABC)-ATPase 51
RAG: recombinase activating gene
RIG-I: retinoic acid-inducible gene I
RPA: replication protein A
ROS: reactive oxygen species
RSS: recombination signal sequences
SARS CoV-2: severe acute respiratory syndrome coronavirus-2
SCID: severe combined immunodeficiency
SHM: somatic hypermutation
SCNAs: somatic copy number alterations
SIDSP: STING-independent DNA sensing pathway
SSBs: single-strand breaks
STING: stimulator of interferon genes
TBK1: TANK-binding kinase 1
TCRβ: T cell receptor beta
TdT: terminal deoxynucleotidyl transferase
TREX1: three prime repair exonuclease 1
WEE1: WEE1 G2 checkpoint kinase
XLF: XRCC4-like factor
XRCC1: x-ray repair cross-complementing group 1
XRCC4: x-ray repair cross-complementing group 4
UNG: uracil DNA glycosylase
V(D)J: variable, diversity, and joining.
2. Bacolla A, Ye Z, Ahmed Z, Tainer JA. Cancer Mutational Burden is Shaped by G4 DNA, Replication Stress and Mitochondrial Dysfunction. Prog Biophys Mol Biol (2019) 147:47–61. doi: 10.1016/j.pbiomolbio.2019.03.004
3. Stratigopoulou M, van Dam TP, Guikema JEJ. Base Excision Repair in the Immune System: Small DNA Lesions With Big Consequences. Front Immunol (2020) 11:1084. doi: 10.3389/fimmu.2020.01084
4. Jackson SP, Bartek J. The DNA-Damage Response in Human Biology and Disease. Nature (2009) 461:1071–8. doi: 10.1038/nature08467
5. Pilger D, Seymour LW, Jackson SP. Interfaces Between Cellular Responses to DNA Damage and Cancer Immunotherapy. Genes Dev (2021) 35:602–18. doi: 10.1101/gad.348314.121
6. Bacolla A, Tainer JA, Vasquez KM, Cooper DN. Translocation and Deletion Breakpoints in Cancer Genomes are Associated With Potential non-B DNA-Forming Sequences. Nucleic Acids Res (2016) 44:5673–88. doi: 10.1093/nar/gkw261
7. Zeman MK, Cimprich KA. Causes and Consequences of Replication Stress. Nat Cell Biol (2014) 16:2–9. doi: 10.1038/ncb2897
8. García-Muse T, Aguilera A. Transcription-Replication Conflicts: How They Occur and How They are Resolved. Nat Rev Mol Cell Biol (2016) 17:553–63. doi: 10.1038/nrm.2016.88
9. Sollier J, Stork CT, García-Rubio ML, Paulsen RD, Aguilera A, Cimprich KA. Transcription-Coupled Nucleotide Excision Repair Factors Promote R-Loop-Induced Genome Instability. Mol Cell (2014) 56:777–85. doi: 10.1016/j.molcel.2014.10.020
10. Ragu S, Matos-Rodrigues G, Lopez BS, Replication Stress DNA. Damage, Inflammatory Cytokines and Innate Immune Response. Genes (Basel) (2020) 11(4):409. doi: 10.3390/genes11040409
11. Reisländer T, Lombardi EP, Groelly FJ, Miar A, Porru M, Di Vito S, et al. BRCA2 Abrogation Triggers Innate Immune Responses Potentiated by Treatment With PARP Inhibitors. Nat Commun (2019) 10:3143. doi: 10.1038/s41467-019-11048-5
12. Schoonen PM, Kok YP, Wierenga E, Bakker B, Foijer F, Spierings DCJ, et al. Premature Mitotic Entry Induced by ATR Inhibition Potentiates Olaparib Inhibition-Mediated Genomic Instability, Inflammatory Signaling, and Cytotoxicity in BRCA2-Deficient Cancer Cells. Mol Oncol (2019) 13:2422–40. doi: 10.1002/1878-0261.12573
13. Syed A, Tainer JA. The MRE11-RAD50-NBS1 Complex Conducts the Orchestration of Damage Signaling and Outcomes to Stress in DNA Replication and Repair. Annu Rev Biochem (2018) 87:263–94. doi: 10.1146/annurev-biochem-062917-012415
14. Li Z, Pearlman AH, Hsieh P. DNA Mismatch Repair and the DNA Damage Response. DNA Repair (Amst) (2016) 38:94–101. doi: 10.1016/j.dnarep.2015.11.019
15. Ciccia A, Elledge SJ. The DNA Damage Response: Making it Safe to Play With Knives. Mol Cell (2010) 40:179–204. doi: 10.1016/j.molcel.2010.09.019
16. Hitomi K, Iwai S, Tainer JA. The Intricate Structural Chemistry of Base Excision Repair Machinery: Implications for DNA Damage Recognition, Removal, and Repair. DNA Repair (Amst) (2007) 6:410–28. doi: 10.1016/j.dnarep.2006.10.004
17. Arcas A, Fernández-Capetillo O, Cases I, Rojas AM. Emergence and Evolutionary Analysis of the Human DDR Network: Implications in Comparative Genomics and Downstream Analyses. Mol Biol Evol (2014) 31:940–61. doi: 10.1093/molbev/msu046
18. Aravind L, Walker DR, Koonin EV. Conserved Domains in DNA Repair Proteins and Evolution of Repair Systems. Nucleic Acids Res (1999) 27:1223–42. doi: 10.1093/nar/27.5.1223
19. Härtlova A, Erttmann SF, Raffi FA, Schmalz AM, Resch U, Anugula S, et al. DNA Damage Primes the Type I Interferon System via the Cytosolic DNA Sensor STING to Promote Anti-Microbial Innate Immunity. Immunity (2015) 42:332–43. doi: 10.1016/j.immuni.2015.01.012
20. Lu C, Guan J, Lu S, Jin Q, Rousseau B, Lu T, et al. DNA Sensing in Mismatch Repair-Deficient Tumor Cells Is Essential for Anti-Tumor Immunity. Cancer Cell (2021) 39:96–108.e6. doi: 10.1016/j.ccell.2020.11.006
21. Wayne J, Brooks T, Landras A, Massey AJ. Targeting DNA Damage Response Pathways to Activate the STING Innate Immune Signaling Pathway in Human Cancer Cells. FEBS J (2021) 288:4507–40. doi: 10.1111/febs.15747
22. Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. Mitotic Progression Following DNA Damage Enables Pattern Recognition Within Micronuclei. Nature (2017) 548:466–70. doi: 10.1038/nature23470
23. James CD, Das D, Bristol ML, Morgan IM. Activating the DNA Damage Response and Suppressing Innate Immunity: Human Papillomaviruses Walk the Line. Pathogens (2020) 9(6):467. doi: 10.3390/pathogens9060467
24. Brodin P, Davis MM. Human Immune System Variation. Nat Rev Immunol (2017) 17:21–9. doi: 10.1038/nri.2016.125
25. Bonilla FA, Oettgen HC. Adaptive Immunity. J Allergy Clin Immunol (2010) 125:S33–40. doi: 10.1016/j.jaci.2009.09.017
26. Carty M, Guy C, Bowie AG. Detection of Viral Infections by Innate Immunity. Biochem Pharmacol (2021) 183:114316. doi: 10.1016/j.bcp.2020.114316
27. Zhang Z, Lu M, Qin Y, Gao W, Tao L, Su W, et al. Neoantigen: A New Breakthrough in Tumor Immunotherapy. Front Immunol (2021) 12:672356. doi: 10.3389/fimmu.2021.672356
28. Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, et al. Checkpoint Blockade Cancer Immunotherapy Targets Tumour-Specific Mutant Antigens. Nature (2014) 515:577–81. doi: 10.1038/nature13988
29. Sharma P, Allison JP. The Future of Immune Checkpoint Therapy. Science (2015) 348:56–61. doi: 10.1126/science.aaa8172
30. Dhatchinamoorthy K, Colbert JD, Rock KL. Cancer Immune Evasion Through Loss of MHC Class I Antigen Presentation. Front Immunol (2021) 12:636568. doi: 10.3389/fimmu.2021.636568
31. Li D, Wu M. Pattern Recognition Receptors in Health and Diseases. Signal Transduct Target Ther (2021) 6(1):291. doi: 10.1038/s41392-021-00687-0
32. Shen YJ, Le Bert N, Chitre AA, Koo CX, Nga XH, Ho SS, et al. Genome-Derived Cytosolic DNA Mediates Type I Interferon-Dependent Rejection of B Cell Lymphoma Cells. Cell Rep (2015) 11:460–73. doi: 10.1016/j.celrep.2015.03.041
33. Gasser S, Orsulic S, Brown EJ, Raulet DH. The DNA Damage Pathway Regulates Innate Immune System Ligands of the NKG2D Receptor. Nature (2005) 436:1186–90. doi: 10.1038/nature03884
34. Wischnewski M, Ablasser A. Interplay of cGAS With Chromatin. Trends Biochem Sci (2021) 46:822–31. doi: 10.1016/j.tibs.2021.05.011
35. Hopfner KP, Hornung V. Molecular Mechanisms and Cellular Functions of cGAS-STING Signalling. Nat Rev Mol Cell Biol (2020) 21:501–21. doi: 10.1038/s41580-020-0244-x
36. Cai X, Chiu YH, Chen ZJ. The cGAS-cGAMP-STING Pathway of Cytosolic DNA Sensing and Signaling. Mol Cell (2014) 54:289–96. doi: 10.1016/j.molcel.2014.03.040
37. Li T, Chen ZJ. The cGAS-cGAMP-STING Pathway Connects DNA Damage to Inflammation, Senescence, and Cancer. J Exp Med (2018) 215:1287–99. doi: 10.1084/jem.20180139
38. Zhou C, Chen X, Planells-Cases R, Chu J, Wang L, Cao L, et al. Transfer of cGAMP Into Bystander Cells via LRRC8 Volume-Regulated Anion Channels Augments STING-Mediated Interferon Responses and Anti-Viral Immunity. Immunity (2020) 52:767–781.e6. doi: 10.1016/j.immuni.2020.03.016
39. Ablasser A, Schmid-Burgk JL, Hemmerling I, Horvath GL, Schmidt T, Latz E, et al. Cell Intrinsic Immunity Spreads to Bystander Cells via the Intercellular Transfer of cGAMP. Nature (2013) 503:530–4. doi: 10.1038/nature12640
40. Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, et al. Phosphorylation of Innate Immune Adaptor Proteins MAVS, STING, and TRIF Induces IRF3 Activation. Science (2015) 347:aaa2630. doi: 10.1126/science.aaa2630
41. Cheng Z, Dai T, He X, Zhang Z, Xie F, Wang S, et al. The Interactions Between cGAS-STING Pathway and Pathogens. Signal Transduct Target Ther (2020) 5(1):91. doi: 10.1038/s41392-020-0198-7
42. Dhanwani R, Takahashi M, Sharma S. Cytosolic Sensing of Immuno-Stimulatory DNA, the Enemy Within. Curr Opin Immunol (2018) 50:82–7. doi: 10.1016/j.coi.2017.11.004
43. Liu H, Zhang H, Wu X, Ma D, Wu J, Wang L, et al. Nuclear cGAS Suppresses DNA Repair and Promotes Tumorigenesis. Nature (2018) 563:131–6. doi: 10.1038/s41586-018-0629-6
44. Bai J, Liu F. Nuclear cGAS: Sequestration and Beyond. Protein Cell (2021). doi: 10.1007/s13238-021-00869-0
45. Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, et al. IFI16 is an Innate Immune Sensor for Intracellular DNA. Nat Immunol (2010) 11:997–1004. doi: 10.1038/ni.1932
46. Meek K. An Antiviral DNA Response Without the STING? Trends Immunol (2020) 41:362–4. doi: 10.1016/j.it.2020.03.010
47. Ghosh R, Roy S, Franco S. PARP1 Depletion Induces RIG-I-Dependent Signaling in Human Cancer Cells. PloS One (2018) 13:e0194611. doi: 10.1371/journal.pone.0194611
48. Kim C, Wang XD, Yu Y. PARP1 Inhibitors Trigger Innate Immunity via PARP1 Trapping-Induced DNA Damage Response. Elife (2020) 9:e60637. doi: 10.7554/eLife.60637
49. Shen J, Zhao W, Ju Z, Wang L, Peng Y, Labrie M, et al. PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer Res (2019) 79:311–9. doi: 10.1158/0008-5472.CAN-18-1003
50. Ding L, Kim HJ, Wang Q, Kearns M, Jiang T, Ohlson CE, et al. PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep (2018) 25:2972–2980.e5. doi: 10.1016/j.celrep.2018.11.054
51. Chabanon RM, Muirhead G, Krastev DB, Adam J, Morel D, Garrido M, et al. PARP Inhibition Enhances Tumor Cell-Intrinsic Immunity in ERCC1-Deficient non-Small Cell Lung Cancer. J Clin Invest (2019) 129:1211–28. doi: 10.1172/JCI123319
52. Pantelidou C, Sonzogni O, De Oliveria Taveira M, Mehta AK, Kothari A, Wang D, et al. PARP Inhibitor Efficacy Depends on CD8(+) T-Cell Recruitment via Intratumoral STING Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer Discovery (2019) 9:722–37. doi: 10.1158/2159-8290.CD-18-1218
53. McLaughlin LJ, Stojanovic L, Kogan AA, Rutherford JL, Choi EY, Yen RC, et al. Pharmacologic Induction of Innate Immune Signaling Directly Drives Homologous Recombination Deficiency. Proc Natl Acad Sci USA (2020) 117:17785–95. doi: 10.1073/pnas.2003499117
54. Sen T, Rodriguez BL, Chen L, Corte CMD, Morikawa N, Fujimoto J, et al. Targeting DNA Damage Response Promotes Antitumor Immunity Through STING-Mediated T-Cell Activation in Small Cell Lung Cancer. Cancer Discovery (2019) 9:646–61. doi: 10.1158/2159-8290.CD-18-1020
55. Dunphy G, Flannery SM, Almine JF, Connolly DJ, Paulus C, Jønsson KL, et al. Non-Canonical Activation of the DNA Sensing Adaptor STING by ATM and IFI16 Mediates NF-κb Signaling After Nuclear DNA Damage. Mol Cell (2018) 71:745–760.e5. doi: 10.1016/j.molcel.2018.07.034
56. Wolf C, Rapp A, Berndt N, Staroske W, Schuster M, Dobrick-Mattheuer M, et al. RPA and Rad51 Constitute a Cell Intrinsic Mechanism to Protect the Cytosol From Self DNA. Nat Commun (2016) 7:11752. doi: 10.1038/ncomms11752
57. Brégnard C, Guerra J, Déjardin S, Passalacqua F, Benkirane M, Laguette N. Upregulated LINE-1 Activity in the Fanconi Anemia Cancer Susceptibility Syndrome Leads to Spontaneous Pro-Inflammatory Cytokine Production. EBioMedicine (2016) 8:184–94. doi: 10.1016/j.ebiom.2016.05.005
58. Moschella F, Torelli GF, Valentini M, Urbani F, Buccione C, Petrucci MT, et al. Cyclophosphamide Induces a Type I Interferon-Associated Sterile Inflammatory Response Signature in Cancer Patients' Blood Cells: Implications for Cancer Chemoimmunotherapy. Clin Cancer Res (2013) 19:4249–61. doi: 10.1158/1078-0432.CCR-12-3666
59. Weichselbaum RR, Ishwaran H, Yoon T, Nuyten DS, Baker SW, Khodarev N, et al. An Interferon-Related Gene Signature for DNA Damage Resistance is a Predictive Marker for Chemotherapy and Radiation for Breast Cancer. Proc Natl Acad Sci USA (2008) 105:18490–5. doi: 10.1073/pnas.0809242105
60. Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, et al. Cancer Cell-Autonomous Contribution of Type I Interferon Signaling to the Efficacy of Chemotherapy. Nat Med (2014) 20:1301–9. doi: 10.1038/nm.3708
61. Yu Q, Katlinskaya YV, Carbone CJ, Zhao B, Katlinski KV, Zheng H, et al. DNA-Damage-Induced Type I Interferon Promotes Senescence and Inhibits Stem Cell Function. Cell Rep (2015) 11:785–97. doi: 10.1016/j.celrep.2015.03.069
62. Hambarde S, Tsai CL, Pandita RK, Bacolla A, Maitra A, Charaka V, et al. EXO5-DNA Structure and BLM Interactions Direct DNA Resection Critical for ATR-Dependent Replication Restart. Mol Cell (2021) 81:2989–3006.e9. doi: 10.1016/j.molcel.2021.05.027
63. Sun Y, Wang J, Ma Y, Li J, Sun X, Zhao X, et al. Radiation Induces NORAD Expression to Promote ESCC Radiotherapy Resistance via EEPD1/ATR/Chk1 Signalling and by Inhibiting pri-miR-199a1 Processing and the Exosomal Transfer of miR-199a-5p. J Exp Clin Cancer Res (2021) 40:306. doi: 10.1186/s13046-021-02084-5
64. Gratia M, Rodero MP, Conrad C, Bou Samra E, Maurin M, Rice GI, et al. Bloom Syndrome Protein Restrains Innate Immune Sensing of Micronuclei by cGAS. J Exp Med (2019) 216:1199–213. doi: 10.1084/jem.20181329
65. Storozynsky Q, Hitt MM. The Impact of Radiation-Induced DNA Damage on cGAS-STING-Mediated Immune Responses to Cancer. Int J Mol Sci (2020) 21(22):8877. doi: 10.3390/ijms21228877
66. Bacolla A, Sengupta S, Ye Z, Yang C, Mitra J, De-Paula RB, et al. Heritable Pattern of Oxidized DNA Base Repair Coincides With Pre-Targeting of Repair Complexes to Open Chromatin. Nucleic Acids Res (2021) 49:221–43. doi: 10.1093/nar/gkaa1120
67. Visnes T, Cázares-Körner A, Hao W, Wallner O, Masuyer G, Loseva O, et al. Small-Molecule Inhibitor of OGG1 Suppresses Proinflammatory Gene Expression and Inflammation. Science (2018) 362:834–9. doi: 10.1126/science.aar8048
68. Perry JJ, Shin DS, Getzoff ED, Tainer JA. The Structural Biochemistry of the Superoxide Dismutases. Biochim Biophys Acta (2010) 1804:245–62. doi: 10.1016/j.bbapap.2009.11.004
69. Putnam CD, Arvai AS, Bourne Y, Tainer JA. Active and Inhibited Human Catalase Structures: Ligand and NADPH Binding and Catalytic Mechanism. J Mol Biol (2000) 296:295–309. doi: 10.1006/jmbi.1999.3458
70. Schlee M, Hartmann G. Discriminating Self From non-Self in Nucleic Acid Sensing. Nat Rev Immunol (2016) 16:566–80. doi: 10.1038/nri.2016.78
71. Fan L, Fuss JO, Cheng QJ, Arvai AS, Hammel M, Roberts VA, et al. XPD Helicase Structures and Activities: Insights Into the Cancer and Aging Phenotypes From XPD Mutations. Cell (2008) 133:789–800. doi: 10.1016/j.cell.2008.04.030
72. Fuss JO, Tsai CL, Ishida JP, Tainer JA. Emerging Critical Roles of Fe-S Clusters in DNA Replication and Repair. Biochim Biophys Acta (2015) 1853:1253–71. doi: 10.1016/j.bbamcr.2015.01.018
73. Thayer MM, Ahern H, Xing D, Cunningham RP, Tainer JA. Novel DNA Binding Motifs in the DNA Repair Enzyme Endonuclease III Crystal Structure. EMBO J (1995) 14:4108–20. doi: 10.1002/j.1460-2075.1995.tb00083.x
74. Brosey CA, Ho C, Long WZ, Singh S, Burnett K, Hura GL, et al. Defining NADH-Driven Allostery Regulating Apoptosis-Inducing Factor. Structure (2016) 24:2067–79. doi: 10.1016/j.str.2016.09.012
75. Bahat A, MacVicar T, Langer T. Metabolism and Innate Immunity Meet at the Mitochondria. Front Cell Dev Biol (2021) 9:720490. doi: 10.3389/fcell.2021.720490
76. Luzwick JW, Boisvert RA, Roy S, Park S, Kunnimalaiyaan S, Goffart S, et al. MRE11-Dependent Instability in Mitochondrial DNA Fork Protection Activates cGAS Inflammation. Sci Adv (in press). doi: 10.1126/sciadv.abf9441
77. Tigano M, Vargas DC, Tremblay-Belzile S, Fu Y, Sfeir A. Nuclear Sensing of Breaks in Mitochondrial DNA Enhances Immune Surveillance. Nature (2021) 591:477–81. doi: 10.1038/s41586-021-03269-w
78. MacDonald KM, Benguerfi S, Harding SM. Alerting the Immune System to DNA Damage: Micronuclei as Mediators. Essays Biochem (2020) 64:753–64. doi: 10.1042/EBC20200016
79. Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, et al. cGAS Surveillance of Micronuclei Links Genome Instability to Innate Immunity. Nature (2017) 548:461–5. doi: 10.1038/nature23449
80. Chen X, Xu X, Chen Y, Cheung JC, Wang H, Jiang J, et al. Structure of an Activated DNA-PK and its Implications for NHEJ. Mol Cell (2021) 81:801–810.e3. doi: 10.1016/j.molcel.2020.12.015
81. Hartley KO, Gell D, Smith GC, Zhang H, Divecha N, Connelly MA, et al. DNA-Dependent Protein Kinase Catalytic Subunit: A Relative of Phosphatidylinositol 3-Kinase and the Ataxia Telangiectasia Gene Product. Cell (1995) 82:849–56. doi: 10.1016/0092-8674(95)90482-4
82. Chen S, Lee L, Naila T, Fishbain S, Wang A, Tomkinson AE, et al. Structural Basis of Long-Range to Short-Range Synaptic Transition in NHEJ. Nature (2021) 593:294–8. doi: 10.1038/s41586-021-03458-7
83. Hepburn M, Saltzberg DJ, Lee L, Fang S, Atkinson C, Strynadka NCJ, et al. The Active DNA-PK Holoenzyme Occupies a Tensed State in a Staggered Synaptic Complex. Structure (2021) 29:467–478.e6. doi: 10.1016/j.str.2020.12.006
84. Hammel M, Rosenberg DJ, Bierma J, Hura GL, Thapar R, Lees-Miller SP, et al. Visualizing Functional Dynamicity in the DNA-Dependent Protein Kinase Holoenzyme DNA-PK Complex by Integrating SAXS With cryo-EM. Prog Biophys Mol Biol (2021) 163:74–86. doi: 10.1016/j.pbiomolbio.2020.09.003
85. Lees-Miller JP, Cobban A, Katsonis P, Bacolla A, Tsutakawa SE, Hammel M, et al. Uncovering DNA-PKcs Ancient Phylogeny, Unique Sequence Motifs and Insights for Human Disease. Prog Biophys Mol Biol (2021) 163:87–108. doi: 10.1016/j.pbiomolbio.2020.09.010
86. Hammel M, Yu Y, Mahaney BL, Cai B, Ye R, Phipps BM, et al. Ku and DNA-Dependent Protein Kinase Dynamic Conformations and Assembly Regulate DNA Binding and the Initial non-Homologous End Joining Complex. J Biol Chem (2010) 285:1414–23. doi: 10.1074/jbc.M109.065615
87. Anisenko A, Kan M, Shadrina O, Brattseva A, Gottikh M. Phosphorylation Targets of DNA-PK and Their Role in HIV-1 Replication. Cells (2020) 9(8):1907. doi: 10.3390/cells9081907
88. Xiao X, Liang J, Huang C, Li K, Xing F, Zhu W, et al. DNA-PK Inhibition Synergizes With Oncolytic Virus M1 by Inhibiting Antiviral Response and Potentiating DNA Damage. Nat Commun (2018) 9:4342. doi: 10.1038/s41467-018-06771-4
89. Justice JL, Kennedy MA, Hutton JE, Liu D, Song B, Phelan B, et al. Systematic Profiling of Protein Complex Dynamics Reveals DNA-PK Phosphorylation of IFI16 En Route to Herpesvirus Immunity. Sci Adv (2021) 7(25):eabg6680. doi: 10.1126/sciadv.abg6680
90. Peters NE, Ferguson BJ, Mazzon M, Fahy AS, Krysztofinska E, Arribas-Bosacoma R, et al. A Mechanism for the Inhibition of DNA-PK-Mediated DNA Sensing by a Virus. PloS Pathog (2013) 9:e1003649. doi: 10.1371/journal.ppat.1003649
91. Schwartz C, Rohr O, Wallet C. Targeting the DNA-PK Complex: Its Rationale Use in Cancer and HIV-1 Infection. Biochem Pharmacol (2019) 160:80–91. doi: 10.1016/j.bcp.2018.12.002
92. Frost JR, Olanubi O, Cheng SK, Soriano A, Crisostomo L, Lopez A, et al. The Interaction of Adenovirus E1A With the Mammalian Protein Ku70/XRCC6. Virology (2017) 500:11–21. doi: 10.1016/j.virol.2016.10.004
93. Sun X, Liu T, Zhao J, Xia H, Xie J, Guo Y, et al. DNA-PK Deficiency Potentiates cGAS-Mediated Antiviral Innate Immunity. Nat Commun (2020) 11:6182. doi: 10.1038/s41467-020-19941-0
94. Karpova AY, Trost M, Murray JM, Cantley LC, Howley PM. Interferon Regulatory Factor-3 is an In Vivo Target of DNA-Pk. Proc Natl Acad Sci USA (2002) 99:2818–23. doi: 10.1073/pnas.052713899
95. Cheradame L, Guerrera IC, Gaston J, Schmitt A, Jung V, Goudin N, et al. STING Protects Breast Cancer Cells From Intrinsic and Genotoxic-Induced DNA Instability via a non-Canonical, Cell-Autonomous Pathway. Oncogene (2021). doi: 10.1038/s41388-021-02037-4
96. Ferguson BJ, Mansur DS, Peters NE, Ren H, Smith GL. DNA-PK is a DNA Sensor for IRF-3-Dependent Innate Immunity. Elife (2012) 1:e00047. doi: 10.7554/eLife.00047
97. Morchikh M, Cribier A, Raffel R, Amraoui S, Cau J, Severac D, et al. HEXIM1 and NEAT1 Long Non-Coding RNA Form a Multi-Subunit Complex That Regulates DNA-Mediated Innate Immune Response. Mol Cell (2017) 67:387–399.e5. doi: 10.1016/j.molcel.2017.06.020
98. Sui H, Zhou M, Imamichi H, Jiao X, Sherman BT, Lane HC, et al. STING is an Essential Mediator of the Ku70-Mediated Production of IFN-λ1 in Response to Exogenous DNA. Sci Signal (2017) 10(488):eaah5054. doi: 10.1126/scisignal.aah5054
99. Zhang X, Brann TW, Zhou M, Yang J, Oguariri RM, Lidie KB, et al. Cutting Edge: Ku70 is a Novel Cytosolic DNA Sensor That Induces Type III Rather Than Type I IFN. J Immunol (2011) 186:4541–5. doi: 10.4049/jimmunol.1003389
100. Scutts SR, Ember SW, Ren H, Ye C, Lovejoy CA, Mazzon M, et al. DNA-PK Is Targeted by Multiple Vaccinia Virus Proteins to Inhibit DNA Sensing. Cell Rep (2018) 25:1953–1965.e4. doi: 10.1016/j.celrep.2018.10.034
101. Burleigh K, Maltbaek JH, Cambier S, Green R, Gale M Jr., James RC, et al. Human DNA-PK Activates a STING-Independent DNA Sensing Pathway. Sci Immunol (2020) 5(43):eaba4219. doi: 10.1126/sciimmunol.aba4219
102. Bañuelos CA, Banáth JP, MacPhail SH, Zhao J, Eaves CA, O'Connor MD, et al. Mouse But Not Human Embryonic Stem Cells are Deficient in Rejoining of Ionizing Radiation-Induced DNA Double-Strand Breaks. DNA Repair (Amst) (2008) 7:1471–83. doi: 10.1016/j.dnarep.2008.05.005
103. Daza P, Reichenberger S, Göttlich B, Hagmann M, Feldmann E, Pfeiffer P. Mechanisms of Nonhomologous DNA End-Joining in Frogs, Mice and Men. Biol Chem (1996) 377:775–86. doi: 10.1515/bchm3.1996.377.12.775
104. Finnie NJ, Gottlieb TM, Blunt T, Jeggo PA, Jackson SP. DNA-Dependent Protein Kinase Activity is Absent in Xrs-6 Cells: Implications for Site-Specific Recombination and DNA Double-Strand Break Repair. Proc Natl Acad Sci USA (1995) 92:320–4. doi: 10.1073/pnas.92.1.320
105. Thapar R, Wang JL, Hammel M, Ye R, Liang K, Sun C, et al. Mechanism of Efficient Double-Strand Break Repair by a Long non-Coding RNA. Nucleic Acids Res (2020) 48:10953–72. doi: 10.1093/nar/gkaa784
106. Williams RS, Williams JS, Tainer JA. Mre11-Rad50-Nbs1 is a Keystone Complex Connecting DNA Repair Machinery, Double-Strand Break Signaling, and the Chromatin Template. Biochem Cell Biol (2007) 85:509–20. doi: 10.1139/O07-069
107. Mariggiò G, Koch S, Zhang G, Weidner-Glunde M, Rückert J, Kati S, et al. Kaposi Sarcoma Herpesvirus (KSHV) Latency-Associated Nuclear Antigen (LANA) Recruits Components of the MRN (Mre11-Rad50-NBS1) Repair Complex to Modulate an Innate Immune Signaling Pathway and Viral Latency. PloS Pathog (2017) 13:e1006335. doi: 10.1371/journal.ppat.1006335
108. Kondo T, Kobayashi J, Saitoh T, Maruyama K, Ishii KJ, Barber GN, et al. DNA Damage Sensor MRE11 Recognizes Cytosolic Double-Stranded DNA and Induces Type I Interferon by Regulating STING Trafficking. Proc Natl Acad Sci USA (2013) 110:2969–74. doi: 10.1073/pnas.1222694110
109. Roth S, Rottach A, Lotz-Havla AS, Laux V, Muschaweckh A, Gersting SW, et al. Rad50-CARD9 Interactions Link Cytosolic DNA Sensing to IL-1β Production. Nat Immunol (2014) 15:538–45. doi: 10.1038/ni.2888
110. Käshammer L, Saathoff JH, Lammens K, Gut F, Bartho J, Alt A, et al. Mechanism of DNA End Sensing and Processing by the Mre11-Rad50 Complex. Mol Cell (2019) 76:382–394.e6. doi: 10.1016/j.molcel.2019.07.035
111. Williams RS, Moncalian G, Williams JS, Yamada Y, Limbo O, Shin DS, et al. Mre11 Dimers Coordinate DNA End Bridging and Nuclease Processing in Double-Strand-Break Repair. Cell (2008) 135:97–109. doi: 10.1016/j.cell.2008.08.017
112. Shibata A, Moiani D, Arvai AS, Perry J, Harding SM, Genois MM, et al. DNA Double-Strand Break Repair Pathway Choice is Directed by Distinct MRE11 Nuclease Activities. Mol Cell (2014) 53:7–18. doi: 10.1016/j.molcel.2013.11.003
113. Tsutakawa SE, Sarker AH, Ng C, Arvai AS, Shin DS, Shih B, et al. Human XPG Nuclease Structure, Assembly, and Activities With Insights for Neurodegeneration and Cancer From Pathogenic Mutations. Proc Natl Acad Sci USA (2020) 117:14127–38. doi: 10.1073/pnas.1921311117
114. Tsutakawa SE, Thompson MJ, Arvai AS, Neil AJ, Shaw SJ, Algasaier SI, et al. Phosphate Steering by Flap Endonuclease 1 Promotes 5'-Flap Specificity and Incision to Prevent Genome Instability. Nat Commun (2017) 8:15855. doi: 10.1038/ncomms15855
115. Trego KS, Chernikova SB, Davalos AR, Perry JJ, Finger LD, Ng C, et al. The DNA Repair Endonuclease XPG Interacts Directly and Functionally With the WRN Helicase Defective in Werner Syndrome. Cell Cycle (2011) 10:1998–2007. doi: 10.4161/cc.10.12.15878
116. Perry JJ, Yannone SM, Holden LG, Hitomi C, Asaithamby A, Han S, et al. WRN Exonuclease Structure and Molecular Mechanism Imply an Editing Role in DNA End Processing. Nat Struct Mol Biol (2006) 13:414–22. doi: 10.1038/nsmb1088
117. Moiani D, Ronato DA, Brosey CA, Arvai AS, Syed A, Masson JY, et al. Targeting Allostery With Avatars to Design Inhibitors Assessed by Cell Activity: Dissecting MRE11 Endo- and Exonuclease Activities. Methods Enzymol (2018) 601:205–41. doi: 10.1016/bs.mie.2017.11.030
118. Ye Z, Xu S, Shi Y, Bacolla A, Syed A, Moiani D, et al. GRB2 Enforces Homology-Directed Repair Initiation by MRE11. Sci Adv (2021) 7(32):eabe9254. doi: 10.1126/sciadv.abe9254
119. Dinur-Schejter Y, Zaidman I, Mor-Shaked H, Stepensky P. The Clinical Aspect of Adaptor Molecules in T Cell Signaling: Lessons Learnt From Inborn Errors of Immunity. Front Immunol (2021) 12:701704. doi: 10.3389/fimmu.2021.701704
120. Houtman JC, Yamaguchi H, Barda-Saad M, Braiman A, Bowden B, Appella E, et al. Oligomerization of Signaling Complexes by the Multipoint Binding of GRB2 to Both LAT and SOS1. Nat Struct Mol Biol (2006) 13:798–805. doi: 10.1038/nsmb1133
121. Prasad M, Brzostek J, Gautam N, Balyan R, Rybakin V, Gascoigne NRJ. Themis Regulates Metabolic Signaling and Effector Functions in CD4. Cell Mol Immunol (2021) 18:2249–61. doi: 10.1038/s41423-020-00578-4
122. Tubbs JL, Latypov V, Kanugula S, Butt A, Melikishvili M, Kraehenbuehl R, et al. Flipping of Alkylated DNA Damage Bridges Base and Nucleotide Excision Repair. Nature (2009) 459:808–13. doi: 10.1038/nature08076
123. Williams RS, Dodson GE, Limbo O, Yamada Y, Williams JS, Guenther G, et al. Nbs1 Flexibly Tethers Ctp1 and Mre11-Rad50 to Coordinate DNA Double-Strand Break Processing and Repair. Cell (2009) 139:87–99. doi: 10.1016/j.cell.2009.07.033
124. Kobayashi J, Tauchi H, Sakamoto S, Nakamura A, Morishima K, Matsuura S, et al. NBS1 Localizes to Gamma-H2AX Foci Through Interaction With the FHA/BRCT Domain. Curr Biol (2002) 12:1846–51. doi: 10.1016/S0960-9822(02)01259-9
125. Deshpande RA, Williams GJ, Limbo O, Williams RS, Kuhnlein J, Lee JH, et al. ATP-Driven Rad50 Conformations Regulate DNA Tethering, End Resection, and ATM Checkpoint Signaling. EMBO J (2014) 33:482–500. doi: 10.1002/embj.201386100
126. Williams GJ, Williams RS, Williams JS, Moncalian G, Arvai AS, Limbo O, et al. ABC ATPase Signature Helices in Rad50 Link Nucleotide State to Mre11 Interface for DNA Repair. Nat Struct Mol Biol (2011) 18:423–31. doi: 10.1038/nsmb.2038
127. Hopfner KP, Craig L, Moncalian G, Zinkel RA, Usui T, Owen BA, et al. The Rad50 Zinc-Hook is a Structure Joining Mre11 Complexes in DNA Recombination and Repair. Nature (2002) 418:562–6. doi: 10.1038/nature00922
128. Dutta D, Dutta S, Veettil MV, Roy A, Ansari MA, Iqbal J, et al. BRCA1 Regulates IFI16 Mediated Nuclear Innate Sensing of Herpes Viral DNA and Subsequent Induction of the Innate Inflammasome and Interferon-β Responses. PloS Pathog (2015) 11:e1005030. doi: 10.1371/journal.ppat.1005030
129. Aglipay JA, Lee SW, Okada S, Fujiuchi N, Ohtsuka T, Kwak JC, et al. A Member of the Pyrin Family, IFI16, is a Novel BRCA1-Associated Protein Involved in the P53-Mediated Apoptosis Pathway. Oncogene (2003) 22:8931–8. doi: 10.1038/sj.onc.1207057
130. Hammel M, Tainer JA. X-Ray Scattering Reveals Disordered Linkers and Dynamic Interfaces in Complexes and Mechanisms for DNA Double-Strand Break Repair Impacting Cell and Cancer Biology. Protein Sci (2021) 30:1735–56. doi: 10.1002/pro.4133
131. Hammel M, Rey M, Yu Y, Mani RS, Classen S, Liu M, et al. XRCC4 Protein Interactions With XRCC4-Like Factor (XLF) Create an Extended Grooved Scaffold for DNA Ligation and Double Strand Break Repair. J Biol Chem (2011) 286:32638–50. doi: 10.1074/jbc.M111.272641
132. Hammel M, Yu Y, Fang S, Lees-Miller SP, Tainer JA. XLF Regulates Filament Architecture of the XRCC4·ligase IV Complex. Structure (2010) 18:1431–42. doi: 10.1016/j.str.2010.09.009
133. Guo G, Gao M, Gao X, Zhu B, Huang J, Tu X, et al. Reciprocal Regulation of RIG-I and XRCC4 Connects DNA Repair With RIG-I Immune Signaling. Nat Commun (2021) 12:2187. doi: 10.1038/s41467-021-22484-7
134. Bednarski JJ, Sleckman BP. At the Intersection of DNA Damage and Immune Responses. Nat Rev Immunol (2019) 19:231–42. doi: 10.1038/s41577-019-0135-6
135. Reisländer T, Groelly FJ, Tarsounas M. DNA Damage and Cancer Immunotherapy: A STING in the Tale. Mol Cell (2020) 80:21–8. doi: 10.1016/j.molcel.2020.07.026
136. O'Driscoll M, Jeggo P. Immunological Disorders and DNA Repair. Mutat Res (2002) 509:109–26. doi: 10.1016/S0027-5107(02)00221-X
137. Davies RC, Pettijohn K, Fike F, Wang J, Nahas SA, Tunuguntla R, et al. Defective DNA Double-Strand Break Repair in Pediatric Systemic Lupus Erythematosus. Arthritis Rheum (2012) 64:568–78. doi: 10.1002/art.33334
138. Figueiredo N, Chora A, Raquel H, Pejanovic N, Pereira P, Hartleben B, et al. Anthracyclines Induce DNA Damage Response-Mediated Protection Against Severe Sepsis. Immunity (2013) 39:874–84. doi: 10.1016/j.immuni.2013.08.039
139. Karakasilioti I, Kamileri I, Chatzinikolaou G, Kosteas T, Vergadi E, Robinson AR, et al. DNA Damage Triggers a Chronic Autoinflammatory Response, Leading to Fat Depletion in NER Progeria. Cell Metab (2013) 18:403–15. doi: 10.1016/j.cmet.2013.08.011
140. Taylor AM, Harnden DG, Arlett CF, Harcourt SA, Lehmann AR, Stevens S, et al. Ataxia Telangiectasia: A Human Mutation With Abnormal Radiation Sensitivity. Nature (1975) 258:427–9. doi: 10.1038/258427a0
141. Bridges BA. Some DNA-Repair-Deficient Human Syndromes and Their Implications for Human Health. Proc R Soc Lond B Biol Sci (1981) 212:263–78. doi: 10.1098/rspb.1981.0038
142. Shiloh Y. ATM and Related Protein Kinases: Safeguarding Genome Integrity. Nat Rev Cancer (2003) 3:155–68. doi: 10.1038/nrc1011
143. Mathieu AL, Verronese E, Rice GI, Fouyssac F, Bertrand Y, Picard C, et al. PRKDC Mutations Associated With Immunodeficiency, Granuloma, and Autoimmune Regulator-Dependent Autoimmunity. J Allergy Clin Immunol (2015) 135:1578–88.e5. doi: 10.1016/j.jaci.2015.01.040
144. Cavazzana I, Ceribelli A, Quinzanini M, Scarsi M, Airò P, Cattaneo R, et al. Prevalence and Clinical Associations of Anti-Ku Antibodies in Systemic Autoimmune Diseases. Lupus (2008) 17:727–32. doi: 10.1177/0961203308089442
145. Schild-Poulter C, Su A, Shih A, Kelly OP, Fritzler MJ, Goldstein R, et al. Association of Autoantibodies With Ku and DNA Repair Proteins in Connective Tissue Diseases. Rheumatol (Oxford) (2008) 47:165–71. doi: 10.1093/rheumatology/kem338
146. Mimori T, Akizuki M, Yamagata H, Inada S, Yoshida S, Homma M. Characterization of a High Molecular Weight Acidic Nuclear Protein Recognized by Autoantibodies in Sera From Patients With Polymyositis-Scleroderma Overlap. J Clin Invest (1981) 68:611–20. doi: 10.1172/JCI110295
147. Brink R, Phan TG. Self-Reactive B Cells in the Germinal Center Reaction. Annu Rev Immunol (2018) 36:339–57. doi: 10.1146/annurev-immunol-051116-052510
148. Bassing CH, Swat W, Alt FW. The Mechanism and Regulation of Chromosomal V(D)J Recombination. Cell (2002) 109 Suppl:S45–55. doi: 10.1016/S0092-8674(02)00675-X
149. Chi X, Li Y, Qiu X. V(D)J Recombination, Somatic Hypermutation and Class Switch Recombination of Immunoglobulins: Mechanism and Regulation. Immunology (2020) 160:233–47. doi: 10.1111/imm.13176
150. Rooney S, Chaudhuri J, Alt FW. The Role of the non-Homologous End-Joining Pathway in Lymphocyte Development. Immunol Rev (2004) 200:115–31. doi: 10.1111/j.0105-2896.2004.00165.x
151. Bednarski JJ, Sleckman BP. Lymphocyte Development: Integration of DNA Damage Response Signaling. Adv Immunol (2012) 116:175–204. doi: 10.1016/B978-0-12-394300-2.00006-5
152. Gao Y, Ferguson DO, Xie W, Manis JP, Sekiguchi J, Frank KM, et al. Interplay of P53 and DNA-Repair Protein XRCC4 in Tumorigenesis, Genomic Stability and Development. Nature (2000) 404:897–900. doi: 10.1038/35009138
153. Bredemeyer AL, Helmink BA, Innes CL, Calderon B, McGinnis LM, Mahowald GK, et al. DNA Double-Strand Breaks Activate a Multi-Functional Genetic Program in Developing Lymphocytes. Nature (2008) 456:819–23. doi: 10.1038/nature07392
154. Bednarski JJ, Pandey R, Schulte E, White LS, Chen BR, Sandoval GJ, et al. RAG-Mediated DNA Double-Strand Breaks Activate a Cell Type-Specific Checkpoint to Inhibit Pre-B Cell Receptor Signals. J Exp Med (2016) 213:209–23. doi: 10.1084/jem.20151048
155. Fistonich C, Zehentmeier S, Bednarski JJ, Miao R, Schjerven H, Sleckman BP, et al. Cell Circuits Between B Cell Progenitors and IL-7(+) Mesenchymal Progenitor Cells Control B Cell Development. J Exp Med (2018) 215:2586–99. doi: 10.1084/jem.20180778
156. Bednarski JJ, Nickless A, Bhattacharya D, Amin RH, Schlissel MS, Sleckman BP. RAG-Induced DNA Double-Strand Breaks Signal Through Pim2 to Promote Pre-B Cell Survival and Limit Proliferation. J Exp Med (2012) 209:11–7. doi: 10.1084/jem.20112078
157. Lee BS, Gapud EJ, Zhang S, Dorsett Y, Bredemeyer A, George R, et al. Functional Intersection of ATM and DNA-Dependent Protein Kinase Catalytic Subunit in Coding End Joining During V(D)J Recombination. Mol Cell Biol (2013) 33:3568–79. doi: 10.1128/MCB.00308-13
158. Ma Y, Pannicke U, Schwarz K, Lieber MR. Hairpin Opening and Overhang Processing by an Artemis/DNA-Dependent Protein Kinase Complex in Nonhomologous End Joining and V(D)J Recombination. Cell (2002) 108:781–94. doi: 10.1016/S0092-8674(02)00671-2
159. Le Deist F, Poinsignon C, Moshous D, Fischer A, de Villartay JP. Artemis Sheds New Light on V(D)J Recombination. Immunol Rev (2004) 200:142–55. doi: 10.1111/j.0105-2896.2004.00169.x
160. Schlissel MS. Does Artemis End the Hunt for the Hairpin-Opening Activity in V(D)J Recombination? Cell (2002) 109:1–4. doi: 10.1016/S0092-8674(02)00694-3
161. Goodarzi AA, Yu Y, Riballo E, Douglas P, Walker SA, Ye R, et al. DNA-PK Autophosphorylation Facilitates Artemis Endonuclease Activity. EMBO J (2006) 25:3880–9. doi: 10.1038/sj.emboj.7601255
162. Niewolik D, Pannicke U, Lu H, Ma Y, Wang LC, Kulesza P, et al. DNA-PKcs Dependence of Artemis Endonucleolytic Activity, Differences Between Hairpins and 5' or 3' Overhangs. J Biol Chem (2006) 281:33900–9. doi: 10.1074/jbc.M606023200
163. Kirchgessner CU, Patil CK, Evans JW, Cuomo CA, Fried LM, Carter T, et al. DNA-Dependent Kinase (P350) as a Candidate Gene for the Murine SCID Defect. Science (1995) 267:1178–83. doi: 10.1126/science.7855601
164. Wiler R, Leber R, Moore BB, VanDyk LF, Perryman LE, Meek K. Equine Severe Combined Immunodeficiency: A Defect in V(D)J Recombination and DNA-Dependent Protein Kinase Activity. Proc Natl Acad Sci USA (1995) 92:11485–9. doi: 10.1073/pnas.92.25.11485
165. Meek K, Kienker L, Dallas C, Wang W, Dark MJ, Venta PJ, et al. SCID in Jack Russell Terriers: A New Animal Model of DNA-PKcs Deficiency. J Immunol (2001) 167:2142–50. doi: 10.4049/jimmunol.167.4.2142
166. Jiang W, Crowe JL, Liu X, Nakajima S, Wang Y, Li C, et al. Differential Phosphorylation of DNA-PKcs Regulates the Interplay Between End-Processing and End-Ligation During Nonhomologous End-Joining. Mol Cell (2015) 58:172–85. doi: 10.1016/j.molcel.2015.02.024
167. van der Burg M, Ijspeert H, Verkaik NS, Turul T, Wiegant WW, Morotomi-Yano K, et al. A DNA-PKcs Mutation in a Radiosensitive T-B- SCID Patient Inhibits Artemis Activation and Nonhomologous End-Joining. J Clin Invest (2009) 119:91–8. doi: 10.1172/JCI37141
168. Woodbine L, Neal JA, Sasi NK, Shimada M, Deem K, Coleman H, et al. PRKDC Mutations in a SCID Patient With Profound Neurological Abnormalities. J Clin Invest (2013) 123:2969–80. doi: 10.1172/JCI67349
169. Li L, Moshous D, Zhou Y, Wang J, Xie G, Salido E, et al. A Founder Mutation in Artemis, an SNM1-Like Protein, Causes SCID in Athabascan-Speaking Native Americans. J Immunol (2002) 168:6323–9. doi: 10.4049/jimmunol.168.12.6323
170. Kobayashi N, Agematsu K, Sugita K, Sako M, Nonoyama S, Yachie A, et al. Novel Artemis Gene Mutations of Radiosensitive Severe Combined Immunodeficiency in Japanese Families. Hum Genet (2003) 112:348–52. doi: 10.1007/s00439-002-0897-x
171. Moshous D, Callebaut I, de Chasseval R, Corneo B, Cavazzana-Calvo M, Le Deist F, et al. Artemis, a Novel DNA Double-Strand Break Repair/V(D)J Recombination Protein, is Mutated in Human Severe Combined Immune Deficiency. Cell (2001) 105:177–86. doi: 10.1016/S0092-8674(01)00309-9
172. Lieber MR. The Polymerases for V(D)J Recombination. Immunity (2006) 25:7–9. doi: 10.1016/j.immuni.2006.07.007
173. Bertocci B, De Smet A, Weill JC, Reynaud CA. Nonoverlapping Functions of DNA Polymerases Mu, Lambda, and Terminal Deoxynucleotidyltransferase During Immunoglobulin V(D)J Recombination In Vivo. Immunity (2006) 25:31–41. doi: 10.1016/j.immuni.2006.04.013
174. Aceytuno RD, Piett CG, Havali-Shahriari Z, Edwards RA, Rey M, Ye R, et al. Structural and Functional Characterization of the PNKP-XRCC4-LigIV DNA Repair Complex. Nucleic Acids Res (2017) 45:6238–51. doi: 10.1093/nar/gkx275
175. Malu S, De Ioannes P, Kozlov M, Greene M, Francis D, Hanna M, et al. Artemis C-Terminal Region Facilitates V(D)J Recombination Through its Interactions With DNA Ligase IV and DNA-PKcs. J Exp Med (2012) 209:955–63. doi: 10.1084/jem.20111437
176. Modesti M, Hesse JE, Gellert M. DNA Binding of Xrcc4 Protein is Associated With V(D)J Recombination But Not With Stimulation of DNA Ligase IV Activity. EMBO J (1999) 18:2008–18. doi: 10.1093/emboj/18.7.2008
177. Grawunder U, Zimmer D, Kulesza P, Lieber MR. Requirement for an Interaction of XRCC4 With DNA Ligase IV for Wild-Type V(D)J Recombination and DNA Double-Strand Break Repair In Vivo. J Biol Chem (1998) 273:24708–14. doi: 10.1074/jbc.273.38.24708
178. Oksenych V, Kumar V, Liu X, Guo C, Schwer B, Zha S, et al. Functional Redundancy Between the XLF and DNA-PKcs DNA Repair Factors in V(D)J Recombination and Nonhomologous DNA End Joining. Proc Natl Acad Sci USA (2013) 110:2234–9. doi: 10.1073/pnas.1222573110
179. Gao Y, Chaudhuri J, Zhu C, Davidson L, Weaver DT, Alt FW. A Targeted DNA-PKcs-Null Mutation Reveals DNA-PK-Independent Functions for KU in V(D)J Recombination. Immunity (1998) 9:367–76. doi: 10.1016/S1074-7613(00)80619-6
180. Chang HHY, Pannunzio NR, Adachi N, Lieber MR. Non-Homologous DNA End Joining and Alternative Pathways to Double-Strand Break Repair. Nat Rev Mol Cell Biol (2017) 18:495–506. doi: 10.1038/nrm.2017.48
181. Chaplin AK, Hardwick SW, Stavridi AK, Buehl CJ, Goff NJ, Ropars V, et al. Cryo-EM of NHEJ Supercomplexes Provides Insights Into DNA Repair. Mol Cell (2021) 81:3400–3409.e3. doi: 10.1016/j.molcel.2021.07.005
182. Zhao B, Watanabe G, Morten MJ, Reid DA, Rothenberg E, Lieber MR. The Essential Elements for the Noncovalent Association of Two DNA Ends During NHEJ Synapsis. Nat Commun (2019) 10:3588. doi: 10.1038/s41467-019-11507-z
183. Carney SM, Moreno AT, Piatt SC, Cisneros-Aguirre M, Lopezcolorado FW, Stark JM, et al. XLF Acts as a Flexible Connector During non-Homologous End Joining. Elife (2020) 9:e61920. doi: 10.7554/eLife.61920
184. Graham TGW, Carney SM, Walter JC, Loparo JJ. A Single XLF Dimer Bridges DNA Ends During Nonhomologous End Joining. Nat Struct Mol Biol (2018) 25:877–84. doi: 10.1038/s41594-018-0120-y
185. Kim MS, Chuenchor W, Chen X, Cui Y, Zhang X, Zhou ZH, et al. Cracking the DNA Code for V(D)J Recombination. Mol Cell (2018) 70:358–370.e4. doi: 10.1016/j.molcel.2018.03.008
186. Chen X, Gellert M, Yang W. Inner Workings of RAG Recombinase and its Specialization for Adaptive Immunity. Curr Opin Struct Biol (2021) 71:79–86. doi: 10.1016/j.sbi.2021.05.014
187. Grundy GJ, Rulten SL, Arribas-Bosacoma R, Davidson K, Kozik Z, Oliver AW, et al. The Ku-Binding Motif is a Conserved Module for Recruitment and Stimulation of non-Homologous End-Joining Proteins. Nat Commun (2016) 7:11242. doi: 10.1038/ncomms11242
188. Barnes DE, Stamp G, Rosewell I, Denzel A, Lindahl T. Targeted Disruption of the Gene Encoding DNA Ligase IV Leads to Lethality in Embryonic Mice. Curr Biol (1998) 8:1395–8. doi: 10.1016/S0960-9822(98)00021-9
189. Frank KM, Sekiguchi JM, Seidl KJ, Swat W, Rathbun GA, Cheng HL, et al. Late Embryonic Lethality and Impaired V(D)J Recombination in Mice Lacking DNA Ligase IV. Nature (1998) 396:173–7. doi: 10.1038/24172
190. Gao Y, Sun Y, Frank KM, Dikkes P, Fujiwara Y, Seidl KJ, et al. A Critical Role for DNA End-Joining Proteins in Both Lymphogenesis and Neurogenesis. Cell (1998) 95:891–902. doi: 10.1016/S0092-8674(00)81714-6
191. Wang XS, Lee BJ, Zha S. The Recent Advances in non-Homologous End-Joining Through the Lens of Lymphocyte Development. DNA Repair (Amst) (2020) 94:102874. doi: 10.1016/j.dnarep.2020.102874
192. Jhappan C, Morse HC 3rd, Fleischmann RD, Gottesman MM, Merlino G. DNA-PKcs: A T-Cell Tumour Suppressor Encoded at the Mouse Scid Locus. Nat Genet (1997) 17:483–6. doi: 10.1038/ng1297-483
193. Li L, Salido E, Zhou Y, Bhattacharyya S, Yannone SM, Dunn E, et al. Targeted Disruption of the Artemis Murine Counterpart Results in SCID and Defective V(D)J Recombination That is Partially Corrected With Bone Marrow Transplantation. J Immunol (2005) 174:2420–8. doi: 10.4049/jimmunol.174.4.2420
194. Li H, Vogel H, Holcomb VB, Gu Y, Hasty P. Deletion of Ku70, Ku80, or Both Causes Early Aging Without Substantially Increased Cancer. Mol Cell Biol (2007) 27:8205–14. doi: 10.1128/MCB.00785-07
195. Fell VL, Schild-Poulter C. The Ku Heterodimer: Function in DNA Repair and Beyond. Mutat Res Rev Mutat Res (2015) 763:15–29. doi: 10.1016/j.mrrev.2014.06.002
196. Rooney S, Sekiguchi J, Zhu C, Cheng HL, Manis J, Whitlow S, et al. Leaky Scid Phenotype Associated With Defective V(D)J Coding End Processing in Artemis-Deficient Mice. Mol Cell (2002) 10:1379–90. doi: 10.1016/S1097-2765(02)00755-4
197. Karanjawala ZE, Adachi N, Irvine RA, Oh EK, Shibata D, Schwarz K, et al. The Embryonic Lethality in DNA Ligase IV-Deficient Mice is Rescued by Deletion of Ku: Implications for Unifying the Heterogeneous Phenotypes of NHEJ Mutants. DNA Repair (Amst) (2002) 1:1017–26. doi: 10.1016/S1568-7864(02)00151-9
198. Mimitou EP, Symington LS. Ku Prevents Exo1 and Sgs1-Dependent Resection of DNA Ends in the Absence of a Functional MRX Complex or Sae2. EMBO J (2010) 29:3358–69. doi: 10.1038/emboj.2010.193
199. Bredemeyer AL, Sharma GG, Huang CY, Helmink BA, Walker LM, Khor KC, et al. ATM Stabilizes DNA Double-Strand-Break Complexes During V(D)J Recombination. Nature (2006) 442:466–70. doi: 10.1038/nature04866
200. Gapud EJ, Sleckman BP. Unique and Redundant Functions of ATM and DNA-PKcs During V(D)J Recombination. Cell Cycle (2011) 10:1928–35. doi: 10.4161/cc.10.12.16011
201. Perlman S, Becker-Catania S, Gatti RA. Ataxia-Telangiectasia: Diagnosis and Treatment. Semin Pediatr Neurol (2003) 10:173–82. doi: 10.1016/S1071-9091(03)00026-3
202. Hu J, Tepsuporn S, Meyers RM, Gostissa M, Alt FW. Developmental Propagation of V(D)J Recombination-Associated DNA Breaks and Translocations in Mature B Cells via Dicentric Chromosomes. Proc Natl Acad Sci USA (2014) 111:10269–74. doi: 10.1073/pnas.1410112111
203. Seifert M, Scholtysik R, Küppers R. Origin and Pathogenesis of B Cell Lymphomas. Methods Mol Biol (2019) 1956:1–33. doi: 10.1007/978-1-4939-9151-8_1
204. Roth DB. V(D)J Recombination: Mechanism, Errors, and Fidelity. Microbiol Spectr (2014) 2(6):10.1128/microbiolspec.MDNA3-0041-2014. doi: 10.1128/microbiolspec.MDNA3-0041-2014
205. Zha S, Guo C, Boboila C, Oksenych V, Cheng HL, Zhang Y, et al. ATM Damage Response and XLF Repair Factor are Functionally Redundant in Joining DNA Breaks. Nature (2011) 469:250–4. doi: 10.1038/nature09604
206. Oksenych V, Alt FW, Kumar V, Schwer B, Wesemann DR, Hansen E, et al. Functional Redundancy Between Repair Factor XLF and Damage Response Mediator 53BP1 in V(D)J Recombination and DNA Repair. Proc Natl Acad Sci USA (2012) 109:2455–60. doi: 10.1073/pnas.1121458109
207. Liu X, Jiang W, Dubois RL, Yamamoto K, Wolner Z, Zha S. Overlapping Functions Between XLF Repair Protein and 53BP1 DNA Damage Response Factor in End Joining and Lymphocyte Development. Proc Natl Acad Sci USA (2012) 109:3903–8. doi: 10.1073/pnas.1120160109
208. DeMicco A, Reich T, Arya R, Rivera-Reyes A, Fisher MR, Bassing CH. Lymphocyte Lineage-Specific and Developmental Stage Specific Mechanisms Suppress Cyclin D3 Expression in Response to DNA Double Strand Breaks. Cell Cycle (2016) 15:2882–94. doi: 10.1080/15384101.2016.1198861
209. Thornton TM, Delgado P, Chen L, Salas B, Krementsov D, Fernandez M, et al. Inactivation of Nuclear GSK3β by Ser(389) Phosphorylation Promotes Lymphocyte Fitness During DNA Double-Strand Break Response. Nat Commun (2016) 7:10553. doi: 10.1038/ncomms10553
210. Jacobs KM, Bhave SR, Ferraro DJ, Jaboin JJ, Hallahan DE, Thotala D. GSK-3β: A Bifunctional Role in Cell Death Pathways. Int J Cell Biol (2012) 2012:930710. doi: 10.1155/2012/930710
211. Hathcock KS, Bowen S, Livak F, Hodes RJ. ATM Influences the Efficiency of Tcrβ Rearrangement, Subsequent Tcrβ-Dependent T Cell Development, and Generation of the Pre-Selection Tcrβ CDR3 Repertoire. PloS One (2013) 8:e62188. doi: 10.1371/journal.pone.0062188
212. Celeste A, Petersen S, Romanienko PJ, Fernandez-Capetillo O, Chen HT, Sedelnikova OA, et al. Genomic Instability in Mice Lacking Histone H2AX. Science (2002) 296:922–7. doi: 10.1126/science.1069398
213. Stavnezer J, Schrader CE. IgH Chain Class Switch Recombination: Mechanism and Regulation. J Immunol (2014) 193:5370–8. doi: 10.4049/jimmunol.1401849
214. Stavnezer J. Complex Regulation and Function of Activation-Induced Cytidine Deaminase. Trends Immunol (2011) 32:194–201. doi: 10.1016/j.it.2011.03.003
215. Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, et al. Activation-Induced Cytidine Deaminase (AID) Deficiency Causes the Autosomal Recessive Form of the Hyper-IgM Syndrome (HIGM2). Cell (2000) 102:565–75. doi: 10.1016/S0092-8674(00)00079-9
216. Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class Switch Recombination and Hypermutation Require Activation-Induced Cytidine Deaminase (AID), a Potential RNA Editing Enzyme. Cell (2000) 102:553–63. doi: 10.1016/S0092-8674(00)00078-7
217. Maul RW, Gearhart PJ. AID and Somatic Hypermutation. Adv Immunol (2010) 105:159–91. doi: 10.1016/S0065-2776(10)05006-6
218. Arudchandran A, Bernstein RM, Max EE. Single-Strand DNA Breaks in Ig Class Switch Recombination That Depend on UNG But Not AID. Int Immunol (2008) 20:1381–93. doi: 10.1093/intimm/dxn097
219. Girelli Zubani G, Zivojnovic M, De Smet A, Albagli-Curiel O, Huetz F, Weill JC, et al. Pms2 and Uracil-DNA Glycosylases Act Jointly in the Mismatch Repair Pathway to Generate Ig Gene Mutations at A-T Base Pairs. J Exp Med (2017) 214:1169–80. doi: 10.1084/jem.20161576
220. Zanotti KJ, Maul RW, Yang W, Gearhart PJ. DNA Breaks in Ig V Regions Are Predominantly Single Stranded and Are Generated by UNG and MSH6 DNA Repair Pathways. J Immunol (2019) 202:1573–81. doi: 10.4049/jimmunol.1801183
221. Twayana S, Bacolla A, Barreto-Galvez A, De-Paula RB, Drosopoulos WC, Kosiyatrakul ST, et al. Translesion Polymerase Eta Both Facilitates DNA Replication and Promotes Increased Human Genetic Variation at Common Fragile Sites. Proc Natl Acad Sci USA (2021) 118(48):e2106477118. doi: 10.1073/pnas.2106477118
222. Jiricny J. Postreplicative Mismatch Repair. Cold Spring Harb Perspect Biol (2013) 5:a012633. doi: 10.1101/cshperspect.a012633
223. Tsutakawa SE, Yan C, Xu X, Weinacht CP, Freudenthal BD, Yang K, et al. Structurally Distinct Ubiquitin- and Sumo-Modified PCNA: Implications for Their Distinct Roles in the DNA Damage Response. Structure (2015) 23:724–33. doi: 10.1016/j.str.2015.02.008
224. Tsutakawa SE, Van Wynsberghe AW, Freudenthal BD, Weinacht CP, Gakhar L, Washington MT, et al. Solution X-Ray Scattering Combined With Computational Modeling Reveals Multiple Conformations of Covalently Bound Ubiquitin on PCNA. Proc Natl Acad Sci USA (2011) 108:17672–7. doi: 10.1073/pnas.1110480108
225. Rada C, Williams GT, Nilsen H, Barnes DE, Lindahl T, Neuberger MS. Immunoglobulin Isotype Switching is Inhibited and Somatic Hypermutation Perturbed in UNG-Deficient Mice. Curr Biol (2002) 12:1748–55. doi: 10.1016/S0960-9822(02)01215-0
226. Methot SP, Di Noia JM. Molecular Mechanisms of Somatic Hypermutation and Class Switch Recombination. Adv Immunol (2017) 133:37–87. doi: 10.1016/bs.ai.2016.11.002
227. Eckelmann BJ, Bacolla A, Wang H, Ye Z, Guerrero EN, Jiang W, et al. XRCC1 Promotes Replication Restart, Nascent Fork Degradation and Mutagenic DNA Repair in BRCA2-Deficient Cells. NAR Cancer (2020) 2:zcaa013. doi: 10.1093/narcan/zcaa013
228. Dutta A, Eckelmann B, Adhikari S, Ahmed KM, Sengupta S, Pandey A, et al. Microhomology-Mediated End Joining is Activated in Irradiated Human Cells Due to Phosphorylation-Dependent Formation of the XRCC1 Repair Complex. Nucleic Acids Res (2017) 45:2585–99. doi: 10.1093/nar/gkw1262
229. Mengwasser KE, Adeyemi RO, Leng Y, Choi MY, Clairmont C, D'Andrea AD, et al. Genetic Screens Reveal FEN1 and APEX2 as BRCA2 Synthetic Lethal Targets. Mol Cell (2019) 73:885–899.e6. doi: 10.1016/j.molcel.2018.12.008
230. Zhou J, Gelot C, Pantelidou C, Li A, Yücel H, Davis RE, et al. A First-in-Class Polymerase Theta Inhibitor Selectively Targets Homologous-Recombination-Deficient Tumors. Nat Cancer (2021) 2:598–610. doi: 10.1038/s43018-021-00203-x
231. Slupphaug G, Mol CD, Kavli B, Arvai AS, Krokan HE, Tainer JA. A Nucleotide-Flipping Mechanism From the Structure of Human Uracil-DNA Glycosylase Bound to DNA. Nature (1996) 384:87–92. doi: 10.1038/384087a0
232. Mol CD, Izumi T, Mitra S, Tainer JA. DNA-Bound Structures and Mutants Reveal Abasic DNA Binding by APE1 and DNA Repair Coordination [Corrected]. Nature (2000) 403:451–6. doi: 10.1038/35000249
233. Nguyen MT, Moiani D, Ahmed Z, Arvai AS, Namjoshi S, Shin DS, et al. An Effective Human Uracil-DNA Glycosylase Inhibitor Targets the Open Pre-Catalytic Active Site Conformation. Prog Biophys Mol Biol (2021) 163:143–59. doi: 10.1016/j.pbiomolbio.2021.02.004
234. Serebrenik AA, Starrett GJ, Leenen S, Jarvis MC, Shaban NM, Salamango DJ, et al. The Deaminase APOBEC3B Triggers the Death of Cells Lacking Uracil DNA Glycosylase. Proc Natl Acad Sci USA (2019) 116:22158–63. doi: 10.1073/pnas.1904024116
235. Boboila C, Yan C, Wesemann DR, Jankovic M, Wang JH, Manis J, et al. Alternative End-Joining Catalyzes Class Switch Recombination in the Absence of Both Ku70 and DNA Ligase 4. J Exp Med (2010) 207:417–27. doi: 10.1084/jem.20092449
236. Han L, Yu K. Altered Kinetics of Nonhomologous End Joining and Class Switch Recombination in Ligase IV-Deficient B Cells. J Exp Med (2008) 205:2745–53. doi: 10.1084/jem.20081623
237. Yan CT, Boboila C, Souza EK, Franco S, Hickernell TR, Murphy M, et al. IgH Class Switching and Translocations Use a Robust non-Classical End-Joining Pathway. Nature (2007) 449:478–82. doi: 10.1038/nature06020
238. Pan-Hammarström Q, Jones AM, Lähdesmäki A, Zhou W, Gatti RA, Hammarström L, et al. Impact of DNA Ligase IV on Nonhomologous End Joining Pathways During Class Switch Recombination in Human Cells. J Exp Med (2005) 201:189–94. doi: 10.1084/jem.20040772
239. Zhao B, Rothenberg E, Ramsden DA, Lieber MR. The Molecular Basis and Disease Relevance of non-Homologous DNA End Joining. Nat Rev Mol Cell Biol (2020) 21:765–81. doi: 10.1038/s41580-020-00297-8
240. Bai W, Zhu G, Xu J, Chen P, Meng F, Xue H, et al. The 3'-Flap Endonuclease XPF-ERCC1 Promotes Alternative End Joining and Chromosomal Translocation During B Cell Class Switching. Cell Rep (2021) 36:109756. doi: 10.1016/j.celrep.2021.109756
241. Liang L, Feng J, Zuo P, Yang J, Lu Y, Yin Y. Molecular Basis for Assembly of the Shieldin Complex and its Implications for NHEJ. Nat Commun (2020) 11:1972. doi: 10.1038/s41467-020-15879-5
242. Noordermeer SM, Adam S, Setiaputra D, Barazas M, Pettitt SJ, Ling AK, et al. The Shieldin Complex Mediates 53BP1-Dependent DNA Repair. Nature (2018) 560:117–21. doi: 10.1038/s41586-018-0340-7
243. Dev H, Chiang TW, Lescale C, de Krijger I, Martin AG, Pilger D, et al. Shieldin Complex Promotes DNA End-Joining and Counters Homologous Recombination in BRCA1-Null Cells. Nat Cell Biol (2018) 20:954–65. doi: 10.1038/s41556-018-0140-1
244. Mirman Z, Lottersberger F, Takai H, Kibe T, Gong Y, Takai K, et al. 53bp1-RIF1-Shieldin Counteracts DSB Resection Through CST- and Polα-Dependent Fill-in. Nature (2018) 560:112–6. doi: 10.1038/s41586-018-0324-7
245. Ghezraoui H, Oliveira C, Becker JR, Bilham K, Moralli D, Anzilotti C, et al. 53BP1 Cooperation With the REV7-Shieldin Complex Underpins DNA Structure-Specific NHEJ. Nature (2018) 560:122–7. doi: 10.1038/s41586-018-0362-1
246. Difilippantonio S, Gapud E, Wong N, Huang CY, Mahowald G, Chen HT, et al. 53BP1 Facilitates Long-Range DNA End-Joining During V(D)J Recombination. Nature (2008) 456:529–33. doi: 10.1038/nature07476
247. Ward IM, Reina-San-Martin B, Olaru A, Minn K, Tamada K, Lau JS, et al. 53BP1 is Required for Class Switch Recombination. J Cell Biol (2004) 165:459–64. doi: 10.1083/jcb.200403021
248. Manis JP, Morales JC, Xia Z, Kutok JL, Alt FW, Carpenter PB. 53BP1 Links DNA Damage-Response Pathways to Immunoglobulin Heavy Chain Class-Switch Recombination. Nat Immunol (2004) 5:481–7. doi: 10.1038/ni1067
249. Feldman S, Wuerffel R, Achour I, Wang L, Carpenter PB, Kenter AL. 53bp1 Contributes to Igh Locus Chromatin Topology During Class Switch Recombination. J Immunol (2017) 198:2434–44. doi: 10.4049/jimmunol.1601947
250. Mirman Z, de Lange T. 53BP1: A DSB Escort. Genes Dev (2020) 34:7–23. doi: 10.1101/gad.333237.119
251. Setiaputra D, Durocher D. Shieldin - the Protector of DNA Ends. EMBO Rep (2019) 20(5):e47560. doi: 10.15252/embr.201847560
252. Ling AK, Munro M, Chaudhary N, Li C, Berru M, Wu B, et al. SHLD2 Promotes Class Switch Recombination by Preventing Inactivating Deletions Within the Igh Locus. EMBO Rep (2020) 21:e49823. doi: 10.15252/embr.201949823
253. Daniel JA, Santos MA, Wang Z, Zang C, Schwab KR, Jankovic M, et al. PTIP Promotes Chromatin Changes Critical for Immunoglobulin Class Switch Recombination. Science (2010) 329:917–23. doi: 10.1126/science.1187942
254. Williams JS, Williams RS, Dovey CL, Guenther G, Tainer JA, Russell P. Gammah2a Binds Brc1 to Maintain Genome Integrity During S-Phase. EMBO J (2010) 29:1136–48. doi: 10.1038/emboj.2009.413
255. Lamont KR, Hasham MG, Donghia NM, Branca J, Chavaree M, Chase B, et al. Attenuating Homologous Recombination Stimulates an AID-Induced Antileukemic Effect. J Exp Med (2013) 210:1021–33. doi: 10.1084/jem.20121258
256. Banday S, Pandita RK, Mushtaq A, Bacolla A, Mir US, Singh DK, et al. Autism-Associated Vigilin Depletion Impairs DNA Damage Repair. Mol Cell Biol (2021) 41:e0008221. doi: 10.1128/MCB.00082-21
257. McLaughlin M, Patin EC, Pedersen M, Wilkins A, Dillon MT, Melcher AA, et al. Inflammatory Microenvironment Remodelling by Tumour Cells After Radiotherapy. Nat Rev Cancer (2020) 20:203–17. doi: 10.1038/s41568-020-0246-1
258. Riaz N, Morris L, Havel JJ, Makarov V, Desrichard A, Chan TA. The Role of Neoantigens in Response to Immune Checkpoint Blockade. Int Immunol (2016) 28:411–9. doi: 10.1093/intimm/dxw019
259. Li T, Fu J, Zeng Z, Cohen D, Li J, Chen Q, et al. TIMER2.0 for Analysis of Tumor-Infiltrating Immune Cells. Nucleic Acids Res (2020) 48:W509–w514. doi: 10.1093/nar/gkaa407
260. Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust Enumeration of Cell Subsets From Tissue Expression Profiles. Nat Methods (2015) 12:453–7. doi: 10.1038/nmeth.3337
261. Houl JH, Ye Z, Brosey CA, Balapiti-Modarage LPF, Namjoshi S, Bacolla A, et al. Selective Small Molecule PARG Inhibitor Causes Replication Fork Stalling and Cancer Cell Death. Nat Commun (2019) 10:5654. doi: 10.1038/s41467-019-13508-4
262. Yang Y. Cancer Immunotherapy: Harnessing the Immune System to Battle Cancer. J Clin Invest (2015) 125:3335–7. doi: 10.1172/JCI83871
263. Wei SC, Duffy CR, Allison JP. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discovery (2018) 8:1069–86. doi: 10.1158/2159-8290.CD-18-0367
264. Wilson DM 3rd, Deacon AM, Duncton MAJ, Pellicena P, Georgiadis MM, Yeh AP, et al. Fragment- and Structure-Based Drug Discovery for Developing Therapeutic Agents Targeting the DNA Damage Response. Prog. Biophys Mol Biol (2021) 163:130–42. doi: 10.1016/j.pbiomolbio.2020.10.005
265. Moiani D, Link TM, Brosey CA, Katsonis P, Lichtarge O, Kim Y, et al. An Efficient Chemical Screening Method for Structure-Based Inhibitors to Nucleic Acid Enzymes Targeting the DNA Repair-Replication Interface and SARS CoV-2. Methods Enzymol (2021) 661:407–431. doi: 10.1016/bs.mie.2021.09.003
266. Noordermeer SM, van Attikum H. PARP Inhibitor Resistance: A Tug-Of-War in BRCA-Mutated Cells. Trends Cell Biol (2019) 29:820–34. doi: 10.1016/j.tcb.2019.07.008
267. Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific Killing of BRCA2-Deficient Tumours With Inhibitors of Poly(ADP-Ribose) Polymerase. Nature (2005) 434:913–7. doi: 10.1038/nature03443
268. Brown JS, O'Carrigan B, Jackson SP, Yap TA. Targeting DNA Repair in Cancer: Beyond PARP Inhibitors. Cancer Discovery (2017) 7:20–37. doi: 10.1158/2159-8290.CD-16-0860
269. Ngoi NYL, Pham MM, Tan DSP, Yap TA. Targeting the Replication Stress Response Through Synthetic Lethal Strategies in Cancer Medicine. Trends Cancer (2021) 7:930–57. doi: 10.1016/j.trecan.2021.06.002
270. Higuchi T, Flies DB, Marjon NA, Mantia-Smaldone G, Ronner L, Gimotty PA, et al. CTLA-4 Blockade Synergizes Therapeutically With PARP Inhibition in BRCA1-Deficient Ovarian Cancer. Cancer Immunol Res (2015) 3:1257–68. doi: 10.1158/2326-6066.CIR-15-0044
271. Wang Z, Sun K, Xiao Y, Feng B, Mikule K, Ma X, et al. Niraparib Activates Interferon Signaling and Potentiates Anti-PD-1 Antibody Efficacy in Tumor Models. Sci Rep (2019) 9:1853. doi: 10.1038/s41598-019-38534-6
272. Pham MM, Ngoi NYL, Peng G, Tan DSP, Yap TA. Development of Poly(ADP-Ribose) Polymerase Inhibitor and Immunotherapy Combinations: Progress, Pitfalls, and Promises. Trends Cancer (2021) 7:958–70. doi: 10.1016/j.trecan.2021.05.004
273. Jiao S, Xia W, Yamaguchi H, Wei Y, Chen MK, Hsu JM, et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin Cancer Res (2017) 23:3711–20. doi: 10.1158/1078-0432.CCR-16-3215
274. Burnette BC, Liang H, Lee Y, Chlewicki L, Khodarev NN, Weichselbaum RR, et al. The Efficacy of Radiotherapy Relies Upon Induction of Type I Interferon-Dependent Innate and Adaptive Immunity. Cancer Res (2011) 71:2488–96. doi: 10.1158/0008-5472.CAN-10-2820
275. Cleary JM, Aguirre AJ, Shapiro GI, D'Andrea AD. Biomarker-Guided Development of DNA Repair Inhibitors. Mol Cell (2020) 78:1070–85. doi: 10.1016/j.molcel.2020.04.035
276. Zhao Y, Thomas HD, Batey MA, Cowell IG, Richardson CJ, Griffin RJ, et al. Preclinical Evaluation of a Potent Novel DNA-Dependent Protein Kinase Inhibitor NU7441. Cancer Res (2006) 66:5354–62. doi: 10.1158/0008-5472.CAN-05-4275
277. Nakamura K, Karmokar A, Farrington PM, James NH, Ramos-Montoya A, Bickerton SJ, et al. Inhibition of DNA-PK With AZD7648 Sensitizes Tumor Cells to Radiotherapy and Induces Type I IFN-Dependent Durable Tumor Control. Clin Cancer Res (2021) 27:4353–66. doi: 10.1158/1078-0432.CCR-20-3701
278. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 Blockade in Tumors With Mismatch-Repair Deficiency. N Engl J Med (2015) 372:2509–20. doi: 10.1056/NEJMoa1500596
279. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch Repair Deficiency Predicts Response of Solid Tumors to PD-1 Blockade. Science (2017) 357:409–13. doi: 10.1126/science.aan6733
280. Germano G, Lamba S, Rospo G, Barault L, Magrì A, Maione F, et al. Inactivation of DNA Repair Triggers Neoantigen Generation and Impairs Tumour Growth. Nature (2017) 552:116–20. doi: 10.1038/nature24673
281. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer Immunology. Mutational Landscape Determines Sensitivity to PD-1 Blockade in non-Small Cell Lung Cancer. Science (2015) 348:124–8. doi: 10.1126/science.aaa1348
282. Davoli T, Uno H, Wooten EC, Elledge SJ. Tumor Aneuploidy Correlates With Markers of Immune Evasion and With Reduced Response to Immunotherapy. Science (2017) 355(6322):eaaf8399. doi: 10.1126/science.aaf8399
283. Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring Function in Exhausted CD8 T Cells During Chronic Viral Infection. Nature (2006) 439:682–7. doi: 10.1038/nature04444
284. Roberts VA, Iverson BL, Iverson SA, Benkovic SJ, Lerner RA, Getzoff ED, et al. Antibody Remodeling: A General Solution to the Design of a Metal-Coordination Site in an Antibody Binding Pocket. Proc Natl Acad Sci USA (1990) 87:6654–8. doi: 10.1073/pnas.87.17.6654
285. Iverson BL, Iverson SA, Roberts VA, Getzoff ED, Tainer JA, Benkovic SJ, et al. Metalloantibodies. Science (1990) 249:659–62. doi: 10.1126/science.2116666
286. Tainer JA, Roberts VA, Getzoff ED. Protein Metal-Binding Sites. Curr Opin Biotechnol (1992) 3:378–87. doi: 10.1016/0958-1669(92)90166-G
287. Nastasi C, Mannarino L, D'Incalci M. DNA Damage Response and Immune Defense. Int J Mol Sci (2020) 21(20):7504. doi: 10.3390/ijms21207504
288. Manolakou T, Verginis P, Boumpas DT. DNA Damage Response in the Adaptive Arm of the Immune System: Implications for Autoimmunity. Int J Mol Sci (2021) 22(11):5842. doi: 10.3390/ijms22115842
289. Taffoni C, Steer A, Marines J, Chamma H, Vila IK, Laguette N. Nucleic Acid Immunity and DNA Damage Response: New Friends and Old Foes. Front Immunol (2021) 12:660560. doi: 10.3389/fimmu.2021.660560
290. Sun W, Zhang Q, Wang R, Li Y, Sun Y, Yang L. Targeting DNA Damage Repair for Immune Checkpoint Inhibition: Mechanisms and Potential Clinical Applications. Front Oncol (2021) 11:648687. doi: 10.3389/fonc.2021.648687
291. Goff PH, Bhakuni R, Pulliam T, Lee JH, Hall ET, Nghiem P. Intersection of Two Checkpoints: Could Inhibiting the DNA Damage Response Checkpoint Rescue Immune Checkpoint-Refractory Cancer? Cancers (Basel) (2021) 13(14):3415. doi: 10.3390/cancers13143415
292. Nakad R, Schumacher B. DNA Damage Response and Immune Defense: Links and Mechanisms. Front Genet (2016) 7:147. doi: 10.3389/fgene.2016.00147
293. Asleh K, Riaz N, Cheng AS, Gao D, Leung SCY, Anurag M, et al. Proteomics-Derived Basal Biomarker DNA-PKcs is Associated With Intrinsic Subtype and Long-Term Clinical Outcomes in Breast Cancer. NPJ Breast Cancer (2021) 7:114. doi: 10.1038/s41523-021-00320-x
294. Hu M, Zhou M, Bao X, Pan D, Jiao M, Liu X, et al. ATM Inhibition Enhances Cancer Immunotherapy by Promoting mtDNA Leakage and cGAS/STING Activation. J Clin Invest (2021) 131(3):e139333. doi: 10.1172/JCI139333
295. Brosey CA, Houl JH, Katsonis P, Balapiti-Modarage LPF, Bommagani S, Arvai A, et al. Targeting SARS-CoV-2 Nsp3 Macrodomain Structure With Insights From Human Poly(ADP-Ribose) Glycohydrolase (PARG) Structures With Inhibitors. Prog Biophys Mol Biol (2021) 163:171–86. doi: 10.1016/j.pbiomolbio.2021.02.002
296. Jiang H, Mei Y-F. SARS–CoV–2 Spike Impairs DNA Damage Repair and Inhibits V(D)J Recombination In Vitro. Viruses (2021) 13:2056. doi: 10.3390/v13102056
297. Dalhus B, Arvai AS, Rosnes I, Olsen ØE, Backe PH, Alseth I, et al. Structures of Endonuclease V With DNA Reveal Initiation of Deaminated Adenine Repair. Nat Struct Mol Biol (2009) 6(2):138-43. doi: 10.1038/nsmb.1538
298. Wu J, Samara NL, Kuraoka I, Yang W. Evolution of Inosine-Specific Endonuclease V from Bacterial DNase to Eukaryotic RNase. Mol Cell (2019) 76(1):44-56.e3. doi: 10.1016/j.molcel.2019.06.046
299. Kong XY, Huse C, Yang K, Øgaard J, Berges N, Vik ES, et al. Endonuclease V Regulates Atherosclerosis Through C-C Motif Chemokine Ligand 2-Mediated Monocyte Infiltration. J Am Heart Assoc (2021) 10(14):e020656.
300. Brosey CA, Tainer JA. Evolving SAXS Versatility: Solution X-Ray Scattering for Macromolecular Architecture, Functional Landscapes, and Integrative Structural Biology. Curr Opin Struct Biol (2019) 58:197–213. doi: 10.1016/j.sbi.2019.04.004
301. Hammel M, Rashid I, Sverzhinsky A, Pourfarjam Y, Tsai MS, Ellenberger T, et al. An Atypical BRCT-BRCT Interaction With the XRCC1 Scaffold Protein Compacts Human DNA Ligase Iiiα Within a Flexible DNA Repair Complex. Nucleic Acids Res (2021) 49:306–21. doi: 10.1093/nar/gkaa1188
Keywords: DNA repair, immune response, DNA damage, cGAS-STING, innate immunity, adaptive immunity, immunomodulatory, cancer therapy
Citation: Ye Z, Shi Y, Lees-Miller SP and Tainer JA (2021) Function and Molecular Mechanism of the DNA Damage Response in Immunity and Cancer Immunotherapy. Front. Immunol. 12:797880. doi: 10.3389/fimmu.2021.797880
Received: 19 October 2021; Accepted: 15 November 2021;
Published: 14 December 2021.
Edited by:
Carlos F. M. Menck, University of São Paulo, BrazilReviewed by:
Kaiyuan Ni, Koch Institute for Integrative Cancer Research at MIT, United StatesCopyright © 2021 Ye, Shi, Lees-Miller and Tainer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John A. Tainer, SlRhaW5lckBtZGFuZGVyc29uLm9yZw==; Susan P. Lees-Miller, bGVlc21pbGxAdWNhbGdhcnkuY2E=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.