 Xiao-xian Li
Xiao-xian Li Feng Zhang
Feng Zhang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Immunol. , 24 December 2021
Sec. Multiple Sclerosis and Neuroimmunology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.795036
Parkinson’s disease (PD) is one of most common neurodegenerative disorders caused by a combination of environmental and genetic risk factors. Currently, numerous population genetic studies have shown that polymorphisms in myeloid cell-triggered receptor II (TREM2) are associated with a variety of neurodegenerative disorders. Recently, TREM2 has been verified to represent a promising candidate gene for PD susceptibility and progression. For example, the expression of TREM2 was apparently increased in the prefrontal cortex of PD patients. Moreover, the rare missense mutations in TREM2 (rs75932628, p.R47H) was confirmed to be a risk factor of PD. In addition, overexpression of TREM2 reduced dopaminergic neurodegeneration in the 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine mouse model of PD. Due to the complex pathogenesis of PD, there is still no effective drug treatment. Thus, TREM2 has received increasing widespread attention as a potential therapeutic target. This review focused on the variation of TREM2 in PD and roles of TREM2 in PD pathogenesis, such as excessive-immune inflammatory response, α-Synuclein aggregation and oxidative stress, to further provide evidence for new immune-related biomarkers and therapies for PD.
Parkinson’s disease (PD) is the second-largest neurodegenerative disease after Alzheimer’s disease (AD) (1). Amounts of studies have shown immune pathway disorders, including changes in cytokine signals, immune cell proliferation and migration, and phagocytosis are closely involved in dopamine (DA) neurodegeneration (2). Microglia, the main cells in brain immune response, come from primitive hematopoietic progenitors in the yolk sac that serve as the first and most important immune defense line of the central nervous system (CNS) (3). Autopsy analysis, positron emission tomography (PET) imaging and molecular and clinical evidence suggest that microglia activation increases and inflammatory mediators accumulate during DA neurodegeneration (4). Thus, microglia-mediated neuroinflammation plays an important role in the pathogenesis of PD (5).
Myeloid cell-triggered receptor II (TREM2), a class of receptors in the immunoglobulin superfamily, is highly expressed on the surface of microglia in CNS and participates in microglial proliferation, phagocytosis, survival, and expression of inflammatory factors (6–10). There is increasing evidence that TREM2 regulates microglia-mediated neuroinflammation and thus play an important role in neurodegenerative diseases, such as AD, PD and amyotrophic lateral sclerosis (ALS) (11–13). At present, TREM2 is considered to be a risk site for PD, and its genetic variation may increase the risk of PD (14). Thus, understanding of the role of TREM2 in PD and the exploration of its mechanism are conducive to provide new therapeutic targets for PD. This review focused on the variation of TREM2 in PD and roles of TREM2 in PD pathogenesis, such as excessive-immune inflammatory response, α-Synuclein (α-Syn) aggregation and oxidative stress, to further provide evidence for new immune-related biomarkers and therapies for PD.
This review focused on the role of TREM2 on pathogenesis of Parkinson’s disease in human, various animal models and in vitro studies. We also summarized the potential regulation of TREM2 on the treatment of Parkinson’s disease. Studies were retrieved from the PubMed and Google Scholar databases using the following search terms: Parkinson’s disease, pathogenesis, TREM2, immunoregulation and treatment. The search terms were applied in different combinations and plural forms, and the search was limited to articles in English. References were screened for additional articles. Retrieval time: from inception to September, 2021.
PD is characterized by aggregation of α-Syn to form Lewy bodies (LB) and degeneration and loss of DA neurons (15), thus affecting other CNS structures and surrounding tissues. The specific pathogenesis of PD is still unclear. Currently, the pathogenesis of PD is recognized to include excessive-immune inflammatory response, α-Syn aggregation, oxidative stress, mitochondrial dysfunction, endoplasmic reticulum stress, iron-induced neurodegeneration, microbial-gut-brain axis disorder, non-coding RNA regulation, and so on (16–21).
Main motor symptoms of PD are rest tremor, rigidity, bradykinesia and loss of postural reflexes. Non-motor symptoms mainly include cognitive dysfunction, sleep disturbance, anosmia, constipation and depression (22). Moreover, PD patients can be divided into three subtypes: 1) mild motor-predominant; 2) diffuse malignant; 3) intermediate (23). Until now, PD is mostly treated by drug therapy. Deep brain stimulation (DBS) of the subthalamic nucleus and levodopa-carbidopa enteral gel preparation are helpful for advanced-complicated patients or for patients that can be selected on the basis of clinical criteria (24, 25). Also, induced pluripotent stem cells (iPSCs) therapy is one of the potential treatments to slow or even prevent PD progression (26). However, the clinical treatment of PD is symptomatic and cannot control the progression of the disease.
TREM2 is a cell-surface glycoprotein with an immunoglobulin-like extracellular domain, transmembrane region and short cytosolic tail region, whose single transmembrane helix interacts with DAP12 to mediate downstream signaling, short cytosolic tail terminates signals that lack signal transduction or trafficking motifs (27). The ectodomain of TREM2, which is susceptible to various post-translational modifications and directly interacts with the environment and regulates microglial function, suggesting that the ectodomain is critical for the function of TREM2 itself (28). Homologues sequences located in the ectodomain of TREM2 might be involved in key functions mediated by TREM2 signaling (29). Moreover, the ectodomain of TREM2 is cleaved by a disintegrin and metalloproteinase (ADAM)-10 and ADAM-17 to produce soluble TREM2 (sTREM2), while C-terminal fragment of TREM2 (TREM2-CTF) is cleaved by γ-secretase into intracellular domain (ICD) (30).
TREM2 is expressed on the membrane surface of myeloid cells, whereas DAP12 requires receptor binding to transport to the cell surface, suggesting that a third protein may drive the binding of the TREM2-DAP12 complex and initiate TREM2-DAP12 signaling (31). In CNS, TREM2-DAP12 leads to phosphorylation of immunoreceptor tyrosine-based activation motif (ITAM), recruitment of spleen tyrosine kinase (SYK), which activates phosphoinositide 3-kinase (PI3K), calcium activation, integrin activation, cytoskeletal rearrangement, mammalian target of rapamycin (mTOR) and mitogen-activated protein kinase (MAPK) signaling activation (32), and thus regulating the survival, phagocytosis and activation of microglia (Figure 1).
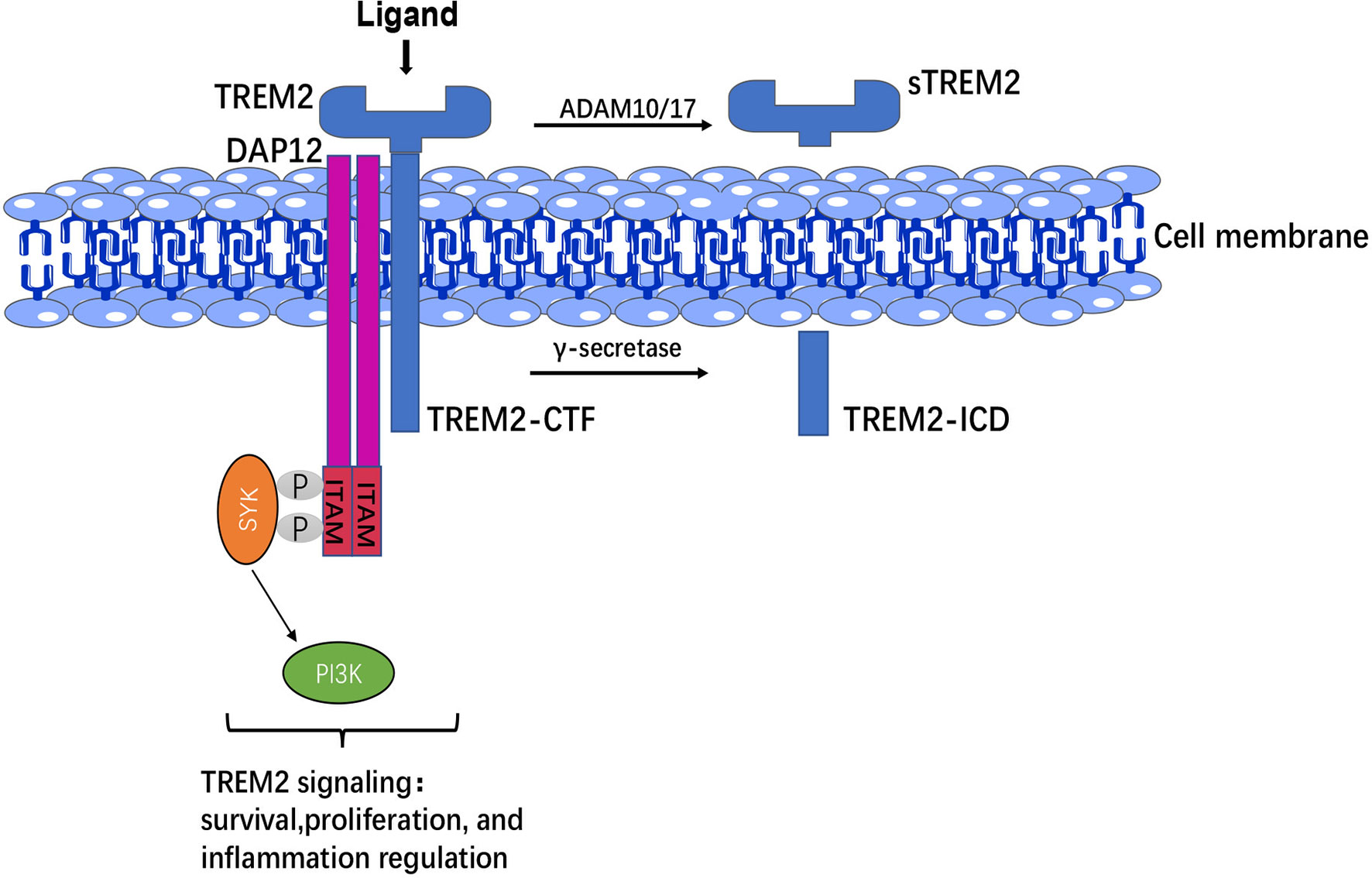
Figure 1 TREM2 structure and signaling. The extracellular domain of TREM2 was cleaved by ADAM 10/17 to produce sTREM2, whereas C-terminal fragment of TREM2 (TREM2-CTF) was cleaved by γ-secretase into intracellular domain (ICD). TREM2-DAP12 led to phosphorylation of ITAM, then recruitment of SYK to activate PI3K signaling and thus regulation of the survival, proliferation, and inflammatory responses of microglia.
TREM2 gene is located on human chromosome 6p21.1. Mutations in TREM2 occur more frequently in the coding sequence and found in the 3’ un-translated region (UTR), the upstream of the transcription start site (33–35). Numerous findings reported TREM2 (rs75932628, p.R47H) is associated with increased PD risk (14, 15). R47H is located on the protein surface and impairs TREM2 binding to lipids by altering the structure of the ligand-binding region (36, 37). In addition, R47H exhibited significant metabolic deficits and impaired precise regulation of TREM2 glycosylation (38, 39). On the other hand, the variation of R47H in the Northern European population was higher than that in the non-Northern European population. Recently, R47H was identified to increase PD susceptibility in American, Polish, Irish and Spanish population and it was unlikely to be a major genetic risk contributor of PD in Greek and Han Chinese population (14, 40, 41). Also, studies confirmed that R47H might increase the risk of PD in North Americans, but not in Europeans (7, 42). However, several studies reported that R47H might increase the risk of PD in European- descended populations (43). These controversial results might be due to the small gene frequency of the R47H polymorphism and the limited sample size of the case-control study experiment.
Additionally, further studies revealed that the biological consequences of TREM2 mutations could be mediated by inhibiting TREM2 receptor signaling or altering the production of other biologically active TREM2 cleavages, such as sTREM2. In addition, TREM2 variants do not affect TREM2 expression levels, suggesting that the effects of TREM2 variants occurred at the post-transcriptional level (44), which might be driven by post-transcription-related regulation. Due to the low mutation rate of TREM2, it is difficult to study in a well-matched population. Meanwhile, wild-type and mutant TREM2 are successfully produced based on expression systems in mammalian cells for structural and biophysical studies, which is critical for understanding the functional consequences of TREM2 mutations associated with the development of neurodegenerative diseases (45).
Autopsy and PET of PD patients exhibited the increased activation of microglia and expression of inflammatory factors on the damaged neurons in the substantia nigra of midbrain (46–48). Same results have been found in various PD animal models. More and more evidence suggests that microglia-mediated neuroinflammation is a key factor and significant feature in the pathogenesis of PD.
TREM2 is expressed only on the surface of microglia in CNS and modulates microglia-mediated neuroinflammation (49). Previous studies have confirmed that microglia have two phenotypes, pro-inflammatory phenotype (M1) and anti-inflammatory phenotype (M2) and TREM2 promotes the transformation of M1 microglia to M2 microglia. On the other hand, TREM2 has been found to be associated with the activation of disease-associated microglia (DAM) (50). Whether DAM are present in PD has not been verified. In MPTP-induced mouse PD models, TREM2 inhibited neuroinflammatory responses and reduced MPTP-induced neuropathic changes by inhibiting toll-like receptor 4(TLR4)/tumor necrosis factor receptor-associated factor 6 (TRAF6)-mediated nuclear factor-κB (NF-κB) and MAPK signaling activation (51). Also, TREM2 inhibited neuroinflammation by down-regulating PI3K/AKT and NF-κB signaling in lipopolysaccharide (LPS)-stimulated BV2 cells (52). Conditioned media from TREM2-siRNA transfected BV2 microglia induced apoptosis of cultured SH-SY5Y cells by inhibiting TREM2 expression and increasing pro-inflammatory factors expressions (53). Moreover, the expression of TREM2 was increased in the inflammatory state in vivo, but decreased in vitro by inflammatory stimulation (54). Likewise, although TREM2 function is classically described as promoting an anti-inflammatory phenotype, several lines of evidence demonstrated a pro-inflammatory role of TREM2 (35), suggesting that its role in inflammation is much more complex and these conflicting results might be related to changes in phenotypes and signaling pathway transduction based on different cell states and activities.
Furthermore, NLRP3 inflammasome in microglia recruited and activated caspase-1, which produced interleukin-1β (IL-1β) via shearing pro-IL-1β and then damaged DA neurons (55). Another evidence indicated that TLR4 mediated NLRP3 inflammasome activation in a NF-κB-dependent manner in CNS (56), whereas TREM2 negatively regulated TLR4-mediated NF-κB signaling pathway activation (51, 57), suggesting that TREM2 might inhibit neuroinflammation by down-regulating NLRP3 signaling activation. In turn, NF-κB signaling also down-regulated the expression of TREM2 (58). On the other hand, DA neurons produced TREM2 ligand (TREM2-L) (59). However, whether DA neurons activated TREM2 through TREM2-L had not been studied. In addition, binding of galactolectin 3(GAL3) to TLR4/TREM2 further activated microglia, and inhibition of GAL3 might be potential benefit for PD treatment (60). Other studies found that interleukin-3 (IL-3) regulated microglial immune responses and acted on downstream pathways in TREM2, and loss of TREM2 reduced the protective effect of IL-3 on neurons (61). Increased interleukin-10 (IL-10) altered TREM2 signaling (62), and TREM2 might act as a negative immunoregulatory molecule through Syk pathway in an IL-10-dependent manner (63). Long-term presence of inflammatory factors also decreased the expression of TREM2 (64), suggesting that TREM2 could interact with inflammatory factors to regulate the inflammatory response in CNS.
Meanwhile, TREM2/iNOS was a marker of increased anti-inflammatory factors, and melatonin could increase the proportion of TREM2/iNOS (65). Additionally, Nilotinib increased TREM2 levels in CSF and treated PD through its anti-inflammatory effects (66). It has also been found that pinitol increased the expression of TREM2 in BV2 microglia and inhibited the expression and secretion of pro-inflammatory cytokines, while silencing of TREM2 abolished the anti-inflammatory effects of pinitol (67). These findings suggested that therapeutic enhancement of TREM2 expression might be a new strategy for the intervention of neuroinflammation-induced PD. Moreover, sTREM2 was reflected in the activation of microglia during neuronal degeneration (68). sTREM2 stimulated the production of inflammatory cytokines through NF-κB (69). The reason might be sTREM2 competed with full-length TREM2 for ligand and inhibiting the anti-inflammatory effects of TREM2. Therefore, inhibition of sTREM2 signaling pathway might also be an effective therapeutic approach for PD (Figure 2).
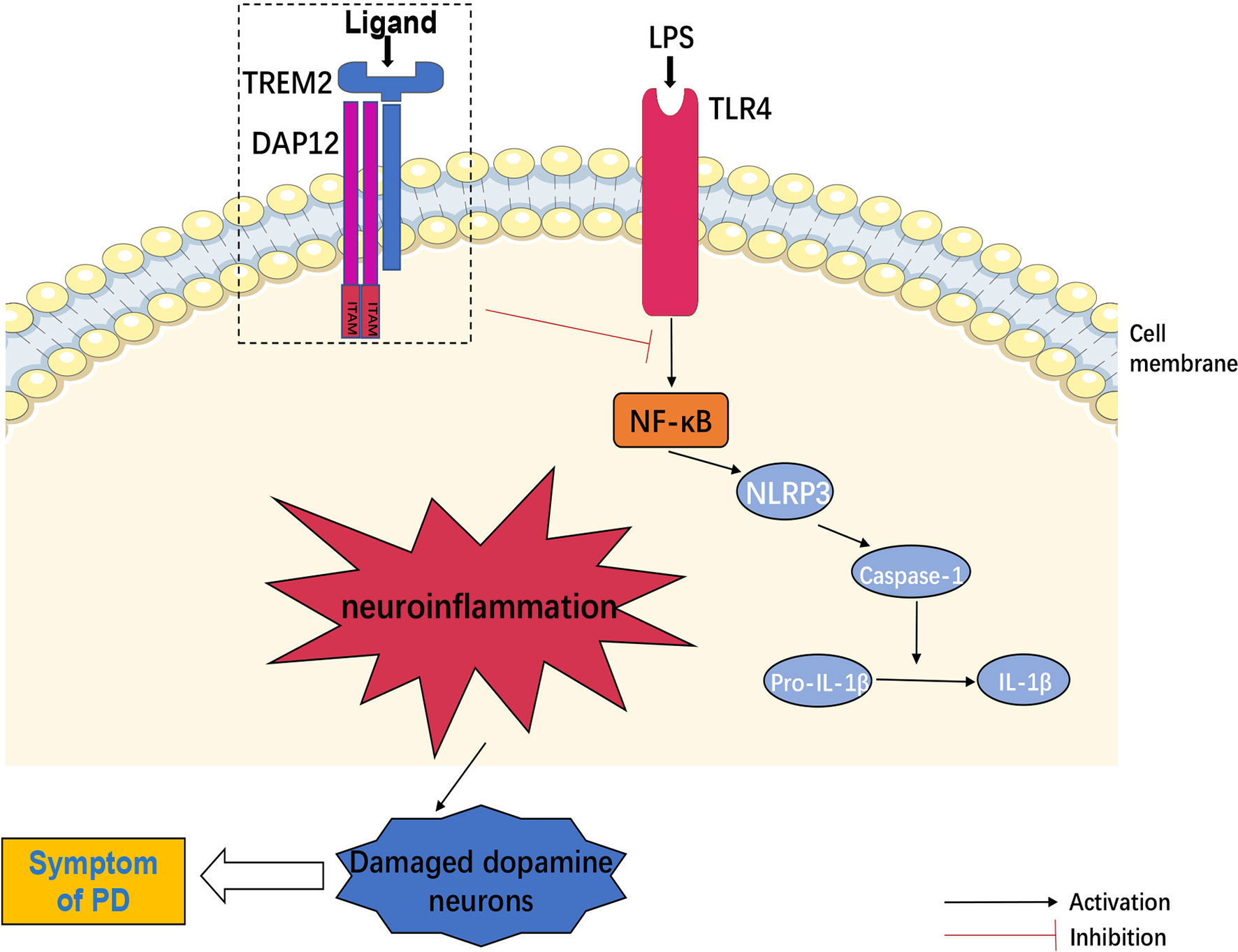
Figure 2 TREM2 and neuroinflammation. TLR4 mediated NLRP3 inflammasome activation in central nervous system (CNS) through regulation of NF-κB signaling pathway. NLRP3 inflammasome recruited and activated caspase-1, produced IL-1β by shearing pro-IL-1β and then induced neuroinflammation, which could damage dopamine neurons. However, TREM2 negatively regulated TLR4-mediated these neuroinflammatory signaling activation.
Aggregation of α -Syn monomer into amyloid fibrils through oligomeric intermediates was found to be the toxic mechanism leading to PD. Oligomers formed at the initial stage of aggregation were potent neurotoxic substances to cause cell death (70). TLR4 played the key role in the neurotoxicity induced by α-Syn oligomers (71). Infection of the midbrain with an adeno-associated virus vector overexpressing α -Syn and inoculation of preformed α -Syn fibrils into the striatum effectively induced DA neurodegeneration (72, 73). Compared with healthy controls, sTREM2 in CSF of PD patients was upregulated and positively correlated with total α-Syn degradation (74). Whether sTREM2 was related to α-Syn degradation remains to be studied. In addition, TREM2 mediated phagocytosis of microglia (75). α -Syn was be recognized by TREM2 on the cell membrane and then phagocytosed. Heat shock protein 60 (HSP60) in the chaperone binded to TREM2 and activated TREM2 and affected phagocytosis in TREM2 (76). During this process, beclin 1 played a critical role in regulating phagocytic receptor function in health and disease, and beclin-1 defects impaired TREM2 recycling (77). Thus, TREM2 might be closely associated with phagocytosis of α -Syn in PD but further studies are needed to determine whether α-Syn could be cleared after phagocytosis.
Oxidative stress was increased in CSF and blood of PD patients (78, 79), and oxidative stress was also found in animal models of PD (80). Oxidative stress products damaged DA neurons through DNA oxidation, protein oxidation and lipid peroxidation. Overexpression of TREM2 alleviated oxidative stress in hippocampal neurons by activating the PI3K/AKT signaling (81). Whether overexpression of TREM2 alleviated oxidative stress in DA neurons remains to be explored. MAPK is a key intermediate molecule in Mn-induced oxidative stress response, and dysregulated peroxisome proliferator-activated receptor γ (PPAR γ)/p38 MAPK signaling underlied the phenotypic deficits in TREM2 variants (38), and TREM2 variants enhanced oxidative stress in the CNS (82). DJ-1(PAPK7) is an oxidative stress sensor, and DJ-1 deficient microglia reduced the expression of TREM2 (83), suggesting that TREM2 was negatively associated with microglia oxidative stress.
Activated microglia are the main source of oxygen-free radical production, while mitochondrial dysfunction is due to disruption of the balance of reactive oxygen species (ROS) accumulation and utilization in cells and tissues. Overexpression of TREM2 inhibited mitochondrial ROS production in macrophages and thus NLRP3/caspase-1 inflammasome activation (84). R47H risk variant lead to mitochondrial dysfunction and stimulated inflammation through NLRP3-dependent inflammatory pathways (38, 85). TREM2 might inhibit NLRP3 activation and neuroinflammation by affecting ROS accumulation in microglia. In addition, TREM2 amplified ROS signaling and promoted osteoclastogenesis in periodontitis (86). Also, TREM2 regulated macrophage immune function by fine-tuning ROS to protect against bacterial infections (84). Oxidative stress was reduced in 24-months-old TREM2 knock-out mice (87). In AD mice, TREM2 activated a downstream pathway leading to mitochondrial damage (88). Thus, TREM2 may be associated differently with oxidative stress in different animal models, and its association with oxidative stress in PD models requires further investigation.
1-methyl-4-phenylpyridinium (MPP+) triggers the transient receptor potential vanilloid 4 (TRPV4) channel and induces ERS (89). Apoptosis induced by ERS overactivation is associated with the decreased DA neurons in PD patients (90). ERS is an abnormal state of lipid metabolism, and TREM2 is a lipid-lipoprotein-binding receptor (91). TREM2 mediated lipid metabolism in pluripotent stem cell-derived microglia-like cells through Phospholipase C gamma2 (PLCγ2) (92). Deficiency of TREM2 resulted in cholesterol ester (CE) overload and fewer lipid droplets, inducing ERS (93–95). ERS mediated changes in inflammatory and apoptotic pathways and was closely associated with cell growth, differentiation, survival and apoptosis. Lack of TREM2 could lead to microglial ERS, laying the foundation for subsequent neurodegenerative mechanisms. In the future, as the negative regulator, overexpression of TREM2 in human or animal models could be considered to observe whether ERS was alleviated.
Iron-induced neurodegeneration is caused by the imbalance between the generation and degradation of intracellular lipid ROS. The iron deposition has been found in the substantia nigra of PD patients and animal models (96). The iron mediated α-Syn aggregation, oxidative stress and toxic effects on DA neurons (97, 98). The capacity of microglia to store iron was three times that of neurons (99), and microglia expressed transporters/molecules involved in brain iron metabolism, inducing ferritin synthesis (100). It has been found that iron overload increased the expression of TREM2 (101), and iron might be involved in various pathogenesis of PD by regulating TREM2 expression in microglia. Whether increased TREM2 expression caused by iron overload could affect ROS production, α-Syn phagocytosis and neuroinflammatory responses is poorly studied and warrants further exploration.
A total of 80% of PD patients is accompanied by gastrointestinal (GI) dysfunction (102). The imbalance of gut microbiota leads to increased intestinal permeability, and inflammatory signals are transmitted to the brain through the gut-brain axis (103). Biopsies of GI tissue from both PD patients and healthy individuals revealed accumulation of α-Syn in the stomach, duodenum and colon (104–106), and α-Syn could be transferred from the enteric nervous system (ENS) to CNS via the vagus nerve (107). Impairment of intestinal barrier function or immune cell activation will promote Th1 cytokines that will overwhelm the ability of TREM2 and cause impaired healing (108). Intestinal dysbiosis may exacerbate neuroinflammation in brain through inhibiting the function of TREM2. It has also been found that LPS, gram-positive and -negative bacteria and fungi could upregulate the expression of TREM2 (109). TREM2 magnified mucosal inflammation during the development of colitis in mice (110). Thus, TREM2 might have different effects on peripheral and central inflammatory responses, affecting intracerebral function by upregulating intestinal inflammatory response (Figure 3).
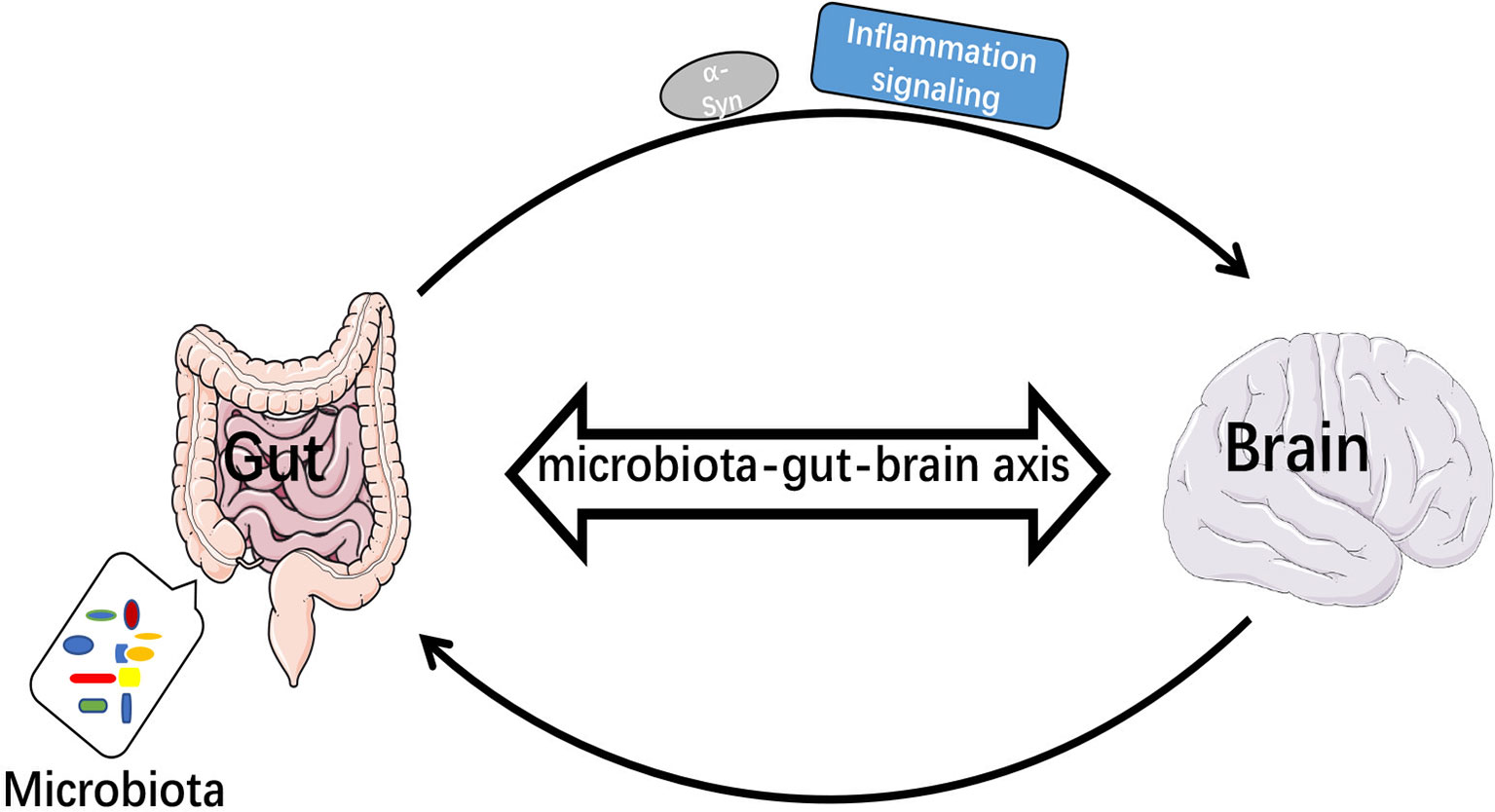
Figure 3 Microbiota-gut-brain axis transmitted inflammatory signals and α-Syn. When intestinal bacteria were imbalanced, inflammatory signals were transmitted to the brain via the bacteria-gut-brain axis and α-Syn was transmitted from enteric nervous system (ENS) to central nervous system (CNS).
In PD, ncRNA regulates the α-Syn expression and LB formation, mitochondrial dysfunction and apoptosis (111). TREM2 is associated with multiple ncRNAs. For example, the level of TREM2 protein was regulated by small nuclear RNA (snRNA) and Mir-665 (112, 113). miR-101a regulated microglia activation and immune responses via TREM2 (114). Epigenetic mechanisms of miRNA-34a-mediated down-regulation of TREM2 expression led to neurodegeneration (115, 116); Specifically, miR-3473b was involved in PD pathogenesis by inhibiting the expression of TREM2 (117). Whether ncRNA is involved in PD development through the regulation of TREM2 is unclear and needs further study. ncRNA are novel and challenging drug targets due to their abilities to affect the expression of other genes. However, it has been difficult to identify whether ncRNA specifically acted on TREM2.
Exosomes are small vesicles about 30-150 nm in diameter secreted by living cells that deliver α-Syn to neurons. The delivered α-Syn induced more neurotoxic aggregation than free α-Syn oligomers. Microglial exosomes were capable of inducing nigrostriatal degeneration (118). Microglia can also transfer neuroinflammatory signals through exosomes (119). Exosomes upregulated TREM2 mRNA expression in LPS-induced THP-1 cells (120). Inflammatory stimuli may upregulated TREM2 expression through exosomes. Moreover, sTREM2 in brain parenchyma could be carried by a subset of microglia, macrophages, or exosomes (121). However, how exosomes regulated TREM2 signaling activation is worthy of further illumination.
In AD mouse models, up-regulation of TREM2 induced microglia to express brain-derived neurotrophic factor (BDNF) (122). Fasudil may target microglial phagocytosis by upregulating the TREM2/DAP12 pathway, with the increased expression of BDNF and glial cell-derived neurotrophic factor (GDNF) (123). Therefore, TREM2 might protect DA neurons by regulating microglia to release neurotrophic factors. However, direct delivery of exogenous BDNF to the brain of patients and enhancement of BDNF expression through gene therapy have not been successful (124). Thus, the possibility of enhancing BDNF expression through TREM2 to treat PD is another field of interest.
Since sTREM2 expression in CSF was increased in PD patients and was positively correlated with total α-Syn in CSF (74), it was suggested that sTREM2 in CSF could be used as a substitute immune biomarker for PD neuron injury. Studies have shown that the misdiagnosis rate of PD was about 25% (125), and correct biomarkers were conducive to the prevention and diagnosis of PD. Moreover, antibody-based radioligands could be used as PET radioligands to track TREM2 changes in vivo and understand the dynamic changes of TREM2 in the PD progress (126). Increased expression of TREM2 reduced pathological microglial responses and neuropathological and behavioral deficits (127). Endogenous small-molecule inhibitors of TREM2 included phosphatidylethanolamine and phosphatidylserine in vivo (128). In a mouse PD model, TREM2 was also down-regulated by LPS/interleukin-34 (IL-34)/interferon-γ (129, 130). In addition, in vitro studies revealed that CELF2 was a novel splicing regulator in exon 3 of TREM2 that regulates species-specific splicing in TREM2 and reduces the expression of TREM2 protein (131), suggesting that inhibition of multiple substances could increase the expression of TREM2 and make it possible to treat PD by increasing the expression of TREM2. The anti-human TREM2 agonistic monoclonal antibody, AL002c, tempered microglial inflammatory response. It was safe and well tolerated in a first-in-human phase I clinical trial, and might be a promising candidate for PD treatment (132). Possible treatments also included agonists of TREM2 signaling in vivo that stabilized TREM2 on the cell surface and reduced its shedding (126). However, whether this affected the immune system, metabolism and fertility and intervention at which stage of PD progression would be beneficial is unclear (133). Crystal packing interfaces analysis using the maltose binding protein (MBP)-TREM2 immunoglobulin (Ig) fusion construct has shown that the surface of the TREM2 Ig domain can bind small molecules, providing potential utility for the discovery of new therapies for TREM2 (134).
In recent years, the role of TREM2 in neurodegenerative diseases has rapidly become an interesting area of active studies. TREM2 adds to the understanding of DAM phenotypes. Currently, there is no effective drug treatment for PD, the reason might be that the etiology and mechanism of PD are still unclear and animal models cannot accurately represent the disease. Until now, there is an increasing focus on single-target therapy for PD. Although TREM2 could affect multiple pathogenesis of PD, including attenuating immune responses, α-Syn aggregation and oxidative stress, multi-target PD-related pathogenesis with TREM2 might be a promising therapeutic option. Importantly, there is a general lack of clinical validation of the correlation between TREM2 and various mechanisms of PD. Thus, further validation is needed in clinical practice. It is now necessary to clarify: 1) TREM2 is a PD susceptibility factor in which populations; 2) In what context does TREM2 function, whether it is affected by time, how does TREM2 function, which pathologies of PD are affected by its function, changes in expression, and signal transduction, and whether it affects multiple PD pathologies simultaneously; 3) ADAM10/17 is an abscission enzyme in TREM2 expression, and its inhibitor might enhance TREM2 expression but has multi-target effects. Could selective competitive inhibitors be found for shedding sites in TREM2? Or is there a viable way to activate TREM2 expression in vivo with fewer side effects, and whether it could be a viable basis for the prevention or treatment of PD. Collectively, a full understanding of role of TREM2 in PD is essential to provide additional insights into the underlying pathology of PD with the ultimate goal of developing new treatments strategies.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
This study was supported by the National Natural Science Foundation of China (No. 82160690), Science and Technology Foundation of Guizhou Province (No. ZK[2021]-014), the foundation for High-level Innovative Talents of Guizhou Province (No. 20164027), the Foundation for Excellent Young Talents of Zunyi Medical University (No. 201603) and Collaborative Innovation Center of Chinese Ministry of Education (No. 2020-39).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Simon DK, Tanner CM, Brundin P. Parkinson Disease Epidemiology, Pathology, Genetics, and Pathophysiology. Clin Geriatr Med (2020) 36:1–12. doi: 10.1016/j.cger.2019.08.002
2. Hammond TR, Marsh SE, Stevens B. Immune Signaling in Neurodegeneration. Immunity (2019) 50:955–74. doi: 10.1016/j.immuni.2019.03.016
3. Tay TL, Savage JC, Hui CW, Bisht K, Tremblay M. Microglia Across the Lifespan: From Origin to Function in Brain Development, Plasticity and Cognition. J Physiol (2017) 595:1929–45. doi: 10.1113/JP272134
4. Ouchi Y, Yoshikawa E, Sekine Y, Futatsubashi M, Kanno T, Ogusu T, et al. Microglial Activation and Dopamine Terminal Loss in Early Parkinson’s Disease. Ann Neurol (2005) 57:168–75. doi: 10.1002/ana.20338
5. Khan AU, Akram M, Daniyal M, Zainab R. Awareness and Current Knowledge of Parkinson’s Disease: A Neurodegenerative Disorder. Int J Neurosci (2019) 129:55–93. doi: 10.1080/00207454.2018.1486837
6. Painter MM, Atagi Y, Liu CC, Rademakers R, Xu H, Fryer JD, et al. TREM2 in CNS Homeostasis and Neurodegenerative Disease. Mol Neurodegener (2015) 10:43. doi: 10.1186/s13024-015-0040-9
7. Zhou SL, Tan CC, Hou XH, Cao XP, Tan L, Yu JT. TREM2 Variants and Neurodegenerative Diseases: A Systematic Review and Meta-Analysis. J Alzheimers Dis (2019) 68:1171–84. doi: 10.3233/JAD-181038
8. Wolfe CM, Fitz NF, Nam KN, Lefterov I, Koldamova R. The Role of APOE and TREM2 in Alzheimer’s Disease-Current Understanding and Perspectives. Int J Mol Sci (2018) 20:81. doi: 10.3390/ijms20010081
9. Ulland TK, Song WM, Huang SC, Ulrich JD, Sergushichev A, Beatty WL, et al. TREM2 Maintains Microglial Metabolic Fitness in Alzheimer’s Disease. Cell (2017) 170:649–63.e13. doi: 10.1016/j.cell.2017.07.023
10. Jiang T, Zhang YD, Chen Q, Gao Q, Zhu XC, Zhou JS, et al. TREM2 Modifies Microglial Phenotype and Provides Neuroprotection in P301S Tau Transgenic Mice. Neuropharmacology (2016) 105:196–206. doi: 10.1016/j.neuropharm.2016.01.028
11. Hou Y, Li X, Liu C, Zhang M, Zhang X, Ge S, et al. Neuroprotective Effects of Short-Chain Fatty Acids in MPTP Induced Mice Model of Parkinson’s Disease. Exp Gerontol (2021) 150:111376. doi: 10.1016/j.exger.2021.111376
12. Bartels T, De Schepper S, Hong S. Microglia Modulate Neurodegeneration in Alzheimer’s and Parkinson’s Diseases. Science (2020) 370:66–9. doi: 10.1126/science.abb8587
13. Lall D, Baloh RH. Microglia and C9orf72 in Neuroinflammation and ALS and Frontotemporal Dementia. J Clin Invest (2017) 127:3250–8. doi: 10.1172/JCI90607
14. Rayaprolu S, Mullen B, Baker M, Lynch T, Finger E, Seeley WW, et al. TREM2 in Neurodegeneration: Evidence for Association of the P.R47H Variant With Frontotemporal Dementia and Parkinson’s Disease. Mol Neurodegener (2013) 8:19. doi: 10.1186/1750-1326-8-19
15. Liu G, Liu Y, Jiang Q, Jiang Y, Feng R, Zhang L, et al. Convergent Genetic and Expression Datasets Highlight TREM2 in Parkinson’s Disease Susceptibility. Mol Neurobiol (2016) 53:4931–8. doi: 10.1007/s12035-015-9416-7
16. Goswami P, Joshi N, Singh S. Neurodegenerative Signaling Factors and Mechanisms in Parkinson’s Pathology. Toxicol In Vitro (2017) 43:104–12. doi: 10.1016/j.tiv.2017.06.008
17. Rocha EM, De Miranda B, Sanders LH. Alpha-Synuclein: Pathology, Mitochondrial Dysfunction and Neuroinflammation in Parkinson’s Disease. Neurobiol Dis (2018) 109:249–57. doi: 10.1016/j.nbd.2017.04.004
18. Costa CAD, Manaa WE, Duplan E, Checler F. The Endoplasmic Reticulum Stress/Unfolded Protein Response and Their Contributions to Parkinson’s Disease Physiopathology. Cells (2020) 9:2495. doi: 10.3390/cells9112495
19. Pickrell AM, Youle RJ. The Roles of PINK1, Parkin, and Mitochondrial Fidelity in Parkinson’s Disease. Neuron (2015) 85:257–73. doi: 10.1016/j.neuron.2014.12.007
20. Perez-Pardo P, Hartog M, Garssen J, Kraneveld AD. Microbes Tickling Your Tummy: The Importance of the Gut-Brain Axis in Parkinson’s Disease. Curr Behav Neurosci Rep (2017) 4:361–8. doi: 10.1007/s40473-017-0129-2
21. Liu H, Wu H, Zhu N, Xu Z, Wang Y, Qu Y, et al. Lactoferrin Protects Against Iron Dysregulation, Oxidative Stress, and Apoptosis in 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine (MPTP)-Induced Parkinson’s Disease in Mice. J Neurochem (2020) 152:397–415. doi: 10.1111/jnc.14857
22. Balestrino R, Schapira AHV. Parkinson Disease. Eur J Neurol (2020) 27:27–42. doi: 10.1111/ene.14108
23. Abbasi N, Fereshtehnejad SM, Zeighami Y, Larcher KM, Postuma RB, Dagher A. Predicting Severity and Prognosis in Parkinson’s Disease From Brain Microstructure and Connectivity. NeuroImage Clin (2020) 25:102111. doi: 10.1016/j.nicl.2019.102111
24. Armstrong MJ, Okun MS. Diagnosis and Treatment of Parkinson Disease: A Review. Jama (2020) 323:548–60. doi: 10.1001/jama.2019.22360
25. Fernandez HH, Boyd JT, Fung VSC, Lew MF, Rodriguez RL, Slevin JT, et al. Long-Term Safety and Efficacy of Levodopa-Carbidopa Intestinal Gel in Advanced Parkinson’s Disease. Mov Disord (2018) 33:928–36. doi: 10.1002/mds.27338
26. Stoddard-Bennett T, Reijo Pera R. Treatment of Parkinson’s Disease Through Personalized Medicine and Induced Pluripotent Stem Cells. Cells (2019) 8:26. doi: 10.3390/cells8010026
27. Kober DL, Brett TJ. TREM2-Ligand Interactions in Health and Disease. J Mol Biol (2017) 429:1607–29. doi: 10.1016/j.jmb.2017.04.004
28. Yang J, Fu Z, Zhang X, Xiong M, Meng L, Zhang Z. TREM2 Ectodomain and its Soluble Form in Alzheimer’s Disease. J Neuroinflamma (2020) 17:204. doi: 10.1186/s12974-020-01878-2
29. Ghani M, Sato C, Kakhki EG, Gibbs JR, Traynor B, St George-Hyslop P, et al. Mutation Analysis of the MS4A and TREM Gene Clusters in a Case-Control Alzheimer’s Disease Data Set. Neurobiol Aging (2016) 42:217.e7–.e13. doi: 10.1016/j.neurobiolaging.2016.03.009
30. Steiner A, Schlepckow K, Brunner B, Steiner H, Haass C, Hagn F. γ-Secretase Cleavage of the Alzheimer Risk Factor TREM2 is Determined by its Intrinsic Structural Dynamics. EMBO J (2020) 39:e104247. doi: 10.15252/embj.2019104247
31. Colonna M, Wang Y. TREM2 Variants: New Keys to Decipher Alzheimer Disease Pathogenesis. Nat Rev Neurosci (2016) 17:201–7. doi: 10.1038/nrn.2016.7
32. Ulland TK, Colonna M. TREM2 - A Key Player in Microglial Biology and Alzheimer Disease. Nat Rev Neurol (2018) 14:667–75. doi: 10.1038/s41582-018-0072-1
33. Cuyvers E, Bettens K, Philtjens S, Van Langenhove T, Gijselinck I, van der Zee J, et al. Investigating the Role of Rare Heterozygous TREM2 Variants in Alzheimer’s Disease and Frontotemporal Dementia. Neurobiol Aging (2014) 35:726.e11–9. doi: 10.1016/j.neurobiolaging.2013.09.009
34. Wang X, Lopez OL, Sweet RA, Becker JT, DeKosky ST, Barmada MM, et al. Genetic Determinants of Disease Progression in Alzheimer’s Disease. J Alzheimers Dis (2015) 43:649–55. doi: 10.3233/JAD-140729
35. Jay TR, von Saucken VE, Landreth GE. TREM2 in Neurodegenerative Diseases. Mol Neurodegener (2017) 12:56. doi: 10.1186/s13024-017-0197-5
36. Sudom A, Talreja S, Danao J, Bragg E, Kegel R, Min X, et al. Molecular Basis for the Loss-of-Function Effects of the Alzheimer’s Disease-Associated R47H Variant of the Immune Receptor TREM2. J Biol Chem (2018) 293:12634–46. doi: 10.1074/jbc.RA118.002352
37. Bailey CC, DeVaux LB, Farzan M. The Triggering Receptor Expressed on Myeloid Cells 2 Binds Apolipoprotein E. J Biol Chem (2015) 290:26033–42. doi: 10.1074/jbc.M115.677286
38. Piers TM, Cosker K, Mallach A, Johnson GT, Guerreiro R, Hardy J, et al. A Locked Immunometabolic Switch Underlies TREM2 R47H Loss of Function in Human iPSC-Derived Microglia. FASEB J (2020) 34:2436–50. doi: 10.1096/fj.201902447R
39. Park JS, Ji IJ, An HJ, Kang MJ, Kang SW, Kim DH, et al. Disease-Associated Mutations of TREM2 Alter the Processing of N-Linked Oligosaccharides in the Golgi Apparatus. Traffic (2015) 16:510–8. doi: 10.1111/tra.12264
40. Dardiotis E, Rikos D, Siokas V, Aloizou AM, Tsouris Z, Sakalakis E, et al. Assessment of TREM2 Rs75932628 Variant’s Association With Parkinson’s Disease in a Greek Population and Meta-Analysis of Current Data. Int J Neurosci (2021) 131:544–8. doi: 10.1080/00207454.2020.1750388
41. Li Z, Zhong L, Gu L, Huang W, Shi X, Zhang X, et al. Association Study of TREM2 Polymorphism Rs75932628 With Leucoaraiosis or Parkinson’s Disease in the Han Chinese Population. BMJ Open (2016) 6:e009499. doi: 10.1136/bmjopen-2015-009499
42. Lill CM, Rengmark A, Pihlstrøm L, Fogh I, Shatunov A, Sleiman PM, et al. The Role of TREM2 R47H as a Risk Factor for Alzheimer’s Disease, Frontotemporal Lobar Degeneration, Amyotrophic Lateral Sclerosis, and Parkinson’s Disease. Alzheimers Dement (2015) 11:1407–16. doi: 10.1016/j.jalz.2014.12.009
43. Feng SJ, Nie K, Gan R, Huang J, Zhang YW, Wang LM, et al. Triggering Receptor Expressed on Myeloid Cells 2 Variants are Rare in Parkinson’s Disease in a Han Chinese Cohort. Neurobiol Aging (2014) 35:1780.e11–2. doi: 10.1016/j.neurobiolaging.2014.01.142
44. Del-Aguila JL, Benitez BA, Li Z, Dube U, Mihindukulasuriya KA, Budde JP, et al. TREM2 Brain Transcript-Specific Studies in AD and TREM2 Mutation Carriers. Mol Neurodegener (2019) 14:18. doi: 10.1186/s13024-019-0319-3
45. Kober DL, Wanhainen KM, Johnson BM, Randolph DT, Holtzman MJ, Brett TJ. Preparation, Crystallization, and Preliminary Crystallographic Analysis of Wild-Type and Mutant Human TREM-2 Ectodomains Linked to Neurodegenerative and Inflammatory Diseases. Protein Expr Purif (2014) 96:32–8. doi: 10.1016/j.pep.2014.01.015
46. Tang Y, Le W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol Neurobiol (2016) 53:1181–94. doi: 10.1007/s12035-014-9070-5
47. Gerhard A, Pavese N, Hotton G, Turkheimer F, Es M, Hammers A, et al. In Vivo Imaging of Microglial Activation With [11C](R)-PK11195 PET in Idiopathic Parkinson’s Disease. Neurobiol Dis (2006) 21:404–12. doi: 10.1016/j.nbd.2005.08.002
48. McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive Microglia Are Positive for HLA-DR in the Substantia Nigra of Parkinson’s and Alzheimer’s Disease Brains. Neurology (1988) 38:1285–91. doi: 10.1212/WNL.38.8.1285
49. Zhang Y, Feng S, Nie K, Li Y, Gao Y, Gan R, et al. TREM2 Modulates Microglia Phenotypes in the Neuroinflammation of Parkinson’s Disease. Biochem Biophys Res Commun (2018) 499:797–802. doi: 10.1016/j.bbrc.2018.03.226
50. Zhou Y, Song WM, Andhey PS, Swain A, Levy T, Miller KR, et al. Human and Mouse Single-Nucleus Transcriptomics Reveal TREM2-Dependent and TREM2-Independent Cellular Responses in Alzheimer’s Disease. Nat Med (2020) 26:131–42. doi: 10.1038/s41591-019-0695-9
51. Ren M, Guo Y, Wei X, Yan S, Qin Y, Zhang X, et al. TREM2 Overexpression Attenuates Neuroinflammation and Protects Dopaminergic Neurons in Experimental Models of Parkinson’s Disease. Exp Neurol (2018) 302:205–13. doi: 10.1016/j.expneurol.2018.01.016
52. Li C, Zhao B, Lin C, Gong Z, An X. TREM2 Inhibits Inflammatory Responses in Mouse Microglia by Suppressing the PI3K/NF-κb Signaling. Cell Biol Int (2019) 43:360–72. doi: 10.1002/cbin.10975
53. Guo Y, Wei X, Yan H, Qin Y, Yan S, Liu J, et al. TREM2 Deficiency Aggravates α-Synuclein-Induced Neurodegeneration and Neuroinflammation in Parkinson’s Disease Models. FASEB J (2019) 33:12164–74. doi: 10.1096/fj.201900992R
54. Wang M, Gao X, Zhao K, Chen H, Xu M, Wang K. Effect of TREM2 on Release of Inflammatory Factor From LPS-Stimulated Microglia and Its Possible Mechanism. Ann Clin Lab Sci (2019) 49:249–56.
55. Wang W, Nguyen LT, Burlak C, Chegini F, Guo F, Chataway T, et al. Caspase-1 Causes Truncation and Aggregation of the Parkinson’s Disease-Associated Protein α-Synuclein. Proc Natl Acad Sci U S A (2016) 113:9587–92. doi: 10.1073/pnas.1610099113
56. Yu R, Jiang S, Tao Y, Li P, Yin J, Zhou Q. Inhibition of HMGB1 Improves Necrotizing Enterocolitis by Inhibiting NLRP3 via TLR4 and NF-κb Signaling Pathways. J Cell Physiol (2019) 234:13431–8. doi: 10.1002/jcp.28022
57. Long H, Zhong G, Wang C, Zhang J, Zhang Y, Luo J, et al. TREM2 Attenuates Aβ1-42-Mediated Neuroinflammation in BV-2 Cells by Downregulating TLR Signaling. Neurochem Res (2019) 44:1830–9. doi: 10.1007/s11064-019-02817-1
58. Zhong L, Zhang ZL, Li X, Liao C, Mou P, Wang T, et al. TREM2/DAP12 Complex Regulates Inflammatory Responses in Microglia via the JNK Signaling Pathway. Front Aging Neurosci (2017) 9:204. doi: 10.3389/fnagi.2017.00204
59. Hsieh CL, Koike M, Spusta SC, Niemi EC, Yenari M, Nakamura MC, et al. A Role for TREM2 Ligands in the Phagocytosis of Apoptotic Neuronal Cells by Microglia. J Neurochem (2009) 109:1144–56. doi: 10.1111/j.1471-4159.2009.06042.x
60. Puigdellívol M, Allendorf DH, Brown GC. Sialylation and Galectin-3 in Microglia-Mediated Neuroinflammation and Neurodegeneration. Front Cell Neurosci (2020) 14:162. doi: 10.3389/fncel.2020.00162
61. McAlpine CS, Park J, Griciuc A, Kim E, Choi SH, Iwamoto Y, et al. Astrocytic Interleukin-3 Programs Microglia and Limits Alzheimer’s Disease. Nature (2021) 595:701–6. doi: 10.1038/s41586-021-03734-6
62. Whittaker GC, Orr SJ, Quigley L, Hughes L, Francischetti IM, Zhang W, et al. The Linker for Activation of B Cells (LAB)/non-T Cell Activation Linker (NTAL) Regulates Triggering Receptor Expressed on Myeloid Cells (TREM)-2 Signaling and Macrophage Inflammatory Responses Independently of the Linker for Activation of T Cells. J Biol Chem (2010) 285:2976–85. doi: 10.1074/jbc.M109.038398
63. Yao Y, Li H, Chen J, Xu W, Yang G, Bao Z, et al. TREM-2 Serves as a Negative Immune Regulator Through Syk Pathway in an IL-10 Dependent Manner in Lung Cancer. Oncotarget (2016) 7:29620–34. doi: 10.18632/oncotarget.8813
64. French T, Düsedau HP, Steffen J, Biswas A, Ahmed N, Hartmann S, et al. Neuronal Impairment Following Chronic Toxoplasma Gondii Infection Is Aggravated by Intestinal Nematode Challenge in an IFN-γ-Dependent Manner. J Neuroinflamma (2019) 16:159. doi: 10.1186/s12974-019-1539-8
65. Azedi F, Mehrpour M, Talebi S, Zendedel A, Kazemnejad S, Mousavizadeh K, et al. Melatonin Regulates Neuroinflammation Ischemic Stroke Damage Through Interactions With Microglia in Reperfusion Phase. Brain Res (2019) 1723:146401. doi: 10.1016/j.brainres.2019.146401
66. Pagan FL, Hebron ML, Wilmarth B, Torres-Yaghi Y, Lawler A, Mundel EE, et al. Pharmacokinetics and Pharmacodynamics of a Single Dose Nilotinib in Individuals With Parkinson’s Disease. Pharmacol Res Perspect (2019) 7:e00470. doi: 10.1002/prp2.470
67. Kong J, Du Z, Dong L. Pinitol Prevents Lipopolysaccharide (LPS)-Induced Inflammatory Responses in BV2 Microglia Mediated by TREM2. Neurotox Res (2020) 38:96–104. doi: 10.1007/s12640-020-00187-z
68. Suárez-Calvet M, Kleinberger G, Araque Caballero M, Brendel M, Rominger A, Alcolea D, et al. Strem2 Cerebrospinal Fluid Levels Are a Potential Biomarker for Microglia Activity in Early-Stage Alzheimer’s Disease and Associate With Neuronal Injury Markers. EMBO Mol Med (2016) 8:466–76. doi: 10.15252/emmm.201506123
69. Zhong L, Chen XF, Wang T, Wang Z, Liao C, Wang Z, et al. Soluble TREM2 Induces Inflammatory Responses and Enhances Microglial Survival. J Exp Med (2017) 214:597–607. doi: 10.1084/jem.20160844
70. Winner B, Jappelli R, Maji SK, Desplats PA, Boyer L, Aigner S, et al. In Vivo Demonstration That Alpha-Synuclein Oligomers are Toxic. Proc Natl Acad Sci U S A (2011) 108:4194–9. doi: 10.1073/pnas.1100976108
71. Rannikko EH, Weber S, SKahle PJ. Exogenous α-Synuclein Induces Toll-Like Receptor 4 Dependent Inflammatory Responses in Astrocytes. BMC Neurosci (2015) 16:57. doi: 10.1186/s12868-015-0192-0
72. Theodore S, Cao S, McLean PJ, Standaert DG. Targeted Overexpression of Human Alpha-Synuclein Triggers Microglial Activation and an Adaptive Immune Response in a Mouse Model of Parkinson Disease. J Neuropathol Exp Neurol (2008) 67:1149–58. doi: 10.1097/NEN.0b013e31818e5e99
73. Tapias V, Hu X, Luk KC, Sanders LH, Lee VM, Greenamyre JT. Synthetic Alpha-Synuclein Fibrils Cause Mitochondrial Impairment and Selective Dopamine Neurodegeneration in Part via iNOS-Mediated Nitric Oxide Production. Cell Mol Life Sci (2017) 74:2851–74. doi: 10.1007/s00018-017-2541-x
74. Peng G, Qiu J, Liu H, Zhou M, Huang S, Guo W, et al. Analysis of Cerebrospinal Fluid Soluble TREM2 and Polymorphisms in Sporadic Parkinson’s Disease in a Chinese Population. J Mol Neurosci (2020) 70:294–301. doi: 10.1007/s12031-019-01424-7
75. Mecca C, Giambanco I, Donato R, Arcuri C. Microglia and Aging: The Role of the TREM2-DAP12 and CX3CL1-CX3CR1 Axes. Int J Mol Sci (2018) 19:318. doi: 10.3390/ijms19010318
76. Stefano L, Racchetti G, Bianco F, Passini N, Gupta RS, Panina Bordignon P, et al. The Surface-Exposed Chaperone, Hsp60, Is an Agonist of the Microglial TREM2 Receptor. J Neurochem (2009) 110:284–94. doi: 10.1111/j.1471-4159.2009.06130.x
77. Lucin KM, O’Brien CE, Bieri G, Czirr E, Mosher KI, Abbey RJ, et al. Microglial Beclin 1 Regulates Retromer Trafficking and Phagocytosis and Is Impaired in Alzheimer’s Disease. Neuron (2013) 79:873–86. doi: 10.1016/j.neuron.2013.06.046
78. Isobe C, Abe T, Terayama Y. Levels of Reduced and Oxidized Coenzyme Q-10 and 8-Hydroxy-2’-Deoxyguanosine in the Cerebrospinal Fluid of Patients With Living Parkinson’s Disease Demonstrate That Mitochondrial Oxidative Damage and/or Oxidative DNA Damage Contributes to the Neurodegenerative Process. Neurosci Lett (2010) 469:159–63. doi: 10.1016/j.neulet.2009.11.065
79. Miletić J, Drakulić D, Pejić S, Petković M, Ilić TV, Miljković M, et al. Prooxidant-Antioxidant Balance, Advanced Oxidation Protein Products and Lipid Peroxidation in Serbian Patients With Parkinson’s Disease. Int J Neurosci (2018) 128:600–7. doi: 10.1080/00207454.2017.1403916
80. Dal Ben M, Bongiovanni R, Tuniz S, Fioriti E, Tiribelli C, Moretti R, et al. Earliest Mechanisms of Dopaminergic Neurons Sufferance in a Novel Slow Progressing Ex Vivo Model of Parkinson Disease in Rat Organotypic Cultures of Substantia Nigra. Int J Mol Sci (2019) 20:2224. doi: 10.3390/ijms20092224
81. Liu AH, Chu M, Wang YP. Up-Regulation of Trem2 Inhibits Hippocampal Neuronal Apoptosis and Alleviates Oxidative Stress in Epilepsy via the PI3K/Akt Pathway in Mice. Neurosci Bull (2019) 35:471–85. doi: 10.1007/s12264-018-0324-5
82. Rohn TT. The Triggering Receptor Expressed on Myeloid Cells 2: "TREM-Ming" the Inflammatory Component Associated With Alzheimer’s Disease. Oxid Med Cell Longev (2013) 2013:860959. doi: 10.1155/2013/860959
83. Trudler D, Weinreb O, Mandel SA, Youdim MB, Frenkel D. DJ-1 Deficiency Triggers Microglia Sensitivity to Dopamine Toward a Pro-Inflammatory Phenotype That is Attenuated by Rasagiline. J Neurochem (2014) 129:434–47. doi: 10.1111/jnc.12633
84. Yang S, Yang Y, Wang F, Luo Q, Zhang Y, Zheng F, et al. TREM2 Dictates Antibacterial Defense and Viability of Bone Marrow-Derived Macrophages During Bacterial Infection. Am J Respir Cell Mol Biol (2021) 65:176–88. doi: 10.1165/rcmb.2020-0521OC
85. He J, Zhu G, Wang G, Zhang F. Oxidative Stress and Neuroinflammation Potentiate Each Other to Promote Progression of Dopamine Neurodegeneration. Oxid Med Cell Longev (2020) 2020:6137521. doi: 10.1155/2020/6137521
86. Weng Y, Wang H, Li L, Feng Y, Xu S, Wang Z. Trem2 Mediated Syk-Dependent ROS Amplification Is Essential for Osteoclastogenesis in Periodontitis Microenvironment. Redox Biol (2021) 40:101849. doi: 10.1016/j.redox.2020.101849
87. Linnartz-Gerlach B, Bodea LG, Klaus C, Ginolhac A, Halder R, Sinkkonen L, et al. TREM2 Triggers Microglial Density and Age-Related Neuronal Loss. Glia (2019) 67:539–50. doi: 10.1002/glia.23563
88. Agrawal I, Jha S. Mitochondrial Dysfunction and Alzheimer’s Disease: Role of Microglia. Front Aging Neurosci (2020) 12:252. doi: 10.3389/fnagi.2020.00252
89. Liu N, Liu J, Wen X, Bai L, Shao R, Bai J. TRPV4 Contributes to ER Stress: Relation to Apoptosis in the MPP(+)-Induced Cell Model of Parkinson’s Disease. Life Sci (2020) 261:118461. doi: 10.1016/j.lfs.2020.118461
90. Ghemrawi R, Khair M. Endoplasmic Reticulum Stress and Unfolded Protein Response in Neurodegenerative Diseases. Int J Mol Sci (2020) 21:6127. doi: 10.3390/ijms21176127
91. Price BR, Sudduth TL, Weekman EM, Johnson S, Hawthorne D, Woolums A, et al. Therapeutic Trem2 Activation Ameliorates Amyloid-Beta Deposition and Improves Cognition in the 5XFAD Model of Amyloid Deposition. J Neuroinflamma (2020) 17:238. doi: 10.1186/s12974-020-01915-0
92. Andreone BJ, Przybyla L, Llapashtica C, Rana A, Davis SS, van Lengerich B, et al. Alzheimer’s-Associated Plcγ2 is a Signaling Node Required for Both TREM2 Function and the Inflammatory Response in Human Microglia. Nat Neurosci (2020) 23:927–38. doi: 10.1038/s41593-020-0650-6
93. Gouna G, Klose C, Bosch-Queralt M, Liu L, Gokce O, Schifferer M, et al. TREM2-Dependent Lipid Droplet Biogenesis in Phagocytes is Required for Remyelination. J Exp Med (2021) 218:e20210227. doi: 10.1084/jem.20210227
94. Nugent AA, Lin K, van Lengerich B, Lianoglou S, Przybyla L, Davis SS, et al. TREM2 Regulates Microglial Cholesterol Metabolism Upon Chronic Phagocytic Challenge. Neuron (2020) 105:837–54.e9. doi: 10.1016/j.neuron.2019.12.007
95. Damisah EC, Rai A, Grutzendler J. TREM2: Modulator of Lipid Metabolism in Microglia. Neuron (2020) 105:759–61. doi: 10.1016/j.neuron.2020.02.008
96. Jiang H, Song N, Jiao Q, Shi L, Du X. Iron Pathophysiology in Parkinson Diseases. Adv Exp Med Biol (2019) 1173:45–66. doi: 10.1007/978-981-13-9589-5_4
97. Abeyawardhane DL, Lucas HR. Iron Redox Chemistry and Implications in the Parkinson’s Disease Brain. Oxid Med Cell Longev (2019) 2019:4609702. doi: 10.1155/2019/4609702
98. Mochizuki H, Choong CJ, Baba K. Parkinson’s Disease and Iron. J Neural Transm (Vienna) (2020) 127:181–7. doi: 10.1007/s00702-020-02149-3
99. Bishop GM, Dang TN, Dringen R, Robinson SR. Accumulation of non-Transferrin-Bound Iron by Neurons, Astrocytes, and Microglia. Neurotox Res (2011) 19:443–51. doi: 10.1007/s12640-010-9195-x
100. Healy S, McMahon J, Owens P, FitzGerald U. Significant Glial Alterations in Response to Iron Loading in a Novel Organotypic Hippocampal Slice Culture Model. Sci Rep (2016) 6:36410. doi: 10.1038/srep36410
101. Liu LL, Cao ZH, He CL, Zhong YC, Liu WY, Zhang P, et al. Ferric Ion Induction of Triggering Receptor Expressed in Myeloid Cells-2 Expression and PI3K/Akt Signaling Pathway in Preosteoclast Cells to Promote Osteoclast Differentiation. Orthop Surg (2020) 12:1304–12. doi: 10.1111/os.12750
102. Frazzitta G, Ferrazzoli D, Folini A, Palamara G, Maestri R. Severe Constipation in Parkinson’s Disease and in Parkinsonisms: Prevalence and Affecting Factors. Front Neurol (2019) 10:621. doi: 10.3389/fneur.2019.00621
103. Liu J, Xu F, Nie Z, Shao L. Gut Microbiota Approach-A New Strategy to Treat Parkinson’s Disease. Front Cell Infect Microbiol (2020) 10:570658. doi: 10.3389/fcimb.2020.570658
104. Sánchez-Ferro Á, Rábano A, Catalán MJ, Rodríguez-Valcárcel FC, Fernández Díez S, Herreros-Rodríguez J, et al. In Vivo Gastric Detection of α-Synuclein Inclusions in Parkinson’s Disease. Mov Disord (2015) 30:517–24. doi: 10.1002/mds.25988
105. Shannon KM, Keshavarzian A, Mutlu E, Dodiya HB, Daian D, Jaglin JA, et al. Alpha-Synuclein in Colonic Submucosa in Early Untreated Parkinson’s Disease. Mov Disord (2012) 27:709–15. doi: 10.1002/mds.23838
106. Challis C, Hori A, Sampson TR, Yoo BB, Challis RC, Hamilton AM, et al. Gut-Seeded α-Synuclein Fibrils Promote Gut Dysfunction and Brain Pathology Specifically in Aged Mice. Nat Neurosci (2020) 23:327–36. doi: 10.1038/s41593-020-0589-7
107. Gershanik OS. Does Parkinson’s Disease Start in the Gut? Arq Neuropsiquiatr (2018) 76:67–70. doi: 10.1590/0004-282x20170188
108. Seno H, Miyoshi H, Brown SL, Geske MJ, Colonna M, Stappenbeck TS. Efficient Colonic Mucosal Wound Repair Requires Trem2 Signaling. Proc Natl Acad Sci U S A (2009) 106:256–61. doi: 10.1073/pnas.0803343106
109. Natale G, Biagioni F, Busceti CL, Gambardella S, Limanaqi F, Fornai F. TREM Receptors Connecting Bowel Inflammation to Neurodegenerative Disorders. Cells (2019) 8:1124. doi: 10.3390/cells8101124
110. Correale C, Genua M, Vetrano S, Mazzini E, Martinoli C, Spinelli A, et al. Bacterial Sensor Triggering Receptor Expressed on Myeloid Cells-2 Regulates the Mucosal Inflammatory Response. Gastroenterology (2013) 144:346–56.e3. doi: 10.1053/j.gastro.2012.10.040
111. Wu YY, Kuo HC. Functional Roles and Networks of non-Coding RNAs in the Pathogenesis of Neurodegenerative Diseases. J BioMed Sci (2020) 27:49. doi: 10.1186/s12929-020-00636-z
112. Yanaizu M, Sakai K, Tosaki Y, Kino Y, Satoh JI. Small Nuclear RNA-Mediated Modulation of Splicing Reveals a Therapeutic Strategy for a TREM2 Mutation and its Post-Transcriptional Regulation. Sci Rep (2018) 8:6937. doi: 10.1038/s41598-018-25204-2
113. Bhattacharjee S, Zhao Y, Dua P, Rogaev EI, Lukiw WJ. microRNA-34a-Mediated Down-Regulation of the Microglial-Enriched Triggering Receptor and Phagocytosis-Sensor TREM2 in Age-Related Macular Degeneration. PloS One (2016) 11:e0150211. doi: 10.1371/journal.pone.0150211
114. Saika R, Sakuma H, Noto D, Yamaguchi S, Yamamura T, Miyake S. MicroRNA-101a Regulates Microglial Morphology and Inflammation. J Neuroinflamma (2017) 14:109. doi: 10.1186/s12974-017-0884-8
115. Liu S, Li XM, Yuan JB, Li LL, Wang C, Lin XM, et al. MiR-665 Inhibits Inflammatory Response in Microglia Following Spinal Cord Injury by Targeting TREM2. Eur Rev Med Pharmacol Sci (2021) 25:65–70. doi: 10.26355/eurrev_202101_24349
116. Alexandrov PN, Zhao Y, Jones BM, Bhattacharjee SLukiw WJ. Expression of the Phagocytosis-Essential Protein TREM2 Is Down-Regulated by an Aluminum-Induced miRNA-34a in a Murine Microglial Cell Line. J Inorg Biochem (2013) 128:267–9. doi: 10.1016/j.jinorgbio.2013.05.010
117. Lv Q, Zhong Z, Hu B, Yan S, Yan Y, Zhang J, et al. MicroRNA-3473b Regulates the Expression of TREM2/ULK1 and Inhibits Autophagy in Inflammatory Pathogenesis of Parkinson Disease. J Neurochem (2021) 157:599–610. doi: 10.1111/jnc.15299
118. Guo M, Wang J, Zhao Y, Feng Y, Han S, Dong Q, et al. Microglial Exosomes Facilitate α-Synuclein Transmission in Parkinson’s Disease. Brain (2020) 143:1476–97. doi: 10.1093/brain/awaa090
119. Paolicelli RC, Bergamini G, Rajendran L. Cell-To-Cell Communication by Extracellular Vesicles: Focus on Microglia. Neuroscience (2019) 405:148–57. doi: 10.1016/j.neuroscience.2018.04.003
120. Li X, He X, Wang J, Wang D, Cong P, Zhu A, et al. The Regulation of Exosome-Derived miRNA on Heterogeneity of Macrophages in Atherosclerotic Plaques. Front Immunol (2020) 11:2175. doi: 10.3389/fimmu.2020.02175
121. Raha-Chowdhury R, Henderson JW, Raha AA, Stott SRW, Vuono R, Foscarin S, et al. Erythromyeloid-Derived TREM2: A Major Determinant of Alzheimer’s Disease Pathology in Down Syndrome. J Alzheimers Dis (2018) 61:1143–62. doi: 10.3233/JAD-170814
122. Fan Y, Ma Y, Huang W, Cheng X, Gao N, Li G, et al. Up-Regulation of TREM2 Accelerates the Reduction of Amyloid Deposits and Promotes Neuronal Regeneration in the Hippocampus of Amyloid Beta1-42 Injected Mice. J Chem Neuroanat (2019) 97:71–9. doi: 10.1016/j.jchemneu.2019.02.002
123. Ding ZB, Han QX, Wang Q, Song LJ, Chu GG, Guo MF, et al. Fasudil Enhances the Phagocytosis of Myelin Debris and the Expression of Neurotrophic Factors in Cuprizone-Induced Demyelinating Mice. Neurosci Lett (2021) 753:135880. doi: 10.1016/j.neulet.2021.135880
124. Palasz E, Wysocka A, Gasiorowska A, Chalimoniuk M, Niewiadomski W, Niewiadomska G. BDNF as a Promising Therapeutic Agent in Parkinson’s Disease. Int J Mol Sci (2020) 21:1170. doi: 10.3390/ijms21031170
125. Tolosa E, Wenning G, Poewe W. The Diagnosis of Parkinson’s Disease. Lancet Neurol (2006) 5:75–86. doi: 10.1016/S1474-4422(05)70285-4
126. Meier SR, Sehlin D, Hultqvist G, Syvänen S. Pinpointing Brain TREM2 Levels in Two Mouse Models of Alzheimer’s Disease. Mol Imaging Biol (2021) 23:665–75. doi: 10.1007/s11307-021-01591-3
127. Lee CYD, Daggett A, Gu X, Jiang LL, Langfelder P, Li X, et al. Elevated TREM2 Gene Dosage Reprograms Microglia Responsivity and Ameliorates Pathological Phenotypes in Alzheimer’s Disease Models. Neuron (2018) 97:1032–1048.e5. doi: 10.1016/j.neuron.2018.02.002
128. Pissarek M. Positron Emission Tomography in the Inflamed Cerebellum: Addressing Novel Targets Among G Protein-Coupled Receptors and Immune Receptors. Pharmaceutics (2020) 12:925. doi: 10.3390/pharmaceutics12100925
129. Schmid CD, Sautkulis LN, Danielson PE, Cooper J, Hasel KW, Hilbush BS, et al. Heterogeneous Expression of the Triggering Receptor Expressed on Myeloid Cells-2 on Adult Murine Microglia. J Neurochem (2002) 83:1309–20. doi: 10.1046/j.1471-4159.2002.01243.x
130. Zuroff LR, Torbati T, Hart NJ, Fuchs DT, Sheyn J, Rentsendorj A, et al. Effects of IL-34 on Macrophage Immunological Profile in Response to Alzheimer’s-Related Aβ(42) Assemblies. Front Immunol (2020) 11:1449. doi: 10.3389/fimmu.2020.01449
131. Yanaizu M, Washizu C, Nukina N, Satoh JI, Kino Y. CELF2 Regulates the Species-Specific Alternative Splicing of TREM2. Sci Rep (2020) 10:17995. doi: 10.1038/s41598-020-75057-x
132. Wang S, Mustafa M, Yuede CM, Salazar SV, Kong P, Long H, et al. Anti-Human TREM2 Induces Microglia Proliferation and Reduces Pathology in an Alzheimer’s Disease Model. J Exp Med (2020) 217:e20200785. doi: 10.1084/jem.20200785
133. Magno L, Bunney TD, Mead E, Svensson F, Bictash MN. Trem2/Plcγ2 Signalling in Immune Cells: Function, Structural Insight, and Potential Therapeutic Modulation. Mol Neurodegener (2021) 16:22. doi: 10.1186/s13024-021-00436-5
Keywords: Parkinson’s disease, pathogenesis, TREM2, immunoregulation, treatment
Citation: Li X-x and Zhang F (2021) Targeting TREM2 for Parkinson’s Disease: Where to Go? Front. Immunol. 12:795036. doi: 10.3389/fimmu.2021.795036
Received: 14 October 2021; Accepted: 13 December 2021;
Published: 24 December 2021.
Edited by:
Cristoforo Comi, University of Eastern Piedmont, ItalyReviewed by:
Giuseppe Cappellano, University of Eastern Piedmont, ItalyCopyright © 2021 Li and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Feng Zhang, emhhbmdmZW5nem1jQDE2My5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.