 Madhuparna Nandi1†
Madhuparna Nandi1† Mitterrand Muamba Moyo2†Sakina Orkhis1†Jeanne Masunga Faida Mobulakani1
Mitterrand Muamba Moyo2†Sakina Orkhis1†Jeanne Masunga Faida Mobulakani1 Marc-André Limoges1
Marc-André Limoges1 Fjolla Rexhepi1Marian Mayhue1Anny Armas Cayarga2Gisela Cofino Marrero2
Fjolla Rexhepi1Marian Mayhue1Anny Armas Cayarga2Gisela Cofino Marrero2 Subburaj Ilangumaran1,3Alfredo Menendez2,3*‡
Subburaj Ilangumaran1,3Alfredo Menendez2,3*‡ Sheela Ramanathan1,3*‡
Sheela Ramanathan1,3*‡- 1Department of Immunology and Cell Biology, Université de Sherbrooke, Sherbrooke, QC, Canada
- 2Department of Microbiology and Infectious Diseases, Université de Sherbrooke, Sherbrooke, QC, Canada
- 3Centre de Recherche du Centre Hospitalier Universitaire de Sherbrooke (CRCHUS), Sherbrooke, QC, Canada
Interleukin-15, produced by hematopoietic and parenchymal cells, maintains immune cell homeostasis and facilitates activation of lymphoid and myeloid cell subsets. IL-15 interacts with the ligand-binding receptor chain IL-15Rα during biosynthesis, and the IL-15:IL-15Rα complex is trans-presented to responder cells that express the IL-2/15Rβγc complex to initiate signaling. IL-15-deficient and IL-15Rα-deficient mice display similar alterations in immune cell subsets. Thus, the trimeric IL-15Rαβγc complex is considered the functional IL-15 receptor. However, studies on the pathogenic role of IL-15 in inflammatory and autoimmune diseases indicate that IL-15 can signal independently of IL-15Rα via the IL-15Rβγc dimer. Here, we compared the ability of mice lacking IL-15 (no signaling) or IL-15Rα (partial/distinct signaling) to control Listeria monocytogenes infection. We show that IL-15-deficient mice succumb to infection whereas IL-15Rα-deficient mice clear the pathogen as efficiently as wildtype mice. IL-15-deficient macrophages did not show any defect in bacterial uptake or iNOS expression in vitro. In vivo, IL-15 deficiency impaired the accumulation of inflammatory monocytes in infected spleens without affecting chemokine and pro-inflammatory cytokine production. The inability of IL-15-deficient mice to clear L. monocytogenes results from impaired early IFNγ production, which was not affected in IL-15Rα-deficient mice. Administration of IFNγ partially enabled IL-15-deficient mice to control the infection. Bone marrow chimeras revealed that IL-15 needed for early bacterial control can originate from both hematopoietic and non-hematopoietic cells. Overall, our findings indicate that IL-15-dependent IL-15Rα-independent signaling via the IL-15Rβγc dimeric complex is necessary and sufficient for the induction of IFNγ from sources other than NK/NKT cells to control bacterial pathogens.
Introduction
Interleukin-15 (IL-15) is a multi-faceted cytokine that facilitates the activation of myeloid and lymphoid components of the innate immune response to pathogens (1–3). IL-15 is expressed by activated macrophages and dendritic cells (DC) as well as stromal cells in the bone marrow and thymus and intestinal epithelial cells (4). Even though IL-15 transcripts are detected in different organs such as skeletal muscle, intestine and kidney at steady state (5), protein expression is not detectable by conventional methods (4). The IL-15 receptor (IL-15R) is a trimeric complex composed of the ligand-binding IL-15Rα chain, the β chain shared by IL-2 and IL-15 (IL-2/15Rβ) and the common γc chain (1). Mice lacking either IL-15 or IL-15Rα display reduced numbers of NK and NKT cells, memory CD8+ T lymphocytes and intestinal epithelial lymphocytes (IEL), indicating that IL-15Rα is crucial for their IL-15-dependent homeostasis (6, 7). Transgenic expression of human IL-15 increases NK and memory CD8+ T cells but not monocytes or B cells (2, 8, 9). Mechanistic studies revealed that the biological activities of IL-15 on lymphoid cells are mediated by IL-15 bound to IL-15Rα during biosynthesis and ‘trans-presentation’ of the IL-15:IL-15Rα (IL15:15Rα) complex to responder cells expressing the IL-2/15Rβγc heterodimeric receptor complex, explaining the similarity in the phenotype of Il15-/- and Il15ra-/- mice (6, 7, 10).
Studying the mechanisms of IL-15 signaling has remained challenging due to post-transcriptional and post-translational regulation of IL-15 expression and the requirement for IL-15 trans-presentation (11, 12). In producer cells, IL-15 protein is synthesized with either a short or a long signal peptide (SSP and LSP, respectively). Whereas the SSP-IL-15 is not released from cells, LSP-IL-15 is inefficiently secreted (13). As transgenic mice expressing LSP-IL-15 mounted a robust immune response following Salmonella infection, whereas mice expressing the non-secreted SSP-IL-15 did not, it was postulated that the over-expressed SSP-IL-15 isoform may compete with endogenous IL-15 for binding to IL-15Rα and inhibit IL-15 signaling (14). While soluble IL-15 can promote activation of NK and memory CD8+ subsets (7), such high concentrations are unlikely to occur under physiological settings. Free IL-15 can bind the IL-2/15Rβγc receptor complex at nanomolar concentration, however, IL-15Rα facilitates ligand binding at picomolar concentrations (15, 16). IL-15 trans-presented by hematopoietic as well as non-hematopoietic cells is the primary mechanism by which IL-15 activates lymphocytes (17). Tissue specific ablation of IL-15Rα has revealed specific, but distinct patterns of requirement for the source of the trans-presented IL-15 (18, 19). We have shown IL-15 trans-presentation by hepatocytes and macrophages is required for the maintenance of NK and NKT cells in the liver (20).
A few studies comparing the immune responses of Il15-/- and Il15ra-/- mice indicated that IL-15 trans-presentation is not the only mechanism of IL-15 signaling and that it can also occur independently of IL-15Rα, although the underlying mechanisms are not yet fully understood. We have shown that the pathogenic potential of IL-15 in autoimmune type 1 diabetes (T1D) does not require IL-15Rα (21, 22). In the dextran sulfate sodium-azoxymethane (DSS-AOM)-induced colitis-associated carcinogenesis model, Il15-/- mice but not Il15ra-/- mice displayed higher incidence of colorectal cancer and reduced survival (23). These differences, which cannot be explained solely by the lack of NK cells that are similarly affected in IL-15 or IL-15Rα deficient mice (6, 24), indicated that IL-15 can mediate certain aspects of the immune responses independently of IL-15Rα. Moreover, an enlarged compartment of IL-17 producing γδ T cells has been observed in Il15ra-/- but not in Il15-/- mice, suggesting IL-15Rα-independent IL-15-mediated regulation (25).
Using IL-15 deficient mice, several studies have established the requirement of IL-15 to efficiently control bacterial, viral and fungal infections (3, 26, 27) presumably through its functions on lymphoid subsets, which require trans-presentation. However, IL-15Rα-independent functions of IL-15 in antimicrobial defenses are poorly characterized. In this work, we compared the requirement of IL-15 and IL-15Rα in controlling Listeria monocytogenes infection and determined that IL-15 signaling independent of IL-15Rα is critical for the induction of IFNγ and bacterial clearance.
Materials and Methods
Mouse Strains
All the mice strains used in this study were bred in our animal facility to avoid major differences in microbiome and environmental influences. Wild type C57Bl/6N (WT) mice, Il15-/-, Il15ra-/- and Ifng-/- mice in the C57Bl/6N background have been described previously (22, 28–30). Mice were housed in micro-isolated sterile cages under specific pathogen-free conditions. All experimental protocols on mice were approved by Université de Sherbrooke Ethics Committee for Animal Care and Use in accordance with guidelines established by the Canadian Council on Animal Care (protocol # 376-2014-2019; 2019-2023-2280).
Bacterial Strains, Infections and Pre/Post Infection Treatments
Frozen glycerol stock of L. monocytogenes strain 10403s and L. monocytogenes expressing green fluorescent protein (GFP) were obtained from Dr. Portnoy (Department of Molecular and Cell Biology, University of California, Berkeley, CA). Bacteria were cultured in BHI (Blood-heart infusion) medium (VWR, Canada). Mice were injected intravenously in the tail vein with 5 000 to 10 000 colony forming units (CFU) of bacteria for low-dose infections and with 100 000 CFU for high-dose infections. Liver and spleen were collected, and tissue lysates were prepared in 1 mL of PBS from weighed pieces using a tissue homogenizer bead mill (MM 400; Retsch, Hann, Germany). Bacterial burden in tissues was determined by plating serial dilutions of the homogenates on BHI agar plates. Indicated groups of mice received intraperitoneal injections of IFNγ (1 x106 units, R&D Systems, on day 0 and day 1), anti-NK1.1 (PK136; 200ug/mouse/injection at day -1 and +1) or anti-IFNγ (H22; 200ug/mouse/injection at day -1 and +1) antibodies (all from BioXCell, USA) relative to the infection day 0.
Bone Marrow-Derived Macrophages (BMDM)
BMDMs were generated following standard methods (31) with some modifications. Briefly, bone marrow cells collected from femurs of the indicated mice were seeded at a density of 5 ×105 cells per well in 24-well plates and at 106 cells per well in 12-well plates in RPMI medium containing 15% fetal bovine serum (FBS) the presence of 50 ng/mL of macrophage colony stimulating factor (M-CSF, Peprotech, USA) for 5 days. M-CSF was supplemented on Day 4. To generate BMDMs grown in granulocyte- macrophage colony stimulating factor (GM-CSF, Peprotech, USA), the cytokine was added to the above cultures at a concentration of 50 ng/mL on day 6 and the cells were used on day 7.
Infection of BMDM
An overnight culture of L. monocytogenes, which contains approximately 2 x 109 CFU/mL, was washed and resuspended in the same volume of PBS. Twenty five μL of washed bacteria were opsonized by mixing with 20 μL of pre-immune mouse serum and 55 uL RPMI (without serum or antibiotics) for 30 min at 25°C. The inoculum was prepared by diluting the opsonized bacteria in RPMI (100 μL of opsonized bacterial suspension in 3ml RPMI) to provide a multiplicity of infection (MOI) of ~25. The MOI was determined for every experiment by plating the inoculum at 10-fold serial dilutions (10-3, 10- 4, 10-5) on BHI agar plates. These plates were incubated at 37°C from 24 hours, and colonies counted to determine the CFU.
Prior to infection, medium was removed from the BMDM culture plates by gentle suction and the wells were washed once with warm RPMI medium without FCS or antibiotics. Opsonized bacteria (400 or 800 ul/well) were added to BMDMs and the infected cells were incubated at 37°C and 5% CO2 for 30 minutes followed by washing the wells with 1 mL of PBS containing Ca2+ and Mg2+. The wells were replenished with RPMI containing 10% FBS and 20 μg/mL gentamicin to kill the extracellular bacteria. After 4 hours, cells were recovered from the plates for flow cytometry analysis. Another batch of culture was washed with PBS and lysed in 200 μL of lysis buffer containing 1% triton X100/0.1% SDS for counting the intracellular bacteria. Numbers of viable intracellular L. monocytogenes were determined by performing 10- fold serial dilutions (10-1, 10-2, 10-3) and plating on BHI agar plates for 14–16 hr at 37°C for CFU quantification.
Preparation of Splenocyte Suspensions
Splenocytes were isolated and dissociated in PBS containing 1% FBS and penicillin and streptomycin. The cells were labeled with fluorochrome-conjugated antibodies for flowcytometry as described (22, 32). The list of antibodies used is given in Supplementary Table S1. Cells were fixed after surface staining and permeabilized for 30 minutes using a wash buffer (BD Biosciences) following manufacturer’s protocols for intracellular staining of iNOS. Cells were analyzed on a Cytoflex 30 flow cytometer (Beckman Coulter, Brea, CA, USA) and data analyzed using the FlowJo software (BD Biosciences).
Data Analysis
Graphpad Prism software (Version 9) was used for data analysis and to generate illustrations. The data are shown as Mean + standard error of mean (SEM). One- or Two-way ANOVA with Tukey’s or Bonferroni’s post-hoc test was used as indicated in figure legends to compare datasets and to calculate statistical significance.
Results
IL-15 Is Crucial but IL-15Rα Is Dispensable for Clearing Low-Dose L. monocytogenes Infection
WT, Il15-/- and Il15ra-/- mice were infected with a low dose of 5 ×103 CFU of L. monocytogenes via the intravenous route. At 5 days post infection (dpi), female WT mice were able to control the infection and showed reduced bacterial load in spleen and the liver, whereas female Il15-/- mice showed high bacterial counts in these tissues (Figure 1A). In contrast to Il15-/- mice, Il15ra-/- animals cleared L. monocytogenes from the spleen and the liver as efficiently as WT mice (Figure 1A). Male mice have been reported to better resist L. monocytogenes infection compared to female mice (33). However, at the infection dose used in our studies, we did not observe appreciable difference between male and female mice in their ability to clear L. monocytogenes (Figure 1B). Similar to females, WT and Il15ra-/- male mice efficiently cleared splenic and hepatic L. monocytogenes within 5 dpi, whereas Il15-/- mice did not. The bacterial load observed in Il15-/- male mice was comparable to that of Ifng-/- male mice, indicating that IL-15 is as important as IFNγ in controlling L. monocytogenes infection (Figure 1B). The dispensability of IL-15Rα but not IL-15 to clear low dose L. monocytogenes infection indicated that IL-15 signaling can occur independently of IL-15Rα and that this signaling plays a key role in the control of bacterial infections.
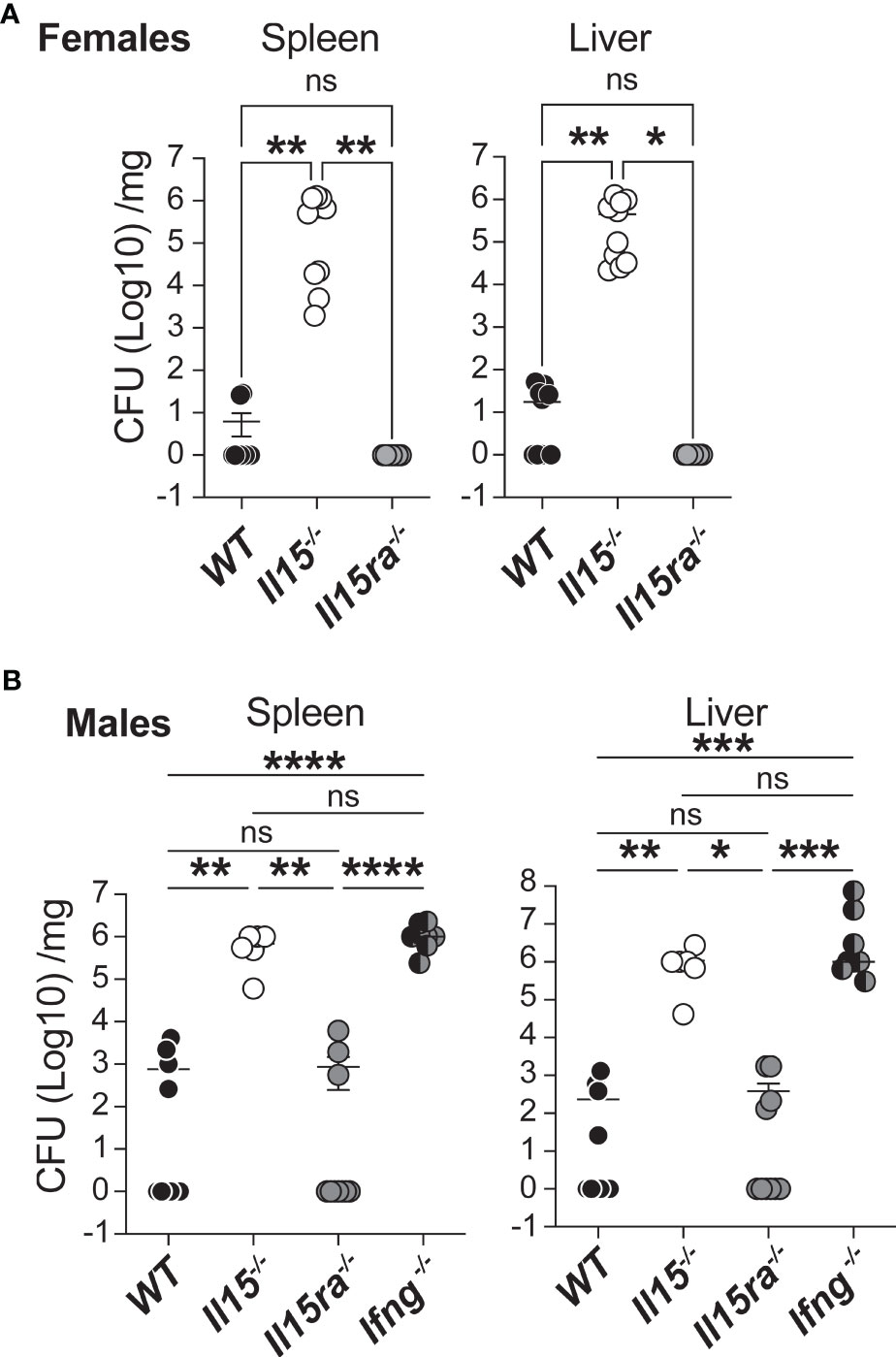
Figure 1 IL-15 is essential and IL-15Rα is dispensable for clearing systemic infection with low dose L. monocytogenes. (A) Female WT, Il15-/- and Il15ra-/- mice were infected i.v. with 5 x103 CFU (colony forming units) of L. monocytogenes. The infected mice were sacrificed 5 days post infection (dpi) and the bacterial load in the spleen and liver was determined by plating on BHI-streptomycin plates. (B) Bacterial load in male WT, Il15-/-, Il15ra-/- and Ifng-/- mice infected i.v. with 5 x103 CFU of L. monocytogenes. Bacterial count is expressed as CFU per mg of tissue. Results are shown as the Mean ± SEM of two independent experiments. n = 5 to 10 mice per group. Statistical significance between groups determined by one-way ANOVA followed by Tukey’s multiple comparisons test, is given as *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001. ns, not significant.
IL-15 Deficiency Does Not Affect the Infection of BMDMs by L. monocytogenes
To investigate whether the IL-15Rα-independent IL-15 signaling impacted bacterial uptake by phagocytic cells, we used bone marrow-derived macrophages (BMDM) established from WT, Il15-/- and Il15ra-/- mice. BMDMs cultured with M-CSF were infected at a MOI of 25 with L. monocytogenes expressing GFP (Lm-GFP) that were opsonized with non-immune mouse serum to facilitate complement-mediated uptake. Thirty min after infection, cells were washed to remove extracellular bacteria and incubated in medium containing gentamycin for additional 4 hours before detection of intracellular bacteria. The fraction of bacteria present within BMDMs was comparable between WT and Il15-/- cells 4 hours post-infection (hpi) (Figure 2A, upper panel). In BMDMs that were activated with GM-CSF for additional 24 hours (31), there was no significant difference among the three groups (Figure 2A, lower panel). Flow cytometry analysis revealed that the proportion of M-CSF-BMDMs harboring Lm-GFP was comparable between the three genotypes (Figure 2B). As BMDMs are diverse in their phenotype (34), it raised the possibility that lack of IL-15 signaling, in the presence or absence of IL-15Rα, might impact the frequencies of BMDM subsets with altered susceptibility to bacterial infection. To determine whether absence of IL-15 altered the infectivity of different subsets of BMDM, the control and infected cultures were labeled for CCR2, Ly6C and MHC class-I (MHC-I) and the GFP+ and GFP- subsets were analyzed. The GFP+ M-CSF-BMDMs from all three genotypes were predominantly Ly6C+CCR2+ (Figure 2C Left panel, bottom row) and upregulated MHC-I expression (Figure 2C Right Panel, bottom row) compared to the GFP- and uninfected cells (middle and top rows, respectively). Macrophages play an essential role in eliminating the L. monocytogenes infected cells by upregulating the expression of inducible NOS2 (iNOS, induced by inflammatory cytokines) for the production of reactive nitrogen species (RNS) (35). Expression of iNOS occurred in a significant proportion of GFP+ BMDM established from all three groups of mice and this proportion was comparable among the three genotypes (Figure 2D). These findings indicated that lack of either IL-15 or IL-15Rα had only a negligible impact on bacterial load in macrophages generated in vitro and their ability to produce iNOS.
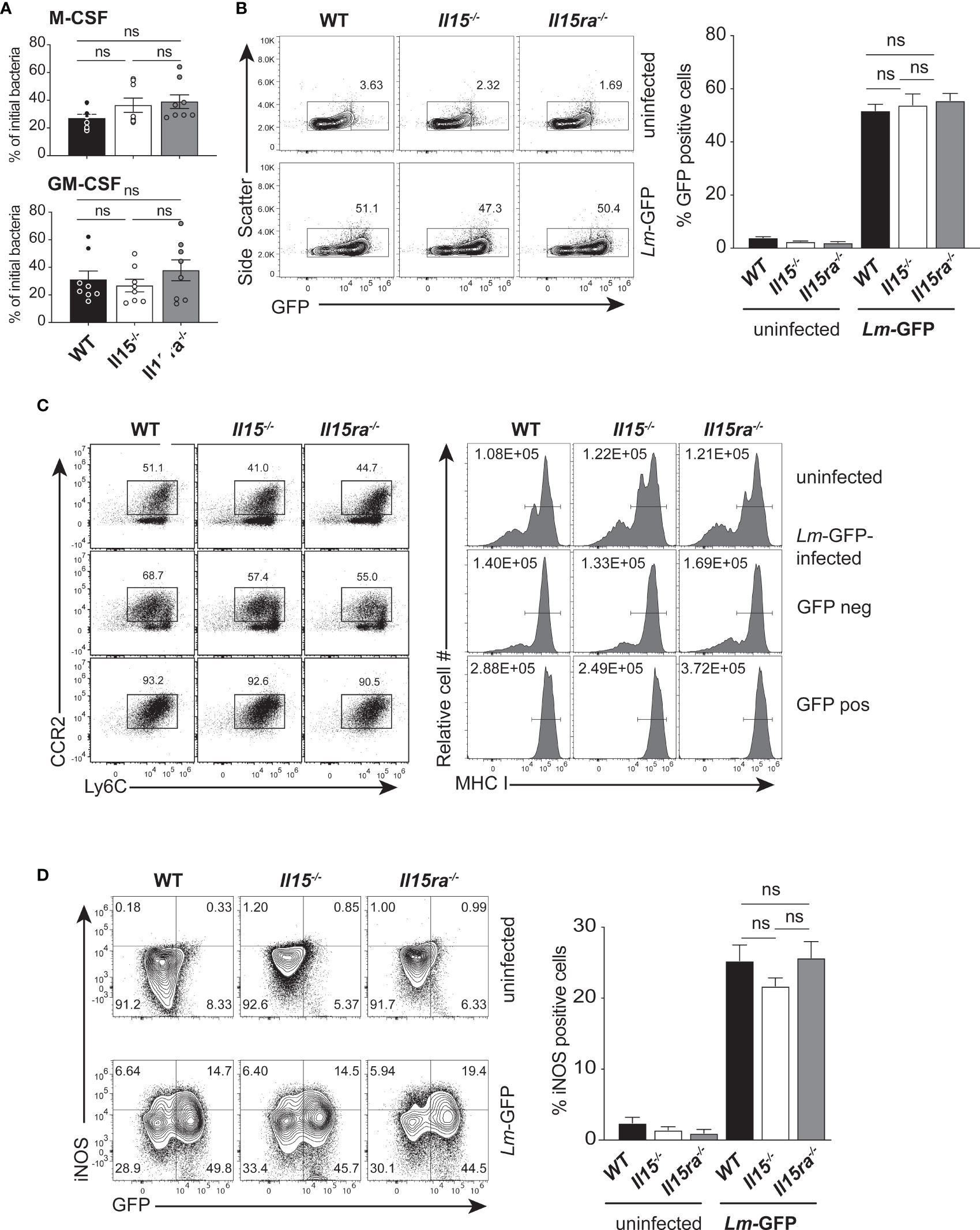
Figure 2 IL-15 deficient BMDMs are infected by L. monocytogenes. (A) BMDMs generated in the presence of M-CSF (upper panel) or treated additionally with GM-CSF for 1 day (lower panel) were infected with opsonized L. monocytogenes expressing GFP (Lm-GFP) at a MOI of 25. The cells were washed 30 minutes after infection and incubated in medium containing gentamycin for additional 4 hours before cell lysis. The number of intracellular bacteria were quantified by plating dilutions of cell lysates on BHI-agar and expressed as a fraction of the bacterial inoculum. (B) The proportion of CD11b+ cells that are positive for GFP 4 hours after infection was determined by flow cytometry. Representative flow cytometry data (left panel) and quantification of GFP+ cells (right panel) are shown. (C) The expression of Ly6C and CCR2 (left Panel) and MHC-I (right Panel) on uninfected controls and GFP+ and GFP-cells from the infected cultures are shown. Representative FACS data from one of the 3 experiments are shown. (D) BMDMs generated using M-CSF were infected with Lm-GFP as in (A) and labelled with CD11b and iNOS Ab for flow cytometry 4 h later. The dot plots (left panel) show iNOS expression in GFP+ and GFP- cells of infected and control cultures. The proportions of iNOS-positive cells among GFP+ cells are shown in the right panel (A) and Right panels in (B, D). Results are shown as the mean ± SEM of 3 independent experiments carried out in triplicates. Statistical significance between groups, determined one-way ANOVA followed by Tukey’s multiple comparisons test, is given. ns, not significant.
Infected IL-15-Deficient Mice Show Accumulation of Inflammatory Monocytes and Elevated Production of Many Chemokines and Pro-Inflammatory Cytokines in Spleen
Next, we determined how IL-15 or IL-15Rα deficiency impacted monocytes/macrophage phenotype in vivo in the spleens of L. monocytogenes-infected WT, Il15-/- and Il15ra-/- mice. The frequency of CD11b+ monocytes was substantially increased in infected Il15-/- mice 3 dpi when compared to infected WT and Il15ra-/- mice (Figure 3A). However, the frequency of Ly6ChiCCR2hiCD11b+ inflammatory monocyte subset showed a moderate but significant reduction in Il15-/- mice (Figures 3B, C). The Ly6CloCCR2loCD11b+ monocyte subset showed a corresponding increase that was not statistically significant. To determine whether the accumulation of Ly6ChiCCR2hiCD11b+ pro-inflammatory monocyte subset in Il15-/- mice was due to the increased bacterial load that occurs in Il15-/- mice (Figure 1A), we used 2 additional groups of mice wherein higher splenic bacterial load can be achieved similar to the levels attained in Il15-/- mice. Ifng-/- mice are susceptible to low dose L. monocytogenes infection (36) and WT mice infected with a high dose of L. monocytogenes (1 x 105 CFU) succumb to disease (37). Even though the bacterial load in Ifng-/- mice and WT mice infected with a high dose inoculum were comparable to Il15-/- mice (Supplementary Figure S1), they did not show an increase in the frequency of CD11b+ cells as in Il15-/- mice (Figure 3A). However, Ifng-/- mice showed an increase in the frequency of the Ly6ChiCCR2hiCD11b+ pro-inflammatory monocyte subset like Il15-/- mice (Figures 3B, C). On the other hand, all groups showed comparable neutrophils infiltration in the spleen, although Il15-/- mice showed a discernible increase (Figure 3D). These results suggest that the lack of IL-15 but not that of IL-15Rα impacts the accumulation of Ly6ChiCCR2hiCD11b+ pro-inflammatory monocyte subset in the infected tissue.
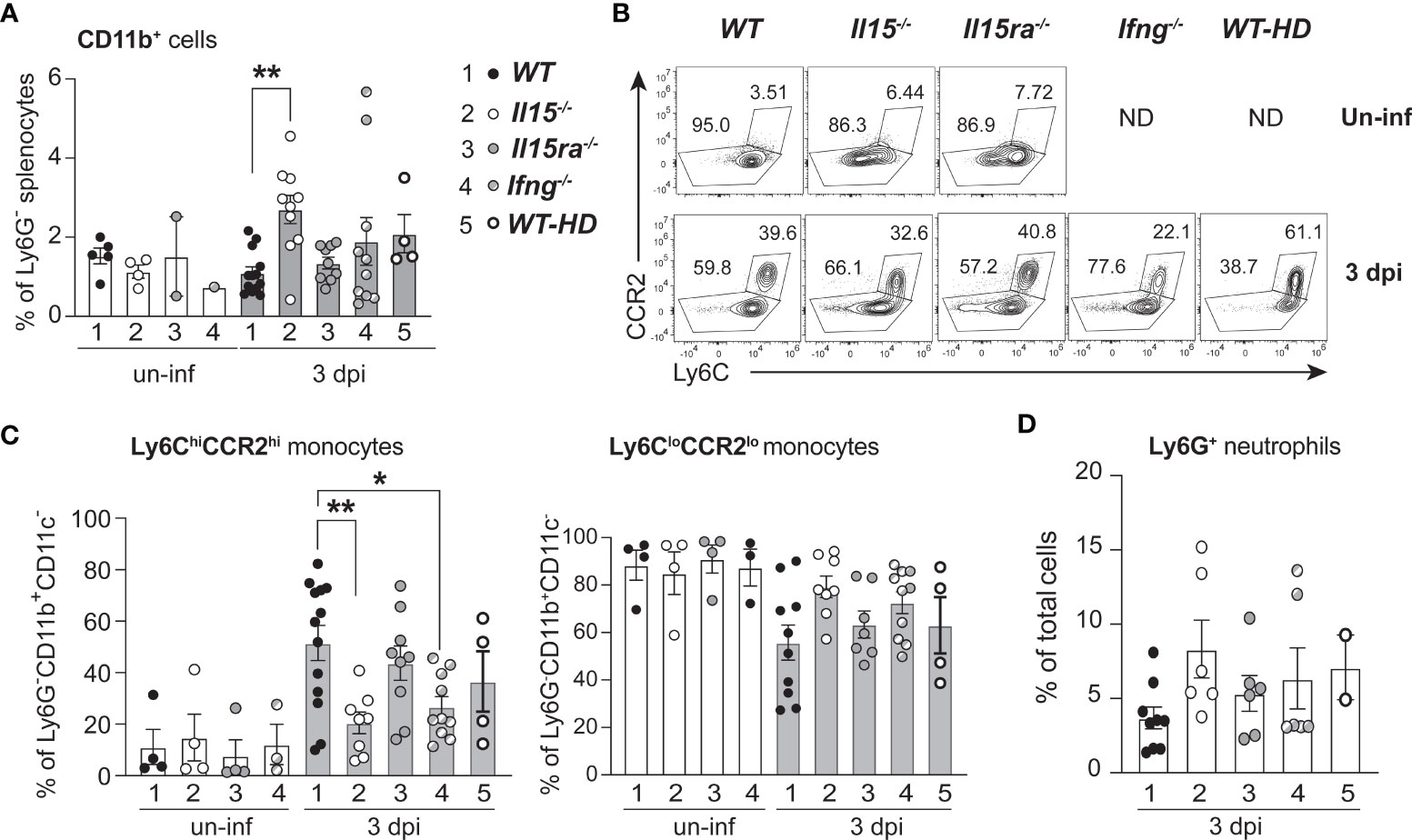
Figure 3 Lack of IL-15 but not IL-15Rα reduces the frequency of Ly6C+CCR2+ pro-inflammatory monocytes in the spleen following L. monocytogenes infection. (A) WT, Il15-/-, Il15ra-/- and Ifng-/- mice were infected i.v. with 5 x103 CFU of L. monocytogenes. A cohort of WT mice were infected i.v. with higher dose (50-100 x103 CFU; WT-HD) of L. monocytogenes to obtain higher bacterial load shown in Supplementary Figure 1. The infected mice were sacrificed at 3 dpi and the splenocytes were analyzed by flow cytometry. The frequency of CD11b+ cells is shown after gating out neutrophils (based on Ly6G positivity) and CD11c+ cells. (B) Representative density plots of Ly6C and CCR2 expression in CD11b+ cells. (C) Frequency of Ly6C+CCR2+ inflammatory monocytes (left panel) and Ly6CloCCR2lo monocytes (right panel) within the Ly6G-CD11b+CD11c- population. (D) Frequency of Ly6G+ neutrophils. Results are shown as the mean ± SEM values from n = 5 to 10 mice per group from of 2-4 independent experiments. Statistics: one-way ANOVA followed by Tukey’s multiple comparisons test. *p < 0.05, **p < 0.01.
Chemokines play a key role in monocyte recruitment to inflammatory sites, of which CCL2 promotes the recruitment of Ly6C+CCR2+ inflammatory monocytes (38). To assess whether IL-15 deficiency compromises the induction of chemokines, we evaluated the chemokine levels in spleen lysates of control and L. monocytogenes-infected mice by multiplex assays. All infected mice groups showed an upregulation of several CCL (CCL2, CCL3, CCL4) and CXCL (CXCL1, CXCL2, CXCL9, CXCL10) chemokines (Figure 4A). However, the spleens of Il15-/- mice showed significantly elevated levels of most of the chemokines tested compared to WT and Il15ra-/- mice, except CXCL9, which showed an inverse trend with a significant reduction in Il15-/- mice (Figure 4A). CXCL9 is also known as monokine-induced by IFNγ (MIG) that recruits macrophages, NK, NKT and cytotoxic lymphocytes (39). The spleens of infected Il15-/- mice also showed high levels of many pro-inflammatory cytokines (TNFα, IL-1α, IL-1β, IL-6, IL-17) and granulocyte/macrophage growth factors (G-CSF, GM-CSF, M-CSF) (Figure 4B). These data indicated that the susceptibility to infection in Il15-/- mice is not due to the lack of production of most of the chemokines and cytokines associated with innate immune responses immediately after infection.
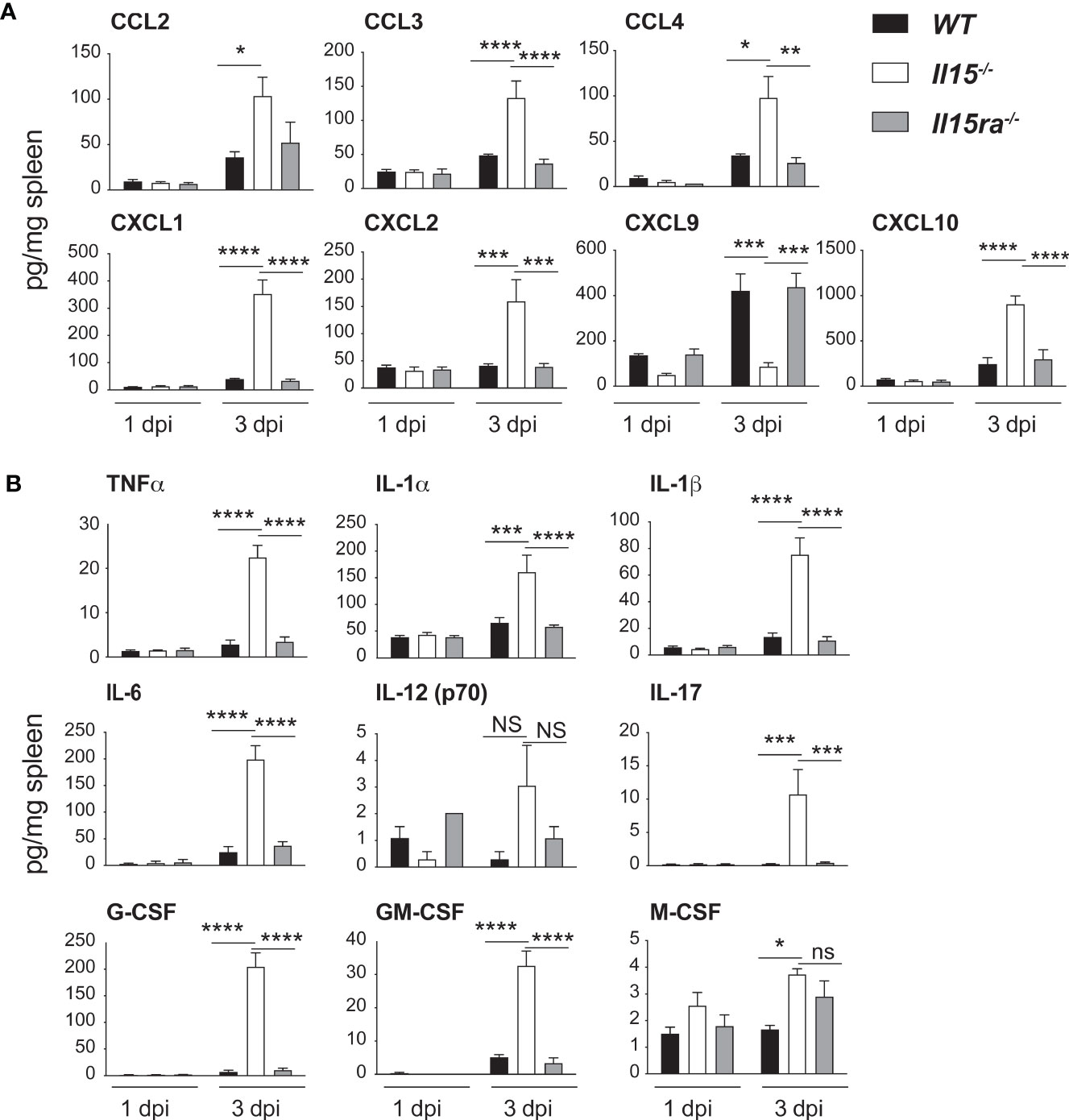
Figure 4 IL-15 deficient mice show increased chemokine and pro-inflammatory cytokine production but reduced CXCL9 expression following L. monocytogenes infection. WT, Il15-/- and Il15ra-/- mice were infected i.v. with 5 x103 CFU of L. monocytogenes. The infected mice were sacrificed at 1 dpi or 3 dpi and spleen tissue lysates were analyzed by multiplex assays for the expression of (A) the indicated CCL and CXCL chemokines and (B) pro-inflammatory cytokines. Results are shown as the mean ± SEM of n = 4 mice per group from 2 independent experiments. Statistical significance between groups was evaluated by Two-way ANOVA followed by Bonferroni’s multiple comparisons test. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, not significant.
To further characterize the response of monocytes/macrophage of L. monocytogenes-infected WT, Il15-/- and Il15ra-/- mice, we assessed the induction of MHC-I, using Ifng-/- mice and WT mice that were infected with high dose L. monocytogenes as controls to mimic the high bacterial load observed in Il15-/- mice. The basal expression of MHC-I on Ly6ChiCCR2hiCD11b+, Ly6CloCCR2loCD11b+ monocytes and CD11b+CD11c+ monocyte subsets was comparable in uninfected mice from the four genotypes, and L. monocytogenes infection induced MHC-I upregulation in all these monocyte subsets (Figure 5A). However, MHC-I expression in Ly6ChiCCR2hiCD11b+ monocytes was significantly lower in Il15-/- and Ifng-/- mice at 3 dpi when compared to WT and Il15ra-/- mice (Figure 5A). The attenuation of MHC-I upregulation in Il15-/- and Ifng-/- mice was also observed in Ly6CloCCR2loCD11b+ but did not show statistical significance (Figure 5B). On the other hand, MHC class I expression in the CD11b+CD11c+ monocyte subsets was comparable between the groups (Figure 5C). The impaired MHC-I upregulation in Ly6ChiCCR2hiCD11b+ monocytes of Il15-/- and Ifng-/- mice could not be attributed to the high bacterial load, as it was not affected in WT-HD mice with high bacterial load achieved by high dose infection (Figure 5A). Moreover, MHC-II expression on the inflammatory monocyte subset was comparable between the mice from different genotypes (Supplementary Figure S2). These findings revealed similarly impaired macrophage responses in Il15-/- and Ifng-/- mice that did not occur in Il15ra-/- mice.
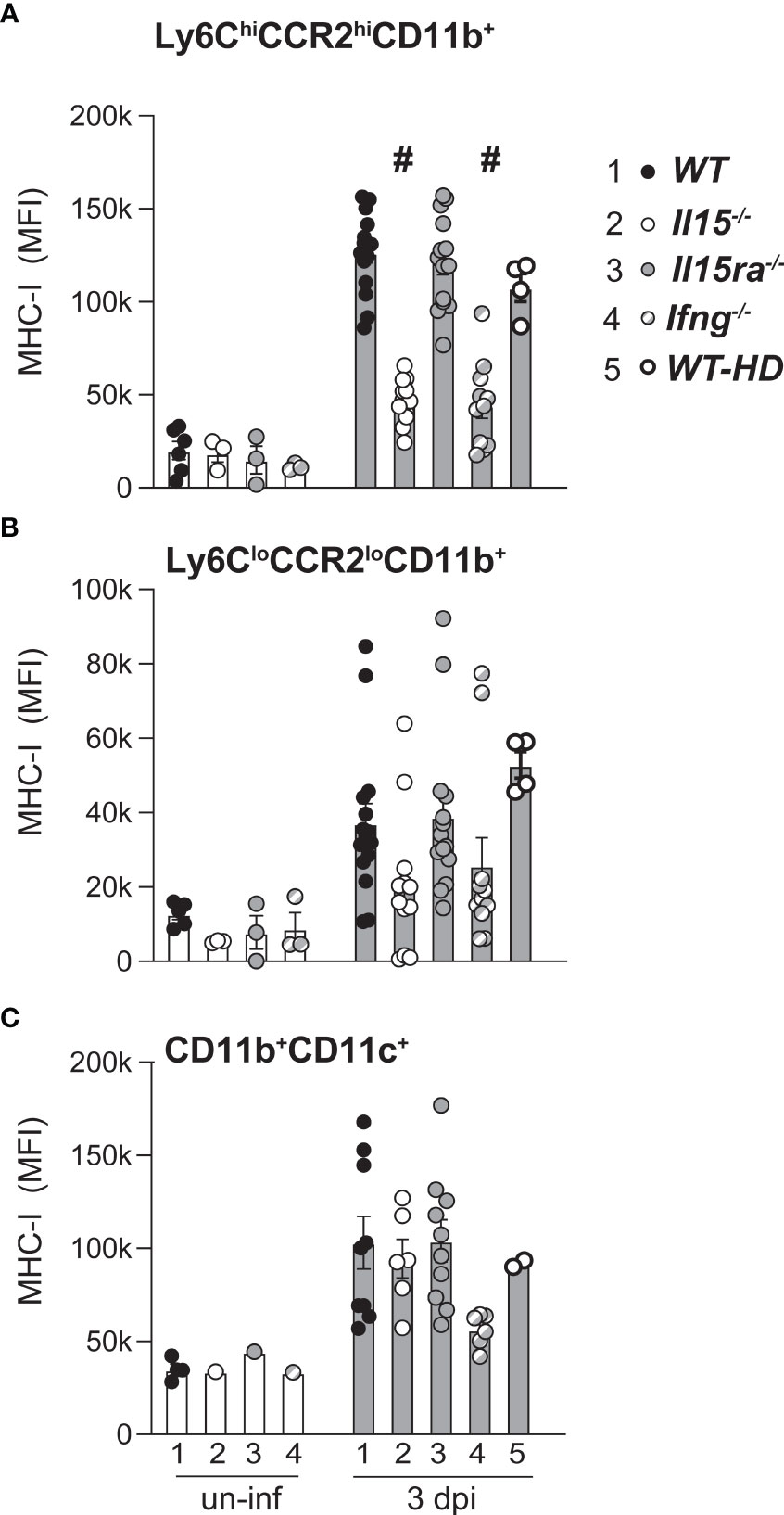
Figure 5 Attenuated MHC Class-I induction in inflammatory monocytes of Il15-/- mice following L. monocytogenes infection. Mean fluorescence intensity (MFI) of MHC class-I expression in Ly6C+CCR2+ inflammatory monocytes (A), Ly6CloCCR2lo monocytes (B) and CD11b+CD11c+ monocytes (C) at 3 dpi. Results are shown as the mean ± SEM of n = 5 to 10 mice per group from 2-4 independent experiments. Statistical significance between groups, one-way ANOVA followed by Tukey’s multiple comparisons test. # p < 0.0001 when compared to groups 1, 3 and 5. Each uninfected group contained 3-4 mice.
Defective Early IFNγ Production in L. monocytogenes Infected Il15-/- but Not Il15ra-/- Mice
The high bacterial load and reduced numbers of Ly6ChiCCR2hiCD11b+ pro-inflammatory monocytes in Il15-/- and Ifng-/- mice but not in Il15ra-/- mice (Figure 3C), and the impaired expression of the IFNγ-inducible CXCL9 in Il15-/- mice (Figure 4A) suggested defective IFNγ responses in the absence of IL-15 but not that of IL-15Rα in the infected mice. Ohteki et al. have reported that heat-killed Propionibacterium acnes (P. acnes) failed to induce IL-12, IFNγ, CCL2 and CCL3 in the livers of Il15-/- mice (40). Similarly, IL-15 deficient mice failed to produce IL-12 during L. monocytogenes infection (41). Low dose L. monocytogenes did not impair the production of IL-12p70, CCL2 or CCL3 in Il15-/- mice (Figure 4A). On the other hand, we observed that IL-15 deficiency but not IL-15Rα deficiency markedly reduced IFNγ expression. Ifng gene expression was induced as early as 1 dpi in the spleens of WT and Il15ra-/- infected mice and peaked around 2 dpi (Figure 6A). However, at these early time points, Ifng expression was very low in the spleen of L. monocytogenes infected Il15-/- mice. Even though Ifng expression in these mice gradually increased at 3 dpi, this level was comparable only to that of uninfected WT and Il15ra-/- mice, which was not sufficient to confer protection, as all infected Il15-/- mice succumbed by 5 dpi. IFNγ protein was detected by 3 dpi in the spleens of WT and Il15ra-/- but was undetectable in Il15-/- mice (Figure 6B).
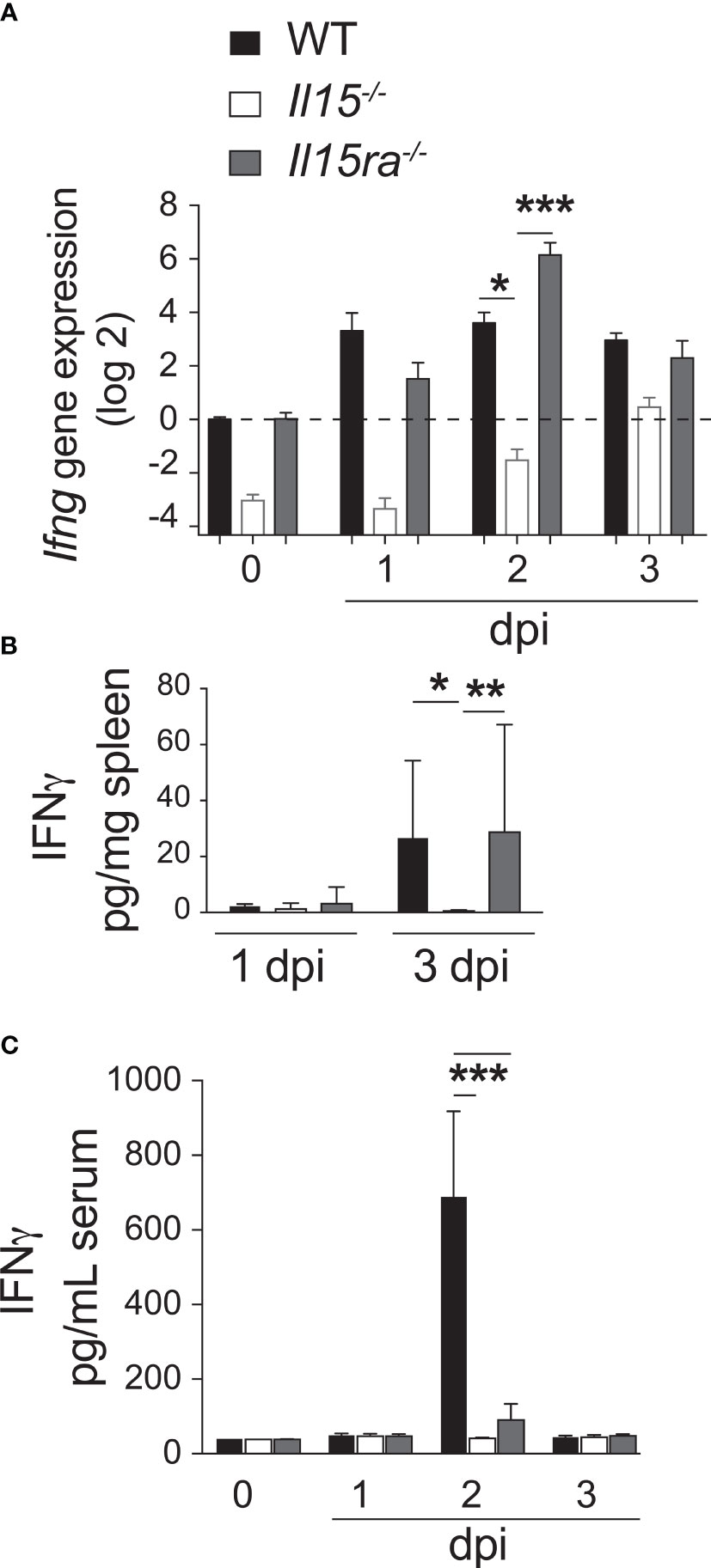
Figure 6 Lack of IL-15 but not IL-15Rα impairs early IFNγ production after L. monocytogenes infection. (A) WT, Il15-/- and Il15ra-/- mice were infected i.v. with 5 x103 CFU of L. monocytogenes. The infected mice were sacrificed at 1, 2 or 3 dpi. (A) Ifng gene expression (fold induction over WT uninfected controls) was determined in the spleen by RT-qPCR. IFNγ protein concentration in spleen lysates (B) and sera (C) from control and infected mice by ELISA. Results are shown as the mean ± SEM for 4 mice per group. Statistical significance between groups, two-way ANOVA followed by Bonferroni’s multiple comparisons test, is given as *p < 0.05, **p < 0.01, ***p < 0.001.
Notably, IFNγ was detected in the sera of infected WT mice and at very low levels in Il15ra-/- mice but not in Il15-/- mice (Figure 6C), suggesting that the source for systemic IFNγ detectable in the sera of WT mice can be NK and NKT cells, which are absent in both Il15ra-/- and Il15-/- mice (7). These observations suggest that early IFNγ production in the infected tissue following exposure to L. monocytogenes is IL-15-dependent but is IL-15Rα independent, and originates from cells other than NK and NKT cells.
Exogenous IFNγ Partially Controls L. monocytogenes Infection in Il15-/- Mice
As IFNγ plays a crucial role in controlling bacterial infections including L. monocytogenes (42), we assessed whether the impaired IFNγ production in Il15-/- mice early during infection was the key defect underlying impaired bacterial clearance despite the elevated production of several inflammatory cytokines in the spleen. To this end, Il15-/- and Ifng-/- mice were treated with 1 x106 units of IFNγ at the time of infection and at 1 dpi. IFNγ treatment reduced the bacterial load in the spleen to a modest level and almost completely in the liver of Il15-/- mice (Figure 7A), suggesting that impaired early IFNγ production plays a crucial role in the control of the infection. Exogenous IFNγ has been shown to limit L. monocytogenes growth in the liver more than in the spleen (43). As we had observed low levels of IFNγ in the circulation of Il15ra-/- mice (Figure 6C), we asked whether depletion of IFNγ prior to infection rendered them susceptible to low dose L. monocytogenes. WT and Il15ra-/- mice received neutralizing anti-IFNγ antibody 18 hours before infection and at 1 dpi and were sacrificed at 3 dpi. Depletion of the low levels of IFNγ present in Il15ra-/- rendered them susceptible to low dose L. monocytogenes and the mice had to be sacrificed 3 dpi (Figure 7B). In line with the observations that lymphocytes including NK cells are not absolutely essential for the early innate immune response in certain situations (44–47), depletion of NK cells in WT or Il15ra-/- mice did not increase their susceptibility to L. monocytogenes (Figure 7C). These observations indicate that IL-15-dependent but IL-15Rα-independent release of IFNγ from sources other than NK cells early during infection is critical for bacterial control.
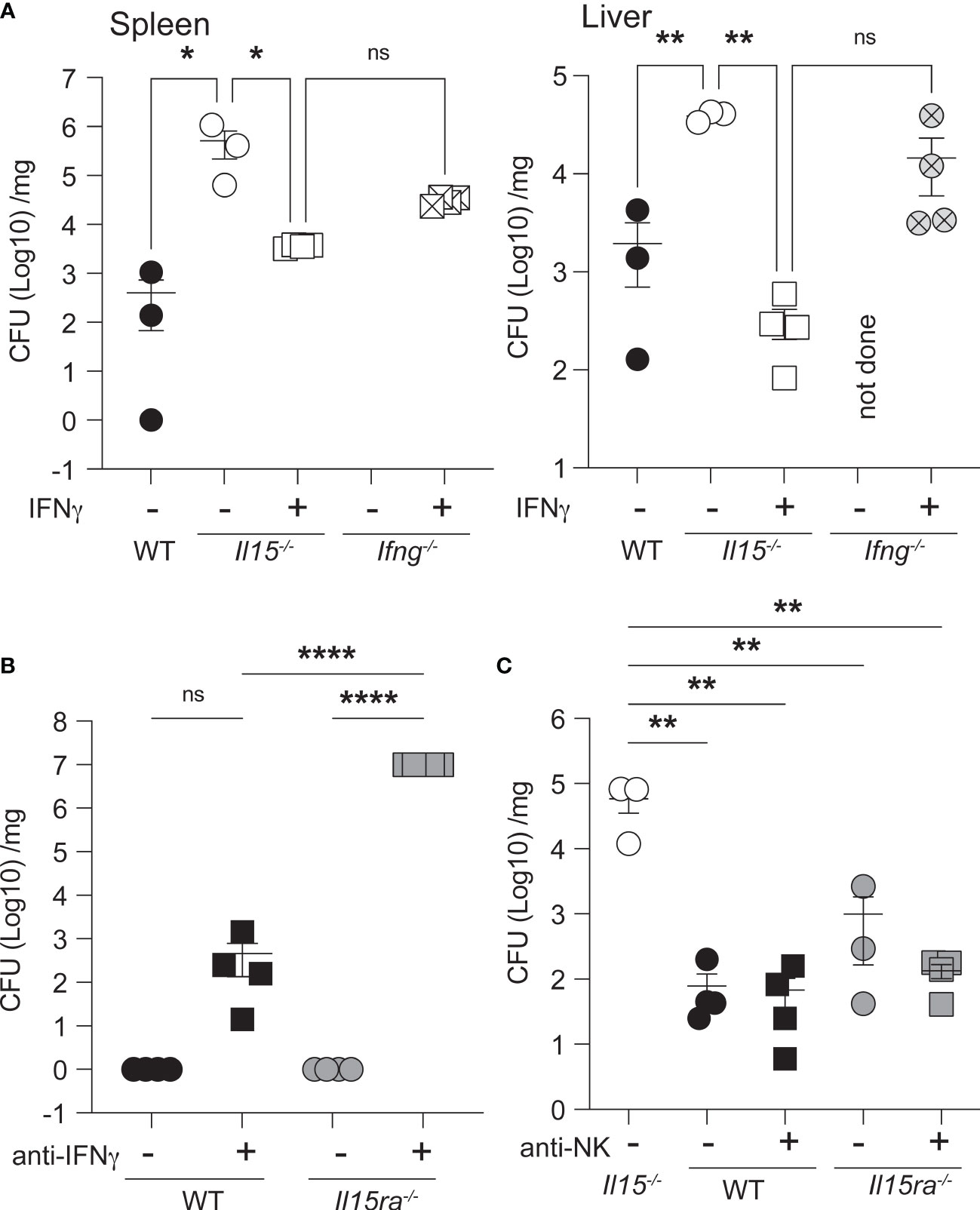
Figure 7 Exogenous IFNγ partially controls the bacterial load in the spleen of Il15-/- mice. (A) Il15-/- and Ifng-/- mice were infected i.v. with 5 x103 CFU of L. monocytogenes. Infected mice received 1 x106 units of IFNγ at the time of infection and at 1 dpi. Bacterial load in the spleens and livers were determined at 3 dpi. (B) IFNγ neutralization increases the susceptibility of Il15ra-/- mice. WT and Il15ra-/- mice received 100μg of anti-IFNγ mAb intraperitoneally on days -1 and +1 and were infected i.v. with 5 x103 CFU of L. monocytogenes on day 0. Bacterial load in the spleen was determined at 3 dpi. (C) WT and Il15ra-/- mice received 100μg of anti-NK1.1 mAb intraperitoneally on days -1 and +1 and were infected i.v. with 5 x103 CFU of L. monocytogenes on day 0. Bacterial load in the spleen was determined at 3 dpi. n = 3-5 mice per group from 2 different experiments. Statistical significance between groups was calculated by one-way ANOVA followed by Tukey’s multiple comparisons test. *p < 0.05, **p < 0.01, ****p < 0.0001, ns, not significant.
Hematopoietic and Non-Hematopoietic IL-15 Contribute to the Control of L. monocytogenes
IL-15 from hematopoietic and non-hematopoietic cells are required for efficient activation of IL-15 dependent immune cells (7, 19, 20, 48). To determine the relative contribution of IL-15 from the hematopoietic and non-hematopoietic compartments, we generated radiation chimeras and infected them with low dose L. monocytogenes two months after lethal radiation and hematopoietic reconstitution. As expected, Il15ra-/- mice reconstituted with Il15ra-/- BM controlled the bacterial load, whereas Il15-/- mice reconstituted with Il15-/- BM failed to control bacterial growth (Figure 8). Reconstitution of Il15-/- mice with WT or Il15ra-/- BM was sufficient to control the bacterial load, and Il15ra-/- mice harboring Il15-/- BM cells still controlled the infection. These observations suggest that IL-15 originating from either the hematopoietic or the non-hematopoietic compartment is sufficient to control L. monocytogenes infection.
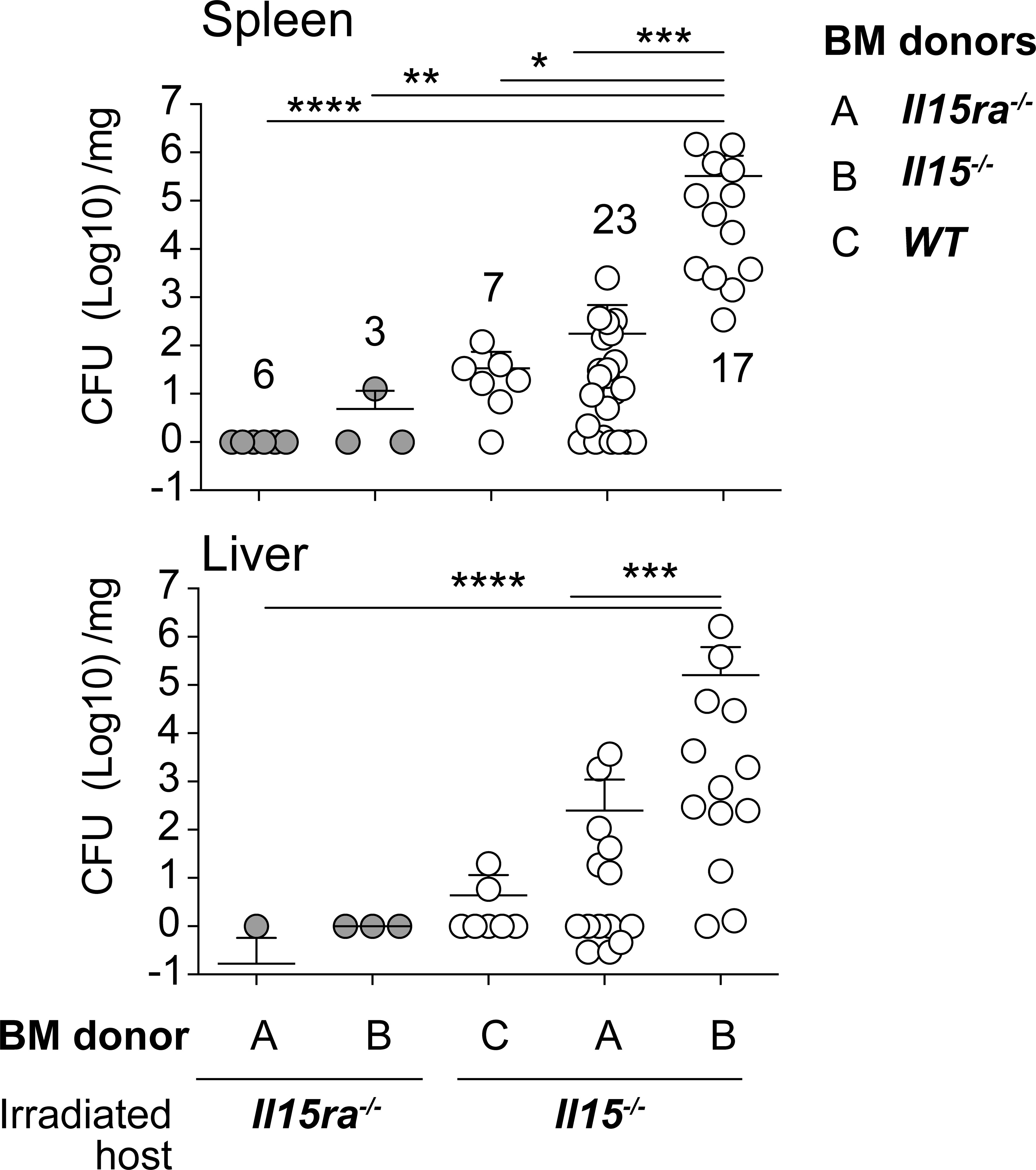
Figure 8 IL-15 from either hematopoietic or non-hematopoietic source is sufficient to control the infection. Two to 3 months old female WT, Il15-/- and Il15ra-/- mice received 10 Grays radiation and were reconstituted with hematopoietic cells from the bone marrow of indicated donors to generate radiation chimeras. The chimeras were infected with low dose L. monocytogenes two months after lethal radiation and hematopoietic reconstitution. The infected mice were sacrificed 5 dpi and the bacterial load in the spleen and liver was determined by plating on BHI-streptomycin plates. Mean ± SEM values from 2-4 independent experiments are shown. The number of recipients in each group is indicated in the figure. Statistical significance between groups, followed by Tukey’s multiple comparisons test, is given as *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001.
Discussion
The finding that IL-15 is trans-presented to neighboring cells by IL-15Rα (15), the strikingly similar impact of genetically ablating IL-15 or IL-15Rα on innate and adaptive immune cell compartments (6, 24) and the dispensability of IL-15Rα on memory CD8 T cells to respond to IL-15 (49) have firmly established the concept that IL-15Rα is essential for IL-15 signaling. However, soluble IL-15 can bind the IL-15Rβγc heterodimeric receptor complex albeit with less affinity than when IL-15 is presented by IL-15Rα (15, 16). A few studies including ours have also reported that IL-15 is more critical than IL-15Rα in the context of antiviral responses, autoimmune disease and antitumor immunity (21–23). It has also been shown that IL-15Rα modulates IL-15 signaling to control the development of γδ-17 T cells and cutaneous psoriasiform inflammation (25, 50). These studies indicate that IL-15Rα-independent IL-15 signaling, presumably via the IL-15Rβ/γc complex is physiologically relevant for certain immune functions. The findings of the present study substantiate this notion by demonstrating that IL-15 can elicit, independently of IL-15Rα, innate immune responses following Listeria infection that involve early IFNγ production necessary for pathogen control by sources other than NK cells.
Even though several studies have established a role for IL-15 in conferring protection against infections (3, 14, 27, 51), few have addressed the role of IL-15Rα in pathogen control. To our knowledge, our study is the first to report the dispensability of IL-15Rα-mediated trans-presentation of IL-15 to mount the early innate responses to Listeria. A striking difference observed in Listeria-infected Il15-/- and Il15ra-/- mice was the profound decrease in the production of early IFNγ in the former but not in the latter. While the levels of IFNγ in the spleen was comparable between WT and Il15ra-/- mice following Listeria infection, WT mice displayed a 4-fold higher level of IFNγ in the serum, which is obviously derived from NK cells but seems dispensable for bacterial control. On the other hand, the induction of IFNγ within the spleen following systemic infection, which is necessary and sufficient for the activation of the early innate immune responses, was not dependent on IL-15Rα. The source of this tissue IFNγ is not yet clear. Alternate sources of the early IFNγ could include neutrophils and innate memory and H2-M3-restricted CD8+ T cells (52–57). Whereas the latter is dependent on IL-15 signaling for development (52) and thus would be unavailable in Il15ra-/- mice, the effect of IL-15 on IFNγ production by neutrophils has not been reported yet. IL-15 signaling has also been shown to elicit IFNγ expression in DCs (58). IL-15 expression in DCs is upregulated immediately after Listeria infection (59). Following Francisella tularensis infection, DCs accounted for 15-50% of innate IFNγ producing cells (60). As depletion of IFNγ in infected Il15ra-/- mice rendered them susceptible to low-dose Listeria infection (Figure 7B), it is possible that the DCs are a potential source of early IFNγ in the spleens of Il15ra-/- mice. Clearly, further studies are needed to identify the cell type involved in IL-15-dependent IL-15Rα-independent early IFNγ production following bacterial infection.
The source of IL-15 that confers protection against Listeria infection can be both hematopoietic and non-hematopoietic, as shown by the bone marrow radiation chimera experiments (Figure 8). Reconstitution of Il15ra-/- mice with Il15-/- bone marrow cells or vice versa conferred the ability to clear Listeria infection. Nevertheless, IL-15 from the non-hematopoietic cells appear to have an important role, as the bacterial load tended to be higher in Il15-/- recipients reconstituted with IL-15Rα bone marrow cells, suggesting that tissue-derived IL-15 has a significant role in pathogen control. IL-15 is expressed in diverse immune and somatic cell types (5, 61) and bone marrow chimeras has shown the distinct roles of hematopoietic and parenchymal sources of IL-15 in controlling lymphoid cell development and homeostasis (48). Such adoptive transfer studies and the use of conditional IL-15Rα deletion in tissues (19, 20) indicated that the dependence on tissue-derived IL-15 on immune functions appears to vary with the niche and the requirement of IL-15Rα-mediated trans-presentation. Whereas IL-15 from intestinal epithelial cells is required for maintaining lymphoid subsets in the intestine (62), lung-derived IL-15 may not be critical for the maintenance of CMV-specific memory T cells (63). As IL-15-mediated control of Listeria does not require IL-15Rα, further work is needed to determine how IL-15 is produced and made available to responder cells that express IFNγ needed for bacterial control.
Besides its role in IL-15 trans-presentation and facilitating ligand interaction with the IL-15Rβγc heterodimer (64, 65), IL-15Rα is implicated in regulating IL-15 availability and in modulating IL-15 signaling. IL-15Rα can be released from the cell surface by proteolytic enzymes, and this soluble form (sIL-15Rα) act as an antagonist of IL-15-dependent functions (66). Even though the trimeric IL-15Rαβγc complex is considered the functional IL-15 receptor, two lines of evidence indicate that IL-15 can signal via the IL-15Rβγc heterodimer. First, we have shown that IL-15 promotes autoimmune Type 1 diabetes (T1D) that does not rely on IL-15Rα but requires IL-15Rβ, as treatment with antibody targeting the IL-15Rβ during the early and late stages of insulitis progression prevented T1D development and the expansion of diabetogenic T cells (21, 22). Similar results were subsequently reported in other models of T1D (67–69). Secondly, two studies have shown that IL-15 signaling elicits divergent signals depending on the availability of IL-15Rα. Comparison of γδ T cells in Il15ra-/- and Il15-/- mice revealed an increased frequency of IL-17 producing γδ T cells in Il15ra-/- mice with respect to Il15-/- or WT mice (25). Mixed bone marrow chimeras revealed that this skewing towards γδ-17 cells arises from a cell-intrinsic phenomenon in IL-15Rα-deficient cells. Bouchaud et al., have shown that IL-15Rα deficiency exacerbated psoriatic lesions by increasing the frequency of IL-17 producing αβ and γδ T cells that was not observed in IL-15-deficient mice (50). Treatment with sIL-15Rα complexes decreased the inflammation in vivo and reduced the frequency of IL-15-induced IL-17 producing αβ and γδ T cells in vitro. Together these two studies indicated that IL-15Rα-dependent IL-15 signaling restrains the skewing of αβ and γδ T cells towards IL-17 producing cells, but did not address whether IL-15 signaling occurred via the IL-15βγc heterodimer. As IL-15Rα can bind IL-15 with higher affinity than that of the IL-15βγc heterodimer, sIL-15Rα may decrease the bioactive IL-15 available to promote inflammatory responses. In support, neutralization of IL-15 decreased psoriatic lesions in mice and in xenografts (70). On the other hand, exogenous IL-15 upregulates Th-17 responses of T cells in psoriasis (71), possibly by saturating the IL-15Rα containing trimeric receptor complexes and engaging IL-15βγc dimeric receptors. Collectively, in line with these studies, our current findings support the idea that IL-15 can elicit productive signaling via the IL-15βγc dimeric receptors, although it remains to be elucidated whether and how the downstream signaling pathways differ from those of the trimeric receptor complex.
The ability of IL-15 that is trans-presented by IL-15Rα to activate NK cells and CD8+ T cells has generated a lot of interest in exploiting it for cancer immunotherapy (72, 73). However excessive activation of NK cells and CD8+ T cells and the associated toxicity following the administration of free IL-15 has dampened its widespread application in the clinics (74). Nonetheless, the use of IL-15 conjugates in conjunction with other therapies have renewed the interest in IL-15 (75). While our knowledge on the mechanisms by which IL-15 activates lymphocytes is extensive, it is not clear how IL-15 signals in macrophages, DC and related cell types. The dispensability of IL-15Rα for eliciting such functions, as shown in our study, should overcome the limitations of trans-presentation-dependent lymphoid cell activation. Besides, dissecting the IL-15 signaling mechanisms via the IL-15βγc heterodimer could identify signaling pathways that could be exploited to achieve the same goals.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by Université de Sherbrooke Ethics Committee for Animal Care and Use.
Author Contributions
MN: Flow cytometry planning, experimentation and analyses, manuscript review and editing. MMM: In vitro infections and analyses, manuscript review and editing; SO: Initial experimentation. JMFM, AAC, GCM, and SO: infections and bacterial counts. FR and M-AL: experimentation for flow cytometry, bacterial counts. MM: generation and maintenance of mice, preparation of tissue lysates, gene expression analyses. SI: Hypothesis generation, manuscript reviewing and editing. AM and SR, Hypothesis generation, Conceptualization, Securing funding, Supervision, Data analysis and interpretation, Manuscript- writing, reviewing and editing. All authors are in agreement with the submitted version. All authors did indeed contribute.
Funding
This work was supported by NSERC Discovery grants to SR (RGPIN-2016-04349) and AM (RGPIN-2018-05404).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
MN is a recipient of FRQS post-doctoral fellowship. AAC is a recipient of scholarship from FMSS, Université de Sherbrooke. MMM and JMFM are recipients of scholarships from Canadian Francophonie Scholarship Program.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.793918/full#supplementary-material
References
1. Tagaya Y, Bamford RN, DeFilippis AP, Waldmann TA. IL-15: A Pleiotropic Cytokine With Diverse Receptor/Signaling Pathways Whose Expression Is Controlled at Multiple Levels. Immunity (1996) 4:329–36. doi: 10.1016/S1074-7613(00)80246-0
2. Fehniger TA, Caligiuri MA. Interleukin 15: Biology and Relevance to Human Disease. Blood (2001) 97:14–32. doi: 10.1182/blood.V97.1.14
3. Verbist KC, Klonowski KD. Functions of IL-15 in Anti-Viral Immunity: Multiplicity and Variety. Cytokine (2012) 59:467–78. doi: 10.1016/j.cyto.2012.05.020
4. Cui G, Hara T, Simmons S, Wagatsuma K, Abe A, Miyachi H, et al. Characterization of the IL-15 Niche in Primary and Secondary Lymphoid Organs In Vivo. Proc Natl Acad Sci USA (2014) 111:1915–20. doi: 10.1073/pnas.1318281111
5. Grabstein KH, Eisenman J, Shanebeck K, Rauch C, Srinivasan S, Fung V, et al. Cloning of a T Cell Growth Factor That Interacts With the Beta Chain of the Interleukin-2 Receptor. Science (1994) 264:965–8. doi: 10.1126/science.8178155
6. Kennedy MK, Glaccum M, Brown SN, Butz EA, Viney JL, Embers M, et al. Reversible Defects in Natural Killer and Memory CD8 T Cell Lineages in Interleukin 15-Deficient Mice. J Exp Med (2000) 191:771–80. doi: 10.1084/jem.191.5.771
7. Lodolce JP, Burkett PR, Boone DL, Chien M, Ma A. T Cell-Independent Interleukin 15Ralpha Signals Are Required for Bystander Proliferation. J Exp Med (2001) 194:1187–94. doi: 10.1084/jem.194.8.1187
8. Marks-Konczalik J, Dubois S, Losi JM, Sabzevari H, Yamada N, Feigenbaum L, et al. IL-2-Induced Activation-Induced Cell Death Is Inhibited in IL-15 Transgenic Mice. Proc Natl Acad Sci USA (2000) 97:11445–50. doi: 10.1073/pnas.200363097
9. Antony PA, Paulos CM, Ahmadzadeh M, Akpinarli A, Palmer DC, Sato N, et al. Interleukin-2-Dependent Mechanisms of Tolerance and Immunity In Vivo. J Immunol (2006) 176:5255–66. doi: 10.4049/jimmunol.176.9.5255
10. Stonier SW, Schluns KS. Trans-Presentation: A Novel Mechanism Regulating IL-15 Delivery and Responses. Immunol Lett (2010) 127:85–92. doi: 10.1016/j.imlet.2009.09.009
11. Bamford RN, DeFilippis AP, Azimi N, Kurys G, Waldmann TA. The 5' Untranslated Region, Signal Peptide, and the Coding Sequence of the Carboxyl Terminus of IL-15 Participate in Its Multifaceted Translational Control. J Immunol (1998) 160:4418–26.
12. Castillo EF, Schluns KS. Regulating the Immune System via IL-15 Transpresentation. Cytokine (2012) 59:479–90. doi: 10.1016/j.cyto.2012.06.017
13. Tagaya Y, Kurys G, Thies TA, Losi JM, Azimi N, Hanover JA, et al. Generation of Secretable and Nonsecretable Interleukin 15 Isoforms Through Alternate Usage of Signal Peptides. Proc Natl Acad Sci USA (1997) 94:14444–9. doi: 10.1073/pnas.94.26.14444
14. Yoshikai Y, Nishimura H. The Role of Interleukin 15 in Mounting an Immune Response Against Microbial Infections. Microbes Infect / Institut Pasteur (2000) 2:381–9. doi: 10.1016/S1286-4579(00)00329-4
15. Dubois S, Mariner J, Waldmann TA, Tagaya Y. IL-15Ralpha Recycles and Presents IL-15 In Trans to Neighboring Cells. Immunity (2002) 17:537–47. doi: 10.1016/S1074-7613(02)00429-6
16. Rao BM, Driver I, Lauffenburger DA, Wittrup KD. High-Affinity CD25-Binding IL-2 Mutants Potently Stimulate Persistent T Cell Growth. Biochemistry (2005) 44:10696–701. doi: 10.1021/bi050436x
17. Colpitts SL, Stonier SW, Stoklasek TA, Root SH, Aguila HL, Schluns KS, et al. Transcriptional Regulation of IL-15 Expression During Hematopoiesis. J Immunol (2013) 191:3017–24. doi: 10.4049/jimmunol.1301389
18. Schluns KS, Stoklasek T, Lefrancois L. The Roles of Interleukin-15 Receptor Alpha: Trans-Presentation, Receptor Component, or Both? Int J Biochem Cell Biol (2005) 37:1567–71. doi: 10.1016/j.biocel.2005.02.017
19. Mortier E, Advincula R, Kim L, Chmura S, Barrera J, Reizis B, et al. Macrophage- and Dendritic-Cell-Derived Interleukin-15 Receptor Alpha Supports Homeostasis of Distinct CD8+ T Cell Subsets. Immunity (2009) 31:811–22. doi: 10.1016/j.immuni.2009.09.017
20. Cepero-Donates Y, Rakotoarivelo V, Mayhue M, Ma A, Chen YG, Ramanathan S. Homeostasis of IL-15 Dependent Lymphocyte Subsets in the Liver. Cytokine (2016) 82:95–101. doi: 10.1016/j.cyto.2015.12.012
21. Bobbala D, Chen XL, Leblanc C, Mayhue M, Stankova J, Tanaka T, et al. Interleukin-15 Plays an Essential Role in the Pathogenesis of Autoimmune Diabetes in the NOD Mouse. Diabetologia (2012) 55:3010–20. doi: 10.1007/s00125-012-2675-1
22. Bobbala D, Mayhue M, Menendez A, Ilangumaran S, Ramanathan S. Trans-Presentation of Interleukin-15 by Interleukin-15 Receptor Alpha Is Dispensable for the Pathogenesis of Autoimmune Type 1 Diabetes. Cell Mol Immunol (2017) 14:590–6. doi: 10.1038/cmi.2015.102
23. Bahri R, Pateras IS, D'Orlando O, Goyeneche-Patino DA, Campbell M, Polansky JK, et al. IL-15 Suppresses Colitis-Associated Colon Carcinogenesis by Inducing Antitumor Immunity. Oncoimmunology (2015) 4:e1002721. doi: 10.1080/2162402X.2014.1002721
24. Lodolce JP, Boone DL, Chai S, Swain RE, Dassopoulos T, Trettin S, et al. IL-15 Receptor Maintains Lymphoid Homeostasis by Supporting Lymphocyte Homing and Proliferation. Immunity (1998) 9:669–76. doi: 10.1016/S1074-7613(00)80664-0
25. Colpitts SL, Puddington L, Lefrancois L. IL-15 Receptor Alpha Signaling Constrains the Development of IL-17-Producing Gammadelta T Cells. Proc Natl Acad Sci USA (2015) 112:9692–7. doi: 10.1073/pnas.1420741112
26. Perera PY, Lichy JH, Waldmann TA, Perera LP. The Role of Interleukin-15 in Inflammation and Immune Responses to Infection: Implications for Its Therapeutic Use. Microbes Infect / Institut Pasteur (2012) 14:247–61. doi: 10.1016/j.micinf.2011.10.006
27. Ashkar AA, Reid S, Verdu EF, Zhang K, Coombes BK. Interleukin-15 and NK1.1+ Cells Provide Innate Protection Against Acute Salmonella Enterica Serovar Typhimurium Infection in the Gut and in Systemic Tissues. Infection Immun (2009) 77:214–22. doi: 10.1128/IAI.01066-08
28. Ramanathan S, Dubois S, Chen XL, Leblanc C, Ohashi PS, Ilangumaran S. Exposure to IL-15 and IL-21 Enables Autoreactive CD8 T Cells to Respond to Weak Antigens and Cause Disease in a Mouse Model of Autoimmune Diabetes. J Immunol (2011) 186:5131–41. doi: 10.4049/jimmunol.1001221
29. Rodriguez GM, D'Urbano D, Bobbala D, Chen XL, Yeganeh M, Ramanathan S, et al. SOCS1 Prevents Potentially Skin-Reactive Cytotoxic T Lymphocytes From Gaining the Ability to Cause Inflammatory Lesions. J Invest Dermatol (2013) 133:2013–22. doi: 10.1038/jid.2013.86
30. Kandhi R, Bobbala D, Yeganeh M, Mayhue M, Menendez A, Ilangumaran S. Negative Regulation of the Hepatic Fibrogenic Response by Suppressor of Cytokine Signaling 1. Cytokine (2016) 82:58–69. doi: 10.1016/j.cyto.2015.12.007
31. Bailey JD, Shaw A, McNeill E, Nicol T, Diotallevi M, Chuaiphichai S, et al. Isolation and Culture of Murine Bone Marrow-Derived Macrophages for Nitric Oxide and Redox Biology. Nitric Oxide (2020) 100-101:17–29. doi: 10.1016/j.niox.2020.04.005
32. Bobbala D, Orkhis S, Kandhi R, Ramanathan S, Ilangumaran S. Interleukin-21-Dependent Modulation of T Cell Antigen Receptor Reactivity Towards Low Affinity Peptide Ligands in Autoreactive CD8(+) T Lymphocytes. Cytokine (2016) 85:83–91. doi: 10.1016/j.cyto.2016.06.011
33. Pasche B, Kalaydjiev S, Franz TJ, Kremmer E, Gailus-Durner V, Fuchs H, et al. Sex-Dependent Susceptibility to Listeria Monocytogenes Infection Is Mediated by Differential Interleukin-10 Production. Infection Immun (2005) 73:5952–60. doi: 10.1128/IAI.73.9.5952-5960.2005
34. Zajd CM, Ziemba AM, Miralles GM, Nguyen T, Feustel PJ, Dunn SM, et al. Bone Marrow-Derived and Elicited Peritoneal Macrophages Are Not Created Equal: The Questions Asked Dictate the Cell Type Used. Front Immunol (2020) 11:269. doi: 10.3389/fimmu.2020.00269
35. Pamer EG. Immune Responses to Listeria Monocytogenes. Nat Rev (2004) 4:812–23. doi: 10.1038/nri1461
36. Harty JT, Bevan MJ. Specific Immunity to Listeria Monocytogenes in the Absence of IFN Gamma. Immunity (1995) 3:109–17. doi: 10.1016/1074-7613(95)90163-9
37. Kuranaga N, Kinoshita M, Kawabata T, Shinomiya N, Seki S. A Defective Th1 Response of the Spleen in the Initial Phase May Explain Why Splenectomy Helps Prevent a Listeria Infection. Clin Exp Immunol (2005) 140:11–21. doi: 10.1111/j.1365-2249.2005.02735.x
38. Soudja SM, Ruiz AL, Marie JC, Lauvau G. Inflammatory Monocytes Activate Memory CD8(+) T and Innate NK Lymphocytes Independent of Cognate Antigen During Microbial Pathogen Invasion. Immunity (2012) 37:549–62. doi: 10.1016/j.immuni.2012.05.029
39. Farber JM. Mig and IP-10: CXC Chemokines That Target Lymphocytes. J Leukoc Biol (1997) 61:246–57. doi: 10.1002/jlb.61.3.246
40. Ohteki T, Tada H, Ishida K, Sato T, Maki C, Yamada T, et al. Essential Roles of DC-Derived IL-15 as a Mediator of Inflammatory Responses In Vivo. J Exp Med (2006) 203:2329–38. doi: 10.1084/jem.20061297
41. Ohteki T, Fukao T, Suzue K, Maki C, Ito M, Nakamura M, et al. Interleukin 12-Dependent Interferon Gamma Production by CD8alpha+ Lymphoid Dendritic Cells. J Exp Med (1999) 189:1981–6. doi: 10.1084/jem.189.12.1981
42. Buchmeier NA, Schreiber RD. Requirement of Endogenous Interferon-Gamma Production for Resolution of Listeria Monocytogenes Infection. Proc Natl Acad Sci USA (1985) 82:7404–8. doi: 10.1073/pnas.82.21.7404
43. Langermans JA, van der Hulst ME, Nibbering PH, van der Meide PH, van Furth R. Intravenous Injection of Interferon-Gamma Inhibits the Proliferation of Listeria Monocytogenes in the Liver But Not in the Spleen and Peritoneal Cavity. Immunology (1992) 77:354–61.
44. Cerny A, Hugin AW, Bazin H, Sutter S, Hengartner H, Zinkernagel RM. Anti-Listeria Monocytogenes Immunity in Mu-Suppressed Mice: A Comparison of Treatment With Conventional Hyperimmune Rabbit Anti-Mouse IgM and Affinity-Purified, Monoclonal Rat Anti-Mouse IgM. Med Microbiol Immunol (1988) 177:123–31. doi: 10.1007/BF00232892
45. Takada H, Matsuzaki G, Hiromatsu K, Nomoto K. Analysis of the Role of Natural Killer Cells in Listeria Monocytogenes Infection: Relation Between Natural Killer Cells and T-Cell Receptor Gamma Delta T Cells in the Host Defence Mechanism at the Early Stage of Infection. Immunology (1994) 82:106–12.
46. Carrero JA, Calderon B, Unanue ER. Lymphocytes are Detrimental During the Early Innate Immune Response Against Listeria Monocytogenes. J Exp Med (2006) 203:933–40. doi: 10.1084/jem.20060045
47. Clark SE, Filak HC, Guthrie BS, Schmidt RL, Jamieson A, Merkel P, et al. Bacterial Manipulation of NK Cell Regulatory Activity Increases Susceptibility to Listeria Monocytogenes Infection. PloS Pathog (2016) 12:e1005708. doi: 10.1371/journal.ppat.1005708
48. Schluns KS, Nowak EC, Cabrera-Hernandez A, Puddington L, Lefrancois L, Aguila HL. Distinct Cell Types Control Lymphoid Subset Development by Means of IL-15 and IL-15 Receptor Alpha Expression. Proc Natl Acad Sci USA (2004) 101:5616–21. doi: 10.1073/pnas.0307442101
49. Sandau MM, Schluns KS, Lefrancois L, Jameson SC. Cutting Edge: Transpresentation of IL-15 by Bone Marrow-Derived Cells Necessitates Expression of IL-15 and IL-15R Alpha by the Same Cells. J Immunol (2004) 173:6537–41. doi: 10.4049/jimmunol.173.11.6537
50. Bouchaud G, Gehrke S, Krieg C, Kolios A, Hafner J, Navarini AA, et al. Epidermal IL-15Ralpha Acts as an Endogenous Antagonist of Psoriasiform Inflammation in Mouse and Man. J Exp Med (2013) 210:2105–17. doi: 10.1084/jem.20130291
51. Waldmann TA, Tagaya Y. The Multifaceted Regulation of Interleukin-15 Expression and the Role of This Cytokine in NK Cell Differentiation and Host Response to Intracellular Pathogens. Annu Rev Immunol (1999) 17:19–49. doi: 10.1146/annurev.immunol.17.1.19
52. Muller JR, Waldmann TA, Dubois S. Selective Dependence of H2-M3-Restricted CD8 Responses on IL-15. J Immunol (2012) 188:2575–82. doi: 10.4049/jimmunol.1102393
53. Yin J, Ferguson TA. Identification of an IFN-Gamma-Producing Neutrophil Early in the Response to Listeria Monocytogenes. J Immunol (2009) 182:7069–73. doi: 10.4049/jimmunol.0802410
54. Rogers HW, Unanue ER. Neutrophils Are Involved in Acute, Nonspecific Resistance to Listeria Monocytogenes in Mice. Infection Immun (1993) 61:5090–6. doi: 10.1128/iai.61.12.5090-5096.1993
55. Berg RE, Cordes CJ, Forman J. Contribution of CD8+ T Cells to Innate Immunity: IFN-Gamma Secretion Induced by IL-12 and IL-18. Eur J Immunol (2002) 32:2807–16. doi: 10.1002/1521-4141(2002010)32:10<2807::AID-IMMU2807>3.0.CO;2-0
56. Berg RE, Crossley E, Murray S, Forman J. Memory CD8+ T Cells Provide Innate Immune Protection Against Listeria Monocytogenes in the Absence of Cognate Antigen. J Exp Med (2003) 198:1583–93. doi: 10.1084/jem.20031051
57. Lertmemongkolchai G, Cai G, Hunter CA, Bancroft GJ. Bystander Activation of CD8+ T Cells Contributes to the Rapid Production of IFN-Gamma in Response to Bacterial Pathogens. J Immunol (2001) 166:1097–105. doi: 10.4049/jimmunol.166.2.1097
58. Ohteki T, Suzue K, Maki C, Ota T, Koyasu S. Critical Role of IL-15-IL-15R for Antigen-Presenting Cell Functions in the Innate Immune Response. Nat Immunol (2001) 2:1138–43. doi: 10.1038/ni729
59. Liu T, Nishimura H, Matsuguchi T, Yoshikai Y. Differences in Interleukin-12 and -15 Production by Dendritic Cells at the Early Stage of Listeria Monocytogenes Infection Between BALB/c and C57 BL/6 Mice. Cell Immunol (2000) 202:31–40. doi: 10.1006/cimm.2000.1644
60. De Pascalis R, Rossi AP, Mittereder L, Takeda K, Akue A, Kurtz SL, et al. Production of IFN-Gamma by Splenic Dendritic Cells During Innate Immune Responses Against Francisella Tularensis LVS Depends on MyD88, But Not TLR2, TLR4, or TLR9. PloS One (2020) 15:e0237034. doi: 10.1371/journal.pone.0237034
61. Giri JG, Ahdieh M, Eisenman J, Shanebeck K, Grabstein K, Kumaki S, et al. Utilization of the Beta and Gamma Chains of the IL-2 Receptor by the Novel Cytokine IL-15. EMBO J (1994) 13:2822–30. doi: 10.1002/j.1460-2075.1994.tb06576.x
62. Zhu Y, Cui G, Miyauchi E, Nakanishi Y, Mukohira H, Shimba A, et al. Intestinal Epithelial Cell-Derived IL-15 Determines Local Maintenance and Maturation of Intraepithelial Lymphocytes in the Intestine. Int Immunol (2019) 32:307–19. doi: 10.1093/intimm/dxz082
63. Baumann NS, Torti N, Welten SPM, Barnstorf I, Borsa M, Pallmer K, et al. Tissue Maintenance of CMV-Specific Inflationary Memory T Cells by IL-15. PloS Pathog (2018) 14:e1006993. doi: 10.1371/journal.ppat.1006993
64. Pistilli EE, Bogdanovich S, Garton F, Yang N, Gulbin JP, Conner JD, et al. Loss of IL-15 Receptor Alpha Alters the Endurance, Fatigability, and Metabolic Characteristics of Mouse Fast Skeletal Muscles. J Clin Invest (2011) 121:3120–32. doi: 10.1172/JCI44945
65. Pistilli EE, Quinn LS. From Anabolic to Oxidative: Reconsidering the Roles of IL-15 and IL-15Ralpha in Skeletal Muscle. Exerc Sport Sci Rev (2013) 41:100–6. doi: 10.1097/JES.0b013e318275d230
66. Mortier E, Bernard J, Plet A, Jacques Y. Natural, Proteolytic Release of a Soluble Form of Human IL-15 Receptor Alpha-Chain That Behaves as a Specific, High Affinity IL-15 Antagonist. J Immunol (2004) 173:1681–8. doi: 10.4049/jimmunol.173.3.1681
67. Brauner H, Hall HT, Flodstrom-Tullberg M, Karre K, Hoglund P, Johansson S. Depletion of IL-2 Receptor Beta-Positive Cells Protects From Diabetes in Non-Obese Diabetic Mice. Immunol Cell Biol (2016) 94:177–84. doi: 10.1038/icb.2015.78
68. Yuan X, Dong Y, Tso JY, Fu W. Restoration of Immune Tolerance in Type 1 Diabetes by Modulating Interleukin-2 Receptor Signaling. J Immunol (2017) 198:80.16.
69. Yuan X, Dong Y, Tsurushita N, Tso JY, Fu W. CD122 Blockade Restores Immunological Tolerance in Autoimmune Type 1 Diabetes via Multiple Mechanisms. JCI Insight (2018) 3:e96600. doi: 10.1172/jci.insight.96600
70. Villadsen LS, Schuurman J, Beurskens F, Dam TN, Dagnaes-Hansen F, Skov L, et al. Resolution of Psoriasis Upon Blockade of IL-15 Biological Activity in a Xenograft Mouse Model. J Clin Invest (2003) 112:1571–80. doi: 10.1172/JCI200318986
71. de Jesus-Gil C, Ruiz-Romeu E, Ferran M, Sagrista M, Chiriac A, Garcia P, et al. IL-15 and IL-23 Synergize to Trigger Th17 Response by CLA(+) T Cells in Psoriasis. Exp Dermatol (2020) 29:630–8. doi: 10.1111/exd.14113
72. Dubois SP, Miljkovic MD, Fleisher TA, Pittaluga S, Hsu-Albert J, Bryant BR, et al. Short-Course IL-15 Given as a Continuous Infusion Led to a Massive Expansion of Effective NK Cells: Implications for Combination Therapy With Antitumor Antibodies. J Immunother Cancer (2021) 9:e002193. doi: 10.1136/jitc-2020-002193
73. Guo Y, Luan L, Patil NK, Sherwood ER. Immunobiology of the IL-15/IL-15Ralpha Complex as an Antitumor and Antiviral Agent. Cytokine Growth Factor Rev (2017) 38:10–21. doi: 10.1016/j.cytogfr.2017.08.002
74. Conlon KC, Lugli E, Welles HC, Rosenberg SA, Fojo AT, Morris JC, et al. Redistribution, Hyperproliferation, Activation of Natural Killer Cells and CD8 T Cells, and Cytokine Production During First-in-Human Clinical Trial of Recombinant Human Interleukin-15 in Patients With Cancer. J Clin Oncol (2015) 33:74–82. doi: 10.1200/JCO.2014.57.3329
Keywords: IL-15, Listeria, IFN-gamma, IL-15 Receptor alpha, infection, spleen, mice
Citation: Nandi M, Moyo MM, Orkhis S, Mobulakani JMF, Limoges M-A, Rexhepi F, Mayhue M, Cayarga AA, Marrero GC, Ilangumaran S, Menendez A and Ramanathan S (2021) IL-15Rα-Independent IL-15 Signaling in Non-NK Cell-Derived IFNγ Driven Control of Listeria monocytogenes. Front. Immunol. 12:793918. doi: 10.3389/fimmu.2021.793918
Received: 12 October 2021; Accepted: 11 November 2021;
Published: 10 December 2021.
Edited by:
Laurel L Lenz, University of Colorado, United StatesReviewed by:
Rance E. Berg, University of North Texas Health Science Center, United StatesRosanne Spolski, National Institutes of Health (NIH), United States
Copyright © 2021 Nandi, Moyo, Orkhis, Mobulakani, Limoges, Rexhepi, Mayhue, Cayarga, Marrero, Ilangumaran, Menendez and Ramanathan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alfredo Menendez, QWxmcmVkby5NZW5lbmRlekBVU2hlcmJyb29rZS5jYQ==; Sheela Ramanathan, c2hlZWxhLnJhbWFuYXRoYW5AdXNoZXJicm9va2UuY2E=
†These authors share first authorship
‡These authors share senior-authorship