 Yaping Chen
Yaping Chen Xiao Zheng1
Xiao Zheng1- 1Department of Tumor Biological Treatment, The Third Affiliated Hospital of Soochow University, Changzhou, China
- 2Department of Oncology, The Third Affiliated Hospital of Soochow University, Changzhou, China
Colorectal cancer (CRC) has the second highest mortality rate among all cancers worldwide. Surgery, chemotherapy, radiotherapy, molecular targeting and other treatment methods have significantly prolonged the survival of patients with CRC. Recently, the emergence of tumor immunotherapy represented by immune checkpoint inhibitors (ICIs) has brought new immunotherapy options for the treatment of advanced CRC. As the efficacy of ICIs is closely related to the tumor immune microenvironment (TME), it is necessary to clarify the relationship between the immune microenvironment of CRC and the efficacy of immunotherapy to ensure that the appropriate drugs are selected. We herein review the latest research progress in the immune microenvironment and strategies related to immunotherapy for CRC. We hope that this review helps in the selection of appropriate treatment strategies for CRC patients.
Introduction
Colorectal cancer (CRC) is a common malignant tumor of the digestive system. Worldwide, CRC ranks third in the incidence rate and second in the mortality rate among malignant tumors (1). The incidence of CRC is related to many factors, such as heredity, a low-fiber diet, smoking, lack of exercise, and obesity. At present, changes in the intestinal microbiome and metabolites are also considered risk factors for CRC (2). Traditionally, the treatment of CRC includes surgery, chemotherapy and radiotherapy. In recent years, along with the continuous research on the embryonic origin, anatomical structure, tumor clinical manifestations and genes of the left and right colon, there have been different targeted therapies for “left and right CRC dispute”, namely, cetuximab and bevacizumab, respectively. Furthermore, the advent of the small molecule anti-vascular oral drug, apatinib mesylate, has diversified the CRC treatment. Nevertheless, the prognosis of CRC depends on the stage of the tumor. The mortality of stage I/II is 8-13%, that of stage III is 11-47%, and that of stage IV is as high as 89% (3). Early detection of tumors and early effective treatment can reduce the mortality of CRC. In addition to the small molecule anti-vascular oral drugs, new therapeutic strategies for the treatment of advanced CRC based on immune checkpoint inhibitors (ICIs) have progressed. Overall, advancements in research on the tumor immune microenvironment (TME) and strategies related to immunotherapy are expected to provide more treatment choices for CRC patients.
CRC is a typical tumor infiltrated by effector memory lymphocytes, yet there have been no major breakthroughs in clinical prognosis and immunotherapy (4). It is essential to elucidate the immune environment of CRC to improve current treatment strategies and prognosis for CRC patients. The TME refers to the environment of tumor cells or tumor stem cells, which is closely related to tumor occurrence, development and metastasis of tumors. In the TME, angiogenesis is induced, and regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs) are recruited, which helps suppress the antitumor immune response and promote tumor progression. Furthermore, cytokines within the TME manipulate immune functions and are involved in muted immune responses that guide tumor progression (5). Therefore, the TME of CRC is an important factor affecting cancer immunotherapy. It is necessary to develop a comprehensive understanding of the TME of CRC and extend this knowledge to current treatment strategies that target dysfunctional components in the TME. This paper reviews the interaction of various components of the CRC microenvironment and related treatment strategies, with the goal of finding more effective treatment methods through a better understanding of the interaction of CRC microenvironment.
The Colorectum Composition and Microenvironment
The colorectum is not only the main digestive organ but also an important immune organ, participating in innate and adaptive immunity. The colorectum includes the intestinal epithelium, intestinal intraepithelial lymphocytes and lamina propria lymphocytes. The intestinal epithelium digests and absorbs nutrients and forms a mucosal barrier, which effectively prevents the invasion of harmful bacteria in the intestine (6, 7). The intestinal epithelium is mainly composed of absorptive columnar epithelial cells, goblet cells and endocrine cells. Goblet cells secrete a variety of mucus proteins to form a mucus barrier to limit the invasion of bacteria into the intestinal mucosa. If the intestinal tract lacks mucus, long-term exposure to the bacterial environment may cause chronic inflammation similar to ulcerative colitis, possibly leading to cancer (8, 9). Intestinal epithelial cells also participate in the immune response. These cells obtain lumen antigens and present them to dendritic cells (DCs) in the intestinal lamina propria, which is called the goblet cell associated antigen channel (gap cells) (10, 11). McDonald et al. found that interfering with the interaction between APCs and intestinal epithelial cells via CCR6- deficiency in mice reduces the transfer of goblet cell products to APCs and induces the mucosal response (12). Goblet cells also regulate the immune response by secreting various cytokines, such as IL25, IL18, IL17, IL15, IL13, IL7 and IL6, as well as the chemokine exotoxins CCL6, CCL9 and CCL20 (10). Therefore, goblet cells play a unique and indispensable role in maintaining intestinal immune homeostasis by interacting with immune cells. Many endocrine cells are distributed in the colon and rectum and act as the sensory sentinels of the intestinal environment and coordinators of mucosal immunity (13). Intestinal endocrine cells can secrete cholecystokinin secretin, somatostatin, and histamine, among others, under the stimulation of chemicals or other molecules. Intestinal endocrine cells are the key receptors of metabolites of intestinal flora. Indeed, endocrine cells recognize pathogen-associated molecular patterns (PAMPs) and release cytokines and peptide hormones, which directly affect the function of the intestinal barrier. In the immune system, peptide hormones such as GLP-1 can regulate the activation of intestinal immune cells (14–16). M cells, also called microfold cells or membranous cells, are located between the lymphoid follicular epithelium and scattered among intestinal mucosal epithelial cells. M cells actively transport a variety of substances, such as soluble antigens and microorganisms, via liquid pinocytosis and receptor-mediated endocytosis. Recently, it was found that M cells play a specialized antigen transport role in the mucosal immune system, transporting antigens from the intestinal cavity to the subepithelial lymphoid tissue, so as to induce an immune mucosal immune response or immune tolerance (17, 18). M cell-dependent antigen uptake is mediated by specific receptors, such as β1 integrin, cellular prion protein and glycoprotein-2 (GP2) (17).
The immune cells involved in colorectal mainly include intestinal intraepithelial lymphocytes (IELs) and lamina propria lymphocytes which play an immunomodulatory role in maintaining colorectal homeostasis. The former cells express a variety of receptors, such as chemokine receptor CCR9 and integrin αEβ7. CCR9 interacts with CCL25 produced by the intestinal epithelial cells to help recruit IELs to the intestinal mucosa. Integrin αEβ7 (αE, also known as CD103) interacts with E-cadherin on intestinal cells to promote entry and retention in the intestinal epithelium (19). Approximately 90% of IELs are T cell receptor (TCR) positive, although a small number are TCR negative (20).
Lamina propria lymphocytes include DCs, intestinal T cells and plasma cells. DCs are antigen-presenting cells that are not evenly distributed in the intestine. CD11c+CD11b-CD103+ DCs are commonly found in the colon of mice, whereas CD11b+CD103+ DCs are commonly found in the small intestine (21). Human intestinal DCs display more complex markers than mouse intestinal DCs. Human CD103+ signal regulatory protein α (SIRPα)- intestinal DCs are associated with mouse intestinal CD11b-CD103+ DCs, whereas human CD103+ SIRPα+ DCs are closely related to mouse intestinal CD103+CD11b+ DCs, which regulate the induction of T cells (22). In recent years, it has been found that DCs and goblet cells can interact and participate in the immune response. CD11c+CD103+ DC subsets present antigens from goblet cells (23). Intestinal T cells include γδ T and αβ T cells. On the one hand, intestinal T cells secrete IFN-γ, TNF-α and other cytokines participating in the immune response against infection and resisting intestinal bacterial immersion. On the other hand, they secrete a variety of factors such as IL-4, IL-5, IL-10, IL-17 and IL-22, to maintain intestinal immune balance. There are also special Treg cells in the intestinal lamina propria that produce IL-10 and TGF-β to negatively regulate the activation of effector T cells and suppress the intestinal inflammation. Plasma cells are distributed in the lamina propria of colorectal tissue and produce different antibodies. In the duodenum/jejunum, 79% of plasma cells express IgA, 18% of plasma cells express IgM and 3% of plasma cells express IgG, while the corresponding numbers in the colon are 90%, 6 and 4%, respectively (24).
TME in CRC
The components of the TME in CRC include tumor cells, blood vessels, the extracellular matrix, fibroblasts, lymphocytes, bone marrow-derived suppressor cells and signaling molecules.
Extracellular Matrix (ECM)
The ECM is composed of glycoprotein, collagen, elastin, proteoglycan and other macromolecules, which support and connect tissues and maintain normal physiological functions (25). Compared with normal tissue, the ECM structure of tumor tissue is disordered, and the process by which the infiltration of fibroblasts/myofibroblasts and the subsequent accumulation of significant amounts of collagenous ECM is observed in the TME is called desmoplasia (26). Desmoplasia is connected with poor prognosis and resistance to therapy (27). Furthermore, an abnormal ECM regulates the epithelial-mesenchymal transition (EMT) and affects cancer progression by directly promoting cell transformation and metastasis (28). Wei et al. found that increasing the stiffness of the surrounding ECM drives the EMT in breast cancer cells by promoting TWIST1 translocalization into the nucleus (29). ECM abnormalities also affect the efficacy of immunotherapy via dense EMC, preventing not only immune cells from reaching the tumor cells but also immunotherapeutic drugs from reaching the tumor. In addition, the shielding diffusion barrier that the ECM forms result in hypoxia, which directly enhances immune escape by upregulating immunomodulatory factors and increasing angiogenic signals (30). In general, ECM abnormalities relieve the behavioral regulation of stromal cells and promote tumor-related angiogenesis and inflammation, resulting in resistance to immunotherapy in the TME (31).
Peptidylarginine deiminase 4 (PAD4) is a member of the PAD family including calcium dependent isozymes (PADs 1-4 and 6) (32). PAD4 overexpression is typically involved in elevated tumor citrullination and hypercitrullination alters cell-matrix adhesion and enhances metastasis (33). Yuzhalin et al. found that citrullination of the ECM and expression of PAD4 promote liver metastasis of human CRC, which may create opportunities for the development of biomarkers and therapeutic targeting (34). Tenascin C (TNC) is a glycoprotein in the extracellular matrix, and plays a role in promoting metastasis, modulating adhesion and motility, developing angiogenesis, and establishing immune tolerance (35). In addition, Murakami et al. reported that the TNC on the CRC interstitial ECM is a factor driving liver metastasis (36), and differences in the expression of ECM-related proteins, such as the upregulated expression of TNC, exist in patients with liver metastasis and CRC recurrence (37).
Angiogenesis
Angiogenesis refers to the production of new blood vessels, while tumor angiogenesis is an endless vicious cycle that cannot be self-regulated. After tumorigenesis, cells proliferate rapidly, and the tumor becomes ischemic and hypoxic. Ischemic and hypoxic cancer cells secrete vascular endothelial growth factor (VEGF), which binds to vascular endothelial growth factor receptors (VEGFRs) on the adjacent vascular endothelium to directly stimulate tumor angiogenesis and promote the migration of endothelial cells (38). The basement membrane cells degrade, and the surrounding vascular endothelial cells proliferate rapidly and migrate to the tumor tissue via angiogenesis. Angiogenesis of tumor tissue is the result of the joint action of cancer cells, various tumor-related cells and their bioactive products, such as cytokines, growth factors and microbubbles. Various immune cells such as macrophages, neutrophils, immature myelocytes, B cells, T cells and peripheral cells interact in tumor angiogenesis (39).
VEGF is the most important growth factor regulating angiogenesis in colon cancer and is expressed in all colon carcinoma surgical specimens, including normal mucosa, primary colon cancers and metastatic tumors, as well as in human colorectal cancer cell lines (40, 41). Colon cancer patients with high VEGF expression had a significantly worse prognosis than those with low VEGF expression (41). Furthermore, VEGF has three receptors on CRC cells. VEGFR-1 is associated with tumor grade, Dukes stage and lymph node involvement, and VEGFR-2 is correlated with lymph node involvement while no correlation with any of the clinicopathological variables was found for VEGFR-3 (42). Witte et al. found that the expression of VEGFR-3 in >25% of colorectal cancer cells was associated with a significantly poorer overall survival, but not with lymph node metastasis or depth of tumor invasion (43). Overall, angiogenesis is an important mechanism for the occurrence and development of CRC. Tumor cells secrete VEGF and promote tumor-related angiogenesis which further promotes proliferation and distant metastasis, seriously affecting the prognosis of tumor patients.
Cancer-Associated Fibroblasts (CAFs)
Fibroblasts are nonepithelial, nonvascular and nonhematopoietic cells in connective tissue that are mainly responsible for the formation of extracellular stroma. Fibroblasts maintain the epithelial homeostasis of normal tissues and play an important role in wound healing. When mechanical injury occurs or radiation, temperature, toxins and pathogens cause acute injury, body cells stimulate the protective system, macrophages produce transformation and growth factor-β (TGF-β) and platelet-derived growth factor (PDGF), and fibroblasts and immune cells proliferate and promote angiogenesis (44). CAFs are important components of the TME and play an essential role in tumorigenesis and development. CAFs have many potential origins, but most are considered to originate from local ancestors (45). Tumor cells are usually derived from fibroblasts in tissues, which are induced and activated by tumor cells in the microenvironment (46–48). CAFs interact with tumor cells to promote tumor growth and maintain their malignant tendency. Tumor cells affect the recruitment of CAF precursors and induce normal fibroblasts to differentiate into CAFs, which secrete a variety of growth factors such as TGF-β, VEGF, chemokines and cytokines, such as CXCL12 (SDF-1), CXCL14, CXCL16, CCL2, CCL5, IL-4, and IL-6, and metalloproteinases, such as MMP-1, MMP-2, MMP-3, MMP-9, MMP-13 and MMP-14. These factors stimulate tumor growth, angiogenesis, invasion and metastasis through a variety of mechanisms, thus affecting tumor prognosis.
Endoglin, which is expressed in CAFs in CRC specimens, metastatic lymph nodes and liver metastases, is a member of the TGF-β family of co-receptors and is involved in CAFs-mediated invasion and metastasis through TGF-β signaling pathway activation (49). Hu et al. found that CAFs can directly secrete exosomes to enhance the cell stemness and epithelial-mesenchymal transformation in CRC cells to promote CRC metastasis and chemotherapy resistance. The mechanism is dependent on increased expression of miR-92a-3p, which directly inhibits Fbxw7 and moap1 and activates Wnt/β-Catenin pathway to inhibit mitochondrial apoptosis and promote stem cell differentiation, the EMT, metastasis and 5-FU/L-OHP resistance in CRC cells (50). According to Heichler et al., CAFs secrete IL-6/IL-11 by activating STAT3 signaling pathway to promote tumor development. Moreover, the expression of pSTAT in CRC correlates with patient survival (51). CAFs are related to resistance in gastrointestinal tumors. The fibroproliferative response induced by CAFs interferes with the delivery of drugs to gastrointestinal cancer cells and causes drug resistance to chemotherapy (52).
Tumor-Associated Macrophages (TAMs)
Macrophages are resident phagocytes in lymphoid and nonlymphoid tissues that participate in steady-state tissue homeostasis by scavenging apoptotic cells and growth factors. Macrophages have a wide range of pathogen recognition receptors, which enable them to effectively phagocytize and induce the production of inflammatory cytokines. It is well known that the TME is rich in macrophages, and TAMs are considered the most abundant immune cell population in solid tumor tissues (53). TAMs are mainly recruited from the periphery by chemokines released from tumor tissues, including CCL2, CCL3, CCL4, CCL5 and CXCL12. These factors bind to corresponding receptors for recruitment of monocytes/macrophages (54). TAMs play an important role in promoting tumor angiogenesis and express a variety of growth factors (such as VEGF, PDGF and bFGF), membrane binding molecules and soluble proteases (including MMPs and cathepsin), inflammatory cytokines (TNF-α, IL-1β, IL-6), cyclooxygenase 2 (COX2) and CXC-chemokine ligand 8 (CXCL8) to promote sustained cell activation and proliferation, promoting ECM remodeling and recruitment and activation of angiogenic cells (55–57).
In CRC, TAMs are enriched in the high incidence site of the epithelial mesenchymal transformation (EMT). TAMs promote the growth and invasion of colon cancer cells through EMT remodeling (58). When HT-29 or HCT116 cells are co-cultured with TAMs (THP-1 cells stimulated by conditioned medium from a CRC cell line), TAM derived IL-6 activates the JAK2/STAT3 pathway, which results in increased FoxQ1 expression, leads to the production of CCL2 and promotes the recruitment of macrophages, thus enhancing the migration and invasion of CRC cells (59). TAMs are the main cells in the tumor EMT (60). TAMs are related to the vascular system of CRC and can be used as markers of angiogenesis-mediated CRC. By studying 76 CRC patients, Marech et al. showed a significant correlation between macrophage infiltration and microvessel density (61). Furthermore, a large total number of TAMs is favorable for the CRC prognosis. Indeed, Nakayama et al. detected high levels of TAMs in patients with a good prognosis (62). Koelzer et al. found that CD68+ TAMs predicted longer OS (63). Similarly, Cavnar et al. reported a significant positive correlation between DFS and CD68+ cells in 188 patients with CRC liver metastasis (64). Compared with the total number of macrophages determined by CD68 markers, the M2-like phenotype of macrophages can better predict the adverse prognosis in CRC. In the study of Wei et al., high-level expression of interstitial CD163 at the front of tumor invasion was significantly correlated with tumor grade, lymphatic vascular invasion, tumor invasion, lymph node metastasis and TNM stage, and was associated with poor recurrence survival rate (RFS), as based on IHC analysis of 81 Chinese CRC patients (59). Yang et al. found that in 81 CRC patients, a high CD163+/CD68+ ratio at the front of tumor invasion (rather than at the tumor stroma) was closely related to enhance lymphatic vascular invasion, tumor invasion, TNM stage, RFS and OS in CRC patients (65).
Myeloid-Derived Suppressor Cells (MDSCs)
Myeloid cells are composed of mononuclear myeloid cells and granulocytes, while mononuclear myeloid cells are mainly composed of monocytes, final differentiated macrophages and DCs. Granulocytes include neutrophils, eosinophils and basophils (66–68). In the early 1980s, these cells were found to be immunosuppressive. Therefore, to unify this group of cells, they were named bone marrow-derived suppressor cells in 2007 (69).
MDSCs interact with the TME, and tumor and stromal cells secrete TGF-β, MMP9, BV8, IL-6, IL-1β, β-FGF and VEFG through autocrine and paracrine mechanisms, mobilizing and expanding MDSCs and further promoting tumor growth (70). The TME can secrete chemokines, cause MDSCs to migrate to the tumor site, inhibit immune function and accelerate tumor progression (71, 72). CCL2 recruits MDSCs to the CRC TME (73), enhancing the immunosuppressive function by inhibiting T cell proliferation and stimulating Treg development (74). In a mouse model, reducing CXCL4 in CRC tumor tissue promoted the recruitment of MDSCs, resulting in an immunosuppressive environment and progression of CRC (75). Ouyang et al. found increased levels of CD33+ CD11b+HLA-DR-MDSCs in primary tumor tissues of CRC patients, which was related to advanced TNM stage and lymph node metastasis. At the same time, it was found that tumor cells induce the expansion of MDSCs through a variety of inflammatory factors. These tumor-derived MDSCs inhibit T cell proliferation and promote tumor cell growth through oxidative metabolism (76).
Tumor-Associated Neutrophils (TANs)
Neutrophils are effector cells of the innate immune system. Unlike macrophages, neutrophils are not antigen-presenting cells but act as killer cells in the blood. Neutrophils are mainly produced in the bone marrow, accounting for 50-70% of human circulating leukocytes, with a half-life of only 5 days; however, they are the only immune cells that can dissolve cells and tissues (77). When the body releases chemokines after infection, neutrophils tend to migrate and recognize pathogens (78). In cancer, tumor cells and TAMs release the chemokines CXCL1/2/3/6/8 and CCL3/5, which induce neutrophils in peripheral blood to enter the TME and polarize into different TANs (79, 80).
A few studies on the relationship between TANs and the survival rate of CRC patients have been conducted (81). Rao et al. found that an increase in neutrophils in tumors is associated with a malignant phenotype and can predict poor prognosis in CRC (82). Galdiero et al. evaluated CRC patients receiving 5-FU chemotherapy and found that a higher TAN concentration was associated with better treatment efficacy. TANs are important immune cell infiltration components in CRC. In fact, evaluating TAN infiltration may help to identify patients who will benefit from 5-FU chemotherapy (83). Berry et al. analyzed the number of neutrophils in CRC tissues. Due to the lack of neutrophil-specific antibodies, neutrophils were counted manually according to their morphology, and high levels of TANs were associated with better overall survival (OS) in patients with stage II CRC (84). Furthermore, the combination of the neutrophil lymphocyte ratio and platelet count is able to predict the future clinical course of CRC (85). A high neutrophil to lymphocyte ratio (NLR) has also been shown to be a poor prognostic factor in CRC patients. Li et al. retrospectively analyzed a cohort of 354 patients with stage I-III CRC and observed a close relationship between dynamic changes in the NLR and the OS rate (86). Additionally, a high NLR has an adverse effect on the OS of CRC patients undergoing radical surgery (87).
Tumor-Infiltrating Lymphocytes (TILs)
Lymphocytes are the main immune cells of tumors, including T, B, NK, and NKT cells, and these subsets can reflect tumor immunotherapy and serve as clinical biomarkers. T cells are the most abundant and characteristic immune cells in the TME and are divided into cytotoxic T cells, helper T cells, inhibitory T cells and NKT cells, in contrast to traditional T cells (88). T cells prevent tumor growth by targeting tumor cells. Tregs are a specific group of CD4+T cells related to the overreaction of immunosuppression, inflammation and allergic diseases (89). In cancers, Tregs are considered to inhibit immunity in most cases, and Treg infiltration is associated with poor prognosis (90–92). Marshall et al. found that Treg cells promote lung cancer metastasis (93). High FoxP3+ Treg infiltration exhibits a significant correlation with shorter OS patients with other solid tumors, including ovarian cancer, gastric cancer, renal cell carcinoma, melanoma, hepatocellular carcinoma, oral squamous cell carcinoma and breast cancer (94–97). However, in some tumors with chronic inflammatory infiltration, the accumulation of Tregs correlates positively with good prognosis. Frey et al. found that patients with mismatch repair deficiency (dMMR) CRC had high-level infiltration of Foxp3+ Tregs, with an increased survival rate (98). According to Hanke et al., high-level infiltration of Foxp3+ Tregs in early lymph node-negative CRC has a good prognosis (99). Vlad et al. also found that an increased Foxp3+ Treg density is associated with improved survival in CRC and is an independent prognostic factor (100). The relationship between Treg infiltration and tumor prognosis seems to be closely related to tumor type. Treg regulation plays the dual or multiple roles in antitumor immunity and the tumor treatment response, maintaining immune homeostasis and preventing autoimmunity (101, 102).
B lymphocytes participate in immune regulation mainly by producing immunoglobulin, presenting antigen secreting cytokines. B lymphocytes produce antibodies in the tumor microenvironment, which promotes tumor development (103, 104). B cells also inhibit tumor growth. Mouse B cells can promote antitumor activity through T cell-mediated immunity, inhibiting tumor development, and CD20-deficient mice show T cell antitumor inhibition (105–107). In malignant melanoma, enhanced patient survival is related to the simultaneous presence of tumor-related CD8+ T cells and CD20+ B cells but not to other clinical features (108). Research on the progression of CRC by B cells is limited, and views are inconsistent. There are differences between the B cell subsets in the peripheral blood, mesenteric lymph nodes and primary tumors of patients with CRC and those of healthy people, and B cells are activated in tumor-related tissues (109). After activation, B cells in patients with CRC differentiate into mature types, resulting in a specific response to tumors. On the other hand, the number of B cells in patients with metastatic CRC (mCRC) is small and the proportion of regulatory B cells is increased, which may be involved in immune escape (110, 111). Nevertheless, Berntsson et al. found that the survival time of CRC patients with B cell infiltration was prolonged (112). Through multiple-regions single-cell sequencing of tumors, normal mucosal tissue, liver metastases, and pairs of noncancerous tissues in CRC patients, a recent study showed that the contradictory effect of B cells on tumors in the past was due to the existence of multiple subtypes of B cells. IgA+IGCL2+ plasma cells are associated with poor prognosis of CRC, whereas highly proliferated GLC2+ plasma cells and circulating B cells are associated with a better prognosis (113).
Natural killer (NK) cells are effector cells of the immune system. When cells are infected or mutated by the virus, the expression of MHC-1 on their surface is lacking or abnormal. NK cells bind to NKG2D interaction ligands through antibody-dependent cell-mediated cytotoxicity (ADCC) or receptors, degranulate and release cytotoxic perforin and granzyme, induce signal transduction, and kill virus-infected cells and tumor cells (114). However, compared with those in adjacent normal tissues, NK cell levels in CRC tissues are low, suggesting that less NK cell infiltration may be one of the mechanisms of TME immune escape (115). The phenotype of peripheral NK cells in CRC patients changes, which promotes tumor progression (116). In CRC patients with curative tumor resection, the expression of NKp44 and NKG2D on circulating NK and NKT cells is increased, suggesting that the primary tumor and TME have an inhibitory effect on the phenotype of NK and NKT cells in CRC (117). In vitro, NK cells can enhance the cytotoxicity of cetuximab and the killing effect on RAS and BRAF mutant CRC cells (118). A phase I clinical trial observed NK cells to be closely related to the therapeutic effect of CRC. Cetuximab was significantly effective in patients with NK cell infiltration, though there was no significant correlation in patients who did not receive cetuximab (119). NK cell therapy has played a key role in hematological diseases and resulted in the use of NK cells in solid tumors. Initial results for chimeric antigen receptor-NK (CAR-NK) in the treatment of CRC patients have been obtained and NKG2D-CAR-modified NK cells showed antitumor effects in mouse models. At the same time, the standard CAR-NK was used in three CRC patients, who reached the safe end point (120). NK cells are also a prognostic factor for CRC recurrence (121).
In addition to NK cells and T cells, there is a special group of cells with the common characteristics of NK and T cells, called NKT cells. NKT cells have CD4+ CD8+ thymocytes, which develop in the thymus and migrate to peripheral organs such as the liver, spleen, lung and intestine (122, 123). Although NKT cells exert cytotoxicity, they mainly secrete a large number of helper T cytokines Th1-, Th2-, Th17-, Treg- or helper follicular cytokine (TFH)-cell related cytokines to play a regulatory role in innate or acquired immunity (124, 125). Type I NKT cells recognize glycosphingolipids α- galactose ceramide or its analogs (126, 127). α-Galactose ceramide (α-galcer) increases NKT expression and PD-1 in combination with α-galce increases the activity of NKT cells, enhances the antitumor response, and significantly reduces the occurrence of small and large intestinal tumors (128). Compared with the normal mucosa, the expression of CD69L and FasL is increased in infiltrating type I NKT cells in CRC tumor tissue, IFN- γ and granzyme B are also increased, and the OS rate of CRC patients with high-level NKT cell infiltration is higher than that of patients with low NKT cell infiltration (129). Intratumoral infiltration of NKT cells can be used as a prognostic factor for CRC.
Exosomes
In the process of tumor cell growth, invasion and metastasis, tumor cells and interstitial fine cells located in the TME can not only secrete various soluble molecules, including cytokines and chemokines, but also release various vesicles. These vesicle structures are extracellular vesicle structures (130) that can be divided into exosomes (20-100 nm), microbubbles (100-1000 nm) and apoptotic bodies (1-5 µm) according to their size. Exosomes are different in size from microbubbles and apoptotic bodies and have specific surface molecular characteristics, such as CD9 and CD63 expression. Exosomes are present in almost all body fluids, including plasma/serum, saliva, breast milk, cerebrospinal fluid, urine and semen (131–141). Exosomes are also distributed in the TME and carry cargo including a variety of proteins, DNA, mRNA, miRNA, long noncoding RNA, and even virus/prion genetic material (142–146). Exosomes play a key role in local and remote intercellular communication in cancer and are an important part of the TME. Almost all kinds of cells in tumors can secrete exosomes, including tumor cells, tumor-associated adipocytes, TAFs, TAMs and vascular endothelial cells. Exosomes can be ingested by recipient cells to participate in intercellular signal exchange (130).
Zeng et al. found that in CRC, cancer-derived exosomal miR-25-3p promotes vascular permeability and angiogenesis by regulating the expression of VEGFR2, ZO-1, occludin and claudin-5 in endothelial cells by targeting KLF2 and KLF4. And miR-25-3p from CRC cells enhances CRC metastasis in the mouse liver and lungs. In addition, the expression of miR-25-3p in circulating exosomes is significantly higher in CRC patients with metastasis than in those without metastasis, and exosomes can be used as blood biomarkers (147). CRC-derived exosomal miR-106b-3p promotes tumor metastasis by downregulating DLC-1 expression (148). Exosomal miR-200c-3p negatively regulates the migration and invasion of CRC stimulated by lipopolysaccharide (LPS) (149). The CRC-derived exosomal circRNA, circPACR can be induced by miR-142-3p/miR-506-3p-TGF-β1 to promote CRC cell proliferation, migration and invasion (150). CAFs are the main components of the TME and promote cancer development through tumor matrix interactions. Bhome et al. compared the exosomes of normal and TAFs in CRC patients and found that CAFs were enriched in microRNAs 329, 181a, 199b, 382, 215 and 21, with microRNA 21 being the most abundant. After establishing the original transplanted tumor model with miR-21-overexpressing fibroblasts, liver metastasis increased (151). Exosomal miR-21 is expressed by stromal fibroblasts and promotes tumor cell metastasis. MiR-21 is involved in the progression of CRC. Exosomes secreted by CAFs are also involved in CRC metastasis and chemoresistance. Hu et al. found that CAFs secrete exosomes, resulting in a significant increase in the level of miR-92a-3p in CRC cells, activating the Wnt/β- catenin pathway and inhibiting mitochondrial apoptosis by directly inhibiting FBXW7 and MOAP1; the effect is to promote stemness, EMT, metastasis and 5-FU/L-OHP resistance in CRC (50). This finding provides an alternative way to predict and treat CRC metastasis and chemoresistance by inhibiting exosomal miR-92a-3p. Exosomes can also be used as diagnostic markers in CRC. Maminezhad et al. detected CRC cell lines and patient serum, and found increased levels of miR19a, miR-20a, miR-150 and let-7a but decreased levels of miR-143 and miR-145, with expression being related to TNM stage (152). Clinically, many miRNAs secreted by exosomes, such as let-7a, miR-1229, miR-1246, miR-150, miR-21, miR-223, and miR-23a have been used as diagnostic and prognostic markers for screening and predicting CRC tumors (153).
Immunosenescence
Immunosenescence is a process of immune dysfunction that tend to cause inflammation and an immunosuppressive microenvironment leading to tumorigenesis and cancer metastasis (154). Senescence might become an obstacle to achieve efficacious immunotherapy in the TME, since senescent cells secret proinflammatory cytokines and growth factors, known as the senescence associated secretory phenotype (SASP), and this secretion has been implicated in both aging and cancer development (155). Giunco et al. found that in elderly CRC patients, senescent CD8 cells, but not CD4, displayed a significant relationship with disease outcome. Furthermore, the CD4/CD8 ratio was a prognostic marker of disease relapse in stage I-III CRC patients (156). In the TME of CRC, immunosenescent cells can influence the therapeutic effect since the majority of CRC patients with microsatellite stability (MSS) do not benefit from current anti-PD-1 therapy. A recent study found that in 18 MSS CRC patients, the number of immunosuppressive/exhausted T-cell phenotypes at tumor lesions were increased and CD8+ CD28- immunosenescent T cells were accumulated according to single-cell mass cytometry analysis. Moreover, the TME of CRC hosts chemokines/cytokines that likely recruit immunosuppressive/exhausted T cell subsets to regulate the immune system (157). It is necessary to comprehensively understand the immunosenescence to help boost the immune response in patients with MSS CRC.
Current Strategies Related to Immunotherapy in CRC
Antiangiogenic Therapy
Bevacizumab, an immunoglobulin G monoclonal antibody against humanized vascular growth factor A, selectively binds to vascular endothelial factor subtype A (VEGF-A), hinders the binding of vascular endothelial growth factor to receptor tyrosine kinases (VEGFR), and initiates signaling to inhibit tumor angiogenesis (158, 159). Bevacizumab has been approved for first-line and second-line treatment of mCRC (160). An Italian randomized, open, multicenter, phase 3 clinical trial (NCT00719797) compared the efficacies of FOLFIRI combined with bevacizumab and FOLFIRI alone, and the median survival time of the FOLFIRI combined with bevacizumab group was greater than that of the FOLFIRI group (29.8 months vs. 25.8 months, HR = 0.80, P = 0.03) (161). Additionally, the combined use of bevacizumab did not significantly increase side effects but did enhance effective PFS and OS (162). The latest study found that bevacizumab combined with capecitabine was also effective as an advanced treatment for previously irinotecan-, oxaliplatin- and fluoropyrimidine-resistant mCRC (163). VEGF plays an important role in the CRC immune microenvironment, which can inhibit DC maturation, reduce T cell tumor infiltration and increase inhibitory cells in the TME (164–167). We found that the level of VEGF was increased in tumors. Moreover, bevacizumab inhibited the VEGF-VEGFR1 binding signal on DCs, NF-κB signaling, and DC cell maturation, prevented the increase in the amount of MHC and other molecules, and suppressed T cell activation. In CRC patients, bevacizumab elevated the number of mature DCs in the peripheral blood (168). Limagne et al. found that the amount pf MDSCs of patients decreased with FOLFOX in combination with bevacizumab, which was related to longer survival (169).
Ramucirumab, a humanized monoclonal antibody, mainly acts on the extracellular region of VEGF receptor 2 and has a beneficial role in gastric cancer, lung cancer and CRC (170–175). In the RAISE trial, after first-line oxaliplatin/fluoropyrimidine chemotherapy combined with bevacizumab for progressed CRC, ramucirumab was added to FOLFIRI, and the OS rate and progression-free survival(PFS) rate of patients were significantly improved (176).
Aflibercept is a monoclonal antibody composed of the extracellular segments of human VEGFR-1 and VEGFR-2 fused with the vascular endothelial growth factor-binding region and human immunoglobulin G1 FC region. Aflibercept β combined with FOLFIRI was approved for second-line treatment of mCRC in 2017 (177). A high-quality double-blind randomized controlled trial (RCT), the VELOUR trial, compared the efficacy of aflibercept plus FOLFIRI with that of placebo plus FOLFIRI, and the median OS, OS and PFS were higher than those in the former group (178). However, aflibercept in elderly patients (> 65 years old) shows a controllable increase in toxicity (179).
In addition to using monoclonal antibodies to inhibit the VEGFA pathway, some small molecule inhibitors have been applied in anti-VEGF therapy, such as regorafenib, sorafenib, sunitinib, pazopanib and axitinib. An international, multicenter, placebo-controlled phase III clinical trial (CORRECT) found that the median survival time of mCRC patients in the regorafenib group was longer than that in the placebo group (6.4 months vs. 5.0 months, HR = 0.77, P = 0.00052) (180). Regorafenib combined with nivolumab also has good applicability for the treatment of MSS chemotherapy-resistant mCRC (181). Sorafenib, a multi-kinase inhibitor that targets serine threonine and tyrosine kinases involved in tumor progression and angiogenesis, including all VEGFRs and PDGFR-β, RET, FLT3 and c-KIT (182), is used to treat advanced renal cell carcinoma, unresectable hepatocellular carcinoma and thyroid cancer (183–186). In CRC, a phase I clinical trial (RESPECT) found that the first-line combined use of sorafenib and oxaliplatin, folic acid and fluorouracil (mFOLFOX6) did not prolong PFS (187). In a multicenter, randomized phase II clinical trial (NEXIRI-2/PRODIGE 27), mCRC patients carrying RAS mutations had a prolonged 2-month no-progression rate and median PFS with the use of sorafenib combined with irinotecan after oxaliplatin, irinotecan, fluoropyrimidines and bevacizumab failed (188). Sunitinib is a small molecule multi-target receptor tyrosine kinase inhibitor. However, for patients with unresectable/advanced mCRC, the first-line combination of sunitinib and FOLFIRI did not lead to significant clinical benefits (189). A randomized, phase III clinical trial found no significant difference in median PFS between sunitinib combined with FOLFIRI and FOLFIRI combined with placebo (190). Fruquintinib, a small molecule selective VEGFR inhibitor independently developed in China, significantly prolonged the median OS of patients after three-line use of fruquintinib compared with that of patients receiving the placebo (9.3 months vs. 6.6 months, HR = 0.65) (191). The major antiangiogenic therapy agents under clinical investigation in CRC are summarized in Table 1.
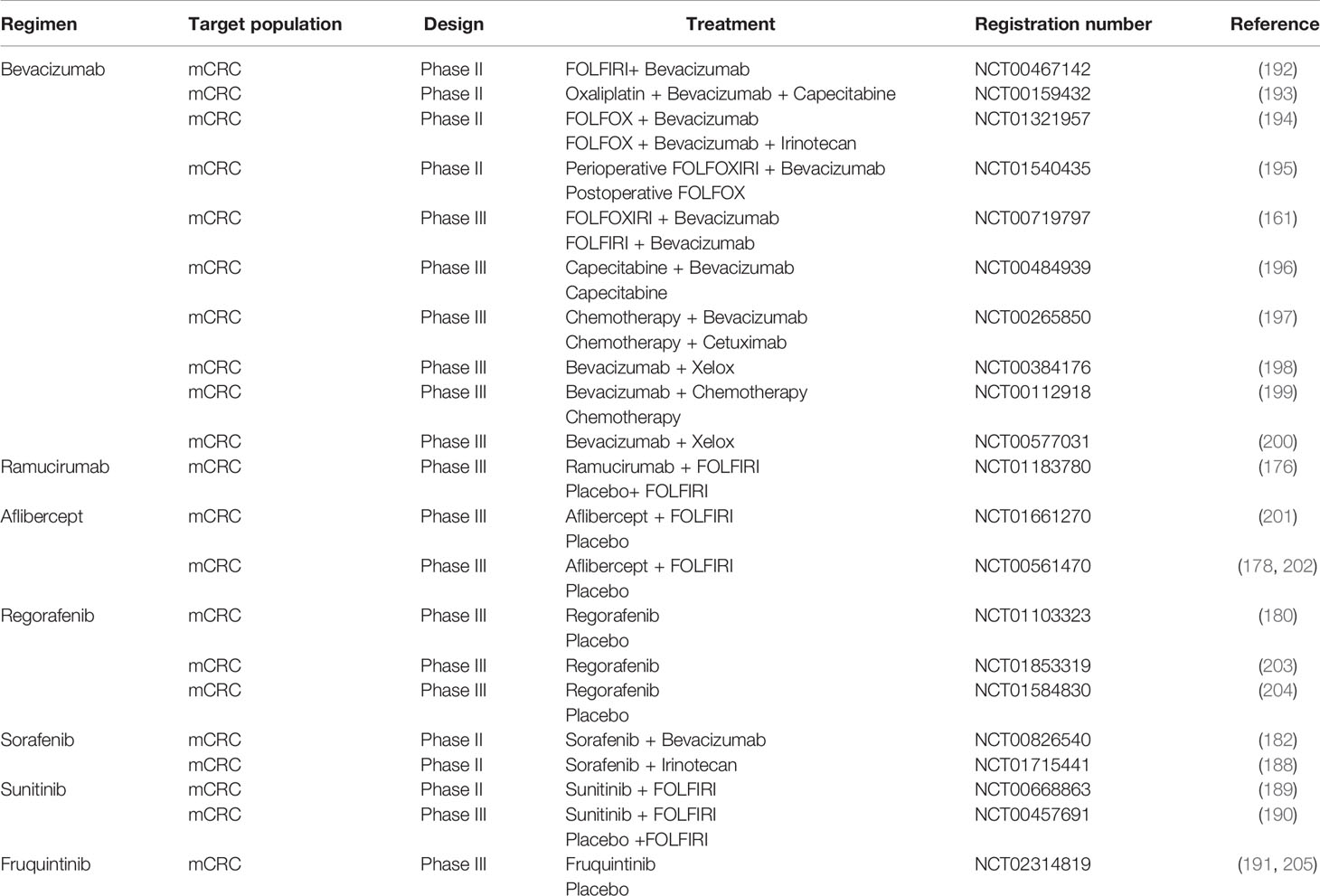
Table 1 Summary of antiangiogenic therapy for colorectal cancer (CRC).
Anti-EGFR Therapy
EGFR is a membrane-bound receptor tyrosine kinase protein that activates downstream intracellular signaling pathways, including MAPK (RAS/RAF/MEK/ERK), PI3K/AKT, and JAK/STAT3 signaling, and plays a role in tumor cell growth, proliferation and differentiation (206, 207). EGFR promotes tumor progression when overexpressed. CRC patients exhibit high-level expression of EGFR. Therefore, targeting EGFR and its downstream pathways has become a new strategy for the treatment of CRC (208). Cetuximab is a human/mouse chimeric IgG1 monoclonal antibody that mainly binds to EGFR on the surface of tumor cells and competitively blocks EGFR signaling to inhibit tumor cell proliferation. Cetuximab also inhibits the development of neovascularization by reducing the production of VEGF and activates the human anti- chimeric antibody (HACA) (209). Initial multiple clinical phase II trials found that among EGFR-positive patients, cetuximab combined with irinotecan had a better clinical effect than chemotherapy alone (210–212). Despite no significant difference between the PFS risk ratios and OS rates of mCRC patients receiving cetuximab combined with FOLFIRI and mCRC patients receiving FOLFIRI alone, cetuximab combined with FOLFIRI benefited KRAS wild-type patients (213). KRAS is an effector molecule responsible for signal transduction from ligand-bound EGFR to the nucleus. Activation of KRAS mutations often leads to CRC resistance to the EGFR targeted monoclonal antibodies (214). Therefore, EGFR-positive wild-type KRAS CRC responds to cetuximab (215). The CEBIFOX study found an ORR of 70.3%, a median PFS of 10.9 months (95% CI 9.0-12.9), and an OS of 33.8 months (95% CI 21.4-45.5) for fortnightly use of cetuximab combined with FOLFOX6 in patients with RAS wild-type mCRC (216).
Panitumumab, the first fully humanized IgG2 monoclonal antibody, displays a high affinity for EGFR, and its mechanism of action in CRC treatment is similar to that of cetuximab. Clinical phase II and III trials have shown that panitumumab can significantly prolong the PFS of patients with refractory CRC, with good tolerance (212, 217). In a randomized phase III trial (PRIME), the PFS of patients with wild-type KRAS was prolonged with mCRC first-line use of panitumumab combined with fluorouracil, folic acid and oxaliplatin (FOLFOX4) compared with that of patients receiving FOLFOX4 alone, though there was no significant difference in OS (218). In the randomized, open, phase II VOLFI study (AIO KRK0109), FOLFOXIRI combined with panitumumab was used as the first-line treatment for Ras wild-type mCRC improving the ORR and secondary surgical resection rate (219). In a phase II trial of locally advanced rectal cancer, FOLFOXIRI combined with panitumumab/cetuximab was used as a new adjuvant chemotherapy for patients with wild-type RAS-BRAF rectal cancer, with good clinical efficacy and tolerance (220). The major agents targeting EGFR therapy under clinical investigation in CRC are summarized in Table 2.

Table 2 Summary of anti-EGFR therapy for colorectal cancer (CRC).
Immune Checkpoint Inhibitors (ICIs)
Immune checkpoints are molecules that express and regulate the activation of immune cells. When the immune response is too strong, the immune checkpoint acts as a key regulator for attenuation (222). However, in cancer, immune checkpoints are highly activated and overexpressed; thus, antigens cannot be presented to T cells, inhibiting their immune function and resulting in malignant cell proliferation and tumor immune escape (223, 224). ICIs restore immune function mainly by targeting and/or blocking immune checkpoint protein ligands on the surface of T cells or other immune cell subsets (225). ICIs constitute a mature monoclonal antibody immunotherapy. The most widely studied immune checkpoint targets are programmed cell death 1 (PD-1) and cytotoxic T -lymphocyte-associated antigen 4 (CTLA-4), which are used for a variety of solid tumors (226–228). There are also studies on the potential role of other checkpoints in tumor immune regulation, such as lymphocyte activation gene-3 (LAG-3), T cell immunoglobulin-3 (Tim-3), T cell immunoglobulin and the ITIM domain (TIGIT) (229–233). CTLA-4 is a transmembrane protein that is mainly expressed on activated T cells and was first cloned in 1987 (234). CTLA-4 binds the B7 molecule to reduce T cell activity and inhibit T cell activation channels, with an immunosuppressive function (235, 236). In 2010, the CTLA-4 inhibitor ipilimumab was demonstrated to improve the long-term prognosis of patients with unresectable malignant melanoma (237). In 2011, ipilimumab became the first ICI approved by the FDA for cancer treatment. PD-1, a new member of the immunoglobulin gene superfamily, is expressed by various immune cells, such as CD4 and CD8 T cells, B cells, macrophages, DCs and tumor-infiltrating lymphocytes (TILs) (238, 239). PD-1 is a negative regulatory molecule that inhibits T cell activation and limits autoimmunity (240, 241). The use of PD-1/PD-L1 pathway inhibitors can restore the function of effector T cells, playing an antitumor role (242). At present, a variety of PD-1 and PD-L1 inhibitors have been approved by the FDA to treat a variety of tumors.
There are currently three PD-1 and CTLA-4 inhibitors approved by the FDA for CRC: pembrolizumab, nivolumab and ipilimumab (Table 3).

Table 3 FDA approved main agents of immune checkpoint inhibitors (ICIs) for colorectal cancer (CRC).
Pembrolizumab (Keytruda®) is the first PD-1 inhibitor approved by the FDA for metastatic malignant melanoma (252). In recent years, pembrolizumab has been used for non-small-cell lung cancer (253, 254), Hodgkin’s lymphoma (255, 256), HNSCC (257), urothelial carcinoma (258, 259), gastric cancer (260) and CRC (243, 244). The landmark clinical trial NCT01876511 for the treatment of CRC with pembrolizumab is noteworthy. The clinical trial included 11 dMMR CRC patients and 21 pMMR CRC patients and 9 patients with dMMR in other cancers. The immune-related objective response rate and immune-related PFS rate were 40% and 78% in dMMR CRC patients, and 0% and 11% in pMMR CRC patients, respectively. The median PFS and OS in the dMMR group were not achieved, and the median PFS and OS in the pMMR CRC group were 2.2 months and 5.0 months, respectively (HR = 0.1, P < 0.001) (245). Based on these data, pembrolizumab (Keytruda®) was approved to treat unresectable or metastatic dMMR and high microsatellite instability (MSI-H) CRC by the FDA on May 23, 2017 (261). KEYNOTE-164 is a phase II clinical trial for evaluating pembrolizumab in the treatment of refractory, MSI-H/dMMR metastatic CRC. At the end of the trial data, the median follow-up time of group A (previously received ≥ 2-line treatment) was 31.3 months (range of 0.2-35.6 months), the objective response rate was 33% (95% CI, 21% - 46%), and the median PFS was 2.3 months (95% CI, 2.1-8.1 months). The median follow-up time of CRC in group B (previous ≥ 1-line treatment) was 24.2 months (range of 0.1-27.1 months), the objective response rate was 33% (95% CI, 22% - 46%) and the median PFS was 4.1 months (95% CI, 2.1-18.9 months). The incidence of treatment-related grade 3-4 adverse events was 16% in group A and 13% in group B. Thus, pembrolizumab can be safely used in patients with MSI-H/dMMR CRC (244). KEYNOTE-177 (NCT02563002) is a phase III clinical trial in which patients with metastatic MSI-H/dMMR CRC were randomly assigned to the pembrolizumab arm, though patients receiving chemotherapy could switch to pembrolizumab if disease progression occurred. The PFSs of the pembrolizumab and chemotherapy groups were 16.5 months and 8.2 months, respectively (HR = 0.60; 95% CI, 0.45-0.80; P = 0.0002) (243).
Another successful PD-1 inhibitor is nivolumab (Opdivo®). Based on the CheckMate 037 and CheckMate 066 trials, nivolumab has also been approved for the first time to treat unresectable or metastatic melanoma (246, 247). Nivolumab showed a good therapeutic effect in mCRC patients with dMMR/MSI-H. CheckMate 142 (NCT02060188) found that 23/74 patients achieved objective remission, and 68.9% (51/74) of patients received > 12 weeks of disease control; the safety of dMMR/MSI-H mCRC was tolerable (248). Nivolumab was approved on August 1, 2017, for dMMR and MSI-H mCRC. Interestingly, the CTLA inhibitor ipilimumab has also shows a certain therapeutic effect in CRC. Ipilimumab combined with nivolumab as the treatment for dMMR/MSI-H mCRC patients was effective at 9 months in 94% of patients; the PFS rates at 12 months were 76% and 71% for dMMR and MSI-H mCRC patients, respectively, and the OS rates at 12 months were 87% and 85% for dMMR and MSI-H mCRC patients, respectively (249). Moreover, ipilimumab combined with nivolumab did not significantly increase toxicity or side effects (250). Therefore, ipilimumab and nivolumab were approved for dMMR and MSI-H mCRC on July 10, 2018. Furthermore, a recent phase II CheckMate 142 study found that first-line nivolumab plus low-dose ipilimumab had robust and durable clinical benefit and was well tolerated as a first-line treatment for MSI-H/dMMR mCRC patients (251).
Adoptive Cell Therapy (ACT)
Adoptive cell therapy utilizes the immune cells, such as T cells, DCs, NK cells or cytokine-induced killer (CIK) cells, of patients or other donors for tumor patients to achieve anti-tumor effects. ACT for CRC treatment includes TILs, CIK cell therapy and chimeric antigen receptor-modified (CAR) T cell therapy (Table 4). In a clinical study on TILs combined with IL-2 in CRC, patients in the ACT group received TILs extracted from metastatic tumors, as stimulated and amplified with high-dose IL-2, whereas the control group received traditional chemotherapy. Although no significant difference in disease-free survival (DFS) was observed between the two groups after 1, 3 and 5 years, TCRϵ chain expression increased significantly in disease-free patients compared with that in patients with recurrence (P = 0.04), suggesting that TILs play a role in the immune response (262). Sentinel lymph node (SLN)-T cells are also used for ACT in the treatment of CRC. A preliminary study found that after 16 CRC patients were injected with SLN T cells, 4 of 9 patients with stage IV CRC experienced complete remission, with a median survival time of 2.6 years, which was much greater than the median survival time of 0.8 years in the control group (263). In another I/II clinical study of 55 patients with CRC with SLN metastasis, the median OS of the experimental group that received expanded lymphocytes was 28 months, whereas that of the control group was 14 months, and no obvious toxicity or side effects was observed (264).
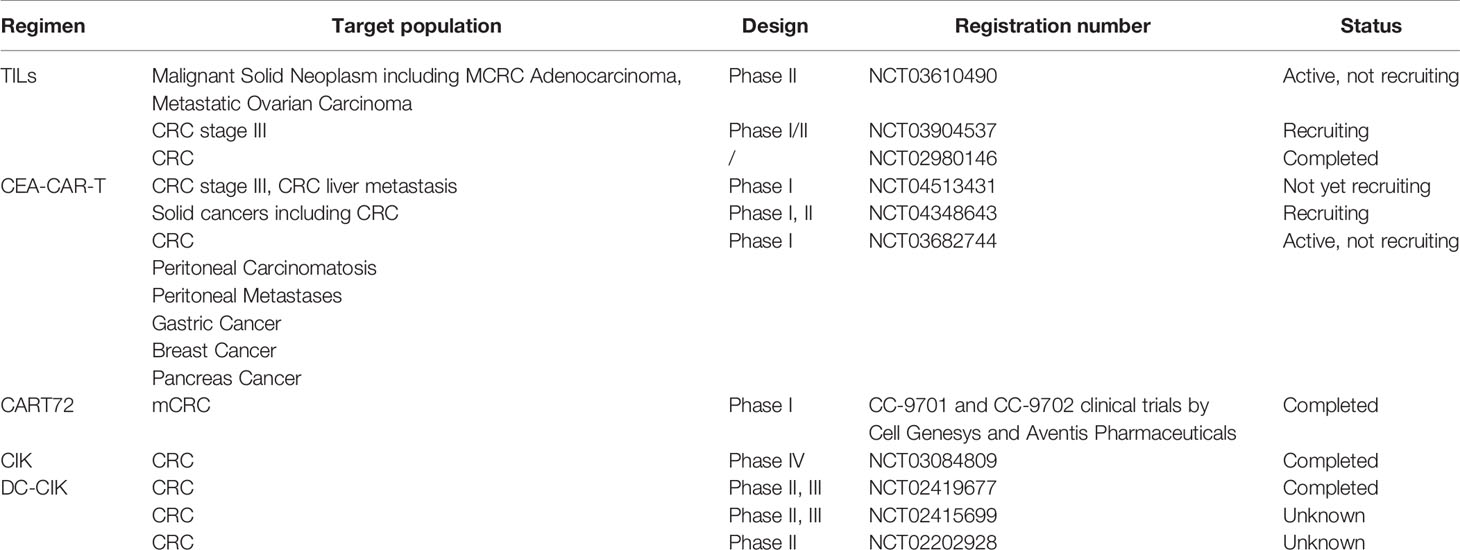
Table 4 Summary of adoptive cell therapy (ACT) for colorectal cancer (CRC).
Many clinical trials using TILs or SLN T cells as a treatment for CRC (NCT03610490, NCT03904537, and NCT02980146) are being performed. NK cells have natural cytotoxicity toward tumor cells, with antibody dependent cytotoxicity (ADCC), and can secrete a variety of cytokines and chemokines for an immunomodulatory role. Therefore, NK cells can also be used for adoptive therapy. In a phase I clinical trial, expanded NK cells combined with IgG1 antibody were used to treat patients with gastric cancer or CRC. Among 6 evaluable patients, 4 were in stable condition (SD), and disease progression occurred in two patients (119). As TILs have the limitation of needing to be expanded from a tumor, exogenous T cell receptors (TCRs) have been expressed on cells by genetic engineering technology. Carcinoembryonic antigen (CEA) levels are often elevated in the tissues and serum of patients with gastrointestinal tumors. Therefore, genetically engineered autologous T lymphocytes that express mouse TCR against human CEA have been used for CRC treatment. In a phase I clinical trial, three patients with refractory mCRC were administered TCR targeting CEA, and their serum CEA levels were significantly decreased (74–99%). One patient showed reduced liver and lung metastases, but all three patients experienced severe transient inflammatory colitis (265). CAR-T cells have achieved remarkable results in B-cell leukemia and lymphoma, although the development of solid tumors is lagging (266, 267). A phase I clinical trial of targeted CEA-CAR-T cells for CRC treatment found that 7 of the 10 CEA+ patients were in stable condition after CAR-T cell treatment, with 2 patients maintaining this state for more than 30 weeks, and 2 patients underwent tumor regression (268). Another study on CAR-T cells targeting tumor-associated glycoprotein (TAG)-72 (CART72 cells) in the treatment of mCRC found that a very short duration of CART72 cells in the blood (≤14 weeks), suggesting that CART72 cells have a limited role in mCRC (269). A CAR-NK study targeting NKG2D found that after three mCRC patients received local infusion of CAR-NK cells, ascites production decreased and tumor cells in ascites samples decreased significantly. In addition, the method using RNA to make CAR can enhance the specificity of NK cells to NKG2DL and their tumor cell killing activity (120).
CIK cells treatment is a part of ACT and is induced by mononuclear cells cultured with CD3 monoclonal antibody and cytokines such as IFN-γ, IL-1 and IL-2. CIK cells include activated NKT cells, CD3-/CD56+ NK cells and CD3+/CD56- CTLs. CIK cells have the characteristics of rapid proliferation, strong antitumor activity, wide spectrum and low toxicity. They have been used in the treatment of various solid tumors, such as hepatocellular carcinoma, renal cell carcinoma, gastric cancer, breast cancer, ovarian cancer, non-small-cell lung cancer and nasopharyngeal carcinoma. The efficacy of somatic CIK cells in patients with mCRC was examined in a phase II clinical trial. MCRC patients in the experimental group received chemotherapy combined with CIK cells, whereas the control group received chemotherapy alone. The median OS rates of the experimental and the control groups were 36 months and 16 months, respectively (P < 0.001), and the PFS rates were 16 months and10 months (P = 0.072), respectively. Although there was no significant difference, there was an increasing trend (270). A retrospective study using CIK cells to treat postoperative CRC patients reported a median PFS and median OS in the CIK group of 25.8 months and 41.3 months, respectively, while the median PFS and median OS in the control group were 12.0 months and 30.8 months, respectively (271). Another retrospective study on the efficacy of postoperative adjuvant infusion of CIK cells combined with chemotherapy for CRC observed a significantly longer DFS in the group than in the control group [HR = 0.28, 95% CI (0.09, 0.91), P = 0.034]. The 2-year DFS rates of the CIK group and control group were 59.65 ± 24.80% and 29.35 ± 6.39%, respectively. Moreover, CIK cell infusion was not associated with immediate adverse reactions (272). Dendritic cytokine-induced killer cells (DC-CIK) were observed in the combined first-line treatment of advanced CRC. The 5-year OS rates of the DC-CIK group and non-DC-CIK group were 41.3% and 19.4% (P = 0.001), and the 5-year PFS rates of the DC-CIK group and non-DC-CIK group were 57.4% and 33.6% (P = 0.022), respectively (273). Overall, DC-CIK immunotherapy combined with first-line treatment can significantly prolong the 5-year OS and PFS rates in patients with advanced CRC.
Cancer Vaccine and Oncolytic Virus Therapy
Cancer vaccines are another method of immunotherapy for CRC (Table 5). Tumor cells express tumor-associated antigen (TAA), and by expressing specific tumor antigens, cancer vaccines can stimulate the body to produce a specific immune response. However, the results obtained for vaccines in the treatment of CRC are not consistent. Initially, a prospective randomized controlled clinical trial was conducted in CRC patients, and an autologous tumor cell BCG vaccine that induced active specific immunotherapy (ASI) was used. The study found that there was no statistically significant difference in the survival rate or disease-free survival rate of 80 eligible patients (279). A randomized phase III clinical trial of adjuvant ASI with autologous tumor cell BCG reported no significant difference in DFS and OS rates between the surgical resection plus ASI group and the simple resection group of stage II and III CRC patients (280). In another study of stage II and III CRC patients, despite no clinical benefit of autologous tumor cell BCG immunotherapy for stage III CRC after surgery, the recurrence-free period of autologous tumor cell BCG adjuvant ASI after surgical resection was significantly longer than that of simple resection (P = 0.011), and recurrence-free survival was significantly prolonged (P = 0.032) (281). As antigen-presenting cells, DCs are also often modified to produce vaccines. A randomized trial for CRC patients on the activation of CD40L by DC vaccines in vitro found that 15 of 24 patients had immune induction reactions. The five-year recurrence-free survival rate (RFS) of those who had tumor-specific T cell proliferation or IFN-γ induced by the vaccine that appeared at one week after vaccination was significantly higher than that of patients without a response (63% vs. 18%, P = 0.037) (282). The randomized phase II clinical trial on administering DC vaccines after complete resection of CRC liver metastasis showed a significantly longer median DFS for the vaccine group was than for the observation group (25.6 months vs. 9.53 months) (283).
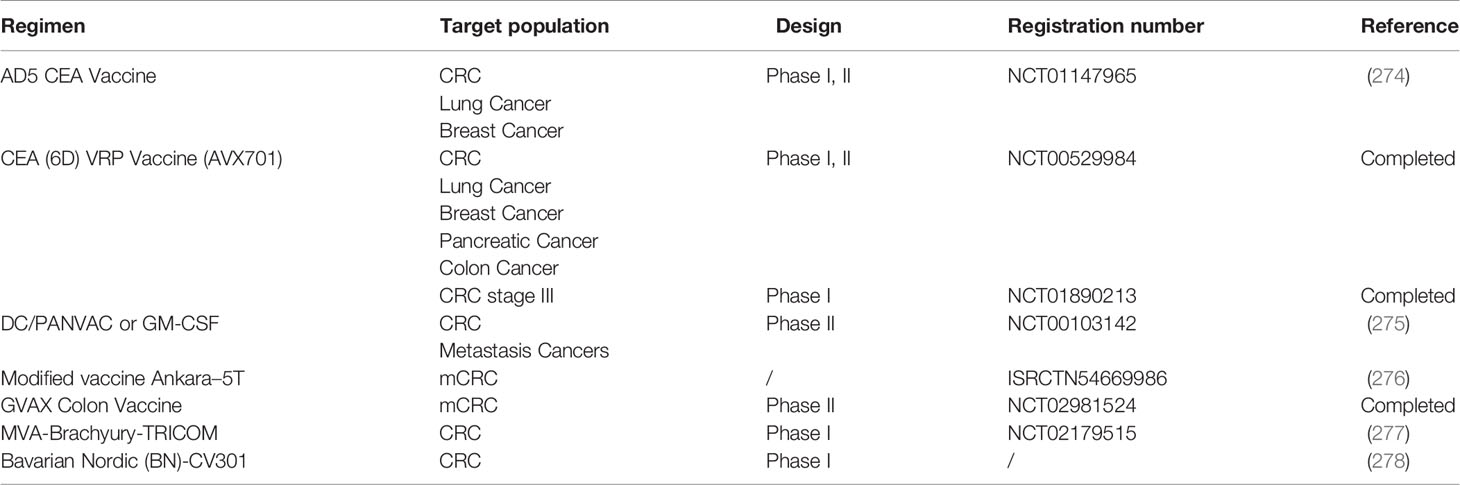
Table 5 Summary of cancer vaccine and oncolytic virus therapies for colorectal cancer (CRC).
Guanylcyclase C (GUCY2C), which is selectively expressed by intestinal epithelial cells and some neurons, is almost universally overexpressed in CRC (284). According to a phase I study using the Ad5-GUCY2C-PADRE vaccine in the treatment of stage I or II (pN0) colon cancer (NCT01972737), the vaccine can stimulate the immune response of T cells and has certain safety (285). CEA is overexpressed in CRC and acts as a tumor antigen marker. A phase I/II trial using the Ad5[E1-, e2b-]-CEA (6D) vaccine for advanced CRC reported a median survival time for the 32 patients included in the study of 11 months, and the Ad5 [E1-, e2b -] - CEA (6D) vaccine was well tolerated and induced an immune response (286). In the phase I study of patients with stage III CRC treated with virus-like replicator particle (VRP)-CEA, 12 CRC patients completed standard postoperative adjuvant chemotherapy and received VRP-CEA immunization 4 times every 3 weeks. The 5-year RFS rate was 75% (95% CI 40-91%), and no deaths were observed during the period. After vaccination, levels of CD8+ TEMs increased (10/12), Foxp3+ Tregs decreased (10/12), and specific CEA and IFN-γ produced by CD8+ granzyme B+ TCM cells increased (287).
Several CRC clinical trials of CEA-modified tumor vaccines have been carried out, such as NCT01147965, NCT00529984, and NCT01890213 (274). MUC1 is abnormally expressed in tumors and is also a tumor-associated antigen. DCs and poxvirus vectors act as immune stimulants against tumor antigens. A randomized phase II trial (NCT00103142) compared whether two vaccines based on DCs and pox vectors encoding CEA and MUC1 (PANVAC) can prolong the survival of mCRC patients after resection (275). Seventy-four mCRC patients after resection and perioperative chemotherapy were randomly treated with autologous modified PANVAC with DC (DC/PANVAC) or GM-CSF (granulocyte macrophage colony-stimulating factor) every time. The 2-year RFS rates of the two groups were similar, and the DC and poxvirus vectors had similar activity. As a treatment for mCRC, the modified vaccine Ankara-5T4 and low-dose cyclophosphamide improved the antitumor immune response and prolonged survival, with no safety problems (276). Furthermore, a phase I trial (NCT02179515) was performed to test the safety and tolerability of a modified vaccinia Ankara (MVA)-based vaccine modified to express brachyury and T-cell costimulatory molecules (MVA-Brachyury-TRICOM) in advanced patients including colon cancer patients. Heery et al. found that the MVA-brachyury-TRICOM vaccine directed against a transcription factor known to mediate EMT can be administered safely in patients with advanced cancer and can activate brachyury-specific T cells in vitro and in patients (277). Recently, a phase I dose-escalation trial of Bavarian Nordic (BN)-CV301, which is a recombinant poxviral vaccine targeting MUC-1 and CEA with costimulatory molecules, was conducted to test the safety and immune response of the vaccine. The trial found that the BN-CV301 vaccine was safely administered to patients with advanced cancer (278).
GVAX is a cellular immunotherapy induced by an allogeneic, whole-cell, granulocyte macrophage colony-stimulating factor that can induce the immune response of T cells to TAAs. A phase 2 study (NCT02981524) of the colon GVAX vaccine, cyclophosphamide and pembrolizumab in 17 patients with pMMR advanced CRC was carried out. The median PFS was 82 days (95% CI 48-97 days), and the median OS was 213 days (95% CI 179-441 days) (288). Although GVAX/Cy plus PD-1 did not achieve the main outcome expected in pMMR CRC, biochemical reactions were observed in patients, providing a certain method to cause insensitivity to PD-1 in pMMR CRC, which still needs to be further explored in combination with other drugs. Although there is no cancer vaccine approved for clinical use, a large number of clinical trials are ongoing and are expected to further improve the therapeutic effect on CRC.
Tumor-Derived Exosomes Therapy
Tumor-derived exosomes have a certain potential antigenicity and can induce a strong antitumor immune response (289). Therefore, in addition to being a potential diagnostic marker, some studies have found that these exosomes can play a role as vaccines in CRC. A phase I clinical trial included 40 patients with HLA-A0201+ CEA+ advanced CRC who were randomly treated with AEX (ascites-derived exosomes) or AEX plus GM-CSF, and both methods were safe and tolerable. The patients in the AEX plus GM-CSF group showed a strong tumor-specific anti-tumor cytotoxic T lymphocyte reaction. These data suggest that immunotherapy with AEX plus GM-CSF can be used as an effective vaccine for mCRC patients (290).
Conclusion and Prospects
The TME is a complicated landscape that is not only closely related to the growth and development of CRC but also affects the treatment and prognosis of patients with colorectal cancer. A variety of cytokines, chemokines, matrix enzymes and immunosuppressive cells, such as Tregs and MDSCs, shape the immunosuppressive environment of CRC. Although immunotherapy has achieved good results in malignant melanoma and lung cancer, its results in CRC are still poor. Therefore, it is particularly important to deeply study the TME, reverse or prevent tumor immune resistance and find a better way to treat CRC. At present, research on anti-PD-1 antibodies, adoptive cell immunotherapy, vaccine therapeutics and oncolytic viruses is being carried out. We need to carry out more clinical experiments, find more biomarkers for CRC, and make rational use of the differences in immune typing and genotyping of CRC such that suitable patients can benefit from immunotherapy.
Author Contributions
The manuscript was conceptualized by YC and CW. YC wrote the majority of the manuscript and produced the tables. XZ helped to write the manuscript. CW critically revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the National Natural Science Foundation of China (No. 31570908).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
CRC, colorectal cancer; ICIs, immune checkpoint inhibitors; TME, tumor immune microenvironment; DCs, dendritic cells; ECM, extracellular matrix; VEGF, vascular endothelial growth factor; VEFGR, vascular endothelial growth factor receptor; TGF-β, transformation and growth factors-β; PDGF, platelet-derived growth factor, CAF, cancer associated fibroblasts; TAM, tumor-associated macrophage; TAN, tumor-associated neutrophil; MDSC, myeloid-derived suppressor cell; TILs, tumor-infiltrating lymphocytes; mCRC, metastatic CRC; NK, Natural killer; OS, overall survival; PD-1, programmed cell death 1; CTLA-4, cytotoxic T-lymphocyte-associated antigen 4; ACT, adoptive cell therapy; MSS, microsatellite stable; dMMR, mismatch repair deficieny; MSI-H, high microsatellite instability; CAR, chimeric antigen receptor-modified; CIK, cytokine-induced killer; OS, overall survival; PFS, progression-free survival; DFS, disease-free survival; RFS, recurrence-free survival rate.
References
1. Keum N, Giovannucci E. Global Burden of Colorectal Cancer: Emerging Trends, Risk Factors and Prevention Strategies. Nat Rev Gastroenterol Hepatol (2019) 16:713–32. doi: 10.1038/s41575-019-0189-8
2. De Almeida CV, De Camargo MR, Russo E, Amedei A. Role of Diet and Gut Microbiota on Colorectal Cancer Immunomodulation. World J Gastroenterol (2019) 25:151–62. doi: 10.3748/wjg.v25.i2.151
3. Koi M, Carethers JM. The Colorectal Cancer Immune Microenvironment and Approach to Immunotherapies. Future Oncol (2017) 13:1633–47. doi: 10.2217/fon-2017-0145
4. Fidelle M, Yonekura S, Picard M, Cogdill A, Hollebecque A, Roberti MP, et al. Resolving the Paradox of Colon Cancer Through the Integration of Genetics, Immunology, and the Microbiota. Front Immunol (2020) 11:600886. doi: 10.3389/fimmu.2020.600886
5. Hinshaw DC, Shevde LA. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res (2019) 79:4557–66. doi: 10.1158/0008-5472.CAN-18-3962
6. Cardoso-Silva D, Delbue D, Itzlinger A, Moerkens R, Withoff S, Branchi F, et al. Intestinal Barrier Function in Gluten-Related Disorders. Nutrients (2019) 11:2325. doi: 10.3390/nu11102325
7. Suzuki T. Regulation of the Intestinal Barrier by Nutrients: The Role of Tight Junctions. Anim Sci J (2020) 91:e13357. doi: 10.1111/asj.13357
8. Johansson ME, Gustafsson JK, Holmen-Larsson J, Jabbar KS, Xia L, Xu H, et al. Bacteria Penetrate the Normally Impenetrable Inner Colon Mucus Layer in Both Murine Colitis Models and Patients With Ulcerative Colitis. Gut (2014) 63:281–91. doi: 10.1136/gutjnl-2012-303207
9. Birchenough GM, Johansson ME, Gustafsson JK, Bergstrom JH, Hansson GC. New Developments in Goblet Cell Mucus Secretion and Function. Mucosal Immunol (2015) 8:712–9. doi: 10.1038/mi.2015.32
10. Knoop KA, Newberry RD. Goblet Cells: Multifaceted Players in Immunity at Mucosal Surfaces. Mucosal Immunol (2018) 11:1551–7. doi: 10.1038/s41385-018-0039-y
11. Mcdole JR, Wheeler LW, Mcdonald KG, Wang B, Konjufca V, Knoop KA, et al. Goblet Cells Deliver Luminal Antigen to CD103+ Dendritic Cells in the Small Intestine. Nature (2012) 483:345–9. doi: 10.1038/nature10863
12. Mcdonald KG, Wheeler LW, Mcdole JR, Joerger S, Gustafsson JK, Kulkarni DH, et al. CCR6 Promotes Steady-State Mononuclear Phagocyte Association With the Intestinal Epithelium, Imprinting and Immune Surveillance. Immunology (2017) 152:613–27. doi: 10.1111/imm.12801
13. Worthington JJ, Reimann F, Gribble FM. Enteroendocrine Cells-Sensory Sentinels of the Intestinal Environment and Orchestrators of Mucosal Immunity. Mucosal Immunol (2018) 11:3–20. doi: 10.1038/mi.2017.73
14. Tsukahara T, Watanabe K, Watanabe T, Yamagami H, Sogawa M, Tanigawa T, et al. Tumor Necrosis Factor Alpha Decreases Glucagon-Like Peptide-2 Expression by Up-Regulating G-Protein-Coupled Receptor 120 in Crohn Disease. Am J Pathol (2015) 185:185–96. doi: 10.1016/j.ajpath.2014.09.010
15. Ellingsgaard H, Hauselmann I, Schuler B, Habib AM, Baggio LL, Meier DT, et al. Interleukin-6 Enhances Insulin Secretion by Increasing Glucagon-Like Peptide-1 Secretion From L Cells and Alpha Cells. Nat Med (2011) 17:1481–9. doi: 10.1038/nm.2513
16. Hadjiyanni I, Siminovitch KA, Danska JS, Drucker DJ. Glucagon-Like Peptide-1 Receptor Signalling Selectively Regulates Murine Lymphocyte Proliferation and Maintenance of Peripheral Regulatory T Cells. Diabetologia (2010) 53:730–40. doi: 10.1007/s00125-009-1643-x
17. Kishikawa S, Sato S, Kaneto S, Uchino S, Kohsaka S, Nakamura S, et al. Allograft Inflammatory Factor 1 Is a Regulator of Transcytosis in M Cells. Nat Commun (2017) 8:14509. doi: 10.1038/ncomms14509
18. Mishima T, Iwabuchi K, Fujii S, Tanaka SY, Ogura H, Watano-Miyata K, et al. Allograft Inflammatory Factor-1 Augments Macrophage Phagocytotic Activity and Accelerates the Progression of Atherosclerosis in ApoE-/- Mice. Int J Mol Med (2008) 21:181–7. doi: 10.3892/ijmm.21.2.181
19. Olivares-Villagomez D, Van Kaer L. Intestinal Intraepithelial Lymphocytes: Sentinels of the Mucosal Barrier. Trends Immunol (2018) 39:264–75. doi: 10.1016/j.it.2017.11.003
20. Bilate AM, London M, Castro TBR, Mesin L, Bortolatto J, Kongthong S, et al. T Cell Receptor Is Required for Differentiation, But Not Maintenance, of Intestinal CD4(+) Intraepithelial Lymphocytes. Immunity (2020) 53:1001–1014 e20. doi: 10.1016/j.immuni.2020.09.003
21. Denning TL, Norris BA, Medina-Contreras O, Manicassamy S, Geem D, Madan R, et al. Functional Specializations of Intestinal Dendritic Cell and Macrophage Subsets That Control Th17 and Regulatory T Cell Responses Are Dependent on the T Cell/APC Ratio, Source of Mouse Strain, and Regional Localization. J Immunol (2011) 187:733–47. doi: 10.4049/jimmunol.1002701
22. Watchmaker PB, Lahl K, Lee M, Baumjohann D, Morton J, Kim SJ, et al. Comparative Transcriptional and Functional Profiling Defines Conserved Programs of Intestinal DC Differentiation in Humans and Mice. Nat Immunol (2014) 15:98–108. doi: 10.1038/ni.2768
23. Knoop KA, Mcdonald KG, Mccrate S, Mcdole JR, Newberry RD. Microbial Sensing by Goblet Cells Controls Immune Surveillance of Luminal Antigens in the Colon. Mucosal Immunol (2015) 8:198–210. doi: 10.1038/mi.2014.58
24. Brandtzaeg P, Farstad IN, Johansen FE, Morton HC, Norderhaug IN, Yamanaka T. The B-Cell System of Human Mucosae and Exocrine Glands. Immunol Rev (1999) 171:45–87. doi: 10.1111/j.1600-065x.1999.tb01342.x
26. Conti JA, Kendall TJ, Bateman A, Armstrong TA, Papa-Adams A, Xu Q, et al. The Desmoplastic Reaction Surrounding Hepatic Colorectal Adenocarcinoma Metastases Aids Tumor Growth and Survival via Alphav Integrin Ligation. Clin Cancer Res (2008) 14:6405–13. doi: 10.1158/1078-0432.CCR-08-0816
27. Schober M, Jesenofsky R, Faissner R, Weidenauer C, Hagmann W, Michl P, et al. Desmoplasia and Chemoresistance in Pancreatic Cancer. Cancers (Basel) (2014) 6:2137–54. doi: 10.3390/cancers6042137
28. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
29. Wei SC, Fattet L, Tsai JH, Guo Y, Pai VH, Majeski HE, et al. Matrix Stiffness Drives Epithelial-Mesenchymal Transition and Tumour Metastasis Through a TWIST1-G3BP2 Mechanotransduction Pathway. Nat Cell Biol (2015) 17:678–88. doi: 10.1038/ncb3157
30. Henke E, Nandigama R, Ergun S. Extracellular Matrix in the Tumor Microenvironment and Its Impact on Cancer Therapy. Front Mol Biosci (2019) 6:160. doi: 10.3389/fmolb.2019.00160
31. Lu P, Weaver VM, Werb Z. The Extracellular Matrix: A Dynamic Niche in Cancer Progression. J Cell Biol (2012) 196:395–406. doi: 10.1083/jcb.201102147
32. Vossenaar ER, Zendman AJ, Van Venrooij WJ, Pruijn GJ. A Growing Family of Citrullinating Enzymes: Genes, Features and Involvement in Disease. Bioessays (2003) 25:1106–18. doi: 10.1002/bies.10357
33. Yuzhalin AE, Lim SY, Kutikhin AG, Gordon-Weeks AN. Dynamic Matrisome: ECM Remodeling Factors Licensing Cancer Progression and Metastasis. Biochim Biophys Acta Rev Cancer (2018) 1870:207–28. doi: 10.1016/j.bbcan.2018.09.002
34. Yuzhalin AE, Gordon-Weeks AN, Tognoli ML, Jones K, Markelc B, Konietzny R, et al. Colorectal Cancer Liver Metastatic Growth Depends on PAD4-Driven Citrullination of the Extracellular Matrix. Nat Commun (2018) 9:4783. doi: 10.1038/s41467-018-07306-7
35. Lowy CM, Oskarsson T. Tenascin C in Metastasis: A View From the Invasive Front. Cell Adh Migr (2015) 9:112–24. doi: 10.1080/19336918.2015.1008331
36. Murakami T, Kikuchi H, Ishimatsu H, Iino I, Hirotsu A, Matsumoto T, et al. Tenascin C in Colorectal Cancer Stroma Is a Predictive Marker for Liver Metastasis and Is a Potent Target of miR-198 as Identified by microRNA Analysis. Br J Cancer (2017) 117:1360–70. doi: 10.1038/bjc.2017.291
37. Voss H, Wurlitzer M, Smit DJ, Ewald F, Alawi M, Spohn M, et al. Differential Regulation of Extracellular Matrix Proteins in Three Recurrent Liver Metastases of a Single Patient With Colorectal Cancer. Clin Exp Metastasis (2020) 37:649–56. doi: 10.1007/s10585-020-10058-8
38. Lagory EL, Giaccia AJ. The Ever-Expanding Role of HIF in Tumour and Stromal Biology. Nat Cell Biol (2016) 18:356–65. doi: 10.1038/ncb3330
39. De Palma M, Biziato D, Petrova TV. Microenvironmental Regulation of Tumour Angiogenesis. Nat Rev Cancer (2017) 17:457–74. doi: 10.1038/nrc.2017.51
40. Ferrarotto R, Hoff PM. Antiangiogenic Drugs for Colorectal Cancer: Exploring New Possibilities. Clin Colorectal Cancer (2013) 12:1–7. doi: 10.1016/j.clcc.2012.06.002
41. Ellis LM, Takahashi Y, Liu W, Shaheen RM. Vascular Endothelial Growth Factor in Human Colon Cancer: Biology and Therapeutic Implications. Oncologist (2000) 5 Suppl 1:11–5. doi: 10.1634/theoncologist.5-suppl_1-11
42. Rmali KA, Puntis MC, Jiang WG. Tumour-Associated Angiogenesis in Human Colorectal Cancer. Colorectal Dis (2007) 9:3–14. doi: 10.1111/j.1463-1318.2006.01089.x
43. Witte D, Thomas A, Ali N, Carlson N, Younes M. Expression of the Vascular Endothelial Growth Factor Receptor-3 (VEGFR-3) and its Ligand VEGF-C in Human Colorectal Adenocarcinoma. Anticancer Res (2002) 22:1463–6.
44. Denton AE, Roberts EW, Fearon DT. Stromal Cells in the Tumor Microenvironment. Adv Exp Med Biol (2018) 1060:99–114. doi: 10.1007/978-3-319-78127-3_6
45. Jungwirth U, Van Weverwijk A, Evans RJ, Jenkins L, Vicente D, Alexander J, et al. Impairment of a Distinct Cancer-Associated Fibroblast Population Limits Tumour Growth and Metastasis. Nat Commun (2021) 12:3516. doi: 10.1038/s41467-021-23583-1
46. Yin C, Evason KJ, Asahina K, Stainier DY. Hepatic Stellate Cells in Liver Development, Regeneration, and Cancer. J Clin Invest (2013) 123:1902–10. doi: 10.1172/JCI66369
47. Kidd S, Spaeth E, Watson K, Burks J, Lu H, Klopp A, et al. Origins of the Tumor Microenvironment: Quantitative Assessment of Adipose-Derived and Bone Marrow-Derived Stroma. PloS One (2012) 7:e30563. doi: 10.1371/journal.pone.0030563
48. Zeisberg EM, Potenta S, Xie L, Zeisberg M, Kalluri R. Discovery of Endothelial to Mesenchymal Transition as a Source for Carcinoma-Associated Fibroblasts. Cancer Res (2007) 67:10123–8. doi: 10.1158/0008-5472.CAN-07-3127
49. Paauwe M, Schoonderwoerd MJA, Helderman R, Harryvan TJ, Groenewoud A, Van Pelt GW, et al. Endoglin Expression on Cancer-Associated Fibroblasts Regulates Invasion and Stimulates Colorectal Cancer Metastasis. Clin Cancer Res (2018) 24:6331–44. doi: 10.1158/1078-0432.CCR-18-0329
50. Hu JL, Wang W, Lan XL, Zeng ZC, Liang YS, Yan YR, et al. CAFs Secreted Exosomes Promote Metastasis and Chemotherapy Resistance by Enhancing Cell Stemness and Epithelial-Mesenchymal Transition in Colorectal Cancer. Mol Cancer (2019) 18:91. doi: 10.1186/s12943-019-1019-x
51. Heichler C, Scheibe K, Schmied A, Geppert CI, Schmid B, Wirtz S, et al. STAT3 Activation Through IL-6/IL-11 in Cancer-Associated Fibroblasts Promotes Colorectal Tumour Development and Correlates With Poor Prognosis. Gut (2020) 69:1269–82. doi: 10.1136/gutjnl-2019-319200
52. Ham IH, Lee D, Hur H. Cancer-Associated Fibroblast-Induced Resistance to Chemotherapy and Radiotherapy in Gastrointestinal Cancers. Cancers (Basel) (2021) 13:1172. doi: 10.3390/cancers13051172
53. Cassetta L, Pollard JW. Tumor-Associated Macrophages. Curr Biol (2020) 30:R246–8. doi: 10.1016/j.cub.2020.01.031
54. Chen C, He W, Huang J, Wang B, Li H, Cai Q, et al. LNMAT1 Promotes Lymphatic Metastasis of Bladder Cancer via CCL2 Dependent Macrophage Recruitment. Nat Commun (2018) 9:3826. doi: 10.1038/s41467-018-06152-x
55. Stockmann C, Doedens A, Weidemann A, Zhang N, Takeda N, Greenberg JI, et al. Deletion of Vascular Endothelial Growth Factor in Myeloid Cells Accelerates Tumorigenesis. Nature (2008) 456:814–8. doi: 10.1038/nature07445
56. Kessenbrock K, Plaks V, Werb Z. Matrix Metalloproteinases: Regulators of the Tumor Microenvironment. Cell (2010) 141:52–67. doi: 10.1016/j.cell.2010.03.015
57. Squadrito ML, De Palma M. Macrophage Regulation of Tumor Angiogenesis: Implications for Cancer Therapy. Mol Aspects Med (2011) 32:123–45. doi: 10.1016/j.mam.2011.04.005
58. Fu XT, Dai Z, Song K, Zhang ZJ, Zhou ZJ, Zhou SL, et al. Macrophage-Secreted IL-8 Induces Epithelial-Mesenchymal Transition in Hepatocellular Carcinoma Cells by Activating the JAK2/STAT3/Snail Pathway. Int J Oncol (2015) 46:587–96. doi: 10.3892/ijo.2014.2761
59. Wei C, Yang C, Wang S, Shi D, Zhang C, Lin X, et al. Crosstalk Between Cancer Cells and Tumor Associated Macrophages Is Required for Mesenchymal Circulating Tumor Cell-Mediated Colorectal Cancer Metastasis. Mol Cancer (2019) 18:64. doi: 10.1186/s12943-019-0976-4
60. Afik R, Zigmond E, Vugman M, Klepfish M, Shimshoni E, Pasmanik-Chor M, et al. Tumor Macrophages Are Pivotal Constructors of Tumor Collagenous Matrix. J Exp Med (2016) 213:2315–31. doi: 10.1084/jem.20151193
61. Marech I, Ammendola M, Sacco R, Sammarco G, Zuccala V, Zizzo N, et al. Tumour-Associated Macrophages Correlate With Microvascular Bed Extension in Colorectal Cancer Patients. J Cell Mol Med (2016) 20:1373–80. doi: 10.1111/jcmm.12826
62. Nakayama Y, Nagashima N, Minagawa N, Inoue Y, Katsuki T, Onitsuka K, et al. Relationships Between Tumor-Associated Macrophages and Clinicopathological Factors in Patients With Colorectal Cancer. Anticancer Res (2002) 22:4291–6.
63. Koelzer VH, Canonica K, Dawson H, Sokol L, Karamitopoulou-Diamantis E, Lugli A, et al. Phenotyping of Tumor-Associated Macrophages in Colorectal Cancer: Impact on Single Cell Invasion (Tumor Budding) and Clinicopathological Outcome. Oncoimmunology (2016) 5:e1106677. doi: 10.1080/2162402X.2015.1106677
64. Cavnar MJ, Turcotte S, Katz SC, Kuk D, Gonen M, Shia J, et al. Tumor-Associated Macrophage Infiltration in Colorectal Cancer Liver Metastases Is Associated With Better Outcome. Ann Surg Oncol (2017) 24:1835–42. doi: 10.1245/s10434-017-5812-8
65. Yang C, Wei C, Wang S, Shi D, Zhang C, Lin X, et al. Elevated CD163(+)/CD68(+) Ratio at Tumor Invasive Front Is Closely Associated With Aggressive Phenotype and Poor Prognosis in Colorectal Cancer. Int J Biol Sci (2019) 15:984–98. doi: 10.7150/ijbs.29836
66. Veglia F, Perego M, Gabrilovich D. Myeloid-Derived Suppressor Cells Coming of Age. Nat Immunol (2018) 19:108–19. doi: 10.1038/s41590-017-0022-x
67. Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of Monocytes, Macrophages, and Dendritic Cells. Science (2010) 327:656–61. doi: 10.1126/science.1178331
68. Veglia F, Gabrilovich DI. Dendritic Cells in Cancer: The Role Revisited. Curr Opin Immunol (2017) 45:43–51. doi: 10.1016/j.coi.2017.01.002
69. Gabrilovich DI, Bronte V, Chen SH, Colombo MP, Ochoa A, Ostrand-Rosenberg S, et al. The Terminology Issue for Myeloid-Derived Suppressor Cells. Cancer Res (2007) 67:425; author reply 426. doi: 10.1158/0008-5472.CAN-06-3037
70. Dysthe M, Parihar R. Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Adv Exp Med Biol (2020) 1224:117–40. doi: 10.1007/978-3-030-35723-8_8
71. Shi H, Zhang J, Han X, Li H, Xie M, Sun Y, et al. Recruited Monocytic Myeloid-Derived Suppressor Cells Promote the Arrest of Tumor Cells in the Premetastatic Niche Through an IL-1beta-Mediated Increase in E-Selectin Expression. Int J Cancer (2017) 140:1370–83. doi: 10.1002/ijc.30538
72. Sasano T, Mabuchi S, Kozasa K, Kuroda H, Kawano M, Takahashi R, et al. The Highly Metastatic Nature of Uterine Cervical/Endometrial Cancer Displaying Tumor-Related Leukocytosis: Clinical and Preclinical Investigations. Clin Cancer Res (2018) 24:4018–29. doi: 10.1158/1078-0432.CCR-17-2472
73. Chun E, Lavoie S, Michaud M, Gallini CA, Kim J, Soucy G, et al. CCL2 Promotes Colorectal Carcinogenesis by Enhancing Polymorphonuclear Myeloid-Derived Suppressor Cell Population and Function. Cell Rep (2015) 12:244–57. doi: 10.1016/j.celrep.2015.06.024
74. Gabrilovich DI, Nagaraj S. Myeloid-Derived Suppressor Cells as Regulators of the Immune System. Nat Rev Immunol (2009) 9:162–74. doi: 10.1038/nri2506
75. Xu P, He H, Gu Y, Wang Y, Sun Z, Yang L, et al. Surgical Trauma Contributes to Progression of Colon Cancer by Downregulating CXCL4 and Recruiting MDSCs. Exp Cell Res (2018) 370:692–8. doi: 10.1016/j.yexcr.2018.07.035
76. Ouyang LY, Wu XJ, Ye SB, Zhang RX, Li ZL, Liao W, et al. Tumor-Induced Myeloid-Derived Suppressor Cells Promote Tumor Progression Through Oxidative Metabolism in Human Colorectal Cancer. J Transl Med (2015) 13:47. doi: 10.1186/s12967-015-0410-7
77. Leliefeld PH, Wessels CM, Leenen LP, Koenderman L, Pillay J. The Role of Neutrophils in Immune Dysfunction During Severe Inflammation. Crit Care (2016) 20:73. doi: 10.1186/s13054-016-1250-4
78. Irimia D, Balazsi G, Agrawal N, Toner M. Adaptive-Control Model for Neutrophil Orientation in the Direction of Chemical Gradients. Biophys J (2009) 96:3897–916. doi: 10.1016/j.bpj.2008.12.3967
79. Ji H, Houghton AM, Mariani TJ, Perera S, Kim CB, Padera R, et al. K-Ras Activation Generates an Inflammatory Response in Lung Tumors. Oncogene (2006) 25:2105–12. doi: 10.1038/sj.onc.1209237
80. Uribe-Querol E, Rosales C. Neutrophils in Cancer: Two Sides of the Same Coin. J Immunol Res (2015) 2015:983698. doi: 10.1155/2015/983698
81. Mizuno R, Kawada K, Itatani Y, Ogawa R, Kiyasu Y, Sakai Y. The Role of Tumor-Associated Neutrophils in Colorectal Cancer. Int J Mol Sci (2019) 20:529. doi: 10.3390/ijms20030529
82. Rao HL, Chen JW, Li M, Xiao YB, Fu J, Zeng YX, et al. Increased Intratumoral Neutrophil in Colorectal Carcinomas Correlates Closely With Malignant Phenotype and Predicts Patients' Adverse Prognosis. PloS One (2012) 7:e30806. doi: 10.1371/journal.pone.0030806
83. Galdiero MR, Bianchi P, Grizzi F, Di Caro G, Basso G, Ponzetta A, et al. Occurrence and Significance of Tumor-Associated Neutrophils in Patients With Colorectal Cancer. Int J Cancer (2016) 139:446–56. doi: 10.1002/ijc.30076
84. Berry RS, Xiong MJ, Greenbaum A, Mortaji P, Nofchissey RA, Schultz F, et al. High Levels of Tumor-Associated Neutrophils Are Associated With Improved Overall Survival in Patients With Stage II Colorectal Cancer. PloS One (2017) 12:e0188799. doi: 10.1371/journal.pone.0188799
85. Ishizuka M, Nagata H, Takagi K, Iwasaki Y, Kubota K. Combination of Platelet Count and Neutrophil to Lymphocyte Ratio Is a Useful Predictor of Postoperative Survival in Patients With Colorectal Cancer. Br J Cancer (2013) 109:401–7. doi: 10.1038/bjc.2013.350
86. Li Z, Zhao R, Cui Y, Zhou Y, Wu X. The Dynamic Change of Neutrophil to Lymphocyte Ratio Can Predict Clinical Outcome in Stage I-III Colon Cancer. Sci Rep (2018) 8:9453. doi: 10.1038/s41598-018-27896-y
87. Borazan E, Balik AA, Bozdag Z, Arik MK, Aytekin A, Yilmaz L, et al. Assessment of the Relationship Between Neutrophil Lymphocyte Ratio and Prognostic Factors in Non-Metastatic Colorectal Cancer. Turk J Surg (2017) 33:185–9. doi: 10.5152/turkjsurg.2017.3528
88. Halle S, Halle O, Forster R. Mechanisms and Dynamics of T Cell-Mediated Cytotoxicity In Vivo. Trends Immunol (2017) 38:432–43. doi: 10.1016/j.it.2017.04.002
89. Dominguez-Villar M, Hafler D. A Regulatory T Cells in Autoimmune Disease. Nat Immunol (2018) 19:665–73. doi: 10.1038/s41590-018-0120-4
90. Stockis J, Roychoudhuri R, Halim TYF. Regulation of Regulatory T Cells in Cancer. Immunology (2019) 157:219–31. doi: 10.1111/imm.13064
91. Shitara K, Nishikawa H. Regulatory T Cells: A Potential Target in Cancer Immunotherapy. Ann N Y Acad Sci (2018) 1417:104–15. doi: 10.1111/nyas.13625
92. Takeuchi Y, Nishikawa H. Roles of Regulatory T Cells in Cancer Immunity. Int Immunol (2016) 28:401–9. doi: 10.1093/intimm/dxw025
93. Marshall EA, Ng KW, Kung SH, Conway EM, Martinez VD, Halvorsen EC, et al. Emerging Roles of T Helper 17 and Regulatory T Cells in Lung Cancer Progression and Metastasis. Mol Cancer (2016) 15:67. doi: 10.1186/s12943-016-0551-1
94. Luen SJ, Salgado R, Dieci MV, Vingiani A, Curigliano G, Gould RE, et al. Prognostic Implications of Residual Disease Tumor-Infiltrating Lymphocytes and Residual Cancer Burden in Triple-Negative Breast Cancer Patients After Neoadjuvant Chemotherapy. Ann Oncol (2019) 30:236–42. doi: 10.1093/annonc/mdy547
95. Shen Z, Zhou S, Wang Y, Li RL, Zhong C, Liang C, et al. Higher Intratumoral Infiltrated Foxp3+ Treg Numbers and Foxp3+/CD8+ Ratio Are Associated With Adverse Prognosis in Resectable Gastric Cancer. J Cancer Res Clin Oncol (2010) 136:1585–95. doi: 10.1007/s00432-010-0816-9
96. Zhou SL, Zhou ZJ, Hu ZQ, Huang XW, Wang Z, Chen EB, et al. Tumor-Associated Neutrophils Recruit Macrophages and T-Regulatory Cells to Promote Progression of Hepatocellular Carcinoma and Resistance to Sorafenib. Gastroenterology (2016) 150:1646–1658.e17. doi: 10.1053/j.gastro.2016.02.040
97. Kouketsu A, Sato I, Oikawa M, Shimizu Y, Saito H, Tashiro K, et al. Regulatory T Cells and M2-Polarized Tumour-Associated Macrophages Are Associated With the Oncogenesis and Progression of Oral Squamous Cell Carcinoma. Int J Oral Maxillofac Surg (2019) 48:1279–88. doi: 10.1016/j.ijom.2019.04.004
98. Frey DM, Droeser RA, Viehl CT, Zlobec I, Lugli A, Zingg U, et al. High Frequency of Tumor-Infiltrating FOXP3(+) Regulatory T Cells Predicts Improved Survival in Mismatch Repair-Proficient Colorectal Cancer Patients. Int J Cancer (2010) 126:2635–43. doi: 10.1002/ijc.24989
99. Hanke T, Melling N, Simon R, Sauter G, Bokemeyer C, Lebok P, et al. High Intratumoral FOXP3(+) T Regulatory Cell (Tregs) Density Is an Independent Good Prognosticator in Nodal Negative Colorectal Cancer. Int J Clin Exp Pathol (2015) 8:8227–35.
100. Vlad C, Kubelac P, Fetica B, Vlad D, Irimie A, Achimas-Cadariu P. The Prognostic Value of FOXP3+ T Regulatory Cells in Colorectal Cancer. J BUON (2015) 20:114–9.
101. Shang B, Liu Y, Jiang SJ, Liu Y. Prognostic Value of Tumor-Infiltrating FoxP3+ Regulatory T Cells in Cancers: A Systematic Review and Meta-Analysis. Sci Rep (2015) 5:15179. doi: 10.1038/srep15179
102. Deng G, Song X, Fujimoto S, Piccirillo CA, Nagai Y, Greene MI. Foxp3 Post-Translational Modifications and Treg Suppressive Activity. Front Immunol (2019) 10:2486. doi: 10.3389/fimmu.2019.02486
103. Tadmor T, Zhang Y, Cho HM, Podack ER, Rosenblatt JD. The Absence of B Lymphocytes Reduces the Number and Function of T-Regulatory Cells and Enhances the Anti-Tumor Response in a Murine Tumor Model. Cancer Immunol Immunother (2011) 60:609–19. doi: 10.1007/s00262-011-0972-z
104. Xiao X, Lao XM, Chen MM, Liu RX, Wei Y, Ouyang FZ, et al. PD-1hi Identifies a Novel Regulatory B-Cell Population in Human Hepatoma That Promotes Disease Progression. Cancer Discov (2016) 6:546–59. doi: 10.1158/2159-8290.CD-15-1408
105. Dilillo DJ, Yanaba K, Tedder TF. B Cells Are Required for Optimal CD4+ and CD8+ T Cell Tumor Immunity: Therapeutic B Cell Depletion Enhances B16 Melanoma Growth in Mice. J Immunol (2010) 184:4006–16. doi: 10.4049/jimmunol.0903009
106. Sarvaria A, Madrigal JA, Saudemont A. B Cell Regulation in Cancer and Anti-Tumor Immunity. Cell Mol Immunol (2017) 14:662–74. doi: 10.1038/cmi.2017.35
107. Sorrentino R, Morello S, Forte G, Montinaro A, De Vita G, Luciano A, et al. B Cells Contribute to the Antitumor Activity of CpG-Oligodeoxynucleotide in a Mouse Model of Metastatic Lung Carcinoma. Am J Respir Crit Care Med (2011) 183:1369–79. doi: 10.1164/rccm.201010-1738OC
108. Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, et al. Tertiary Lymphoid Structures Improve Immunotherapy and Survival in Melanoma. Nature (2020) 577:561–5. doi: 10.1038/s41586-019-1914-8
109. Zirakzadeh AA, Marits P, Sherif A, Winqvist O. Multiplex B Cell Characterization in Blood, Lymph Nodes, and Tumors From Patients With Malignancies. J Immunol (2013) 190:5847–55. doi: 10.4049/jimmunol.1203279
110. Shimabukuro-Vornhagen A, Schlosser HA, Gryschok L, Malcher J, Wennhold K, Garcia-Marquez M, et al. Characterization of Tumor-Associated B-Cell Subsets in Patients With Colorectal Cancer. Oncotarget (2014) 5:4651–64. doi: 10.18632/oncotarget.1701
111. Guo L, Wang C, Qiu X, Pu X, Chang P. Colorectal Cancer Immune Infiltrates: Significance in Patient Prognosis and Immunotherapeutic Efficacy. Front Immunol (2020) 11:1052. doi: 10.3389/fimmu.2020.01052
112. Berntsson J, Nodin B, Eberhard J, Micke P, Jirstrom K. Prognostic Impact of Tumour-Infiltrating B Cells and Plasma Cells in Colorectal Cancer. Int J Cancer (2016) 139:1129–39. doi: 10.1002/ijc.30138
113. Wang W, Zhong Y, Zhuang Z, Xie J, Lu Y, Huang C, et al. Multiregion Single-Cell Sequencing Reveals the Transcriptional Landscape of the Immune Microenvironment of Colorectal Cancer. Clin Transl Med (2021) 11:e253. doi: 10.1002/ctm2.253
114. Reid FSW, Egoroff N, Pockney PG, Smith SR. A Systematic Scoping Review on Natural Killer Cell Function in Colorectal Cancer. Cancer Immunol Immunother (2021) 70:597–606. doi: 10.1007/s00262-020-02721-6
115. Halama N, Braun M, Kahlert C, Spille A, Quack C, Rahbari N, et al. Natural Killer Cells Are Scarce in Colorectal Carcinoma Tissue Despite High Levels of Chemokines and Cytokines. Clin Cancer Res (2011) 17:678–89. doi: 10.1158/1078-0432.CCR-10-2173
116. Rocca YS, Roberti MP, Arriaga JM, Amat M, Bruno L, Pampena MB, et al. Altered Phenotype in Peripheral Blood and Tumor-Associated NK Cells From Colorectal Cancer Patients. Innate Immun (2013) 19:76–85. doi: 10.1177/1753425912453187
117. Krijgsman D, Roelands J, Andersen MN, Wieringa C, Tollenaar R, Hendrickx W, et al. Expression of NK Cell Receptor Ligands in Primary Colorectal Cancer Tissue in Relation to the Phenotype of Circulating NK- and NKT Cells, and Clinical Outcome. Mol Immunol (2020) 128:205–18. doi: 10.1016/j.molimm.2020.10.012
118. Veluchamy JP, Spanholtz J, Tordoir M, Thijssen VL, Heideman DA, Verheul HM, et al. Combination of NK Cells and Cetuximab to Enhance Anti-Tumor Responses in RAS Mutant Metastatic Colorectal Cancer. PloS One (2016) 11:e0157830. doi: 10.1371/journal.pone.0157830
119. Ishikawa T, Okayama T, Sakamoto N, Ideno M, Oka K, Enoki T, et al. Phase I Clinical Trial of Adoptive Transfer of Expanded Natural Killer Cells in Combination With IgG1 Antibody in Patients With Gastric or Colorectal Cancer. Int J Cancer (2018) 142:2599–609. doi: 10.1002/ijc.31285
120. Xiao L, Cen D, Gan H, Sun Y, Huang N, Xiong H, et al. Adoptive Transfer of NKG2D CAR mRNA-Engineered Natural Killer Cells in Colorectal Cancer Patients. Mol Ther (2019) 27:1114–25. doi: 10.1016/j.ymthe.2019.03.011
121. Marechal R, De Schutter J, Nagy N, Demetter P, Lemmers A, Deviere J, et al. Putative Contribution of CD56 Positive Cells in Cetuximab Treatment Efficacy in First-Line Metastatic Colorectal Cancer Patients. BMC Cancer (2010) 10:340. doi: 10.1186/1471-2407-10-340
122. Van Kaer L. NKT cells: T Lymphocytes With Innate Effector Functions. Curr Opin Immunol. (2007) 19:354–64. doi: 10.1016/j.coi.2007.03.001
123. Coles MC, Raulet DH. NK1.1+ T Cells in the Liver Arise in the Thymus and Are Selected by Interactions With Class I Molecules on CD4+CD8+ Cells. J Immunol (2000) 164:2412–8. doi: 10.4049/jimmunol.164.5.2412
124. Gerth E, Mattner J. The Role of Adaptor Proteins in the Biology of Natural Killer T (NKT) Cells. Front Immunol (2019) 10:1449. doi: 10.3389/fimmu.2019.01449
125. Brennan PJ, Brigl M, Brenner MB. Invariant Natural Killer T Cells: An Innate Activation Scheme Linked to Diverse Effector Functions. Nat Rev Immunol (2013) 13:101–17. doi: 10.1038/nri3369
126. Kinjo Y, Illarionov P, Vela JL, Pei B, Girardi E, Li X, et al. Invariant Natural Killer T Cells Recognize Glycolipids From Pathogenic Gram-Positive Bacteria. Nat Immunol (2011) 12:966–74. doi: 10.1038/ni.2096
127. Kumar V, Delovitch TL. Different Subsets of Natural Killer T Cells may Vary in Their Roles in Health and Disease. Immunology (2014) 142:321–36. doi: 10.1111/imm.12247
128. Wang Y, Bhave MS, Yagita H, Cardell SL. Natural Killer T-Cell Agonist Alpha-Galactosylceramide and PD-1 Blockade Synergize to Reduce Tumor Development in a Preclinical Model of Colon Cancer. Front Immunol (2020) 11:581301. doi: 10.3389/fimmu.2020.581301
129. Tachibana T, Onodera H, Tsuruyama T, Mori A, Nagayama S, Hiai H, et al. Increased Intratumor Valpha24-Positive Natural Killer T Cells: A Prognostic Factor for Primary Colorectal Carcinomas. Clin Cancer Res (2005) 11:7322–7. doi: 10.1158/1078-0432.CCR-05-0877
130. Xu R, Rai A, Chen M, Suwakulsiri W, Greening DW, Simpson RJ. Extracellular Vesicles in Cancer - Implications for Future Improvements in Cancer Care. Nat Rev Clin Oncol (2018) 15:617–38. doi: 10.1038/s41571-018-0036-9
131. Khan S, Jutzy JM, Valenzuela MM, Turay D, Aspe JR, Ashok A, et al. Plasma-Derived Exosomal Survivin, a Plausible Biomarker for Early Detection of Prostate Cancer. PloS One (2012) 7:e46737. doi: 10.1371/journal.pone.0046737
132. Lasser C, Alikhani VS, Ekstrom K, Eldh M, Paredes PT, Bossios A, et al. Human Saliva, Plasma and Breast Milk Exosomes Contain RNA: Uptake by Macrophages. J Transl Med (2011) 9:9. doi: 10.1186/1479-5876-9-9
133. Admyre C, Johansson SM, Qazi KR, Filen JJ, Lahesmaa R, Norman M, et al. Exosomes With Immune Modulatory Features Are Present in Human Breast Milk. J Immunol (2007) 179:1969–78. doi: 10.4049/jimmunol.179.3.1969
134. Yun B, Kim Y, Park DJ, Oh S. Comparative Analysis of Dietary Exosome-Derived microRNAs From Human, Bovine and Caprine Colostrum and Mature Milk. J Anim Sci Technol (2021) 63:593–602. doi: 10.5187/jast.2021.e39
135. Street JM, Barran PE, Mackay CL, Weidt S, Balmforth C, Walsh TS, et al. Identification and Proteomic Profiling of Exosomes in Human Cerebrospinal Fluid. J Transl Med (2012) 10:5. doi: 10.1186/1479-5876-10-5
136. Malenica M, Vukomanovic M, Kurtjak M, Masciotti V, Dal Zilio S, Greco S, et al. Perspectives of Microscopy Methods for Morphology Characterisation of Extracellular Vesicles From Human Biofluids. Biomedicines (2021) 9:603. doi: 10.3390/biomedicines9060603
137. Hassanzadeh A, Rahman HS, Markov A, Endjun JJ, Zekiy AO, Chartrand MS, et al. Mesenchymal Stem/Stromal Cell-Derived Exosomes in Regenerative Medicine and Cancer; Overview of Development, Challenges, and Opportunities. Stem Cell Res Ther (2021) 12:297. doi: 10.1186/s13287-021-02378-7
138. Pisitkun T, Shen RF, Knepper M. A Identification and Proteomic Profiling of Exosomes in Human Urine. Proc Natl Acad Sci USA (2004) 101:13368–73. doi: 10.1073/pnas.0403453101
139. Gonzales PA, Pisitkun T, Hoffert JD, Tchapyjnikov D, Star RA, Kleta R, et al. Large-Scale Proteomics and Phosphoproteomics of Urinary Exosomes. J Am Soc Nephrol (2009) 20:363–79. doi: 10.1681/ASN.2008040406
140. Sung CC, Chen MH, Lin YC, Lin YC, Lin YJ, Yang SS, et al. Urinary Extracellular Vesicles for Renal Tubular Transporters Expression in Patients With Gitelman Syndrome. Front Med (Lausanne) (2021) 8:679171. doi: 10.3389/fmed.2021.679171
141. Morup N, Stakaitis R, Golubickaite I, Riera M, Dalgaard MD, Schierup MH, et al. Small RNAs in Seminal Plasma as Novel Biomarkers for Germ Cell Tumors. Cancers (Basel) (2021) 13:2346. doi: 10.3390/cancers13102346
142. Silva J, Garcia V, Rodriguez M, Compte M, Cisneros E, Veguillas P, et al. Analysis of Exosome Release and its Prognostic Value in Human Colorectal Cancer. Genes Chromosomes Cancer (2012) 51:409–18. doi: 10.1002/gcc.21926
143. Que R, Ding G, Chen J, Cao L. Analysis of Serum Exosomal microRNAs and Clinicopathologic Features of Patients With Pancreatic Adenocarcinoma. World J Surg Oncol (2013) 11:219. doi: 10.1186/1477-7819-11-219
144. Xue X, Wang C, Xue Z, Wen J, Han J, Ma X, et al. Exosomal miRNA Profiling Before and After Surgery Revealed Potential Diagnostic and Prognostic Markers for Lung Adenocarcinoma. Acta Biochim Biophys Sin (Shanghai) (2020) 52:281–93. doi: 10.1093/abbs/gmz164
145. Lessi F, Aretini P, Rizzo M, Morelli M, Menicagli M, Franceschi S, et al. Analysis of Exosome-Derived microRNAs Reveals Insights of Intercellular Communication During Invasion of Breast, Prostate and Glioblastoma Cancer Cells. Cell Adh Migr (2021) 15:180–201. doi: 10.1080/19336918.2021.1935407
146. Fan X, Zou X, Liu C, Cheng W, Zhang S, Geng X, et al. MicroRNA Expression Profile in Serum Reveals Novel Diagnostic Biomarkers for Endometrial Cancer. Biosci Rep (2021) 41:BSR20210111. doi: 10.1042/BSR20210111
147. Zeng Z, Li Y, Pan Y, Lan X, Song F, Sun J, et al. Cancer-Derived Exosomal miR-25-3p Promotes Pre-Metastatic Niche Formation by Inducing Vascular Permeability and Angiogenesis. Nat Commun (2018) 9:5395. doi: 10.1038/s41467-018-07810-w
148. Liu H, Liu Y, Sun P, Leng K, Xu Y, Mei L, et al. Colorectal Cancer-Derived Exosomal miR-106b-3p Promotes Metastasis by Down-Regulating DLC-1 Expression. Clin Sci (Lond) (2020) 134:419–34. doi: 10.1042/CS20191087
149. Jiang Y, Ji X, Liu K, Shi Y, Wang C, Li Y, et al. Exosomal miR-200c-3p Negatively Regulates the Migraion and Invasion of Lipopolysaccharide (LPS)-Stimulated Colorectal Cancer (CRC). BMC Mol Cell Biol (2020) 21:48. doi: 10.1186/s12860-020-00291-0
150. Shang A, Gu C, Wang W, Wang X, Sun J, Zeng B, et al. Exosomal circPACRGL Promotes Progression of Colorectal Cancer via the miR-142-3p/miR-506-3p- TGF-Beta1 Axis. Mol Cancer (2020) 19:117. doi: 10.1186/s12943-020-01235-0
151. Bhome R, Goh RW, Bullock MD, Pillar N, Thirdborough SM, Mellone M, et al. Exosomal microRNAs Derived From Colorectal Cancer-Associated Fibroblasts: Role in Driving Cancer Progression. Aging (Albany NY) (2017) 9:2666–94. doi: 10.18632/aging.101355
152. Maminezhad H, Ghanadian S, Pakravan K, Razmara E, Rouhollah F, Mossahebi-Mohammadi M, et al. A Panel of Six-Circulating miRNA Signature in Serum and its Potential Diagnostic Value in Colorectal Cancer. Life Sci (2020) 258:118226. doi: 10.1016/j.lfs.2020.118226
153. Wan Z, Gao X, Dong Y, Zhao Y, Chen X, Yang G, et al. Exosome-Mediated Cell-Cell Communication in Tumor Progression. Am J Cancer Res (2018) 8:1661–73.
154. Fane M, Weeraratna AT. How the Ageing Microenvironment Influences Tumour Progression. Nat Rev Cancer (2020) 20:89–106. doi: 10.1038/s41568-019-0222-9
155. Larbi A, Fulop T. From "Truly Naive" to "Exhausted Senescent" T Cells: When Markers Predict Functionality. Cytometry A (2014) 85:25–35. doi: 10.1002/cyto.a.22351
156. Giunco S, Petrara MR, Bergamo F, Del Bianco P, Zanchetta M, Carmona F, et al. Immune Senescence and Immune Activation in Elderly Colorectal Cancer Patients. Aging (Albany NY) (2019) 11:3864–75. doi: 10.18632/aging.102022
157. Di J, Liu M, Fan Y, Gao P, Wang Z, Jiang B, et al. Phenotype Molding of T Cells in Colorectal Cancer by Single-Cell Analysis. Int J Cancer (2020) 146:2281–95. doi: 10.1002/ijc.32856
158. Rosen LS, Jacobs IA, Burkes RL. Bevacizumab in Colorectal Cancer: Current Role in Treatment and the Potential of Biosimilars. Target Oncol (2017) 12:599–610. doi: 10.1007/s11523-017-0518-1
159. Modest DP, Pant S, Sartore-Bianchi A. Treatment Sequencing in Metastatic Colorectal Cancer. Eur J Cancer (2019) 109:70–83. doi: 10.1016/j.ejca.2018.12.019
160. Roviello G, Bachelot T, Hudis CA, Curigliano G, Reynolds AR, Petrioli R, et al. The Role of Bevacizumab in Solid Tumours: A Literature Based Meta-Analysis of Randomised Trials. Eur J Cancer (2017) 75:245–58. doi: 10.1016/j.ejca.2017.01.026
161. Cremolini C, Loupakis F, Antoniotti C, Lupi C, Sensi E, Lonardi S, et al. FOLFOXIRI Plus Bevacizumab Versus FOLFIRI Plus Bevacizumab as First-Line Treatment of Patients With Metastatic Colorectal Cancer: Updated Overall Survival and Molecular Subgroup Analyses of the Open-Label, Phase 3 TRIBE Study. Lancet Oncol (2015) 16:1306–15. doi: 10.1016/S1470-2045(15)00122-9
162. Botrel TEA, Clark LGO, Paladini L, Clark OAC. Efficacy and Safety of Bevacizumab Plus Chemotherapy Compared to Chemotherapy Alone in Previously Untreated Advanced or Metastatic Colorectal Cancer: A Systematic Review and Meta-Analysis. BMC Cancer (2016) 16:677. doi: 10.1186/s12885-016-2734-y
163. Bang YH, Kim JE, Lee JS, Kim SY, Kim KP, Kim TW, et al. Bevacizumab Plus Capecitabine as Later-Line Treatment for Patients With Metastatic Colorectal Cancer Refractory to Irinotecan, Oxaliplatin, and Fluoropyrimidines. Sci Rep (2021) 11:7118. doi: 10.1038/s41598-021-86482-x
164. Ramjiawan RR, Griffioen AW, Duda DG. Anti-Angiogenesis for Cancer Revisited: Is There a Role for Combinations With Immunotherapy? Angiogenesis (2017) 20:185–204. doi: 10.1007/s10456-017-9552-y
165. Comunanza V, Bussolino F. Therapy for Cancer: Strategy of Combining Anti-Angiogenic and Target Therapies. Front Cell Dev Biol (2017) 5:101. doi: 10.3389/fcell.2017.00101
166. Chen DS, Hurwitz H. Combinations of Bevacizumab With Cancer Immunotherapy. Cancer J (2018) 24:193–204. doi: 10.1097/PPO.0000000000000327
167. Wallin JJ, Bendell JC, Funke R, Sznol M, Korski K, Jones S, et al. Atezolizumab in Combination With Bevacizumab Enhances Antigen-Specific T-Cell Migration in Metastatic Renal Cell Carcinoma. Nat Commun (2016) 7:12624. doi: 10.1038/ncomms12624
168. Osada T, Chong G, Tansik R, Hong T, Spector N, Kumar R, et al. The Effect of Anti-VEGF Therapy on Immature Myeloid Cell and Dendritic Cells in Cancer Patients. Cancer Immunol Immunother (2008) 57:1115–24. doi: 10.1007/s00262-007-0441-x
169. Limagne E, Euvrard R, Thibaudin M, Rebe C, Derangere V, Chevriaux A, et al. Accumulation of MDSC and Th17 Cells in Patients With Metastatic Colorectal Cancer Predicts the Efficacy of a FOLFOX-Bevacizumab Drug Treatment Regimen. Cancer Res (2016) 76:5241–52. doi: 10.1158/0008-5472.CAN-15-3164
170. Fuchs CS, Tomasek J, Yong CJ, Dumitru F, Passalacqua R, Goswami C, et al. Ramucirumab Monotherapy for Previously Treated Advanced Gastric or Gastro-Oesophageal Junction Adenocarcinoma (REGARD): An International, Randomised, Multicentre, Placebo-Controlled, Phase 3 Trial. Lancet (2014) 383:31–9. doi: 10.1016/S0140-6736(13)61719-5
171. Buyukkaramikli NC, Blommestein HM, Riemsma R, Armstrong N, Clay FJ, Ross J, et al. Ramucirumab for Treating Advanced Gastric Cancer or Gastro-Oesophageal Junction Adenocarcinoma Previously Treated With Chemotherapy: An Evidence Review Group Perspective of a NICE Single Technology Appraisal. Pharmacoeconomics (2017) 35:1211–21. doi: 10.1007/s40273-017-0528-y
172. Murahashi S, Takahari D, Wakatsuki T, Fukuda N, Ichimura T, Ogura M, et al. A Retrospective Analysis of Ramucirumab Monotherapy in Previously Treated Japanese Patients With Advanced or Metastatic Gastric Adenocarcinoma. Int J Clin Oncol (2018) 23:92–7. doi: 10.1007/s10147-017-1192-0
173. Garon EB, Ciuleanu TE, Arrieta O, Prabhash K, Syrigos KN, Goksel T, et al. Ramucirumab Plus Docetaxel Versus Placebo Plus Docetaxel for Second-Line Treatment of Stage IV non-Small-Cell Lung Cancer After Disease Progression on Platinum-Based Therapy (REVEL): A Multicentre, Double-Blind, Randomised Phase 3 Trial. Lancet (2014) 384:665–73. doi: 10.1016/S0140-6736(14)60845-X
174. Ren S, Xiong X, You H, Shen J, Zhou P. The Combination of Immune Checkpoint Blockade and Angiogenesis Inhibitors in the Treatment of Advanced Non-Small Cell Lung Cancer. Front Immunol (2021) 12:689132. doi: 10.3389/fimmu.2021.689132
175. Verdaguer H, Tabernero J, Macarulla T. Ramucirumab in Metastatic Colorectal Cancer: Evidence to Date and Place in Therapy. Ther Adv Med Oncol (2016) 8:230–42. doi: 10.1177/1758834016635888
176. Tabernero J, Yoshino T, Cohn AL, Obermannova R, Bodoky G, Garcia-Carbonero R, et al. Ramucirumab Versus Placebo in Combination With Second-Line FOLFIRI in Patients With Metastatic Colorectal Carcinoma That Progressed During or After First-Line Therapy With Bevacizumab, Oxaliplatin, and a Fluoropyrimidine (RAISE): A Randomised, Double-Blind, Multicentre, Phase 3 Study. Lancet Oncol (2015) 16:499–508. doi: 10.1016/S1470-2045(15)70127-0
177. Denda T, Sakai D, Hamaguchi T, Sugimoto N, Ura T, Yamazaki K, et al. Phase II Trial of Aflibercept With FOLFIRI as a Second-Line Treatment for Japanese Patients With Metastatic Colorectal Cancer. Cancer Sci (2019) 110:1032–43. doi: 10.1111/cas.13943
178. Tabernero J, Van Cutsem E, Lakomy R, Prausova J, Ruff P, Van Hazel GA, et al. Aflibercept Versus Placebo in Combination With Fluorouracil, Leucovorin and Irinotecan in the Treatment of Previously Treated Metastatic Colorectal Cancer: Prespecified Subgroup Analyses From the VELOUR Trial. Eur J Cancer (2014) 50:320–31. doi: 10.1016/j.ejca.2013.09.013
179. Ruff P, Van Cutsem E, Lakomy R, Prausova J, Van Hazel GA, Moiseyenko VM, et al. Observed Benefit and Safety of Aflibercept in Elderly Patients With Metastatic Colorectal Cancer: An Age-Based Analysis From the Randomized Placebo-Controlled Phase III VELOUR Trial. J Geriatr Oncol (2018) 9:32–9. doi: 10.1016/j.jgo.2017.07.010
180. Grothey A, Van Cutsem E, Sobrero A, Siena S, Falcone A, Ychou M, et al. Regorafenib Monotherapy for Previously Treated Metastatic Colorectal Cancer (CORRECT): An International, Multicentre, Randomised, Placebo-Controlled, Phase 3 Trial. Lancet (2013) 381:303–12. doi: 10.1016/S0140-6736(12)61900-X
181. Wang C, Chevalier D, Saluja J, Sandhu J, Lau C, Fakih M. Regorafenib and Nivolumab or Pembrolizumab Combination and Circulating Tumor DNA Response Assessment in Refractory Microsatellite Stable Colorectal Cancer. Oncologist (2020) 25:e1188–94. doi: 10.1634/theoncologist.2020-0161
182. Xie H, Lafky JM, Morlan BW, Stella PJ, Dakhil SR, Gross GG, et al. Dual VEGF Inhibition With Sorafenib and Bevacizumab as Salvage Therapy in Metastatic Colorectal Cancer: Results of the Phase II North Central Cancer Treatment Group Study N054C (Alliance). Ther Adv Med Oncol (2020) 12:1758835920910913. doi: 10.1177/1758835920910913
183. Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Staehler M, et al. Sorafenib for Treatment of Renal Cell Carcinoma: Final Efficacy and Safety Results of the Phase III Treatment Approaches in Renal Cancer Global Evaluation Trial. J Clin Oncol (2009) 27:3312–8. doi: 10.1200/JCO.2008.19.5511
184. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al. Sorafenib in Advanced Hepatocellular Carcinoma. N Engl J Med (2008) 359:378–90. doi: 10.1056/NEJMoa0708857
185. Bruix J, Takayama T, Mazzaferro V, Chau GY, Yang J, Kudo M, et al. Adjuvant Sorafenib for Hepatocellular Carcinoma After Resection or Ablation (STORM): A Phase 3, Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Oncol (2015) 16:1344–54. doi: 10.1016/S1470-2045(15)00198-9
186. Brose MS, Nutting CM, Sherman SI, Shong YK, Smit JW, Reike G, et al. Rationale and Design of Decision: A Double-Blind, Randomized, Placebo-Controlled Phase III Trial Evaluating the Efficacy and Safety of Sorafenib in Patients With Locally Advanced or Metastatic Radioactive Iodine (RAI)-Refractory, Differentiated Thyroid Cancer. BMC Cancer (2011) 11:349. doi: 10.1186/1471-2407-11-349
187. Tabernero J, Garcia-Carbonero R, Cassidy J, Sobrero A, Van Cutsem E, Kohne CH, et al. Sorafenib in Combination With Oxaliplatin, Leucovorin, and Fluorouracil (Modified FOLFOX6) as First-Line Treatment of Metastatic Colorectal Cancer: The RESPECT Trial. Clin Cancer Res (2013) 19:2541–50. doi: 10.1158/1078-0432.CCR-13-0107
188. Samalin E, Fouchardiere C, Thezenas S, Boige V, Senellart H, Guimbaud R, et al. Sorafenib Plus Irinotecan Combination in Patients With RAS-Mutated Metastatic Colorectal Cancer Refractory To Standard Combined Chemotherapies: A Multicenter, Randomized Phase 2 Trial (NEXIRI-2/PRODIGE 27). Clin Colorectal Cancer (2020) 19:301–310 e1. doi: 10.1016/j.clcc.2020.04.008
189. Tsuji Y, Satoh T, Tsuji A, Muro K, Yoshida M, Nishina T, et al. First-Line Sunitinib Plus FOLFIRI in Japanese Patients With Unresectable/Metastatic Colorectal Cancer: A Phase II Study. Cancer Sci (2012) 103:1502–7. doi: 10.1111/j.1349-7006.2012.02320.x
190. Carrato A, Swieboda-Sadlej A, Staszewska-Skurczynska M, Lim R, Roman L, Shparyk Y, et al. Fluorouracil, Leucovorin, and Irinotecan Plus Either Sunitinib or Placebo in Metastatic Colorectal Cancer: A Randomized, Phase III Trial. J Clin Oncol (2013) 31:1341–7. doi: 10.1200/JCO.2012.45.1930
191. Li J, Qin S, Xu RH, Shen L, Xu J, Bai Y, et al. Effect of Fruquintinib vs Placebo on Overall Survival in Patients With Previously Treated Metastatic Colorectal Cancer: The FRESCO Randomized Clinical Trial. JAMA (2018) 319:2486–96. doi: 10.1001/jama.2018.7855
192. Becouarn Y, Cany L, Pulido M, Beyssac R, Texereau P, Le Morvan V, et al. FOLFIRI(R) and Bevacizumab in First-Line Treatment for Colorectal Cancer Patients: Safety, Efficacy and Genetic Polymorphisms. BMC Res Notes (2014) 7:260. doi: 10.1186/1756-0500-7-260
193. Hurwitz H, Mitchell EP, Cartwright T, Kwok A, Hu S, Mckenna E, et al. A Randomized, Phase II Trial of Standard Triweekly Compared With Dose-Dense Biweekly Capecitabine Plus Oxaliplatin Plus Bevacizumab as First-Line Treatment for Metastatic Colorectal Cancer: XELOX-A-DVS (Dense Versus Standard). Oncologist (2012) 17:937–46. doi: 10.1634/theoncologist.2012-0071
194. Cremolini C, Antoniotti C, Stein A, Bendell J, Gruenberger T, Rossini D, et al. Individual Patient Data Meta-Analysis of FOLFOXIRI Plus Bevacizumab Versus Doublets Plus Bevacizumab as Initial Therapy of Unresectable Metastatic Colorectal Cancer. J Clin Oncol (2020) 38:3314–24. doi: 10.1200/JCO.20.01225
195. Stein A, Glockzin G, Wienke A, Arnold D, Edelmann T, Hildebrandt B, et al. Treatment With Bevacizumab and FOLFOXIRI in Patients With Advanced Colorectal Cancer: Presentation of Two Novel Trials (CHARTA and PERIMAX) and Review of the Literature. BMC Cancer (2012) 12:356. doi: 10.1186/1471-2407-12-356
196. Cunningham D, Lang I, Marcuello E, Lorusso V, Ocvirk J, Shin DB, et al. Bevacizumab Plus Capecitabine Versus Capecitabine Alone in Elderly Patients With Previously Untreated Metastatic Colorectal Cancer (AVEX): An Open-Label, Randomised Phase 3 Trial. Lancet Oncol (2013) 14:1077–85. doi: 10.1016/S1470-2045(13)70154-2
197. Venook AP, Niedzwiecki D, Lenz HJ, Innocenti F, Fruth B, Meyerhardt JA, et al. Effect of First-Line Chemotherapy Combined With Cetuximab or Bevacizumab on Overall Survival in Patients With KRAS Wild-Type Advanced or Metastatic Colorectal Cancer: A Randomized Clinical Trial. JAMA (2017) 317:2392–401. doi: 10.1001/jama.2017.7105
198. Robertson JD, Botwood NA, Rothenberg ML, Schmoll HJ. Phase III Trial of FOLFOX Plus Bevacizumab or Cediranib (AZD2171) as First-Line Treatment of Patients With Metastatic Colorectal Cancer: HORIZON III. Clin Colorectal Cancer (2009) 8:59–60. doi: 10.3816/CCC.2009.n.010
199. De Gramont A, Van Cutsem E, Schmoll HJ, Tabernero J, Clarke S, Moore MJ, et al. Bevacizumab Plus Oxaliplatin-Based Chemotherapy as Adjuvant Treatment for Colon Cancer (AVANT): A Phase 3 Randomised Controlled Trial. Lancet Oncol (2012) 13:1225–33. doi: 10.1016/S1470-2045(12)70509-0
200. Antonuzzo L, Giommoni E, Pastorelli D, Latiano T, Pavese I, Azzarello D, et al. Bevacizumab Plus XELOX as First-Line Treatment of Metastatic Colorectal Cancer: The OBELIX Study. World J Gastroenterol (2015) 21:7281–8. doi: 10.3748/wjg.v21.i23.7281
201. Li J, Xu R, Qin S, Liu T, Pan H, Xu J, et al. Aflibercept Plus FOLFIRI in Asian Patients With Pretreated Metastatic Colorectal Cancer: A Randomized Phase III Study. Future Oncol (2018) 14:2031–44. doi: 10.2217/fon-2017-0669
202. Van Cutsem E, Tabernero J, Lakomy R, Prenen H, Prausova J, Macarulla T, et al. Addition of Aflibercept to Fluorouracil, Leucovorin, and Irinotecan Improves Survival in a Phase III Randomized Trial in Patients With Metastatic Colorectal Cancer Previously Treated With an Oxaliplatin-Based Regimen. J Clin Oncol (2012) 30:3499–506. doi: 10.1200/JCO.2012.42.8201
203. Dane F, Ozgurdal K, Yalcin S, Benekli M, Aykan NF, Yucel I, et al. Safety and Efficacy of Regorafenib in Patients With Treatment-Refractory Metastatic Colorectal Cancer in Turkey: The Single-Arm, Open-Label REGARD Study. BMJ Open (2020) 10:e027665. doi: 10.1136/bmjopen-2018-027665
204. Li J, Qin S, Xu R, Yau TC, Ma B, Pan H, et al. Regorafenib Plus Best Supportive Care Versus Placebo Plus Best Supportive Care in Asian Patients With Previously Treated Metastatic Colorectal Cancer (CONCUR): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol (2015) 16:619–29. doi: 10.1016/S1470-2045(15)70156-7
205. Qin S, Xu RH, Shen L, Xu J, Bai Y, Yang L, et al. Subgroup Analysis by Liver Metastasis in the FRESCO Trial Comparing Fruquintinib Versus Placebo Plus Best Supportive Care in Chinese Patients With Metastatic Colorectal Cancer. Onco Targets Ther (2021) 14:4439–50. doi: 10.2147/OTT.S307273
206. Yarden Y. The EGFR Family and its Ligands in Human Cancer. Signalling Mechanisms and Therapeutic Opportunities. Eur J Cancer (2001) 37 (Suppl 4):S3–8. doi: 10.1016/s0959-8049(01)00230-1
207. Yamaoka T, Ohba M, Ohmori T. Molecular-Targeted Therapies for Epidermal Growth Factor Receptor and Its Resistance Mechanisms. Int J Mol Sci (2017) 18:2420. doi: 10.3390/ijms18112420
208. Chan DLH, Segelov E, Wong RS, Smith A, Herbertson RA, Li BT, et al. Epidermal Growth Factor Receptor (EGFR) Inhibitors for Metastatic Colorectal Cancer. Cochrane Database Syst Rev (2017) 6:CD007047. doi: 10.1002/14651858.CD007047.pub2
209. Fornasier G, Francescon S, Baldo P. An Update of Efficacy and Safety of Cetuximab in Metastatic Colorectal Cancer: A Narrative Review. Adv Ther (2018) 35:1497–509. doi: 10.1007/s12325-018-0791-0
210. Herbst RS, Hong WK. IMC-C225, an Anti-Epidermal Growth Factor Receptor Monoclonal Antibody for Treatment of Head and Neck Cancer. Semin Oncol (2002) 29:18–30. doi: 10.1053/sonc.2002.35644
211. Personeni N, Hendlisz A, Gallez J, Galdon MG, Larsimont D, Van Laethem JL, et al. Correlation Between the Response to Cetuximab Alone or in Combination With Irinotecan and the Activated/Phosphorylated Epidermal Growth Factor Receptor in Metastatic Colorectal Cancer. Semin Oncol (2005) 32:S59–62. doi: 10.1053/j.seminoncol.2005.04.029
212. Saltz LB, Meropol NJ, Loehrer PJ Sr., Needle MN, Kopit J, Mayer RJ. Phase II Trial of Cetuximab in Patients With Refractory Colorectal Cancer That Expresses the Epidermal Growth Factor Receptor. J Clin Oncol (2004) 22:1201–8. doi: 10.1200/JCO.2004.10.182
213. Van Cutsem E, Kohne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, et al. Cetuximab and Chemotherapy as Initial Treatment for Metastatic Colorectal Cancer. N Engl J Med (2009) 360:1408–17. doi: 10.1056/NEJMoa0805019
214. Yarla NS, Madka V, Pathuri G, Rao CV. Molecular Targets in Precision Chemoprevention of Colorectal Cancer: An Update From Pre-Clinical to Clinical Trials. Int J Mol Sci (2020) 21:9609. doi: 10.3390/ijms21249609
215. Vangala D, Ladigan S, Liffers ST, Noseir S, Maghnouj A, Gotze TM, et al. Secondary Resistance to Anti-EGFR Therapy by Transcriptional Reprogramming in Patient-Derived Colorectal Cancer Models. Genome Med (2021) 13:116. doi: 10.1186/s13073-021-00926-7
216. Kasper S, Meiler J, Knipp H, Hohler T, Reimer P, Steinmetz T, et al. Biweekly Cetuximab Plus FOLFOX6 as First-Line Therapy in Patients With RAS Wild-Type Metastatic Colorectal Cancer: The CEBIFOX Trial. Clin Colorectal Cancer (2020) 19:236–247 e6. doi: 10.1016/j.clcc.2020.03.003
217. Van Cutsem E, Peeters M, Siena S, Humblet Y, Hendlisz A, Neyns B, et al. Open-Label Phase III Trial of Panitumumab Plus Best Supportive Care Compared With Best Supportive Care Alone in Patients With Chemotherapy-Refractory Metastatic Colorectal Cancer. J Clin Oncol (2007) 25:1658–64. doi: 10.1200/JCO.2006.08.1620
218. Douillard JY, Siena S, Cassidy J, Tabernero J, Burkes R, Barugel M, et al. Randomized, Phase III Trial of Panitumumab With Infusional Fluorouracil, Leucovorin, and Oxaliplatin (FOLFOX4) Versus FOLFOX4 Alone as First-Line Treatment in Patients With Previously Untreated Metastatic Colorectal Cancer: The PRIME Study. J Clin Oncol (2010) 28:4697–705. doi: 10.1200/JCO.2009.27.4860
219. Modest DP, Martens UM, Riera-Knorrenschild J, Greeve J, Florschutz A, Wessendorf S, et al. FOLFOXIRI Plus Panitumumab As First-Line Treatment of RAS Wild-Type Metastatic Colorectal Cancer: The Randomized, Open-Label, Phase II VOLFI Study (AIO Krk0109). J Clin Oncol (2019) 37:3401–11. doi: 10.1200/JCO.19.01340
220. De Felice F, D'ambrosio G, Iafrate F, Gelibter A, Magliocca FM, Musio D, et al. Intensified Total Neoadjuvant Therapy in Patients With Locally Advanced Rectal Cancer: A Phase II Trial. Clin Oncol (R Coll Radiol) (2021) 33:788–94. doi: 10.1016/j.clon.2021.06.006
221. Tol J, Koopman M, Cats A, Rodenburg CJ, Creemers GJ, Schrama JG, et al. Chemotherapy, Bevacizumab, and Cetuximab in Metastatic Colorectal Cancer. N Engl J Med (2009) 360:563–72. doi: 10.1056/NEJMoa0808268
222. Ljunggren HG, Jonsson R, Hoglund P. Seminal Immunologic Discoveries With Direct Clinical Implications: The 2018 Nobel Prize in Physiology or Medicine Honours Discoveries in Cancer Immunotherapy. Scand J Immunol (2018) 88:e12731. doi: 10.1111/sji.12731
223. Makkouk A, Weiner GJ. Cancer Immunotherapy and Breaking Immune Tolerance: New Approaches to an Old Challenge. Cancer Res (2015) 75:5–10. doi: 10.1158/0008-5472.CAN-14-2538
224. Vinay DS, Ryan EP, Pawelec G, Talib WH, Stagg J, Elkord E, et al. Immune Evasion in Cancer: Mechanistic Basis and Therapeutic Strategies. Semin Cancer Biol (2015) 35 Suppl:S185–98. doi: 10.1016/j.semcancer.2015.03.004
225. Gu L, Khadaroo PA, Su H, Kong L, Chen L, Wang X, et al. The Safety and Tolerability of Combined Immune Checkpoint Inhibitors (Anti-PD-1/PD-L1 Plus Anti-CTLA-4): A Systematic Review and Meta-Analysis. BMC Cancer (2019) 19:559. doi: 10.1186/s12885-019-5785-z
226. Seidel JA, Otsuka A, Kabashima K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front Oncol (2018) 8:86. doi: 10.3389/fonc.2018.00086
227. Li X, Shao C, Shi Y, Han W. Lessons Learned From the Blockade of Immune Checkpoints in Cancer Immunotherapy. J Hematol Oncol (2018) 11:31. doi: 10.1186/s13045-018-0578-4
228. Ouyang T, Cao Y, Kan X, Chen L, Ren Y, Sun T, et al. Treatment-Related Serious Adverse Events of Immune Checkpoint Inhibitors in Clinical Trials: A Systematic Review. Front Oncol (2021) 11:621639. doi: 10.3389/fonc.2021.621639
229. Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: Co-Inhibitory Receptors With Specialized Functions in Immune Regulation. Immunity (2016) 44:989–1004. doi: 10.1016/j.immuni.2016.05.001
230. Dougall WC, Kurtulus S, Smyth MJ, Anderson AC. TIGIT and CD96: New Checkpoint Receptor Targets for Cancer Immunotherapy. Immunol Rev (2017) 276:112–20. doi: 10.1111/imr.12518
231. Maruhashi T, Sugiura D, Okazaki IM, Okazaki T. LAG-3: From Molecular Functions to Clinical Applications. J Immunother Cancer (2020) 8:e001014. doi: 10.1136/jitc-2020-001014
232. Das M, Zhu C, Kuchroo VK. Tim-3 and its Role in Regulating Anti-Tumor Immunity. Immunol Rev (2017) 276:97–111. doi: 10.1111/imr.12520
233. De Mingo Pulido A, Gardner A, Hiebler S, Soliman H, Rugo HS, Krummel MF, et al. TIM-3 Regulates CD103(+) Dendritic Cell Function and Response to Chemotherapy in Breast Cancer. Cancer Cell (2018) 33:60–74 e6. doi: 10.1016/j.ccell.2017.11.019
234. Brunet JF, Denizot F, Luciani MF, Roux-Dosseto M, Suzan M, Mattei MG, et al. A New Member of the Immunoglobulin Superfamily–CTLA-4. Nature (1987) 328:267–70. doi: 10.1038/328267a0
235. Corse E, Allison JP. Cutting Edge: CTLA-4 on Effector T Cells Inhibits in Trans. J Immunol (2012) 189:1123–7. doi: 10.4049/jimmunol.1200695
236. De Sousa Linhares A, Leitner J, Grabmeier-Pfistershammer K, Steinberger P. Not All Immune Checkpoints Are Created Equal. Front Immunol (2018) 9:1909. doi: 10.3389/fimmu.2018.01909
237. Hodi FS, O'day SJ, Mcdermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved Survival With Ipilimumab in Patients With Metastatic Melanoma. N Engl J Med (2010) 363:711–23. doi: 10.1056/NEJMoa1003466
238. Zoroquiain P, Esposito E, Logan P, Aldrees S, Dias AB, Mansure JJ, et al. Programmed Cell Death Ligand-1 Expression in Tumor and Immune Cells Is Associated With Better Patient Outcome and Decreased Tumor-Infiltrating Lymphocytes in Uveal Melanoma. Mod Pathol (2018) 31:1201–10. doi: 10.1038/s41379-018-0043-5
239. Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, et al. Tumor Antigen-Specific CD8 T Cells Infiltrating the Tumor Express High Levels of PD-1 and Are Functionally Impaired. Blood (2009) 114:1537–44. doi: 10.1182/blood-2008-12-195792
240. Salmaninejad A, Khoramshahi V, Azani A, Soltaninejad E, Aslani S, Zamani MR, et al. PD-1 and Cancer: Molecular Mechanisms and Polymorphisms. Immunogenetics (2018) 70:73–86. doi: 10.1007/s00251-017-1015-5
241. Zamani MR, Aslani S, Salmaninejad A, Javan MR, Rezaei N. PD-1/PD-L and Autoimmunity: A Growing Relationship. Cell Immunol (2016) 310:27–41. doi: 10.1016/j.cellimm.2016.09.009
242. Ren M, Dai B, Kong YY, Lv JJ, Cai X. PD-L1 Expression in Tumour-Infiltrating Lymphocytes Is a Poor Prognostic Factor for Primary Acral Melanoma Patients. Histopathology (2018) 73:386–96. doi: 10.1111/his.13527
243. Andre T, Shiu KK, Kim TW, Jensen BV, Jensen LH, Punt C, et al. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N Engl J Med (2020) 383:2207–18. doi: 10.1056/NEJMoa2017699
244. Le DT, Kim TW, Van Cutsem E, Geva R, Jager D, Hara H, et al. Phase II Open-Label Study of Pembrolizumab in Treatment-Refractory, Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: KEYNOTE-164. J Clin Oncol (2020) 38:11–9. doi: 10.1200/JCO.19.02107
245. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 Blockade in Tumors With Mismatch-Repair Deficiency. N Engl J Med (2015) 372:2509–20. doi: 10.1056/NEJMoa1500596
246. Weber JS, D'angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, et al. Nivolumab Versus Chemotherapy in Patients With Advanced Melanoma Who Progressed After Anti-CTLA-4 Treatment (CheckMate 037): A Randomised, Controlled, Open-Label, Phase 3 Trial. Lancet Oncol (2015) 16:375–84. doi: 10.1016/S1470-2045(15)70076-8
247. Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in Previously Untreated Melanoma Without BRAF Mutation. N Engl J Med (2015) 372:320–30. doi: 10.1056/NEJMoa1412082
248. Overman MJ, Mcdermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA, et al. Nivolumab in Patients With Metastatic DNA Mismatch Repair-Deficient or Microsatellite Instability-High Colorectal Cancer (CheckMate 142): An Open-Label, Multicentre, Phase 2 Study. Lancet Oncol (2017) 18:1182–91. doi: 10.1016/S1470-2045(17)30422-9
249. Overman MJ, Lonardi S, Wong KYM, Lenz HJ, Gelsomino F, Aglietta M, et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J Clin Oncol (2018) 36:773–9. doi: 10.1200/JCO.2017.76.9901
250. Morse MA, Overman MJ, Hartman L, Khoukaz T, Brutcher E, Lenz HJ, et al. Safety of Nivolumab Plus Low-Dose Ipilimumab in Previously Treated Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer. Oncologist (2019) 24:1453–61. doi: 10.1634/theoncologist.2019-0129
251. Lenz HJ, Van Cutsem E, Luisa Limon M, Wong KYM, Hendlisz A, Aglietta M, et al. First-Line Nivolumab Plus Low-Dose Ipilimumab for Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: The Phase II CheckMate 142 Study. J Clin Oncol (2021), JCO2101015. doi: 10.1200/JCO.21.01015
252. Barone A, Hazarika M, Theoret MR, Mishra-Kalyani P, Chen H, He K, et al. FDA Approval Summary: Pembrolizumab for the Treatment of Patients With Unresectable or Metastatic Melanoma. Clin Cancer Res (2017) 23:5661–5. doi: 10.1158/1078-0432.CCR-16-0664
253. Leighl NB, Hellmann MD, Hui R, Carcereny E, Felip E, Ahn MJ, et al. Pembrolizumab in Patients With Advanced Non-Small-Cell Lung Cancer (KEYNOTE-001): 3-Year Results From an Open-Label, Phase 1 Study. Lancet Respir Med (2019) 7:347–57. doi: 10.1016/S2213-2600(18)30500-9
254. Herbst RS, Baas P, Kim DW, Felip E, Perez-Gracia JL, Han JY, et al. Pembrolizumab Versus Docetaxel for Previously Treated, PD-L1-Positive, Advanced non-Small-Cell Lung Cancer (KEYNOTE-010): A Randomised Controlled Trial. Lancet (2016) 387:1540–50. doi: 10.1016/S0140-6736(15)01281-7
255. Chen R, Zinzani PL, Fanale MA, Armand P, Johnson NA, Brice P, et al. Phase II Study of the Efficacy and Safety of Pembrolizumab for Relapsed/Refractory Classic Hodgkin Lymphoma. J Clin Oncol (2017) 35:2125–32. doi: 10.1200/JCO.2016.72.1316
256. Kuruvilla J, Ramchandren R, Santoro A, Paszkiewicz-Kozik E, Gasiorowski R, Johnson NA, et al. Pembrolizumab Versus Brentuximab Vedotin in Relapsed or Refractory Classical Hodgkin Lymphoma (KEYNOTE-204): An Interim Analysis of a Multicentre, Randomised, Open-Label, Phase 3 Study. Lancet Oncol (2021) 22:512–24. doi: 10.1016/S1470-2045(21)00005-X
257. Bauml J, Seiwert TY, Pfister DG, Worden F, Liu SV, Gilbert J, et al. Pembrolizumab for Platinum- and Cetuximab-Refractory Head and Neck Cancer: Results From a Single-Arm, Phase II Study. J Clin Oncol (2017) 35:1542–9. doi: 10.1200/JCO.2016.70.1524
258. Bellmunt J, De Wit R, Vaughn DJ, Fradet Y, Lee JL, Fong L, et al. Pembrolizumab as Second-Line Therapy for Advanced Urothelial Carcinoma. N Engl J Med (2017) 376:1015–26. doi: 10.1056/NEJMoa1613683
259. Balar AV, Castellano D, O'donnell PH, Grivas P, Vuky J, Powles T, et al. First-Line Pembrolizumab in Cisplatin-Ineligible Patients With Locally Advanced and Unresectable or Metastatic Urothelial Cancer (KEYNOTE-052): A Multicentre, Single-Arm, Phase 2 Study. Lancet Oncol (2017) 18:1483–92. doi: 10.1016/S1470-2045(17)30616-2
260. Janjigian YY, Maron SB, Chatila WK, Millang B, Chavan SS, Alterman C, et al. First-Line Pembrolizumab and Trastuzumab in HER2-Positive Oesophageal, Gastric, or Gastro-Oesophageal Junction Cancer: An Open-Label, Single-Arm, Phase 2 Trial. Lancet Oncol (2020) 21:821–31. doi: 10.1016/S1470-2045(20)30169-8
261. Marcus L, Lemery SJ, Keegan P, Pazdur R. FDA Approval Summary: Pembrolizumab for the Treatment of Microsatellite Instability-High Solid Tumors. Clin Cancer Res (2019) 25:3753–8. doi: 10.1158/1078-0432.CCR-18-4070
262. Gardini A, Ercolani G, Riccobon A, Ravaioli M, Ridolfi L, Flamini E, et al. Adjuvant, Adoptive Immunotherapy With Tumor Infiltrating Lymphocytes Plus Interleukin-2 After Radical Hepatic Resection for Colorectal Liver Metastases: 5-Year Analysis. J Surg Oncol (2004) 87:46–52. doi: 10.1002/jso.20066
263. Karlsson M, Marits P, Dahl K, Dagoo T, Enerback S, Thorn M, et al. Pilot Study of Sentinel-Node-Based Adoptive Immunotherapy in Advanced Colorectal Cancer. Ann Surg Oncol (2010) 17:1747–57. doi: 10.1245/s10434-010-0920-8
264. Zhen YH, Liu XH, Yang Y, Li B, Tang JL, Zeng QX, et al. Phase I/II Study of Adjuvant Immunotherapy With Sentinel Lymph Node T Lymphocytes in Patients With Colorectal Cancer. Cancer Immunol Immunother (2015) 64:1083–93. doi: 10.1007/s00262-015-1715-3
265. Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DA, Feldman SA, et al. T Cells Targeting Carcinoembryonic Antigen can Mediate Regression of Metastatic Colorectal Cancer But Induce Severe Transient Colitis. Mol Ther (2011) 19:620–6. doi: 10.1038/mt.2010.272
266. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric Antigen Receptor-Modified T Cells in Chronic Lymphoid Leukemia. N Engl J Med (2011) 365:725–33. doi: 10.1056/NEJMoa1103849
267. Maude SL, Teachey DT, Porter DL, Grupp SA. CD19-Targeted Chimeric Antigen Receptor T-Cell Therapy for Acute Lymphoblastic Leukemia. Blood (2015) 125:4017–23. doi: 10.1182/blood-2014-12-580068
268. Zhang C, Wang Z, Yang Z, Wang M, Li S, Li Y, et al. Phase I Escalating-Dose Trial of CAR-T Therapy Targeting CEA(+) Metastatic Colorectal Cancers. Mol Ther (2017) 25:1248–58. doi: 10.1016/j.ymthe.2017.03.010
269. Hege KM, Bergsland EK, Fisher GA, Nemunaitis JJ, Warren RS, Mcarthur JG, et al. Safety, Tumor Trafficking and Immunogenicity of Chimeric Antigen Receptor (CAR)-T Cells Specific for TAG-72 in Colorectal Cancer. J Immunother Cancer (2017) 5:22. doi: 10.1186/s40425-017-0222-9
270. Zhao H, Wang Y, Yu J, Wei F, Cao S, Zhang X, et al. Autologous Cytokine-Induced Killer Cells Improves Overall Survival of Metastatic Colorectal Cancer Patients: Results From a Phase II Clinical Trial. Clin Colorectal Cancer (2016) 15:228–35. doi: 10.1016/j.clcc.2016.02.005
271. Zhang J, Zhu L, Zhang Q, He X, Yin Y, Gu Y, et al. Effects of Cytokine-Induced Killer Cell Treatment in Colorectal Cancer Patients: A Retrospective Study. BioMed Pharmacother (2014) 68:715–20. doi: 10.1016/j.biopha.2014.07.010
272. Lin T, Song C, Chuo DY, Zhang H, Zhao J. Clinical Effects of Autologous Dendritic Cells Combined With Cytokine-Induced Killer Cells Followed by Chemotherapy in Treating Patients With Advanced Colorectal Cancer: A Prospective Study. Tumour Biol (2016) 37:4367–72. doi: 10.1007/s13277-015-3957-2
273. Xie Y, Huang L, Chen L, Lin X, Chen L, Zheng Q. Effect of Dendritic Cell-Cytokine-Induced Killer Cells in Patients With Advanced Colorectal Cancer Combined With First-Line Treatment. World J Surg Oncol (2017) 15:209. doi: 10.1186/s12957-017-1278-1
274. Morse MA, Chaudhry A, Gabitzsch ES, Hobeika AC, Osada T, Clay TM, et al. Novel Adenoviral Vector Induces T-Cell Responses Despite Anti-Adenoviral Neutralizing Antibodies in Colorectal Cancer Patients. Cancer Immunol Immunother (2013) 62:1293–301. doi: 10.1007/s00262-013-1400-3
275. Morse MA, Niedzwiecki D, Marshall JL, Garrett C, Chang DZ, Aklilu M, et al. A Randomized Phase II Study of Immunization With Dendritic Cells Modified With Poxvectors Encoding CEA and MUC1 Compared With the Same Poxvectors Plus GM-CSF for Resected Metastatic Colorectal Cancer. Ann Surg (2013) 258:879–86. doi: 10.1097/SLA.0b013e318292919e
276. Scurr M, Pembroke T, Bloom A, Roberts D, Thomson A, Smart K, et al. Effect of Modified Vaccinia Ankara-5T4 and Low-Dose Cyclophosphamide on Antitumor Immunity in Metastatic Colorectal Cancer: A Randomized Clinical Trial. JAMA Oncol (2017) 3:e172579. doi: 10.1001/jamaoncol.2017.2579
277. Heery CR, Palena C, Mcmahon S, Donahue RN, Lepone LM, Grenga I, et al. Phase I Study of a Poxviral TRICOM-Based Vaccine Directed Against the Transcription Factor Brachyury. Clin Cancer Res (2017) 23:6833–45. doi: 10.1158/1078-0432.CCR-17-1087
278. Gatti-Mays ME, Strauss J, Donahue RN, Palena C, Del Rivero J, Redman JM, et al. A Phase I Dose-Escalation Trial of BN-CV301, a Recombinant Poxviral Vaccine Targeting MUC1 and CEA With Costimulatory Molecules. Clin Cancer Res (2019) 25:4933–44. doi: 10.1158/1078-0432.CCR-19-0183
279. Hoover HC Jr., Brandhorst JS, Peters LC, Surdyke MG, Takeshita Y, Madariaga J, et al. Adjuvant Active Specific Immunotherapy for Human Colorectal Cancer: 6.5-Year Median Follow-Up of a Phase III Prospectively Randomized Trial. J Clin Oncol (1993) 11:390–9. doi: 10.1200/JCO.1993.11.3.390
280. Harris JE, Ryan L, Hoover HC Jr., Stuart RK, Oken MM, Benson AB 3rd, et al. Adjuvant Active Specific Immunotherapy for Stage II and III Colon Cancer With an Autologous Tumor Cell Vaccine: Eastern Cooperative Oncology Group Study E5283. J Clin Oncol (2000) 18:148–57. doi: 10.1200/JCO.2000.18.1.148
281. Vermorken JB, Claessen AM, Van Tinteren H, Gall HE, Ezinga R, Meijer S, et al. Active Specific Immunotherapy for Stage II and Stage III Human Colon Cancer: A Randomised Trial. Lancet (1999) 353:345–50. doi: 10.1016/S0140-6736(98)07186-4
282. Barth RJ Jr., Fisher DA, Wallace PK, Channon JY, Noelle RJ, Gui J, et al. A Randomized Trial of Ex Vivo CD40L Activation of a Dendritic Cell Vaccine in Colorectal Cancer Patients: Tumor-Specific Immune Responses Are Associated With Improved Survival. Clin Cancer Res (2010) 16:5548–56. doi: 10.1158/1078-0432.CCR-10-2138
283. Rodriguez J, Castanon E, Perez-Gracia JL, Rodriguez I, Viudez A, Alfaro C, et al. A Randomized Phase II Clinical Trial of Dendritic Cell Vaccination Following Complete Resection of Colon Cancer Liver Metastasis. J Immunother Cancer (2018) 6:96. doi: 10.1186/s40425-018-0405-z
284. Frick GS, Pitari GM, Weinberg DS, Hyslop T, Schulz S, Waldman SA. Guanylyl Cyclase C: A Molecular Marker for Staging and Postoperative Surveillance of Patients With Colorectal Cancer. Expert Rev Mol Diagn (2005) 5:701–13. doi: 10.1586/14737159.5.5.701
285. Snook AE, Baybutt TR, Xiang B, Abraham TS, Flickinger JC Jr., Hyslop T, et al. Split Tolerance Permits Safe Ad5-GUCY2C-PADRE Vaccine-Induced T-Cell Responses in Colon Cancer Patients. J Immunother Cancer (2019) 7:104. doi: 10.1186/s40425-019-0576-2
286. Balint JP, Gabitzsch ES, Rice A, Latchman Y, Xu Y, Messerschmidt GL, et al. Extended Evaluation of a Phase 1/2 Trial on Dosing, Safety, Immunogenicity, and Overall Survival After Immunizations With an Advanced-Generation Ad5 [E1-, E2b-]-CEA(6D) Vaccine in Late-Stage Colorectal Cancer. Cancer Immunol Immunother (2015) 64:977–87. doi: 10.1007/s00262-015-1706-4
287. Crosby EJ, Hobeika AC, Niedzwiecki D, Rushing C, Hsu D, Berglund P, et al. Long-Term Survival of Patients With Stage III Colon Cancer Treated With VRP-CEA(6D), an Alphavirus Vector That Increases the CD8+ Effector Memory T Cell to Treg Ratio. J Immunother Cancer (2020) 8:e001662. doi: 10.1136/jitc-2020-001662
288. Yarchoan M, Huang CY, Zhu Q, Ferguson AK, Durham JN, Anders RA, et al. A Phase 2 Study of GVAX Colon Vaccine With Cyclophosphamide and Pembrolizumab in Patients With Mismatch Repair Proficient Advanced Colorectal Cancer. Cancer Med (2020) 9:1485–94. doi: 10.1002/cam4.2763
289. Santos P, Almeida F. Exosome-Based Vaccines: History, Current State, and Clinical Trials. Front Immunol (2021) 12:711565. doi: 10.3389/fimmu.2021.711565
Keywords: immunotherapy, immune microenvironment, bevacizumab, PD-1, colorectal cancer
Citation: Chen Y, Zheng X and Wu C (2021) The Role of the Tumor Microenvironment and Treatment Strategies in Colorectal Cancer. Front. Immunol. 12:792691. doi: 10.3389/fimmu.2021.792691
Received: 11 October 2021; Accepted: 15 November 2021;
Published: 02 December 2021.
Edited by:
Elena Ciaglia, University of Salerno, ItalyReviewed by:
Valentina Lopardo, University of Salerno, ItalyBiao Yang, Sichuan University, China
José Medina-Echeverz, Affimed Therapeutics, Germany
Copyright © 2021 Chen, Zheng and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Changping Wu, d2Nwamp0QDE2My5jb20=