
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 16 December 2021
Sec. Autoimmune and Autoinflammatory Disorders
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.790962
 Jiawei Yang
Jiawei Yang Xueyan Liu*
Xueyan Liu*Autoimmune encephalitis (AE) is an immune-mediated disease involving the central nervous system, usually caused by antigen-antibody reactions. With the advent of autoantibody-associated diseases, AE has become a hot research frontier in neuroimmunology. The first-line conventional treatments of autoimmune encephalitis consist of steroids, intravenous immunoglobulin (IVIG), plasma exchange (PLEX), and second-line therapy includes rituximab. Despite considerable research and expanding clinical experience, current treatments are still ineffective for a significant number of patients. Although there is no clear consensus, clinical trial evidence limited, and the level of evidence for some of the drugs based on single reports, third-line therapy is a viable alternative for refractory encephalitis patients. With the current rapid research progress, a breakthrough in the treatment of AE is critical. This article aims to review the third-line therapy for refractory AE
Autoimmune encephalitis (AE) was first reported in the 1960s (1). It was initially described as limbic encephalitis (LE), characterized by seizures, movement disorders, behavioral changes, mood disorders, cognitive impairment, autonomic dysfunction, and altered level of consciousness. The majority of LE was initially diagnosed with paraneoplastic syndromes commonly associated with lung, breast, ovarian, or testicular malignancies. The autoantibodies target nuclear and cytoplasmic proteins. The vast majority of these patients have a seemingly poor prognosis and do not respond to immunotherapy (2). A previous study has found that tumor treatment often improves neurological function, suggesting that autoantibodies play indirect pathogenic roles and changes in autoantibody levels may not achieve a complete cure (3). Therefore, LE is known as “paraneoplastic LE”. In recent years, autoantibodies targeting cell surfaces or ion channels, such as Anti-N-methyl-d-aspartate receptor (NMDAR) antibodies, have been identified. These autoantibodies affect not only the limbic system but also various brain structures (4). Most of these patients do not have malignancies and respond well to immunotherapy. The term AE gradually replaces autoimmune LE and specifically refers to the disease entity. In this context, AE refers to autoantibodies targeting cell surfaces or ion channels.
To date, the treatment of AE is mostly based on data from retrospective cohort and clinical cases, and prospective randomized controlled studies on immunotherapy are rare (5). The dosage and treatment course of AE are generally modeled on the treatment of rheumatism or other immune-mediated central nervous system diseases and refer to the clinical experience of experts. Unlike paraneoplastic LE, antibodies targeting cell surface receptors can be pathogenic, bind to receptors, and directly affect neuronal function rather than causing neuronal death. Animal models replicating features of AE by passive transfer of cerebrospinal fluid (CSF) or immunoglobulins from anti-NMDAR encephalitis patients to mice provided more direct evidence of autoantibody pathogenicity (6). Therefore, the pathogenic key is the continuous production of autoantibodies by self-reactive B cells and their subsequent proliferation and differentiation into autoantigen reactive memory B cells and autoantibody-secreting plasma cells. Autoantibodies against neuronal surfaces mediate pathogenic effects via multiple mechanisms. Observing the incubation of neuron culture with anti-NMDAR encephalitis patients’ autoantibody, patients’ antibodies cross-linking and internalizing NMDA receptors result in a selective and reversible decrease in NMDAR surface density and synaptic localization (7, 8). This is one of the main pathogenic pathways of autoantibodies. Another pathogenic pathway is complement activation, demonstrated in a case study of contactin-associated protein-like 2 (CASPR2) antibody-associated encephalitis (9).
Based on in-depth tumor screening, the immunotherapy framework for AE can be divided into first-line therapy and second-line therapy. First-line therapies include steroids, IVIG, and PLEX. They have a rapid onset of action with lower risk and could suppress autoimmune diseases systemically and non-specific. Thus, they are widely used as the first choice of treatment against AE. Steroids and IVIG are usually combined, which exhibit good efficacy in more than half of patients and are well tolerated, with few adverse effects that make treatment impossible to continue (10). PLEX is another option for patients with incomplete response to steroids or IVIG; however, it is limited due to being perceived as more invasive. PLEX would be more difficult to accept in children and in patients with poor co-operation (11). Previous studies have shown that timely delivery of immunotherapies is crucial (12). Delayed treatment due to negative autoantibodies can lead to poor prognosis and even death (13). Therefore, immunotherapy should be started once the clinical symptoms highly suspect AE and exclude infectious encephalitis (14). If first-line therapies are ineffective, physicians will choose second-line therapies, yet there is still controversy about the observation time limit. Second-line therapy includes the monoclonal antibody against CD20, i.e., rituximab, and is also preferentially considered by most clinical specialists (14). In a cohort study that included 27 cases of anti-NMDAR encephalitis and 3 cases of anti-leucine-rich glioma inactivated-1 (LGI1) encephalitis, mRS scores improved more in the rituximab treated group, both in the group that responded to first-line therapy (88% vs 83.3%) and in the group that did not respond to first-line therapy (60% vs 22.2%) (15). Moreover, adverse reactions caused by rituximab are mostly mild, and life-threatening severe adverse reactions are rare (16). The remainder includes cyclophosphamide, an alkylating agent that inhibits cell proliferation, as well as the oral antimetabolite drugs mycophenolate mofetil and azathioprine (17–19). Cyclophosphamide is often used as a monotherapy for rheumatic diseases affecting the CNS, such as mental lupus and primary CNS vasculitis (20). However, given its slow mechanism of action and significant side effects, it is less frequently used than rituximab to treat AE (21). Particularly, it is rarely used alone and mostly used in combination with rituximab. However, 19%-33% of patients with AE still do not respond well to first-line and second-line therapy, and there are persistent neurological and psychological problems (10, 22–24). New immunotherapy drugs, known as “third-line therapy”, are increasingly emerging for patients with refractory AE. This article is a narrative review of immunotherapy drugs for treating refractory AE (Table 1).
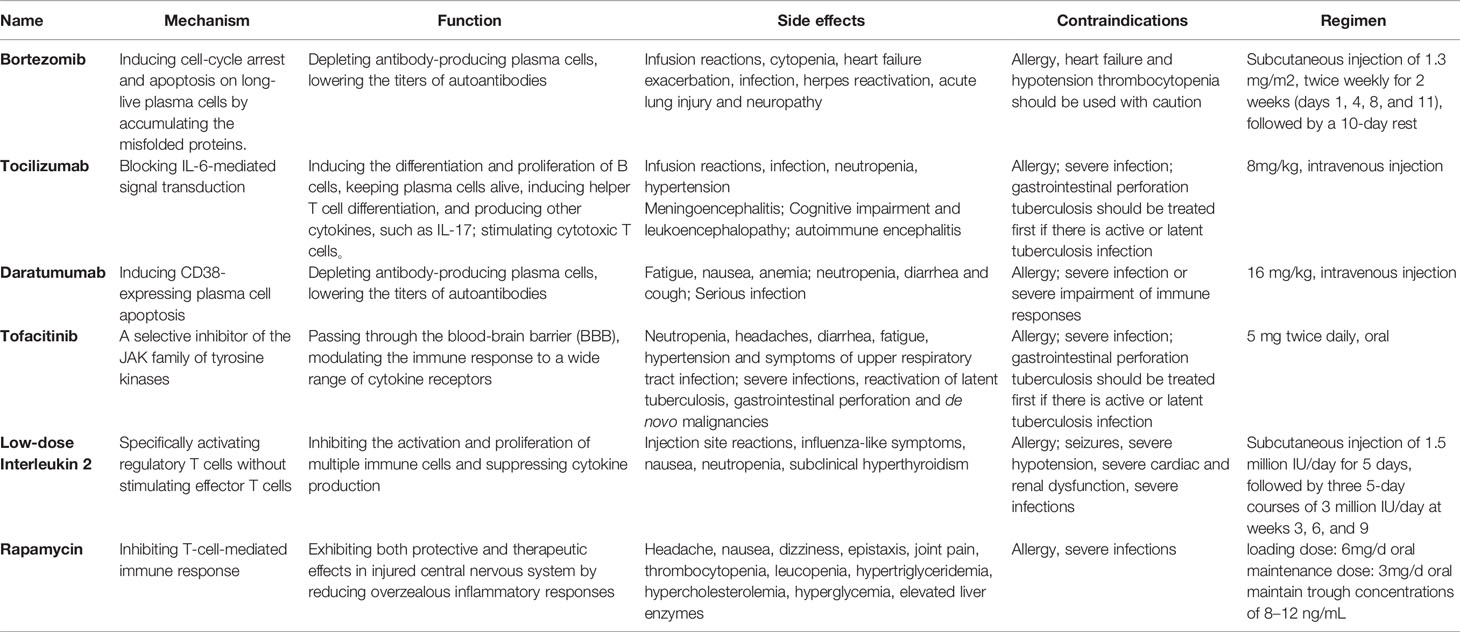
Table 1 Overview of third-line therapy drugs.
Because refractory AE is uncommon, much of the data included in this review is low-level evidence, such as case reports and observational cohort studies. For further investigation, a higher level of evidence is required. The evidence of relevant articles has been summarized in Table 2.
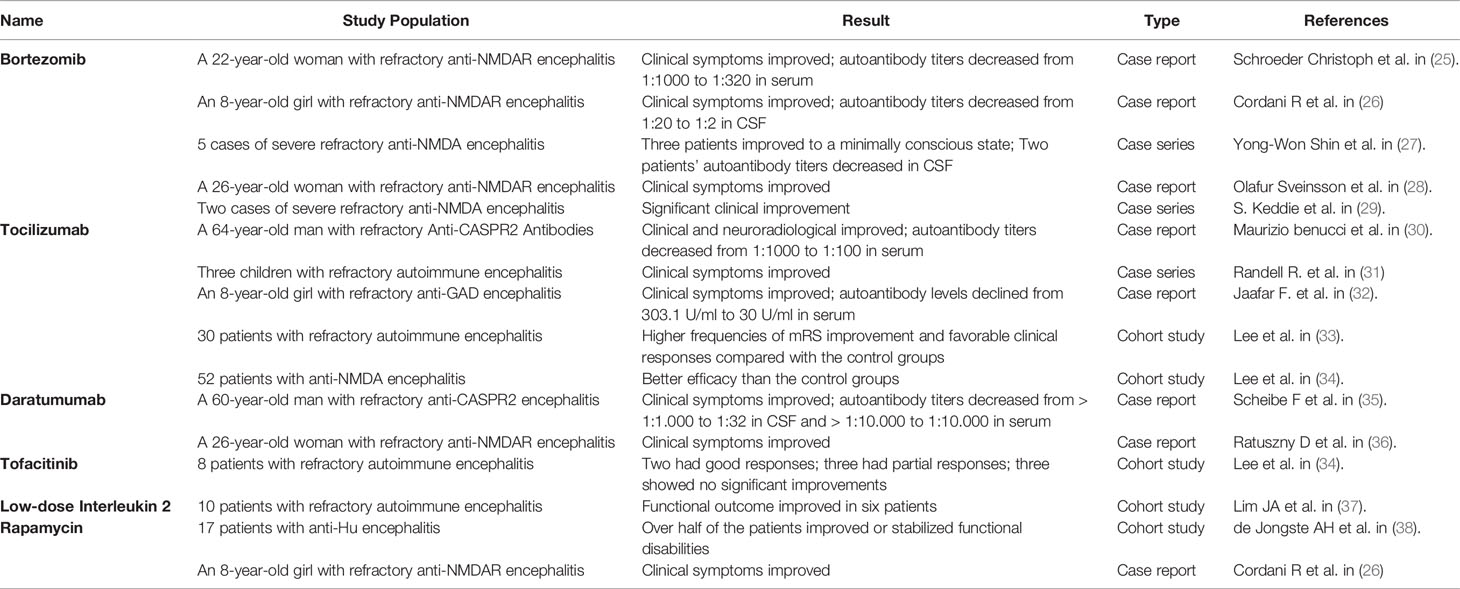
Table 2 Studies on third-line therapy for refractory autoimmune encephalitis.
Refractory AE is defined as cases that are ineffective with conventional first-line and second-line therapy. Rituximab is the representative drug of second-line therapy; however, the failure of rituximab provokes the consideration of third-line therapy for such refractory cases. Since long-lived plasma cells do not express CD20, are unaffected by rituximab, and continue to produce autoantibodies after depleting pre-B cell and mature B cell pools, they are thought to be the main reason for the failure of AE therapies. In a case report of a young woman who died of refractory anti-NMDAR encephalitis, CD19 + lymphocytes comprised only 0.1% of the blood cells in patients 33 and 56 days following rituximab treatment. Autoantibodies were still present in serum after 66 days of rituximab treatment. The autopsy report revealed no B cells, plasma cells, or plasmablasts in any brain region, whereas examination of lymph node tissue revealed many plasma cells aligned along the sinusoidal line. These plasma cells are considered the best explanation for the continued production of antibodies (39). Moreover, peripherally activated B cells can cross the blood–brain barrier (BBB) (40), undergo clonal proliferation, and differentiate between daughter cells into antibody-secreting plasma cells. In contrast, rituximab cannot cross the BBB, which is considered another reason for the failure of rituximab. Consequently, third-line therapies for refractory AE can be divided into three categories (Figure 1): (i) treatments targeting long-lived plasma cells, such as daratumumab (targeting CD38, a type II glycoprotein highly expressed on long-lived plasma cells), bortezomib and tocilizumab (negatively affecting plasma cell survival; (ii) immunotherapeutic drugs that can pass through the BBB, such as tofacitinib; (iii) drugs that alleviate clinical symptoms by regulating immune balance and ameliorating neuronal damage, such as low-dose IL-2, and rapamycin.
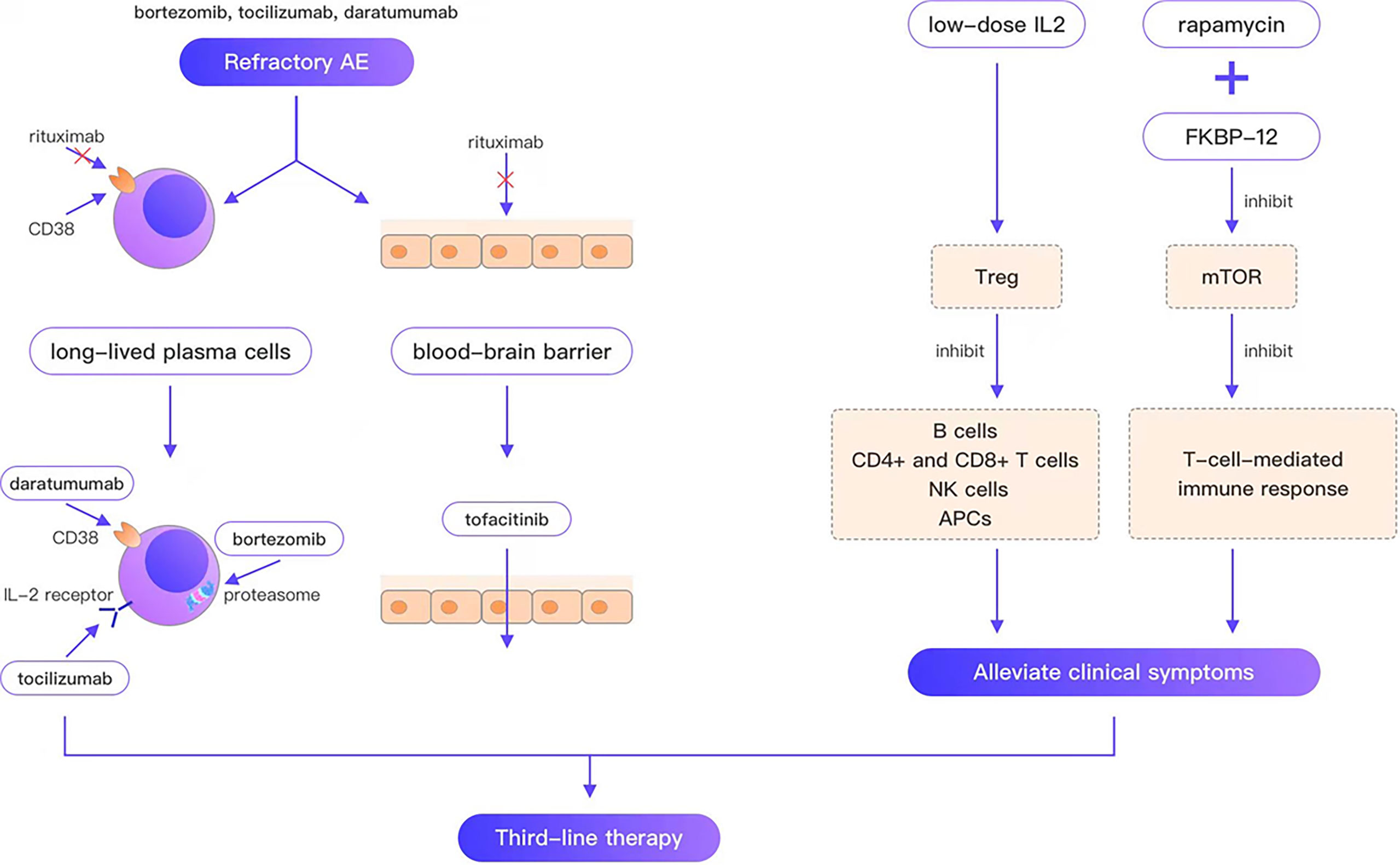
Figure 1 Bortezomib, tocilizumab, and daratumumab overcome the weakness that rituximab cannot target plasma cells; tofacitinib overcomes the weakness that rituximab cannot cross the blood-brain barrier; low-dose IL2, rapamycin can alleviate the clinical symptom.
Bortezomib acts as a reversible inhibitor of the 26S proteasome, the endpoint of the ubiquitin-proteasome system and plays a key regulatory role in cellular homeostasis through the degradation of intracellular proteins. It was first used for the treatment of myeloma (41, 42). Once the Bortezomib mechanism of action is established, it is used to treat many other diseases, such as post-transplant desensitization, refractory hematologic diseases, inflammatory injury in intracerebral hemorrhage, and severe viral encephalitis (43–48). The proteasome inhibitors can induce cell-cycle arrest and apoptosis in plasma cells by accumulating the misfolded proteins. Therefore, they can be used to prevent the production of autoantibodies secreted by plasma cells and improve the therapy of autoimmune diseases, such as autoimmune hemolytic anemia (AIHA), immune thrombocytopenia, antineutrophil cytoplasmic antibodies (ANCA) associated vasculitis, immunologic thrombocytopenic purpura (ITP), systemic lupus, neuromyelitis Optica, and particularly refractory AE.
The therapeutic effect of bortezomib to treat refractory AE has been reported in several cases. A previous study reported that a 22-year-old woman with refractory anti-NMDAR encephalitis failed to respond to PLEX and a high dose of rituximab but recovered swiftly after bortezomib treatment (25). Another study described the case of an 8-year-old child with refractory anti-NMDAR encephalitis, despite high-dose of intravenous steroids, intravenous immunoglobulins, and rituximab, who was treated successfully with bortezomib administration (26). After the fourth bortezomib dose, the patient showed successful control of status epilepticus and progressive improvement of consciousness. Eight months later, the child was able to go to school as usual and return to his normal activities. Five severe refractory anti-NMDA encephalitis cases were reported, where patients were in a vegetative state (27). Three patients moved from the vegetative state into minimally conscious states. One patient’s stereotypic movements and rigid behaviors decreased despite no obvious signs of progress into a minimally conscious state. Another patient failed to recover from a vegetative state after two months of bortezomib treatment. The clinical symptoms of a 26-year-old woman showed signs of improvement after receiving bortezomib in combination with tocilizumab and cyclophosphamide to treat refractory anti-NMDA encephalitis (28). In addition, other cases of refractory AE were successfully treated with bortezomib (29).
Bortezomib is well tolerated with minimum adverse reactions. Acute lung injury (49) and bortezomib-associated neuropathy (50) are reported in only a few cases. However, a preclinical study has shown that bortezomib can affect testicular function for a long time (51); that’s why we should pay attention to the reproductive health of male children.
IL-6 is a pleiotropic cytokine that can be synthesized by various cells, including monocytes, macrophages, lymphocytes, keratinocytes, fibroblasts, endothelial cells, and certain kinds of tumor cells (52). IL-6 can induce the differentiation and proliferation of B cells, keep plasma cells alive, induce helper T cell differentiation, and produce other cytokines, such as IL-17 (a cytokine closely related to autoimmune diseases). Furthermore, it can also stimulate cytotoxic T cells, and promote excitotoxicity-induced neuronal damage (53). Tocilizumab, developed in 1993 by a Japanese group led by Tadamitsu Kishimoto of Osaka University, is an anti-interleukin (IL)-6 receptor monoclonal antibody that blocks IL-6-mediated signal transduction (54). Tocilizumab is a humanized anti-IL-6R antibody engineered by grafting the complementarily determining regions of a mouse antihuman IL-6R antibody into a human IgG1k to create a human antibody with a human IL-6R binding site. Tocilizumab binds and neutralizes IL-6R, resulting in the inhibition of various IL-6-mediated biological activities.
Tocilizumab has shown to be effective in treating autoimmune disorders in both adults and children, such as rheumatoid arthritis, neuromyelitis Optica, and multiple sclerosis (55, 56). Moreover, a few cases of treating AE have also been reported, such as a 64-year-old patient with anti-CASPR2 AE switching to tocilizumab after the failure of high-dose steroid and IVIg therapy (30). After one month of treatment, her abnormal behaviors were reduced, and seizures subsided. She exhibited significantly better results in several cognitive assessments, and the titer of anti-CASPR2 antibodies was reduced from 1:1000 to 1:100. After 4 months of treatment, the modified Rankin Scale score of the patient was 0 and she returned to work. A previous study reported three children with refractory AE who showed significant improvement in clinical symptoms with no significant adverse reactions after being treated with tocilizumab (31). In a case report of an 8-year-old girl with anti- glutamic acid decarboxylase (GAD) encephalitis, tocilizumab significantly improved clinical symptoms, while immune globulin, high dose corticosteroids, and plasmapheresis had limited effects (32). In a retrospective study of 91 patients with AE who failed to respond to first-line immunotherapy and rituximab, Lee et al. (33) divided the patients into the tocilizumab, additional rituximab, and the observation groups based on their response to immunotherapy. Their results showed that the MRS score of the tocilizumab group was higher than the other two groups after 2 months of treatment and the last follow-up. After 1 month of treatment, sustained clinical improvement was observed in 89.5% of the patients. They (34) conducted another cohort study and confirmed that the regimen with tocilizumab was more effective than conventional regimens without tocilizumab.
The serious adverse reactions caused by tocilizumab are mostly found in other autoimmune disease cases. This may be related to the fewer cases of AE treated with tocilizumab. For example, in treating a patient with rheumatoid arthritis and another patient with juvenile idiopathic arthritis, meningoencephalitis was induced (57); in treating a patient with refractory giant cell arteritis, tocilizumab was considered to be a possible cause of Pseudomonas meningitis (58). Patients with rheumatoid arthritis who were given tocilizumab also had cognitive impairment and leukoencephalopathy (59). Even so, the treatment itself has the risk of inducing autoimmune encephalitis (60).
Despite the above risks, tocilizumab should be carefully considered in patients who do not respond to first-line and second-line therapy.
Daratumumab is a human monoclonal IgGkappa antibody targeting CD38, a 45 kDa type II transmembrane glycoprotein expressed on plasma cells. Daratumumab inhibits the progression of CD38-expressing multiple myeloma tumor cells, inducing apoptosis directly through Fc-mediated crosslinking and triggering immune-mediated tumor cell lysis through complement-dependent cytotoxicity, antibody-dependent cell-mediated cytotoxicity, and antibody-dependent cellular phagocytosis. Therefore, daratumumab could effectively inhibit the growth of CD38-expressing tumor cells and has been approved for treating multiple myeloma since 2015 (61). Since then, it has been applied to treat other tumor diseases such as leukemia, AL amyloidosis, blastic plasmacytoid dendritic cell neoplasm, etc. (62–64). Because CD38 is also utilized on nonmalignant plasma cells, T cells, and NK cells, thus daratumumab is also used to treat other diseases, such as post-transplant treatment and AE, triggered by plasma cells or T cell- and NK cell-mediated immune response (65–68).
The traditional second-line treatment drug rituximab targets CD20, expressed on pre-B cells, immature B cells, mature B cells, and activated B cells but not on plasma cells. Rituximab indirectly inhibits plasma cells’ production by killing B cells and blocking the differentiation of B cells into plasma cells. A substantial fraction of plasma cells, such as long-lived plasma cells, can survive in the absence of B-cell precursors and can continue to produce antibodies that may lead to treatment failure or disease recurrences. Compared with rituximab, the target protein of daratumumab, namely CD38, is expressed on plasma cells, and thus daratumumab can deplete antibody-producing plasma cells to achieve the desired therapeutic effects. In a case report of a 60-year-old patient with anti-CASPR2 encephalitis, the patient started 13 cycles of daratumumab treatment after not responding to methylprednisolone, immunoglobulins, plasma exchange, immunoadsorption, rituximab, and bortezomib for more than five months (35). After 8 treatment cycles, the patient recovered from disorientation, behavioral problems, breathing disorders and went into partial remission. After 12 treatment cycles, the antibody titers in the serum and cerebrospinal fluid of the patients significantly decreased. However, during the 13th treatment cycle, the patient died due to Gram-negative bacterial septic shock. Another study (36) reported a case of a 20-year-old woman with refractory anti-NMDA encephalitis. She received daratumumab after the failure of first-line and second-line therapy and showed clinical improvement. Although only two single cases were reported, it still suggested that daratumumab has potential therapeutic value for AE, but its value should be carefully examined due to its serious adverse events (69).
Tofacitinib is a specific inhibitor of Janus kinase (JAK) 1 and 3. They play an important role in regulating cytokine receptors, pro-inflammatory cytokines, and T-helper cells (70). Tofacitinib was primarily used to treat refractory rheumatoid arthritis (RA) and was approved for medical use by the Food and Drug Administration (FDA) in 2012 (71, 72). Recently, tofacitinib has been employed to treat many other refractory immune-mediated diseases, including psoriasis and ulcerative colitis (73, 74).
In the CNS, the JAK/STAT signaling pathway is mainly associated with gene regulation during development, inflammation, or hormone release. Therefore, tofacitinib is considered a therapeutic candidate for immune-related CNS disorders. However, Experimental autoimmune encephalomyelitis (EAE) models and animal models of multiple sclerosis showed contradictory results (75).
In EAE models, tofacitinib activates tolerogenic dendritic cells that lead to the amelioration of EAE, suggesting a potential therapeutic role in MS (76). However, a low dose of tofacitinib accelerated EAE by activating T17 cells and produced excess interleukin-17 (77). This mechanism can explain the potential for iatrogenic demyelination using this drug (78). However, its ability to pass through the BBB and modulate the immune response to a wide range of cytokine receptors makes it a potential therapeutic candidate to be applied for treating refractory AE (79). In a retrospective study, tofacitinib was administered orally in eight patients with refractory AE. Complete response was observed in two patients, partial response in three patients, and no response in another three patients. It is noteworthy that one of the patients with chronic meningoencephalitis, achieved complete remission after tofacitinib therapy, and magnetic resonance imaging also showed improvement in his brain function. In addition, none of these eight patients treated with tofacitinib experienced serious adverse events, one patient had mild nausea, and one had transient neutropenia (80).
IL-2 was discovered in the supernatants of mice splenocytes cell culture by Morgan et al. in 1976 (81). IL-2 plays an important role in the differentiation, survival, and maintenance of regulatory T cells (also called Tregs). It can activate Tregs without stimulating effector T cells, restoring the balance between regulatory and effector T cells (82).
As the name suggests, regulatory T cells are T cells that regulate or suppress other cells in the immune system. Tregs control the immune response to self and foreign particles (antigens) and prevent autoimmune diseases. Tregs suppress activation, proliferation, and cytokine production of CD4+ T cells and CD8+ T cells, natural killer cells, B cells, and antigen-presenting cells (83).
Initially, IL-2 was administered intravenously in high doses to treat cancer. However, the high dose results in serious adverse events, such as vascular leak syndrome and severe bacteremia. Nevertheless, Tregs have a low activation threshold for IL-2, and the number and function of Tregs are dysregulated in most autoimmune diseases, such as type 1 diabetes, multiple sclerosis (MS), systemic lupus erythematosus (SLE), RA, inflammatory bowel disease, and psoriasis, making low-dose IL-2 a new therapeutic candidate for many autoimmune diseases with low toxic effects (84–86).
A retrospective study demonstrated the efficacy and tolerability of low-dose IL-2 therapy for refractory AE (37). In the study, patients with refractory AE received low-dose of IL-2 at Seoul National University Hospital from October 2015 to March 2016. All four patients with anti-NMDA encephalitis showed good responses to low-dose IL-2 therapy, improving gait speed, memory, language function, and mental status. Among the six patients who are negative for antibodies, two showed a decrease in seizure frequency. Only one serious adverse event was observed, manifested as a severe reduction in neutrophil count, and the patient recovered after subcutaneous injection of colony-stimulating factor. Mild adverse events included two cases of injection site reactions, one case of influenza-like symptoms, one case of nausea, one case of neutropenia, and three cases of subclinical hyperthyroidism.
Rapamycin was initially developed as a macrolide antifungal drug, which was banned due to its potent immunosuppressive effect. This situation did not change until mammalian target of rapamycin (mTOR) was discovered. As a natural inhibitor of mTOR, rapamycin’s therapeutic effectiveness was again recognized. Rapamycin inhibits the mammalian target of rapamycin-regulated kinase by forming immunosuppressive complexes with a family of an intracellular protein termed FK506-binding protein 12 (FKBP-12) (87). The Rapamycin-FKBP12 complexes limit the cytokine-mediated proliferation of T cells following antigen stimulation, thus inhibiting T-cell-mediated immune responses. In 1997, rapamycin was approved by the US FDA as an immunosuppressive agent, and effectively prevents acute rejection in kidney transplant recipients. Later, it was also used to treat other immune-related diseases.
In recent studies, rapamycin exhibited protective and therapeutic effects in the injured CNS by reducing overzealous inflammatory responses. A previous study reported that the survival rates of mice treated with rapamycin and valacyclovir were higher than those injected with valacyclovir alone, despite similar viral loads in their brain tissues (88). Rapamycin reduced brain lesions, improved motor function, and significantly reduced the secretion of pro-inflammatory cytokines and chemokines in a rat stroke model. These results suggest that rapamycin can alleviate post-stroke inflammation and neuroprotective brain injury (89).
Although rare cases have been reported, rapamycin has been used to treat AE. In a retrospective study, rapamycin was administered in 17 patients with anti-HU-associated encephalitis. Rapamycin improved or stabilized functional disabilities in more than half of patients and managed neurological impairments in two patients (38). In a case of anti-NMDAR encephalitis, rapamycin was used for maintaining treatment because of its long-lasting immunosuppressive effect (26).
There are many challenges that need to be tackled. The most appropriate timing for initiating third-line therapies, the best choice of doses, and schedules for each drug are not specified. There is still a lack of unified and well-considered guidelines for treating refractory AE, and thus further studies are needed to more precisely and comprehensively assess the advantages and disadvantages of AE therapies and drugs. To date, differentiated antibody therapeutics for the treatment of AE has not been developed. AE is associated with a wide range of antibodies and represents a complex syndrome with diverse clinical manifestations. Different types of AE respond differently to therapeutic agents; for example, patients with LGI1 antibodies respond faster and better to steroids than patients with NMDA antibodies (90). However, uniform treatment (including the same drugs and doses) in differentiated AE patients is still used. Third-line therapy for refractory encephalitis is even more “indistinguishable” for different types of encephalitis. The “effectiveness “exhibited by third-line therapy is controversial due to the paucity of high-level evidence, such as double-blind randomized controlled trials. On the one hand, the patients who used third-line therapy were mostly mixed with multiple immunotherapy drugs, and it is difficult to completely exclude the effect of other immunotherapy on the outcome; On the other hand, part of the symptoms of AE may be treatment-independent, they have independent natural pathological processes, and the effectiveness of third-line therapy may only be coincidental with the natural pathological process (91). Moreover, if the effectiveness of third-line therapy is difficult to confirm, weighing the benefits and risks of applying this drug becomes challenging. Therefore, comprehensive evidence is needed to clarify the effectiveness of third-line therapy. In addition, the clinical presentation of AE patients at different ages varies significantly: the younger the child, the more difficult it is to recognize specific AE syndromes, and its diagnosis will rely on antibodies and other ancillary tests. Whereas elderly patients are often comorbid with multiple diseases related to their age, causing interference with their clinical manifestations and the diagnosis (90). Developing treatment strategies for different ages that are more suitable for their disease characteristics is critical. The study of genetic susceptibility to AE has been unfolded, and the role of genes in refractory AE deserves more attention (92). If specific genotypes exist, through specific pathways, render patients with AE refractory to first - and second-line therapies, then targeted therapies for specific genotypes become possible. Given the low incidence of encephalitis and difficulty in its early diagnosis, more effort is required to established animal models (93). Unfortunately, only a few therapeutic options are available for AE. Therefore, further clinical studies need to be conducted to identify other possible therapeutic targets and explore more therapeutic strategies for AE.
Efforts are, in fact, underway. Basiliximab, a chimeric mouse-human monoclonal antibody to the α chain (CD25) of the IL-2 receptor of T-lymphocytes, has been used to treat patients with anti-GAD65 AE and achieved promising clinical outcomes, including seizure free (94). Although anti-GAD65 encephalitis as a subclass of autoantibodies against intracellular antigens, differs in terms of pathogenesis and therapeutic focus from the AE discussed in this review, it remains possible that the therapeutic effects of basiliximab, which selectively inhibits activated cytotoxic T cells, may benefit patients with AE who have autoantibodies against cell membranes and ion channels, and maybe a potential therapeutic option.
CD19, as one of the hallmark antigens of B cells similar to CD20, has long been linked to AE and is more significantly expressed in antibody-negative AE (95). Expression of CD19 begins at the pre-B cell stage and is stable throughout B cell development. CD19 is also expressed on all plasma cells and most plasma cells of the bone marrow and secondary lymphoid organs. Unlike CD20, which is expressed on a subset of CD4 + T cells, CD19 is expressed only on B cells. Thus, compared to CD20, CD19 has a broader and more specific pattern of B-cell expression. In animal experiment, Anti-CD19 antibodies showed superior depleting activity than traditional CD20 antibodies against a broad subset of B cells, including autoantibody secreting plasma cells (96). However, whether anti-CD19 antibody can be one of the options for treating AE, needed further exploration.
Furthermore, studies have been carried out to explore the role of IL-1 and IL-17 in the pathogenesis of AE (97–99), to treat febrile seizures in AE patients by IL-1 antibodies (100), or to identify the potential role of tramadol, a non-competitive NMDAR antagonist, in reducing movement disorders (101). These drugs alone may not cure encephalitis, but they may be used as complementary treatment options.
To sum up, the timing of upgrade to third-line therapy is very challenging and clinicians need to balance the risk of severe disease with the risk of treatment side effects (102); third-line therapy still provides a therapeutic option for patients with refractory AE and has shown beneficial results in some of the case reports or cohort studies listed in the current study. With ever-growing attention on refractory AE, we can expect that future studies could potentially elucidate these problems and find more effective therapies for AE.
XL: Revise and guide the work. JY: draft the article. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Corsellis JA, Goldberg GJ, Norton AR. “Limbic Encephalitis” and Its Association With Carcinoma. Brain (1968) 91(3):481–96. doi: 10.1093/brain/91.3.481
2. Graus F, Saiz A, Dalmau J. Antibodies and Neuronal Autoimmune Disorders of the CNS. J Neurol (2010) 257:509–17. doi: 10.1007/s00415-009-5431-9
3. Gultekin SH, Rosenfeld MR, Voltz R, Eichen J, Posner JB, Dalmau J. Paraneoplastic Limbic Encephalitis: Neurological Symptoms, Immunological Findings and Tumour Association in 50 Patients. Brain (2000) 123:1481–94. doi: 10.1093/brain/123.7.1481
4. Saraya AW, Worachotsueptrakun K, Vutipongsatorn K, Sonpee C, Hemachudha T. Differences and Diversity of Autoimmune Encephalitis in 77 Cases From a Single Tertiary Care Center. BMC Neurol (2019) 19:273. doi: 10.1186/s12883-019-1501-5
5. Dubey D, Britton J, McKeon A, Gadoth A, Zekeridou A, Lopez Chiriboga SA, et al. Randomized Placebo-Controlled Trial of Intravenous Immunoglobulin in Autoimmune LGI1/CASPR2 Epilepsy. Ann Neurol (2020) 87(2):313–23. doi: 10.1002/ana.25655
6. Taraschenko O, Fox HS, Pittock SJ, Zekeridou A, Gafurova M, Eldridge E, et al. A Mouse Model of Seizures in Anti-N-Methyl-D-Aspartate Receptor Encephalitis. Epilepsia (2019) 60(3):452–63. doi: 10.1111/epi.14662
7. Hughes EG, Peng X, Gleichman AJ, Lai M, Zhou L, Tsou R, et al. Cellular and Synaptic Mechanisms of Anti-NMDA Receptor Encephalitis. J Neurosci (2010) 30(17):5866–75. doi: 10.1523/JNEUROSCI.0167-10.2010
8. Moscato EH, Peng X, Jain A, Parsons TD, Dalmau J, Balice-Gordon RJ. Acute Mechanisms Underlying Antibody Effects in Anti-N-Methyl-D-Aspartate Receptor Encephalitis. Ann Neurol (2014) 76(1):108–19. doi: 10.1002/ana.24195
9. Körtvelyessy P, Bauer J, Stoppel CM, Brück W, Gerth I, Vielhaber S, et al. Complement-Associated Neuronal Loss in a Patient With CASPR2 Antibody-Associated Encephalitis. Neurol Neuroimmunol Neuroinflamm (2015) 2(2):e75. doi: 10.1212/NXI.0000000000000075
10. Titulaer MJ, McCracken L, Gabilondo I, Armangué T, Glaser C, Iizuka T, et al. Treatment and Prognostic Factors for Long-Term Outcome in Patients With Anti-NMDA Receptor Encephalitis: An Observational Cohort Study. Lancet Neurol (2013) 12(2):157–65. doi: 10.1016/S1474-4422(12)70310-1
11. Dalmau J, Lancaster E, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon R. Clinical Experience and Laboratory Investigations in Patients With Anti-NMDAR Encephalitis. Lancet Neurol (2011) 10(1):63–74. doi: 10.1016/S1474-4422(10)70253-2
12. Wingfield T, McHugh C, Vas A, Richardson A, Wilkins E, Bonington A, et al. Autoimmune Encephalitis: A Case Series and Comprehensive Review of the Literature. QJM (2011) 104(11):921–31. doi: 10.1093/qjmed/hcr111
13. Ben Achour N, Ben Younes T, Rebai I, Ben Ahmed M, Kraoua I, Ben Youssef-Turki I. Severe Dysautonomia as a Main Feature of Anti-GAD Encephalitis: Report of a Paediatric Case and Literature Review. Eur J Paediatr Neurol (2018) 22:548–51. doi: 10.1016/j.ejpn.2018.01.004
14. Abboud H, Probasco JC, Irani S, Ances B, Benavides DR, Bradshaw M, et al. Autoimmune Encephalitis: Proposed Best Practice Recommendations for Diagnosis and Acute Management. J Neurol Neurosurg Psychiatry (2021) 92(7):757–68. doi: 10.1136/jnnp-2020-325300
15. Lee WJ, Lee ST, Byun JI, Sunwoo JS, Kim TJ, Lim JA, et al. Rituximab Treatment for Autoimmune Limbic Encephalitis in an Institutional Cohort. Neurology (2016) 86:1683–91. doi: 10.1212/WNL.0000000000002635
16. Nepal G, Shing YK, Yadav JK, Rehrig JH, Ojha R, Huang DY, et al. Efficacy and Safety of Rituximab in Autoimmune Encephalitis: A Meta-Analysis. Acta Neurol Scand (2020) 142(5):449–59. doi: 10.1111/ane.13291
17. Wu XP, Xie GM, Wang HF, Sun Q, Zhuang LP. Etoposide and Immunotherapy can Improve the Outcome of Severe Anti-GABAB R Encephalitis Presenting With Delta Brush: A Case Report. Med (Baltimore) (2020) 99(37):e22087. doi: 10.1097/MD.0000000000022087
18. Rutatangwa A, Mittal N, Francisco C, Nash K, Waubant E. Autoimmune Encephalitis in Children: A Case Series at a Tertiary Care Center. J Child Neurol (2020) 35(9):591–9. doi: 10.1177/0883073820923834
19. Suthar R, Saini AG, Sankhyan N, Sahu JK, Singhi P. Childhood Anti-NMDA Receptor Encephalitis. Indian J Pediatr (2016) 83(7):628–33. doi: 10.1007/s12098-015-1988-8
20. Brummaier T, Pohanka E, Studnicka-Benke A, Pieringer H. Using Cyclophosphamide in Inflammatory Rheumatic Diseases. Eur J Intern Med (2013) 24(7):590–6. doi: 10.1016/j.ejim.2013.02.008
21. Bartolini L, Muscal E. Differences in Treatment of Anti-NMDA Receptor Encephalitis: Results of a Worldwide Survey. J Neurol (2017) 264(4):647–53. doi: 10.1007/s00415-017-8407-1
22. Pruetarat N, Netbaramee W, Pattharathitikul S, Veeravigrom M. Clinical Manifestations, Treatment Outcomes, and Prognostic Factors of Pediatric Anti-NMDAR Encephalitis in Tertiary Care Hospitals: A Multicenter Retrospective/Prospective Cohort Study. Brain Dev (2019) 41(5):436–42. doi: 10.1016/j.braindev.2018.12.009
23. Shim Y, Kim SY, Kim H, Hwang H, Chae JH, Choi J, et al. Clinical Outcomes of Pediatric Anti-NMDA Receptor Encephalitis. Eur J Paediatr Neurol (2020) 29:87–91. doi: 10.1016/j.ejpn.2020.10.001
24. Wright SK, Wood AG. Neurodevelopmental Outcomes in Paediatric Immune-Mediated and Autoimmune Epileptic Encephalopathy. Eur J Paediatr Neurol (2020) 24:53–7. doi: 10.1016/j.ejpn.2019.12.010
25. Schroeder C, Back C, Koc Ü, Strassburger-Krogias K, Reinacher-Schick A, Gold R, et al. Breakthrough Treatment With Bortezomib for a Patient With Anti-NMDAR Encephalitis. Clin Neurol Neurosurg (2018) 172:24–26. doi: 10.1016/j.clineuro.2018.06.005
26. Cordani R, Micalizzi C, Giacomini T, Gastaldi M, Franciotta D, Fioredda F, et al. Bortezomib-Responsive Refractory Anti-N-Methyl-D-Aspartate Receptor Encephalitis. Pediatr Neurol (2020) 103:61–4. doi: 10.1016/j.pediatrneurol.2019.09.004
27. Shin YW, Lee ST, Kim TJ, Jun JS, Chu K. Bortezomib Treatment for Severe Refractory Anti-NMDA Receptor Encephalitis. Ann Clin Transl Neurol (2018) 5(5):598–605. doi: 10.1002/acn3.557
28. Sveinsson O, Granqvist M, Forslin Y, Blennow K, Zetterberg H, Piehl F. Successful Combined Targeting of B- and Plasma Cells in Treatment Refractory Anti-NMDAR Encephalitis. J Neuroimmunol (2017) 312:15–8. doi: 10.1016/j.jneuroim.2017.08.011
29. Keddie S, Crisp SJ, Blackaby J, Cox A, Coles A, Hart M, et al. Plasma Cell Depletion With Bortezomib in the Treatment of Refractory N-Methyl-D-Aspartate (NMDA) Receptor Antibody Encephalitis. Rational Developments in Neuroimmunological Treatment. Eur J Neurol (2018) 25(11):1384–8. doi: 10.1111/ene.13759
30. Benucci M, Tramacere L, Infantino M, Manfredi M, Grossi V, Damiani A. Efficacy of Tocilizumab in Limbic Encephalitis With Anti-CASPR2 Antibodies. Case Rep Neurol Med (2020) 2020:5697670. doi: 10.1155/2020/5697670
31. Randell RL, Adams AV, Van Mater H. Tocilizumab in Refractory Autoimmune Encephalitis: A Series of Pediatric Cases. Pediatr Neurol (2018) 86:66–68. doi: 10.1016/j.pediatrneurol.2018.07.016
32. Jaafar F, Haddad L, Koleilat N, Sharara-Chami R, Shbarou R. Super Refractory Status Epilepticus Secondary to Anti-GAD Antibody Encephalitis Successfully Treated With Aggressive Immunotherapy. Epilepsy Behav Rep (2020) 14:100396. doi: 10.1016/j.ebr.2020.100396
33. Lee WJ, Lee ST, Moon J, Sunwoo JS, Byun JI, Lim JA, et al. Tocilizumab in Autoimmune Encephalitis Refractory to Rituximab: An Institutional Cohort Study. Neurotherapeutics (2016) 13(4):824–32. doi: 10.1007/s13311-016-0442-6
34. Lee WJ, Lee ST, Shin YW, Lee HS, Shin HR, Kim DY, et al. Teratoma Removal, Steroid, IVIG, Rituximab and Tocilizumab (T-SIRT) in Anti-NMDAR Encephalitis. Neurotherapeutics (2021) 18(1):474–87. doi: 10.1007/s13311-020-00921-7
35. Scheibe F, Ostendorf L, Reincke SM, Prüss H, von Brünneck AC, Köhnlein M, et al. Daratumumab Treatment for Therapy-Refractory Anti-CASPR2 Encephalitis. J Neurol (2020) 267(2):317–23. doi: 10.1007/s00415-019-09585-6
36. Ratuszny D, Skripuletz T, Wegner F, Groß M, Falk C, Jacobs R, et al. Case Report: Daratumumab in a Patient With Severe Refractory Anti-NMDA Receptor Encephalitis. Front Neurol (2020) 11:602102. doi: 10.3389/fneur.2020.602102
37. Lim JA, Lee ST, Moon J, Jun JS, Park BS, Byun JI, et al. New Feasible Treatment for Refractory Autoimmune Encephalitis: Low-Dose Interleukin-2. J Neuroimmunol (2016) 299:107–11. doi: 10.1016/j.jneuroim.2016.09.001
38. de Jongste AH, van Gelder T, Bromberg JE, de Graaf MT, Gratama JW, Schreurs MW. A Prospective Open-Label Study of Sirolimus for the Treatment of Anti-Hu Associated Paraneoplastic Neurological Syndromes. Neuro Oncol (2015) 17(1):145–50. doi: 10.1093/neuonc/nou126
39. Nauen DW. Extra-Central Nervous System Target for Assessment and Treatment in Refractory Anti-N-Methyl-D-Aspartate Receptor Encephalitis. J Crit Care (2017) 37:234–6. doi: 10.1016/j.jcrc.2016.09.016
40. Martinez-Hernandez E, Horvath J, Shiloh-Malawsky Y, Sangha N, Martinez-Lage M, Dalmau J. Analysis of Complement and Plasma Cells in the Brain of Patients With Anti-NMDAR Encephalitis. Neurology (2011) 77(6):589–93. doi: 10.1212/WNL.0b013e318228c136
41. Yimer H, Melear J, Faber E, Bensinger WI, Burke JM, Narang M, et al. Daratumumab, Bortezomib, Cyclophosphamide and Dexamethasone in Newly Diagnosed and Relapsed Multiple Myeloma: LYRA Study. Br J Haematol (2019) 185(3):492–502. doi: 10.1111/bjh.15806
42. Kapoor P, Ramakrishnan V, Rajkumar SV. Bortezomib Combination Therapy in Multiple Myeloma. Semin Hematol (2012) 49(3):228–42. doi: 10.1053/j.seminhematol.2012.04.010
43. Chedid MF, Rodrigo E. Bortezomib: A New Promising Therapy for Early Antibody-Mediated Rejection After Liver Transplantation? Transplant Direct (2019) 5(10):e492. doi: 10.1097/TXD.0000000000000931
44. Zhou J, Cheng H, Wang Z, Chen H, Suo C, Zhang H, et al. Bortezomib Attenuates Renal Interstitial Fibrosis in Kidney Transplantation via Regulating the EMT Induced by TNF-α-Smurf1-Akt-mTOR-P70S6K Pathway. J Cell Mol Med (2019) 23(8):5390–402. doi: 10.1111/jcmm.14420
45. Beckman JD, Rollins-Raval MA, Raval JS, Park YA, Mazepa M, Ma A. Bortezomib for Refractory Immune-Mediated Thrombocytopenia Purpura. Am J Ther (2018) 25(2):e270–2. doi: 10.1097/MJT.0000000000000517
46. Ratnasingam S, Walker PA, Tran H, Kaplan ZS, McFadyen JD, Tran H, et al. Bortezomib-Based Antibody Depletion for Refractory Autoimmune Hematological Diseases. Blood Adv (2016) 1(1):31–5. doi: 10.1182/bloodadvances.2016001412
47. Amaya M, Keck F, Lindquist M, Voss K, Scavone L, Kehn-Hall K, et al. The Ubiquitin Proteasome System Plays a Role in Venezuelan Equine Encephalitis Virus Infection. PloS One (2015) 10(4):e0124792. doi: 10.1371/journal.pone.0124792
48. Sinn DI, Lee ST, Chu K, Jung KH, Kim EH, Kim JM, et al. Proteasomal Inhibition in Intracerebral Hemorrhage: Neuroprotective and Anti-Inflammatory Effects of Bortezomib. Neurosci Res (2007) 58(1):12–8. doi: 10.1016/j.neures.2007.01.006
49. O’Riordan D, Houghton J, Chisholm R, O’Driscoll BR. Breathless on Bortezomib. BMJ Case Rep (2011) 2011:bcr0320114028. doi: 10.1136/bcr.03.2011.4028
50. Scheibe F, Prüss H, Mengel AM, Kohler S, Nümann A, Köhnlein M, et al. Bortezomib for Treatment of Therapy-Refractory Anti-NMDA Receptor Encephalitis. Neurology (2017) 88(4):366–70. doi: 10.1212/WNL.0000000000003536
51. Hou M, Eriksson E, Svechnikov K, Jahnukainen K, Söder O, Meinhardt A, et al. Bortezomib Treatment Causes Long-Term Testicular Dysfunction in Young Male Mice. Mol Cancer (2014) 13:155. doi: 10.1186/1476-4598-13-155
52. Yao X, Huang J, Zhong H, Shen N, Faggioni R, Fung M, et al. Targeting Interleukin-6 in Inflammatory Autoimmune Diseases and Cancers. Pharmacol Ther (2014) 141(2):125–39. doi: 10.1016/j.pharmthera.2013.09.004
53. Mihara M, Kasutani K, Okazaki M, Nakamura A, Kawai S, Sugimoto M, et al. Tocilizumab Inhibits Signal Transduction Mediated by Both mIL-6R and sIL-6R, But Not by the Receptors of Other Members of IL-6 Cytokine Family. Int Immunopharmacol (2005) 5(12):1731–40. doi: 10.1016/j.intimp.2005.05.010
54. Sato K, Tsuchiya M, Saldanha J, Koishihara Y, Ohsugi Y, Kishimoto T, et al. Reshaping a Human Antibody to Inhibit the Interleukin 6-Dependent Tumor Cell Growth. Cancer Res (1993) 53(4):851–6.
55. Araki M, Matsuoka T, Miyamoto K, Kusunoki S, Okamoto T, Murata M, et al. Efficacy of the Anti-IL-6 Receptor Antibody Tocilizumab in Neuromyelitis Optica: A Pilot Study. Neurology (2014) 82(15):1302–6. doi: 10.1212/WNL.0000000000000317
56. Hoshino H, Shirai Y, Konishi H, Yamamura T, Shimizu N. Efficacy of Tocilizumab for Fulminant Multiple Sclerosis With a Tumefactive Cervical Lesion: A 12-Year-Old Boy. Mult Scler Relat Disord (2020) 37:101460. doi: 10.1016/j.msard.2019.101460
57. Richebé P, Bailly F, Mariani LL, Pena PS, Pedespan JM, Fautrel B. Report of Two Cases of Tocilizumab Induced Recurrent Meningitis or Meningoencephalitis. Joint Bone Spine (2018) 85(5):643–4. doi: 10.1016/j.jbspin.2018.01.002
58. Williams CJ, Foote A, Choi P. Pseudomonas Meningoencephalitis Masquerading as a Stroke in a Patient on Tocilizumab. BMJ Case Rep (2019) 12(1):bcr–2018-227296. doi: 10.1136/bcr-2018-227296
59. Kobayashi K, Okamoto Y, Inoue H, Usui T, Ihara M, Kawamata J, et al. Leukoencephalopathy With Cognitive Impairment Following Tocilizumab for the Treatment of Rheumatoid Arthritis (RA). Intern Med (2009) 48(15):1307–9. doi: 10.2169/internalmedicine.48.1926
60. Yamaguchi Y, Furukawa K, Yamamoto T, Takahashi Y, Tanaka K, Takahashi M. Multifocal Encephalopathy and Autoimmune-Mediated Limbic Encephalitis Following Tocilizumab Therapy. Intern Med (2014) 53(8):879–82. doi: 10.2169/internalmedicine.53.0615
61. Nooka AK, Kaufman JL, Hofmeister CC, Joseph NS, Heffner TL, Gupta VA, et al. Daratumumab in Multiple Myeloma. Cancer (2019) 125(14):2364–82. doi: 10.1002/cncr.32065
62. Mirgh S, Ahmed R, Agrawal N, Khushoo V, Garg A, Francis S, et al. Will Daratumumab be the Next Game Changer in Early Thymic Precursor-Acute Lymphoblastic Leukaemia? Br J Haematol (2019) 187(2):e33–5. doi: 10.1111/bjh.16154
63. Schwotzer R, Manz MG, Pederiva S, Waibel C, Caspar C, Lerch E, et al. Daratumumab for Relapsed or Refractory AL Amyloidosis With High Plasma Cell Burden. Hematol Oncol (2019) 37(5):595–600. doi: 10.1002/hon.2677
64. Iversen KF, Holdgaard PC, Preiss B, Nyvold CG, Plesner T. Daratumumab for Treatment of Blastic Plasmacytoid Dendritic Cell Neoplasm. A Single-Case Report. Haematologica (2019) 104(9):e432–3. doi: 10.3324/haematol.2018.214635
65. Bathini S, Holtzman NG, Koka R, Singh Z, Wilding E, Zou Y, et al. Refractory Postallogeneic Stem Cell Transplant Pure Red Cell Aplasia in Remission After Treatment With Daratumumab. Am J Hematol (2019) 94(8):E216–9. doi: 10.1002/ajh.25515
66. Spica D, Junker T, Dickenmann M, Schaub S, Steiger J, Rüfli T, et al. Daratumumab for Treatment of Antibody-Mediated Rejection After ABO-Incompatible Kidney Transplantation. Case Rep Nephrol Dial (2019) 9(3):149–57. doi: 10.1159/000503951
67. Even-Or E, Naser Eddin A, Shadur B, Dinur Schejter Y, Najajreh M, Zelig O, et al. Successful Treatment With Daratumumab for Post-HSCT Refractory Hemolytic Anemia. Pediatr Blood Cancer (2020) 67(1):e28010. doi: 10.1002/pbc.28010
68. Blennerhassett R, Sudini L, Gottlieb D, Bhattacharyya A. Post-Allogeneic Transplant Evans Syndrome Successfully Treated With Daratumumab. Br J Haematol (2019) 187(2):e48–51. doi: 10.1111/bjh.16171
69. Woods A, Wooten M, Thompson Heffner L Jr, Waller E. Daratumumab-Associated Hemophagocytic Lymphohistiocytosis. Ann Hematol (2020) 99(1):181–2. doi: 10.1007/s00277-019-03877-7
70. Jamilloux Y, El Jammal T, Vuitton L, Gerfaud-Valentin M, Kerever S, Sève P. JAK Inhibitors for the Treatment of Autoimmune and Inflammatory Diseases. Autoimmun Rev (2019) 18(11):102390. doi: 10.1016/j.autrev.2019.102390
71. Bethesda. Tofacitinib. In: LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases. (2020).
72. Dhillon S. Tofacitinib: A Review in Rheumatoid Arthritis. Drugs (2017) 77(18):1987–2001. doi: 10.1007/s40265-017-0835-9
73. Gratacós Masmitjà J, González Fernández CM, Gómez Castro S, Rebollo Laserna FJ. Efficacy of Tofacitinib in the Treatment of Psoriatic Arthritis: A Systematic Review. Adv Ther (2021) 38(2):868–84. doi: 10.1007/s12325-020-01585-7
74. Panés J, Gisbert JP. Efficacy of Tofacitinib Treatment in Ulcerative Colitis. Gastroenterol Hepatol (2019) 42(6):403–12. doi: 10.1016/j.gastrohep.2019.03.002
75. Rawlings JS, Rosler KM, Harrison DA. The JAK/STAT Signaling Pathway. J Cell Sci (2004) 117(Pt 8):1281–3. doi: 10.1242/jcs.00963
76. Zhou Y, Leng X, Luo S, Su Z, Luo X, Guo H, et al. Tolerogenic Dendritic Cells Generated With Tofacitinib Ameliorate Experimental Autoimmune Encephalomyelitis Through Modulation of Th17/Treg Balance. J Immunol Res (2016) 2016:5021537. doi: 10.1155/2016/5021537
77. Yoshida H, Kimura A, Fukaya T, Sekiya T, Morita R, Shichita T, et al. Low Dose CP-690,550 (Tofacitinib), a Pan-JAK Inhibitor, Accelerates the Onset of Experimental Autoimmune Encephalomyelitis by Potentiating Th17 Differentiation. Biochem Biophys Res Commun (2012) 418(2):234–40. doi: 10.1016/j.bbrc.2011.12.156
78. Massoud F, Ismail II, Al-Hashel JY, Abboud H. CNS Demyelination During Tofacitinib Therapy: First Report. Mult Scler Relat Disord (2020) 46:102568. doi: 10.1016/j.msard.2020.102568
79. Fukuyama T, Tschernig T, Qi Y, Volmer DA, Bäumer W. Aggression Behaviour Induced by Oral Administration of the Janus-Kinase Inhibitor Tofacitinib, But Not Oclacitinib, Under Stressful Conditions. Eur J Pharmacol (2015) 764:278–82. doi: 10.1016/j.ejphar.2015.06.060
80. Jang Y, Lee WJ, Lee HS, Chu K, Lee SK, Lee ST. Tofacitinib Treatment for Refractory Autoimmune Encephalitis. Epilepsia (2021) 62(4):e53–9. doi: 10.1111/epi.16848
81. Morgan DA, Ruscetti FW, Gallo R. Selective In Vitro Growth of T Lymphocytes From Normal Human Bone Marrows. Science (1976) 193(4257):1007–8. doi: 10.1126/science.181845
82. Mizui M, Tsokos GC. Low-Dose IL-2 in the Treatment of Lupus. Curr Rheumatol Rep (2016) 18(11):68. doi: 10.1007/s11926-016-0617-5
83. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T Cells and Immune Tolerance. Cell (2008) 133(5):775–87. doi: 10.1016/j.cell.2008.05.009
84. He J, Zhang X, Wei Y, Sun X, Chen Y, Deng J, et al. Low-Dose Interleukin-2 Treatment Selectively Modulates CD4(+) T Cell Subsets in Patients With Systemic Lupus Erythematosus. Nat Med (2016) 22(9):991–3. doi: 10.1038/nm.4148
85. Przepiorka D, Jaeckle KA, Birdwell RR, Fuller GN, Kumar AJ, Huh YO, et al. Successful Treatment of Progressive Multifocal Leukoencephalopathy With Low-Dose Interleukin-2. Bone Marrow Transplant (1997) 20(11):983–7. doi: 10.1038/sj.bmt.1701010
86. Humrich JY, Riemekasten G. Low-Dose Interleukin-2 Therapy for the Treatment of Systemic Lupus Erythematosus. Curr Opin Rheumatol (2019) 31(2):208–12. doi: 10.1097/BOR.0000000000000575
87. Powell JD, Delgoffe GM. The Mammalian Target of Rapamycin: Linking T Cell Differentiation, Function, and Metabolism. Immunity (2010) 33(3):301–11. doi: 10.1016/j.immuni.2010.09.002
88. Canivet C, Menasria R, Rhéaume C, Piret J, Boivin G. Valacyclovir Combined With Artesunate or Rapamycin Improves the Outcome of Herpes Simplex Virus Encephalitis in Mice Compared to Antiviral Therapy Alone. Antiviral Res (2015) 123:105–13. doi: 10.1016/j.antiviral.2015.09.007
89. Li D, Wang C, Yao Y, Chen L, Liu G, Zhang R, et al. Mtorc1 Pathway Disruption Ameliorates Brain Inflammation Following Stroke via a Shift in Microglia Phenotype From M1 Type to M2 Type. FASEB J (2016) 30(10):3388–99. doi: 10.1096/fj.201600495R
90. Graus F, Titulaer MJ, Balu R, Benseler S, Bien CG, Cellucci T, et al. A Clinical Approach to Diagnosis of Autoimmune Encephalitis. Lancet Neurol (2016) 15(4):391–404. doi: 10.1016/S1474-4422(15)00401-9
91. Hansen N, Ernst L, Rüber T, Widman G, Becker AJ, Elger CE, et al. Pre- and Long-Term Postoperative Courses of Hippocampus-Associated Memory Impairment in Epilepsy Patients With Antibody-Associated Limbic Encephalitis and Selective Amygdalohippocampectomy. Epilepsy Behav (2018) 79:93–9. doi: 10.1016/j.yebeh.2017.10.033
92. Mueller SH, Färber A, Prüss H, Melzer N, Golombeck KS, Kümpfel T, et al. Genetic Predisposition in Anti-LGI1 and Anti-NMDA Receptor Encephalitis. Ann Neurol (2018) 83(4):863–9. doi: 10.1002/ana.25216
93. Wagnon I, Hélie P, Bardou I, Regnauld C, Lesec L, Leprince J, et al. Autoimmune Encephalitis Mediated by B-Cell Response Against N-Methyl-D-Aspartate Receptor. Brain (2020) 143(10):2957–72. doi: 10.1093/brain/awaa250
94. Widman G, Golombeck K, Hautzel H, Gross CC, Quesada CM, Witt JA, et al. Treating a GAD65 Antibody-Associated Limbic Encephalitis With Basiliximab: A Case Study. Front Neurol (2015) 6:167. doi: 10.3389/fneur.2015.00167
95. Hansen N, Önder D, Schwing K, Widman G, Leelaarporn P, Prusseit I, et al. CD19+ B-Cells in Autoantibody-Negative Limbic Encephalitis. Epilepsy Behav (2020) 106:107016. doi: 10.1016/j.yebeh.2020.107016
96. Chen D, Gallagher S, Monson NL, Herbst R, Wang Y. Inebilizumab, a B Cell-Depleting Anti-CD19 Antibody for the Treatment of Autoimmune Neurological Diseases: Insights From Preclinical Studies. J Clin Med (2016) 5(12):107. doi: 10.3390/jcm5120107
97. Peng Y, Liu B, Pei S, Zheng D, Wang Z, Ji T, et al. Higher CSF Levels of NLRP3 Inflammasome Is Associated With Poor Prognosis of Anti-N-Methyl-D-Aspartate Receptor Encephalitis. Front Immunol (2019) 10:905. doi: 10.3389/fimmu.2019.00905
98. Ai P, Zhang X, Xie Z, Liu G, Liu X, Pan S, et al. The HMGB1 is Increased in CSF of Patients With an Anti-NMDAR Encephalitis. Acta Neurol Scand (2018) 137(2):277–82. doi: 10.1111/ane.12850
99. Zeng C, Chen L, Chen B, Cai Y, Li P, Yan L, et al. Th17 Cells Were Recruited and Accumulated in the Cerebrospinal Fluid and Correlated With the Poor Prognosis of Anti-NMDAR Encephalitis. Acta Biochim Biophys Sin (Shanghai) (2018) 50(12):1266–73. doi: 10.1093/abbs/gmy137
100. Novroski AR, Baldwin KJ. Chronic Autoimmune Meningoencephalitis and Periodic Fever Syndrome Treated With Anakinra. Case Rep Neurol (2017) 9(1):91–7. doi: 10.1159/000472147
101. Lazzarin SM, Vabanesi M, Cecchetti G, Fazio R, Fanelli GF, Volonté MA, et al. Refractory Anti-NMDAR Encephalitis Successfully Treated With Bortezomib and Associated Movements Disorders Controlled With Tramadol: A Case Report With Literature Review. J Neurol (2020) 267(8):2462–8. doi: 10.1007/s00415-020-09988-w
Keywords: autoimmune encephalitis, refractory, third-line therapy, rituximab, neuroimmunology
Citation: Yang J and Liu X (2021) Immunotherapy for Refractory Autoimmune Encephalitis. Front. Immunol. 12:790962. doi: 10.3389/fimmu.2021.790962
Received: 07 October 2021; Accepted: 02 December 2021;
Published: 16 December 2021.
Edited by:
Nancy Agmon-Levin, Sheba Medical Center, IsraelReviewed by:
Sudarshini Ramanathan, The University of Sydney, AustraliaCopyright © 2021 Yang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xueyan Liu, bGl1X3h1ZXlhbjIwMDRAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.