 Jianan Zhao
Jianan Zhao Shicheng Guo
Shicheng Guo Steven J. Schrodi
Steven J. Schrodi Dongyi He
Dongyi He- 1Guanghua Clinical Medical College, Shanghai University of Traditional Chinese Medicine, Shanghai, China
- 2Department of Rheumatology, Shanghai Guanghua Hospital, Shanghai University of Traditional Chinese Medicine, Shanghai, China
- 3Computation and Informatics in Biology and Medicine, University of Wisconsin-Madison, Madison, WI, United States
- 4Department of Medical Genetics, School of Medicine and Public Health, University of Wisconsin-Madison, Madison, WI, United States
- 5Arthritis Institute of Integrated Traditional and Western Medicine, Shanghai Chinese Medicine Research Institute, Shanghai, China
Rheumatoid arthritis is an autoimmune disease that exhibits significant clinical heterogeneity. There are various treatments for rheumatoid arthritis, including disease-modifying anti-rheumatic drugs (DMARDs), glucocorticoids, non-steroidal anti-inflammatory drugs (NSAIDs), and inflammatory cytokine inhibitors (ICI), typically associated with differentiated clinical effects and characteristics. Personalized responsiveness is observed to the standard treatment due to the pathophysiological heterogeneity in rheumatoid arthritis, resulting in an overall poor prognosis. Understanding the role of individual variation in cellular and molecular mechanisms related to rheumatoid arthritis will considerably improve clinical care and patient outcomes. In this review, we discuss the source of pathophysiological heterogeneity derived from genetic, molecular, and cellular heterogeneity and their possible impact on precision medicine and personalized treatment of rheumatoid arthritis. We provide emphasized description of the heterogeneity derived from mast cells, monocyte cell, macrophage fibroblast-like synoviocytes and, interactions within immune cells and with inflammatory cytokines, as well as the potential as a new therapeutic target to develop a novel treatment approach. Finally, we summarize the latest clinical trials of treatment options for rheumatoid arthritis and provide a suggestive framework for implementing preclinical and clinical experimental results into clinical practice.
Introduction
Rheumatoid arthritis (RA) is an autoimmune disease characterized by chronic inflammation of the synovial joints, pannus formation, progressive bone erosion, and joint destruction. Patients typically present joint swelling and tenderness, which can progress to serious disability, severely affecting the quality of the patient’s physical and mental life (1). RA affects approximately 1% of the world’s population and disproportionally affects female population (2). RA development is a continuous, progressive, systemic pathology process and multiple autoantibodies, including rheumatoid factor (RF) and anti-citrullinated protein antibodies (ACPA), are detectable in serum before the onset or in the early stage of RA (3–5). With the gradual interaction of various immnue and fibroblast cells and cytokines, synovial tissue gradually produces chronic inflammation accompanied by bone erosion and destruction, resulting in various clinical symptoms and injuries (3–5). Further, a number of organ systems can be damaged by the systemic inflammation, such as cardiac tissue, vascular system, kidneys, lung tissue, and the nervous system (6–8).
Ideally, chronic disease management approaches include preventive strategies. Modern medicine utilizes therapies that target the disease mechanism, the so-called “precision individualized diagnosis and treatment.” Discerning the specific environmental, cellular and molecular mechanisms suitable for early intervention is challenging given the complexity of etiological factors that give rise to RA. However, there is evidence that clinical monitoring of RA symptomology can improve the physical and mental health of patients (9, 10).
The pathogenesis of RA is thought to be involved with the interaction of genetic, epigenetics (11), environmental, metabolic, immune, and microbial factors. The relationship between genetic, epigenetic, metabolic, and microbial factors and RA has been widely reviewed (12–14). The progression of the disease is also influenced by the crosstalk among a variety of immune cells, such as T cells, B cells, monocytes, macrophages, neutrophils, mast cells, dendritic cells, T-reg cells, and fibroblast-like synoviocytes (FLS). These immune cells show plasticity in the disease microenvironment and heterogeneity in their roles depending on the context of the disease. Importantly, the mechanisms for immune cell-mediated synovial inflammation and cartilage destruction may not be active in all patients, and the extent of these effects varies from patient to patient and across the disease stages. Patients may show resistance (poor to no response) to one treatment and significant recovery with another. The considerable degree of clinical heterogeneity in RA affects the accuracy of a patient’s prognosis. Therefore, our most urgent challenge is to evaluate the heterogeneity inherent in the pathophysiology of RA and to identify the mechanisms of action in important cell subsets. Here, we summarize and comment on a variety of heterogeneous features to inform the development of precision and personalized medicine to RA which includes genetic variation, the various expression patterns in the synovium, and the heterogeneity of RA relevant cells, such as FLS, monocytes, macrophages, and mast cells.
Genetic Heterogeneity in Rheumatoid Arthritis: Susceptibility and Clinical Implications
RA is a heritable autoimmnune disease mediated by genetic (15), epigenetic (11, 16), genetic-epigenetic (17) and genetic-environment (18) interactions while treatment usually take epigenetics and immnue factors as therapeutic targets (19). These inherited factors influence the heterogeneity of disease progression and determine the underlying set of cellular and molecular mechanisms. The role of heritability has been dissected in linkage and association studies, revealing the molecular variation underlying RA risk variability. The most evident aspects include (i) class II HLA genes, primarily HLA-DRB1, segregate variants that elevate RA risk 2–3-fold through aberrant antigen presentation (20–22); (ii) the 620W allele of the protein tyrosine phosphatase nonreceptor 22 (PTPN22) gene, which generates various effects across numerous immune cell types and confers elevated risk of autoimmunity (15, 23); (iii) chemokine receptor genes, including C-C chemokine receptor 6 (CCR6) (15, 24, 25); (iv) peptidyl arginine deiminase type IV (PADI4), which encodes peptidylarginine deiminase type 4 (involved in protein citrullination) (26, 27); (v) the transcription factor-encoding gene signal transducer and activator of transcription 4 protein (STAT4) (15, 25), (vi) cytotoxic T-lymphocyte antigen 4 (CTLA4), encoding for the cytotoxic T-cell associated protein (25, 28), and (vii) the B-cell cell surface receptor gene, CD40 (29, 30). Although these genetic factors predispose individuals to RA, patients exhibit a wide variety of combinations of these variants. This genetic variation across patients is considered to partially drive the heterogeneity in RA pathophysiology, clinical presentation, and response to therapies (31) (See Table 1).

Table 1 Main molecular variation underlying the RA risk variability.
RA genetic risk factors, which underlie molecular and cellular perturbations, exhibit substantial differences across ethnic backgrounds. Indeed, this is a feature of many autoimmune diseases, such as multiple sclerosis, systemic lupus erythematosus, and type 1 diabetes. There are variations in the molecular pathophysiology of RA that vary across ancestral genetic backgrounds. For example, the 620W allele of the PTPN22 gene displays a remarkable frequency gradient across Europe and is largely absent outside the continent (37). Therefore, the impact of the allele on critically important autoimmune mechanisms (e.g., T-cell activation and B cell pruning) has this geographical variation. The effects of PADI4 on RA susceptibility, which may be driven by citrullination of arginine residues and/or leukocyte development, mostly affects East Asian patents. As clinical treatments progress toward more individualized approaches, a thorough understanding of these molecular effects will aid both therapeutic development and clinical implementation by medical practitioners. Seropositive RA (ACPA-positive and/or RF-positive) carries a different genetic susceptibility profile compared to seronegative RA. This fundamental difference in RA subtypes may be partly due to differences in genetic factors within the patient population. For example, HLA-DRB1 haplotypes and PTPN22 R620W are primarily correlated with seropositive disease (23, 38), whereas other susceptibility variants are similar between seropositive and seronegative diseases.
The set of RA susceptibility variants is likely to play a role in disease severity. A genome-wide study of 384 autoantibody-positive RA patients showed that the single nucleotide polymorphism (SNP) at position 2q34 of sperm-associated antigen 16 (SPAG16) in monocytes, rs7607479, has a protective effect on joints. This SNP primarily regulates the secretion of matrix metalloproteinases (MMPs), the expression of SPAG16 protein, and mRNA in the RA synovium and FLS (32). Baseline ACPA positivity is associated with increased radiological damage, and the genetic polymorphism rs2900180 in the TRAF1/C5 locus is associated with aggressive disease (33). In addition, a genome-wide study of 262 ACPA-negative RA patients showed that 33 SNPs were associated with joint destruction. Among them, the rs2833522 region contains H3K4me3 histone markers, transcription factors, and long non-coding RNAs, which are related to the degree of bone destruction in ACPA-negative RA patients (34). A modern and more accurate method of predicting the impact of SNPs on RA-related radiological characteristics involves the combination of genome-wide association study (GWAS) with bioinformatic analysis and repeated machine learning (39).
Although the treatment for RA has advanced considerably over the past few decades, nonsteroidal anti-inflammatory drugs (NSAIDs), DMARDs, corticosteroids, and biologics have shown an unequally efficacy cross some patients (40). This variation in the response to therapy also has a genetic component. Some studies have explored the effect of TNF-α gene polymorphism on anti-TNF-a therapy. Among them, single nucleotide polymorphisms (SNPs)-308 G/G, -857 C/T, +489 GG and GA, HLA-DRB1-encoding SE (allele *0404 and allele *0101) is beneficial to etanercept, while SNP-308 A/A and TNFR1A AA has the opposite effect (41, 42). Employing the dialogue on reverse engineering assessment and methods (DREAM), a Gaussian regression model was created to predict the changes in the DAS28 scores of patients with RA receiving tumor necrosis factor-α (TNF-α) inhibitor therapy, by using the expression patterns of genes involved in insulin secretion (PDZD2), immune response (CD84), and eicosanoid synthesis (PLA2G4A). Complex geographical variation has caused obstacles in the modeling of genetic markers. However, using extensive genetic subtype modeling, it was found that specific genetic markers were shared by particular populations and their identification could improve the prediction of efficacy of anti-TNF-α therapeutics (43). The SNP at position 1q23 (rs6427528) is associated with changes in RA’s disease activity scores in patients treated with etanercept, an anti-TNF-α drug. The SNP may disrupt transcription factor binding site motifs of the 3’ UTR of an immune-related gene, CD84 (an immune-related gene), and the allele related to a better response to etanercept was linked to higher gene expression levels of CD84 in peripheral blood mononuclear cells (35). In addition, studies have shown that the PTPRC SNP (rs10919563) was associated with an excellent response to anti-TNF-α therapy in patients with RA (36), and the rs7195994 variant at the FTO locus has been related to a clinical response with infliximab (44). In the genome-wide data for the RA population’s response to methotrexate (MTX), ten new risk sites were associated with poor MTX response. The genes that have been verified include GHFR, FGPS, and TYMS (all three related to purine and nucleotide metabolism) and ENOSF1 (which interacts with TYMS at the transcriptional level) (45).
Heterogeneous Expression Pattern of Synovial Tissue in Rheumatoid Arthritis
The structure of healthy synovial tissues has been well described (46). Understanding the difference between healthy and abnormal synovial tissue structure is essential for exploring the heterogeneity of RA. Healthy synovium consists of two parts: (i) the intima layer composed of macrophage-like synoviocytes (MLSs) and FLSs and (ii) the vascularized sublining layer comprising fibroblasts, macrophages, and the collagenous extracellular matrix (46, 47). Maintaining a healthy synovium is essential for optimal joint function and structure. By supplying nutrients for chondrocytes and preserving various cellular components of the synovial fluid, the synovium also plays a vital role in the homeostasis of the joint microenvironment.
The FLSs (unlike synovial macrophages) in the synovial intima express CD55; this expression can be used to discriminate FLSs from synovial macrophages (47, 48). Intimal FLSs also produce hyaluronic acid to inhibit adhesion and promote lubrication of joints, and the rate of secretion of hyaluronic acid depends on the mechanical stimulation of FLS. In addition, intimal FLSs coregulate immune responses with macrophages derived from monocytes (46).
In RA, the number of cells in the synovial intima substantially increases, primarily because of the abnormal proliferation of FLS. Macrophages flow into the vascular compartment, induced by a variety of cytokines and chemokines, and release various pro-inflammatory factors. These immune cells cooperate with FLSs and B cells to facilitate inflammation (49, 50). The salient feature of RA synovial tissue is the formation of a pannus (a source of matrix-degrading enzymes), which exacerbates cartilage erosion, inflammation, and joint destruction (51); Multiple histological studies have shown that the pannus is primarily composed of fibroblasts, macrophages, multinucleated neutrophils, and mesenchymal cells, though the roles of these cells in RA vary (52–60). With synovial biopsy technology, researchers can evaluate the cartilage-pannus junction (CPJ) and non-CPJ sites in human synovial tissue to identify the mechanisms and characteristics of cartilage and bone erosion in RA. Cell populations vary between the CPJ and non-CPJ. The most common cell populations in the CPJ include macrophages, FLSs, osteoclasts, chondrocytes, and mast cells. In the non-CPJ, the sublining layer has T cells, B cells, differentiated plasma cells, antigen-presenting cells, natural killer cells, and macrophages, and is characterized by synovial neovascularization. The dominant cell populations are FLSs and macrophages (61). Pannocytes, a unique type of rhombohedral cells, exist in the CPJ site and express high levels of transcriptional proto-oncogenes c-FOS, c-MYC, and c-JUN and express MMP-1, cathepsin B, and cathepsin L. Such expression may represent the early stage of mesenchymal cell differentiation; however, the role of pannocytes in bone destruction in RA remains to be elucidated (62, 63).
Some studies have described four modes of synovial histology in RA, including lymphoid, myeloid, pauci-immune, and fibroid variants (64, 65). All four models have unique gene expression characteristics. Based on such characteristics, the corresponding biomarkers should be identified to have reference values for different therapies. Lymphoid variants express high levels of genes related to the activation and differentiation of lymphoid B cells and lymphoid T cells, immunoglobulin production, antigen presentation, and cytokine signaling (Jak/STAT, interleukin [IL]-17), including CD19, CD20, XBP-1, CD38, CXCL13, and down-regulate the genes expression of TGF-α, and down-regulate the genes expression related to Wnt signaling, mesenchymal cell proliferation, proteolysis, cell transport, and ribosome metabolic processing (64). The presence of autoimmune B cell antigens and other lymphoid factors may promote the progression of synovitis and lead to a poor response to anti-TNF-α therapy (64). In addition, accumulation of cell aggregates reflects the proliferation of B and T cells in the lymphoid synovium. The existence of a large number of such aggregates may be one of the reasons for poor responses to anti-TNF-α therapy in RA (66).
Myeloid variants express high levels of genes related to chemotaxis, TNFα and IL-1β production, Toll-like receptor and nucleotide-binding oligomerization domain (NOD)-like receptor signaling, Fcγ-receptor-meditated phagocytosis, and proliferation of mononuclear cells. They highly activate nuclear factor kappa-light-chain enhancer of activated B cells (NF-κB) pathway genes, including TNFα, IL-1β, IL-1Rα, ICAM1, and MyD88, the inflammatory chemokines CC-chemokine ligand (CCL)-2 and IL-8, granulocyte and inflammatory macrophage lineage genes such as S100A12, CD14 and OSCAR. In contrast, they downregulate the expression of genes related to transcription and splicing. The myeloid synovial tissue has fewer cell aggregates (primarily containing pro-inflammatory M1 mononuclear macrophages), and it highly expresses a variety of inflammatory genes; therefore, such tissues are more responsive to anti-TNF-α therapy (64). Studies have shown that RA synovium across joints that express high levels of inflammatory genes, have high levels of inflammation (i.e., high DAS28 score, high CRP level, high erythrocyte sedimentation rate, high platelet count, and a shorter course of disease) (28);
The pauci-immune synovium is primarily enriched in genes involved in the process of inflammation and wound healing. It shows gene expression patterns related to several other phenotypes, and expresses high levels of IL-6, the IL-6 receptor (IL-6R) components IL-6ST/gp130 and STAT3. Such expression patterns are consistent with previous studies on the biological role of IL-6 in RA. The pauci-immune synovium is primarily characterized by low levels of inflammation and accumulation of anti-inflammatory M2 monocytes/macrophages, which increases the potential for a poor response to anti-TNF-α therapy and B cell depletion therapy (64, 67–70).
Fibroid variants are primarily enriched in genes involved in the regulation of FLSs and osteoclasts/osteoblasts, such as transforming growth factor (TGF)-β signaling, bone morphogenetic protein (BMP) signaling together with associated Sma Mothers Against Decapentaplegic (SMAD) binding, as well as endocytosis and cell projection process-related genes. They express high levels of Wnt pathway genes while significantly downregulating multiple immune-system processes (associated with B cells, immunoglobulins, myeloid cells), the innate immune response (including NOD-like receptor signaling), and chemotactic processes. The expression levels of angiogenesis-related genes in fibroid variant synovium is high, which is correlated with reduced response to anti-TNF-α therapy (64).
Importantly, RA seropositive patients are limited to lymphoid, myeloid, and pauci-immune types, and both soluble intercellular adhesion molecule-1 (sICAM-1) and CXCL13 levels are elevated in these patients. The sICAM-1 and CXCL13 levels can represent the two synovial phenotypes of myeloid lymphoid cells, respectively, and can be used as a biomarker for predicting anti-TNF-α and anti-IL-6R therapies. Synovial migration of inflammatory cells can be induced by sICAM1 binding to receptors of neutrophils and monocytes in response to TNF-α stimulation (64, 71). CXCL13 is a B-cell-specific chemokine that plays a key role in secondary lymphoid tissues and germinal center tissues (72). IL-6/IL-6R and lymphoid B cell-driven synovitis are highly correlated (a consistent result in multiple studies), but in myeloid synovial tissue dominated by activated monocytes, anti-TNF-α therapy is most effective. A major limitation is the necessity for multiple biomarkers to accurately apply RA therapy. Predicting the efficacy of different therapies for RA based on the heterogeneity of synovial tissue is a promising direction for future research (64, 71, 73, 74).
Mast Cells, a Novel Target for Treating Rheumatoid Arthritis, Which Contributes to Therapeutic Heterogeneity
Mast cells (MCs) consist primarily of two subtypes, tryptase-positive (MCT) cells and tryptase/chymase double-positive (MCTC) cells, and play a role in RA by promoting inflammation, osteoclast differentiation, and angiogenesis (75). Tetlow et al. studied the activation and degranulation of MCs at the CPJ. Levels of stromelysin-1, TNF-α, IL-1β, extracellular MC tryptase, and matrix degradation mediators at the CPJ site are all significantly elevated, suggesting that MC degranulation is related to local matrix degradation and inflammation (76). In RA synovial tissue, the number and proportion of MC subpopulations are characterized by significant modifications. In normal synovium, mast cells account for approximately 5% of synovial cells and primarily contain MCTC cell subpopulations. Gotis-Graham et al. found that the number of synovial MCT cells in early RA patients increased to three times the number of MCTC cells and this ratio strongly correlated with histological inflammation (77). The researchers found that RA patients in the active stage of the disease have a higher number of MCs than patients with end-stage RA (78). In addition, MCs can promote inflammation by releasing various chemokines and pro-inflammatory factors in cooperation with other cells. Mast cells mediate the release of IL-6, IL-8, TNF-α, histamine, heparin, monocyte chemoattractant protein-1 (MCP-1), MIP-1α, and RANTES, and they stimulate monocytes/macrophages to produce/release IL-1 family members and TNF-α, which exacerbate inflammation (79–82). MCs can also participate in bone destruction by promoting the release of proteases, histamine, TNF-α, IL-6, IL-11, IFN-γ, RANKL, and other mediators. MCs increase the expression level of TNF-α, IL-1β, IL-6, IL-17, RANKL, and MMP-9 genes in response to IL-33 stimulation and stimulate human CD14+ monocytes to differentiate into TRAP-positive osteoclasts (83). Shin et al. found that secretion of tryptase β by MCs was abundant in experimental models of arthritic mice, and the tryptase β/heparin complex promoted FLS to express inflammation-mediating neutrophil chemokines CXCL1/KC, CXCL5/LIX, and CXCL8/IL-8; additionally, tryptase β-deficient mice showed lower disease activity and bone destruction (84). Another study found that experimental models of arthritic mice lacking mouse MC protease-4 had lower clinical scores, as well as lower levels of cartilage destruction and pannus formation (85). Studies have shown that histamine released by MCs can stimulate the expression of the histamine H4 receptor and RANKL in monocytes and induce osteoclast differentiation (86). Vascular endothelial growth factor (VEGF), tryptase, and fibroblast growth factor-2 (FGF-2) released by MCs can stimulate FLS proliferation and promote the formation of pannus (87). Studies have also found that CXCR3 is strongly expressed on the surface of mast cells in synovial tissues, leading to increased recruitment of MCs and increased levels of chemokines such as CXCL9 and CXCL10, exacerbating RA (88).
Monocyte Heterogeneity and Plasticity in Rheumatoid Arthritis Pathogenesis
The origin and typing of monocytes have been extensively studied across multiple species, including humans, mice, pigs, monkeys, and horses (89). Monocytes were first defined in a classic study conducted by Ehrlich et al. in the 20th century (90). Monocytes are circulating blood cells that develop in human bone marrow (BM) from the common myeloid progenitor (CMP). Together with macrophages and dendritic cells, they are members of the mononuclear phagocyte system (91, 92). The widely used monocyte subpopulation classification standard is based on the expression of CD14 and CD16. The monocyte population is primarily divided into three subpopulations: the classic monocyte population (CD14++), which accounts for approximately 80% of the blood mononuclear cells in healthy individuals, the intermediate monocyte population (CD14++CD16++), and the non-classical monocyte population (CD14+CD16++). The latter two types of CD16-expressing monocytes account for a relatively small proportion in healthy individuals, but their number can increase in different pathological conditions (93). Changes in monocyte subpopulations can be used as biomarkers of RA disease activity and to assess the clinical response. Studies have reported irregularities of monocyte subsets in patients with RA. The proportion of classical monocytes in patients with early RA is relatively low, and the proportions of intermediate and non-classical monocytes are relatively high (94). Similarly, Tsukamoto et al. found that the proportion of intermediate monocyte subpopulation was positively correlated with RA disease activity and negatively correlated with the prevalence of the classical monocyte subpopulation; intermediate monocytes express CD16 under the stimulation of IL-10 (95). In addition, studies have found that classic monocytes and intermediate monocyte subpopulations can predict the clinical response to MTX (96). Increased expression levels of CD16 on CD14++ monocytes in RA patients leads to an intensified response of IgG-containing immune complexes (IC) and the excessive production of pro-inflammatory factors. This change may cause a lack of responsiveness to MTX therapy (97).
Monocytes display phenotypic heterogeneity and plasticity, expressing a variety of cell receptors and secreting various cytokines, including CD14, CD16, HLA-DR, Toll-like receptor, B1 integrins, B2 integrins, a proliferation-inducing ligand (APRIL), CCR2, CX3CR1, siglec-1, and interleukin family cytokines (e.g., IL-18, IL-1β, IL-6, IL-1, IL-32, IL-33, IL-10, and IL-11); depending on the microenvironmental stimuli, they can differentiate into macrophages and dendritic cells (98). Further clarification of the different mechanisms of expressed receptors and secreted cytokines can inform targeted clinical treatment plans. In response to LPS stimulation and binding to the LPS-binding protein in the plasma, the toll-like receptor (TLR)-4 binds to CD14 on the cell membrane, inducing the release of different mediators, including pro-inflammatory chemokines (e.g., IP10) and cytokines (e.g., TNF-α, IL-6, and IL-1) (99, 100). CD16 (Fc gamma RIII) is another receptor for the lgG Fc fragment expressed in the monocyte population (101). Compared with that in a control group, the number of monocytes expressing CD16+ increased sharply in RA patients (102). McGarry et al. found that CD14+ monocytes displayed mitochondrial respiration, biogenesis, enhanced glucose consumption and mitochondrial morphological changes, and they enhanced gene expression of key glycolytic enzymes, such as HIFIα, HK2, and PEKFB. Blocking of STAT3 inhibits this forced glycolytic flux along with inflammation and may represent a potential therapeutic approach for preclinical RA (103). Belge et al. found that, compared with classic monocytes, CD14++CD16++ monocytes expressed higher levels of HLA-DR antigens and can produce higher levels of TNF-α. The mechanism involves an interaction of CD14 and Pam3Cys with TLR2 (104, 105). Yoon et al. showed that in the presence of TGF-β, IFN-γ induces the expression of HLA-DR and CD80 and CD276 in synovial intermediate monocyte subpopulations, which act as signal one and signal two, respectively, to induce CD4+ T cell polarization to Th1 and Th17 cells, which secrete more IFN-γ and IL-17. CD16+ monocytes can also promote the release of TGF-β, which promotes synovial inflammation through positive feedback (106). Iwahashi et al. showed that the human mitochondrial protein HSP60, which is widely expressed in RA synovium, can induce CD16+ monocytes to produce TNF-α, possibly through cascade activation of MyD88-NF-κB-mitogen-activated protein kinase (MAPK) (107). The classic and intermediate monocyte subpopulations in the synovium and blood of RA patients express TLR2 in large quantities, and all three monocyte subpopulations express TLR9; and the subpopulation of classical monocytes is triggered produces pro-inflammatory cytokines in response to TLR2 and TLR9 agonist stimulation (108). FLS expresses TLR1–6 (TLR3 and TLR4 are highly expressed), which may be involved in the activation of pathways related to inflammation and joint destruction in early RA (109). Thwaites et al. showed that the activation of monocyte surface receptors TLR1 and TLR2 in patients with RA mediated the increase in the levels of IL-6 and TNF-α, and the activation of TLR5 mediated the increase in the levels of IL-6 and IL-10. IL-6 induced by TLR1/2 is related to the DAS28 score, a commonly used score to measure disease activity in RA (110). Alpha-enolase (ENO-1) can be produced by FLSs, promoting the proliferation and survival of the cells and can combine with TLR4 on monocytes to produce pro-inflammatory factors and chemokines, such as TNF-α, IL-1β, IL-6, CCL3, IL-8, and CXCL1 (111, 112). In addition, the levels of surface expression on the monocyte subpopulations for transmembrane TNF (tmTNF) are high in all RA patients. Anti-TNF antibodies can act as ligands, binding to tmTNF and mediating reverse signal transmission (113). Anti-TNF binding to tmTNF can also exert anti-inflammatory effects by inducing the release of the decoy receptors sTNFR1, sIL-1R1, and sIL-1R2 and increasing the production of IL-10. In addition, patients whose monocytes express TNFR1 have a lower disease activity score, which may be due to inhibited inflammation caused by sTNFR1 neutralizing TNF or through the apoptosis of pro-inflammatory monocyte subpopulations by the activation of the TNFR1 exogenous death receptor pathway (114). Monocytes can act as antigen-presenting cells to activate lymphoid T cells. Monocyte-derived dendritic cells (Mo-DCs) can be produced under IL-4/granulocyte-macrophage colony-stimulating factor (GM-CSF) conditions to induce CD4+ T cell polarization to Th17 and Treg cells and increase the release of IL-6 and IL-23 (115). Jongbloed et al. found that the extent of two heterogeneous subsets of dendritic cells, myeloid DCs (mDCs) and plasmacytoid DCs (pDCs), were significantly increased in the synovial fluid of patients with RA (116), pDCs can activate B cells to differentiate into plasma cells and secrete antibodies in T cell-dependent and T cell-independent manners. The number of pDCs showed a significant positive correlation with the serum level of ACPA (117–120). Compared with normal monocytes, monocytes in RA also express higher levels of various adhesion proteins, including fibronectin and the B2 integrin complex CD11b, which promote adhesion and migration (121). The monocytes in RA express proliferation-inducing ligands on the cell surface and release soluble forms, which are significantly related to disease activity. B cell activating factor (BAFF) combined with APRIL sustains the survival of autoimmune B cells and promotes an autoimmune response (122). By exploring the changes in serum APRIL in anti-CCP positive and negative patients treated with MTX and hydroxychloroquine for 6 months, it was found that the serum concentrations of APRIL increased compared with that of the control group, and they were positively correlated with disease activity, joint swelling, visual analog score, and a simplified disease activity index. In addition, the levels of APRIL in anti-CCP-positive RA patients were significantly lower than that in anti-CCP-negative RA patients after treatment. APRIL is highly expressed on the surface of all circulating monocyte subsets in patients with RA and is related to disease activity (123, 124). A high concentration of APRIL was detected in the synovial homogenate of rats with adjuvant-induced arthritis (AA); APRIL stimulated FLS proliferation, migration, and secretion of pro-inflammatory factors (125). Siglec-1 levels in RA peripheral blood mononuclear cells and monocyte subpopulations were significantly increased in response to TNF-α, IFN-γ, and type II collagen stimulation and was positively correlated with clinical disease indicators (e.g., DAS28, ESR, CRP, and IgM-RF) (126). Monocytes, macrophages, and FLSs in RA increase the expression levels of protease-activated receptor-2 (PAR2); they promote the production of IL-6 and TNF-α and enable the proliferation and invasion of FLSs (127, 128).
Macrophage Heterogeneity in the Personalized Treatment of Rheumatoid Arthritis
There are primarily two macrophage subsets in the peritoneal cavity of the arthritic mice, namely large peritoneal macrophages (LPMs) and small peritoneal macrophages (SPMs). LPMs are derived from fetal liver macrophages, and their major markers are CD11bhigh, CD11clow, F4/80high, CD209-, GATA-6+, MHC IIlow/-, CD62L-,and TIM4+. SPMs develop from bone marrow-derived monocytes, and their major markers are mainly CD11blow, CD11c-, F4/80low, CD209+, GATA-6+, MHC II high, CD62L+, and TIM4- (129–132). Two subsets of synovial macrophages were observed: F4/80+CD11b- (releasing anti-inflammatory mediators such as IL-4 and IL-10) and F4/80-CD11b+ (bone marrow-derived; releasing pro-inflammatory mediators such as IL-1 β and TNF- α) (133). In addition, it has been found that CX3CR1+ tissue-resident macrophages subsets can isolate joints and inhibit the inflammation by forming tight junction barriers with membrane-like structures (134).
Many macrophages can be found in the synovial tissue in the active stage of RA, and the number of macrophages after clinical remission is reduced, indicating that macrophages are and the number of macrophages decrease after clinical remission, indicating that macrophages are highly plastic and can respond to microenvironmental stimuli (e.g., via polarization). Many studies have shown that changes in macrophage prevalence in synovial tissue can predict the effect of treatment. The changes in the number of macrophages in the sublining layer are significantly different among good responders, moderate responders, and non-responders after clinical treatment; these changes are strongly related to changes in DAS28 scores, with excellent sensitivity (135, 136). Activated macrophages recruit immune cells and FLSs by producing IL-1β, IL-6, TNF-α, IL-12, and other cytokines to promote inflammation, while chronic inflammation and cytokines secreted by other cells assist in the activation, polarization, and apoptosis of macrophages (132). The expression levels of macrophages at the CPJ and non-CPJ sites were significantly different. Youssef et al. discovered the RA synovial expression pattern of myeloid related proteins (MRPs), which are macrophage activation markers. Macrophages expressed MRP8, MRP14, and MRP8/14 dimers in the lining layer of the CPJ only in the active stage of the disease, indicating that the macrophages were activated and polarized at the cartilage destruction site in RA (137).
The concept of macrophage polarization helps distinguish the biological functions of macrophages in different states and their characteristics in the pathophysiology of disease. Studies have tested the expression of macrophage populations in multiple locations associated with RA. The synovial lining layer macrophages primarily display an IL-10 polarized-like phenotype, indicating the dominance of M2 macrophages. The macrophages in the synovial sublining layer co-express M1 and M2 markers, indicating a more heterogeneous phenotype (138). Palacios et al. found that macrophages in RA synovial fluid and synovium highly express pro-inflammatory polarizing genes, including INHBA, MMP12, EGLN3, and CCR2, and show low levels of expression of anti-inflammatory genes, including IGF1, HTR2B, FOLR2, SERPINB2, and CD36 (139). ACPA can induce the activity of interferon regulatory factor-5 (IRF5) to promote polarization toward M1 macrophages and increase the ratio of M1 to M2 cells (140). M1 macrophages were observed to be predominant among peritoneal macrophages in collagen-induced arthritis (CIA) mice; this finding is consistent with the previous result (141). Therefore, there is an imbalance between the number of M1 and M2 macrophage subpopulations in RA. M1 is predominant and promotes inflammation. Although the polarization mechanisms of M1 and M2 have not been fully elucidated, both M1 and M2 macrophages secrete different cytokines and mediators that cooperate with other cells to affect disease progression by responding to corresponding stimuli (Figure 1).
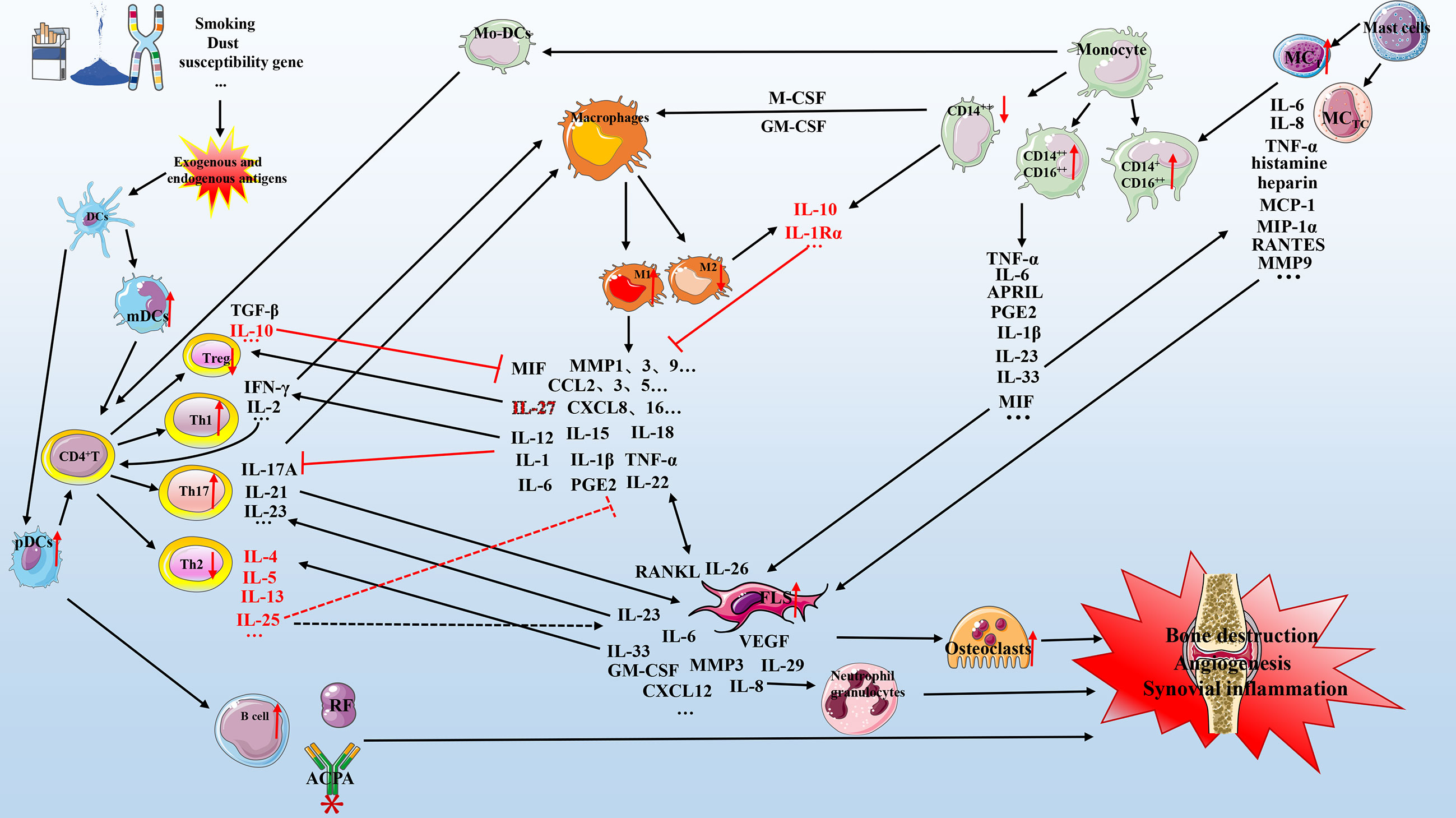
Figure 1 Mechanism of cell-cytokine interaction in RA. Various factors (e.g., smoking, dust, genetic factors, and microorganisms) lead to the production of exogenous and endogenous antigens. Antigen-presenting cells (primarily dendritic cells) present exogenous and endogenous antigens to CD4+ T cells that differentiate into T cells with different functions, including Th1, Th2, and Th17. These cells cooperate with mast cells, macrophages, and monocytes to secrete multiple pro-inflammatory mediators that act on FLSs and osteoclasts, which in turn can secrete various biological mediators to aggravate the circulation. The anti-inflammatory mechanism (red represents anti-inflammatory mediators) is active but insufficient to inhibit the pro-inflammatory process (black represents pro-inflammatory mediators). Briefly, the interaction of various cell subgroups and cell mediators forms a complex network that promotes the development of RA, including bone destruction, angiogenesis, and synovial inflammation. DC, dendritic cells; IL, interleukin; TNF-α, tumor necrosis factor-α; mDCs, myeloid DCs; pDCs, plasmacytoid DCs; GM-CSF, granulocyte-macrophage colony-stimulating factor; M-CSF, macrophage colony-stimulating factor; CXCL, chemokine CXC ligand; CCL, CC-chemokine ligand; MMP, matrix metalloproteinase; MCP-1, monocyte chemoattractant protein-1; PGE2, prostaglandin E2; APRIL, a proliferation-inducing ligand; RF, rheumatoid factor; ACPA, anti-citrullinated protein antibody; FLS, fibroblast-like synoviocyte; TGF, transforming growth factor; VEGF, vascular endothelial growth factor; MC, mast cell.
Interaction of Cell Subsets and Cytokines in Rheumatoid Arthritis
The RA pathogenesis is complex and involves the interaction of various innate and adaptive immune cells, which secrete various chemokines, inflammatory mediators, pro-inflammatory factors, anti-inflammatory factors, and other substances that act on the patient’s synovial tissue and joints. At the initial stage of RA, DCs present exogenous and endogenous antigens to CD4+ T cells by expressing HLA-DR molecules. CD4+ T cells can differentiate into Th1, Th2, and Th17 cells secreting different cytokines, which have different functions in RA. The levels of IL-17, IL-21, and IL23 secreted by Th17 are significantly increased in the synovial fluid of RA patients and are positively correlated with DAS28, CRP, and ACPA. IL17A has a variety of functions. It can stimulate FLS and macrophages to produce VEGF, IL-1, IL-6, TNF-α, and prostaglandin E-2 (PGE-2), and can promote RANKL expression to stimulate synovial inflammation, angiogenesis, and osteoclast formation. In addition, IL-17, IL-1, and TNF-α upregulate the levels of IL-6, MMP, and type I collagen and participate in bone destruction (142). Different subsets of monocytes can secrete various cytokines, including IL-1β, IL-6, IL-18, IL-32, TNF-α, IL-33, IL-23, IL-10, and IL-1Rα and express various cell receptors that cooperate with other cells in RA in different ways. Monocytes can act as antigen-presenting cells to activate CD4+ T cells to differentiate into Th1 and Th17 cells. IL-18 is a key factor in Th1 response and cooperates with IL-12 and IL-15 to induce IFN-γ production in synovial tissue and promotes the production of pro-inflammatory factors, such as TNF-α and IL-1, in an IFN-γ-independent manner. IL-10 can inhibit the production of TNF-α and IFN-γ, and IL-1β and TNF-α promote IL-18 release by FLSs to strengthen this effect (143–145). IL-32 is highly expressed in RA synovial tissue and can induce PGE2 release from mouse macrophages and human blood monocytes. Synovial IL-32 staining was positively correlated with the indices of synovial inflammation, TNF-α, IL-1β, and IL-18 (146). Monocytes exposed to the synovial fluid of RA express lower levels of IL-10 and higher levels of IL-1Rα, which may result from the anti-inflammatory mechanism; however, this effect does not offset the rest of the pro-inflammatory process (147). IL-6 and IL-1β produced by monocytes can promote inflammation and increase adhesion (121). Monocytes and FLSs can also secrete IL-23 and IL-33. IL-23 is essential for Th17 cell differentiation and IL-17A production (148, 149). IL-33 can induce Th2 cell differentiation; stimulate the production of IL-4, IL-5, and IL-13; and promote inflammation by binding to ST2 receptors on MCs to release related cytokines (150–152). Th1, Th17, FLSs, and monocytes can secrete GM-CSF, induce polarization of macrophages into M1 phenotypes through IRF4 or IRF5, and promote the production of IL-6, IL-23, and CCL17 to induce inflammation (153). They can also secrete M-CSF, which induces macrophage polarization toward M2 type, thereby exerting an anti-inflammatory mechanism (154).
M1/M2 macrophages can secrete a variety of biological mediators, including IL-1, IL-1β, IL-6, IL-26, IL-27, IL-29, IFN-γ, IL-22, TNF-α, IL-12, IL-15, IL-18, IL-20, macrophage migration inhibitory factor (MIF), MMP-1, MMP-2, MMP-9, MMP-12, MMP-13, various CXCL chemokines, CCL2, CCL3, CCL5, ROS, IL-10, IL-1RII, and TGF-β. Serum IL-12 levels are positively correlated with the number of swollen joints, levels of RF, and other disease activity markers in patients with RA, and can be used as a predictor of disease activity (155). Vitamin D receptors expressed on the surface of macrophages. Also, vitamin D receptors are widely expressed on the surface of many immune cells, such as CD4+ T cell, CD8+ T cell, B cell, and DC cell. Vitamin D has roles in calcium metabolism, bone health, and anti-inflammatory, which has been widely reviewed in many diseases. It may be possible to partially alleviate various autoimmune responses by regulating maladjusted microbial populations in RA. The specific mechanism needs further study (156–160). IL-12 can also induce Th1 cell differentiation (161). In the presence of a large amount of TNF-α, Th1 cells release IFN-γ and IL-2, and IFN-γ further induces macrophages to polarize toward M1 phenotypes. IL-2 is an important regulator of T cell-dependent responses and plays a crucial role in T cell proliferation and survival (132, 162). The cytokine IL-22, produced by macrophages and Th22 cell subsets, is related to disease activity and promotes osteoclast production by inducing RANKL production in FLSs, while IL-25 produced by Th2 cells can inhibit this effect via the STAT3/P38/MAPK/IkBa pathway (163, 164). In addition, IL-25 attenuates the development of CIA by inhibiting the differentiation of CD4+ T cells into Th17 cells (165, 166). However, some studies have shown that IL-25 is involved in the aberrant proliferation of FLSs (167). M1 macrophages promote synovial inflammation by producing a large number of pro-inflammatory factors, including IL-1, IL-6, IL-1β, IL-12, and TNF-α (168). The level of IL-15 is increased in the serum and synovial fluid in the early stage of RA, and promotes the release of TNF-α and IL-17 to participate in T lymphatic activation and subsequent bone destruction. It can be used as an independent biomarker for the detection of early RA (169–172). IL-27 secreted by macrophages has dual anti-inflammatory and pro-inflammatory functions. It induces FLSs to release a variety of pro-inflammatory mediators, including IL-6, vascular cell adhesion protein 1 (VCAM1), CCL2, CXCL9, CXCL10, and MMP-1. On the other hand, IL-27 binding to the IL-27 receptor of T cells induces their differentiation into Treg cells that secrete IL-10, which inhibits Th17 differentiation as an anti-inflammatory mechanism (173–176). FLSs and macrophages produce IL-26 and IL-29, respectively. IL-26 can promote the polarization of M1 macrophages by activating the c-JUN/NF-κB/STAT1 signaling pathway and produce IL-6 and TNF-α pro-inflammatory factors (177). IL-29 is elevated in the serum of RA patients and participates in the chemotaxis of peripheral blood neutrophils, inhibits T follicular helper (Tfh) cell differentiation by reducing STAT3/BCL6 activity, and induces RANKL expression in FLSs through the MAPK pathway to participate in bone destruction. IL-29 upregulates the levels of TLRs in FLS to mediate the expression of IL-6 and IL-8, which promotes inflammation (178–182). IL-18 levels in the synovium and serum are related to RA disease activity. Inhibition of IL-18 can inhibit the secretion of pro-inflammatory factors such as IL-6, IL-18, TNF-α, and IFN-γ. Compared with those of other cytokines, IL-18 levels were significantly reduced after leflunomide treatment; therefore, the IL-18 serum levels can be used as a potential biomarker to measure the efficacy of leflunomide (183, 184). TNF-α inhibitor treatment can significantly reduce bone loss in patients with RA, which is related to decreased IL-20 and RANKL expression levels (185). MIF secreted by monocytes and macrophages promotes the secretion by Th1 and Th17 cells. Pro-inflammatory cytokines, including TNF-α, IFN-γ, IL-1β, IL-6, and IL-17A, induce the production of MMP-1, MMP-3, MMP-9, MMP-13, phospholipase A2, and cyclooxygenase-2, which induce FLS to produce VEGF, IL-8, and RANKL, and promote the differentiation of monocytes into osteoclasts, all of which mediate bone destruction in RA (186, 187). CXCL1–8 can combine with CXCR2 in ECs cells to participate in angiogenesis. Inhibition of CXCL4 may improve inflammation in CIA mice. The inhibition of CXCL8 reduces angiogenesis in RA, and the release of CXCL12 from FLS could inhibit the pro-angiogenic activity of other chemokines and VEFG by binding to CXCR4 or CXCR7 (188, 189). CCL2/3/4/5 and CXCL9/10 levels are increased in RA, of which CCL2/5 and CXCL10 promote osteoclastogenesis. The production of CCL2/MCP-1 from FLS and macrophages through the PI3K/ERK/JNK pathway could result in IL-17-induced monocyte migration from the blood to the synovium (190, 191). Briefly, the interaction of various cell subgroups in RA promotes inflammation and bone destruction. Although many anti-inflammatory mediators are secreted, the level of anti-inflammatory mediators is not sufficient to inhibit the process of chronic inflammation and bone destruction. About other existing cell subsets in autoimmune diseases, we intend to clarify the roles and their mechanisms for RA in the future, such as NK cells, neutrophils (192).
Future Perspective: Biologics and Non-Biologic Drugs for Rheumatoid Arthritis
The common drugs used to treat RA include MTX and leflunomide, among others. Based on the heterogeneity of multiple cell subsets in different stages of RA, it becomes apparent why some patients with RA have poor or no response to treatment. Therefore, researchers have focused on developing biological therapies that target different cells or cytokines and have achieved some therapeutic success. When MTX and leflunomide show poor efficacy, the combined use of biologics may improve the response. There have been many notable reviews summarizing the progress of pharmacological research in this field (193, 194), which we have updated and briefly described in Table 2.
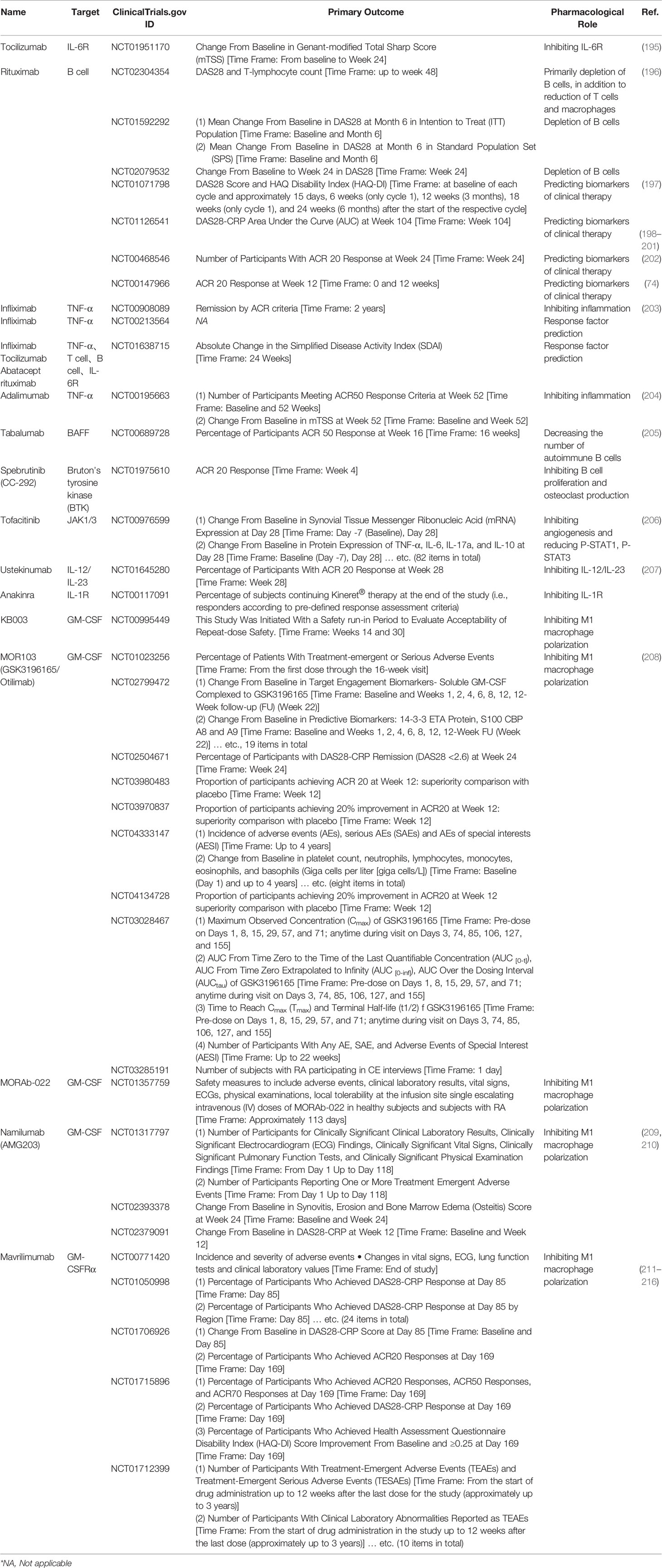
Table 2 RA-related biologic therapy and clinical trials.
Anti-TNF-α therapy is currently the most widely used targeted biological agent. As mentioned above, sICAM1 may represent the myeloid subtype in synovial tissue and predict the therapeutic response to TNF-α therapy. Research has shown that lymphoid aggregates in synovial tissue are an independent negative predictive factor for anti-TNF-α therapy (66). A prospective study of the effect of infliximab on lymphoid aggregates after 16 weeks of treatment in RA patients, consistent with the above, also showed that patients with lymphocyte aggregates show a lower treatment response (217). By examining the expression profile of infliximab in the treatment of RA by arthroscopy, it was found that high-level tissue inflammation responds better to anti-TNF-α therapy (218). An evaluation of the effect of anti-TNF-α monoclonal antibody cA2 on RA showed that cA2 significantly reduced the migration of neutrophils, the number of infiltrated immune cells, and the expression levels of IL-8 and MCP-1, which relieve synovial inflammation (219). RA patients were injected with 10 mg/kg infliximab and underwent synovial biopsy. The results showed that the extents of ACR20, ACR50, and TNF-α, IL-1α, and IL-1β synthesis were all reduced (220). Studies have shown that high resistin is associated with high disease inflammation in RA and can be used as a biomarker to distinguish the early activity of RA from the increased risk of erosive diseases and to predict the treatment response of DMARD and infliximab (203). A clinical study predicting the response of RA to infliximab by measuring biochemical, immune, and bone markers in serum and using transcriptome analysis to identify gene expression characteristics in PBMCs is currently ongoing (NCT00213564). In addition, a randomized, multi-center biological experiment predicting the biomarkers of the effects of four biological agents (infliximab, tocilizumab, rituximab, abatacept) on RA (NCT01638715) has been coordinated. Similar clinical trials should be designed to predict the treatment response of different treatment modalities and to screen appropriate biomarkers. Baseline DAS28-CRP can predict the response of early RA patients to MTX treatment, and when MTX monotherapy fails, adding another TNF-α inhibitor, adalimumab, can achieve better clinical prognostic results (204). Another study determined the gene expression profile of RA patients with poor response to adalimumab and found 439 marker genes, primarily related to cell division and immune regulation pathways. The high expression levels of IL-7R, CXCL11, IL-18, IL- 18 receptor accessory (IL-18rap), and MK167 were related to absence of response to adalimumab treatment in the synovial membrane (221).
Possible indications for using rituximab in RA, including poor response to anti-TNF-α therapy, associated lymphoproliferative disease, RA that overlaps with systemic lupus erythematosus, Sjogren’s syndrome, and undifferentiated connective tissue disease have previously been reviewed (222). Given the heterogeneity of cells expressed in the pathological model of synovial tissue, among which autoimmune lymphoid B cells play an important role, the importance of targeting B cells to treat RA is self-evident. Rituximab can cause CD20+ B cell depletion. Some clinical studies have found that the use of rituximab alone or in conjunction with MTX has a higher rate of patient recovery than MTX alone (223, 224). The study found that a normal level of CD19+B cells and an increase in CD19+CD27-lgD-double negative B cells can predict RA patients’ response to rituximab (197). Serum IL-33 can be used as a predictor of response to rituximab in treating RA independently or in combination with RF or anti-CCP and high serum IgG (198, 199). Elevated levels of IgM-RF, IgG-RF, IgA-RF, anti-CCP, and CRP increase the sensitivity of RA patients with poor anti-TNF-α responses to rituximab response prediction (202). Biopsy analysis of the synovial tissue of RA patients receiving rituximab showed that DAS scores, RF, ACPA, T cells, macrophages, peripheral blood, and synovial B cells were significantly reduced after the treatment; the reduction of plasma cells indicated the therapeutic effect of rituximab (225). CXCL13 is a B cell chemokine and a serum biomarker of RA. By detecting the levels of B cells, CXCL13, RF-IgM, and anti-CCP in 20 RA patients treated with rituximab for six months, it was found that patients with detectable B cells had higher levels of CXCL13 and RF-IgM than those with undetectable B cells. The synovial tissue with high expression level of serum CXCL13 has increased levels of IL-1β, IL-8, MMP-1, and MMP3, suggesting that the level of serum CXCL13 can be used as a predictor of B cell depletion therapy in RA. In addition, the expression of CXCL13 is related to many markers of synovitis (74). CCL19 is also a B cell chemokine. Studies have shown that the serum levels of CXCL12, CXCL13, and CCL19 are increased and positively correlated with a variety of B cell markers, such as serum immunoglobulin IgG, IgA, BAFF, free light chain, and RF. It can also be used as a predictor of the response to rituximab (200). In addition, studies of rituximab treatment have shown that the low frequency of CD27+ memory B cells may be more beneficial in the B cell-driven RA subtypes (201). Studies have found that elevated levels of IgM-RF, IgG-RF, IgA-RF, anti-CCP, and CRP increase the sensitivity of prediction to rituximab therapy for RA patients who have a poor response to anti-TNF-α therapy (202). DAS28 scores, IgM-RF, anti-ACPA, synovial B cells, synovial T cells, and synovial macrophages were significantly reduced after rituximab treatment (225). Rituximab almost completely exhausted the number of circulating B cells in RA patients who failed anti-TNF-α therapy, and all achieved varying degrees of ACR response, which was accompanied by a decrease in synovial immunoglobulin synthesis (226). Another study used high-throughput PCR to detect the clinical response to rituximab in RA and found that the high expression level of inflammation-related genes in T cells and macrophages in the synovium were appropriate predictors for treatment response. The higher expression levels of interferon-α and remodeling-related genes indicated a poor response to rituximab (70). Combined with another clinical study to evaluate the therapeutic characteristics of rituximab on lymphatic T cells in RA patients, it was found that rituximab accidentally depleted a large number of CD4+ T cells, implying that B cells, T cells, and macrophages are interrelated in RA (196). Two other clinical trials to observe rituximab in RA patients with insufficient response or intolerance to anti-TNF-α therapy are underway, awaiting the release of the corresponding results (NCT01592292, NCT02079532). Also, RA may accelerate the progression of atherosclerosis, and the TNF-α inhibitor can alleviate atherosclerosis by regulating free radicals and endothelial dysfunction, involving the regulation of vascular endothelial growth factor and endothelial nitric oxide synthase (227, 228).
GM-CSF promotes monocyte differentiation into M1 pro-inflammatory macrophages in RA. A randomized, multi-center clinical trial for RA has revealed that MOR103, a human monoclonal antibody against GM-CSF, is well tolerated, safe, and effective for patients with RA. Patients in the 1.0 and 1.5 mg/kg MOR103 groups had significantly improved DAS28 and joint counts; the 1.0 mg/kg dose was the best correlated with the reduction in disease severity (208). A phase 1b, randomized clinical study evaluated the safety and tolerability to receive AMG203, a GM-CSF ligand with a high-affinity monoclonal antibody, after MTX treatment for ≥12 weeks and revealed that 150 mg or 300 mg of AMG203 exhibited good tolerance and improvement of disease activity indicators, such as DAS28-CRP (209). A phase II randomized, double-blind, placebo-controlled clinical trial studied the efficacy of AMG203 (20, 80, or 150 mg) in RA patients with insufficient response to MTX or anti-TNF antibody treatment. The results showed that the DAS28-CRP level for 12 weeks with AMG203 150 mg was significantly reduced at baseline. However, serious adverse events (myocardial infarction) occurred without a dose–response relationship (210). By evaluating the response of a total of 1145 RA patients in five studies with mavrilimumab, a human monoclonal antibody against GM-CSF receptor (GM-CSFR)-α, it was found that DAS28-CRP was significantly reduced after 12 weeks of treatment, and there were no significant adverse events, indicating good treatment efficacy and tolerability (229). The therapeutic intervention effect for ACR20 response to mavrilimumab 150 mg + MTX, mavrilimumab 100 mg + MTX, tofacitinib 10 mg + MTX, and tofacitinib 5 mg + MTX on RA patients with insufficient DMARD response was successively reduced, among which mavrilimumab 150 mg + MTX and mavrilimumab 100 mg + MTX had the most significant ACR20 response (230). In another study, mavrilimumab and golimumab, an anti-TNF monoclonal antibody, were combined with MTX to treat RA patients with insufficient DMARD response. Evidently, the ACR20, ACR50, and ACR70 response rates were not significantly different, which may be due to the non-optimal therapeutic dose (mavrilimumab 100 mg + MTX). Although there was no direct comparison between the two therapies, the effect of mavrilimumab is clinically promising (211). Similar research results showed that mavrilimumab could reduce the DAS28-CRP of RA patients and achieve the ACR20 remission rate, both of which indicate the potential therapeutic effect of mavrilimumab for the inhibition of subsets of monocytes and macrophages in patients with RA (212–215).
Increased IL-6 activity in patients with RA is significantly related to synovial inflammation and autoantibody production. As mentioned above, CXCL13 is used as a biomarker to predict the efficacy of anti-IL-6R therapy (tocilizumab) in RA with a synovial lymphoid phenotype. An open-label, multi-center, prospective, single-arm study used hand-foot radiography and hand magnetic resonance imaging to detect the efficacy of a tocilizumab subcutaneous injection for 24 weeks on joint damage in RA patients. The results showed synovial inflammation, the DAS28 score was lessened, and the treatment was well-tolerated (195). By comparing the gene expression data of tocilizumab, MTX, rituximab, and adalimumab in synovial biopsy tissue of RA patients, it was shown that IL-6R, chemokines, and T cell activation-related gene expression level was reduced after treatment with tocilizumab, and showed a similar gene expression pattern to that of treatment with rituximab and MTX, which implies that B cells and IL-6 have a molecular synergistic effect (231). In addition, after tocilizumab treatment of RA patients, the ERK in the synovial tissue was significantly increased, the number of lymphatic B cells (CD20) was significantly reduced, and IL-6 was blocked entirely, which was comparable to MTX treatment (232). Another study on 65 RA synovial biopsy tissues found that TNF-α-induced disease activity-related transcripts were highly enriched in patients with early RA, which is associated with higher disease activity and predicts a poor response to other therapies, such as MTX and tocilizumab (233). BAFF protects autoimmune B cells by binding to APRIL. Using 30 mg and 80 mg doses of tabalumab (anti-BAFF monoclonal antibody) to treat patients with insufficient TNF-α therapy, some therapeutic effects were observed earlier, including changes in ACR20, ACR50, and DAS28-CPR (205). Spebrutinib (CC-292), a Bruton’s tyrosine kinase inhibitor, can inhibit B cell proliferation and osteoclast production in vitro and can reduce the number of CD19B cells, serum CXCL13, macrophage inflammatory protein-1β (MIP-1β), carboxy-terminal collagen cross-linking telopeptide, and C-telopeptide of type I collagen (CTx-1), among which CD19B cells and CTx-1 can predict the response of RA patients to spebrutinib (234). Tofacitinib, a JAK inhibitor, can inhibit the angiogenesis and migration ability of endothelial cells (ECs) in vitro and reduce VEGF levels in CIA mice, suggesting that its efficacy could be related to angiogenic inhibition (235). A randomized, double-blind, phase II clinical trial tested the effect of 10 mg of tofacitinib twice daily on patients with poor MTX response for 28 days. The results showed that the gene expression level in the synovial membrane is decreased with the well EULAR response, such as the reduction of MMP-1, MMP-3, CCL2, CXCL10, and CXCL13 levels, which may be related to the decrease in phosphorylation of STAT1 and STAT3 (206). The mechanism of abatacept is to block cytotoxic T lymphocyte associated antigen 4 (CTLA4) to prevent T cell costimulatory signals and T cell activation. A prospective, multi-center, observational clinical trial studied the efficacy of abatacept in elderly RA patients and found that after abatacept treatment, more patients improved compared with DMARD. Compared with patients with no response or poor response to DMARD, abatacept was more effective (UMIN000014913) (236). After abatacept treatment, no significant changes in synovial immune infiltration were found, but the expression levels of pro-inflammatory genes (IFN-γ and IL-1β), MMP-1, and MMP-3 was significantly reduced (237). Several other biological agents (anakinra, ustekinumab, and guselkumab) are targeted at IL-1R, IL-12/IL-23, and IL-23, respectively, and several related clinical trials are in progress (207).
Conclusions
Although various treatment options exist for RA, many of these options show little to no efficacy, even when used in combination. Understanding the heterogeneity in the cellular and molecular features of RA could aid the development of safe and effective treatments. Many clinical studies on RA are underway; however, it remains unclear how to accurately find biomarkers that predict the patient’s response to a certain therapy. Finding such biomarkers is a major goal of precise treatment. Treatment should be combined with procedures like synovial biopsy and modern techniques such as arthroscopic surgery, assisted computer imaging, and multi-omics to better evaluate the disease activity/progression and the patient’s response. Inflammation in RA is a complex process and not an independent and discrete set of events. There are typically multiple synovial tissue disease expression patterns in patients with RA with many different subsets of corresponding cells. One of these subsets may be dominant at a specific disease stage; however, it may not appear independently. Well-designed clinical trials that explore the impact of various cell subgroups on different RA therapies need to be urgently conducted for clinical application. The goal is to support the beneficial cell subsets and to inhibit the cell subsets that hinder a specific treatment. A certain strategy may inhibit the activity of a cytokine but is typically accompanied by serious adverse reactions possibly related to the breakdown of the physiological balance of cytokines. Therefore, targeting the activity of common mediators of multiple cells and maintaining the corresponding balance based on disease activity and biomarker response is essential for drug therapy development.
Author Contributions
JZ is responsible for the collection, collation, and writing of the original manuscript. SG, SS, and DH are responsible for the concept development, revision, and review of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by the National Natural Science Funds of China (81774114), Shanghai Chinese Medicine Development Office, Shanghai Chinese and Western Medicine Clinical Pilot Project (ZY(2018–2020)-FWTX-1010), Shanghai Chinese Medicine Development Office, Shanghai Traditional Chinese Medicine Specialty Alliance Project (ZY(2018-2020)-FWTX-4017), National Administration of Traditional Chinese Medicine, Regional Chinese Medicine (Specialist) Diagnosis and Treatment Center Construction Project-Rheumatology.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary

References
1. Scott D, Wolfe F, Huizinga T. Rheumatoid Arthritis. Lancet (London England) (2010) 376(9746):1094–108. doi: 10.1016/S0140-6736(10)60826-4
2. Smolen JS, Aletaha D IBM. Rheumatoid Arthritis. Lancet (2016) 388:2023–38. doi: 10.1016/S0140-6736(16)30173-8
3. Klareskog L, Rönnelid J, Lundberg K, Padyukov L, Alfredsson L. Immunity to Citrullinated Proteins in Rheumatoid Arthritis. Annu Rev Immunol (2008) 26:651–75. doi: 10.1146/annurev.immunol.26.021607.090244
4. Deane K, Norris J, Holers V. Preclinical Rheumatoid Arthritis: Identification, Evaluation, and Future Directions for Investigation. Rheum Dis Clin North Am (2010) 36(2):213–41. doi: 10.1016/j.rdc.2010.02.001
5. Smolen J. Insights Into the Treatment of Rheumatoid Arthritis: A Paradigm in Medicine. J Autoimmun (2020) 110:102425. doi: 10.1016/j.jaut.2020.102425
6. Weyand CM, Goronzy JJ. The Immunology of Rheumatoid Arthritis. Nat Immunol (2021) 22(1):10–8. doi: 10.1038/s41590-020-00816-x
7. Hansildaar R, Vedder D, Baniaamam M, Tausche AK, Gerritsen M, Nurmohamed MT. Cardiovascular Risk in Inflammatory Arthritis: Rheumatoid Arthritis and Gout. Lancet Rheumatol (2021) 3(1):e58–70. doi: 10.1016/S2665-9913(20)30221-6
8. Nerurkar L, Siebert S, McInnes IB, Cavanagh J. Rheumatoid Arthritis and Depression: An Inflammatory Perspective. Lancet Psychiatry (2019) 6(2):164–73. doi: 10.1016/S2215-0366(18)30255-4
9. Hensvold A, Klareskog L. Towards Prevention of Autoimmune Diseases: The Example of Rheumatoid Arthritis. Eur J Immunol (2021) 51(8):1921–33. doi: 10.1002/eji.202048952
10. Yin J, He D, Jiang L, Cheng F, Guo Q, Huang S, et al. Influence of Cigarette Smoking on Rheumatoid Arthritis Risk in the Han Chinese Population. Front Med (Lausanne) (2017) 4:76. doi: 10.3389/fmed.2017.00076
11. Guo S, Zhu Q, Jiang T, Wang R, Shen Y, Zhu X, et al. Genome-Wide DNA Methylation Patterns in CD4+ T Cells From Chinese Han Patients With Rheumatoid Arthritis. Mod Rheumatol (2017) 27(3):441–7. doi: 10.1080/14397595.2016.1218595
12. Plant D, Wilson A, Barton A. Genetic and Epigenetic Predictors of Responsiveness to Treatment in RA. Nat Rev Rheumatol (2014) 10(6):329–37. doi: 10.1038/nrrheum.2014.16
13. Mei F, Xie M, Huang X, Long Y, Lu X, Wang X, et al. Porphyromonas Gingivalis and Its Systemic Impact: Current Status. Pathog (Basel Switzerland) (2020) 9(11):944. doi: 10.3390/pathogens9110944
14. Smolen J, Aletaha D, Barton A, Burmester G, Emery P, Firestein G, et al. Rheumatoid Arthritis. Nat Rev Dis Primers (2018) 4:18001. doi: 10.1038/nrdp.2018.1
15. Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K, et al. Genetics of Rheumatoid Arthritis Contributes to Biology and Drug Discovery. Nature (2014) 506(7488):376–81. doi: 10.1038/nature12873
16. Guo S, Xu L, Chang C, Zhang R, Jin Y, He D. Epigenetic Regulation Mediated by Methylation in the Pathogenesis and Precision Medicine of Rheumatoid Arthritis. Front Genet (2020) 11:811. doi: 10.3389/fgene.2020.00811
17. Guo S, Jin Y, Zhou J, Zhu Q, Jiang T, Bian Y, et al. MicroRNA Variants and HLA-miRNA Interactions Are Novel Rheumatoid Arthritis Susceptibility Factors. Front Genet (2021) 12(747274). doi: 10.3389/fgene.2021.747274
18. Karlson EW, Chang SC, Cui J, Chibnik LB, Fraser PA, De Vivo I, et al. Gene-Environment Interaction Between HLA-DRB1 Shared Epitope and Heavy Cigarette Smoking in Predicting Incident Rheumatoid Arthritis. Ann Rheum Dis (2010) 69(1):54–60. doi: 10.1136/ard.2008.102962
19. Guo S, Liu J, Jiang T, Lee D, Wang R, Zhou X, et al. (5r)-5-Hydroxytriptolide (LLDT-8) Induces Substantial Epigenetic Mediated Immune Response Network Changes in Fibroblast-Like Synoviocytes From Rheumatoid Arthritis Patients. Sci Rep (2019) 9(1):11155. doi: 10.1038/s41598-019-47411-1
20. Gregersen P, Silver J, Winchester R. The Shared Epitope Hypothesis. An Approach to Understanding the Molecular Genetics of Susceptibility to Rheumatoid Arthritis. Arthritis Rheum (1987) 30(11):1205–13. doi: 10.1002/art.1780301102
21. Hill J, Southwood S, Sette A, Jevnikar A, Bell D, Cairns E. Cutting Edge: The Conversion of Arginine to Citrulline Allows for a High-Affinity Peptide Interaction With the Rheumatoid Arthritis-Associated HLA-DRB1*0401 MHC Class II Molecule. J Immunol (Baltimore Md: 1950) (2003) 171(2):538–41. doi: 10.4049/jimmunol.171.2.538
22. Eyre S, Bowes J, Diogo D, Lee A, Barton A, Martin P, et al. High-Density Genetic Mapping Identifies New Susceptibility Loci for Rheumatoid Arthritis. Nat Genet (2012) 44(12):1336–40. doi: 10.1038/ng.2462
23. Begovich AB, Carlton VE, Honigberg LA, Schrodi SJ, Chokkalingam AP, Alexander HC, et al. A Missense Single-Nucleotide Polymorphism in a Gene Encoding a Protein Tyrosine Phosphatase (PTPN22) Is Associated With Rheumatoid Arthritis. Am J Hum Genet (2004) 75(2):330–7. doi: 10.1086/422827
24. Kim K, Bang S, Lee H, Cho S, Choi C, Sung Y, et al. High-Density Genotyping of Immune Loci in Koreans and Europeans Identifies Eight New Rheumatoid Arthritis Risk Loci. Ann Rheum Dis (2015) 74(3):e13. doi: 10.1136/annrheumdis-2013-204749
25. Laufer V, Tiwari H, Reynolds R, Danila M, Wang J, Edberg J, et al. Genetic Influences on Susceptibility to Rheumatoid Arthritis in African-Americans. Hum Mol Genet (2019) 28(5):858–74. doi: 10.1093/hmg/ddy395
26. Suzuki A, Yamada R, Chang X, Tokuhiro S, Sawada T, Suzuki M, et al. Functional Haplotypes of PADI4, Encoding Citrullinating Enzyme Peptidylarginine Deiminase 4, Are Associated With Rheumatoid Arthritis. Nat Genet (2003) 34(4):395–402. doi: 10.1038/ng1206
27. Ikari K, Kuwahara M, Nakamura T, Momohara S, Hara M, Yamanaka H, et al. Association Between PADI4 and Rheumatoid Arthritis: A Replication Study. Arthritis Rheum (2005) 52(10):3054–7. doi: 10.1002/art.21309
28. Márquez A, Kerick M, Zhernakova A, Gutierrez-Achury J, Chen W, Onengut-Gumuscu S, et al. Meta-Analysis of Immunochip Data of Four Autoimmune Diseases Reveals Novel Single-Disease and Cross-Phenotype Associations. Genome Med (2018) 10(1):97. doi: 10.1186/s13073-018-0604-8
29. Raychaudhuri S, Remmers E, Lee A, Hackett R, Guiducci C, Burtt N, et al. Common Variants at CD40 and Other Loci Confer Risk of Rheumatoid Arthritis. Nat Genet (2008) 40(10):1216–23. doi: 10.1038/ng.233
30. van der Linden M, Feitsma A, le Cessie S, Kern M, Olsson L, Raychaudhuri S, et al. Association of a Single-Nucleotide Polymorphism in CD40 With the Rate of Joint Destruction in Rheumatoid Arthritis. Arthritis Rheum (2009) 60(8):2242–7. doi: 10.1002/art.24721
31. Viatte S, Plant D, Raychaudhuri S. Genetics and Epigenetics of Rheumatoid Arthritis. Nat Rev Rheumatol (2013) 9(3):141–53. doi: 10.1038/nrrheum.2012.237
32. Knevel R, Klein K, Somers K, Ospelt C, Houwing-Duistermaat J, van Nies J, et al. Identification of a Genetic Variant for Joint Damage Progression in Autoantibody-Positive Rheumatoid Arthritis. Ann Rheum Dis (2014) 73(11):2038–46. doi: 10.1136/annrheumdis-2013-204050
33. Plant D, Thomson W, Lunt M, Flynn E, Martin P, Eyre S, et al. The Role of Rheumatoid Arthritis Genetic Susceptibility Markers in the Prediction of Erosive Disease in Patients With Early Inflammatory Polyarthritis: Results From the Norfolk Arthritis Register. Rheumatol (Oxford England) (2011) 50(1):78–84. doi: 10.1093/rheumatology/keq032
34. de Rooy D, Tsonaka R, Andersson M, Forslind K, Zhernakova A, Frank-Bertoncelj M, et al. Genetic Factors for the Severity of ACPA-Negative Rheumatoid Arthritis in 2 Cohorts of Early Disease: A Genome-Wide Study. J Rheumatol (2015) 42(8):1383–91. doi: 10.3899/jrheum.140741
35. Cui J, Stahl E, Saevarsdottir S, Miceli C, Diogo D, Trynka G, et al. Genome-Wide Association Study and Gene Expression Analysis Identifies CD84 as a Predictor of Response to Etanercept Therapy in Rheumatoid Arthritis. PloS Genet (2013) 9(3):e1003394. doi: 10.1371/journal.pgen.1003394
36. Plant D, Prajapati R, Hyrich K, Morgan A, Wilson A, Isaacs J, et al. Replication of Association of the PTPRC Gene With Response to Anti-Tumor Necrosis Factor Therapy in a Large UK Cohort. Arthritis Rheum (2012) 64(3):665–70. doi: 10.1002/art.33381
37. Coenen M, Gregersen P. Rheumatoid Arthritis: A View of the Current Genetic Landscape. Genes Immun (2009) 10(2):101–11. doi: 10.1038/gene.2008.77
38. Frisell T, Hellgren K, Alfredsson L, Raychaudhuri S, Klareskog L, Askling J. Familial Aggregation of Arthritis-Related Diseases in Seropositive and Seronegative Rheumatoid Arthritis: A Register-Based Case-Control Study in Sweden. Ann Rheum Dis (2016) 75(1):183–9. doi: 10.1136/annrheumdis-2014-206133
39. Joo Y, Kim Y, Park Y, Kim K, Ryu J, Lee S, et al. Biological Function Integrated Prediction of Severe Radiographic Progression in Rheumatoid Arthritis: A Nested Case Control Study. Arthritis Res Ther (2017) 19(1):244. doi: 10.1186/s13075-017-1414-x
40. Yamamoto K, Okada Y, Suzuki A, Kochi Y. Genetics of Rheumatoid Arthritis in Asia–Present and Future. Nat Rev Rheumatol (2015) 11(6):375–9. doi: 10.1038/nrrheum.2015.7
41. Murdaca G, Spanò F, Contatore M, Guastalla A, Magnani O, Puppo F. Pharmacogenetics of Etanercept: Role of TNF-α Gene Polymorphisms in Improving Its Efficacy. Expert Opin Drug Metab Toxicol (2014) 10(12):1703–10. doi: 10.1517/17425255.2014.970165
42. Murdaca G, Gulli R, Spanò F, Lantieri F, Burlando M, Parodi A, et al. TNF-α Gene Polymorphisms: Association With Disease Susceptibility and Response to Anti-TNF-α Treatment in Psoriatic Arthritis. J Invest Dermatol (2014) 134(10):2503–9. doi: 10.1038/jid.2014.123
43. Guan Y, Zhang H, Quang D, Wang Z, Parker S, Pappas D, et al. Machine Learning to Predict Anti-Tumor Necrosis Factor Drug Responses of Rheumatoid Arthritis Patients by Integrating Clinical and Genetic Markers. Arthritis Rheumatol (Hoboken NJ) (2019) 71(12):1987–96. doi: 10.1002/art.41056
44. Massey J, Plant D, Hyrich K, Morgan A, Wilson A, Spiliopoulou A, et al. Genome-Wide Association Study of Response to Tumour Necrosis Factor Inhibitor Therapy in Rheumatoid Arthritis. Pharmacogenomics J (2018) 18(5):657–64. doi: 10.1038/s41397-018-0040-6
45. Senapati S, Singh S, Das M, Kumar A, Gupta R, Kumar U, et al. Genome-Wide Analysis of Methotrexate Pharmacogenomics in Rheumatoid Arthritis Shows Multiple Novel Risk Variants and Leads for TYMS Regulation. Pharmacogenet Genomics (2014) 24(4):211–9. doi: 10.1097/FPC.0000000000000036
47. Smith M, Barg E, Weedon H, Papengelis V, Smeets T, Tak P, et al. Microarchitecture and Protective Mechanisms in Synovial Tissue From Clinically and Arthroscopically Normal Knee Joints. Ann Rheum Dis (2003) 62(4):303–7. doi: 10.1136/ard.62.4.303
48. Stevens C, Mapp P, Revell P. A Monoclonal Antibody (Mab 67) Marks Type B Synoviocytes. Rheumatol Int (1990) 10(3):103–6. doi: 10.1007/BF02274823
49. Singh JA, Arayssi T, Duray P, Schumacher HR. Immunohistochemistry of Normal Human Knee Synovium: A Quantitative Study. Ann Rheum Dis (2004) 63(7):785–90. doi: 10.1136/ard.2003.013383
50. Henderson B, Revell PA, Edwards JC. Synovial Lining Cell Hyperplasia in Rheumatoid Arthritis: Dogma and Fact. Ann Rheum Dis (1988) 47(4):348–9. doi: 10.1136/ard.47.4.348
51. Bromley M, Woolley DE. Histopathology of the Rheumatoid Lesion. Identification of Cell Types at Sites of Cartilage Erosion. Arthritis Rheum (1984) 27(8):857–63. doi: 10.1002/art.1780270804
52. Fassbender H. Histomorphological Basis of Articular Cartilage Destruction in Rheumatoid Arthritis. Coll Relat Res (1983) 3(2):141–55. doi: 10.1016/S0174-173X(83)80040-5
53. Kobayashi I, Ziff M. Electron Microscopic Studies of the Cartilage-Pannus Junction in Rheumatoid Arthritis. Arthritis Rheum-US (1975) 18(5):475–83. doi: 10.1002/art.1780180507
54. Muirden K. Electron Microscopic Studies of the Synovial-Cartilage Junction in Rheumatoid Arthritis. Eur J Rheumatol Inflamm (1982) 5(1):30–8.
55. Shiozawa S, Shiozawa K, Fujita T. Morphologic Observations in the Early Phase of the Cartilage-Pannus Junction. Light and Electron Microscopic Studies of Active Cellular Pannus. Arthritis Rheum (1983) 26(4):472–8. doi: 10.1002/art.1780260404
56. Mills K. Pathology of the Knee Joint in Rheumatoid Arthritis. A Contribution to the Understanding of Synovectomy. J Bone Joint Surg Br (1970) 52(4):746–56. doi: 10.1302/0301-620X.52B4.746
57. Tateishi H. Ultrastructure of Synovio-Cartilage Junction in Rheumatoid Arthritis. Kobe J Med Sci (1973) 19(2):51–66.
58. Mohr W, Menninger H. Polymorphonuclear Granulocytes at the Pannus-Cartilage Junction in Rheumatoid Arthritis. Arthritis Rheum (1980) 23(12):1413–14. doi: 10.1002/art.1780231224
59. Mohr W, Wessinghage D. The Relationship Between Polymorphonuclear Granulocytes and Cartilage Destruction in Rheumatoid Arthritis. Z Rheumatol (1978) 37(3-4):81–6.
60. Fassbender HG, Simmling-Annefeld M. The Potential Aggressiveness of Synovial Tissue in Rheumatoid Arthritis. J Pathol (1983) 139(3):399–406. doi: 10.1002/path.1711390314
61. Tak P, Bresnihan B. The Pathogenesis and Prevention of Joint Damage in Rheumatoid Arthritis: Advances From Synovial Biopsy and Tissue Analysis. Arthritis Rheum-US (2000) 43(12):2619–33. doi: 10.1002/1529-0131(200012)43:12<2619::AID-ANR1>3.0.CO;2-V
62. Zvaifler N, Tsai V, Alsalameh S, von Kempis J, Firestein G, Lotz M. Pannocytes: Distinctive Cells Found in Rheumatoid Arthritis Articular Cartilage Erosions. Am J Pathol (1997) 150(3):1125–38.
63. Xue C, Takahashi M, Hasunuma T, Aono H, Yamamoto K, Yoshino S, et al. Characterisation of Fibroblast-Like Cells in Pannus Lesions of Patients With Rheumatoid Arthritis Sharing Properties of Fibroblasts and Chondrocytes. Ann Rheum Dis (1997) 56(4):262–7. doi: 10.1136/ard.56.4.262
64. Dennis G, Holweg C, Kummerfeld S, Choy D, Setiadi A, Hackney J, et al. Synovial Phenotypes in Rheumatoid Arthritis Correlate With Response to Biologic Therapeutics. Arthritis Res Ther (2014) 16(2):R90. doi: 10.1186/ar4555
65. Pitzalis C, Kelly S, Humby F. New Learnings on the Pathophysiology of RA From Synovial Biopsies. Curr Opin Rheumatol (2013) 25(3):334–44. doi: 10.1097/BOR.0b013e32835fd8eb
66. Cañete J, Celis R, Moll C, Izquierdo E, Marsal S, Sanmartí R, et al. Clinical Significance of Synovial Lymphoid Neogenesis and Its Reversal After Anti-Tumour Necrosis Factor Alpha Therapy in Rheumatoid Arthritis. Ann Rheum Dis (2009) 68(5):751–6. doi: 10.1136/ard.2008.089284
67. Kishimoto T. Interleukin-6: From Basic Science to Medicine–40 Years in Immunology. Annu Rev Immunol (2005) 23:1–21. doi: 10.1146/annurev.immunol.23.021704.115806
68. Ohshima S, Saeki Y, Mima T, Sasai M, Nishioka K, Nomura S, et al. Interleukin 6 Plays a Key Role in the Development of Antigen-Induced Arthritis. Proc Natl Acad Sci USA (1998) 95(14):8222–6. doi: 10.1073/pnas.95.14.8222
69. Hirano T, Matsuda T, Turner M, Miyasaka N, Buchan G, Tang B, et al. Excessive Production of Interleukin 6/B Cell Stimulatory Factor-2 in Rheumatoid Arthritis. Eur J Immunol (1988) 18(11):1797–801. doi: 10.1002/eji.1830181122
70. Hogan V, Holweg C, Choy D, Kummerfeld S, Hackney J, Teng Y, et al. Pretreatment Synovial Transcriptional Profile Is Associated With Early and Late Clinical Response in Rheumatoid Arthritis Patients Treated With Rituximab. Ann Rheum Dis (2012) 71(11):1888–94. doi: 10.1136/annrheumdis-2011-201115
71. Zhao J, Ye X, Zhang Z. The Predictive Value of Serum Soluble ICAM-1 and CXCL13 in the Therapeutic Response to TNF Inhibitor in Rheumatoid Arthritis Patients Who Are Refractory to csDMARDs. Clin Rheumatol (2020) 39(9):2573–81. doi: 10.1007/s10067-020-05043-1
72. Aloisi F, Pujol-Borrell R. Lymphoid Neogenesis in Chronic Inflammatory Diseases. Nat Rev Immunol (2006) 6(3):205–17. doi: 10.1038/nri1786
73. Tuckwell K, Gabay C, Sornasse T, Laubender R, Wang J, Townsend M. Levels of CXCL13 and sICAM-1 Correlate With Disease Activity Score in Patients With Rheumatoid Arthritis Treated With Tocilizumab. Adv Rheumatol (2019) 59(1):54. doi: 10.1186/s42358-019-0097-1
74. Rosengren S, Wei N, Kalunian K, Kavanaugh A, Boyle D. CXCL13: A Novel Biomarker of B-Cell Return Following Rituximab Treatment and Synovitis in Patients With Rheumatoid Arthritis. Rheumatol (Oxford) (2011) 50(3):603–10. doi: 10.1093/rheumatology/keq337
75. Maruotti N, Crivellato E, Cantatore F, Vacca A, Ribatti D. Mast Cells in Rheumatoid Arthritis. Clin Rheumatol (2007) 26(1):1–4. doi: 10.1007/s10067-006-0305-3
76. Tetlow L, Woolley D. Mast Cells, Cytokines, and Metalloproteinases at the Rheumatoid Lesion: Dual Immunolocalisation Studies. Ann Rheum Dis (1995) 54(11):896–903. doi: 10.1136/ard.54.11.896
77. Gotis-Graham I, Smith M, Parker A, McNeil H. Synovial Mast Cell Responses During Clinical Improvement in Early Rheumatoid Arthritis. Ann Rheum Dis (1998) 57(11):664–71. doi: 10.1136/ard.57.11.664
78. Godfrey H, Ilardi C, Engber W, Graziano F. Quantitation of Human Synovial Mast Cells in Rheumatoid Arthritis and Other Rheumatic Diseases. Arthritis Rheum-US (1984) 27(8):852–6. doi: 10.1002/art.1780270803
79. Ragipoglu D, Dudeck A, Haffner-Luntzer M, Voss M, Kroner J, Ignatius A, et al. The Role of Mast Cells in Bone Metabolism and Bone Disorders. Front Immunol (2020) 11:163. doi: 10.3389/fimmu.2020.00163
80. Wernersson S, Pejler G. Mast Cell Secretory Granules: Armed for Battle. Nat Rev Immunol (2014) 14(7):478–94. doi: 10.1038/nri3690
81. Nigrovic P, Binstadt B, Monach P, Johnsen A, Gurish M, Iwakura Y, et al. Mast Cells Contribute to Initiation of Autoantibody-Mediated Arthritis via IL-1. Proc Natl Acad Sci USA (2007) 104(7):2325–30. doi: 10.1073/pnas.0610852103
82. Sandler C, Lindstedt K, Joutsiniemi S, Lappalainen J, Juutilainen T, Kolah J, et al. Selective Activation of Mast Cells in Rheumatoid Synovial Tissue Results in Production of TNF-Alpha, IL-1beta and IL-1ra. Inflamm Res (2007) 56(6):230–9. doi: 10.1007/s00011-007-6135-1
83. Kim K, Kim B, Won J, Min H, Lee K, Lee S, et al. Regulation of Osteoclastogenesis by Mast Cell in Rheumatoid Arthritis. Arthritis Res Ther (2021) 23(1):124. doi: 10.1186/s13075-021-02491-1
84. Shin K, Nigrovic P, Crish J, Boilard E, McNeil H, Larabee K, et al. Mast Cells Contribute to Autoimmune Inflammatory Arthritis via Their Tryptase/Heparin Complexes. J Immunol (2009) 182(1):647–56. doi: 10.4049/jimmunol.182.1.647
85. Magnusson S, Pejler G, Kleinau S, Abrink M. Mast Cell Chymase Contributes to the Antibody Response and the Severity of Autoimmune Arthritis. FASEB J (2009) 23(3):875–82. doi: 10.1096/fj.08-120394
86. Kim K, Kim B, Lee K, Lee S, Firestein G, Kim H. Histamine and Histamine H4 Receptor Promotes Osteoclastogenesis in Rheumatoid Arthritis. Sci Rep (2017) 7(1):1197. doi: 10.1038/s41598-017-01101-y
87. Hiromatsu Y, Toda S. Mast Cells and Angiogenesis. Microsc Res Tech (2003) 60(1):64–9. doi: 10.1002/jemt.10244
88. Ruschpler P, Lorenz P, Eichler W, Koczan D, Hänel C, Scholz R, et al. High CXCR3 Expression in Synovial Mast Cells Associated With CXCL9 and CXCL10 Expression in Inflammatory Synovial Tissues of Patients With Rheumatoid Arthritis. Arthritis Res Ther (2003) 5(5):R241–52. doi: 10.1186/ar783
89. Ziegler-Heitbrock L. Monocyte Subsets in Man and Other Species. Cell Immunol (2014) 289:135–9. doi: 10.1016/j.cellimm.2014.03.019
90. Guilliams M, Mildner A, Yona S. Developmental and Functional Heterogeneity of Monocytes. Immunity (2018) 49(4):595–613. doi: 10.1016/j.immuni.2018.10.005
91. Guilliams M, Ginhoux F, Jakubzick C, Naik S, Onai N, Schraml B, et al. Dendritic Cells, Monocytes and Macrophages: A Unified Nomenclature Based on Ontogeny. Nat Rev Immunol (2014) 14(8):571–8. doi: 10.1038/nri3712
92. Ginhoux F, Guilliams M. Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity (2016) 44(3):439–49. doi: 10.1016/j.immuni.2016.02.024
93. Kapellos T, Bonaguro L, Gemünd I, Reusch N, Saglam A, Hinkley E, et al. Human Monocyte Subsets and Phenotypes in Major Chronic Inflammatory Diseases. Front Immunol (2019) 10:2035. doi: 10.3389/fimmu.2019.02035
94. Prajzlerová K, Kryštůfková O, Komarc M, Mann H, Hulejová H, Petrovská N, et al. The Dysregulation of Monocyte Subpopulations in Individuals at Risk of Developing Rheumatoid Arthritis. Rheumatol (Oxford) (2021) 60(4):1823–31. doi: 10.1093/rheumatology/keaa518
95. Tsukamoto M, Seta N, Yoshimoto K, Suzuki K, Yamaoka K, Takeuchi T. CD14CD16+ Intermediate Monocytes Are Induced by Interleukin-10 and Positively Correlate With Disease Activity in Rheumatoid Arthritis. Arthritis Res Ther (2017) 19(1):28. doi: 10.1186/s13075-016-1216-6
96. Chara L, Sánchez-Atrio A, Pérez A, Cuende E, Albarrán F, Turrión A, et al. The Number of Circulating Monocytes as Biomarkers of the Clinical Response to Methotrexate in Untreated Patients With Rheumatoid Arthritis. J Transl Med (2015) 13:2. doi: 10.1186/s12967-014-0375-y
97. Cooper D, Martin S, Robinson J, Mackie S, Charles C, Nam J, et al. Fcγriiia Expression on Monocytes in Rheumatoid Arthritis: Role in Immune-Complex Stimulated TNF Production and Non-Response to Methotrexate Therapy. PloS One (2012) 7(1):e28918. doi: 10.1371/journal.pone.0028918
98. Rana A, Li Y, Dang Q, Yang F. Monocytes in Rheumatoid Arthritis: Circulating Precursors of Macrophages and Osteoclasts and, Their Heterogeneity and Plasticity Role in RA Pathogenesis. Int Immunopharmacol (2018) 65:348–59. doi: 10.1016/j.intimp.2018.10.016
99. Akira S, Takeda K, Kaisho T. Toll-Like Receptors: Critical Proteins Linking Innate and Acquired Immunity. Nat Immunol (2001) 2(8):675–80. doi: 10.1038/90609
100. Lohmann K, Vandenplas M, Barton M, Bryant C, Moore J. The Equine TLR4/MD-2 Complex Mediates Recognition of Lipopolysaccharide From Rhodobacter Sphaeroides as an Agonist. J Endotoxin Res (2007) 13(4):235–42. doi: 10.1177/0968051907083193
101. Unkeless J. Human Fc Gamma Receptors. Curr Opin Immunol (1989) 2(1):63–7. doi: 10.1016/0952-7915(89)90098-8
102. Laurent L, Clavel C, Lemaire O, Anquetil F, Cornillet M, Zabraniecki L, et al. Fcγ Receptor Profile of Monocytes and Macrophages From Rheumatoid Arthritis Patients and Their Response to Immune Complexes Formed With Autoantibodies to Citrullinated Proteins. Ann Rheum Dis (2011) 70(6):1052–9. doi: 10.1136/ard.2010.142091
103. McGarry T, Hanlon M, Marzaioli V, Cunningham C, Krishna V, Murray K, et al. Rheumatoid Arthritis CD14 Monocytes Display Metabolic and Inflammatory Dysfunction, a Phenotype That Precedes Clinical Manifestation of Disease. Clin Transl Immunol (2021) 10(1):e1237. doi: 10.1002/cti2.1237
104. Belge K, Dayyani F, Horelt A, Siedlar M, Frankenberger M, Frankenberger B, et al. The Proinflammatory CD14+CD16+DR++ Monocytes Are a Major Source of TNF. J Immunol (2002) 168(7):3536–42. doi: 10.4049/jimmunol.168.7.3536
105. Ziegler-Heitbrock H, Fingerle G, Ströbel M, Schraut W, Stelter F, Schütt C, et al. The Novel Subset of CD14+/CD16+ Blood Monocytes Exhibits Features of Tissue Macrophages. Eur J Immunol (1993) 23(9):2053–8. doi: 10.1002/eji.1830230902
106. Yoon B, Yoo S, Choi Y, Chung Y, Kim J, Yoo I, et al. Functional Phenotype of Synovial Monocytes Modulating Inflammatory T-Cell Responses in Rheumatoid Arthritis (RA). PloS One (2014) 9(10):e109775. doi: 10.1371/journal.pone.0109775
107. Iwahashi M, Yamamura M, Aita T, Okamoto A, Ueno A, Ogawa N, et al. Expression of Toll-Like Receptor 2 on CD16+ Blood Monocytes and Synovial Tissue Macrophages in Rheumatoid Arthritis. Arthritis Rheum-US (2004) 50(5):1457–67. doi: 10.1002/art.20219
108. Lacerte P, Brunet A, Egarnes B, Duchêne B, Brown J, Gosselin J. Overexpression of TLR2 and TLR9 on Monocyte Subsets of Active Rheumatoid Arthritis Patients Contributes to Enhance Responsiveness to TLR Agonists. Arthritis Res Ther (2016) 18:10. doi: 10.1186/s13075-015-0901-1
109. Ospelt C, Brentano F, Rengel Y, Stanczyk J, Kolling C, Tak P, et al. Overexpression of Toll-Like Receptors 3 and 4 in Synovial Tissue From Patients With Early Rheumatoid Arthritis: Toll-Like Receptor Expression in Early and Longstanding Arthritis. Arthritis Rheum-US (2008) 58(12):3684–92. doi: 10.1002/art.24140
110. Thwaites R, Unterberger S, Chamberlain G, Walker-Bone K, Davies K, Sacre S. TLR1/2 and 5 Induce Elevated Cytokine Levels From Rheumatoid Arthritis Monocytes Independent of ACPA or RF Autoantibody Status. Rheumatol (Oxford) (2020) 59(11):3533–9. doi: 10.1093/rheumatology/keaa220
111. Fan S, Zong M, Zhang H, Lu Y, Lu T, Fan L. Decreased Expression of Alpha-Enolase Inhibits the Proliferation of Hypoxia-Induced Rheumatoid Arthritis Fibroblasts-Like Synoviocytes. Mod Rheumatol (2015) 25(5):701–7. doi: 10.3109/14397595.2015.1014141
112. Guillou C, Fréret M, Fondard E, Derambure C, Avenel G, Golinski M, et al. Soluble Alpha-Enolase Activates Monocytes by CD14-Dependent TLR4 Signalling Pathway and Exhibits a Dual Function. Sci Rep (2016) 6:23796. doi: 10.1038/srep23796
113. Meusch U, Rossol M, Baerwald C, Hauschildt S, Wagner U. Outside-To-Inside Signaling Through Transmembrane Tumor Necrosis Factor Reverses Pathologic Interleukin-1beta Production and Deficient Apoptosis of Rheumatoid Arthritis Monocytes. Arthritis Rheum-US (2009) 60(9):2612–21. doi: 10.1002/art.24778
114. Meusch U, Krasselt M, Rossol M, Baerwald C, Klingner M, Wagner U. In Vitro Response Pattern of Monocytes After tmTNF Reverse Signaling Predicts Response to Anti-TNF Therapy in Rheumatoid Arthritis. J Transl Med (2015) 13:256. doi: 10.1186/s12967-015-0620-z
115. Estrada-Capetillo L, Hernández-Castro B, Monsiváis-Urenda A, Alvarez-Quiroga C, Layseca-Espinosa E, Abud-Mendoza C, et al. Induction of Th17 Lymphocytes and Treg Cells by Monocyte-Derived Dendritic Cells in Patients With Rheumatoid Arthritis and Systemic Lupus Erythematosus. Clin Dev Immunol (2013) 2013:584303. doi: 10.1155/2013/584303
116. Jongbloed S, Lebre M, Fraser A, Gracie J, Sturrock R, Tak P, et al. Enumeration and Phenotypical Analysis of Distinct Dendritic Cell Subsets in Psoriatic Arthritis and Rheumatoid Arthritis. Arthritis Res Ther (2006) 8(1):R15. doi: 10.1186/ar1864
117. Lebre M, Tak P. Dendritic Cells in Rheumatoid Arthritis: Which Subset Should be Used as a Tool to Induce Tolerance? Hum Immunol (2009) 70(5):321–4. doi: 10.1016/j.humimm.2009.02.006
118. Jego G, Palucka AK, Blanck JP, Chalouni C, Pascual V, Banchereau J. Plasmacytoid Dendritic Cells Induce Plasma Cell Differentiation Through Type I Interferon and Interleukin 6. Immunity (2003) 19(2):225–34. doi: 10.1016/S1074-7613(03)00208-5
119. Poeck H, Wagner M, Battiany J, Rothenfusser S, Wellisch D, Hornung V, et al. Plasmacytoid Dendritic Cells, Antigen, and CpG-C License Human B Cells for Plasma Cell Differentiation and Immunoglobulin Production in the Absence of T-Cell Help. Blood (2004) 103(8):3058–64. doi: 10.1182/blood-2003-08-2972
120. Lebre MC, Jongbloed SL, Tas SW, Smeets TJ, McInnes IB, Tak PP. Rheumatoid Arthritis Synovium Contains Two Subsets of CD83-DC-LAMP- Dendritic Cells With Distinct Cytokine Profiles. Am J Pathol (2008) 172(4):940–50. doi: 10.2353/ajpath.2008.070703
121. Lioté F, Boval-Boizard B, Weill D, Kuntz D, Wautier J. Blood Monocyte Activation in Rheumatoid Arthritis: Increased Monocyte Adhesiveness, Integrin Expression, and Cytokine Release. Clin Exp Immunol (1996) 106(1):13–9. doi: 10.1046/j.1365-2249.1996.d01-820.x
122. Samy E, Wax S, Huard B, Hess H, Schneider P. Targeting BAFF and APRIL in Systemic Lupus Erythematosus and Other Antibody-Associated Diseases. Int Rev Immunol (2017) 36(1):3–19. doi: 10.1080/08830185.2016.1276903
123. Boghdadi G, El-Sokkary R, Elewa E, Abbas S. APRIL Level as a Marker of Disease Activity in Treated Rheumatoid Arthritis Patients: Association With Disease Activity and Anti-CCP Antibody. Egypt J Immunol (2015) 22(2):31–9.
124. Weldon A, Moldovan I, Cabling M, Hernandez E, Hsu S, Gonzalez J, et al. Surface APRIL Is Elevated on Myeloid Cells and Is Associated With Disease Activity in Patients With Rheumatoid Arthritis. J Rheumatol (2015) 42(5):749–59. doi: 10.3899/jrheum.140630
125. Chang Y, Jia X, Sun X, Xu S, Wu Y, Zhang L, et al. APRIL Promotes Proliferation, Secretion and Invasion of Fibroblast-Like Synoviocyte From Rats With Adjuvant Induced Arthritis. Mol Immunol (2015) 64(1):90–8. doi: 10.1016/j.molimm.2014.11.002
126. Xiong Y, Cheng Y, Lin Q, Wu A, Yu J, Li C, et al. Increased Expression of Siglec-1 on Peripheral Blood Monocytes and Its Role in Mononuclear Cell Reactivity to Autoantigen in Rheumatoid Arthritis. Rheumatol (Oxford) (2014) 53(2):250–9. doi: 10.1093/rheumatology/ket342
127. McCulloch K, McGrath S, Huesa C, Dunning L, Litherland G, Crilly A, et al. Rheumatic Disease: Protease-Activated Receptor-2 in Synovial Joint Pathobiology. Front Endocrinol (Lausanne) (2018) 9:257. doi: 10.3389/fendo.2018.00257
128. Xue M, Chan Y, Shen K, Dervish S, March L, Sambrook P, et al. Protease-Activated Receptor 2, Rather Than Protease-Activated Receptor 1, Contributes to the Aggressive Properties of Synovial Fibroblasts in Rheumatoid Arthritis. Arthritis Rheum-US (2012) 64(1):88–98. doi: 10.1002/art.33323
129. Bain C, Hawley C, Garner H, Scott C, Schridde A, Steers N, et al. Long-Lived Self-Renewing Bone Marrow-Derived Macrophages Displace Embryo-Derived Cells to Inhabit Adult Serous Cavities. Nat Commun (2016) 7:ncomms11852. doi: 10.1038/ncomms11852
130. Imperatore F, Maurizio J, Vargas Aguilar S, Busch C, Favret J, Kowenz-Leutz E, et al. SIRT1 Regulates Macrophage Self-Renewal. EMBO J (2017) 36(16):2353–72. doi: 10.15252/embj.201695737
131. Ghosn E, Cassado A, Govoni G, Fukuhara T, Yang Y, Monack D, et al. Two Physically, Functionally, and Developmentally Distinct Peritoneal Macrophage Subsets. Proc Natl Acad Sci USA (2010) 107(6):2568–73. doi: 10.1073/pnas.0915000107
132. Yang X, Chang Y, Wei W. Emerging Role of Targeting Macrophages in Rheumatoid Arthritis: Focus on Polarization, Metabolism and Apoptosis. Cell Proliferat (2020) 53(7):e12854. doi: 10.1111/cpr.12854
133. Tu J, Hong W, Guo Y, Zhang P, Fang Y, Wang X, et al. Ontogeny of Synovial Macrophages and the Roles of Synovial Macrophages From Different Origins in Arthritis. Front Immunol (2019) 10:1146. doi: 10.3389/fimmu.2019.01146
134. Culemann S, Grüneboom A, Nicolás-Ávila J, Weidner D, Lämmle K, Rothe T, et al. Locally Renewing Resident Synovial Macrophages Provide a Protective Barrier for the Joint. Nature (2019) 572(7771):670–5. doi: 10.1038/s41586-019-1471-1
135. Haringman J, Gerlag D, Zwinderman A, Smeets T, Kraan M, Baeten D, et al. Synovial Tissue Macrophages: A Sensitive Biomarker for Response to Treatment in Patients With Rheumatoid Arthritis. Ann Rheum Dis (2005) 64(6):834–8. doi: 10.1136/ard.2004.029751
136. Mulherin D, Fitzgerald O, Bresnihan B. Synovial Tissue Macrophage Populations and Articular Damage in Rheumatoid Arthritis. Arthritis Rheum-US (1996) 39(1):115–24. doi: 10.1002/art.1780390116
137. Youssef P, Roth J, Frosch M, Costello P, Fitzgerald O, Sorg C, et al. Expression of Myeloid Related Proteins (MRP) 8 and 14 and the MRP8/14 Heterodimer in Rheumatoid Arthritis Synovial Membrane. J Rheumatol (1999) 26(12):2523–8.
138. Ambarus C, Noordenbos T, de Hair M, Tak P, Baeten D. Intimal Lining Layer Macrophages But Not Synovial Sublining Macrophages Display an IL-10 Polarized-Like Phenotype in Chronic Synovitis. Arthritis Res Ther (2012) 14(2):R74. doi: 10.1186/ar3796
139. Soler Palacios B, Estrada-Capetillo L, Izquierdo E, Criado G, Nieto C, Municio C, et al. Macrophages From the Synovium of Active Rheumatoid Arthritis Exhibit an Activin A-Dependent Pro-Inflammatory Profile. J Pathol (2015) 235(3):515–26. doi: 10.1002/path.4466
140. Zhu W, Li X, Fang S, Zhang X, Wang Y, Zhang T, et al. Anti-Citrullinated Protein Antibodies Induce Macrophage Subset Disequilibrium in RA Patients. Inflammation (2015) 38(6):2067–75. doi: 10.1007/s10753-015-0188-z
141. Yang X, Li S, Zhao Y, Li S, Zhao T, Tai Y, et al. GRK2 Mediated Abnormal Transduction of PGE2-EP4-cAMP-CREB Signaling Induces the Imbalance of Macrophages Polarization in Collagen-Induced Arthritis Mice. Cells (2019) 8(12):1596. doi: 10.3390/cells8121596
142. Yang P, Qian F, Zhang M, Xu A, Wang X, Jiang B, et al. Th17 Cell Pathogenicity and Plasticity in Rheumatoid Arthritis. J Leukoc Biol (2019) 106(6):1233–40. doi: 10.1002/JLB.4RU0619-197R
143. Joosten L, van De Loo F, Lubberts E, Helsen M, Netea M, van der Meer J, et al. An IFN-Gamma-Independent Proinflammatory Role of IL-18 in Murine Streptococcal Cell Wall Arthritis. J Immunol (2000) 165(11):6553–8. doi: 10.4049/jimmunol.165.11.6553
144. Gracie J, Forsey R, Chan W, Gilmour A, Leung B, Greer M, et al. A Proinflammatory Role for IL-18 in Rheumatoid Arthritis. J Clin Invest (1999) 104(10):1393–401. doi: 10.1172/JCI7317
145. Zhang W, Cong XL, Qin YH, He ZW, He DY, Dai SM. IL-18 Upregulates the Production of Key Regulators of Osteoclastogenesis From Fibroblast-Like Synoviocytes in Rheumatoid Arthritis. Inflammation (2013) 36(1):103–9. doi: 10.1007/s10753-012-9524-8
146. Joosten L, Netea M, Kim S, Yoon D, Oppers-Walgreen B, Radstake T, et al. IL-32, a Proinflammatory Cytokine in Rheumatoid Arthritis. Proc Natl Acad Sci USA (2006) 103(9):3298–303. doi: 10.1073/pnas.0511233103
147. Lopa S, Leijs MJ, Moretti M, Lubberts E, van Osch GJ, Bastiaansen-Jenniskens YM. Arthritic and Non-Arthritic Synovial Fluids Modulate IL10 and IL1RA Gene Expression in Differentially Activated Primary Human Monocytes. Osteoarthritis Cartilage (2015) 23(11):1853–7. doi: 10.1016/j.joca.2015.06.003
148. Stamp L, Easson A, Pettersson L, Highton J, Hessian P. Monocyte Derived Interleukin (IL)-23 Is an Important Determinant of Synovial IL-17A Expression in Rheumatoid Arthritis. J Rheumatol (2009) 36(11):2403–8. doi: 10.3899/jrheum.081304
149. Bettelli E, Carrier Y, Gao W, Korn T, Strom T, Oukka M, et al. Reciprocal Developmental Pathways for the Generation of Pathogenic Effector TH17 and Regulatory T Cells. Nature (2006) 441(7090):235–8. doi: 10.1038/nature04753
150. Xu D, Jiang HR, Kewin P, Li Y, Mu R, Fraser AR, et al. IL-33 Exacerbates Antigen-Induced Arthritis by Activating Mast Cells. Proc Natl Acad Sci USA (2008) 105(31):10913–8. doi: 10.1073/pnas.0801898105
151. Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an Interleukin-1-Like Cytokine That Signals via the IL-1 Receptor-Related Protein ST2 and Induces T Helper Type 2-Associated Cytokines. Immunity (2005) 23(5):479–90. doi: 10.1016/j.immuni.2005.09.015
152. Nile C, Barksby E, Jitprasertwong P, Preshaw P, Taylor J. Expression and Regulation of Interleukin-33 in Human Monocytes. Immunology (2010) 130(2):172–80. doi: 10.1111/j.1365-2567.2009.03221.x
153. Lee K, Achuthan A, Hamilton J. GM-CSF: A Promising Target in Inflammation and Autoimmunity. Immunotargets Ther (2020) 9:225–40. doi: 10.2147/ITT.S262566
154. Erwig L, Kluth D, Walsh G, Rees A. Initial Cytokine Exposure Determines Function of Macrophages and Renders Them Unresponsive to Other Cytokines. J Immunol (1998) 161(4):1983–8.
155. El-Hakeim E, Abd Elhameed Z, Nouby F, Abdel Razek M, El-Moneum O, Goma S. Association of Interleukin-12b Polymorphism and Serum Level of Interleukin-12 in a Sample of Egyptian Patients With Rheumatoid Arthritis. Egypt J Immunol (2020) 27(1):19–28.
156. Yamamoto E, Jørgensen T. Relationships Between Vitamin D, Gut Microbiome, and Systemic Autoimmunity. Front Immunol (2019) 10:3141. doi: 10.3389/fimmu.2019.03141
157. Murdaca G, Gerosa A, Paladin F, Petrocchi L, Banchero S, Gangemi S. Vitamin D and Microbiota: Is There a Link With Allergies? Int J Mol Sci (2021) 22(8):4288. doi: 10.3390/ijms22084288
158. Murdaca G, Paladin F, Gangemi S. Role of Vitamin D in the Clinical Course of Nasal Polyposis. Biomedicines (2021) 9(8):855. doi: 10.3390/biomedicines9080855
159. Murdaca G, Tonacci A, Negrini S, Greco M, Borro M, Puppo F, et al. Emerging Role of Vitamin D in Autoimmune Diseases: An Update on Evidence and Therapeutic Implications. Autoimmun Rev (2019) 18(9):102350. doi: 10.1016/j.autrev.2019.102350
160. Murdaca G, Greco M, Negrini S, Casciaro M, Gangemi S. The Role of Skin and Gut Microbiome and Epigenetic Modifications in Skin-Autoimmune Disorders. Curr Mol Med (2021) 21(4):283–90. doi: 10.2174/1566524020666200812222324
161. Hsieh C, Macatonia S, Tripp C, Wolf S, O’Garra A, Murphy K. Development of TH1 CD4+ T Cells Through IL-12 Produced by Listeria-Induced Macrophages. Sci (New York NY) (1993) 260(5107):547–9. doi: 10.1126/science.8097338
162. Ottolenghi A, Bolel P, Sarkar R, Greenshpan Y, Iraqi M, Ghosh S, et al. Life-Extended Glycosylated IL-2 Promotes Treg Induction and Suppression of Autoimmunity. Sci Rep (2021) 11(1):7676. doi: 10.1038/s41598-021-87102-4
163. Min H, Won J, Kim B, Lee K, Lee S, Lee S, et al. Interleukin (IL)-25 Suppresses IL-22-Induced Osteoclastogenesis in Rheumatoid Arthritis via STAT3 and P38 MAPK/Iκbα Pathway. Arthritis Res Ther (2020) 22(1):222. doi: 10.1186/s13075-020-02315-8
164. Zhao L, Jiang Z, Jiang Y, Ma N, Zhang Y, Feng L, et al. IL-22+ CD4+ T Cells in Patients With Rheumatoid Arthritis. Int J Rheum Dis (2013) 16(5):518–26. doi: 10.1111/1756-185X.12099
165. Liu D, Cao T, Wang N, Liu C, Ma N, Tu R, et al. IL-25 Attenuates Rheumatoid Arthritis Through Suppression of Th17 Immune Responses in an IL-13-Dependent Manner. Sci Rep (2016) 6:36002. doi: 10.1038/srep36002
166. Lavocat F, Ndongo-Thiam N, Miossec P. Interleukin-25 Produced by Synoviocytes Has Anti-Inflammatory Effects by Acting As a Receptor Antagonist for Interleukin-17a Function. Front Immunol (2017) 8:647. doi: 10.3389/fimmu.2017.00647
167. Su X, Huang Q, Chen J, Wang M, Pan H, Wang R, et al. Calycosin Suppresses Expression of Pro-Inflammatory Cytokines via the Activation of P62/Nrf2-Linked Heme Oxygenase 1 in Rheumatoid Arthritis Synovial Fibroblasts. Pharmacol Res (2016) 113:695–704. doi: 10.1016/j.phrs.2016.09.031
168. Littlewood-Evans A, Sarret S, Apfel V, Loesle P, Dawson J, Zhang J, et al. GPR91 Senses Extracellular Succinate Released From Inflammatory Macrophages and Exacerbates Rheumatoid Arthritis. J Exp Med (2016) 213(9):1655–62. doi: 10.1084/jem.20160061
169. Kurowska W, Przygodzka M, Jakubaszek M, Kwiatkowska B, Maslinski W. Interleukin-15 as a Biomarker Candidate of Rheumatoid Arthritis Development. J Clin Med (2020) 9(5):1555. doi: 10.3390/jcm9051555
170. Ziolkowska M, Koc A, Luszczykiewicz G, Ksiezopolska-Pietrzak K, Klimczak E, Chwalinska-Sadowska H, et al. High Levels of IL-17 in Rheumatoid Arthritis Patients: IL-15 Triggers In Vitro IL-17 Production via Cyclosporin A-Sensitive Mechanism. J Immunol (2000) 164(5):2832–8. doi: 10.4049/jimmunol.164.5.2832
171. Ortiz A, Laffon A, Gonzalez-Alvaro I. CD69 Expression on Lymphocytes and Interleukin-15 Levels in Synovial Fluids From Different Inflammatory Arthropathies. Rheumatol Int (2002) 21(5):182–8. doi: 10.1007/s00296-001-0161-z
172. McInnes I, al-Mughales J, Field M, Leung B, Huang F, Dixon R, et al. The Role of Interleukin-15 in T-Cell Migration and Activation in Rheumatoid Arthritis. Nat Med (1996) 2(2):175–82. doi: 10.1038/nm0296-175
173. Chen Z, Bozec A, Ramming A, Schett G. Anti-Inflammatory and Immune-Regulatory Cytokines in Rheumatoid Arthritis. Nat Rev Rheumatol (2019) 15(1):9–17. doi: 10.1038/s41584-018-0109-2
174. Wong CK, Chen DP, Tam LS, Li EK, Yin YB, Lam CW. Effects of Inflammatory Cytokine IL-27 on the Activation of Fibroblast-Like Synoviocytes in Rheumatoid Arthritis. Arthritis Res Ther (2010) 12(4):R129. doi: 10.1186/ar3067
175. Awasthi A, Carrier Y, Peron JP, Bettelli E, Kamanaka M, Flavell RA, et al. A Dominant Function for Interleukin 27 in Generating Interleukin 10-Producing Anti-Inflammatory T Cells. Nat Immunol (2007) 8(12):1380–9. doi: 10.1038/ni1541
176. Hirahara K, Ghoreschi K, Yang XP, Takahashi H, Laurence A, Vahedi G, et al. Interleukin-27 Priming of T Cells Controls IL-17 Production in Trans via Induction of the Ligand PD-L1. Immunity (2012) 36(6):1017–30. doi: 10.1016/j.immuni.2012.03.024
177. Lin Y, Wang Y, Peng Y, Liu F, Lin G, Huang S, et al. Interleukin 26 Skews Macrophage Polarization Towards M1 Phenotype by Activating cJUN and the NF-κb Pathway. Cells (2020) 9(4):938. doi: 10.3390/cells9040938
178. Xu T, Jia S, Li P. Interleukin-29 and Interleukin-28A Induce Migration of Neutrophils in Rheumatoid Arthritis. Clin Rheumatol (2021) 40(1):369–75. doi: 10.1007/s10067-020-05211-3
179. Xu T, Yan T, Li P. Interleukin-29 Regulates T Follicular Helper Cells by Repressing BCL6 in Rheumatoid Arthritis Patients. Clin Rheumatol (2020) 39(12):3797–804. doi: 10.1007/s10067-020-05151-y
180. da Rocha Junior L, Branco Pinto Duarte A, de Melo Rêgo M, de Almeida A, de Melo Vilar K, de Lima H, et al. Sensitivity and Specificity of Interleukin 29 in Patients With Rheumatoid Arthritis and Other Rheumatic Diseases. Immunol Lett (2020) 220:38–43. doi: 10.1016/j.imlet.2020.01.004
181. Xu L, Feng X, Shi Y, Wang X, Kong X, Zhang M, et al. Interleukin-29 Induces Receptor Activator of NF-κb Ligand Expression in Fibroblast-Like Synoviocytes via MAPK Signaling Pathways. Int J Rheum Dis (2015) 18(8):842–9. doi: 10.1111/1756-185X.12747
182. Xu L, Feng X, Tan W, Gu W, Guo D, Zhang M, et al. IL-29 Enhances Toll-Like Receptor-Mediated IL-6 and IL-8 Production by the Synovial Fibroblasts From Rheumatoid Arthritis Patients. Arthritis Res Ther (2013) 15(5):R170. doi: 10.1186/ar4357
183. Vasilev G, Manolova I, Ivanova M, Stanilov I, Miteva L, Stanilova S. The Role of IL-18 in Addition to Th17 Cytokines in Rheumatoid Arthritis Development and Treatment in Women. Sci Rep (2021) 11(1):15391. doi: 10.1038/s41598-021-94841-x
184. Gualberto Cardoso P, Diniz Lopes Marques C, de Melo Vilar K, Dantas A, Branco Pinto Duarte A, Pitta I, et al. Interleukin-18 in Brazilian Rheumatoid Arthritis Patients: Can Leflunomide Reduce it? Autoimmun Dis (2021) 2021:6672987. doi: 10.1155/2021/6672987
185. Al-Bogami M, Bystrom J, Clanchy F, Taher T, Mangat P, Williams R, et al. Tnfα Inhibitors Reduce Bone Loss in Rheumatoid Arthritis Independent of Clinical Response by Reducing Osteoclast Precursors and IL-20. Rheumatol (Oxford) (2021) 60(2):947–57. doi: 10.1093/rheumatology/keaa551
186. García-Arellano S, Hernández-Palma L, Bucala R, Hernández-Bello J, de la Cruz-Mosso U, García-Iglesias T, et al. Th1/Th17 Cytokine Profile Is Induced by Macrophage Migration Inhibitory Factor in Peripheral Blood Mononuclear Cells From Rheumatoid Arthritis Patients. Curr Mol Med (2018) 18(10):679–88. doi: 10.2174/1566524019666190129123240
187. Kim K, Kim H. Macrophage Migration Inhibitory Factor: A Potential Therapeutic Target for Rheumatoid Arthritis. Korean J Intern Med (2016) 31(4):634–42. doi: 10.3904/kjim.2016.098
188. Yang X, Chang Y, Wei W. Endothelial Dysfunction and Inflammation: Immunity in Rheumatoid Arthritis. Mediat Inflamm (2016) 2016:6813016. doi: 10.1155/2016/6813016
189. Li M, Ransohoff R. The Roles of Chemokine CXCL12 in Embryonic and Brain Tumor Angiogenesis. Semin Cancer Biol (2009) 19(2):111–5. doi: 10.1016/j.semcancer.2008.11.001
190. Sucur A, Jajic Z, Artukovic M, Matijasevic M, Anic B, Flegar D, et al. Chemokine Signals Are Crucial for Enhanced Homing and Differentiation of Circulating Osteoclast Progenitor Cells. Arthritis Res Ther (2017) 19(1):142. doi: 10.1186/s13075-017-1337-6
191. Shahrara S, Pickens S, Mandelin A, Karpus W, Huang Q, Kolls J, et al. IL-17-Mediated Monocyte Migration Occurs Partially Through CC Chemokine Ligand 2/Monocyte Chemoattractant Protein-1 Induction. J Immunol (2010) 184(8):4479–87. doi: 10.4049/jimmunol.0901942
192. Costa P, Rusconi S, Mavilio D, Fogli M, Murdaca G, Pende D, et al. Differential Disappearance of Inhibitory Natural Killer Cell Receptors During HAART and Possible Impairment of HIV-1-Specific CD8 Cytotoxic T Lymphocytes. AIDS (London England) (2001) 15(8):965–74. doi: 10.1097/00002030-200105250-00004
193. Hamilton J. GM-CSF as a Target in Inflammatory/Autoimmune Disease: Current Evidence and Future Therapeutic Potential. Expert Rev Clin Immunol (2015) 11(4):457–65. doi: 10.1586/1744666X.2015.1024110
194. Crotti C, Agape E, Becciolini A, Biggioggero M, Favalli E. Targeting Granulocyte-Monocyte Colony-Stimulating Factor Signaling in Rheumatoid Arthritis: Future Prospects. Drugs (2019) 79(16):1741–55. doi: 10.1007/s40265-019-01192-z
195. Bird P, Peterfy C, Countryman P, Griffiths H, Barrett R, Youssef P, et al. AC-CUTE: An Open-Label Study to Evaluate Progression of Structural Joint Damage and Inflammation in Subjects With Moderate to Severe Rheumatoid Arthritis. Int J Rheumatol (2018) 2018:8721753. doi: 10.1155/2018/8721753
196. Mélet J, Mulleman D, Goupille P, Ribourtout B, Watier H, Thibault G. Rituximab-Induced T Cell Depletion in Patients With Rheumatoid Arthritis: Association With Clinical Response. Arthritis Rheum-US (2013) 65(11):2783–90. doi: 10.1002/art.38107
197. Tony HP, Roll P, Mei HE, Blümner E, Straka A, Gnuegge L, et al. Combination of B Cell Biomarkers as Independent Predictors of Response in Patients With Rheumatoid Arthritis Treated With Rituximab. Clin Exp Rheumatol (2015) 33(6):887–94.
198. Sellam J, Rivière E, Courties A, Rouzaire P, Tolusso B, Vital E, et al. Serum IL-33, a New Marker Predicting Response to Rituximab in Rheumatoid Arthritis. Arthritis Res Ther (2016) 18(1):294. doi: 10.1186/s13075-016-1190-z
199. Sellam J, Hendel-Chavez H, Rouanet S, Abbed K, Combe B, Le Loët X, et al. B Cell Activation Biomarkers as Predictive Factors for the Response to Rituximab in Rheumatoid Arthritis: A Six-Month, National, Multicenter, Open-Label Study. Arthritis Rheum (2011) 63(4):933–8. doi: 10.1002/art.30233
200. Sellam J, Rouanet S, Hendel-Chavez H, Miceli-Richard C, Combe B, Sibilia J, et al. CCL19, a B Cell Chemokine, Is Related to the Decrease of Blood Memory B Cells and Predicts the Clinical Response to Rituximab in Patients With Rheumatoid Arthritis. Arthritis Rheum (2013) 65(9):2253–61. doi: 10.1002/art.38023
201. Sellam J, Rouanet S, Hendel-Chavez H, Abbed K, Sibilia J, Tebib J, et al. Blood Memory B Cells Are Disturbed and Predict the Response to Rituximab in Patients With Rheumatoid Arthritis. Arthritis Rheum (2011) 63(12):3692–701. doi: 10.1002/art.30599
202. Lal P, Su Z, Holweg C, Silverman G, Schwartzman S, Kelman A, et al. Inflammation and Autoantibody Markers Identify Rheumatoid Arthritis Patients With Enhanced Clinical Benefit Following Rituximab Treatment. Arthritis Rheum-US (2011) 63(12):3681–91. doi: 10.1002/art.30596
203. Vuolteenaho K, Tuure L, Nieminen R, Laasonen L, Leirisalo-Repo M, Moilanen E. Pretreatment Resistin Levels Are Associated With Erosive Disease in Early Rheumatoid Arthritis Treated With Disease-Modifying Anti-Rheumatic Drugs and Infliximab. Scand J Rheumatol (2021) 15:1–6. doi: 10.1080/03009742.2021.1929456
204. Smolen J, van Vollenhoven R, Florentinus S, Chen S, Suboticki J, Kavanaugh A. Predictors of Disease Activity and Structural Progression After Treatment With Adalimumab Plus Methotrexate or Continued Methotrexate Monotherapy in Patients With Early Rheumatoid Arthritis and Suboptimal Response to Methotrexate. Ann Rheum Dis (2018) 77(11):1566–72. doi: 10.1136/annrheumdis-2018-213502
205. Genovese M, Fleischmann R, Greenwald M, Satterwhite J, Veenhuizen M, Xie L, et al. Tabalumab, an Anti-BAFF Monoclonal Antibody, in Patients With Active Rheumatoid Arthritis With an Inadequate Response to TNF Inhibitors. Ann Rheum Dis (2013) 72(9):1461–8. doi: 10.1136/annrheumdis-2012-202775
206. Boyle D, Soma K, Hodge J, Kavanaugh A, Mandel D, Mease P, et al. The JAK Inhibitor Tofacitinib Suppresses Synovial JAK1-STAT Signalling in Rheumatoid Arthritis. Ann Rheum Dis (2015) 74(6):1311–6. doi: 10.1136/annrheumdis-2014-206028
207. Smolen JS, Agarwal SK, Ilivanova E, Xu XL, Miao Y, Zhuang Y, et al. A Randomised Phase II Study Evaluating the Efficacy and Safety of Subcutaneously Administered Ustekinumab and Guselkumab in Patients With Active Rheumatoid Arthritis Despite Treatment With Methotrexate. Ann Rheum Dis (2017) 76(5):831–9. doi: 10.1136/annrheumdis-2016-209831
208. Behrens F, Tak P, Østergaard M, Stoilov R, Wiland P, Huizinga T, et al. MOR103, a Human Monoclonal Antibody to Granulocyte-Macrophage Colony-Stimulating Factor, in the Treatment of Patients With Moderate Rheumatoid Arthritis: Results of a Phase Ib/IIa Randomised, Double-Blind, Placebo-Controlled, Dose-Escalation Trial. Ann Rheum Dis (2015) 74(6):1058–64. doi: 10.1136/annrheumdis-2013-204816
209. Huizinga T, Batalov A, Stoilov R, Lloyd E, Wagner T, Saurigny D, et al. Phase 1b Randomized, Double-Blind Study of Namilumab, an Anti-Granulocyte Macrophage Colony-Stimulating Factor Monoclonal Antibody, in Mild-to-Moderate Rheumatoid Arthritis. Arthritis Res Ther (2017) 19(1):53. doi: 10.1186/s13075-017-1267-3
210. Taylor P, Saurigny D, Vencovsky J, Takeuchi T, Nakamura T, Matsievskaia G, et al. Efficacy and Safety of Namilumab, a Human Monoclonal Antibody Against Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF) Ligand in Patients With Rheumatoid Arthritis (RA) With Either an Inadequate Response to Background Methotrexate Therapy or an Inadequate Response or Intolerance to an Anti-TNF (Tumour Necrosis Factor) Biologic Therapy: A Randomized, Controlled Trial. Arthritis Res Ther (2019) 21(1):101. doi: 10.1186/s13075-019-1879-x
211. Weinblatt M, McInnes I, Kremer J, Miranda P, Vencovsky J, Guo X, et al. A Randomized Phase IIb Study of Mavrilimumab and Golimumab in Rheumatoid Arthritis. Arthritis Rheumatol (2018) 70(1):49–59. doi: 10.1002/art.40323
212. Takeuchi T, Tanaka Y, Close D, Godwood A, Wu C, Saurigny D. Efficacy and Safety of Mavrilimumab in Japanese Subjects With Rheumatoid Arthritis: Findings From a Phase IIa Study. Mod Rheumatol (2015) 25(1):21–30. doi: 10.3109/14397595.2014.896448
213. Burmester G, Weinblatt M, McInnes I, Porter D, Barbarash O, Vatutin M, et al. Efficacy and Safety of Mavrilimumab in Subjects With Rheumatoid Arthritis. Ann Rheum Dis (2013) 72(9):1445–52. doi: 10.1136/annrheumdis-2012-202450
214. Burmester G, McInnes I, Kremer J, Miranda P, Korkosz M, Vencovsky J, et al. A Randomised Phase IIb Study of Mavrilimumab, a Novel GM-CSF Receptor Alpha Monoclonal Antibody, in the Treatment of Rheumatoid Arthritis. Ann Rheum Dis (2017) 76(6):1020–30. doi: 10.1136/annrheumdis-2016-210624
215. Burmester GR, McInnes IB, Kremer JM, Miranda P, Vencovský J, Godwood A, et al. Mavrilimumab, a Fully Human Granulocyte-Macrophage Colony-Stimulating Factor Receptor α Monoclonal Antibody: Long-Term Safety and Efficacy in Patients With Rheumatoid Arthritis. Arthritis Rheumatol (2018) 70(5):679–89. doi: 10.1002/art.40420
216. Burmester G, Feist E, Sleeman M, Wang B, White B, Magrini F. Mavrilimumab, a Human Monoclonal Antibody Targeting GM-CSF Receptor-α, in Subjects With Rheumatoid Arthritis: A Randomised, Double-Blind, Placebo-Controlled, Phase I, First-in-Human Study. Ann Rheum Dis (2011) 70(9):1542–9. doi: 10.1136/ard.2010.146225
217. Klaasen R, Thurlings RM, Wijbrandts CA, van Kuijk AW, Baeten D, Gerlag DM, et al. The Relationship Between Synovial Lymphocyte Aggregates and the Clinical Response to Infliximab in Rheumatoid Arthritis: A Prospective Study. Arthritis Rheum (2009) 60(11):3217–24. doi: 10.1002/art.24913
218. van der Pouw Kraan T, Wijbrandts C, van Baarsen L, Rustenburg F, Baggen J, Verweij C, et al. Responsiveness to Anti-Tumour Necrosis Factor Alpha Therapy Is Related to Pre-Treatment Tissue Inflammation Levels in Rheumatoid Arthritis Patients. Ann Rheum Dis (2008) 67(4):563–6. doi: 10.1136/ard.2007.081950
219. Taylor P, Peters A, Paleolog E, Chapman P, Elliott M, McCloskey R, et al. Reduction of Chemokine Levels and Leukocyte Traffic to Joints by Tumor Necrosis Factor Alpha Blockade in Patients With Rheumatoid Arthritis. Arthritis Rheum-US (2000) 43(1):38–47. doi: 10.1002/1529-0131(200001)43:1<38::AID-ANR6>3.0.CO;2-L
220. Ulfgren AK, Andersson U, Engström M, Klareskog L, Maini RN, Taylor PC. Systemic Anti-Tumor Necrosis Factor Alpha Therapy in Rheumatoid Arthritis Down-Regulates Synovial Tumor Necrosis Factor Alpha Synthesis. Arthritis Rheum (2000) 43(11):2391–6. doi: 10.1002/1529-0131(200011)43:11<2391::AID-ANR3>3.0.CO;2-F
221. Badot V, Galant C, Nzeusseu Toukap A, Theate I, Maudoux A, Van den Eynde B, et al. Gene Expression Profiling in the Synovium Identifies a Predictive Signature of Absence of Response to Adalimumab Therapy in Rheumatoid Arthritis. Arthritis Res Ther (2009) 11(2):R57. doi: 10.1186/ar2678
222. De Vita S, Quartuccio L. Treatment of Rheumatoid Arthritis With Rituximab: An Update and Possible Indications. Autoimmun Rev (2006) 5(7):443–8. doi: 10.1016/j.autrev.2006.02.007
223. Edwards J, Szczepanski L, Szechinski J, Filipowicz-Sosnowska A, Emery P, Close D, et al. Efficacy of B-Cell-Targeted Therapy With Rituximab in Patients With Rheumatoid Arthritis. N Engl J Med (2004) 350(25):2572–81. doi: 10.1056/NEJMoa032534
224. Emery P, Deodhar A, Rigby WF, Isaacs JD, Combe B, Racewicz AJ, et al. Efficacy and Safety of Different Doses and Retreatment of Rituximab: A Randomised, Placebo-Controlled Trial in Patients Who Are Biological Naive With Active Rheumatoid Arthritis and an Inadequate Response to Methotrexate (Study Evaluating Rituximab’s Efficacy in MTX Inadequate Responders (SERENE)). Ann Rheum Dis (2010) 69(9):1629–35. doi: 10.1136/ard.2009.119933
225. Thurlings R, Vos K, Wijbrandts C, Zwinderman A, Gerlag D, Tak P. Synovial Tissue Response to Rituximab: Mechanism of Action and Identification of Biomarkers of Response. Ann Rheum Dis (2008) 67(7):917–25. doi: 10.1136/ard.2007.080960
226. Kavanaugh A, Rosengren S, Lee S, Hammaker D, Firestein G, Kalunian K, et al. Assessment of Rituximab’s Immunomodulatory Synovial Effects (ARISE Trial). 1: Clinical and Synovial Biomarker Results. Ann Rheum Dis (2008) 67(3):402–8. doi: 10.1136/ard.2007.074229
227. Murdaca G, Spanò F, Cagnati P, Puppo F. Free Radicals and Endothelial Dysfunction: Potential Positive Effects of TNF-α Inhibitors. Redox Report: Commun Free Radical Res (2013) 18(3):95–9. doi: 10.1179/1351000213Y.0000000046
228. Murdaca G, Colombo B, Cagnati P, Gulli R, Spanò F, Puppo F. Endothelial Dysfunction in Rheumatic Autoimmune Diseases. Atherosclerosis (2012) 224(2):309–17. doi: 10.1016/j.atherosclerosis.2012.05.013
229. Shamseldin L, Shawqi M, Al Hashem N, Alhyari M, Abd Elazeem H, Elghazaly S, et al. Safety and Efficacy of Mavrilimumab For Rheumatoid Arthritis: A Systematic Review and Meta-Analysis. Curr Rheumatol Rev (2021) 17(2):184–92. doi: 10.2174/1573397116666201113085444
230. Sung Y, Lee Y. Comparison of the Efficacy and Safety of Tofacitinib and Mavrilimumab in Patients With Active Rheumatoid Arthritis: A Bayesian Network Meta-Analysis of Randomized Controlled Trials. Int J Clin Pharmacol Ther (2021) 59(8):557–65. doi: 10.5414/CP203979
231. Ducreux J, Durez P, Galant C, Nzeusseu Toukap A, Van den Eynde B, Houssiau F, et al. Global Molecular Effects of Tocilizumab Therapy in Rheumatoid Arthritis Synovium. Arthritis Rheumatol (2014) 66(1):15–23. doi: 10.1002/art.38202
232. Kanbe K, Chen Q, Nakamura A, Hobo K. Inhibition of MAP Kinase in Synovium by Treatment With Tocilizumab in Rheumatoid Arthritis. Clin Rheumatol (2011) 30(11):1407–13. doi: 10.1007/s10067-011-1833-z
233. De Groof A, Ducreux J, Humby F, Nzeusseu Toukap A, Badot V, Pitzalis C, et al. Higher Expression of Tnfα-Induced Genes in the Synovium of Patients With Early Rheumatoid Arthritis Correlates With Disease Activity, and Predicts Absence of Response to First Line Therapy. Arthritis Res Ther (2016) 18:19. doi: 10.1186/s13075-016-0919-z
234. Schafer P, Kivitz A, Ma J, Korish S, Sutherland D, Li L, et al. Spebrutinib (CC-292) Affects Markers of B Cell Activation, Chemotaxis, and Osteoclasts in Patients With Rheumatoid Arthritis: Results From a Mechanistic Study. Rheumatol Ther (2020) 7(1):101–19. doi: 10.1007/s40744-019-00182-7
235. Di Benedetto P, Ruscitti P, Berardicurti O, Panzera N, Grazia N, Di Vito Nolfi M, et al. Blocking Jak/STAT Signalling Using Tofacitinib Inhibits Angiogenesis in Experimental Arthritis. Arthritis Res Ther (2021) 23(1):213. doi: 10.1186/s13075-021-02587-8
236. Muraoka S, Yamada Z, Kawazoe M, Hirose W, Kono H, Yasuda S, et al. Abatacept Is Efficacious in the Treatment of Older Patients With csDMARD-Refractory Rheumatoid Arthritis: A Prospective, Multicenter, Observational Study. Rheumatol Ther (2021) 8(4):1585–601. doi: 10.1007/s40744-021-00356-2
237. Buch M, Boyle D, Rosengren S, Saleem B, Reece R, Rhodes L, et al. Mode of Action of Abatacept in Rheumatoid Arthritis Patients Having Failed Tumour Necrosis Factor Blockade: A Histological, Gene Expression and Dynamic Magnetic Resonance Imaging Pilot Study. Ann Rheum Dis (2009) 68(7):1220–7. doi: 10.1136/ard.2008.091876
Keywords: rheumatoid arthritis, heterogeneity, pathophysiology, interaction, genetics, mechanism, precision medicine, responsiveness
Citation: Zhao J, Guo S, Schrodi SJ and He D (2021) Molecular and Cellular Heterogeneity in Rheumatoid Arthritis: Mechanisms and Clinical Implications. Front. Immunol. 12:790122. doi: 10.3389/fimmu.2021.790122
Received: 06 October 2021; Accepted: 08 November 2021;
Published: 25 November 2021.
Edited by:
Haitao Wang, National Cancer Institute, United StatesReviewed by:
George E. Fragoulis, Laiko General Hospital of Athens, GreeceGiuseppe Murdaca, University of Genoa, Italy
Mark Kaddumukasa, Makerere University, Uganda
Copyright © 2021 Zhao, Guo, Schrodi and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Steven J. Schrodi, U2Nocm9kaUB3aXNjLmVkdQ==; Shicheng Guo, U2hpY2hlbmcuR3VvQHdpc2MuZWR1; Dongyi He, ZG9uZ3lpaGVAbWVkbWFpbC5jb20uY24=