 Surekha Nimma
Surekha Nimma Weixi Gu
Weixi Gu Natsumi Maruta
Natsumi Maruta Yan LiMengqi PanForhad Karim SaikotBryan Y. J. LimHelen Ying McGuinness
Yan LiMengqi PanForhad Karim SaikotBryan Y. J. LimHelen Ying McGuinness Zannati Ferdous ZaotiSulin LiSneha DesaMohammad Kawsar Manik
Zannati Ferdous ZaotiSulin LiSneha DesaMohammad Kawsar Manik Jeffrey D. Nanson
Jeffrey D. Nanson Bostjan Kobe*
Bostjan Kobe*- School of Chemistry and Molecular Biosciences, Institute for Molecular Bioscience and Australian Infectious Diseases Research Centre, University of Queensland, Brisbane, QLD, Australia
TIR (Toll/interleukin-1 receptor/resistance protein) domains are cytoplasmic domains widely found in animals and plants, where they are essential components of the innate immune system. A key feature of TIR-domain function in signaling is weak and transient self-association and association with other TIR domains. An additional new role of TIR domains as catalytic enzymes has been established with the recent discovery of NAD+-nucleosidase activity by several TIR domains, mostly involved in cell-death pathways. Although self-association of TIR domains is necessary in both cases, the functional specificity of TIR domains is related in part to the nature of the TIR : TIR interactions in the respective signalosomes. Here, we review the well-studied TIR domain-containing proteins involved in eukaryotic immunity, focusing on the structures, interactions and their corresponding functional roles. Structurally, the signalosomes fall into two separate groups, the scaffold and enzyme TIR-domain assemblies, both of which feature open-ended complexes with two strands of TIR domains, but differ in the orientation of the two strands. We compare and contrast how TIR domains assemble and signal through distinct scaffolding and enzymatic roles, ultimately leading to distinct cellular innate-immunity and cell-death outcomes.
Introduction
TIR (Toll/interleukin-1 receptor/resistance protein) domains are cytoplasmic domains found in both eukaryotic and prokaryotic proteins that are involved in innate-immunity and cell-death pathways. They consist of 135–160 residues and typically display a five-stranded parallel β-sheet (strands βA–βE) surrounded by five α-helices (αA–αE) (Figure 1) (1, 2). TIR-domain functions are governed by weak and transient interactions. They predominantly function through homotypic interactions, including self-association or association with other TIR domains, to create scaffolds that facilitate signal transduction, leading to immune and cell-death responses (3, 4). The mechanism of signaling employed has been described as SCAF (signaling by cooperative assembly formation) (5–7). SCAF involves the assembly of higher-order complexes - signalosomes or “supramolecular organizing centers” (SMOCs) (8). In the case of SCAF, receptor activation through activating ligand binding induces receptor oligomerization, which in turn nucleates recruitment and oligomerization of adaptor proteins, and subsequently the recruitment and oligomerization of effector enzymes that can be activated through proximity-induced mechanisms in the resulting signalosome.
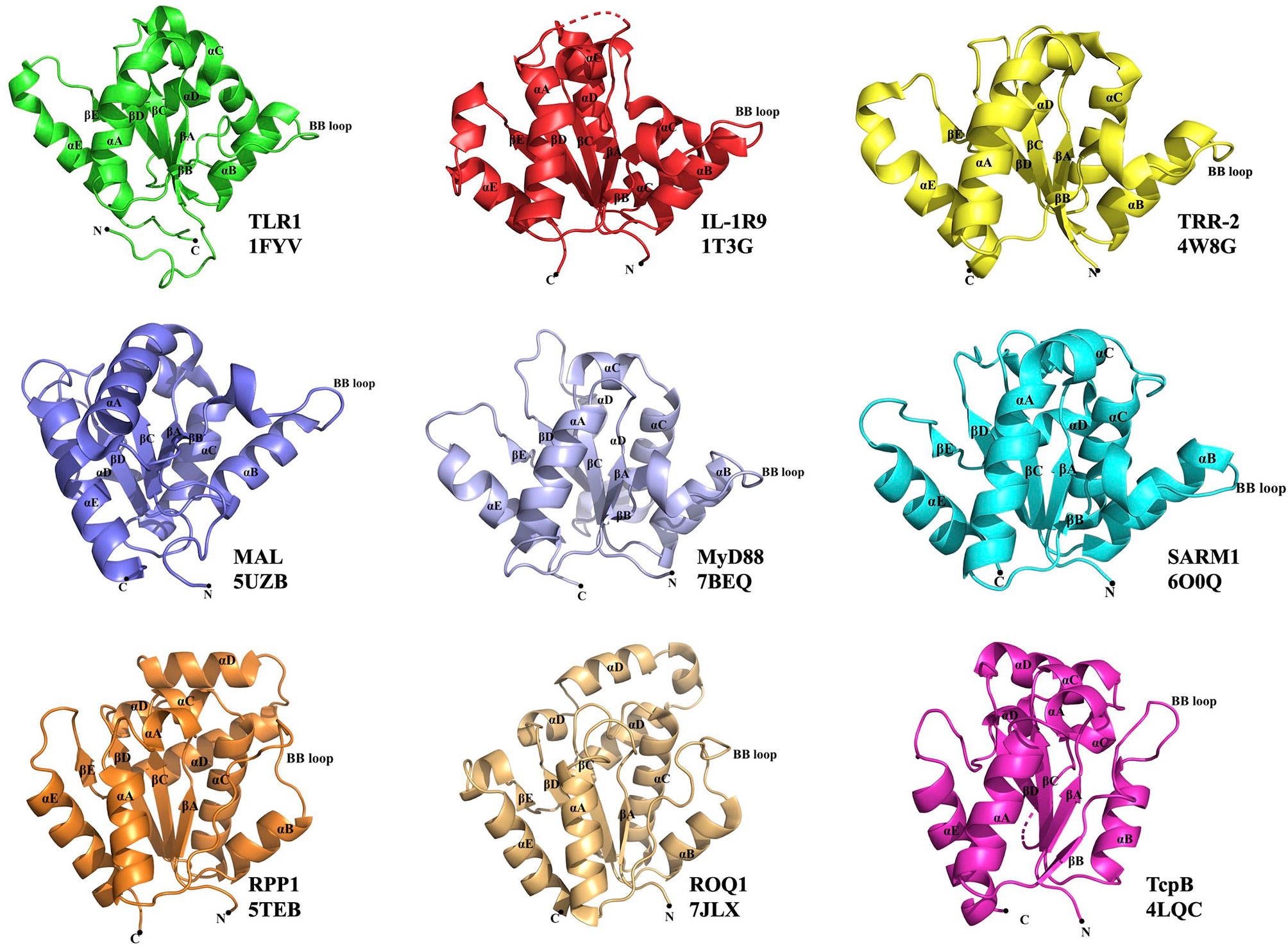
Figure 1 Representative TIR domain structures. TIR domains of the human (TLR1, IL-1R9, MAL, MyD88 and SARM1), lower metazoan Hydra magnipapillata (TRR-2), plant (RPP1 and ROQ1) and bacterial (TcpB) proteins with their corresponding PDB IDs are shown. All the TIR domains show a central core of five β-strands (βA–βE) surrounded by five α-helices (αA–αE). The functionally important BB-loop in each TIR is labeled.
In mammals, TIR domains are found in Toll-like receptors (TLRs), interleukin-1 receptors (IL-1Rs) and cytoplasmic adaptor proteins, such as MyD88 (myeloid differentiation primary response gene 88) and MAL (MyD88 adaptor-like protein) (Figures 1, 2) (2). TLRs and IL-1Rs are pattern recognition receptors (PRRs) that recognize evolutionarily conserved pathogen-associated molecular patterns (PAMPs) and endogenous danger-associated molecular patterns (DAMPs) released by dying or damaged cells (3, 4). Upon activation, TLR TIR domains dimerize, creating an intracellular TIR-domain signaling scaffold, which then recruits TIR domain-containing adaptor proteins that activate further downstream signaling (e.g. recruiting IL-1R-associated kinases (IRAKs) and activating the transcription factor NF-κB [nuclear factor kappa-light-chain-enhancer of activated B cells]), to induce inflammatory responses through the production of proinflammatory cytokines and programmed cell-death responses (3, 4).
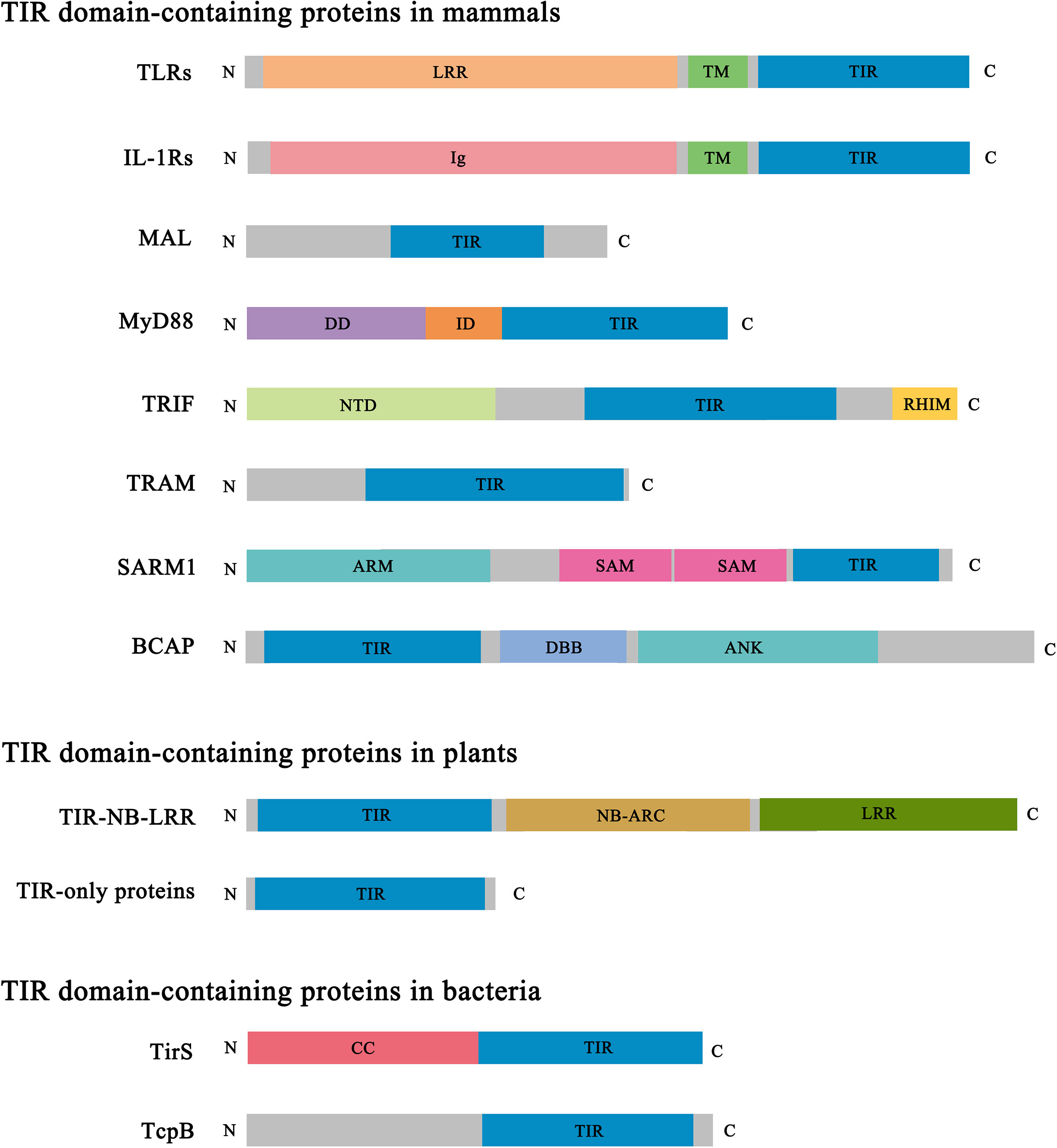
Figure 2 Domain architecture of representative TIR domain-containing proteins. TIR, Toll/interleukin-1 receptor/resistance protein; LRR, leucine-rich repeat; TM, transmembrane; Ig, immunoglobulin; DD, death domain; ID, intermediate domain; NTD, N-terminal domain; RHIM, RIP (receptor-interacting protein) homotypic interaction motif; ARM, armadillo-repeat motif; SAM, sterile alpha motif; DBB, Dof (Drosophila downstream of fibroblast growth factor receptor)/BCAP/BANK (B cell scaffold protein with ankyrin repeats); ANK, ankyrin repeat; NB-ARC, nucleotide-binding adaptor shared by APAF-1 (apoptotic protease-activating factor 1), R proteins and CED-4 (cell death protein 4); CC, coiled coil.
In contrast to the scaffolding protein-protein interaction function described above, the TIR domain-containing protein SARM1 (sterile alpha and TIR motif containing 1) has been found to cleave NAD+ (nicotinamide adenine dinucleotide) and NADP+ (nicotinamide adenine dinucleotide phosphate) and initiate axon degeneration, in a process also dependent on the self-assembly of SARM1 TIR domains (Figure 2) (9–11). The dual role of TIR domains, serving as scaffolds or as enzymes, relies in part on the different assembly mechanisms of the proteins, leading to functional specificity.
Similar to mammals, plants also have a complex immune system to protect themselves from pathogen invasion. To restrict pathogen infection, the intracellular innate immune receptors called NLRs (nucleotide-binding domain (NBD)/leucine-rich repeat (LRR) receptors) directly or indirectly detect pathogen effector proteins via their C-terminal LRR domains and activate defense responses, including localized programmed cell death, in a process termed hypersensitive response (HR) (Figure 2) (12). The central NBD (usually called NB-ARC – see Figure 2) is important for the oligomerization of these proteins into a signalosome called the “resistosome” upon activation. TIR domains are found at the N-termini of a large group of NLRs (TIR domain-containing NLRs, TNLs), as well as TIR-only proteins, and truncated NLRs lacking LRR domains (Figure 2) (12, 13). It is unclear if some plant TIR domains serve scaffolding functions like the canonical mammalian TIR domains; however, plant TIR domains self-associate and display the enzymatic function of cleaving NAD+, sharing similarities with the function of SARM1 (10, 14). In agreement, plant TIR domains exhibit similar assembly mechanisms as SARM1 (10, 15, 16).
TIR domains are also found in bacteria and archaea. Structures and functions of bacterial and archaeal TIR domains and the corresponding proteins are poorly characterized, compared to their animal and plant counterparts, and are not the focus of the current review. Bacterial TIR domains are found in a wide range of domain architectures and domain types, which indicates diverse functional roles (Figure 2) (17). Some bacterial TIR domains, including TcpC (Escherichia coli), TirS (Staphylococcus aureus), PumA (Pseudomonas aeruginosa) and TcpB (Brucella melitensis), are linked to bacterial pathogenicity (18, 19). TirS and TcpC possess NAD+-nucleosidase activity (20), but so do bacterial TIR domains from non-pathogenic bacteria, suggesting roles in bacterial physiology (20, 21). Recent studies have also shown that the NAD+-nucleosidase activity of the bacterial TIR domains is linked to bacterial antiviral defenses (22–24).
Here, we review structural information available for various TIR domains, focusing on the nature of the different assemblies they form and their corresponding functional roles. Based on the available structural and functional evidence, we observe a correlation of TIR-domain assembly with their specific functional roles. In this respect, they fall into two different groups, the “scaffold” assemblies involved in innate-immunity signaling, and “enzyme” assemblies leading to NAD+ cleavage associated with cell-death signaling.
Scaffold TIR-Domain Assemblies
This group comprises the TIR domains that undergo self-association to form a scaffold, which facilitates nucleation-controlled cooperative recruitment of other TIR domain-containing proteins and signal transduction (employing a SCAF mechanism) (5–7, 25). The group includes TIR domains from mammalian membrane receptors (TLRs and IL-1Rs), as well as those from the cytoplasmic adaptor proteins. A number of crystal structures of TIR domains from this group have been determined, but the structural basis of their self-assembly only became clear through the structural studies of higher-order structures reconstituted for the adaptors MAL and MyD88 (26, 27).
TLRs and IL-1Rs
Ten TLRs are present in humans and found either on the cell surface (TLR1, TLR2, TLR4, TLR5, TLR6 and TLR10) or in intracellular endosomal compartments (TLR3, TLR4, TLR7, TLR8 and TLR9) (28). TLRs are characterized by an extra-cytoplasmic LRR domain, a transmembrane domain, and an intracellular TIR domain (Figure 2). When activated by their ligands, they mostly function as homodimers, but TLR2 functions as a heterodimer with either TLR1 or TLR6. A considerable amount of structural information is available on their LRR domains (29, 30), and even on the full-length TLR3 and TLR7 in complex with the membrane chaperone UNC93B1 involved in TLR trafficking (although the TIR domains could not be visualized in this case) (31). Crystal structures of the TIR domains of TLR1 (1), TLR2 (1, 32), TLR6 (33) and TLR10 (34) have been reported. These TIR domains, when expressed as separate proteins, are all monomeric in solution under the conditions tested. The characteristic BB-loop (connecting the βB strand and the αB helix) has been shown to play an important role in signaling, as a naturally occurring mutation (P712H) in this loop in TLR4 makes it non-responsive to the PAMP lipopolysaccharide (LPS) (35). Other mutations in this loop were also shown to abolish signaling in numerous TIR domains, including TLR4 (36) and TLR7 (37). Downstream signaling involves the recruitment of the cytoplasmic adaptors MyD88 (for all TLRs except TLR3), MAL (as a bridging adaptor for MyD88 in the case of TLR4 and TLR1/2/6), TRIF (TIR domain-containing adaptor protein-inducing interferon β; for TLR3) and TRAM (TRIF-related adaptor molecule; as a bridging adaptor for TRIF in the case of TLR4).
There are also ten IL-1Rs (IL-1R1 to IL-1R10) in humans, characterized by extracellular immunoglobulin domains, a transmembrane domain, and an intracellular TIR domain (except for IL-1R2) (38) (Figure 2). IL-1Rs recognize and bind specific IL-1 family cytokines, which leads to recruitment of an accessory receptor chain. The downstream signaling mechanism is similar to that of TLRs, involving recruitment of MyD88, IRAKs and activating NF-κB to induce inflammatory responses (39). The IL-1R TIR domains are structurally less well characterized, with the majority of the available structural data limited to the complexes involving the ectodomains and their corresponding ligands (40). The only available structure of a TIR domain corresponds to that from IL-1R9 (IL-1RAPL1, IL-1R accessory protein like 1) (41). However, IL-1R9 is not a classical signaling IL-1R; it does not activate NF-κB, but has been reported to be involved in trans-synaptic signaling (42).
Cytoplasmic TLR Adaptor Proteins
TLRs and IL-1Rs require adaptor proteins for signaling. Six adaptor proteins have been identified in humans: MyD88, MAL, TRIF, TRAM, SARM1 and BCAP (B-cell adaptor for phosphoinositide 3-kinase) (28) (Figure 2). MyD88, MAL, TRIF and TRAM are the principal signaling adaptors in this pathway (43), while SARM1 and BCAP have been reported to negatively regulate TLR signaling (44–46). Crystal and/or NMR structures are available for the TIR domains from all these proteins (2, 5, 10).
Structures of Higher-Order Assemblies of MAL and MyD88 TIR Domains
Reconstitution of higher-order assemblies of MAL and MyD88 TIR domains yielded filamentous and micro-crystalline complexes, respectively (26, 27). These reconstitution experiments correlated with the functional signaling pathway, as the TLR4 TIR domain seeded the assembly of MAL TIR domains, while MAL TIR domains seeded the assembly of MyD88 TIR domains. Structure determination of these higher-order assemblies, combined with mutagenesis and signaling assays, has provided clarity on the biologically relevant association of scaffold TIR domains (26, 27). These structures feature two parallel strands of TIR domains, each showcasing a head-to-tail arrangement of TIR domains, held together through a BB-loop-mediated “BE” intrastrand interface. The two strands are offset and held together through a “BCD” interstrand interface (Figures 3, 4).
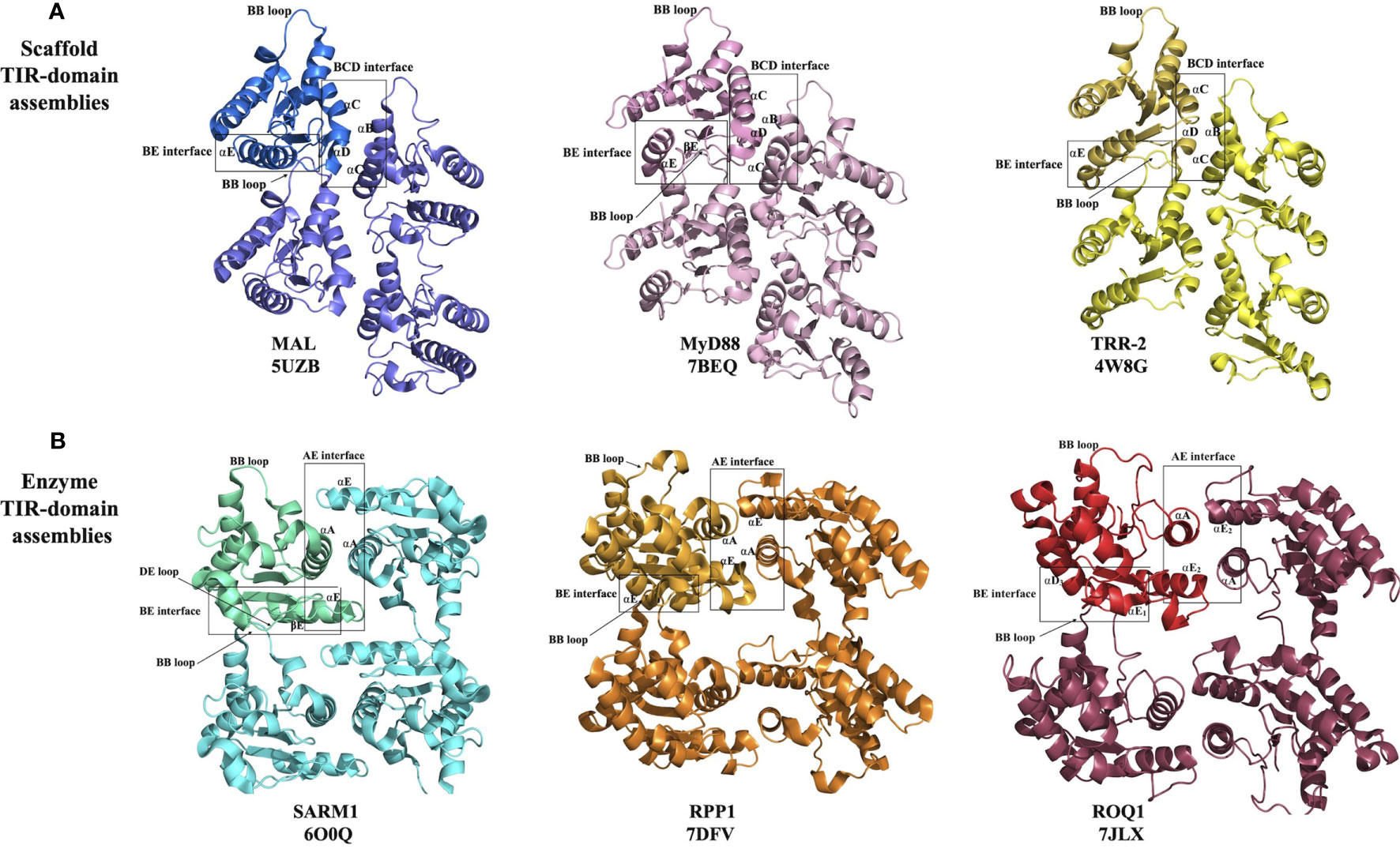
Figure 3 Structural basis of eukaryotic TIR-domain assembly formation. (A) Scaffold TIR-domain assemblies, held together by the BE and BCD interfaces, represented by the TIR domains from MAL, MyD88 and TRR-2. (B) Enzyme TIR-domain assemblies held together by the BE and AE interfaces, represented by the TIR domains from SARM1, RPP1 and ROQ1.
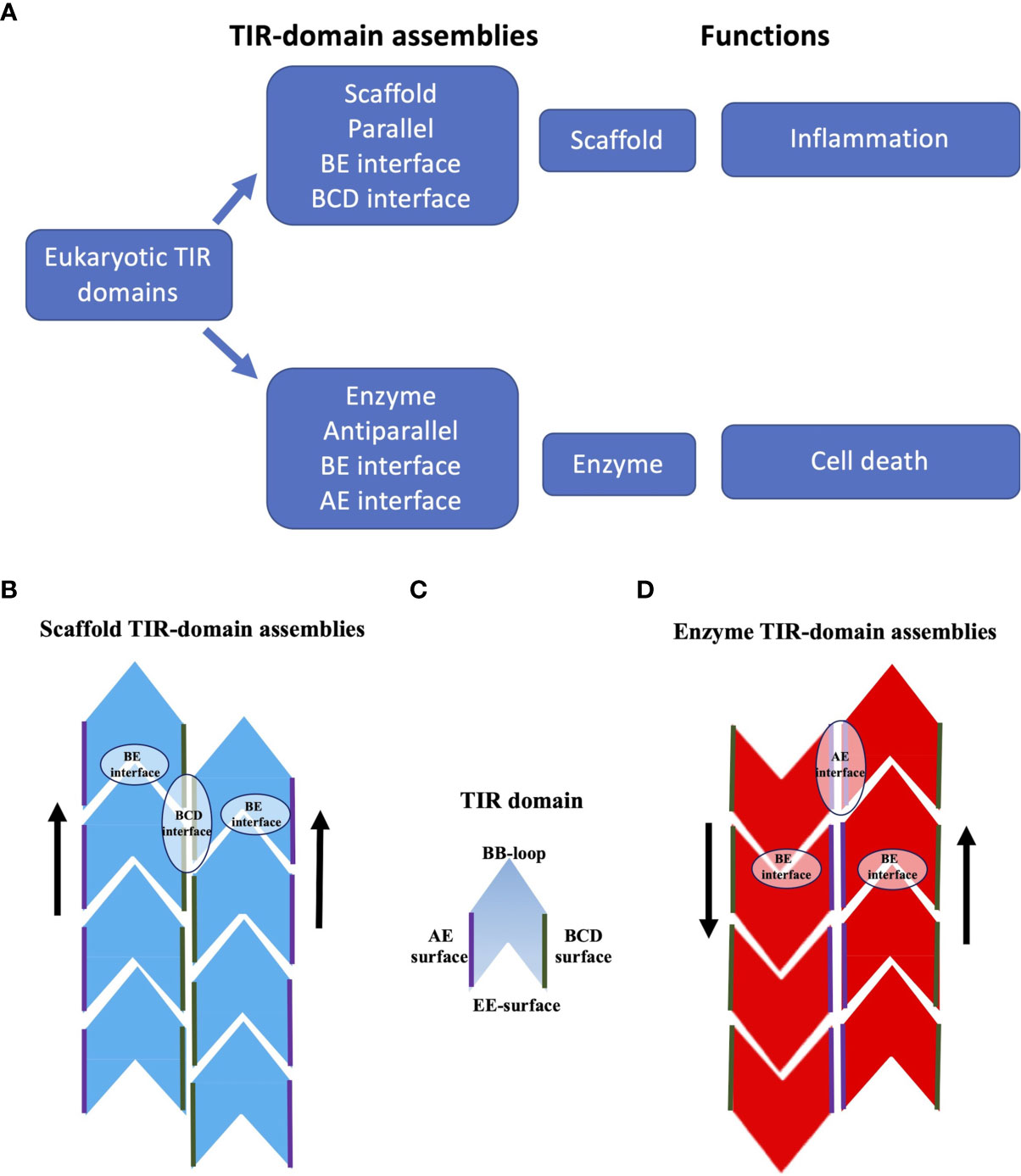
Figure 4 Schematic representation of TIR-domain assemblies. (A) Overview of TIR-domain assemblies. (B) Scaffold TIR-domain assemblies, represented by the structures formed by the TIR domains from human MAL and MyD88. These parallel two-stranded assemblies are held together by the intrastrand BE interfaces, and the interstrand BCD interfaces. (C) Schematic diagram of a single TIR domain, to highlight the BB-loop, BCD, EE and AE surfaces (the diagram does not differentiate the structures of TIR domains in monomeric and signalosome forms). (D) Enzyme TIR-domain assemblies, represented by the structures formed by the TIR domains from human SARM1 and the plant NLRs ROQ1 and RPP1. These antiparallel two-stranded assemblies are held together by the intrastrand BE interfaces and the interstrand AE interfaces.
The structure of the filament formed by MAL TIR domains was determined by helical reconstruction cryo-electron microscopy (cryo-EM) (26). It revealed a hollow tube consisting of 12 protofilaments, with each protofilament corresponding to a two-stranded assembly of TIR domains described above. The intrastrand BE interface includes the area around the BB-loop of one subunit and the EE surface (the βD and βE strands and the αE helix) of the interacting subunit (Figure 3A). The interstrand interaction connecting the two strands involves the residues on the BC surface (αB and αC helices) of one subunit from strand 1 and the CD surface (αD helix and the CD loop) of another subunit from strand 2 (Figure 3A). Structure-based mutagenesis studies confirmed that key residues in both interfaces (e.g., P125A in the intrastrand interface; L162A, L165A, W156A, Y159A and F193A in the interstrand interface) are necessary for the activation of downstream signaling through NF-κB, whereas mutations of residues mediating interactions between protofilaments were not found to have consequences for function (26).
The structure of the MAL TIR domain-nucleated microcrystals of MyD88 TIR domain was determined using two approaches, microcrystal electron diffraction (microED) and serial femtosecond crystallography (SFX) with an X-ray free electron laser (X-FEL) source (both yielding nearly identical structures) (27). The structure shows that the TIR domains of MyD88 assemble in a fashion analogous to MAL TIR-domain protofilaments (Figure 3A). The interfaces responsible for this association were again found to be relevant to signaling, with the mutations R196A, W284A, I253D and R288A in the intrastrand interface, and mutations K238A, L241A, F270A and F270E in the interstrand interface abolishing TLR4-induced NF-κB activation (27). The analogous structural arrangement of TIR domains in the MAL and MyD88 higher-order assemblies suggests a hierarchical, nucleation-controlled and cooperative mechanism for TLR signal transduction, in which the receptor and adaptor TIR domains assemble via the inter- and intrastrand interactions observed in the MyD88 and MAL TIR-domain higher-order assemblies, leading to formation of a TIR-domain signalosome. This in turn promotes clustering of the MyD88 death domains (DDs) to form a signalosome termed the “Myddosome” (47), recruiting and activating IRAKs (26, 27), thereby facilitating signaling through a SCAF mechanism.
Several mutations in the TIR domains of TLRs and adaptors are associated with disease. In MyD88, the R196C polymorphism, which is associated with susceptibility to pyogenic bacterial infection during childhood (48), maps to the intrastrand interface of the TIR-domain signalosome. Similarly, the L252P gain-of-function variant of MyD88, which is found in diffuse large B cell lymphoma and promotes tumor survival through enhanced NF-κB activation (49), also maps to the intrastrand interface. The corresponding mutant forms extremely stable oligomers, compared to the wild-type protein, explaining the molecular basis of its phenotype (50).
Interestingly, in one of the crystal structures of the TRR-2 TIR domain from the lower metazoan Hydra magnipapillata (PDB ID 4W8G), a parallel two-strand arrangement analogous to MAL and MyD88 TIR-domain signalosomes is observed (Figures 1, 3A). These signalosomes are therefore likely to be structurally conserved in a range of eukaryotes.
The interactions observed in the MyD88 and MAL TIR-domain signalosomes were not captured in the crystal structures of any of the mammalian TLR or adaptor TIR domains. However, surfaces equivalent to the ones mediating interstrand interactions (involving residues in αB, αC and αD helices and the BB-loop; “BCD surface”) are found to mediate symmetric dimer formation in the crystals of a number of TLR and IL-1R TIR domains (TLR1, TLR2, TLR6, TLR10 and IL-1R9) (5). While it is possible that functional surfaces simply find alternative, biologically irrelevant binding partners when forming crystals, it could be that such symmetric interaction contributes to the regulation of signaling by representing an inactive, auto-inhibited state, especially in the case of TLRs that always exist as dimers; such an interaction would presumably be broken when the receptor is activated by ligand binding and the signalosome formation would be allowed.
In summary, the scaffold TIR-domain signalosomes correspond to parallel two-stranded open-ended assemblies of TIR domains, held together by two asymmetric interactions, the intrastrand and interstrand interactions, mediated by the BE and BCD interfaces, respectively (Figures 3, 4). The nucleation-controlled cooperative assembly of these signalosomes is responsible for the SCAF mechanism of signaling.
Enzyme TIR-Domain Assemblies
This group comprises the TIR domains that undergo self-association, which facilitates NAD+-nucleosidase activity and eventually cell death. It includes TIR domains from the mammalian protein SARM1 and plant TIR domain-containing proteins. While some bacterial and archaeal TIR domains have also been shown to have NAD+-nucleosidase activity, the structural basis of their self-association and enzymatic activity is not well characterized and may be different from their eukaryotic counterparts discussed in this group (10, 20). On the other hand, the TIR domains from SARM1 and plant NLRs assemble in an analogous fashion, forming two-stranded assemblies different from that of the scaffold TIR-domain assemblies (Figures 3, 4). The association of TIR domains in each individual strand resembles the one observed in scaffold TIR-domain signalosomes, featuring a head-to-tail arrangement mediated by the BB-loop-containing BE interface. However, the two strands in enzyme TIR-domain assemblies are associated in an antiparallel, rather than parallel, fashion. The interstrand interface corresponds to a symmetric “AE interface”, involving the αA and αE helices. This structural information is based on three key structures: the crystal structure of the SARM1 TIR domain (10), and the cryo-EM structures of activated resistosome complexes of the TNLs ROQ1 and RPP1 (15, 16).
SARM1
While SARM1 has a number of suggested roles in the regulation of innate immunity, the central function appears to be to serve as the executioner of Wallerian or programmed axon degeneration, a highly conserved pathway of injury-induced axon degeneration (51–54). SARM1 facilitates rapid depletion of NAD+ in response to axon injury, leading to subsequent axon demise (10, 11, 55). SARM1 deletion mutants lacking the TIR domain show a dominant-negative phenotype, delaying axon degeneration (52). In addition to its C-terminal TIR domain, SARM1 contains an N-terminal ARM (armadillo repeat motif) domain and central tandem SAM (sterile alpha motif) domains. SAM domains form an octameric ring, and the formation of such an oligomeric state is essential for axon degeneration (10, 52, 56). The SARM1 TIR domain has intrinsic NAD+-nucleosidase activity, cleaving NAD+ into nicotinamide and either ADPR (ADP-ribose) or cyclic ADPR (cADPR) (10, 11, 57). The active site has similarities to that of the NAD+ glycohydrolase CD38, including a catalytic glutamate residue (10, 11). Mutation of this residue is sufficient to abolish NAD+-nucleosidase activity, indicating similar mechanisms of NAD+-nucleosidase activity among these enzymes (10, 11). Self-association of SARM1 TIR domains is essential for enzyme activity (10, 11). The crystal structure of the SARM1 TIR domain revealed the two-stranded antiparallel arrangement described above, and mutational analysis of the BE (D594A, E596K, and G601P) and AE interfaces (L579A and H685A) revealed that these interactions are functionally important for stabilization of the active conformation of the TIR domains (10).
In the inactive SARM1 octamer, interaction of the ARM and TIR domains is responsible for preventing TIR-domain self-association, directly inhibiting NAD+-nucleosidase activity (58–60). The ARM-TIR inhibitory interaction is regulated by the cellular ratios of NAD+ and its metabolites NMN (nicotinamide mononucleotide) and NaMN (nicotinic acid mononucleotide), through binding to an allosteric site in the ARM domain; NMN as an activator, and NAD+ and NaMN as inhibitors (59–63). Injury-associated increase in NMN results in the release of this ARM-TIR autoinhibition and subsequent assembly of TIR domains (59).
Plant TIR Domains
TIR domains from several plant NLRs and TIR-only plant proteins can also cleave NAD+, dependent on the TIR domain self-association and the conserved glutamate residue at the catalytic site; this activity is essential for cell-death signaling (10, 14). Crystal structures of numerous plant TIR domains [including those from RPS4 and RPS4-RRS1 heterodimer, AtTIR, SNC1, RPP1 and RPV1 (64–67)] feature a symmetrical AE interface analogous to the one observed in SARM1, and mutagenesis of residues in this interface was shown to abolish cell-death signaling in plants (10, 65–68). The AE interface often features conserved residues, such as the SH (serine-histidine) motif in the αA helices of the TIR domains, which allows stacking and hydrogen-bonding interactions across the interface (65). Charged residues surrounding the SH motif further stabilize the AE interface (67). However, the structure of a functional TIR-domain assembly only became clear based on the cryo-EM structures of ROQ1 and RPP1 NLRs (15, 16).
Activated ROQ1 and RPP1 form tetrameric structures, largely through the interactions of their NBDs (15, 16). This NBD-mediated tetramer exhibits four-fold symmetry; however, the TIR domains arranged on top of this tetramer exhibit only two-fold symmetry, featuring a dimer of dimers. The arrangement of TIR domains is identical to the arrangement of SARM1 TIR domains observed in the corresponding crystals. Therefore, each dimer features the symmetrical AE interface, but the two dimers are held together through BE interfaces (Figure 3B). Such a BE interface was never observed in any of the crystal structures of plant TIR domains; however, the DE surface mediating this interaction has been implicated previously in the function of these domains through site-directed mutagenesis (67–69).
The BE interface-mediated interaction is crucial for NAD+-nucleosidase activity, because in the absence of this interaction, the positively charged lysine and arginine residues in the BB-loop block NAD+ binding to the active site. In the presence of BE-interface interaction, the BB-loop moves under the DE surface and allows access by the substrate to the NAD+-binding site. Jointly, the AE and BE interface-mediated interactions facilitate the appropriate conformation of the active site to enable enzymatic activity (16). The oligomerization of plant NLRs, driven largely by the NBDs, is presumed to nucleate assembly of the TIR domains.
In summary, the enzyme TIR-domain signalosomes involve anti parallel head-to-tail asymmetric intrastrand and symmetric interstrand interactions, mediated by the BE and AE interfaces, respectively (Figures 3, 4). Such an assembly is required for configuring the NAD+-nucleosidase active site, and consequently enzymatic activity and biological function (axon degeneration or cell death).
Signaling by Cooperative Assembly Formation
Both the scaffold and enzyme types of TIR-domain signalosomes function through a signaling mechanism termed SCAF (5–7). Compared to a more gradual signal amplification in a classical signaling pathway, SCAF enables a rapid and strong response to minute amounts of stimulus, resembling a switch; it is therefore well suited to innate-immunity and cell-death pathways. An important aspect of regulation involves the nucleation barriers to oligomerization (25). In the case of scaffold TIR-domain assemblies, the effector enzymes correspond to proteins kinases (IRAKs), which can activate themselves through phosphorylation when brought together. In the case of enzyme TIR-domain assemblies, the TIR domains themselves serve as effector enzymes, as they require self-association for activity. In both scaffold and enzyme groups, other domains play key roles to create a functional pathway, for example LRR domains as receptors (also called sensors) in TLRs and plant NLRs, ARM domains as receptors in SARM1, and DDs and NBDs as adaptors in TLR and plant NLR pathways. The presence of more than one self-associating domain provides further opportunities for regulation of the system (e.g. auto-inhibition between the DD and TIR domains in MyD88), which ultimately creates a large range of concentrations where the system is poised for activation (50, 70, 71).
Conclusions and Future Directions
TIR domains are found in animals, plants, bacteria and archaea, and often have functions associated with innate immunity and cell death. Self-association of TIR domains is key to their function, but weak affinities prevent association until aided by activating ligands, other domains in TIR domain-containing proteins, or adaptor proteins. The differences in the structures of scaffold and enzyme TIR-domain assemblies correlate with their distinct functional roles. Scaffold TIR-domain signalosomes, represented by MAL and MyD88 TIR-domain complexes, correspond to parallel two-stranded assemblies that are used in nucleation-controlled pathways that are activated by PAMPs or DAMPs and result in the activation of protein kinases (such as IRAKs). Enzyme TIR-domain signalosomes, represented by TIR-domain complexes from SARM1 and plant NLRs, correspond to antiparallel two-stranded assemblies with NAD+-nucleosidase activity that form in response to NMN or plant pathogen effector protein binding, respectively. Both groups of assemblies feature an analogous head-to-tail arrangement of TIR domains in individual strands, mediated by the BE interface, but differ in their interstrand association. In line with their analogous interstrand interactions, the functionally important BB-loop adopts a similar conformation in the assembled state in both cases.
Our classification into the two groups of TIR-domain signalosomes is based on a limited number of signalosome structures, and further work will be required to establish how generally applicable the current models are to TIR-domain signaling, and what variations exist in different pathways. However, in support of our general conclusions, molecular, mutational, and functional data on a large variety of TIR domains are consistent with our proposed models. For example, the TIR domain of the TLR adaptor TRAM only has limited sequence identity to those from MAL and MyD88, yet it is compatible with filaments with the same arrangement of TIR domains as seen in the latter two cases (26). Similarly, the Arabidopsis plant TIR-only immune receptor RBA1 and the TIR-only protein BdTIR of unknown function from the monocot plant Brachypodium distachyon display self-association-dependent NAD+-nucleosidase activity (14). Nevertheless, further studies on different TIR domain-containing proteins will be required to understand the breadth of similarities and differences in their molecular basis of function, in particular the less characterized proteins from bacteria and archaea. Another important but poorly characterized aspect involves the molecular details of the reaction catalyzed by different enzyme TIR domains. While all characterized enzyme TIR domains cleave the nicotinamide moiety off the substrate NAD+ molecule, many produce a cyclic variant of the remaining ADPR, rather than just the linear ADPR molecule. Human SARM1 produces a small proportion of the classical cADPR, but several bacterial and plant TIR domains instead produce variants with a different cyclic linkage (11, 14, 20). The chemical structures of these variants remain uncharacterized, as do their roles in the corresponding biological pathways.
Author Contributions
All authors contributed to the article and approved the submitted version.
Funding
This work was supported by funding from the National Health and Medical Research Council (NHMRC) (Project Grant 1160570 to BK) and the Australian Research Council (ARC) (Discovery Project DP190102526 and Laureate Fellowship FL180100109 to BK).
Conflict of Interest
BK is a shareholder of Disarm Therapeutics, a wholly owned subsidiary of Eli Lilly & Company. BK is a consultant to Disarm Therapeutics. BK and WG receive research funding from Disarm Therapeutics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank Dr Gayle Petersen for proofreading, editing, and constructive criticism of the manuscript.
References
1. Xu Y, Tao X, Shen B, Horng T, Medzhitov R, Manley JL, et al. Structural Basis for Signal Transduction by the Toll/interleukin-1 Receptor Domains. Nature (2000) 408:111–5. doi: 10.1038/35040600
2. Ve T, Williams SJ, Kobe B. Structure and Function of Toll/interleukin-1 Receptor/Resistance Protein (TIR) Domains. Apoptosis (2015) 20:250–61. doi: 10.1007/s10495-014-1064-2
3. Dinarello CA. Interleukin-1 in the Pathogenesis and Treatment of Inflammatory Diseases. Blood (2011) 117:3720–32. doi: 10.1182/blood-2010-07-273417
4. Kawai T, Akira S. The Role of Pattern-Recognition Receptors in Innate Immunity: Update on Toll-Like Receptors. Nat Immunol (2010) 11:373–84. doi: 10.1038/ni.1863
5. Nimma S, Ve T, Williams SJ, Kobe B. Towards the Structure of the TIR-Domain Signalosome. Curr Opin Struct Biol (2017) 43:122–30. doi: 10.1016/j.sbi.2016.12.014
6. Vajjhala PR, Ve T, Bentham A, Stacey KJ, Kobe B. The Molecular Mechanisms of Signaling by Cooperative Assembly Formation in Innate Immunity Pathways. Mol Immunol (2017) 86:23–37. doi: 10.1016/j.molimm.2017.02.012
7. Nanson JD, Kobe B, Ve T. Death TIR, and RHIM: Self-Assembling Domains Involved in Innate Immunity and Cell-Death Signaling. J Leukoc Biol (2019) 105:363–75. doi: 10.1002/JLB.MR0318-123R
8. Kagan JC, Magupalli VG, Wu H. SMOCs: Supramolecular Organizing Centres That Control Innate Immunity. Nat Rev Immunol (2014) 14:821–6. doi: 10.1038/nri3757
9. Zhao ZY, Xie XJ, Li WH, Liu J, Chen Z, Zhang B, et al. A Cell-Permeant Mimetic of NMN Activates SARM1 to Produce Cyclic ADP-Ribose and Induce Non-Apoptotic Cell Death. iScience (2019) 15:452–66. doi: 10.1016/j.isci.2019.05.001
10. Horsefield S, Burdett H, Zhang X, Manik MK, Shi Y, Chen J, et al. NAD(+) Cleavage Activity by Animal and Plant TIR Domains in Cell Death Pathways. Science (2019) 365:793–9. doi: 10.1126/science.aax1911
11. Essuman K, Summers DW, Sasaki Y, Mao X, DiAntonio A, Milbrandt J. The SARM1 Toll/Interleukin-1 Receptor Domain Possesses Intrinsic NAD+ Cleavage Activity That Promotes Pathological Axonal Degeneration. Neuron (2017) 93:1334–43.e1335. doi: 10.1016/j.neuron.2017.02.022
12. Tamborski J, Krasileva KV. Evolution of Plant NLRs: From Natural History to Precise Modifications. Annu Rev Plant Biol (2020) 71:355–78. doi: 10.1146/annurev-arplant-081519-035901
13. Meyers BC, Morgante M, Michelmore RW. TIR-X and TIR-NBS Proteins: Two New Families Related to Disease Resistance TIR-NBS-LRR Proteins Encoded in Arabidopsis and Other Plant Genomes. Plant J (2002) 32:77–92. doi: 10.1046/j.1365-313X.2002.01404.x
14. Wan L, Essuman K, Anderson RG, Sasaki Y, Monteiro F, Chung E-H, et al. TIR Domains of Plant Immune Receptors Are NAD+-Cleaving Enzymes That Promote Cell Death. Science (2019) 365:799–803. doi: 10.1126/science.aax1771
15. Ma S, Lapin D, Liu L, Sun Y, Song W, Zhang X, et al. Direct Pathogen-Induced Assembly of an NLR Immune Receptor Complex to Form a Holoenzyme. Science (2020) 370(6521):eabe3069. doi: 10.1126/science.abe3069
16. Martin R, Qi T, Zhang H, Liu F, King M, Toth C, et al. Structure of the Activated ROQ1 Resistosome Directly Recognizing the Pathogen Effector XopQ. Science (2020) 370(6521):eabd9993. doi: 10.1126/science.abd9993
17. Spear AM, Loman NJ, Atkins HS, Pallen MJ. Microbial TIR Domains: Not Necessarily Agents of Subversion? Trends Microbiol (2009) 17:393–8. doi: 10.1016/j.tim.2009.06.005
18. Sengupta D, Koblansky A, Gaines J, Brown T, West AP, Zhang D, et al. Subversion of Innate Immune Responses by Brucella Through the Targeted Degradation of the TLR Signaling Adapter, MAL. J Immunol (2010) 184:956–64. doi: 10.4049/jimmunol.0902008
19. Snyder GA, Deredge D, Waldhuber A, Fresquez T, Wilkins DZ, Smith PT, et al. Crystal Structures of the Toll/Interleukin-1 Receptor (TIR) Domains From the Brucella Protein TcpB and Host Adaptor TIRAP Reveal Mechanisms of Molecular Mimicry. J Biol Chem (2014) 289:669–79. doi: 10.1074/jbc.M113.523407
20. Essuman K, Summers DW, Sasaki Y, Mao X, Yim AKY, DiAntonio A, et al. TIR Domain Proteins Are an Ancient Family of NAD+-Consuming Enzymes. Curr Biol (2018) 28:421–30.e424. doi: 10.1016/j.cub.2017.12.024
21. Coronas-Serna JM, Louche A, Rodríguez-Escudero M, Roussin M, Imbert PR, Rodríguez-Escudero I, et al. The TIR-Domain Containing Effectors BtpA and BtpB From Brucella Abortus Impact NAD Metabolism. PloS Pathog (2020) 16:e1007979. doi: 10.1371/journal.ppat.1007979
22. Ka D, Oh H, Park E, Kim J-H, Bae E. Structural and Functional Evidence of Bacterial Antiphage Protection by Thoeris Defense System via NAD+ Degradation. Nat Comm (2020) 11:1–8. doi: 10.1038/s41467-020-16703-w
23. Doron S, Melamed S, Ofir G, Leavitt A, Lopatina A, Keren M, et al. Systematic Discovery of Antiphage Defense Systems in the Microbial Pangenome. Science (2018) 359(6739):eaar4120. doi: 10.1126/science.aar4120
24. Ofir G, Herbst E, Baroz M, Cohen D, Millman A, Doron S, et al. Antiviral Activity of Bacterial TIR Domains via Signaling Molecules That Trigger Cell Death. bioRxiv (2021). doi: 10.1101/2021.01.06.425286. 2021.2001.2006.425286.
25. Rodriguez Gama A, Miller T, Halfmann R. Mechanics of a Molecular Mousetrap-Nucleation-Limited Innate Immune Signaling. Biophys J (2021) 120:1150–60. doi: 10.1016/j.bpj.2021.01.007
26. Ve T, Vajjhala PR, Hedger A, Croll T, DiMaio F, Horsefield S, et al. Structural Basis of TIR-Domain-Assembly Formation in MAL- and MyD88-Dependent TLR4 Signaling. Nat Struct Mol Biol (2017) 24:743–51. doi: 10.1038/nsmb.3444
27. Clabbers MT, Holmes S, Muusse TW, Vajjhala PR, Thygesen SJ, Malde AK, et al. MyD88 TIR Domain Higher-Order Assembly Interactions Revealed by Microcrystal Electron Diffraction and Serial Femtosecond Crystallography. Nat Comm (2021) 12:1–14. doi: 10.1038/s41467-021-22590-6
28. Ve T, Gay NJ, Mansell A, Kobe B, Kellie S. Adaptors in Toll-Like Receptor Signaling and Their Potential as Therapeutic Targets. Curr Drug Targets (2012) 13:1360–74. doi: 10.2174/138945012803530260
29. Behzadi P, Garcia-Perdomo HA, Karpinski TM. Toll-Like Receptors: General Molecular and Structural Biology. J Immunol Res (2021) 2021:9914854. doi: 10.1155/2021/9914854
30. Asami J, Shimizu T. Structural and Functional Understanding of the Toll-Like Receptors. Protein Sci (2021) 30:761–72. doi: 10.1002/pro.4043
31. Ishida H, Asami J, Zhang Z, Nishizawa T, Shigematsu H, Ohto U, et al. Cryo-EM Structures of Toll-Like Receptors in Complex With UNC93B1. Nat Struct Mol Biol (2021) 28:173–80. doi: 10.1038/s41594-020-00542-w
32. Tao X, Xu Y, Zheng Y, Beg AA, Tong L. An Extensively Associated Dimer in the Structure of the C713S Mutant of the TIR Domain of Human TLR2. Biochem Biophys Res Commun (2002) 299:216–21. doi: 10.1016/s0006-291x(02)02581-0
33. Jang T-h, Park HH. Crystal Structure of TIR Domain of TLR6 Reveals Novel Dimeric Interface of TIR–TIR Interaction for Toll-Like Receptor Signaling Pathway. J Mol Biol (2014) 426:3305–13. doi: 10.1016/j.jmb.2014.07.024
34. Nyman T, Stenmark P, Flodin S, Johansson I, Hammarström M, Nordlund P. The Crystal Structure of the Human Toll-Like Receptor 10 Cytoplasmic Domain Reveals a Putative Signaling Dimer. J Biol Chem (2008) 283:11861–5. doi: 10.1074/jbc.C800001200
35. Poltorak A, He X, Smirnova I, Liu M-Y, Van Huffel C, Du X, et al. Defective LPS Signaling in C3H/HeJ and C57BL/10ScCr Mice: Mutations in Tlr4 Gene. Science (1998) 282:2085–8. doi: 10.1126/science.282.5396.2085
36. Nunez Miguel R, Wong J, Westoll JF, Brooks HJ, O'Neill LA, Gay NJ, et al. A Dimer of the Toll-Like Receptor 4 Cytoplasmic Domain Provides a Specific Scaffold for the Recruitment of Signalling Adaptor Proteins. PloS One (2007) 2:e788. doi: 10.1371/journal.pone.0000788
37. Lech M, Skuginna V, Kulkarni OP, Gong J, Wei T, Stark RW, et al. Lack of SIGIRR/TIR8 Aggravates Hydrocarbon Oil-Induced Lupus Nephritis. J Pathol (2010) 220:596–607. doi: 10.1002/path.2678
38. Boraschi D, Tagliabue A. The Interleukin-1 Receptor Family. Semin Immunol (2013) 25:394–407. doi: 10.1016/j.smim.2013.10.023
39. Boraschi D, Italiani P, Weil S, Martin MU. The Family of the Interleukin-1 Receptors. Immunol Rev (2018) 281:197–232. doi: 10.1111/imr.12606
40. Fields JK, Günther S, Sundberg EJ. Structural Basis of IL-1 Family Cytokine Signaling. Front Immunol (2019) 10:1412. doi: 10.3389/fimmu.2019.01412
41. Khan JA, Brint EK, O'Neill LA, Tong L. Crystal Structure of the Toll/interleukin-1 Receptor Domain of Human IL-1rapl. J Biol Chem (2004) 279:31664–70. doi: 10.1074/jbc.M403434200
42. Montani C, Gritti L, Beretta S, Verpelli C, Sala C. The Synaptic and Neuronal Functions of the X-Linked Intellectual Disability Protein Interleukin-1 Receptor Accessory Protein Like 1 (Il1rapl1). Dev Neurobiol (2019) 79:85–95. doi: 10.1002/dneu.22657
43. Gay NJ, Symmons MF, Gangloff M, Bryant CE. Assembly and Localization of Toll-Like Receptor Signalling Complexes. Nat Rev Immunol (2014) 14:546–58. doi: 10.1038/nri3713
44. Carty M, Goodbody R, Schröder M, Stack J, Moynagh PN, Bowie AG. The Human Adaptor SARM Negatively Regulates Adaptor Protein TRIF–dependent Toll-Like Receptor Signaling. Nat Immunol (2006) 7:1074–81. doi: 10.1038/ni1382
45. Halabi S, Sekine E, Verstak B, Gay NJ, Moncrieffe MC. Structure of the Toll/interleukin-1 Receptor (TIR) Domain of the B-Cell Adaptor That Links Phosphoinositide Metabolism With the Negative Regulation of the Toll-Like Receptor (TLR) Signalosome. J Biol Chem (2017) 292:652–60. doi: 10.1074/jbc.M116.761528
46. Troutman TD, Hu W, Fulenchek S, Yamazaki T, Kurosaki T, Bazan JF, et al. Role for B-Cell Adapter for PI3K (BCAP) as a Signaling Adapter Linking Toll-Like Receptors (TLRs) to Serine/Threonine Kinases PI3K/Akt. Proc Natl Acad Sci USA (2012) 109:273–8. doi: 10.1073/pnas.1118579109
47. Lin SC, Lo YC, Wu H. Helical Assembly in the MyD88-IRAK4-IRAK2 Complex in TLR/IL-1R Signalling. Nature (2010) 465:885–90. doi: 10.1038/nature09121
48. Von Bernuth H, Picard C, Jin Z, Pankla R, Xiao H, Ku C-L, et al. Pyogenic Bacterial Infections in Humans With MyD88 Deficiency. Science (2008) 321:691–6. doi: 10.1126/science.1158298
49. Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim K-H, et al. Oncogenically Active MYD88 Mutations in Human Lymphoma. Nature (2011) 470:115–9. doi: 10.1038/nature09671
50. O'Carroll A, Chauvin B, Brown JWP, Meagher A, Coyle J, Schill J, et al. Pathological Mutations Differentially Affect the Self-Assembly and Polymerisation of the Innate Immune System Signalling Adaptor Molecule Myd88. BMC Biol (2018) 16:149. doi: 10.1186/s12915-018-0611-7
51. Osterloh JM, Yang J, Rooney TM, Fox AN, Adalbert R, Powell EH, et al. Dsarm/Sarm1 is Required for Activation of an Injury-Induced Axon Death Pathway. Science (2012) 337:481–4. doi: 10.1126/science.1223899
52. Gerdts J, Summers DW, Sasaki Y, DiAntonio A, Milbrandt J. Sarm1-Mediated Axon Degeneration Requires Both SAM and TIR Interactions. J Neurosci (2013) 33:13569–80. doi: 10.1523/JNEUROSCI.1197-13.2013
53. Hopkins EL, Gu W, Kobe B, Coleman MP. A Novel NAD Signaling Mechanism in Axon Degeneration and Its Relationship to Innate Immunity. Front Mol Biosci (2021) 8:703532. doi: 10.3389/fmolb.2021.703532
54. Carty M, Bowie AG. SARM: From Immune Regulator to Cell Executioner. Biochem Pharmacol (2019) 161:52–62. doi: 10.1016/j.bcp.2019.01.005
55. Angeletti C, Amici A, Gilley J, Loreto A, Trapanotto AG, Antoniou C, et al. Programmed Axon Death Executor SARM1 is a Multi-Functional NAD (P) Ase With Prominent Base Exchange Activity, All Regulated by Physiological Levels of NMN, NAD, NADP and Other Metabolites. bioRxiv (2021). doi: 10.1101/2021.07.14.451805. 2021.07.14.451805.
56. Sporny M, Guez-Haddad J, Lebendiker M, Ulisse V, Volf A, Mim C, et al. Structural Evidence for an Octameric Ring Arrangement of SARM1. J Mol Biol (2019) 431:3591–605. doi: 10.1016/j.jmb.2019.06.030
57. Summers DW, Gibson DA, DiAntonio A, Milbrandt J. SARM1-Specific Motifs in the TIR Domain Enable NAD+ Loss and Regulate Injury-Induced SARM1 Activation. Proc Natl Acad Sci USA (2016) 113:E6271–80. doi: 10.1073/pnas.1601506113
58. Bratkowski M, Xie T, Thayer DA, Lad S, Mathur P, Yang Y-S, et al. Structural and Mechanistic Regulation of the Pro-Degenerative NAD Hydrolase SARM1. Cell Rep (2020) 32:107999. doi: 10.1016/j.celrep.2020.107999
59. Figley MD, Gu W, Nanson JD, Shi Y, Sasaki Y, Cunnea K, et al. SARM1 is a Metabolic Sensor Activated by an Increased NMN/NAD(+) Ratio to Trigger Axon Degeneration. Neuron (2021) 109:1118–36.e1111. doi: 10.1016/j.neuron.2021.02.009
60. Jiang Y, Liu T, Lee CH, Chang Q, Yang J, Zhang Z. The NAD(+)-Mediated Self-Inhibition Mechanism of Pro-Neurodegenerative SARM1. Nature (2020) 588:658–63. doi: 10.1038/s41586-020-2862-z
61. Sasaki Y, Zhu J, Shi Y, Gu W, Kobe B, Ve T, et al. Nicotinic Acid Mononucleotide is an Allosteric SARM1 Inhibitor Promoting Axonal Protection. Exp Neurol (2021) 345:113842. doi: 10.1016/j.expneurol.2021.113842
62. Di Stefano M, Nascimento-Ferreira I, Orsomando G, Mori V, Gilley J, Brown R, et al. A Rise in NAD Precursor Nicotinamide Mononucleotide (NMN) After Injury Promotes Axon Degeneration. Cell Death Differ (2015) 22:731–42. doi: 10.1038/cdd.2014.164
63. Di Stefano M, Loreto A, Orsomando G, Mori V, Zamporlini F, Hulse RP, et al. NMN Deamidase Delays Wallerian Degeneration and Rescues Axonal Defects Caused by NMNAT2 Deficiency In Vivo. Curr Biol (2017) 27:784–94. doi: 10.1016/j.cub.2017.01.070
64. Chan SL, Mukasa T, Santelli E, Low LY, Pascual J. The Crystal Structure of a TIR Domain From Arabidopsis Thaliana Reveals a Conserved Helical Region Unique to Plants. Protein Sci (2010) 19:155–61. doi: 10.1002/pro.275
65. Williams SJ, Sohn KH, Wan L, Bernoux M, Sarris PF, Segonzac C, et al. Structural Basis for Assembly and Function of a Heterodimeric Plant Immune Receptor. Science (2014) 344:299–303. doi: 10.1126/science.1247357
66. Williams SJ, Yin L, Foley G, Casey LW, Outram MA, Ericsson DJ, et al. Structure and Function of the TIR Domain From the Grape NLR Protein RPV1. Front Plant Sci (2016) 7:1850. doi: 10.3389/fpls.2016.01850
67. Zhang X, Bernoux M, Bentham AR, Newman TE, Ve T, Casey LW, et al. Multiple Functional Self-Association Interfaces in Plant TIR Domains. Proc Natl Acad Sci USA (2017) 114:E2046–52. doi: 10.1073/pnas.1621248114
68. Bernoux M, Ve T, Williams S, Warren C, Hatters D, Valkov E, et al. Structural and Functional Analysis of a Plant Resistance Protein TIR Domain Reveals Interfaces for Self-Association, Signaling, and Autoregulation. Cell Host Microbe (2011) 9:200–11. doi: 10.1016/j.chom.2011.02.009
69. Nishimura MT, Anderson RG, Cherkis KA, Law TF, Liu QL, Machius M, et al. TIR-Only Protein RBA1 Recognizes a Pathogen Effector to Regulate Cell Death in Arabidopsis. Proc Natl Acad Sci USA (2017) 114:E2053–62. doi: 10.1073/pnas.1620973114
70. Uno M, Watanabe-Nakayama T, Konno H, Akagi KI, Tsutsumi N, Fukao T, et al. Intramolecular Interaction Suggests an Autosuppression Mechanism for the Innate Immune Adaptor Protein Myd88. Chem Commun (Camb) (2018) 54:12318–21. doi: 10.1039/c8cc06480f
Keywords: protein structure, protein-protein interactions, axon degeneration, cell-death signaling, signaling by cooperative assembly formation (SCAF), innate immunity, plant disease resistance, toll/interleukin-1 receptor/resistance protein
Citation: Nimma S, Gu W, Maruta N, Li Y, Pan M, Saikot FK, Lim BYJ, McGuinness HY, Zaoti ZF, Li S, Desa S, Manik MK, Nanson JD and Kobe B (2021) Structural Evolution of TIR-Domain Signalosomes. Front. Immunol. 12:784484. doi: 10.3389/fimmu.2021.784484
Received: 27 September 2021; Accepted: 25 October 2021;
Published: 17 November 2021.
Edited by:
Pedro Jose Esteves, Centro de Investigacao em Biodiversidade e Recursos Geneticos (CIBIO-InBIO), PortugalReviewed by:
Iva Hafner Bratkovic, National Institute of Chemistry, SloveniaTakaki Maekawa, Max Planck Institute for Plant Breeding Research, Germany
Copyright © 2021 Nimma, Gu, Maruta, Li, Pan, Saikot, Lim, McGuinness, Zaoti, Li, Desa, Manik, Nanson and Kobe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bostjan Kobe, Yi5rb2JlQHVxLmVkdS5hdQ==