 Nikolaos Skartsis1,2
Nikolaos Skartsis1,2 Yani Peng1
Yani Peng1 Leonardo M. R. Ferreira1
Leonardo M. R. Ferreira1 Vinh Nguyen1
Vinh Nguyen1 Emilie Ronin1†
Emilie Ronin1† Yannick D. Muller1
Yannick D. Muller1 Flavio Vincenti1,2
Flavio Vincenti1,2 Qizhi Tang1,3*
Qizhi Tang1,3*- 1Department of Surgery, University of California San Francisco, San Francisco, CA, United States
- 2Division of Nephrology, Department of Medicine, University of California San Francisco, San Francisco, CA, United States
- 3Diabetes Center, University of California San Francisco, San Francisco, CA, United States
Treg therapies are being tested in clinical trials in transplantation and autoimmune diseases, however, the impact of inflammation on Tregs remains controversial. We challenged human Tregs ex-vivo with pro-inflammatory cytokines IL-6 and TNFα and observed greatly enhanced proliferation stimulated by anti-CD3 and anti-CD28 (aCD3/28) beads or CD28 superagonist (CD28SA). The cytokine-exposed Tregs maintained high expression of FOXP3 and HELIOS, demethylated FOXP3 enhancer, and low IFNγ, IL-4, and IL-17 secretion. Blocking TNF receptor using etanercept or deletion of TNF receptor 2 using CRISPR/Cas9 blunted Treg proliferation and attenuated FOXP3 and HELIOS expression. These results prompted us to consider using CD28SA together with IL-6 and TNFα without aCD3/28 beads (beadless) as an alternative protocol for therapeutic Treg manufacturing. Metabolomics profiling revealed more active glycolysis and oxidative phosphorylation, increased energy production, and higher antioxidant potential during beadless Treg expansion. Finally, beadless expanded Tregs maintained suppressive functions in vitro and in vivo. These results demonstrate that human Tregs positively respond to proinflammatory cytokines with enhanced proliferation without compromising their lineage identity or function. This property can be harnessed for therapeutic Treg manufacturing.
Introduction
Tregs are a subset of CD4+ T cells that prevent unwanted immune activation in steady state and promote resolution of immune response at the site of inflammation (1–3). In mouse models of type 1 diabetes, Tregs isolated from inflamed islets, but not from lymphoid organs, are able to suppress anti-islet autoimmune attacks (4). In humans, higher FOXP3 in graft biopsies predicts reversibility of T cell-mediated rejection of kidney transplant whereas higher Tregs in the tumor microenvironment is associated with worse prognosis (5, 6). However, it has been suggested that Tregs may be destabilized under certain inflammatory conditions and there has been debate on the impact of proinflammatory signals on Treg function (7). During immune activation, proinflammatory cytokines such as TNFa and IL-6 are produced by macrophages, neutrophils, T cells, B cells, and stromal cells (8–11). a and IL-6 promote T cell activation (12, 13) and increase resistance to Treg-mediated suppression (14–16), both of these functions contribute to mounting an effective immune response. While there is consensus that TNFa and IL-6 heighten effector T cell (Teff) resistance to suppression by Tregs (17, 18), their direct impact on Tregs, especially on human Tregs, is unclear.
The effect of TNFa on mouse Tregs has been extensively studied. While TNFa impairs the differentiation and function of TGFβ-induced Tregs (19), it is generally accepted that TNFα exerts positive effects on thymic derived Tregs (20–22). TNFα optimally activates Tregs via upregulating TNFR2, 4-1BB, and OX40 expression (23, 24). TNFR2-deficient Tregs lose Foxp3 expression and cannot protect against colitis when co-transferred with naïve CD4+ T cells into Rag1KO mice (25). TNFR2-expressing Tregs are critical to suppressing EAE by limiting T cell activation in the CNS during overt inflammation (26). Finally, TNFa-primed mouse Tregs have enhanced efficacy against graft-versus-host disease (GVHD) when compared with unprimed Tregs (27, 28). Overall, these results suggest that TNFα negatively affects TGFβ-induced Tregs, but exerts a positive effect on thymic derived Tregs in mice.
The effects of TNFa on human Tregs has been more controversial. Tregs isolated from rheumatoid arthritis patients have been reported to lose FOXP3 expression and convert into pathogenic T cells when exposed to TNFa in vitro (29, 30). TNFα partially abrogated the suppressive function of CD4+CD25+ T cells isolated from chronic HBV-infected patients (31). Activation of the canonical NF-κB pathway in CD45RA- Tregs by TNFa-TNFR2 interaction downmodulated their suppressive function (32). However, these findings have been challenged by others who report a pro-regulatory role of TNFa in optimally activating Tregs isolated from healthy donors, without negatively affecting their in vitro suppressive function (33). Similar to studies in mouse Tregs, TNFR2 expression identifies a subset of highly suppressive Tregs isolated from PBMC (34). TNFR2 signaling has been shown to enhance human Treg proliferation in response to IL-2 via activation of the non-canonical NF-κB pathway (35). As a result, TNFR2 agonists are used in ex-vivo human Treg expansion protocols to facilitate homogeneous high purity cellular products (36, 37). Multiple studies have shown that TNFα blockade therapy paradoxically exacerbate autoimmune pathologies (38, 39), suggesting a regulatory role of TNFa in these disease settings.
Multiple studies reported a negative role of IL-6 on mouse Tregs. IL-6 prevents the differentiation of naïve CD4+ T cells into peripherally induced Tregs in a dominant and non-redundant manner (40). IL-6 has been reported to destabilize mouse thymic derived Tregs and drive them to produce IL-17 in vitro (41). However, Tregs isolated from inflamed CNS lesions in EAE-affected mice are insensitive to IL-6-driven Th17 conversion due to a downregulation of IL-6Ra and gp130 (42). IL-6 has been found to increase the rate of Foxp3 proteasomal degradation via downregulation of the expression of deubiquitinase USP7, which is normally upregulated and associated with Foxp3 in the nucleus of Tregs (43). In contrast, IL-6 Tg mice harbor increased numbers of Tregs in their lymphoid organs with intact functional capacity against naïve T cells in vitro (44). While IL-6 enhances the generation of Th17 cells, classic signaling via IL-6Ra induces double positive RORγt+Foxp3+ Tregs with potent immunoregulatory properties in a mouse model of glomerulonephritis (45). The prevailing view is that IL-6 negatively impacts mouse Tregs, but emerging data suggest that differential mode of signaling (classic versus trans-signaling) may have divergent impacts on mouse Tregs.
Similar to findings in mouse Tregs, IL-6 prevents in-vitro induction of human Tregs from conventional T cells (Tconvs) (46). Peripheral blood CD4+CD25+ Tregs downregulate FOXP3 and lose their suppressive function following IL-6 exposure (47). Retinoic acid downregulates IL-6Ra and renders Tregs insensitive to the destabilizing effect of IL-6 (47). gp130hi Tregs are enriched amongst naïve CD45RA+ Tregs isolated from human PBMC and have lower in vitro suppressive capacity when compared to gp130lo Tregs (48). Another group has reported the presence of a subpopulation of human IL-6RahiCD45RA- Tregs in PBMC that have potent suppressive capacity in vitro, demethylated Treg-specific demethylated region (TSDR) and a Th17 transcriptional signature (49). These studies highlight the heterogeneity of circulating human Tregs. However, there is paucity of studies measuring the direct impact of IL-6 on human Tregs.
In this study, we examined the direct impact of TNFa and IL-6 on human Tregs in an ex-vivo culture system and evaluated the in-vivo function of TNFa and IL-6 exposed human Tregs in a xenogeneic GVHD model in humanized mice. Our data show a positive role of both IL-6 and TNFa on Tregs in promoting their proliferation without lineage destabilization, suggesting that Tregs can respond to proinflammatory signals to increase their presence at sites of inflammation without compromising their lineage stability.
Materials and Methods
Human Peripheral Blood Samples
De-identified peripheral blood units were purchased from StemCell Technologies (Vancouver, Canada). Peripheral blood samples from 25 healthy donors (11 male and 14 female) between the age of 21 and 72 were used.
Ex-Vivo Treg Culture
Human blood samples were processed the same day after collection using ficoll (Cytiva, Marlborough MA) density gradient to isolate PBMC. PBMC were stained with anti-CD4 FITC (clone OKT4), anti-CD25 APC (clone 4E3) and anti-CD127 PE (clone HIL-7R-M21, all from BD Biosciences, San Jose, CA). CD4+CD25+CD127lo/- Tregs and CD4+CD25-CD127+ Tconvs were purified using FACS. Post sort analyses confirmed >99% purity. Purified Tregs were plated in 48-well plate at a density of 1x105 cells/well in 250μL medium consisting of 10% heat-inactivated fetal bovine serum (Biosource International), nonessential amino acids, 0.5 mM sodium pyruvate, 5 mM Hepes and 1 mM glutaMax I (all from Invitrogen) in an RPMI-1640 base. Human anti-CD3 and anti-CD28-conjugated dynabeads (aCD3/CD28 beads; Fischer Scientific, Hampton, NH) were added to the T cells at 1:1 bead to cell ratio. Alternatively, CD28 superagonist (CD28SA) (5mg/mL; clone anti-CD28.1; Ancell, Stillwater, MN) were used to stimulate the T cells. Recombinant human IL-2 (either 300IU/mL or 15IU/mL, as indicated in the result section; Proleukin, Prometheus laboratories, Switzerland) were added at concentration indicated. Recombinant human IL-6 (15 to 150ng/ml as indicated in the result section; Peprotech, Rocky Hill, NJ) and recombinant human TNFa (50ng/ml; Peprotech, Rocky Hill, NJ) were also added in selected wells. Etanercept (5μg/mL; ETN-Enbrel, Pfizer) was added in selected experiments. Ex-vivo expansion cultures were supplemented with fresh media containing IL-2 on day 0, 2, 5, and every other day thereafter, until the cells returned quiescent state (typically, day 9 for aCD3/CD28 bead-stimulated Tregs and day 14 for CD28SA stimulated Tregs). IL-6 and TNFα were supplemented on days 0, 2, and 5 of culture. aCD3/28 bead stimulated CD4+ Tconvs were included in some experiments as a reference cell population.
Flow Cytometry
Single-cell suspensions were recovered from ex-vivo expanded Treg cultures. The cellular suspensions were first stained with surface antibodies to CD4 (Per-CP; clone SK3), CD25 (PE-Cy7; clone 4E3), TNFR2 (APC; clone hTNFR-M1), and fixable viability dye (APC-e780, all from purchased from BD Biosciences) prior to fixation and permeabilization using kit according to manufacturer protocol (catalog number 88-8824-00; Thermofisher) and intracellular staining for FOXP3 (e450; clone PCH101) and HELIOS (FITC; clone 22F6, both from BD Biosciences). The samples were analyzed on a FACS LSR-II flow cytometer (BD Biosciences) and analyzed using FACSDiva (BD Biosciences) or Flow Jo software (Tree Star, Ashland, OR).
In-Vitro Treg Suppression Assay
Ex-vivo expanded human Tregs were harvested on day 7 of culture and varying numbers of Tregs were then co-cultured with 1x 105 ex-vivo expanded Tconv at Treg/Tconv ratios of 1:1, 1:2, 1:4 and 1:8 in the presence of aCD3/CD28 beads (1:10 bead to Tconv ratio). 0.5μCi [3H]- Thymidine was added 16hrs before the cells were harvested for analysis on day 4 of co-culture. Triplicate wells were established for each condition.
TSDR Analysis
Ex-vivo expanded Tregs (2x106 from each condition) were harvested on day 7 of culture and then cryopreserved in FBS containing 10% DMSO at -80°C. The samples were submitted to Epigendx (Boston, MA) for analysis of DNA methylation of the TSDR in FOXP3 intron 1 using the ADS783FS2 assay.
Cytokine Assay
Supernatants of Treg and Tconv cultures were aspirated on day 7 and sent for analysis using a 42-plexed Luminex human cytokine detection kit (EveTechnologies, Vancouver, Canada). The cytokine concentrations were normalized per 106 cells for each condition. For all culture conditions, Cells were plated at 1x105/well in one well of 48-well plates in 250mL medium supplemented with cytokines as indicated; culture media were doubled on day 2; then cells were counted and resuspended at 2.5x105 cells per 1mL on day 5; and the supernatant was collected on day 7 for multiplex Luminex analysis on day 7 and the cell counts on day 7 were used to normalize the cytokine concentrations.
Humanized NSG Mouse Graft-Versus-Host-Disease Models
Eight to twelve-week-old NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG, Jackson Laboratory, Stock No 005557) male mice were bred in our animal facilities in specific pathogen-free conditions. To induce GVHD, NSG mice were irradiated (2.5Gy) one day prior to retroorbital vein infusion of either 4 x106 fresh PBMCs or ex-vivo expanded Tconvs. In GVHD prevention experiments, some mice receive co-infusion of ex-vivo expanded Tregs. In GVHD treatment experiments, Tregs were infused 4 days after PBMC infusion. Xenogeneic GVHD development was evaluated by clinical examination and body weight measurements for either 60 days (prevention experiments) or 100 days (treatment experiments). To assess the phenotypical stability of ex vivo expanded Tregs after adoptive transfer, irradiated NSG mice were injected with 2x106 fresh autologous PBMCs 9 days before receiving 4x106 CFSE-labeled Tregs. On day 5 after Treg infusion, peripheral blood and lung tissue were obtained for analyses using flow cytometry.
TNFR2 Gene Deletion Using CRISPR
Ribonucleoprotein complexes were made by complexing crRNAs and tracrRNAs chemically synthesized (Integrated DNA Technologies (IDT), Coralville, IA) with recombinant Cas9NLS (QB3 Macrolab, UC Berkley, CA) as previously described (50). Guide RNA sequences used for gene editing were (1) GGTTCTTGACTACCGTAATT (scrambled gRNA) and (2) GGCAUUUACACCCUACGCCC (TNFR2 gene). Briefly, Lyophilized RNAs were resuspended at 160μM in 10mM Tris-HCL with 150mM KCL and stored in aliquots at minus 80. The day of electroporation, CrRNA and tracrRNA aliquots were thawed and mixed at a 1:1 volume and incubated 30 minutes at 37°C for annealing. The 80µM guide RNA complex was mixed at 37°C with Cas9 NLs at a 2:1 gRNA to Cas9 molar ratio for another 15 minutes and the mixture is defined as RNP at 20µM. RNP were electroporated using a Lonza 4D 96-well electroporation system (code EH115). Genome editing efficiency was measured by assessing cell surface TNFR2 expression using flow cytometry on days 4 and 8 of culture.
Metabolomics
At least 2x 106 FACS sorted fresh and ex-vivo expanded Tregs were collected on days 0 and 7 of culture, respectively. The cells were stored according to manufacturer protocol prior to being sent for analysis using capillary electrophoresis- and liquid chromatography- mass spectrometry platforms to generate their global metabolic profile (Human Metabolome Technologies, Boston, MA).
Statistics
Kaplan-Meier survival graphs were constructed, and a log-rank comparison of the groups was used to calculate p-values. The paired t test was used for comparison of experimental groups. Differences were considered significant for p less than 0.05. Prism software (GraphPad Software, San Diego, CA) version 9 was used for data analysis and graphing data.
Study Approval
All experiments were approved (IACUC protocol No. AN183959-02) and conducted in accordance with UCSF IACUC regulations.
Results
IL-6 and TNFα Promoted Proliferation of Lineage-Committed Tregs
To determine the direct effects of TNFα and IL-6 on human Tregs, we challenged FACS purified CD4+CD25+CD127lo/- Tregs (sorting strategy shown in Supplemental Figure 1) with IL-6 and/or TNFα, during in vitro activation. We stimulated the cells with anti-CD3 and anti-CD28 (aCD3/28) beads in the presence of 300 IU/ml recombinant human IL-2 to simulate antigen activation of Tregs. Activated Tregs formed multicellular clusters, which was followed by cell proliferation and return to a single-celled quiescent state by day 9 of culture as previously reported (51). FACS purified CD4+CD25-CD127+ Tconv were also stimulated using the same conditions and proliferated more intensely than Tregs (Supplemental Figure 2). The addition of IL-6, TNFα, or both in the Treg cultures resulted in persistent cell clustering (Figure 1A). Moreover, combination of IL-6 and TNFα resulted in significantly improved Treg expansion, which mainly manifested during the later stage of culture (Figure 1B).
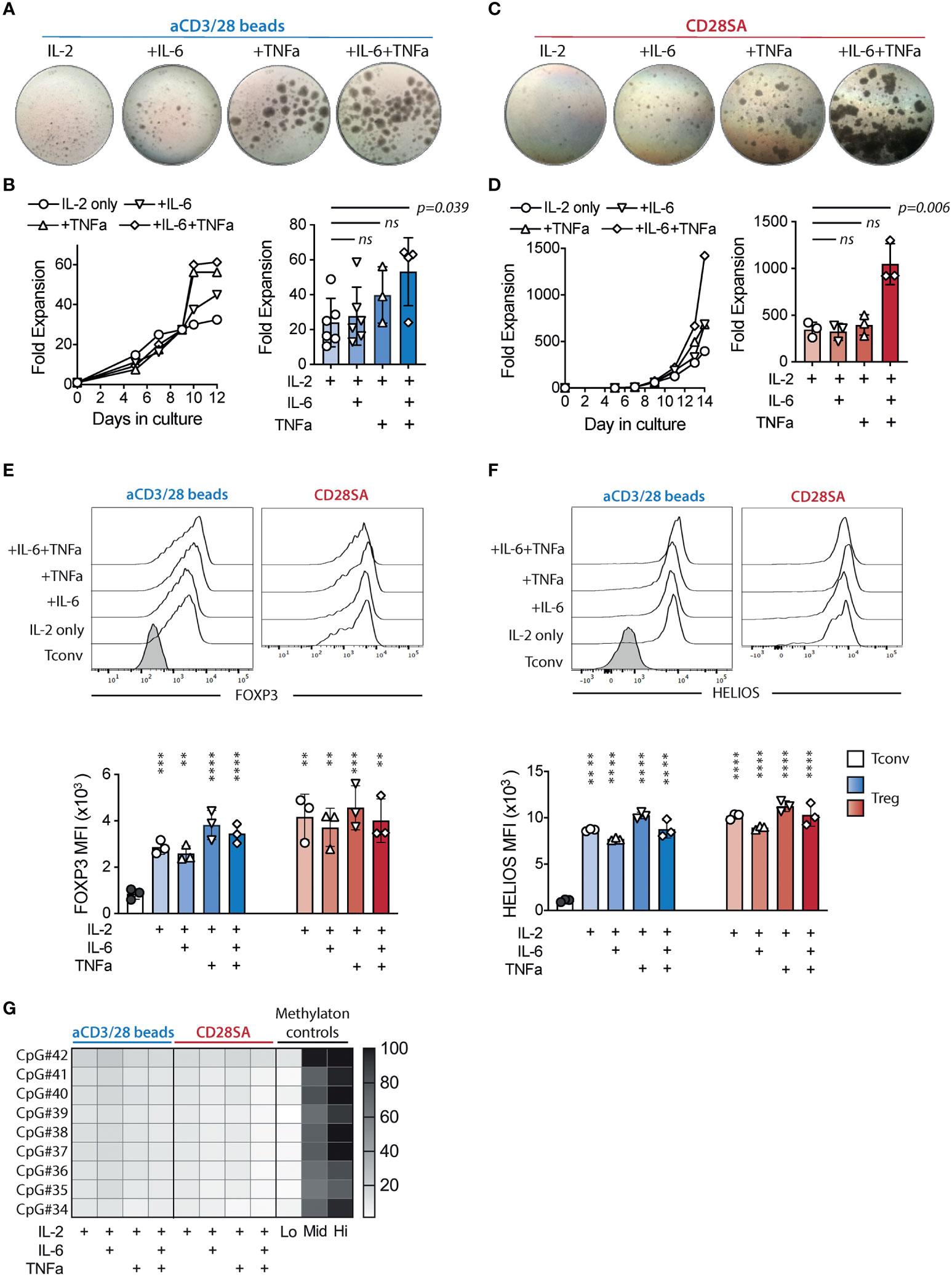
Figure 1 IL-6 and TNFα augmented ex-vivo proliferation of lineage committed human Tregs. FACS purified human Tregs were stimulated with either aCD3/28 beads or CD28SA and cultured in the presence of 300 IU/ml rhIL-2 with or without TNFα and IL-6 for 12 and 14 days as indicated (A) Microscopic pictures of aCD3/28 bead-stimulated Treg cultures on day 8 of culture. Original magnification was 10x. (B) An example of aCD3/28 bead-stimulated Treg expansion kinetics from one representative experiment (left) and a summary of fold expansion on day 12 of 3 to 6 independent experiments (right) are shown. (C, D) Same as panel (A, B), except the Tregs were stimulated with CD28SA and overall expansion (right panel) was assessed on day 14. Statistical significance was assessed using one-way ANOVA and Dunnett’s multiple comparisons posttest using IL-2 alone as reference. p values are stated. Results shown used cells from 3 distinct donors and are representative of 6 independent experiments using cells from 6 distinct donors. (E, F) Flow cytometric analysis of FOXP3 (E) and HELIOS (F) expression in Tregs on day 9 after stimulation. Representative histograms (left) and summaries of mean fluorescent intensities (MFI) from 3 independent experiments (right) are shown. Statistical significance was assessed using one-way ANOVA with Geisser-Greenhouse’s correction and Dunnett’s multiple comparisons test using either Tconv or Treg treated with IL-2 alone (E, F) as a reference. p values are marked as ns, not significant, p > 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. (G) Heatmap summary of TSDR demethylation of Tregs expanded in various conditions. Results shown are averages of Treg cultures using 2 unrelated male donors in 2 independent experiments.
It has been previously reported that CD28 superagonist antibodies (CD28SA) can also induce ex-vivo expansion of human Tregs (52). We thus replaced the aCD3/28 beads with CD28SA and evaluated the impact of IL-6 and TNFα in the context of a different mitogenic stimulation. While CD28SA-stimulated Tregs showed delayed kinetics in proliferation when compared to their aCD3/CD28-stimulated counterparts, the cells proliferated more persistently, resulting in more overall expansion (Figures 1C, D). Exposure to both TNFa and IL-6 resulted in robust cell clustering over a prolonged period and synergistic enhancement of cell expansion that was far more pronounced than seen with aCD3/CD28 bead-stimulated Tregs (Figures 1B, D).
Since FOXP3 had been reported to limit Treg proliferation and metabolism (53), we wondered if the enhanced proliferation in the presence of TNFa and IL-6 was due to destabilization of Tregs. We measured the expression of FOXP3 and HELIOS, the two lineage-defining transcription factors for Tregs (54), on days 9 and 14 during the ex vivo expansion (Supplemental Figure 3). The results showed that Tregs exposed to IL-6 and/or TNFa maintained high levels of FOXP3 and HELIOS expression (Figures 1E, F). Furthermore, we analyzed TSDR methylation status in Tregs cultured in various conditions. The results showed a high degree of TSDR demethylation in all expanded Tregs (Figure 1G), supporting the flow cytometric data to show that the exposure to IL-6 and/or TNFa did not alter Treg identity.
To further assess if IL-6 and/or TNFa induced effector functions in Tregs, we analyzed the supernatant of Treg and Tconv cultures on day 7 after stimulation using a 42-plex Luminex assay. Among the 42 cytokines in the panel, 21 were not present in any of the T cell cultures, 3 were added (IL-2, IL-6, and TNFa), and 18 were detected in Treg cultures. Among the 18 de novo cytokines detected, the amounts found in Treg cultures were markedly lower than those seen in Tconv cultures (Figure 2A). Particularly, no increase in IFNγ (Th1), IL-4 (Th2), or IL-17A (Th17) were seen in aCD3/28 bead or CD28SA stimulated cultures by the exposures to IL-6 and/or TNFα (Figure 2B). In fact, IL-6 did not induce increased secretion of any of the cytokines. On the other hand, exposure to TNFa, alone or in the presence of IL-6, led to a consistent trend of increase of CCL3 and CCL5 (Figure 2C). Taken together, these experiments revealed that IL-6 and TNFa exposure resulted in robust human Treg proliferation without lineage destabilization.
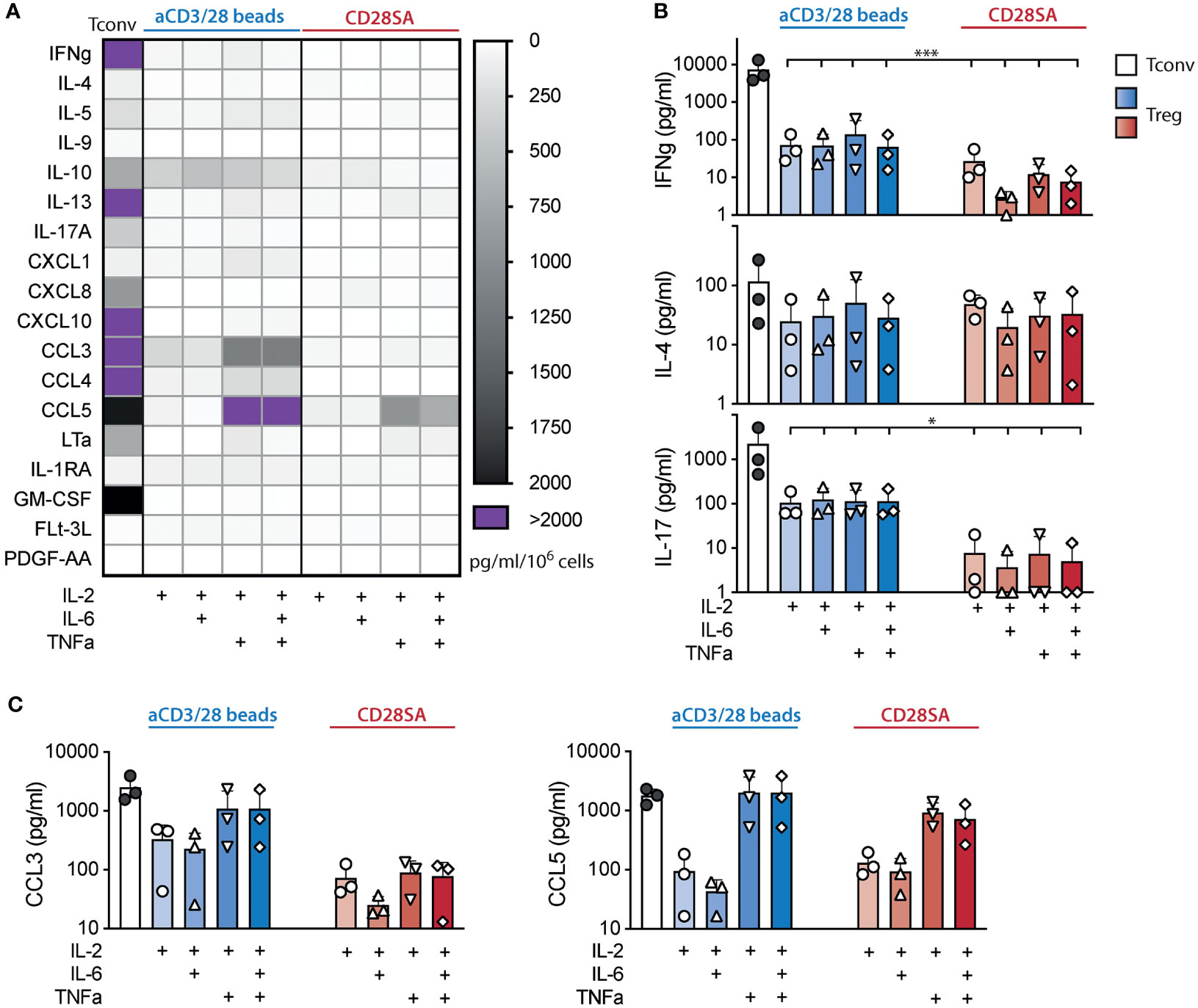
Figure 2 Tregs expanded in IL-6 and TNFα did not secrete more proinflammatory cytokines. FACS purified human Tregs were stimulated with either aCD3/28 beads or CD28SA and cultured in the presence of 300 IU/ml rhIL-2 with or without TNFα and IL-6 as indicated. Cytokine and chemokine secretion in the culture supernatant of various Treg cultures was assessed using a multiplex Luminex panel. Supernatant of aCD3/28 bead stimulated Tconv cultures is included as a reference. (A) Heatmap summary of cytokines and chemokines that were present in any of the culture condition is shown. (B) IFNγ, IL-4, and IL-17 concentrations in the Day 7 culture supernatants are shown. (C) CCL3 and CCL5 concentrations in the Day 7 culture supernatant are shown. Results shown are summaries of 3 independent experiments using cells from 3 unrelated donors. Statistical significance was assessed using one-way ANOVA with Geisser-Greenhouse’s correction and Dunnett’s multiple comparisons posttest using Treg treated with IL-2 alone (B, C) as reference. p values are marked as *p < 0.05, ***p < 0.001.
These results contradicted previous reports on the destabilizing effect of proinflammatory cytokines on Tregs (29, 30, 55, 56). Many of these studies stimulated Tregs in the presence of lower concentration of IL-2 than we used. We hypothesized that the high concentration of IL-2 in our culture condition may have protected Tregs from the destabilizing effect of TNFa and IL-6. Therefore, we challenged the Tregs by reducing IL-2 concentration from 300 IU/ml to 15 IU/ml. Treg expansion was minimal with reduced IL-2, even when they were exposed to IL-6 and/or TNFα. FOXP3 expression was reduced when compared to cells cultured in 300 IU/ml of IL-2 but remained significantly elevated when compared to that expressed by similarly expanded Tconvs (Supplemental Figure 4A). HELIOS expression was comparably high in Tregs cultured in 15 IU/ml and 300 IU/ml of IL-2 (Supplemental Figure 4B). More importantly, Tregs exposed to TNFa and IL-6 in the presence of low IL-2 maintained demethylated TSDR (Supplemental Figure 4C) and did not produce more effector cytokines (Supplemental Figure 5A), including IFNγ, IL-4, or IL-17 (Supplemental Figure 5B) when compared to Tregs stimulated without these cytokines. Similar to cultures stimulated in the presence of high IL-2, co-culture with TNFα stimulated higher secretion of CCL5 in Tregs (Supplementary Figure 5C). Thus, IL-6 and TNFa did not induce Treg destabilization even when IL-2 was limited.
Ex-Vivo Expanded Human Tregs Produce TNFα and Depend on TNFR2 Signaling to Proliferate and Safeguard Their Identity
While analyzing cytokine secretion in Treg cultures, we noticed that TNFα was consistently detected in all cultures in higher amounts in aCD3/28 bead-stimulated cultures than in CD28SA-stimulated cultures (Figure 3A). The amount of TNFa in the supernatant was not affected by the concentration of IL-2 or the addition of IL-6. Moreover, another TNF superfamily member, lymphotoxin a (LTa), was also consistently detected (Figure 3B). The presence of TNFa, but not IL-6, resulted in significantly increased level of LTa in the aCD3/28 bead-stimulated Treg culture supernatant and a trend of increased LTa in CD28SA-stimulated cultures (Figure 3B). Both of TNFa and LTa bind to TNFR1 and TNFR2 (57). It has been well-established that Tregs preferentially express TNFR2 (24). In vitro stimulation with either aCD3/28 beads or CD28SA uniformly increased TNFR2 expression, although CD28SA induced upregulation was delayed compared to aCD3/CD28 beads (Figure 3C). Thus, activated Tregs produced TNFa and LTa and also have increased expression of TNFR2, suggesting that TNFa and LTa may function as autocrine or paracrine factors for activated Tregs.
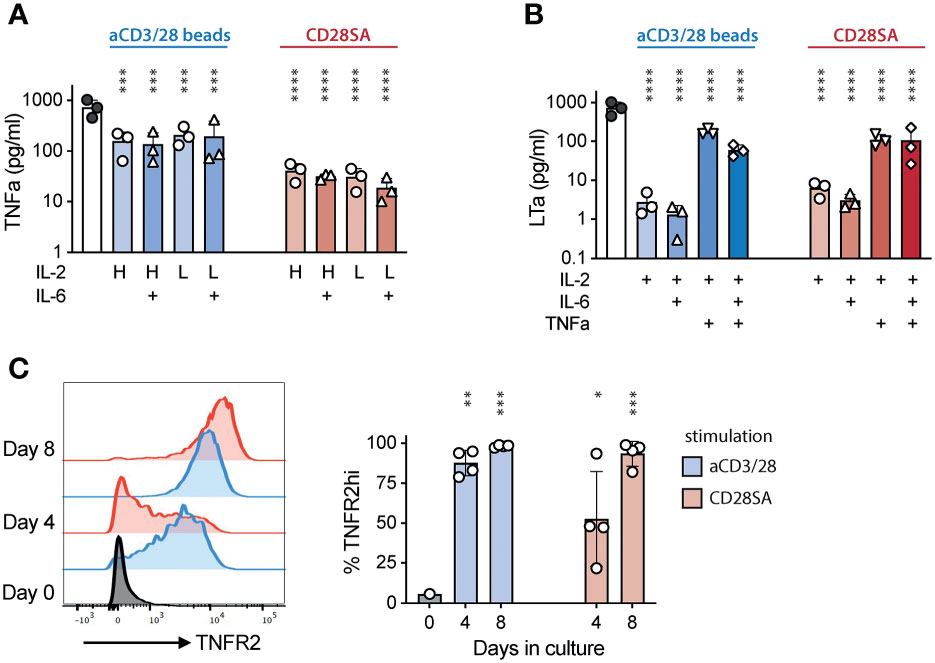
Figure 3 Activated Tregs produced TNFα and LTa and increased TNFR2 expression. FACS purified human Tregs were stimulated with either aCD3/28 beads or CD28SA and cultured in the presence of 300 IU/mL (H) or 15 IU/mL (L) rhIL-2 with or without IL-6 as indicated. (A) TNFα and (B) LTa concentration in the Day 7 culture supernatant of various T cell cultures. (C) Flow cytometric analysis of TNFR2 expression in Tregs stimulated with either aCD3/28 beads or CD28SA on day 0, 4 and 8 after stimulation. Representative histograms (left) and summary of percentage of TNFR2+ Tregs from 3 independent experiments (right) are shown. Statistical significance was assessed using one-way ANOVA and Dunnett’s multiple comparisons posttest using Treg treated with IL-2 alone (A, B) or Day 0 (C) as reference. p values are marked as ns, not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
To investigate a potential role of TNFR2 in human Treg activation, we evaluated the impact of etanercept, a soluble TNFR2-Fc fusion protein, in ex-vivo Treg proliferation. Addition of etanercept significantly reduced Treg proliferation induced by either aCD3/CD28 beads or CD28SA (Figure 4A). The inhibitory effect of etanercept was most pronounced during the later stage of Treg culture, suggesting that TNFR2 signaling did not affect initial Treg proliferation likely due to delayed induction of TNFR2 or TNFa and LTa. Furthermore, etanercept significantly decreased TNFR2 expression and the expanded Tregs showed a trend of moderate reduction in CD25, FOXP3, and HELIOS expression (Supplemental Figure 6).
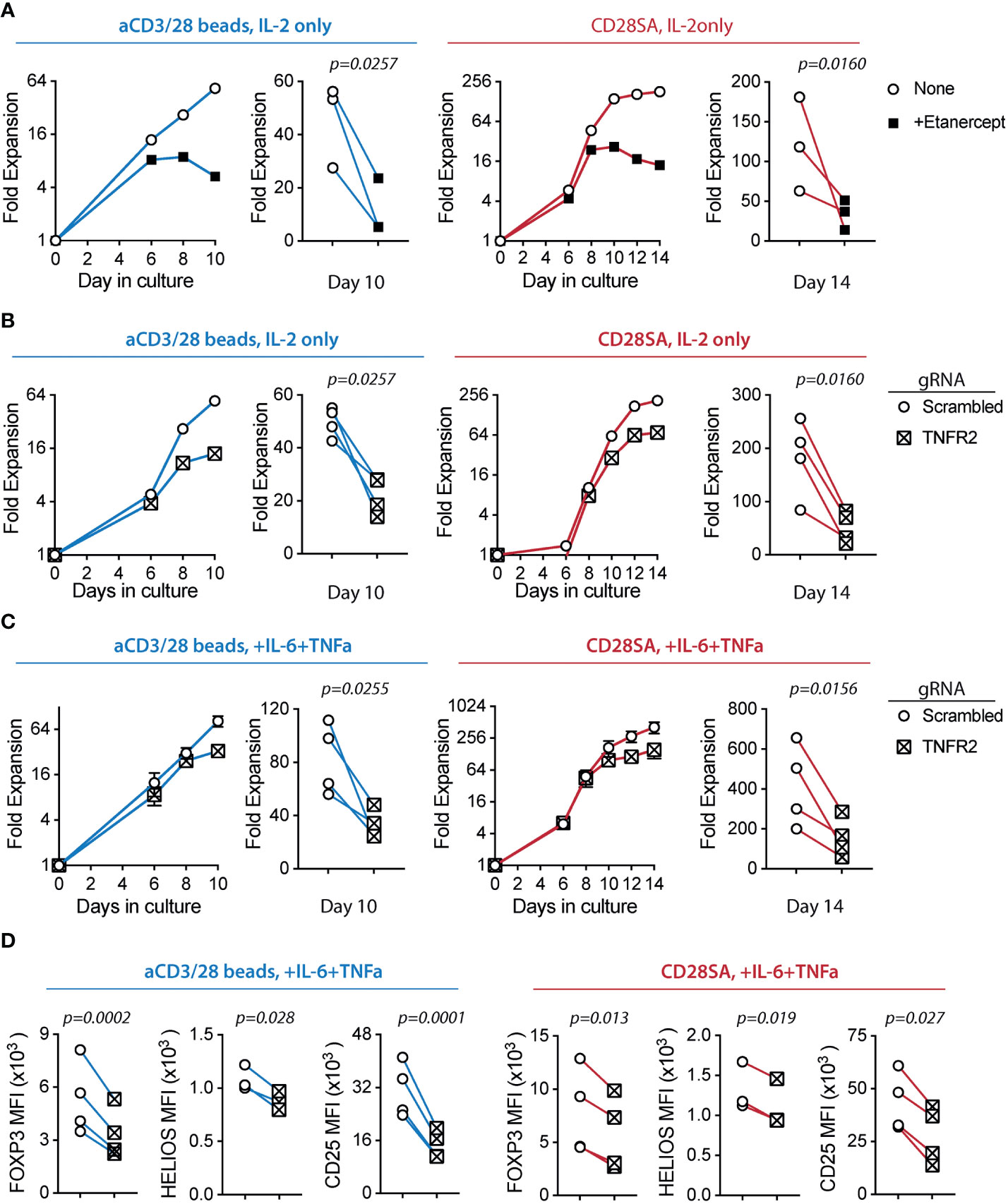
Figure 4 Ex-vivo proliferation of human Tregs is dependent on TNFR2 expression. FACS purified human Tregs were stimulated with either aCD3/28 beads or CD28SA and cultured in the presence of 300 IU/ml rhIL-2 with or without TNFα and IL-6 for 10 and 14 days, as indicated. (A) Representative aCD3/28 bead stimulated Treg expansion kinetics and summary of final fold expansion on day 10 of Treg expanded in the presence or absence of etanercept (left). Similarly, CD28SA stimulated Tregs were expanded for 14 days (right). Results of 3 independent experiments using 3 unrelated donors are shown. (B) Tregs were gene edited to delete TNFR2 gene using CRISPR-Cas9 and then stimulated with either aCD3/28 beads (left) or CD28SA (right). Results shown are from 4 independent experiments using 4 unrelated donors. (C) Same as panel (B), except the Tregs were cultured in the presence of IL-6 and TNFα. Results shown are from 4 independent donors in 4 independent experiments. (D) Tregs were expanded as shown in panel (C) and flow cytometric analysis of FOXP3, HELIOS and CD25 expression was performed on day 8 after stimulation. Results show summary of MFI of FOXP3, HELIOS and CD25 from 4 independent donors in 4 independent experiments. Paired t-test was used to determine statistical significance. p values are stated.
To directly examine the role of TNFR2 in human Treg proliferation, we deleted TNFR2 gene in purified Tregs using CRISPR/Cas9 technology. On day 4 after activation, only 27.3% of aCD3/CD28 bead stimulated cells electroporated with gRNA targeting the TNFR2 gene were TNFR2+ when compared to 87.75% of cells that were electroporated with scrambled gRNA (Supplemental Figure 7A). Similarly, 14.5% of CD28SA stimulated cells were TNFR2+ when compared to 52.6% of cells that were electroporated with scrambled gRNA (Supplemental Figure 7B), indicating successful TNFR2 deletion in the majority of Tregs. We observed that TNFR2KO Tregs have significantly reduced ex-vivo expansion with either aCD3/CD28 bead or CD28SA stimulation (Figure 4B), demonstrating a positive role of TNFR2 signaling in ex-vivo proliferation of human Tregs. It is worth noting that, on day 8 post activation, 44.2% of aCD3/CD28 bead-stimulated and 44.6% of CD28SA-stimulated cells were TNFR2+, increased from those detected on day 4 after stimulation. This suggested that the few TNFR2-sufficient Tregs after CRISPR editing had a proliferative advantage over their TNFR2KO counterparts (Supplemental Figures 7A, B). Similar to our observation using etanercept, TNFR2KO Tregs showed a trend of decreased expression of CD25, FOXP3, and HELIOS when compared to TNFR2+ Tregs (Supplemental Figures 7C, D). Together, these results demonstrated a role of TNFR2 in promoting human Treg ex-vivo proliferation and a potential role in preserving Treg lineage by sustaining FOXP3, HELIOS, and CD25 expression.
Finally, we exposed TNFR2KO Tregs to IL-6 and TNFa to assess the requirement for TNFR2 in Treg proliferative boost by these cytokines shown in Figure 1. We observed that TNFR2KO Tregs had a diminished proliferative response to IL-6 and TNFα when compared to control Tregs (Figure 4C). Moreover, TNFR2KO Tregs had significantly decreased expression of key Treg lineage markers CD25, FOXP3, and HELIOS (Figure 4D). Together, these results suggest that TNFR2 signaling enhanced human Treg proliferation and safeguarded their identity during exposure to proinflammatory cytokines.
Beadless Protocol for Ex-Vivo Expansion of Human Tregs
Ex-vivo expanded human Tregs are currently being evaluated in clinical trials in transplantation and autoimmune diseases (58). Current Treg manufacturing processes rely on multiple rounds of stimulation with aCD3/28 beads (59). Our results of highly efficient expansion of stable human Tregs using one cycle of CD28SA stimulation in the presence of IL-2, IL-6, and TNFa prompted us to consider this protocol as an alternative approach to expand Tregs for clinical use. We thus compared rates of Treg expansion induced with 1 or 2 rounds of aCD3/28 bead stimulation versus those achieved with single round of CD28SA stimulation with or without the addition of IL-6 and TNFa. Tregs stimulated with aCD3/28 beads entered cell expansion more rapidly than that induced by CD28SA, but the cells rested by day 9 and required restimulation to proliferate again (Figure 5A). In contrast, Tregs stimulated with CD28SA continue to proliferate and began to rest by 14 days after stimulation. Addition of IL-6, TNFa, or both did not lead more rapid entry into cell division, but more persistent proliferation resulting in more total cell yields at the end of the two-week expansion (Figure 5A). Given the concern of the reported negative impact of IL-6 on Tregs, we determined if the persistent Treg proliferation can be achieved using less IL-6. Altering IL-6 concentrations from 15 ng/ml to 150 ng/ml in the context of the beadless protocol resulted in similar Treg expansion yields (Figure 5B).
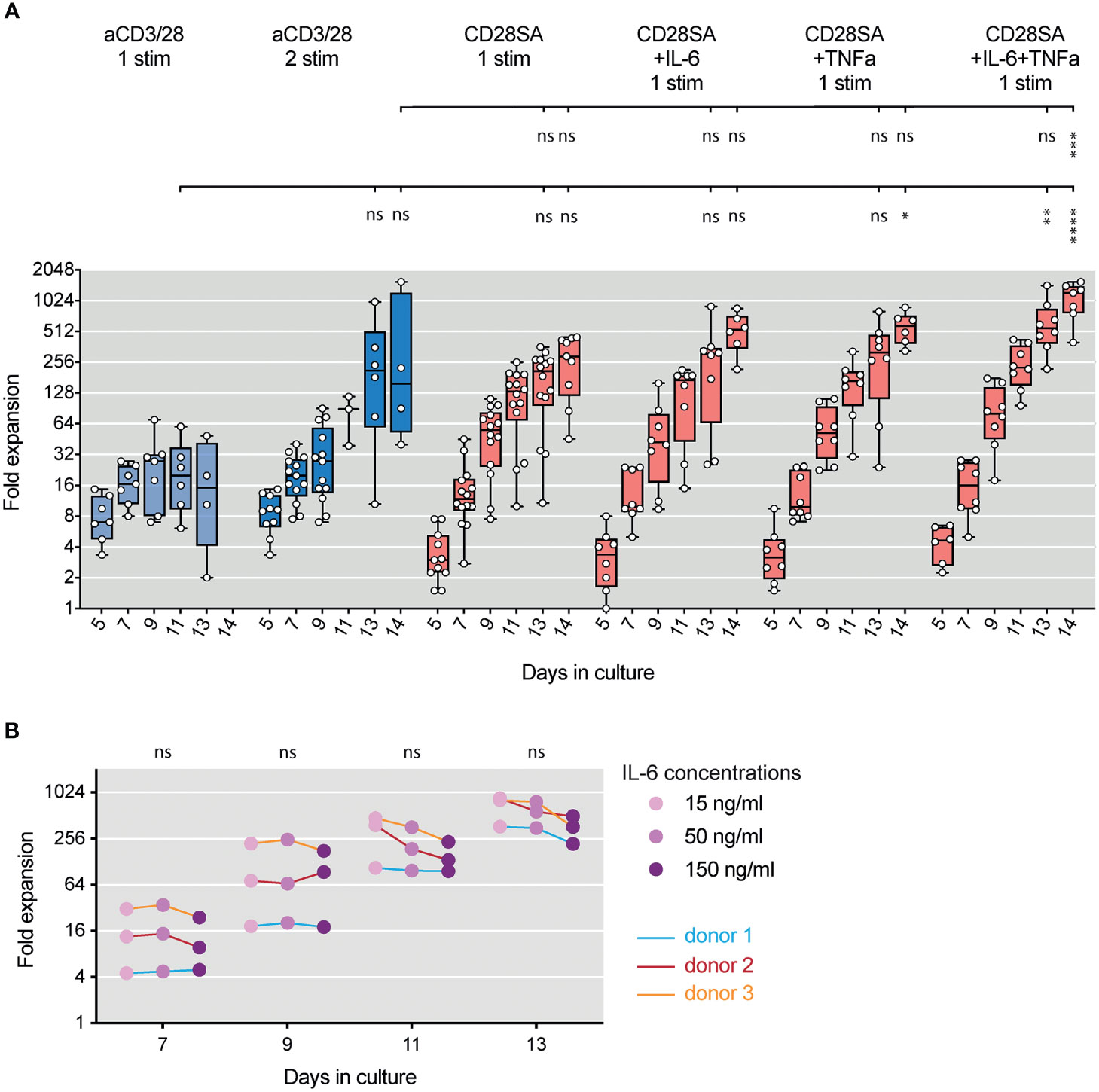
Figure 5 Beadless ex-vivo expansion of human Tregs. (A) FACS purified human Tregs were stimulated with either aCD3/28 beads or CD28SA and cultured in the presence of 300 IU/ml rhIL-2 with or without 150 ng/ml IL-6 and/or 50 ng/ml TNFα for up to 14 days. Expansion kinetics of Tregs from 7 to 14 unrelated donors in independent experiments are shown. Statistical significance was assessed using one-way ANOVA and Dunnett’s multiple comparisons posttest using aCD3/28 1 stim Day 13 and aCD3/28 2 stim Day 14 as reference. (B) FACS purified human Tregs were stimulated with CD28SA and cultured in the presence of 300 IU/mL rhIL-2, 50 ng/ml TNFα and varying concentrations of IL-6 (15 ng/ml, 50 ng/ml, and 150 ng/ml) as indicated. Expansion kinetics over 14 days of Tregs of from 3 unrelated donors in 3 independent experiments are shown. Data connected by the same line are from the same donor. Statistical significance was assessed using one-way ANOVA with Geisser-Greenhouse’s correction and Dunnett’s multiple comparisons posttest using 150 ng/ml IL-6 as reference. p values are marked as ns, not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
The pattern of persistent proliferation after CD28SA stimulation in the presence of TNFa and IL-6 when compared with aCD3/28 bead stimulation suggested that Tregs cultured in these two protocols were in distinct metabolic states. We thus performed metabolomic profiling of aCD3/28 bead-stimulated versus CD28SA+IL-6+TNFa beadless-stimulated Tregs by using capillary electrophoresis mass spectrometry. Intracellular concentrations of 116 metabolites involved in glycolysis, pentose phosphate pathway, TCA cycle, lipid metabolism, urea cycle, and polyamine, creatine, purine, glutathione, nicotinamide, choline, and amino acid metabolisms were captured (Supplemental Table 1). We selected day 7 for the comparison because the Tregs in both protocols were briskly proliferating and had comparable fold expansion at that time, but about to diverge in the rate of proliferation.
Activated Tregs with beads or the beadless protocol had significantly higher concentrations of all 20 amino acids than freshly purified Tregs (Figure 6A). All the amino acids, except aspartate, were present at equal or higher concentration in Tregs expanded with the beadless protocol when compared to those expanded with aCD3/28 beads. It was also notable that cysteine was only detected in Tregs stimulated with the beadless protocol and not detected in fresh or bead-stimulated Tregs. Both aspartate and cysteine are non-essential amino acids, and their altered levels may indicate altered metabolism of these amino acids in Tregs under different culture conditions.
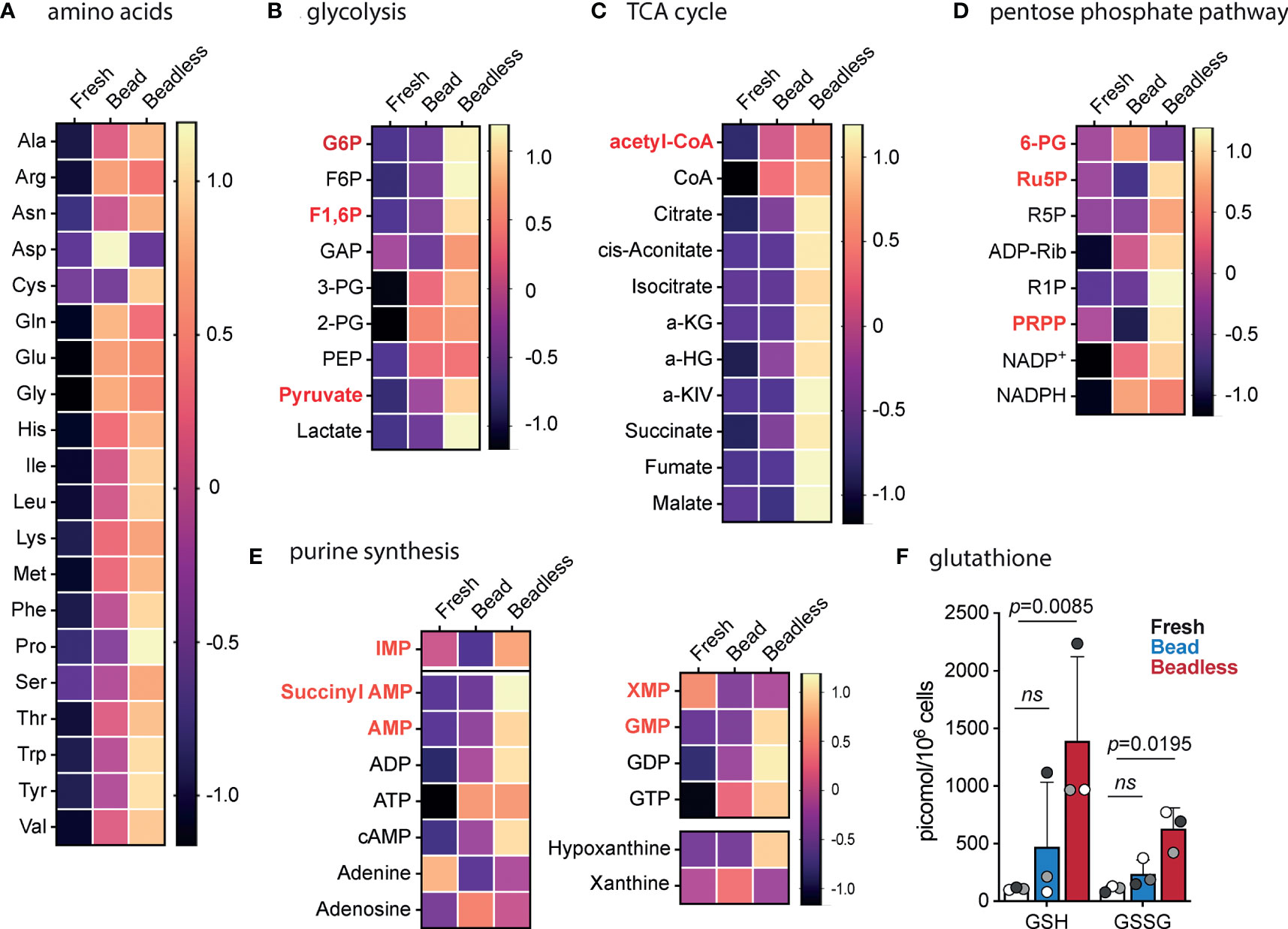
Figure 6 Metabolomic profile of Tregs before and after bead or beadless activation. Tregs were purified from 3 unrelated healthy donors. Tregs from each donor were divided into three parts: fresh Tregs without in vitro stimulation, Tregs expanded with aCD3/28 beads and IL-2 for 7 days (Bead), and Tregs expanded with CD28SA, IL-6, TNFα, and IL-2 for 7 days (Beadless). Intracellular metabolites were extracted and subjected to capillary electrophoresis mass spectrometry to profile 116 metabolites. The amount of each metabolite was normalized to the cell number and expressed as pmol/106 cells. The data for each metabolite were then normalized to the mean of all the samples and log2 transformed so that differences among the three experimental conditions can be compared across different metabolites. Non-detected metabolites were given a value of 2-52. The transformed data for amino acids (A), glycolysis (B), TCA cycle (C), pentose phosphate pathway (D), and purine synthesis (E) are summarized as heatmaps. Products of rate-limiting steps or key metabolites are highlighted with bold red text. (F) Intracellular concentration of reduced (GSH) and oxidized (GSSG) glutathione are shown. Circles represent individual data and data from the same Tregs donor are represented by the same fill color. Statistical significance was determined using ratio paired t test. p values are as marked. ns, not significant, p > 0.05.
Glycolysis was activated by both protocols with a trend of higher concentrations of glycolytic intermediates in the beadless-stimulated Tregs (Figure 6B). Notably, Tregs in the beadless protocol contained 10 times higher concentration of lactic acid compared to bead stimulated Tregs, indicating highly active glycolytic activities. Concurrently, beadless-stimulated Tregs had higher oxidative phosphorylation (OXPHOS) activities indicated by the higher concentrations of TCA intermediates when compared to bead stimulated Tregs, which were nearly depleted of TCA intermediates of cis-aconitate, isocitrate, alpha-ketoglutarate, fumarate and malate (Figure 6C). Thus, Tregs in the beadless protocol were in a high energy state with concurrent activation of both glycolysis and OXPHOS whereas bead stimulated Tregs mostly relied on glycolysis for energy production.
The pentose phosphate pathway also utilizes glycolysis intermediates for purine and pyrimidine synthesis. Intermediates in the pentose phosphate pathway were elevated in the beadless protocol expanded Tregs when compared to fresh and bead stimulated Tregs (Figure 6D). This correlated with higher concentrations of intermediates of purine biosynthesis (Figure 6E). The pentose phosphate pathway also converts NADP+ to NADPH, thus an important regulator of the redox state of the cell. Moreover, NADPH also supports de novo fatty acid synthesis. NADP+ and NADPH were present at comparable elevated concentrations in bead versus beadless stimulated Tregs (Figure 6D) whereas the NADPH to NADP+ ratios were both comparably reduced from the fresh Treg baseline (Supplemental Table 1), suggesting higher antioxidant demand and/or higher fatty acid synthesis in activated Tregs. NADPH protects mitochondria against oxidative stress by transferring its reductive power to oxidized glutathione disulfide (GSSG) to generate reduced glutathione (GSH). GSH and GSSG concentrations were consistently low in fresh Tregs, variably and moderately increased in bead stimulated Tregs, and significantly increased in Tregs activated with the beadless protocol (Figure 6F). Beadless protocol Tregs had higher levels of total glutathione, suggestive of greater buffering ability for reactive oxygen species (ROS). The increased energy production and de novo nucleotide synthesis combined with higher antioxidant potential may underscore the better metabolic fitness and more persistent proliferation of Tregs stimulated with the beadless protocol when compared to those stimulated with aCD3/28 beads.
TNFα and IL-6-Exposed Tregs Maintained Their Function In Vitro and In Vivo
Lastly, we compared the function of Tregs expanded using aCD3/28 beads versus the beadless protocol. For the in vitro suppression assay, we used aCD3/28 beads expanded CD4+CD25-CD127hi Tconv as responders and mixed in titrated numbers of Tregs expanded with either aCD3/28 beads or CD28SA, with or without IL-6 and TNFa. Tregs expanded with various protocols maintained high suppressive capacity, suggesting that ex-vivo exposure to IL-6 and TNFa did not negatively affect Treg suppressive function (Figure 7A).
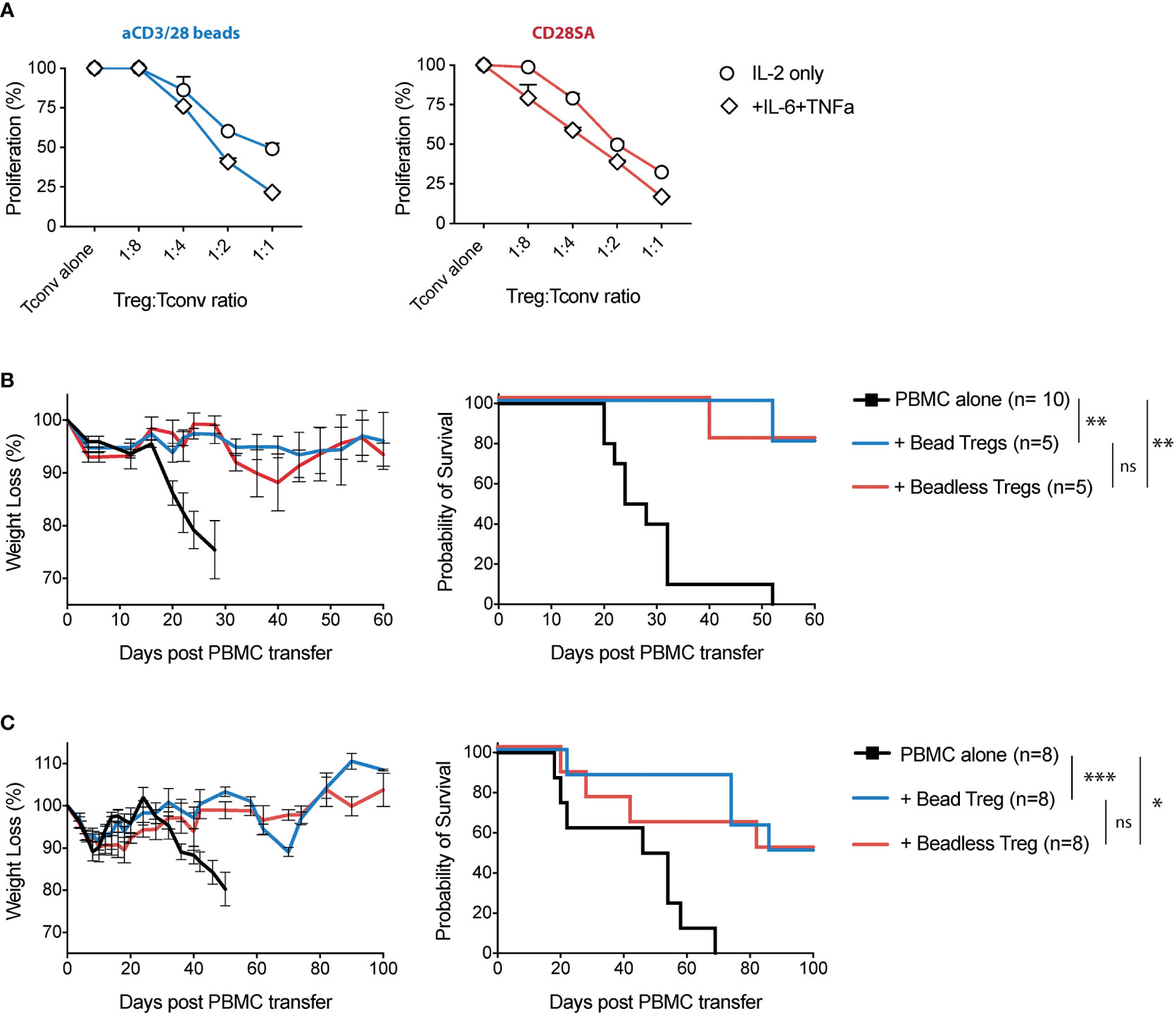
Figure 7 Tregs expanded with the beadless protocol maintained their in-vitro and in-vivo suppressive function. (A) FACS purified human Tregs were stimulated with either aCD3/28 beads or CD28SA and cultured in the presence of 300 IU/ml rhIL-2 with or without TNFα and/or IL-6 for 9 days, as indicated. In addition, FACS purified human Tconvs were stimulated with aCD3/28 beads and cultured in parallel to autologous Tregs. At day 7 of culture, expanded Tregs were harvested and then co-cultured with 5x 104 ex-vivo expanded Tconv at Treg/Tconv ratios of 1:1, 1:2, 1:4 and 1:8 in the presence of aCD3/CD28 beads (1:10 bead to Tconv ratio). Proliferation was measured using 3[H]-thymidine incorporation in triplicate wells on day 4 of co-culture. Results shown are representative of 2 independent experiments using 2 unrelated donors. (B) At day 9 of culture, either standard protocol (aCD3/28 beads and IL-2) or beadless protocol (CD28SA, IL-2, IL-6 and TNFα) expanded Tregs were adoptively co-transferred into sub lethally irradiated NSG mice along with autologous PBMC. Results shown are from 1 experiment with a total of at least 5 mice per group. (C) Same as panel (B), except the Tregs were injected 4 days after the PBMC infusion. Weight loss and survival were monitored every other day. Results shown are summary of 2 independent experiments using 2 unrelated donors with a total of 8 mice per group. Kaplan-Meier survival graphs are shown, and a log-rank comparison of the groups was used to calculate p values. p values are marked as ns, not significant, p > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001.
To measure the in-vivo suppressive function of Tregs, we used models of xenogeneic GVHD in the NSG mice. Intravenous injection of 5 x 106 PBMC’s into sub lethally irradiated NSG mice resulted in the development of GVHD that was lethal in 20 to 30 days (Figure 7B). Co-transfer of 5 x 106 Tregs expanded with standard aCD3/28 bead protocol or the beadless protocol significantly attenuated weight loss and improved survival from GVHD in most of the mice (Figure 7B). To compare the function of the Tregs expanded with the standard versus beadless protocols in a more challenging condition, we infused Treg 4 days after PBMC injection. It has been previously reported that co-infusion of Treg with GVHD-inducing PBMCs effectively prevents Tconv proliferation and improves survival, while delayed Treg infusion requires higher doses of Tregs to reverse disease that has already been initiated (60). Indeed, an increased percentage of humanized NSG mice treated with late Treg infusion eventually succumbed to GVHD compared to the GVHD prevention model with PBMC and Treg co-infusions. However, no differences were identified between Tregs expanded with the standard protocol versus the beadless protocol in their ability to prevent weight loss and reduce mortality (Figure 7C).
To assess the phenotypic stability of Tregs after infusion, we injected 2x106 autologous PBMCs into sub lethally irradiated NSG mice 9 days prior to adoptively transferring CFSE-labeled Tregs expanded with either the standard bead-based or the beadless protocol. We collected blood and lung tissues 5 days after Treg infusion to analyze the Treg expression of FOXP3. Beadless expanded Tregs maintained their FOXP3 expression when compared to the CD8+ T cells among the PBMCs which were largely FOXP3- (Supplemental Figure 8A). We observed slightly lower levels of FOXP3 expression in beadless protocol expanded Tregs when compared to bead-expanded Tregs (Supplemental Figures 8B, C) and this was associated with significantly more proliferation of the beadless Tregs (Supplemental Figure 8D). In summary, these results demonstrated that ex-vivo expanded Tregs exposed to IL-6 and TNFa maintained their function in vitro and in vivo in humanized mouse models of GVHD.
Discussion
In this study, we challenged purified human peripheral blood Tregs ex-vivo with IL-6 and TNFa to determine their direct effect on Tregs. Our data collectively demonstrate that human Tregs respond to these cytokines positively by increasing their proliferation while preserving their lineage identity.
A Treg-enhancing role of TNFa and TNFR2 has been reported in various mouse models and with human Tregs ex vivo. In this study, we have identified 3 effects of TNFa on human Tregs. First, by using TNFR blockers and CRISPR/Cas 9 mediated TNFR2 gene deletion, we further show that activated human Tregs engage TNFa-TNFR2 in an autocrine or paracrine fashion for persistent proliferation. Exogenous provision of TNFa and IL-6 further boosted this effect, suggesting the endogenous production of TNFa and LTa does not maximally activate this pathway. Second, TNFa helps to preserve Treg lineage identity by sustaining high expression of FOXP3 and HELIOS. This finding is consistent with a previous report that TNFR2 signaling in human Tregs induces EZH2 to preserve Treg lineage identity (61). Lastly, TNFa increases Treg secretion of CCL3, CCL4, and CCL5. It has been shown previously that mouse Tregs produce CCL3 and CCL4 to attract activated T cells to suppress them (62). Together, these findings point to multiple mechanisms of TNFa-TNFR2 in promoting Treg numerical dominance lineage stability, and function at sites of inflammation.
In contrast to TNFa, IL-6 has mostly been thought to subvert Treg functions. Our finding of a lack of destabilizing effect of IL-6 on human Tregs despite the use of supraphysiological concentration of IL-6 and reduced IL-2 is thus unexpected. Further review of the literature shows a unanimous conclusion that IL-6 antagonizes in vitro induction of Tregs from Tconvs by driving the cells to differentiation into Th17 effectors (15). Moreover, there is consensus that IL-6 enhances Teff activation to resist suppression by Tregs (13, 16). The body of literature on a direct role of IL-6 on lineage committed Tregs, especially on human Tregs, is limited and more controversial. While it is difficult to pinpoint the cause of divergent findings between this study and previous studies that reported Treg destabilizing effects of IL-6, we suspect that differences in experimental setup may have contributed. An uncertainty when working with human Tregs is the identity of the cells at the start of the experiments depending on the markers used for Treg isolation. In this study, we isolated human Tregs from peripheral blood using FACS with markers of CD4+CD25+CD127lo/-. The addition of CD127 allows isolation of human Tregs with higher purity and higher yield (63, 64). Isolation of human Tregs based on the expression of CD4 and CD25 using magnetic activated cell sorting often results in less pure Tregs and outgrowth of Tconvs that may be mistaken for destabilized Tregs. Our findings with Tregs from many unrelated donors over the span of 4 years show that acute high dose exposure to IL-6 does not cause destabilization of human Tregs.
In this study, we noted significantly more robust proliferation of Tregs stimulated with CD28SA when compared to aCD3/28 bead stimulation. Furthermore, IL-6 and TNFa synergistically enhanced the proliferation stimulated by CD28SA. It has been reported that IL-6 promotes TNFR2 expression by Tregs in malignant ascites (65), which may explain the synergy we have observed. It is worth noting that aCD3/28 bead stimulated faster entry into cell cycle but earlier rest akin to logarithmic growth; whereas CD28SA stimulated Tregs showed slower start but more robust and persistent cell cycling as seen in exponential growth, especially when IL-6 and TNFα are added. This suggests that Tregs are capable of exponential growth and the pattern of expansion stimulated by aCD3/28 beads may be a result of negative feedback that limits proliferation. The biochemical basis of the distinct proliferation patterns remains to be elucidated. In Tconvs, TCR stimulation in the absence of CD28 signaling activate nuclear factor of activated T-cells that is not balanced with concurrent activation of NF-κB and activator protein 1, which leads induction of E3 ubiquitin ligases Cbl-b and cell cycle arrest (66). CD28-mediated costimulation complement TCR signaling by activating NF-κB and AP1 while releasing the cells from the inhibitory effect of Cbl-b (66). We speculate that stimulation of Tregs with strong TCR agonist such as bead-bound aCD3/28 induces imbalanced activation of transcription factors that triggers negative feedback to limit Treg proliferation, resulting in short bursts of proliferation followed by stagnation (67). On the other hand, CD28SA stimulated T cell activation depends on the expression of TCR but does not ostensibly induce activation of signaling intermediates immediately downstream of the TCR such as CD3 chains and ZAP70 (68, 69). Thus, CD28SA delivers a strong costimulatory signal in the context of a weak TCR signal (70–72), which may avoid the engagement of negative feedback loops that leads to earlier cell cycle arrest. The addition of IL-6 and TNFa may further engage complementary signaling pathways to boost Treg proliferation. Ongoing work in our laboratory is actively testing these predications.
Another non-mutually exclusive explanation for the distinct Treg proliferation patterns is the divergent metabolic programs induced by aCD3/28 bead versus CD28SA plus IL-6 and TNFa (beadless protocol). Tregs undergo metabolic reprogramming after activation to accommodate the anabolic and energy demands for cell proliferation (73). Previous studies have reported either preferential fatty acid oxidation (FAO)-fueled oxidative phosphorylation over glycolysis by Tregs (74–76) or reliance on both glycolysis and FAO to support Treg proliferation and suppressive function (77, 78). In our metabolomic analysis, both bead and beadless stimulations led to increased intracellular amino acid concentrations, energy production, and nucleotide synthesis when compared to freshly isolated Tregs, consistent with their anabolic state. We noted three major distinctions in the metabolic state of Tregs stimulated with aCD3/28 beads versus the beadless protocol. First, while both protocols increased glycolytic flux, the beadless protocol Tregs had a slightly higher concentration of pyruvate, but 10 times more intracellular lactate. The conversion of pyruvate to lactate is coupled with oxidation of NADH to replenish the NAD+ pool in the cytosol, which is essential in preventing stagnation of glycolysis from NAD+ shortage (79). This suggests that Tregs in the beadless protocol had more active glycolysis. Second, the beadless protocol stimulated Tregs had higher concentrations of all TCA intermediates measured, whereas intermediates downstream of citrate were almost completely depleted in aCD3/28 bead stimulated Tregs. This suggests that the aCD3/28 bead stimulated Tregs have very low level of OXPHOS and relied mostly on glycolysis for energy production. Third, concentrations of total and reduced glutathione were consistently higher in the beadless protocol stimulated Tregs, suggestive of greater buffering capability for ROS. Intense OXPHOS can increase the generation of ROS leading to cell death. Therefore, reduced glutathione and other antioxidants are essential for maintaining cells in a high energetic state by scavenging ROS (80). GSH deficiency in Tregs results in an imbalanced intracellular redox state and impaired suppressive function (81). Overall, our data indicate that the beadless protocol stimulated Tregs have better metabolic fitness that may contribute to their persistent proliferation.
It is unclear at this point of our investigations which component of the beadless protocol contributed to the more favorable metabolic program in Tregs. CD28 costimulation is required for inducing glycolysis in activated T cells during proliferation (82, 83), and can also prime mitochondria for increased OXPHOS demand (84). CD28SA has been shown to drive T effector memory cells into an adaptable metabolic state that can flexibly maximize glycolysis and OXPHOS potentials depending on glucose and oxygen availability (85), but the impact of CD28SA on metabolic programming of Tregs remains uncertain. Another component of the beadless protocol, TNFa, may act through TNFR2, to induce a glycolytic switch in Tregs coupled with shunting of intermediates into the TCA cycle, thereby promoting anabolic biosynthetic processes (86). Lastly, it has been reported that STAT3 can localize in the inner membrane of mitochondria and enhance the efficiency of electron transport chain and reduce the generation of ROS (87). Further research is needed to dissect the role of the individual components of the beadless protocol on Treg metabolism, which is currently ongoing in our laboratory.
A practical implication of our data is a new protocol for ex-vivo human Treg expansion. One of the challenges facing Treg therapy is the ability to reliably manufacture enough Tregs without the need for repeated stimulations that negatively affect Treg stability (88–90). The beadless protocol promotes highly efficient Treg expansion with only one cycle of CD28SA stimulation. Another advantage of the beadless protocol is the use of all soluble reagents, thus harvesting of Treg products at the end of the expansion is simplified. Thus, the beadless protocol offers several improvements in the Treg manufacturing process.
Taken together, our findings show that human Tregs positively respond to TNFa and IL-6 by increased proliferation while safeguarding their lineage stability. With proper stimulation and the right cytokine milieu, human Tregs can grow exponentially, which may be a result of balanced transcription and metabolic programing. We speculate that increased proliferation in response to inflammatory cytokines allows Tregs to scale to inflammation to restore immune homeostasis. These properties of Tregs may be harnessed to improve the manufacturing of therapeutic Tregs for autoimmune diseases and transplantation.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics Statement
The animal study was reviewed and approved by UCSF IACUC protocol No. AN183959-02.
Author Contributions
Designed the project: QT, NS. Supervised the project: QT, FV. Designed experiments: NS, QT. Performed experiments: NS, YP, LF, VN, ER. Analyzed data: NS, YP, QT. Provided reagents and advice: YDM, FV. Wrote the manuscript: NS, YP, QT. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from NIAID (Clinical Trials in Organ Transplant, 0255-B011-4609 and 0255-B012-4609, which were ancillary to a NIAID grant U01 AI113362), The Leona M. and Harry B. Helmsley Charitable Trust (2018PG-T1D042), NIDDK (UC4 DK116264). This works used the UCSF Flow Cytometry Core, which is supported in part by the Diabetes Research Center Grant from NIDDK (P30 DK063720 and P30 DK063720). NS was supported by a fellowship grant from the American Society of Nephrology. YDM was supported by the Swiss National Science Foundation (Mobility Grant no. P300PB_174500) and a fellowship grant from the University Hospital of Geneva. LF was the Jeffrey G. Klein Family Diabetes Fellow.
Conflict of Interest
QT is a co-founder and scientific advisor of Sonoma Biotherapeutics. QT, NS, and FV are co-inventors of a patent on manufacturing Tregs based on results from this work.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank Juan Du for the technical support and lab management. We would also like to thank the Marson lab for technical support. Finally, we would like to thank Jeffrey A. Bluestone for critical review of this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.783282/full#supplementary-material
References
1. Tang Q, Bluestone JA. The Foxp3+ Regulatory T Cell: A Jack of All Trades, Master of Regulation. Nat Immunol (2008) 9(3):239–44. doi: 10.1038/ni1572
2. Vignali DA. Mechanisms of T(reg) Suppression: Still a Long Way to Go. Front Immunol (2012) 3:191. doi: 10.3389/fimmu.2012.00191
3. Smigiel KS, Srivastava S, Stolley JM, Campbell DJ. Regulatory T-Cell Homeostasis: Steady-State Maintenance and Modulation During Inflammation. Immunol Rev (2014) 259(1):40–59. doi: 10.1111/imr.12170
4. Spence A, Purtha W, Tam J, Dong S, Kim Y, Ju CH, et al. Revealing the Specificity of Regulatory T Cells in Murine Autoimmune Diabetes. Proc Natl Acad Sci USA (2018) 115(20):5265–70. doi: 10.1073/pnas.1715590115
5. Luan D, Dadhania DM, Ding R, Muthukumar T, Lubetzky M, Lee JR, et al. FOXP3 mRNA Profile Prognostic of Acute T Cell-Mediated Rejection and Human Kidney Allograft Survival. Transplantation (2021) 105(8):1825–39. doi: 10.1097/TP.0000000000003478
6. Shang B, Liu Y, Jiang SJ, Liu Y. Prognostic Value of Tumor-Infiltrating FoxP3+ Regulatory T Cells in Cancers: A Systematic Review and Meta-Analysis. Sci Rep (2015) 5:15179. doi: 10.1038/srep15179
7. Sakaguchi S, Vignali DA, Rudensky AY, Niec RE, Waldmann H. The Plasticity and Stability of Regulatory T Cells. Nat Rev Immunol (2013) 13(6):461–7. doi: 10.1038/nri3464
8. Papadakis KA, Targan SR. Tumor Necrosis Factor: Biology and Therapeutic Inhibitors. Gastroenterology (2000) 119(4):1148–57. doi: 10.1053/gast.2000.18160
9. Grivennikov SI, Tumanov AV, Liepinsh DJ, Kruglov AA, Marakusha BI, Shakhov AN, et al. Distinct and Nonredundant In Vivo Functions of TNF Produced by T Cells and Macrophages/Neutrophils: Protective and Deleterious Effects. Immunity (2005) 22(1):93–104. doi: 10.1016/j.immuni.2004.11.016
10. Apostolaki M, Armaka M, Victoratos P, Kollias G. Cellular Mechanisms of TNF Function in Models of Inflammation and Autoimmunity. Curr Dir Autoimmun (2010) 11:1–26. doi: 10.1159/000289195
11. Tanaka T, Narazaki M, Kishimoto T. IL-6 in Inflammation, Immunity, and Disease. Cold Spring Harb Perspect Biol (2014) 6(10):a016295. doi: 10.1101/cshperspect.a016295
12. Yokota S, Geppert TD, Lipsky PE. Enhancement of Antigen- and Mitogen-Induced Human T Lymphocyte Proliferation by Tumor Necrosis Factor-Alpha. J Immunol (1988) 140(2):531–6.
13. Li B, Jones LL, Geiger TL. IL-6 Promotes T Cell Proliferation and Expansion Under Inflammatory Conditions in Association With Low-Level RORgammat Expression. J Immunol (2018) 201(10):2934–46. doi: 10.4049/jimmunol.1800016
14. Chen X, Oppenheim JJ. Contrasting Effects of TNF and Anti-TNF on the Activation of Effector T Cells and Regulatory T Cells in Autoimmunity. FEBS Lett (2011) 585(23):3611–8. doi: 10.1016/j.febslet.2011.04.025
15. Chen X, Howard OM, Oppenheim JJ. Pertussis Toxin by Inducing IL-6 Promotes the Generation of IL-17-Producing CD4 Cells. J Immunol (2007) 178(10):6123–9. doi: 10.4049/jimmunol.178.10.6123
16. Pasare C, Medzhitov R. Toll Pathway-Dependent Blockade of CD4+CD25+ T Cell-Mediated Suppression by Dendritic Cells. Science (2003) 299(5609):1033–6. doi: 10.1126/science.1078231
17. Mercadante ER, Lorenz UM. Breaking Free of Control: How Conventional T Cells Overcome Regulatory T Cell Suppression. Front Immunol (2016) 7:193. doi: 10.3389/fimmu.2016.00193
18. Wohlfert EA, Clark RB. ‘Vive La Resistance!’–the PI3K-Akt Pathway can Determine Target Sensitivity to Regulatory T Cell Suppression. Trends Immunol (2007) 28(4):154–60. doi: 10.1016/j.it.2007.02.003
19. Zhang Q, Cui F, Fang L, Hong J, Zheng B, Zhang JZ. TNF-Alpha Impairs Differentiation and Function of TGF-Beta-Induced Treg Cells in Autoimmune Diseases Through Akt and Smad3 Signaling Pathway. J Mol Cell Biol (2013) 5(2):85–98. doi: 10.1093/jmcb/mjs063
20. Housley WJ, Adams CO, Nichols FC, Puddington L, Lingenheld EG, Zhu L, et al. Natural But Not Inducible Regulatory T Cells Require TNF-Alpha Signaling for In Vivo Function. J Immunol (2011) 186(12):6779–87. doi: 10.4049/jimmunol.1003868
21. Ni X, Kou W, Gu J, Wei P, Wu X, Peng H, et al. TRAF6 Directs FOXP3 Localization and Facilitates Regulatory T-Cell Function Through K63-Linked Ubiquitination. EMBO J (2019) 38(9):e99766. doi: 10.15252/embj.201899766
22. Grinberg-Bleyer Y, Saadoun D, Baeyens A, Billiard F, Goldstein JD, Gregoire S, et al. Pathogenic T Cells Have a Paradoxical Protective Effect in Murine Autoimmune Diabetes by Boosting Tregs. J Clin Invest (2010) 120(12):4558–68. doi: 10.1172/JCI42945
23. Hamano R, Huang J, Yoshimura T, Oppenheim JJ, Chen X. TNF Optimally Activatives Regulatory T Cells by Inducing TNF Receptor Superfamily Members TNFR2, 4-1BB and OX40. Eur J Immunol (2011) 41(7):2010–20. doi: 10.1002/eji.201041205
24. Chen X, Baumel M, Mannel DN, Howard OM, Oppenheim JJ. Interaction of TNF With TNF Receptor Type 2 Promotes Expansion and Function of Mouse CD4+CD25+ T Regulatory Cells. J Immunol (2007) 179(1):154–61. doi: 10.4049/jimmunol.179.1.154
25. Chen X, Wu X, Zhou Q, Howard OM, Netea MG, Oppenheim JJ. TNFR2 Is Critical for the Stabilization of the CD4+Foxp3+ Regulatory T. Cell Phenotype in the Inflammatory Environment. J Immunol (2013) 190(3):1076–84. doi: 10.4049/jimmunol.1202659
26. Ronin E, Pouchy C, Khosravi M, Hilaire M, Gregoire S, Casrouge A, et al. Tissue-Restricted Control of Established Central Nervous System Autoimmunity by TNF Receptor 2-Expressing Treg Cells. Proc Natl Acad Sci USA (2021) 118(13):e2014043118. doi: 10.1073/pnas.2014043118
27. Pierini A, Strober W, Moffett C, Baker J, Nishikii H, Alvarez M, et al. TNF-Alpha Priming Enhances CD4+FoxP3+ Regulatory T-Cell Suppressive Function in Murine GVHD Prevention and Treatment. Blood (2016) 128(6):866–71. doi: 10.1182/blood-2016-04-711275
28. Leclerc M, Naserian S, Pilon C, Thiolat A, Martin GH, Pouchy C, et al. Control of GVHD by Regulatory T Cells Depends on TNF Produced by T Cells and TNFR2 Expressed by Regulatory T Cells. Blood (2016) 128(12):1651–9. doi: 10.1182/blood-2016-02-700849
29. Nie H, Zheng Y, Li R, Guo TB, He D, Fang L, et al. Phosphorylation of FOXP3 Controls Regulatory T Cell Function and Is Inhibited by TNF-Alpha in Rheumatoid Arthritis. Nat Med (2013) 19(3):322–8. doi: 10.1038/nm.3085
30. Valencia X, Stephens G, Goldbach-Mansky R, Wilson M, Shevach EM, Lipsky PE, et al. TNF Downmodulates the Function of Human CD4+CD25hi T-Regulatory Cells. Blood (2006) 108(1):253–61. doi: 10.1182/blood-2005-11-4567
31. Stoop JN, Woltman AM, Biesta PJ, Kusters JG, Kuipers JEJ, Janssen HL, et al. Tumor Necrosis Factor Alpha Inhibits the Suppressive Effect of Regulatory T Cells on the Hepatitis B Virus-Specific Immune Response. Hepatology (2007) 46(3):699–705. doi: 10.1002/hep.21761
32. Nagar M, Jacob-Hirsch J, Vernitsky H, Berkun Y, Ben-Horin S, Amariglio N, et al. TNF Activates a NF-kappaB-Regulated Cellular Program in Human CD45RA- Regulatory T Cells That Modulates Their Suppressive Function. J Immunol (2010) 184(7):3570–81. doi: 10.4049/jimmunol.0902070
33. Zaragoza B, Chen X, Oppenheim JJ, Baeyens A, Gregoire S, Chader D, et al. Suppressive Activity of Human Regulatory T Cells is Maintained in the Presence of TNF. Nat Med (2016) 22(1):16–7. doi: 10.1038/nm.4019
34. Chen X, Subleski JJ, Hamano R, Howard OM, Wiltrout RH, Oppenheim JJ. Co-Expression of TNFR2 and CD25 Identifies More of the Functional CD4+FOXP3+ Regulatory T Cells in Human Peripheral Blood. Eur J Immunol (2010) 40(4):1099–106. doi: 10.1002/eji.200940022
35. Wang J, Ferreira R, Lu W, Farrow S, Downes K, Jermutus L, et al. TNFR2 Ligation in Human T Regulatory Cells Enhances IL2-Induced Cell Proliferation Through the Non-Canonical NF-kappaB Pathway. Sci Rep (2018) 8(1):12079. doi: 10.1038/s41598-018-30621-4
36. Okubo Y, Mera T, Wang L, Faustman DL. Homogeneous Expansion of Human T-Regulatory Cells via Tumor Necrosis Factor Receptor 2. Sci Rep (2013) 3:3153. doi: 10.1038/srep03153
37. He X, Landman S, Bauland SC, van den Dolder J, Koenen HJ, Joosten I, et al. A TNFR2-Agonist Facilitates High Purity Expansion of Human Low Purity Treg Cells. PloS One (2016) 11(5):e0156311. doi: 10.1371/journal.pone.0156311
38. Brown G, Wang E, Leon A, Huynh M, Wehner M, Matro R, et al. Tumor Necrosis Factor-Alpha Inhibitor-Induced Psoriasis: Systematic Review of Clinical Features, Histopathological Findings, and Management Experience. J Am Acad Dermatol (2017) 76(2):334–41. doi: 10.1016/j.jaad.2016.08.012
39. Kemanetzoglou E, Andreadou E. CNS Demyelination With TNF-Alpha Blockers. Curr Neurol Neurosci Rep (2017) 17(4):36. doi: 10.1007/s11910-017-0742-1
40. Korn T, Mitsdoerffer M, Croxford AL, Awasthi A, Dardalhon VA, Galileos G, et al. IL-6 Controls Th17 Immunity In Vivo by Inhibiting the Conversion of Conventional T Cells Into Foxp3+ Regulatory T Cells. Proc Natl Acad Sci USA (2008) 105(47):18460–5. doi: 10.1073/pnas.0809850105
41. Xu L, Kitani A, Fuss I, Strober W. Cutting Edge: Regulatory T Cells Induce CD4+CD25-Foxp3- T Cells or Are Self-Induced to Become Th17 Cells in the Absence of Exogenous TGF-Beta. J Immunol (2007) 178(11):6725–9. doi: 10.4049/jimmunol.178.11.6725
42. O’Connor RA, Floess S, Huehn J, Jones SA, Anderton SM. Foxp3(+) Treg Cells in the Inflamed CNS are Insensitive to IL-6-Driven IL-17 Production. Eur J Immunol (2012) 42(5):1174–9. doi: 10.1002/eji.201142216
43. van Loosdregt J, Fleskens V, Fu J, Brenkman AB, Bekker CP, Pals CE, et al. Stabilization of the Transcription Factor Foxp3 by the Deubiquitinase USP7 Increases Treg-Cell-Suppressive Capacity. Immunity (2013) 39(2):259–71. doi: 10.1016/j.immuni.2013.05.018
44. Fujimoto M, Nakano M, Terabe F, Kawahata H, Ohkawara T, Han Y, et al. The Influence of Excessive IL-6 Production In Vivo on the Development and Function of Foxp3+ Regulatory T Cells. J Immunol (2011) 186(1):32–40. doi: 10.4049/jimmunol.0903314
45. Hagenstein J, Melderis S, Nosko A, Warkotsch MT, Richter JV, Ramcke T, et al. A Novel Role for IL-6 Receptor Classic Signaling: Induction of RORgammat(+)Foxp3(+) Tregs With Enhanced Suppressive Capacity. J Am Soc Nephrol (2019) 30(8):1439–53. doi: 10.1681/ASN.2019020118
46. Samanta A, Li B, Song X, Bembas K, Zhang G, Katsumata M, et al. TGF-Beta and IL-6 Signals Modulate Chromatin Binding and Promoter Occupancy by Acetylated FOXP3. Proc Natl Acad Sci USA (2008) 105(37):14023–7. doi: 10.1073/pnas.0806726105
47. Lu L, Lan Q, Li Z, Zhou X, Gu J, Li Q, et al. Critical Role of All-Trans Retinoic Acid in Stabilizing Human Natural Regulatory T Cells Under Inflammatory Conditions. Proc Natl Acad Sci USA (2014) 111(33):E3432–40. doi: 10.1073/pnas.1408780111
48. Bin Dhuban K, Bartolucci S, d'Hennezel E, Piccirillo CA. Signaling Through Gp130 Compromises Suppressive Function in Human FOXP3(+) Regulatory T Cells. Front Immunol (2019) 10:1532. doi: 10.3389/fimmu.2019.01532
49. Ferreira RC, Rainbow DB, Rubio Garcia A, Pekalski ML, Porter L, Oliveira JJ, et al. Human IL-6R(Hi)TIGIT(-) CD4(+)CD127(low)CD25(+) T Cells Display Potent In Vitro Suppressive Capacity and a Distinct Th17 Profile. Clin Immunol (2017) 179:25–39. doi: 10.1016/j.clim.2017.03.002
50. Roth TL, Puig-Saus C, Yu R, Shifrut E, Carnevale J, Li PJ, et al. Reprogramming Human T Cell Function and Specificity With Non-Viral Genome Targeting. Nature (2018) 559(7714):405–9. doi: 10.1038/s41586-018-0326-5
51. Putnam AL, Brusko TM, Lee MR, Liu W, Szot GL, Ghosh T, et al. Expansion of Human Regulatory T-Cells From Patients With Type 1 Diabetes. Diabetes (2009) 58(3):652–62. doi: 10.2337/db08-1168
52. He X, Smeets RL, van Rijssen E, Boots AM, Joosten I, Koenen HJ, et al. Single CD28 Stimulation Induces Stable and Polyclonal Expansion of Human Regulatory T Cells. Sci Rep (2017) 7:43003. doi: 10.1038/srep43003
53. Gerriets VA, Kishton RJ, Johnson MO, Cohen S, Siska PJ, Nichols AG, et al. Foxp3 and Toll-Like Receptor Signaling Balance Treg Cell Anabolic Metabolism for Suppression. Nat Immunol (2016) 17(12):1459–66. doi: 10.1038/ni.3577
54. Kim HJ, Barnitz RA, Kreslavsky T, Brown FD, Moffett H, Lemieux ME, et al. Stable Inhibitory Activity of Regulatory T Cells Requires the Transcription Factor Helios. Science (2015) 350(6258):334–9. doi: 10.1126/science.aad0616
55. Yang XO, Nurieva R, Martinez GJ, Kang HS, Chung Y, Pappu BP, et al. Molecular Antagonism and Plasticity of Regulatory and Inflammatory T Cell Programs. Immunity (2008) 29(1):44–56. doi: 10.1016/j.immuni.2008.05.007
56. Li L, Kim J, Boussiotis VA. IL-1beta-Mediated Signals Preferentially Drive Conversion of Regulatory T Cells But Not Conventional T Cells Into IL-17-Producing Cells. J Immunol (2010) 185(7):4148–53. doi: 10.4049/jimmunol.1001536
57. Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF Receptor Superfamilies: Integrating Mammalian Biology. Cell (2001) 104(4):487–501. doi: 10.1016/S0092-8674(01)00237-9
58. Esensten JH, Muller YD, Bluestone JA, Tang Q. Regulatory T-Cell Therapy for Autoimmune and Autoinflammatory Diseases: The Next Frontier. J Allergy Clin Immunol (2018) 142(6):1710–8. doi: 10.1016/j.jaci.2018.10.015
59. Tang Q, Bluestone JA. Regulatory T-Cell Therapy in Transplantation: Moving to the Clinic. Cold Spring Harb Perspect Med (2013) 3(11):a015552. doi: 10.1101/cshperspect.a015552
60. Nguyen VH, Zeiser R, Dasilva DL, Chang DS, Beilhack A, Contag CH, et al. In Vivo Dynamics of Regulatory T-Cell Trafficking and Survival Predict Effective Strategies to Control Graft-Versus-Host Disease Following Allogeneic Transplantation. Blood (2007) 109(6):2649–56. doi: 10.1182/blood-2006-08-044529
61. Urbano PCM, Koenen H, Joosten I, He X. An Autocrine TNFalpha-Tumor Necrosis Factor Receptor 2 Loop Promotes Epigenetic Effects Inducing Human Treg Stability In Vitro. Front Immunol (2018) 9:573. doi: 10.3389/fimmu.2018.00573
62. Patterson SJ, Pesenacker AM, Wang AY, Gillies J, Mojibian M, Morishita K, et al. T Regulatory Cell Chemokine Production Mediates Pathogenic T Cell Attraction and Suppression. J Clin Invest (2016) 126(3):1039–51. doi: 10.1172/JCI83987
63. Liu W, Putnam AL, Xu-Yu Z, Szot GL, Lee MR, Zhu S, et al. CD127 Expression Inversely Correlates With FoxP3 and Suppressive Function of Human CD4+ T Reg Cells. J Exp Med (2006) 203(7):1701–11. doi: 10.1084/jem.20060772
64. Seddiki N, Santner-Nanan B, Martinson J, Zaunders J, Sasson S, Landay A, et al. Expression of Interleukin (IL)-2 and IL-7 Receptors Discriminates Between Human Regulatory and Activated T Cells. J Exp Med (2006) 203(7):1693–700. doi: 10.1084/jem.20060468
65. Kampan NC, Madondo MT, McNally OM, Stephens AN, Quinn MA, Plebanski M. Interleukin 6 Present in Inflammatory Ascites From Advanced Epithelial Ovarian Cancer Patients Promotes Tumor Necrosis Factor Receptor 2-Expressing Regulatory T Cells. Front Immunol (2017) 8:1482. doi: 10.3389/fimmu.2017.01482
66. Paolino M, Penninger JM. Cbl-B in T-Cell Activation. Semin Immunopathol (2010) 32(2):137–48. doi: 10.1007/s00281-010-0197-9
67. Hippen KL, Merkel SC, Schirm DK, Sieben CM, Sumstad D, Kadidlo DM, et al. Massive Ex Vivo Expansion of Human Natural Regulatory T Cells (T(regs)) With Minimal Loss of In Vivo Functional Activity. Sci Transl Med (2011) 3(83):83ra41. doi: 10.1126/scitranslmed.3001809
68. Tacke M, Hanke G, Hanke T, Hunig T. CD28-Mediated Induction of Proliferation in Resting T Cells In Vitro and In Vivo Without Engagement of the T Cell Receptor: Evidence for Functionally Distinct Forms of CD28. Eur J Immunol (1997) 27(1):239–47. doi: 10.1002/eji.1830270136
69. Hunig T, Dennehy K. CD28 Superagonists: Mode of Action and Therapeutic Potential. Immunol Lett (2005) 100(1):21–8. doi: 10.1016/j.imlet.2005.06.012
70. Bischof A, Hara T, Lin CH, Beyers AD, Hunig T. Autonomous Induction of Proliferation, JNK and NF-alphaB Activation in Primary Resting T Cells by Mobilized CD28. Eur J Immunol (2000) 30(3):876–82. doi: 10.1002/1521-4141(200003)30:3<876::AID-IMMU876>3.0.CO;2-M
71. Waibler Z, Sender LY, Merten C, Hartig R, Kliche S, Gunzer M, et al. Signaling Signatures and Functional Properties of Anti-Human CD28 Superagonistic Antibodies. PloS One (2008) 3(3):e1708. doi: 10.1371/journal.pone.0001708
72. Dennehy KM, Kerstan A, Bischof A, Park JH, Na SY, Hunig T. Mitogenic Signals Through CD28 Activate the Protein Kinase Ctheta-NF-kappaB Pathway in Primary Peripheral T Cells. Int Immunol (2003) 15(5):655–63. doi: 10.1093/intimm/dxg063
73. Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling Immunity: Insights Into Metabolism and Lymphocyte Function. Science (2013) 342(6155):1242454. doi: 10.1126/science.1242454
74. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting Edge: Distinct Glycolytic and Lipid Oxidative Metabolic Programs are Essential for Effector and Regulatory CD4+ T Cell Subsets. J Immunol (2011) 186(6):3299–303. doi: 10.4049/jimmunol.1003613
75. Angelin A, Gil-de-Gomez L, Dahiya S, Jiao J, Guo L, Levine MH, et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab (2017) 25(6):1282–93.e7. doi: 10.1016/j.cmet.2016.12.018
76. Howie D, Cobbold SP, Adams E, Ten Bokum A, Necula AS, Zhang W, et al. Foxp3 Drives Oxidative Phosphorylation and Protection From Lipotoxicity. JCI Insight (2017) 2(3):e89160. doi: 10.1172/jci.insight.89160
77. Procaccini C, Carbone F, Di Silvestre D, Brambilla F, De Rosa V, Galgani M, et al. The Proteomic Landscape of Human Ex Vivo Regulatory and Conventional T Cells Reveals Specific Metabolic Requirements. Immunity (2016) 44(2):406–21. doi: 10.1016/j.immuni.2016.01.028
78. Cluxton D, Petrasca A, Moran B, Fletcher JM. Differential Regulation of Human Treg and Th17 Cells by Fatty Acid Synthesis and Glycolysis. Front Immunol (2019) 10:115. doi: 10.3389/fimmu.2019.00115
79. Liberti MV, Locasale JW. The Warburg Effect: How Does It Benefit Cancer Cells? Trends Biochem Sci (2016) 41(3):211–8. doi: 10.1016/j.tibs.2015.12.001
80. Sena LA, Chandel NS. Physiological Roles of Mitochondrial Reactive Oxygen Species. Mol Cell (2012) 48(2):158–67. doi: 10.1016/j.molcel.2012.09.025
81. Kurniawan H, Franchina DG, Guerra L, Bonetti L, Baguet LS, Grusdat M, et al. Glutathione Restricts Serine Metabolism to Preserve Regulatory T Cell Function. Cell Metab (2020) 31(5):920–36.e7. doi: 10.1016/j.cmet.2020.03.004
82. Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, et al. The CD28 Signaling Pathway Regulates Glucose Metabolism. Immunity (2002) 16(6):769–77. doi: 10.1016/S1074-7613(02)00323-0
83. Jacobs SR, Herman CE, Maciver NJ, Wofford JA, Wieman HL, Hammen JJ, et al. Glucose Uptake Is Limiting in T Cell Activation and Requires CD28-Mediated Akt-Dependent and Independent Pathways. J Immunol (2008) 180(7):4476–86. doi: 10.4049/jimmunol.180.7.4476
84. Klein Geltink RI, O'Sullivan D, Corrado M, Bremser A, Buck MD, Buescher JM, et al. Mitochondrial Priming by CD28. Cell (2017) 171(2):385–97.e11. doi: 10.1016/j.cell.2017.08.018
85. Thaventhiran T, Wong W, Alghanem AF, Alhumeed N, Aljasir MA, Ramsey S, et al. CD28 Superagonistic Activation of T Cells Induces a Tumor Cell-Like Metabolic Program. Monoclon Antib Immunodiagn Immunother (2019) 38(2):60–9. doi: 10.1089/mab.2018.0042
86. de Kivit S, Mensink M, Hoekstra AT, Berlin I, Derks RJE, Both D, et al. Stable Human Regulatory T Cells Switch to Glycolysis Following TNF Receptor 2 Costimulation. Nat Metab (2020) 2(10):1046–61. doi: 10.1038/s42255-020-00271-w
87. Rincon M, Pereira FV. A New Perspective: Mitochondrial Stat3 as a Regulator for Lymphocyte Function. Int J Mol Sci (2018) 19(6):1656. doi: 10.3390/ijms19061656
88. Hoffmann P, Boeld TJ, Eder R, Huehn J, Floess S, Wieczorek G, et al. Loss of FOXP3 Expression in Natural Human CD4+CD25+ Regulatory T Cells Upon Repetitive In Vitro Stimulation. Eur J Immunol (2009) 39(4):1088–97. doi: 10.1002/eji.200838904
89. Tang Q, Vincenti F. Transplant Trials With Tregs: Perils and Promises. J Clin Invest (2017) 127(7):2505–12. doi: 10.1172/JCI90598
Keywords: Tregs, inflammation, IL-6, TNFα, CD28 signaling, metabolomics, GVHD
Citation: Skartsis N, Peng Y, Ferreira LMR, Nguyen V, Ronin E, Muller YD, Vincenti F and Tang Q (2021) IL-6 and TNFα Drive Extensive Proliferation of Human Tregs Without Compromising Their Lineage Stability or Function. Front. Immunol. 12:783282. doi: 10.3389/fimmu.2021.783282
Received: 25 September 2021; Accepted: 29 November 2021;
Published: 23 December 2021.
Edited by:
Magdalena Plebanski, RMIT University, AustraliaReviewed by:
Adam Waickman, Upstate Medical University, United StatesDipayan Rudra, ShanghaiTech University, China
Xuehui He, Radboud University Nijmegen Medical Centre, Netherlands
Copyright © 2021 Skartsis, Peng, Ferreira, Nguyen, Ronin, Muller, Vincenti and Tang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qizhi Tang, UWl6aGkuVGFuZ0B1Y3NmLmVkdQ==
†ORCID: Emilie Ronin, orcid.org/0000-0003-0295-7933