 Łukasz Przykaza
Łukasz Przykaza
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 15 November 2021
Sec. Inflammation
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.782569
Despite the enormous progress in the understanding of the course of the ischemic stroke over the last few decades, a therapy that effectively protects neurovascular units (NVUs) and significantly improves neurological functions in stroke patients has still not been achieved. The reasons for this state are unclear, but it is obvious that the cerebral ischemia and reperfusion cascade is a highly complex phenomenon, which includes the intense neuroinflammatory processes, and comorbid stroke risk factors strongly worsen stroke outcomes and likely make a substantial contribution to the pathophysiology of the ischemia/reperfusion, enhancing difficulties in searching of successful treatment. Common concomitant stroke risk factors (arterial hypertension, diabetes mellitus and hyperlipidemia) strongly drive inflammatory processes during cerebral ischemia/reperfusion; because these factors are often present for a long time before a stroke, causing low-grade background inflammation in the brain, and already initially disrupting the proper functions of NVUs. Broad consideration of this situation in basic research may prove to be crucial for the success of future clinical trials of neuroprotection, vasculoprotection and immunomodulation in stroke. This review focuses on the mechanism by which coexisting common risk factors for stroke intertwine in cerebral ischemic/reperfusion cascade and the dysfunction and disintegration of NVUs through inflammatory processes, principally activation of pattern recognition receptors, alterations in the expression of adhesion molecules and the subsequent pathophysiological consequences.
Ischemic stroke is a serious clinical and socioeconomic problem, especially among the aging populations of industrialized countries. Despite the enormous progress in the understanding of the course of the ischemic cascade over the last few decades, a neuroprotective therapy that effectively protects neurovascular units (NVUs) and significantly improves neurological functions in stroke patients has still not been achieved. The reasons for this state are still far from fully clear. Admittedly, the ischemia and reperfusion cascade is a highly complex process that consists of many elements of a diverse biological nature, and comorbid stroke risk factors strongly worsen stroke outcomes and likely make a substantial contribution to the pathophysiology of the ischemic/reperfusion cascade, enhancing difficulties in treatment (1, 2). The most common risk factors found in stroke patients are arterial hypertension, diabetes mellitus and hyperlipidemia. All of these factors cause low-grade inflammation and microcirculatory disturbances in many organs, and also – in the brain. Therefore, in this paper, a strong emphasis was placed on the perspective that the main link unifying the ischemia/reperfusion cascade and these risk factors are inflammatory processes that already initially disrupt the proper functions of NVUs, before a stroke onset, and participate in the course of a stroke pathomechanism.
For many years, evidence has increased for an ambiguous, destructive, protective and repairing role of some inflammatory cells in the course of a stroke, although many early studies in this field unequivocally showed that the infiltration of the ischemic brain by inflammatory cells enlarged the extent of post-ischemic damage through subsequent edema or intracranial hemorrhage. This ambiguity does not undermine the possibility of therapeutic targeting of the ischemic inflammatory process; on the contrary, in the future, appropriate immunomodulation may halt damaging processes and/or enhance the protective or repair functions of some of their effectors. However, despite ambiguous details, the negative role of inflammatory processes in focal and global cerebral ischemia/reperfusion, intracranial hemorrhages, or mechanical brain injuries is brought to the fore. Clinically, in the course of ischemic stroke, a higher level of inflammation is a significant prognostic factor for worse treatment outcomes (3). In contrast, animal experimental studies on neuroprotection still do not consider the coexisting risk factors for stroke in humans as an experimental standard. This is probably an important cause of the failure of attempts to introduce neuroprotection into clinical practice. Accompanying risk factors already presensitize neurovascular units to the ischemic/reperfusion cascade. When the cascade occurs, it encounters NVUs with their already initially impaired functionality and consistently worsens cerebral perfusion at the level of the microcirculation in the ischemic penumbra. Considering this situation in basic research may prove to be crucial for the success of future clinical trials of neuroprotection and immunomodulation.
This review of the literature focuses on the mechanism by which coexisting common risk factors for stroke intertwine in ischemic/reperfusion and the dysfunction and disintegration of NVUs through inflammatory processes, principally activation of pattern recognition receptors, alterations in the expression of adhesion molecules and the subsequent pathophysiological consequences.
For the review, the PubMed electronic database was searched using the following keywords: adhesion molecules, inflammation, ischemic stroke, pattern recognition receptors, penumbra, reperfusion, stroke risk factors. Experimental, clinical and review publications from 1971 to 2021 were cited, with a predominance of those published after the year 2000.
Arterial hypertension, diabetes mellitus, and hyperlipidemia are well-known modifiable risk factors for the development of ischemic stroke in humans. Currently, an epidemic increase in the incidence of these diseases is observed in the Western world. In many countries, approximately 50% of the population over 60 years of age has been diagnosed with hypertension, and this prevalence is actually increasing (4). Elevated diastolic or systolic blood pressure, or both parameters, is a major risk factor for stroke development (5, 6). Similarly, diabetes mellitus shows a worrying trend of increasing morbidity, despite the presence of many pro-health social campaigns. Type 2 diabetes, until recently considered mainly as a disease of elderly age, is now responsible for approximately 4-5% of premature deaths (7). Approximately 50% of people with diabetes signs delayed end-organ/system damages and as many as 40% of subjects have vascular complications, both result from the silent progression of the disease before its diagnosis (8). Hyperlipidemia (elevated blood lipid/lipoprotein levels) also severely affects various age groups in wealthy Western societies. As an example in the United States of America, ~33.5% of adults aged ≥20 years have elevated low-density lipoprotein cholesterol levels above 160 mg/dL (9). In adults, hyperlipidemia is an important risk factor for the development of cardiovascular diseases and stroke (10, 11).
Molecular mechanisms involved in inflammation, e.g., in general, an activation of the pro-inflammatory genes and an increase in the expression of adhesion molecules, in addition to their well-documented role in the ischemic cascade in the brain, can now also be reasonably considered an important connecting bridge between the above risk factors for ischemic stroke and the unfavorable prognosis in the event of its occurrence. Hypertension and diabetic hyperglycemia can worsen the prognosis of stroke through endothelial dysfunction and increased leukocyte adhesion to blood vessels and other pathologies affecting the functions of NVUs. Moreover, chronically elevated blood cholesterol levels also result in endothelial disturbances and increased adhesion of leukocytes and platelets during ischemia/reperfusion (12–14). These observations constitute important clinical indications for research in the field of ischemic stroke neuroprotection, vasculoprotection and immunomodulation, and for widespread preclinical studies with animal models with modeled risk factors for stroke in humans.
Based on the current knowledge, to understand the mechanistic links how the common risk factors for stroke may worsen its course through amplification of the disintegration and dysfunction of NVUs, we first need to review the mechanisms of ischemic/reperfusion cascade and ischemic inflammation in light of this regard.
During an ischemic stroke, a decrease in the patency of the plugged artery lumen leads to a local decrease in the volume of blood flow in the brain area supplied by it. This decline is not uniform across the entire ischemic region. On the basis of the degree of blood flow reduction, it is possible to distinguish the infarct core (< 20% of basal blood flow) and the penumbra, where the collateral circulation maintains the blood flow at the level of ~40% of the basal blood flow (15). During ischemic oxygen-glucose deprivation in the cells located in the infarct core, there is a strong decrease in the rate of oxidative phosphorylation, a decrease in intracellular ATP (adenosine 5′-triphosphate) concentration, and consequently a decrease in the activity of Na+/K+-ATPase and a disturbance in the ATP-dependent transport of K+ and Na+ ions. The ion homeostasis of the cells in the infarct core is lost. The outflow of K+ from cells and the inflow of Na+ into the cytosol causes cellular depolarization. High cytosolic concentrations of Na+ and Cl- result in the development of strong cytotoxic edema (16, 17). Depolarized neurons in the infarct core are not able to restore their resting potentials, and they die within a few minutes as a result of energetic collapse, a loss of ion homeostasis, cytotoxic edema, proteolysis, cytoskeleton disintegration, lipolysis and cell membrane fragmentation (18). The loss of energy/ion homeostasis disrupts the communication of neurons with glial and endothelial cells. Astrocytes exposed to glutamate toxicity, similar to neurons, suffer from cytotoxic edema, calcium overload and mitochondrial depolarization, followed by free radical damage (19, 20).
Currently, it is commonly accepted that damage to NVUs in the penumbra occurs as a consequence of a combination of numerous factors of a diverse nature – such as excitotoxicity, peri-infarct depolarizations (PIDs), free radical stress, apoptosis, inflammatory processes, cerebral blood vessel damage, and microcirculatory disturbances during ischemia and reperfusion. It seems, however, that glutamate excitotoxicity and peri-infarct depolarizations are the most primary (21, 22). Within 6–8 hours, the penumbra is recruited to the infarct core (23). According to the excitotoxicity hypothesis, a high concentration of glutamate diffuses from the infarct core to its edge and hyperactivates postsynaptic N-methyl-D-aspartate receptors (NMDA-Rs) in penumbra neurons (24, 25). In response to excessive, long-term stimulation of NMDA-Rs, there is an excessive intracellular influx of Na+ and Ca2+ ions. Highly elevated intracellular concentrations of Ca2+ initiate a number of pathways, leading to the death of neurons (26). At the same time, astrocytes internalize glutamate transporter-1 (GLT-1) – transporters most importantly involved in the reuptake of glutamate from the synaptic cleft, which leads to a high concentration of glutamate in the interstitial fluid. The downregulation of GLT-1 enhances and prolongs the excitation of postsynaptic neurons (27, 28). Moreover, a sharp increase in the activity of astrocytic cAMP/PKA (3′,5′-cyclic adenosine monophosphate/protein kinase A) signaling leads to the phosphorylation of aquaporin-4 (AQP4), which ultimately increases the permeability of the cell membrane to water and promotes cytotoxic edema of astrocytes and mechanical pressure on microvessels (29). These astrocytic metabolic disorders (with calcium overload) are the initiation of the astrogliosis. In turn, PIDs are initiated mainly by a high concentration of K+ ions diffusing in the extracellular space. These ions come from depolarized/dead neurons of the infarct core and reach high concentrations there as a result of astrocyte dysfunction. PIDs, due to insufficient blood supply and the decoupling of the relationship between the metabolism of neurons and the reactivity of microcirculation in the penumbra, lead to episodes of hypoxia and energetic depletion of neurons in this zone and a loss of their ion homeostasis, which may also cause anoxic depolarizations. The number of PIDs positively correlates with the size of ischemic damage (30). With regard to astrocytic upregulation AQP4, it was shown that this protein significantly contributed to the propagation of depolarization waves by increasing the extracellular concentration of K+ (31). Additionally, the downregulation of GLT-1 protein is involved in the propagation of depolarizing waves as well (32). PIDs also damage NVUs as a result of the activation of metalloproteinases (e.g., metalloproteinase-9, MMP-9) and the subsequent weakening of the blood–brain barrier (BBB). The severity of ischemic BBB damage and the degree of vasogenic edema are positively correlated with the number of PID episodes (33).
When a reopening of an arterial vessel occurs, reperfusion takes place, restoring blood flow through the ischemic area. Depending on the duration of ischemia and/or the patient’s health history, reperfusion may bring clinically beneficial as well as unfavorable effects (34). However, reperfusion carries risks, especially after a long episode of ischemia. Some experimental studies have shown that focal ischemia with subsequent reperfusion can cause more extensive brain damage than focal ischemia without reperfusion (35, 36). During reperfusion, disturbances in the regulation of blood flow in the penumbra and oligemic areas are revealed, and reactive oxygen species are produced by the NADPH oxidase (NOX) system, the xanthine oxidase (XO) system, and the mitochondrial enzymatic systems damaged by ischemia (37). The significant presence of the NOX and XO systems has been confirmed in blood vessels (endothelial cells). Hypoxic/reoxygenated endothelial cells show an increase in NOX expression/activity, reactive oxygen species (ROS) production, and adhesion molecule expression, causing vascular injuries. All of these responses can be prevented by treating cells with inhibitors of NOX-activating signaling pathways (38–40). Likewise, for XO, various cytokines released during reperfusion, including IL-1 (interleukin 1), IL-6 (interleukin 6), TNF-α (tumor necrosis factor alpha), and IFN-γ (interferon gamma), increase the expression/activity of the enzyme in various cell types, and inhibitors of XO prevents against vascular injuries (37, 41).
During the reperfusion phase, inflammatory processes evidently begin; activated peripheral leukocytes migrate to brain tissues, contributing to brain tissue damage, the aggravation of BBB disruption and vasogenic edema, as well as hemorrhagic transformation (HT); and blood cellular elements create intravascular conglomerates that impair microcirculation (42, 43).
The dead and dying cells of the brain ischemic area induce inflammatory processes by releasing into their surroundings molecules that activate microglial cells, endothelial cells of the brain blood vessels, astrocytes and, in subsequent stages, infiltrating leukocytes. These molecules are collectively known as DAMPs (danger-associated molecular patterns) and include, e.g., ATP, UTP (uridine 5’-triphosphate), nicotinamide adenine dinucleotide, peroxiredoxins, HMGB1 (high mobility group box 1) and HSP60 (heat shock protein 60) (44). Microglial cells, leukocytes, astrocytes and neurons, and endothelial cells present receptors in their cell membranes that can be stimulated by these molecules. The activation of these receptors triggers a series of processes leading to the expression of pro-inflammatory genes and changes the resting phenotype of microglia into the amoeboid M1. Microglia transform, proliferate, rearrange the cytoskeleton, and acquire the ability to migrate and phagocytose (45). ATP and UTP stimulate P2X7 (P2X purinoceptor 7) purinergic receptors. HMGB1 stimulates CD36 (cluster of differentiation 36), TLR4 (Toll-like receptor 4) and TLR2 (Toll-like receptor 2), RAGE (receptor for advanced glycation end products), and receptors for HSP60. Peroxiredoxins stimulate TLR2 and TLR4 receptors (46–49). As a result of the activation of the aforementioned receptors, the transcription factor NF-κB (nuclear factor kappa B) triggers the expression of genes encoding pro-inflammatory molecules, including IL-1β (interleukin-1β), IL-6, IL-8 (interleukin 8), IL-18 (interleukin 18), and TNF-α. Another activated transcription factor involved in the expression of pro-inflammatory genes is AP-1 (activator protein 1), an important activator of IL-1 and TNF-α expression and the promotion of the expression of some adhesion molecules (50–52). The path from TLR4 receptors to NF-κB and AP-1 activation is mainly through the MyD88 protein (myeloid differentiation primary response protein) (53, 54). Apart from cytokines, chemokines such as: MCP-1 (CCL2; monocyte chemoattractant protein-1), MIP-1a (CCL3; macrophage inflammatory-1 alpha), CXCL2 (MIP-2, macrophage inflammatory protein 2), RANTES (CCL5; regulated on activation, normal T-cell expressed and secreted), CINC-1 (cytokine-induced neutrophil chemoattractant 1), fractalkine (CX3CL1; C-X3-C motif chemokine ligand 1), and CXCL10 (IP-10; C-X-C motif chemokine ligand 10) are expressed in the ischemic area (55–57). The activation of microglial cells occurs in the first minutes of a stroke. Microglial cells can damage neurons, oligodendrocytes, astrocytes and blood vessels in the early phase, releasing, e.g., ROS. However, in later stages, they may play a protective or repair role (M2 population) due to the release of TGF-beta1 (transforming growth factor beta 1), GDNF (glial cell derived neurotrophic factor) and the anti-inflammatory IL-10 (interleukin 10) (58–60).
Pro-inflammatory processes make the ischemic environment conducive to the migration of immune cells from peripheral blood over time. The most abundant leukocytes to first make the passage from the blood to the brain parenchyma are neutrophils. Experimental studies have shown that neutrophils contribute to the enlargement of the area of damage after infiltration by releasing ROS and metalloproteinases, and by activating own iNOS (inducible nitric oxide synthase) in response to the presence of specific cytokines (61–63). Secondly, circulating monocytes attracted by specific chemokines, penetrate the brain damage through the weakness of BBB. Peripheral monocytes, considering their role in post-ischemic inflammatory processes, can be divided into pro-inflammatory, with the phenotype Ly-6Chigh/CCR2+, releasing IL-1β and TNF-α, and anti-inflammatory Ly-6Clow/CCR2-, releasing IL-10 (64, 65). Lymphocytes, another class of leukocytes playing a complex role in the pathogenesis of ischemic damage, appear in the ischemic area 24 hours after the start of reperfusion and reach a peak after seven days. There are several types of lymphocytes involved, of which, for example, CD8+ T cells play a negative role, and γδ T cells play a negative role in the late but not the early phase of stroke, substantially through the production of pro-inflammatory IL-17 (interleukin 17) and thus supporting the influx of neutrophils (66, 67).
CD8+ T lymphocytes and Natural Killer cells (NK) enhance the infiltration of the brain parenchyma by recognizing the IL-15 (interleukin 15) signal produced by activated astrocytes. The increase in IL-15 expression during ischemia significantly contributes to inflammatory processes and enlarges the area of post-ischemic brain damage (68). The activation of astrocytes is important and one of the first processes during ischemic inflammation. During this activation astrocytes undergo morphological changes, principally hyperplasia and hypertrophy, and one of the molecular markers of this process is increased expression of GFAP (glial fibrillary acidic protein) (69). Moreover, changes in AQP4 expression are observed, whose directivity depends on the timing of the ischemic cascade and on the ischemic region location. There is a strong increase in the expression of this protein at 1 hour of ischemia in the infarct core and at 1 and 48 hour of ischemia in the penumbra, which correlates with cytotoxic edema (70). In the core area of the striatum 24 hours after ischemia/reperfusion, the perivascular AQP-4 level was shown to be significantly decreased and had no recovery tendency. In the ischemic core of the cortex, however, the presence of AQP-4 is obviously reduced at 24 h and gradually recovers at 72 h after reperfusion (71). Overall, an increase in AQP4 expression is correlated with the cytotoxic edema of astrocytes, and a decrease in AQP4 expression is correlated with vasogenic cerebral edema (72).
From the above data, a very complex landscape of the inflammation involved in the pathophysiology of ischemic stroke emerges. In general, a clinically useful marker of the advancement of the ongoing inflammatory processes is CRP (C-reactive protein), and in stroke patients the concentration of this marker is significantly elevated. On the one hand, this increase in CRP indicates the presence of a serious disease, and on the other hand, it is an important prognostic marker of the outcomes of stroke treatment, probably also because CRP can directly cause changes in the endothelium – such as increasing of the expression of adhesion molecules, mediating the invasion of peripheral leukocytes into the brain tissues (73–75).
Inflammatory processes are a double-edged sword and play both a positive and negative role in the development of ischemic cerebral damage, depending on their phase, the type of actually involved inflammatory cells, and the patient’s health history. This presents a challenge for research into the mechanisms and future therapy of ischemic stroke. However, there are valid immunological goals for research on neuroprotection and vasculoprotection. The penetration of leukocytes from blood into the damaged brain involves several stages, mainly determined by: 1) the appearance of pro-inflammatory cytokines and chemokines in damaged tissues, and 2) the expression of adhesion molecules in microvascular endothelial cells and their presentation on the surface of the cell membranes.
The negative effects of leukocyte invasion into the ischemic brain may cause secondary damage within the penumbra. Several mechanisms of secondary brain injury have been proposed. Three of them that appear to be the most important are a secondary reduction in cerebral blood flow during reperfusion, the permeabilization of blood vessels, and the production of oxygen (ROS) and nitrogen (RNS – reactive nitrogen species) free radicals causing general tissue damage. Secondary reduction of blood flow in the vessels of the microcirculation (no-reflow effect) may be, on the one hand, an effect of cytotoxic swelling of astrocytes and the compression of their foot-ends on arterioles, and, on the other hand, a result of leukocyte adhesion to the inner surfaces of blood vessel walls, in particular the endothelial cells of venules. The adherence of leukocytes and platelets to each other causes the formation of a specific network inside the lumen of the vessel, which is additionally strengthened by fibrin, which immobilizes erythrocytes. In this way, conglomerates/aggregates of blood cells, or platelet-leukocyte aggregates (PLAs), are formed, and their adherence to the inner walls of the microvessels narrows the vascular lumen, disrupting blood microcirculation as a result. PLAs may also detach from the site of their formation and enter the general bloodstream (76). Therefore, it seems that when the therapy achieves only the elimination of the occlusion of a larger, blood-supplying vessel, which is the cause of ischemia, incomplete restoration of penumbral microcirculation may significantly weaken the treatment.
Activated inflammatory cells, mainly neutrophils and macrophages, release large amounts of metalloproteinases, in particular, gelatinases: MMP-9, MMP-2 (metalloproteinase-2), which is also released by activated astrocytes, and collagenase: MMP-13 (metalloproteinase-13). These enzymes have neurotoxic properties, and they degrade the integrity of the BBB, among other ways, by digesting the basal membrane of the vessels. This can lead to HT during reperfusion, contribute to the enlargement of the developing vasogenic cerebral edema, and further increase the influx of leukocytes into the brain. These processes significantly reduce the clinical capabilities of using tissue plasminogen activators and the mechanical recanalization of the plugged artery (77, 78).
Free radicals, including ROS, are produced in high concentrations by leukocytes to fight microorganisms. Although the post-ischemic inflammatory process develops under sterile conditions, stimulated leukocytes produce ROS, similar to their native response to pathogens. One of the most reactive free radicals is the superoxide anion radical . The major producing enzyme system found in inflammatory cells such as neutrophils, lymphocytes and macrophages is NOX (79, 80). As mentioned, NOX is also widely abundant in the endothelial cells of blood vessels and may be stimulated by cytokines. Superoxide anion radicals and NO (nitric oxide) show strong mutual chemical affinity. During ischemia/reperfusion, iNOS is stimulated because of the presence of inflammatory IL-1β and TNF-α molecules in ischemic tissues – which increases the expression of the iNOS isoform in endothelium and infiltrating neutrophils, producing high concentrations of NO (62, 81). Other types of NO synthases are additionally stimulated during a stroke, e.g., endothelial isoform (eNOS) is stimulated by an increase in the intracellular concentration of Ca2+ ions, which is considered a protective phenomenon as it may improve the blood flow in the penumbra (82). NO and react with each other to form peroxynitrite (ONOO-), a very chemically reactive free radical with a long half-life. Similar to other free radicals, peroxynitrite damages, e.g., nucleic acids, proteins and lipids. It has been observed that microvessels located in the ischemia/reperfusion region have numerous zones containing products of reaction with ONOO-, such as 3-nitrotyrosine, a marker of protein nitrosative damage (this type of damage is also observed in other brain tissues affected by ischemia/reperfusion). Moreover, the formation of ONOO- reduces the bioavailability of NO, which is the reason for the disturbance in the NO-dependent mechanism of regulation of the tension of the vascular wall and is a marker of endothelial dysfunction (83, 84).
In response to ischemia/reperfusion, activated endothelial cells begin to express adhesion molecules and, within a few hours, display them in large numbers on their cellular membrane; as a result, they start to recruit large numbers of myeloid cells to their surface, mainly neutrophils as well as, in a smaller number, macrophages, which then pass into post-ischemic tissues during reperfusion. The endothelium can be activated by IL-1β and TNF-α molecules (85, 86). Leukocyte capture and rolling on the endothelial surface, firm adhesion and transmigration through the blood vessel wall (diapedesis) constitute three stages of the process of entering the tissues, which is determined by the expression of adhesion molecules on the membranes of endothelial cells and white blood cells.
Among the adhesion molecules, three classes can be distinguished: the selectins, which include, e.g., P-selectin, E-selectin and L-selectin; the integrins, which include, e.g., CD18/CD11a (LFA-1, lymphocyte function associated antigen 1), CD18/CD11b (Mac-1, macrophage-1 antigen), CD18/CD11c (integrin alpha X beta 2), α4ß1 (VLA-4, very late antigen-4) and integrin α6ß4; and the immunoglobulin superfamily, which include, e.g., ICAM-1 (intercellular adhesion molecule 1), ICAM-2 (intercellular adhesion molecule 2), VCAM-1 (vascular cell adhesion molecule 1), PECAM-1 (platelet endothelial cell adhesion molecule-1), and MAdCAM-1 (mucosal vascular addressin cell adhesion molecule 1) (87).
Selectins mediate non-tight interactions (mainly rolling) between the endothelium and leukocytes. P-selectin is expressed in endothelium and platelets. In endothelial cells, P-selectin is deposited in Weibel-Palade bodies, and in platelets, it forms cytoplasmic alpha granules. E-selectin is expressed only in the endothelium, while L-selectin is expressed in the endothelium and leukocytes. Leukocytes express the SLeX oligosaccharide antigen (Lewis-X sialyl), which binds to P- and E-selectin, conditioning the rolling stage (88). Experimental work on models of focal cerebral ischemia has shown that blocking P-selectin or E-selectin with monoclonal antibodies or the knockout of the genes of these molecules reduced post-ischemic brain damage and improved the motor skills of tested animals. P-selectin blockade with specific monoclonal antibodies also reduced the number of no-reflow phenomena during reperfusion. In addition, the overexpression of P-selectin or E-selectin magnified post-stroke injury (89, 90). However, in the global cerebral ischemia model, the blockage of P-selectin with specific monoclonal antibodies increased the mortality of the animals used in the experiment (91). These inconsistencies perhaps result from differences between experimental stroke models. Leukocytes have a specific antigen for P-selectin – PSGL-1 (P-selectin glycoprotein ligand-1) deployed to their cell membranes. This interaction likely participates in the formation of PLA aggregates and the secondary activation of both types of cells (92). The use of sCRsLex, an inhibitor of platelet-leukocyte adhesion interactions, reduced the volume of post-stroke cerebral infarction in experimental animals (93). Very promising observations of a reduction in post-stroke damage have been made by administering E-selectin intranasally to spontaneously hypertensive stroke-prone rats prior to focal cerebral stroke with reperfusion. In addition, a decrease in the incidence of spontaneous strokes has been demonstrated in this strain of rats after the intranasal administration of E-selectin (94, 95). Thus, there is expectation for the development of a specific vaccine that could, for example, improve prognosis in patients at risk. However, in some studies of patients with ischemic stroke to date, inconsistent results in measurements of serum E-selectin levels have been obtained. In the study of patients with stroke symptoms, an increase in the concentration of E-selectin was observed for 24 hours, while in other studies, no such phenomenon was observed (96). Inconsistencies also appear in experimental research. Experiments in rats with spontaneous hypertension have shown that functional blocking of the E-selectin molecule reduced post-stroke damage in a model of transient but not permanent focal cerebral ischemia (97). The role of L-selectin in stroke is unclear. L-selectin has been shown to mediate the process of neutrophil rolling, but the blockade of L-selectin with monoclonal antibodies did not result in neuroprotection in a focal stroke model in rabbits (98, 99).
Another class of adhesion molecules are integrins, which are heterodimeric cell membrane glycoproteins consisting of two subunits: α and β, bounded by weak interactions. There are several β subunit subtypes, e.g., β1, β2, β3, and β4, which represent a specific subclass of molecules within which the α subunit changes (100). Examples of integrins containing β1 and β4 subunits are α1β1, expressed in microvascular endothelia and astrocytes, and α6β4, found at the perivascular end-feet of astrocytes. These integrins play a very important role in the brain, taking part in the formation of neurovascular units. α1β1 integrins contact the endothelium with its extracellular matrix; similarly, α6β4 integrins stabilize the contact of astrocyte projections with the laminin-5 of the microvascular matrix. β2 integrins mainly mediate the adhesive interaction of leukocytes, while β3 integrins (cytoadhesins), including platelet glycoprotein αIIbβ3 (glycoprotein IIb-IIIa), are involved in the formation and stabilization of a blood clot. In response to brain ischemia, α1β1 and α6β4 integrins disappear very quickly in the ischemic region, which is explained by a decrease in their biosynthesis. Neurovascular units in this situation disintegrate (101, 102). According to the main structural pattern, integrins found in leukocytes consist mainly of those with the α and β2 subunits. The β2 subunit (also referred to as CD18) is homogenous, but there are a number of types of α subunits, e.g. 11a, 11b, and 11c. Thus, we can distinguish diverse types of CD18 integrins on the basis of their α-subunit. CD18/CD11a integrin is expressed in all leukocytes, and CD18/CD11b is expressed in neutrophils, monocytes and NK cells (100, 103, 104). These are the two leukocyte integrins most studied for their role in cerebral ischemia/reperfusion. Leukocyte activation increases the affinity of integrins for specific ligands on the surface of activated endothelia. Factors activating the expression of leukocyte integrins are IL-8 and MCP-1. The binding of an integrin to a ligand causes a conformational change in its intracellular domain, which interacts with elements of the cytoskeleton, giving the cell the opportunity for tight adhesion and migration through the vessel wall (100). In experimental studies on animals, it has been shown that the administration of antibodies against CD11a and CD18 reduced the extent of post-stroke damage and reduced the number of infiltrating neutrophils (105). It has also been shown that the expression of CD11a and CD18 in leukocytes was significantly increased in patients with stroke and transient ischemic attack. On the other hand, some studies have shown no increase in CD11b expression in stroke patients, and that the use of integrin-targeting therapy (anti-CD18/CD11b antibodies) in patients was not effective (103, 106, 107). Similar results were also obtained in studies concerning alpha-4 integrins (α4), which are crucial for the transvascular egress of lymphocytes T. Initially, using antibody blockade of α4, animal experiments showed promising outcomes, including a significant reduction in the size of the brain post-ischemic damage in normotensive and hypertensive rats (108). However, subsequent experimental studies yielded negative results (109). Finally, clinical trials (ACTION and ACTION II) with the use of monoclonal antibodies targeting α4 within the VLA-4 molecule (natalizumab) showed no therapeutic effect in patients with ischemic stroke (110, 111).
Immunoglobulin superfamily (IgSF) molecules mediate tight interactions between leukocytes and endothelia. Among them, the role of ICAM-1 has been the relatively most researched in the pathophysiology of brain ischemia/reperfusion. ICAM-1 is a molecule constitutively expressed in the endothelium, but its expression significantly increases during stroke in response to ischemic conditions and the stimulation of the endothelium with cytokines (IL-1, TNF-α, gamma interferon), reaching peak concentrations 12–24 hours after reperfusion (112, 113). Clinical tests showed an increase in ICAM-1 concentration in the blood serum and cerebrospinal fluid of patients after stroke (114). Experiments in mice in which the ICAM-1 gene has been knocked out showed an improvement in cerebral microcirculation, a decrease in the volume of post-stroke necrotic tissue, and a decrease in the number of infiltrating leukocytes after focal cerebral stroke with reperfusion compared to mice in the control group. The blockade of ICAM-1 with specific antibodies also reduced infarction and leukocyte infiltration of brain tissues in rats and rabbits following experimental stroke (112, 115, 116). Specific antigens for ICAM-1, such as LFA-1 and Mac-1, are found on the surface of leukocytes (117). In brain ischemia/reperfusion experiments on mice with a null mutation in the LFA-1 and Mac-1 genes, no effect on the adhesion process of leukocytes 4 hours after reperfusion was shown, while such an effect appeared after 24 hours, which correlated with the time of ICAM-1 peak density at the cell membranes of endothelial cells (92, 118). Despite promising results from preclinical studies, clinical trials using antibodies against ICAM-1 (Enlimomab) and Mac-1 (LeukArrest) were unsuccessful (119, 120). The roles of other molecules in the immunoglobulin superfamily in ischemic stroke are very poorly understood, and experimental studies have produced inconsistent results. In patients with stroke, the concentration of VCAM-1 in blood and its expression in endothelial cells and astrocytes in the ischemic region of the brain were significantly increased (121). However, experiments in rats and mice have shown that blocking VCAM-1 molecules with specific antibodies did not reduce the magnitude of post-stroke damage (122). Likewise, the role of ICAM-2 in brain ischemia/reperfusion is unknown. This molecule is expressed in activated endothelia and in nonactivated and activated platelets. It is speculated that ICAM-2 may be involved in the adhesion of platelets with leukocytes and the formation of PLAs during reperfusion (123). The last considered IgSF molecule – PECAM-1 is expressed in endothelial cells and most leukocytes. It can be reasonably postulated, that this molecule may be involved in the processes of adhesion and the transmigration of leukocytes through the endothelium during a ischemic stroke, as has been observed that the concentration of PECAM-1 increased significantly in the blood serum and cerebrospinal fluid of patients 24 hours after stroke insult (124). However, to date, there are few data on the role of PECAM-1 in experimental ischemia/reperfusion models. In one experimental study, PECAM-1 was found to control the transendothelial migration of neutrophils in a experimental mouse model of ischemic stroke, and antibody blockade of PECAM-1 during reperfusion ameliorated stroke severity in these mice (125).
Hypertension, defined as chronically elevated arterial blood pressure, leads to numerous changes in the blood vessels of the whole organism. Adaptive remodeling of the vascular walls or their hypertrophy or stiffness, a reduction of vessel caliber and changes in functional physiology, such as increased vascular resistance and circulatory perturbations, occur (126). The brain is an organ particularly affected by circulatory changes caused by hypertension. Several key mechanisms regulating cerebral blood microflow and maintaining brain energy homeostasis are disturbed, and therefore, the functions of NVUs are affected (127).
The dysfunction of the endothelium in the course of hypertension may be of various origins. One of the pro-dysfunctional pathways may be the production of oxygen free radicals by vascular systems: NOX (especially the NOX2 isoform), XO and the mitochondrial respiratory chain. It has been shown that the activity of NOX2 and XO is increased in hypertensive vessels (128). ROS react with NO to form peroxynitrite, thereby reducing the bioavailability of NO and causing an imbalance between this important vasodilator and the vasoconstrictors; disturbing the regulation of vascular wall tone and causing significant circulatory/energy deficits; and damaging proteins, lipids and nucleic acids (129). Systemic inflammation and neuroinflammation also play an important role in the pathophysiology of hypertension. Ongoing processes of ROS production and neuroinflammation likely damage various cells, which therefore release DAMPs signals (130). These molecules are well-known activators of TLR4. TLR4 receptors were the first TLRs proposed in the etiology of vascular inflammatory damage in hypertension (131). The activation of endothelial TLR4 leads, through the MyD88 protein, to the activation of the transcription factors AP-1 and NF-κB, which further enhance ongoing inflammation (132).
Although, it is difficult to state whether inflammatory processes are one of the primary or secondary causes of hypertension, it can now be assumed that inflammatory processes result from both primary and secondary causes of hypertension. The inhibition of NF-κB counters the increase in blood pressure that normally occurs in spontaneously hypertensive rats (SHRs), and decline high blood pressure in a neurogenic model of hypertension induced by deoxycorticosterone acetate administration in rats fed a high-sodium diet (133, 134). It is known that hypertension-linked inflammation most often occurs at a low-grade level, notwithstanding causing a number of unfavorable changes in the brain.
The inflammation marker considered to be significantly correlated with hypertension is CRP, and it has been confirmed that prehypertensive and hypertensive patients have elevated levels of CRP in blood serum (135). CRP molecule can stimulate circulating monocytes to express IL-6, IL-13 (interleukin 13) and TNF-α, and the expression of ICAM-1 and VCAM-1 adhesion molecules in the endothelium, including via activated NF-κB and AP-1. It has been shown that CRP can directly stimulate endothelial and smooth muscle cells to increase activation of NF-κB and AP-1 (136, 137). Moreover, elevated levels of chemokines, e.g., RANTES, were found in the serum of hypertensive patients (138, 139). The expression of some other adhesion molecules, e.g., P-selectin in platelets and endothelium and its antigen, PSGL-1 in the endothelium, may also take place through the action of the pro-hypertensive angiotensin II on the AT1R receptor (angiotensin II type 1 receptor) (140). Also immune cells residing in the brain – microglia/macrophages are sensitive to ongoing hypertension. As a result of the action of hypertension on microvessels in the brain, microglia are activated, expressing IL-1β and other pro-inflammatory molecules, such as IL-6 and TNF-α (141). Additionally, the specific class of macrophages residing in the vicinity of arterioles and venules, called perivascular macrophages (PVMs), plays a very important role in NVU dysfunction. Many studies on this issue have shown that the deletion of AT1R in PVMs partially attenuates BBB dysfunction, which results from NOX2 activity elevation. In contrast, the downregulation of AT1R in cerebral endothelial cells completely prevented BBB disruption. The results indicate that while endothelial AT1R, mainly in arterioles and venules, initiates BBB disruption in hypertension, PVMs are required for the full expression of the dysfunction (142). Another important molecule involved in hypertension and BBB damage is the CD36 receptor. In stroke-prone SHRs, increased expression of CD36 in microglia and associated BBB lesions were demonstrated, indicating a pro-inflammatory role of CD36 in the brain under hypertensive conditions (143). Importantly, CD36 receptor may be involved in the activation of astrocytes following brain ischemia, contributing significantly to the progression of inflammatory phenomena after a stroke onset (144).
The pro-inflammatory environment, formed in the brain as a result of the above processes (i.e., the accumulation of ROS, DAMPs, pro-inflammatory cytokines and chemokines; BBB weakening; and the activation of resident and systemic immune cells), promotes the formation of subsequent changes in brain tissues. Under the described conditions, the transmigration of systemic leukocytes may be promoted by an overestimated expression of PECAM-1, which in turn distinctly intensifies the weakening of the BBB (145). In hypertensive patients and in animal hypertension models, elevated concentrations of MMP-9 were also found, which indicates ongoing processes of leukocyte infiltration into the tissues (146). Inflammatory processes are also often associated with vasogenic edema. However, during low-grade neuroinflammation, the activation of astrocytes, astrocytic AQP4 overexpression and the development of mild cytotoxic edema should be taken into account – as has been confirmed that astrocytes in SHRs have been shown to increased expression of GFAP (147). Moreover, overexpresion of AQP4 in SHR brains was also detected (148).
All the above changes indicate a significant activation of astrocytes, a tendency toward cytotoxic edema, possible pressure on microvessels and disturbed communication with neurons. In this situation, the functions of NVUs (i.e., neurovascular coupling, NVC) will undoubtedly be disturbed, and there is numerous experimental evidence confirming this assumption; for a detailed review, see (149). Several experimental studies also indicate a significant increase in post-ischemic brain damage in hypertensive animals (150–153).
Diabetes mellitus is a chronic metabolic disease caused by insufficient insulin production (type 1 diabetes mellitus, T1DM) and/or a decrease in the tissue response to available insulin (type 2 diabetes mellitus, T2DM). This results in an elevation of blood glucose concentration, and in T2DM, also of insulin. The role of insulin resistance in the course of T2DM and the resulting hyperglycemia and hyperinsulinemia have been studied intensively in terms of causing dysfunction and damage to blood vessels at the level of the endothelium. Diabetic vascular complications affect many organs, including the brain-cerebral circulatory system and also the cerebral parenchyma (154). The brain is an organ that is significantly sensitive to inadequate blood glucose levels and to circulatory disturbances (155). A remodeling, stiffening of the vascular walls, and reduction of vessel caliber with changes in cerebral perfusion occur during diabetes (156).
The etiology of these diabetic complications is complex, alike as it is in the case of arterial hypertension and hyperlipidemia. Possible pathways of endothelial damage may involve ROS production and inflammatory processes. The production of ROS can take place in blood vessels through the activity of NOX, XO and the mitochondrial respiratory chain (157, 158). The decrease in NO bioavailability due to the generation of ROS/peroxynitrite and damage to biomolecules are important consequences leading to endothelial dysfunction. Oxidative stress and low-grade inflammation are recognized as important factors in the progression of T2DM and its vascular complications. Affected endothelial cells can release DAMP molecules, activating TLR4 and further potentiating inflammation (159). The specific DAMP signal – the peculiar effect of hyperglycemia is the formation of AGEs (advanced glycation end products), including proteins or lipids that, when exposed to an elevated glucose concentration, become covalently glycated. AGEs can stimulate RAGE, CD36 and TLR4 receptors, the pathways of which significantly contribute to the progression of inflammation in diabetes, particularly through NF-κB activity (160, 161). Additionally, the activation of MMP-9 in the brain and osmotic disturbances resulting from abnormal blood glucose levels weaken the BBB (162).
Inflammatory processes during diabetes are manifested by an increase in the CRP concentration in blood serum, the production of pro-inflammatory molecules, and the activation of immune system cells. CRP exerts a direct pro-inflammatory effect on endothelial and smooth muscle cells, inducing NF-κB and the expression of adhesion molecules (136, 137, 163). During hyperglycemia, NF-κB is significantly active in both types of tissues, resulting in the transcription of pro-inflammatory cytokines and an increase in leukocyte adhesion (164, 165). The expression of adhesion molecules in the endothelium in animal models of diabetes and in diabetic patients was found to be enhanced compared to nondiabetic control groups. Additionally, pro-inflammatory proteins such as IL-6 and MCP-1 show higher serum levels in diabetic patients (166, 167). It has been demonstrated in excellent study targeting neuroinflammation and BBB disorders (using T1DM and T2DM murine models) that diabetic hyperglycemia enhances BBB permeability and memory loss (Y maze and water maze tests) and elevated expression of inflammatory molecules in the brain, a.o.: MMP-9, VCAM-1, E-selectin, TNF-α, and CXCL2. Activation of microglia has also been shown (168). Brain microglia play an important role in diabetic cerebral injury. In murine models of diabetes mellitus, the activation and proliferation of microglia were observed in the brain, and activated microglia largely contributed to neuroinflammation and oxidative stress (169).
As in the case of hypertension, diabetes also causes the activation of cerebral astrocytes, which is manifested by the increased expression of GFAP by these cells. However, unlike in hypertension and hyperlipidemia, astrocytic AQP4 levels are lower in diabetic animals than in normoglycemic ones (170). Both of these results indicate the possibility of communication disorders between astrocytes, blood vessels and neurons, which, combined with the abovementioned processes of ROS and DAMPs production, endothelial damage, the release of inflammatory molecules, microglial activation, indicates the creation of an environment that strongly promotes NVUs dysfunction in the brain. NVC abnormalities in the brain have been detected in both an animal models of diabetes and in diabetic patients (171). The initial diabetic NVUs dysfunctions, may be a significant cause of a worsen stroke outcomes, which has been shown in several studies in experimental models of cerebral ischemia (150, 172–174).
Hyperlipidemia is most commonly defined as elevated plasma cholesterol levels, which may be associated with elevated plasma triglycerides. Lipids are transported by the plasma in the form of lipoproteins (e.g., LDL – low-density lipoprotein); hence, hyperlipidemia is most commonly diagnosed in patients based on elevated levels of plasma lipoproteins. Both hypercholesterolemia and hypertriglyceridemia can cause the dysfunction of the whole body vasculature, including cerebral blood vessels (10, 175).
In experimental research, one of the frequently used animal models of hyperlipidemia is the apolipoprotein E-deficient murine model (Apoe-/-, apolipoprotein E is one of the protein components of lipoproteins such as LDL and is involved in lipid transport to tissues). In interesting studies on these mice fed a high-fat diet (HFD), elevated plasma lipid levels disturbed the regulation of cerebral blood flow. Cerebral microcirculation responses to hypercapnia and functional stimulation (NVC) were impaired, while no atherosclerotic plaques were found in these animals (176). In other experiments in the same mouse model, a decrease in the microcirculatory response to acetylcholine was shown, which was improved with the use of a free radical scavenger or NOX inhibitor (177). In another hyperlipidemia model, after 10 weeks of HFD feeding in mice, both cerebral macrovessels and microvessels underwent remodeling, including an increased cerebrovascular tortuosity index and decreased arterial inner diameters. This remodeling could be mediated, at least in part, by MMP-9, as an HFD induced similar levels of hyperlipidemia in MMP-9-deficient mice, but there was no cerebrovascular remodeling (178). Nevertheless, the exact mechanistic insights into how elevated plasma lipids damage cerebral blood vessels and impair neurovascular coupling are not fully clear.
Some studies have shown an increased level of CRP in the blood plasma of patients with hypercholesterolemia (179). This result may indicate that these individuals have low-grade background inflammation, similar to hypertension and diabetes. Likewise, oxidative processes can also occur. There is evidence of oxidative modification of LDL mediated by lipoxygenase (expressed by macrophages) and/or myeloperoxidase (expressed by neutrophils and monocytes), which generate the product of LDL oxidation – oxidized LDL molecule (oxLDL) (180). It should be emphasized that some authors assign oxLDL to the category of DAMP molecules, there is also evidence pointing to a pro-HMGBl action of oxLDL in cerebral vessels, which indicate on the generation of an alarm signal to excite the effectors of the immune system (181). OxLDL induces the assembly of the TLR 4/6 heterodimer, involves CD36, and consequently induces the NF-κB and AP-1 pathways and the secretion of pro-inflammatory molecules, e.g., cytokines and chemokines (182).
Low-grade inflammation in the course of hyperlipidemia is a likely cause of NVUs dysfunction in the brain. Excellent experimental studies in wild-type C57/BL6J mice fed a high-cholesterol diet (HCD) showed a tremendous increase in rolling and adherent leukocytes and in platelets in the cerebral venules. P-selectin immunoneutralization attenuated the interactions of leukocytes and platelets with endothelia. The same study also showed that in mice in which one of the subunits of NOX had been knocked out and fed an HCD diet, the number of rolling and adherent leukocytes in the cerebral venules decreased significantly compared to that in wild-type mice fed an HCD diet (14). Moreover, it has been shown that oxLDL can induce the expression of ICAM-1 and VCAM-1 adhesion molecules in the human endothelium (183). Studies with HCD-fed Apoe-/- and wild-type control mice showed a significant increase in IL-6 expression in the hippocampus and cerebral cortex in both strains (184). In studies carried out on rats fed hypercholesterolemic diets, an increase in the concentration of IL-1 and TNF-a in blood plasma was detected after the first week, and an increase in GFAP expression in the brain, while after the third week, a decrease in expression of claudin-5 in the brain was detected (185). In other studies carried out on rats fed an HFD, an increase in GFAP expression as well as AQP4 and microglial activation were detected (186). However, in studies in wild-type mice fed an HCD diet, no significant changes in GFAP expression in the brain were detected after three months. The same result was obtained in transgenic mice overexpressing transforming growth factor-ß1; however, in these mice, the HCD diet caused spatial learning deficits and increased the number of thinned blood vessels in the internal capsule. The effects of the HCD on spatial learning and the studied vascular morphology changes in these mice were reduced when simvastatin, a drug that lowers blood cholesterol, was administered during the diet (187). In other studies, HCD-fed C57/BL6J wild-type mice showed an increase in GFAP expression in the hippocampus and the cerebral cortex and AQP4 in the hippocampus. There was no microglial activation (188). All of the above studies were conducted on males, interesting results of the influence of an HFD on inflammatory markers in the brains of wild-type C57BL/6J female mice (10–12 weeks old) were provided by the study of Peterson TC., et al., which showed that 6 weeks of the HFD did not induce changes in GFAP expression or microglial activation in the cerebral cortex (189). The above differences in the results of studies on the impact of an HFD on GFAP expression and microglia status may result from differences between the experimental procedures and animal models used. In the case of the study by Peterson TC., et al., one of the results was that the HFD had no effect on GFAP expression, possibly due to the protective effect of female sex hormones counteracting low-grade neuroinflammation. Estrogens have both neuroprotective and anti-inflammatory effects, e.g., 17β-estradiol has been shown to have an inhibitory effect on GFAP expression in mice after traumatic brain injury (190). Nevertheless, extensive studies in rats and mice fed an HFD and observations of obese patients found that rodents had increased GFAP expression in the hypothalamus and obese patients had features of gliosis in the hypothalamus (191).
In summary, all the above study results strongly suggest that processes that damage NVUs in the brain take place in the course of hyperlipidemia. This initial disturbance in NVUs may impart an important contribution to the pathophysiology of ischemic stroke, as hyperlipidemia has been shown to increase post-ischemic brain damage in experimental animal models of cerebral ischemia (174, 192–194).
The insufficient blood supply to the brain initiates the ischemic cascade. In a focal stroke, tissues located in the ischemic core are the first to be destroyed, as neurons located in this area undergo necrosis within a few minutes due to the disruption to energy and ion homeostasis, and the neurovascular units in this zone decay. At the same time, harmful factors such as glutamic acid and potassium ions, released in excessive concentrations from the infarct core, reach the surrounding penumbral area and cause gradual recruitment of the penumbra to the infarct core through excitotoxicity processes and recurrent peri-infarct depolarizations, which cause the production of ROS and RNS, increased expression MMP-9 and MMP-2, disturbances in communication between components of the neurovascular units, and further hemodynamic disturbances during reperfusion. Dead/dying cells release DAMPs signals, leading to the mobilization of the innate immune system through the activation of pattern recognition receptors (TLR4, RAGE, and CD36) in microglia, astrocytes, endothelia, and peripheral leukocytes recruited to the infarction. Intracranial edema, resulting from changes in AQP4 expression (upregulation and/or downregulation) and BBB weakening, strongly contributes to the progression of cerebral damage and inflammation.
Concomitant stroke risk factors (as dealt with here: arterial hypertension, diabetes mellitus and hyperlipidemia) strongly drive these inflammatory processes; because these factors are often present for a long time before a stroke, causing low-grade background inflammation throughout the body and, as we have seen, in the brain, negatively affecting the functioning of neurovascular units. Figure 1 summarizes proposed pathways along which the risk factors for stroke, clinically coexisting severally or in clusters, contribute to the course of the cerebral ischemic/reperfusion cascade, enhancing its progression and destructive effects on the neurovascular units in the ischemic penumbra. The key bridging factors proposed here are inflammatory processes. Low-grade inflammation in the brain before a stroke insult occurs through the activation of mostly the same inflammatory pathways that are activated by the ischemia/reperfusion cascade, i.e., the production of ROS/RNS, DAMPs and the activation of TLR4, RAGE and CD36 receptors, leading to an increase in the expression of MMP-9 and GFAP and changes in AQP4 expression, which are important markers of ongoing NVU-damaging processes. Hence, presensitized neurovascular units are more easily disintegrated and become dysfunctional in the face of damaging factors in the ischemic/reperfusion cascade in the penumbra. Initial sensitization of NVUs as well as driving and strengthening the processes of the ischemia/reperfusion cascade are very likely causes of the great difficulties in the treatment of acute stroke patients. What is equally important is that these factors are also likely the cause of the failure of attempts to introduce neuroprotectants into clinical practice, as most of the in vivo studies were conducted on healthy and young animals that did not show initial sensitization of NVUs to the ischemia/reperfusion cascade.
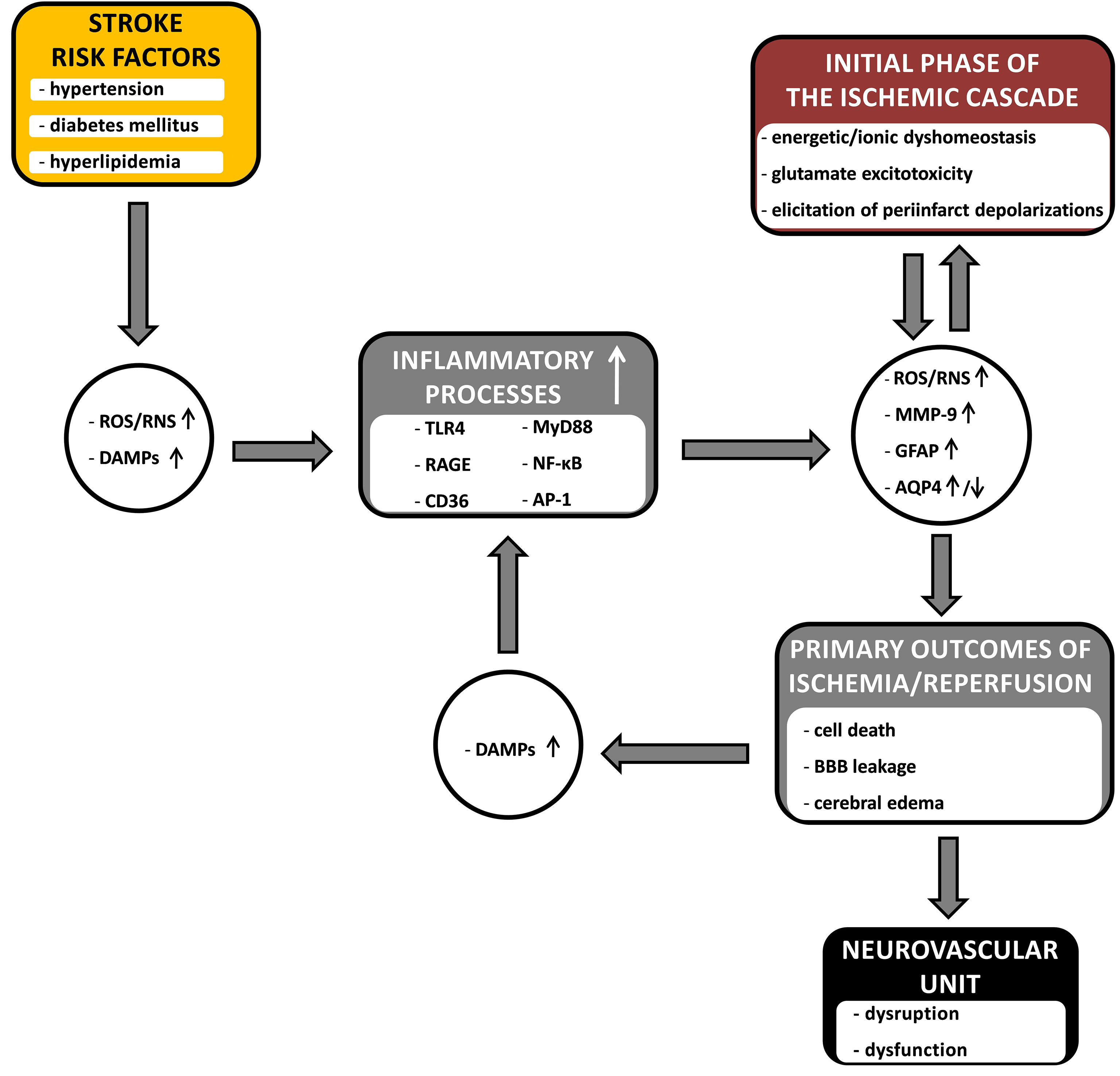
Figure 1 The diagram showing the hypothetical links between common stroke comorbidities (arterial hypertension, diabetes mellitus and hyperlipidemia), inflammatory processes and the course of cerebral ischemia/reperfusion cascade. The comorbid risk factors enhance the influence of the inflammatory processes on the course of the ischemia/reperfusion cascade, contributing to the processes leading to the damage and dysfunction of the neurovascular unit in the penumbra. Inflammatory processes, together with the primary outcomes of the ischemic cascade, form a vicious cycle – where they constitute an amplifying point, and the connecting bridge point, between risk factors and the ischemic cascade. The detailed description is provided in the text. ROS, reactive oxygen species; RNS, reactive nitrogen species; DAMPs, danger associated molecular patterns; TLR4, toll like receptor 4; RAGE, receptor for advanced glycation end products; CD36, platelet glycoprotein 4; MyD88, myeloid differentiation primary response protein (innate immune signal transduction adaptor); NF-κB, nuclear factor kappa B; AP-1, activator protein 1; MMP-9, matrix metalloproteinase-9; GFAP, glial fibrillary acidic protein; AQP4, aquaporin-4; BBB, blood-brain barrier; ↑, upregulation; ↓, downregulation.
As a conclusion, targets for immunomodulatory stroke treatment that can be proposed in the context of the issues reviewed include the pattern recognition receptors that recognize DAMPs. This is because, as was suggested, they may constitute the main bridge between the common stroke risk factors and the ischemic cascade in the brain, and its activation is one of the first stages of neuroinflammatory processes in the course of a stroke. Therefore, reduction of their activation may be crucial for the therapy. Such management may also prove to be safer than typical immunosuppression strictly targeted cellular response, due to the fact that it may increase potential infections, e.g. in the lungs. Arguably, an effective treatment of a ischemic stroke should consist of three elements progressing in succession: neuroprotection and immunomodulation applied as soon as possible before recanalization of the plugged vessel, effective recanalization, and then neuroprotective and immunomodulating treatment during reperfusion (195). Neuroprotection and immunomodulation are both important considering the classic concept of the hemodynamic penumbra and the concept of the “inflammatory penumbra” (196, 197). The inflammatory penumbra spreads beyond the hemodynamic penumbra, increasing the probability of more brain damage by acting through inflammation in the brain.
In the context of the above-mentioned conclusions, there is also a need for preclinical research using animal models that are more relevant to the general health background-condition of patients. Animal models should emphasize the coexistence of several risk factors concerning a single patient, as they often occur jointly (e.g. hypertension-diabetes-obesity), even if mild, and on the duration of their impact on the body. Furthermore, theoretical considerations when designing a therapy should also take into account this type of background and should be directed at several components of the whole neurovascular unit (195). Presumably, the treatment effectiveness confirmed on such experimental models gives a greater chance of its success in the clinic.
ŁP wrote the manuscript and approved it for publication.
This article was financially supported from the Mossakowski Medical Research Institute, Polish Academy of Sciences funds.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Brouns R, De Deyn PP. The Complexity of Neurobiological Processes in Acute Ischemic Stroke. Clin Neurol Neurosurg (2009) 111:483–95. doi: 10.1016/j.clineuro.2009.04.001
2. Gallacher KI, Jani BD, Hanlon P, Nicholl BI, Mair FS. Multimorbidity in Stroke. Stroke (2019) 50:1919–26. doi: 10.1161/STROKEAHA.118.020376
3. den Hertog HM, van Rossum JA, van der Worp HB, van Gemert HMA, de Jonge R, Koudstaal PJ, et al. PAIS Investigators. C-Reactive Protein in the Very Early Phase of Acute Ischemic Stroke: Association With Poor Outcome and Death. J Neurol (2009) 256:2003–8. doi: 10.1007/s00415-009-5228-x
4. Wright JT Jr., Fine LJ, Lackland DT, Ogedegbe G, Dennison Himmelfarb CR. Evidence Supporting a Systolic Blood Pressure Goal of Less Than 150 Mm Hg in Patients Aged 60 Years or Older: The Minority View. Ann Internal Med (2014) 160(7):499–503. doi: 10.7326/M13-2981
5. Lawes CMM, Bennett DA, Feigin VL, Rodgers A. Blood Pressure and Stroke: An Overview of Published Reviews. Stroke (2004) 35:776–85. doi: 10.1161/01.STR.0000116869.64771.5A
6. Fields LE, Burt VL, Cutler JA, Hughes J, Roccella EJ, Sorlie P. The Burden of Adult Hypertension in the United States 1999 to 2000: A Rising Tide. Hypertension (2004) 44:398–404. doi: 10.1161/01.HYP.0000142248.54761.56
7. Roglic G, Unwin N, Bennett PH, Mathers C, Tuomilehto J, Nag S, et al. The Burden of Mortality Attributable to Diabetes: Realistic Estimates for the Year 2000. Diabetes Care (2005) 28:2130–5. doi: 10.2337/diacare.28.9.2130
8. Bonora E, Trombetta M, Dauriz M, Travia D, Cacciatori V, Brangani C, et al. Chronic Complications in Patients With Newly Diagnosed Type 2 Diabetes: Prevalence and Related Metabolic and Clinical Features: The Verona Newly Diagnosed Type 2 Diabetes Study (VNDS) 9. BMJ Open Diabetes Res Care (2020) 8:e001549. doi: 10.1136/bmjdrc-2020-001549
9. Centers for Disease Control and Prevention (CDC). Vital Signs: Prevalence, Treatment, and Control of High Levels of Low-Density Lipoprotein Cholesterol–United States, 1999-2002 and 2005-200. MMWR Morb Mortal Wkly Rep (2011) 60:109–14.
10. Nelson RH. Hyperlipidemia as a Risk Factor for Cardiovascular Disease. Prim Care (2013) 40:195–211. doi: 10.1016/j.pop.2012.11.003
11. Yaghi S, Elkind MSV. Lipids and Cerebrovascular Disease: Research and Practice. Stroke (2015) 46:3322–8. doi: 10.1161/STROKEAHA.115.011164
12. Ishikawa M, Sekizuka E, Yamaguchi N, Nakadate H, Terao S, Granger DN, et al. Angiotensin II Type 1 Receptor Signaling Contributes to Platelet-Leukocyte-Endothelial Cell Interactions in the Cerebral Microvasculature. Am J Physiol Heart Circ Physiol (2007) 292:H2306–15. doi: 10.1152/ajpheart.00601.2006
13. Ding C, He Q, Li P-A. Diabetes Increases Expression of ICAM After a Brief Period of Cerebral Ischemia. J Neuroimmunol (2005) 161:61–7. doi: 10.1016/j.jneuroim.2004.12.011
14. Ishikawa M, Stokes KY, Zhang JH, Nanda A, Granger DN. Cerebral Microvascular Responses to Hypercholesterolemia: Roles of NADPH Oxidase and P-Selectin. Circ Res (2004) 94:239–44. doi: 10.1161/01.RES.0000111524.05779.60
15. Zhao W, Belayev L, Ginsberg MD. Transient Middle Cerebral Artery Occlusion by Intraluminal Suture: II. Neurological Deficits, and Pixel-Based Correlation of Histopathology With Local Blood Flow and Glucose Utilization. J Cereb Blood Flow Metab (1997) 17:1281–90. doi: 10.1097/00004647-199712000-00003
16. Yushmanov VE, Kharlamov A, Yanovski B, LaVerde G, Boada FE, Jones SC. Correlated Sodium and Potassium Imbalances Within the Ischemic Core in Experimental Stroke: A 23Na MRI and Histochemical Imaging Study. Brain Res (2013) 1527:199–208. doi: 10.1016/j.brainres.2013.06.012
17. Rungta RL, Choi HB, Tyson JR, Malik A, Dissing-Olesen L, Lin PJC, et al. The Cellular Mechanisms of Neuronal Swelling Underlying Cytotoxic Edema. Cell (2015) 161:610–21. doi: 10.1016/j.cell.2015.03.029
18. Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of Ischaemic Stroke: An Integrated View. Trends Neurosci (1999) 22:391–7. doi: 10.1016/s0166-2236(99)01401-0
19. Matute C, Domercq M, Sánchez-Gómez M-V. Glutamate-Mediated Glial Injury: Mechanisms and Clinical Importance. Glia (2006) 53:212–24. doi: 10.1002/glia.20275
20. Matute C, Alberdi E, Ibarretxe G, Sánchez-Gómez MV. Excitotoxicity in Glial Cells. Eur J Pharmacol (2002) 447:239–46. doi: 10.1016/s0014-2999(02)01847-2
21. Simon RP, Swan JH, Griffiths T, Meldrum BS. Blockade of N-Methyl-D-Aspartate Receptors may Protect Against Ischemic Damage in the Brain. Science (1984) 226:850–2. doi: 10.1126/science.6093256
22. Hartings JA, Rolli ML, Lu X-CM, Tortella FC. Delayed Secondary Phase of Peri-Infarct Depolarizations After Focal Cerebral Ischemia: Relation to Infarct Growth and Neuroprotection. J Neurosci (2003) 23:11602–10. doi: 10.1523/JNEUROSCI.23-37-11602.2003
23. Heiss W-D. The Ischemic Penumbra: How Does Tissue Injury Evolve? Ann N Y Acad Sci (2012) 1268:26–34. doi: 10.1111/j.1749-6632.2012.06668.x
24. Choi DW. Excitotoxicity: Still Hammering the Ischemic Brain in 2020. Front Neurosci (2020) 14:579953. doi: 10.3389/fnins.2020.579953
25. Choi DW, Koh JY, Peters S. Pharmacology of Glutamate Neurotoxicity in Cortical Cell Culture: Attenuation by NMDA Antagonists. J Neurosci (1988) 8:185–96. doi: 10.1523/JNEUROSCI.08-01-00185.1988
26. Abramov AY, Duchen MR. Mechanisms Underlying the Loss of Mitochondrial Membrane Potential in Glutamate Excitotoxicity. Biochim Biophys Acta (2008) 1777:953–64. doi: 10.1016/j.bbabio.2008.04.017
27. Mitani A, Tanaka K. Functional Changes of Glial Glutamate Transporter GLT-1 During Ischemia: An In Vivo Study in the Hippocampal CA1 of Normal Mice and Mutant Mice Lacking GLT-1. J Neurosci (2003) 23:7176–82. doi: 10.1523/JNEUROSCI.23-18-07176.2003
28. Nishizaki T, Nagai K, Nomura T, Tada H, Kanno T, Tozaki H, et al. A New Neuromodulatory Pathway With a Glial Contribution Mediated via A(2a) Adenosine Receptors. Glia (2002) 39:133–47. doi: 10.1002/glia.10100
29. Kitchen P, Salman MM, Halsey AM, Clarke-Bland C, MacDonald JA, Ishida H, et al. Targeting Aquaporin-4 Subcellular Localization to Treat Central Nervous System Edema. Cell (2020) 181:784–99.e19. doi: 10.1016/j.cell.2020.03.037
30. Strong AJ, Anderson PJ, Watts HR, Virley DJ, Lloyd A, Irving EA, et al. Peri-Infarct Depolarizations Lead to Loss of Perfusion in Ischaemic Gyrencephalic Cerebral Cortex. Brain (2007) 130:995–1008. doi: 10.1093/brain/awl392
31. Yao X, Smith AJ, Jin B-J, Zador Z, Manley GT, Verkman AS. Aquaporin-4 Regulates the Velocity and Frequency of Cortical Spreading Depression in Mice. Glia (2015) 63:1860–9. doi: 10.1002/glia.22853
32. Aizawa H, Sun W, Sugiyama K, Itou Y, Aida T, Cui W, et al. Glial Glutamate Transporter GLT-1 Determines Susceptibility to Spreading Depression in the Mouse Cerebral Cortex. Glia (2020) 68:2631–42. doi: 10.1002/glia.23874
33. Gursoy-Ozdemir Y, Qiu J, Matsuoka N, Bolay H, Bermpohl D, Jin H, et al. Cortical Spreading Depression Activates and Upregulates MMP-9. J Clin Invest (2004) 113:1447–55. doi: 10.1172/JCI21227
34. Farooq MU, Goshgarian C, Min J, Gorelick PB. Pathophysiology and Management of Reperfusion Injury and Hyperperfusion Syndrome After Carotid Endarterectomy and Carotid Artery Stenting. Exp Transl Stroke Med (2016) 8:7. doi: 10.1186/s13231-016-0021-2
35. Granger DN, Kvietys PR. Reperfusion Therapy-What’s With the Obstructed, Leaky and Broken Capillaries? Pathophysiology (2017) 24:213–28. doi: 10.1016/j.pathophys.2017.09.003
36. Aronowski J, Strong R, Grotta JC. Reperfusion Injury: Demonstration of Brain Damage Produced by Reperfusion After Transient Focal Ischemia in Rats. J Cereb Blood Flow Metab (1997) 17:1048–56. doi: 10.1097/00004647-199710000-00006
37. Granger DN, Kvietys PR. Reperfusion Injury and Reactive Oxygen Species: The Evolution of a Concept. Redox Biol (2015) 6:524–51. doi: 10.1016/j.redox.2015.08.020
38. De Silva TM, Brait VH, Drummond GR, Sobey CG, Miller AA. Nox2 Oxidase Activity Accounts for the Oxidative Stress and Vasomotor Dysfunction in Mouse Cerebral Arteries Following Ischemic Stroke. PloS One (2011) 6:e28393. doi: 10.1371/journal.pone.0028393
39. Kinuta Y, Kimura M, Itokawa Y, Ishikawa M, Kikuchi H. Changes in Xanthine Oxidase in Ischemic Rat Brain. J Neurosurg (1989) 71:417–20. doi: 10.3171/jns.1989.71.3.0417
40. Kim JY, Park J, Lee JE, Yenari MA. NOX Inhibitors - A Promising Avenue for Ischemic Stroke. Exp Neurobiol (2017) 26:195–205. doi: 10.5607/en.2017.26.4.195
41. Granger DN. Role of Xanthine Oxidase and Granulocytes in Ischemia-Reperfusion Injury. Am J Physiol (1988) 255:H1269–75. doi: 10.1152/ajpheart.1988.255.6.H1269
42. Enzmann G, Kargaran S, Engelhardt B. Ischemia-Reperfusion Injury in Stroke: Impact of the Brain Barriers and Brain Immune Privilege on Neutrophil Function. Ther Adv Neurol Disord (2018) 11:1756286418794184. doi: 10.1177/1756286418794184
43. Ritter LS, Stempel KM, Coull BM, McDonagh PF. Leukocyte-Platelet Aggregates in Rat Peripheral Blood After Ischemic Stroke and Reperfusion. Biol Res Nurs (2005) 6:281–8. doi: 10.1177/1099800405274579
44. Gülke E, Gelderblom M, Magnus T. Danger Signals in Stroke and Their Role on Microglia Activation After Ischemia. Ther Adv Neurol Disord (2018) 11:1756286418774254. doi: 10.1177/1756286418774254
45. Masuda T, Croom D, Hida H, Kirov SA. Capillary Blood Flow Around Microglial Somata Determines Dynamics of Microglial Processes in Ischemic Conditions. Glia (2011) 59:1744–53. doi: 10.1002/glia.21220
46. Boddaert J, Bielen K, Jongers B, Manocha E, Yperzeele L, Cras P, et al. CD8 Signaling in Microglia/Macrophage M1 Polarization in a Rat Model of Cerebral Ischemia. PloS One (2018) 13:e0186937. doi: 10.1371/journal.pone.0186937
47. Chu K, Yin B, Wang J, Peng G, Liang H, Xu Z, et al. Inhibition of P2X7 Receptor Ameliorates Transient Global Cerebral Ischemia/Reperfusion Injury via Modulating Inflammatory Responses in the Rat Hippocampus. J Neuroinflamm (2012) 9:69. doi: 10.1186/1742-2094-9-69
48. Gesuete R, Kohama SG, Stenzel-Poore MP. Toll-Like Receptors and Ischemic Brain Injury. J Neuropathol Exp Neurol (2014) 73:378–86. doi: 10.1097/NEN.0000000000000068
49. Wang L, Zhang X, Liu L, Yang R, Cui L, Li M. Atorvastatin Protects Rat Brains Against Permanent Focal Ischemia and Downregulates HMGB1, HMGB1 Receptors (RAGE and TLR4), NF-kappaB Expression. Neurosci Lett (2010) 471:152–6. doi: 10.1016/j.neulet.2010.01.030
50. Wang Y, Li L, Deng S, Liu F, He Z. Ursolic Acid Ameliorates Inflammation in Cerebral Ischemia and Reperfusion Injury Possibly via High Mobility Group Box 1/Toll-Like Receptor 4/NFκB Pathway. Front Neurol (2018) 9:253. doi: 10.3389/fneur.2018.00253
51. Li W, Dong M, Chu L, Feng L, Sun X. MicroRNA-451 Relieves Inflammation in Cerebral Ischemia-Reperfusion via the Toll-like Receptor 4/MyD88/NF-κB Signaling Pathway. Mol Med Rep (2019) 20:3043–54. doi: 10.3892/mmr.2019.10587
52. Xie X-K, Xu Z-K, Xu K, Xiao Y-X. DUSP19 Mediates Spinal Cord Injury-Induced Apoptosis and Inflammation in Mouse Primary Microglia Cells via the NF-kB Signaling Pathway. Neurol Res (2020) 42:31–8. doi: 10.1080/01616412.2019.1685068
53. Zhu S, Tang S, Su F. Dioscin Inhibits Ischemic Stroke-Induced Inflammation Through Inhibition of the TLR4/MyD88/NF-κB Signaling Pathway in a Rat Model. Mol Med Rep (2018) 17:660–6. doi: 10.3892/mmr.2017.7900
54. Fitzgerald KA, Palsson-McDermott EM, Bowie AG, Jefferies CA, Mansell AS, Brady G, et al. Mal (MyD88-Adapter-Like) Is Required for Toll-Like Receptor-4 Signal Transduction. Nature (2001) 413:78–83. doi: 10.1038/35092578
55. Ramos CDL, Canetti C, Souto JT, Silva JS, Hogaboam CM, Ferreira SH, et al. MIP-1alpha[CCL3] Acting on the CCR1 Receptor Mediates Neutrophil Migration in Immune Inflammation via Sequential Release of TNF-Alpha and LTB4. J Leukoc Biol (2005) 78:167–77. doi: 10.1189/jlb.0404237
56. Wang J, Gan Y, Han P, Yin J, Liu Q, Ghanian S, et al. Ischemia-Induced Neuronal Cell Death Is Mediated by Chemokine Receptor CX3CR1. Sci Rep (2018) 8:556. doi: 10.1038/s41598-017-18774-0
57. Landreneau MJ, Mullen MT, Messé SR, Cucchiara B, Sheth KN, McCullough LD, et al. Serum Markers After Spontaneous Cerebral Hemorrhage (SMASCH) Investigators. CCL2 and CXCL10 Are Associated With Poor Outcome After Intracerebral Hemorrhage. Ann Clin Transl Neurol (2018) 5:962–70. doi: 10.1002/acn3.595
58. Li T, Zhao J, Xie W, Yuan W, Guo J, Pang S, et al. Specific Depletion of Resident Microglia in the Early Stage of Stroke Reduces Cerebral Ischemic Damage. J Neuroinflamm (2021) 18:81. doi: 10.1186/s12974-021-02127-w
59. Lee GA, Lin C-H, Jiang H-H, Chao H-J, Wu C-L, Hsueh C-M. Microglia-Derived Glial Cell Line-Derived Neurotrophic Factor Could Protect Sprague-Dawley Rat Astrocyte From In Vitro Ischemia-Induced Damage. Neurosci Lett (2004) 356:111–4. doi: 10.1016/j.neulet.2003.11.030
60. Colton CA. Heterogeneity of Microglial Activation in the Innate Immune Response in the Brain. J Neuroimmune Pharmacol (2009) 4:399–418. doi: 10.1007/s11481-009-9164-4
61. Price CJS, Menon DK, Peters AM, Ballinger JR, Barber RW, Balan KK, et al. Cerebral Neutrophil Recruitment, Histology, and Outcome in Acute Ischemic Stroke: An Imaging-Based Study. Stroke (2004) 35:1659–64. doi: 10.1161/01.STR.0000130592.71028.92
62. Garcia-Bonilla L, Moore JM, Racchumi G, Zhou P, Butler JM, Iadecola C, et al. Inducible Nitric Oxide Synthase in Neutrophils and Endothelium Contributes to Ischemic Brain Injury in Mice. J Immunol (2014) 193:2531–7. doi: 10.4049/jimmunol.1400918
63. Gidday JM, Gasche YG, Copin J-C, Shah AR, Perez RS, Shapiro SD, et al. Leukocyte-Derived Matrix Metalloproteinase-9 Mediates Blood-Brain Barrier Breakdown and is Proinflammatory After Transient Focal Cerebral Ischemia. Am J Physiol Heart Circ Physiol (2005) 289:H558–68. doi: 10.1152/ajpheart.01275.2004
64. Gliem M, Mausberg AK, Lee J-I, Simiantonakis I, van Rooijen N, Hartung H-P, et al. Macrophages Prevent Hemorrhagic Infarct Transformation in Murine Stroke Models. Ann Neurol (2012) 71:743–52. doi: 10.1002/ana.23529
65. Bao Y, Kim E, Bhosle S, Mehta H, Cho S. A Role for Spleen Monocytes in Post-Ischemic Brain Inflammation and Injury. J Neuroinflamm (2010) 7:92. doi: 10.1186/1742-2094-7-92
66. Shichita T, Sugiyama Y, Ooboshi H, Sugimori H, Nakagawa R, Takada I, et al. Pivotal Role of Cerebral Interleukin-17-Producing γδt Cells in the Delayed Phase of Ischemic Brain Injury. Nat Med (2009) 15:946–50. doi: 10.1038/nm.1999
67. Gelderblom M, Weymar A, Bernreuther C, Velden J, Arunachalam P, Steinbach K, et al. Neutralization of the IL-17 Axis Diminishes Neutrophil Invasion and Protects From Ischemic Stroke. Blood (2012) 120:3793–802. doi: 10.1182/blood-2012-02-412726
68. Lee GA, Lin T-N, Chen C-Y, Mau S-Y, Huang W-Z, Kao Y-C, et al. Interleukin 15 Blockade Protects the Brain From Cerebral Ischemia-Reperfusion Injury. Brain Behav Immun (2018) 73:562–70. doi: 10.1016/j.bbi.2018.06.021
69. Liu Z, Chopp M. Astrocytes, Therapeutic Targets for Neuroprotection and Neurorestoration in Ischemic Stroke. Prog Neurobiol (2016) 144:103–20. doi: 10.1016/j.pneurobio.2015.09.008
70. Ribeiro MdC, Hirt L, Bogousslavsky J, Regli L, Badaut J. Time Course of Aquaporin Expression After Transient Focal Cerebral Ischemia in Mice. J Neurosci Res (2006) 83:1231–40. doi: 10.1002/jnr.20819
71. Li Q, Li Z, Mei Y, Guo Y. Neuregulin Attenuated Cerebral Ischemia-Creperfusion Injury via Inhibiting Apoptosis and Upregulating Aquaporin-4. Neurosci Lett (2008) 443:155–9. doi: 10.1016/j.neulet.2008.07.064
72. Frydenlund DS, Bhardwaj A, Otsuka T, Mylonakou MN, Yasumura T, Davidson KGV, et al. Temporary Loss of Perivascular Aquaporin-4 in Neocortex After Transient Middle Cerebral Artery Occlusion in Mice. Proc Natl Acad Sci U S A (2006) 103:13532–6. doi: 10.1073/pnas.0605796103
73. Rocco A, Ringleb PA, Grittner U, Nolte CH, Schneider A, Nagel S. Follow-Up C-Reactive Protein Level is More Strongly Associated With Outcome in Stroke Patients Than Admission Levels. Neurol Sci (2015) 36:2235–41. doi: 10.1007/s10072-015-2342-7
74. Yu B, Yang P, Xu X, Shao L. C-Reactive Protein for Predicting All-Cause Mortality in Patients With Acute Ischemic Stroke: A Meta-Analysis. Biosci Rep (2019) 39:BSR20181135. doi: 10.1042/BSR20181135
75. Pasceri V, Willerson JT, Yeh ET. Direct Proinflammatory Effect of C-Reactive Protein on Human Endothelial Cells. Circulation (2000) 102:2165–8. doi: 10.1161/01.cir.102.18.2165
76. Iadecola C, Anrather J. The Immunology of Stroke: From Mechanisms to Translation. Nat Med (2011) 17:796–808. doi: 10.1038/nm.2399
77. Romanic AM, White RF, Arleth AJ, Ohlstein EH, Barone FC. Matrix Metalloproteinase Expression Increases After Cerebral Focal Ischemia in Rats: Inhibition of Matrix Metalloproteinase-9 Reduces Infarct Size. Stroke (1998) 29:1020–30. doi: 10.1161/01.str.29.5.1020
78. Rosell A, Alvarez-Sabín J, Arenillas JF, Rovira A, Delgado P, Fernández-Cadenas I, et al. A Matrix Metalloproteinase Protein Array Reveals a Strong Relation Between MMP-9 and MMP-13 With Diffusion-Weighted Image Lesion Increase in Human Stroke. Stroke (2005) 36:1415–20. doi: 10.1161/01.STR.0000170641.01047.cc
79. Babior BM, Lambeth JD, Nauseef W. The Neutrophil NADPH Oxidase. Arch Biochem Biophys (2002) 397:342–4. doi: 10.1006/abbi.2001.2642
80. Robinson JM. Reactive Oxygen Species in Phagocytic Leukocytes. Histochem Cell Biol (2008) 130:281–97. doi: 10.1007/s00418-008-0461-4
81. Licinio J, Prolo P, McCann SM, Wong ML. Brain iNOS: Current Understanding and Clinical Implications. Mol Med Today (1999) 5:225–32. doi: 10.1016/S1357-4310(99)01453-7
82. Veltkamp R, Rajapakse N, Robins G, Puskar M, Shimizu K, Busija D. Transient Focal Ischemia Increases Endothelial Nitric Oxide Synthase in Cerebral Blood Vessels. Stroke (2002) 33:2704–10. doi: 10.1161/01.str.0000033132.85123.6a
83. Forman LJ, Liu P, Nagele RG, Yin K, Wong PY. Augmentation of Nitric Oxide, Superoxide, and Peroxynitrite Production During Cerebral Ischemia and Reperfusion in the Rat. Neurochem Res (1998) 23:141–8. doi: 10.1023/a:1022468522564
84. Gürsoy-Ozdemir Y, Bolay H, Saribaş O, Dalkara T. Role of Endothelial Nitric Oxide Generation and Peroxynitrite Formation in Reperfusion Injury After Focal Cerebral Ischemia. Stroke (2000) 31:1974–1980; discussion 1981. doi: 10.1161/01.str.31.8.1974
85. Stanimirovic DB, Wong J, Shapiro A, Durkin JP. Increase in Surface Expression of ICAM-1, VCAM-1 and E-Selectin in Human Cerebromicrovascular Endothelial Cells Subjected to Ischemia-Like Insults. Acta Neurochir Suppl (1997) 70:12–6. doi: 10.1007/978-3-7091-6837-0_4
86. Clark WM, Coull BM, Corliss L, Bearner N, Austin T, de Garmo P, et al. Role of Leukocyte Adhesion in Clinical Stroke. J Stroke Cerebrovasc Dis (1992) 2:80–4. doi: 10.1016/S1052-3057(10)80210-1
87. Emsley HCA, Tyrrell PJ. Inflammation and Infection in Clinical Stroke. J Cereb Blood Flow Metab (2002) 22:1399–419. doi: 10.1097/01.WCB.0000037880.62590.28
88. McEver RP, Beckstead JH, Moore KL, Marshall-Carlson L, Bainton DF. GMP-140, a Platelet Alpha-Granule Membrane Protein, is Also Synthesized by Vascular Endothelial Cells and is Localized in Weibel-Palade Bodies. J Clin Invest (1989) 84:92–9. doi: 10.1172/JCI114175
89. Connolly ES, Winfree CJ, Prestigiacomo CJ, Kim SC, Choudhri TF, Hoh BL, et al. Exacerbation of Cerebral Injury in Mice That Express the P-Selectin Gene: Identification of P-Selectin Blockade as a New Target for the Treatment of Stroke. Circ Res (1997) 81:304–10. doi: 10.1161/01.res.81.3.304
90. Huang J, Choudhri TF, Winfree CJ, McTaggart RA, Kiss S, Mocco J, et al. Postischemic Cerebrovascular E-Selectin Expression Mediates Tissue Injury in Murine Stroke. Stroke (2000) 31:3047–53. doi: 10.1161/01.str.31.12.3047
91. Lehmberg J, Beck J, Baethmann A, Uhl E. Effect of P-Selectin Inhibition on Leukocyte-Endothelium Interaction and Survival After Global Cerebral Ischemia. J Neurol (2006) 253:357–63. doi: 10.1007/s00415-005-0996-4
92. Tailor A, Cooper D, Granger DN. Platelet-Vessel Wall Interactions in the Microcirculation. Microcirculation (2005) 12:275–85. doi: 10.1080/10739680590925691
93. Huang J, Kim LJ, Mealey R, Marsh HC, Zhang Y, Tenner AJ, et al. Neuronal Protection in Stroke by an Slex-Glycosylated Complement Inhibitory Protein. Science (1999) 285:595–9. doi: 10.1126/science.285.5427.595
94. Chen Y, Ruetzler C, Pandipati S, Spatz M, McCarron RM, Becker K, et al. Mucosal Tolerance to E-Selectin Provides Cell-Mediated Protection Against Ischemic Brain Injury. Proc Natl Acad Sci U S A (2003) 100:15107–12. doi: 10.1073/pnas.2436538100
95. Takeda H, Spatz M, Ruetzler C, McCarron R, Becker K, Hallenbeck J. Induction of Mucosal Tolerance to E-Selectin Prevents Ischemic and Hemorrhagic Stroke in Spontaneously Hypertensive Genetically Stroke-Prone Rats. Stroke (2002) 33:2156–63. doi: 10.1161/01.str.0000029821.82531.8b
96. Shyu KG, Chang H, Lin CC. Serum Levels of Intercellular Adhesion Molecule-1 and E-Selectin in Patients With Acute Ischaemic Stroke. J Neurol (1997) 244:90–3. doi: 10.1007/s004150050055
97. Morikawa E, Zhang SM, Seko Y, Toyoda T, Kirino T. Treatment of Focal Cerebral Ischemia With Synthetic Oligopeptide Corresponding to Lectin Domain of Selectin. Stroke (1996) 27:951–955; discussion 956. doi: 10.1161/01.str.27.5.951
98. von Andrian UH, Chambers JD, Berg EL, Michie SA, Brown DA, Karolak D, et al. L-Selectin Mediates Neutrophil Rolling in Inflamed Venules Through Sialyl LewisX-Dependent and -Independent Recognition Pathways. Blood (1993) 82:182–91. doi: 10.1182/blood.V82.1.182.bloodjournal821182
99. Yenari MA, Sun GH, Kunis DM, Onley D, Vexler V. L-Selectin Inhibition Does Not Reduce Injury in a Rabbit Model of Transient Focal Cerebral Ischemia. Neurol Res (2001) 23:72–8. doi: 10.1179/016164101101198154
100. Arnaout MA. Structure and Function of the Leukocyte Adhesion Molecules CD11/CD18. Blood (1990) 75:1037–50. doi: 10.1182/blood.V75.5.1037.1037
101. Albelda SM. Endothelial and Epithelial Cell Adhesion Molecules. Am J Respir Cell Mol Biol (1991) 4:195–203. doi: 10.1165/ajrcmb/4.3.195
102. Tagaya M, Haring HP, Stuiver I, Wagner S, Abumiya T, Lucero J, et al. Rapid Loss of Microvascular Integrin Expression During Focal Brain Ischemia Reflects Neuron Injury. J Cereb Blood Flow Metab (2001) 21:835–46. doi: 10.1097/00004647-200107000-00009
103. Soriano SG, Coxon A, Wang YF, Frosch MP, Lipton SA, Hickey PR, et al. Mice Deficient in Mac-1 (CD11b/CD18) Are Less Susceptible to Cerebral Ischemia/Reperfusion Injury. Stroke (1999) 30:134–9. doi: 10.1161/01.str.30.1.134
104. Muto S, Větvicka V, Ross GD. CR3 (CD11b/CD18) Expressed by Cytotoxic T Cells and Natural Killer Cells is Upregulated in a Manner Similar to Neutrophil CR3 Following Stimulation With Various Activating Agents. J Clin Immunol (1993) 13:175–84. doi: 10.1007/BF00919970
105. Matsuo Y, Onodera H, Shiga Y, Shozuhara H, Ninomiya M, Kihara T, et al. Role of Cell Adhesion Molecules in Brain Injury After Transient Middle Cerebral Artery Occlusion in the Rat. Brain Res (1994) 656:344–52. doi: 10.1016/0006-8993(94)91478-8
106. Kim JS, Chopp M, Chen H, Levine SR, Carey JL, Welch KM. Adhesive Glycoproteins CD11a and CD18 Are Upregulated in the Leukocytes From Patients With Ischemic Stroke and Transient Ischemic Attacks. J Neurol Sci (1995) 128:45–50. doi: 10.1016/0022-510x(94)00203-z
107. Krams M, Lees KR, Hacke W, Grieve AP, Orgogozo J-M, Ford GA. ASTIN Study Investigators. Acute Stroke Therapy by Inhibition of Neutrophils (ASTIN): An Adaptive Dose-Response Study of UK-279,276 in Acute Ischemic Stroke. Stroke (2003) 34:2543–8. doi: 10.1161/01.STR.0000092527.33910.89
108. Relton JK, Sloan KE, Frew EM, Whalley ET, Adams SP, Lobb RR. Inhibition of Alpha4 Integrin Protects Against Transient Focal Cerebral Ischemia in Normotensive and Hypertensive Rats. Stroke (2001) 32:199–205. doi: 10.1161/01.str.32.1.199
109. Langhauser F, Kraft P, Göb E, Leinweber J, Schuhmann MK, Lorenz K, et al. Blocking of α4 Integrin Does Not Protect From Acute Ischemic Stroke in Mice. Stroke (2014) 45:1799–806. doi: 10.1161/STROKEAHA.114.005000
110. Elkins J, Veltkamp R, Montaner J, Johnston SC, Singhal AB, Becker K, et al. Safety and Efficacy of Natalizumab in Patients With Acute Ischaemic Stroke (ACTION): A Randomised, Placebo-Controlled, Double-Blind Phase 2 Trial. Lancet Neurol (2017) 16:217–26. doi: 10.1016/S1474-4422(16)30357-X
111. Elkind MSV, Veltkamp R, Montaner J, Johnston SC, Singhal AB, Becker K, et al. Natalizumab in Acute Ischemic Stroke (ACTION II). Neurology (2020) 95:e1091–104. doi: 10.1212/WNL.0000000000010038
112. Kitagawa K, Matsumoto M, Mabuchi T, Yagita Y, Ohtsuki T, Hori M, et al. Deficiency of Intercellular Adhesion Molecule 1 Attenuates Microcirculatory Disturbance and Infarction Size in Focal Cerebral Ischemia. J Cereb Blood Flow Metab (1998) 18:1336–45. doi: 10.1097/00004647-199812000-00008
113. Zhang RL, Chopp M, Zaloga C, Zhang ZG, Jiang N, Gautam SC, et al. The Temporal Profiles of ICAM-1 Protein and mRNA Expression After Transient MCA Occlusion in the Rat. Brain Res (1995) 682:182–8. doi: 10.1016/0006-8993(95)00346-r
114. Bitsch A, Klene W, Murtada L, Prange H, Rieckmann P. A Longitudinal Prospective Study of Soluble Adhesion Molecules in Acute Stroke. Stroke (1998) 29:2129–35. doi: 10.1161/01.str.29.10.2129
115. Zhang RL, Chopp M, Li Y, Zaloga C, Jiang N, Jones ML, et al. Anti-ICAM-1 Antibody Reduces Ischemic Cell Damage After Transient Middle Cerebral Artery Occlusion in the Rat. Neurology (1994) 44:1747–51. doi: 10.1212/wnl.44.9.1747
116. Bowes MP, Zivin JA, Rothlein R. Monoclonal Antibody to the ICAM-1 Adhesion Site Reduces Neurological Damage in a Rabbit Cerebral Embolism Stroke Model. Exp Neurol (1993) 119:215–9. doi: 10.1006/exnr.1993.1023
117. Frijns CJM, Kappelle LJ. Inflammatory Cell Adhesion Molecules in Ischemic Cerebrovascular Disease. Stroke (2002) 33:2115–22. doi: 10.1161/01.str.0000021902.33129.69
118. Lindsberg PJ, Carpén O, Paetau A, Karjalainen-Lindsberg ML, Kaste M. Endothelial ICAM-1 Expression Associated With Inflammatory Cell Response in Human Ischemic Stroke. Circulation (1996) 94:939–45. doi: 10.1161/01.cir.94.5.939
119. Schneider D, Berrouschot J, Brandt T, Hacke W, Ferbert A, Norris SH, et al. Safety, Pharmacokinetics and Biological Activity of Enlimomab (Anti-ICAM-1 Antibody): An Open-Label, Dose Escalation Study in Patients Hospitalized for Acute Stroke. Eur Neurol (1998) 40:78–83. doi: 10.1159/000007962
120. Becker KJ. Anti-Leukocyte Antibodies: LeukArrest (Hu23F2G) and Enlimomab (R6.5) in Acute Stroke. Curr Med Res Opin (2002) 18 Suppl 2:s18–22. doi: 10.1185/030079902125000688
121. Blann A, Kumar P, Krupinski J, McCollum C, Beevers DG, Lip GY. Soluble Intercelluar Adhesion Molecule-1, E-Selectin, Vascular Cell Adhesion Molecule-1 and Von Willebrand Factor in Stroke. Blood Coagul Fibrinolysis (1999) 10:277–84. doi: 10.1097/00001721-199907000-00009
122. Justicia C, Martín A, Rojas S, Gironella M, Cervera A, Panés J, et al. Anti-VCAM-1 Antibodies did Not Protect Against Ischemic Damage Either in Rats or in Mice. J Cereb Blood Flow Metab (2006) 26:421–32. doi: 10.1038/sj.jcbfm.9600198
123. Diacovo TG, deFougerolles AR, Bainton DF, Springer TA. A Functional Integrin Ligand on the Surface of Platelets: Intercellular Adhesion Molecule-2. J Clin Invest (1994) 94:1243–51. doi: 10.1172/JCI117442
124. Zaremba J, Losy J. sPECAM-1 in Serum and CSF of Acute Ischaemic Stroke Patients. Acta Neurol Scand (2002) 106:292–8. doi: 10.1034/j.1600-0404.2002.01339.x
125. Winneberger J, Schöls S, Lessmann K, Rández-Garbayo J, Bauer AT, Mohamud Yusuf A, et al. Platelet Endothelial Cell Adhesion Molecule-1 is a Gatekeeper of Neutrophil Transendothelial Migration in Ischemic Stroke. Brain Behav Immun (2021) 93:277–87. doi: 10.1016/j.bbi.2020.12.026
126. Lee RM, Dickhout JG, Sandow SL. Vascular Structural and Functional Changes: Their Association With Causality in Hypertension: Models, Remodeling and Relevance. Hypertens Res (2017) 40:311–23. doi: 10.1038/hr.2016.145
127. Meissner A. Hypertension and the Brain: A Risk Factor for More Than Heart Disease. Cerebrovasc Dis (2016) 42:255–62. doi: 10.1159/000446082
128. Datla SR, Griendling KK. Reactive Oxygen Species, NADPH Oxidases, and Hypertension. Hypertension (2010) 56:325–30. doi: 10.1161/HYPERTENSIONAHA.109.142422
129. Touyz RM, Rios FJ, Alves-Lopes R, Neves KB, Camargo LL, Montezano AC. Oxidative Stress: A Unifying Paradigm in Hypertension. Can J Cardiol (2020) 36:659–70. doi: 10.1016/j.cjca.2020.02.081
130. McCarthy CG, Goulopoulou S, Wenceslau CF, Spitler K, Matsumoto T, Webb RC. Toll-Like Receptors and Damage-Associated Molecular Patterns: Novel Links Between Inflammation and Hypertension. Am J Physiol Heart Circ Physiol (2014) 306:H184–196. doi: 10.1152/ajpheart.00328.2013
131. Bomfim GF, Dos Santos RA, Oliveira MA, Giachini FR, Akamine EH, Tostes RC, et al. Toll-Like Receptor 4 Contributes to Blood Pressure Regulation and Vascular Contraction in Spontaneously Hypertensive Rats. Clin Sci (Lond) (2012) 122:535–43. doi: 10.1042/CS20110523
132. Yang Y, Lv J, Jiang S, Ma Z, Wang D, Hu W, et al. The Emerging Role of Toll-Like Receptor 4 in Myocardial Inflammation. Cell Death Dis (2016) 7:e2234. doi: 10.1038/cddis.2016.140
133. Rodríguez-Iturbe B, Ferrebuz A, Vanegas V, Quiroz Y, Mezzano S, Vaziri ND. Early and Sustained Inhibition of Nuclear factor-kappaB Prevents Hypertension in Spontaneously Hypertensive Rats. J Pharmacol Exp Ther (2005) 315:51–7. doi: 10.1124/jpet.105.088062
134. Anthony Beswick RA, Dorrance AN, Rajagopalan S, Webb RC. Nf-Kb Inhibition Lowers Blood Pressure in Mineralocorticoid Hypertensive Rats. Hypertension (2000) 36:692.
135. Sesso HD, Buring JE, Rifai N, Blake GJ, Gaziano JM, Ridker PM. C-Reactive Protein and the Risk of Developing Hypertension. JAMA (2003) 290:2945–51. doi: 10.1001/jama.290.22.2945
136. Cirillo P, Golino P, Calabrò P, Calì G, Ragni M, De Rosa S, et al. C-Reactive Protein Induces Tissue Factor Expression and Promotes Smooth Muscle and Endothelial Cell Proliferation. Cardiovasc Res (2005) 68:47–55. doi: 10.1016/j.cardiores.2005.05.010
137. Hattori Y, Matsumura M, Kasai K. Vascular Smooth Muscle Cell Activation by C-Reactive Protein. Cardiovasc Res (2003) 58:186–95. doi: 10.1016/s0008-6363(02)00855-6
138. Seo HS. The Role and Clinical Significance of High-Sensitivity C-Reactive Protein in Cardiovascular Disease. Korean Circ J (2012) 42:151–3. doi: 10.4070/kcj.2012.42.3.151
139. Ryu J, Lee CW, Shin J-A, Park C-S, Kim JJ, Park S-J, et al. FcgammaRIIa Mediates C-Reactive Protein-Induced Inflammatory Responses of Human Vascular Smooth Muscle Cells by Activating NADPH Oxidase 4. Cardiovasc Res (2007) 75:555–65. doi: 10.1016/j.cardiores.2007.04.027
140. Wang Q, Wang H, Wang J, Venugopal J, Kleiman K, Guo C, et al. Angiotensin II-Induced Hypertension is Reduced by Deficiency of P-Selectin Glycoprotein Ligand-1. Sci Rep (2018) 8:3223. doi: 10.1038/s41598-018-21588-3
141. Shi P, Diez-Freire C, Jun JY, Qi Y, Katovich MJ, Li Q, et al. Brain Microglial Cytokines in Neurogenic Hypertension. Hypertension (2010) 56:297–303. doi: 10.1161/HYPERTENSIONAHA.110.150409
142. Santisteban MM, Ahn SJ, Lane D, Faraco G, Garcia-Bonilla L, Racchumi G, et al. Endothelium-Macrophage Crosstalk Mediates Blood-Brain Barrier Dysfunction in Hypertension. Hypertension (2020) 76:795–807. doi: 10.1161/HYPERTENSIONAHA.120.15581
143. Ueno M, Nakagawa T, Nagai Y, Nishi N, Kusaka T, Kanenishi K, et al. The Expression of CD36 in Vessels With Blood-Brain Barrier Impairment in a Stroke-Prone Hypertensive Model. Neuropathol Appl Neurobiol (2011) 37:727–37. doi: 10.1111/j.1365-2990.2011.01172.x
144. Bao Y, Qin L, Kim E, Bhosle S, Guo H, Febbraio M, et al. CD36 is Involved in Astrocyte Activation and Astroglial Scar Formation. J Cereb Blood Flow Metab (2012) 32:1567–77. doi: 10.1038/jcbfm.2012.52
145. Wimmer I, Tietz S, Nishihara H, Deutsch U, Sallusto F, Gosselet F, et al. PECAM-1 Stabilizes Blood-Brain Barrier Integrity and Favors Paracellular T-Cell Diapedesis Across the Blood-Brain Barrier During Neuroinflammation. Front Immunol (2019) 10:711. doi: 10.3389/fimmu.2019.00711
146. Flamant M, Placier S, Dubroca C, Esposito B, Lopes I, Chatziantoniou C, et al. Role of Matrix Metalloproteinases in Early Hypertensive Vascular Remodeling. Hypertension (2007) 50:212–8. doi: 10.1161/HYPERTENSIONAHA.107.089631
147. Tomassoni D, Avola R, Di Tullio MA, Sabbatini M, Vitaioli L, Amenta F. Increased Expression of Glial Fibrillary Acidic Protein in the Brain of Spontaneously Hypertensive Rats. Clin Exp Hypertens (2004) 26:335–50. doi: 10.1081/ceh-120034138
148. Tomassoni D, Bramanti V, Amenta F. Expression of Aquaporins 1 and 4 in the Brain of Spontaneously Hypertensive Rats. Brain Res (2010) 1325:155–63. doi: 10.1016/j.brainres.2010.02.023
149. Iadecola C, Gottesman RF. Neurovascular and Cognitive Dysfunction in Hypertension. Circ Res (2019) 124:1025–44. doi: 10.1161/CIRCRESAHA.118.313260
150. Duverger D, MacKenzie ET. The Quantification of Cerebral Infarction Following Focal Ischemia in the Rat: Influence of Strain, Arterial Pressure, Blood Glucose Concentration, and Age. J Cereb Blood Flow Metab (1988) 8:449–61. doi: 10.1038/jcbfm.1988.86
151. Chen S, Li G, Zhang W, Wang J, Sigmund CD, Olson JE, et al. Ischemia-Induced Brain Damage is Enhanced in Human Renin and Angiotensinogen Double-Transgenic Mice. Am J Physiol Regul Integr Comp Physiol (2009) 297:R1526–1531. doi: 10.1152/ajpregu.91040.2008
152. Garcia J, Dang J, Habib P, Beyer C, Kipp M. Comparison of Infarct Volume and Behavioral Deficit in Wistar Kyoto and Spontaneously Hypertensive Rat After Transient Occlusion of the Middle Cerebral Artery. Springerplus (2013) 2:414. doi: 10.1186/2193-1801-2-414
153. Bhuiyan MIH, Song S, Yuan H, Begum G, Kofler J, Kahle KT, et al. WNK-Cab39-NKCC1 Signaling Increases the Susceptibility to Ischemic Brain Damage in Hypertensive Rats. J Cereb Blood Flow Metab (2017) 37:2780–94. doi: 10.1177/0271678X16675368
154. Love KM, Barrett EJ, Malin SK, Reusch JEB, Regensteiner JG, Liu Z. Diabetes Pathogenesis and Management: The Endothelium Comes of Age. J Mol Cell Biol (2021) 13(7):500–12. doi: 10.1093/jmcb/mjab024
155. Mergenthaler P, Lindauer U, Dienel GA, Meisel A. Sugar for the Brain: The Role of Glucose in Physiological and Pathological Brain Function. Trends Neurosci (2013) 36:587–97. doi: 10.1016/j.tins.2013.07.001
156. Spinetti G, Kraenkel N, Emanueli C, Madeddu P. Diabetes and Vessel Wall Remodelling: From Mechanistic Insights to Regenerative Therapies. Cardiovasc Res (2008) 78:265–73. doi: 10.1093/cvr/cvn039
157. Guo Y, Wang S, Liu Y, Fan L, Booz GW, Roman RJ, et al. Accelerated Cerebral Vascular Injury in Diabetes is Associated With Vascular Smooth Muscle Cell Dysfunction. Geroscience (2020) 42:547–61. doi: 10.1007/s11357-020-00179-z
158. Elbatreek MH, Pachado MP, Cuadrado A, Jandeleit-Dahm K, Schmidt HHHW. Reactive Oxygen Comes of Age: Mechanism-Based Therapy of Diabetic End-Organ Damage. Trends Endocrinol Metab (2019) 30:312–27. doi: 10.1016/j.tem.2019.02.006
159. Li C, Che L-H, Ji T-F, Shi L, Yu J-L. Effects of the TLR4 Signaling Pathway on Apoptosis of Neuronal Cells in Diabetes Mellitus Complicated With Cerebral Infarction in a Rat Model. Sci Rep (2017) 7:43834. doi: 10.1038/srep43834
160. Goh S-Y, Cooper ME. Clinical Review: The Role of Advanced Glycation End Products in Progression and Complications of Diabetes. J Clin Endocrinol Metab (2008) 93:1143–52. doi: 10.1210/jc.2007-1817
161. Ott C, Jacobs K, Haucke E, Navarrete Santos A, Grune T, Simm A. Role of Advanced Glycation End Products in Cellular Signaling. Redox Biol (2014) 2:411–29. doi: 10.1016/j.redox.2013.12.016
162. Aggarwal A, Khera A, Singh I, Sandhir R. S-Nitrosoglutathione Prevents Blood-Brain Barrier Disruption Associated With Increased Matrix Metalloproteinase-9 Activity in Experimental Diabetes. J Neurochem (2015) 132:595–608. doi: 10.1111/jnc.12939
163. Singh TP, Morris DR, Smith S, Moxon JV, Golledge J. Systematic Review and Meta-Analysis of the Association Between C-Reactive Protein and Major Cardiovascular Events in Patients With Peripheral Artery Disease. Eur J Vasc Endovasc Surg (2017) 54:220–33. doi: 10.1016/j.ejvs.2017.05.009
164. Hattori Y, Hattori S, Sato N, Kasai K. High-Glucose-Induced Nuclear Factor kappaB Activation in Vascular Smooth Muscle Cells. Cardiovasc Res (2000) 46:188–97. doi: 10.1016/s0008-6363(99)00425-3
165. Kassan M, Choi SK, Galán M, Bishop A, Umezawa K, Trebak M, et al. Enhanced NF-κB Activity Impairs Vascular Function Through PARP-1-, SP-1-, and COX-2-Dependent Mechanisms in Type 2 Diabetes. Diabetes (2013) 62:2078–87. doi: 10.2337/db12-1374
166. Salas A, Panés J, Elizalde JI, Casadevall M, Anderson DC, Granger DN, et al. Mechanisms Responsible for Enhanced Inflammatory Response to Ischemia-Reperfusion in Diabetes. Am J Physiol (1998) 275:H1773–81. doi: 10.1152/ajpheart.1998.275.5.H1773
167. Yoshimura T, Sonoda K, Sugahara M, Mochizuki Y, Enaida H, Oshima Y, et al. Comprehensive Analysis of Inflammatory Immune Mediators in Vitreoretinal Diseases. PloS One (2009) 4:e8158. doi: 10.1371/journal.pone.0008158
168. Rom S, Zuluaga-Ramirez V, Gajghate S, Seliga A, Winfield M, Heldt NA, et al. Hyperglycemia-Driven Neuroinflammation Compromises BBB Leading to Memory Loss in Both Diabetes Mellitus (DM) Type 1 and Type 2 Mouse Models. Mol Neurobiol (2019) 56:1883–96. doi: 10.1007/s12035-018-1195-5
169. Jackson L, Dumanli S, Johnson MH, Fagan SC, Ergul A. Microglia Knockdown Reduces Inflammation and Preserves Cognition in Diabetic Animals After Experimental Stroke. J Neuroinflamm (2020) 17:137. doi: 10.1186/s12974-020-01815-3
170. Zhang L, Chopp M, Zhang Y, Xiong Y, Li C, Sadry N, et al. Diabetes Mellitus Impairs Cognitive Function in Middle-Aged Rats and Neurological Recovery in Middle-Aged Rats After Stroke. Stroke (2016) 47:2112–8. doi: 10.1161/STROKEAHA.115.012578
171. Zhang Y, Zhang X, Ma G, Qin W, Yang J, Lin J, et al. Neurovascular Coupling Alterations in Type 2 Diabetes: A 5-Year Longitudinal MRI Study. BMJ Open Diabetes Res Care (2021) 9:e001433. doi: 10.1136/bmjdrc-2020-001433
172. Kusaka I, Kusaka G, Zhou C, Ishikawa M, Nanda A, Granger DN, et al. Role of AT1 Receptors and NAD(P)H Oxidase in Diabetes-Aggravated Ischemic Brain Injury. Am J Physiol Heart Circ Physiol (2004) 286:H2442–2451. doi: 10.1152/ajpheart.01169.2003
173. Tureyen K, Bowen K, Liang J, Dempsey RJ, Vemuganti R. Exacerbated Brain Damage, Edema and Inflammation in Type-2 Diabetic Mice Subjected to Focal Ischemia. J Neurochem (2011) 116:499–507. doi: 10.1111/j.1471-4159.2010.07127.x
174. Kalani A, Kamat PK, Tyagi N. Diabetic Stroke Severity: Epigenetic Remodeling and Neuronal, Glial, and Vascular Dysfunction. Diabetes (2015) 64:4260–71. doi: 10.2337/db15-0422
175. Fredrickson DS. An International Classification of Hyperlipidemias and Hyperlipoproteinemias. Ann Intern Med (1971) 75:471–2. doi: 10.7326/0003-4819-75-3-471
176. Ayata C, Shin HK, Dileköz E, Atochin DN, Kashiwagi S, Eikermann-Haerter K, et al. Hyperlipidemia Disrupts Cerebrovascular Reflexes and Worsens Ischemic Perfusion Defect. J Cereb Blood Flow Metab (2013) 33:954–62. doi: 10.1038/jcbfm.2013.38
177. Kitayama J, Faraci FM, Lentz SR, Heistad DD. Cerebral Vascular Dysfunction During Hypercholesterolemia. Stroke (2007) 38:2136–41. doi: 10.1161/STROKEAHA.107.481879
178. Deng J, Zhang J, Feng C, Xiong L, Zuo Z. Critical Role of Matrix Metalloprotease-9 in Chronic High Fat Diet-Induced Cerebral Vascular Remodelling and Increase of Ischaemic Brain Injury in Mice†. Cardiovasc Res (2014) 103:473–84. doi: 10.1093/cvr/cvu154
179. Greenfield JR, Samaras K, Jenkins AB, Kelly PJ, Spector TD, Gallimore JR, et al. Obesity is an Important Determinant of Baseline Serum C-Reactive Protein Concentration in Monozygotic Twins, Independent of Genetic Influences. Circulation (2004) 109:3022–8. doi: 10.1161/01.CIR.0000130640.77501.79
180. Poznyak AV, Nikiforov NG, Markin AM, Kashirskikh DA, Myasoedova VA, Gerasimova EV, et al. Overview of OxLDL and Its Impact on Cardiovascular Health: Focus on Atherosclerosis. Front Pharmacol (2020) 11:613780. doi: 10.3389/fphar.2020.613780
181. Bai Y, Du S, Li F, Huang F, Deng R, Zhou J, et al. Histone Deacetylase-High Mobility Group Box-1 Pathway Targeted by Hypaconitine Suppresses the Apoptosis of Endothelial Cells. Exp Biol Med (Maywood) (2017) 242:527–35. doi: 10.1177/1535370216685433
182. Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, et al. CD36 Ligands Promote Sterile Inflammation Through Assembly of a Toll-Like Receptor 4 and 6 Heterodimer. Nat Immunol (2010) 11:155–61. doi: 10.1038/ni.1836
183. Zhao W, Wu C, Chen X. Cryptotanshinone Inhibits Oxidized LDL-Induced Adhesion Molecule Expression via ROS Dependent NF-κB Pathways. Cell Adh Migr (2016) 10:248–58. doi: 10.1080/19336918.2015.1119361
184. Rahman SMA, Van Dam A-M, Schultzberg M, Crisby M. High Cholesterol Diet Results in Increased Expression of Interleukin-6 and Caspase-1 in the Brain of Apolipoprotein E Knockout and Wild Type Mice. J Neuroimmunol (2005) 169:59–67. doi: 10.1016/j.jneuroim.2005.07.018
185. Yang W, Shi H, Zhang J, Shen Z, Zhou G, Hu M. Effects of the Duration of Hyperlipidemia on Cerebral Lipids, Vessels and Neurons in Rats. Lipids Health Dis (2017) 16:26. doi: 10.1186/s12944-016-0401-6
186. Micioni Di Bonaventura MV, Martinelli I, Moruzzi M, Micioni Di Bonaventura E, Giusepponi ME, Polidori C, et al. Brain Alterations in High Fat Diet Induced Obesity: Effects of Tart Cherry Seeds and Juice. Nutrients (2020) 12:E623. doi: 10.3390/nu12030623
187. Tong X-K, Trigiani LJ, Hamel E. High Cholesterol Triggers White Matter Alterations and Cognitive Deficits in a Mouse Model of Cerebrovascular Disease: Benefits of Simvastatin. Cell Death Dis (2019) 10:89. doi: 10.1038/s41419-018-1199-0
188. Chen YL, Wang LM, Chen Y, Gao JY, Marshall C, Cai ZY, et al. Changes in Astrocyte Functional Markers and β-Amyloid Metabolism-Related Proteins in the Early Stages of Hypercholesterolemia. Neuroscience (2016) 316:178–91. doi: 10.1016/j.neuroscience.2015.12.039
189. Peterson TC, Lechtenberg KJ, Piening BD, Lucas TA, Wei E, Chaib H, et al. Obesity Drives Delayed Infarct Expansion, Inflammation, and Distinct Gene Networks in a Mouse Stroke Model. Transl Stroke Res (2021) 12:331–46. doi: 10.1007/s12975-020-00826-9
190. Day NL, Floyd CL, D’Alessandro TL, Hubbard WJ, Chaudry IH. 17β-Estradiol Confers Protection After Traumatic Brain Injury in the Rat and Involves Activation of G Protein-Coupled Estrogen Receptor 1. J Neurotrauma (2013) 30:1531–41. doi: 10.1089/neu.2013.2854
191. Thaler JP, Yi C-X, Schur EA, Guyenet SJ, Hwang BH, Dietrich MO, et al. Obesity is Associated With Hypothalamic Injury in Rodents and Humans. J Clin Invest (2012) 122:153–62. doi: 10.1172/JCI59660
192. Deutsch C, Portik-Dobos V, Smith AD, Ergul A, Dorrance AM. Diet-Induced Obesity Causes Cerebral Vessel Remodeling and Increases the Damage Caused by Ischemic Stroke. Microvasc Res (2009) 78:100–6. doi: 10.1016/j.mvr.2009.04.004
193. ElAli A, Doeppner TR, Zechariah A, Hermann DM. Increased Blood-Brain Barrier Permeability and Brain Edema After Focal Cerebral Ischemia Induced by Hyperlipidemia: Role of Lipid Peroxidation and Calpain-1/2, Matrix Metalloproteinase-2/9, and RhoA Overactivation. Stroke (2011) 42:3238–44. doi: 10.1161/STROKEAHA.111.615559
194. Cao X-L, Du J, Zhang Y, Yan J-T, Hu X-M. Hyperlipidemia Exacerbates Cerebral Injury Through Oxidative Stress, Inflammation and Neuronal Apoptosis in MCAO/reperfusion Rats. Exp Brain Res (2015) 233:2753–65. doi: 10.1007/s00221-015-4269-x
195. Przykaza Ł, Kozniewska E. Ligands of the Neuropeptide Y Y2 Receptors as a Potential Multitarget Therapeutic Approach for the Protection of the Neurovascular Unit Against Acute Ischemia/Reperfusion: View From the Perspective of the Laboratory Bench. Transl Stroke Res (2021). doi: 10.1007/s12975-021-00930-4
196. Gauberti M, Montagne A, Marcos-Contreras OA, Le Béhot A, Maubert E, Vivien D. Ultra-Sensitive Molecular MRI of Vascular Cell Adhesion Molecule-1 Reveals a Dynamic Inflammatory Penumbra After Strokes. Stroke (2013) 44:1988–96. doi: 10.1161/STROKEAHA.111.000544
Keywords: adhesion molecules, cerebral ischemia/reperfusion, inflammation, neurovascular unit, stroke comorbidities
Citation: Przykaza Ł (2021) Understanding the Connection Between Common Stroke Comorbidities, Their Associated Inflammation, and the Course of the Cerebral Ischemia/Reperfusion Cascade. Front. Immunol. 12:782569. doi: 10.3389/fimmu.2021.782569
Received: 24 September 2021; Accepted: 29 October 2021;
Published: 15 November 2021.
Edited by:
Beate E. Kehrel, University Hospital Münster, GermanyReviewed by:
Sven Meuth, University Hospital of Düsseldorf, GermanyCopyright © 2021 Przykaza. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Łukasz Przykaza, bHByenlrYXphQGltZGlrLnBhbi5wbA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.