 Heather A. Parker
Heather A. Parker Lorna Forrester
Lorna Forrester Christopher D. Kaldor
Christopher D. Kaldor Nina Dickerhof
Nina Dickerhof Mark B. Hampton
Mark B. Hampton- Centre for Free Radical Research, Department of Pathology and Biomedical Science, University of Otago, Christchurch, New Zealand
The mycobacterium genus contains a broad range of species, including the human pathogens M. tuberculosis and M. leprae. These bacteria are best known for their residence inside host cells. Neutrophils are frequently observed at sites of mycobacterial infection, but their role in clearance is not well understood. In this review, we discuss how neutrophils attempt to control mycobacterial infections, either through the ingestion of bacteria into intracellular phagosomes, or the release of neutrophil extracellular traps (NETs). Despite their powerful antimicrobial activity, including the production of reactive oxidants such as hypochlorous acid, neutrophils appear ineffective in killing pathogenic mycobacteria. We explore mycobacterial resistance mechanisms, and how thwarting neutrophil action exacerbates disease pathology. A better understanding of how mycobacteria protect themselves from neutrophils will aid the development of novel strategies that facilitate bacterial clearance and limit host tissue damage.
Introduction
Mycobacterium is a diverse genus comprising almost 200 species (1). The most well-known members are the human pathogens Mycobacterium tuberculosis and Mycobacterium leprae, which are the causative agents of tuberculosis and leprosy, respectively. Tuberculosis is a pulmonary disease that has plagued humans for thousands of years, and while global prevalence was reduced in the early 20th century due to the development of vaccines and antibiotics, the incidence has increased again such that it is estimated that a quarter of the world’s population is currently infected with M. tuberculosis with more than 4,000 deaths per day (2). The prevalence of leprosy is still of significant concern in endemic areas (3), and while curable the age-old stigma associated with leprosy still persists, creating fear and a reluctance to seek medical help. The mycobacterium genus also contains obligate and opportunistic pathogenic mycobacteria, which are grouped together as non-tuberculous mycobacteria (NTM). The incidence of NTM infection is increasing, such that in the USA the prevalence of pulmonary disease due to NTM is now greater than that of tuberculosis (4). The appearance of multi-drug resistant mycobacteria is of major concern, and new treatments are urgently required.
Mycobacteria can be remarkably successful intracellular pathogens that not only survive the initial assault of the innate immune system, but eventually take up residence within macrophages (5, 6). Neutrophils are also prominent in the lungs of patients with active pulmonary tuberculosis (7). They migrate to sites of infection in response to chemotactic signals, where they ingest pathogens into intracellular phagosomes (Figure 1). Neutrophil cytoplasmic granules fuse with the phagosomal membrane and empty antimicrobial peptides and proteins onto the pathogen, in a process termed degranulation. At the same time, an NADPH oxidase (NOX) complex assembles on the phagosomal membrane, transferring electrons from cytosolic NADPH to molecular oxygen in the phagosome. The initial product is superoxide, which dismutates to hydrogen peroxide and is converted to the potent bactericidal oxidant hypochlorous acid (HOCl) by another granule constituent, myeloperoxidase (MPO) (8). Pathogens that survive the initial oxidative burst may take up residence inside neutrophils. However, the neutrophil is a short-lived cell, providing a transport route into resident macrophages that are charged with clearing apoptotic neutrophils.
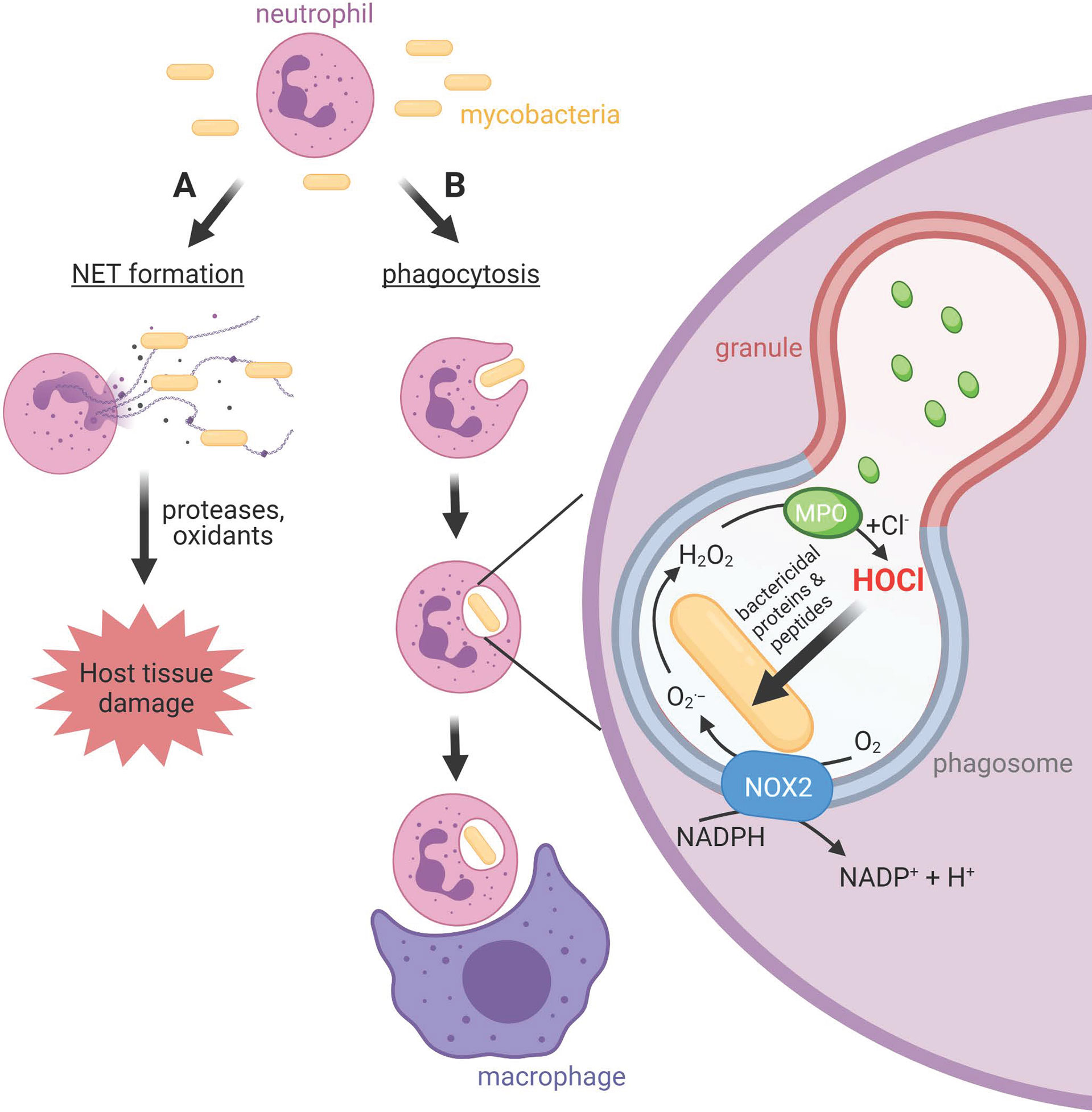
Figure 1 Neutrophil activities at sites of mycobacterial infections. (A) Neutrophils release neutrophil extracellular traps (NETs), chromatin structures decorated with neutrophil bactericidal peptides and proteins, in response to mycobacteria. Whether NETs contribute to bacterial clearance or predominantly promote host tissue damage is unclear. (B) Neutrophils phagocytose mycobacteria, albeit slower than other bacteria. The phagosomal membrane fuses with cytoplasmic granules to release antimicrobial peptides and proteins including myeloperoxidase (MPO) into the phagosome. The NOX2 assembles on the phagosomal membrane resulting in the production of superoxide and hydrogen peroxide, which MPO uses to produce the strong antimicrobial oxidant HOCl. Unlike other bacteria, and for reasons as yet unknown, mycobacteria do not succumb to HOCl produced in the phagosome. Neutrophils and their resident mycobacteria are ingested by macrophages. This may augment macrophage killing of mycobacteria via delivery of neutrophil antimicrobial agents, or provide transfer of live bacteria to a longer-lived host cell.
While the internalization of pathogens limits exposure of host tissue to the toxic compounds produced by neutrophils, extracellular release can occur (Figure 1). This includes the ejection of strands of chromatin coated with neutrophil proteins, which form a meshwork termed neutrophil extracellular traps (NETs) (9–11). NETs have been shown to trap bacteria and fungi and are thought to contribute towards containment of infection (9, 12) and antimicrobial activity (9, 13, 14). However, NETs can cause damage to host cells and tissue, and NETs are linked with various pathological conditions and diseases (15, 16). Unresolved inflammation will damage lung tissue and provide further opportunity for bacterial expansion.
In recent years it has become clear that neutrophils have more complex roles in immune regulation, with their ability to produce and modify cytokines, and release extracellular vesicles (17), enabling significant crosstalk with adaptive immune cells (18). This review focuses, however, on the early interactions between mycobacteria and neutrophils, and we ask the question of how pathogenic mycobacteria avoid destruction by neutrophils. Insight into their underlying survival mechanisms may provide therapeutic strategies that tilt this balance in favour of the neutrophil, and enable the early resolution of infection.
Neutrophils in Mycobacterial Infection and Disease
Neutrophils in Tuberculosis
Lung resident macrophages are the first immune cells to encounter inhaled M. tuberculosis, and they contribute towards bacterial clearance (19, 20). Neutrophils are subsequently recruited to the site of infection. The number of circulating neutrophils increases in patients with active tuberculosis (21–23), and a rise in neutrophil-derived transcriptional signatures has been observed in blood from patients with active tuberculosis (24). A study of close contacts of patients with active pulmonary tuberculosis showed an inverse correlation between peripheral blood neutrophil counts and risk of M. tuberculosis infection (21), and depletion of neutrophils from whole blood in vitro increased M. tuberculosis growth (25), suggesting that neutrophils can play an active role in limiting infection. Indeed, more neutrophils than macrophages were observed to have intracellular M. tuberculosis in sputum, bronchoalveolar lavage (BAL) fluid and granulomas from patients with active pulmonary tuberculosis (7). However, the fate of M. tuberculosis phagocytosed by neutrophils is not clear.
M. tuberculosis can survive and replicate within macrophages (26–28), where they are hidden from the immune system (29). Neutrophils have been proposed to play a similar “Trojan horse” role for M. tuberculosis (30, 31). Some bacteria and parasites, including Yersinia pestis (32), Chlamydia pneumoniae (33) and Leishmania major (34) can survive within neutrophils, and the length of M. tuberculosis bacilli observed in BAL fluid and sputum from patients with active tuberculosis was noted to be similar to lengths observed in logarithmic phase cultures (7), consistent with bacterial survival.
Neutrophils undergo apoptosis at sites of infection and can be cleared by macrophages. In these cases, any viable intracellular bacteria in neutrophils will be transferred to macrophages, where they can replicate and modulate macrophage responses and function. While it has been shown that neutrophils provide a source of antimicrobial agents that potentiate macrophage killing of M. tuberculosis by delivery of their granule contents (35), the survival of M. tuberculosis within macrophages has been shown to be enhanced after ingestion of neutrophils containing the bacterium (36). Further evidence that neutrophils play a permissive role in M. tuberculosis transmission was recently demonstrated in a report showing that dead neutrophils have the capacity to mediate short range, aerosol transmission of viable M. tuberculosis aggregates (37).
Disease severity in tuberculosis-sensitive mice has been linked to the survival of M. tuberculosis within neutrophils (31, 38). Neutrophil phagocytosis of M. tuberculosis was greater in genetically-susceptible mice than in those more resistant to tuberculosis, but the majority of the bacteria remained viable within the neutrophils (31). Furthermore, in a study of M. bovis BCG infection following inoculation of bacteria into the ears of C57BL/6 mice, neutrophils were observed to transport BCG to the auricular draining lymph nodes early in infection (30). BCG-laden neutrophils penetrated the paracortex, an area that is rich in T-cells (39). Notably, the bacilli load at the infection site did not decrease, indicating that in this model neutrophils do not clear the infection. Neutrophils are reported to be capable of functioning as antigen presenting cells (40), and others have suggested that while neutrophils may not directly control M. tuberculosis growth, they promote migration of dendritic cells to the lung draining mediastinal lymph node, facilitating priming of the adaptive immune response (41). However, neutrophils may protect the phagocytosed bacteria from recognition by the immune system thus delaying the adaptive immune response. In a study by Abadie et al. live BCG were recovered from the lymph nodes in numbers that remained stable over two weeks, lending support to this scenario (30).
The host response to pulmonary M. tuberculosis infection involves formation of nodule-like structures in the lung called granulomas, which comprise an assemblage of various immune cells, including neutrophils (42). Granulomas contain infection by preventing dissemination of bacteria; however, they may also provide a niche for M. tuberculosis survival (43–45). Neutrophils in zebrafish infected with M. marinum have been shown to work in conjunction with macrophages in developing granulomas to contain infection (46). However, in active disease the granulomas develop into large inflammatory lesions, and neutrophil accumulation may play an important part of this process. In a non-human primate model of tuberculosis, greater neutrophil accumulation was observed in the larger granulomas associated with active tuberculosis compared with the smaller granulomas of a latent infection (47). In a mouse model, interleukin (IL)-17 neutralization decreased neutrophil accumulation in lung granulomas but there was no difference in bacterial burden (47), while neutrophil depletion in C57BL/6J mice during the chronic phase of infection improved control of M. tuberculosis (48).
Several studies suggest that neutrophil accumulation is associated with a dysregulated immune response and a poor prognosis in tuberculosis. Even after bacterial clearance, neutrophil accumulation is linked with post-TB lung disease (49). Levels of calprotectin, the most abundant neutrophil cytoplasmic protein, and neutrophil chemokines are increased in patients with active tuberculosis compared to healthy controls and those with latent infection, and this is correlated with lung damage (47). Increased neutrophil accumulation was shown to be mediated, at least in part, via calprotectin-dependent upregulation of the neutrophil integrin CD11b (48). Calprotectin has antimicrobial activity, however improved control of infection was observed when a subunit of calprotectin was knocked out (48). Mice infected by aerosol with a low dose of M. tuberculosis were grouped into three different classes: resistant, susceptible and super-susceptible mice; reflecting the variation observed in humans (50). Lungs of super-susceptible mice showed the highest numbers of infiltrating neutrophils, had the largest granulomas with areas of necrosis, and the highest bacterial burden. Another mouse study found that neutrophil depletion significantly increased survival following M. tuberculosis infection (51). Using several different models, Mishra et al. concluded that neutrophil accumulation in the lungs correlated with increased bacterial burden and animal weight loss, and that inhibition of neutrophil recruitment decreased bacterial numbers (52).
In summary, current evidence indicates that neutrophils play an important role during in bacterial clearance the acute stages of human TB. This is supported by several animal studies (53, 54). However, if the infection continues, accumulating evidence indicates that neutrophils contribute towards disease pathology and a negative prognosis. Investigation of the suitability of neutrophils as a marker for poor outcome in TB is ongoing (55).
Neutrophils in Leprosy
M. leprae infects macrophages (56) and Schwann cells (57), and it is these cells that are traditionally thought to be important players in infection (58, 59). Peripheral blood neutrophils from lepromatous leprosy patients have, however, also been shown to harbor M. leprae (60). The course of lepromatous leprosy can involve periods of acute inflammation called reactions, the most common of which is erythema nodosum leprosum (ENL). Neutrophil infiltration is a feature of the skin lesions associated with this reaction, although neutrophils are not always present (61–63). ENL can occur as a single acute episode, several discrete acute episodes or as a chronic condition, and can occur before, during or after multi-drug therapy (64, 65). Importantly, ENL is a major contributor towards nerve damage in leprosy patients and contributes significantly towards mortality (66). Despite the fact that neutrophils are the hallmark of ENL in histological samples (67), few studies have examined their role, but the evidence indicates neutrophils may make a significant contribution to the pathogenesis of ENL [reviewed in (63, 68, 69)].
Two early studies examined neutrophil activation in ENL by measuring reduction of nitroblue tetrazolium (NBT) (70, 71), which is reduced by superoxide to dark formazan precipitates (72). In the first study, spontaneous reduction of NBT was significantly increased in blood from patients with reactional lepromatous leprosy (RLL), of which ENL is a major subset, compared with blood from healthy controls and patients with other forms of leprosy (70). RLL patient sera did not increase neutrophil activation in blood from healthy controls, suggesting that the activating stimulus was not present in the circulation (70). In contrast, the second study that used isolated neutrophils reported no increase in spontaneous NBT reduction in neutrophils from ENL patients, and ENL patient sera induced a large increase in NBT reduction in neutrophils from healthy controls and ENL patients (71). The difference between studies may have been due to the use of heparin at higher concentrations, which can form particles with NBT that activate neutrophils (73), and/or the presence of an inhibitory factor that is absent from ENL sera.
Activated neutrophils express the Fcγ-receptor I (Fcγ-R1) (also known as CD64), a high affinity receptor for IgG (74). Fcγ-R1 expression was observed on neutrophils within ENL skin lesions and expression was significantly higher in the peripheral blood of ENL patients compared to lepromatous leprosy patients without ENL (62). The presence of Fcγ-R1 expressing neutrophils in peripheral blood increased with disease severity, and treatment of ENL with thalidomide, which improved symptoms, decreased Fcγ-R1 expression and the level of neutrophils in skin lesions (62). In addition, the neutrophil granule protein pentraxin-3 (PTX-3) was found to be increased in the blood of multibacillary leprosy patients, in particular, levels were higher in those that went on to develop ENL (75). PTX-3 levels correlated with Fcγ-R1 expression in the circulation and PTX-3 was also increased in ENL skin lesions and correlated with neutrophils (myeloperoxidase) (75). More studies on the relationship of neutrophils to more severe forms of leprosy are needed.
Neutrophils in Other Mycobacterial Infections
M. avium complex (MAC) are the most common cause of NTM-induced pulmonary disease, predominately, but not exclusively, in those with pre-existing lung conditions (76). They are also a leading cause of disseminated NTM infection (76). Mouse studies examining the role of neutrophils in MAC infection show differences in their role dependent on the form of infection, pulmonary or systemic. C57BL/6 mice with the beige mutation, whose neutrophils are defective in chemotaxis and killing (77), have increased susceptibility to MAC infection (78, 79). In a study of disseminated infection, neutrophil transfusion from WT mice improved the resistance of beige mice to intravenous M. avium infection while neutrophil depletion in WT mice increased their susceptibility (78). This implies a protective role for neutrophils in systemic MAC infection in this mouse model. However, in lung infection of C57BL/6 mice, Saunders et al. found that a 95% decrease in neutrophils in the lungs had no effect on bacterial numbers, indicating neutrophils are dispensable in controlling MAC lung infection in these mice (79). Further support that neutrophils are ineffective in controlling MAC infection was provided by a study of mice over-expressing the transcription factor RAR-related orphan receptor gamma t (RORγt), which regulates Th17 responses and increased pulmonary neutrophil infiltration, yet bacterial burden was similar to that in WT mice (80).
In humans, a study of MAC-infected patients with no pre-existing lung disease found that the level of neutrophils in patient BAL fluid was significantly higher than that of control patients (81), with higher neutrophil counts subsequently correlated with worsening disease (82). Similarly, in a more recent retrospective study of pulmonary MAC infection, BAL fluid from patients whose disease progressed had higher numbers of neutrophils than those who had stable infection (83). Together these studies provide evidence that neutrophils are ineffective in preventing MAC-induced pulmonary disease.
Slow growing M. kansasii are considered to be the most pathogenic of the NTM, as when isolated they are almost always associated with disease (84, 85). M. kansasii most frequently cause pulmonary disease that is clinically similar to tuberculosis (86), but infections at other sites are also reported (87). In humans, abundant neutrophils have been observed at sites of M. kansasii infection (87–89). In CD-1 mice, peritoneal inoculation with M. kansasii or M. avium was found to lead to chronic neutrophil infiltration, with bacterial numbers gradually decreased during this time (90). Neutrophil ingestion of M. kansasii and M. avium was not examined, however neutrophil uptake of non-pathogenic M. aurum only occurred during the first two days (90). Macrophages were present at the infection site and ingested dying neutrophils (90). Lactoferrin, present in neutrophils but not macrophages, was detected within peritoneal macrophages suggesting transfer from neutrophils to macrophages either by neutrophil degranulation and subsequent uptake of granule components by resident macrophages, or through uptake of the intact neutrophil. Macrophage antibacterial activity in vitro was enhanced when the macrophages were pre-incubated with neutrophils, leading the authors to conclude that neutrophils do not directly control NTM infection but participate indirectly via transfer of macromolecules that enhance macrophage killing of these bacteria (90).
M. abscessus are rapidly growing NTM that cause pulmonary disease in both healthy individuals and those with underlying lung disease, disseminated infections, and skin and soft tissue infections (91–93). Of note, M. abscessus are particularly recalcitrant to antibiotic therapy (94, 95). Extensive numbers of neutrophils are reported within patient granulomas or at the site of infection (96–98). In addition, human lung tissue infected with M. abscessus ex vivo showed bacteria within neutrophils at the site of infection (99). M. abscessus, like several other NTM (91), exist in one of two colony morphological forms. A smooth, non-cording form and a rough cording form, that differ in their concentration of cell wall glycopeptidolipid (100). The rough form is associated with more severe pulmonary disease (101). Greater neutrophil numbers were measured in BAL fluid from C57BL/6 mice infected with the rough form (102). In a zebrafish model of M. abscessus infection, ingestion of the rough form by macrophages was associated with increased macrophage apoptosis, release of viable bacteria and intense cording growth, which neither macrophages nor neutrophils could engulf because of the size of the cords (103). However, in a subsequent study both rough and smooth forms induced a large influx of neutrophils early at the infection site that was dependent on IL-8 and macrophage-secreted TNF (104). Both forms were engulfed equally by neutrophils, and neutrophil depletion resulted in uncontrolled bacterial growth and zebrafish larvae death with either (104). Neutrophils were found to be essential for the development and maintenance of protective granuloma in the infected zebrafish (104).
M. smegmatis is a rapidly growing NTM that is ubiquitous in the environment and is generally considered to be non-pathogenic. Due to its fast replication rate (relative to other mycobacteria), amenability to genetic manipulation, and the fact that it can be grown under normal Biosafety Level 2 laboratory conditions, M. smegmatis is often used as a model to study M. tuberculosis infection and virulence factors. Occasionally M. smegmatis causes skin and soft tissue infections, and very rarely disseminated infection (105–108). Hospital-acquired infections also occur, resulting from a variety of procedures including catheterization, cardiac and plastic surgery (109). Although infrequent, these infections can be difficult to treat requiring surgical debridement and long term antibiotic therapy (105). Neutrophils are recruited to the site of infection in humans (106, 110), and in mice infected with M. smegmatis intratracheally (111). M. smegmatis has been shown to induce neutrophil exocytosis of gelatinase granules releasing active matrix metalloproteinase-9 that degrades the extracellular matrix (112). Release of these granules may contribute towards the tissue degradation observed in soft tissue infections caused by this bacteria. Neutrophil exocytosis of azurophilic granules has also been reported in response to M. smegmatis and constituents of these granules can also cause host tissue damage (113).
Much work remains to be done to gain a better understanding of the role of neutrophils in mycobacterial infection and disease. The plethora of studies implicating neutrophils in the pathogenesis of mycobacterial infections strongly suggests that the capacity of neutrophils to control infection by intra- and extra-cellular killing mechanisms is either insufficient, defective or thwarted by mycobacteria. In the next sections of this review we discuss the current evidence for neutrophil phagocytosis and killing of mycobacteria (both phagosomal and NET-mediated) and evidence for resistance of mycobacteria to neutrophil oxidants.
Neutrophil Phagocytosis of Mycobacteria
The major antimicrobial strategy employed by neutrophils involves the ingestion of pathogens into phagosomes, followed by the degranulation of antimicrobial peptides and proteins and the production of toxic reactive oxygen species inside the phagosome (8, 114). Neutrophils are known to phagocytose both pathogenic and non-pathogenic mycobacteria (7, 30, 36, 112, 115–121), with ingestion of mycobacteria increasing in the presence of serum (112, 115–117). The mechanism of phagocytosis differs depending on whether neutrophils bind opsonized or non-opsonized mycobacteria. Neutrophil complement receptor 3 (CR3) binds M. leprae phenolic glycolipid-I resulting in bacterial phagocytosis and activation of the Syk tyrosine kinase, which leads to activation of the transcription factor NFATc and Il-10 production (122). Phagocytosis of non-opsonized M. kansasii has also been shown to occur via CR3 in a cholesterol-dependent and glycosylphosphatidylinositol (GPI) anchored protein-dependent manner, while cholesterol was not required with opsonized bacteria (123). Phagocytosis of non-opsonized M. smegmatis was also dependent on CR3 and cholesterol (113). GPI-anchored proteins and cholesterol accumulate in lipid rafts (124) suggesting that the localization of CR3 to lipid rafts is required for neutrophil internalization of non-opsonized mycobacteria. The glycosphingolipid lactosylceramide (LacCer), enriched in lipid rafts (125), is also required for non-opsonic internalization of mycobacteria (126). Binding to LacCer is mediated by lipoarabinomannan (LAM) on the mycobacterial surface. Interestingly, the mannose cap on LAM (ManLAM) of pathogenic mycobacteria, but not the phosphoinositol cap (PILAM) of non-pathogenic mycobacteria, appears to prevent fusion of azurophil granules with the phagosome (126), which will have a significant impact on the antimicrobial activity of neutrophils.
While most studies report that neutrophils phagocytose mycobacteria, a few have reported impaired phagocytosis. In zebrafish infected with M. marium, neutrophils were absent from the initial infection site but were recruited to the developing granuloma by dying macrophages (46). Neutrophils then phagocytosed M. marium indirectly by taking them up from macrophages. The investigators observed a small increase in direct neutrophil phagocytosis of M. marium when inoculum numbers were increased (46). Neutrophil uptake in BALB/c mice was shown to be negligible after intravenous injection of a relatively low dose of M. tuberculosis (53). However, Abadie et al. observed abundant neutrophil phagocytosis of BCG in C57BL/6 mice after inoculation with a similar low dose of bacteria as present in the BCG vaccine (30).
Recently we measured the rate of phagocytosis of M. smegmatis and found that it was five times slower than the phagocytosis of Staphylococcus aureus and 3.5 times slower than that for Escherichia coli (127, 128). Phagocytosis of M. abscessus was also found to be slower than that of S. aureus when examined by counting the number of neutrophils containing fluorescently labelled bacteria (129). In another study, approximately half of a population of M. fortuitum was phagocytosed within 30 min (130), only slightly faster than the 43 min we measured for M. smegmatis, and still considerably slower than the 9 min for S. aureus and 12 min for E. coli (127, 131). As far as we are aware no in vitro studies have directly compared phagocytosis of different mycobacteria by the same neutrophils; however, neutrophils were found to ingest M. abscessus more frequently than either M. tuberculosis or M. avium in an ex vivo infection of human lung tissue (99).
Evidence from tuberculosis patients indicates that patient neutrophils are primed for phagocytosis. Surface expression of the Ig receptor FcγR1 (CD64) was found to be increased on peripheral blood neutrophils from patients with tuberculosis pleuritis compared to healthy controls, and neutrophils obtained from pleural fluid showed further enhanced FcγR1 expression and increased expression of the pattern recognition receptor TLR2 (132). An increase in expression of FcγR1, TLR2 and TLR4 was also observed in neutrophils from the peripheral blood of patients with active pulmonary tuberculosis prior to treatment (133). Despite the evidence for increased receptor expression, studies examining the phagocytic activity of neutrophils from patients with active pulmonary tuberculosis generally show their capacity for phagocytosis is decreased. Hilda et al. measured significantly reduced phagocytic activity in blood neutrophils from patients prior to treatment (133), and Shalekoff et al. also found peripheral blood neutrophil function impaired in patients with active pulmonary tuberculosis (134). Patient neutrophils showed a reduced capacity for phagocytosis compared to healthy controls and this was observed both soon after the start of treatment and when treatment had been undertaken for almost 30 weeks (134). Similarly, another study found peripheral blood neutrophils from tuberculosis patients had reduced phagocytic activity compared to healthy controls (135). In these studies, phagocytic capacity was measured in patient neutrophils by assessing phagocytosis of E. coli or yeast to rule out M. tuberculosis factors that may interfere with neutrophil uptake.
Mycobacteria are able to directly inhibit neutrophil phagocytosis. M. abscessus rough morphotypes prevent phagocytosis by formation of serpentine cords that are too large for neutrophils to ingest, with these large aggregates linked to pathogenesis (103). M. leprae cell wall lipids inhibit macrophage phagocytic activity (136), but to our knowledge there are no reports on whether these lipids affect neutrophil phagocytosis. Exposure of M. tuberculosis to human alveolar lining fluid results in changes to the bacterial cell wall and release of cell wall fragments (137). Macrophage phagocytosis of M. tuberculosis decreased when the bacteria were pre-exposed to alveolar lining fluid, however, neutrophil phagocytosis was found to increase (138, 139). This increase in phagocytosis was due to bacterial alveolar lining fluid exposure rather than M. tuberculosis cell wall fragments released by treatment with alveolar lining fluid (139).
Phagosomal Killing of Mycobacteria
The ability of neutrophils to kill M. tuberculosis is controversial with some studies observing killing (116, 140–142) while others do not (115, 143–145). Neutrophils have been reported to kill 50-70% of M. tuberculosis within 90-120 minutes (116, 140); however, Corleis et al. found neutrophils did not kill M. tuberculosis even after six hours co-incubation (115) and others found no killing after incubation with neutrophils for 24 hours (144, 145). While Hartman et al. reported near complete killing of M. avium by neutrophils in two hours (120), only around 40% of populations of M. abscessus and M. fortuitum were killed after two hours incubation with neutrophils (146). We have previously reported half-lives for E. coli and S. aureus inside the neutrophil phagosome of 2 min and 6 min, respectively (128), and recently measured a half-life for M. smegmatis of 30 min inside the neutrophil phagosome, which indicates that even though killing occurs, it is slow (127).
In terms of bactericidal mechanisms, the neutrophil oxidative burst is activated upon uptake of various mycobacteria, including M. tuberculosis, M. canettii, M. abscessus, M. kansaii, M. phlei, M. fortuitum and M. smegmatis (115, 117, 130, 144, 146–148). Contrary to this, M. gordonae did not stimulate neutrophil oxidant production (144) and M. bovis induced a weak oxidative burst in comparison to Listeria monocytogenes (118). Interestingly, M. tuberculosis were found to induce a stronger oxidative burst than M. smegmatis, yet M. smegmatis was killed while M. tuberculosis was not (115). Rough morphotypes of M. abscessus induced a stronger oxidative burst in comparison to smooth morphotypes, yet neutrophil killing of both was unaffected by inhibition of oxidant production (146).
The purified MPO/H2O2/Cl- system is capable of killing M. tuberculosis and M. leprae (149, 150); however, it has been reported that the complete system did not augment killing of M. tuberculosis over that observed by H2O2 alone (141). Reagent HOCl killed M. smegmatis, but approximately seven times more HOCl was required to kill M. smegmatis than S. aureus suggesting mycobacteria may be innately more resistant to HOCl (127). Further studies are required to determine if other mycobacteria are similarly resistant to HOCl. We recently sought to examine whether HOCl plays a role in killing of M. smegmatis in the neutrophil phagosome. The amount of reagent HOCl required to kill a bacterium cannot be directly translated to the phagosome as neutrophil oxidants are produced in a flux in the phagosome, and a flux of HOCl may be less harmful to the bacteria than a single high dose. Additionally, the neutrophil phagosome contains many proteins and amines that can react with phagosomal HOCl before it reaches the bacterium (151, 152). Using a fluorescent probe we observed HOCl production in the phagosome and that MPO inhibition abrogated HOCl production (127). By modelling the data obtained in our study we estimated that it would take 30-40 minutes at full MPO capacity for sufficient HOCl to be produced to kill a single ingested M. smegmatis. Sustained MPO activity for that length of time is unlikely and therefore we concluded that insufficient HOCl is produced in the neutrophil phagosome to directly kill this bacterium. In support of this conclusion, inhibition of MPO had no effect on neutrophil killing of M. smegmatis (127).
Inhibition of the NADPH oxidase had no effect on the ability of human neutrophils to kill M. tuberculosis and M. abscessus (142, 146). Defective killing by neutrophils from patients with chronic granulomatous disease (CGD), who have mutations that lead to a non-functional NADPH oxidase (153), is often used as evidence for the requirement for oxidants in bacterial killing. Jones et al. showed that CGD neutrophils killed M. tuberculosis as effectively as neutrophils from healthy donors (140). However, in countries where tuberculosis is endemic, the incidence of tuberculosis is greater in CGD patients than the rest of the population (154–156), though it is important to consider that NOX2 has other roles during inflammation (157). X-linked CGD mice showed increased bacterial growth and an increase in granuloma size in their lungs compared to C57BL/6 mice (158). A high incidence of complications due to BCG vaccination has also been reported in CGD patients (155, 156, 159–161). Neutrophil killing of M. marinum was found to depend on an active NADPH oxidase in a zebrafish model of early tuberculosis disease using morphant larvae with neutrophils deficient in two subunits of the NADPH oxidase (gp91phox and pg22phox) (46). In a zebrafish model of cystic fibrosis, transmembrane conductance regulator morphants showed reduced neutrophil oxidant production and reduced intracellular control of M. abscessus that was linked to a reduction in NADPH oxidase activity (147).
Neutrophil killing of mycobacteria can occur via non-oxidative processes. Defensins, components of azurophil granules, have been shown to have anti-mycobacterial activity (21, 162, 163). Defensin-depleted granules have also been shown to kill M. tuberculosis, M. bovis BCG and M. smegmatis although M. tuberculosis were more resistant to killing (164). The azurophil granule proteins elastase, azurocidin, and lysozyme, and the specific granule protein lactoferrin were shown to kill M. smegmatis (164). In addition, conditioned media from neutrophils incubated with M. abscessus for 90 minutes showed bactericidal activity towards this bacterium (146). This was most likely due to degranulation of cytotoxic agents. In vitro, M. tuberculosis induced neutrophils to release MPO and elastase (165) and neutrophils were found to release elastase and cathepsin G, another major azurophil granule protein, into the bronchoalveolar space in mice infected with M. bovis BCG (166). Released neutrophil granule proteins have been shown to be taken up by infected macrophages and to increase macrophage killing of M. tuberculosis and M. bovis BCG (35, 164). Efferocytosis also potentiates macrophage killing of ingested M. tuberculosis through delivery of granule proteins to early endosomes and fusion of these with the phagosome (35). Augmented macrophage killing due to uptake of neutrophil granule proteins has been demonstrated in other bacteria (167, 168). When neutrophils struggle to control a mycobacterial infection through intracellular killing, degranulation and efferocytosis could be an important mechanism by which neutrophils contribute towards host defense.
In the early stages of M. tuberculosis infection, and when they escape from phagocytic cells, M. tuberculosis are exposed to a variety of agents in alveolar lining fluid (ALF) that can alter the bacterial cell wall (137). Exposure of neutrophils to M. tuberculosis pre-treated with human ALF increased phagocytosis and neutrophil killing of M. tuberculosis while dampening the oxidative burst response (138). The increase in intracellular killing corresponded with an increase in granule/phagosome fusion and was mediated by a protein component in ALF, as the observed increase in killing was lost when ALF was heat-inactivated (138). Incubation with ALF also reduced extracellular degranulation in response to M. tuberculosis (138). Of note, neutrophils infected with ALF-treated M. tuberculosis did not activate macrophages and infection with ALF-treated M. tuberculosis had no significant effect on neutrophil apoptosis or necrosis. This study shows that exposure of M. tuberculosis to ALF alters the interaction of the bacteria with neutrophils in a way that facilitates neutrophils to kill the bacteria intracellularly without release of damaging neutrophil proteins.
Taken together, the evidence so far indicates that neutrophil oxidants are dispensable for killing of some mycobacteria. By dispensable we mean that oxidants are likely to contribute when they are being produced, but in their absence the non-oxidative mechanisms are able to compensate. More studies are required, ideally using neutrophils from CGD patients, to examine neutrophil killing of a wider range of mycobacteria. To our knowledge the question of whether neutrophils kill M. leprae is unanswered, and more studies are also needed to examine neutrophil killing of the more significant NTM, particularly MAC, M. kansaii and M. abscessus.
Neutrophil Extracellular Trap-Mediated Killing of Mycobacteria
NETs contain proteins with antimicrobial activities, including MPO, calprotectin and elastase (12, 13, 169). Histones on NETs have also been shown to mediate microbial killing as has NET-DNA (9, 14, 170). The prolonged presence of cytotoxic NET constituents as a result of excessive NET release or impaired clearance can also be detrimental to the host.
In vivo, NET markers have been measured in the plasma of patients with active tuberculosis (171, 172) and observed to decrease with antibiotic therapy (171). NETs have also been observed in BALF samples from mice 3 – 4 weeks after aerosol challenge with M. tuberculosis (119) and in skin samples from guinea pigs within several hours following intradermal M. tuberculosis inoculation (173), indicating that NETs may participate in both early and late stages of infection. Whether their presence is important in control of M. tuberculosis infection or if they contribute towards pathogenesis remains to be determined. Recently, NETs have been observed in lung lesions resected from patients with persistent pulmonary TB and from TB-susceptible mice lending support to their role in TB pathogenesis (174). Increased human DNA-histone complexes associated with NETs have been measured in ENL sera compared with lepromatous and borderline lepromatous leprosy patients (175). NET components (DNA/histone/MPO) were observed in ENL skin lesions, and DNA-histone complexes in patient sera were significantly higher than those with lepromatous and borderline lepromatous leprosy and healthy controls (176). This data is suggestive of NETs contributing towards more severe leprosy disease. A recent review discusses the potential for NETs as a putative prognostic tool in ENL to direct medical treatment (177).
Several mycobacteria, including M. tuberculosis, M. bovis BCG, M. abscessus, M. avium subsp. paratuberculosis and M. leprae, have been shown to induce NETs in vitro (121, 129, 146, 148, 176, 178–181). How the interaction of mycobacteria with neutrophils leads to the formation of NETs appears to differ depending on the bacterial species. The early secreted antigen-6 (ESAT-6) of M. tuberculosis has been shown to induce neutrophils to release NETs (148, 181, 182), as has secreted sphingomyelinase Rv088 (182, 183). Rv088 has both sphingomyelinase (184) and nuclease (185) activity but it is the sphingomyelinase activity that is required for neutrophils to form NETs (183). Phagocytosis is also a prerequisite for the induction of NETs in response to M. tuberculosis and M. abscessus, as NET formation did not occur when phagocytosis was inhibited with cytochalasin D (146, 181). In contrast, Branzk et al. found with M. bovis BCG that NETs only occurred in response to small non-phagocytosed aggregates (179). Single bacteria were phagocytosed and neutrophils containing these bacteria did not go on to form NETs, at least not over the four hours of this study (179).
Neutrophil oxidants are required for NET formation in response to various stimuli (186–188), and M. bovis BCG NET induction was shown to be dependent on neutrophil oxidants (189). In addition, NETs were not formed under hypoxia in response to M. tuberculosis (190). Both rough and smooth morphotypes of M. abscessus were found to induce NETs, though the mechanism differed between morphotypes (146). Early NET induction (up to one hour) was independent of neutrophil oxidant production, while NETs formed after four hours co-incubation with bacteria did depend on oxidants, similar to that which has been reported for S. aureus (191).
Despite increasing evidence that mycobacteria induce neutrophils to form NETs, there is relatively little information on whether NETs contribute to killing of mycobacteria or control of infection. Evidence suggests that M. abscessus are killed by NETs whereas M. tuberculosis are not (146, 148, 173). In a study by Ramos-Kichik et al., M. tuberculosis was not killed by NETs formed by PMA (148). NET constituents as well as their post-translation modifications can vary depending on the stimulus (11, 181) potentially altering their bactericidal activity. Therefore, it is important to examine NETs induced either by the bacterium studied or host/bacterial factors that may be present at the infection site. In a guinea pig model of extra-pulmonary tuberculosis, NETs induced by M. tuberculosis in vivo were not bactericidal (173). This provides good evidence that NETs do not contribute towards killing of M. tuberculosis, but further studies are required to corroborate this in human disease.
NET-mediated killing of M. abscessus, as with NET induction, appears to differ with the morphotype studied. Neutrophil killing of the smooth morphotype was found to occur predominately via NETs, as removal with DNase resulted in only slight killing (146). In contrast, approximately half the rough morphotype were still killed when NETs were degraded, indicating other neutrophil killing mechanisms are more important for this morphotype. Notably in this study, neutrophil killing was only measured over the first hour of co-incubation when NET release was relatively low making it difficult to gain a full appreciation of the effect of NETs on M. abscessus viability. As the mechanism of NET formation differed between early and later formed NETs, it is conceivable that later-formed NETs may contain different constituents and therefore different antimicrobial activity.
There is evidence that NETs may increase the anti-mycobacterial capacity of macrophages. In one study, NETs produced in response to M. tuberculosis were shown to activate macrophages, leading to production of pro-inflammatory cytokines (181). In another study, M. bovis BCG was shown to induce NETs that contain the antimicrobial cathelicidin LL37, and macrophages were observed to ingest NET fragments containing this cathelicidin (189). By creating DNA : LL37 complexes to mimic NET fragments, Stephan et al. monitored the intracellular localization of these complexes in macrophages that had already phagocytosed BCG. Following uptake, the DNA was degraded in lysosomes releasing LL37 in close proximity to the internalized bacteria and inhibiting bacterial growth (189). Interestingly, significantly greater growth inhibition was observed when infected macrophages were incubated with DNA : LL37 complexes rather than LL37 alone. The authors speculated that the binding and internalization of DNA may increase killing by activating intrinsic antimicrobial pathways within the macrophage.
NETs may play a beneficial role in mycobacterial infection simply by capturing bacteria and preventing dissemination, and by facilitating activation of macrophages and other immune cells. Indeed, NETs have been shown to prime T cells (192). NETs are observed in the leprosy reaction ENL. However, evidence of free bacteria in ENL patient sera suggests that NETs are not capable of containing M. leprae. Moreover, NETs may also be detrimental. They may bind other immune cells inhibiting their function, and may also directly damage host tissue. NETs are linked to lung injury in respiratory conditions such as cystic fibrosis, chronic obstructive pulmonary disease, and pneumonia-associated acute respiratory distress syndrome (193–195). They have also been linked to lung injury in mice infected with M. smegmatis expressing the sphingomyelinase/nuclease Rv088 from M. tuberculosis (183). Lung injury was largely mediated by MPO (183), which is present and active on NETs (13). If NETs are proven ineffective at limiting infection, then there may be an advantage in blocking production or removing them to help protect damage to lung tissue.
Mycobacterial Resistance to Neutrophil Oxidants
Killing of mycobacteria by neutrophils appears to be much slower than other pathogens, suggesting that these bacteria possess an innate resistance to the fast-acting oxidative killing mechanisms in the phagosome. In support of this, we have shown that M. smegmatis can cope with relatively high doses of the most bactericidal oxidant produced in the neutrophil phagosome, HOCl (LD50 of 90 nmol/108 CFU) (127). HOCl reacts rapidly with a wide range of biomolecules, and the relative resistance may be related to the larger size of these microbes i.e. more HOCl is required to damage enough critical targets in the bacterium. A rod-shape M. smegmatis 10 µm in length and 0.8 µm in diameter has a volume of 5x10-12 mL. In contrast, S. aureus, P. aeruginosa and E. coli (rod- or sphere-shaped with 1-2 µm in length and 0.5–1 µm diameter) have approximately 10% that volume. The susceptibility of these bacteria to HOCl ranges from 2.5-3 nmol/108 CFU for P. aeruginosa and E. coli (196, 197), to approximately 10 nmol/108 CFU for S. aureus (151, 197, 198).
The presence of protective compounds will also contribute to HOCl resistance. Mycothiol (MSH), the main low molecular weight thiol (LMWT) in mycobacteria, has been proposed to play a role in defending against neutrophil oxidants. Mycothiol carries out many of the cellular functions performed by glutathione in eukaryotic cells and gram-negative bacteria, including the detoxification of electrophilic compounds and maintaining redox homeostasis (199). Interestingly, MSH levels in M. tuberculosis and M. smegmatis are much higher than those of the main LMWT in other bacteria such as bacillithiol (BSH) in S. aureus or glutathione in Pseudomonas aeruginosa (9–19, 0.7 and 1.1 µmol/g residual dry weight, respectively) (200). Since HOCl reacts very rapidly with thiol moieties (201), the higher LMWT content in mycobacteria might protect them from killing by HOCl. Consistent with a role for mycothiol in HOCl resistance, mycobacteria lacking the ability to synthesize this LMWT are significantly more sensitive to reagent HOCl and its secondary oxidants, chloramines (127, 202, 203). However, the amount of HOCl required to kill M. smegmatis (200-300 nmol/108 bacteria) greatly exceeds that of mycothiol (5 nmol/108 bacteria) (127), suggesting that mycothiol exerts its protective role not by scavenging the oxidant directly, but by forming mixed disulfides with critical cysteine residues in proteins in a process termed S-mycothiolation. Because S-mycothiolated proteins can be reduced by mycoredoxin-1 (203), this modification protects cysteine residues from irreversible oxidation. S-mycothiolation occurs in M. smegmatis exposed to hypochlorous acid (202). We recently observed that neutrophils killed mycothiol-deficient M. smegmatis at the same rate as wild type bacteria, indicating that mycothiol itself is not responsible for the ability of M. smegmatis to cope with HOCl or other oxidants produced in the phagosome (127).
Mycobacteria also contain other LMWTs such as gamma-glutamylcysteine, coenzyme A, cysteine and ergothioneine, albeit at much lower levels than mycothiol (127, 204). Interestingly, unlike mycothiol, ergothioneine is known to be actively exported suggesting an extracellular function for this LMWT (205), which may provide greater protection against HOCl. Also, ergothioneine, which is upregulated in mycothiol-deficient mutants, can compensate for the loss of mycothiol in protecting against organic hydroperoxides and is essential for survival of M. tuberculosis in macrophages and mice (205). The contribution of ergothioneine and other LMWTs to surviving neutrophil phagocytosis remains unexplored and future investigations are needed to establish their role in mycobacterial resistance to neutrophil oxidants. Apart from thiol groups, HOCl also reacts rapidly with methionine residues on proteins (201) resulting in the formation of methionine sulfoxide. Methionine sulfoxides are reduced by methionine sulfoxide reductases, the lack of which in M. tuberculosis made the bacteria more susceptible to HOCl (206). Whether or not methionine sulfoxide reductase activity protects mycobacteria from oxidative killing by neutrophils remains to be investigated.
Bacterial superoxide dismutase (SOD), which catalyzes the conversion of superoxide to hydrogen peroxide, is another candidate for conferring resistance of mycobacteria to neutrophil killing. SOD is exported in large amounts by M. tuberculosis and production increases further under hydrogen peroxide stress (207, 208). A role for SOD in the virulence of M. tuberculosis was established in a mouse infection model (207). Superoxide is the most-upstream of the oxidants produced by the neutrophil, and while SOD will facilitate conversion to hydrogen peroxide, the superoxide itself can modulate MPO activity (209). The addition of SOD to the surface of S. aureus slowed the rate at which they were killed by neutrophils (210). Non-pathogenic M. smegmatis express almost 100-fold less SOD than M. tuberculosis and export a smaller fraction (208). However, we could not slow the killing of M. smegmatis by genetically-modifying them to express large amounts of M. tuberculosis SOD, albeit as noted above, wild-type M. smegmatis are already killed very slowly (127).
Conclusions and Future Perspectives
While neutrophils attempt to control mycobacterial infection, the bulk of the evidence indicates that the effectiveness of their phagosomal and extracellular killing mechanisms is thwarted by the microbes. Rather than destroy mycobacteria, the neutrophils appear to provide a safe haven and transport them into macrophages, while retaining the potential to damage host tissue. With regards to the latter, it will be valuable to determine if NETs play a significant role in the control of infection by preventing the spread or directly killing mycobacteria, or if they simply cause tissue damage and contribute towards a pro-inflammatory environment.
It is important to note the limitations of current experimental models (211). In vitro studies of neutrophil and mycobacteria interactions occur using neutrophils isolated from circulating blood; they have not been exposed to the plethora of signals and cell interactions that occur during migration and upon arrival at a site of infection. Human tuberculosis granulomas are highly hypoxic (212), which will suppress the oxidative burst, yet little is known about how hypoxia affects the response of neutrophils to mycobacteria (213). There is considerable heterogeneity in neutrophil populations, and certain subpopulations may be more effective at ingesting and destroying mycobacteria. Also, a significant amount of information has been derived from animal models, but neutrophils function differently between species, particularly mouse and human (214).
The current challenge is to apply our knowledge of neutrophil-mycobacteria interactions to improving treatments. The appearance of multi-drug resistance strains of M. tuberculosis is of major concern. Treatment for drug-susceptible M. tuberculosis involves six months of antibiotic therapy provided by four front line drugs: isoniazid, rifampicin, ethambutol and pyrazinamide. Treatment of infections with multi-drug resistant (MDR) strains takes longer, costs considerably more and uses second generation drugs with greater side effects and more complex drug delivery (2). Extensively drug-resistant strains have also arisen that are resistant to at least one second-line drug used to treat MDR (fluoroquinolone) and one second-line injectable drug (2). Infections with NTM can also be difficult to treat (215, 216).
In terms of bacterial resistance, the mycobacteria tested so far appear to be phagocytosed and killed more slowly than other pathogenic bacteria, with resistance to neutrophil oxidants likely to be an important factor. While a number of mechanisms have been shown to underwrite the resistance of mycobacteria to individual oxidants, none of these have yet been conclusively demonstrated to play a role in resistance to neutrophils. Better understanding of how these microbes survive oxidant exposure in the phagosome may provide therapeutic targets for sensitizing pathogenic mycobacteria to killing by the immune system. Pharmacological interventions might also be useful in limiting any adverse effects of NETs during mycobacterial infection.
Author Contributions
HP compiled the first draft of the review, using additional content provided by LF, CK, and ND. MH edited the article and generated the final version. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Canterbury Medical Research Foundation, the Health Research Council of New Zealand (15/479), and the Travis Trust of New Zealand.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Tortoli E, Meehan CJ, Grottola A, Serpini GF, Fabio A, Trovato A, et al. Genome-Based Taxonomic Revision Detects a Number of Synonymous Taxa in the Genus Mycobacterium. Infect Genet Evol (2019) 75:103983. doi: 10.1016/j.meegid.2019.103983
2. World Health Organisation. Global Tuberculosis Report 2020. Geneva: World Health Organization (2020). p. 1–232.
3. World Health Organisation. Global Leprosy (Hansen Disease) Update, 2019: Time to Step-Up Prevention Initiatives. Wkly Epidemiol Rec. (2020) 95:417–40.
4. Daniel-Wayman S, Abate G, Barber DL, Bermudez LE, Coler RN, Cynamon MH, et al. Advancing Translational Science for Pulmonary Nontuberculous Mycobacterial Infections. A Road Map for Research. Am J Respir Crit Care Med (2019) 199(8):947–51. doi: 10.1164/rccm.201807-1273PP
5. Hmama Z, Pena-Diaz S, Joseph S, Av-Gay Y. Immunoevasion and Immunosuppression of the Macrophage by Mycobacterium Tuberculosis. Immunol Rev (2015) 264(1):220–32. doi: 10.1111/imr.12268
6. Houben EN, Nguyen L, Pieters J. Interaction of Pathogenic Mycobacteria With the Host Immune System. Curr Opin Microbiol (2006) 9(1):76–85. doi: 10.1016/j.mib.2005.12.014
7. Eum S-Y, Kong J-H, Hong M-S, Lee Y-J, Kim J-H, Hwang S-H, et al. Neutrophils Are the Predominant Infected Phagocytic Cells in the Airways of Patients With Active Pulmonary TB. Chest (2010) 137(1):122–8. doi: 10.1378/chest.09-0903
8. Winterbourn CC, Kettle AJ, Hampton MB. Reactive Oxygen Species and Neutrophil Function. Annu Rev Biochem (2016) 85(1):765–92. doi: 10.1146/annurev-biochem-060815-014442
9. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil Extracellular Traps Kill Bacteria. Science (2004) 303(5663):1532–5. doi: 10.1126/science.1092385
10. Chapman EA, Lyon M, Simpson D, Mason D, Beynon RJ, Moots RJ, et al. Caught in a Trap? Proteomic Analysis of Neutrophil Extracellular Traps in Rheumatoid Arthritis and Systemic Lupus Erythematosus. Front Immunol (2019) 10:423. doi: 10.3389/fimmu.2019.00423
11. Petretto A, Bruschi M, Pratesi F, Croia C, Candiano G, Ghiggeri G, et al. Neutrophil Extracellular Traps (NET) Induced by Different Stimuli: A Comparative Proteomic Analysis. PloS One (2019) 14(7):e0218946. doi: 10.1371/journal.pone.0218946
12. Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, et al. Neutrophil Extracellular Traps Contain Calprotectin, a Cytosolic Protein Complex Involved in Host Defense Against Candida Albicans. PloS Pathog (2009) 5(10):e1000639. doi: 10.1371/journal.ppat.1000639
13. Parker H, Albrett AM, Kettle AJ, Winterbourn CC. Myeloperoxidase Associated With Neutrophil Extracellular Traps Is Active and Mediates Bacterial Killing in the Presence of Hydrogen Peroxide. J Leukoc Biol (2012) 91(3):369–76. doi: 10.1189/jlb.0711387
14. Guimarães-Costa AB, Nascimento MT, Froment GS, Soares RP, Morgado FN, Conceição-Silva F, et al. Leishmania Amazonensis Promastigotes Induce and Are Killed by Neutrophil Extracellular Traps. Proc Natl Acad Sci USA (2009) 106(16):6748–53. doi: 10.1073/pnas.0900226106
15. Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G, Galuska SP, et al. Neutrophil Extracellular Traps Directly Induce Epithelial and Endothelial Cell Death: A Predominant Role of Histones. PloS One (2012) 7:e32366. doi: 10.1371/journal.pone.0032366
16. Wang L, Zhou X, Yin Y, Mai Y, Wang D, Zhang X. Hyperglycemia Induces Neutrophil Extracellular Traps Formation Through an NADPH Oxidase-Dependent Pathway in Diabetic Retinopathy. Front Immunol (2019) 9:3076. doi: 10.3389/fimmu.2018.03076
17. Alvarez-Jimenez VD, Leyva-Paredes K, Garcia-Martinez M, Vazquez-Flores L, Garcia-Paredes VG, Campillo-Navarro M, et al. Extracellular Vesicles Released From Mycobacterium Tuberculosis-Infected Neutrophils Promote Macrophage Autophagy and Decrease Intracellular Mycobacterial Survival. Front Immunol (2018) 9:272. doi: 10.3389/fimmu.2018.00272
18. Rosales C. Neutrophils at the Crossroads of Innate and Adaptive Immunity. J Leukoc Biol (2020) 108(1):377–96. doi: 10.1002/JLB.4MIR0220-574RR
19. Cambier C, O’Leary SM, O’Sullivan MP, Keane J, Ramakrishnan L. Phenolic Glycolipid Facilitates Mycobacterial Escape From Microbicidal Tissue-Resident Macrophages. Immunity (2017) 47(3):552–65. doi: 10.1016/j.immuni.2017.08.003
20. Huang L, Nazarova EV, Tan S, Liu Y, Russell DG. Growth of Mycobacterium tuberculosis In Vivo Segregates With Host Macrophage Metabolism and Ontogeny. J Exp Med (2018) 215(4):1135–52. doi: 10.1084/jem.20172020
21. Martineau AR, Newton SM, Wilkinson KA, Kampmann B, Hall BM, Nawroly N, et al. Neutrophil-Mediated Innate Immune Resistance to Mycobacteria. J Clin Invest (2007) 117(7):1988–94. doi: 10.1172/jci31097
22. Estévez O, Anibarro L, Garet E, Martínez A, Pena A, Barcia L, et al. Multi-Parameter Flow Cytometry Immunophenotyping Distinguishes Different Stages of Tuberculosis Infection. J Infect (2020) 81:57–71. doi: 10.1016/j.jinf.2020.03.064
23. Olaniyi J, Aken’Ova Y. Haematological Profile of Patients With Pulmonary Tuberculosis in Ibadan, Nigeria. Afr J Med Med Sci (2003) 32(3):239–42.
24. Berry MP, Graham CM, McNab FW, Xu Z, Bloch SA, Oni T, et al. An Interferon-Inducible Neutrophil-Driven Blood Transcriptional Signature in Human Tuberculosis. Nature (2010) 466(7309):973–7. doi: 10.1038/nature09247
25. Lowe DM, Demaret J, Bangani N, Nakiwala JK, Goliath R, Wilkinson KA, et al. Differential Effect of Viable Versus Necrotic Neutrophils on Mycobacterium Tuberculosis Growth and Cytokine Induction in Whole Blood. Front Immunol (2018) 9:903. doi: 10.3389/fimmu.2018.00903
26. Jayachandran R, Sundaramurthy V, Combaluzier B, Mueller P, Korf H, Huygen K, et al. Survival of Mycobacteria in Macrophages Is Mediated by Coronin 1-Dependent Activation of Calcineurin. Cell (2007) 130(1):37–50. doi: 10.1016/j.cell.2007.04.043
27. Sundaramurthy V, Korf H, Singla A, Scherr N, Nguyen L, Ferrari G, et al. Survival of Mycobacterium Tuberculosis and Mycobacterium Bovis BCG in Lysosomes In Vivo. Microbes Infect (2017) 19(11):515–26. doi: 10.1016/j.micinf.2017.06.008
28. Levitte S, Adams KN, Berg RD, Cosma CL, Urdahl KB, Ramakrishnan L. Mycobacterial Acid Tolerance Enables Phagolysosomal Survival and Establishment of Tuberculous Infection. vivo. Cell Host Microbe (2016) 20(2):250–8. doi: 10.1016/j.chom.2016.07.007
29. BoseDasgupta S, Pieters J. Macrophage-Microbe Interaction: Lessons Learned From the Pathogen Mycobacterium Tuberculosis. Sem Immunopath (2018) 40(6):577–91. doi: 10.1007/s00281-018-0710-0.
30. Abadie V, Badell E, Douillard P, Ensergueix D, Leenen PJ, Tanguy M, et al. Neutrophils Rapidly Migrate via Lymphatics After Mycobacterium Bovis BCG Intradermal Vaccination and Shuttle Live Bacilli to the Draining Lymph Nodes. Blood (2005) 106(5):1843–50. doi: 10.1182/blood-2005-03-1281
31. Eruslanov EB, Lyadova IV, Kondratieva TK, Majorov KB, Scheglov IV, Orlova MO, et al. Neutrophil Responses to Mycobacterium Tuberculosis Infection in Genetically Susceptible and Resistant Mice. Infect Immun (2005) 73(3):1744–53. doi: 10.1128/iai.73.3.1744-1753.2005
32. Spinner JL, Winfree S, Starr T, Shannon JG, Nair V, Steele-Mortimer O, et al. Yersinia Pestis Survival and Replication Within Human Neutrophil Phagosomes and Uptake of Infected Neutrophils by Macrophages. J Leukoc Biol (2014) 95(3):389–98. doi: 10.1189/jlb.1112551
33. Rupp J, Pfleiderer L, Jugert C, Moeller S, Klinger M, Dalhoff K, et al. Chlamydia Pneumoniae Hides Inside Apoptotic Neutrophils to Silently Infect and Propagate in Macrophages. PloS One (2009) 4:e6020. doi: 10.1371/journal.pone.0006020
34. van Zandbergen G, Klinger M, Mueller A, Dannenberg S, Gebert A, Solbach W, et al. Cutting Edge: Neutrophil Granulocyte Serves as a Vector for Leishmania Entry Into Macrophages. J Immunol (2004) 173(11):6521–5. doi: 10.4049/jimmunol.173.11.6521
35. Tan BH, Meinken C, Bastian M, Bruns H, Legaspi A, Ochoa MT, et al. Macrophages Acquire Neutrophil Granules for Antimicrobial Activity Against Intracellular Pathogens. J Immunol (2006) 177(3):1864–71. doi: 10.4049/jimmunol.177.3.1864
36. Dallenga T, Repnik U, Corleis B, Eich J, Reimer R, Griffiths GW, et al. M. Tuberculosis-Induced Necrosis of Infected Neutrophils Promotes Bacterial Growth Following Phagocytosis by Macrophages. Cell Host Microbe (2017) 22(4):519–30. doi: 10.1016/j.chom.2017.09.003
37. Pfrommer E, Dreier C, Gabriel G, Dallenga T, Reimer R, Schepanski K, et al. Enhanced Tenacity of Mycobacterial Aerosols From Necrotic Neutrophils. Sci Rep (2020) 10(1):1–14. doi: 10.1038/s41598-020-65781-9
38. Sánchez F, Radaeva TV, Nikonenko BV, Persson A-S, Sengul S, Schalling M, et al. Multigenic Control of Disease Severity After Virulent Mycobacterium Tuberculosis Infection in Mice. Infect Immun (2003) 71(1):126–31. doi: 10.1128/IAI.71.1.126-131.2003
39. Bogle G, Dunbar PR. Simulating T-Cell Motility in the Lymph Node Paracortex With a Packed Lattice Geometry. Immunol Cell Biol (2008) 86(8):676–87. doi: 10.1038/icb.2008.60
40. Vono M, Lin A, Norrby-Teglund A, Koup RA, Liang F, Loré K. Neutrophils Acquire the Capacity for Antigen Presentation to Memory CD4+ T Cells. Vitro ex vivo. Blood (2017) 129(14):1991–2001. doi: 10.1182/blood-2016-10-744441
41. Blomgran R, Ernst JD. Lung Neutrophils Facilitate Activation of Naive Antigen-Specific CD4+ T Cells During Mycobacterium Tuberculosis Infection. J Immunol (2011) 186(12):7110–9. doi: 10.4049/jimmunol.1100001
42. Chai Q, Lu Z, Liu CH. Host Defense Mechanisms Against Mycobacterium Tuberculosis. Cell Mol Life Sci (2019) 77:1859–78. doi: 10.1007/s00018-019-03353-5
43. Cambier C, Falkow S, Ramakrishnan L. Host Evasion and Exploitation Schemes of Mycobacterium Tuberculosis. Cell (2014) 159(7):1497–509. doi: 10.1016/j.cell.2014.11.024
44. Cosma CL, Humbert O, Ramakrishnan L. Superinfecting Mycobacteria Home to Established Tuberculous Granulomas. Nat Immunol (2004) 5(8):828–35. doi: 10.1038/ni1091
45. Saunders BM, Cooper AM. Restraining Mycobacteria: Role of Granulomas in Mycobacterial Infections. Immunol Cell Biol (2000) 78(4):334–41. doi: 10.1046/j.1440-1711.2000.00933.x
46. Yang C-T, Cambier CJ, Davis JM, Hall CJ, Crosier PS, Ramakrishnan L. Neutrophils Exert Protection in the Early Tuberculous Granuloma by Oxidative Killing of Mycobacteria Phagocytosed From Infected Macrophages. Cell Host Microbe (2012) 12(3):301–12. doi: 10.1016/j.chom.2012.07.009
47. Gopal R, Monin L, Torres D, Slight S, Mehra S, McKenna KC, et al. S100A8/A9 Proteins Mediate Neutrophilic Inflammation and Lung Pathology During Tuberculosis. Am J Respir Crit Care Med (2013) 188(9):1137–46. doi: 10.1164/rccm.201304-0803OC
48. Scott NR, Swanson RV, Al-Hammadi N, Domingo-Gonzalez R, Rangel-Moreno J, Kriel BA, et al. S100A8/A9 Regulates CD11b Expression and Neutrophil Recruitment During Chronic Tuberculosis. J Clin Invest (2020) 130:3098–112. doi: 10.1172/JCI130546
49. Jones TPW, Dabbaj S, Mandal I, Cleverley J, Cash C, Lipman MCI, et al. The Blood Neutrophil Count After 1 Month of Treatment Predicts the Radiologic Severity of Lung Disease at Treatment End. Chest (2021) 160(6):2030–41.. doi: 10.1016/j.chest.2021.07.041
50. Niazi MK, Dhulekar N, Schmidt D, Major S, Cooper R, Abeijon C, et al. Lung Necrosis and Neutrophils Reflect Common Pathways of Susceptibility to Mycobacterium Tuberculosis in Genetically Diverse, Immune-Competent Mice. Dis Model Mech (2015) 8(9):1141–53. doi: 10.1242/dmm.020867
51. Nandi B, Behar SM. Regulation of Neutrophils by Interferon-γ Limits Lung Inflammation During Tuberculosis Infection. J Exp Med (2011) 208(11):2251–62. doi: 10.1084/jem.20110919
52. Mishra BB, Lovewell RR, Olive AJ, Zhang G, Wang W, Eugenin E, et al. Nitric Oxide Prevents a Pathogen-Permissive Granulocytic Inflammation During Tuberculosis. Nat Microbiol (2017) 2(7):1–11. doi: 10.1038/nmicrobiol.2017.72
53. Pedrosa J, Saunders BM, Appelberg R, Orme IM, Silva MT, Cooper AM. Neutrophils Play a Protective Nonphagocytic Role in Systemic Mycobacterium Tuberculosis Infection of Mice. Infect Immun (2000) 68(2):577–83. doi: 10.1128/IAI.68.2.577-583.2000
54. Sugawara I, Udagawa T, Yamada H. Rat Neutrophils Prevent the Development of Tuberculosis. Infect Immun (2004) 72(3):1804–6. doi: 10.1128/IAI.72.3.1804-1806.2004
55. Carvalho ACC, Amorim G, Melo MGM, Silveira AKA, Vargas PHL, Moreira ASR, et al. Pre-Treatment Neutrophil Count as a Predictor of Antituberculosis Therapy Outcomes: A Multicenter Prospective Cohort Study. Front Immunol (2021) 12:661934. doi: 10.3389/fimmu.2021.661934
56. McDougall A, Rees R, Weddell A, Wajdi Kanan M. The Histopathology of Lepromatous Leprosy in the Nose. J Path (1975) 115(4):215–26. doi: 10.1002/path.1711150406
57. Kumar V, Sachan T, Natrajan M, Sharma A. High Resolution Structural Changes of Schwann Cell and Endothelial Cells in Peripheral Nerves Across Leprosy Spectrum. Ultrastruct Path (2014) 38(2):86–92. doi: 10.3109/01913123.2013.870273
58. Montoya D, Cruz D, Teles RM, Lee DJ, Ochoa MT, Krutzik SR, et al. Divergence of Macrophage Phagocytic and Antimicrobial Programs in Leprosy. Cell Host Microbe (2009) 6(4):343–53. doi: 10.1016/j.chom.2009.09.002
59. Spierings E, De Boer T, Zulianello L, Ottenhoff T. The Role of Schwann Cells, T Cells and Mycobacterium Leprae in the Immunopathogenesis of Nerve Damage in Leprosy. Lepr Rev (2000) 71(Suppl):S121–S9.
60. Drutz DJ, Chen TS, Lu W-H. The Continuous Bacteremia of Lepromatous Leprosy. N Eng J Med (1972) 287(4):159–64. doi: 10.1056/NEJM197207272870402
61. Sarita S, Muhammed K, Najeeba R, Rajan GN, Anza K, Binitha MP, et al. A Study on Histological Features of Lepra Reactions in Patients Attending the Dermatology Department of the Government Medical College, Calicut, Kerala, India. Lepr Rev (2013) 84(1):51–64.
62. Schmitz V, Prata R, Barbosa M, Mendes MA, Brandão SS, Amadeu TP, et al. Expression of CD64 on Circulating Neutrophils Favoring Systemic Inflammatory Status in Erythema Nodosum Leprosum. PloS Negl Trop Dis (2016) 10(8):e0004955. doi: 10.1371/journal.pntd.0004955
63. Polycarpou A, Walker SL, Lockwood DN. A Systematic Review of Immunological Studies of Erythema Nodosum Leprosum. Front Immunol (2017) 8:233. doi: 10.3389/fimmu.2017.00233
64. Pocaterra L, Jain S, Reddy R, Muzaffarullah S, Torres O, Suneetha S, et al. Clinical Course of Erythema Nodosum Leprosum: An 11-Year Cohort Study in Hyderabad, India. Am J Trop Med Hyg (2006) 74(5):868–79.
65. Walker SL, Balagon M, Darlong J, Doni SN, Hagge DA, Halwai V, et al. ENLIST 1: An International Multi-Centre Cross-Sectional Study of the Clinical Features of Erythema Nodosum Leprosum. PloS Negl Trop Dis (2015) 9(9):e0004065. doi: 10.1371/journal.pntd.0004065
66. Walker SL, Lebas E, Doni SN, Lockwood DN, Lambert SM. The Mortality Associated With Erythema Nodosum Leprosum in Ethiopia: A Retrospective Hospital-Based Study. PloS Negl Trop Dis (2014) 8(3):e2690. doi: 10.1371/journal.pntd.0002690
67. Mabalay M, Helwig E, Tolentino J, Binford C. The Histopathology and Histochemistry of Erythema Nodosum Leprosum. Int J Lepr (1965) 33(1):28–49.
68. Schmitz V, Tavares IF, Pacheco F, dos Santos JB, dos Santos CO, Sarno EN. Neutrophils in Leprosy. Front Immunol (2019) 10:495. doi: 10.3389/fimmu.2019.00495
69. Pinheiro RO, Schmitz V, Silva B, Dias AA, De Souza BJ, de Mattos Barbosa MG, et al. Innate Immune Responses in Leprosy. Front Immunol (2018) 9:518. doi: 10.3389/fimmu.2018.00518
70. Goihman-Yahr M, Rodriguez-Ochoa G, Aranzazu N, Convit J. Polymorphonuclear Activation in Leprosy. I. Spontaneous and Endotoxin-Stimulated Reduction of Nitroblue Tetrazolium: Effects of Serum and Plasma on Endotoxin-Induced Activation. Clin Exp Immunol (1975) 20(2):257.
71. Sher R, Anderson R, Glover A, Wadee AA. Polymorphonuclear Cell Function in the Various Polar Types of Leprosy and Erythema Nodosum Leprosum. Infect Immun (1978) 21(3):959–65. doi: 10.1128/iai.21.3.959-965.1978
72. Park BH, Fikrig SM, Smithwick EM. Infection and Nitroblue-Tetrazolium Reduction by Neutrophils: A Diagnostic Aid. Lancet (1968) 292(7567):532–4. doi: 10.1016/S0140-6736(68)92406-9
73. Hohn DC, Lehrer RI. Mechanism of the Heparin Effect on the Nitroblue-Tetrazolium Slide Test. Infect Immun (1974) 10(4):772–5. doi: 10.1128/iai.10.4.772-775.1974
74. Schiff DE, Rae J, Martin TR, Davis BH, Curnutte JT. Increased Phagocyte Fc gammaRI Expression and Improved Fc Gamma-Receptor-Mediated Phagocytosis After in Vivo Recombinant Human Interferon-Gamma Treatment of Normal Human Subjects. Blood (1997) 90(8):3187–94. doi: 10.1182/blood.V90.8.3187
75. Mendes MA, de Carvalho DS, Amadeu TP, Silva B, Prata R, da Silva CO, et al. Elevated Pentraxin-3 Concentrations in Patients With Leprosy: Potential Biomarker of Erythema Nodosum Leprosum. J Infect Dis (2017) 216(12):1635–43. doi: 10.1093/infdis/jix267
76. Daley C. Mycobacterium Avium Complex Disease. Microbiol Spectr (2017) 5:TNMI7–0045-2017. doi: 10.1128/microbiolspec.TNMI7-0045-2017
77. Gallin JI, Bujak JS, Patten E, Wolff SM. Granulocyte Function in the Chediak-Higashi Syndrome of Mice. Blood (1974) 43(2):201–6. doi: 10.1182/blood.V43.2.201.201
78. Appelberg R, Castro AG, Gomes S, Pedrosa J, Silva MT. Susceptibility of Beige Mice to Mycobacterium Avium: Role of Neutrophils. Infect Immun (1995) 63(9):3381–7. doi: 10.1128/iai.63.9.3381-3387.1995
79. Saunders BM, Cheers C. Intranasal Infection of Beige Mice With Mycobacterium Avium Complex: Role of Neutrophils and Natural Killer Cells. Infect Immun (1996) 64(10):4236–41. doi: 10.1128/iai.64.10.4236-4241.1996
80. Matsuyama M, Ishii Y, Sakurai H, Ano S, Morishima Y, Yoh K, et al. Overexpression of Rorγt Enhances Pulmonary Inflammation After Infection With Mycobacterium Avium. PloS One (2016) 11(1):e0147064. doi: 10.1371/journal.pone.0147064
81. Yamazaki Y, Kubo K, Sekiguchi M, Honda T. Analysis of BAL Fluid in M. Avium-Intracellulare Infection in Individuals Without Predisposing Lung Disease. Eur Respir J (1998) 11(6):1227–31. doi: 10.1183/09031936.98.11061227
82. Yamazaki Y, Kubo K, Takamizawa A, Yamamoto H, Honda T, Sone S. Markers Indicating Deterioration of Pulmonary Mycobacterium Avium-Intracellulare Infection. Am J Respir Crit Care Med (1999) 160(6):1851–5. doi: 10.1164/ajrccm.160.6.9902019
83. Inomata T, Konno S, Nagai K, Suzuki M, Nishimura M. Neutrophil Predominance in Bronchoalveolar Lavage Fluid Is Associated With Disease Severity and Progression of HRCT Findings in Pulmonary Mycobacterium Avium Infection. PloS One (2018) 13(2):e0190189. doi: 10.1371/journal.pone.0190189
84. Gommans EPAT, Even P, Linssen CFM, van Dessel H, van Haren E, de Vries GJ, et al. Risk Factors for Mortality in Patients With Pulmonary Infections With Non-Tuberculous Mycobacteria: A Retrospective Cohort Study. Respir Med (2015) 109(1):137–45. doi: 10.1016/j.rmed.2014.10.013
85. Van Ingen J, Bendien SA, De Lange WCM, Hoefsloot W, Dekhuijzen PNR, Boeree MJ, et al. Clinical Relevance of Non-Tuberculous Mycobacteria Isolated in the Nijmegen-Arnhem Region, The Netherlands. Thorax (2009) 64(6):502–6. doi: 10.1136/thx.2008.110957
86. Johnston JC CL, Elwood K. Mycobacterium Kansasii. Microbiol Spectr (2017) 5:TNMI7–0011-2016. doi: 10.1128/microbiolspec.TNMI7-0011-2016
87. Smith MB, Molina CP, Schnadig VJ, Boyars MC, Aronson JF. Pathologic Features of Mycobacterium Kansasii Infection in Patients With Acquired Immunodeficiency Syndrome. Arch Path Lab Med (2003) 127(5):554–60. doi: 10.5858/2003-127-0554-PFOMKI
88. Domfeh AB, Nodit L, Gradowski JF, Bastacky S. Mycobacterium Kansasii Infection Diagnosed by Pleural Fluid Cytology. Acta Cytol (2007) 51(4):627–30. doi: 10.1159/000325813
89. Schnadig VJ, Quadri SF, Boyvat F, Borucki M. Mycobacterium Kansasii Osteomyelitis Presenting as a Solitary Lytic Lesion of the Ulna: Fine-Needle Aspiration Findings and Morphologic Comparison With Other Mycobacteria. Diagn Cytopathol (1998) 19(2):94–7. doi: 10.1002/(SICI)1097-0339(199808)19:2<94::AID-DC4>3.0.CO;2-N
90. Silva MT, Silva MNT, Appelberg R. Neutrophil-Macrophage Cooperation in the Host Defence Against Mycobacterial Infections. Microb Pathog (1989) 6(5):369–80. doi: 10.1016/0882-4010(89)90079-X
91. Johansen MD, Herrmann J-L, Kremer L. Non-Tuberculous Mycobacteria and the Rise of Mycobacterium Abscessus. Nat Rev Microbiol (2020) 18:392–407. doi: 10.1038/s41579-020-0331-1
92. Dedrick RM, Guerrero-Bustamante CA, Garlena RA, Russell DA, Ford K, Harris K, et al. Engineered Bacteriophages for Treatment of a Patient With a Disseminated Drug-Resistant Mycobacterium Abscessus. Nat Med (2019) 25(5):730–3. doi: 10.1038/s41591-019-0437-z
93. Griffith DE, Aksamit T, Brown-Elliott BA, Catanzaro A, Daley C, Gordin F, et al. An Official ATS/IDSA Statement: Diagnosis, Treatment, and Prevention of Nontuberculous Mycobacterial Diseases. Am J Respir Crit Care Med (2007) 175(4):367–416. doi: 10.1164/rccm.200604-571ST
94. Kwak N, Dalcolmo MP, Daley CL, Eather G, Gayoso R, Hasegawa N, et al. Mycobacterium Abscessus Pulmonary Disease: Individual Patient Data Meta-Analysis. Eur Respir J (2019) 54(1):1801991. doi: 10.1183/13993003.01991-2018
95. Nessar R, Cambau E, Reyrat JM, Murray A, Gicquel B. Mycobacterium Abscessus: A New Antibiotic Nightmare. J Antimicrob Chemother (2012) 67(4):810–8. doi: 10.1093/jac/dkr578
96. Chuang A-Y, Tsou M-H, Chang S-J, Yang L-Y, Shih C-C, Tsai M-P, et al. Mycobacterium Abscessus Granulomatous Prostatitis. Am J Surg Pathol (2012) 36(3):418–22. doi: 10.1097/PAS.0b013e31823dafad
97. Rodriguez G, Ortegon M, Camargo D, Orozco L. Iatrogenic Mycobacterium Abscessus Infection: Histopathology of 71 Patients. Brit J Dermatol (1997) 137(2):214–8. doi: 10.1046/j.1365-2133.1997.18081891.x
98. Choi H, Kim YI, Na CH, Kim MS, Shin BS. Mycobacterium Abscessus Skin Infection Associated With Shaving Activity in a 75-Year-Old Man. Ann Geriatr Med Res (2018) 22(4):204–7. doi: 10.4235/agmr.18.0034
99. Ganbat D, Seehase S, Richter E, Vollmer E, Reiling N, Fellenberg K, et al. Mycobacteria Infect Different Cell Types in the Human Lung and Cause Species Dependent Cellular Changes in Infected Cells. BMC Pulmon Med (2016) 16(1):1–16. doi: 10.1186/s12890-016-0185-5
100. Pawlik A, Garnier G, Orgeur M, Tong P, Lohan A, Le Chevalier F, et al. Identification and Characterization of the Genetic Changes Responsible for the Characteristic Smooth-to-Rough Morphotype Alterations of Clinically Persistent Mycobacterium Abscessus. Mol Microbiol (2013) 90(3):612–29. doi: 10.1111/mmi.12387
101. Catherinot E, Roux A-L, Macheras E, Hubert D, Matmar M, Dannhoffer L, et al. Acute Respiratory Failure Involving an R Variant of Mycobacterium Abscessus. J Clin Microbiol (2009) 47(1):271–4. doi: 10.1128/JCM.01478-08
102. Caverly LJ, Caceres SM, Fratelli C, Happoldt C, Kidwell KM, Malcolm KC, et al. Mycobacterium Abscessus Morphotype Comparison in a Murine Model. PloS One (2015) 10(2):e0117657. doi: 10.1371/journal.pone.0117657
103. Bernut A, Herrmann J-L, Kissa K, Dubremetz J-F, Gaillard J-L, Lutfalla G, et al. Mycobacterium Abscessus Cording Prevents Phagocytosis and Promotes Abscess Formation. Proc Natl Acad Sci USA (2014) 111(10):E943–52. doi: 10.1073/pnas.1321390111
104. Bernut A, Nguyen-Chi M, Halloum I, Herrmann J-L, Lutfalla G, Kremer L. Mycobacterium Abscessus-Induced Granuloma Formation Is Strictly Dependent on TNF Signaling and Neutrophil Trafficking. PloS Pathog (2016) 12(11):1005986. doi: 10.1371/journal.ppat.1005986
105. Best CA, Best TJ. Mycobacterium Smegmatis Infection of the Hand. Hand (2009) 4(2):165–6. doi: 10.1007/s11552-008-9147-6
106. Newton JA Jr., Weiss PJ, Bowler WA, Oldfield EC III. Soft-Tissue Infection Due to Mycobacterium Smegmatis: Report of Two Cases. Clin Infect Dis (1993) 16(4):531–3. doi: 10.1093/clind/16.4.531
107. Wallace RJ Jr., Nash DR, Tsukamura M, Blacklock ZM, Silcox VA. Human Disease Due to Mycobacterium Smegmatis. J Infect Dis (1988) 158(1):52–9. doi: 10.1093/infdis/158.1.52
108. Sevrin A, Reboli AC. Disseminated Mycobacterium Smegmatis Infection Associated With an Implantable Cardioverter Defibrillator. Infect Dis Clin Pract (2009) 17(5):349–51. doi: 10.1097/IPC.0b013e31819b8a9b
109. Brown-Elliott BA, Wallace RJ. Clinical and Taxonomic Status of Pathogenic Nonpigmented or Late-Pigmenting Rapidly Growing Mycobacteria. Clin Microbiol Rev (2002) 15(4):716–46. doi: 10.1128/CMR.15.4.716-746.2002
110. Saffo Z, Ognjan A. Mycobacterium Smegmatis Infection of a Prosthetic Total Knee Arthroplasty. IDCases (2016) 5:80–2. doi: 10.1016/j.idcr.2016.07.007
111. Wang J, Zhu X, Peng Y, Zhu T, Liu H, Zhu Y, et al. Mycobacterium Tuberculosis YrbE3A Promotes Host Innate Immune Response by Targeting NF-κb/JNK Signaling. Microorganisms (2020) 8(4):584. doi: 10.3390/microorganisms8040584
112. Miralda I, Klaes CK, Graham JE, Uriarte SM. Human Neutrophil Granule Exocytosis in Response to Mycobacterium Smegmatis. Pathogens (2020) 9(2):123. doi: 10.3390/pathogens9020123
113. Cougoule C, Constant P, Etienne G, Daffé M, Maridonneau-Parini I. Lack of Fusion of Azurophil Granules With Phagosomes During Phagocytosis of Mycobacterium Smegmatis by Human Neutrophils Is Not Actively Controlled by the Bacterium. Infect Immun (2002) 70(3):1591–8. doi: 10.1128/iai.70.3.1591-1598.2002
114. Nauseef WM. How Human Neutrophils Kill and Degrade Microbes: An Integrated View. Immunol Rev (2007) 219(1):88–102. doi: 10.1111/j.1600-065X.2007.00550.x
115. Corleis B, Korbel D, Wilson R, Bylund J, Chee R, Schaible UE. Escape of Mycobacterium Tuberculosis From Oxidative Killing by Neutrophils. Cell Microbiol (2012) 14(7):1109–21. doi: 10.1111/j.1462-5822.2012.01783.x
116. Majeed M, Perskvist N, Ernst JD, Orselius K, Stendahl O. Roles of Calcium and Annexins in Phagocytosis and Elimination of an Attenuated Strain of Mycobacterium Tuberculosis in Human Neutrophils. Microb Pathog (1998) 24(5):309–20. doi: 10.1006/mpat.1997.0200
117. N’Diaye E-N, Darzacq X, Astarie-Dequeker C, Daffé M, Calafat J, Maridonneau-Parini I. Fusion of Azurophil Granules With Phagosomes and Activation of the Tyrosine Kinase Hck Are Specifically Inhibited During Phagocytosis of Mycobacteria by Human Neutrophils. J Immunol (1998) 161(9):4983–91.
118. Seiler P, Aichele P, Raupach B, Odermatt B, Steinhoff U, Kaufmann SHE. Rapid Neutrophil Response Controls Fast-Replicating Intracellular Bacteria But Not Slow-Replicating Mycobacterium Tuberculosis. J Infect Dis (2000) 181(2):671–80. doi: 10.1086/315278
119. Repasy T, Lee J, Marino S, Martinez N, Kirschner DE, Hendricks G, et al. Intracellular Bacillary Burden Reflects a Burst Size for Mycobacterium Tuberculosis In Vivo. PloS Pathog (2013) 9(2):e1003190. doi: 10.1371/journal.ppat.1003190
120. Hartmann P, Becker R, Franzen C, Schell-Frederick E, Römer J, Jacobs M, et al. Phagocytosis and Killing of Mycobacterium Avium Complex by Human Neutrophils. J Leuk Biol (2001) 69(3):397–404. doi: 10.1189/jlb.69.3.397
121. Ladero-Aunon I, Molina E, Holder A, Kolakowski J, Harris H, Urkitza A, et al. Bovine Neutrophils Release Extracellular Traps and Cooperate With Macrophages in Mycobacterium Avium Subsp. Paratuberculosis Clearance In Vitro. Front Immunol (2021) 12:645304. doi: 10.3389/fimmu.2021.645304
122. Doz-Deblauwe E, Carreras F, Arbues A, Remot A, Epardaud M, Malaga W, et al. CR3 Engaged by PGL-I Triggers Syk-Calcineurin-NFATc to Rewire the Innate Immune Response in Leprosy. Front Immunol (2019) 10:2913. doi: 10.3389/fimmu.2019.02913
123. Peyron P, Bordier C, Elsa-Noah N, Maridonneau-Parini I. Nonopsonic Phagocytosis of Mycobacterium Kansasii by Human Neutrophils Depends on Cholesterol and is Mediated by CR3 Associated With Glycosylphosphatidylinositol-Anchored Proteins. J Immunol (2000) 165(9):5186–91. doi: 10.4049/jimmunol.165.9.5186
124. Garner AE, Smith DA, Hooper NM. Sphingomyelin Chain Length Influences the Distribution of GPI-Anchored Proteins in Rafts in Supported Lipid Bilayers. Mol Memb Biol (2007) 24(3):233–42. doi: 10.1080/09687860601127770
125. Iwabuchi K. Involvement of Glycosphingolipid-Enriched Lipid Rafts in Inflammatory Responses. Front Biosci (2015) 20(2):325–34. doi: 10.2741/4312
126. Nakayama H, Kurihara H, Morita YS, Kinoshita T, Mauri L, Prinetti A, et al. Lipoarabinomannan Binding to Lactosylceramide in Lipid Rafts Is Essential for the Phagocytosis of Mycobacteria by Human Neutrophils. Sci Signal (2016) 9(449)::ra101. doi: 10.1126/scisignal.aaf1585
127. Parker HA, Dickerhof N, Forrester L, Ryburn H, Smyth L, Messens J, et al. Mycobacterium Smegmatis Resists the Bactericidal Activity of Hypochlorous Acid Produced in Neutrophil Phagosomes. J Immunol (2021) 206(8):1901–12. doi: 10.4049/jimmunol.2001084
128. Hampton MB, Vissers MCM, Winterbourn CC. A Single Assay for Measuring the Rates of Phagocytosis and Bacterial Killing by Neutrophils. J Leukoc Biol (1994) 55(2):147–52. doi: 10.1002/jlb.55.2.147
129. Malcolm KC, Nichols EM, Caceres SM, Kret JE, Martiniano SL, Sagel SD, et al. Mycobacterium Abscessus Induces a Limited Pattern of Neutrophil Activation That Promotes Pathogen Survival. PloS One (2013) 8(2):e57402. doi: 10.1371/journal.pone.0057402
130. Geertsma MF, Nibbering PH, Pos O, Van Furth R. Interferon-γ-Activated Human Granulocytes Kill Ingested Mycobacterium Fortuitum More Efficiently Than Normal Granulocytes. Eur J Immunol (1990) 20(4):869–73. doi: 10.1002/eji.1830200423
131. Hampton MB, Winterbourn CC. Modification of Neutrophil Oxidant Production With Diphenyleneiodonium and Its Effect on Bacterial Killing. Free Radic Biol Med (1995) 18(4):633–9. doi: 10.1016/0891-5849(94)00181-i
132. Alemán M, de la Barrera SS, Schierloh PL, Alves L, Yokobori N, Baldini M, et al. In Tuberculous Pleural Effusions, Activated Neutrophils Undergo Apoptosis and Acquire a Dendritic Cell–Like Phenotype. J Infect Dis (2005) 192(3):399–409. doi: 10.1086/431680
133. Hilda JN, Das S. Neutrophil CD64, TLR2 and TLR4 Expression Increases But Phagocytic Potential Decreases During Tuberculosis. Tuberculosis (2018) 111:135–42. doi: 10.1016/j.tube.2018.06.010
134. Shalekoff S, Tiemessen CT, Gray CM, Martin DJ. Depressed Phagocytosis and Oxidative Burst in Polymorphonuclear Leukocytes From Individuals With Pulmonary Tuberculosis With or Without Human Immunodeficiency Virus Type 1 Infection. Clin Diagn Lab Immunol (1998) 5(1):41–4. doi: 10.1128/CDLI.5.1.41-44
135. da Glória Bonecini-Almeida M, Werneck-Barroso E, Carvalho PB, de Moura CP, Andrade EF, Hafner A, et al. Functional Activity of Alveolar and Peripheral Cells in Patients With Human Acquired Immunodeficiency Syndrome and Pulmonary Tuberculosis. Cell Immunol (1998) 190(2):112–20. doi: 10.1006/cimm.1998.1399
136. Moura ACN, Modolell M, Mariano M. Down-Regulatory Effect of Mycobacterium Leprae Cell Wall Lipids on Phagocytosis, Oxidative Respiratory Burst and Tumour Cell Killing by Mouse Bone Marrow Derived Macrophages. Scand J Immunol (1997) 46(5):500–5. doi: 10.1046/j.1365-3083.1997.d01-158.x
137. Arcos J, Sasindran SJ, Fujiwara N, Turner J, Schlesinger LS, Torrelles JB. Human Lung Hydrolases Delineate Mycobacterium Tuberculosis–Macrophage Interactions and the Capacity to Control Infection. J Immunol (2011) 187(1):372–81. doi: 10.4049/jimmunol.1100823
138. Arcos J, Diangelo LE, Scordo JM, Sasindran SJ, Moliva JI, Turner J, et al. Lung Mucosa Lining Fluid Modification of Mycobacterium Tuberculosis to Reprogram Human Neutrophil Killing Mechanisms. J Infect Dis (2015) 212:948–58. doi: 10.1093/infdis/jiv146
139. Scordo JM, Arcos J, Kelley HV, Diangelo L, Sasindran SJ, Youngmin E, et al. Mycobacterium Tuberculosis Cell Wall Fragments Released Upon Bacterial Contact With the Human Lung Mucosa Alter the Neutrophil Response to Infection. Front Immunol (2017) 8:307. doi: 10.3389/fimmu.2017.00307
140. Jones GS, Amirault HJ, Andersen BR. Killing of Mycobacterium Tuberculosis by Neutrophils: A Nonoxidative Process. J Infect Dis (1990) 162(3):700–4. doi: 10.1093/infdis/162.3.700
141. Brown AE, Holzer TJ, Andersen BR. Capacity of Human Neutrophils to Kill Mycobacterium Tuberculosis. J Infect Dis (1987) 156(6):985–9. doi: 10.1093/infdis/156.6.985
142. Kisich KO, Higgins M, Diamond G, Heifets L. Tumor Necrosis Factor Alpha Stimulates Killing of Mycobacterium Tuberculosis by Human Neutrophils. Infect Immun (2002) 70(8):4591–9. doi: 10.1128/iai.70.8.4591-4599.2002
143. Denis M. Human Neutrophils, Activated With Cytokines or Not, do Not Kill Virulent Mycobacterium Tuberculosis. J Infect Dis (1991) 163(4):919–20. doi: 10.1093/infdis/163.4.919
144. González-Cortés C, Reyes-Ruvalcaba D, Diez-Tascón C, Rivero-Lezcano OM. Apoptosis and Oxidative Burst in Neutrophils Infected With Mycobacterium Spp. Immunol Lett (2009) 126(1–2):16–21. doi: 10.1016/j.imlet.2009.07.006
145. Reyes-Ruvalcaba D, González-Cortés C, Rivero-Lezcano OM. Human Phagocytes Lack the Ability to Kill Mycobacterium Gordonae, a Non-Pathogenic Mycobacteria. Immunol Lett (2008) 116(1):72–8. doi: 10.1016/j.imlet.2007.11.010
146. Malcolm KC, Caceres SM, Pohl K, Poch KR, Bernut A, Kremer L, et al. Neutrophil Killing of Mycobacterium Abscessus by Intra-and Extracellular Mechanisms. PloS One (2018) 13(4):e0196120. doi: 10.1371/journal.pone.0196120
147. Bernut A, Dupont C, Ogryzko NV, Neyret A, Herrmann J-L, Floto RA, et al. CFTR Protects Against Mycobacterium Abscessus Infection by Fine-Tuning Host Oxidative Defenses. Cell Rep (2019) 26(7):1828–40. doi: 10.1016/j.celrep.2019.01.071
148. Ramos-Kichik V, Mondragón-Flores R, Mondragón-Castelán M, Gonzalez-Pozos S, Muñiz-Hernandez S, Rojas-Espinosa O, et al. Neutrophil Extracellular Traps Are Induced by Mycobacterium Tuberculosis. Tuberculosis (2009) 89(1):29–37. doi: 10.1016/j.tube.2008.09.009
149. Borelli V, Banfi E, Perrotta MG, Zabucchi G. Myeloperoxidase Exerts Microbicidal Activity Against Mycobacterium Tuberculosis. Infect Immun (1999) 67(8):4149–52. doi: 10.1128/IAI.67.8.4149-4152.1999
150. Klebanoff SJ, Shepard CC. Toxic Effect of the Peroxidase-Hydrogen Peroxide-Halide Antimicrobial System on Mycobacterium Leprae. Infect Immun (1984) 44(2):534. doi: 10.1128/iai.44.2.534-536.1984
151. Chapman ALP, Hampton MB, Senthilmohan R, Winterbourn CC, Kettle AJ. Chlorination of Bacterial and Neutrophil Proteins During Phagocytosis and Killing of Staphylococcus Aureus. J Biol Chem (2002) 277(12):9757–62. doi: 10.1074/jbc.M106134200
152. Green JN, Kettle AJ, Winterbourn CC. Protein Chlorination in Neutrophil Phagosomes and Correlation With Bacterial Killing. Free Radic Biol Med (2014) 77:49–56. doi: 10.1016/j.freeradbiomed.2014.08.013
153. Segal BH, Leto TL, Gallin JI, Malech HL, Holland SM. Genetic, Biochemical, and Clinical Features of Chronic Granulomatous Disease. Medicine (2000) 79(3):170–200. doi: 10.1097/00005792-200005000-00004
154. Lau Y, Chan G, Ha S, Hui Y, Yuen K. The Role of Phagocytic Respiratory Burst in Host Defense. Clin Infect Dis (1998) 26(1):226–7. doi: 10.1086/517036
155. Conti F, Lugo-Reyes SO, Galicia LB, He J, Aksu G, de Oliveira EB Jr., et al. Mycobacterial Disease in Patients With Chronic Granulomatous Disease: A Retrospective Analysis of 71 Cases. J Allergy Clin Immunol (2016) 138(1):241–8. doi: 10.1016/j.jaci.2015.11.041
156. Fattahi F, Badalzadeh M, Sedighipour L, Movahedi M, Fazlollahi MR, Mansouri SD, et al. Inheritance Pattern and Clinical Aspects of 93 Iranian Patients With Chronic Granulomatous Disease. J Clin Immunol (2011) 31(5):792. doi: 10.1007/s10875-011-9567-x
157. Giardino G, Cicalese MP, Delmonte O, Migliavacca M, Palterer B, Loffredo L, et al. NADPH Oxidase Deficiency: A Multisystem Approach. Oxid Med Cell Longev (2017) 2017:4590127. doi: 10.1155/2017/4590127
158. Adams LB, Dinauer MC, Morgenstern DE, Krahenbuhl JL. Comparison of the Roles of Reactive Oxygen and Nitrogen Intermediates in the Host Response to Mycobacterium Tuberculosis Using Transgenic Mice. Tubercle Lung Dis (1997) 78(5):237–46. doi: 10.1016/S0962-8479(97)90004-6
159. Lee PPW, Chan K-W, Jiang L, Chen T, Li C, Lee T-L, et al. Susceptibility to Mycobacterial Infections in Children With X-Linked Chronic Granulomatous Disease: A Review of 17 Patients Living in a Region Endemic for Tuberculosis. Pediatr Infect Dis J (2008) 27(3):224–30. doi: 10.1097/INF.0b013e31815b494c
160. AlKhater SA, Deswarte C, Casanova JL, Bustamante J. A Novel Variant in the Neutrophil Cytosolic Factor 2 (NCF2) Gene Results in Severe Disseminated BCG Infectious Disease: A Clinical Report and Literature Review. Mol Genet Genomic Med (2020) 2020:e1237. doi: 10.1002/mgg3.1237
161. Li T, Zhou X, Ling Y, Jiang N, Ai J, Wu J, et al. Genetic and Clinical Profiles of Disseminated Bacillus Calmette-Guérin Disease and Chronic Granulomatous Disease in China. Front Immunol (2019) 10:73. doi: 10.3389/fimmu.2019.00073
162. Kalita A, Verma I, Khuller GK. Role of Human Neutrophil Peptide-1 as a Possible Adjunct to Antituberculosis Chemotherapy. J Infect Dis (2004) 190(8):1476–80. doi: 10.1086/424463
163. Ogata K, Linzer BA, Zuberi RI, Ganz T, Lehrer RI, Catanzaro A. Activity of Defensins From Human Neutrophilic Granulocytes Against Mycobacterium Avium-Mycobacterium Intracellulare. Infect Immun (1992) 60(11):4720–5. doi: 10.1128/iai.60.11.4720-4725.1992
164. Jena P, Mohanty S, Mohanty T, Kallert S, Morgelin M, Lindstrøm T, et al. Azurophil Granule Proteins Constitute the Major Mycobactericidal Proteins in Human Neutrophils and Enhance the Killing of Mycobacteria in Macrophages. PloS One (2012) 7(12):e50345. doi: 10.1371/journal.pone.0050345
165. Hilda JN, Narasimhan M, Das SD. Mycobacterium Tuberculosis Strains Modify Granular Enzyme Secretion and Apoptosis of Human Neutrophils. Mol Immunol (2015) 68(2):325–32. doi: 10.1016/j.molimm.2015.09.019
166. Steinwede K, Maus R, Bohling J, Voedisch S, Braun A, Ochs M, et al. Cathepsin G and Neutrophil Elastase Contribute to Lung-Protective Immunity Against Mycobacterial Infections in Mice. J Immunol (2012) 188(9):4476–87. doi: 10.4049/jimmunol.1103346
167. Ribeiro-Gomes FL, Moniz-de-Souza MCA, Alexandre-Moreira MS, Dias WB, Lopes MF, Nunes MP, et al. Neutrophils Activate Macrophages for Intracellular Killing of Leishmania Major Through Recruitment of TLR4 by Neutrophil Elastase. J Immunol (2007) 179(6):3988–94. doi: 10.4049/jimmunol.179.6.3988
168. Mathy-Hartert M, Deby-Dupont G, Melin P, Lamy M, Deby C. Bactericidal Activity Against Pseudomonas Aeruginosa Is Acquired by Cultured Human Monocyte-Derived Macrophages After Uptake of Myeloperoxidase. Experientia (1996) 52(2):167–74. doi: 10.1007/BF01923364
169. Barrientos L, Marin-Esteban V, de Chaisemartin L, Sandre C, Bianchini E, Nicolas V, et al. An Improved Strategy to Recover Large Fragments of Functional Human Neutrophil Extracellular Traps. Front Immunol (2013) 4:166. doi: 10.3389/fimmu.2013.00166
170. Halverson TWR, Wilton M, Poon KKH, Petri B, Lewenza S. DNA Is an Antimicrobial Component of Neutrophil Extracellular Traps. PloS Pathog (2015) 11(1):e1004593. doi: 10.1371/journal.ppat.1004593
171. Schechter MC, Buac K, Adekambi T, Cagle S, Celli J, Ray SM, et al. Neutrophil Extracellular Trap (NET) Levels in Human Plasma Are Associated With Active TB. PloS One (2017) 12(8):e0182587. doi: 10.1371/journal.pone.0182587
172. van der Meer AJ, Zeerleder S, Blok DC, Kager LM, Lede IO, Rahman W, et al. Neutrophil Extracellular Traps in Patients With Pulmonary Tuberculosis. Respir Res (2017) 18(1):181. doi: 10.1186/s12931-017-0663-1
173. Filio-Rodríguez G, Estrada-García I, Arce-Paredes P, Moreno-Altamirano MM, Islas-Trujillo S, Ponce-Regalado MD, et al. In Vivo Induction of Neutrophil Extracellular Traps by Mycobacterium Tuberculosis in a Guinea Pig Model. Innate Immun (2017) 23(7):625–37. doi: 10.1177/1753425917732406
174. Moreira-Teixeira L, Stimpson PJ, Stavropoulos E, Hadebe S, Chakravarty P, Ioannou M, et al. Type I IFN Exacerbates Disease in Tuberculosis-Susceptible Mice by Inducing Neutrophil-Mediated Lung Inflammation and NETosis. Nat Commun (2020) 11(1):5566. doi: 10.1038/s41467-020-19412-6
175. Dias AA, Silva CO, Santos JPS, Batista-Silva LR, Acosta CCD, Fontes AN, et al. DNA Sensing via TLR-9 Constitutes a Major Innate Immunity Pathway Activated During Erythema Nodosum Leprosum. J Immunol (2016) 197(5):1905–13. doi: 10.4049/jimmunol.1600042
176. Da Silva CO, Dias AA, da Costa Nery JA, de Miranda Machado A, Ferreira H, Rodrigues TF, et al. Neutrophil Extracellular Traps Contribute to the Pathogenesis of Leprosy Type 2 Reactions. PloS Negl Trop Dis (2019) 13(9):e0007368. doi: 10.1371/journal.pntd.0007368
177. Sahu S, Sharma K, Sharma M, Narang T, Dogra S, Minz RW, et al. Neutrophil NETworking in ENL: Potential as a Putative Biomarker: Future Insights. Front Med (Lausanne) (2021) 8:697804. doi: 10.3389/fmed.2021.697804
178. Liu K, Sun E, Lei M, Li L, Gao J, Nian X, et al. BCG-Induced Formation of Neutrophil Extracellular Traps Play an Important Role in Bladder Cancer Treatment. Clin Immunol (2019) 201:4–14. doi: 10.1016/j.clim.2019.02.005
179. Branzk N, Lubojemska A, Hardison SE, Wang Q, Gutierrez MG, Brown GD, et al. Neutrophils Sense Microbe Size and Selectively Release Neutrophil Extracellular Traps in Response to Large Pathogens. Nat Immunol (2014) 15(11):1017–25. doi: 10.1038/ni.2987
180. Tenland E, Håkansson G, Alaridah N, Lutay N, Rönnholm A, Hallgren O, et al. Innate Immune Responses After Airway Epithelial Stimulation With Mycobacterium Bovis Bacille-Calmette Guérin. PloS One (2016) 11(10):e0164431. doi: 10.1371/journal.pone.0164431
181. Braian C, Hogea V, Stendahl O. Mycobacterium Tuberculosis-Induced Neutrophil Extracellular Traps Activate Human Macrophages. J Innate Immun (2013) 5(6):591–602. doi: 10.1159/000348676
182. Francis RJ, Butler RE, Stewart GR. Mycobacterium Tuberculosis ESAT-6 Is a Leukocidin Causing Ca(2+) Influx, Necrosis and Neutrophil Extracellular Trap Formation. Cell Death Dis (2014) 5(10):e1474. doi: 10.1038/cddis.2014.394
183. Dang G, Cui Y, Wang L, Li T, Cui Z, Song N, et al. Extracellular Sphingomyelinase Rv0888 of Mycobacterium Tuberculosis Contributes to Pathological Lung Injury of Mycobacterium Smegmatis in Mice via Inducing Formation of Neutrophil Extracellular Traps. Front Immunol (2018) 9:677. doi: 10.3389/fimmu.2018.00677
184. Speer A, Sun J, Danilchanka O, Meikle V, Rowland JL, Walter K, et al. Surface Hydrolysis of Sphingomyelin by the Outer Membrane Protein R V0888 Supports Replication of Mycobacterium Tuberculosis in Macrophages. Mol Microbiol (2015) 97(5):881–97. doi: 10.1111/mmi.13073
185. Dang G, Cao J, Cui Y, Song N, Chen L, Pang H, et al. Characterization of Rv0888, a Novel Extracellular Nuclease From Mycobacterium Tuberculosis. Sci Rep (2016) 6(1):1–11. doi: 10.1038/srep19033
186. Parker H, Dragunow M, Hampton MB, Kettle AJ, Winterbourn CC. Requirements for NADPH Oxidase and Myeloperoxidase in Neutrophil Extracellular Trap Formation Differ Depending on the Stimulus. J Leuk Biol (2012) 92:841–9. doi: 10.1189/jlb.1211601
187. Bianchi M, Hakkim A, Brinkmann V, Siler U, Seger RA, Zychlinsky A, et al. Restoration of NET Formation by Gene Therapy in CGD Controls Aspergillosis. Blood (2009) 114:2619–22. doi: 10.1182/blood-2009-05-221606
188. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil Extracellular Traps Enriched in Oxidized Mitochondrial DNA Are Interferogenic and Contribute to Lupus-Like Disease. Nat Med (2016) 22:146–53. doi: 10.1038/nm.4027
189. Stephan A, Batinica M, Steiger J, Hartmann P, Zaucke F, Bloch W, et al. LL37: DNA Complexes Provide Antimicrobial Activity Against Intracellular Bacteria in Human Macrophages. Immunol (2016) 148(4):420–32. doi: 10.1111/imm.12620
190. Ong CWM, Fox K, Ettorre A, Elkington PT, Friedland JS. Hypoxia Increases Neutrophil-Driven Matrix Destruction After Exposure to Mycobacterium Tuberculosis. Sci Rep (2018) 8(1):1–11. doi: 10.1038/s41598-018-29659-1
191. Pilsczek FH, Salina D, Poon KKH, Fahey C, Yipp BG, Sibley CD, et al. A Novel Mechanism of Rapid Nuclear Neutrophil Extracellular Trap Formation in Response to Staphylococcus Aureus. J Immunol (2010) 185(12):7413–25. doi: 10.4049/jimmunol.1000675
192. Tillack K, Breiden P, Martin R, Sospedra M. T Lymphocyte Priming by Neutrophil Extracellular Traps Links Innate and Adaptive Immune Responses. J Immunol (2012) 188(7):3150–9. doi: 10.4049/jimmunol.1103414
193. Marcos V, Zhou-Suckow Z, Önder Yildirim A, Bohla A, Hector A, Vitkov L, et al. Free DNA in Cystic Fibrosis Airway Fluids Correlates With Airflow Obstruction. Mediat Inflamm (2015) 2015:408935. doi: 10.1155/2015/408935
194. Grabcanovic-Musija F, Obermayer A, Stoiber W, Krautgartner W-D, Steinbacher P, Winterberg N, et al. Neutrophil Extracellular Trap (NET) Formation Characterises Stable and Exacerbated COPD and Correlates With Airflow Limitation. Respir Res (2015) 16(1):59. doi: 10.1186/s12931-015-0221-7
195. Adrover JM, Aroca-Crevillén A, Crainiciuc G, Ostos F, Rojas-Vega Y, Rubio-Ponce A, et al. Programmed ‘Disarming’of the Neutrophil Proteome Reduces the Magnitude of Inflammation. Nat Immunol (2020) 21:135–44. doi: 10.1038/s41590-019-0571-2
196. Bonvillain RW, Painter RG, Ledet EM, Wang G. Comparisons of Resistance of CF and Non-CF Pathogens to Hydrogen Peroxide and Hypochlorous Acid Oxidants In Vitro. BMC Microbiol (2011) 11(1):112. doi: 10.1186/1471-2180-11-112
197. Dickerhof N, Isles V, Pattemore P, Hampton MB, Kettle AJ. Exposure of Pseudomonas Aeruginosa to Bactericidal Hypochlorous Acid During Neutrophil Phagocytosis Is Compromised in Cystic Fibrosis. J Biol Chem (2019) 294(36):13502–14. doi: 10.1074/jbc.RA119.009934
198. Coker MS, Forbes LV, Plowman-Holmes M, Murdoch DR, Winterbourn CC, Kettle AJ. Interactions of Staphyloxanthin and Enterobactin With Myeloperoxidase and Reactive Chlorine Species. Arch Biochem Biophys (2018) 646:80–9. doi: 10.1016/j.abb.2018.03.039
199. Reyes AM, Pedre B, De Armas MI, Tossounian M-A, Radi R, Messens J, et al. Chemistry and Redox Biology of Mycothiol. Antioxid Redox Signal (2018) 28(6):487–504. doi: 10.1089/ars.2017.7074
200. Newton GL, Arnold K, Price MS, Sherrill C, Delcardayre SB, Aharonowitz Y, et al. Distribution of Thiols in Microorganisms: Mycothiol Is a Major Thiol in Most Actinomycetes. J Bacteriol (1996) 178(7):1990–5. doi: 10.1128/jb.178.7.1990-1995.1996
201. Storkey C, Davies MJ, Pattison DI. Reevaluation of the Rate Constants for the Reaction of Hypochlorous Acid (HOCl) With Cysteine, Methionine, and Peptide Derivatives Using a New Competition Kinetic Approach. Free Radic Biol Med (2014) 73:60–6. doi: 10.1016/j.freeradbiomed.2014.04.024
202. Hillion M, Bernhardt J, Busche T, Rossius M, Maaß S, Becher D, et al. Monitoring Global Protein Thiol-Oxidation and Protein S-Mycothiolation in Mycobacterium Smegmatis Under Hypochlorite Stress. Sci Rep (2017) 7(1):1195. doi: 10.1038/s41598-017-01179-4
203. Van Laer K, Buts L, Foloppe N, Vertommen D, Van Belle K, Wahni K, et al. Mycoredoxin-1 Is One of the Missing Links in the Oxidative Stress Defence Mechanism of Mycobacteria. Mol Microbiol (2012) 86(4):787–804. doi: 10.1111/mmi.12030
204. Sao Emani C, Williams MJ, Van Helden PD, Taylor MJC, Wiid IJ, Baker B. Gamma-Glutamylcysteine Protects Ergothioneine-Deficient Mycobacterium Tuberculosis Mutants Against Oxidative and Nitrosative Stress. Biochem Biophys Res Comm (2018) 495(1):174–8. doi: 10.1016/j.bbrc.2017.10.163
205. Sao Emani C, Williams MJ, Wiid IJ, Hiten NF, Viljoen AJ, Pietersen R-DD, et al. Ergothioneine Is a Secreted Antioxidant in Mycobacterium Smegmatis. Antimicrob Agents Chemother (2013) 57:3202–7. doi: 10.1128/AAC.02572-12
206. Lee WL, Gold B, Darby C, Brot N, Jiang X, De Carvalho LPS, et al. Mycobacterium Tuberculosis Expresses Methionine Sulphoxide Reductases A and B That Protect From Killing by Nitrite and Hypochlorite. Mol Microbiol (2009) 71:583–93. doi: 10.1111/j.1365-2958.2008.06548.x
207. Edwards KM, Cynamon MH, Voladri RK, Hager CC, DeSTEFANO MS, Tham KT, et al. Iron-Cofactored Superoxide Dismutase Inhibits Host Responses to Mycobacterium Tuberculosis. Am J Respir Crit Care Med (2001) 164(12):2213–9. doi: 10.1164/ajrccm.164.12.2106093
208. Harth G, Horwitz MA. Export of Recombinant Mycobacterium Tuberculosis Superoxide Dismutase Is Dependent Upon Both Information in the Protein and Mycobacterial Export Machinery. A Model for Studying Export of Leaderless Proteins by Pathogenic Mycobacteria. J Biol Chem (1999) 274(7):4281–92. doi: 10.1074/jbc.274.7.4281
209. Kettle AJ, Anderson RF, Hampton MB, Winterbourn CC. Reactions of Superoxide With Myeloperoxidase. Biochemistry (2007) 46(16):4888–97. doi: 10.1021/bi602587k
210. Hampton MB, Kettle AJ, Winterbourn CC. Involvement of Superoxide and Myeloperoxidase in Oxygen-Dependent Killing of Staphylococcus Aureus by Neutrophils. Infect Immun (1996) 64(9):3512–7. doi: 10.1128/iai.64.9.3512-3517.1996
211. Fonseca KL, Rodrigues PNS, Olsson IAS, Saraiva M. Experimental Study of Tuberculosis: From Animal Models to Complex Cell Systems and Organoids. PloS Pathog (2017) 13(8):e1006421. doi: 10.1371/journal.ppat.1006421
212. Belton M, Brilha S, Manavaki R, Mauri F, Nijran K, Hong YT, et al. Hypoxia and Tissue Destruction in Pulmonary TB. Thorax (2016) 71(12):1145–53. doi: 10.1136/thoraxjnl-2015-207402
213. Remot A, Doz E, Winter N. Neutrophils and Close Relatives in the Hypoxic Environment of the Tuberculous Granuloma: New Avenues for Host-Directed Therapies? Front Immunol (2019) 10:417. doi: 10.3389/fimmu.2019.00417
214. Mestas J, Hughes CC. Of Mice and Not Men: Differences Between Mouse and Human Immunology. J Immunol (2004) 172(5):2731–8. doi: 10.4049/jimmunol.172.5.2731
215. van der Werf TS, Stienstra Y, Johnson RC, Phillips R, Adjei O, Fleischer B, et al. Mycobacterium Ulcerans Disease. Bull World Health Organ (2005) 83:785–91.
Keywords: phagosomes, neutrophil extracellular traps, oxidative stress, tuberculosis, leprosy
Citation: Parker HA, Forrester L, Kaldor CD, Dickerhof N and Hampton MB (2021) Antimicrobial Activity of Neutrophils Against Mycobacteria. Front. Immunol. 12:782495. doi: 10.3389/fimmu.2021.782495
Received: 24 September 2021; Accepted: 06 December 2021;
Published: 23 December 2021.
Edited by:
Clare Hawkins, University of Copenhagen, DenmarkReviewed by:
William Michael Nauseef, The University of Iowa, United StatesNathalie Winter, Institut National de recherche pour l’agriculture, l’alimentation et l’environnement (INRAE), France
Copyright © 2021 Parker, Forrester, Kaldor, Dickerhof and Hampton. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mark B. Hampton, bWFyay5oYW1wdG9uQG90YWdvLmFjLm56