 Tomonori Kadowaki
Tomonori Kadowaki Saori Kadowaki
Saori Kadowaki Hidenori Ohnishi
Hidenori Ohnishi
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 26 November 2021
Sec. Primary Immunodeficiencies
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.780689
This article is part of the Research TopicCreating Awareness for Primary Immunodeficiencies in the Southeast and East Asia RegionsView all 19 articles
A20, encoded by the TNFAIP3 gene, is a negative regulator of tumor necrosis factor (TNF)-nuclear factor-κB signaling. It was recently demonstrated that A20 haploinsufficiency (HA20), caused by a heterozygous mutation in the TNFAIP3 gene, can present as an early onset autoinflammatory disease resembling Behçet’s disease (BD). In addition to autoinflammatory symptoms, HA20 was also reported to be associated with autoimmune diseases and immunodeficiency. Because the phenotypes associated with HA20 are broad, with different severities observed even among individuals in the same family with identical mutations, it has been assumed that the symptoms of HA20 may depend on genetic background and environmental factors. In this review, we summarize the characteristics of patients with HA20 in East Asia and compare these with patients in other regions, mainly the USA and Europe. Patients with HA20 in East Asia developed recurrent fever more frequently than patients in other regions, but were less likely to develop typical BD symptoms such as skin rashes and genital ulcers. In addition, patients with HA20 in East Asia had low rates of complication with autoimmune diseases and low autoantibody detection rates. While anti-TNF-α agents were the primary treatments for severe HA20 in East Asia, anti-interleukin-1 agents and Janus kinase inhibitors were also administered in other regions. Future studies will need to establish methods for analyzing the pathophysiology of HA20 and determining optimal treatment strategies for each patient.
Behçet’s disease (BD) is a chronic inflammatory disease first described by Hulusi Behçet in 1937. BD is characterized by recurrent aphthous stomatitis, skin lesions, uveitis, and genital ulcers (1). Gastrointestinal, cardiovascular, and central nervous system symptoms can also occur in patients with BD. BD is also called the Silk Road disease and has higher prevalence in the Mediterranean, the Middle East, and East Asia compared with other regions. BD shows familial clustering, and thus it has been proposed that genetic predisposition might be a major factor in pathogenesis. Associations between BD and human leukocyte antigen (HLA) genes and polymorphisms, especially HLA-B51, are well established. Recent genome-wide association studies (GWAS) reported that polymorphisms in IL-10, IL-23R, and IL-12RB2 were associated with risk of developing BD; however, none contributed directly to the onset of BD (2) with high penetrance. In 2016, A20 haploinsufficiency (HA20), caused by heterozygous mutation of the TNFAIP3 gene, was reported to cause an early-onset autoinflammatory disease presenting with BD-like features such as recurrent aphthous stomatitis, genital ulcers, and gastrointestinal symptoms (3). Since that time, numerous cases with varied clinical presentation have been reported all over the world, especially in East Asia including China and Japan. Several studies have investigated the function of A20, the pathophysiology of HA20, and optimal treatment strategies. Furthermore, patients with HA20 can present with both autoinflammatory BD-like symptoms as well as autoimmune symptoms and/or immunodeficiency. Variation in symptoms, disease severity, and response to treatment occurs even in individuals within a single family carrying identical mutations. To describe the characteristics of HA20 in East Asia and future prospects for treatment of HA20, we reviewed the published literature on HA20 in East Asia and other regions, focusing on disease manifestations, complications, treatments, and distribution of TNFAIP3 mutations.
A20 is encoded by the TNFAIP3 gene located on chromosome 6q23.3. A20 is a negative regulator of tumor necrosis factor (TNF)-nuclear factor (NF)-κB signaling. In the NF-κB signaling pathway, signals transmitted from TNF-α, interleukin (IL)-1 family members, Toll-like receptors (TLRs), B-cell receptors (BCRs), and T-cell receptors result in ubiquitination of factors in each pathway, NF-κB activation, and production of inflammatory cytokines (4). A20 has an ovarian tumor (OTU) domain in the N-terminal region and seven zinc finger (ZnF) domains in the C-terminal region. The OTU domain deubiquitinates K63 polyubiquitin chains from receptor interacting protein 1, TNF receptor-associated factor (TRAF) 6, and IκB kinase (IKK) γ (5–7) resulting in suppression of signaling. In addition, the ZnF4 domain has E3 ligase function and supports polyubiquitination with K48 polyubiquitin chains, inducing degradation by the proteasome (6). The ZnF7 domain binds the TNF receptor complex via linear polyubiquitin chains and inhibits the formation of the linear ubiquitin chain assembly complex and the IKK complex (8). Posttranslational modifications of A20 have suppressive effects on this pathway: for instance, IKKβ-mediated serine phosphorylation near the ZnF domains (notably at Ser381) promotes A20-mediated cleavage of K63-linked polyubiquitin chains and enhances the inhibitory activity of A20 (9). These functions of A20 contribute to inhibition of NF-κB signaling from each receptor.
A20 also functions to regulate the interferon regulatory factor (IRF) pathway following pathogen recognition. In this pathway, the pattern recognition receptors retinoic acid-inducible gene I and melanoma differentiation-associated protein 5 recognize viral nucleic acids and activate mitochondrial antiviral signaling protein and TRAF3. Subsequently, they combine with TANK-binding kinase 1 (TBK1) and IKKi and are activated by ubiquitination with K63 polyubiquitin chains. Activation of these kinases results in phosphorylation, dimerization, and nuclear translocation of the transcription factor IRF3, followed by transcriptional activation of the type 1 interferon (IFN) gene. Type 1 IFN binds to the IFN-α/β receptor, activates Janus kinase (JAK)-signal transducer and activator of transcription signaling, and induces transcription of IFN-inducible-genes (ISGs). A20 inhibits the IRF pathway and IFN responses by cleaving K63 polyubiquitin chains from TBK1/IKKi (4). In patients with HA20, disrupted suppression of signal transduction results in increased production of inflammatory cytokines and type 1 IFNs followed by autoinflammatory symptoms.
We summarized the literature on patients with HA20 resulting from TNFAIP3 mutations starting from the first report of this disease in 2016 until August 2021. A total of 20 studies in East Asia and 15 studies in other regions, mainly the USA and Europe, were published during this period (10–44). Case reports from East Asia were analyzed separately from those in other regions. We focused on major BD-like symptoms, development of autoimmune diseases, non-autoimmune complications, and treatments administered. The following East Asian patients with HA20 were excluded: one patient’s gender was not stated, three patients did not have detailed symptom information, and two of these three patients were not investigated for autoimmune diseases and/or autoantibodies. A single patient described by multiple studies was counted as one case. The prevalence of HA20 symptoms was compared between East Asia and other regions using Fisher’s exact test in Prism 7 (GraphPad Software, San Diego, CA, USA). Values of p < 0.05 were considered statistically significant.
Table 1 shows the symptoms of HA20 patients in East Asia and other regions. A total of 74 patients in 39 families were affected by HA20 in East Asia, while a total of 51 patients in 23 families were affected by HA20 in other regions. The age of onset ranged from neonatal to around 30 years in both groups. Thus, the age of onset of HA20 appears to be younger than that of BD.

Table 1 The comparison of symptoms and treatment between East Asia versus other countries.
The domain structure of A20 and sites of mutations in TNFAIP3 among East Asian patients with HA20 are shown in Figure 1. A20 mutations in patients with HA20 in East Asia were widely distributed from the OTU to the ZnF7 domains. Most mutations were truncating and included frameshift mutations, nonsense mutations, splice site mutations, and large deletions. However, some missense mutations were also reported. Three missense mutations that were judged as pathogenic via functional analyses are shown in blue squares. TNFAIP3 Cys243Tyr (45) and Glu192Lys (18) were reported in Japan, while Met476Ile (20) was reported in China. Among previously reported mutations, the pathogenic significance of four potential splice site mutations or duplication mutations (shown in red squares) has not been evaluated using functional analyses. One of these mutations was reported in Japan (16), while four were reported in China (25, 27). Among the mutations in East Asia, Arg183X was found in two families (22, 27). The only mutation overlapping between East Asia and other regions was Arg87X, which was found in one family in each case (21, 42).
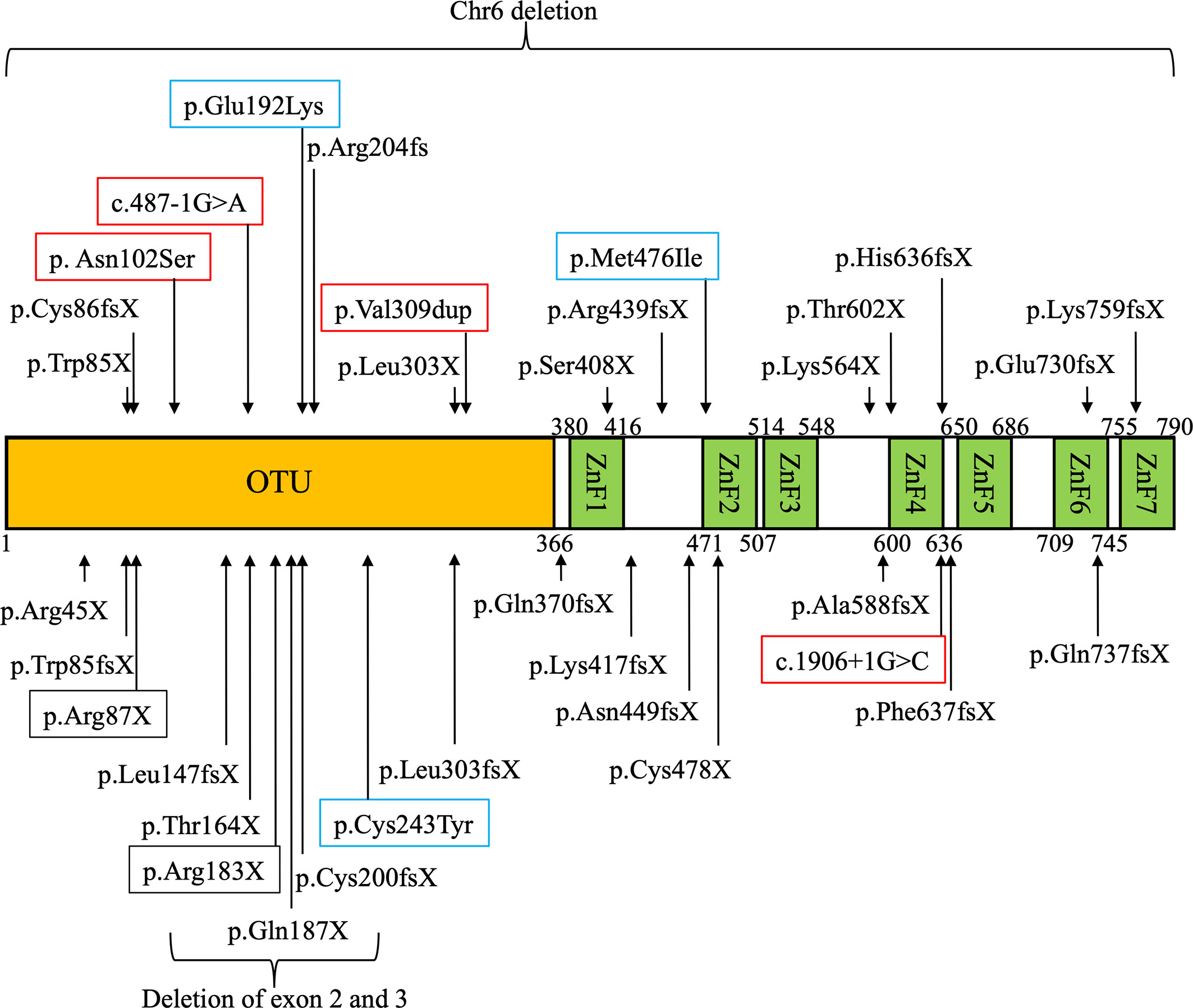
Figure 1 Domain structure of A20 and locations of TNFAIP3 gene mutations in East Asian patients with A20 haploinsufficiency (HA20). Mutations of TNFAIP3 reported in East Asia are indicated with arrows on the domain structure of A20. Three mutations whose pathogenic functions were evaluated by in vitro functional analysis are shown in blue squares. Four mutations whose pathogenic functions were not evaluated by in vitro functional analysis are shown in red squares. The mutations overlapping between different families are shown in black squares.
Table 1 shows the symptoms of patients with HA20 in East Asia and other regions. Recurrent stomatitis was the most common symptom in both groups of patients with a prevalence of 80.2% and 82.6%, respectively. In East Asia, the next most common symptoms of HA20 were recurrent fever, abdominal symptoms, genital ulcers, and skin rashes. In other regions, these symptoms were also common, albeit with different frequencies. Recurrent fever was significantly more common among East Asian patients compared with those in other regions (70.4% vs. 37.3%, p < 0.001). Interestingly, the major BD-like symptoms of genital ulcers and skin rash, including erythema nodules, folliculitis-like eruption, and thrombophlebitis, were more common among patients with HA20 in other regions compared with East Asia. There was no significant difference in the proportion of HA20 patients in East Asia and other regions who fulfilled the International Study Group for Behçet’s disease (ISGFBD) 1990 criteria (46). However, there was a trend toward higher frequency of meeting these criteria in other regions compared with East Asia (25.4% vs. 37.3%, p = 0.169).
HA20 has been reported to often lead to autoimmune diseases such as systemic lupus erythematosus (SLE) and autoimmune thyroid disease. We compared the autoimmune disease development rate and/or autoantibody positive rate in East Asia and other regions and found that these rates were significantly lower in East Asia (Table 1). Non-autoimmune complications were diverse and included IgA vasculitis, chronic hepatitis, and nephrotic syndrome. One patient with HA20 in East Asia developed Hodgkin lymphoma (10). Interestingly, persistent Epstein-Barr virus (EBV) infection occurred in patients with HA20 in both East Asia and other regions (20, 40). Furthermore, some patients in other regions developed immunodeficiency symptoms such as IgG2 and IgG4 deficiency, pancytopenia, and recurrent infection (30, 40, 41).
The number of patients with HA20 treated with medications and administration rates in East Asia and other regions are shown in Table 1. In both groups of patients, colchicine, systemic corticosteroids, disease-modifying drugs, and molecular targeted therapies were relatively commonly administered. In East Asia, anti-TNF-α agents (infliximab, adalimumab, and etanercept) were administered in most severe cases, while anti-IL-6 agents (tocilizumab) were also administered in some cases (10, 13, 16, 21, 24). Only one patient was treated with anti-IL-1 agents (canakinumab) in East Asia (47). In other regions, patients with severe HA20 who did not respond to anti-TNF-α agents were commonly treated with anti-IL-1 agents (anakinra, canakinumab, and rilonacept), anti-IL-6 agents, and JAK inhibitors (tofacitinib and baricitinib) (30, 32, 35, 36, 38–40). Rituximab was administered in a few patients with nephrotic syndrome or autoimmune diseases including SLE in East Asia and other regions (10, 31, 41). In addition, allogeneic hematopoietic cell transplantation (HCT) was also performed for one refractory patient in East Asia and one refractory patient in other regions (31, 47), and autologous HCT was performed for one patient in other regions (30).
This review summarizes the current status of HA20, including regional differences in clinical features, evaluation of the pathogenic significance of TNFAIP3 variants, and treatment strategies.
We found that the proportions of patients with typical BD symptoms, such as skin rash and genital ulcers, and of patients who met the ISGFBD diagnostic criteria for BD, tended to be lower in East Asia compared with other regions. Additionally, the proportions of patients who were autoantibody-positive and/or who were developed autoimmune diseases were also significantly lower in East Asia compared with other regions. A multicenter study in Japan demonstrated that increased frequencies of double-negative T cells and follicular T cells contributed to the development of autoimmune diseases in patients with HA20 (48). Differences in autoimmune disease development rates between regions might be related to immunological differences associated with racial background. In addition, activation of type 1 IFN was reported to cause autoimmune diseases (49). HA20 patients in other regions were reported to show overexpression of ISGs (36, 38, 40, 43), while only one patient in East Asia showed elevated ISG expression (47). In this study, it is unclear whether the overexpression of ISGs is associated with regional differences in autoimmune disease because few patients were investigated for ISG expression. Future work should investigate the relationship between the expression of ISGs in HA20 and the development of autoimmune diseases. There are insufficient data to account for regional differences in the clinical features of HA20. Variation in the clinical features of HA20 were not completely explained by differences in mutation sites. It has been reported that musculoskeletal disorders were significantly more frequent among patients with mutations disrupting the ZnF domain of A20, whereas other symptoms were not affected by mutation site (50). Because genotype-phenotype correlations have not been demonstrated in HA20, variation in clinical features might be affected by other modifier genes and/or environmental factors. Some HA20 patients present with immunodeficiency symptoms such as hypogammaglobulinemia and persistent EBV infection (20, 30, 40). It was reported that mice with selective loss of A20 in B cells had fewer memory B cell and diminished levels of IgG1 and IgG3 (51). Moreover, in HA20 patients, it was suggested that over-activation of NF-κB signaling may cause T cell exhaustion and senescence and thereby decrease naïve Th cell frequency and facilitate persistent EBV infection (48). The composition of lymphocyte subsets may also be affected by age. We were unable to analyze age of onset in patients with HA20 because age of onset in some patients were not stated. Future studies will need to conduct detailed analyses of lymphocyte subsets in patients with HA20 and immunodeficiency to clarify the underlying pathophysiology.
Most HA20 mutations are truncating and include nonsense, frameshift, and splice site mutations. Three missense mutations have been reported in East Asia: TNFAIP3 Glu192Lys, Cys243Tyr, and Met476Ile. While these missense mutations have demonstrated pathogenicity in in vitro functional analyses (18, 52), the pathogenic significance of other variants, such as p.Asn102Ser, c.487-1G>A, and p.Val309dup, has yet to be demonstrated via functional analyses. In particular, the frequency of the Asn102Ser allele is 0.01394 in East Asian according to the Genome Aggregation Database (gnomAD) dataset v.2.1.1 (https://gnomad.broadinstitute.org). Because HA20 is an autosomal dominant disease with high penetrance, in vitro functional analysis is essential to demonstrate that this variant with high allele frequency represents a pathogenic mutation. The analysis of NF-κB reporter gene activity has been commonly used as in vitro functional analysis to demonstrate the pathological significance of TNFAIP3 variants, especially for truncating mutations (3, 10). Furthermore, it was reported that disruption of the inhibitory effects of TNFAIP3 missense variants could be more sensitively distinguished from wild type TNFAIP3 function by analyzing NF-κB reporter gene activity in response to TLR or BCR signaling (18, 52). However, some missense variants did not show significant difference from wild type TNFAIP3 using the methods mentioned above (18). One must carefully interpret whether such variants represent low-penetrance mutations, polymorphisms that contribute mildly to the development of HA20, or polymorphisms that contribute to other diseases. TNFAIP3 Phe127Cys (rs2230926) is a single-nucleotide polymorphism associated with development of autoimmune diseases such as rheumatoid arthritis, SLE, and type 1 diabetes mellitus (DM) (53, 54). However, this variant showed no significant disruption of the inhibitory effects of A20, and it has not been evaluated as a pathogenic mutation for onset of HA20 in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/). It cannot be concluded that TNFAIP3 variants identified by comprehensive genetic analysis are pathogenic based solely on allele frequencies and patient symptoms; validation by functional analysis is required. Furthermore, more sensitive and specific methods for functional analysis of TNFAIP3 variants of unknown significance are required for future studies.
There is no standard treatment protocol for HA20. Various treatments are used in both East Asia and other regions, including colchicine, systemic corticosteroids, disease-modifying drugs, and molecular targeted therapies. The clinical severity of HA20 varies from mild to severe; responses to treatment also vary significantly. It has been reported that colchicine is effective to some extent in mild cases of HA20 (3, 10, 30, 37). In severe cases with poor response to colchicine, biological drugs are administered such as anti-TNF-α agents, anti-IL-1 agents, and anti-IL-6 agents. The rationale for such interventions is based on the increased production of proinflammatory cytokines such as TNF-α, IL-1β, IL-18, and IFN-γ-induced protein 10 in sera and stimulated peripheral blood mononuclear cells from patients with HA20 (3, 10). These therapies are effective in suppressing systemic inflammation in patients with other diseases. Anti-IL-1 agents were administered to one patient with HA20 in East Asia and were not effective; however, these agents were reported to be effective in some patients in other regions (3, 30, 35, 39, 40). Anti-IL-6 agents have been used less frequently to treat HA20 than anti-TNF-α and anti-IL-1 agents, but have been reported to be effective in a subset of patients (32). The efficacy of anti-IL-6 agents may relate to elevation of serum IL-6 during the active phase of disease (10). The possibility of the secondary ineffectiveness of biological drugs due to production of antibodies for them, especially anti-TNF-α agents, should be considered because severe HA20 patients complicated with autoimmune diseases are expected to have both autoinflammatory conditions and increased antibody production. Among molecular targeted therapies, JAK inhibitors were reported to be effective in patients with HA20 whose ISG expression was elevated in the active disease phase (38). Thus, administration of JAK inhibitors may become a more prominent treatment strategy. Although it has been conducted in only two patients with HA20 to date, HCT was effective in patients resistant to various treatments including several biological agents and led to remission of their inflammatory symptoms (31, 47). By contrast, a patient who received autologous HCT because of central nervous system vasculitis relapsed after 18 months of remission and various immunosuppressive agents were reinitiated (30). The first case of allogeneic HCT was a 14-year-old English boy. He had complex clinical features including development of insulin-dependent DM, cell depletion, hepatitis, enteropathy, and interstitial lung disease. Because his symptoms were refractory to treatment with prednisolone, sirolimus, tacrolimus, infliximab, and rituximab, he received HCT. Following HCT, he entered complete remission from all autoimmune disorders except DM. The second case was a 6-year-old Japanese boy. He had frequent fever, arthritis, psoriasis, aortic regurgitation, bowel disease, and genital ulcers. His symptoms were refractory to treatment with immunosuppressive drugs including prednisolone, cyclosporine, tocilizumab, anti-TNF-α agents, canakinumab, and tofacitinib. He underwent HCT and his autoinflammatory symptoms improved. However, some of his symptoms, such as severe aortic regurgitation and adrenal insufficiency, persisted because of long term administration of steroids. HCT might be an effective strategy for patients with treatment resistant HA20. However, it should be note that not all symptoms improve following HCT and that pre-existing organ damage will remain, potentially leading to symptom relapse. Because HA20 presents with various symptoms and severity, information on more cases is needed to establish appropriate treatment strategies.
We summarized the symptoms and treatments of patients with HA20 in East Asia and other regions. Patients in East Asia had significantly higher rates of recurrent fever and lower rates of typical BD symptoms including genital ulcers and skin rashes. Patients in East Asia were less likely to have autoimmune disease complications. Differences between patients with HA20 in East Asia and other regions may relate to variation in the frequencies of other modifier genes and/or environmental factors. Future studies will need to establish functional analysis methods to validate the pathogenicity of TNFAIP3 variants to enable rapid diagnosis of HA20 and development of appropriate treatment strategies.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This study was supported by MEXT KAKENHI (grant number JP21K07770), Health and Labour Science Research Grants for Research on Intractable Diseases from the Ministry of Health, Labour and Welfare of Japan (grant numbers 20316700 and 20317089), and AMED (grant number JP20ek0109480).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a past collaboration with the authors TK, SK, and HO.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript.
BD, Behçet’s disease; BCR, B-cell receptor; DM, diabetes mellitus; EBV, Epstein–Barr virus; HA20, A20 haploinsufficiency; HCT, hematopoietic cell transplantation; HLA, Human Leukocyte Antigen; IKK, IκB kinase; IL, interleukin; IRF, interferon regulatory factor; ISGFBD, International Study Group for Behçet’s disease; ISGs, IFN-inducible-genes; JAK, Janus kinase; NF, nuclear factor; OTU, ovarian tumor; SLE, systemic lupus erythematosus; TBK1, TANK-binding kinase 1; TLR, Toll-like receptors; TNF, tumor necrosis factor; TRAF, TNF receptor associated factor; ZnF, zinc finger.
1. Sakane T, Takeno M, Suzuki N, Inaba G. Behçet’s Disease. N Engl J Med (1999) 341:1284–91. doi: 10.1056/nejm199910213411707
2. Takeuchi M, Mizuki N, Meguro A, Ombrello MJ, Kirino Y, Satorius C, et al. Dense Genotyping of Immune-Related Loci Implicates Host Responses to Microbial Exposure in Behçet’s Disease Susceptibility. Nat Genet (2017) 49:438–43. doi: 10.1038/ng.3786
3. Zhou Q, Wang H, Schwartz DM, Stoffels M, Park YH, Zhang Y, et al. Loss-of-Function Mutations in TNFAIP3 Leading to A20 Haploinsufficiency Cause an Early-Onset Autoinflammatory Disease. Nat Genet (2016) 48:67–73. doi: 10.1038/ng.3459
4. Catrysse L, Vereecke L, Beyaert R, van Loo G. A20 in Inflammation and Autoimmunity. Trends Immunol (2014) 35:22–31. doi: 10.1016/j.it.2013.10.005
5. Boone DL, Turer EE, Lee EG, Ahmad RC, Wheeler MT, Tsui C, et al. The Ubiquitin-Modifying Enzyme A20 Is Required for Termination of Toll-Like Receptor Responses. Nat Immunol (2004) 5:1052–60. doi: 10.1038/ni1110
6. Wertz IE, O’Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, et al. De-Ubiquitination and Ubiquitin Ligase Domains of A20 Downregulate NF-kappaB Signalling. Nature (2004) 430:694–9. doi: 10.1038/nature02794
7. Mauro C, Pacifico F, Lavorgna A, Mellone S, Iannetti A, Acquaviva R, et al. ABIN-1 Binds to NEMO/IKKgamma and Co-Operates With A20 in Inhibiting NF-Kappab. J Biol Chem (2006) 281:18482–8. doi: 10.1074/jbc.M601502200
8. Tokunaga F, Nishimasu H, Ishitani R, Goto E, Noguchi T, Mio K, et al. Specific Recognition of Linear Polyubiquitin by A20 Zinc Finger 7 is Involved in NF-κb Regulation. EMBO J (2012) 31:3856–70. doi: 10.1038/emboj.2012.241
9. Wertz IE, Newton K, Seshasayee D, Kusam S, Lam C, Zhang J, et al. Phosphorylation and Linear Ubiquitin Direct A20 Inhibition of Inflammation. Nature (2015) 528:370–5. doi: 10.1038/nature16165
10. Kadowaki T, Ohnishi H, Kawamoto N, Hori T, Nishimura K, Kobayashi C, et al. Haploinsufficiency of A20 Causes Autoinflammatory and Autoimmune Disorders. J Allergy Clin Immunol (2018) 141:1485–8.e1411. doi: 10.1016/j.jaci.2017.10.039
11. Sato S, Fujita Y, Shigemura T, Matoba H, Agematsu K, Sumichika Y, et al. Juvenile Onset Autoinflammatory Disease Due to a Novel Mutation in TNFAIP3 (A20). Arthritis Res Ther (2018) 20:274. doi: 10.1186/s13075-018-1766-x
12. Tsuchida N, Kirino Y, Soejima Y, Onodera M, Arai K, Tamura E, et al. Haploinsufficiency of A20 Caused by a Novel Nonsense Variant or Entire Deletion of TNFAIP3 Is Clinically Distinct From Behçet’s Disease. Arthritis Res Ther (2019) 21:137. doi: 10.1186/s13075-019-1928-5
13. Shimizu M, Matsubayashi T, Ohnishi H, Nakama M, Izawa K, Honda Y, et al. Haploinsufficiency of A20 With a Novel Mutation of Deletion of Exons 2-3 of TNFAIP3. Mod Rheumatol (2021) 31:493–7. doi: 10.1080/14397595.2020.1719595
14. Imai T, Shiraishi A, Nishiyama K, Ishimura M, Ohga S. Lipopolysaccharide-Induced Monocyte Death in a Novel ZnF7 Domain Mutation of TNFAIP3. J Allergy Clin Immunol Pract (2020) 8:2071–4.e2075. doi: 10.1016/j.jaip.2020.01.026
15. Endo Y, Funakoshi Y, Koga T, Furukawa K, Sasaki D, Miura K, et al. Paediatric-Onset Haploinsufficiency of A20 Associated With a Novel and De Novo Nonsense TNFAIP3 Mutation. Rheumatology (Oxford) (2020) 59:e85–7. doi: 10.1093/rheumatology/keaa206
16. Uchida T, Suzuki T, Kikuchi A, Kakuta F, Ishige T, Nakayama Y, et al. Comprehensive Targeted Sequencing Identifies Monogenic Disorders in Patients With Early-Onset Refractory Diarrhea. J Pediatr Gastroenterol Nutr (2020) 71:333–9. doi: 10.1097/mpg.0000000000002796
17. Taniguchi K, Inoue M, Arai K, Uchida K, Migita O, Akemoto Y, et al. Novel TNFAIP3 Microdeletion in a Girl With Infantile-Onset Inflammatory Bowel Disease Complicated by a Severe Perianal Lesion. Hum Genome Var (2021) 8:1. doi: 10.1038/s41439-020-00128-4
18. Kadowaki S, Hashimoto K, Nishimura T, Kashimada K, Kadowaki T, Kawamoto N, et al. Functional Analysis of Novel A20 Variants in Patients With Atypical Inflammatory Diseases. Arthritis Res Ther (2021) 23:52. doi: 10.1186/s13075-021-02434-w
19. Zheng C, Huang Y, Ye Z, Wang Y, Tang Z, Lu J, et al. Infantile Onset Intractable Inflammatory Bowel Disease Due to Novel Heterozygous Mutations in TNFAIP3 (A20). Inflamm Bowel Dis (2018) 24:2613–20. doi: 10.1093/ibd/izy165
20. Dong X, Liu L, Wang Y, Yang X, Wang W, Lin L, et al. Novel Heterogeneous Mutation of TNFAIP3 in a Chinese Patient With Behçet-Like Phenotype and Persistent EBV Viremia. J Clin Immunol (2019) 39:188–94. doi: 10.1007/s10875-019-00604-9
21. Li GM, Liu HM, Guan WZ, Xu H, Wu BB, Sun L. Expanding the Spectrum of A20 Haploinsufficiency in Two Chinese Families: Cases Report. BMC Med Genet (2019) 20:124. doi: 10.1186/s12881-019-0856-1
22. Liang J, Zhang H, Guo Y, Yang K, Ni C, Yu H, et al. Coinheritance of Generalized Pustular Psoriasis and Familial Behçet-Like Autoinflammatory Syndrome With Variants in IL36RN and TNFAIP3 in the Heterozygous State. J Dermatol (2019) 46:907–10. doi: 10.1111/1346-8138.15034
23. Duan R, Liu Q, Li J, Bian X, Yuan Q, Li Y, et al. A De Novo Frameshift Mutation in TNFAIP3 Impairs A20 Deubiquitination Function to Cause Neuropsychiatric Systemic Lupus Erythematosus. J Clin Immunol (2019) 39:795–804. doi: 10.1007/s10875-019-00695-4
24. He T, Huang Y, Luo Y, Xia Y, Wang L, Zhang H, et al. Haploinsufficiency of A20 Due to Novel Mutations in TNFAIP3. J Clin Immunol (2020) 40:741–51. doi: 10.1007/s10875-020-00792-9
25. Chen Y, Huang H, He Y, Chen M, Seidler U, Tian D, et al. A20 Haploinsufficiency in a Chinese Patient With Intestinal Behcet’s Disease-Like Symptoms: A Case Report. Front Immunol (2020) 11:1414. doi: 10.3389/fimmu.2020.01414
26. Kim HY, Song JY, Kim WI, Ko HC, Park SE, Jang JH, et al. The First Case of an Infant With Familial A20 Haploinsufficiency in Korea. J Korean Med Sci (2020) 35:e252. doi: 10.3346/jkms.2020.35.e252
27. Zhang D, Su G, Zhou Z, Lai J. Clinical Characteristics and Genetic Analysis of A20 Haploinsufficiency. Pediatr Rheumatol Online J (2021) 19:75. doi: 10.1186/s12969-021-00558-6
28. Zou D, Zhou S, Wang H, Gou J, Wang S. Knee Joint Swelling at Presentation: A Case of Pediatric Crohn Disease With a TNFAIP3 Mutation. Pediatrics (2020) 146:e20193416. doi: 10.1542/peds.2019-3416
29. Yan M, Li D, Aknai S, Zhu H, Abudureyim M. Mutation Analysis of the TNFAIP3 in A20 Haploinsufficiency: A Case Report. Medicine (Baltimore) (2021) 100:e25954. doi: 10.1097/md.0000000000025954
30. Aeschlimann FA, Batu ED, Canna SW, Go E, Gül A, Hoffmann P, et al. A20 Haploinsufficiency (HA20): Clinical Phenotypes and Disease Course of Patients With a Newly Recognised NF-kB-Mediated Autoinflammatory Disease. Ann Rheum Dis (2018) 77:728–35. doi: 10.1136/annrheumdis-2017-212403
31. Duncan CJA, Dinnigan E, Theobald R, Grainger A, Skelton AJ, Hussain R, et al. Early-Onset Autoimmune Disease Due to a Heterozygous Loss-of-Function Mutation in TNFAIP3 (A20). Ann Rheum Dis (2018) 77:783–6. doi: 10.1136/annrheumdis-2016-210944
32. Lawless D, Pathak S, Scambler TE, Ouboussad L, Anwar R, Savic S. A Case of Adult-Onset Still’s Disease Caused by a Novel Splicing Mutation in TNFAIP3 Successfully Treated With Tocilizumab. Front Immunol (2018) 9:1527. doi: 10.3389/fimmu.2018.01527
33. Franco-Jarava C, Wang H, Martin-Nalda A, Alvarez SD, García-Prat M, Bodet D, et al. TNFAIP3 Haploinsufficiency Is the Cause of Autoinflammatory Manifestations in a Patient With a Deletion of 13Mb on Chromosome 6. Clin Immunol (2018) 191:44–51. doi: 10.1016/j.clim.2018.03.009
34. Viel S, Cheyssac E, Pescarmona R, Besson L, Till M, Viremouneix L, et al. Large Deletion in 6q Associated to A20 Haploinsufficiency and Thoracoabdominal Heterotaxy. Ann Rheum Dis (2018) 77:1697–8. doi: 10.1136/annrheumdis-2018-213300
35. Berteau F, Rouviere B, Delluc A, Nau A, Le Berre R, Sarrabay G, et al. Autosomic Dominant Familial Behçet Disease and Haploinsufficiency A20: A Review of the Literature. Autoimmun Rev (2018) 17:809–15. doi: 10.1016/j.autrev.2018.02.012
36. Mulhern CM, Hong Y, Omoyinmi E, Jacques TS, D’Arco F, Hemingway C, et al. Janus Kinase 1/2 Inhibition for the Treatment of Autoinflammation Associated With Heterozygous TNFAIP3 Mutation. J Allergy Clin Immunol (2019) 144:863–866.e865. doi: 10.1016/j.jaci.2019.05.026
37. Horita N, Gül A, Aksentijevich I, Kastner D, Remmers EF. Pseudodominance of Autoinflammatory Disease in a Single Turkish Family Explained by Co-Inheritance of Haploinsufficiency of A20 and Familial Mediterranean Fever. Clin Exp Rheumatol (2019) 37 Suppl 121:89–92.
38. Schwartz DM, Blackstone SA, Sampaio-Moura N, Rosenzweig S, Burma AM, Stone D, et al. Type I Interferon Signature Predicts Response to JAK Inhibition in Haploinsufficiency of A20. Ann Rheum Dis (2020) 79:429–31. doi: 10.1136/annrheumdis-2019-215918
39. Hautala T, Vähäsalo P, Kuismin O, Keskitalo S, Rajamäki K, Väänänen A, et al. A Family With A20 Haploinsufficiency Presenting With Novel Clinical Manifestations and Challenges for Treatment. J Clin Rheumatol (2020). doi: 10.1097/rhu.0000000000001268
40. Gans MD, Wang H, Moura NS, Aksentijevich I, Rubinstein A. A20 Haploinsufficiency Presenting With a Combined Immunodeficiency. J Clin Immunol (2020) 40:1041–4. doi: 10.1007/s10875-020-00823-5
41. Shaheen ZR, Williams SJA, Binstadt BA. Case Report: A Novel TNFAIP3 Mutation Causing Haploinsufficiency of A20 With a Lupus-Like Phenotype. Front Immunol (2021) 12:629457. doi: 10.3389/fimmu.2021.629457
42. Deshayes S, Bazille C, El Khouri E, Kone-Paut I, Giurgea I, Georgin-Lavialle S, et al. Chronic Hepatic Involvement in the Clinical Spectrum of A20 Haploinsufficiency. Liver Int (2021) 41:1894–900. doi: 10.1111/liv.14935
43. Girardelli M, Valencic E, Moressa V, Margagliotta R, Tesser A, Pastore S, et al. Genetic and Immunologic Findings in Children With Recurrent Aphthous Stomatitis With Systemic Inflammation. Pediatr Rheumatol Online J (2021) 19:70. doi: 10.1186/s12969-021-00552-y
44. Baxter SK, Walsh T, Casadei S, Eckert MM, Allenspach EJ, Hagin D, et al. Molecular Diagnosis of Childhood Immune Dysregulation, Polyendocrinopathy, and Enteropathy, and Implications for Clinical Management. J Allergy Clin Immunol (2021). doi: 10.1016/j.jaci.2021.04.005
45. Shigemura T, Kaneko N, Kobayashi N, Kobayashi K, Takeuchi Y, Nakano N, et al. Novel Heterozygous C243Y A20/TNFAIP3 Gene Mutation Is Responsible for Chronic Inflammation in Autosomal-Dominant Behçet’s Disease. RMD Open (2016) 2:e000223. doi: 10.1136/rmdopen-2015-000223
46. Criteria for Diagnosis of Behcet’s Disease. International Study Group for Behcet’s Disease. Lancet (1990) 335:1078–80.
47. Shiraki M, Williams E, Yokoyama N, Shinoda K, Nademi Z, Matsumoto K, et al. Hematopoietic Cell Transplantation Ameliorates Autoinflammation in A20 Haploinsufficiency. J Clin Immunol (2021). doi: 10.1007/s10875-021-01124-1
48. Kadowaki T, Ohnishi H, Kawamoto N, Kadowaki S, Hori T, Nishimura K, et al. Immunophenotyping of A20 Haploinsufficiency by Multicolor Flow Cytometry. Clin Immunol (2020) 216:108441. doi: 10.1016/j.clim.2020.108441
49. Chasset F, Dayer JM, Chizzolini C. Type I Interferons in Systemic Autoimmune Diseases: Distinguishing Between Afferent and Efferent Functions for Precision Medicine and Individualized Treatment. Front Pharmacol (2021) 12:633821. doi: 10.3389/fphar.2021.633821
50. Chen Y, Ye Z, Chen L, Qin T, Seidler U, Tian D, et al. Association of Clinical Phenotypes in Haploinsufficiency A20 (HA20) With Disrupted Domains of A20. Front Immunol (2020) 11:574992. doi: 10.3389/fimmu.2020.574992
51. Chu Y, Vahl JC, Kumar D, Heger K, Bertossi A, Wójtowicz E, et al. B Cells Lacking the Tumor Suppressor TNFAIP3/A20 Display Impaired Differentiation and Hyperactivation and Cause Inflammation and Autoimmunity in Aged Mice. Blood (2011) 117:2227–36. doi: 10.1182/blood-2010-09-306019
52. Escudero-Ibarz L, Wang M, Du MQ. Significant Functional Difference Between TNFAIP3 Truncation and Missense Mutants. Haematologica (2016) 101:e382–4. doi: 10.3324/haematol.2016.148346
53. Ma A, Malynn BA. A20: Linking a Complex Regulator of Ubiquitylation to Immunity and Human Disease. Nat Rev Immunol (2012) 12:774–85. doi: 10.1038/nri3313
Keywords: autoimmune disease, autoinflammatory disease, A20 haploinsufficiency, East Asia, TNFAIP3
Citation: Kadowaki T, Kadowaki S and Ohnishi H (2021) A20 Haploinsufficiency in East Asia. Front. Immunol. 12:780689. doi: 10.3389/fimmu.2021.780689
Received: 21 September 2021; Accepted: 11 November 2021;
Published: 26 November 2021.
Edited by:
Hirokazu Kanegane, Tokyo Medical and Dental University, JapanReviewed by:
Mario Abinun, Newcastle University, United KingdomCopyright © 2021 Kadowaki, Kadowaki and Ohnishi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hidenori Ohnishi, b2huaXNoaWhAZ2lmdS11LmFjLmpw
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.