 Modjtaba Emadi-Baygi
Modjtaba Emadi-Baygi Mahsa Ehsanifard
Mahsa Ehsanifard Najmeh Afrashtehpour
Najmeh Afrashtehpour Mahnaz Norouzi
Mahnaz Norouzi Zahra Joz-Abbasalian
Zahra Joz-Abbasalian- 1Department of Genetics, Faculty of Basic Sciences, Shahrekord University, Shahrekord, Iran
- 2Department of Research and Development, Erythrogen Medical Genetics Lab, Isfahan, Iran
- 3Clinical Laboratory, Sina Hospital, Tehran University of Medical Sciences, Tehran, Iran
The current global pandemic of the Severe Acute Respiratory Syndrome CoronaVirus 2 (SARS-CoV-2) causing COVID-19, has infected millions of people and continues to pose a threat to many more. Angiotensin-Converting Enzyme 2 (ACE2) is an important player of the Renin-Angiotensin System (RAS) expressed on the surface of the lung, heart, kidney, neurons, and endothelial cells, which mediates SARS-CoV-2 entry into the host cells. The cytokine storms of COVID-19 arise from the large recruitment of immune cells because of the dis-synchronized hyperactive immune system, lead to many abnormalities including hyper-inflammation, endotheliopathy, and hypercoagulability that produce multi-organ dysfunction and increased the risk of arterial and venous thrombosis resulting in more severe illness and mortality. We discuss the aberrated interconnectedness and forthcoming crosstalks between immunity, the endothelium, and coagulation, as well as how sex disparities affect the severity and outcome of COVID-19 and harm men especially. Further, our conceptual framework may help to explain why persistent symptoms, such as reduced physical fitness and fatigue during long COVID, may be rooted in the clotting system.
Introduction
Corona Virus Disease 2019 (COVID-19) is an infectious disease caused by SARS-CoV-2, an RNA virus with a crown-like appearance, and spreads rapidly all over the world. It transmits from human-to-human mainly via respiratory system (1). COVID-19, known to be a heterogeneous disease that manifests a varying range of symptoms from asymptomatic to severe disease. As an RNA virus, SARS-CoV-2 is a highly mutable virus with a rapidly evolving rate that leads to a various subtypes of the virus. Some characteristics of the SARS-CoV-2 have changed significantly in some evolved subtypes of the virus. Rapid evolution of the virus creates critical changes in SARS-CoV-2 behaviors, including an altered transmission or more severe disease (2). Considering the high rate of alterations in SARS-CoV-2 genome that cause various types of clinical and laboratory manifestations that in turn leads to the progression of COVID-19 towards severe and fatal forms of the disease in some cases, it calls for an urgent need for the identification of aberrated interconnectedness of biological levels of organization of humans and crosstalks between pathways that establish a malicious circuitry involving in COVID-19 pathogenesis. Interconnected cell signaling pathways (interconnectedness) may effectively and precisely transmit innumerable diverse signals, despite an intrinsic potential for improper levels of cross-talk. Network-level mechanisms insulate pathways from crosstalk and allow cells to process information from their environment and respond in ways to input signals. Evolutionarily, metazoan signaling networks are intricate, with incredible levels of crosstalk between pathways where proteins shared between two pathways provoke one pathway’s activity to be modulated by the activity of another. Indeed, crosstalk allows different cell types, each expressing a specific subset of signaling proteins, to trigger distinct outputs when dealt with the same inputs, reacting distinctly to the same environment. To point out, the tissue-specific networks mainly respond distinctly to the inhibition of individual proteins. These findings imply that the intricate interaction between network topology and gene expression that allows various cell types to respond distinctly to the same signals has significant implications for the development of drugs that target signaling processes (3, 4). As COVID-19 clinical manifestations indicate the symptoms, severity of the disease, and corresponding care settings, vary amongst the infected patients (5). One crucial determining factor in COVID-19 severity is the interaction between the virus and the host cells. It is known that the Spike (S) protein of CoronaViruses (CoVs) mediates the binding of the virus to the cell receptors and enables the fusion of the virus with the host cell membrane (6). Of particular interest, SARS-CoV-2 interacts with the RAS via ACE2, which was also identified as a functional receptor for Severe Acute Respiratory Syndrome CoronaVirus (SARS-CoV) (7–9). It has been indicated that RAS activation during SARS-CoV-2 infection leads to a number of unfavorable effects, which include vasoconstriction and hypertension, cellular differentiation and growth, endothelial dysfunction, and the formation of Reactive Oxidative Species (ROS) that may ultimately lead to organ damage (10, 11).
In human, ACE2 is expressed in nearly all human organs, such as the upper respiratory tract, alveolar epithelial cells, vascular endothelial cells and macrophages. In addition to acting as the receptor for SARS-CoV-2, ACE2 is a component of RAS which regulates several pathological processes like fibrosis, inflammation, oxidative stress and vasoconstriction (12, 13). Currently, there is a lack of evidence on how aberrated interconnectedness of RAS with the other systems contributed in COVID-19 pathophysiology. It has been hypothesized that the RAS may be involved in the COVID-19 pathogenesis via activation of the classic pathway. The broad distribution of ACE2 receptors in the endothelium could potentially allow for widespread effects outside the lung, following SARS-CoV-2 infection (14).
COVID-19 appears as asymptomatic disease or shows mild symptoms in the majority of patients (about 80%) (15, 16). In clinical evaluation, fever, cough, dyspnea, myalgia, and fatigue are the most common symptoms among mildly symptomatic patients. Moreover, uncommon symptoms, including headache, sputum production, hemoptysis, and diarrhea, have been reported in SARS-CoV-2 infection (17, 18). However, the remained proportion of the patients experience severe complications within a short time after infection, such as Acute Respiratory Distress Syndrome (ARDS), Disseminated Intravascular Coagulation (DIC), sepsis followed by organ failure, and death (19, 20). Notably, studies reported elder people are at a greater risk for developing severe forms of COVID-19, and a higher mortality rate was reported among older adults (21). COVID-19 is a complicated multi-system disease that greatly affects the vascular system and hemostasis maintenance. The correlation between the immune system dysfunction and impairment of general hemostasis is well-known and has been addressed in different contexts (1). The innate immune response is the first-line defense of the human body and has a determinant role in protective and/or destructive responses to any infections (22). An effective immune response upon viral infection includes type I InterFeroN (IFN-I) responses and its downstream cascades (23). In SARS-CoV-2, at the first contact with the respiratory mucosa and following SARS-CoV-2 entrance, the production of structural and non-structural proteins of the virus is triggered, subsequently blocking of the interferon response is promoted via SARS-CoV-2 N-protein (24). Failed IFN-I response curtails the early viral control and induced the penetration of hyper-inflammatory neutrophils, monocytes and macrophages, which lead to extensive production of pro-inflammatory cytokines and may evoke a cytokine storm that is correlated to the severe manifestations of COVID-19 (25–27).
Clinical evidence of severe patients of COVID-19 indicate that hyper-inflammation, and particular forms of vasculopathy, including Thrombotic MicroAngiopathy (TMA) and intravascular coagulopathy, are frequent features among them. In these cases, an uncontrolled increase of inflammatory cytokines induces vascular hyperpermeability and Multi-Organ Dysfunction Syndrome (MODS) leading to cardiac, hepatic, renal systems’ failure, and eventually death (1). Consistent with a state of hypercoagulability linked with a severe inflammatory response, coagulopathy is an important pathophysiological feature of COVID-19, characterized by the elevated fibrinogen levels, Von Willebrand Factor (VWF), and the fibrin degradation product (D-dimer, a fibrin degradation product that its increases are frequently reported in COVID-19 patients) (28). In fact, a failure to retain hemostasis due to pulmonary injury and MODS creates a critical condition in COVID-19 severe patients (29). A more complicated situation has been reported in the presence of coagulopathy, which is rather a prothrombotic character with a high chance of Venous ThromboEmbolism (VTE) among COVID-19 patients in Intensive Care Units (ICUs) (30). It seems that endothelial dysfunction, microvascular thrombosis, and occlusion, or autoimmune mechanisms may contribute to developing coagulopathy in severe SARS-CoV-2 pneumonia (31). In general, COVID-19 severity is not limited to the respiratory tract and shows age and sex tendencies. Besides the age bias, sex bias with higher numbers of cases, greater disease severity, and higher death rates among men compared to women is one of the interesting features of this disease that might occur due to male-specific factors that increase men’s susceptibility to the SARS-CoV-2 infection (32, 33). A growing body of evidence implies that we need to consider COVID-19 as a system-level infectious disease in which aberrated interconnectedness of immune system dysfunction, aberrant inflammatory responses, and prothrombotic conditions and crosstalks between some pathways occurred during COVID-19 pathogenesis that lead to the various manifestations and severity of the disease. Interconnected biological levels of organization of humans are capable of transmiting a multitude of different signals efficiently and accurately, regardless of a built-in potential for unenviable levels of crosstalks (28, 34, 35). With this in mind, we aimed to throw more light on pivotal aberrated interconnectedness of biological levels of organizations and pathways crosstalks in progression of COVID-19, to provide a well-defined insight to recognize COVID-19-associated pathophysiology as a system-level infectious disease.
The Role of Renin-Angiotensin System
When SARS-CoV-2 infects cells expressing the surface receptors ACE2 and transmembrane serine protease 2 (TMPRSS2), the active replication and release of the virus causes the host cell to undergo pyroptosis, an inflammatory cell death, and release Damage-Associated Molecular Patterns (DAMPs). In contrast to Angiotensin-Converting Enzyme (ACE), a zinc metalloproteinase and a key regulator of the RAS, ACE2 is an enzyme identified in rodents and humans with a more restricted distribution than ACE (36). In humans, ACE2 is mainly expressed in lung epithelial cells, enterocytes, arterial endothelial cells, and smooth muscle cells in cardiovascular tissues (37). In ACE2/TMPRSS2 dependent cell entry, S protein of SARS-CoV-2 binds to ACE2 to initiate virus entry (38, 39). Once bound, TMPRSS2 cleaves the S protein to allow for membrane fusion. Upon endocytosis, the viral RNA genome is released into the cytoplasm of the host by fusion of the virions with the endosomal membrane (6). This process is the most decisive phase in defining host compatibility and transmissibility of SARS-CoV-2.
The RAS has two pathways, the ACE-dependent pathway (vasoconstrictive side) and the ACE-independent pathway (vasodilative side). Both pathways begin with renin, which is produced by the kidney, converting angiotensinogen from the liver into ANGiotensin I (ANG I). In the ACE-dependent pathway, ANG I is converted to Angiotensin II (ANG II) by ACE. ANG II attaches to its receptor, ANG II Type 1 Receptor (AT1R). This increases Blood Pressure (BP) by causing vasoconstriction and sodium retention. Notwithstanding, in ACE-independent pathway a different enzyme, ACE2, converts ANG I to Angiotensin-1-9 (ANG-1-9) and ANG II to Angiotensin-1-7 (ANG-1-7). ANG-1-7 interacts with two different receptors, Mas and ANG II Type 2 Receptor (AT2R) receptor (40, 41). This pathway works to oppose the actions of the ACE-dependent pathway by causing vasodilation, thereupon lowering BP and providing other cardioprotective effects (40).
As SARS-CoV infection on lung cells leads to the decreased levels of ACE2 (42), it is postulated that SARS-CoV-2 works in the same vein in individuals without pre-existing conditions to reduce ACE2 expression in lung tissue. Markedly, ACE2 is highly expressed in the lung parenchyma, especially in type II pneumocytes (Alveolar Type II (AT2) cells) (43). Type II cells synthesize and release pulmonary surfactant, enriched with a rather unique phospholipid and four surfactant-associated proteins, which is necessary to maintain alveolar structure (44). Furthermore, Type II cells can differentiate to become Alveolar Type I (AT1) cells, a mechanism for replacement of type I cells that are damaged. The SARS-CoV-2 and SARS-CoV-1 viruses perturb alveoli to cause the main pathology in the lung, with increased fluid entry, cell death and inflammation along with reduction in gas exchange and levels of surfactant (45, 46). Moreover, viral infection triggers ACE2 endocytosis, leading to reduced cell surface expression of ACE2 (Figure 1A). Conversely, in individuals with comorbidities including diabetes, cardiovascular disease and hypertension, the ACE independent pathway is activated after SARS-CoV-2 infection due to the administrated drugs in these patients that block RAS. Indeed, pharmacological research concerning the SARS-Cov-2 patients and animal studies revealed frequent use of ACE inhibitor and/or Angiotensin II type I receptor blockers lead to a significant increase in ACE2 expression due to rise of angiotensin- (1–7) levels and decline in Ang-II levels in the plasma (47, 48) (Figure 1B). In addition, Gottschalk G et al. reported that ACE2 receptor was strongly upregulated in lungs during SARS-CoV-2 infection. As a result, ACE2 is seen as a double-edged sword in which in normal people the activation of ACE-dependent pathway upon SARS-CoV-2 infection leads to reduced expression of ACE2 that in turn results in ARDS and endothelial dysfunction, while in individuals with comorbidities leads to coagulation, inflammation, and damage to lungs and brain (49–51).
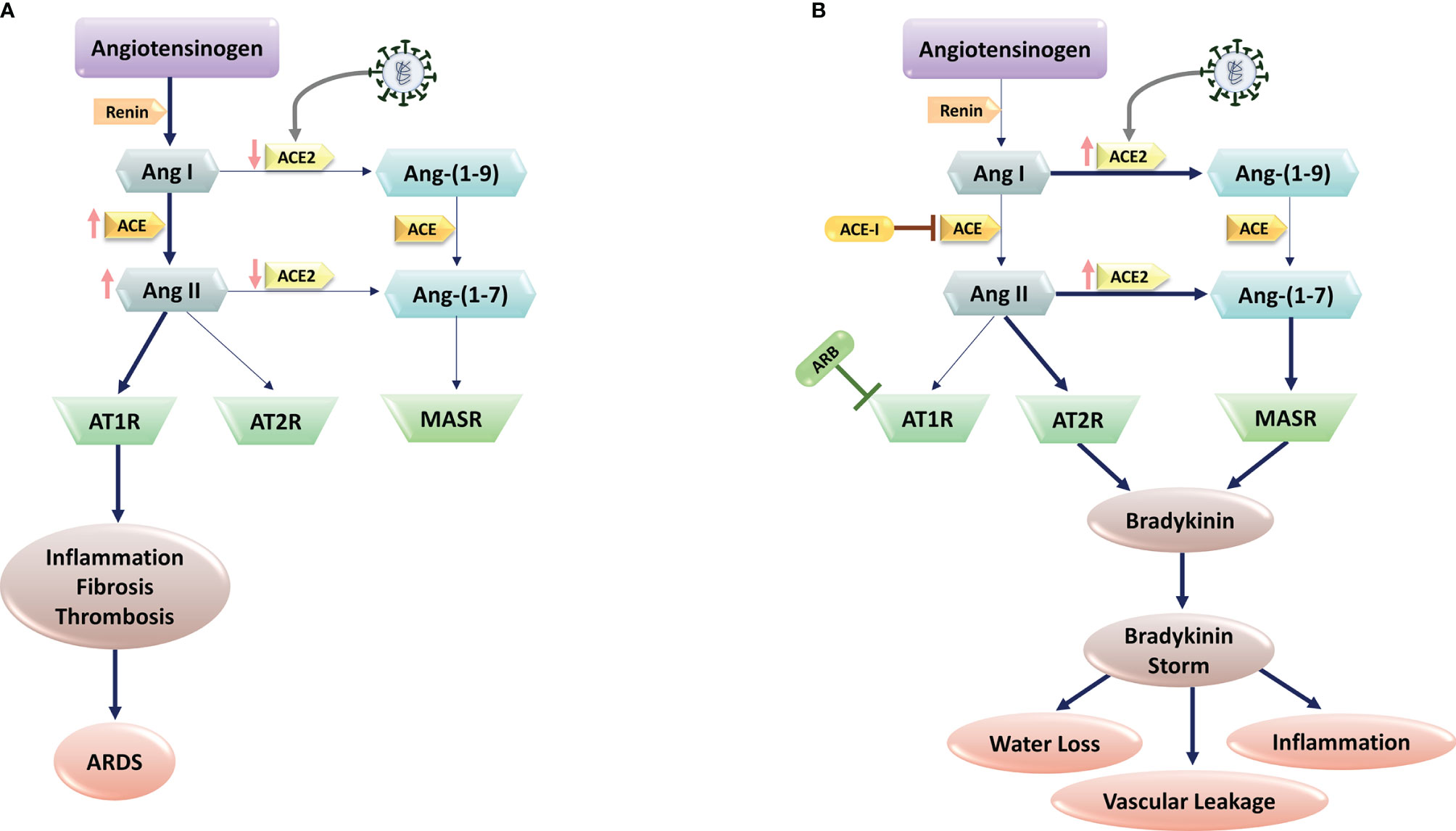
Figure 1 SARS-CoV-2 and Renin-Angiotensin System (RAS) interactions. (A) SARS-CoV-2 infection is mediated via the human Angiotensin-Converting Enzyme 2 (ACE2), which would cause ACE2 downregulation. Subsequent block of Ang II/Ang–(1-7) metabolism lead to the high levels of Ang II production through ACE upregulation. The imbalanced Ang II/Ang 1–7 causes the suppression of AT2 and Mas receptors with anti-inflammatory properties. In contrast, Ang II and its receptor AT1R activations induce pro-inflammatory effects, fibrosis, and thrombosis, leading to Acute Respiratory Distress Syndrome (ARDS) in lungs. (B) In patients with comorbidities, including hypertension, administration of RAS blockers (e.g., ACE-I, and ARBs) suppresses the ACE pathway. In these patients, SARS-CoV-2 infection increase ACE2 expression. Upregulated ACE-2 converts Ang I to Ang–(1-9) and Ang–(1-7), activating the bradykinin pathway via MAS and AT2R receptors. Upregulated bradykinin (bradykinin storm) in the lungs of COVID-19 patients would increase SARS-CoV-2 virulence by inducing hypotension, vascular permeability, water loss, and inflammation.
Using RNA sequencing of BronchoAlveolar Lavage (BAL) samples from patients with severe COVID-19 and comparing the results with control samples, Garvin et al. provided further detail into dysregulation of the RAS (52). Of note, the BAL fluid showed profound dysregulation of the RAS. Angiotensinogen and renin increased significantly. The degradation of Inhibitor of nuclear factor Kappa-B Kinase subunit gamma (IKK- γ) by the virus-encoded protein results in blocking production of interferon and ACE transcription. Without ACE, and thereby ANG II, ACE2 is upregulated in the lavage samples that in turn provide exceptionally more entry points for the virus. Furthermore, ACE2 converts ANG I to the fragment ANG-1-9. This fragment activates bradykinin receptor signaling that leads to bradykinin storm (52). In the lavage samples, bradykinin receptors 1 and bradykinin receptors 2 were substantially upregulated (53). Meanwhile, because of the reduction of ACE2 expression in response to SARS-CoV-2 infection ostensively in normal individuals, Ang II/AT1 activation via ACE-dependent pathway results in inflammation through an increase in ROS level that in turn leads to a rise in InterLeukin 6 (IL-6) and C-reactive protein levels. Furthermore, Ang II/AT1 activation results in endothelial dysfunction, and in the context of a viral infection, increased endothelial signaling may be the catalyst for initiation of the coagulation cascade in certain individuals. In this pathway, Ang2/AT1 activation results in multiplicative effects on vasoconstriction, BP, endothelial dysfunction, ROS formation, and finally damage to organs (Figure 1) (54).
Considering the role of RAS in COVID-19 pathogenesis, the key mechanisms associated with lower COVID-19 severity and mortality in women are: I) Decreased ACE2 methylation in women, resulting in increased ACE2 expression; II) ACE2 is on the short arm of the X chromosome, where up to 30% of genes undergo X inactivation Escape; III) estrogen promotes ACE2 expression. Higher levels of ACE2 could supply a better source to protect tissue after viral entry. Evidence shows ACE2 plays a protective role in chronic pathologies including hypertension, cardiovascular diseases, and acute respiratory distress syndrome, which are the comorbidities representing the risk of worse prognosis in COVID-19. Studies in mouse models support the protective role of ACE2 by showing more severe lung failure upon ACE2 down-regulation that results in overactivation of the Angiotensin (Ang) II/AT1R axis that may explain the multi-organ dysfunction seen in patients (Figure 1) (55, 56). However, low estrogens in men lead to the absence of higher levels of ACE2, supporting the ACE pathway in the RAS axis that facilitates disease severity in men with the same viral load as women. In men, androgens increased the expression of TMPRSS2 that supports viral entry, resulting in male increased susceptibility. In contrast, decreased levels of androgens in women may keep TMPRSS2 expression at lower levels, providing an additional defensive determinant against the development of COVID-19 infection (57).
Immune System and Inflammatory Responses
The human immune system protects us against life-threatening and pathogenic agents. As the first line of immune defense, the innate immune system plays an indispensable role against viruses (58, 59). Germline-encoded Pattern Recognition Receptors (PRRs) are essential immune receptors that trigger antiviral innate immune responses through sensing the conservative structures of viruses. PRRs have a crucial role in immune responses by recognizing Pathogen-Associated Molecular Patterns (PAMPs) that are unique microbial molecules and DAMPs that are self-derived molecules elicited from damaged cells (60, 61). Two PRRs, Toll-Like Receptors (TLRs) and Retinoic acid-Inducible Gene-I [RIG-I Like Receptors (RLRs)], are identified to have a crucial function in sensing viral ssRNA genome and dsRNA replication intermediates. TLRs and RLRs recognize these nucleic acid species and bind certain intracellular adaptor proteins, which activate NF-κB, mitogen-activated protein kinases, and interferon regulatory factors, which control the transcription of genes encoding IFN-I and other inflammatory cytokines, which are critical for virus elimination (62).
PRRs can identify SARS-CoV-2, like other RNA viruses, leading to the activation of IFN-I response (29, 63, 64). To point out, multiple viral structural and non-structural proteins of SARS-CoV impair IFN-I response, resulting in a hyper-inflammatory response. To enumerate, SARS-CoV is able to antagonize the TLR signaling pathway via its papain-like protease (65). Indeed, antagonized IFN responses in SARS-CoV-2 infection is likely to occur at various pathways involved in immune responses (Figure 2) (1). Like SARS-CoV and Middle East Respiratory Syndrome CoronaVirus (MERS-CoV), vesicle-dependent replication of SARS-CoV-2 genome may cause protecting RNA genome from host detection by cytosolic (e.g., RIG-I) and endosomal (e.g., TLR3/7) PRRs (66, 67). Furthermore, it is possible that INF-I inhibitions cause the antiviral response delay (e.g., by inhibiting TLR3 and TLR7 signaling pathways, virus-encoded antagonists to the IFN responses and auto-antibodies) that in turn it facilitates replication of virus particles and extensive virus cytopathic effects at the early stages of the COVID-19 disease (29, 63, 65, 68–70). Moreover, the infection of ACE2-positive ATII pneumocytes would lead to a significant decrease in the production of pulmonary surfactant and exposing the TLR4. In addition, direct or indirect SARS-CoV-2 binding to TLR4 causes an increase in the expression of ACE2 via IFNs and interferon-stimulated genes. Therefore, the virus may directly enter the cell using TLR4 and cause aberrant TLR4 signaling (64). However, Onabajo et al. show that the ACE2 induced by IFN is a truncated isoform of ACE2, deltaACE2, which is nonfunctional in binding the SARS-CoV-2 spike protein (71). Correspondingly, the activity of SARS-CoV-2 non-structural protein 14, which has a (guanine-N7)-methyltransferase activity, results in the efficient escape of viral RNA from detection by the RIG-I receptor. RIG-I is a critical cytosolic RNA sensor that interacts with the mitochondrial antiviral signaling proteins to activate the downstream programs such as type I/III IFN responses (72). This suboptimal innate immune operation by host PRRs during SARS-CoV-2 infection induces non-productive inflammatory responses, resulting in a cytokine storm and disseminated damage to the host. Generally, cytokine storm is a hyperactive immune response characterized by the release of different cytokines, chemokines, and other immune mediators that may hurt host cells. Notably, the majority of cytokine storm mediators demonstrate pleiotropic downstream effects mostly interdependent in their biological activity. Therefore discovering the precise dysregulated inflammatory response implies pathogenesis of the disease has been a major challenge in critical illness like COVID-19 (73). Although reports suggest that like SARS-CoV, SARS-CoV-2 have subversion strategies against innate immune signaling and antiviral interferon response, the exact aberrations of interconnectedness and crosstalks remain poorly understood (66, 72, 74). Commonly, cytokine storm is considered a critical determining factor associated with adverse outcomes of the COVID-19 disease; however, there is variation and sometimes-even discrepancies between studies about the exact detail of this phenomenon. Despite the poorly defined pathophysiology of cytokine storm, widespread acceptance of the term in COVID-19 has motivated to apply potent immunomodulatory therapies such as IL-6 inhibitors and high-dose corticosteroids. Nevertheless,in comparison with the other causes of ARDS (median IL-6 level is 10- to 200-fold higher than COVID-19) the lower levels of circulating cytokines in COVID-19 may not be representative of lung inflammation and need to be determined whether COVID-19 related ARDS phenotype is associated with the cytokine storm (73, 75).
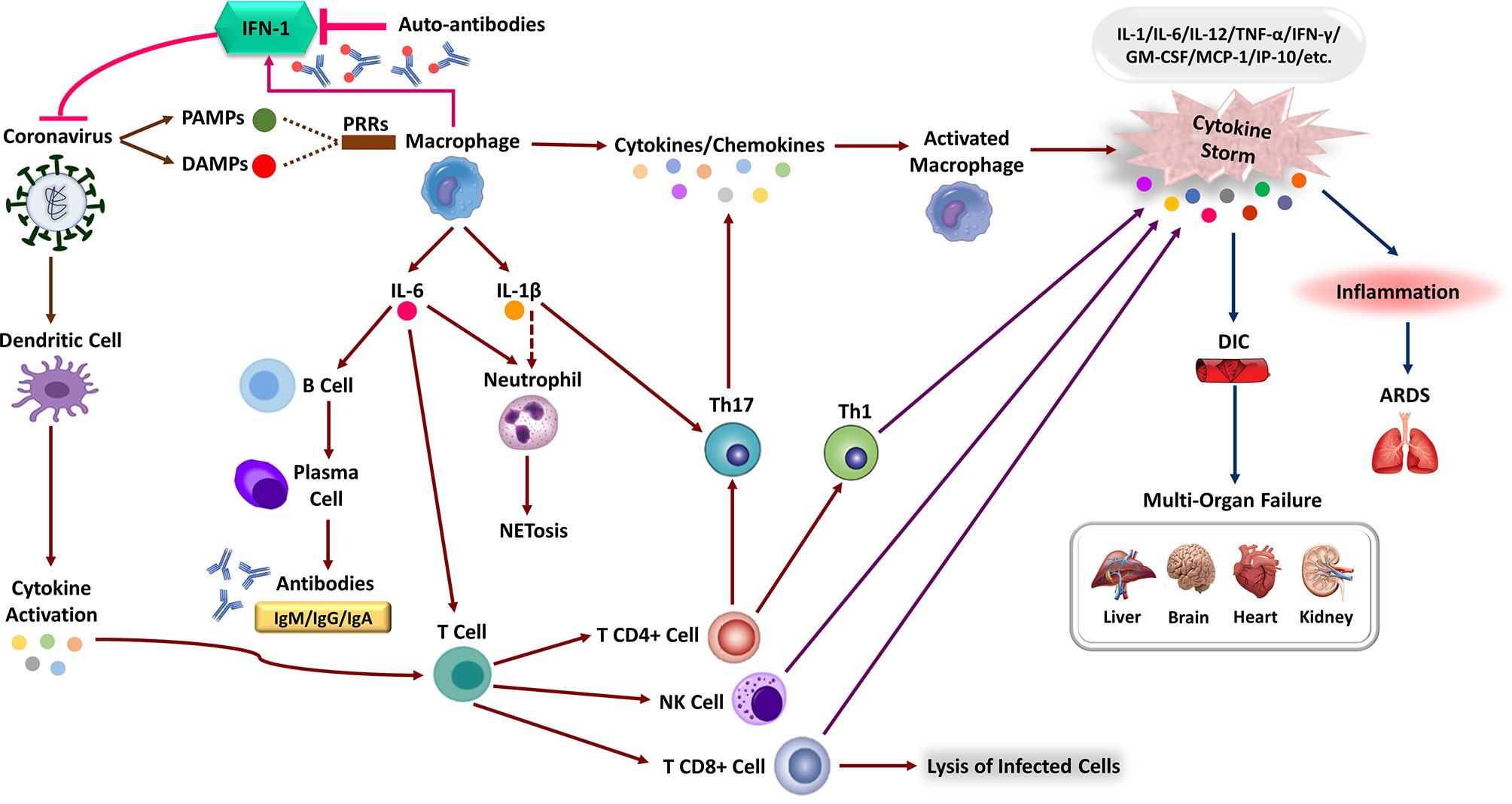
Figure 2 The immune response corporation in covid-19 disease. Type 1 interferon (IFN-I) immune response plays a pivotal role in effective immunity against SARS-CoV-2 infection and rapid viral clearance. Following SARS-CoV-2 infection, the IFN-1 initiates via recognizing PAMPs/DAMPs by PRRs of the human immune cells and releasing inflammatory cytokines (e.g., IL-1 beta, IL-6). The expression of numerous inflammatory cytokines leads to activation of neutrophils (NET formation) and attraction of different immune cells toward the site of infection. Macrophages activation, Dendritic Cell (DC) maturation and inflammatory cytokines stimulate the adaptive immune response to join the fight against SARS-CoV-2. Activation of T-cells via cytokines promotes multiple T-cell differentiation (i.e., CD4 +, CD8 +, NK cell, Th1) that directly kills virus-infected cells. Activated B-cells produce virus-specific antibodies that would participate in a successive immune response. In patients with severe forms of COVID-19, the delayed IFN-I pathways or neutralizing auto-Abs will lead to immune system overreaction and the generation of high inflammatory cytokine levels. The aberrant induction of the immune system and the production of various pro-inflammatory cytokines (e.g., IL-1, IL-6, MCP-1, TNF-α, and etc.), the so-called “cytokine storm,” leads to severe COVID-19 immunopathology. These can cause severe local damage to the lungs (e.g., ARDS) and other organs (e.g., DIC), and in the worst case, can lead to Multi-Organ Failures (MOF) and even death.
SARS-CoV-2 infection in pulmonary airway epithelial cells triggers local and systemic pathological responses through recruiting macrophages and monocytes, expressing inflammatory cytokines, and inducing adaptive immune responses. Although immune response resolves the infection in most COVID-19 patients, immune system dysfunction leads to hyper-inflammation resulting in a cytokine storm that causes severe lung injury and multi-system damages (1, 76, 77). These pro-inflammatory cytokines stimulate an influx of neutrophils and other myeloid cells into the lung, creating a local hyper-inflammatory response and significant immunopathology (78). The association of hyper-inflammatory response with lung injury, high rate of progression to ARDS, Multi-Organ Failure (MOF), and unfavorable prognosis of severe COVID-19 has been described in several studies (15, 79, 80). In the same way, overproduction of pro-inflammatory cytokines (Tumor Necrosis Factor alpha (TNF-α), IL-1, IL-6, and IL-1β), monocytes, and neutrophils, followed by a sharp decrease in lymphocytes, have been shown in various studies (15, 45, 81).
Clinical studies show major differences in the expression of inflammatory markers and immune phenotypes between moderate and severe cases, along with disease progression (82). An elevated level of immunomodulatory cytokines, including IL-1α, IL-1β, IFN-α, IL-17A, and IL-12p70, constitute COVID-19 signature that exhibits dynamic features associated with clinical manifestations (Figure 2) (15, 45). However, a considerable extent of other cytokines and chemokines, including IFN-γ, thrombopoietin (associated with blood clotting aberrations), IL-1, IL-6, IL-8, IL-2, IL-7, IL-10, Granulocyte Colony-Stimulating Factor (G-CSF), Interferon-inducible Protein 10 (IP-10), Monocyte Chemoattractant Protein 1 (MCP-1), Macrophage Inflammatory Protein 1α (MIP-1α) and TNF, C-X-C Ligand 10 (CXCL10), C-C motif Ligand 2 (CCL2) and CCL3, have been reported in severe patients (15). IL-6 is thought to play a pivotal role in pathology of COVID-19, considering its highest levels in non-survivors and critically ill patients. Notwithstanding that the IL-6 levels in these patients continue to increase over time, the mechanism leading to its elevation in severe COVID-19 is not currently clear. It is possible that aberrant activation of virus-specific PRRs like TLR4 triggers some sorts of crosstalks, which then drives IL-6 production (1, 83, 84).
The high levels of IL-6 observed in COVID-19 patients are apt to elicit Neutrophil Extracellular Traps (NETs) which include extracellular DNA fibers, histones, microbicidal proteins, proteases and oxidant enzymes to be released by neutrophils (Figure 2) (85, 86). Pathological effects of NETs have been indicated in propagated inflammation and respiratory failure. Although neutrophils are early indicators of SARS-CoV-2 infection, their excessive recruitment causes an unregulated NETs release that contributed to organ damage and mortality in COVID-19 patients (87). A significant increase in the blood neutrophil counts has been indicated in the COVID-19 patients admitted to the ICU. Consistent with SARS-CoV and MERS-CoV, increased levels of pro-inflammatory cytokines may drive an influx of neutrophils and other myeloid cells into the lung. The elevated neutrophil/lymphocyte ratio is considered an early prognostic marker in SARS-CoV-2 infection, which increases in parallel with the severity of the disease. Transcriptional analysis of BAL fluid and peripheral blood mononuclear cells showed that elevated CXCL2 and CXCL8 chemokines contribute to neutrophils recruitment and aggravate the inflammatory response in COVID-19 patients (66, 76). Of note, besides neutrophils, inflammatory chemokines cause the recruitment of other innate immune cells like monocytes, Dendritic Cells (DCs), and Natural Killer (NK) cells. These innate immune cells respond to tissue damage via producing several cytokines, including IL-1, IL-6, and TNF. Consequently, various immune cells such as neutrophils, macrophages, and T cells mobilize from the blood circulation into the infected tissue that leads to diffuse alveolar damage, capillary damage, vascular barrier damage, MOF and ultimately death (88–90).
Considering that the association of COVID-19 severity with aberrant inflammatory response has been demonstrated in various studies, the exact molecular mechanisms causing innate immune response dysregulation are yet to be elucidated (1). In fact, activation of innate immunity against pathogens would benefit the host in two ways. First, effective initiation of innate immune responses would directly kill pathogens. If those responses failed to eliminate the infection, the adaptive immune responses would initiate to provide a second layer of immune protection (61). In SARS-CoV-2 infection, however, inadequate and delayed intracellular innate immune responses failed to prime adaptive immune response for a long time, leading to severe lung disease and MODS. This particular condition in SARS-CoV-2 infection leads to a dis-synchronized innate and adaptive immune response (74), a kind of aberrated interconnedness.
In general, the high specificity of the adaptive immune system enables a highly regulated and targeted immune response to eliminate the infected cells and neutralizing free virions (91, 92). The effective adaptive immune response contains both humoral (B-cells) and cellular (T-cells) responses. The activity of CD8+ T cells and B cells is predominantly regulated by CD4+ T cells. In contrast, the targeted killing of virus-infected cells is accomplished by CD8+ T cells. B cells Antibodies (Abs) block surface proteins and agglutinate virions and thereby prevent infection (Figure 2) (93). Humoral immune system produces specific Abs that are expected to neutralize different antigens, limit virus viability and also elicit T-cells to the location of infection by delivering them. But some produced Abs against CoVs result in more complicated situations. Neutralizing antigen-specific Abs against CoVs is mostly targeting spike domain (94). A group of anti-spike Immunoglobulin G (IgG) in acute phase of infection before viral clearance can end in some inflammatory hyper-reactions (95). This phenomenon is due to a shift in polarization of alveolar macrophages and their related cytokines to a pro-inflammatory state. This alteration is mostly because of the binding affinity of anti-spike IgG and receptors presented on macrophages (91). Equally important, some infected individuals with SARS-CoV-2 exhibit a range of B cell population producing auto-antibodies similar to those observed in Systemic Lupus Erythromatose (SLE) (96). These Abs are mostly antinuclear Abs, rheumatoid factor (anti-IgG-Fc Abs), AntiPhosphoLipid Antibodies (APLAbs), and Abs against IFN-I. These Abs suppress the innate immune response via IFN-I antagonizing and directly contributes to pathophysiology of COVID-19 (97). Furthermore, auto-antibodies target immune-related proteins including those involved in lymphocyte function and activation, leukocyte trafficking, the type I and type III IFN responses, type II immunity and the acute phase response increase in patients with severe COVID-19. With this in mind, patients with auto-antibodies against IFN-1 experience extended durations of hospital admission due to impairment of virological clearance (98).
Equally important, like SARS-CoV and MERS-CoV infection, a drastic diminish in the number of T cells would ultimately cause an inefficient T cell response in severe COVID-19 patients (99). The critical role of CD4+ and CD8+ T cells in virus clearance has been demonstrated in the immunodeficient mouse model of SARS-CoV (100). Considering the pivotal function of T cell immunity in SARS-CoV infection, T cells appear to have a protective role against SARS-CoV-2 infection. However, the function of T cells in the resolution, and long-term protection against SARS-CoV-2 remains debated. The significant dysregulation of T-cell response has been indicated in severe COVID-19 patients; However, it has not been clear which T-cell activities contribute to the development of the disease severity (101, 102). Lymphopenia is one of the prominent features of severe COVID-19 that have been reported to affect CD4+, CD8+, B, and NK cells in some patients. Coupled with the high IL-6, IL-10, and TNF levels, lymphopenia was observed in severe COVID-19 manifestations (103–105). In particular, different studies have demonstrated that lymphopenia harbors T cells biased in COVID-19, which would lead to hyperactivation of these cells (101). While activation of T cells seems to have positive effects, SARS-CoV-2 may have mechanisms to restrict T cell activation. Regarding the peripheral lymphopenia observed in patients with COVID-19, the autopsy studies showed that the lymphocytic infiltration to the respiratory tract or adhesion to inflamed respiratory vascular endothelium is not excessive (106, 107). Observed lymphopenia in adult COVID-19 patients is possibly multifactorial (108). Lymphopenia in patients with severe disease may be related to cytokine over-production possibly by a direct effect of the cytokines on T cell populations (109, 110) and/or indirect effects through other cell types including dendritic (111) cells and neutrophils (112, 113). Augmented expression of pro-apoptotic molecules (apoptotic loss) (114), probably decreased mobilization of lymphocytes from bone marrow, and immunosenescence may also partake in T cell depletion (108, 115). Moreover, with unknown mechanisms, limited MHC I and MHC II antigen presentation was demonstrated in association with T cell dysregulation in these patients (116). In contrast to the mild SARS-CoV-2 infection that harbors a successful lymphocyte-mediated virus clearance, T cells are functionally exhausted, a state that arises during many chronic infections (117), and express a high level of Programmed cell Death protein 1 (PD-1) and T-cell immunoglobulin mucin-3 in severe COVID-19 patients (117, 118). Besides, the implication of the expression of these markers in acute viral infection relates rather with the activation state than with functional exhaustion (119). Moreover, the observed decrease in IFN-γ and IL-21 production would support CD8+ and CD4+ T exhaustion in SARS-CoV-2 patients. In addition, the functional exhaustion of NK cells has been demonstrated in the persistence COVID-19 condition as seen in cancer and chronic viral infection (99, 120). Nevertheless, this exhaustion is likely to be transient, as the decreased expression of PD-1 has been observed in recovered patients from ICU compared to the severe ICU ones (121).
Eventually, a sex-specific immune transcriptome is documented in humans, resulting in females exhibiting an enhanced adaptive immune response, while males show enhanced aspects of innate immunity (122). Equally important, in the context of the COVID-19, there is a difference in immune responses and in turn in severity between males and females. Male patients had substantial induction of non-classical monocytes and higher plasma levels of innate immune cytokines such as IL-8 and IL-18. By contrast, female patients during SARS-CoV-2 infection had substantial T cell activation compared to male patients. In particular, poor T cell response was exclusively associated with worse prognosis in male patients, which negatively correlated with patients’ age. By contrast, higher levels of innate immune cytokines were associated with worse disease progression in female patients, but not in male patients (123). In general, since an extra X chromosome exists in women, cellular mosaicism created by X inactivation in females may contribute partly to the more efficient immune response against SARS-CoV-2. Besides, more ICU admissions and mortality rates are observed in men with COVID-19. To point out, there are many X-linked genes that are related to the immune system, including PRR (for instance TLR7 and TLR8) and the major TLR signaling regulators (like IL-1 receptor-associated kinase 1), that may have a higher copy number in females (124). To emphasize, the biallelic expression of TLR7, an endosomal innate immune receptor, through X chromosome, may potentially cause a more substantial IFN-I response in the early stages of COVID-19 in female patients. Furthermore, increased IFN-α production triggered by TLR7 ligands as shown by in vitro experimental observations are detected more in females and subsequently lead to a more rapid antiviral response (29, 124). Moreover, Female reproductive steroids are anti-inflammatory, reshape competence of immune cells, stimulate Ab production, and promote proliferation and repair of respiratory epithelial cells, suggesting they may protect against COVID-19 symptoms (125). Equally important, steroid hormone receptors play a pivotal role in sex-dependent immunity, as defined by lower T lymphocytes percentage in men compared to women. Meanwhile, multiple pieces of evidence imply that there are associations between testosterone levels (normal male level vs age-related hypogonadism) in COVID-19 pathogenesis, indicating its function as a double-edged sword. Testosterone and dihydrotestosterone mediate their actions via the Androgen Receptor (AR), a ligand-dependent nuclear transcription factor (126). High testosterone impact on COVID-19 severity through the TMPRSS2 Connection (Figure 3) (127). Taken together, epidemiological data emerging from the COVID-19 pandemic, backed by animal studies and further by preliminary clinical studies in diverse clinical settings, support the notion that high testosterone levels acting via the AR attune TMPRSS2 function positively to enhance SARS-CoV-2 S proteins and eventually increase COVID-19 infectivity and severity. Additionally, AR mutations or other gene polymorphisms along the pathway of SARS-CoV-2 pathogenesis may further lead to COVID-19 progression and deterioration. This concept ought to be further explored in future studies (127). However, low testosterone, a characteristic biomarker of aging males with functional hypogonadism, impact on COVID-19 severity through the ACE2 Connection (Figure 3) (127). To clarify, in COVID-19, SARS-CoV-2 infection may impact the testes via binding to ACE2 expressed in the Sertoli and Leydig cells, provoking infertility and inhibiting testosterone production (128). It emphasized that low testosterone serum level is associated with SARS-CoV-2 infections and COVID-19 severity in critically ill patients through lower immunomodulatory properties of androgen antiviral effects (129). Altogether, low testosterone levels appear to be a major factor for poor prognosis and mortality in COVID-19 male patients. This is likely to be significantly exacerbated in men with co-morbid conditions admitted to the ICU. Further research is needed to prove this concept (127).
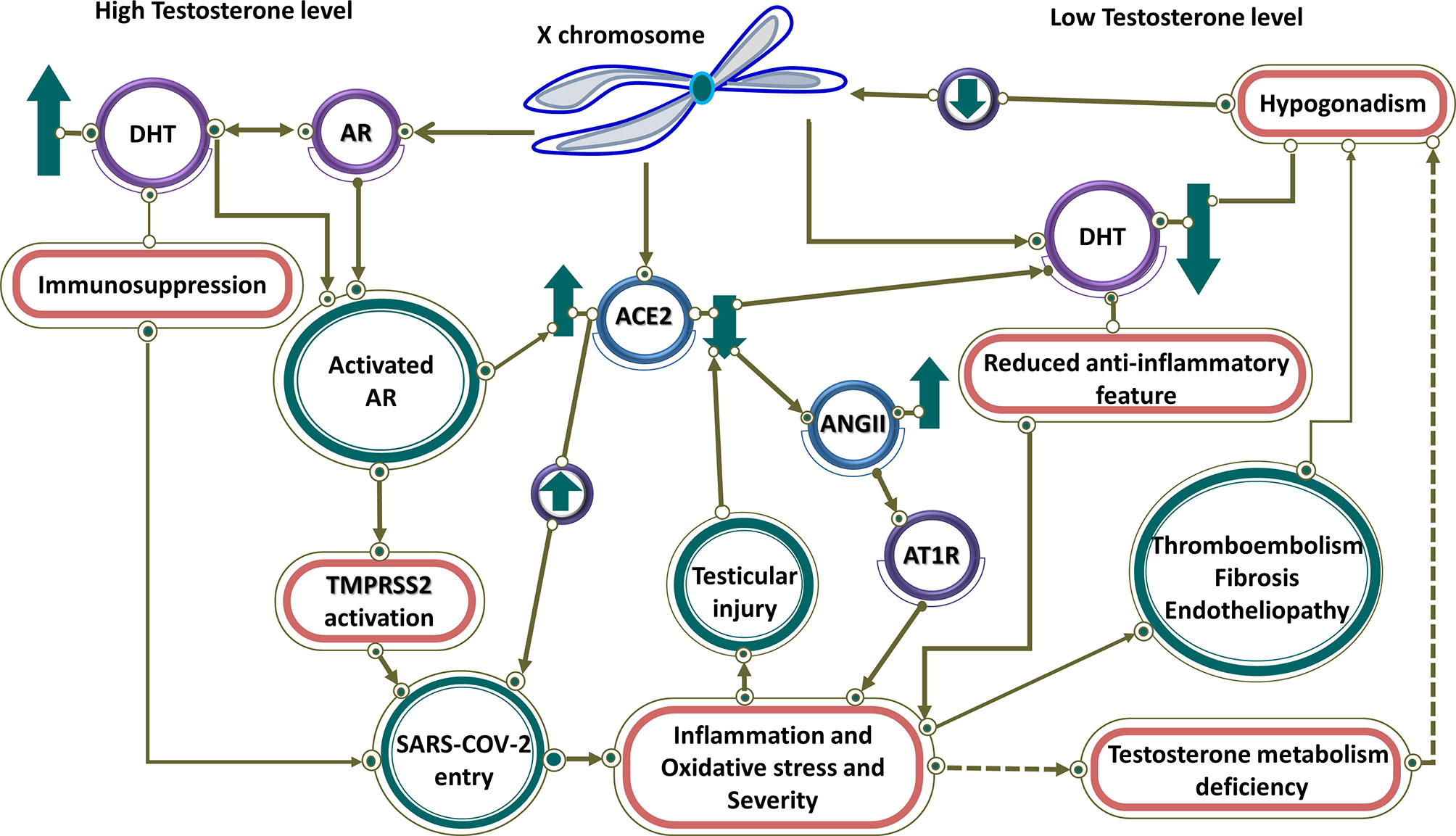
Figure 3 High and low testosterone impact on COVID-19 severity. Controlled by androgen, both TMPRSS2 and ACE2 are regulated by genes on the X chromosome. At high testosterone levels, both receptors are up-regulated, resulting in SARS-CoV-2 entry. Testosterone has an immunosuppressive feature, which leads to more virus entry and disease severity. In contrast, it has linked low testosterone level caused by hypogonadism in men to increased infection of COVID-19. Therefore, in men with hypogonadism, caused by various factors such as reduction of ACE2, testicular damage, and thromboembolism through hyperinflammation caused by the viral infection, the level of testosterone is reduced and the anti-inflammatory effect of testosterone is blocked, resulting in disease severity. Low testosterone results in downregulation of ACE2 that through activation of AR1T by ANG II leads to inflammation and disease severity.
By all means, COVID-19 is a complicated multi-system disease that greatly affects the immune system and homeostasis maintenance of the body (Figure 2) (130, 131). A serious clinical implication and involvement of aberrated interconnectedness and possibly triggered crosstalks have been observed in COVID-19 pathogenesis. The correlation between the immune system dysfunction and impairment of homeostasis is well-known and has been addressed in different contexts (132). Clinical evidence indicates that hyper-inflammation, and particular forms of vasculopathy, including TMA and intravascular coagulopathy, are frequent features of COVID-19 among severe patients (29). In these cases, an uncontrolled increase of inflammatory cytokines induces vascular hyperpermeability and MODS, leading to failure of cardiac, hepatic, renal systems, and eventually death (1). In fact, a failure to retain hemostasis due to pulmonary injury and MODS creates a critical condition in COVID-19 patients (29, 130). A more complicated situation has been reported in the presence of coagulopathy, which is rather a prothrombotic character with a high chance of VTE among COVID-19 patients in ICUs (30). As a result, the overproduction of inflammatory cytokines and the over-activation of immune cells during SARS-CoV-2 infection promote endothelial dysfunction and vascular permeability. Further investigations are mandatory to explore possible mechanisms behind these aberrated interconnectedness and possibly triggered crosstalks (29, 133).
Coagulopathy and endothelial dysfunction
Coagulation is a tightly regulated process that involves interactions between numerous blood components called coagulation factors and mechanisms that prevent thrombosis (134, 135). Currently, the so-called ‘COVID-19 associated coagulopathy’, is considered to be a critical player in the pathophysiology of SARS-CoV-2 infection, especially in its severe form (136–138). Coagulation and inflammation are highly integrated and delicately balanced biological systems with extensive crosstalk to optimize the body’s response to any damage and invasion. The impaired interplay of these systems affects both homeostasis and hemostasis, leading to various conditions with varying degrees of excessive inflammation, thrombosis, or bleeding that may lead to tissue damage and MOF (Figure 4) (34, 139–141). The hyper-coagulation state in COVID-19 is triggered by the deep and complex inflammatory response to the virus via the activation of tissue factors (TFs) on the surface of activated endothelial cells. Bidirectionally, coagulation and inflammation drive an intensifying circle of events by inducing Protease-Activated Receptors (PARs) mediated inflammatory signaling, participation of innate immune pathways, the engaging of the PC–thrombomodulin mechanism as negative regulatory systems, and the playing roles of NETs, and the fibrinolytic system (34, 142). PARs express in many cell types, including immune cells, platelets, endothelial cells, and smooth muscle cells. After proteolytic cleavage by serine proteases such as factor X and thrombi, they activate and induce the production of cytokines and chemokines. PAR-mediated activation of adhesion molecules in endothelial cells results in the production of IL-6 and IL-8 by fibroblasts and monocytes and increased platelet effects via a positive feedback loop, amplifying inflammation and procoagulant processes (34, 138, 142).
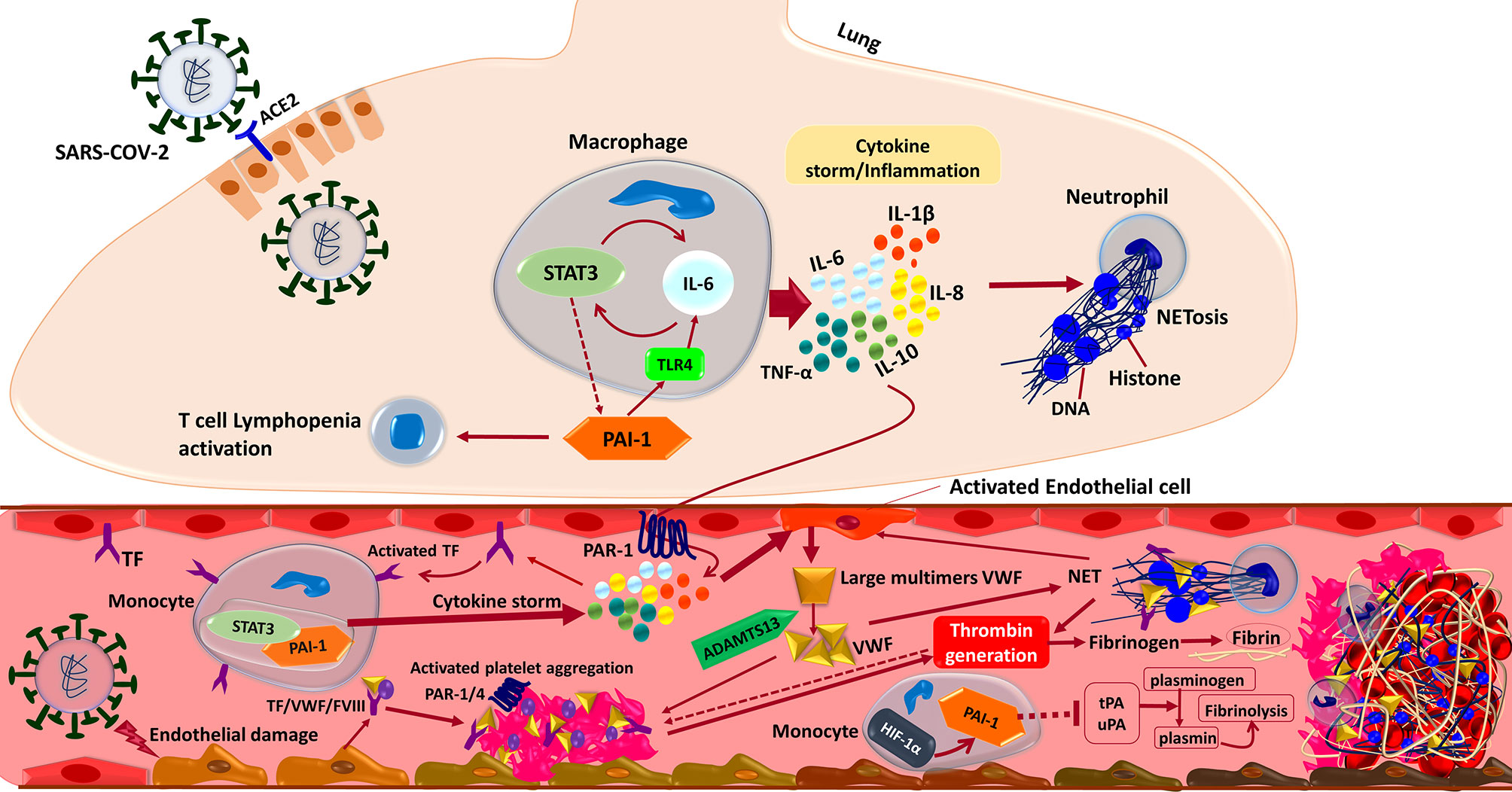
Figure 4 Interactions between the components of the immune system and coagulation cascades that induce thrombosis in COVID-19. In brief, the SARS-CoV-2 infection triggers an inflammatory response in the alveolar lumen by macrophages and neutrophils. Overexpression of PAI-1 and its interaction with TLR4 induce the IL-6 expression and STAT3 activation in the lung of COVID-19 patients. Subsequent production of inflammatory cytokines such as IL-1β, IL-8, IL-6, and TNF-α causes neutrophils to be recruited and release Neutrophil Extracellular Traps (NETs). NETs directly activate factor XII, thereby activating the contact-dependent clotting pathway. The PAR-1 receptor mediates cytokine-dependent Tissue Factor (TF) activation, which stimulates the STAT3/PAI-1 complex in blood’s monocytes and enhances inflammatory cytokine production leading to extrinsic coagulation cascade activation and thrombosis formation. Moreover, inflammatory cytokine in combination with the direct binding of SARS-CoV-2 to the endothelial cells triggers Von Willebrand Factor (VWF) secretion cleaved by ADAMTS13 to the regular size. Activated TF/VWF/FVIII complex in cooperation with NETs recruits platelets to adhere to endothelial surfaces and provoke intrinsic coagulation cascades. Altogether these events ultimately driving thrombin formation from circulating prothrombin, which cleaves fibrinogen to fibrin and stimulates thrombosis formation. Besides, suppression of endothelial enzymes and Plasminogen Activators (tPA/uPA) would aggravate the coagulopathy state by preventing effective fibrinolysis. Both tPA and uPA participate in the normal coagulation-plasmin-fibrin pathway, can be inhibited by PAI-1/HIF-1α in severe COVID-19 patients with ARDS.
Mounting evidence shows that coagulopathy events, including immunothrombosis and thromboinflammation, have been observed in COVID-19 (28, 143). Although pieces of literature are controversial and show conflicting findings (144–146), severe COVID-19 patients specially critically ill ones not responding to shock management may show features of systemic hyper-inflammation called Macrophage Activation Syndrome (MAS) or cytokine storm, also known as secondary Hemophagocytic LymphoHistiocytosis (sHLH). It should be noted that the cytokine profile similar to MAS/sHLH has also been observed in COVID-19 patients, especially the increase of IL1β, IL2, IL6, IL17, IL8, TNF and CCL2. Furthermore, like DIC associated with MAS/sHLH, there is evidence that D-dimer levels are elevated in COVID-19 pneumonia which may suggest that the virus-induced hyper-inflammatory pulmonary immunopathology is spreading to the adjacent microcirculation with a broad secondary fibrinolytic activation (Figure 4) (144, 147). An increase in D-dimer indicates that COVID-19 patients are in a hypercoagulable state, which can be attributed to the following reasons. First, viral infections are often accompanied by a threatening pro-inflammatory response and inadequate control of the anti-inflammatory response. It can cause endothelial cell dysfunction, leading to excessive thrombin production. Second, the hypoxia found in severe COVID-19 can stimulate thrombosis through both increasing blood viscosity and a hypoxia-inducible transcription factor-dependent signaling pathway. Third, hospitalized severe COVID-19 patients are more likely to have risk factors such as advanced age, underlying diseases, long-term bed rest, and receiving invasive treatment, which are risk factors for hypercoagulability or thrombosis. To point out, the dissection of the lungs of critically ill patients with COVID-19 shows occlusion of small pulmonary vessels and microthrombosis (148). Fourth, some patients may develop septic coagulopathy or even DIC. Elevated D-dimer is always associated with adverse events. In some clinical patients with SARS-CoV-2 infection, acral gangrene in hypercoagulation state has been described pathophysiologically, indicating that coagulation is the most significant cause of vascular thrombosis and venous gangrene in COVID-19 cases (149). Of note, the serological observations of these COVID-19 patients revealed increased D-dimers associated with antithrombin III deficiency, illustrating wide vascular microthrombi and necrosis in skin biopsy (148, 149). Finally, Similar to COVID-19 non-survivors, DIC is one of the major complications reported in fatal MERS-CoV cases. Markedly, a case of MERS-CoV induced DIC, intracerebral hemorrhage, and MOF appeared two weeks’ post-admission in an otherwise stable patient. Furthermore, a fatal case of MERS-CoV was associated with DIC, hyperkalemia, ventricular tachycardia, and cardiac arrest (150–152).
Most compelling evidence shows that endothelial cells can be infected by SARS-CoV-2, leading to endothelialitis. The initiation of inflammation-induced coagulation is mostly mediated by the expression of the TF pathway (CD142). TF is expressed in both mononuclear cells in response to pro-inflammatory cytokines (mainly IL-6) and vascular endothelial cells to promote the conversion of prothrombin into thrombin resulting in fibrin-based blood clots due to the conversion of circulating fibrinogen into fibrin (Figure 4) (153–156). To point out, evidence reveals that severe cases of COVID-19 are commonly dependent on a positive feedback loop established between Signal Transducer and Activator of Transcription-3 (STAT3) and Plasminogen Activator Inhibitor-1 (PAI-1) that results in the over-stimulation of the STAT3/PAI-1 signaling network that is shared among diverse disease manifestations and leads to catastrophic consequences (157). To enumerate, PAI-1 is highly expressed in lungs and plasma of COVID-19 patients (158, 159). Furthermore, PAI-1 interacts with TLR4 and triggers the expression of IL-6 that activates STAT3 (157, 158). In turn, STAT3 and PAI-1 augmented thrombosis and coagulopathy in COVID-19 possibly by effectively suppressing urokinase-type Plasminogen Activator (uPA) and tissue-type Plasminogen Activator (tPA) (Figure 4) (158–160).
Neutrophils kill and clear invading microorganisms through phagocytosis and degranulation. NETs are a novel antimicrobial strategy of neutrophils that are released from neutrophils into extracellular space to catch and kill invading bacteria to protect the host from infection (161). Regarding the pathophysiology of COVID-19, immunothrombosis and release of NET lead to inflammatory lung/organ damage and thrombosis via enhanced NETosis in which neutrophils are more prone to spontaneously form NETs by adopting the low-density phenotype (28, 162–165). Notably, isolated neutrophils from patients with COVID-19 displayed increased NET release at baseline, similar to neutrophils stimulated by phorbol myristate acetate from healthy donors, implying that the environment of the COVID-19 plasma provokes NET formation (Figure 4) (28, 165). In the same vein, high levels of NETs detected in the sera of COVID-19 patients can trigger NETs formation in vitro in neutrophils collected from healthy volunteers (166, 167). Further, direct stimulation of NETosis by APLAbs of COVID-19 patients (28, 168), as well as the involvement of infectious diseases in the AntiPhospholipid Syndrome (APS) (28, 169), indicates that SARS-CoV-2 infection possibly synergize with APLAbs to provoke the immunothrombotic process. To clarify, Higher APLAbs titers are associated with increased neutrophil and platelet activity and more serious respiratory disease (28, 170). NETs also contribute to acute lung injury by inducing macrophage release of IL-1β, which in turn can reinforce the formation of NET (25, 28, 171). Furthermore, it is known that most of the inflammatory mediators increased in COVID-19 patients regulate the activity of neutrophils through the expression of chemotactic factors (15, 28, 172). Altogether, the evidence indicate that dysregulation of cytokine release might be maintained by NET-mediated crosstalk between neutrophils and macrophages, leading to disordered or disproportionate immunothrombotic status (28).
As a key mediator of hemostasis and plasma glycoprotein, the VWF takes circulating platelets at the vascular injury sites and mediates activation and aggregation of platelets (166, 173). VWF is a multimer that exists in the endothelial Weibel-Palade bodies and releases under the stimulation of cytokines. Proteolytically cleaved by A Disintegrin And Metalloproteinase with ThromboSpondin type 1 motifs, member 13 (ADAMTS13) to regulate the size and activity of VWF multimers, ultra-large VWF multimers are processed into smaller VWF forms to prevent the formation of thrombus (166, 174). To clarify, high molecular weight forms of excessively released VWF may provoke microcirculatory dysfunction through spontaneous binding to platelets resulting in TMA (175, 176). Considering COVID-19, VWF antigen levels (VWF : Ag) and VWF : Ristocetin cofactor activity (VWF : RCo) have significantly increased in moderate and severe cases than in normal controls possibly without change in ADAMTS13 activity (Figure 4) (175).
Overall, NETs and VWF/ADAMTS13 axis are essential for thrombosis and inflammation. Given these points, recent evidence shows that the axis is associated with and contributes to the poor prognosis of COVID-19 patients manifested initially by ARDS and then developed to more complex clinical phenotypes, including thrombotic thrombocytopenic purpura-like syndrome, hepatic coagulopathy, MODS, and extensive micro- and macrovascular thrombosis (163, 166, 177). There are increasing global reports considering APLAbs in COVID-19–associated coagulopathy (178). APS is an acquired thrombophilia in which patients develop pathogenic auto-antibodies against phospholipids and phospholipid-binding proteins. Of note, patients in a subtype of APS develop multi-organ thromboses over the shortest periods (Catastrophic APS (CAPS)) like patients with severe COVID-19–associated coagulopathy. Both conditions show acute inflammatory response, cytokine storm, and highly elevated ferritin levels (179, 180). Furthermore, both conditions develop retiform purpura and livedoid rashes representing thrombotic microvascular injury with endothelial damage and cytokine reaction, spread by activation and deposition of complement that predisposes to thrombosis (181). However, thrombocytopenia is not as common as CAPS in COVID-19 (182). Therefore, there are clinical and laboratory similarities between severe COVID-19 and CAPS. However, we need more studies to delineate APL-related mechanisms of thrombosis in COVID-19 (178).
As aforementioned, males and females respond differently to MERS- and SARS-CoV infections in which the conditions are more threatening for males (183–185). Comparatively, there are important differences between men and women for cardiovascular diseases. To point out, men have a higher risk of first and recurrent venous thrombosis than do women (186). Considering COVID-19, different genetic and endocrine mechanisms might influence the mechanisms of coagulopathy and thrombosis in COVID-19 (187). Most compelling evidence shows genetic mechanisms of hemostasis do not differ in men and women. Henceforth, the greater risk of thromboembolism in men, even in the context of COVID-19, does not attribute to any sex-linked difference in genetic predisposition (186, 187). However, there are some pieces of evidence indicating the different role of endocrine mechanisms in regulating hemostasis in males and females. In humans, the plausible general rule is that under normal conditions of exposure to at least normal sex hormone levels, estrogens play a possible positive role and androgens play a negative role in hemostasis (187). Of note, testosterone deficiency in men correlated well with an increase in procoagulant factors and a decrease in anticoagulant factors contributes to the higher risk of thromboembolism in men than in women at any age including a greater frequency in men during elderly and a decreased frequency in women during fertile periods (188–192). To point out, the estrogen deficiency possibly explains why the incidence of thrombosis observed in the elderly is higher than in young women (187, 193). Given these points, a growing body of evidence seems to imply that deficiencies of sex hormones are a detrimental factor in the immune and inflammatory response or predisposition to thrombosis, upon which they present an unfavorable prognostic factor in the severity and outcome of COVID-19 (187, 194). To clarify, evidence imply that primary hypogonadism in males and secondary hypogonadism in both sexes may have a direct or indirect role in the development of systemic inflammation, endotheliopathy, and thromboembolism in patients with COVID-19 (129, 187, 194, 195). Altogether, pieces of evidence imply that the severity of COVID-19 deteriorates in older ages for both sexes, in men more than in women, possibly because of inflammaging and endothelial dysfunction with aging. However, it is not quite clear if sex hormones have different effects on immunity, inflammation, and thrombotic status, and if different reductions in sex hormones in both sexes affect the severity and outcome of COVID-19 (183–185, 187, 196, 197).
Besides susceptibility to age-dependent diseases, epidemiological studies indicated sexdifferences in incidence and Case Fatality Rates (CFRs) after SARS-CoV infection in humans in which males experience higher CFRs than females (183, 184). Similarly, data from recent MERS-CoV outbreaks showed high incidence and CFRs among men (185).
Conclusion
COVID-19, especially in its severe form, is a multisystem syndrome with sex disparities in severity and outcome characterized by a state of immunothrombosis in which a complex interaction of SARS-CoV-2 virus invasion with platelets, leukocytes, endothelial cells, immune response, and the possible involvement of megakaryocytes resulting in endotheliopathy, coagulopathy and thromboinflammation that culminate in more severe complications and mortality. With this in mind, we may claim that the capability of interconnected biological levels of the organization is aberrated significantly by SARS-CoV-2 infection during COVID-19 progression, resulting in pathologically and fatally enhanced levels of crosstalks. A recent systematic review and meta-analysis of over 50 long-term effects of COVID-19 show that fatigue, headache, attention disorder, hair loss, and dyspnea are the five most common symptoms (198). Correspondingly, a more recent study (199) revealed that clotting markers elevated in patients with long COVID syndrome while inflammation markers had returned to normal, showing that the clotting system may be involved in the root cause of long COVID syndrome that helps to explain persistent symptoms such as reduced physical fitness and fatigue. To clarify, we propose that a deeper understanding of the coagulation and inflammation in acute COVID-19 may also be relevant for a better understanding of certain cases of long COVID.
Author Contributions
All authors wrote the first draft of the manuscript. MEB and MN critically reviewed and edited the manuscript. ME and NA created figures. MEB supervised all aspects of the work. All authors contributed to the article and approved the submitted version.
Funding
This project is supported in part by the Shahrekord university grant to MEB (grant number 1399).
Conflict of Interest
MN was employed by Erythrogen Medical Genetics Lab.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank all individuals who cooperated in this study.
References
1. Tay MZ, Poh CM, Rénia L, MacAry PA, Ng LFP. The Trinity of COVID-19: Immunity, Inflammation and Intervention. Nat Rev Immunol (2020) 20(6):363–74. doi: 10.1038/s41577-020-0311-8
2. Giovanetti M, Benedetti F, Campisi G, Ciccozzi A, Fabris S, Ceccarelli G, et al. Evolution Patterns of SARS-CoV-2: Snapshot on Its Genome Variants. Biochem Biophys Res Commun (2021) 538:88–91. doi: 10.1016/j.bbrc.2020.10.102
3. Haney S, Bardwell L, Nie Q. Ultrasensitive Responses and Specificity in Cell Signaling. BMC Syst Biol (2010) 4:119. doi: 10.1186/1752-0509-4-119
4. Rowland MA, Greenbaum JM, Deeds EJ. Crosstalk and the Evolvability of Intracellular Communication. Nat Commun (2017) 8(1):1–8. doi: 10.1038/ncomms16009
5. Lin L, Lu L, Cao W, Li T. Hypothesis for Potential Pathogenesis of SARS-CoV-2 Infection–A Review of Immune Changes in Patients With Viral Pneumonia. Emerg Microbes Infect (2020) 9(1):727–32. doi: 10.1080/22221751.2020.1746199
6. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell (2020) 181(2):271–80.e8. doi: 10.1016/j.cell.2020.02.052
7. Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A Pneumonia Outbreak Associated With a New Coronavirus of Probable Bat Origin. Nature (2020) 579(7798):270–3. doi: 10.1038/s41586-020-2012-7
8. Amraei R, Rahimi N. COVID-19, Renin-Angiotensin System and Endothelial Dysfunction. Cells (2020) 9(7):1652. doi: 10.3390/cells9071652
9. Wulandari L, Hamidah B, Pakpahan C, Damayanti NS, Kurniati ND, Adiatmaja CO, et al. Initial Study on TMPRSS2 P.Val160Met Genetic Variant in COVID-19 Patients. Hum Genomics (2021) 15(1):1–9. doi: 10.1186/s40246-021-00330-7
10. Dhanachandra Singh K, Karnik SS. Angiotensin Receptors: Structure, Function, Signaling and Clinical Applications. J Cell Signal (2017) 01(02):1–2. doi: 10.4172/2576-1471.1000111
11. Ni W, Yang X, Yang D, Bao J, Li R, Xiao Y, et al. Role of Angiotensin-Converting Enzyme 2 (ACE2) in COVID-19. Crit Care (2020) 24(1):1–10. doi: 10.1186/s13054-020-03120-0
12. Li W, Moore MJ, Vasllieva N, Sui J, Wong SK, Berne MA, et al. Angiotensin-Converting Enzyme 2 Is a Functional Receptor for the SARS Coronavirus. Nature (2003) 426(6965):450–4. doi: 10.1038/nature02145
13. Shukla AK, Banerjee M. Angiotensin-Converting-Enzyme 2 and Renin-Angiotensin System Inhibitors in COVID-19: An Update. High Blood Press Cardiovasc Prev (2021) 28(2):129–39. doi: 10.1007/s40292-021-00439-9
14. Wiese OJ, Allwood BW, Zemlin AE. COVID-19 and the Renin-Angiotensin System (RAS): A Spark That Sets the Forest Alight? Med Hypotheses (2020) 144:110231. doi: 10.1016/j.mehy.2020.110231
15. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical Features of Patients Infected With 2019 Novel Coronavirus in Wuhan, China. Lancet (2020) 395(10223):497–506. doi: 10.1016/S0140-6736(20)30183-5
16. Wu Z, McGoogan JM. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72314 Cases From the Chinese Center for Disease Control and Prevention. JAMA - J Am Med Assoc (2020) 323(13):1239–42. doi: 10.1001/jama.2020.2648
17. Rodriguez-Morales AJ, Cardona-Ospina JA, Gutiérrez-Ocampo E, Villamizar-Peña R, Holguin-Rivera Y, Escalera-Antezana JP, et al. Clinical, Laboratory and Imaging Features of COVID-19: A Systematic Review and Meta-Analysis. Travel Med Infect Dis (2020) 34:101623. doi: 10.1016/j.tmaid.2020.101623
18. Yang W, Cao Q, Qin L, Wang X, Cheng Z, Pan A, et al. Clinical Characteristics and Imaging Manifestations of the 2019 Novel Coronavirus Disease (COVID-19):A Multi-Center Study in Wenzhou City, Zhejiang, China. J Infect (2020) 80(4):388–93. doi: 10.1016/j.jinf.2020.02.016
19. Im Kampe EO, Lehfeld AS, Buda S, Buchholz U, Haas W. Surveillance of COVID-19 School Outbreaks, Germany, March to August 2020. Eurosurveillance (2020) 25(38):2001645. doi: 10.2807/1560-7917.ES.2020.25.38.2001645
20. Miesbach W, Makris M. COVID-19: Coagulopathy, Risk of Thrombosis, and the Rationale for Anticoagulation. Clin Appl Thromb (2020) 26:1–7. doi: 10.1177/1076029620938149
21. Farshbafnadi M, Kamali Zonouzi S, Sabahi M, Dolatshahi M, Aarabi MH. Aging & COVID-19 Susceptibility, Disease Severity, and Clinical Outcomes: The Role of Entangled Risk Factors. Exp Gerontol (2021) 154:111507. doi: 10.1016/j.exger.2021.111507
22. Kindler E, Thiel V, Weber F. Interaction of SARS and MERS Coronaviruses With the Antiviral Interferon Response. Adv Virus Res (2016) 96:219–43. doi: 10.1016/bs.aivir.2016.08.006
23. Prompetchara E, Ketloy C, Palaga T. Immune Responses in COVID-19 and Potential Vaccines: Lessons Learned From SARS and MERS Epidemic. Asian Pac J Allergy Immunol (2020) 38(1):1–9. doi: 10.12932/AP-200220-0772
24. Larenas-Linnemann D, Rodríguez-Pérez N, Arias-Cruz A, Blandón-Vijil MV, Del Río-Navarro BE, Estrada-Cardona A, et al. Enhancing Innate Immunity Against Virus in Times of COVID-19: Trying to Untangle Facts From Fictions. World Allergy Organ J (2020) 13(11):100476. doi: 10.1016/j.waojou.2020.100476
25. Channappanavar R, Perlman S. Pathogenic Human Coronavirus Infections: Causes and Consequences of Cytokine Storm and Immunopathology. Semin Immunopathol (2017) 39(5):529–39. doi: 10.1007/s00281-017-0629-x
26. Alipoor SD, Mortaz E, Jamaati H, Tabarsi P, Bayram H, Varahram M, et al. COVID-19: Molecular and Cellular Response. Front Cell Infect Microbiol (2021) 11:563085. doi: 10.3389/fcimb.2021.563085
27. Lazzaroni MG, Piantoni S, Masneri S, Garrafa E, Martini G, Tincani A, et al. Coagulation Dysfunction in COVID-19: The Interplay Between Inflammation, Viral Infection and the Coagulation System. Blood Rev (2021) 46:100745. doi: 10.1016/j.blre.2020.100745
28. Bonaventura A, Vecchié A, Dagna L, Martinod K, Dixon DL, Van Tassell BW, et al. Endothelial Dysfunction and Immunothrombosis as Key Pathogenic Mechanisms in COVID-19. Nat Rev Immunol (2021) 21(5):319–29. doi: 10.1038/s41577-021-00536-9
29. Henry BM, Vikse J, Benoit S, Favaloro EJ, Lippi G. Hyperinflammation and Derangement of Renin-Angiotensin-Aldosterone System in COVID-19: A Novel Hypothesis for Clinically Suspected Hypercoagulopathy and Microvascular Immunothrombosis. Clin Chim Acta (2020) 507:167–73. doi: 10.1016/j.cca.2020.04.027
30. Kollias A, Kyriakoulis KG, Dimakakos E, Poulakou G, Stergiou GS, Syrigos K. Thromboembolic Risk and Anticoagulant Therapy in COVID-19 Patients: Emerging Evidence and Call for Action. Br J Haematol (2020) 189(5):846–7. doi: 10.1111/bjh.16727
31. Zhang Y, Xiao M, Zhang S, Xia P, Cao W, Jiang W, et al. Coagulopathy and Antiphospholipid Antibodies in Patients With Covid-19. N Engl J Med (2020) 382(17):e38. doi: 10.1056/NEJMc2007575
32. Farghaly S, Makboul M. Correlation Between Age, Sex, and Severity of Coronavirus Disease-19 Based on Chest Computed Tomography Severity Scoring System. Egypt J Radiol Nucl Med (2021) 52(1):23. doi: 10.1186/s43055-021-00408-1
33. Gadi N, Wu SC, Spihlman AP, Moulton VR. What’s Sex Got to Do With COVID-19? Gender-Based Differences in the Host Immune Response to Coronaviruses. Front Immunol (2020) 11:2147. doi: 10.3389/fimmu.2020.02147
34. Foley JH, Conway EM. Cross Talk Pathways Between Coagulation and Inflammation. Circ Res (2016) 118(9):1392–408. doi: 10.1161/CIRCRESAHA.116.306853
35. Kohansal Vajari M, Shirin M, Pourbagheri-Sigaroodi A, Akbari ME, Abolghasemi H, Bashash D. COVID-19-Related Coagulopathy: A Review of Pathophysiology and Pharmaceutical Management. Cell Biol Int (2021) 45(9):1832–50. doi: 10.1002/cbin.11623
36. Burrell LM, Johnston CI, Tikellis C, Cooper ME. ACE2, A New Regulator of the Renin-Angiotensin System. Trends Endocrinol Metab (2004) 15(4):166–9. doi: 10.1016/j.tem.2004.03.001
37. Hamming I, Timens W, Bulthuis MLC, Lely AT, Navis GJ, van Goor H. Tissue Distribution of ACE2 Protein, the Functional Receptor for SARS Coronavirus. A First Step in Understanding SARS Pathogenesis. J Pathol (2004) 203(2):631–7. doi: 10.1002/path.1570
38. Letko M, Marzi A, Munster V. Functional Assessment of Cell Entry and Receptor Usage for SARS-CoV-2 and Other Lineage B Betacoronaviruses. Nat Microbiol (2020) 5(4):562–9. doi: 10.1038/s41564-020-0688-y
39. Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Structural Basis for the Recognition of SARS-CoV-2 by Full-Length Human ACE2. Science (80-) (2020) 367(6485):1444–8. doi: 10.1126/science.abb2762
40. Santos RAS, Oudit GY, Verano-Braga T, Canta G, Steckelings UM, Bader M. The Renin-Angiotensin System: Going Beyond the Classical Paradigms. Am J Physiol - Hear Circ Physiol (2019) 316(5):H958–70. doi: 10.1152/ajpheart.00723.2018
41. Underwood PC, Adler GK. The Renin Angiotensin Aldosterone System and Insulin Resistance in Humans. Curr Hypertens Rep (2013) 15(1):59–70. doi: 10.1007/s11906-012-0323-2
42. Kuba K, Imai Y, Rao S, Jiang C, Penninger JM. Lessons From SARS: Control of Acute Lung Failure by the SARS Receptor ACE2. J Mol Med (2006) 84(10):814–20. doi: 10.1007/s00109-006-0094-9
43. Zou L, Ruan F, Huang M, Liang L, Huang H, Hong Z, et al. SARS-CoV-2 Viral Load in Upper Respiratory Specimens of Infected Patients. N Engl J Med (2020) 382(12):1177–9. doi: 10.1056/NEJMc2001737
44. Han SH, Mallampalli RK. The Role of Surfactant in Lung Disease and Host Defense Against Pulmonary Infections. Ann Am Thorac Soc (2015) 12(5):765–74. doi: 10.1513/AnnalsATS.201411-507FR
45. Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, et al. Pathological Findings of COVID-19 Associated With Acute Respiratory Distress Syndrome. Lancet Respir Med (2020) 8(4):420–2. doi: 10.1016/S2213-2600(20)30076-X
46. Gralinski LE, Baric RS. Molecular Pathology of Emerging Coronavirus Infections. J Pathol (2015) 235(2):185–95. doi: 10.1002/path.4454
47. Parit R, Jayavel S. Association of ACE Inhibitors and Angiotensin Type II Blockers With ACE2 Overexpression in COVID-19 Comorbidities: A Pathway-Based Analytical Study. Eur J Pharmacol (2021) 896:173899. doi: 10.1016/j.ejphar.2021.173899
48. Ferrario CM, Jessup J, Chappell MC, Averill DB, Brosnihan KB, Tallant EA, et al. Effect of Angiotensin-Converting Enzyme Inhibition and Angiotensin II Receptor Blockers on Cardiac Angiotensin-Converting Enzyme 2. Circulation (2005) 111(20):2605–10. doi: 10.1161/CIRCULATIONAHA.104.510461
49. Pedrosa MA, Valenzuela R, Garrido-Gil P, Labandeira CM, Navarro G, Franco R, et al. Experimental Data Using Candesartan and Captopril Indicate No Double-Edged Sword Effect in COVID-19. Clin Sci (2021) 135(3):465–81. doi: 10.1042/CS20201511
50. Wang K, Gheblawi M, Oudit GY. Angiotensin Converting Enzyme 2: A Double-Edged Sword. Circulation (2020) 142(5):426–8. doi: 10.1161/CIRCULATIONAHA.120.047049
51. Onweni CL, Zhang YS, Caulfield T, Hopkins CE, Fairweather DL, Freeman WD. ACEI/ARB Therapy in COVID-19: The Double-Edged Sword of ACE2 and SARS-CoV-2 Viral Docking. Crit Care (2020) 24(1):475. doi: 10.1186/s13054-020-03195-9
52. Garvin MR, Alvarez C, Miller JI, Prates ET, Walker AM, Amos BK, et al. A Mechanistic Model and Therapeutic Interventions for Covid-19 Involving a Ras-Mediated Bradykinin Storm. Elife (2020) 9:1–16. doi: 10.7554/eLife.59177
53. Simoneaux R, Shafer SL. A RAS and Bradykinin-Mediated Mechanism for COVID-19. ASA Monit (2020) 84(11):1–11. doi: 10.1097/01.ASM.0000722064.35978.f6
54. Liu F, Li L, Xu M, Wu J, Luo D, Zhu YS, et al. Prognostic Value of Interleukin-6, C-Reactive Protein, and Procalcitonin in Patients With COVID-19. J Clin Virol (2020) 127:104370. doi: 10.1016/j.jcv.2020.104370
55. Hanff TC, Harhay MO, Brown TS, Cohen JB, Mohareb AM. Is There an Association Between COVID-19 Mortality and the Renin-Angiotensin System? A Call for Epidemiologic Investigations. Clin Infect Dis (2020) 71(15):870–4. doi: 10.1093/cid/ciaa329
56. Banu N, Panikar SS, Leal LR, Leal AR. Protective Role of ACE2 and Its Downregulation in SARS-CoV-2 Infection Leading to Macrophage Activation Syndrome: Therapeutic Implications. Life Sci (2020) 256:117905. doi: 10.1016/j.lfs.2020.117905
57. Foresta C, Rocca MS, Di Nisio A. Gender Susceptibility to COVID-19: A Review of the Putative Role of Sex Hormones and X Chromosome. J Endocrinol Invest (2021) 44(5):951–6. doi: 10.1007/s40618-020-01383-6
58. Chumakov K, Avidan MS, Benn CS, Bertozzi SM, Blatt L, Chang AY, et al. Old Vaccines for New Infections: Exploiting Innate Immunity to Control COVID-19 and Prevent Future Pandemics. Proc Natl Acad Sci USA (2021) 118(21):e2101718118. doi: 10.1073/pnas.2101718118
59. Marshall JS, Warrington R, Watson W, Kim HL. An Introduction to Immunology and Immunopathology. Allergy Asthma Clin Immunol (2018) 14(2):1–10. doi: 10.1186/s13223-018-0278-1
60. Amarante-Mendes GP, Adjemian S, Branco LM, Zanetti LC, Weinlich R, Bortoluci KR. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front Immunol (2018) 9:2379. doi: 10.3389/fimmu.2018.02379
61. Suresh R, Mosser DM. Pattern Recognition Receptors in Innate Immunity, Host Defense, and Immunopathology. Am J Physiol - Adv Physiol Educ (2013) 37(4):284–91. doi: 10.1152/advan.00058.2013
62. Jensen S, Thomsen AR. Sensing of RNA Viruses: A Review of Innate Immune Receptors Involved in Recognizing RNA Virus Invasion. J Virol (2012) 86(6):2900–10. doi: 10.1128/JVI.05738-11
63. Siu KL, Kok KH, Ng MHJ, Poon VKM, Yuen KY, Zheng BJ, et al. Severe Acute Respiratory Syndrome Coronavirus M Protein Inhibits Type I Interferon Production by Impeding Theformation of TRAF3·TANK·Tbk1/Ikkϵ Complex. J Biol Chem (2009) 284(24):16202–9. doi: 10.1074/jbc.M109.008227
64. Aboudounya MM, Heads RJ. COVID-19 and Toll-Like Receptor 4 (TLR4): SARS-CoV-2 May Bind and Activate TLR4 to Increase ACE2 Expression, Facilitating Entry and Causing Hyperinflammation. Mediators Inflamm (2021) 2021:8874339. doi: 10.1155/2021/8874339
65. Li SW, Wang CY, Jou YJ, Huang SH, Hsiao LH, Wan L, et al. SARS Coronavirus Papain-Like Protease Inhibits the TLR7 Signaling Pathway Through Removing Lys63-Linked Polyubiquitination of TRAF3 and TRAF6. Int J Mol Sci (2016) 17(5):678. doi: 10.3390/ijms17050678
66. Taefehshokr N, Taefehshokr S, Hemmat N, Heit B. Covid-19: Perspectives on Innate Immune Evasion. Front Immunol (2020) 11:580641. doi: 10.3389/fimmu.2020.580641
67. Knoops K, Kikkert M, Van Den Worm SHE, Zevenhoven-Dobbe JC, van der Meer Y, Koster AJ, et al. SARS-Coronavirus Replication Is Supported by a Reticulovesicular Network of Modified Endoplasmic Reticulum. PloS Biol (2008) 6(9):1957–74. doi: 10.1371/journal.pbio.0060226
68. Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Møller R, et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell (2020) 181(5):1036–45.e9. doi: 10.1016/j.cell.2020.04.026
69. Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Smith N, et al. Impaired Type I Interferon Activity and Inflammatory Responses in Severe COVID-19 Patients. Science (80-) (2020) 369(6504):718–24. doi: 10.1126/science.abc6027
70. Zhou W, Wang W. Auto-Antibodies Against Type I IFNs Are Associated With Severe COVID-19 Pneumonia. Signal Transduct Target Ther (2021) 6(1):1–2. doi: 10.1038/s41392-021-00514-6
71. Onabajo OO, Banday AR, Stanifer ML, Yan W, Obajemu A, Santer DM, et al. Interferons and Viruses Induce a Novel Truncated ACE2 Isoform and Not the Full-Length SARS-CoV-2 Receptor. Nat Genet (2020) 52(12):1283–93. doi: 10.1038/s41588-020-00731-9
72. Yamada T, Sato S, Sotoyama Y, Orba Y, Sawa H, Yamauchi H, et al. RIG-I Triggers a Signaling-Abortive Anti-SARS-CoV-2 Defense in Human Lung Cells. Nat Immunol (2021) 22(7):820–8. doi: 10.1038/s41590-021-00942-0
73. Sinha P, Matthay MA, Calfee CS. Is a “Cytokine Storm” Relevant to COVID-19? JAMA Intern Med (2020) 180(9):1152–4. doi: 10.1001/jamainternmed.2020.3313
74. Siu KL, Chan CP, Kok KH, Chiu-Yat Woo P, Jin DY. Suppression of Innate Antiviral Response by Severe Acute Respiratory Syndrome Coronavirus M Protein Is Mediated Through the First Transmembrane Domain. Cell Mol Immunol (2014) 11(2):141–9. doi: 10.1038/cmi.2013.61
75. The REMAP-CAP Investigators, Derde LPG. Effectiveness of Tocilizumab, Sarilumab, and Anakinra for Critically Ill Patients With COVID-19 The REMAP-CAP COVID-19 Immune Modulation Therapy Domain Randomized Clinical Trial. medRxiv (2021) 2021.06.18.21259133. doi: 10.1101/2021.06.18.21259133
76. Narasaraju T, Tang BM, Herrmann M, Muller S, Chow VTK, Radic M. Neutrophilia and NETopathy as Key Pathologic Drivers of Progressive Lung Impairment in Patients With COVID-19. Front Pharmacol (2020) 11:870. doi: 10.3389/fphar.2020.00870
77. Chauhan AJ, Wiffen LJ, Brown TP. COVID-19: A Collision of Complement, Coagulation and Inflammatory Pathways. J Thromb Haemost (2020) 18(9):2110–7. doi: 10.1111/jth.14981
78. Vabret N, Britton GJ, Gruber C, Hegde S, Kim J, Kuksin M, et al. Immunology of COVID-19: Current State of the Science. Immunity (2020) 52(6):910–41. doi: 10.1016/j.immuni.2020.05.002
79. Ruan Q, Yang K, Wang W, Jiang L, Song J. Clinical Predictors of Mortality Due to COVID-19 Based on an Analysis of Data of 150 Patients From Wuhan, China. Intensive Care Med (2020) 46(5):846–8. doi: 10.1007/s00134-020-05991-x
80. Gao Y, Li T, Han M, Li X, Wu D, Xu Y, et al. Diagnostic Utility of Clinical Laboratory Data Determinations for Patients With the Severe COVID-19. J Med Virol (2020) 92(7):791–6. doi: 10.1002/jmv.25770
81. Qin C, Zhou L, Hu Z, Zhang S, Yang S, Tao Y, et al. Dysregulation of Immune Response in Patients With Coronavirus 2019 (COVID-19) in Wuhan, China. Clin Infect Dis (2020) 71(15):762–8. doi: 10.1093/cid/ciaa248
82. Lucas C, Wong P, Klein J, Castro TBR, Silva J, Sundaram M, et al. Longitudinal Analyses Reveal Immunological Misfiring in Severe COVID-19. Nature (2020) 584(7821):463–9. doi: 10.1038/s41586-020-2588-y
83. Chen X, Zhao B, Qu Y, Chen Y, Xiong J, Feng Y, et al. Detectable Serum Severe Acute Respiratory Syndrome Coronavirus 2 Viral Load (RNAemia) Is Closely Correlated With Drastically Elevated Interleukin 6 Level in Critically Ill Patients With Coronavirus Disease 2019. Clin Infect Dis (2020) 71(8):1937–42. doi: 10.1093/cid/ciaa449
84. Zhang X, Wu K, Wang D, Yue X, Song D, Zhu Y, et al. Nucleocapsid Protein of SARS-CoV Activates Interleukin-6 Expression Through Cellular Transcription Factor NF-κb. Virology (2007) 365(2):324–35. doi: 10.1016/j.virol.2007.04.009
85. Merza M, Hartman H, Rahman M, Hwaiz R, Zhang E, Renström E, et al. Neutrophil Extracellular Traps Induce Trypsin Activation, Inflammation, and Tissue Damage in Mice With Severe Acute Pancreatitis. Gastroenterology (2015) 149(7):1920–31.e8. doi: 10.1053/j.gastro.2015.08.026
86. Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ. COVID-19: Consider Cytokine Storm Syndromes and Immunosuppression. Lancet (2020) 395(10229):1033–4. doi: 10.1016/S0140-6736(20)30628-0
87. Barnes BJ, Adrover JM, Baxter-Stoltzfus A, Borczuk A, Cools-Lartigue J, Crawford JM, et al. Targeting Potential Drivers of COVID-19: Neutrophil Extracellular Traps. J Exp Med (2020) 217(6):e20200652. doi: 10.1084/jem.20200652
88. Mangalmurti N, Hunter CA. Cytokine Storms: Understanding COVID-19. Immunity (2020) 53(1):19–25. doi: 10.1016/j.immuni.2020.06.017
89. Hosseini A, Hashemi V, Shomali N, Asghari F, Gharibi T, Akbari M, et al. Innate and Adaptive Immune Responses Against Coronavirus. BioMed Pharmacother (2020) 132:110859. doi: 10.1016/j.biopha.2020.110859
90. Nile SH, Nile A, Qiu J, Li L, Jia X, Kai G. COVID-19: Pathogenesis, Cytokine Storm and Therapeutic Potential of Interferons. Cytokine Growth Factor Rev (2020) 53:66–70. doi: 10.1016/j.cytogfr.2020.05.002
91. Hasan A, Al-Ozairi E, Al-Baqsumi Z, Ahmad R, Al-Mulla F. Cellular and Humoral Immune Responses in Covid-19 and Immunotherapeutic Approaches. ImmunoTargets Ther (2021) 10:63–85. doi: 10.2147/ITT.S280706
92. Christiaansen A, Varga SM, Spencer JV. Viral Manipulation of the Host Immune Response. Curr Opin Immunol (2015) 36:54–60. doi: 10.1016/j.coi.2015.06.012
93. Channappanavar R, Zhao J, Perlman S. T Cell-Mediated Immune Response to Respiratory Coronaviruses. Immunol Res (2014) 59(1–3):118–28. doi: 10.1007/s12026-014-8534-z
94. Cerutti G, Guo Y, Zhou T, Gorman J, Lee M, Rapp M, et al. Potent SARS-CoV-2 Neutralizing Antibodies Directed Against Spike N-Terminal Domain Target a Single Supersite. Cell Host Microbe (2021) 29(5):819–33.e7. doi: 10.1016/j.chom.2021.03.005
95. Fu Y, Cheng Y, Wu Y. Understanding SARS-CoV-2-Mediated Inflammatory Responses: From Mechanisms to Potential Therapeutic Tools. Virol Sin (2020) 35(3):266–71. doi: 10.1007/s12250-020-00207-4
96. Chang SE, Feng A, Meng W, Apostolidis SA, Mack E, Artandi M, et al. New-Onset IgG Autoantibodies in Hospitalized Patients With COVID-19. Nat Commun (2021) 12(1):1–15. doi: 10.1038/s41467-021-25509-3
97. Bastard P, Gervais A, Le Voyer T, Rosain J, Philippot Q, Manry J, et al. Autoantibodies Neutralizing Type I IFNs Are Present in ~4% of Uninfected Individuals Over 70 Years Old and Account for ~20% of COVID-19 Deaths. Sci Immunol (2021) 6(62):eabl4340. doi: 10.1126/sciimmunol.abl4340
98. Wang EY, Mao T, Klein J, Dai Y, Huck JD, Jaycox JR, et al. Diverse Functional Autoantibodies in Patients With COVID-19. Nature (2021) 595(7866):283–8. doi: 10.1038/s41586-021-03631-y
99. Taefehshokr N, Taefehshokr S, Heit B. Mechanisms of Dysregulated Humoral and Cellular Immunity by SARS-CoV-2. Pathogens (2020) 9(12):1–21. doi: 10.3390/pathogens9121027
100. Zhao J, Zhao J, Perlman S. T Cell Responses Are Required for Protection From Clinical Disease and for Virus Clearance in Severe Acute Respiratory Syndrome Coronavirus-Infected Mice. J Virol (2010) 84(18):9318–25. doi: 10.1128/JVI.01049-10
101. Chen Z, John Wherry E. T Cell Responses in Patients With COVID-19. Nat Rev Immunol (2020) 20(9):529–36. doi: 10.1038/s41577-020-0402-6
102. Kalfaoglu B, Almeida-Santos J, Tye CA, Satou Y, Ono M. T-Cell Dysregulation in COVID-19. Biochem Biophys Res Commun (2021) 538:204–10. doi: 10.1016/j.bbrc.2020.10.079
103. Kuri-Cervantes L, Pampena MB, Meng W, Rosenfeld AM, Ittner CAG, Weisman AR, et al. Comprehensive Mapping of Immune Perturbations Associated With Severe COVID-19. Sci Immunol (2020) 5(49):eabd7114. doi: 10.1126/sciimmunol.abd7114
104. Giamarellos-Bourboulis EJ, Netea MG, Rovina N, Akinosoglou K, Antoniadou A, Antonakos N, et al. Complex Immune Dysregulation in COVID-19 Patients With Severe Respiratory Failure. Cell Host Microbe (2020) 27(6):992–1000.e3. doi: 10.1016/j.chom.2020.04.009
105. Mazzoni A, Salvati L, Maggi L, Capone M, Vanni A, Spinicci M, et al. Impaired Immune Cell Cytotoxicity in Severe COVID-19 Is IL-6 Dependent. J Clin Invest (2020) 130(9):4694–703. doi: 10.1172/JCI138554
106. Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, et al. Single-Cell Landscape of Bronchoalveolar Immune Cells in Patients With COVID-19. Nat Med (2020) 26(6):842–4. doi: 10.1038/s41591-020-0901-9
107. Wichmann D, Sperhake JP, Lütgehetmann M, Steurer S, Edler C, Heinemann A, et al. Autopsy Findings and Venous Thromboembolism in Patients With COVID-19: A Prospective Cohort Study. Ann Intern Med (2020) 173(4):268–77. doi: 10.7326/M20-2003
108. Shrotri M, van Schalkwyk MCI, Post N, Eddy D, Huntley C, Leeman D, et al. T Cell Response to SARS-Cov-2 Infection in Humans: A Systematic Review. PloS One (2021) 16(1 January):e0245532. doi: 10.1371/journal.pone.0245532
109. Böttcher JP, Schanz O, Garbers C, Zaremba A, Hegenbarth S, Kurts C, et al. IL-6 Trans-Signaling-Dependent Rapid Development of Cytotoxic CD8+ T Cell Function. Cell Rep (2014) 8(5):1318–27. doi: 10.1016/j.celrep.2014.07.008
110. Taga K, Tosato G. IL-10 Inhibits Human T Cell Proliferation and IL-2 Production. J Immunol (Baltimore Md : 1950) (1992) 148:1143–8.
111. Waal Malefyt R, Haanen J, Spits H, Koncarolo MG, Te Velde A, Figdor C, et al. Interleukin 10 (Il-10) and Viral Il-10 Strongly Reduce Antigen-Specific Human T Cell Proliferation by Diminishing the Antigen-Presenting Capacity of Monocytes via Dowm’egulation of Class H Major Histocompatibility Complex Expression. J Exp Med (1991) 174(4):915–24. doi: 10.1084/jem.174.4.915
112. Liu Y, Du X, Chen J, Jin Y, Peng L, Wang HHX, et al. Neutrophil-To-Lymphocyte Ratio as an Independent Risk Factor for Mortality in Hospitalized Patients With COVID-19. J Infect (2020) 81(1):e6–12. doi: 10.1016/j.jinf.2020.04.002
113. Ma Y, Shi N, Fan Y, Wang J, Zhao C, Li G, et al. Predictive Value of the Neutrophil-To-Lymphocyte Ratio(NLR) for Diagnosis and Worse Clinical Course of the COVID-19: Findings From Ten Provinces in China. SSRN Electron J (2020). doi: 10.2139/ssrn.3569838
114. Adamo S, Chevrier S, Cervia C, Zurbuchen Y, Raeber ME, Yang L, et al. Lymphopenia-Induced T Cell Proliferation Is a Hallmark of Severe COVID-19. bioRxiv (2020) 2020.08.04.236521. doi: 10.1101/2020.08.04.236521
115. Vardhana SA, Wolchok JD. The Many Faces of the Anti-COVID Immune Response. J Exp Med (2020) 217(6):e20200678. doi: 10.1084/jem.20200678
116. Zhang Y, Zhang J, Chen Y, Luo B, Yuan Y, Huang F, et al. The ORF8 Protein of SARS-CoV-2 Mediates Immune Evasion Through Potently Downregulating MHC-I. bioRxiv (2020) 2020.05.24.111823. doi: 10.1101/2020.05.24.111823v1
117. Diao B, Wang C, Tan Y, Chen X, Liu Y, Ning L, et al. Reduction and Functional Exhaustion of T Cells in Patients With Coronavirus Disease 2019 (COVID-19). Front Immunol (2020) 11:827. doi: 10.3389/fimmu.2020.00827
118. Yang X, Dai T, Zhou X, Qian H, Guo R, Lei L, et al. Analysis of Adaptive Immune Cell Populations and Phenotypes in the Patients Infected by SARS-CoV-2. medRxiv (2020) 2020.03.23.20040675. doi: 10.1101/2020.03.23.20040675v2
119. Rha MS, Jeong HW, Ko JH, Choi SJ, Seo IH, Lee JS, et al. PD-1-Expressing SARS-CoV-2-Specific CD8+ T Cells Are Not Exhausted, But Functional in Patients With COVID-19. Immunity (2021) 54(1):44–52.e3. doi: 10.1016/j.immuni.2020.12.002
120. Zheng M, Gao Y, Wang G, Song G, Liu S, Sun D, et al. Functional Exhaustion of Antiviral Lymphocytes in COVID-19 Patients. Cell Mol Immunol (2020) 17(5):533–5. doi: 10.1038/s41423-020-0402-2
121. Oja AE, Saris A, Ghandour CA, Kragten NAM, Hogema BM, Nossent EJ, et al. Divergent SARS-CoV-2-Specific T and B Cell Responses in Severe But Not Mild COVID-19. bioRxiv (2020) 2020.06.18.159202. doi: 10.1101/2020.06.18.159202v1
122. Bongen E, Lucian H, Khatri A, Fragiadakis GK, Bjornson ZB, Nolan GP, et al. Sex Differences in the Blood Transcriptome Identify Robust Changes in Immune Cell Proportions With Aging and Influenza Infection. Cell Rep (2019) 29(7):1961–73.e4. doi: 10.1016/j.celrep.2019.10.019
123. Takahashi T, Ellingson MK, Wong P, Israelow B, Lucas C, Klein J, et al. Sex Differences in Immune Responses That Underlie COVID-19 Disease Outcomes. Nature (2020) 588(7837):315–20. doi: 10.1038/s41586-020-2700-3
124. Viveiros A, Rasmuson J, Vu J, Mulvagh SL, Yip CYY, Norris CM, et al. Sex Differences in COVID-19: Candidate Pathways, Genetics of ACE2, and Sex Hormones. Am J Physiol - Hear Circ Physiol (2021) 320(1):H296–304. doi: 10.1152/ajpheart.00755.2020
125. Pinna G. Sex and COVID-19: A Protective Role for Reproductive Steroids. Trends Endocrinol Metab (2021) 32(1):3–6. doi: 10.1016/j.tem.2020.11.004
126. Chang C, Saltzman A, Yeh S, Young W, Keller E, Lee HJ, et al. Androgen Receptor: An Overview. Crit Rev Eukaryot Gene Expr (1995) 5(2):97–125. doi: 10.1615/CritRevEukarGeneExpr.v5.i2.10
127. Younis JS, Skorecki K, Abassi Z. The Double Edge Sword of Testosterone’s Role in the COVID-19 Pandemic. Front Endocrinol (Lausanne) (2021) 12:607179. doi: 10.3389/fendo.2021.607179
128. Abobaker A, Raba AA. Does COVID-19 Affect Male Fertility? World J Urol (2021) 39(3):975–6. doi: 10.1007/s00345-020-03208-w
129. Schroeder M, Schaumburg B, Mueller Z, Parplys A, Jarczak D, Nierhaus A, et al. The Majority of Male Patients With COVID-19 Present Low Testosterone Levels on Admission to Intensive Care in Hamburg, Germany: A Retrospective Cohort Study. medRxiv (2020) 2020.05.07.20073817.
130. Wang J, Saguner AM, An J, Ning Y, Yan Y, Li G. Dysfunctional Coagulation in COVID-19: From Cell to Bedside. Adv Ther (2020) 37(7):3033–9. doi: 10.1007/s12325-020-01399-7
131. Terpos E, Ntanasis-Stathopoulos I, Elalamy I, Kastritis E, Sergentanis TN, Politou M, et al. Hematological Findings and Complications of COVID-19. Am J Hematol (2020) 95(7):834–47. doi: 10.1002/ajh.25829
132. Levi M, van der Poll T. Inflammation and Coagulation. Crit Care Med (2010) 38(SUPPL 2):S26–34. doi: 10.1097/CCM.0b013e3181c98d21
133. Smargiassi A, Soldati G, Borghetti A, Scoppettuolo G, Tamburrini E, Testa AC, et al. Lung Ultrasonography for Early Management of Patients With Respiratory Symptoms During COVID-19 Pandemic. J Ultrasound (2020) 23(4):449–56. doi: 10.1007/s40477-020-00501-7
134. Palta S, Saroa R, Palta A. Overview of the Coagulation System. Indian J Anaesth (2014) 58(5):515–23. doi: 10.4103/0019-5049.144643
135. Ezihe-Ejiofor JA, Hutchinson N. Anticlotting Mechanisms 1: Physiology and Pathology. Contin Educ Anaesth Crit Care Pain (2013) 13(3):87–92. doi: 10.1093/bjaceaccp/mks061
136. Loo J, Spittle DA, Newnham M. COVID-19, Immunothrombosis and Venous Thromboembolism: Biological Mechanisms. Thorax (2021) 76(4):412–20. doi: 10.1136/thoraxjnl-2020-216243
137. Lorini FL, Di Matteo M, Gritti P, Grazioli L, Benigni A, Zacchetti L, et al. Coagulopathy and COVID-19. Eur Hear J Suppl (2021) 23(Supplement_E):E95–8. doi: 10.1093/eurheartj/suab100
138. Frantzeskaki F, Armaganidis A, Orfanos SE. Immunothrombosis in Acute Respiratory Distress Syndrome: Cross Talks Between Inflammation and Coagulation. Respiration (2017) 93(3):212–25. doi: 10.1159/000453002
139. Engelmann B, Massberg S. Thrombosis as an Intravascular Effector of Innate Immunity. Nat Rev Immunol (2013) 13(1):34–45. doi: 10.1038/nri3345
140. Schulz C, Engelmann B, Massberg S. Crossroads of Coagulation and Innate Immunity: The Case of Deep Vein Thrombosis. J Thromb Haemost (2013) 11(SUPPL.1):233–41. doi: 10.1111/jth.12261
141. Newton K, Dixit VM. Signaling in Innate Immunity and Inflammation. Cold Spring Harb Perspect Biol (2012) 4(3):a006049. doi: 10.1101/cshperspect.a006049
142. Stark K, Massberg S. Interplay Between Inflammation and Thrombosis in Cardiovascular Pathology. Nat Rev Cardiol (2021) 18(9):666–82. doi: 10.1038/s41569-021-00552-1
143. Jayarangaiah A, Kariyanna PT, Chen X, Jayarangaiah A, Kumar A. COVID-19-Associated Coagulopathy: An Exacerbated Immunothrombosis Response. Clin Appl Thromb (2020) 26:1076029620943293. doi: 10.1177/1076029620943293
144. McGonagle D, Sharif K, O’Regan A, Bridgewood C. The Role of Cytokines Including Interleukin-6 in COVID-19 Induced Pneumonia and Macrophage Activation Syndrome-Like Disease. Autoimmun Rev (2020) 19(6):102537. doi: 10.1016/j.autrev.2020.102537
145. Schnaubelt S, Tihanyi D, Strassl R, Schmidt R, Anders S, Laggner AN, et al. Hemophagocytic Lymphohistiocytosis in COVID-19: Case Reports of a Stepwise Approach. Med (Baltimore) (2021) 100(12):e25170. doi: 10.1097/MD.0000000000025170
146. Lorenz G, Moog P, Bachmann Q, La Rosée P, Schneider H, Schlegl M, et al. Title: Cytokine Release Syndrome Is Not Usually Caused by Secondary Hemophagocytic Lymphohistiocytosis in a Cohort of 19 Critically Ill COVID-19 Patients. Sci Rep (2020) 10(1):1–11. doi: 10.1038/s41598-020-75260-w
147. Wan S, Yi Q, Fan S, Lv J, Zhang X, Guo L, et al. Characteristics of Lymphocyte Subsets and Cytokines in Peripheral Blood of 123 Hospitalized Patients With 2019 Novel Coronavirus Pneumonia (NCP). medRxiv (2020) 2020.02.10.20021832. doi: 10.1101/2020.02.10.20021832v1
148. Bamgboje A, Hong J, Mushiyev S, Pekler G. A 61-Year-Old Man With Sars-Cov-2 Infection and Venous Thrombosis Presenting With Painful Swelling and Gangrene of the Lower Limb Consistent With Phlegmasia Cerulea Dolens. Am J Case Rep (2020) 21:1–6. doi: 10.12659/AJCR.928342
149. Wang JS, Pasieka HB, Petronic-Rosic V, Sharif-Askary B, Evans KK. Digital Gangrene as a Sign of Catastrophic Coronavirus Disease 2019-Related Microangiopathy. Plast Reconstr Surg - Glob Open (2020) 8(7):e3025. doi: 10.1097/GOX.0000000000003025
150. Zhang L, Yan X, Fan Q, Liu H, Liu X, Liu Z, et al. D-Dimer Levels on Admission to Predict in-Hospital Mortality in Patients With Covid-19. J Thromb Haemost (2020) 18(6):1324–9. doi: 10.1111/jth.14859
151. Giannis D, Ziogas IA, Gianni P. Coagulation Disorders in Coronavirus Infected Patients: COVID-19, SARS-CoV-1, MERS-CoV and Lessons From the Past. J Clin Virol (2020) 127:104362. doi: 10.1016/j.jcv.2020.104362
152. Iba T, Levi M, Levy JH. Sepsis-Induced Coagulopathy and Disseminated Intravascular Coagulation. Semin Thromb Hemost (2020) 46(1):89–95. doi: 10.1055/s-0039-1694995
153. Simmons J, Pittet JF. The Coagulopathy of Acute Sepsis. Curr Opin Anaesthesiol (2015) 28(2):227–36. doi: 10.1097/ACO.0000000000000163
154. Iba T, Levy J, Raj A, Warkentin T. Advance in the Management of Sepsis-Induced Coagulopathy and Disseminated Intravascular Coagulation. J Clin Med (2019) 8(5):728. doi: 10.3390/jcm8050728
155. Van Der Poll T, Van De Veerdonk FL, Scicluna BP, Netea MG. The Immunopathology of Sepsis and Potential Therapeutic Targets. Nat Rev Immunol (2017) 17(7):407–20. doi: 10.1038/nri.2017.36
156. Merad M, Martin JC. Pathological Inflammation in Patients With COVID-19: A Key Role for Monocytes and Macrophages. Nat Rev Immunol (2020) 20(6):355–62. doi: 10.1038/s41577-020-0331-4
157. Matsuyama T, Kubli SP, Yoshinaga SK, Pfeffer K, Mak TW. An Aberrant STAT Pathway Is Central to COVID-19. Cell Death Differ (2020) 27(12):3209–25. doi: 10.1038/s41418-020-00633-7
158. Jafarzadeh A, Nemati M, Jafarzadeh S. Contribution of STAT3 to the Pathogenesis of COVID-19. Microb Pathog (2021) 154:104836. doi: 10.1016/j.micpath.2021.104836
159. Zuo Y, Warnock M, Harbaugh A, Yalavarthi S, Gockman K, Zuo M, et al. Plasma Tissue Plasminogen Activator and Plasminogen Activator Inhibitor-1 in Hospitalized COVID-19 Patients. Sci Rep (2021) 11(1):2020.08.29.20184358. doi: 10.1101/2020.08.29.20184358v4
160. Cimmino G, Cirillo P. Tissue Factor: Newer Concepts in Thrombosis and Its Role Beyond Thrombosis and Hemostasis. Cardiovasc Diagn Ther (2018) 8(5):581–93. doi: 10.21037/cdt.2018.10.14
161. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil Extracellular Traps Kill Bacteria. Science (80-) (2004) 303(5663):1532–5. doi: 10.1126/science.1092385
162. Leppkes M, Knopf J, Naschberger E, Lindemann A, Singh J, Herrmann I, et al. Vascular Occlusion by Neutrophil Extracellular Traps in COVID-19. EBioMedicine (2020) 58:102925. doi: 10.1016/j.ebiom.2020.102925
163. Fernández-Pérez MP, Águila S, Reguilón-Gallego L, los Reyes-García AM, Miñano A, Bravo-Pérez C, et al. Neutrophil Extracellular Traps and Von Willebrand Factor Are Allies That Negatively Influence COVID-19 Outcomes. Clin Transl Med (2021) 11(1):e268. doi: 10.1002/ctm2.268
164. Goshua G, Pine AB, Meizlish ML, Chang CH, Zhang H, Bahel P, et al. Endotheliopathy in COVID-19-Associated Coagulopathy: Evidence From a Single-Centre, Cross-Sectional Study. Lancet Haematol (2020) 7(8):e575–82. doi: 10.1016/S2352-3026(20)30216-7
165. Middleton EA, He XY, Denorme F, Campbell RA, Ng D, Salvatore SP, et al. Neutrophil Extracellular Traps Contribute to Immunothrombosis in COVID-19 Acute Respiratory Distress Syndrome. Blood (2020) 136(10):1169–79. doi: 10.1182/blood.2020007008
166. Yang J, Wu Z, Long Q, Huang J, Hong T, Liu W, et al. Insights Into Immunothrombosis: The Interplay Among Neutrophil Extracellular Trap, Von Willebrand Factor, and ADAMTS13. Front Immunol (2020) 11:610696. doi: 10.3389/fimmu.2020.610696
167. Zuo Y, Yalavarthi S, Shi H, Gockman K, Zuo M, Madison JA, et al. Neutrophil Extracellular Traps in COVID-19. JCI Insight (2020) 5(11):e138999. doi: 10.1172/jci.insight.138999
168. Yalavarthi S, Gould TJ, Rao AN, Mazza LF, Morris AE, Núñez-Álvarez C, et al. Release of Neutrophil Extracellular Traps by Neutrophils Stimulated With Antiphospholipid Antibodies: A Newly Identified Mechanism of Thrombosis in the Antiphospholipid Syndrome. Arthritis Rheumatol (2015) 67(11):2990–3003. doi: 10.1002/art.39247
169. Mendoza-Pinto C, García-Carrasco M, Cervera R. Role of Infectious Diseases in the Antiphospholipid Syndrome (Including Its Catastrophic Variant). Curr Rheumatol Rep (2018) 20(10):62. doi: 10.1007/s11926-018-0773-x
170. Zuo Y, Estes SK, Ali RA, Gandhi AA, Yalavarthi S, Shi H, et al. Prothrombotic Autoantibodies in Serum From Patients Hospitalized With COVID-19. Sci Transl Med (2020) 12(570):3876. doi: 10.1126/scitranslmed.abd3876
171. Chousterman BG, Swirski FK, Weber GF. Cytokine Storm and Sepsis Disease Pathogenesis. Semin Immunopathol (2017) 39(5):517–28. doi: 10.1007/s00281-017-0639-8
172. Bösmüller H, Traxler S, Bitzer M, Häberle H, Raiser W, Nann D, et al. The Evolution of Pulmonary Pathology in Fatal COVID-19 Disease: An Autopsy Study With Clinical Correlation. Virchows Arch (2020) 477(3):349–57. doi: 10.1007/s00428-020-02881-x
173. Löf A, Müller JP, Brehm MA. A Biophysical View on Von Willebrand Factor Activation. J Cell Physiol (2018) 233(2):799–810. doi: 10.1002/jcp.25887
174. South K, Lane DA. ADAMTS-13 and Von Willebrand Factor: A Dynamic Duo. J Thromb Haemost (2018) 16(1):6–18. doi: 10.1111/jth.13898
175. Francischetti IMB, Toomer K, Zhang Y, Jani J, Siddiqui Z, Brotman DJ, et al. Upregulation of Pulmonary Tissue Factor, Loss of Thrombomodulin and Immunothrombosis in SARS-CoV-2 Infection. EClinicalMedicine (2021) 39:101069. doi: 10.1016/j.eclinm.2021.101069
176. Masias C, Cataland SR. The Role of ADAMTS13 Testing in the Diagnosis and Management of Thrombotic Microangiopathies and Thrombosis. Blood (2018) 132(9):903–10. doi: 10.1182/blood-2018-02-791533
177. Chang JC. Disseminated Intravascular Coagulation: New Identity as Endotheliopathy-Associated Vascular Microthrombotic Disease Based on In Vivo Hemostasis and Endothelial Molecular Pathogenesis. Thromb J (2020) 18(1):1–21. doi: 10.1186/s12959-020-00231-0
178. Gkrouzman E, Barbhaiya M, Erkan D, Lockshin MD. Reality Check on Antiphospholipid Antibodies in COVID-19–Associated Coagulopathy. Arthritis Rheumatol (2021) 73(1):173–4. doi: 10.1002/art.41472
179. Rosário C, Zandman-Goddard G, Meyron-Holtz EG, D’Cruz DP, Shoenfeld Y. The Hyperferritinemic Syndrome: Macrophage Activation Syndrome, Still’s Disease, Septic Shock and Catastrophic Antiphospholipid Syndrome. BMC Med (2013) 11(1):185. doi: 10.1186/1741-7015-11-185
180. Henderson LA, Canna SW, Schulert GS, Volpi S, Lee PY, Kernan KF, et al. On the Alert for Cytokine Storm: Immunopathology in COVID-19. Arthritis Rheumatol (2020) 72(7):1059–63. doi: 10.1002/art.41285
181. Magro C, Mulvey JJ, Berlin D, Nuovo G, Salvatore S, Harp J, et al. Complement Associated Microvascular Injury and Thrombosis in the Pathogenesis of Severe COVID-19 Infection: A Report of Five Cases. Transl Res (2020) 220:1–13. doi: 10.1016/j.trsl.2020.04.007
182. Connors JM, Levy JH. COVID-19 and Its Implications for Thrombosis and Anticoagulation. Blood (2020) 135(23):2033–40. doi: 10.1182/blood.2020006000
183. Karlberg J, Chong DSY, Lai WYY. Do Men Have a Higher Case Fatality Rate of Severe Acute Respiratory Syndrome Than Women Do? Am J Epidemiol (2004) 159(3):229–31. doi: 10.1093/aje/kwh056
184. Leong: SARS in Singapore-Predictors of Disease Severity - Google Scholar. Available at: https://scholar.google.com/scholar_lookup?title=SARSinSingapore—predictorsofdiseaseseverity&author=HNLeong&author=AEarnest&author=HHLim&publication_year=2006&journal=AnnAcadMedSingapore&volume=35&pages=326-31.
185. Alghamdi IG, Hussain II, Almalki SS, Alghamdi MS, Alghamdi MM, El-Sheemy MA. The Pattern of Middle East Respiratory Syndrome Coronavirus in Saudi Arabia: A Descriptive Epidemiological Analysis of Data From the Saudi Ministry of Health. Int J Gen Med (2014) 7:417–23. doi: 10.2147/IJGM.S67061
186. Roach REJ, Cannegieter SC, Lijfering WM. Differential Risks in Men and Women for First and Recurrent Venous Thrombosis: The Role of Genes and Environment. J Thromb Haemost (2014) 12(10):1593–600. doi: 10.1111/jth.12678
187. Pivonello R, Auriemma RS, Pivonello C, Isidori AM, Corona G, Colao A, et al. Sex Disparities in COVID-19 Severity and Outcome: Are Men Weaker or Women Stronger? Neuroendocrinology (2021) 111(11):1066–85. doi: 10.1159/000513346
188. Erem C, Kocak M, Hacihasanoglu A, Yilmaz M. Blood Coagulation and Fibrinolysis in Male Patient With Hypogonadotropic Hypogonadism: Plasma Factor V and Factor X Activities Increase in Hypogonadotropic Hypogonadism. J Endocrinol Invest (2008) 31(6):537–41. doi: 10.1007/BF03346404
189. Roy-O’Reilly M, McCullough LD. Sex Differences in Stroke: The Contribution of Coagulation. Exp Neurol (2014) 259:16–27. doi: 10.1016/j.expneurol.2014.02.011
190. Næss IA, Christiansen SC, Romundstad P, Cannegieter SC, Rosendaal FR, Hammerstrøm J. Incidence and Mortality of Venous Thrombosis: A Population-Based Study. J Thromb Haemost (2007) 5(4):692–9. doi: 10.1111/j.1538-7836.2007.02450.x
191. Kyrle PA, Minar E, Bialonczyk C, Hirschl M, Weltermann A, Eichinger S. The Risk of Recurrent Venous Thromboembolism in Men and Women. N Engl J Med (2004) 350(25):2558–63. doi: 10.1056/NEJMoa032959
192. Salzano A, Demelo-Rodriguez P, Marra AM, Proietti M. A Focused Review of Gender Differences in Antithrombotic Therapy. Curr Med Chem (2017) 24(24):2576–88. doi: 10.2174/0929867323666161029223512
193. Previtali E, Bucciarelli P, Passamonti SM, Martinelli I. Risk Factors for Venous and Arterial Thrombosis. Blood Transfus (2011) 9(2):120–38. doi: 10.2450/2010.0066-10
194. Rastrelli G, Di Stasi V, Inglese F, Beccaria M, Garuti M, Di Costanzo D, et al. Low Testosterone Levels Predict Clinical Adverse Outcomes in SARS-CoV-2 Pneumonia Patients. Andrology (2021) 9(1):88–98. doi: 10.1111/andr.12821
195. Wang Z, Xu X. scRNA-Seq Profiling of Human Testes Reveals the Presence of the ACE2 Receptor, A Target for SARS-CoV-2 Infection in Spermatogonia, Leydig and Sertoli Cells. Cells (2020) 9(4):3474. doi: 10.3390/cells9040920
196. Gemmati D, Bramanti B, Serino ML, Secchiero P, Zauli G, Tisato V. COVID-19 and Individual Genetic Susceptibility/Receptivity: Role of ACE1/ACE2 Genes, Immunity, Inflammation and Coagulation. Might the Double X-Chromosome in Females be Protective Against SARS-COV-2 Compared to the Single X-Chromosome in Males? Int J Mol Sci (2020) 21(10):3474. doi: 10.3390/ijms21103474
197. Tzoran I, Hoffman R, Monreal M. Hemostasis and Thrombosis in the Oldest Old. Semin Thromb Hemost (2018) 44(7):624–31. doi: 10.1055/s-0038-1657779
198. Lopez-Leon S, Wegman-Ostrosky T, Perelman C, Sepulveda R, Rebolledo PA, Cuapio A, et al. More Than 50 Long-Term Effects of COVID-19: A Systematic Review and Meta-Analysis. Sci Rep (2021) 11(1):1–12. doi: 10.1038/s41598-021-95565-8
Keywords: COVID-19, interconnectedness, immunity, coagulation, thromboembolism
Citation: Emadi-Baygi M, Ehsanifard M, Afrashtehpour N, Norouzi M and Joz-Abbasalian Z (2021) Corona Virus Disease 2019 (COVID-19) as a System-Level Infectious Disease With Distinct Sex Disparities. Front. Immunol. 12:778913. doi: 10.3389/fimmu.2021.778913
Received: 17 September 2021; Accepted: 11 November 2021;
Published: 29 November 2021.
Edited by:
Federica Eduati, Eindhoven University of Technology, NetherlandsReviewed by:
Darragh Duffy, Institut Pasteur, FranceMark E Snyder, University of Pittsburgh, United States
Copyright © 2021 Emadi-Baygi, Ehsanifard, Afrashtehpour, Norouzi and Joz-Abbasalian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Modjtaba Emadi-Baygi, ZW1hZGktbUBza3UuYWMuaXI=
†These authors have contributed equally to this work