 Adiratna Mat Ripen
Adiratna Mat Ripen Mei Yee Chiow
Mei Yee Chiow Prakash Rao Rama Rao
Prakash Rao Rama Rao Saharuddin Bin Mohamad
Saharuddin Bin Mohamad- 1Primary Immunodeficiency Unit, Allergy and Immunology Research Centre, Institute for Medical Research, National Institutes of Health, Ministry of Health Malaysia, Selangor, Malaysia
- 2Institute of Biological Sciences, Faculty of Science, University of Malaya, Kuala Lumpur, Malaysia
- 3Pediatrics Department, Keningau Hospital, Ministry of Health Malaysia, Sabah, Malaysia
- 4Centre of Research in Systems Biology, Structural Bioinformatics and Human Digital Imaging (CRYSTAL), University of Malaya, Kuala Lumpur, Malaysia
Blended phenotypes exhibited by a patient may present a challenge to the establishment of diagnosis. In this study, we report a seven-year-old Murut girl with unusual features of Williams-Beuren syndrome (WBS), including recurrent infections and skin abscesses. Considering the possibility of a second genetic disorder, a mutation screening for genes associated with inborn errors of immunity (IEI) was conducted using whole exome sequencing (WES). Analysis of copy number variations (CNVs) from the exome data revealed a 1.53Mb heterozygous deletion on chromosome 7q11.23, corresponding to the known WBS. We also identified a biallelic loss of NCF1, which indicated autosomal recessive chronic granulomatous disease (CGD). Dihydrorhodamine (DHR) flow cytometric assay demonstrated abnormally low neutrophil oxidative burst activity. Coamplification of NCF1 and its pseudogenes identified a GT-deletion (ΔGT) at the start of exon 2 in NCF1 (NM_000265.7: c.75_76delGT: p.Tyr26Hisfs*26). Estimation of NCF1-to-NCF1 pseudogenes ratio using ΔGT and 20-bp gene scans affirmed nil copies of NCF1 in the patient. While the father had a normal ratio of 2:4, the mother had a ratio of 1:5, implicating the carrier of ΔGT-containing NCF1. Discovery of a 7q11.23 deletion involving one NCF1 allele and a ΔGT in the second NCF1 allele explained the coexistence of WBS and CGD in our patient. This study highlights the capability of WES to establish a molecular diagnosis for a case with blended phenotypes, enabling the provision of appropriate prophylactic treatment.
Introduction
Williams-Beuren syndrome (WBS) (OMIM ID: 194050) is a contiguous gene deletion syndrome inherited in autosomal dominant pattern. Most cases occurred sporadically with an estimated prevalence of 1/7,500 to 1/20,000 live births (1, 2). Patients with WBS typically exhibited distinct facial appearance, cardiovascular abnormalities and developmental delay (3). Other features including idiopathic infantile hypercalcemia, gastrointestinal problems, musculoskeletal defects, hypothyroidism and hernias were noted in some patients (4). This genetic disorder is caused by a heterozygous deletion of 1.5-1.8Mb on chromosome 7q11.23 that encodes approximately 26 to 28 genes (5). Phenotypic variability between WBS patients may complicate the establishment of diagnosis based on clinical symptoms. Hence, a comprehensive genetic testing is necessary to confirm the diagnosis, particularly in cases with unusual manifestations.
Our study involved a seven-year-old patient with blended phenotypes. The patient manifested typical WBS features such as aortic stenosis, inguinal hernia, subclinical hypothyroidism and learning difficulty with developmental delay. Nevertheless, she also had recurrent infections, diarrhea and skin abscesses since the age of one year. As WBS patients are not commonly associated with immune disorders, we suspected the possibility of a primary immunodeficiency. Primary immunodeficiencies, recently termed as inborn errors of immunity (IEI) are characterized by increased susceptibility to infections, autoimmune diseases, autoinflammatory disorders, allergies and malignancies (6). These disorders are caused by monogenic mutations in at least 430 genes (6, 7). Diagnosing IEI based on the clinical and immunological abnormalities can be challenging owing to their phenotypic and genetic heterogeneity (8). Since the application of next-generation sequencing (NGS), the molecular diagnostics of IEI was revolutionized, facilitating the continuous discovery of IEI-associated genes (6, 9).
Previously, we utilized whole exome sequencing (WES) to diagnose patients suspected with IEI and obtained molecular diagnoses in 46.7% of the cohort (10). Among the three NGS approaches, whole genome sequencing (WGS) has the highest diagnostic capacity compared to targeted gene panel and WES. Still, WGS is minimally used in the clinical settings because of its high cost and large data output. Although a targeted gene panel generates more manageable data, it restricts the identification of novel disease-causing genes. In light of the affordable cost and computational requirements, WES is reasonable for both diagnostic and research purposes of IEI (11). The likelihood of encountering incidental findings and variants of uncertain significance increases as the NGS coverage expands. Thus, NGS data should be interpreted meticulously to ensure accurate reporting of deleterious mutations. Herein, we identified the genetic etiology responsible for the unexpected features of WBS in our patient using WES.
Materials and Methods
Study Subject
A seven-year-old Murut girl with WBS caused by a heterozygous deletion (cytogenetic analysis not shown) was recruited by the Institute for Medical Research. The patient was presented with unexpected phenotypes of WBS, i.e., multiple severe infections and abscess formation, raising clinical suspicion of a cooccurring genetic disorder. Written informed consent was collected from the patient and her parents upon blood sample collection. The study received approval from the Medical Research and Ethics Committee, Ministry of Health Malaysia (KKM/NIHSEC/P16-837) and adhered to the Declaration of Helsinki.
Exome Sequencing and Bioinformatics Analysis
Genomic DNA was isolated from peripheral blood mononuclear cells using QIAamp DNA Blood Mini Kit (Qiagen, Germany) according to the manufacturer’s protocol. Exome capture was performed using Agilent SureSelect Human All Exon V5 (Agilent, USA) with target size of 50Mb. Paired-end reads of 101 base pairs (bp) were generated from a HiSeq 4000 sequencer (Illumina, USA) at mean coverage of 100x. Burrows-Wheeler Aligner-maximal exact matches (BWA-MEM) was used to align the sequencing reads to the human reference genome GRCh38 (12). Mapped reads were subjected to soft clipping of adapter sequences, duplicate marking and read sorting using Picard tools. Genome Analysis Toolkit (GATK) was used for base quality score recalibration followed by variant calling. Variants including single nucleotide variants (SNVs) and short insertions or deletions (indels) were annotated using web-based ANNOVAR.
Variant Prioritization
Annotated variants were primarily filtered against the known IEI-associated genes documented by the International Union of Immunological Societies in 2019 and 2021 (6, 7). Exonic and splice site variants resulting in missense, nonsense, frameshift and nonframeshift mutations were retained. Rare variants with allele frequency of 0.0001 or less as reported by Genome Aggregation Database (gnomAD) were further analyzed. The functional impact of the variants was evaluated using in silico pathogenicity prediction tools, namely Sorting Intolerant From Tolerant (SIFT), Polymorphism Phenotyping v2 (PolyPhen-2) and MutationTaster (13–15). Variants with tolerated or benign impact predicted by more than one variant effect predictor were excluded from analysis. Mode of inheritance and genotype-phenotype correlation were determined through extensive literature search. The variants were interpreted according to the standards and guidelines of American College of Medical Genetics and Genomics (ACMG) (16).
Copy Number Variation (CNV) Analysis
ExomeDepth was used to detect CNVs from WES data via read depth approach (17). Read counts were generated from the BAM file containing aligned reads. An optimized reference set was built by selecting the most correlated samples with the study subject from the in-house exomes. Each exon was assigned with a likelihood value that designates one of the copy number variable states, namely deletion, normal and duplication. The likelihood values across multiple exons were merged using hidden Markov model. Bayes factor, which represents the likelihood ratio of CNV to normal copy number was calculated. In this regard, CNV with a high Bayes factor is more likely to be true positive. CNVs shared by the patient and control were excluded from analysis. CNVs on autosomes were interpreted based on findings reported in Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources (DECIPHER) and Database of Genomic Variants (DGV) (18, 19). The potential disease-causing CNV was plotted to show the ratio between the observed and expected read depth.
Dihydrorhodamine (DHR) Flow Cytometric Assay
Neutrophil oxidative burst activity was evaluated using Phagoburst™ kit (Orpegen Pharma, Germany) according to the manufacturer’s instructions. The heparinized whole blood sample was incubated with a stimulant, protein kinase C activator phorbol 12-myristate 13-acetate (PMA) at 37°C. A negative control was prepared by substituting the stimulant with wash solution. After 10 minutes of incubation, nonfluorescent dihydrorhodamine 123 substrate solution was added to every sample. Oxidation of dihydrorhodamine 123 to fluorescent rhodamine 123 upon reaction with reactive oxygen species (ROS) was measured by a BD FACSCanto™ II flow cytometer (Becton Dickinson, USA) using FlowJo™ v10. Neutrophils were gated to analyze the percentage of cells producing ROS during unstimulated and PMA-stimulated conditions.
Genomic DNA Amplification and Sequencing
Polymerase chain reaction using published primers 2LB2 and 2RB2 was performed to screen for the GT-deletion (ΔGT) at the beginning of exon 2 in NCF1 (20). The amplicons were sequenced bidirectionally using BigDye™ Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, USA) on a 3730xl DNA Analyzer (Applied Biosystems, USA). FinchTV was used to visualize the sequence chromatograms.
ΔGT and 20-bp Gene Scans
Gene scans were performed to estimate the ratio of NCF1 and its two pseudogenes (21). ΔGT gene scan targets the ΔGT in exon 2, while 20-bp gene scan targets both ΔGT in exon 2 and 20-bp copy in intron 2. The primers used for ΔGT gene scan were 6-FAM-labeled p47-ΔGT-fwd and p47-ΔGT-rev primers. Whereas for 20-bp gene scan, primers p47-ΔGT-fwd and HEX-labeled p47-20bp-rev were used. Separation of fluorescently labeled fragments was performed on a 3730xl DNA Analyzer (Applied Biosystems, USA). The ratio of NCF1 and its pseudogenes was determined by dividing the peak heights displayed on PeakScanner.
Results
Case Presentation
The patient was the fifth child born to nonconsanguineous parents. Both parents and her four elder siblings were well with no known medical illnesses. Cytogenetic analysis was prompted before enrolment considering the characteristic facial features, revealing the diagnosis of WBS. However, this diagnosis did not explain the recurrence of infections and abscesses experienced by the patient since early childhood. Hence, she was recruited for further genetic analysis. She initially presented with fever, abdominal distension, diarrhea and limb stiffening at the age of one year. Blood culture and sensitivity test detected Escherichia coli, while cerebrospinal fluid fungal culture grew non-encapsulated yeast. She was treated as Escherichia coli sepsis and meningitis with ampicillin, metronidazole, cefepime, vancomycin, meropenem and amphotericin B. She was also noted to have abscesses on her left distal forearm and right popliteal area which were treated with cloxacillin. At the age of three years, she had prolonged fever with diarrhea and vomiting caused by Salmonella typhi, requiring a two-week course of cefuroxime. Until the age of five years, she was admitted for infectious diarrhea and treated with meropenem. One month later, she presented with a left knee abscess and right forearm cellulitis. Blood culture grew Streptococcus sp. In between the admissions, she also had multiple episodes of bronchopneumonia, requiring oxygen support and intravenous antibiotics. Laboratory screening tests encompassing full blood count assessment, lymphocyte subset enumeration and quantitation of immunoglobulin and complement levels were performed (Table 1). The test results showed no abnormalities except elevated IgE.
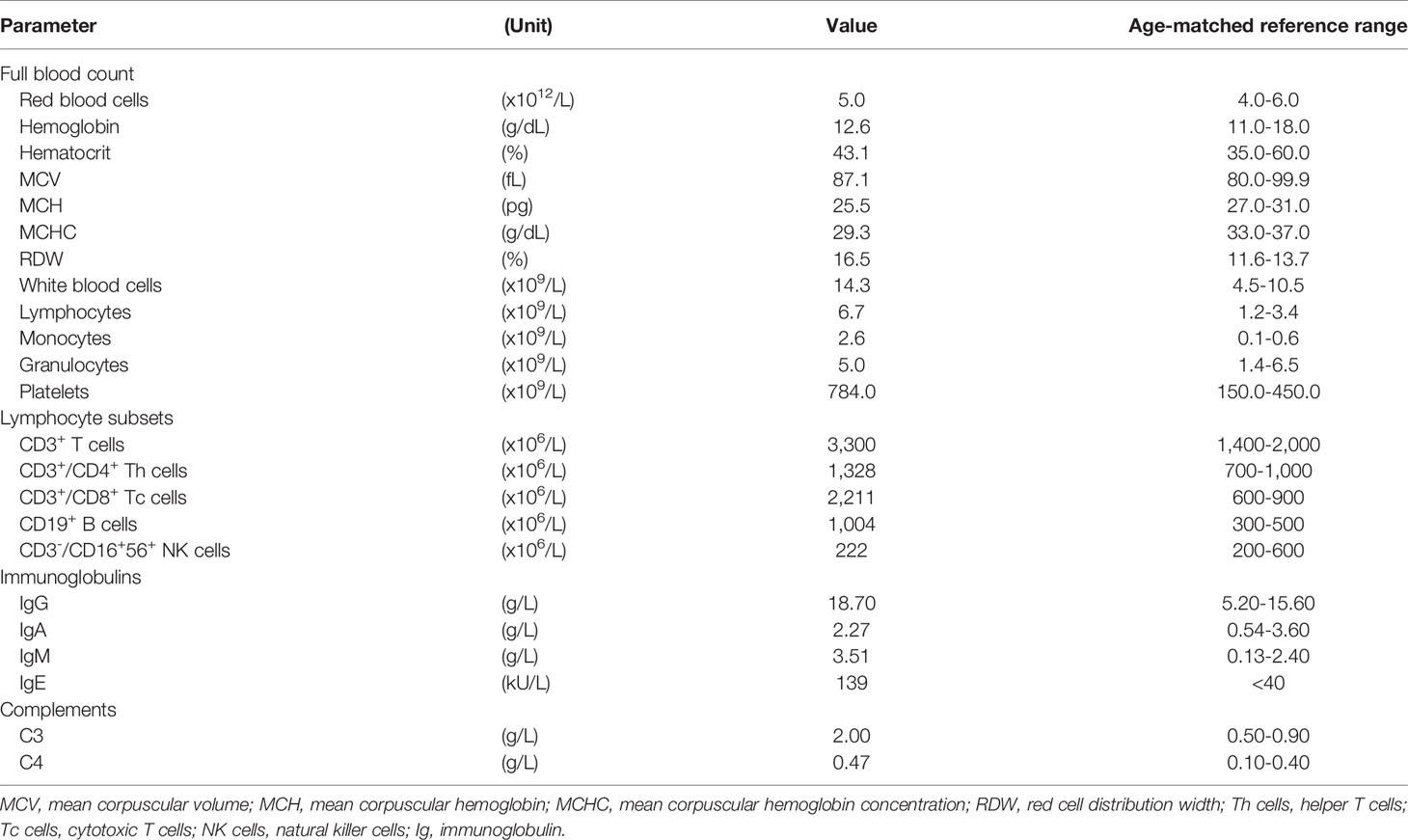
Table 1 Blood test results of the patient.
Bioinformatics Interpretation of Exome Sequencing
Exome sequencing generated 67,606,668 paired-end reads with 99.5% of them properly paired and mapped to the human reference genome GRCh38. A total of 23,364 variants were detected in the exons (n=23,249) and splice sites (n=115) (Figure 1). These variants comprised 97.5% of SNVs and 2.5% of indels. After filtering against stringent criteria, the total variants were reduced to five potential disease-causing point mutations (Table 2). One SNV was identified in each of the five genes, namely CFHR4, IL7R, CFTR, ERCC4 and G6PD. However, the inheritance pattern and associated features of these monogenic disorders were inconsistent with the patient’s zygosity and clinical presentation respectively.
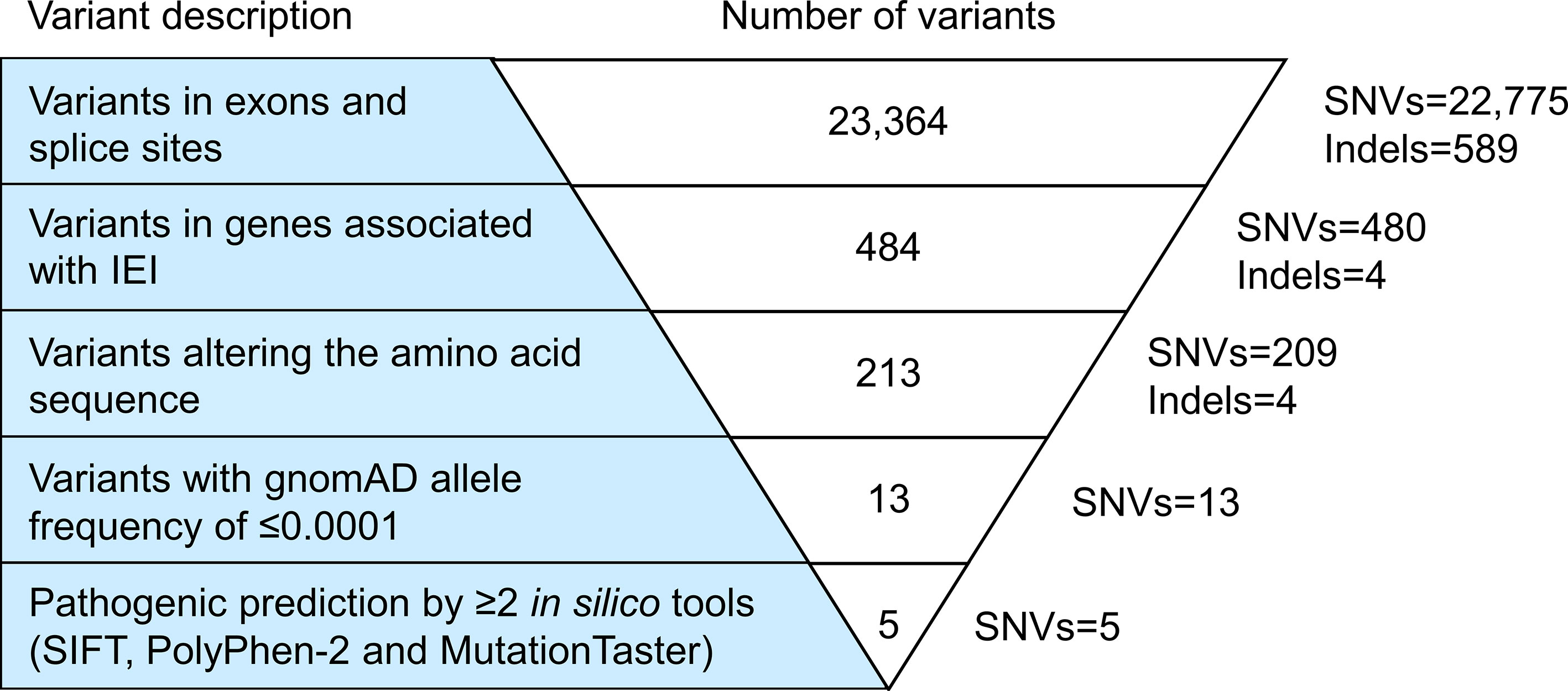
Figure 1 Variant filtering strategy. All variants harbored in the exonic regions and splice sites were filtered based on the known IEI-associated genes, variant class, allele frequency and variant functional impact. Five potential causative single nucleotide variants (SNVs) fulfilled the filtering criteria. However, the inheritance pattern and associated features of these genetic disorders did not match the zygosity and clinical symptoms of the patient respectively. IEI, inborn errors of immunity; SNVs, single nucleotide variants; Indels, small insertions or deletions.
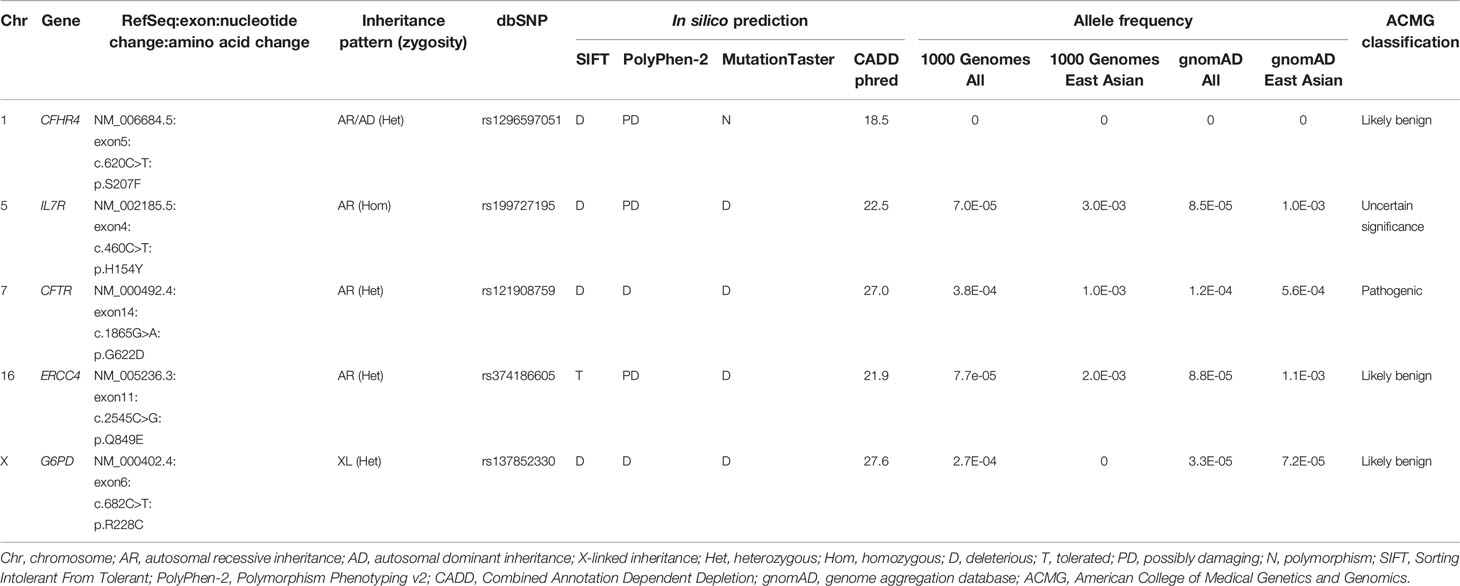
Table 2 List of nonsynonymous single nucleotide variants (SNVs) found in the inborn errors of immunity (IEI) genes after variant filtering.
Biallelic Loss of NCF1
ExomeDepth identified a total of 195 CNV calls in the patient, of which 19 were shared with the control and two were localized on sex chromosomes. The CNVs retained for further analysis encompassed 94 deletions and 80 duplications. The proportion of predicted CNVs with a size larger than 1 kilobases (kb) (63.8%) was higher compared to those shorter than 1kb (36.2%). A high Bayes factor of 1,100 was computed for the heterozygous deletion on chromosome 7q11.23 ranging from 73,303,201 to 74,836,526, corresponding to the WBS region (Figure 2A). The 1.53Mb deletion involved genes from NSUN5 to GTF2IRD2, including NCF1. The second allele of NCF1 was found to be deleted, resulting in biallelic loss of NCF1.
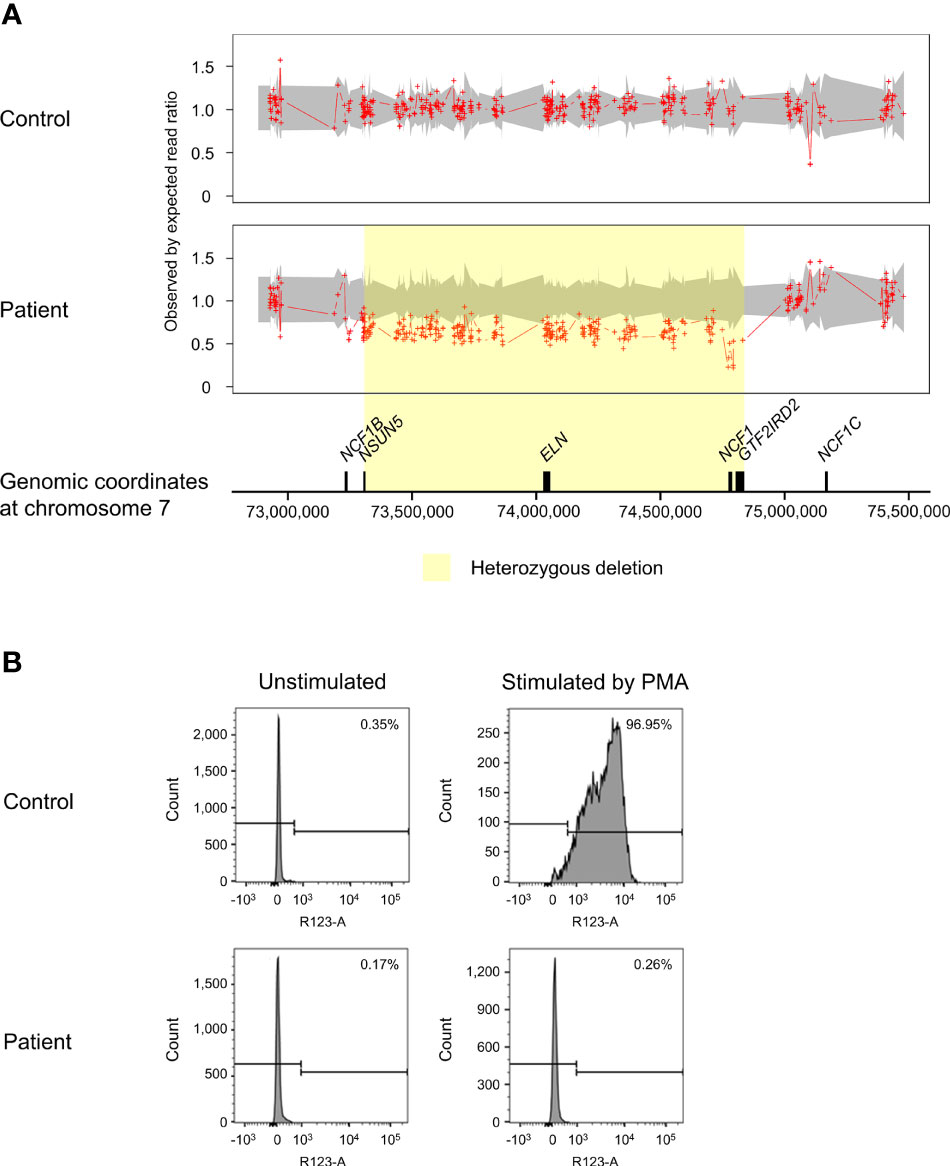
Figure 2 Copy number variation (CNV) analysis on chromosome 7q11.23 and assessment of NADPH oxidase activity. (A) The patient was found to have a biallelic deletion of NCF1 localized within the 1.53Mb heterozygous deletion associated with Williams-Beuren syndrome (WBS) (shaded in yellow). The breakpoints of WBS deletion occurred from chromosome 7:73,303,201-74,836,526, affecting 27 genes. Biallelic loss of NCF1 leads to autosomal recessive chronic granulomatous disease (CGD). Aortic stenosis observed in the patient is consistent with the haploinsufficiency of ELN. Ratio of 1.5 indicates one-copy duplication, 1.0 indicates normal copy state, 0.5 indicates one-copy deletion and 0 indicates two-copy deletion. Grey shade represents the 95% confidence interval of expected read ratio. Each red plus symbol indicates the genomic position being probed. Size of genes (black-filled boxes) was approximate and not drawn to scale. (B) Assessment of neutrophil oxidative burst activity using dihydrorhodamine (DHR) flow cytometry assay. Upon stimulation by phorbol 12-myristate 13-acetate (PMA), the patient showed no change in the fluorescence intensity of rhodamine 123 (right panel). This result is consistent with CGD that lacks normal neutrophil oxidative burst activity. The percentage in the histograms indicates the proportion of neutrophils that produced reactive oxygen species (ROS). PMA, phorbol 12-myristate 13-acetate.
Impaired Neutrophil Oxidative Burst Activity
In response to PMA, the patient showed profoundly low intensity of rhodamine 123 as compared to a distinct shift in the control (Figure 2B). Decreased oxidation of DHR in the patient indicated defective oxidative burst activity of NADPH oxidase.
Validation of ΔGT in NCF1
Nonallele-specific amplification displayed overlapping traces of GTGT-containing NCF1 and ΔGT-containing pseudogenes in the control and parents (Figure 3A). Conversely, only a sequence of NCF1 pseudogenes was shown in the patient, indicating a ΔGT in NCF1 (NM_000265.7: c.75_76delGT: p.Tyr26Hisfs*26). ΔGT gene scan detected fragments of two sizes, i.e., 196-bp fragment reflected NCF1 pseudogenes with ΔGT and 198-bp fragment reflected NCF1 with GTGT. The proportion of GTGT and ΔGT was found to be 2:4 in the control and father (Figure 3B). However, the patient had a single peak of NCF1 pseudogenes, confirming the absence of wild-type NCF1. The mother had a ratio of 1:5, signifying the carrier of ΔGT-containing NCF1. In 20-bp gene scan, 411-bp fragment denoted the product of NCF1 with GTGT and a single 20-bp stretch while 429-bp fragment denoted the product of pseudogenes with ΔGT and a duplication of 20-bp stretch. Ratios of NCF1 and its pseudogenes obtained from ΔGT gene scan were consistent with that of 20-bp gene scan (Figure 3C).
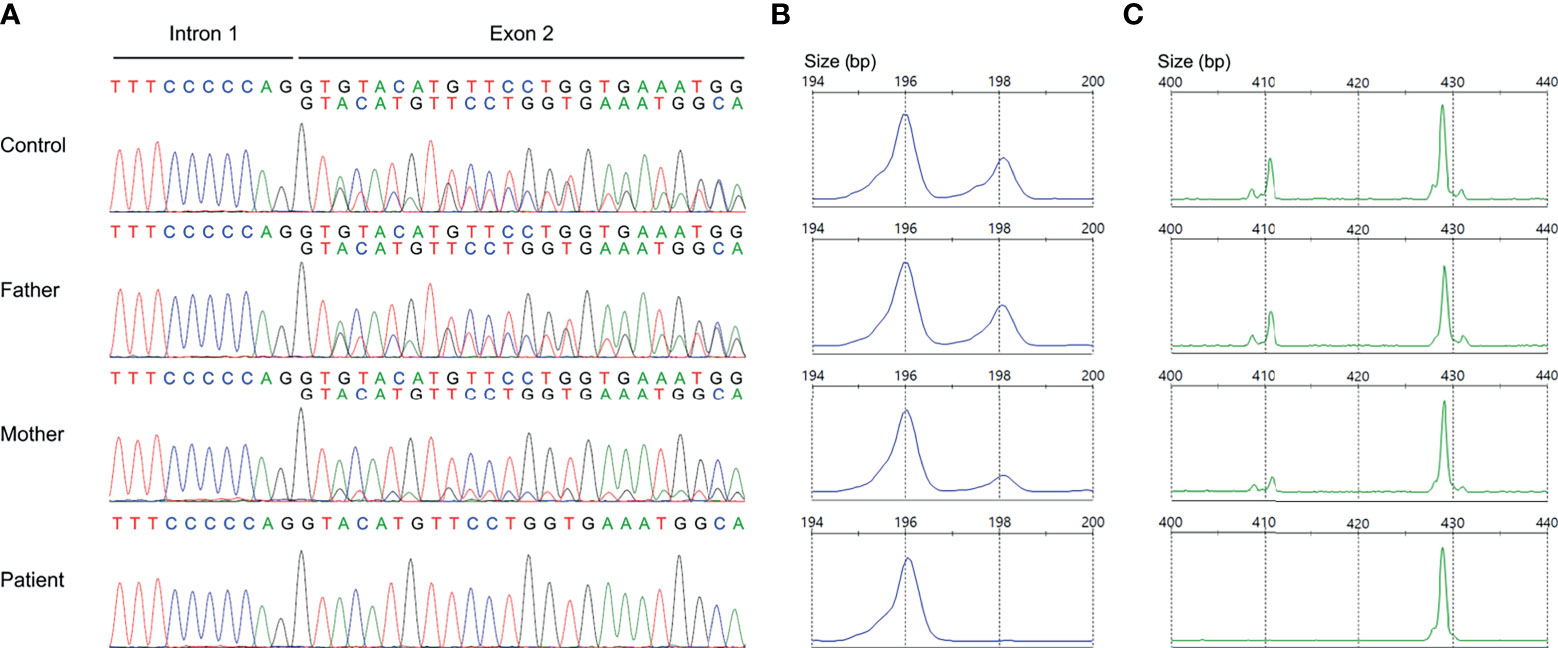
Figure 3 Validation of GT-deletion (ΔGT) in NCF1. (A) Coamplification of NCF1 and its pseudogenes at intron 1/exon 2. Overlapping peaks of GTGT-containing NCF1 and ΔGT-containing pseudogenes were observed in the control and parents. The patient only had a product of NCF1 pseudogenes, indicating a ΔGT in NCF1 (NM_000265.7: exon 2: c.75_76delGT: p.Tyr26Hisfs*26). (B) ΔGT gene scan. The fragments of NCF1 and its pseudogenes were depicted by the 198-bp peak and 196-bp peak respectively. A normal ratio of 2:4 was identified in the control and father, reflecting two copies of NCF1 per four copies of pseudogenes. Meanwhile, the mother had a ratio of 1:5, signifying the carrier of ΔGT in NCF1. Only a single peak of NCF1 pseudogenes was observed in the patient. (C) 20-bp gene scan. The 411-bp-peak and 429-bp-peak represented the fragments of NCF1 and its pseudogenes respectively. Results from 20-bp gene scan corroborated the ratios from ΔGT gene scan. The patient was confirmed to have a biallelic loss of NCF1, in which one allele was included in the Williams-Beuren syndrome (WBS) deletion while another allele harbors a ΔGT. bp, base pair.
Discussion
In this study, we reported a case with dual molecular diagnosis of WBS and chronic granulomatous disease (CGD). Repeated infections and formation of skin abscesses in our patient raised the suspicion of an IEI. We performed initial screening for germline short variants associated with IEI using WES, but detected no causative variants matching the clinical presentation. Further analysis involving CNV detection from exome data uncovered a biallelic loss of NCF1, suggesting CGD. This finding was corroborated by DHR assay that showed remarkably low neutrophil oxidative burst activity. DHR assay is commonly used to diagnose CGD owing to its quantitative measurement of NADPH oxidase activity and high sensitivity (22). However, abnormal DHR oxidation in complete myeloperoxidase deficiency and Rac2 deficiency may lead to false-positive CGD diagnosis (23, 24). Thus, genetic analysis is useful to confirm the diagnosis of CGD.
Oxidative burst activity of NADPH oxidase, which involves ROS production is essential for the elimination of invading pathogens (25). Defects in one of the five subunits of NADPH oxidase will impair the release of ROS, resulting in CGD (26). Due to ineffective clearance of microbes, CGD patients are predisposed to recurrent infections and excessive inflammation (22). This IEI was estimated to affect 1/200,000 to 1/250,000 newborns, with majority of the cases caused by X-linked CYBB mutations (27). Monogenic defects in CYBA, CYBC1, NCF1, NCF2 and NCF4 led to the less common form of CGD inherited in autosomal recessive pattern (28, 29).
Mutation detection in NCF1 can be tricky due to the presence of two pseudogenes that share at least 98% of DNA sequence similarity with the gene (30). Prominent differences, including GTGT at exon 2 and single 20-bp stretch at intron 2 distinguished NCF1 from its pseudogenes that contain a ΔGT and duplicated 20-bp stretch respectively (31). Taking these two features into account, Dekker et al. had established a reliable gene scan method to determine the ratio of NCF1 and its pseudogenes (21). The ΔGT and 20-bp gene scans revealed NCF1 deficiency caused by a ΔGT in our patient as opposed to a normal ratio of 2:4 and a carrier ratio of 1:5 in the father and mother respectively. Deletion of GT in NCF1 led to a frameshift followed by premature termination at amino acid residue 51, generating a truncated p47phox. Unusually high incidence of ΔGT in unrelated patients was thought to arise from unequal crossing-over between NCF1 and its highly homologous pseudogenes (31, 32). Clinical symptoms of WBS and CGD in the patient can be explained by a WBS deletion involving one NCF1 allele and a ΔGT in the second NCF1 allele. As the mother was found to be the carrier of ∆GT-containing NCF1, we hypothesized that the patient inherited the de novo 7q11.23 deletion including NCF1 from her father (Figure 4). The genetic diagnosis has enabled our patient to be treated with continuous prophylactic antibiotics and antifungal to prevent severe infections.
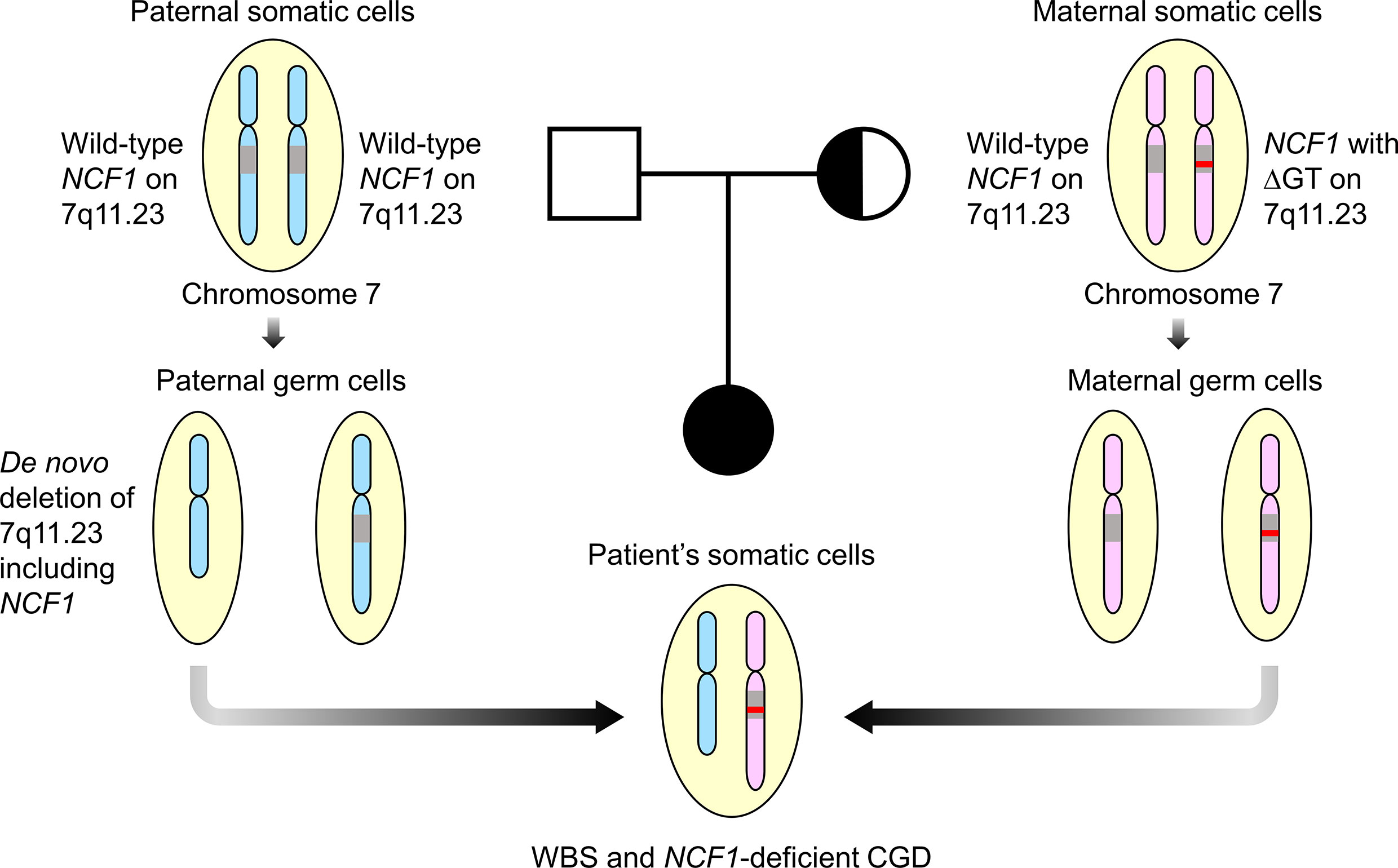
Figure 4 Familial segregation analysis. The patient was postulated to inherit a paternal allele with de novo 7q11.23 deletion involving NCF1 and a maternal allele with ΔGT-containing NCF1, resulting in Williams-Beuren syndrome (WBS) and NCF1-deficient chronic granulomatous disease (CGD). Both parents were well with no features of WBS and CGD. Square symbol indicates male, half-filled circle indicates female carrier of NCF1 with ΔGT and filled circle indicates female affected by WBS and CGD. The diagram was not drawn to scale. ΔGT, GT-deletion; WBS, Williams-Beuren syndrome; CGD, chronic granulomatous disease.
Patients with defective NADPH oxidase are increasingly susceptible to infections caused by catalase-positive microorganisms (27). This is because microbes with catalase are able to break down both host and microbial hydrogen peroxide and escape from the oxidative killing. On the contrary, catalase-negative microorganisms seldom caused infections in CGD patients as they were killed by their self-produced hydrogen peroxide. Interestingly, we detected the growth of catalase-negative Streptococcus sp. in our patient in addition to catalase-positive bacteria, i.e., Escherichia coli and Salmonella typhi. The pathogenicity of Streptococci in CGD can be explained by some strains that produced no hydrogen peroxide (33, 34). Hence, the possibility of getting infected by catalase-negative microbes should be taken into consideration despite being rare in CGD patients.
Misalignment of segmental duplications on chromosome 7q11.23 mediates nonallelic homologous recombination during meiosis, resulting in WBS deletion (35). Cases with a 1.5Mb deletion are more common than those with a larger deletion of 1.8Mb (36). Diagnosing WBS using fluorescence in situ hybridization with an ELN-targeting probe hampers the detection of deletion size (37). This limitation was then overcome by chromosomal microarray and multiplex ligation-dependent probe amplification (38, 39). Alternatively, we used read depth data from WES to call CNVs, revealing a heterozygous deletion of 1.53Mb on chromosome 7q11.23. The deletion breakpoints were predicted within NSUN5 and GTF2IRD2, affecting 27 genes, including ELN and NCF1. Phenotypic consequences were identified in some of the deleted genes. As reported by Curran et al., haploinsufficiency of ELN was responsible for the pathogenesis of aortic stenosis (40). This heart abnormality was noted in our patient, whom had single copy of ELN. Arterial stiffening, possibly due to elastin insufficiency, may increase the risk for cardiovascular events (41, 42). Contrastingly, hemizygosity for NCF1 generated lower oxidative stress, serving as a protective factor for hypertension (43). Biallelic pathogenic mutations in NCF1 will lead to autosomal recessive CGD (20). Stenton et al. recently revealed a causal link between homozygous mutations in DNAJC30 and autosomal recessive Leber’s hereditary optic neuropathy in humans (44). This feature has yet to be reported in WBS patients. Understanding the potential impacts of genetic variants help to predict the disease progression and tailor a proper therapeutic treatment.
Dual molecular diagnosis caused by two disease loci within a patient was less widely recognized before the adoption of NGS. According to a retrospective analysis of exome by Smith et al., patients with clinically complex phenotypes were more likely to have multiple genetic diagnoses (45). This finding suggests the need for reevaluation of existing NGS data and additional laboratory screening in patients with phenotypic complexity. Our study described the first case from Southeast Asia, whom was diagnosed with WBS and NCF1-deficient CGD using WES. Until now, only six patients were reported with similar dual diagnosis (46–50). Despite NCF1 hemizygosity conferred a lower risk for hypertension, Stasia et al. reported one NCF1-deficient patient suffering from high blood pressure (49). Hence, regular medical evaluation is crucial to detect the unexpected medical conditions and initiate a timely regimen.
There are several limitations in this study. Firstly, WES impedes the detection of true positive variants in genes with pseudogenes and highly repetitive sequence due to poor mapping in these regions. Although WES may not be an ideal approach for mutation screening in NCF1, it is useful for the identification of exonic and splicing variants in other CGD genes. Additional genetic testing, particularly WGS is required to detect disease-causing deep intronic variants that will be missed by WES. Secondly, uneven coverage resulted from amplification bias in WES may reduce the sensitivity and accuracy of CNV detection. Wide coverage of WGS made it more superior to detect CNVs that affect both coding and noncoding regions. Chromosomal microarray and multiplex ligation-dependent probe amplification can be used to validate CNVs identified from NGS data.
Conclusion
Our study revealed a case with dual diagnosis of WBS and NCF1-deficient CGD. Blended phenotypes of two genetic conditions can get misinterpreted as atypical presentation of a single disease, resulting in misdiagnosis or underdiagnosis. The use of NGS has facilitated the elucidation of multiple molecular diagnoses caused by multilocus genomic variations. Still, NGS has a low variant detection rate in genes with homologous pseudogenes, which can be complemented by Sanger sequencing. This study demonstrated the capacity of WES to detect both germline short variants and CNVs that may provide insights into pathogenesis and optimal treatment options.
Data Availability Statement
The dataset presented in this study can be found in online repository. The name of the repository and accession number are as follows: https://www.ncbi.nlm.nih.gov/, PRJNA763322.
Ethics Statement
The studies involving human participants were reviewed and approved by Medical Research and Ethics Committee, Ministry of Health Malaysia. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
AMR and SBM designed the study framework and supervised the study. MYC performed the experiments and data analysis. MYC and AMR drafted the manuscript. RRPR recruited the patient, provided clinical treatments and contributed critical views to the study. All co-authors critically reviewed and approved the final version of the manuscript.
Funding
This study was funded by a grant from the Ministry of Health Malaysia (NMRR-16-892-31023) awarded to AMR.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the Director General of Health Malaysia for his permission to publish this article. We appreciate the constructive discussion with Koay BT, Chear CT and Mohd Farid B. We are indebted to the patient and her parents for their participation in this study.
References
1. Davies JH, Moon RJ. Hypercalcemia. In: Huhtaniemi I, Martini L, editors. Encyclopedia of Endocrine Diseases, 2nd ed. Oxford: Academic Press (2019). p. 366–77.
2. Strømme P, Bjømstad PG, Ramstad K. Prevalence Estimation of Williams Syndrome. J Child Neurol (2002) 17(4):269–71. doi: 10.1177/088307380201700406
3. Morris CA, Demsey SA, Leonard CO, Dilts C, Blackburn BL. Natural History of Williams Syndrome: Physical Characteristics. J Pediatr (1988) 113(2):318–26. doi: 10.1016/s0022-3476(88)80272-5
4. Morris CA, Braddock SR, GENETICS CO. Health Care Supervision for Children With Williams Syndrome. Pediatrics (2020) 145(2):e20193761. doi: 10.1542/peds.2019-3761
5. Pober BR. Williams-Beuren Syndrome. N Engl J Med (2010) 362(3):239–52. doi: 10.1056/NEJMra0903074
6. Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human Inborn Errors of Immunity: 2019 Update on the Classification From the International Union of Immunological Societies Expert Committee. J Clin Immunol (2020) 40(1):24–64. doi: 10.1007/s10875-019-00737-x
7. Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. The Ever-Increasing Array of Novel Inborn Errors of Immunity: An Interim Update by the IUIS Committee. J Clin Immunol (2021) 41(3):666–79. doi: 10.1007/s10875-021-00980-1
8. Notarangelo LD, Bacchetta R, Casanova JL, Su HC. Human Inborn Errors of Immunity: An Expanding Universe. Sci Immunol (2020) 5(49):eabb1662. doi: 10.1126/sciimmunol.abb1662
9. Yska HAF, Elsink K, Kuijpers TW, Frederix GWJ, van Gijn ME, van Montfrans JM. Diagnostic Yield of Next Generation Sequencing in Genetically Undiagnosed Patients With Primary Immunodeficiencies: A Systematic Review. J Clin Immunol (2019) 39(6):577–91. doi: 10.1007/s10875-019-00656-x
10. Ripen AM, Chear CT, Baharin MF, Nallusamy R, Chan KC, Kassim A, et al. A Single-Center Pilot Study in Malaysia on the Clinical Utility of Whole-Exome Sequencing for Inborn Errors of Immunity. Clin Exp Immunol (2021) 206(2):119–28. doi: 10.1111/cei.13626
11. Meyts I, Bosch B, Bolze A, Boisson B, Itan Y, Belkadi A, et al. Exome and Genome Sequencing for Inborn Errors of Immunity. J Allergy Clin Immunol (2016) 138(4):957–69. doi: 10.1016/j.jaci.2016.08.003
12. Heng L. Aligning Sequence Reads, Clone Sequences and Assembly Contigs With BWA-MEM. arXiv (2013) 1303.3997v2:1–3.
13. Adzhubei I, Jordan DM, Sunyaev SR. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr Protoc Hum Genet (2013) Chapter 7:Unit7 20. doi: 10.1002/0471142905.hg0720s76
14. Ng PC, Henikoff S. SIFT: Predicting Amino Acid Changes That Affect Protein Function. Nucleic Acids Res (2003) 31(13):3812–4. doi: 10.1093/nar/gkg509
15. Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: Mutation Prediction for the Deep-Sequencing Age. Nat Methods (2014) 11(4):361–2. doi: 10.1038/nmeth.2890
16. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
17. Plagnol V, Curtis J, Epstein M, Mok KY, Stebbings E, Grigoriadou S, et al. A Robust Model for Read Count Data in Exome Sequencing Experiments and Implications for Copy Number Variant Calling. Bioinformatics (2012) 28(21):2747–54. doi: 10.1093/bioinformatics/bts526
18. Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D, et al. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am J Hum Genet (2009) 84(4):524–33. doi: 10.1016/j.ajhg.2009.03.010
19. MacDonald JR, Ziman R, Yuen RK, Feuk L, Scherer SW. The Database of Genomic Variants: A Curated Collection of Structural Variation in the Human Genome. Nucleic Acids Res (2014) 42(Database issue):D986–92. doi: 10.1093/nar/gkt958
20. Noack D, Rae J, Cross AR, Ellis BA, Newburger PE, Curnutte JT, et al. Autosomal Recessive Chronic Granulomatous Disease Caused by Defects in NCF-1, the Gene Encoding the Phagocyte P47-Phox: Mutations Not Arising in the NCF-1 Pseudogenes. Blood (2001) 97(1):305–11. doi: 10.1182/blood.V97.1.305
21. Dekker J, de Boer M, Roos D. Gene-Scan Method for the Recognition of Carriers and Patients With P47(Phox)-Deficient Autosomal Recessive Chronic Granulomatous Disease. Exp Hematol (2001) 29(11):1319–25. doi: 10.1016/s0301-472x(01)00731-7
22. Anjani G, Vignesh P, Joshi V, Shandilya JK, Bhattarai D, Sharma J, et al. Recent Advances in Chronic Granulomatous Disease. Genes Dis (2020) 7(1):84–92. doi: 10.1016/j.gendis.2019.07.010
23. Accetta D, Syverson G, Bonacci B, Reddy S, Bengtson C, Surfus J, et al. Human Phagocyte Defect Caused by a Rac2 Mutation Detected by Means of Neonatal Screening for T-Cell Lymphopenia. J Allergy Clin Immunol (2011) 127(2):535–8.e1-2. doi: 10.1016/j.jaci.2010.10.013
24. Milligan KL, Mann D, Rump A, Anderson VL, Hsu AP, Kuhns DB, et al. Complete Myeloperoxidase Deficiency: Beware the “False-Positive” Dihydrorhodamine Oxidation. J Pediatr (2016) 176:204–6. doi: 10.1016/j.jpeds.2016.05.047
25. Nguyen GT, Green ER, Mecsas J. Neutrophils to the ROScue: Mechanisms of NADPH Oxidase Activation and Bacterial Resistance. Front Cell Infect Microbiol (2017) 7:373(373). doi: 10.3389/fcimb.2017.00373
26. Panday A, Sahoo MK, Osorio D, Batra S. NADPH Oxidases: An Overview From Structure to Innate Immunity-Associated Pathologies. Cell Mol Immunol (2015) 12(1):5–23. doi: 10.1038/cmi.2014.89
27. Winkelstein JA, Marino MC, Johnston RBJ, Boyle J, Curnutte J, Gallin JI, et al. Chronic Granulomatous Disease: Report on a National Registry of 368 Patients. Medicine (2000) 79(3):155–69. doi: 10.1097/00005792-200005000-00003
28. Arnadottir GA, Norddahl GL, Gudmundsdottir S, Agustsdottir AB, Sigurdsson S, Jensson BO, et al. A Homozygous Loss-of-Function Mutation Leading to CYBC1 Deficiency Causes Chronic Granulomatous Disease. Nat Commun (2018) 9(1):4447. doi: 10.1038/s41467-018-06964-x
29. Roos D, van Leeuwen K, Hsu AP, Priel DL, Begtrup A, Brandon R, et al. Hematologically Important Mutations: The Autosomal Forms of Chronic Granulomatous Disease (Third Update). Blood Cells Mol Dis (2021) 92:102596. doi: 10.1016/j.bcmd.2021.102596
30. Chanock SJ, Roesler J, Zhan S, Hopkins P, Lee P, Barrett DT, et al. Genomic Structure of the Human P47-Phox (NCF1) Gene. Blood Cells Mol Dis (2000) 26(1):37–46. doi: 10.1006/bcmd.2000.0274
31. Görlach A, Lee PL, Roesler J, Hopkins PJ, Christensen B, Green ED, et al. A P47-Phox Pseudogene Carries the Most Common Mutation Causing P47-Phox- Deficient Chronic Granulomatous Disease. J Clin Invest (1997) 100(8):1907–18. doi: 10.1172/JCI119721
32. Roesler J, Curnutte JT, Rae J, Barrett D, Patino P, Chanock SJ, et al. Recombination Events Between the P47-Phoxgene and Its Highly Homologous Pseudogenes Are the Main Cause of Autosomal Recessive Chronic Granulomatous Disease. Blood (2000) 95(6):2150–6. doi: 10.1182/blood.V95.6.2150
33. Falcone EL, Hanses S, Stock F, Holland SM, Zelazny AM, Uzel G. Streptococcal Infections in Patients With Chronic Granulomatous Disease: Case Report and Review of the Literature. J Clin Immunol (2012) 32(4):649–52. doi: 10.1007/s10875-012-9661-8
34. Whiley RA, Fraser H, Hardie JM, Beighton D. Phenotypic Differentiation of Streptococcus Intermedius, Streptococcus Constellatus, and Streptococcus Anginosus Strains Within the “Streptococcus Milleri Group”. J Clin Microbiol (1990) 28(7):1497–501. doi: 10.1128/jcm.28.7.1497-1501.1990
35. Cuscó I, Corominas R, Bayés M, Flores R, Rivera-Brugués N, Campuzano V, et al. Copy Number Variation at the 7q11.23 Segmental Duplications Is a Susceptibility Factor for the Williams-Beuren Syndrome Deletion. Genome Res (2008) 18(5):683–94. doi: 10.1101/gr.073197.107
36. Bayés M, Magano LF, Rivera N, Flores R, A. Pérez Jurado L. Mutational Mechanisms of Williams-Beuren Syndrome Deletions. Am J Hum Genet (2003) 73(1):131–51. doi: 10.1086/376565
37. Brewer CM, Morrison N, Tolmie JL. Clinical and Molecular Cytogenetic (FISH) Diagnosis of Williams Syndrome. Arch Dis Child (1996) 74(1):59–61. doi: 10.1136/adc.74.1.59
38. Fusco C, Micale L, Augello B, Teresa Pellico M, Menghini D, Alfieri P, et al. Smaller and Larger Deletions of the Williams Beuren Syndrome Region Implicate Genes Involved in Mild Facial Phenotype, Epilepsy and Autistic Traits. Eur J Hum Genet (2014) 22(1):64–70. doi: 10.1038/ejhg.2013.101
39. Li L, Huang L, Luo Y, Huang X, Lin S, Fang Q. Differing Microdeletion Sizes and Breakpoints in Chromosome 7q11.23 in Williams-Beuren Syndrome Detected by Chromosomal Microarray Analysis. Mol Syndromol (2016) 6(6):268–75. doi: 10.1159/000443942
40. Curran ME, Atkinson DL, Ewart AK, Morris CA, Leppert MF, Keating MT. The Elastin Gene Is Disrupted by a Translocation Associated With Supravalvular Aortic Stenosis. Cell (1993) 73(1):159–68. doi: 10.1016/0092-8674(93)90168-P
41. Kozel BA, Danback JR, Waxler JL, Knutsen RH, de Las Fuentes L, Reusz GS, et al. Williams Syndrome Predisposes to Vascular Stiffness Modified by Antihypertensive Use and Copy Number Changes in NCF1. Hypertension (2014) 63(1):74–9. doi: 10.1161/HYPERTENSIONAHA.113.02087
42. Mitchell GF, Hwang S-J, Vasan RS, Larson MG, Pencina MJ, Hamburg NM, et al. Arterial Stiffness and Cardiovascular Events: The Framingham Heart Study. Circulation (2010) 121(4):505–11. doi: 10.1161/CIRCULATIONAHA.109.886655
43. Del Campo M, Antonell A, Magano LF, Muñoz FJ, Flores R, Bayés M, et al. Hemizygosity at the NCF1 Gene in Patients With Williams-Beuren Syndrome Decreases Their Risk of Hypertension. Am J Hum Genet (2006) 78(4):533–42. doi: 10.1086/501073
44. Stenton SL, Sheremet NL, Catarino CB, Andreeva NA, Assouline Z, Barboni P, et al. Impaired Complex I Repair Causes Recessive Leber's Hereditary Optic Neuropathy. J Clin Invest (2021) 131(6):e138267. doi: 10.1172/JCI138267
45. Smith ED, Blanco K, Sajan SA, Hunter JM, Shinde DN, Wayburn B, et al. A Retrospective Review of Multiple Findings in Diagnostic Exome Sequencing: Half Are Distinct and Half Are Overlapping Diagnoses. Genet Med (2019) 21(10):2199–207. doi: 10.1038/s41436-019-0477-2
46. Gilbert-Barness E, Fox T, Morrow G, Luquette M, Pomerance HH. Williams Syndrome Associated With Crohn Disease, Multiple Infections, and Chronic Granulomatous Disease. Fetal Pediatr Pathol (2004) 23(1):29–37. doi: 10.1080/15227950490423016
47. Kabuki T, Kawai T, Kin Y, Joh K, Ohashi H, Kosho T, et al. A Case of Williams Syndrome With P47-Phox-Deficient Chronic Granulomatous Disease. Nihon Rinsho Meneki Gakkai Kaishi (2003) 26(5):299–303. doi: 10.2177/jsci.26.299
48. Soukup D, Kuechler A, Roesler J, Pichlmaier L, Eckerland M, Olivier M, et al. Genetic Diagnostic Elucidation of a Patient With Multiorgan Granulomas, Facial Peculiarities, and Psychomotor Retardation. Front Genet (2018) 9:355. doi: 10.3389/fgene.2018.00355
49. Stasia MJ, Mollin M, Martel C, Satre V, Coutton C, Amblard F, et al. Functional and Genetic Characterization of Two Extremely Rare Cases of Williams-Beuren Syndrome Associated With Chronic Granulomatous Disease. Eur J Hum Genet (2013) 21(10):1079–84. doi: 10.1038/ejhg.2012.310
Keywords: whole exome sequencing (WES), chronic granulomatous disease (CGD), Williams-Beuren syndrome (WBS), copy number variation (CNV), blended phenotypes, dual molecular diagnosis
Citation: Ripen AM, Chiow MY, Rama Rao PR and Mohamad SB (2021) Revealing Chronic Granulomatous Disease in a Patient With Williams-Beuren Syndrome Using Whole Exome Sequencing. Front. Immunol. 12:778133. doi: 10.3389/fimmu.2021.778133
Received: 16 September 2021; Accepted: 11 October 2021;
Published: 04 November 2021.
Edited by:
Hirokazu Kanegane, Tokyo Medical and Dental University, JapanReviewed by:
Dirk Roos, Sanquin Research, NetherlandsToshinao Kawai, National Center for Child Health and Development (NCCHD), Japan
Copyright © 2021 Ripen, Chiow, Rama Rao and Mohamad. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adiratna Mat Ripen, YWRpcmF0bmFAbW9oLmdvdi5teQ==; Saharuddin Bin Mohamad, c2FoYXJ1ZGRpbkB1bS5lZHUubXk=
†These authors have contributed equally to this work and share first authorship