 Erica A. Steen
Erica A. Steen Michelle L. Hermiston
Michelle L. Hermiston Kim E. Nichols
Kim E. Nichols Lauren K. Meyer
Lauren K. Meyer
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 17 November 2021
Sec. Primary Immunodeficiencies
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.777851
This article is part of the Research Topic Deciphering the Landscape of Immunohematology: Enhancing our Understanding and Management of Hematological Disorders Through Advances in Immunology and Genetics View all 15 articles
Hemophagocytic lymphohistiocytosis (HLH) is a hyperinflammatory disorder characterized by the inability to properly terminate an immune response. Familial HLH (FHLH) and related immune dysregulation syndromes are associated with mutations in the genes PRF1, UNC13D, STX11, STXBP2, LYST, AP3B1, and RAB27A, all of which are required for the assembly, exocytosis, and function of cytotoxic granules within CD8+ T cells and natural killer (NK) cells. Loss-of-function mutations in these genes render the cytotoxicity pathway ineffective, thereby failing to eradicate immune stimuli, such as infectious pathogens or malignant cells. The resulting persistent immune system stimulation drives hypercytokinemia, ultimately leading to severe tissue inflammation and end-organ damage. Traditionally, a diagnosis of FHLH requires the identification of biallelic loss-of-function mutations in one of these degranulation pathway genes. However, this narrow definition fails to encompass patients with other genetic mechanisms underlying degranulation pathway dysfunction. In particular, mounting clinical evidence supports a potential digenic mode of inheritance of FHLH in which single loss-of-function mutations in two different degranulation pathway genes cooperate to impair pathway activity. Here, we review the functions of the FHLH-associated genes within the degranulation pathway and summarize clinical evidence supporting a model in which cumulative defects along this mechanistic pathway may underlie HLH.
Hemophagocytic lymphohistiocytosis (HLH) is a hyperinflammatory syndrome mediated by an ineffective yet hyperactive immune response. Patients with HLH appropriately initiate an immune response in the presence of an immunogenic stimulus, but they display impaired eradication of these stimuli, resulting in persistent immune cell activation and excessive cytokine secretion. This in turn can lead to fatal end-organ damage in the absence of treatment aimed at controlling hyperinflammation (1). Based on diagnostic criteria proposed by the Histiocyte Society in conjunction with the HLH-2004 clinical trial, HLH should be considered in patients presenting with at least five of eight clinical features including fever, splenomegaly, multi-lineage cytopenias, hypofibrinogenemia or hypertriglyceridemia, hyperferritinemia, hemophagocytosis, elevated soluble CD25, and low or absent natural killer (NK) cell activity (2).
Historically, HLH has been subdivided into primary, or familial HLH (FHLH), and secondary, nonfamilial HLH (3, 4). FHLH most commonly presents during infancy or early childhood, with approximately 70% of patients presenting before one year of age (3). Inherited in an autosomal recessive manner, FHLH is characterized by the presence of germline homozygous or compound heterozygous loss-of-function (LOF) mutations in a defined set of FHLH-related genes consisting of PRF1, UNC13D, STX11, and STXBP2, which comprise FHLH subtypes 2-5, respectively. Related immunologic disorders characterized by germline biallelic LOF mutations in genes such as LYST, AP3B1, and RAB27A similarly underlie the development of HLH. Each of these genes is essential to the cytolytic activity of cytotoxic T-lymphocytes (CD8+ T-cells, hereafter referred to as CTLs) and NK cells (2). In contrast, secondary HLH has historically been diagnosed in patients without a clear genetic predisposition to immune dysregulation. Most commonly presenting in later childhood or adulthood, these patients develop hyperinflammation following exposure to a strong immunogenic stimulus such as an infection or malignancy (4). Many reports have demonstrated that homozygous LOF variants affecting FHLH genes result in early disease presentation, sometimes without an identifiable trigger (5). However, patients harboring less damaging variants, such as missense alterations that impair but do not eliminate protein expression or function, may also develop disease at a later age or in response to a more significant immunogenic stimulus (5). Thus, familial and secondary HLH are often challenging to differentiate in the absence of germline genetic testing.
In addition to the classic monogenic model of autosomal recessive inheritance as a cause for FHLH, there is increasing evidence in favor of a mechanism mediated by digenic inheritance (DI), defined broadly as germline genetic variation at two distinct loci that cooperate to mediate disease (6). Practically, this definition is more nuanced, as there is a spectrum of the extent to which two co-inherited variants interact. As a result, it has been proposed that DI be subclassified into pseudo-DI and true DI. In pseudo-DI, the inheritance of one pathogenic variant is itself sufficient to cause disease. However, the phenotype may be modified by co-inheritance of a second pathogenic variant that when present, enhances disease severity. In this scenario, the disease may be considered monogenic but with a variable phenotype determined by the presence of another variant that functions as a genetic modifier. True DI, in contrast, occurs when mutations in two different genes are required for disease to occur, representing oligogenic inheritance (7). In this review, we discuss the functions of genes associated with FHLH and related immunologic disorders and describe the consequences resulting from LOF mutations in those genes, providing a framework for considering how LOF mutations in two distinct genes within the same functional pathway may cooperate to mediate disease. We then review the clinical data suggesting potential DI and consider areas in which additional research is necessary to better understand the functional implications of this genetic mechanism.
CTLs and NK cells respond to and eliminate infected or malignant cells via the release of cytotoxic granules (CGs) at the immunological synapse (IS) formed at the site of target cell engagement (8). These CGs contain serine proteases, known as granzymes, which enter the target cell and induce caspase-dependent and -independent apoptosis (9). Successful target cell killing via the degranulation pathway requires the coordinated activity of numerous proteins involved in granule biogenesis, trafficking, and exocytosis (Figure 1). Homozygous or compound heterozygous LOF mutations in any one of these respective genes underlie HLH and related immune dysregulation syndromes by impeding the ability to terminate the immune response (Table 1). Here, we briefly review the molecular and biochemical functions of these genes, focusing on the consequences of LOF mutations. For more detailed information about protein function, we refer the reader to several excellent reviews on this topic (9–12).
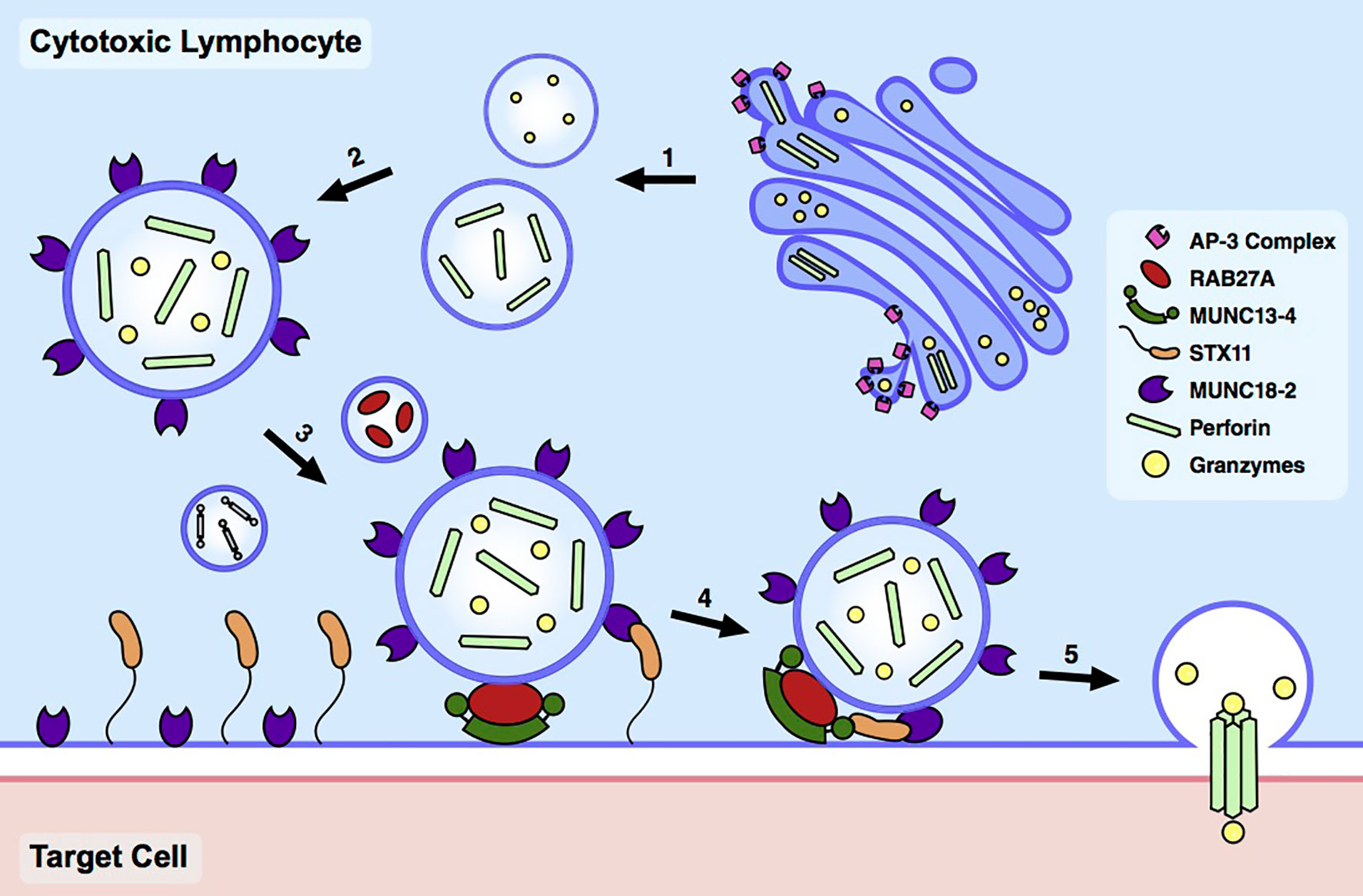
Figure 1 Cytotoxic lymphocyte degranulation pathway. Upon target cell engagement, the degranulation pathway in cytotoxic lymphocytes undergoes a series of coordinated steps consisting of (1-2) cytotoxic granule biogenesis, (3-4) docking and priming, and (5) fusion and pore formation.

Table 1 Degranulation Pathway Genes in HLH and Related Immune Dysregulation Syndromes.
CGs are specialized secretory lysosomes that are assembled in the cytoplasm of CTLs and NK cells. Granzymes and other granule contents are trafficked from the Golgi network to the core of these lysosomes, where an acidic microenvironment maintains the cytotoxic proteins in an inactive state prior to their release at the IS. Two key effectors of proper CG biogenesis are the lysosomal trafficking regulator (LYST) and the clathrin adaptor protein 3 (AP-3) complex (9).
Biallelic LOF mutations in LYST underlie Chediak-Higashi syndrome (CHS), a rare inherited immune disorder in which patients can develop the signs and symptoms of HLH. The clinical features of CHS, including oculocutaneous albinism, prolonged bleeding, neurodegeneration, and immune dysregulation, are all attributable to widespread secretory granule dysfunction in multiple cell types. In particular, the impact of these mutations on immune cell function predisposes to hyperinflammation, which is the primary cause of mortality for patients with CHS (13). Functionally, CTLs and NK cells from patients with CHS are characterized by the presence of fewer but significantly enlarged CGs relative to wild-type (WT) cells (9). In WT CTLs, CGs are undetectable prior to exposure to an immune stimulus. Upon CTL activation, cytotoxic protein expression is rapidly upregulated, concomitant with the assembly of numerous, small CGs in the cytoplasm (14). In contrast, in CHS CTLs, CGs initially form identically to those in WT CTLs, but at the later stages of CTL activation, can be seen forming giant intracellular structures (14).
LYST-deficient CTLs and NK cells display impaired target cell killing, suggesting that these giant CGs are unable to successfully release cytotoxic proteins at the IS. Gil-Krzewska et al. studied NK cells from patients with CHS carrying mutations in the ARM/HEAT (armadillo/huntingin/elongation factor 3, protein phosphatase 2A, TOR1) domain of LYST, a functional domain involved in vesicular trafficking. These cells had fewer and larger CGs relative to WT NK cells. While these large CGs appropriately localized to the IS following target cell engagement, they were unable to fuse with the plasma membrane, thereby preventing the release of cytotoxic proteins toward the target cell (15). To further study the functional consequences of these ARM/HEAT domain mutations, the same authors used CRISPR-Cas9 genome editing to mutate LYST in a human NK cell line. As observed in the patient cells, these knockout cells had a small number of large CGs. In order for CGs to fuse with the plasma membrane to release their contents, they must traverse a network of cortical actin that accumulates at the IS. These authors demonstrated that the formation of this network was identical in WT and knockout cells, however the enlarged granules from the LYST-deficient cells were excluded from the small openings within the actin meshwork, impeding their localization to the plasma membrane. Treatment of these cells with compounds that interfere with actin polymerization potentiated degranulation and restored cytotoxicity, indicating that CGs in LYST-deficient cells retain functionality but are prevented from reaching the cell surface as a result of their excessively large size (16).
The AP-3 protein complex is involved in the formation of clathrin-coated vesicles that traffic to and from lysosome-related organelles (17), a process that is essential for CG biogenesis. Hermansky-Pudlak syndrome (HPS) is a collection of ten autosomal recessive immune disorders mediated by LOF mutations in the subunits of this protein complex. HPS shares a number of clinical features with CHS, including albinism, a bleeding diathesis, and immune dysregulation, reflecting a shared underlying pathophysiology related to secretory lysosome dysfunction. In particular, the immune dysregulation in patients with HPS type 2 (HPS2), caused by biallelic LOF mutations in the β3A subunit (AP3B1) of the AP-3 complex, has been associated with the development of HLH (18).
Cells from patients with HPS2 have decreased protein expression of all AP-3 complex subunits relative to WT cells, as the β3A subunit is thought to stabilize this multi-protein complex. LOF mutations in this subunit alter the conformation of the complex and increase its susceptibility to proteolytic degradation (19). As a result, cells from patients with HPS2 lack the function of the entire AP-3 complex. While LYST regulates the size of CGs, the AP-3 complex is necessary for the appropriate localization of key lysosomal proteins to these granules, which are in turn required for their function. Specifically, this complex mediates the shuttling of proteins from the Golgi network to the lysosome (9). Studies using fibroblasts (20), CTLs (21), and NK cells (22) have consistently demonstrated increased mis-localization of lysosomal proteins to the plasma membrane in cells from patients with HPS2.
Following their transport along microtubule pathways to the IS, CGs must dock at the plasma membrane and undergo a priming step prior to their fusion with the membrane and subsequent exocytosis of their contents. Critical mediators of these processes include RAB27A and MUNC13-4, respectively (9).
Griscelli syndrome type 2 (GS2) is an autosomal recessive immune disorder characterized by biallelic LOF mutations in RAB27A, a gene encoding a small GTPase. As in CHS and HPS2, patients with GS2 present with albinism, neurologic sequelae, and immune dysregulation frequently associated with the development of HLH (23). In regards to the latter, CTLs from patients with GS2 produce CGs with appropriate contents, but these granules are unable to be released via exocytosis, correlating with impaired cytotoxicity (24).
Using CTLs from ashen mice, a murine model of human GS2 in which animals lack Rab27a expression, Stinchcombe et al. studied the mechanistic basis for the cytotoxicity defect in this disease. The authors demonstrated that upon target cell engagement, CGs in RAB27A-deficient CTLs appropriately migrated along microtubules to re-localize to the IS. However, using electron microscopy to obtain detailed images of the IS, they found that these properly localized granules failed to dock to the membrane, with a subsequent failure to release their contents into the IS (25).
Similar to the phenotype in cells from patients with GS2, CTLs and NK cells from patients with FHLH3 demonstrate impaired cytotoxicity despite appropriate localization of CGs to the IS following cell activation. FHLH3 is characterized by biallelic LOF mutations in UNC13D, which encodes the MUNC13-4 protein. Like RAB27A, the vesicular distribution of MUNC13-4 in resting CTLs is largely distinct from that of granzymes. Following cell activation and formation of the IS, vesicles containing RAB27A, vesicles containing MUNC13-4, and CGs co-localize at the IS and fuse into a common vesicular structure (26). While RAB27A mediates the docking of these CGs, MUNC13-4, itself tethered to the membrane via a required protein-protein interaction with the small GTPase RhoG (27), is required for the final step prior to granule fusion with the plasma membrane. This final process, known as priming, makes these granules competent for exocytosis (9). As a result, CGs in MUNC13-4-deficient CTLs can be seen docking at the plasma membrane, but failing to undergo exocytic fusion (28).
Much of what is known about the priming function of MUNC13-4 at the IS comes from studies of the role of other MUNC13 family members in regulating neurotransmitter release at neurological synapses. Using total internal reflection fluorescence microscopy (TIRFM) in neuroendocrine cells, it has been shown that following MUNC13-mediated priming, vesicles docked at the plasma membrane have significantly reduced mobility (29). Similarly, in activated MUNC13-4-deficient murine CTLs, the docked CGs are significantly more mobile than those in WT CTLs, and this phenotype can be rescued with ectopic expression of MUNC13-4 (30).
After docking and priming, CGs are finally able to fuse with the plasma membrane, enabling the release of their cytotoxic contents across the IS toward the target cell. This fusion process is mediated by the activity of soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNAREs), a family of proteins that are ubiquitous through the immune system, where they function to mediate the fusion of docked vesicles with their target membranes. While the requirement for specific SNARE proteins differs according to cell type, the mechanism is shared for all vesicular fusion events. Specifically, SNARE proteins on the vesicular membrane lock together with SNAREs on the target cell membrane to form a protein bridge between the two structures. This interaction then pulls the two compartments into close proximity, and a subsequent conformational change in the SNARE protein complex generates a force that is sufficient to fuse the lipid bilayers of the two membrane compartments (31). These SNARE protein interactions are further regulated by the activity of SNARE accessory proteins, as binding to these accessory proteins is required for SNARE-mediated vesicle fusion with the plasma membrane (9).
LOF mutations in the genes STX11, encoding the SNARE protein syntaxin 11, and STXBP2, encoding the SNARE accessory protein MUNC18-2, underlie FHLH4 (32) and FHLH5 (33), respectively. Like patients with FHLH3, patients harboring biallelic STX11 and STXBP2 mutations demonstrate impaired CTL and NK cell degranulation with an associated cytotoxicity defect, despite appropriate mobilization of CGs to the IS following cell activation (33, 34). Analyzing lymphocytes from patients with FHLH4 and FHLH5, Côte et al. highlighted the importance of interactions between these two proteins. Specifically, in cells lacking MUNC18-2, they found markedly reduced expression of STX11, suggesting that MUNC18-2 is required for STX11 stabilization. The converse was not true, as MUNC18-2 expression was the same in WT and FHLH4 lymphocytes (33). This finding is further supported by work from Dieckmann et al. demonstrating a role for MUNC18-2 as a chaperone of STX11 at the plasma membrane. Using healthy human CTLs, they demonstrated that MUNC18-2 localizes primarily to CGs, traveling with them to the IS upon target cell engagement. In contrast, STX11 was found primarily at the plasma membrane, and became concentrated at the IS after cell activation. While this localization of MUNC18-2 was unchanged in the absence of STX11, STX11 was lost from the plasma membrane in cells lacking MUNC18-2 expression (35), suggesting that MUNC18-2 is required both for the stability and the proper subcellular localization of STX11. Intriguingly, in addition to its chaperone capacity, MUNC18-2 may also play a functional role in the fusion of CGs with the plasma membrane. Spessott et al. demonstrated that STX11, while anchored to the plasma membrane, can support the exchange of lipids between vesicular compartments, but cannot independently facilitate the exchange of vesicular contents. With the addition of WT MUNC18-2, but not a mutant incapable of binding to STX11, complete fusion could be induced (36). These data indicate the importance of an intact molecular interaction between STX11 and MUNC18-2 to facilitate CG fusion, thereby underscoring the defective cytotoxicity in cells from patients with either FHLH4 or FHLH5.
Once CGs are released from the CTL or NK cell, they traverse the IS to induce target cell apoptosis. While the granzyme contents of the CGs directly mediate apoptosis, their activity is dependent on perforin (PRF1), a pore-forming protein that is required for the delivery of granzymes into the target cell cytoplasm (37). The importance of perforin for the cytotoxic activity of CTLs and NK cells is reflected in the severity of the clinical phenotype associated with LOF mutations in PRF1, which was the first identified FHLH gene. Patients with biallelic PRF1 mutations, now known as FHLH2, commonly present early in life with severe immune dysregulation (38).
Given the clinical significance of impaired PRF1 function, there has been considerable interest in understanding how PRF1 mediates granzyme access to the target cell cytoplasm. It is well-established that the pore-forming capabilities of PRF1 are dependent on its oligomerization into a complex that has structural similarity to the membrane attack complex formed in the innate immune system (39). Using human cell lines, Keefe et al. demonstrated that exposing cells to PRF1 induces rapid and profound changes to the plasma membrane, with the formation of membrane blebs and a loss of membrane integrity that facilitates the translocation of cell impermeable dyes into the cytoplasm. Intriguingly, when they tracked the localization of these dyes, they observed that, rather than disseminating into the cytosol, they remained contained within membrane-proximal blebs. Similarly, when cells were incubated in the presence of granzyme B and exposed to PRF1, granzyme B could be found within vesicular structures in the target cell cytoplasm, suggesting that PRF1 mediates entry into target cells via an endocytic process (40). These same authors went on to demonstrate that these endocytic structures, termed “gigantosomes”, contain both PRF1 and granzymes. Approximately 15 minutes after their formation, these structures rupture due to PRF1-induced pore formation in the endosome membrane. This then releases endosomal contents, thereby exposing the target cell cytoplasm to apoptosis-inducing granzymes (41).
FHLH and related immune disorders associated with an HLH phenotype have traditionally been defined as autosomal recessive diseases characterized by germline biallelic LOF mutations in one of the degranulation pathway genes, which in turn confer defective lymphocyte cytotoxicity. This is in contrast to secondary HLH, which is diagnosed in patients meeting the clinical diagnostic criteria for HLH in the absence of a clear genetic predisposition. However, there is increasing evidence that this dichotomous definition is likely an oversimplification, as it does not adequately address the complex interactions between genetic predisposition and environmental influence. A more comprehensive model considers a threshold for disease development, in which severe inherited dysfunction in the degranulation pathway is sufficient to mediate disease with little or no identifiable environmental trigger, while more subtle insults to pathway function require a profound immunogenic stimulus to induce hyperinflammation (42–44). This model suggests that a multitude of genetic mechanisms may contribute to the risk of developing HLH by mediating some degree of underlying dysfunction in the lymphocyte degranulation pathway (Figure 2).
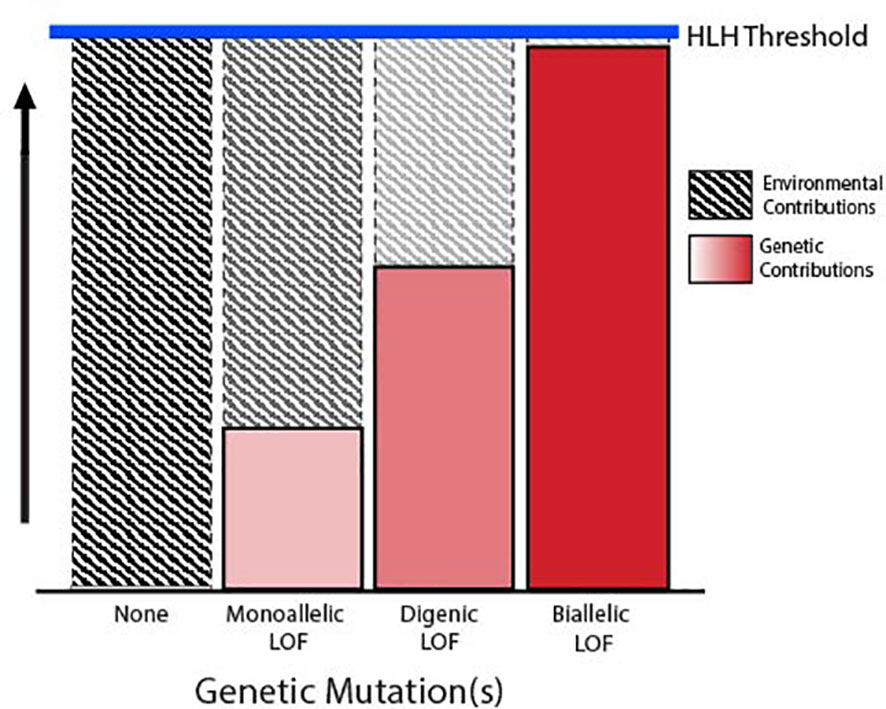
Figure 2 Threshold model of HLH. Schematic depicting the inverse relationship between the degree of degranulation pathway dysfunction conferred by germline variants and the environmental trigger required to reach the threshold for development of HLH.
Even amongst those patients with a classic autosomal recessive pattern of inheritance, clinical and experimental data suggest that LOF mutations in different degranulation pathway genes have non-equivalent consequences, with loss of some proteins resulting in a more severe phenotype than others. In a cohort of patients with complete LOF of PRF1, RAB27A, or STX11, it was found that the severity of disease differed as a function of the involved gene. At one extreme, patients with PRF1 mutations presented with severe, early-onset disease, while those with STX11 mutations had milder disease and presented at a later age (45). These clinical data are recapitulated in murine models of degranulation pathway dysfunction. When Jessen et al. infected multiple HLH-prone murine models with lymphocytic choriomeningitis virus (LCMV) to stimulate the development of HLH, they observed a spectrum of disease severity that correlated with the extent of the cytotoxicity defect conferred by the respective gene knockout. Based on this analysis, they proposed a hierarchy of degranulation pathway genes, with mutations in Ap3b1 conferring the mildest phenotype followed by Lyst, Stx11, Rab27a, and finally Prf1. Interestingly, when they analyzed a cohort of patients with complete LOF of these same genes, they found the same hierarchy reflected in the average age of disease onset (46), suggesting that even amongst patients with biallelic LOF mutations, a threshold model of disease may apply.
Beyond this autosomal recessive mechanism, clinical evidence also suggests that inheritance of a single heterozygous mutation in a degranulation pathway gene may be sufficient to predispose to HLH. Zhang et al. reported on two unrelated teenagers with HLH who were found to have the same heterozygous missense mutation in RAB27A. When this mutation was expressed in an NK cell line, the authors found impaired degranulation and cytotoxic activity as well as decreased capacity for binding to MUNC13-4. Interestingly, clinical NK cell studies in one of these patients similarly revealed impaired NK cell function. Her father was found to be the carrier of the mutation and had a comparable degree of NK cell dysfunction but was otherwise healthy (47), suggesting that this RAB27A mutation may predispose to HLH via cooperation with other currently unknown genetic modifiers and environmental triggers. Heterozygous mutations in RAB27A and STXBP2 have also been shown to mediate degranulation pathway dysfunction by acting in a dominant-negative fashion to impair CG docking and fusion (47, 48). Similarly, monoallelic mutations in STXBP2 and LYST have been reported to occur with significantly greater frequency in patients with systemic juvenile idiopathic arthritis (sJIA) who develop clinical manifestations of HLH relative to those patients with sJIA who do not show signs of hyperinflammation (49). In another cohort of 175 patients with adult-onset HLH, 25 were found to have single heterozygous missense or splice-site mutations in PRF1, MUNC13-4, or STXBP2. Out of the 14 of these patients who had NK cell studies performed, nine had low or absent function. Interestingly, 12 carried the A91V allele of PRF1, an allele that is found at a frequency of up to 4-7% in healthy control populations (50). However, functional studies have demonstrated that expression of this allele on an otherwise WT background does confer cytotoxic dysfunction (51), and that this allele is significantly more prevalent in patients with HLH relative to healthy controls (52), suggesting that it may be a hypomorphic allele that predisposes to HLH.
Together, these data suggest that even partial genetic impairments of the degranulation pathway can be sufficient to predispose to HLH in cooperation with an adequately strong environmental stimulus and/or other genetic modifiers. Similarly, cumulative monoallelic “hits” affecting different genes within the degranulation pathway might also negatively impact immune cell function and increase the risk for HLH. In this way, patients who are doubly heterozygous for mutations in two distinct degranulation pathway genes, also known as DI, could manifest an HLH phenotype that is intermediate between one resulting from a biallelic LOF versus a single heterozygous LOF mutation.
At least one functional study suggests that multiple genetic hits to the degranulation pathway may contribute to a predisposition to HLH. Sepulveda et al. used murine models to study the functional consequences of combinations of heterozygous LOF in Rab27a/Stx11, Rab27a/Prf1, and Rab27a/Stx11/Prf1. In each of these mouse strains, the respective protein expression was reduced by 50%, concomitant with reduced cytotoxicity and manifestations of HLH, though the phenotypic severity differed across the combinations. Mice with the Rab27a/Prf1 combination had a more severe phenotype than did mice with the Rab27a/Stx11 combination, while mice with LOF of all three had the most profound NK cell dysfunction and disease severity (53), suggesting that cumulative effects on the degranulation pathway mediate hyperinflammation.
Next, we summarize available clinical data describing patients with DI of degranulation pathway genes, providing when available information regarding age of HLH onset and functional studies as correlates of the degree of degranulation pathway dysfunction.
At least two case reports have described patients with HLH who were found to have monoallelic variants affecting two distinct degranulation pathway genes. One report describes a 32-year-old male who presented with fever, rash, joint pain, nausea, and vomiting and was subsequently diagnosed with adult-onset Still’s disease (54). After initial management with prednisone and antibiotics, he re-presented with fever, splenomegaly, hyperferritinemia, and hypertriglyceridemia, fulfilling four HLH diagnostic criteria. Despite further treatment with prednisone, the patient subsequently died of progressive multi-organ dysfunction. Later genetic evaluation revealed a heterozygous variant in UNC13D and another in AP3B1, though no NK cell functional studies were performed.
A second report presents the case of a 30-year-old female with chronic active Epstein Barr virus (CAEBV) infection (55). The patient fulfilled diagnostic criteria for HLH, including fever, pancytopenia, hypofibrinogenemia, hyperferritinemia, elevated soluble CD25, decreased NK cell function, and impaired CD107a mobilization, an indicator of degranulation pathway dysfunction. Treatment with etoposide and dexamethasone per the HLH-2004 protocol successfully ameliorated her hyperinflammation. Whole exome sequencing (WES) identified a heterozygous variant in STXBP2 and another in LYST. Intriguingly, the patient’s mother also carried these two variants and had severely impaired NK cell function, but had never developed HLH, again suggesting that additional environmental and genetic factors may have cooperated with underlying degranulation pathway dysfunction to trigger HLH in this patient. Specifically, while the patient had a long history of CAEBV, the mother was noted to be negative for EBV, which is a well-established trigger of HLH in patients with underlying genetic predisposition (56). Through whole exome sequencing (WES), the patient was also found to carry three additional heterozygous variants affecting LRBA (encoding LPS-responsive beige-like anchor protein), AIRE (encoding autoimmune regulator protein), and IRF8 (encoding interferon regulator factor 8). Importantly, all three of these genes have demonstrated functions in immune regulation (57–59), and the latter two variants were inherited from the patient’s father, suggesting that they may have modified the patient’s risk for developing HLH in response to an adequate immunogenic stimulus. These data underscore the importance of WES for the identification of potential genetic modifiers of underlying degranulation pathway dysfunction.
In addition to these reports, multiple groups have performed retrospective studies of cohorts of patients with HLH who underwent clinical genetic testing, most commonly via targeted sequencing of HLH-associated genes. Several of these studies have identified a small number of patients with heterozygous variants in two different degranulation pathway genes. In one cohort of 24 patients who were heterozygous for the A91V (c.272C>T) allele of PRF1, three were found to also carry heterozygous variants in UNC13D. Two of the three had confirmed NK cell dysfunction and presented with HLH prior to one year of age (60). Analysis of 94 Vietnamese patients identified a child presenting with HLH prior to one year of age who had single variants in both UNC13D and STX11 (61). In a cohort of 14 patients with sJIA and HLH, one was found have variants in LYST and STXBP2, and had absent NK cell function and the presence of hemophagocytosis on bone marrow biopsy (49). In two cohorts of adult Chinese patients, two patients with DI of degranulation pathway variants were identified. One was found to have heterozygous variants in both STX11 and LYST (62). Another had variants in UNC13D and LYST, and corresponding functional studies showed significantly impaired NK cell function and CD107a mobilization (63). Additional cohort studies presenting patients with DI of degranulation pathway genes are summarized in Table 2.
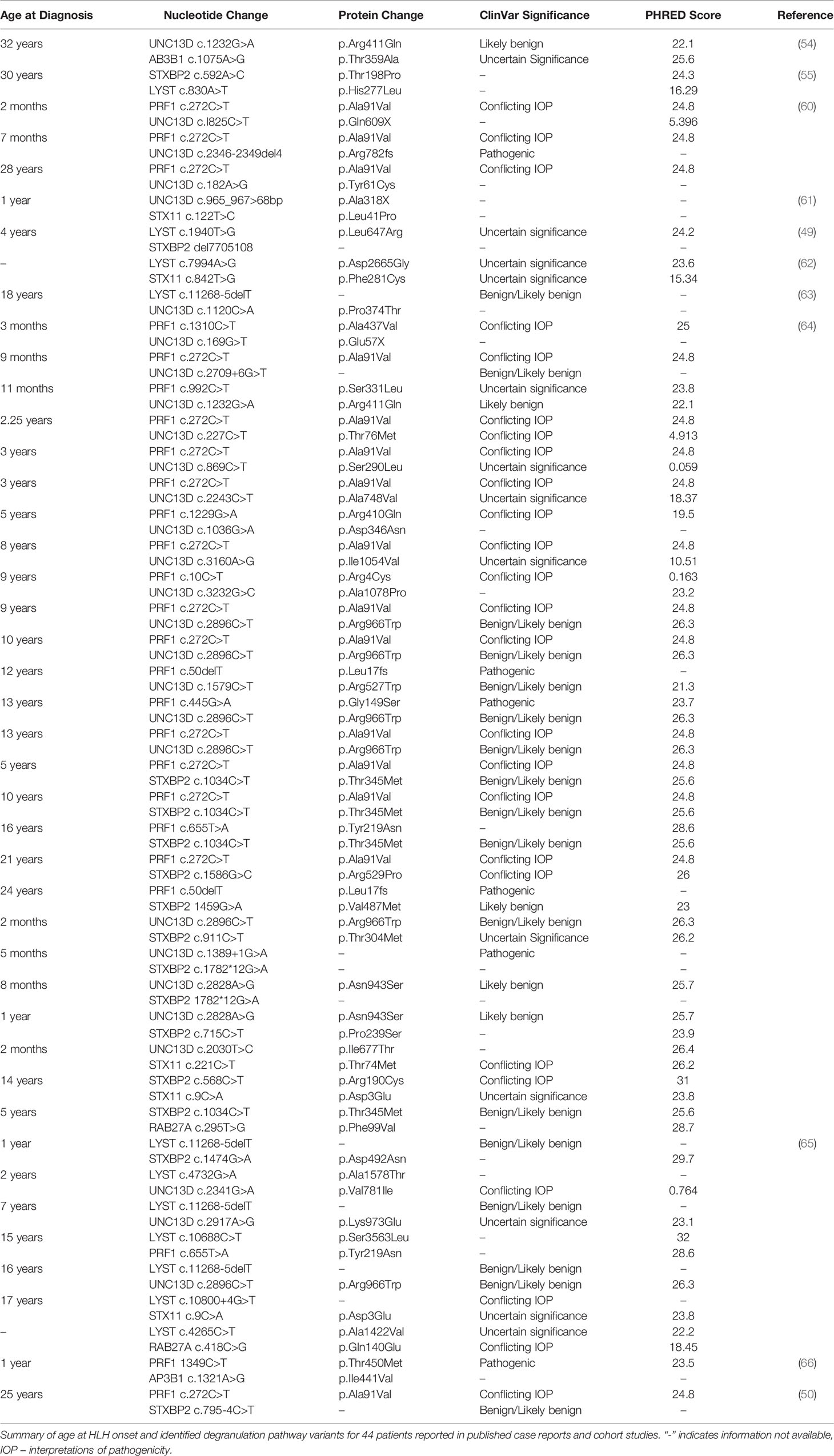
Table 2 Summary of Clinical Data of Patients with DI of Degranulation Pathway Gene Mutations.
In the largest study to date directly evaluating the plausibility of DI in HLH, Zhang et al. analyzed targeted sequencing data from 2,701 patients with HLH and identified 28 with heterozygous variants in two different degranulation pathway genes. Twenty-one of these patients had variants in PRF1 plus another gene, while the remaining seven had variants in two other genes in the degranulation pathway. These seven tended to have an earlier age of onset, comparable to that of patients with classic biallelic LOF mutations. Of the four who had corresponding functional studies, three had impaired CD107a mobilization. Based on these data, the authors conclude that cooperation between two distinct variants likely mediates immune dysregulation that is sufficient to cause HLH, though they acknowledge the caveat of incomplete functional data (64).
Finally, in a cohort of 48 pediatric patients with HLH who underwent WES, Chinn et al. identified heterozygous variants in two degranulation pathway genes in seven (14.5%) patients. To determine the significance of these findings, the authors compared the genotypes observed in their patient cohort to genotypes present in the Baylor-Hopkins CMG database. Intriguingly, they concluded that these combinations were unlikely to occur more commonly in patients with HLH than in the general population, and based on this result, cautioned that other studies reporting on the clinical significance of DI should be interpreted carefully (65).
The data from these case reports and cohort studies are summarized in Table 2. To enable comparisons between these studies, we used ClinVar (67) and CADD scores (68), reported as PHRED-scaled scores, to determine the significance of the reported variants as known at the time of publication of this article.
Here, we review the consequences of LOF mutations in genes encoding several key components of the lymphocyte degranulation pathway, providing a mechanistic basis for interpreting the clinical significance of variants in one or more of these genes. Additionally, we summarize the clinical and experimental data that support revisiting the historical division of HLH into its familial and secondary forms. These data instead favor a more comprehensive model that takes into account both genetic and environmental factors, resulting in a threshold over which the combined effect of these two factors can lead to the development of HLH in the context of an appropriately strong antigenic stimulus. In support of this model, we present an analysis of 44 patients with HLH lacking the classic biallelic LOF mutations but found instead to harbor heterozygous variants in two different genes within the degranulation pathway (Table 2).
While the clinical data presented here suggest the plausibility of DI in HLH, several caveats limit the interpretation of these data. First, of the variants identified in these studies that have information available in the ClinVar database, 33% are reported as “benign” or “likely benign” and 58% have “uncertain significance” or “conflicting interpretations of pathogenicity”. Without corresponding functional data, it is not possible to determine their true impact on protein expression or activity. In addition, clinical NK cell function and CD107a mobilization studies are lacking for many of these patients, further complicating the interpretation of any putative functional impacts of the reported genotypes. Second, most of the studies summarized here relied on targeted sequencing of HLH-associated genes rather than on WES, precluding the ability to identify other potential genetic causes of immune dysregulation. Finally, as noted by Chinn et al. (65), many of the reported digenic combinations are also present at some frequency in healthy populations, confounding the attribution of these genotypes to the development of HLH in these patients. Taken together, the data presented here suggest that co-inheritance of variants in degranulation pathway genes does occur in a subset of patients who develop HLH. However, more comprehensive studies consisting of WES in combination with functional and biochemical analyses of the associated variants and correlation with environmental triggers will be required to better understand the impact of these variant combinations on predisposition to HLH and the extent to which these combinations manifest clinically as pseudo-DI or true DI.
ES and LM wrote the manuscript. MH and KN reviewed and edited the manuscript. All authors approved the submitted version.
This work was supported by grants from the Immune Deficiency Foundation (MH) and the University of California, San Francisco Sandler Program for Breakthrough Biomedical Research (MH). LM is supported by the NIGMS Medical Scientist Training Program Grant T32GM141323.
MH is a consultant for Novartis and Sobi. KN receives research funding from Incyte.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Dr. Claudio Giraudo and Dr. Amanda Marinoff for their careful review of the manuscript and contributions to the ideas presented here. This work is dedicated to and inspired by Celeste Leonhart, as well as her loving parents, Juliana and Andrew, and her sister, Avalon. Celeste died at the age of 8 years after a long battle with an aggressive T-cell lymphoma and HLH. Throughout the time her medical team knew her, Celeste was enveloped in the fierce love of her parents amid a room decorated with rainbows, unicorns, books, and her own artwork. Celeste was a fighter; she overcame T-ALL as a young child and endured innumerable obstacles during her course with lymphoma and HLH. Despite receiving all established and multiple experimental therapies currently available for these diseases, her disease processes ultimately proved smarter than our current best medicine and science. Nevertheless, Celeste showed off her exuberant spirit throughout her course: She created art. She often woke up singing songs about Avalon. She went outside to soak up life outside the hospital even when she was critically ill in the Intensive Care Unit. Her medical team will never forget the time she uttered her first words after being unable to speak for several weeks: “I want a strawberry donut.” Our hope is this review will shed light on the mechanistic basis underlying Celeste’s presentation and offer a more comprehensive model for defining HLH.
1. Canna SW, Marsh RA. Pediatric Hemophagocytic Lymphohistiocytosis. Blood (2020) 135:1332–43. doi: 10.1182/blood.2019000936
2. Henter J-I, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: Diagnostic and Therapeutic Guidelines for Hemophagocytic Lymphohistiocytosis. Pediatr Blood Cancer (2007) 48:124–31. doi: 10.1002/pbc.21039
3. Henter JI, Aricò M, Elinder G, Imashuku S, Janka G. Familial Hemophagocytic Lymphohistiocytosis. Primary Hemophagocytic Lymphohistiocytosis. Hematol Oncol Clin North Am (1998) 12:417–33. doi: 10.1016/s0889-8588(05)70520-7
4. Janka G, Imashuku S, Elinder G, Schneider M, Henter JI. Infection- and Malignancy-Associated Hemophagocytic Syndromes. Secondary Hemophagocytic Lymphohistiocytosis. Hematol Oncol Clin North Am (1998) 12:435–44. doi: 10.1016/s0889-8588(05)70521-9
5. Jordan MB, Allen CE, Greenberg J, Henry M, Hermiston ML, Kumar A, et al. Challenges in the Diagnosis of Hemophagocytic Lymphohistiocytosis: Recommendations From the North American Consortium for Histiocytosis (NACHO). Pediatr Blood Cancer (2019) 66:e27929. doi: 10.1002/pbc.27929
6. Schäffer AA. Digenic Inheritance in Medical Genetics. J Med Genet (2013) 50:641–52. doi: 10.1136/jmedgenet-2013-101713
7. Deltas C. Digenic Inheritance and Genetic Modifiers. Clin Genet (2018) 93:429–38. doi: 10.1111/cge.13150
8. Barry M, Bleackley RC. Cytotoxic T Lymphocytes: All Roads Lead to Death. Nat Rev Immunol (2002) 2:401–9. doi: 10.1038/nri819
9. de Saint Basile G, Ménasché G, Fischer A. Molecular Mechanisms of Biogenesis and Exocytosis of Cytotoxic Granules. Nat Rev Immunol (2010) 10:568–79. doi: 10.1038/nri2803
10. Brisse E, Wouters CH, Matthys P. Hemophagocytic Lymphohistiocytosis (HLH): A Heterogeneous Spectrum of Cytokine-Driven Immune Disorders. Cytokine Growth Factor Rev (2015) 26:263–80. doi: 10.1016/j.cytogfr.2014.10.001
11. Sieni E, Cetica V, Hackmann Y, Coniglio ML, Da Ros M, Ciambotti B, et al. Familial Hemophagocytic Lymphohistiocytosis: When Rare Diseases Shed Light on Immune System Functioning. Front Immunol (2014) 5:167. doi: 10.3389/fimmu.2014.00167
12. Zhang K, Astigarraga I, Bryceson Y, Lehmberg K, Machowicz R, Marsh R, et al. “Familial Hemophagocytic Lymphohistiocytosis”, in: GeneReviews®. Seattle (WA): University of Washington, Seattle. Available at: http://www.ncbi.nlm.nih.gov/books/NBK1444/ (Accessed October 14, 2021).
13. Sharma P, Nicoli E-R, Serra-Vinardell J, Morimoto M, Toro C, Malicdan MCV, et al. Chediak-Higashi Syndrome: A Review of the Past, Present, and Future. Drug Discov Today Dis Models (2020) 31:31–6. doi: 10.1016/j.ddmod.2019.10.008
14. Stinchcombe JC, Page LJ, Griffiths GM. Secretory Lysosome Biogenesis in Cytotoxic T Lymphocytes From Normal and Chediak Higashi Syndrome Patients. Traffic Cph Den (2000) 1:435–44. doi: 10.1034/j.1600-0854.2000.010508.x
15. Gil-Krzewska A, Wood SM, Murakami Y, Nguyen V, Chiang SCC, Cullinane AR, et al. Chediak-Higashi Syndrome: Lysosomal Trafficking Regulator Domains Regulate Exocytosis of Lytic Granules But Not Cytokine Secretion by Natural Killer Cells. J Allergy Clin Immunol (2016) 137:1165–77. doi: 10.1016/j.jaci.2015.08.039
16. Gil-Krzewska A, Saeed MB, Oszmiana A, Fischer ER, Lagrue K, Gahl WA, et al. An Actin Cytoskeletal Barrier Inhibits Lytic Granule Release From Natural Killer Cells in Patients With Chediak-Higashi Syndrome. J Allergy Clin Immunol (2018) 142:914–27.e6. doi: 10.1016/j.jaci.2017.10.040
17. Peden AA, Oorschot V, Hesser BA, Austin CD, Scheller RH, Klumperman J. Localization of the AP-3 Adaptor Complex Defines a Novel Endosomal Exit Site for Lysosomal Membrane Proteins. J Cell Biol (2004) 164:1065–76. doi: 10.1083/jcb.200311064
18. Huizing M, Malicdan MCV, Wang JA, Pri-Chen H, Hess RA, Fischer R, et al. Hermansky-Pudlak Syndrome: Mutation Update. Hum Mutat (2020) 41:543–80. doi: 10.1002/humu.23968
19. Huizing M, Scher CD, Strovel E, Fitzpatrick DL, Hartnell LM, Anikster Y, et al. Nonsense Mutations in ADTB3A Cause Complete Deficiency of the Beta3a Subunit of Adaptor Complex-3 and Severe Hermansky-Pudlak Syndrome Type 2. Pediatr Res (2002) 51:150–8. doi: 10.1203/00006450-200202000-00006
20. Dell’Angelica EC, Shotelersuk V, Aguilar RC, Gahl WA, Bonifacino JS. Altered Trafficking of Lysosomal Proteins in Hermansky-Pudlak Syndrome Due to Mutations in the Beta 3A Subunit of the AP-3 Adaptor. Mol Cell (1999) 3:11–21. doi: 10.1016/s1097-2765(00)80170-7
21. Clark RH, Stinchcombe JC, Day A, Blott E, Booth S, Bossi G, et al. Adaptor Protein 3-Dependent Microtubule-Mediated Movement of Lytic Granules to the Immunological Synapse. Nat Immunol (2003) 4:1111–20. doi: 10.1038/ni1000
22. Fontana S, Parolini S, Vermi W, Booth S, Gallo F, Donini M, et al. Innate Immunity Defects in Hermansky-Pudlak Type 2 Syndrome. Blood (2006) 107:4857–64. doi: 10.1182/blood-2005-11-4398
23. Meeths M, Bryceson YT, Rudd E, Zheng C, Wood SM, Ramme K, et al. Clinical Presentation of Griscelli Syndrome Type 2 and Spectrum of RAB27A Mutations. Pediatr Blood Cancer (2010) 54:563–72. doi: 10.1002/pbc.22357
24. Ménasché G, Pastural E, Feldmann J, Certain S, Ersoy F, Dupuis S, et al. Mutations in RAB27A Cause Griscelli Syndrome Associated With Haemophagocytic Syndrome. Nat Genet (2000) 25:173–6. doi: 10.1038/76024
25. Stinchcombe JC, Barral DC, Mules EH, Booth S, Hume AN, Machesky LM, et al. Rab27a Is Required for Regulated Secretion in Cytotoxic T Lymphocytes. J Cell Biol (2001) 152:825–34. doi: 10.1083/jcb.152.4.825
26. Ménager MM, Ménasché G, Romao M, Knapnougel P, Ho C-H, Garfa M, et al. Secretory Cytotoxic Granule Maturation and Exocytosis Require the Effector Protein Hmunc13-4. Nat Immunol (2007) 8:257–67. doi: 10.1038/ni1431
27. Kalinichenko A, Perinetti Casoni G, Dupré L, Trotta L, Huemer J, Galgano D, et al. RhoG Deficiency Abrogates Cytotoxicity of Human Lymphocytes and Causes Hemophagocytic Lymphohistiocytosis. Blood (2021) 137:2033–45. doi: 10.1182/blood.2020008738
28. Feldmann J, Callebaut I, Raposo G, Certain S, Bacq D, Dumont C, et al. Munc13-4 Is Essential for Cytolytic Granules Fusion and Is Mutated in a Form of Familial Hemophagocytic Lymphohistiocytosis (FHL3). Cell (2003) 115:461–73. doi: 10.1016/s0092-8674(03)00855-9
29. Nofal S, Becherer U, Hof D, Matti U, Rettig J. Primed Vesicles can be Distinguished From Docked Vesicles by Analyzing Their Mobility. J Neurosci (2007) 27:1386–95. doi: 10.1523/JNEUROSCI.4714-06.2007
30. Dudenhöffer-Pfeifer M, Schirra C, Pattu V, Halimani M, Maier-Peuschel M, Marshall MR, et al. Different Munc13 Isoforms Function as Priming Factors in Lytic Granule Release From Murine Cytotoxic T Lymphocytes. Traffic Cph Den (2013) 14:798–809. doi: 10.1111/tra.12074
31. Stow JL, Manderson AP, Murray RZ. SNAREing Immunity: The Role of SNAREs in the Immune System. Nat Rev Immunol (2006) 6:919–29. doi: 10.1038/nri1980
32. zur Stadt U, Schmidt S, Kasper B, Beutel K, Diler AS, Henter J-I, et al. Linkage of Familial Hemophagocytic Lymphohistiocytosis (FHL) Type-4 to Chromosome 6q24 and Identification of Mutations in Syntaxin 11. Hum Mol Genet (2005) 14:827–34. doi: 10.1093/hmg/ddi076
33. Côte M, Ménager MM, Burgess A, Mahlaoui N, Picard C, Schaffner C, et al. Munc18-2 Deficiency Causes Familial Hemophagocytic Lymphohistiocytosis Type 5 and Impairs Cytotoxic Granule Exocytosis in Patient NK Cells. J Clin Invest (2009) 119:3765–73. doi: 10.1172/JCI40732
34. Bryceson YT, Rudd E, Zheng C, Edner J, Ma D, Wood SM, et al. Defective Cytotoxic Lymphocyte Degranulation in Syntaxin-11 Deficient Familial Hemophagocytic Lymphohistiocytosis 4 (FHL4) Patients. Blood (2007) 110:1906–15. doi: 10.1182/blood-2007-02-074468
35. Dieckmann NMG, Hackmann Y, Aricò M, Griffiths GM. Munc18-2 Is Required for Syntaxin 11 Localization on the Plasma Membrane in Cytotoxic T-Lymphocytes. Traffic Cph Den (2015) 16:1330–41. doi: 10.1111/tra.12337
36. Spessott WA, Sanmillan ML, McCormick ME, Kulkarni VV, Giraudo CG. SM Protein Munc18-2 Facilitates Transition of Syntaxin 11-Mediated Lipid Mixing to Complete Fusion for T-Lymphocyte Cytotoxicity. Proc Natl Acad Sci USA (2017) 114:E2176–85. doi: 10.1073/pnas.1617981114
37. Chowdhury D, Lieberman J. Death by a Thousand Cuts: Granzyme Pathways of Programmed Cell Death. Annu Rev Immunol (2008) 26:389–420. doi: 10.1146/annurev.immunol.26.021607.090404
38. Stepp SE, Dufourcq-Lagelouse R, Le Deist F, Bhawan S, Certain S, Mathew PA, et al. Perforin Gene Defects in Familial Hemophagocytic Lymphohistiocytosis. Science (1999) 286:1957–9. doi: 10.1126/science.286.5446.1957
39. Baran K, Dunstone M, Chia J, Ciccone A, Browne KA, Clarke CJP, et al. The Molecular Basis for Perforin Oligomerization and Transmembrane Pore Assembly. Immunity (2009) 30:684–95. doi: 10.1016/j.immuni.2009.03.016
40. Keefe D, Shi L, Feske S, Massol R, Navarro F, Kirchhausen T, et al. Perforin Triggers a Plasma Membrane-Repair Response That Facilitates CTL Induction of Apoptosis. Immunity (2005) 23:249–62. doi: 10.1016/j.immuni.2005.08.001
41. Thiery J, Keefe D, Boulant S, Boucrot E, Walch M, Martinvalet D, et al. Perforin Pores in the Endosomal Membrane Trigger the Release of Endocytosed Granzyme B Into the Cytosol of Target Cells. Nat Immunol (2011) 12:770–7. doi: 10.1038/ni.2050
42. Kim YR, Kim D-Y. Current Status of the Diagnosis and Treatment of Hemophagocytic Lymphohistiocytosis in Adults. Blood Res (2021) 56:S17–25. doi: 10.5045/br.2021.2020323
43. de Saint Basile G, Sepulveda FE, Maschalidi S, Fischer A. Cytotoxic Granule Secretion by Lymphocytes and Its Link to Immune Homeostasis. F1000Research (2015) 4:930. doi: 10.12688/f1000research.6754.1
44. Schulert GS, Cron RQ. The Genetics of Macrophage Activation Syndrome. Genes Immun (2020) 21:169–81. doi: 10.1038/s41435-020-0098-4
45. Sepulveda FE, Debeurme F, Ménasché G, Kurowska M, Côte M, Pachlopnik Schmid J, et al. Distinct Severity of HLH in Both Human and Murine Mutants With Complete Loss of Cytotoxic Effector PRF1, RAB27A, and STX11. Blood (2013) 121:595–603. doi: 10.1182/blood-2012-07-440339
46. Jessen B, Kögl T, Sepulveda FE, de Saint Basile G, Aichele P, Ehl S. Graded Defects in Cytotoxicity Determine Severity of Hemophagocytic Lymphohistiocytosis in Humans and Mice. Front Immunol (2013) 4:448. doi: 10.3389/fimmu.2013.00448
47. Zhang M, Bracaglia C, Prencipe G, Bemrich-Stolz CJ, Beukelman T, Dimmitt RA, et al. A Heterozygous RAB27A Mutation Associated With Delayed Cytolytic Granule Polarization and Hemophagocytic Lymphohistiocytosis. J Immunol Baltim Md 1950 (2016) 196:2492–503. doi: 10.4049/jimmunol.1501284
48. Spessott WA, Sanmillan ML, McCormick ME, Patel N, Villanueva J, Zhang K, et al. Hemophagocytic Lymphohistiocytosis Caused by Dominant-Negative Mutations in STXBP2 That Inhibit SNARE-Mediated Membrane Fusion. Blood (2015) 125:1566–77. doi: 10.1182/blood-2014-11-610816
49. Kaufman KM, Linghu B, Szustakowski JD, Husami A, Yang F, Zhang K, et al. Whole-Exome Sequencing Reveals Overlap Between Macrophage Activation Syndrome in Systemic Juvenile Idiopathic Arthritis and Familial Hemophagocytic Lymphohistiocytosis. Arthritis Rheumatol Hoboken NJ (2014) 66:3486–95. doi: 10.1002/art.38793
50. Zhang K, Jordan MB, Marsh RA, Johnson JA, Kissell D, Meller J, et al. Hypomorphic Mutations in PRF1, MUNC13-4, and STXBP2 Are Associated With Adult-Onset Familial HLH. Blood (2011) 118:5794–8. doi: 10.1182/blood-2011-07-370148
51. House IG, Thia K, Brennan AJ, Tothill R, Dobrovic A, Yeh WZ, et al. Heterozygosity for the Common Perforin Mutation, P.A91V, Impairs the Cytotoxicity of Primary Natural Killer Cells From Healthy Individuals. Immunol Cell Biol (2015) 93:575–80. doi: 10.1038/icb.2015.1
52. Busiello R, Fimiani G, Miano MG, Aricò M, Santoro A, Ursini MV, et al. A91V Perforin Variation in Healthy Subjects and FHLH Patients. Int J Immunogenet (2006) 33:123–5. doi: 10.1111/j.1744-313X.2006.00582.x
53. Sepulveda FE, Garrigue A, Maschalidi S, Garfa-Traore M, Ménasché G, Fischer A, et al. Polygenic Mutations in the Cytotoxicity Pathway Increase Susceptibility to Develop HLH Immunopathology in Mice. Blood (2016) 127:2113–21. doi: 10.1182/blood-2015-12-688960
54. Gao L, Zhu L, Huang L, Zhou J. Synergistic Defects of UNC13D and AP3B1 Leading to Adult Hemophagocytic Lymphohistiocytosis. Int J Hematol (2015) 102:488–92. doi: 10.1007/s12185-015-1807-z
55. Sheng L, Zhang W, Gu J, Shen K, Luo H, Yang Y. Novel Mutations of STXBP2 and LYST Associated With Adult Haemophagocytic Lymphohistiocytosis With Epstein-Barr Virus Infection: A Case Report. BMC Med Genet (2019) 20:34. doi: 10.1186/s12881-019-0765-3
56. El-Mallawany NK, Curry CV, Allen CE. Haemophagocytic Lymphohistiocytosis and Epstein-Barr Virus: A Complex Relationship With Diverse Origins, Expression and Outcomes. Br J Haematol (2021). doi: 10.1111/bjh.17638
57. Kardelen AD, Kara M, Güller D, Ozturan EK, Abalı ZY, Ceylaner S, et al. LRBA Deficiency: A Rare Cause of Type 1 Diabetes, Colitis, and Severe Immunodeficiency. Horm Athens Greece (2021) 20:389–94. doi: 10.1007/s42000-020-00257-z
58. Adams NM, Lau CM, Fan X, Rapp M, Geary CD, Weizman O-E, et al. Transcription Factor IRF8 Orchestrates the Adaptive Natural Killer Cell Response. Immunity (2018) 48:1172–82.e6. doi: 10.1016/j.immuni.2018.04.018
59. Zhao B, Chang L, Fu H, Sun G, Yang W. The Role of Autoimmune Regulator (AIRE) in Peripheral Tolerance. J Immunol Res (2018) 2018:3930750. doi: 10.1155/2018/3930750
60. Zhang K, Johnson JA, Biroschak J, Villanueva J, Lee SM, Bleesing JJ, et al. Familial Haemophagocytic Lymphohistiocytosis in Patients Who Are Heterozygous for the A91V Perforin Variation Is Often Associated With Other Genetic Defects. Int J Immunogenet (2007) 34:231–3. doi: 10.1111/j.1744-313X.2007.00679.x
61. Xinh PT, Chuong HQ, Diem TPH, Nguyen TM, Van ND, Mai Anh NH, et al. Spectrum Mutations of PRF1, UNC13D, STX11, and STXBP2 Genes in Vietnamese Patients With Hemophagocytic Lymphohistiocytosis. Int J Lab Hematol (2021). doi: 10.1111/ijlh.13674
62. Miao Y, Zhu H-Y, Qiao C, Xia Y, Kong Y, Zou Y-X, et al. Pathogenic Gene Mutations or Variants Identified by Targeted Gene Sequencing in Adults With Hemophagocytic Lymphohistiocytosis. Front Immunol (2019) 10:395. doi: 10.3389/fimmu.2019.00395
63. Jin Z, Wang Y, Wang J, Zhang J, Wu L, Gao Z, et al. Primary Hemophagocytic Lymphohistiocytosis in Adults: The Utility of Family Surveys in a Single-Center Study From China. Orphanet J Rare Dis (2018) 13:17. doi: 10.1186/s13023-017-0753-7
64. Zhang K, Chandrakasan S, Chapman H, Valencia CA, Husami A, Kissell D, et al. Synergistic Defects of Different Molecules in the Cytotoxic Pathway Lead to Clinical Familial Hemophagocytic Lymphohistiocytosis. Blood (2014) 124:1331–4. doi: 10.1182/blood-2014-05-573105
65. Chinn IK, Eckstein OS, Peckham-Gregory EC, Goldberg BR, Forbes LR, Nicholas SK, et al. Genetic and Mechanistic Diversity in Pediatric Hemophagocytic Lymphohistiocytosis. Blood (2018) 132:89–100. doi: 10.1182/blood-2017-11-814244
66. Mukda E, Trachoo O, Pasomsub E, Tiyasirichokchai R, Iemwimangsa N, Sosothikul D, et al. Exome Sequencing for Simultaneous Mutation Screening in Children With Hemophagocytic Lymphohistiocytosis. Int J Hematol (2017) 106:282–290. doi: 10.1007/s12185-017-2223-3
67. Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, et al. ClinVar: Improving Access to Variant Interpretations and Supporting Evidence. Nucleic Acids Res (2018) 46:D1062–7. doi: 10.1093/nar/gkx1153
Keywords: hemophagocytic lymphohistiocytosis, digenic, degranulation, variants, cytotoxic lymphocyte, natural killer cell
Citation: Steen EA, Hermiston ML, Nichols KE and Meyer LK (2021) Digenic Inheritance: Evidence and Gaps in Hemophagocytic Lymphohistiocytosis. Front. Immunol. 12:777851. doi: 10.3389/fimmu.2021.777851
Received: 15 September 2021; Accepted: 19 October 2021;
Published: 17 November 2021.
Edited by:
Shanmuganathan Chandrakasan, Emory University, United StatesReviewed by:
Hirokazu Kanegane, Tokyo Medical and Dental University, JapanCopyright © 2021 Steen, Hermiston, Nichols and Meyer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lauren K. Meyer, TGF1cmVuLk1leWVyQHVjc2YuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.