 Tony Marchand1,2,3*
Tony Marchand1,2,3* Sandra Pinho4
Sandra Pinho4- 1Service d’Hématologie Clinique, Centre Hospitalier Universitaire de Rennes, Rennes, France
- 2Faculté de médecine, Université Rennes 1, Rennes, France
- 3Institut National de la Santé et de la Recherche Médicale (INSERM) U1236, Rennes, France
- 4Department of Pharmacology & Regenerative Medicine, University of Illinois at Chicago, Chicago, IL, United States
Acute myeloid leukemia (AML) is one of the most common types of leukemia in adults. While complete remission can be obtained with intensive chemotherapy in young and fit patients, relapse is frequent and prognosis remains poor. Leukemic cells are thought to arise from a pool of leukemic stem cells (LSCs) which sit at the top of the hierarchy. Since their discovery, more than 30 years ago, LSCs have been a topic of intense research and their identification paved the way for cancer stem cell research. LSCs are defined by their ability to self-renew, to engraft into recipient mice and to give rise to leukemia. Compared to healthy hematopoietic stem cells (HSCs), LSCs display specific mutations, epigenetic modifications, and a specific metabolic profile. LSCs are usually considered resistant to chemotherapy and are therefore the drivers of relapse. Similar to their HSC counterpart, LSCs reside in a highly specialized microenvironment referred to as the “niche”. Bidirectional interactions between leukemic cells and the microenvironment favor leukemic progression at the expense of healthy hematopoiesis. Within the niche, LSCs are thought to be protected from genotoxic insults. Improvement in our understanding of LSC gene expression profile and phenotype has led to the development of prognosis signatures and the identification of potential therapeutic targets. In this review, we will discuss LSC biology in the context of their specific microenvironment and how a better understanding of LSC niche biology could pave the way for new therapies that target AML.
Introduction
Acute myeloid leukemia (AML) is the most common type of acute leukemia in adults. AML is characterized by the clonal proliferation of abnormal hematopoietic progenitors leading to blood and bone marrow infiltration and consequently hematopoietic failure (1). Over the past decades, intensive research has significantly improved our understanding of AML biology, highlighting the role of clonal evolution and identifying potential therapeutic targets based on recurrent molecular abnormalities (2, 3). However, therapeutic progress has been limited (4). Despite a promising initial response to intensive chemotherapy, relapse occurs in the majority of patients and prognosis remains poor with a long-term overall survival of 40-50% in patients younger than 60 years old (5–8). In older patients not able to endure intensive chemotherapy, therapeutic options are limited, and long-term overall survival remains low at 15% (9, 10).
Leukemic stem cells (LCSs), also sometimes referred to as leukemic initiating cells, were first described 25 years ago, when Lapidot et al. showed that a small subset of leukemic cells could be transplanted and give rise to leukemia in immunocompromised recipient mice (11). The same group latter identified the CD34posCD38neg phenotype as a way to enrich the LSC population. Similar to normal hematopoietic stem cells (HSCs), LSCs are able to differentiate and self-renew suggesting a leukemic hierarchy (12–16).
Like their normal counterpart, LSCs reside in the bone marrow in a specialized microenvironment termed “niche”. Schofield first described the concept of niche in 1978 and defined it as a limited specific anatomical site where stem cells could be maintained, undergo self-renewal, and where differentiation is inhibited (17). Over the past 20 years, the development of transgenic mice and the improvement of imaging techniques has led to several breakthrough discoveries suggesting that the bone marrow microenvironment plays a central role in normal and pathological hematopoiesis (18). Within the niche, LSCs are thought to be protected from chemotherapy (19–22). Therefore, targeting the LSCs niche represents a promising option to cure AML.
Leukemic Stem Cells Ontogeny And Phenotype
The concept of LSCs is based on the idea that a small subset of cells is able to continually replenish the bulk of leukemic cells. Leukemic stem cells are defined by their capacity to self-renew, incompletely differentiate, and reinitiate leukemia upon serial transplantation in immunocompromised mice (11, 23). Initially thought to originate from the healthy HSC compartment, recent studies have shown that LSCs may instead emerge from committed progenitors (24, 25). Most of human AMLs have at least two molecularly hierarchically ordered distinct LSCs populations (24). Interestingly, the more mature LSC population most closely mirrors normal granulocyte-macrophages progenitors (GMP) whereas the immature LSC population is functionally similar to lymphoid-primed multipotent progenitors (LMPPs). Leukemia originates from the acquisition of driver mutations by HSC or early progenitors (26–28). Identification of clonal hematopoiesis of indeterminate potential (CHIP) has recently generated a significant interest (29). The sequential acquisition of mutations in HSCs and progenitors over a lifetime is suspected to favor hematological malignancies. However, given the high frequency of CHIP in the general population, the exact significance of these mutations and implication in leukemogenesis still needs clarification. To add more complexity, LSCs ontogeny seems to be reversible as opposed to the previously accepted idea that LSCs unidirectionally differentiate into mature AML cells. Indeed, PU.1 gene suppression in differentiated AML-derived cells has been shown to revert AML cells to an immature, clonogenic leukemogenic state (30).
Following the pioneering work done by John Dick’s group, showing that LSCs are enriched within the CD34posCD38neg fraction, several surface markers have been described. Indeed, studies showed that when compared to normal HSCs, LSCs displayed a higher expression of CD25 (31), CD32 (31), CD44 (32), CD96 (33), CD123 (34–36), GPR56 (37), C-type lectin-like molecule-1 (38), IL1RAP (Interleukin 1 Receptor Accessory Protein) (39, 40), N-cadherin, and Tie2 (41). However, a high intra and inter-patients’ heterogeneity prevents the use of a single surface marker to easily isolate LSCs.
The Healthy Hematopoietic Niche
Hematopoietic stem cells reside in a highly specialized microenvironment or niche within the bone marrow (18). Cellular and molecular interactions between niche constituents and HSCs tightly control their self-renewal, proliferation, and differentiation properties. The development of reporter mice and the improvement of imaging techniques has led to a better understanding of the niche since the concept was first proposed in 1978 (17). Studies have identified several cell populations, sometimes redundant, implicated in homeostatic and pathologic hematopoiesis. Similar to the heterogeneity of the hematopoietic system, niche cells are also highly heterogeneous (42–46).
Early studies have suggested a major role of osteoblasts in hematopoiesis by showing hematopoietic stem and progenitor cells (HSPCs) and osteolineage cells in close proximity at steady state and after bone marrow transplantation, additionally osteoblasts have the capacity to support HSPCs in vitro (47–50). Other studies showed a correlation between the number of osteoblasts and LinnegSca1posc-Kitpos HSPCs (51, 52). However, the specific genetic deletion in osteoblast of two key cytokines required for HSC maintenance, stem cell factor (Scf) and CXC-chemokine ligand 12 (Cxcl12), did not have a major effect on HSCs (53–55). In addition, 3-D imaging of the bone marrow revealed that HSCs were preferentially localized close to the vascular network but not to the endosteal surface (56, 57). However, osteolineage cells form a niche for early lymphoid progenitors (53, 54, 58), and are implicated in the development and progression of several hematological malignancies like leukemia (54, 58–61).
The identification of the SLAM cell surface markers allowed the imaging of purified HSCs in their native niche (62). This study and others revealed the close proximity of HSCs and blood vessels suggesting the existence of a vascular niche composed by different types of blood vessels and associated perivascular cells (18). Bone marrow mesenchymal stem cells (BM-MSCs) represent a rare and heterogeneous population of stromal cells characterized by their ability to self-renew and differentiate into osteoblasts, chrondrocytes and adipocytes (63). In the bone marrow, MSCs are located around the blood vessels where they closely interact with HSCs and support hematopoiesis. The development of new transgenic mice models led to the identification of several MSC subsets with significant overlap between the different populations identified (53, 55, 64–68). BM-MSCs are major sources of key niche factors important for the maintenance, proliferation and retention in the mouse bone marrow of HSCs (69). Deletion of Scf or Cxcl12 in stromal cells directly affects HSC number and localization (67, 70, 71). Recent single cell RNA sequencing-based studies have confirmed the high heterogeneity among stromal cells in the bone marrow in particular within the MSC compartment at an unprecedented resolution (42, 44, 45).
The bone marrow is highly vascularized which provides nutrients and oxygen and furthermore allows HSCs and newly generated hematopoietic cells to leave the bone marrow and circulate throughout the body. Bone marrow vascularization is composed of thin-walled arterioles paralleled to the long bone axis and mostly closed to the endosteal region. Arteriolar vessels are connected to the dense network of highly branched sinusoids by type-H vessels at the proximity of the bone (72). Endothelial cells are also key regulators of HSC maintenance and function, and most HSCs localize within 5µm of a bone marrow vessel (56, 62). Indeed, endothelial cells express several factors that regulate HSC function such as SCF, CXCL12, and Notch ligands among others. Depletion of these factors has a dramatic effect on HSC number at steady state and hematopoietic recovery following myeloablative treatment (53–55, 73, 74).
The nervous system plays a crucial role in bone and bone marrow homeostasis (75). Whereas parasympathetic fibers only innervate the compact bone, the bone marrow cavity is innervated by both sympathetic and sensory nerves (76, 77). Although sympathetic nerves do not regulate HSC directly, they are important regulators of HSC mobilization from the bone marrow in response to G-CSF (78). HSCs are also released into the circulation in a circadian manner in response to adrenergic signals from the sympathetic nervous system (SNS) that regulate the synthesis of MSC derived CXCL12, critical for the retention of HSCs inside the bone marrow (65, 78, 79). Interestingly, nociceptive nerves collaborate with the SNS in HSC maintenance and G-CSF-induced mobilization via the secretion of calcitonine gene-related peptide (80). Bone marrow neuropathy observed in aging or after the administration of genotoxic drugs induced a profound remodeling of the HSC niche and affected bone marrow regeneration (81–83). Non-myelinating Schwann cells are also involved in HSCs maintenance by converting the latent Transforming Growth Factor β (TGFβ) into the active form inducing HSCs quiescence (84).
In addition to bone marrow stromal cells, healthy HSCs are also directly and indirectly regulated by their own hematopoietic progeny including megakaryocytes, macrophages, regulatory T cells, neutrophils and other myeloid cells, reviewed elsewhere (18).
The Leukemic Niche
Although the exact location of LSCs within the bone marrow niche still needs to be clarified, it is now clear that the microenvironment plays a role in leukemogenesis and that leukemic cells can also alter the bone marrow at the expense of physiological hematopoiesis.
A Potential Role of the Microenvironment in Leukemogenesis
Leukemogenesis was long regarded as a cell autonomous process. This dogma was challenged by the early description of donor cell derived leukemia in bone marrow transplanted patients (85). These observations supported the “seed and soil” theory proposed by Paget in 1889 who suggested that tumor metastasis required favorable interactions between tumor cells (the “seed”) and their microenvironment (the “soil”) (86). The role of non-hematopoietic cells in leukemogenesis was first demonstrated by the development of transgenic mice and the capacity to delete genes in a cell-specific manner. In the context of hematological malignancies, the proof of concept came from the description of a myeloproliferative disorder induced by deregulated expression of Jagged 1 in IκBα deficient hepatocytes. In contrast, mice with a conditional deletion of IκBα specifically in the myeloid lineage did not develop any myeloproliferative neoplasm (MPN) (87), suggesting that premalignant hematopoietic disorders can be initiated by nonhematopoietic cells. Walkley, et al. demonstrated the role of the retinoic acid receptor-γ (RARγ) in niche-driven MPN. Mice deficient in RARγ developed a MPN-like phenotype even when transplanted with wild-type cells (88). The same group investigated the role of the retinoblastoma protein (RB) in hematopoiesis and demonstrated that the deletion of Rb induced a MPN-like phenotype only when deleted in both the hematopoietic and non-hematopoietic compartments (89). These studies support the role of the interaction between hematopoietic cells and their microenvironment in the development of hematological malignancies.
Bone marrow MSCs play a central role in the regulation of HSCs during homeostatic hematopoiesis while also involved in the development of myelodysplasia and leukemia. Indeed, specific deletion of the gene encoding Dicer 1, an enzyme involved in micro-RNA processing in osteoprogenitors induces myelodysplasia and sporadic secondary leukemia (59). This phenotype was not observed when Dicer1 was deleted in the hematopoietic cells demonstrating that the myelodysplasia was environmentally induced. Deletion of Dicer1 induced the downregulation of Sbds, a gene mutated in Schwachman-Bodian-Diamond syndrome, which is a rare human disease characterized by bone marrow failure and a predisposition to leukemia. Specific deletion of Sbds in MSCs induced mitochondrial dysfunction, oxidative stress, and activation of the DNA damage response in HSPCs ultimately impairing hematopoiesis and favoring leukemogenesis (90). This effect is a consequence of the secretion of the pro-inflammatory molecules, S100A8 and S100A9, by MSCs. Conditional expression of a mutated form of Ptpn11, the gene encoding for the protein tyrosine phosphatase SHP2, in MSCs and osteoprogenitors also induced a MPN-like phenotype (91). To further support the role of the osteolineage compartment in leukemogenesis, activating mutation of beta-catenin in osteoblasts induced AML by activation of Notch signaling in HSPCs (92). By contrast, the defective activation of Notch in the microenvironment leads to myeloproliferative disease (93). This effect is attributed to a Notch-dependent repression of the micro-RNA miR-155, regulating the inflammatory state of the bone marrow niche (94).
Healthy hematopoiesis is the consequence of close and highly regulated interactions between HSPCs and their microenvironment. Overall, cumulative evidence suggests that niche constituents can also drive hematopoietic malignancy.
Remodeling of the Hematopoietic Niche by Leukemic Cells
As our knowledge of the normal hematopoietic niche improved in the past 20 years, the role of the microenvironment in leukemia development captured the attention of the field. Leukemic cells can remodel the niche creating a favorable microenvironment at the expense of the normal hematopoiesis (Figure 1) (95). Imaging studies in mice have shown that chemotherapy resistant human LSCs primarily home to and engraft close to the endosteal region where they closely interact with different microenvironmental structures (19).
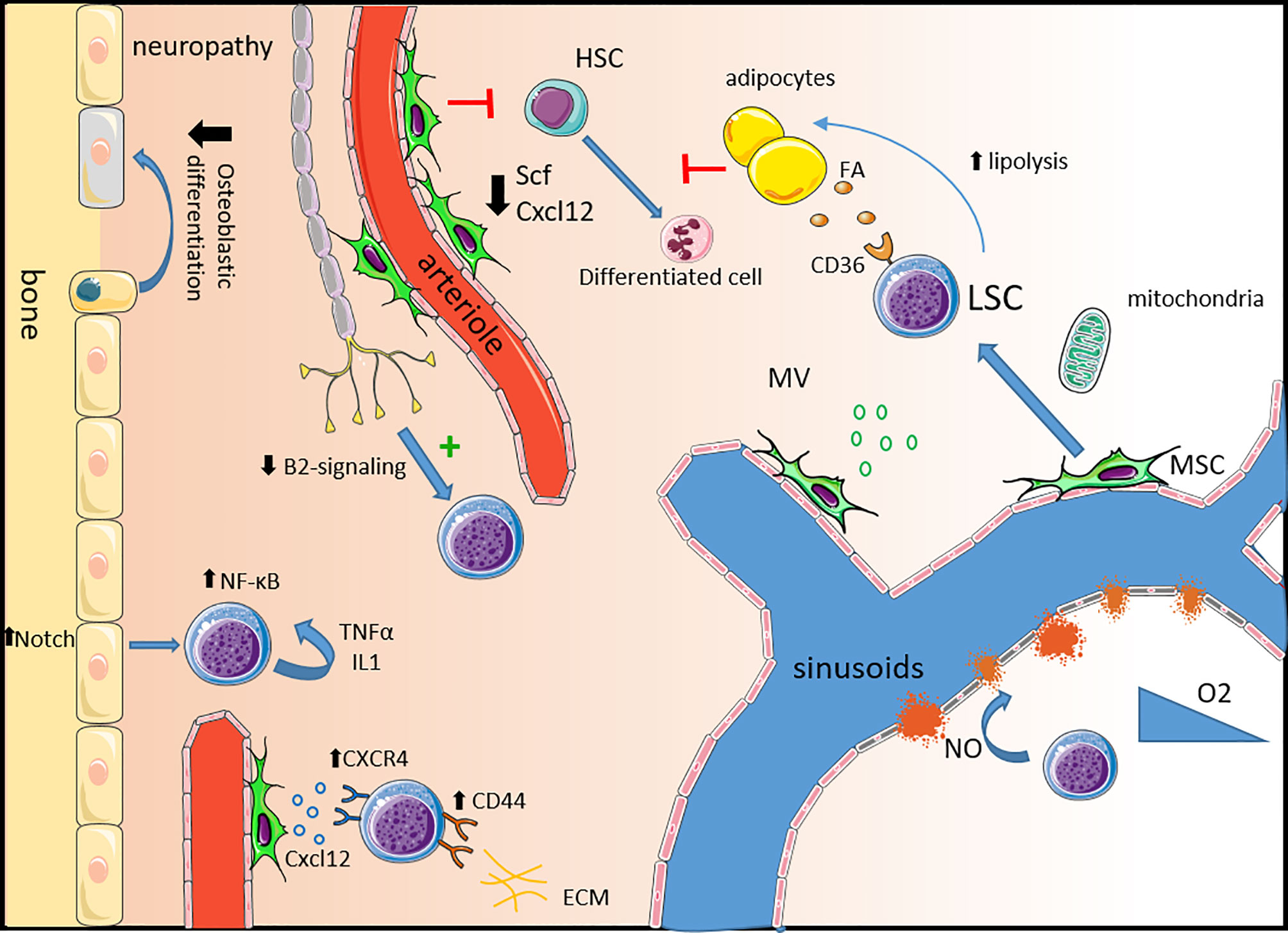
Figure 1 Remodeling of the healthy niche into a permissive leukemic niche. Neuropathy: Leukemic progression is associated with sympathetic neuropathy. Loss of β2-adrenergic signaling directly promotes leukemic progression and triggers the expansion of MSCs primed for osteoblastic differentiation but with a defect in terminal maturation leading to a reduction in mineralized trabecular bone. Mesenchymal stem cells: In leukemia, MSCs are dysfunctional expressing lower levels of key healthy HSC niche factors such as Scf and Cxcl12 impairing healthy hematopoiesis. LSCs express high levels of the CXCL12 receptor CXCR4 and other adhesion molecules such as CD44 and VLA-4 to usurp the adhesion mechanisms of healthy HSCs. MSC also contribute to LSC survival by the production of microvesicules and via mitochondria transfer, providing energy support. Alteration of the vascular niche: The expression of VEGF in the leukemic niche induces an increase in vascular density and the production of NO by endothelial cells increases vascular leakiness contributing to hypoxia. In leukemia, endosteal blood vessels are more disrupted than the central bone marrow ones. Adipocytes: Leukemic cells support their own metabolism and survival by stimulating lipolysis which fuels fatty acid oxidation in chemotherapy resistant LSCs expressing the fatty acid transporter CD36. Inflammatory niche: Activation of Notch signaling in osteolineage cells leads to the activation of the NF-κB pathway in leukemic cells supporting their survival and proliferation. An autocrine secretion of pro-inflammatory molecules like IL-1 and TNF-α also activates the NF-κB pathway. HSC, hematopoietic cells; SCF, stem cell factor; FA, fatty acid; LSC, leukemic stem cell; MSC, mesenchymal stem cell; MV, microvesicule; NO, nitric oxide; ECM, extracellular matrix.
The bone marrow vascularization is altered in AML with an increased micro-vessel density consequence of the production of pro-angiogenic factors like vascular endothelial growth factor (VEGF) (96–99). AML progression induces the production of nitric oxide (NO) which increases vascular permeability and maintains overall hypoxia (100). Interestingly AML leads to a differential remodeling of vasculature in central and endosteal regions (101). A preferential disruption of the endosteal blood vessels leads to progressive remodeling of the endosteal stroma and the progressive loss of stromal cells. Inhibition of the AML-driven vascular remodeling was shown to improve chemotherapy efficiency in mice (100, 101).
Leukemic cells can reprogram MSCs to create a pro-tumoral niche. MSCs reprogramming can occur following direct cell-to-cell contact, via secreted factors, or via exosomes (102–104). In addition, human MSCs isolated from AML patients (AML-MSC) displayed in-vitro reduced proliferative potential and increased levels of apoptosis (105). Compared to MSCs isolated from healthy donors, AML- MSCs have a lower expression of several niche factors such as SCF, THPO, ANGPT1, VCAM1 and BMI1 (106). In mice, MSCs support AML cells by transferring mitochondria to provide additional energy (107, 108). This transfer is enhanced by some chemotherapies and provides a survival advantage to leukemic blasts and LSCs. This transfer occurs through AML-derived nanotubes. Study in mice showed that superoxide produced by AML cells NADPH oxidase-2 (NOX2) stimulates the nanotubes formation in MSCs. Interestingly, inhibition of NOX2 was able to prevent mitochondrial transfer and improved survival in a xenograft model (107). MSCs also help LSCs to cope with increased reactive oxygen species (ROS) levels, consequence of the mitochondrial transfer by providing increased bioenergetics and detoxifying enzymes (109). Furthermore, MSCs protect AML from chemotherapy through increased Notch and Wnt signaling and inhibition of apoptosis (110–113). Dysregulation of the cytokine profile is suspected to create a pro-tumoral niche in AML (114, 115). LSCs reside in a pro-inflammatory environment known to favor LSCs survival and proliferation. As opposed to normal HSCs and differentiated blasts, LSCs exhibit constitutive NF-κB activity. This activity is partly the consequence of an autocrine tumor necrosis factor-α (TNF-α) secretion, formed by an NF-κB/TNF-α positive feedback loop (116). Activation of Notch signaling also contributes to the activation of the NF-κB pathway (111). Similarly, LSCs aberrantly express the co-receptor for interleukine-1 (IL-1), IL1RAP. Downregulation of IL1RAP inhibits the clonogenic activity of AML cells and leads to increased apoptosis (39). Interestingly, LSCs express IL-1 suggesting another pro-inflammatory autocrine loop. Within the leukemic niche, cytokines can be produced by either immune or leukemic cells. Several cytokines and soluble factors have been shown to affect leukemic cells survival and growth in-vitro (117). While pro-inflammatory cytokines such as IL-1β, GM-CSF, IL-3, TNF-α seem to promote AML cells growth, anti-inflammatory molecules such as IL-1Rα, TGF-β and IL-10 have an inhibitory effect (117–119). The function of a specific cytokine is dependent on multiple complex molecular interactions within the microenvironment. Therefore, despite a major improvement in our understanding over the past decade, further studies are needed to clarify the cytokine network in AML.
Adipocytes are classically considered negative regulators of normal hematopoiesis (120). However, this negative action seems to depend on adipocytes anatomical location. Indeed, adipocytes in the active red bone marrow support blood regeneration and myelo-erythroid maturation (121, 122). In the context of AML, leukemic cells repress bone marrow adipocyte maturation impairing myelo-erythoid differentiation (122). Leukemic cells induce the lipolysis of triglyceride to free fatty acids supporting their proliferation and survival (123). Interestingly, outside the bone marrow, gonadal adipose tissue represents a reservoir for LSCs. Within this adipose tissue, leukemic cells create an inflammatory environment triggering lipolysis and the released of fatty acids that fuel LSCs expressing the fatty acid transporter CD36, contributing to chemo-resistance (124).
The sympathetic nervous system is a critical regulatory component of the bone marrow microenvironment that controls the plasticity of bone marrow stromal cells under homeostatic conditions (78, 79, 125). Aging, a condition associated with myeloid biased hematopoiesis and an increased risk of myelodysplastic syndromes and leukemia is associated with sympathetic neuropathy and decreased β3-adrenergic signaling (82, 83). In a MLL-AF9 mouse model, AML infiltration induced sympathetic neuropathy which further promoted AML (60). This neuropathy was associated with an expansion of Nestin-GFPpos MSCs primed for osteolineage differentiation, and HSC exhaustion. Loss of β2-adrenergic signals directly promotes an expansion of LSCs expressing the β2-adrenergic receptor. Studies using primary AML cells from patients showed that leukemic cells altered adipogenesis in favor of osteolineage differentiation (122, 126). However, sympathetic neuropathy impairs terminal osteoblastic lineage differentiation leading to a reduction in mineralized bone density (60). Sympathetic neuropathy was also induced by the pro-inflammatory environment observed in a JAK2V617F MPN mouse model (127). In this context, Nestin-GFPpos MSCs are reduced, which in turn led to the expansion of altered HSPCs and disease progression.
Similar to their healthy counterpart, LSC localization is dependent on the expression of cytokines and adhesion receptors. Leukemic cells adhere to the bone marrow through three main receptors: CXCR4, Very Late Antigen-4 (VLA-4) and CD44 (128). The high expression of these adhesion molecules facilitates the homing and retention of leukemic cells in the niche impairing chemosensitivity (32, 129–131). In addition, interactions between VLA-4 expressed by leukemic cells and VCAM1 expressed at the surface of BM-MSC mediates chemoresistance via activation of the NF-κB pathway in stromal cells (20).
Leukemic Stem Cells: A Therapeutic Opportunity
LSCs as a Prognostic Marker
Patients with AML are treated according to a risk stratification aiming to identify the patients with low, intermediate, and high risk of relapse based on the disease characteristics at diagnosis (9). Since LSCs have been implicated in treatment resistance and relapse, quantification of the LSC pool could be an additional prognostic factor beside the traditional genetic and molecular abnormalities. As we discussed before, a clear definition of the LSC phenotype does not exist, and different approaches have been used to estimate the LSC pool in patients. Using flow cytometry, Zeijlemaker W. et al. showed that CD34-positive AML blasts were associated with an increased incidence of relapse compared to CD34-negative AML (132). More recently, the prognostic impact of LSC frequency defined by the CD34posCD38neg phenotype combined with minimal residual disease (MRD) evaluation was demonstrated in a prospective study (133). High level of CD34posCD38low/CD123pos blasts at diagnosis is predictive of an adverse outcome (134). Interestingly, a recent study performed in older AML patients showed that this predictive impact is only seen in patients treated by intensive chemotherapy but not by hypomethylating agents (36). Leukemic stem cells frequency seems to be correlated with a lower white blood cell count, an adverse cytogenetic risk, and less frequent NPM1 mutation (36, 135).
Stem cell gene expression signatures have been shown to have a prognostic impact in AML, also highlighting the potential role of leukemia stemness in treatment response (25). Based on this observation, a 17 genes score (LSC17) that compared the gene expression profiles between 138 LSCpos and 89 LSCneg isolated from 78 AML patients was developed (136). A high score is associated with a poor outcome after standard treatment including HSC transplantation (136). The LSC17 was recently challenged by the newly developed AML prognostic score (APS), a 16 gene expression signature score, derived from RNA-sequencing and whole exome sequencing results (137). Interestingly, the authors hypothesized that APS can outperform the LSC17 because of its capacity to capture signal from the microenvironment.
How to Target the Leukemic Stem Cell Niche
Compared with other hematological malignancies, therapeutic progresses have been limited in AML highlighting the need for new strategies. The microenvironment shelters LSCs, protects them from genotoxic drugs and therefore represents a possible cause of treatment failure and relapse. Different strategies have attempted to target the LSC-niche interactions and several studies are currently ongoing (Figure 2). LSCs can also be directly targeted based on their phenotypic and functional differences compared to healthy HSCs. These strategies are beyond the scope of this article and have been reviewed elsewhere (31, 138–140).
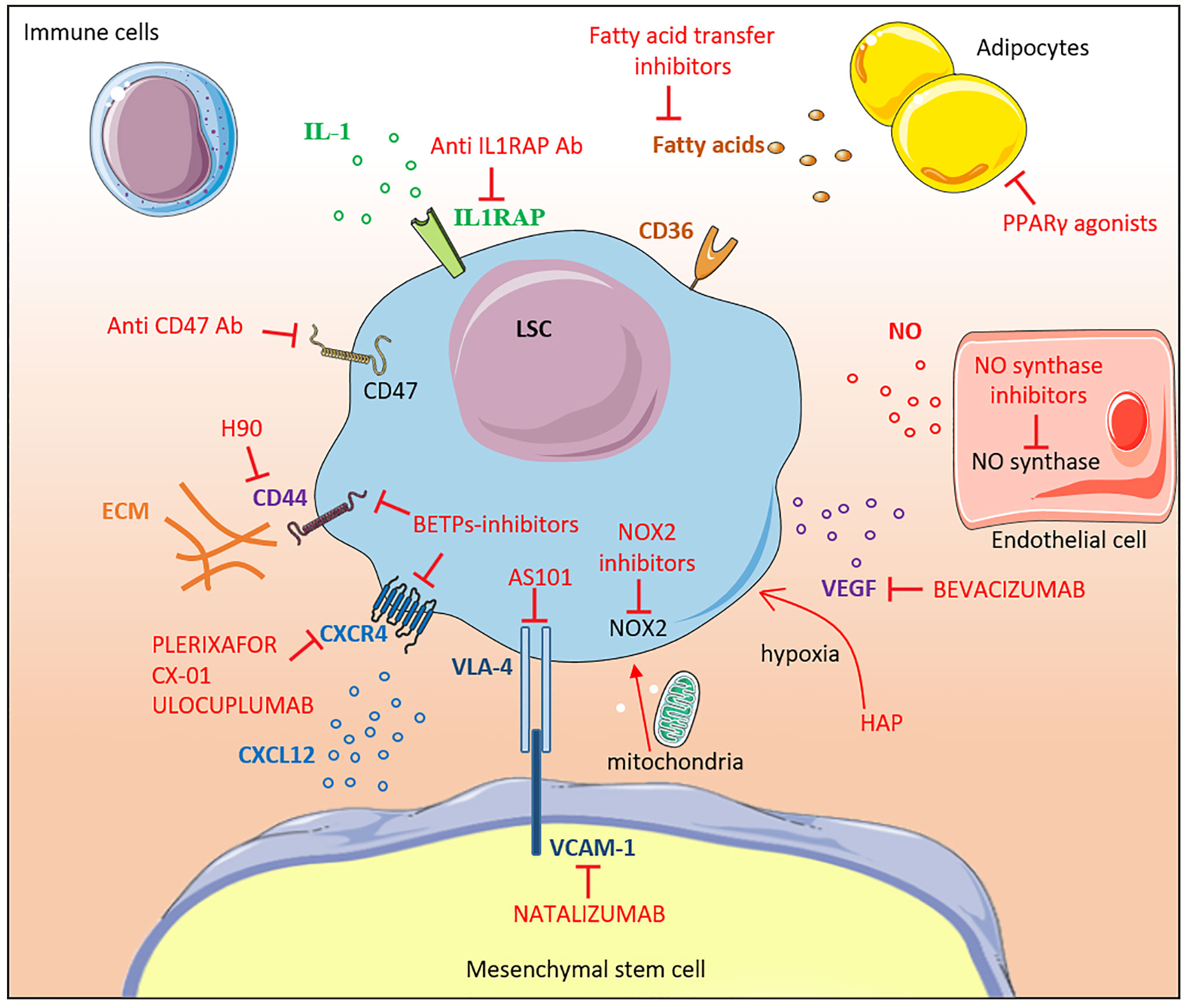
Figure 2 Therapeutic targeting of the leukemic niche. The different molecular interactions between LSCs and the bone marrow niche constituents are shown. Inhibitors are labeled in red. Most of the drugs shown in the figure are under pre-clinical or early clinical development. IL-1, interleukine-1; Ab, antibody; CD, cluster of differentiation, FA, fatty acid; LSC, leukemic stem cell; MSC, mesenchymal stem cell; NOX2, NADPH oxidase 2; NO, nitric oxide; ECM, extracellular matrix; VEGF, vascular endothelial growth factor; HAP, Hypoxia-activated prodrugs; PPARγ, Peroxisome Proliferator-activated Receptor gamma; VCAM-1, Vascular Cell Adhesion Molecule-1; BETPs, Bromodomain Extra-Terminal Protein.
Adhesion Molecules
Adhesion molecules maintain LSCs in the hypoxic niche protecting them from cycling-dependent chemotherapies. Targeting adhesion molecules aims to mobilize LSCs out of their protective niche in order to expose them to chemotherapy. LSCs express the receptor CXCR4 and migrate in response to CXCL12 (141). Moreover, high levels of CXCR4 expression are associated with relapse and poor overall survival in patients (142). Plerixafor, a potent inhibitor of CXCR4, is currently used in association with G-CSF to induce HSCs mobilization (143). In an acute promyelocytic leukemia murine model, treatment with plerixafor in combination with cytarabine and daunorubicine improved chemosensibility and overall survival (144). Since this early study, plerixafor has been tested in phase I-II studies, in combination with various chemotherapies and hypomethylating agents with promising results (145–147). Other CXCR4-CXCL12 axis inhibitors are under clinical development like CX-01, BL-8040 and ulocuplumab. These drugs showed encouraging results in combination with chemotherapy in phase I-II studies (148–151). However, larger phase III studies are needed to confirm the benefit and the exact place of the CXCR4-CXCL12 axis inhibition in AML treatment strategy.
Bromodomain and extra-terminal domain-containing (BET-containing) proteins (BETPs)-inhibitors, can also target adhesion molecules. Sustained degradation of BETPs induced the downregulation of CXCR4 and CD44 expression, decreased the LSC population, and improved overall survival in a patient-derived xenotransplantation model (152). Importantly, BETPs inhibition significantly reduced the number of LSCs when used alone or in combination with chemotherapy. CD44 represents an exciting target since it is differentially expressed between LSCs and normal HSCs (130, 131). Administration of H90, a monoclonal antibody directed to CD44, in immunocompromised mice transplanted with human AML reduced the leukemic burden. Interestingly, H90 seemed to specifically target the LSCs population since no leukemia was observed in serially transplanted mice (32).
Vascularization Remodeling and Hypoxia
VEGF was early identified as a promising target given its pro-angiogenic and anti-apoptotic effects on leukemic cells (153). However, results of clinical studies using bevacizumab, a humanized recombinant monoclonal antibody directed against VEGF have proven disappointing (154, 155). A recent study in mice suggests that inhibition of NO production by endothelial cells could restore the normal vascularization and improve response to cytarabine (100). Targeting NO production by inhibiting the NO synthase could therefore represent a new therapeutic target. The niche represents a hypoxic environment that maintains LSCs in a quiescent state. Moreover, hypoxia inducible factor-1 (HIF-1α) expression induced by hypoxia upregulates CXCR4 expression at the membrane surface of LSCs (19). However, the exact impact of HIF-1α inhibition is still debated (156, 157). Another way to target the hypoxic microenvironment is to use hypoxia-activated prodrugs (HAPs) (158) specifically designed to form cytotoxic agents under hypoxic conditions while limiting the toxicity on normal tissues. Evofosfamide (also known as TH-302) is a 2-nitroimidazole-linked prodrug. In vitro, evofosfamide treatment promotes a dose- and hypoxia-dependent apoptosis and cell death in AML cells. Interestingly, in a xenograft model, evofosfamide reduces LSC pool with limited toxicity on normal hematopoiesis (159, 160). However, a phase I study conducted in 49 patients with advanced leukemia showed disappointing results with an overall response rate of only 6% only (161). Other HAPs are currently under development.
Cytokines and Soluble Factors
Targeting the pro-inflammatory environment represents another interesting strategy considering the importance of cytokines like IL-1, IL-6 and TNFα for LSC survival and proliferation. IL-1 and IL-6 inhibitors are already commercially available for the treatment of autoimmune disease and cytokine released syndromes (162, 163). It would be interesting to test these inhibitors in combination with chemotherapy even if caution is needed regarded the risk of infections. Given the higher expression of IL1RAP at the surface of LSCs compared with normal HSCs, targeting IL1RAP is an attractive option. Indeed, in a preclinical study, targeting IL1RAP using a monoclonal antibody induced selective killing of AML CD34posCD38pos, and CD34posCD38neg cells both in vitro and in a xenograft model (164).
Since leukemic cells trigger lipolysis and use fatty acids as a source of energy, targeting the adipose tissue represents another possible strategy. Studies in mice have shown that restoring normal adipocyte maturation using PPARγ agonists inhibits leukemic growth. Similarly, inhibiting fatty acids transfer to leukemic cells improved survival in a xenograft model (123). However, further studies in human are warranted.
Conclusion
According to the cancer stem cell theory, LSCs sit at the top of the hierarchy and are the source of the more differentiated leukemic blasts. Even if these cells represent an attractive target, eradicating LSCs is highly complex, notably due to the lack of specific markers. AML is associated with a remodeling of the hematopoietic niche where HSCs and LSCs reside, however, modifications of the microenvironment also contribute to leukemia development at the expense of normal hematopoiesis. Since the first description of LSCs more than 25 years ago, our understanding of this small subset of leukemic cells has greatly improved with the identification of potential therapeutic targets paving the way for the development of new treatment strategies in a still deadly disease.
Author Contributions
TM and SP conceptualized and finalized the manuscript. All authors contributed to the article and approved the submitted version.
Funding
TM is supported by the “association pour le développement de l’hématologie oncologie/ADHO”.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors apologize to investigators whose work could not be cited owing to space limitations. The authors thank Anna Di Staulo and Charles Ayemoba for helpful discussions.
References
1. Dohner H, Weisdorf DJ, Bloomfield CD. Acute Myeloid Leukemia. N Engl J Med (2015) 373(12):1136–52. doi: 10.1056/NEJMra1406184
2. Cancer Genome Atlas Research N, Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, et al. Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia. N Engl J Med (2013) 368(22):2059–74. doi: 10.1056/NEJMoa1301689
3. DiNardo CD, Wei AH. How I Treat Acute Myeloid Leukemia in the Era of New Drugs. Blood (2020) 135(2):85–96. doi: 10.1182/blood.2019001239
4. Dohner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Buchner T, et al. Diagnosis and Management of AML in Adults: 2017 ELN Recommendations From an International Expert Panel. Blood (2017) 129(4):424–47. doi: 10.1182/blood-2016-08-733196
5. Burnett A, Wetzler M, Lowenberg B. Therapeutic Advances in Acute Myeloid Leukemia. J Clin Oncol (2011) 29(5):487–94. doi: 10.1200/JCO.2010.30.1820
6. Ganzel C, Sun Z, Cripe LD, Fernandez HF, Douer D, Rowe JM, et al. Very Poor Long-Term Survival in Past and More Recent Studies for Relapsed AML Patients: The ECOG-ACRIN Experience. Am J Hematol (2018) 00:1–8. doi: 10.1002/ajh.25162
7. Marcucci G, Haferlach T, Dohner H. Molecular Genetics of Adult Acute Myeloid Leukemia: Prognostic and Therapeutic Implications. J Clin Oncol (2011) 29(5):475–86. doi: 10.1200/JCO.2010.30.2554
8. Breems DA, Van Putten WL, Huijgens PC, Ossenkoppele GJ, Verhoef GE, Verdonck LF, et al. Prognostic Index for Adult Patients With Acute Myeloid Leukemia in First Relapse. J Clin Oncol (2005) 23(9):1969–78. doi: 10.1200/JCO.2005.06.027
9. Dombret H, Seymour JF, Butrym A, Wierzbowska A, Selleslag D, Jang JH, et al. International Phase 3 Study of Azacitidine vs Conventional Care Regimens in Older Patients With Newly Diagnosed AML With >30% Blasts. Blood (2015) 126(3):291–9. doi: 10.1182/blood-2015-01-621664
10. Song X, Peng Y, Wang X, Chen Y, Jin L, Yang T, et al. Incidence, Survival, and Risk Factors for Adults With Acute Myeloid Leukemia Not Otherwise Specified and Acute Myeloid Leukemia With Recurrent Genetic Abnormalities: Analysis of the Surveillance, Epidemiology, and End Results (SEER) Database, 2001-2013. Acta Haematol (2018) 139(2):115–27. doi: 10.1159/000486228
11. Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A Cell Initiating Human Acute Myeloid Leukaemia After Transplantation Into SCID Mice. Nature (1994) 367(6464):645–8. doi: 10.1038/367645a0
12. Farge T, Saland E, De Toni F, Aroua N, Hosseini M, Perry R, et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells But Require Oxidative Metabolism. Cancer Discovery (2017) 7(7):716–35. doi: 10.1158/2159-8290.CD-16-0441
13. Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, et al. BCL-2 Inhibition Targets Oxidative Phosphorylation and Selectively Eradicates Quiescent Human Leukemia Stem Cells. Cell Stem Cell (2013) 12(3):329–41. doi: 10.1016/j.stem.2012.12.013
14. Ma J, Liu B, Yu D, Zuo Y, Cai R, Yang J, et al. SIRT3 Deacetylase Activity Confers Chemoresistance in AML via Regulation of Mitochondrial Oxidative Phosphorylation. Br J Haematol (2019) 187(1):49–64. doi: 10.1111/bjh.16044
15. Shih A, Jiang Y, Meydan C, Shank K, Pandey S, Barreyro L, et al. Mutational Cooperativity Linked to Combinatorial Epigenetic Gain of Function in Acute Myeloid Leukemia. Cancer Cell (2015) 27(4):502–15. doi: 10.1016/j.ccell.2015.03.009
16. Jung N, Dai B, Gentles AJ, Majeti R, Feinberg AP. An LSC Epigenetic Signature is Largely Mutation Independent and Implicates the HOXA Cluster in AML Pathogenesis. Nat Commun (2015) 6(1):8489. doi: 10.1038/ncomms9489
17. Schofield R. The Relationship Between the Spleen Colony-Forming Cell and the Haemopoietic Stem Cell. Blood Cells (1978) 4(1-2):7–25.
18. Pinho S, Frenette PS. Haematopoietic Stem Cell Activity and Interactions With the Niche. Nat Rev Mol Cell Biol (2019) 20(5):303–20. doi: 10.1038/s41580-019-0103-9
19. Ishikawa F, Yoshida S, Saito Y, Hijikata A, Kitamura H, Tanaka S, et al. Chemotherapy-Resistant Human AML Stem Cells Home to and Engraft Within the Bone-Marrow Endosteal Region. Nat Biotechnol (2007) 25(11):1315–21. doi: 10.1038/nbt1350
20. Jacamo R, Chen Y, Wang Z, Ma W, Zhang M, Spaeth EL, et al. Reciprocal Leukemia-Stroma VCAM-1/VLA-4-Dependent Activation of NF-κb Mediates Chemoresistance. Blood (2014) 123(17):2691–702. doi: 10.1182/blood-2013-06-511527
21. Schelker RC, Iberl S, Müller G, Hart C, Herr W, Grassinger J. TGF-β1 and CXCL12 Modulate Proliferation and Chemotherapy Sensitivity of Acute Myeloid Leukemia Cells Co-Cultured With Multipotent Mesenchymal Stromal Cells. Hematology (2018) 23(6):337–45. doi: 10.1080/10245332.2017.1402455
22. Mendez-Ferrer S, Bonnet D, Steensma DP, Hasserjian RP, Ghobrial IM, Gribben JG, et al. Bone Marrow Niches in Haematological Malignancies. Nat Rev Cancer (2020) 20(5):285–98. doi: 10.1038/s41568-020-0245-2
23. Bonnet D, Dick JE. Human Acute Myeloid Leukemia is Organized as a Hierarchy That Originates From a Primitive Hematopoietic Cell. Nat Med (1997) 3(7):730–7. doi: 10.1038/nm0797-730
24. Goardon N, Marchi E, Atzberger A, Quek L, Schuh A, Soneji S, et al. Coexistence of LMPP-Like and GMP-Like Leukemia Stem Cells in Acute Myeloid Leukemia. Cancer Cell (2011) 19(1):138–52. doi: 10.1016/j.ccr.2010.12.012
25. Eppert K, Takenaka K, Lechman ER, Waldron L, Nilsson B, van Galen P, et al. Stem Cell Gene Expression Programs Influence Clinical Outcome in Human Leukemia. Nat Med (2011) 17(9):1086–93. doi: 10.1038/nm.2415
26. Jan M, Snyder TM, Corces-Zimmerman MR, Vyas P, Weissman IL, Quake SR, et al. Clonal Evolution of Preleukemic Hematopoietic Stem Cells Precedes Human Acute Myeloid Leukemia. Sci Transl Med (2012) 4(149):149ra18. doi: 10.1126/scitranslmed.3004315
27. Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V, et al. Identification of Pre-Leukaemic Haematopoietic Stem Cells in Acute Leukaemia. Nature (2014) 506(7488):328–33. doi: 10.1038/nature13038
28. Corces-Zimmerman MR, Hong WJ, Weissman IL, Medeiros BC, Majeti R. Preleukemic Mutations in Human Acute Myeloid Leukemia Affect Epigenetic Regulators and Persist in Remission. Proc Natl Acad Sci U.S.A. (2014) 111(7):2548–53. doi: 10.1073/pnas.1324297111
29. Jaiswal S, Ebert BL. Clonal Hematopoiesis in Human Aging and Disease. Sci (2019) 366(6465):eaan4673. doi: 10.1126/science.aan4673
30. McKenzie MD, Ghisi M, Oxley EP, Ngo S, Cimmino L, Esnault C, et al. Interconversion Between Tumorigenic and Differentiated States in Acute Myeloid Leukemia. Cell Stem Cell (2019) 25(2):258–72 e9. doi: 10.1016/j.stem.2019.07.001
31. Saito Y, Kitamura H, Hijikata A, Tomizawa-Murasawa M, Tanaka S, Takagi S, et al. Identification of Therapeutic Targets for Quiescent, Chemotherapy-Resistant Human Leukemia Stem Cells. Sci Transl Med (2010) 2(17):17ra9. doi: 10.1126/scitranslmed.3000349
32. Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 Eradicates Human Acute Myeloid Leukemic Stem Cells. Nat Med (2006) 12(10):1167–74. doi: 10.1038/nm1483
33. Hosen N, Park CY, Tatsumi N, Oji Y, Sugiyama H, Gramatzki M, et al. CD96 is a Leukemic Stem Cell-Specific Marker in Human Acute Myeloid Leukemia. Proc Natl Acad Sci U.S.A. (2007) 104(26):11008–13. doi: 10.1073/pnas.0704271104
34. Jordan CT, Upchurch D, Szilvassy SJ, Guzman ML, Howard DS, Pettigrew AL, et al. The Interleukin-3 Receptor Alpha Chain is a Unique Marker for Human Acute Myelogenous Leukemia Stem Cells. Leukemia (2000) 14(10):1777–84. doi: 10.1038/sj.leu.2401903
35. Jin L, Lee EM, Ramshaw HS, Busfield SJ, Peoppl AG, Wilkinson L, et al. Monoclonal Antibody-Mediated Targeting of CD123, IL-3 Receptor Alpha Chain, Eliminates Human Acute Myeloid Leukemic Stem Cells. Cell Stem Cell (2009) 5(1):31–42. doi: 10.1016/j.stem.2009.04.018
36. Vergez F, Nicolau-Travers ML, Bertoli S, Rieu JB, Tavitian S, Bories P, et al. CD34(+)CD38(-)CD123(+) Leukemic Stem Cell Frequency Predicts Outcome in Older Acute Myeloid Leukemia Patients Treated by Intensive Chemotherapy But Not Hypomethylating Agents. Cancers (Basel) (2020) 12(5):1174. doi: 10.3390/cancers12051174
37. Pabst C, Bergeron A, Lavallee VP, Yeh J, Gendron P, Norddahl GL, et al. GPR56 Identifies Primary Human Acute Myeloid Leukemia Cells With High Repopulating Potential In Vivo. Blood (2016) 127(16):2018–27. doi: 10.1182/blood-2015-11-683649
38. van Rhenen A, van Dongen GA, Kelder A, Rombouts EJ, Feller N, Moshaver B, et al. The Novel AML Stem Cell Associated Antigen CLL-1 Aids in Discrimination Between Normal and Leukemic Stem Cells. Blood (2007) 110(7):2659–66. doi: 10.1182/blood-2007-03-083048
39. Barreyro L, Will B, Bartholdy B, Zhou L, Todorova TI, Stanley RF, et al. Overexpression of IL-1 Receptor Accessory Protein in Stem and Progenitor Cells and Outcome Correlation in AML and MDS. Blood (2012) 120(6):1290–8. doi: 10.1182/blood-2012-01-404699
40. Mitchell K, Barreyro L, Todorova TI, Taylor SJ, Antony-Debré I, Narayanagari SR, et al. IL1RAP Potentiates Multiple Oncogenic Signaling Pathways in AML. J Exp Med (2018) 215(6):1709–27. doi: 10.1084/jem.20180147
41. Qiu S, Jia Y, Xing H, Yu T, Yu J, Yu P, et al. N-Cadherin and Tie2 Positive CD34(+)CD38(-)CD123(+) Leukemic Stem Cell Populations can Develop Acute Myeloid Leukemia More Effectively in NOD/SCID Mice. Leuk Res (2014) 38(5):632–7. doi: 10.1016/j.leukres.2014.03.007
42. Baccin C, Al-Sabah J, Velten L, Helbling PM, Grünschläger F, Hernández-Malmierca P, et al. Combined Single-Cell and Spatial Transcriptomics Reveal the Molecular, Cellular and Spatial Bone Marrow Niche Organization. Nat Cell Biol (2020) 22(1):38–48. doi: 10.1038/s41556-019-0439-6
43. Wolock SL, Krishnan I, Tenen DE, Matkins V, Camacho V, Patel S, et al. Mapping Distinct Bone Marrow Niche Populations and Their Differentiation Paths. Cell Rep (2019) 28(2):302–11.e5. doi: 10.1016/j.celrep.2019.06.031
44. Tikhonova AN, Dolgalev I, Hu H, Sivaraj KK, Hoxha E, Cuesta-Dominguez A, et al. The Bone Marrow Microenvironment at Single-Cell Resolution. Nature (2019) 569(7755):222–8. doi: 10.1038/s41586-019-1104-8
45. Baryawno N, Przybylski D, Kowalczyk MS, Kfoury Y, Severe N, Gustafsson K, et al. A Cellular Taxonomy of the Bone Marrow Stroma in Homeostasis and Leukemia. Cell (2019) 177(7):1915–32 e16. doi: 10.1016/j.cell.2019.04.040
46. Severe N, Karabacak NM, Gustafsson K, Baryawno N, Courties G, Kfoury Y, et al. Stress-Induced Changes in Bone Marrow Stromal Cell Populations Revealed Through Single-Cell Protein Expression Mapping. Cell Stem Cell (2019) 25(4):570–83 e7. doi: 10.1016/j.stem.2019.06.003
47. Ellis SL, Grassinger J, Jones A, Borg J, Camenisch T, Haylock D, et al. The Relationship Between Bone, Hemopoietic Stem Cells, and Vasculature. Blood (2011) 118(6):1516–24. doi: 10.1182/blood-2010-08-303800
48. Lord BI, Testa NG, Hendry JH. The Relative Spatial Distributions of CFUs and CFUc in the Normal Mouse Femur. Blood (1975) 46(1):65–72. doi: 10.1182/blood.V46.1.65.65
49. Gong JK. Endosteal Marrow: A Rich Source of Hematopoietic Stem Cells. Science (1978) 199(4336):1443–5. doi: 10.1126/science.75570
50. Taichman RS, Reilly MJ, Emerson SG. Human Osteoblasts Support Human Hematopoietic Progenitor Cells In Vitro Bone Marrow Cultures. Blood (1996) 87(2):518–24. doi: 10.1182/blood.V87.2.518.bloodjournal872518
51. Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, et al. Osteoblastic Cells Regulate the Haematopoietic Stem Cell Niche. Nature (2003) 425(6960):841–6. doi: 10.1038/nature02040
52. Zhang J, Niu C, Ye L, Huang H, He X, Tong WG, et al. Identification of the Haematopoietic Stem Cell Niche and Control of the Niche Size. Nature (2003) 425(6960):836–41. doi: 10.1038/nature02041
53. Greenbaum A, Hsu YM, Day RB, Schuettpelz LG, Christopher MJ, Borgerding JN, et al. CXCL12 in Early Mesenchymal Progenitors is Required for Haematopoietic Stem-Cell Maintenance. Nature (2013) 495(7440):227–30. doi: 10.1038/nature11926
54. Ding L, Morrison SJ. Haematopoietic Stem Cells and Early Lymphoid Progenitors Occupy Distinct Bone Marrow Niches. Nature (2013) 495(7440):231–5. doi: 10.1038/nature11885
55. Ding L, Saunders TL, Enikolopov G, Morrison SJ. Endothelial and Perivascular Cells Maintain Haematopoietic Stem Cells. Nature (2012) 481(7382):457–62. doi: 10.1038/nature10783
56. Kunisaki Y, Bruns I, Scheiermann C, Ahmed J, Pinho S, Zhang D, et al. Arteriolar Niches Maintain Haematopoietic Stem Cell Quiescence. Nature (2013) 502(7473):637–43. doi: 10.1038/nature12612
57. Nombela-Arrieta C, Pivarnik G, Winkel B, Canty KJ, Harley B, Mahoney JE, et al. Quantitative Imaging of Haematopoietic Stem and Progenitor Cell Localization and Hypoxic Status in the Bone Marrow Microenvironment. Nat Cell Biol (2013) 15(5):533–43. doi: 10.1038/ncb2730
58. Zhu J, Garrett R, Jung Y, Zhang Y, Kim N, Wang J, et al. Osteoblasts Support B-Lymphocyte Commitment and Differentiation From Hematopoietic Stem Cells. Blood (2007) 109(9):3706–12. doi: 10.1182/blood-2006-08-041384
59. Raaijmakers MH, Mukherjee S, Guo S, Zhang S, Kobayashi T, Schoonmaker JA, et al. Bone Progenitor Dysfunction Induces Myelodysplasia and Secondary Leukaemia. Nature (2010) 464(7290):852–7. doi: 10.1038/nature08851
60. Hanoun M, Zhang D, Mizoguchi T, Pinho S, Pierce H, Kunisaki Y, et al. Acute Myelogenous Leukemia-Induced Sympathetic Neuropathy Promotes Malignancy in an Altered Hematopoietic Stem Cell Niche. Cell Stem Cell (2014) 15(3):365–75. doi: 10.1016/j.stem.2014.06.020
61. Asada N, Katayama Y. Regulation of Hematopoiesis in Endosteal Microenvironments. Int J Hematol (2014) 99(6):679–84. doi: 10.1007/s12185-014-1583-1
62. Kiel MJ, Yilmaz ÖH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM Family Receptors Distinguish Hematopoietic Stem and Progenitor Cells and Reveal Endothelial Niches for Stem Cells. Cell (2005) 121(7):1109–21. doi: 10.1016/j.cell.2005.05.026
63. Frenette PS, Pinho S, Lucas D, Scheiermann C. Mesenchymal Stem Cell: Keystone of the Hematopoietic Stem Cell Niche and a Stepping-Stone for Regenerative Medicine. Annu Rev Immunol (2013) 31:285–316. doi: 10.1146/annurev-immunol-032712-095919
64. Zhou BO, Yue R, Murphy MM, Peyer JG, Morrison SJ. Leptin-Receptor-Expressing Mesenchymal Stromal Cells Represent the Main Source of Bone Formed by Adult Bone Marrow. Cell Stem Cell (2014) 15(2):154–68. doi: 10.1016/j.stem.2014.06.008
65. Mendez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, et al. Mesenchymal and Haematopoietic Stem Cells Form a Unique Bone Marrow Niche. Nature (2010) 466(7308):829–34. doi: 10.1038/nature09262
66. Mizoguchi T, Pinho S, Ahmed J, Kunisaki Y, Hanoun M, Mendelson A, et al. Osterix Marks Distinct Waves of Primitive and Definitive Stromal Progenitors During Bone Marrow Development. Dev Cell (2014) 29(3):340–9. doi: 10.1016/j.devcel.2014.03.013
67. Asada N, Kunisaki Y, Pierce H, Wang Z, Fernandez NF, Birbrair A, et al. Differential Cytokine Contributions of Perivascular Haematopoietic Stem Cell Niches. Nat Cell Biol (2017) 19(3):214–23. doi: 10.1038/ncb3475
68. Pinho S, Lacombe J, Hanoun M, Mizoguchi T, Bruns I, Kunisaki Y, et al. PDGFRalpha and CD51 Mark Human Nestin+ Sphere-Forming Mesenchymal Stem Cells Capable of Hematopoietic Progenitor Cell Expansion. J Exp Med (2013) 210(7):1351–67. doi: 10.1084/jem.20122252
69. Crippa S, Bernardo ME. Mesenchymal Stromal Cells: Role in the BM Niche and in the Support of Hematopoietic Stem Cell Transplantation. Hemasphere (2018) 2(6):e151. doi: 10.1097/HS9.0000000000000151
70. Omatsu Y, Sugiyama T, Kohara H, Kondoh G, Fujii N, Kohno K, et al. The Essential Functions of Adipo-Osteogenic Progenitors as the Hematopoietic Stem and Progenitor Cell Niche. Immunity (2010) 33(3):387–99. doi: 10.1016/j.immuni.2010.08.017
71. Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the Hematopoietic Stem Cell Pool by CXCL12-CXCR4 Chemokine Signaling in Bone Marrow Stromal Cell Niches. Immunity (2006) 25(6):977–88. doi: 10.1016/j.immuni.2006.10.016
72. Kusumbe AP, Ramasamy SK, Adams RH. Coupling of Angiogenesis and Osteogenesis by a Specific Vessel Subtype in Bone. Nature (2014) 507(7492):323–8. doi: 10.1038/nature13145
73. Poulos MG, Guo P, Kofler NM, Pinho S, Gutkin MC, Tikhonova A, et al. Endothelial Jagged-1 is Necessary for Homeostatic and Regenerative Hematopoiesis. Cell Rep (2013) 4(5):1022–34. doi: 10.1016/j.celrep.2013.07.048
74. Xu C, Gao X, Wei Q, Nakahara F, Zimmerman SE, Mar J, et al. Stem Cell Factor is Selectively Secreted by Arterial Endothelial Cells in Bone Marrow. Nat Commun (2018) 9(1):2449. doi: 10.1038/s41467-018-04726-3
75. Maryanovich M, Takeishi S, Frenette PS. Neural Regulation of Bone and Bone Marrow. Cold Spring Harb Perspect Med (2018) 8(9). doi: 10.1101/cshperspect.a031344
76. Mach DB, Rogers SD, Sabino MC, Luger NM, Schwei MJ, Pomonis JD, et al. Origins of Skeletal Pain: Sensory and Sympathetic Innervation of the Mouse Femur. Neuroscience (2002) 113(1):155–66. doi: 10.1016/S0306-4522(02)00165-3
77. Bellinger DL, Lorton D, Felten SY, Felten DL. Innervation of Lymphoid Organs and Implications in Development, Aging, and Autoimmunity. Int J Immunopharmacol (1992) 14(3):329–44. doi: 10.1016/0192-0561(92)90162-E
78. Katayama Y, Battista M, Kao WM, Hidalgo A, Peired AJ, Thomas SA, et al. Signals From the Sympathetic Nervous System Regulate Hematopoietic Stem Cell Egress From Bone Marrow. Cell (2006) 124(2):407–21. doi: 10.1016/j.cell.2005.10.041
79. Mendez-Ferrer S, Lucas D, Battista M, Frenette PS. Haematopoietic Stem Cell Release is Regulated by Circadian Oscillations. Nature (2008) 452(7186):442–7. doi: 10.1038/nature06685
80. Gao X, Zhang D, Xu C, Li H, Caron KM, Frenette PS. Nociceptive Nerves Regulate Haematopoietic Stem Cell Mobilization. Nature (2021) 589(7843):591–6. doi: 10.1038/s41586-020-03057-y
81. Lucas D, Scheiermann C, Chow A, Kunisaki Y, Bruns I, Barrick C, et al. Chemotherapy-Induced Bone Marrow Nerve Injury Impairs Hematopoietic Regeneration. Nat Med (2013) 19(6):695–703. doi: 10.1038/nm.3155
82. Maryanovich M, Zahalka AH, Pierce H, Pinho S, Nakahara F, Asada N, et al. Adrenergic Nerve Degeneration in Bone Marrow Drives Aging of the Hematopoietic Stem Cell Niche. Nat Med (2018) 24(6):782–91. doi: 10.1038/s41591-018-0030-x
83. Ho YH, Del Toro R, Rivera-Torres J, Rak J, Korn C, Garcia-Garcia A, et al. Remodeling of Bone Marrow Hematopoietic Stem Cell Niches Promotes Myeloid Cell Expansion During Premature or Physiological Aging. Cell Stem Cell (2019) 25(3):407–18 e6. doi: 10.1016/j.stem.2019.06.007
84. Yamazaki S, Ema H, Karlsson G, Yamaguchi T, Miyoshi H, Shioda S, et al. Nonmyelinating Schwann Cells Maintain Hematopoietic Stem Cell Hibernation in the Bone Marrow Niche. Cell (2011) 147(5):1146–58. doi: 10.1016/j.cell.2011.09.053
85. Fialkow PJ, Thomas ED, Bryant JI, Neiman PE. Leukaemic Transformation of Engrafted Human Marrow Cells In Vivo. Lancet (1971) 1(7693):251–5. doi: 10.1016/S0140-6736(71)90998-6
86. Paget S. The Distribution of Secondary Growths in Cancer of the Breast. 1889. Cancer Metastasis Rev (1989) 8(2):98–101.
87. Rupec RA, Jundt F, Rebholz B, Eckelt B, Weindl G, Herzinger T, et al. Stroma-Mediated Dysregulation of Myelopoiesis in Mice Lacking I Kappa B Alpha. Immunity (2005) 22(4):479–91. doi: 10.1016/j.immuni.2005.02.009
88. Walkley CR, Olsen GH, Dworkin S, Fabb SA, Swann J, McArthur GA, et al. A Microenvironment-Induced Myeloproliferative Syndrome Caused by Retinoic Acid Receptor Gamma Deficiency. Cell (2007) 129(6):1097–110. doi: 10.1016/j.cell.2007.05.014
89. Walkley CR, Shea JM, Sims NA, Purton LE, Orkin SH. Rb Regulates Interactions Between Hematopoietic Stem Cells and Their Bone Marrow Microenvironment. Cell (2007) 129(6):1081–95. doi: 10.1016/j.cell.2007.03.055
90. Zambetti NA, Ping Z, Chen S, Kenswil KJG, Mylona MA, Sanders MA, et al. Mesenchymal Inflammation Drives Genotoxic Stress in Hematopoietic Stem Cells and Predicts Disease Evolution in Human Pre-Leukemia. Cell Stem Cell (2016) 19(5):613–27. doi: 10.1016/j.stem.2016.08.021
91. Dong L, Yu WM, Zheng H, Loh ML, Bunting ST, Pauly M, et al. Leukaemogenic Effects of Ptpn11 Activating Mutations in the Stem Cell Microenvironment. Nature (2016) 539(7628):304–8. doi: 10.1038/nature20131
92. Kode A, Manavalan JS, Mosialou I, Bhagat G, Rathinam CV, Luo N, et al. Leukaemogenesis Induced by an Activating Beta-Catenin Mutation in Osteoblasts. Nature (2014) 506(7487):240–4. doi: 10.1038/nature12883
93. Kim YW, Koo BK, Jeong HW, Yoon MJ, Song R, Shin J, et al. Defective Notch Activation in Microenvironment Leads to Myeloproliferative Disease. Blood (2008) 112(12):4628–38. doi: 10.1182/blood-2008-03-148999
94. Wang L, Zhang H, Rodriguez S, Cao L, Parish J, Mumaw C, et al. Notch-Dependent Repression of miR-155 in the Bone Marrow Niche Regulates Hematopoiesis in an NF-kappaB-Dependent Manner. Cell Stem Cell (2014) 15(1):51–65. doi: 10.1016/j.stem.2014.04.021
95. Boyd AL, Campbell CJ, Hopkins CI, Fiebig-Comyn A, Russell J, Ulemek J, et al. Niche Displacement of Human Leukemic Stem Cells Uniquely Allows Their Competitive Replacement With Healthy HSPCs. J Exp Med (2014) 211(10):1925–35. doi: 10.1084/jem.20140131
96. Chand R, Chandra H, Chandra S, Verma SK. Role of Microvessel Density and Vascular Endothelial Growth Factor in Angiogenesis of Hematological Malignancies. Bone Marrow Res (2016) 2016:5043483. doi: 10.1155/2016/5043483
97. Hussong JW, Rodgers GM, Shami PJ. Evidence of Increased Angiogenesis in Patients With Acute Myeloid Leukemia. Blood (2000) 95(1):309–13. doi: 10.1182/blood.V95.1.309.001k17_309_313
98. Kampen KR, Ter Elst A, de Bont ES. Vascular Endothelial Growth Factor Signaling in Acute Myeloid Leukemia. Cell Mol Life Sci (2013) 70(8):1307–17. doi: 10.1007/s00018-012-1085-3
99. Poulos MG, Gars EJ, Gutkin MC, Kloss CC, Ginsberg M, Scandura JM, et al. Activation of the Vascular Niche Supports Leukemic Progression and Resistance to Chemotherapy. Exp Hematol (2014) 42(11):976–86.e3. doi: 10.1016/j.exphem.2014.08.003
100. Passaro D, Di Tullio A, Abarrategi A, Rouault-Pierre K, Foster K, Ariza-McNaughton L, et al. Increased Vascular Permeability in the Bone Marrow Microenvironment Contributes to Disease Progression and Drug Response in Acute Myeloid Leukemia. Cancer Cell (2017) 32(3):324–41 e6. doi: 10.1016/j.ccell.2017.08.001
101. Duarte D, Hawkins ED, Akinduro O, Ang H, De Filippo K, Kong IY, et al. Inhibition of Endosteal Vascular Niche Remodeling Rescues Hematopoietic Stem Cell Loss in AML. Cell Stem Cell (2018) 22(1):64–77 e6. doi: 10.1016/j.stem.2017.11.006
102. Borella G, Da Ros A, Borile G, Porcu E, Tregnago C, Benetton M, et al. Targeting Mesenchymal Stromal Cells Plasticity to Reroute Acute Myeloid Leukemia Course of Acute Myeloid Leukemia. Blood (2021) 138(7):557–70. doi: 10.1182/blood.2020009845
103. Kumar B, Garcia M, Weng L, Jung X, Murakami JL, Hu X, et al. Acute Myeloid Leukemia Transforms the Bone Marrow Niche Into a Leukemia-Permissive Microenvironment Through Exosome Secretion. Leukemia (2018) 32(3):575–87. doi: 10.1038/leu.2017.259
104. Waclawiczek A, Hamilton A, Rouault-Pierre K, Abarrategi A, Albornoz MG, Miraki-Moud F, et al. Mesenchymal Niche Remodeling Impairs Hematopoiesis via Stanniocalcin 1 in Acute Myeloid Leukemia. J Clin Invest (2020) 130(6):3038–50. doi: 10.1172/JCI133187
105. Corradi G, Baldazzi C, Ocadlikova D, Marconi G, Parisi S, Testoni N, et al. Mesenchymal Stromal Cells From Myelodysplastic and Acute Myeloid Leukemia Patients Display In Vitro Reduced Proliferative Potential and Similar Capacity to Support Leukemia Cell Survival. Stem Cell Res Ther (2018) 9(1):271. doi: 10.1186/s13287-018-1013-z
106. Desbourdes L, Javary J, Charbonnier T, Ishac N, Bourgeais J, Iltis A, et al. Alteration Analysis of Bone Marrow Mesenchymal Stromal Cells From De Novo Acute Myeloid Leukemia Patients at Diagnosis. Stem Cells Dev (2017) 26(10):709–22. doi: 10.1089/scd.2016.0295
107. Marlein CR, Zaitseva L, Piddock RE, Robinson SD, Edwards DR, Shafat MS, et al. NADPH Oxidase-2 Derived Superoxide Drives Mitochondrial Transfer From Bone Marrow Stromal Cells to Leukemic Blasts. Blood (2017) 130(14):1649–60. doi: 10.1182/blood-2017-03-772939
108. Moschoi R, Imbert V, Nebout M, Chiche J, Mary D, Prebet T, et al. Protective Mitochondrial Transfer From Bone Marrow Stromal Cells to Acute Myeloid Leukemic Cells During Chemotherapy. Blood (2016) 128(2):253–64. doi: 10.1182/blood-2015-07-655860
109. Forte D, Garcia-Fernandez M, Sanchez-Aguilera A, Stavropoulou V, Fielding C, Martin-Perez D, et al. Bone Marrow Mesenchymal Stem Cells Support Acute Myeloid Leukemia Bioenergetics and Enhance Antioxidant Defense and Escape From Chemotherapy. Cell Metab (2020) 32(5):829–43 e9. doi: 10.1016/j.cmet.2020.09.001
110. Lane SW, Wang YJ, Lo Celso C, Ragu C, Bullinger L, Sykes SM, et al. Differential Niche and Wnt Requirements During Acute Myeloid Leukemia Progression. Blood (2011) 118(10):2849–56. doi: 10.1182/blood-2011-03-345165
111. Takam Kamga P, Bassi G, Cassaro A, Midolo M, Di Trapani M, Gatti A, et al. Notch Signalling Drives Bone Marrow Stromal Cell-Mediated Chemoresistance in Acute Myeloid Leukemia. Oncotarget (2016) 7(16):21713–27. doi: 10.18632/oncotarget.7964
112. Carter BZ, Mak PY, Chen Y, Mak DH, Mu H, Jacamo R, et al. Anti-Apoptotic ARC Protein Confers Chemoresistance by Controlling Leukemia-Microenvironment Interactions Through a Nfκb/Il1β Signaling Network. Oncotarget (2016) 7(15):20054–67. doi: 10.18632/oncotarget.7911
113. Carter BZ, Mak PY, Wang X, Tao W, Ruvolo V, Mak D, et al. An ARC-Regulated Il1β/Cox-2/Pge2/β-Catenin/ARC Circuit Controls Leukemia-Microenvironment Interactions and Confers Drug Resistance in AML. Cancer Res (2019) 79(6):1165–77. doi: 10.1158/0008-5472.CAN-18-0921
114. Karimdadi Sariani O, Eghbalpour S, Kazemi E, Rafiei Buzhani K, Zaker F. Pathogenic and Therapeutic Roles of Cytokines in Acute Myeloid Leukemia. Cytokine (2021) 142:155508. doi: 10.1016/j.cyto.2021.155508
115. Binder S, Luciano M, Horejs-Hoeck J. The Cytokine Network in Acute Myeloid Leukemia (AML): A Focus on Pro- and Anti-Inflammatory Mediators. Cytokine Growth Factor Rev (2018) 43:8–15. doi: 10.1016/j.cytogfr.2018.08.004
116. Kagoya Y, Yoshimi A, Kataoka K, Nakagawa M, Kumano K, Arai S, et al. Positive Feedback Between NF-κb and TNF-α Promotes Leukemia-Initiating Cell Capacity. J Clin Invest (2014) 124(2):528–42. doi: 10.1172/JCI68101
117. Carey A, Edwards DK, Eide CA, Newell L, Traer E, Medeiros BC, et al. Identification of Interleukin-1 by Functional Screening as a Key Mediator of Cellular Expansion and Disease Progression in Acute Myeloid Leukemia. Cell Rep (2017) 18(13):3204–18. doi: 10.1016/j.celrep.2017.03.018
118. Westermann F, Kube D, Haier B, Bohlen H, Engert A, Zuehlsdorf M, et al. Interleukin 10 Inhibits Cytokine Production of Human AML Cells. Ann Oncol (1996) 7(4):397–404. doi: 10.1093/oxfordjournals.annonc.a010607
119. Wu Y, Su M, Zhang S, Cheng Y, Liao XY, Lin BY, et al. Abnormal Expression of TGF-Beta Type II Receptor Isoforms Contributes to Acute Myeloid Leukemia. Oncotarget (2017) 8(6):10037–49. doi: 10.18632/oncotarget.14325
120. Naveiras O, Nardi V, Wenzel PL, Hauschka PV, Fahey F, Daley GQ. Bone-Marrow Adipocytes as Negative Regulators of the Haematopoietic Microenvironment. Nature (2009) 460(7252):259–63. doi: 10.1038/nature08099
121. Zhou BO, Yu H, Yue R, Zhao Z, Rios JJ, Naveiras O, et al. Bone Marrow Adipocytes Promote the Regeneration of Stem Cells and Haematopoiesis by Secreting SCF. Nat Cell Biol (2017) 19(8):891–903. doi: 10.1038/ncb3570
122. Boyd AL, Reid JC, Salci KR, Aslostovar L, Benoit YD, Shapovalova Z, et al. Acute Myeloid Leukaemia Disrupts Endogenous Myelo-Erythropoiesis by Compromising the Adipocyte Bone Marrow Niche. Nat Cell Biol (2017) 19(11):1336–47. doi: 10.1038/ncb3625
123. Shafat MS, Oellerich T, Mohr S, Robinson SD, Edwards DR, Marlein CR, et al. Leukemic Blasts Program Bone Marrow Adipocytes to Generate a Protumoral Microenvironment. Blood (2017) 129(10):1320–32. doi: 10.1182/blood-2016-08-734798
124. Ye H, Adane B, Khan N, Sullivan T, Minhajuddin M, Gasparetto M, et al. Leukemic Stem Cells Evade Chemotherapy by Metabolic Adaptation to an Adipose Tissue Niche. Cell Stem Cell (2016) 19(1):23–37. doi: 10.1016/j.stem.2016.06.001
125. Hanoun M, Maryanovich M, Arnal-Estape A, Frenette PS. Neural Regulation of Hematopoiesis, Inflammation, and Cancer. Neuron (2015) 86(2):360–73. doi: 10.1016/j.neuron.2015.01.026
126. Battula VL, Le PM, Sun JC, Nguyen K, Yuan B, Zhou X, et al. AML-Induced Osteogenic Differentiation in Mesenchymal Stromal Cells Supports Leukemia Growth. JCI Insight (2017) 2(13). doi: 10.1172/jci.insight.90036
127. Arranz L, Sanchez-Aguilera A, Martin-Perez D, Isern J, Langa X, Tzankov A, et al. Neuropathy of Haematopoietic Stem Cell Niche is Essential for Myeloproliferative Neoplasms. Nature (2014) 512(7512):78–81. doi: 10.1038/nature13383
128. Becker PS. Dependence of Acute Myeloid Leukemia on Adhesion Within the Bone Marrow Microenvironment. Sci World J (2012) 2012:856467. doi: 10.1100/2012/856467
129. Matsunaga T, Takemoto N, Sato T, Takimoto R, Tanaka I, Fujimi A, et al. Interaction Between Leukemic-Cell VLA-4 and Stromal Fibronectin is a Decisive Factor for Minimal Residual Disease of Acute Myelogenous Leukemia. Nat Med (2003) 9(9):1158–65. doi: 10.1038/nm909
130. Krause DS, Lazarides K, von Andrian UH, Van Etten RA. Requirement for CD44 in Homing and Engraftment of BCR-ABL-Expressing Leukemic Stem Cells. Nat Med (2006) 12(10):1175–80. doi: 10.1038/nm1489
131. Avigdor A, Goichberg P, Shivtiel S, Dar A, Peled A, Samira S, et al. CD44 and Hyaluronic Acid Cooperate With SDF-1 in the Trafficking of Human CD34+ Stem/Progenitor Cells to Bone Marrow. Blood (2004) 103(8):2981–9. doi: 10.1182/blood-2003-10-3611
132. Zeijlemaker W, Kelder A, Wouters R, Valk PJM, Witte BI, Cloos J, et al. Absence of Leukaemic CD34(+) Cells in Acute Myeloid Leukaemia is of High Prognostic Value: A Longstanding Controversy Deciphered. Br J Haematol (2015) 171(2):227–38. doi: 10.1111/bjh.13572
133. Zeijlemaker W, Grob T, Meijer R, Hanekamp D, Kelder A, Carbaat-Ham JC, et al. CD34(+)CD38(-) Leukemic Stem Cell Frequency to Predict Outcome in Acute Myeloid Leukemia. Leukemia (2019) 33(5):1102–12. doi: 10.1038/s41375-018-0326-3
134. Vergez F, Green AS, Tamburini J, Sarry JE, Gaillard B, Cornillet-Lefebvre P, et al. High Levels of CD34+CD38low/-CD123+ Blasts are Predictive of an Adverse Outcome in Acute Myeloid Leukemia: A Groupe Ouest-Est Des Leucemies Aigues Et Maladies Du Sang (GOELAMS) Study. Haematologica (2011) 96(12):1792–8. doi: 10.3324/haematol.2011.047894
135. Khan N, Freeman SD, Virgo P, Couzens S, Richardson P, Thomas I, et al. An Immunophenotypic Pre-Treatment Predictor for Poor Response to Induction Chemotherapy in Older Acute Myeloid Leukaemia Patients: Blood Frequency of CD34+ CD38 Low Blasts. Br J Haematol (2015) 170(1):80–4. doi: 10.1111/bjh.13398
136. Ng SW, Mitchell A, Kennedy JA, Chen WC, McLeod J, Ibrahimova N, et al. A 17-Gene Stemness Score for Rapid Determination of Risk in Acute Leukaemia. Nature (2016) 540(7633):433–7. doi: 10.1038/nature20598
137. Docking TR, Parker JDK, Jadersten M, Duns G, Chang L, Jiang J, et al. A Clinical Transcriptome Approach to Patient Stratification and Therapy Selection in Acute Myeloid Leukemia. Nat Commun (2021) 12(1):2474. doi: 10.1038/s41467-021-22625-y
138. van Gils N, Denkers F, Smit L. Escape From Treatment; the Different Faces of Leukemic Stem Cells and Therapy Resistance in Acute Myeloid Leukemia. Front Oncol (2021) 11:659253. doi: 10.3389/fonc.2021.659253
139. O'Reilly E, Zeinabad HA, Szegezdi E. Hematopoietic Versus Leukemic Stem Cell Quiescence: Challenges and Therapeutic Opportunities. Blood Rev (2021) 100850. doi: 10.1016/j.blre.2021.100850
140. Vetrie D, Helgason GV, Copland M. The Leukaemia Stem Cell: Similarities, Differences and Clinical Prospects in CML and AML. Nat Rev Cancer (2020) 20(3):158–73. doi: 10.1038/s41568-019-0230-9
141. Möhle R, Bautz F, Rafii S, Moore MA, Brugger W, Kanz L. The Chemokine Receptor CXCR-4 is Expressed on CD34+ Hematopoietic Progenitors and Leukemic Cells and Mediates Transendothelial Migration Induced by Stromal Cell-Derived Factor-1. Blood (1998) 91(12):4523–30. doi: 10.1182/blood.V91.12.4523
142. Spoo AC, Lübbert M, Wierda WG, Burger JA. CXCR4 is a Prognostic Marker in Acute Myelogenous Leukemia. Blood (2007) 109(2):786–91. doi: 10.1182/blood-2006-05-024844
143. DiPersio JF, Micallef IN, Stiff PJ, Bolwell BJ, Maziarz RT, Jacobsen E, et al. Phase III Prospective Randomized Double-Blind Placebo-Controlled Trial of Plerixafor Plus Granulocyte Colony-Stimulating Factor Compared With Placebo Plus Granulocyte Colony-Stimulating Factor for Autologous Stem-Cell Mobilization and Transplantation for Patients With non-Hodgkin's Lymphoma. J Clin Oncol (2009) 27(28):4767–73. doi: 10.1200/JCO.2008.20.7209
144. Nervi B, Ramirez P, Rettig MP, Uy GL, Holt MS, Ritchey JK, et al. Chemosensitization of Acute Myeloid Leukemia (AML) Following Mobilization by the CXCR4 Antagonist AMD3100. Blood (2009) 113(24):6206–14. doi: 10.1182/blood-2008-06-162123
145. Cooper TM, Sison EAR, Baker SD, Li L, Ahmed A, Trippett T, et al. A Phase 1 Study of the CXCR4 Antagonist Plerixafor in Combination With High-Dose Cytarabine and Etoposide in Children With Relapsed or Refractory Acute Leukemias or Myelodysplastic Syndrome: A Pediatric Oncology Experimental Therapeutics Investigators' Consortium Study (POE 10-03). Pediatr Blood Cancer (2017) 64(8). doi: 10.1002/pbc.26414
146. Uy GL, Rettig MP, Motabi IH, McFarland K, Trinkaus KM, Hladnik LM, et al. A Phase 1/2 Study of Chemosensitization With the CXCR4 Antagonist Plerixafor in Relapsed or Refractory Acute Myeloid Leukemia. Blood (2012) 119(17):3917–24. doi: 10.1182/blood-2011-10-383406
147. Roboz GJ, Ritchie EK, Dault Y, Lam L, Marshall DC, Cruz NM, et al. Phase I Trial of Plerixafor Combined With Decitabine in Newly Diagnosed Older Patients With Acute Myeloid Leukemia. Haematologica (2018) 103(8):1308–16. doi: 10.3324/haematol.2017.183418
148. Becker PS, Foran JM, Altman JK, Yacoub A, Castro JE, Sabbatini P, et al. Targeting the CXCR4 Pathway: Safety, Tolerability and Clinical Activity of Ulocuplumab (BMS-936564), an Anti-CXCR4 Antibody, in Relapsed/Refractory Acute Myeloid Leukemia. Blood (2014) 124(21):386–. doi: 10.1182/blood.V124.21.386.386
149. Kovacsovics TJ, Mims A, Salama ME, Pantin J, Rao N, Kosak KM, et al. Combination of the Low Anticoagulant Heparin CX-01 With Chemotherapy for the Treatment of Acute Myeloid Leukemia. Blood Adv (2018) 2(4):381–9. doi: 10.1182/bloodadvances.2017013391
150. Borthakur G, Ofran Y, Tallman MS, Foran J, Uy GL, DiPersio JF, et al. BL-8040 CXCR4 Antagonist is Safe and Demonstrates Antileukemic Activity in Combination With Cytarabine for the Treatment of Relapsed/Refractory Acute Myelogenous Leukemia: An Open-Label Safety and Efficacy Phase 2a Study. Cancer (2021) 127(8):1246–59. doi: 10.1002/cncr.33338
151. Kovacsovics T, Levy MY, Cook RJ, Kolitz JE, Westervelt P, Donnellan WB, et al. A Randomized Phase II Trial of CX-01 With Standard Therapy in Elderly Patients With Acute Myeloid Leukemia (AML). J Clin Oncol (2019) 37(15_suppl):7001–. doi: 10.1200/JCO.2019.37.15_suppl.7001
152. Piya S, Mu H, Bhattacharya S, Lorenzi PL, Davis RE, McQueen T, et al. BETP Degradation Simultaneously Targets Acute Myelogenous Leukemia Stem Cells and the Microenvironment. J Clin Invest (2019) 129(5):1878–94. doi: 10.1172/JCI120654
153. Dias S, Shmelkov SV, Lam G, Rafii S. VEGF(165) Promotes Survival of Leukemic Cells by Hsp90-Mediated Induction of Bcl-2 Expression and Apoptosis Inhibition. Blood (2002) 99(7):2532–40. doi: 10.1182/blood.V99.7.2532
154. Zahiragic L, Schliemann C, Bieker R, Thoennissen NH, Burow K, Kramer C, et al. Bevacizumab Reduces VEGF Expression in Patients With Relapsed and Refractory Acute Myeloid Leukemia Without Clinical Antileukemic Activity. Leukemia (2007) 21(6):1310–2. doi: 10.1038/sj.leu.2404632
155. Ossenkoppele GJ, Stussi G, Maertens J, van Montfort K, Biemond BJ, Breems D, et al. Addition of Bevacizumab to Chemotherapy in Acute Myeloid Leukemia at Older Age: A Randomized Phase 2 Trial of the Dutch-Belgian Cooperative Trial Group for Hemato-Oncology (HOVON) and the Swiss Group for Clinical Cancer Research (SAKK). Blood (2012) 120(24):4706–11. doi: 10.1182/blood-2012-04-420596
156. Velasco-Hernandez T, Hyrenius-Wittsten A, Rehn M, Bryder D, Cammenga J. HIF-1α can Act as a Tumor Suppressor Gene in Murine Acute Myeloid Leukemia. Blood (2014) 124(24):3597–607. doi: 10.1182/blood-2014-04-567065
157. Wang Y, Liu Y, Malek SN, Zheng P, Liu Y. Targeting Hif1α Eliminates Cancer Stem Cells in Hematological Malignancies. Cell Stem Cell (2011) 8(4):399–411. doi: 10.1016/j.stem.2011.02.006
158. Li Y, Zhao L, Li XF. Targeting Hypoxia: Hypoxia-Activated Prodrugs in Cancer Therapy. Front Oncol (2021) 11:700407. doi: 10.3389/fonc.2021.700407
159. Portwood S, Lal D, Hsu YC, Vargas R, Johnson MK, Wetzler M, et al. Activity of the Hypoxia-Activated Prodrug, TH-302, in Preclinical Human Acute Myeloid Leukemia Models. Clin Cancer Res (2013) 19(23):6506–19. doi: 10.1158/1078-0432.CCR-13-0674
160. Benito J, Ramirez MS, Millward NZ, Velez J, Harutyunyan KG, Lu H, et al. Hypoxia-Activated Prodrug TH-302 Targets Hypoxic Bone Marrow Niches in Preclinical Leukemia Models. Clin Cancer Res (2016) 22(7):1687–98. doi: 10.1158/1078-0432.CCR-14-3378
161. Badar T, Handisides DR, Benito JM, Richie MA, Borthakur G, Jabbour E, et al. Phase I Study of Evofosfamide, an Investigational Hypoxia-Activated Prodrug, in Patients With Advanced Leukemia. Am J Hematol (2016) 91(8):800–5. doi: 10.1002/ajh.24415
162. Tanaka T, Narazaki M, Kishimoto T. IL-6 in Inflammation, Immunity, and Disease. Cold Spring Harb Perspect Biol (2014) 6(10):a016295. doi: 10.1101/cshperspect.a016295
163. Fajgenbaum DC, June CH. Cytokine Storm. N Engl J Med (2020) 383(23):2255–73. doi: 10.1056/NEJMra2026131
Keywords: leukemic stem cell (LSC), acute myeloid leukemia, stem cell niche, genetic heterogeneity, therapeutic targets
Citation: Marchand T and Pinho S (2021) Leukemic Stem Cells: From Leukemic Niche Biology to Treatment Opportunities. Front. Immunol. 12:775128. doi: 10.3389/fimmu.2021.775128
Received: 13 September 2021; Accepted: 28 September 2021;
Published: 15 October 2021.
Edited by:
Dominique Bonnet, Francis Crick Institute, United KingdomReviewed by:
Andre Larochelle, National Institutes of Health (NIH), United StatesRobert Welner, University of Alabama at Birmingham, United States
Copyright © 2021 Marchand and Pinho. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tony Marchand, dG9ueS5tYXJjaGFuZEBjaHUtcmVubmVzLmZy