 Rebecca L. Watkinson1,2
Rebecca L. Watkinson1,2
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 29 November 2021
Sec. Molecular Innate Immunity
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.773600
The epithelium is integral to the protection of many different biological systems and for the maintenance of biochemical homeostasis. Emerging evidence suggests that particular children have epithelial vulnerabilities leading to dysregulated barrier function and integrity, that resultantly contributes to disease pathogenesis. These epithelial vulnerabilities likely develop in utero or in early life due to various genetic, epigenetic and environmental factors. Although various epithelia are uniquely structured with specific function, prevalent allergic-type epithelial diseases in children potentially have common or parallel disease processes. These include inflammation and immune response dysregulation stemming from atypical epithelial barrier function and integrity. Two diseases where aetiology and pathogenesis are potentially linked to epithelial vulnerabilities include Paediatric Asthma and Eosinophilic Oesophagitis (EoE). For example, rhinovirus C (RV-C) is a known risk factor for paediatric asthma development and is known to disrupt respiratory epithelial barrier function causing acute inflammation. In addition, EoE, a prevalent atopic condition of the oesophageal epithelium, is characterised by similar innate immune and epithelial responses to viral injury. This review examines the current literature and identifies the gaps in the field defining viral-induced effects on a vulnerable respiratory epithelium and resulting chronic inflammation, drawing from knowledge generated in acute wheezing illness, paediatric asthma and EoE. Besides highlighting the importance of epithelial structure and barrier function in allergic disease pathogenesis regardless of specific epithelial sub-types, this review focuses on the importance of examining other parallel allergic-type disease processes that may uncover commonalities driving disease pathogenesis. This in turn may be beneficial in the development of common therapeutics for current clinical management and disease prevention in the future.
Asthma is a heterogeneous, multifaceted respiratory disorder often emerging in early childhood (1–4). It is considered a symptomatic respiratory disorder, ranging from mild to severe. A review of several birth cohorts and risk factors for asthma development that encompassed 122 paediatric studies identified over 60 individual asthma definitions with different parameters for diagnosis (5). Thus, a clear definition has not been established (6). Clinically, paediatric asthma is diagnosed as having onset between the ages of 0 months up to 18 years, although approximately 80% of paediatric asthma cases begin between the ages of 0 months and 6 years (7). To further improve asthma diagnosis in the paediatric population, the Asthma Predictive Index (API) was developed to assist diagnosis of asthma under 3 years. However, observations from several cohort studies have indicated that asthma diagnosis in children under five years is difficult, with most standard testing regimens being variable or inaccurate under the age of seven (5, 8, 9). Many children, in particular, infants, have episodes of wheezing, which is often associated with respiratory viral illness (10) and has been shown to be a strong predictor for asthma diagnosis (1, 11). However, there is often heterogeneity in asthma onset particularly between gender and age. Males are more likely to be diagnosed pre-puberty and to have a heritable component to their disease (1, 12, 13). Females, in contrast, are often diagnosed with asthma later in life. Irrespective of such gender differences, children with wheezing illness exhibit clinical pathological features including smooth muscle hyper-constriction, immune responses such as inflammation, chronic remodelling such as mucous metaplasia and the resultant symptoms. A large proportion of children with early onset asthma have these innate immune system pathologies of allergic disease, and are also found to have elevated adaptive immune cells such as eosinophils in both blood and inflamed tissue (14, 15). Collectively, these pathological changes are triggered by insults such as acute viral infection or allergens which then contribute to a progressive loss of lung function through repeated damage to the airways. This is particularly evident in children who experience more intense and frequent asthma exacerbations (1, 3, 4).
Children exhibiting severe symptoms or respiratory distress caused by several of these pathologies may be hospitalised as their illness requires intervention (16–20) and in rare cases may be life-threatening (21–23). The frequency and severity of wheezing illnesses in infancy and early childhood may determine the likelihood of paediatric asthma development. Paediatric asthma is characterised by multiple phenotypes that have been identified in several different paediatric birth cohort studies (2–4, 24, 25). The Tucson Arizona birth cohort (1) has examined factors affecting presentation of wheezing illness before three years of age in relation to wheezing illnesses at six years and identified four specific phenotypes, (A) non-wheezing illness, (B) intermittent wheezing illness, (C) late-onset wheezing illness and (D) persistent wheezing illness (Table 1). This seminal study (1) suggests inherent differences in likelihood of asthma development disease phenotype and the likely mechanisms of disease progression.

Table 1 Asthma Phenotypes identified in the tucson arizona birth cohort and corresponding lung function findings (1).
To address this further, Oksel et al. (25) used latent class analysis to assess five other birth cohorts and found that the ‘persistent wheeze’ phenotype (26) has the strongest association with asthma development (25). In addition, all asthma phenotypes in which wheezing illness was present had significantly diminished lung function by 4 to 5 years of age when compared to non-wheezing children (25). However, observations between groups suggest that some children are uniquely susceptible to asthma development, and that in addition to symptoms such as wheezing, other factors from the prenatal to early childhood period contribute to the persistent wheezing phenotype and asthma susceptibility. For example, using multivariate analysis, Hallit and colleagues (4) found that early persistent wheezing at one year of age was independently associated with respiratory distress, excess bronchial secretions, reflux and nocturnal cough at two months of age (4). Others have found that maternal smoking during pregnancy and maternal history of asthma are also associated with the early persistent wheezing phenotype in children from two months to one year of age (4, 13, 27). Furthermore, the incidence rate of wheezing is increased when these same risk factors are paired with paternal history of asthma and cutaneous rash at two months of age (4, 27). In addition, another contributing factor for asthma development is allergic sensitisation to at least one allergen. Rubner and colleagues (28) have reported that 65% of children that were sensitised under twelve months, ended up being diagnosed with asthma by 13 years (28), and others have found that allergic sensitisation by the age of three is pivotal in asthma development (1, 3). Through the Urban Environment Childhood Asthma (URECA) birth cohort (24), a study characterising patterns of wheezing and allergic sensitisation in early life, five wheeze and atopy phenotypes were identified: (1) low wheeze/low atopy; (2) low wheeze/high atopy; (3) transient wheeze/low atopy; (4) high wheeze/low atopy; and (5) high wheeze/high atopy (24). Although asthma was overrepresented in the high wheeze phenotypes, most cases of respiratory morbidity were observed in children with both frequent (persistent) wheezing and allergic sensitisation (high atopy) (24). Children that are pre-disposed to airway vulnerabilities through a variety of factors may have more severe and frequent wheezing illnesses in response to an environmental trigger in early life that often leads to an asthma diagnosis. Therefore, recurrent wheezing illnesses in early childhood are a potential indicator of eventual paediatric asthma diagnosis. Furthermore, the combination of exogenous insults to the airway in early life, and the child’s specific innate and adaptive immune responses to these may further potentiate any vulnerabilities, and likely contribute to asthma development.
Collectively, numerous heterogeneous risk factors contribute towards asthma susceptibility as well as wheezing illness in children. With regards to asthma development, parental history of asthma, host genetics, wheezing illnesses and wheeze phenotype all play a pivotal role as well as exposure to allergens, pathogens and exogenous particulates (1, 4, 19, 20, 29–31). Additional risk factors that contribute to wheezing illnesses and asthma exacerbations in susceptible children include exhaust fumes, cold air, and respiratory viruses known to cause acute respiratory infections (ARI). Respiratory viral infections such as respiratory syncytial virus (RSV), influenza, adenovirus, coronavirus and rhinovirus (RV) have also been highlighted as key triggers of wheezing illnesses and asthma exacerbations in children (18–20, 32–37). Furthermore, various studies have suggested a potential nexus between the airway epithelium, respiratory viral insults and its association with wheezing illness. However, the mechanism for this remains unclear (20, 37–39) but there is cogent evidence (40–45) to support the pivotal and contributory role of the airway epithelium to disease progression.
The airway epithelium is a pseudostratified structure whose complex functions provides protection through structural, mucociliary and innate-immunological barriers. These barriers work synergistically in maintaining epithelial homeostasis and providing a dynamic response to pathogens, allergens and particulate matter. As reviewed by Knight and Holgate (46) and also identified by Garcia and colleagues (47), there are various types of epithelial cells, including basal, club, ciliated columnar and goblet cells (46–48). In addition, there are other lesser known epithelial cells involved in innate-immunological epithelial function including neuroendocrine cells, ionocytes expressing CFTR that contribute significantly to the muco-viscosity of airway surface liquid, and solitary chemosensory cells which are involved in the detection and release of neurotransmitters and ion channel function, among other functions (49–51). The heterogeneity of epithelial cell populations is further highlighted when comparing between the proximal and distal airway epithelium, each having similar yet distinct roles and functional processes (Figure 1).
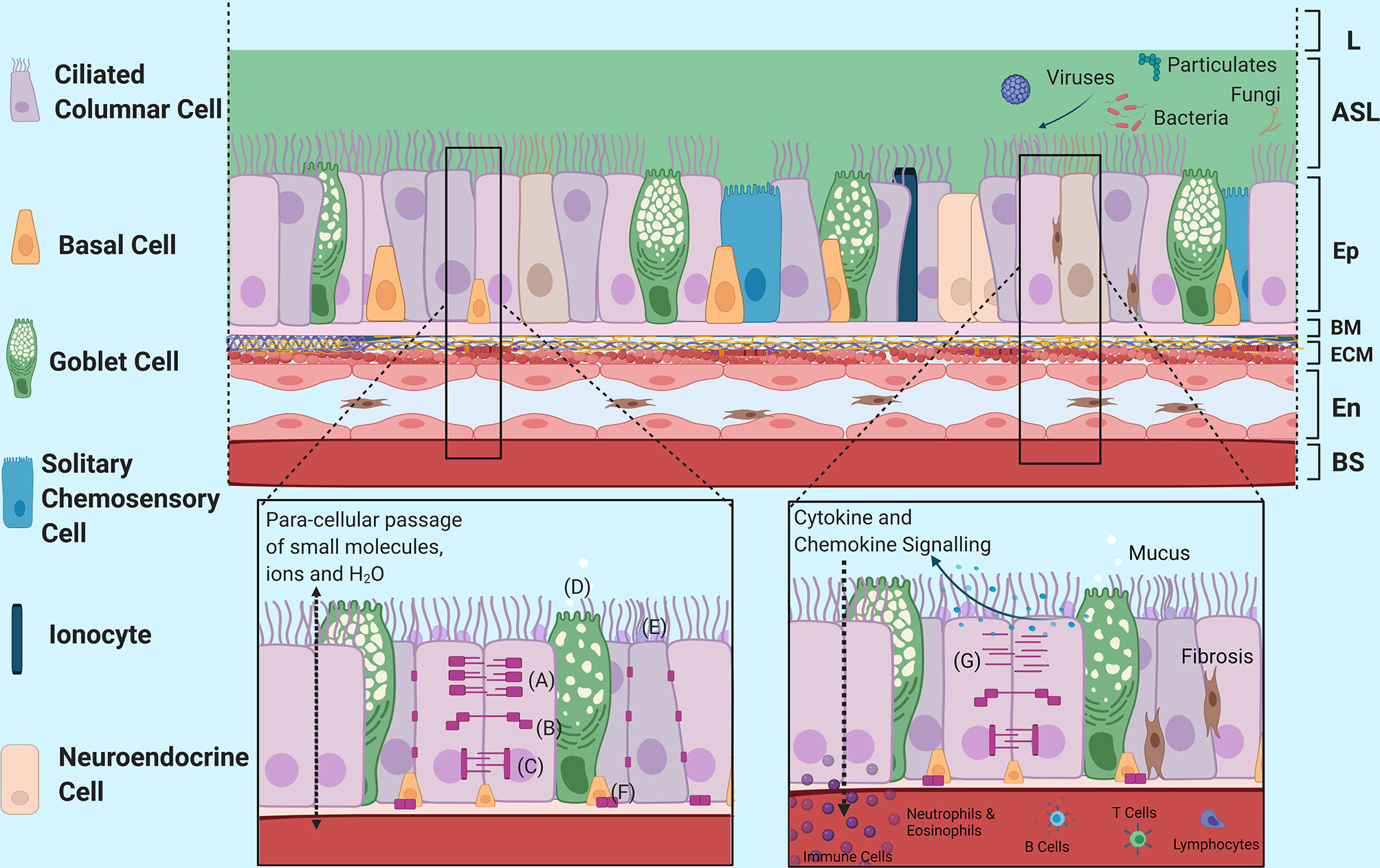
Figure 1 Structural Morphology and Barrier Functions of the Airway Epithelium. An overview of the structural morphology of the airway epithelium and it’s multifaceted barrier functions including structural, mucociliary and immunomodulatory barrier functions in response to injury. (A) Tight Junction Complex, (B) Adherence Junction Complex, (C) Desmosomes, (D) Mucin Release into Airway Surface Liquid, (E) Structural Mucins, (F) Hemidesmosomes, (G) Disruption of Junctional Complexes due to epithelial vulnerabilities and injury. L, Lumen; ASL, Airway Surface Liquid; Ep, Epithelium; BM, Basement Membrane; ECM, Extracellular Matrix; En, Endothelium; BS, Bloodstream. Created with BioRender.com.
The airway epithelium was once regarded as inert but is now known to be a dynamic barrier which actively regulates the passage of smaller molecules, ions and water while remaining impermeable to macromolecules (Figure 1). This is achieved via a myriad of junctional complexes (52, 53), which provides anchorage not only between adjacent cells but also to the basement layer and works to prevent exogenous molecules from passing through to the systemic circulation (52–55). In addition, O-glycosylated linear glycoproteins, which are mucins produced and secreted by goblet cells onto the airways, provide further defence against external insults. Structural mucins exist as brush-like structures attached to epithelial cells adjacent to the lumen in between cilia to form an additional structural barrier. These mucins bind to or block pathogens and other exogenous stimuli to prevent them from reaching the cell surface (56–58). Goblet cells, along with non-epithelial submucosal glands, also secrete non-structural polymeric mucins such as MUC5AC and MUC5B that form interconnected strands and sheets as part of airway mucus (59, 60). Airway mucus forms a percentage of the airway surface liquid layer atop the epithelium and helps to capture exogenous matter including pathogens and particulates (59, 60). Mucociliary clearance of the trapped pathogens and particulates are then unidirectionally propelled away from the distal and proximal airways towards the oropharyngeal regions for expectoration.
Adding to the protective capacity provided by mucins, basal epithelial cells have also been demonstrated to have a reparative capacity (61–63). Following wounding or injury to the epithelium by airborne pathogens, initiation of the cell repair process commences with the leading edge basal cells surrounding the injury site migrating into the wound to begin restitution of barrier integrity. This then follows by proliferation and differentiation as part of the wound repair process (61).
Complementing the physical defence of the airway epithelium, airway epithelial cells have also been shown to have an innate-immunological function that often leads to the recruitment of the adaptive immune system. This encompasses epithelial derived cytokines (Table 2) that define the pro-inflammatory responses that comprise part of the innate immune system and are released in response to stimuli. These have been comprehensively reviewed in numerous studies assessing adult airway epithelium (106–108) and will not be covered within the scope of this review.
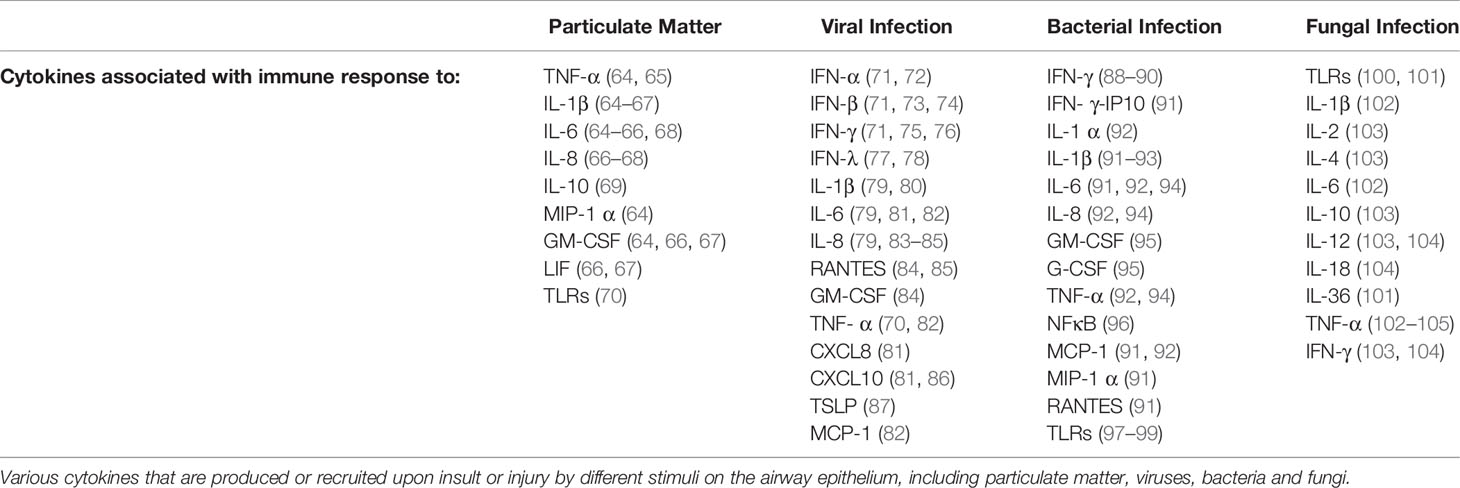
Table 2 Various cytokines involved in immune response to different stimuli.
The paediatric upper airway epithelium has been shown to have 91% transcriptional homology with the lower airway epithelium and a similar gene expression profile particularly in children with atopy (109). This conservation between the upper and lower respiratory airways lends support to the unified airway hypothesis that disease manifestation at both sites is likely a consequence of similar processes (109). However, it is acknowledged that this may not always translate functionally in both upper and lower respiratory airways. Foxman and colleagues (110) have shown that there is a temperature dependent innate immune response in murine airway epithelial cells infected with RV-A1. At 37°C, which is equivalent of the lower airways, there were increased interferon (IFN) type I and III genes, along with IFN stimulated genes compared to at 33°C, the equivalent of upper airways. This suggests a higher level of anti-viral defence or a more potent host response to infection at the higher temperature and in the lower airway. Furthermore, Lopez-Souza and colleagues (111) identified that RV-A16-infected human bronchial epithelial cells at air-liquid interface had lower trans-epithelial resistance, increased viral load (20-30 times), increased anti-viral and inflammatory cytokine production such as regulated on activation, normal T-cell expressed and secreted (RANTES), interleukin (IL) -8, IP-10, and IL-1α, compared to RV-A16-infected human nasal epithelial cells irrespective of asthma diagnosis (111). This suggests that the nasal epithelium has a greater protective capacity than the bronchial epithelium, and that asthmatic children with epithelial vulnerability may have a greater chance of RV infection spreading from the upper to the lower respiratory tract. Foxman et al. also found that genetic deficiency in mitochondrial antiviral-signalling protein (MAVS) and IFN type 1 receptor allowed for higher levels of RV replication at 37°C (110), suggesting that children with innate immunity deficiencies such as inadequate IFN response in the airway may allow for greater severity of RV infection. However, not all children with asthma are deficient in IFN production, as shown by Miller et al., where some children were able to produce sufficient IFN even while exacerbating (112). It was also demonstrated that IFN-λ was increased in nasal lavage fluid from wheezing asthmatic children with an RV infection compared to non-wheezing asthmatic children with an RV infection (112). Therefore, this again suggests that there are immunological and mechanistic differences between asthma phenotypes in different children, as well as potential respiratory epithelial differences, resulting in varying levels of vulnerability towards exogenous stimuli.
It is evident that despite similarities between the upper and lower airways of a child, there may be innate differences compared to other children. Factors that can cause this heterogeneity include host genetics (29, 113–117), epigenetic modifications throughout infancy and early childhood (117–119) as well as environmental risk factors (61, 120–122), that together, result in variable gene expression patterns and clinical characteristics. These may be hereditable or caused by environmental modification during the in utero or early childhood period. Although children with a healthy, respiratory epithelium functioning at full capacity are unlikely to develop vulnerability, those who are at risk or predisposed to disease may be more susceptible to external insults such as viral infection, allergens, or particulate matter by exhibiting an atypical epithelial barrier and immune response. These insults may then contribute to further epithelial dysregulation.
Studies have observed differences in individual genes involved in innate immune responses activated during viral respiratory illness including JAK2, STAT4, MX1, DDX58, VDR and EIF2AK2 (29, 114). Interestingly, genetic polymorphisms on these genes have been significantly associated with asthma exacerbations (29, 114). Variations in the ORMDL3/GSDMB locus, and GSDMB, CD14, CC16, CYSLTR1, ST2, GSTP1, and IL1RL1 genes among others (123–129), some of which may be epithelium and innate immunity specific have also been shown to be associated with childhood onset asthma. In addition, single nucleotide polymorphisms (SNP) have been shown to occur in mucin genes, potentially causing alterations in airway mucus (130). SNPs such as the MUC5AC coding rs1132440 G/C (C risk allele variant) have been associated with increased MUC5AC expression during respiratory viral illnesses (130). Jackson and colleagues found that this particular risk allele enhanced expression of a gene hub containing MUC5AC and other genes related to mucus hypersecretion and activation of eosinophils resulting in clinical outcomes such as airway-hyper-responsiveness, mucus plugging and increased inflammatory processes (130).
Other genetic variations in cellular viral receptors are also likely to contribute to epithelial vulnerability. SNPs such as rs5498 and rs688 have been found in RV species A and B receptors - the intracellular adhesion molecule (ICAM-1) and the low density lipoprotein receptor (LDLR) respectively (131, 132). However, the most clinically relevant viral receptor SNP rs6967330 A/G (Cys529Tyr) is located in the cadherin related family member 3 (CDHR3), the viral receptor for RV-C, the species most prevalent in paediatric wheezing illness (19, 20, 35–37, 115, 116). The CDHR3 receptor protein is present on all ciliated epithelial cells with its extracellular domains 1-3 mediating the interaction of epithelial cells with RV-C (133). The CDHR3 gene risk allele is associated with a ten-fold increase in cellular CDHR3 expression (134) and has been shown to increase RV-C infection levels and protein surface localisation of the receptor on epithelial cells potentially leading to more severe infection (134). This risk allele has also been associated with earlier, faster ciliogenesis and a ten-fold increase in FOXJ transcription factor expression, a known driver of basal epithelial cell differentiation into ciliated cells (116, 117). Despite this, it appears to have no effect on ciliary beat frequency or the integrity of tight junctions (117). Furthermore, cleaved cytoplasmic domains of some cadherins are able to self-activate their own gene expression which decreases as cells mature and protein interactions increase (135). This suggests that epithelial cell damaged due to infection may trigger the repair process, which in turn may lead to an increase of immature cells and increased CDHR3. RV-C infection has been observed to decrease CDHR3 mRNA expression in wheezing children compared to the control group (136). Clinically, the CDHR3 risk allele has been associated with increased risk of wheezing illness leading to hospitalisation. It was found to be overrepresented in wheezing children, and children with this risk allele were observed to need an increased amount of respiratory medical care during wheezing illness (136).
There are various genetic risk factors that may cause variability in the amount gene encoding or gene function as recently identified by Khoo and colleagues in their Mechanisms of Acute Viral Respiratory Infection in Children (MAVRIC) acute wheezing cohort (113). They found that some children, in response to an acute viral-induced wheezing illness, have different upper airway gene network expression patterns in their anti-viral Interferon Regulatory factor 7 (IRF7) network. These network patterns, named “IRF7hi” and “IRF7lo”, are likely to represent different immune responses to respiratory viral infection. Children characterised as “IRF7hi” exhibited a gene expression pattern associated Th1 and type 1 interferon responses to viral-induced asthma exacerbations. Alternatively, children characterised as “IRF7lo” exhibited a gene expression pattern associated with epithelial cytokine signalling, upregulated growth factor signalling and downregulated anti-viral interferon gamma. The study also reported that children with “IRF7lo” expression exhibited symptoms twice as long prior to hospital presentation from initial symptoms and had cough three times as long compared to children with “IRF7hi” expression. In conjunction, the odds ratio for hospital admission for children with “IRF7lo” was increased by a factor of four and a much shorter time until wheezing illness recurrence. Thus, children with the “IRF7lo” phenotype may have more frequent wheezing illnesses which may potentiate asthma development. These molecular sub-phenotypes may impact on each child’s epithelial response due to the genes and cytokines associated with them.
In addition to genetic risk factors, each child will likely incur epigenetic modifications, due to environmental factors such as diet and exposure to allergens and exogenous pathogens to their airway epithelium during development. These epigenetic modifications have the potential to change epithelial functionality and integrity, as shown by Lund et al., where RV infection causes DNA methylation (118) and has been found to occur in genes including SMAD3, that encodes for the cell signalling protein SMAD3, as well as DDO and METTL24, genes that encode for peroxisomal flavoprotein and methyltransferase 24 respectively (118, 137). Furthermore, Pech and colleagues found that DNA methylation and resultant changes in mRNA expression in genes occurred in nasal cells from children with asthma following infection with RV (119) And that these altered genes are associated with host immune response to viral infection as well as asthma pathogenesis (119).
In addition to genetic and epigenetic risk factors, there are also environmental risk factors. An important subset of environmental risk factors for epithelial vulnerability are those imparted by pre-term birth (<32 weeks) and its associated medical stresses such as oxygenation, steroids use and mechanical ventilation (120). Hillas and colleagues observed that cultured primary nasal epithelial cells from healthy full-term children were able to complete in vitro wound closure by 60 hours (120). In contrast, pre-term infants appear to have either (1) delayed but complete (>80%); (2) significant but incomplete (50-80%); or (3) fully incomplete (20-50%) epithelial wound closure (120). Moreover, epithelial vulnerability in pre-term children may develop in utero during maternal exposure to pathogens, particulates and resultant inflammation. Chorioamnionitis, an inflammatory in utero risk factor for pre-term birth (121, 122), has been associated with increased airway epithelial apoptosis (121) with further studies also showing that epithelial cells treated with bronchoalveolar lavage fluid from infants born pre-term with chorioamnionitis have a reduced capacity for epithelial wound repair after mechanical wounding (122).
A dysregulation in epithelial wound repair potentially leaves children more susceptible to exogenous insults and further epithelial vulnerability. Defective epithelial wound repair has been found to be highly prevalent in children with asthma (38, 39). Deficient production of fibronectin, an epithelial extracellular matrix protein, may contribute to aberrant wound repair (138) although treating in vitro cell cultures with exogenous fibronectin only partially restored wound repair (138). Iosifidis et al., using paediatric asthmatic primary airway epithelial cells, demonstrated an aberrant wound migration pattern associated with decreased integrin α5β1 expression (61) that is regulated by the PI3K/Akt pathway (61). The transcriptomic signature associated with aberrant wound repair and the PI3K/Akt pathway was associated with viral-induced wheezing illness, suggesting that RV infection could disrupt the PI3K/Akt pathway particularly in children susceptible to asthma. Importantly, they showed that Akt restoration with a repurposed drug resulted in improve epithelial repair capacity and integrin expression, thus providing proof of principle that a dysregulated airway epithelium can be therapeutically targeted (61).
There are many other risk factors that increases epithelial vulnerability and eventually leading to the development of persistent wheeze and ultimately, asthma. Epithelial vulnerability has the potential to cause a dysregulated response to an environmental insult which may potentiate disease progression. Furthermore, it is possible that RV infections, particularly RV-C, may infect the vulnerable epithelium opportunistically. The consequence of this may be a repetitive cycle of infection, dysregulated epithelial barrier function, repair, leading to persistent wheezing exacerbations and potential asthma pathogenesis. Although there has been progress in research on epithelial vulnerability and its potential to contribute to airway disease pathogenesis, elucidating the role of the vulnerable epithelium in disease progression and in particular, its association with respiratory viral infections such as rhinovirus, remains significantly inadequate.
Rhinovirus (RV) is the most common virus found in children, particularly in those admitted to hospital with wheezing (19, 139). As reviewed by Palmenberg and Gern (140), RVs are viruses that are part of the enterovirus genus and the picornaviridae family. These viruses are known to have a five prime (5’) virus encoded protein (VP) and a three prime (3’) poly adenosine tail like that of messenger RNA (140). They are small, approximately 30nm in diameter irrespective of RV species. Three known species of RV exist: RV-A, RV-B and RV-C. There are three host viral receptors for RVs; major genotypes of RV-A and RV-B use the intracellular adhesion molecule 1 (ICAM-1), and minor genotypes use the low density lipoprotein receptor (LDLR) (140), however, RV-C uses CDHR3 as its receptor (134, 141, 142). There are many RV strains with more than 150 RV sub-types identified and extensive antigenic diversity and presently, there are no therapeutics capable of mitigating its effects and the development of serious respiratory disease (143). Interestingly, the increase in RV infections in spring and autumn correlate with hospital admission rates for paediatric asthma exacerbations (144, 145) and the virus is present in the majority of paediatric asthma exacerbations (11, 19, 20, 23, 35–37, 144, 146–150). The anti-viral immune response triggered by ARIs is likely to contribute to the overall inflammatory load that is in turn capable of triggering an asthma exacerbation.
The inter-relationships between RV infections, epithelial vulnerability and why some children have persistent wheeze, leading to the development of asthma, is still largely unclear (146, 151). Rhinovirus is commonly detected in children with ARIs (11, 139, 152–160), with ARIs leading to symptoms in the lower respiratory tract being a major cause of paediatric morbidity and mortality worldwide (151). ARIs, particularly those caused by RV may contribute towards epithelial vulnerability, further wheezing illnesses and asthma development. RVs are ubiquitous in the community, and it is estimated that they cause ~50% of all upper respiratory tract infections in humans, as well as being associated with acute asthma exacerbations in both children and adults (11, 19, 20, 23, 35–37, 144, 147–150). This is due to RV being identified as the most common virus in wheezing children admitted to the hospital emergency department, associated with 20 - 87.5% of hospitalisations (19, 20, 35–37, 144, 146, 150) and 10 - 15% of admissions into the paediatric intensive care unit (18, 19). The highest instances of RV infection have been reported in children with a history of asthma related symptoms (144). Moreover, Jackson and colleagues utilising the COAST high-risk birth cohort of children with parental history of asthma or allergy, also found that 90% of wheezing illnesses were of viral origin, and that by the age of three years, RV-associated wheezing had a three-fold stronger association with increased asthma risk by six years than aeroallergen sensitisation or RSV (11). Almost 90% of the children that had RV induced wheezing illness at the age of three years were diagnosed with asthma at the age of six (11). They concluded that among all outpatient viral wheezing illnesses in infancy and early childhood, those caused by RV were the most significant predictors of asthma development in their high-risk birth cohort (11). Wheezing illnesses and their relation to house dust mite allergy and RV was also examined in another study, which found that the probability of acute wheezing in children was positively correlated with increasing IgE titres to house dust mite. This was also significantly further potentiated when an RV infection was present during exacerbation (161).
The effect of RV on the paediatric airway epithelium - and its contribution to wheezing illness and eventual asthma development - is not yet fully understood. One study suggests that receptor specificity determined by the major or minor genotype of RV- A and RV-B can influence host response to the virus (162). Other studies have shown that RVs can dysregulate epithelial barrier function and integrity, potentially contributing to asthma development by altering epithelial barrier function and integrity in several ways including by disruption of homeostatic and dynamic cytokine production, tight junction complexes, as well as dysregulating wound repair (38, 79, 144, 163).
An example of one of the effects that RV has on the epithelium is that RV is able to dysregulate the production of anti-viral and pro-inflammatory cytokines and biochemical signalling molecules during infection, particularly in asthmatic children (144, 164–169) potentially leading to systemic inflammation and the switch from the innate immune system of the epithelial response to the adaptive immune system. Contradictory findings have been made around the interferon response in this setting. For example, some have found that IFN type I, II and III genes are induced by the airway epithelium during RV infection (71). Interestingly, this response appears to be dysregulated particularly in asthmatics (170). IFN-α levels have been found to be upregulated in PBMCs from asthmatic children infected with RV, while IFN-λ is already upregulated at baseline in these children (171), which, when combined, suggest a hyperactive immune response. In addition, Miller and colleagues (112) found that IFN-λ is further increased in RV-infected-wheezing children with asthma compared to RV-infected-non-wheezing children with asthma (112). Conversely, others have shown that primary airway epithelial cells from asthmatic children infected with RV-A1 and RV-A16 produce less IFN-β and IFN-λ than cells from their healthy non-asthmatic counterparts (38, 162, 170, 172). In addition, Edwards and colleagues (170), found that deficient IFN β and λ was positively correlated with asthma severity, with high levels of IFN deficiency found in severe therapy resistant atopic asthmatics (170). However, the controversy surrounding these findings is potentially due to children having different interferon molecular sub-phenotypes (“IRF7hi” and “IRF7lo”) such as was identified in the MAVRIC acute wheezing cohort by Khoo and colleagues (113). These studies collectively suggest a dysregulated anti-viral response to RV infection. In conjunction with epithelial IFN production in response to RV, there are other antiviral mechanisms that appear to be dysregulated during infection, such as the signal transducer and activator of transcription 1 (STAT1) signalling pathway. STAT1 is typically activated in response to RV in epithelial cells and activates a signalling cascade that promotes the expression of anti-viral genes (71) as part of the multifaceted epithelial response to infection.
The airway epithelium’s multifaceted response to RV infection has been shown to upregulate gene expression and ultimately the release of innate immune system pro-inflammatory cytokines, chemokines (79, 81, 83–86, 173), including eotaxins, IL-17C, IL-4, IL-5, IL-13, IL-33, NFκB, TSLP (71, 85, 173–175) and others (Table 2). Nasal washes collected from children during confirmed RV infection, have increased thymic stromal lymphopoietin (TSLP) levels at the time of infection which is also linked to atopy and plays a role in many allergic diseases (176). Kennedy and colleagues (173), observed an increase in TSLP gene expression and protein as well as gene expression of IL-25 and IL-33, in asthmatic donor lungs compared to non-asthmatic controls following RV-A39 infection (173). They also observed that there was only a difference in carbachol-induced airway constriction between the two cohorts post RV-A39 infection, suggesting an altered immune response even in the lower airway (173). In addition, Subauste and colleagues (177) demonstrated that RV-B14 was able to induce TNF-α, IL-6 and IL-8 release in human bronchial epithelial cells, and that prior exposure to TNF-α, increased susceptibility to RV-B14 infections suggesting the potential for cytokines to potentiate further RV infection (177). Another study examining the nasal cytokine profiles of children hospitalised with respiratory wheeze found that RV-C-induced wheezing was identified to have a characteristic Th2 type cytokine release profile in both non-asthmatic children and asthmatic children (17). Interestingly, the same study also found that cytokines IL-17 and IL-1β (characteristic of Th17) were increased in children with pre-existing asthma and not in non-asthmatic children, irrespective of both being diagnosed with wheeze and viral infection symptoms (17). In addition, the pro-Th2 inflammatory profile inducing cytokine IL-33, is shown to be downregulated in children with the “IRF7hi” phenotype defined in the paper by Khoo and colleagues (113) but not in the “IRF7lo” phenotype and thus could potentially be a driver of wheezing illnesses in children with the “IRF7lo” phenotype. IL-33 has been associated with paediatric asthma in other studies also (17, 113, 175, 178). Moreover, in response to RV infection by adult bronchial biopsy specimens, there is an increased number of sub-epithelial inflammatory cells that express IFN, as well as epithelial and sub-epithelial pattern recognition receptors (PRR) (169). It is important to note that RV infection is recognised by PRRs (86, 179, 180) that activate MAPK signalling pathways that in turn induce inflammatory gene production. MAPK pathways p38 and JNK have pivotal roles in the epithelial inflammatory response to RV infection (181). Duel Specificity Phosphatase 10 (DUSP10) also plays a pivotal role by regulating it inflammatory cytokine production (e.g. IL-1β) and has been observed to be downregulated by RV infection, thus weakening the anti-viral response and perpetuating uncontrolled inflammation (181). The overproduction or upregulation of pro-inflammatory cytokines and chemokines potentially confers a hyper-active response which may damage the airway epithelium further and go on to impair barrier function and integrity. Interestingly, even without RV infection present at the time, asthmatic airway epithelial cells from children have been identified to have increased IL-6, epidermal growth factor and prostaglandin-E2 as well as decreased TGF-β1 (39). This suggests intrinsic differences in the airway epithelium between asthmatic and non-asthmatic children, and thus a different response to pathogens such as RV.
RV infection also disrupts the barrier function of the airway epithelium by dissociating tight junction proteins such as zonula-occludens 1 from the tight junction complex in both asthmatic and non-asthmatic children (163, 182). Despite upregulation of the basal gene expression of the tight junctions claudin-1 and occludin in children with asthma compared to non-asthmatics, protein levels are significantly reduced (163). This suggests that although genes are being transcribed, translation into protein may not be occurring. Furthermore, in vitro air-liquid interface cultures of epithelial cells established from asthmatic children show a sustained decrease in tight junction protein staining, decreased trans-epithelial resistance, (TEER) and a consequent increase in permeability when infected with RV-A1 (163). Dysregulated tight junction expression and resulting function as seen in asthmatic epithelial cells would potentially allow pathogens and particulates to pass through the epithelial layer and into the bloodstream resulting in a heightened host response.
In addition, RV has also been observed to be able to disrupt epithelial wound repair and increase cellular cytotoxicity (38, 183–185). RV-A1 has been shown to delay wound repair capacity and inhibit apoptotic processes by epithelial cells, exaggerating the already defective repair in the asthmatic airway (38). RV infection of a bronchial epithelial cell line in vitro has been able to stimulate mRNA expression and release of basic fibroblast growth factor (bFGF), leading to fibroblastic repair rather than normal epithelial repair processes (184). The release of bFGF is associated with RV-induced cytotoxicity and resultant epithelial necrosis as opposed to apoptosis. Furthermore, RV infection also causes an increase in matrix metalloproteinase (MMP) activity in vitro, and may affect proteins of the extracellular matrix to which the epithelium is attached (184). Levels of bFGF and MMP are also induced following RV infection of epithelial cells in vitro (184). This is further evidence that epithelial wound repair is dysregulated in asthmatics, and potentiated further by RV. Furthermore, Altman and colleagues (186) examined cellular transcriptome networks and found that peak upregulation of epithelial SMAD3 and type one IFN signalling occurs at day two of a viral-induced asthma exacerbation, followed by peak upregulation of epidermal growth factor, and extracellular matrix at day three to four (186). Deficient or dysregulated wound repair of the epithelium likely leaves the epithelium susceptible to further infection or damage from exogenous insults.
Of the three species of RV, RV-C is of particular interest as it is associated with more severe acute wheezing illnesses (20). In addition, several studies have shown that RV-C accounts for the majority of RV positive cases in children (Table 3) (17, 19, 20, 35–37, 188, 189) and coincide with higher asthma severity scores as evidenced in children presenting to the emergency department with a wheezing illness (20). Furthermore, wheezing illnesses induced by RV-C are often more commonly associated with typical asthma symptoms such as wheezing and cyanosis when compared to other RV species (36, 37, 190). It is possible that RV-C may further potentiate epithelial vulnerability, yet this is currently unknown.
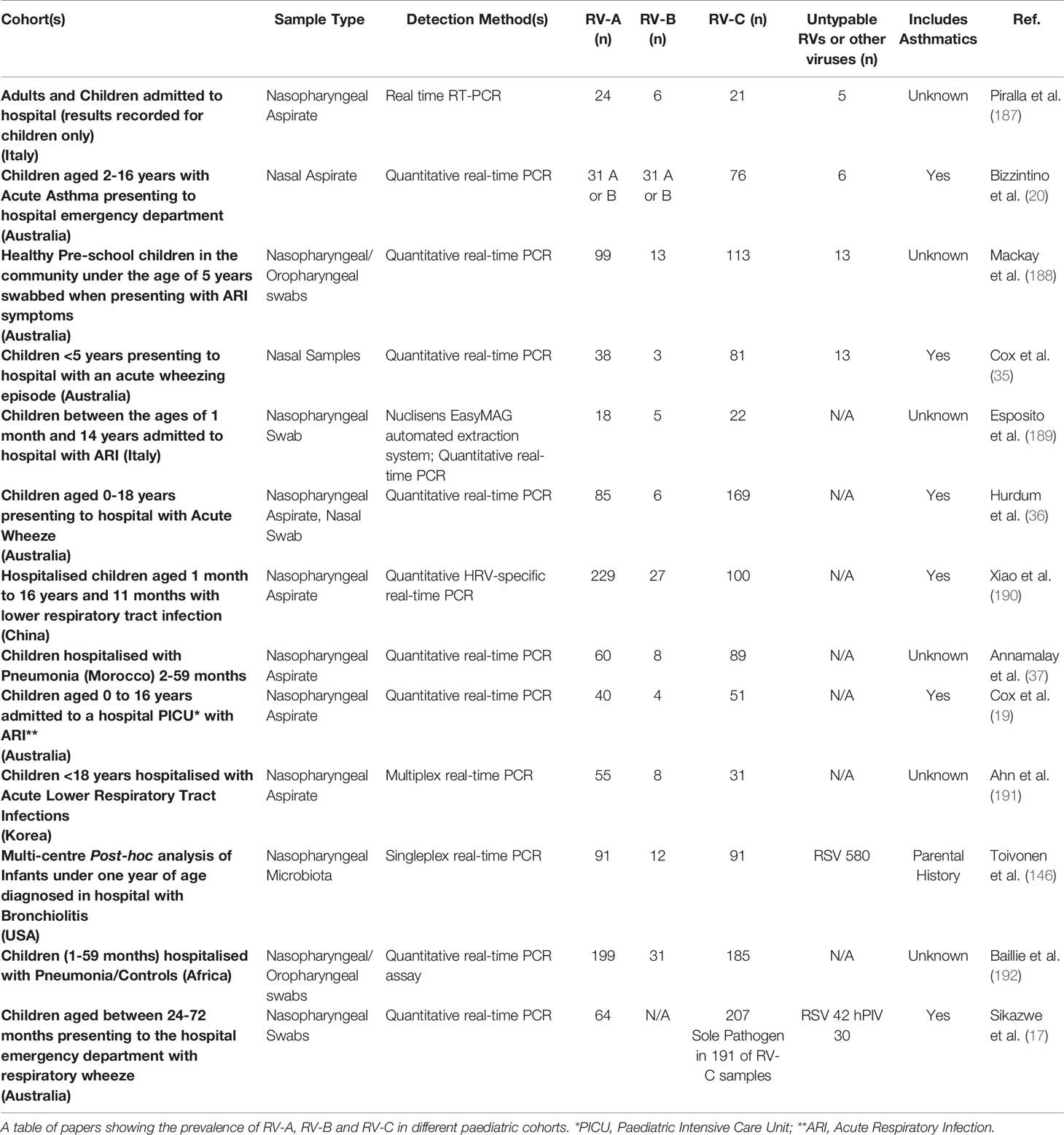
Table 3 Different species of RV in paediatric patients.
Despite anatomical differences, other epithelial surfaces are damaged following insult, which when dysregulated can contribute towards disease manifestation and progression. One example, eosinophilic oesophagitis (EoE), exhibits similar disease characteristics to paediatric asthma including atypical epithelial barrier integrity and dysregulated innate and adaptive immunity (193). Although the intricate interplay between allergic sensitization and airway inflammation has been studied in these diseases (28, 187), their association with epithelial barrier dysregulation remains relatively unknown, however, there is evidence to suggest that systemic inflammation could be a major contributor (191, 192). Therefore, examining parallel epithelial diseases processes may further the understanding of their pathogenesis. For example, to understand the role of the vulnerable epithelium and its dysregulation in acute wheezing illness and paediatric asthma, it is important to examine what is known about the epithelial response to injury in EoE.
Eosinophilic Oesophagitis (EoE) is an increasingly prevalent atopic condition defined by eosinophilia of at least 15 eosinophils per high power field (eos/hpf) and symptoms of oesophageal dysfunction (194, 195). As an auto-immune disease, EoE is potentiated by abnormal host response to a trigger, yet still exhibits typical allergy symptoms similar to asthma (Figure 2). Although not inherently linked, EoE and asthma are two distinct allergic diseases that have parallel disease processes. Thus, the similarities and differences identified can be drawn upon to expand the knowledge of each disease and further the understanding of the mechanisms behind them. Increasing evidence suggests that genetic predisposition and environmental triggers contribute to disrupted oesophageal epithelial integrity, initiate an innate pro-Th2 immune response and lead to Th2 chronic inflammation with consequent oesophageal dysmotility and fibrosis (196). Airways dysmotility and fibrosis are common features in asthma as well, and therefore EoE may be a representative model for possible similar pathogenic pathways.
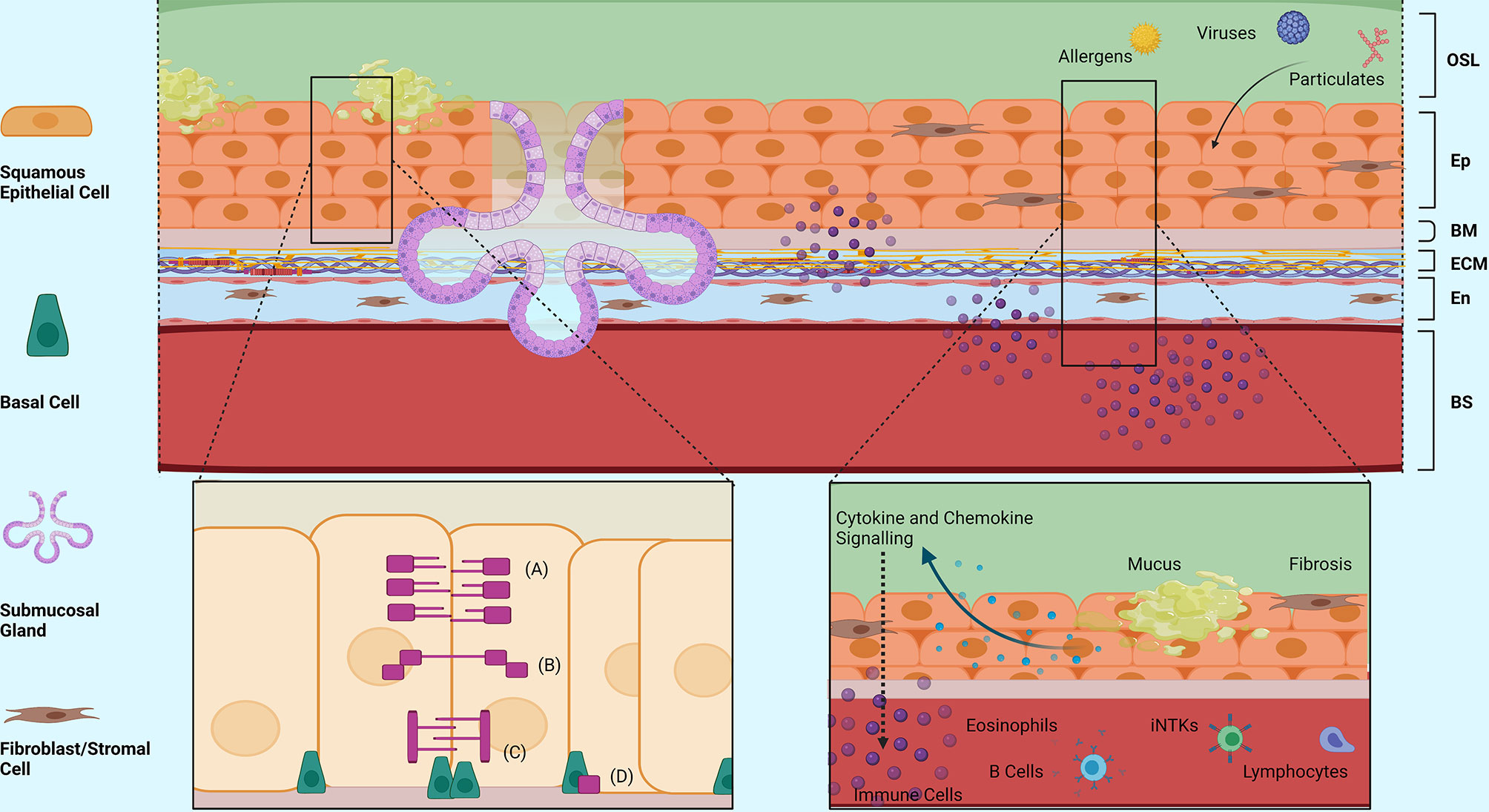
Figure 2 Structural Morphology and Barrier Functions of the Oesophageal Epithelium. An overview of the structural morphology of the oesophageal epithelium and it’s multifaceted barrier functions including structural, mucosal and immunomodulatory barrier functions in response to injury. (A) Tight Junction Complex, (B) Adherence Junction Complex, (C) Desmosomes, (D) Hemidesmosomes. OSL, Oesophageal Surface Liquid; Ep, Epithelium; BM, Basement Membrane; ECM, Extracellular Matrix; En, Endothelium; BS, Bloodstream. Created with BioRender.com.
Fibrosis is responsible for EoE symptoms such as dysphagia and food impaction. However, children tend to present with less specific symptoms related with oesophageal inflammation and oesophageal dysmotility such as feeding difficulties, gagging and vomiting (196, 197). Confirming what is suspected by examining the differences in symptoms in adult and children, prospective studies suggest that inflammation progresses to fibrosis in the majority of untreated patients (198). Food allergens drive the majority of cases of EoE, but food allergy (FA) in EoE presents differently from IgE mediated FA, in that it appears to start not in infancy but instead seems to be due to a break in tolerance of previously well tolerated common foods such as milk and wheat (199–202). Many lines of evidence suggest that such disruption is due to the epithelial barrier insults and consequent induction of a Pro-Th2 inflammation in genetically predisposed individuals. What such an insult is still largely unknown. Viral infections have often been suspected, but difficult to prove in a disease that needs an endoscopy to be diagnosed. However, a high incidence of EoE (30%) in immunocompetent atopic individuals recovering from Herpes Virus Simplex esophagitis, suggests that viral infection may indeed play a role in EoE (203). If the initial insult has not been clarified, the last 10 years of research have clearly demonstrated the central role of the oesophageal epithelium in driving and maintaining chronic pro inflammatory inflammation in EoE. Eosinophils define the disease in EoE, but the inflammation in EoE is more complex and all the other cell types of atopic inflammation such as basophils, mast cells, IgE producing B cells, innate Lymphocytes type 2 (IL2), invariant natural killer cells (iNKTs) have been shown to be important players (197, 204). This is not surprising considering that like many other diseases, EoE is highly associated with atopic comorbidities and may just be the last step on a slow evolving atopic march (205–208). Food allergens such as milk and wheat and likely environmental allergens are the major drivers of EoE (202, 208–210). Allergens may stimulate Th2 lymphocytes directly, as demonstrated in a small group of patients with milk allergy (209). Although a large proportion of asthmatics have elevated eosinophils and concurrent allergic disease, similar to suffers of EoE, the response to treatments targeting these factors have had difference levels of response in each of these diseases. Differently from other atopic diseases, IgE and eosinophils appear not to be central in the pathogenesis of EoE (208). Indeed, biologics directed against IgE (omalizumab) and eosinophils (anti-IL-5 reslizumab and mepolizumab) have been shown to control asthma in patients with allergic sensitisation and an eosinophilic endotype (211, 212) but have not been proven to be helpful in EoE (213, 214). On the other hand, antibodies with broader Th2 inhibition like antiIL-4/IL-13 or anti-IL-13 seem to be more effective in controlling EoE (215, 216). Due to its genetic predisposition, studies, particularly those involving sibling and twins, have helped to understand how such inflammation develops in EoE (217). Over the years, several genetic loci have been linked to EoE, which confirm the central role of the epithelium in driving EoE and are grouped into three major gene categories: Th2 atopic inflammation, epithelial barrier and fibrosis (218–221).
Several single nucleotide polymorphisms (SNPs) of epithelial genes promoting Th2 inflammation such as eotaxin-3 (CCL26) on 7q11.23, thymic stromal lymphopoietin (TSLP) on 5q22, and Toll-like receptor 3 (TLR3) on 4q35.1, have been connected to EoE by several independent groups (218, 221, 222). Blanchard et al., found a genetic polymorphism in CCL-26 (eotaxin-3), a gene important for eosinophil chemotaxis, present in about 13% of patients with EoE, who overexpressed CCL-26 by 50-foldcompared with healthy controls (218, 221, 223). Similarly, a risk allele (AA) on the TSLP gene has been shown to be correlated with increased epithelial TSLP expression, EoE development and increased mucosal basophils (218, 221, 224). Animal studies have also shown that TSLP is pathogenetic for EoE development as its inhibition prevents EoE development (224). Therefore, TSLP, an epithelial derived cytokine that can strongly induce a Th2 effector response from adaptive and innate cells, appears to be important in EoE pathophysiology like other atopic diseases (225). Other epithelial signalling related polymorphisms such as the TLR3 SNP (CC or CG at rs3775292), are found more frequently in allergic EoE patients (222).
Th2 inflammation leads to fibrosis in asthmatic patients as well in EoE patients. In EoE, the pro-fibrotic factor transforming growth factor-beta (TGF-β) on 19q13 has been connected with EoE (226, 227). EoE is highly prevalent in patients with connective tissue disorders such as Ehlers- Danlos or Loeys-Dietz or Maran’s Syndromes where dysregulation in TGF-β signalling is well known. Similarly, EoE patients with reduced fibrosis after steroid therapy are more likely to have a certain SNP (the CC genotype at the -509 position) in the TGF-β promoter (226–228). Autosomal dominant Hyper-IgE Syndrome due to dominant-negative STAT3 mutations in which there is an upregulation of TGF-β have higher incidence of EoE (229, 230).
Inflammation is also known to lead to epithelial barrier dysfunction and genes that build the epithelial barrier have been implicated in EoE pathogenesis. These include calpain 14 (CAPN14) on chr2p23.1, Filaggrin (FLG) on 1q21 and epithelial serine protease inhibitor SPINK5 on 5q32. Like other atopic diseases such as asthma or eczema, genetic polymorphisms may predispose the epithelium to be more permeable and more vulnerable to damage by the Th2 inflammation. This is thought to increase the amount of contact between antigens and the immune system, and predispose to inappropriate loss of tolerance for these antigens (231). Calpain 14 (CAPN14) is a protease in the calpain family that is expressed at the highest level in the esophageal epithelium and upper GI tract. IL-13 induces CAPN14 expression in the esophageal epithelium with consequent loss of barrier function (218). Netherton’s syndrome, which is caused by a defect in the epithelial serine protease inhibitor SPINK5, has also been described as an EoE risk as well as for severe atopic dermatitis (232, 233).
Although at the moment there is no data that RV or any other virus may induce the epithelium changes that eventually lead to food sensitisation and EoE, it is possible that viruses such as herpes virus or others could act similarly to RV in initiating EoE after infection directly or indirectly through upper airways infection. Indeed, the common cold can involve one or all the sinuses, nasopharynx, oropharynx and larynx. EoE is commonly associated with Eosinophilic Laryngitis and Aerodigestive Dysfunction in children (234). At the molecular level, CAPN14 is highly expressed in the oesophagus and pharyngeal cells (218). In asthma models, viruses like RV can induce TSLP, and by doing so antagonise tolerance to inhaled antigen (235), or create steroid resistance (176, 236, 237). The cellular constituents are vastly different between the airway epithelium and the oesophageal/gastro-intestinal (GI) epithelium, particularly as the GI epithelium secretes digestive molecules. Nevertheless, they are both endoderm-derived epithelia and both exhibit characteristic allergic features such as inflammation, mucus production, and eosinophil recruitment in response to injury. As a result, it is possible that both the airway epithelium and oesophageal (GI) epithelium may react in the same way to the same stimuli and therefore may also respond to similar therapies. More studies are needed to find any possible connection between viruses and EoE, but these studies and studies on epithelial host response similarities between the two disease processes may be essential and lead to novel common pathogenic mechanism for the induction of multiple inflammatory centered comorbidities.
It is inherently clear that RV infection is of critical importance in wheezing illnesses leading to asthma development and diagnosis. RV has been shown to have a dysregulatory effect on the barrier function and integrity of the paediatric airway epithelium, particularly in asthmatic children who may already have intrinsic epithelial vulnerabilities. Many of the effects of RV are related to the child’s epithelial host response which is unable to effectively fight the virus (23). Moreover, the plethora of anti-viral and pro-inflammatory responses to RV have been shown to overlap with atopic mechanisms and play a role in other severe allergic diseases including EoE. This highlights the importance of ‘epithelial vulnerability’ with dysregulated epithelial barrier function and integrity at the forefront of allergic disease, and many of them presenting as comorbidities. For example, in one cohort of children with EoE, 59.8% of them also had asthma (205). It also suggests commonality in elucidating effective therapeutics to tackle not only the symptoms of these diseases but their root cause. There are various pharmacological therapies used in the management of wheezing and asthma, EoE and other allergic diseases, although the search continues for more effective therapeutics with improved efficacy, especially considering the health burden for these diseases is large. Particularly, asthma and wheezing exacerbations may still occur regardless of maintenance with ongoing treatment or treatment type, with this echoed in EoE. Most current asthma therapeutics are aimed at treating the visible symptoms to reduce frequency and severity of exacerbations (238, 239). Similarly, as previously mentioned, the focus of treatment for EoE targets resultant fibrosis, but does not prevent or target the underlying cause of the disease. Preventing exacerbations in allergic diseases is challenging as there is a need to first understand the underlying mechanisms. The biological mechanisms as to why allergic diseases such as asthma and EoE occur in children remains to be fully understood and thus, broad therapies including short acting beta agonists, long acting beta agonists, and corticosteroids for asthma; and steroids, biologics directed against IgE and eosinophils and elimination diets for EoE cannot yet be replaced in favour of much more targeted treatment. In addition, some children respond less rapidly to these types of treatment and thus they are less effective (240). Excitingly, studies that have identified novel and re-purposed therapeutics as well as targetable biological pathways that potentially have anti-viral and epithelial barrier integrity aiding effects (61, 241–244). Two in particular include celecoxib, a COX-2 inhibiting non-steroidal anti-inflammatory drug, and azithromycin, a macrolide class antibiotic (61, 243). Celecoxib has been shown to restore wound repair capacity to the airway epithelium and azithromycin has shown to decrease the frequency of asthma exacerbations in adults (61, 243). The effects and full potential of these drugs are still being investigated. Nevertheless, it is postulated that as well as being useful in treating asthma exacerbations they may also be useful in other allergic diseases such as EoE due to their epithelial barrier function-restoring abilities. Furthermore, as TSLP plays an important role in both EoE and RV induced wheezing, Tezepelumab, a TSLP inhibiting agent may help attenuate exacerbations of both diseases though more evidence for this is needed (245). Although these emerging therapeutics are promising, RV infection, particularly RV-C infection in asthma, and epithelial dysregulation and vulnerability in allergic diseases need to be characterised fully in paediatric patients to determine the most effect strategy to improve the quality of life for these children, and thus are of active interest.
This review highlights the growing significance and clinical relevance of an innately vulnerable epithelium in different prevalent allergic-type epithelial diseases, and the effect of viral infection, particularly RV-C infection on airway epithelial barrier function and integrity in paediatric asthma, and viral and injury-induced effects on the oesophageal epithelium in EoE. In asthma specifically, it is postulated that innate epithelial vulnerability further potentiates RV-C infection, due to dysregulated host response to the virus. In addition, RV-C may further potentiate this vulnerability and barrier dysregulation compared to other RV species, leading to a persistent cycle of infection. Furthermore, it is suggested that consecutive or repeated insults to the airway of a child with a vulnerable epithelium culminates in persistent wheezing illnesses and eventually asthma development and diagnosis. This is one current research focus. Subsequently, once this has been identified, therapeutic development pipelines can be developed and potentially be extrapolated to and used in other allergic diseases such as EoE. It is important to compare diseases such as paediatric asthma and EoE as they have parallel disease pathologies and there are lessons that can be learnt from identifying commonalities such as epithelial vulnerabilities in children.
RW is the Primary Author of this manuscript. KL, IL, AC, and AK all contributed in equal proportions and have joint Senior Authorship over this manuscript. All authors contributed to the article and approved the submitted version.
AK is a Rothwell Family Fellow. Dr Ingrid Laing is funded by NHMRC App#1147630.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Martinez FD, Wright AL, Taussig LM, Holberg CJ, Halonen M, Morgan WJ. Asthma and Wheezing in the First Six Years of Life. The Group Health Medical Associates. N Engl J Med (1995) 332(3):133–8. doi: 10.1056/NEJM199501193320301
2. Fitzpatrick AM, Bacharier LB, Guilbert TW, Jackson DJ, Szefler SJ, Beigelman A, et al. Phenotypes of Recurrent Wheezing in Preschool Children: Identification by Latent Class Analysis and Utility in Prediction of Future Exacerbation. J Allergy Clin Immunol Pract (2019) 7(3):915–24.e7. doi: 10.1016/j.jaip.2018.09.016
3. Lowe LA, Simpson A, Woodcock A, Morris J, Murray CS, Custovic A. Wheeze Phenotypes and Lung Function in Preschool Children. Am J Respir Crit Care Med (2005) 171(3):231–7. doi: 10.1164/rccm.200406-695OC
4. Hallit S, Leynaert B, Delmas MC, Rocchi S, De Blic J, Marguet C, et al. Correction: Wheezing Phenotypes and Risk Factors in Early Life: The ELFE Cohort. PloS One (2018) 13(8):e0201863. doi: 10.1371/journal.pone.0201863
5. Castro-Rodriguez JA. The Asthma Predictive Index: A Very Useful Tool for Predicting Asthma in Young Children. J Allergy Clin Immunol (2010) 126(2):212–6. doi: 10.1016/j.jaci.2010.06.032
6. Van Wonderen KE, van der Mark LB, Mohrs J, Bindels PJ, Van Aalderen WM, Ter Riet G. Different Definitions in Childhood Asthma: How Dependable is the Dependent Variable? Eur Respir J (2010) 36(1):48–56. doi: 10.1183/09031936.00154409
7. Yunginger JW, Reed CE, O’Connell EJ, Melton LJ 3rd, O’Fallon WM, Silverstein MD. A Community-Based Study of the Epidemiology of Asthma. Incidence Rates, 1964-1983. Am Rev Respir Dis (1992) 146(4):888–94. doi: 10.1164/ajrccm/146.4.888
8. Busse W. Expert Panel Report 3 (EPR-3): Guidelines for the Diagnosis and Management of Asthma-Summary Report 2007. J Allergy Clin Immunol (2007) 120(5 Suppl):S94–138. doi: 10.1016/j.jaci.2007.09.029
9. Murray C, Foden P, Lowe L, Durrington H, Custovic A, Simpson A. Diagnosis of Asthma in Symptomatic Children Based on Measures of Lung Function: An Analysis of Data From a Population-Based Birth Cohort Study. Lancet Child Adolesc Health (2017) 1(2):114–23. doi: 10.1016/S2352-4642(17)30008-1
10. Taussig LM, Wright AL, Holberg CJ, Halonen M, Morgan WJ, Martinez FD. Tucson Children’s Respiratory Study: 1980 to Present. J Allergy Clin Immunol (2003) 111(4):661–75; quiz 76. doi: 10.1067/mai.2003.162
11. Jackson DJ, Gangnon RE, Evans MD, Roberg KA, Anderson EL, Pappas TE, et al. Wheezing Rhinovirus Illnesses in Early Life Predict Asthma Development in High-Risk Children. Am J Respir Crit Care Med (2008) 178(7):667–72. doi: 10.1164/rccm.200802-309OC
12. Ober C, Loisel DA, Gilad Y. Sex-Specific Genetic Architecture of Human Disease. Nat Rev Genet (2008) 9(12):911–22. doi: 10.1038/nrg2415
13. de Gouveia Belinelo P, Collison AM, Murphy VE, Robinson PD, Jesson K, Hardaker K, et al. Maternal Asthma Is Associated With Reduced Lung Function in Male Infants in a Combined Analysis of the BLT and BILD Cohorts. Thorax (2021) 76(10):996–1001. doi: 10.1136/thoraxjnl-2020-215526
14. Just J, Fournier L, Momas I, Zambetti C, Sahraoui F, Grimfeld A. Clinical Significance of Bronchoalveolar Eosinophils in Childhood Asthma. J Allergy Clin Immunol (2002) 110(1):42–4. doi: 10.1067/mai.2002.123304
15. Koller DY, Halmerbauer G, Frischer T, Roithner B. Assessment of Eosinophil Granule Proteins in Various Body Fluids: Is There a Relation to Clinical Variables in Childhood Asthma? Clin Exp Allergy (1999) 29(6):786–93. doi: 10.1046/j.1365-2222.1999.00546.x
16. Henderson J, Granell R, Heron J, Sherriff A, Simpson A, Woodcock A, et al. Associations of Wheezing Phenotypes in the First 6 Years of Life With Atopy, Lung Function and Airway Responsiveness in Mid-Childhood. Thorax (2008) 63(11):974–80. doi: 10.1136/thx.2007.093187
17. Sikazwe CT, Laing IA, Imrie A, Smith David W. Nasal Cytokine Profiles of Patients Hospitalised With Respiratory Wheeze Associated With Rhinovirus C. MDPI Viruses (2019) 11(11):1038. doi: 10.3390/v11111038
18. Purcell K, Fergie J. Driscoll Children’s Hospital Respiratory Syncytial Virus Database: Risk Factors, Treatment and Hospital Course in 3308 Infants and Young Children, 1991 to 2002. Pediatr Infect Dis J (2004) 23(5):418–23. doi: 10.1097/01.inf.0000126273.27123.33
19. Cox DW, Khoo SK, Zhang G, Lindsay K, Keil AD, Knight G, et al. Rhinovirus Is the Most Common Virus and Rhinovirus-C Is the Most Common Species in Paediatric Intensive Care Respiratory Admissions. Eur Respir J (2018) 52(2):1800207. doi: 10.1183/13993003.00207-2018
20. Bizzintino J, Lee WM, Laing IA, Vang F, Pappas T, Zhang G, et al. Association Between Human Rhinovirus C and Severity of Acute Asthma in Children. Eur Respir J (2011) 37(5):1037–42. doi: 10.1183/09031936.00092410
21. Calvo C, Garcia ML, Pozo F, Reyes N, Pérez-Breña P, Casas I. Role of Rhinovirus C in Apparently Life-Threatening Events in Infants, Spain. Emerg Infect Dis (2009) 15(9):1506–8. doi: 10.3201/1509.090493
22. McGovern MC, Smith MB. Causes of Apparent Life Threatening Events in Infants: A Systematic Review. Arch Dis Child (2004) 89(11):1043–8. doi: 10.1136/adc.2003.031740
23. Busse WW, Lemanske RF Jr., Gern JE. Role of Viral Respiratory Infections in Asthma and Asthma Exacerbations. Lancet (2010) 376(9743):826–34. doi: 10.1016/S0140-6736(10)61380-3
24. Bacharier LB, Beigelman A, Calatroni A, Jackson DJ, Gergen PJ, O’Connor GT, et al. Longitudinal Phenotypes of Respiratory Health in a High-Risk Urban Birth Cohort. Am J Respir Crit Care Med (2019) 199(1):71–82. doi: 10.1164/rccm.201801-0190OC
25. Oksel C, Granell R, Haider S, Fontanella S, Simpson A, Turner S, et al. Distinguishing Wheezing Phenotypes From Infancy to Adolescence. A Pooled Analysis of Five Birth Cohorts. Ann Am Thorac Soc (2019) 16(7):868–76. doi: 10.1513/AnnalsATS.201811-837OC
26. Leffler J, Jones AC, Hollams EM, Prastanti F, Le Souëf PN, Holt PG, et al. Basophil Counts in PBMC Populations During Childhood Acute Wheeze/Asthma Are Associated With Future Exacerbations. J Allergy Clin Immunol (2018) 142(5):1639–41.e5. doi: 10.1016/j.jaci.2018.07.003
27. Paaso EM, Jaakkola MS, Lajunen TK, Hugg TT, Jaakkola JJ. The Importance of Family History in Asthma During the First 27 Years of Life. Am J Respir Crit Care Med (2013) 188(5):624–6. doi: 10.1164/rccm.201212-2236LE
28. Rubner FJ, Jackson DJ, Evans MD, Gangnon RE, Tisler CJ, Pappas TE, et al. Early Life Rhinovirus Wheezing, Allergic Sensitization, and Asthma Risk at Adolescence. J Allergy Clin Immunol (2017) 139(2):501–7. doi: 10.1016/j.jaci.2016.03.049
29. Loisel DA, Du G, Ahluwalia TS, Tisler CJ, Evans MD, Myers RA, et al. Genetic Associations With Viral Respiratory Illnesses and Asthma Control in Children. Clin Exp Allergy (2016) 46(1):112–24. doi: 10.1111/cea.12642
30. Lemanske RF Jr. The Childhood Origins of Asthma (COAST) Study. Pediatr Allergy Immunol (2002) 13(s15):38–43. doi: 10.1034/j.1399-3038.13.s.15.8.x
31. Loss GJ, Depner M, Hose AJ, Genuneit J, Karvonen AM, Hyvärinen A, et al. The Early Development of Wheeze. Environmental Determinants and Genetic Susceptibility at 17q21. Am J Respir Crit Care Med (2016) 193(8):889–97. doi: 10.1164/rccm.201507-1493OC
32. Perez L, Declercq C, Iñiguez C, Aguilera I, Badaloni C, Ballester F, et al. Chronic Burden of Near-Roadway Traffic Pollution in 10 European Cities (APHEKOM Network). Eur Respir J (2013) 42(3):594–605. doi: 10.1183/09031936.00031112
33. Stoltz DJ, Jackson DJ, Evans MD, Gangnon RE, Tisler CJ, Gern JE, et al. Specific Patterns of Allergic Sensitization in Early Childhood and Asthma & Rhinitis Risk. Clin Exp Allergy (2013) 43(2):233–41. doi: 10.1111/cea.12050
34. Dasaraju PV, Liu C. Infections of the Respiratory System. In: Baron S, editor. Medical Microbiology. Galveston (TX: University of Texas Medical Branch at Galveston (1996). Copyright © 1996, The University of Texas Medical Branch at Galveston.
35. Cox DW, Bizzintino J, Ferrari G, Khoo SK, Zhang G, Whelan S, et al. Human Rhinovirus Species C Infection in Young Children With Acute Wheeze is Associated With Increased Acute Respiratory Hospital Admissions. Am J Respir Crit Care Med (2013) 188(11):1358–64. doi: 10.1164/rccm.201303-0498OC
36. Halmø Hürdum S, Zhang G, Khoo SK, Bizzintino J, Franks KM, Lindsay K, et al. Recurrent Rhinovirus Detections in Children Following a Rhinovirus-Induced Wheezing Exacerbation: A Retrospective Study. Int J Pediatr Child Health (2015) 3(1):10–8. doi: 10.12974/2311-8687.2015.03.01.2
37. Annamalay AA, Jroundi I, Bizzintino J, Khoo SK, Zhang G, Lehmann D, et al. Rhinovirus C is Associated With Wheezing and Rhinovirus A is Associated With Pneumonia in Hospitalized Children in Morocco. J Med Virol (2017) 89(4):582–8. doi: 10.1002/jmv.24684
38. Kicic A, Stevens PT, Sutanto EN, Kicic-Starcevich E, Ling KM, Looi K, et al. Impaired Airway Epithelial Cell Responses From Children With Asthma to Rhinoviral Infection. Clin Exp Allergy (2016) 46(11):1441–55. doi: 10.1111/cea.12767
39. Kicic A, Sutanto EN, Stevens PT, Knight DA, Stick SM. Intrinsic Biochemical and Functional Differences in Bronchial Epithelial Cells of Children With Asthma. Am J Respir Crit Care Med (2006) 174(10):1110–8. doi: 10.1164/rccm.200603-392OC
40. Loxham M, Smart DE, Bedke NJ, Smithers NP, Filippi I, Blume C, et al. Allergenic Proteases Cleave the Chemokine CX3CL1 Directly From the Surface of Airway Epithelium and Augment the Effect of Rhinovirus. Mucosal Immunol (2018) 11(2):404–14. doi: 10.1038/mi.2017.63
41. Holgate ST. Epithelium Dysfunction in Asthma. J Allergy Clin Immunol (2007) 120(6):1233–44; quiz 45-6. doi: 10.1016/j.jaci.2007.10.025
42. Holgate ST. The Airway Epithelium is Central to the Pathogenesis of Asthma. Allergol Int (2008) 57(1):1–10. doi: 10.2332/allergolint.R-07-154
43. Holgate ST. The Epithelium Takes Centre Stage in Asthma and Atopic Dermatitis. Trends Immunol (2007) 28(6):248–51. doi: 10.1016/j.it.2007.04.007
44. Holgate ST, Roberts G, Arshad HS, Howarth PH, Davies DE. The Role of the Airway Epithelium and Its Interaction With Environmental Factors in Asthma Pathogenesis. Proc Am Thorac Soc (2009) 6(8):655–9. doi: 10.1513/pats.200907-072DP
45. Holgate ST. Innate and Adaptive Immune Responses in Asthma. Nat Med (2012) 18(5):673–83. doi: 10.1038/nm.2731
46. Knight DA, Holgate ST. The Airway Epithelium: Structural and Functional Properties in Health and Disease. Respirology (2003) 8(4):432–46. doi: 10.1046/j.1440-1843.2003.00493.x
47. Ruiz García S, Deprez M, Lebrigand K, Cavard A, Paquet A, Arguel MJ, et al. Novel Dynamics of Human Mucociliary Differentiation Revealed by Single-Cell RNA Sequencing of Nasal Epithelial Cultures. Development (2019) 146(20):dev177428. doi: 10.1242/dev.177428
48. Mercer RR, Russell ML, Roggli VL, Crapo JD. Cell Number and Distribution in Human and Rat Airways. Am J Respir Cell Mol Biol (1994) 10(6):613–24. doi: 10.1165/ajrcmb.10.6.8003339
49. Montoro DT, Haber AL, Biton M, Vinarsky V, Lin B, Birket SE, et al. A Revised Airway Epithelial Hierarchy Includes CFTR-Expressing Ionocytes. Nature (2018) 560(7718):319–24. doi: 10.1038/s41586-018-0393-7
50. Youngson C, Nurse C, Yeger H, Cutz E. Oxygen Sensing in Airway Chemoreceptors. Nature (1993) 365(6442):153–5. doi: 10.1038/365153a0
51. Gosney JR, Sissons MC, Allibone RO. Neuroendocrine Cell Populations in Normal Human Lungs: A Quantitative Study. Thorax (1988) 43(11):878–82. doi: 10.1136/thx.43.11.878
52. Hartsock A, Nelson WJ. Adherens and Tight Junctions: Structure, Function and Connections to the Actin Cytoskeleton. Biochim Biophys Acta (2008) 1778(3):660–9. doi: 10.1016/j.bbamem.2007.07.012
53. Sparrow MP, Omari TI, Mitchell HW. The Epithelial Barrier and Airway Responsiveness. Can J Physiol Pharmacol (1995) 73(2):180–90. doi: 10.1139/y95-027
54. Churg A. The Uptake of Mineral Particles by Pulmonary Epithelial Cells. Am J Respir Crit Care Med (1996) 154(4 Pt 1):1124–40. doi: 10.1164/ajrccm.154.4.8887617
55. Farquhar MG, Palade GE. Junctional Complexes in Various Epithelia. J Cell Biol (1963) 17(2):375–412. doi: 10.1083/jcb.17.2.375
56. Button B, Cai LH, Ehre C, Kesimer M, Hill DB, Sheehan JK, et al. A Periciliary Brush Promotes the Lung Health by Separating the Mucus Layer From Airway Epithelia. Science (2012) 337(6097):937–41. doi: 10.1126/science.1223012
57. Stonebraker JR, Wagner D, Lefensty RW, Burns K, Gendler SJ, Bergelson JM, et al. Glycocalyx Restricts Adenoviral Vector Access to Apical Receptors Expressed on Respiratory Epithelium In Vitro and In Vivo: Role for Tethered Mucins as Barriers to Lumenal Infection. J Virol (2004) 78(24):13755–68. doi: 10.1128/JVI.78.24.13755-13768.2004
58. McAuley JL, Corcilius L, Tan HX, Payne RJ, McGuckin MA, Brown LE. The Cell Surface Mucin MUC1 Limits the Severity of Influenza A Virus Infection. Mucosal Immunol (2017) 10(6):1581–93. doi: 10.1038/mi.2017.16
59. Kirkham S, Sheehan JK, Knight D, Richardson PS, Thornton DJ. Heterogeneity of Airways Mucus: Variations in the Amounts and Glycoforms of the Major Oligomeric Mucins MUC5AC and MUC5B. Biochem J (2002) 361(Pt 3):537–46. doi: 10.1042/bj3610537
60. Ermund A, Meiss LN, Rodriguez-Pineiro AM, Bähr A, Nilsson HE, Trillo-Muyo S, et al. The Normal Trachea is Cleaned by MUC5B Mucin Bundles From the Submucosal Glands Coated With the MUC5AC Mucin. Biochem Biophys Res Commun (2017) 492(3):331–7. doi: 10.1016/j.bbrc.2017.08.113
61. Iosifidis T, Sutanto EN, Buckley AG, Coleman L, Gill EE, Lee AH, et al. Aberrant Cell Migration Contributes to Defective Airway Epithelial Repair in Childhood Wheeze. JCI Insight (2020) 5(7):e133125. doi: 10.1172/jci.insight.133125
62. Boers JE, Ambergen AW, Thunnissen FB. Number and Proliferation of Basal and Parabasal Cells in Normal Human Airway Epithelium. Am J Respir Crit Care Med (1998) 157(6 Pt 1):2000–6. doi: 10.1164/ajrccm.157.6.9707011
63. Hackett NR, Shaykhiev R, Walters MS, Wang R, Zwick RK, Ferris B, et al. The Human Airway Epithelial Basal Cell Transcriptome. PloS One (2011) 6(5):e18378. doi: 10.1371/journal.pone.0018378
64. van Eeden SF, Tan WC, Suwa T, Mukae H, Terashima T, Fujii T, et al. Cytokines Involved in the Systemic Inflammatory Response Induced by Exposure to Particulate Matter Air Pollutants (PM(10)). Am J Respir Crit Care Med (2001) 164(5):826–30.
65. Tsai DH, Amyai N, Marques-Vidal P, Wang JL, Riediker M, Mooser V, et al. Effects of Particulate Matter on Inflammatory Markers in the General Adult Population. Part Fibre Toxicol (2012) 9:24. doi: 10.1186/1743-8977-9-24
66. Sakamoto N, Hayashi S, Gosselink J, Ishii H, Ishimatsu Y, Mukae H, et al. Calcium Dependent and Independent Cytokine Synthesis by Air Pollution Particle-Exposed Human Bronchial Epithelial Cells. Toxicol Appl Pharmacol (2007) 225(2):134–41. doi: 10.1016/j.taap.2007.07.006
67. Fujii T, Hayashi S, Hogg JC, Vincent R, Van Eeden SF. Particulate Matter Induces Cytokine Expression in Human Bronchial Epithelial Cells. Am J Respir Cell Mol Biol (2001) 25(3):265–71. doi: 10.1165/ajrcmb.25.3.4445
68. Veranth JM, Moss TA, Chow JC, Labban R, Nichols WK, Walton JC, et al. Correlation of In Vitro Cytokine Responses With the Chemical Composition of Soil-Derived Particulate Matter. Environ Health Perspect (2006) 114(3):341–9. doi: 10.1289/ehp.8360
69. Gruzieva O, Merid SK, Gref A, Gajulapuri A, Lemonnier N, Ballereau S, et al. Exposure to Traffic-Related Air Pollution and Serum Inflammatory Cytokines in Children. Environ Health Perspect (2017) 125(6):067007. doi: 10.1289/EHP460
70. Hammad H, Chieppa M, Perros F, Willart MA, Germain RN, Lambrecht BN. House Dust Mite Allergen Induces Asthma via Toll-Like Receptor 4 Triggering of Airway Structural Cells. Nat Med (2009) 15(4):410–6. doi: 10.1038/nm.1946
71. Veerati PC, Troy NM, Reid AT, Li NF, Nichol KS, Kaur P, et al. Airway Epithelial Cell Immunity Is Delayed During Rhinovirus Infection in Asthma and COPD. Front Immunol (2020) 11:974. doi: 10.3389/fimmu.2020.00974
72. Francis ML, Meltzer MS. Induction of IFN-Alpha by HIV-1 in Monocyte-Enriched PBMC Requires Gp120-CD4 Interaction But Not Virus Replication. J Immunol (1993) 151(4):2208–16.
73. Merika M, Williams AJ, Chen G, Collins T, Thanos D. Recruitment of CBP/p300 by the IFN Beta Enhanceosome is Required for Synergistic Activation of Transcription. Mol Cell (1998) 1(2):277–87. doi: 10.1016/S1097-2765(00)80028-3
74. Wathelet MG, Lin CH, Parekh BS, Ronco LV, Howley PM, Maniatis T. Virus Infection Induces the Assembly of Coordinately Activated Transcription Factors on the IFN-Beta Enhancer In Vivo. Mol Cell (1998) 1(4):507–18. doi: 10.1016/S1097-2765(00)80051-9
75. Orange JS, Biron CA. An Absolute and Restricted Requirement for IL-12 in Natural Killer Cell IFN-Gamma Production and Antiviral Defense. Studies of Natural Killer and T Cell Responses in Contrasting Viral Infections. J Immunol (1996) 156(3):1138–42.
76. Sareneva T, Matikainen S, Kurimoto M, Julkunen I. Influenza A Virus-Induced IFN-Alpha/Beta and IL-18 Synergistically Enhance IFN-Gamma Gene Expression in Human T Cells. J Immunol (1998) 160(12):6032–8.
77. Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK, et al. IFN-Lambdas Mediate Antiviral Protection Through a Distinct Class II Cytokine Receptor Complex. Nat Immunol (2003) 4(1):69–77. doi: 10.1038/ni875
78. Fox JM, Crabtree JM, Sage LK, Tompkins SM, Tripp RA. Interferon Lambda Upregulates IDO1 Expression in Respiratory Epithelial Cells After Influenza Virus Infection. J Interferon Cytokine Res (2015) 35(7):554–62. doi: 10.1089/jir.2014.0052
79. Terajima M, Yamaya M, Sekizawa K, Okinaga S, Suzuki T, Yamada N, et al. Rhinovirus Infection of Primary Cultures of Human Tracheal Epithelium: Role of ICAM-1 and IL-1beta. Am J Physiol (1997) 273(4):L749–59. doi: 10.1152/ajplung.1997.273.4.L749
80. Peiró T, Patel DF, Akthar S, Gregory LG, Pyle CJ, Harker JA, et al. Neutrophils Drive Alveolar Macrophage IL-1β Release During Respiratory Viral Infection. Thorax (2018) 73(6):546–56. doi: 10.1136/thoraxjnl-2017-210010
81. Ravi A, Koster J, Dijkhuis A, Bal SM, Sabogal Piñeros YS, Bonta PI, et al. Interferon-Induced Epithelial Response to Rhinovirus 16 in Asthma Relates to Inflammation and FEV(1). J Allergy Clin Immunol (2019) 143(1):442–7.e10. doi: 10.1016/j.jaci.2018.09.016
82. Zheng J, Shi Y, Xiong L, Zhang W, Li Y, Gibson PG, et al. The Expression of IL-6, TNF-α, and MCP-1 in Respiratory Viral Infection in Acute Exacerbations of Chronic Obstructive Pulmonary Disease. J Immunol Res (2017) 2017:8539294. doi: 10.1155/2017/8539294
83. Johnston SL, Papi A, Bates PJ, Mastronarde JG, Monick MM, Hunninghake GW. Low Grade Rhinovirus Infection Induces a Prolonged Release of IL-8 in Pulmonary Epithelium. J Immunol (1998) 160(12):6172–81.
84. Schroth MK, Grimm E, Frindt P, Galagan DM, Konno SI, Love R, et al. Rhinovirus Replication Causes RANTES Production in Primary Bronchial Epithelial Cells. Am J Respir Cell Mol Biol (1999) 20(6):1220–8. doi: 10.1165/ajrcmb.20.6.3261
85. Papadopoulos NG, Papi A, Meyer J, Stanciu LA, Salvi S, Holgate ST, et al. Rhinovirus Infection Up-Regulates Eotaxin and Eotaxin-2 Expression in Bronchial Epithelial Cells. Clin Exp Allergy (2001) 31(7):1060–6. doi: 10.1046/j.1365-2222.2001.01112.x
86. Ganesan S, Pham D, Jing Y, Farazuddin M, Hudy MH, Unger B, et al. TLR2 Activation Limits Rhinovirus-Stimulated CXCL-10 by Attenuating IRAK-1-Dependent IL-33 Receptor Signaling in Human Bronchial Epithelial Cells. J Immunol (2016) 197(6):2409–20. doi: 10.4049/jimmunol.1502702
87. Kato A, Favoreto S Jr, Avila PC, Schleimer RP. TLR3- and Th2 Cytokine-Dependent Production of Thymic Stromal Lymphopoietin in Human Airway Epithelial Cells. J Immunol (2007) 179(2):1080–7. doi: 10.4049/jimmunol.179.2.1080
88. Green AM, Difazio R, Flynn JL. IFN-γ From CD4 T Cells is Essential for Host Survival and Enhances CD8 T Cell Function During Mycobacterium Tuberculosis Infection. J Immunol (2013) 190(1):270–7. doi: 10.4049/jimmunol.1200061
89. Beekhuizen H, van de Gevel JS. Gamma Interferon Confers Resistance to Infection With Staphylococcus Aureus in Human Vascular Endothelial Cells by Cooperative Proinflammatory and Enhanced Intrinsic Antibacterial Activities. Infect Immun (2007) 75(12):5615–26. doi: 10.1128/IAI.00530-07
90. Flynn JL, Chan J, Triebold KJ, Dalton DK, Stewart TA, Bloom BR. An Essential Role for Interferon Gamma in Resistance to Mycobacterium Tuberculosis Infection. J Exp Med (1993) 178(6):2249–54. doi: 10.1084/jem.178.6.2249
91. Krakauer T. Stimulant-Dependent Modulation of Cytokines and Chemokines by Airway Epithelial Cells: Cross Talk Between Pulmonary Epithelial and Peripheral Blood Mononuclear Cells. Clin Diagn Lab Immunol (2002) 9(1):126–31. doi: 10.1128/cdli.9.1.126-131
92. Clemans DL, Bauer RJ, Hanson JA, Hobbs MV, St Geme JW 3rd, Marrs CF, et al. Induction of Proinflammatory Cytokines From Human Respiratory Epithelial Cells After Stimulation by Nontypeable Haemophilus Influenzae. Infect Immun (2000) 68(8):4430–40. doi: 10.1128/IAI.68.8.4430-4440.2000
93. van der Meer JW, Barza M, Wolff SM, Dinarello CA. A Low Dose of Recombinant Interleukin 1 Protects Granulocytopenic Mice From Lethal Gram-Negative Infection. Proc Natl Acad Sci USA (1988) 85(5):1620–3. doi: 10.1073/pnas.85.5.1620
94. Khair OA, Devalia JL, Abdelaziz MM, Sapsford RJ, Tarraf H, Davies RJ. Effect of Haemophilus Influenzae Endotoxin on the Synthesis of IL-6, IL-8, TNF-Alpha and Expression of ICAM-1 in Cultured Human Bronchial Epithelial Cells. Eur Respir J (1994) 7(12):2109–16. doi: 10.1183/09031936.94.07122109
95. Saba S, Soong G, Greenberg S, Prince A. Bacterial Stimulation of Epithelial G-CSF and GM-CSF Expression Promotes PMN Survival in CF Airways. Am J Respir Cell Mol Biol (2002) 27(5):561–7. doi: 10.1165/rcmb.2002-0019OC
96. Fallahi-Sichani M, Kirschner DE, Linderman JJ. NF-κB Signaling Dynamics Play a Key Role in Infection Control in Tuberculosis. Front Physiol (2012) 3:170. doi: 10.3389/fphys.2012.00170
97. Regueiro V, Moranta D, Campos MA, Margareto J, Garmendia J, Bengoechea JA. Klebsiella Pneumoniae Increases the Levels of Toll-Like Receptors 2 and 4 in Human Airway Epithelial Cells. Infect Immun (2009) 77(2):714–24. doi: 10.1128/IAI.00852-08
98. Raoust E, Balloy V, Garcia-Verdugo I, Touqui L, Ramphal R, Chignard M. Pseudomonas Aeruginosa LPS or Flagellin Are Sufficient to Activate TLR-Dependent Signaling in Murine Alveolar Macrophages and Airway Epithelial Cells. PloS One (2009) 4(10):e7259. doi: 10.1371/journal.pone.0007259
99. Adamo R, Sokol S, Soong G, Gomez MI, Prince A. Pseudomonas Aeruginosa Flagella Activate Airway Epithelial Cells Through Asialogm1 and Toll-Like Receptor 2 as Well as Toll-Like Receptor 5. Am J Respir Cell Mol Biol (2004) 30(5):627–34. doi: 10.1165/rcmb.2003-0260OC
100. Mambula SS, Sau K, Henneke P, Golenbock DT, Levitz SM. Toll-Like Receptor (TLR) Signaling in Response to Aspergillus Fumigatus. J Biol Chem (2002) 277(42):39320–6. doi: 10.1074/jbc.M201683200
101. Gresnigt MS, Rösler B, Jacobs CW, Becker KL, Joosten LA, van der Meer JW, et al. The IL-36 Receptor Pathway Regulates Aspergillus Fumigatus-Induced Th1 and Th17 Responses. Eur J Immunol (2013) 43(2):416–26. doi: 10.1002/eji.201242711
102. Wang JE, Warris A, Ellingsen EA, Jørgensen PF, Flo TH, Espevik T, et al. Involvement of CD14 and Toll-Like Receptors in Activation of Human Monocytes by Aspergillus Fumigatus Hyphae. Infect Immun (2001) 69(4):2402–6. doi: 10.1128/IAI.69.4.2402-2406.2001
103. Cenci E, Mencacci A, FdO C, Del Sero G, Mosci P, Montagnoli C, et al. Cytokine- and T Helper-Dependent Lung Mucosal Immunity in Mice With Invasive Pulmonary Aspergillosis. J Infect Dis (1998) 178(6):1750–60. doi: 10.1086/314493
104. Brieland JK, Jackson C, Menzel F, Loebenberg D, Cacciapuoti A, Halpern J, et al. Cytokine Networking in Lungs of Immunocompetent Mice in Response to Inhaled Aspergillus Fumigatus. Infect Immun (2001) 69(3):1554–60. doi: 10.1128/IAI.69.3.1554-1560.2001
105. Mehrad B, Strieter RM, Standiford TJ. Role of TNF-Alpha in Pulmonary Host Defense in Murine Invasive Aspergillosis. J Immunol (1999) 162(3):1633–40.
106. Frey A, Lunding LP, Ehlers JC, Weckmann M, Zissler UM, Wegmann M. More Than Just a Barrier: The Immune Functions of the Airway Epithelium in Asthma Pathogenesis. Front Immunol (2020) 11:761. doi: 10.3389/fimmu.2020.00761
107. Hellings PW, Steelant B. Epithelial Barriers in Allergy and Asthma. J Allergy Clin Immunol (2020) 145(6):1499–509. doi: 10.1016/j.jaci.2020.04.010
108. Michi AN, Love ME, Proud D. Rhinovirus-Induced Modulation of Epithelial Phenotype: Role in Asthma. Viruses (2020) 12(11):1328. doi: 10.3390/v12111328
109. Kicic A, de Jong E, Ling KM, Nichol K, Anderson D, Wark PAB, et al. Assessing the Unified Airway Hypothesis in Children via Transcriptional Profiling of the Airway Epithelium. J Allergy Clin Immunol (2020) 145(6):1562–73. doi: 10.1016/j.jaci.2020.02.018
110. Foxman EF, Storer JA, Fitzgerald ME, Wasik BR, Hou L, Zhao H, et al. Temperature-Dependent Innate Defense Against the Common Cold Virus Limits Viral Replication at Warm Temperature in Mouse Airway Cells. Proc Natl Acad Sci USA (2015) 112(3):827–32. doi: 10.1073/pnas.1411030112
111. Lopez-Souza N, Favoreto S, Wong H, Ward T, Yagi S, Schnurr D, et al. In Vitro Susceptibility to Rhinovirus Infection is Greater for Bronchial Than for Nasal Airway Epithelial Cells in Human Subjects. J Allergy Clin Immunol (2009) 123(6):1384–90.e2. doi: 10.1016/j.jaci.2009.03.010
112. Miller EK, Hernandez JZ, Wimmenauer V, Shepherd BE, Hijano D, Libster R, et al. A Mechanistic Role for Type III IFN-λ1 in Asthma Exacerbations Mediated by Human Rhinoviruses. Am J Respir Crit Care Med (2012) 185(5):508–16. doi: 10.1164/rccm.201108-1462OC
113. Khoo SK, Read J, Franks K, Zhang G, Bizzintino J, Coleman L, et al. Upper Airway Cell Transcriptomics Identify a Major New Immunological Phenotype With Strong Clinical Correlates in Young Children With Acute Wheezing. J Immunol (2019) 202(6):1845–58. doi: 10.4049/jimmunol.1800178
114. Hutchinson K, Kerley CP, Faul J, Greally P, Coghlan D, Louw M, et al. Vitamin D Receptor Variants and Uncontrolled Asthma. Eur Ann Allergy Clin Immunol (2018) 50(3):108–16. doi: 10.23822/EurAnnACI.1764-1489.46
115. Bønnelykke K, Sleiman P, Nielsen K, Kreiner-Møller E, Mercader JM, Belgrave D, et al. A Genome-Wide Association Study Identifies CDHR3 as a Susceptibility Locus for Early Childhood Asthma With Severe Exacerbations. Nat Genet (2014) 46(1):51–5. doi: 10.1038/ng.2830
116. O’Neill MB, Laval G, Teixeira JC, Palmenberg AC, Pepperell CS. Genetic Susceptibility to Severe Childhood Asthma and Rhinovirus-C Maintained by Balancing Selection in Humans for 150 000 Years. Hum Mol Genet (2020) 29(5):736–44. doi: 10.1093/hmg/ddz304
117. Basnet S, Bochkov YA, Brockman-Schneider RA, Kuipers I, Aesif SW, Jackson DJ, et al. CDHR3 Asthma-Risk Genotype Affects Susceptibility of Airway Epithelium to Rhinovirus C Infections. Am J Respir Cell Mol Biol (2019) 61(4):450–8. doi: 10.1165/rcmb.2018-0220OC
118. Lund RJ, Osmala M, Malonzo M, Lukkarinen M, Leino A, Salmi J, et al. Atopic Asthma After Rhinovirus-Induced Wheezing Is Associated With DNA Methylation Change in the SMAD3 Gene Promoter. Allergy (2018) 73(8):1735–40. doi: 10.1111/all.13473
119. Pech M, Weckmann M, König IR, Franke A, Heinsen FA, Oliver B, et al. Rhinovirus Infections Change DNA Methylation and mRNA Expression in Children With Asthma. PloS One (2018) 13(11):e0205275. doi: 10.1371/journal.pone.0205275
120. Hillas J ED, Hemy N, Iosifidis T, Looi K, Garratt L, Simpson S, et al. Neonatal Predictors of Aberrant Wound Repair in Very Preterm Infants. Respirology (2019) 24(S1):TP024. doi: 10.1111/resp.13492
121. May M, Marx A, Seidenspinner S, Speer CP. Apoptosis and Proliferation in Lungs of Human Fetuses Exposed to Chorioamnionitis. Histopathology (2004) 45(3):283–90. doi: 10.1111/j.1365-2559.2004.01936.x
122. Been JV, Zimmermann LJ, Debeer A, Kloosterboer N, van Iwaarden JF. Bronchoalveolar Lavage Fluid From Preterm Infants With Chorioamnionitis Inhibits Alveolar Epithelial Repair. Respir Res (2009) 10(1):116. doi: 10.1186/1465-9921-10-116
123. Tulah AS, Holloway JW, Sayers I. Defining the Contribution of SNPs Identified in Asthma GWAS to Clinical Variables in Asthmatic Children. BMC Med Genet (2013) 14:100. doi: 10.1186/1471-2350-14-100
124. Martin AC, Laing IA, Khoo SK, Zhang G, Rueter K, Teoh L, et al. Acute Asthma in Children: Relationships Among CD14 and CC16 Genotypes, Plasma Levels, and Severity. Am J Respir Crit Care Med (2006) 173(6):617–22. doi: 10.1164/rccm.200509-1367OC
125. Bizzintino JA, Khoo SK, Zhang G, Martin AC, Rueter K, Geelhoed GC, et al. Leukotriene Pathway Polymorphisms Are Associated With Altered Cysteinyl Leukotriene Production in Children With Acute Asthma. Prostaglandins Leukot Essent Fatty Acids (2009) 81(1):9–15. doi: 10.1016/j.plefa.2009.05.022
126. Ali M, Zhang G, Thomas WR, McLean CJ, Bizzintino JA, Laing IA, et al. Investigations Into the Role of ST2 in Acute Asthma in Children. Tissue Antigens (2009) 73(3):206–12. doi: 10.1111/j.1399-0039.2008.01185.x
127. Schultz EN, Devadason SG, Khoo SK, Zhang G, Bizzintino JA, Martin AC, et al. The Role of GSTP1 Polymorphisms and Tobacco Smoke Exposure in Children With Acute Asthma. J Asthma (2010) 47(9):1049–56. doi: 10.1080/02770903.2010.508856
128. Savenije OE, Kerkhof M, Reijmerink NE, Brunekreef B, de Jongste JC, Smit HA, et al. Interleukin-1 Receptor-Like 1 Polymorphisms Are Associated With Serum IL1RL1-A, Eosinophils, and Asthma in Childhood. J Allergy Clin Immunol (2011) 127(3):750–6.e1-5. doi: 10.1016/j.jaci.2010.12.014
129. Moffatt MF, Gut IG, Demenais F, Strachan DP, Bouzigon E, Heath S, et al. A Large-Scale, Consortium-Based Genomewide Association Study of Asthma. N Engl J Med (2010) 363(13):1211–21. doi: 10.1056/NEJMoa0906312
130. Jackson D FK, Rosasco M, Gill M, Liu A, Gruchalla R, O G, et al. The Influence of MUC5AC SNPs on Expression of MUC5AC and Mucus Hypersecretion Genes During Asthma Exacerbations. J Allergy Clin Immunol (2020) 145(2):SAB176. doi: 10.1016/j.jaci.2019.12.371
131. Puthothu B, Krueger M, Bernhardt M, Heinzmann A. ICAM1 Amino-Acid Variant K469E is Associated With Paediatric Bronchial Asthma and Elevated Sicam1 Levels. Genes Immun (2006) 7(4):322–6. doi: 10.1038/sj.gene.6364302
132. Steba GS, Koekkoek SM, Tanck MWT, Vanhommerig JW, van der Meer JTM, Kwa D, et al. SNP Rs688 Within the Low-Density Lipoprotein Receptor (LDL-R) Gene Associates With HCV Susceptibility. Liver Int (2019) 39(3):463–9. doi: 10.1111/liv.13978
133. Watters K, Palmenberg AC. CDHR3 Extracellular Domains EC1-3 Mediate Rhinovirus C Interaction With Cells and as Recombinant Derivatives, Are Inhibitory to Virus Infection. PloS Pathog (2018) 14(12):e1007477. doi: 10.1371/journal.ppat.1007477
134. Bochkov YA, Watters K, Ashraf S, Griggs TF, Devries MK, Jackson DJ, et al. Cadherin-Related Family Member 3, a Childhood Asthma Susceptibility Gene Product, Mediates Rhinovirus C Binding and Replication. Proc Natl Acad Sci U S A (2015) 112(17):5485–90. doi: 10.1073/pnas.1421178112
135. Haas IG, Frank M, Véron N, Kemler R. Presenilin-Dependent Processing and Nuclear Function of Gamma-Protocadherins. J Biol Chem (2005) 280(10):9313–9. doi: 10.1074/jbc.M412909200
136. Stenberg Hammar K, Niespodziana K, van Hage M, Kere J, Valenta R, Hedlin G, et al. Reduced CDHR3 Expression in Children Wheezing With Rhinovirus. Pediatr Allergy Immunol (2018) 29(2):200–6. doi: 10.1111/pai.12858
137. Liu L, Liu X, Ren X, Tian Y, Chen Z, Xu X, et al. Smad2 and Smad3 Have Differential Sensitivity in Relaying TGFβ Signaling and Inversely Regulate Early Lineage Specification. Sci Rep (2016) 6:21602. doi: 10.1038/srep21602
138. Kicic A, Hallstrand TS, Sutanto EN, Stevens PT, Kobor MS, Taplin C, et al. Decreased Fibronectin Production Significantly Contributes to Dysregulated Repair of Asthmatic Epithelium. Am J Respir Crit Care Med (2010) 181(9):889–98. doi: 10.1164/rccm.200907-1071OC
139. Louie JK, Roy-Burman A, Guardia-Labar L, Boston EJ, Kiang D, Padilla T, et al. Rhinovirus Associated With Severe Lower Respiratory Tract Infections in Children. Pediatr Infect Dis J (2009) 28(4):337–9. doi: 10.1097/INF.0b013e31818ffc1b
140. Palmenberg AC, Gern JE. Classification and Evolution of Human Rhinoviruses. Methods Mol Biol (2015) 1221:1–10. doi: 10.1007/978-1-4939-1571-2_1
141. Palmenberg AC, Rhinovirus C. Asthma, and Cell Surface Expression of Virus Receptor CDHR3. J Virol (2017) 91(7):e00072–17. doi: 10.1128/JVI.00072-17
142. Basta HA, Sgro JY, Palmenberg AC. Modeling of the Human Rhinovirus C Capsid Suggests a Novel Topography With Insights on Receptor Preference and Immunogenicity. Virology (2014) 448:176–84. doi: 10.1016/j.virol.2013.10.006
143. Adler FR, Stockmann C, Ampofo K, Pavia AT, Byington CL. Transmission of Rhinovirus in the Utah BIG-LoVE Families: Consequences of Age and Household Structure. PloS One (2018) 13(7):e0199388. doi: 10.1371/journal.pone.0199388
144. Heymann PW, Carper HT, Murphy DD, Platts-Mills TA, Patrie J, McLaughlin AP, et al. Viral Infections in Relation to Age, Atopy, and Season of Admission Among Children Hospitalized for Wheezing. J Allergy Clin Immunol (2004) 114(2):239–47. doi: 10.1016/j.jaci.2004.04.006
145. Lee WM, Lemanske RF Jr., Evans MD, Vang F, Pappas T, Gangnon R, et al. Human Rhinovirus Species and Season of Infection Determine Illness Severity. Am J Respir Crit Care Med (2012) 186(9):886–91. doi: 10.1164/rccm.201202-0330OC
146. Toivonen L, Camargo CA, Gern JE, Bochkov YA, Mansbach JM, Piedra PA, et al. Association Between Rhinovirus Species and Nasopharyngeal Microbiota in Infants With Severe Bronchiolitis. J Allergy Clin Immunol (2019) 143(5):1925–8.e7. doi: 10.1016/j.jaci.2018.12.1004
147. Miller EK, Lu X, Erdman DD, Poehling KA, Zhu Y, Griffin MR, et al. Rhinovirus-Associated Hospitalizations in Young Children. J Infect Dis (2007) 195(6):773–81. doi: 10.1086/511821
148. Stenberg-Hammar K, Hedlin G, Söderhäll C. Rhinovirus and Preschool Wheeze. Pediatr Allergy Immunol (2017) 28(6):513–20. doi: 10.1111/pai.12740
149. Corne JM, Marshall C, Smith S, Schreiber J, Sanderson G, Holgate ST, et al. Frequency, Severity, and Duration of Rhinovirus Infections in Asthmatic and Non-Asthmatic Individuals: A Longitudinal Cohort Study. Lancet (2002) 359(9309):831–4. doi: 10.1016/S0140-6736(02)07953-9
150. Zheng SY, Wang LL, Ren L, Luo J, Liao W, Liu EM. Epidemiological Analysis and Follow-Up of Human Rhinovirus Infection in Children With Asthma Exacerbation. J Med Virol (2018) 90(2):219–28. doi: 10.1002/jmv.24850
151. Tregoning JS, Schwarze J. Respiratory Viral Infections in Infants: Causes, Clinical Symptoms, Virology, and Immunology. Clin Microbiol Rev (2010) 23(1):74–98. doi: 10.1128/CMR.00032-09
152. Chonmaitree T, Revai K, Grady JJ, Clos A, Patel JA, Nair S, et al. Viral Upper Respiratory Tract Infection and Otitis Media Complication in Young Children. Clin Infect Dis (2008) 46(6):815–23. doi: 10.1086/528685
153. Chung JY, Han TH, Kim SW, Kim CK, Hwang ES. Detection of Viruses Identified Recently in Children With Acute Wheezing. J Med Virol (2007) 79(8):1238–43. doi: 10.1002/jmv.20926
154. Fabbiani M, Terrosi C, Martorelli B, Valentini M, Bernini L, Cellesi C, et al. Epidemiological and Clinical Study of Viral Respiratory Tract Infections in Children From Italy. J Med Virol (2009) 81(4):750–6. doi: 10.1002/jmv.21457
155. Jartti T, Lee WM, Pappas T, Evans M, Lemanske RF Jr., Gern JE. Serial Viral Infections in Infants With Recurrent Respiratory Illnesses. Eur Respir J (2008) 32(2):314–20. doi: 10.1183/09031936.00161907
156. Jartti T, Lehtinen P, Vuorinen T, Osterback R, van den Hoogen B, Osterhaus AD, et al. Respiratory Picornaviruses and Respiratory Syncytial Virus as Causative Agents of Acute Expiratory Wheezing in Children. Emerg Infect Dis (2004) 10(6):1095–101. doi: 10.3201/eid1006.030629
157. Kusel MM, de Klerk NH, Holt PG, Kebadze T, Johnston SL, Sly PD. Role of Respiratory Viruses in Acute Upper and Lower Respiratory Tract Illness in the First Year of Life: A Birth Cohort Study. Pediatr Infect Dis J (2006) 25(8):680–6. doi: 10.1097/01.inf.0000226912.88900.a3
158. Lehtinen P, Jartti T, Virkki R, Vuorinen T, Leinonen M, Peltola V, et al. Bacterial Coinfections in Children With Viral Wheezing. Eur J Clin Microbiol Infect Dis (2006) 25(7):463–9. doi: 10.1007/s10096-006-0166-3
159. Regamey N, Kaiser L, Roiha HL, Deffernez C, Kuehni CE, Latzin P, et al. Viral Etiology of Acute Respiratory Infections With Cough in Infancy: A Community-Based Birth Cohort Study. Pediatr Infect Dis J (2008) 27(2):100–5. doi: 10.1097/INF.0b013e31815922c8
160. Talbot HK, Crowe JE Jr., Edwards KM, Griffin MR, Zhu Y, Weinberg GA, et al. Coronavirus Infection and Hospitalizations for Acute Respiratory Illness in Young Children. J Med Virol (2009) 81(5):853–6. doi: 10.1002/jmv.21443
161. Soto-Quiros M, Avila L, Platts-Mills TA, Hunt JF, Erdman DD, Carper H, et al. High Titers of IgE Antibody to Dust Mite Allergen and Risk for Wheezing Among Asthmatic Children Infected With Rhinovirus. J Allergy Clin Immunol (2012) 129(6):1499–505.e5. doi: 10.1016/j.jaci.2012.03.040
162. Wark PA, Grissell T, Davies B, See H, Gibson PG. Diversity in the Bronchial Epithelial Cell Response to Infection With Different Rhinovirus Strains. Respirology (2009) 14(2):180–6. doi: 10.1111/j.1440-1843.2009.01480.x
163. Looi K, Buckley AG, Rigby PJ, Garratt LW, Iosifidis T, Zosky GR, et al. Effects of Human Rhinovirus on Epithelial Barrier Integrity and Function in Children With Asthma. Clin Exp Allergy (2018) 48(5):513–24. doi: 10.1111/cea.13097
164. Steinke JW, Borish L. Immune Responses in Rhinovirus-Induced Asthma Exacerbations. Curr Allergy Asthma Rep (2016) 16(11):78. doi: 10.1007/s11882-016-0661-2
165. Durrani SR, Montville DJ, Pratt AS, Sahu S, DeVries MK, Rajamanickam V, et al. Innate Immune Responses to Rhinovirus Are Reduced by the High-Affinity IgE Receptor in Allergic Asthmatic Children. J Allergy Clin Immunol (2012) 130(2):489–95. doi: 10.1016/j.jaci.2012.05.023
166. Kennedy JL, Turner RB, Braciale T, Heymann PW, Borish L. Pathogenesis of Rhinovirus Infection. Curr Opin Virol (2012) 2(3):287–93. doi: 10.1016/j.coviro.2012.03.008
167. Kennedy JL, Shaker M, McMeen V, Gern J, Carper H, Murphy D, et al. Comparison of Viral Load in Individuals With and Without Asthma During Infections With Rhinovirus. Am J Respir Crit Care Med (2014) 189(5):532–9. doi: 10.1164/rccm.201310-1767OC
168. Oliver BG, Johnston SL, Baraket M, Burgess JK, King NJ, Roth M, et al. Increased Proinflammatory Responses From Asthmatic Human Airway Smooth Muscle Cells in Response to Rhinovirus Infection. Respir Res (2006) 7(1):71. doi: 10.1186/1465-9921-7-71
169. Zhu J, Message SD, Mallia P, Kebadze T, Contoli M, Ward CK, et al. Bronchial Mucosal IFN-α/β and Pattern Recognition Receptor Expression in Patients With Experimental Rhinovirus-Induced Asthma Exacerbations. J Allergy Clin Immunol (2019) 143(1):114–25.e4. doi: 10.1016/j.jaci.2018.04.003
170. Edwards MR, Regamey N, Vareille M, Kieninger E, Gupta A, Shoemark A, et al. Impaired Innate Interferon Induction in Severe Therapy Resistant Atopic Asthmatic Children. Mucosal Immunol (2013) 6(4):797–806. doi: 10.1038/mi.2012.118
171. Monneret D. IFN-α/IFN-λ Responses to Respiratory Viruses in Paediatric Asthma. Eur Respir J (2017) 49(2):1600969. doi: 10.1183/13993003.00969-2016
172. Wark PA, Johnston SL, Bucchieri F, Powell R, Puddicombe S, Laza-Stanca V, et al. Asthmatic Bronchial Epithelial Cells Have a Deficient Innate Immune Response to Infection With Rhinovirus. J Exp Med (2005) 201(6):937–47. doi: 10.1084/jem.20041901
173. Kennedy JL, Koziol-White CJ, Jeffus S, Rettiganti MR, Fisher P, Kurten M, et al. Effects of Rhinovirus 39 Infection on Airway Hyperresponsiveness to Carbachol in Human Airways Precision Cut Lung Slices. J Allergy Clin Immunol (2018) 141(5):1887–90.e1. doi: 10.1016/j.jaci.2017.11.041
174. Jamieson KC, Wiehler S, Michi AN, Proud D. Rhinovirus Induces Basolateral Release of IL-17C in Highly Differentiated Airway Epithelial Cells. Front Cell Infect Microbiol (2020) 10:103. doi: 10.3389/fcimb.2020.00103
175. Jackson DJ, Makrinioti H, Rana BM, Shamji BW, Trujillo-Torralbo MB, Footitt J, et al. IL-33-Dependent Type 2 Inflammation During Rhinovirus-Induced Asthma Exacerbations In Vivo. Am J Respir Crit Care Med (2014) 190(12):1373–82. doi: 10.1164/rccm.201406-1039OC
176. Perez GF, Pancham K, Huseni S, Preciado D, Freishtat RJ, Colberg-Poley AM, et al. Rhinovirus Infection in Young Children is Associated With Elevated Airway TSLP Levels. Eur Respir J (2014) 44(4):1075–8. doi: 10.1183/09031936.00049214
177. Subauste MC, Jacoby DB, Richards SM, Proud D. Infection of a Human Respiratory Epithelial Cell Line With Rhinovirus. Induction of Cytokine Release and Modulation of Susceptibility to Infection by Cytokine Exposure. J Clin Invest (1995) 96(1):549–57. doi: 10.1172/JCI118067
178. Bosco A, Wiehler S, Proud D. Interferon Regulatory Factor 7 Regulates Airway Epithelial Cell Responses to Human Rhinovirus Infection. BMC Genomics (2016) 17:76. doi: 10.1186/s12864-016-2405-z
179. Wang Q, Nagarkar DR, Bowman ER, Schneider D, Gosangi B, Lei J, et al. Role of Double-Stranded RNA Pattern Recognition Receptors in Rhinovirus-Induced Airway Epithelial Cell Responses. J Immunol (2009) 183(11):6989–97. doi: 10.4049/jimmunol.0901386
180. Slater L, Bartlett NW, Haas JJ, Zhu J, Message SD, Walton RP, et al. Co-Ordinated Role of TLR3, RIG-I and MDA5 in the Innate Response to Rhinovirus in Bronchial Epithelium. PloS Pathog (2010) 6(11):e1001178. doi: 10.1371/journal.ppat.1001178
181. Manley GCA, Stokes CA, Marsh EK, Sabroe I, Parker LC. DUSP10 Negatively Regulates the Inflammatory Response to Rhinovirus Through Interleukin-1β Signaling. J Virol (2019) 93(2):e01659–18. doi: 10.1128/JVI.01659-18
182. Sajjan U, Wang Q, Zhao Y, Gruenert DC, Hershenson MB. Rhinovirus Disrupts the Barrier Function of Polarized Airway Epithelial Cells. Am J Respir Crit Care Med (2008) 178(12):1271–81. doi: 10.1164/rccm.200801-136OC
183. Bossios A, Psarras S, Gourgiotis D, Skevaki CL, Constantopoulos AG, Saxoni-Papageorgiou P, et al. Rhinovirus Infection Induces Cytotoxicity and Delays Wound Healing in Bronchial Epithelial Cells. Respir Res (2005) 6:114–. doi: 10.1186/1465-9921-6-114
184. Skevaki CL, Psarras S, Volonaki E, Pratsinis H, Spyridaki IS, Gaga M, et al. Rhinovirus-Induced Basic Fibroblast Growth Factor Release Mediates Airway Remodeling Features. Clin Trans Allergy (2012) 2(1):1–11. doi: 10.1186/2045-7022-2-14
185. Peters M. Actions of Cytokines on the Immune Response and Viral Interactions: An Overview. Hepatology (1996) 23(4):909–16. doi: 10.1002/hep.510230436
186. Altman MC, Gill MA, Whalen E, Babineau DC, Shao B, Liu AH, et al. Transcriptome Networks Identify Mechanisms of Viral and Nonviral Asthma Exacerbations in Children. Nat Immunol (2019) 20(5):637–51. doi: 10.1038/s41590-019-0347-8
187. Piralla A, Rovida F, Campanini G, Rognoni V, Marchi A, Locatelli F, et al. Clinical Severity and Molecular Typing of Human Rhinovirus C Strains During a Fall Outbreak Affecting Hospitalized Patients. J Clin Virol (2009) 45(4):311–7. doi: 10.1016/j.jcv.2009.04.016
188. Mackay IM, Lambert SB, Faux CE, Arden KE, Nissen MD, Sloots TP, et al. Community-Wide, Contemporaneous Circulation of a Broad Spectrum of Human Rhinoviruses in Healthy Australian Preschool-Aged Children During a 12-Month Period. J Infect Dis (2013) 207(9):1433–41. doi: 10.1093/infdis/jis476
189. Esposito S, Daleno C, Scala A, Castellazzi L, Terranova L, Sferrazza Papa S, et al. Impact of Rhinovirus Nasopharyngeal Viral Load and Viremia on Severity of Respiratory Infections in Children. Eur J Clin Microbiol Infect Dis (2014) 33(1):41–8. doi: 10.1007/s10096-013-1926-5
190. Xiao Q, Zheng S, Zhou L, Ren L, Xie X, Deng Y, et al. Impact of Human Rhinovirus Types and Viral Load on the Severity of Illness in Hospitalized Children With Lower Respiratory Tract Infections. Pediatr Infect Dis J (2015) 34(11):1187–92. doi: 10.1097/INF.0000000000000879
191. Ahn JG, Kim DS, Kim KH. Clinical Characteristics and Cytokine Profiles of Children With Acute Lower Respiratory Tract Infections Caused by Human Rhinovirus. PloS One (2018) 13(7):1–9. doi: 10.1371/journal.pone.0198624
192. Baillie VL, Moore DP, Mathunjwa A, Morailane P, Simões EAF, Madhi SA. Molecular Subtyping of Human Rhinovirus in Children From Three Sub-Saharan African Countries. J Clin Microbiol (2019) 57(9):e00723–19. doi: 10.1128/JCM.00723-19
193. Sugita K, Kabashima K. Tight Junctions in the Development of Asthma, Chronic Rhinosinusitis, Atopic Dermatitis, Eosinophilic Esophagitis, and Inflammatory Bowel Diseases. J Leukoc Biol (2020) 107(5):749–62. doi: 10.1002/JLB.5MR0120-230R
194. Dellon ES, Jensen ET, Martin CF, Shaheen NJ, Kappelman MD. Prevalence of Eosinophilic Esophagitis in the United States. Clin Gastroenterol Hepatol (2014) 12(4):589–96.e1. doi: 10.1016/j.cgh.2013.09.008
195. Liacouras CA, Furuta GT, Hirano I, Atkins D, Attwood SE, Bonis PA, et al. Eosinophilic Esophagitis: Updated Consensus Recommendations for Children and Adults. J Allergy Clin Immunol (2011) 128(1):3–20 e6; quiz 1-2. doi: 10.1016/j.jaci.2011.04.016
196. Nurko S, Rosen R. Esophageal Dysmotility in Patients Who Have Eosinophilic Esophagitis. Gastrointest Endosc Clin N Am (2008) 18(1):73–89; ix. doi: 10.1016/j.giec.2007.09.006
197. Cianferoni A, Spergel J. Eosinophilic Esophagitis: A Comprehensive Review. Clin Rev Allergy Immunol (2016) 50(2):159–74. doi: 10.1007/s12016-015-8501-z
198. Schoepfer AM, Safroneeva E, Bussmann C, Kuchen T, Portmann S, Simon HU, et al. Delay in Diagnosis of Eosinophilic Esophagitis Increases Risk for Stricture Formation, in a Time-Dependent Manner. Gastroenterology (2013) 145(6):1230–6.e1–2. doi: 10.1053/j.gastro.2013.08.015
199. Gonsalves N, Yang GY, Doerfler B, Ritz S, Ditto AM, Hirano I. Elimination Diet Effectively Treats Eosinophilic Esophagitis in Adults; Food Reintroduction Identifies Causative Factors. Gastroenterology (2012) 142(7):1451–9 e1; quiz e14-5. doi: 10.1053/j.gastro.2012.03.001
200. Spergel JM. Eosinophilic Esophagitis in Adults and Children: Evidence for a Food Allergy Component in Many Patients. Curr Opin Allergy Clin Immunol (2007) 7(3):274–8. doi: 10.1097/ACI.0b013e32813aee4a
201. Spergel JM, Brown-Whitehorn TF, Beausoleil JL, Franciosi J, Shuker M, Verma R, et al. 14 Years of Eosinophilic Esophagitis: Clinical Features and Prognosis. J Pediatr Gastroenterol Nutr (2009) 48(1):30–6. doi: 10.1097/MPG.0b013e3181788282
202. Spergel JM, Brown-Whitehorn TF, Cianferoni A, Shuker M, Wang ML, Verma R, et al. Identification of Causative Foods in Children With Eosinophilic Esophagitis Treated With an Elimination Diet. J Allergy Clin Immunol (2012) 130(2):461–7.e5. doi: 10.1016/j.jaci.2012.05.021
203. Fritz J, Lerner D, Suchi M. Herpes Simplex Virus Esophagitis in Immunocompetent Children: A Harbinger of Eosinophilic Esophagitis? J Pediatr Gastroenterol Nutr (2018) 66(4):609–13. doi: 10.1097/MPG.0000000000001748
204. Abonia JP, Spergel JM, Cianferoni A. Eosinophilic Esophagitis: A Primary Disease of the Esophageal Mucosa. J Allergy Clin Immunol Pract (2017) 5(4):951–5. doi: 10.1016/j.jaip.2017.02.004
205. Capucilli P, Cianferoni A, Grundmeier RW, Spergel JM. Comparison of Comorbid Diagnoses in Children With and Without Eosinophilic Esophagitis in a Large Population. Ann Allergy Asthma Immunol (2018) 121(6):711–6. doi: 10.1016/j.anai.2018.08.022
206. Cianferoni A, Spergel JM. From Genetics to Treatment of Eosinophilic Esophagitis. Curr Opin Allergy Clin Immunol (2015) 15(5):417–25. doi: 10.1097/ACI.0000000000000200
207. Cianferoni A, Spergel JM, Muir A. Recent Advances in the Pathological Understanding of Eosinophilic Esophagitis. Expert Rev Gastroenterol Hepatol (2015) 9(12):1501–10. doi: 10.1586/17474124.2015.1094372
208. Simon D, Cianferoni A, Spergel JM, Aceves S, Holbreich M, Venter C, et al. Eosinophilic Esophagitis is Characterized by a Non-IgE-Mediated Food Hypersensitivity. Allergy (2016) 71(5):611–20. doi: 10.1111/all.12846
209. Cianferoni A, Ruffner MA, Guzek R, Guan S, Brown-Whitehorn T, Muir A, et al. Elevated Expression of Activated TH2 Cells and Milk-Specific TH2 Cells in Milk-Induced Eosinophilic Esophagitis. Ann Allergy Asthma Immunol (2018) 120(2):177–83 e2. doi: 10.1016/j.anai.2017.11.006
210. Ram G, Lee J, Ott M, Brown-Whitehorn TF, Cianferoni A, Shuker M, et al. Seasonal Exacerbation of Esophageal Eosinophilia in Children With Eosinophilic Esophagitis and Allergic Rhinitis. Ann Allergy Asthma Immunol (2015) 115(3):224–8.e1. doi: 10.1016/j.anai.2015.07.004
211. Ortega HG, Liu MC, Pavord ID, Brusselle GG, FitzGerald JM, Chetta A, et al. Mepolizumab Treatment in Patients With Severe Eosinophilic Asthma. N Engl J Med (2014) 371(13):1198–207. doi: 10.1056/NEJMoa1403290
212. Wang E, Hoyte FC. Traditional Therapies for Severe Asthma. Immunol Allergy Clin North Am (2016) 36(3):581–608. doi: 10.1016/j.iac.2016.03.013
213. Spergel JM, Rothenberg ME, Collins MH, Furuta GT, Markowitz JE, Fuchs G 3rd, et al. Reslizumab in Children and Adolescents With Eosinophilic Esophagitis: Results of a Double-Blind, Randomized, Placebo-Controlled Trial. J Allergy Clin Immunol (2012) 129(2):456–63, 63.e1-3. doi: 10.1016/j.jaci.2011.11.044
214. Straumann A, Conus S, Grzonka P, Kita H, Kephart G, Bussmann C, et al. Anti-Interleukin-5 Antibody Treatment (Mepolizumab) in Active Eosinophilic Oesophagitis: A Randomised, Placebo-Controlled, Double-Blind Trial. Gut (2010) 59(1):21–30. doi: 10.1136/gut.2009.178558
215. Hirano I, Dellon ES, Hamilton JD, Collins MH, Peterson K, Chehade M, et al. Efficacy of Dupilumab in a Phase 2 Randomized Trial of Adults With Active Eosinophilic Esophagitis. Gastroenterology (2020) 158(1):111–22 e10. doi: 10.1053/j.gastro.2019.09.042
216. Gann PH, Deaton RJ, McMahon N, Collins MH, Dellon ES, Hirano I, et al. An Anti-IL-13 Antibody Reverses Epithelial-Mesenchymal Transition Biomarkers in Eosinophilic Esophagitis: Phase 2 Trial Results. J Allergy Clin Immunol (2020) 146(2):367–76 e3. doi: 10.1016/j.jaci.2020.03.045
217. Alexander ES, Martin LJ, Collins MH, Kottyan LC, Sucharew H, He H, et al. Twin and Family Studies Reveal Strong Environmental and Weaker Genetic Cues Explaining Heritability of Eosinophilic Esophagitis. J Allergy Clin Immunol (2014) 134(5):1084–92.e1. doi: 10.1016/j.jaci.2014.07.021
218. Kottyan LC, Davis BP, Sherrill JD, Liu K, Rochman M, Kaufman K, et al. Genome-Wide Association Analysis of Eosinophilic Esophagitis Provides Insight Into the Tissue Specificity of This Allergic Disease. Nat Genet (2014) 46(8):895–900. doi: 10.1038/ng.3033
219. Kottyan LC, Maddox A, Braxton JR, Stucke EM, Mukkada V, Putnam PE, et al. Genetic Variants at the 16p13 Locus Confer Risk for Eosinophilic Esophagitis. Genes Immun (2019) 20(4):281–92. doi: 10.1038/s41435-018-0034-z
220. Martin LJ, He H, Collins MH, Abonia JP, Biagini Myers JM, Eby M, et al. Eosinophilic Esophagitis (EoE) Genetic Susceptibility is Mediated by Synergistic Interactions Between EoE-Specific and General Atopic Disease Loci. J Allergy Clin Immunol (2018) 141(5):1690–8. doi: 10.1016/j.jaci.2017.09.046
221. Sleiman PM, Wang ML, Cianferoni A, Aceves S, Gonsalves N, Nadeau K, et al. GWAS Identifies Four Novel Eosinophilic Esophagitis Loci. Nat Commun (2014) 5:5593. doi: 10.1038/ncomms6593
222. Avila-Castellano R, Garcia-Lozano JR, Cimbollek S, Lucendo AJ, Bozada JM, Quiralte J. Genetic Variations in the TLR3 Locus Are Associated With Eosinophilic Esophagitis. United Eur Gastroenterol J (2018) 6(3):349–57. doi: 10.1177/2050640617732643
223. Blanchard C, Wang N, Stringer KF, Mishra A, Fulkerson PC, Abonia JP, et al. Eotaxin-3 and a Uniquely Conserved Gene-Expression Profile in Eosinophilic Esophagitis. J Clin Invest (2006) 116(2):536–47. doi: 10.1172/JCI26679
224. Noti M, Wojno ED, Kim BS, Siracusa MC, Giacomin PR, Nair MG, et al. Thymic Stromal Lymphopoietin-Elicited Basophil Responses Promote Eosinophilic Esophagitis. Nat Med (2013) 19(8):1005–13. doi: 10.1038/nm.3281
225. Cianferoni A, Spergel J. The Importance of TSLP in Allergic Disease and Its Role as a Potential Therapeutic Target. Expert Rev Clin Immunol (2014) 10(11):1463–74. doi: 10.1586/1744666X.2014.967684
226. Aceves SS, Ackerman SJ. Relationships Between Eosinophilic Inflammation, Tissue Remodeling, and Fibrosis in Eosinophilic Esophagitis. Immunol Allergy Clin North Am (2009) 29(1):197–211, xiii-xiv. doi: 10.1016/j.iac.2008.10.003
227. Aceves SS, Broide DH. Airway Fibrosis and Angiogenesis Due to Eosinophil Trafficking in Chronic Asthma. Curr Mol Med (2008) 8(5):350–8. doi: 10.2174/156652408785161023
228. Aceves S, Hirano I, Furuta GT, Collins MH. Eosinophilic Gastrointestinal Diseases–Clinically Diverse and Histopathologically Confounding. Semin Immunopathol (2012) 34(5):715–31. doi: 10.1007/s00281-012-0324-x
229. Lyons JJ, Liu Y, Ma CA, Yu X, O’Connell MP, Lawrence MG, et al. ERBIN Deficiency Links STAT3 and TGF-Beta Pathway Defects With Atopy in Humans. J Exp Med (2017) 214(3):669–80. doi: 10.1084/jem.20161435
230. Arora M, Bagi P, Strongin A, Heimall J, Zhao X, Lawrence MG, et al. Gastrointestinal Manifestations of STAT3-Deficient Hyper-IgE Syndrome. J Clin Immunol (2017) 37(7):695–700. doi: 10.1007/s10875-017-0429-z
231. Ruffner MA, Brown-Whitehorn TF, Verma R, Cianferoni A, Gober L, Shuker M, et al. Clinical Tolerance in Eosinophilic Esophagitis. J Allergy Clin Immunol Pract (2018) 6(2):661–3. doi: 10.1016/j.jaip.2017.06.035
232. Paluel-Marmont C, Bellon N, Barbet P, Leclerc-Mercier S, Hadj-Rabia S, Dupont C, et al. Eosinophilic Esophagitis and Colonic Mucosal Eosinophilia in Netherton Syndrome. J Allergy Clin Immunol (2017) 139(6):2003–5.e1. doi: 10.1016/j.jaci.2016.10.045
233. Henderson CJ, Ngeow J, Collins MH, Martin LJ, Putnam PE, Abonia JP, et al. Increased Prevalence of Eosinophilic Gastrointestinal Disorders in Pediatric PTEN Hamartoma Tumor Syndromes. J Pediatr Gastroenterol Nutr (2014) 58(5):553–60. doi: 10.1097/MPG.0000000000000253
234. Yawn RJ, Acra S, Goudy SL, Flores R, Wootten CT. Eosinophilic Laryngitis in Children With Aerodigestive Dysfunction. Otolaryngol Head Neck Surg (2015) 153(1):124–9. doi: 10.1177/0194599815577568
235. Mehta AK, Duan W, Doerner AM, Traves SL, Broide DH, Proud D, et al. Rhinovirus Infection Interferes With Induction of Tolerance to Aeroantigens Through OX40 Ligand, Thymic Stromal Lymphopoietin, and IL-33. J Allergy Clin Immunol (2016) 137(1):278–88.e6. doi: 10.1016/j.jaci.2015.05.007
236. Perez GF, Rodriguez-Martinez CE, Nino G. Rhinovirus-Induced Airway Disease: A Model to Understand the Antiviral and Th2 Epithelial Immune Dysregulation in Childhood Asthma. J Investig Med (2015) 63(6):792–5. doi: 10.1097/JIM.0000000000000209
237. Liu S, Verma M, Michalec L, Liu W, Sripada A, Rollins D, et al. Steroid Resistance of Airway Type 2 Innate Lymphoid Cells From Patients With Severe Asthma: The Role of Thymic Stromal Lymphopoietin. J Allergy Clin Immunol (2018) 141(1):257–68.e6. doi: 10.1016/j.jaci.2017.03.032
238. Calhoun WJ, Haselkorn T, Mink DR, Miller DP, Dorenbaum A, Zeiger RS. Clinical Burden and Predictors of Asthma Exacerbations in Patients on Guideline-Based Steps 4-6 Asthma Therapy in the TENOR Cohort. J Allergy Clin Immunol Pract (2014) 2(2):193–200. doi: 10.1016/j.jaip.2013.11.013
239. Esquivel A, Busse WW, Calatroni A, Togias AG, Grindle KG, Bochkov YA, et al. Effects of Omalizumab on Rhinovirus Infections, Illnesses, and Exacerbations of Asthma. Am J Respir Crit Care Med (2017) 196(8):985–92. doi: 10.1164/rccm.201701-0120OC
240. Martin AC, Zhang G, Rueter K, Khoo SK, Bizzintino J, Hayden CM, et al. Beta2-Adrenoceptor Polymorphisms Predict Response to Beta2-Agonists in Children With Acute Asthma. J Asthma (2008) 45(5):383–8. doi: 10.1080/02770900801971792
241. Yang Z, Bochkov YA, Voelker DR, Foster MW, Que LG. Identification of a Novel Inhibitor of Human Rhinovirus Replication and Inflammation in Airway Epithelial Cells. Am J Respir Cell Mol Biol (2019) 60(1):58–67. doi: 10.1165/rcmb.2018-0058OC
242. Nance DM, Sanders VM. Autonomic Innervation and Regulation of the Immune System (1987-2007). Brain Behav Immun (2007) 21(6):736–45. doi: 10.1016/j.bbi.2007.03.008
243. Gibson PG, Yang IA, Upham JW, Reynolds PN, Hodge S, James AL, et al. Effect of Azithromycin on Asthma Exacerbations and Quality of Life in Adults With Persistent Uncontrolled Asthma (AMAZES): A Randomised, Double-Blind, Placebo-Controlled Trial. Lancet (2017) 390(10095):659–68. doi: 10.1016/S0140-6736(17)31281-3
244. Matthews DA, Dragovich PS, Webber SE, Fuhrman SA, Patick AK, Zalman LS, et al. Structure-Assisted Design of Mechanism-Based Irreversible Inhibitors of Human Rhinovirus 3C Protease With Potent Antiviral Activity Against Multiple Rhinovirus Serotypes. Proc Natl Acad Sci USA (1999) 96(20):11000–7. doi: 10.1073/pnas.96.20.11000
Keywords: rhinovirus (RV), epithelium, asthma, wheezing, allergic, airway, paediatric, EoE
Citation: Watkinson RL, Looi K, Laing IA, Cianferoni A and Kicic A (2021) Viral Induced Effects on a Vulnerable Epithelium; Lessons Learned From Paediatric Asthma and Eosinophilic Oesophagitis . Front. Immunol. 12:773600. doi: 10.3389/fimmu.2021.773600
Received: 10 September 2021; Accepted: 05 November 2021;
Published: 29 November 2021.
Edited by:
Janos G. Filep, Université de Montréal, CanadaReviewed by:
Leyla Pur Ozyigit, University Hospitals of Leicester NHS Trust, United KingdomCopyright © 2021 Watkinson, Looi, Laing, Cianferoni and Kicic. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anthony Kicic, Anthony.Kicic@telethonkids.org.au
†These authors share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.