 Nicole Schäfer1,2
Nicole Schäfer1,2 Anas Rasras3,4
Anas Rasras3,4 Delia M. Ormenisan1
Delia M. Ormenisan1 Sabine Amslinger4
Sabine Amslinger4 Volker Enzmann5Herbert Jägle1
Volker Enzmann5Herbert Jägle1 Diana Pauly1,6*
Diana Pauly1,6*- 1Department of Ophthalmology, University Hospital Regensburg, Regensburg, Germany
- 2Department of Orthopaedic Surgery, Experimental Orthopaedics, Centre for Medical Biotechnology (ZMB), University of Regensburg, Regensburg, Germany
- 3Chemistry Department, Al-Balqa Applied University, Al-Salt, Jordan
- 4Institute of Organic Chemistry, University of Regensburg, Regensburg, Germany
- 5Department of Ophthalmology, University Hospital of Bern and Department of Biomedical Research, University of Bern, Bern, Switzerland
- 6Experimental Ophthalmology, Philipps-University Marburg, Marburg, Germany
Complement Factor H-Related 3 (FHR-3) is a major regulator of the complement system, which is associated with different diseases, such as age-related macular degeneration (AMD). However, the non-canonical local, cellular functions of FHR-3 remained poorly understood. Here, we report that FHR-3 bound to oxidative stress epitopes and competed with FH for interaction. Furthermore, FHR-3 was internalized by viable RPE cells and modulated time-dependently complement component (C3, FB) and receptor (C3aR, CR3) expression of human RPE cells. Independently of any external blood-derived proteins, complement activation products were detected. Anaphylatoxin C3a was visualized in treated cells and showed a translocation from the cytoplasm to the cell membrane after FHR-3 exposure. Subsequently, FHR-3 induced a RPE cell dependent pro-inflammatory microenvironment. Inflammasome NLRP3 activation and pro-inflammatory cytokine secretion of IL-1ß, IL-18, IL-6 and TNF-α were induced after FHR-3-RPE interaction. Our previously published monoclonal anti-FHR-3 antibody, which was chimerized to reduce immunogenicity, RETC-2-ximab, ameliorated the effect of FHR-3 on ARPE-19 cells. Our studies suggest FHR-3 as an exogenous trigger molecule for the RPE cell “complosome” and as a putative target for a therapeutic approach for associated degenerative diseases.
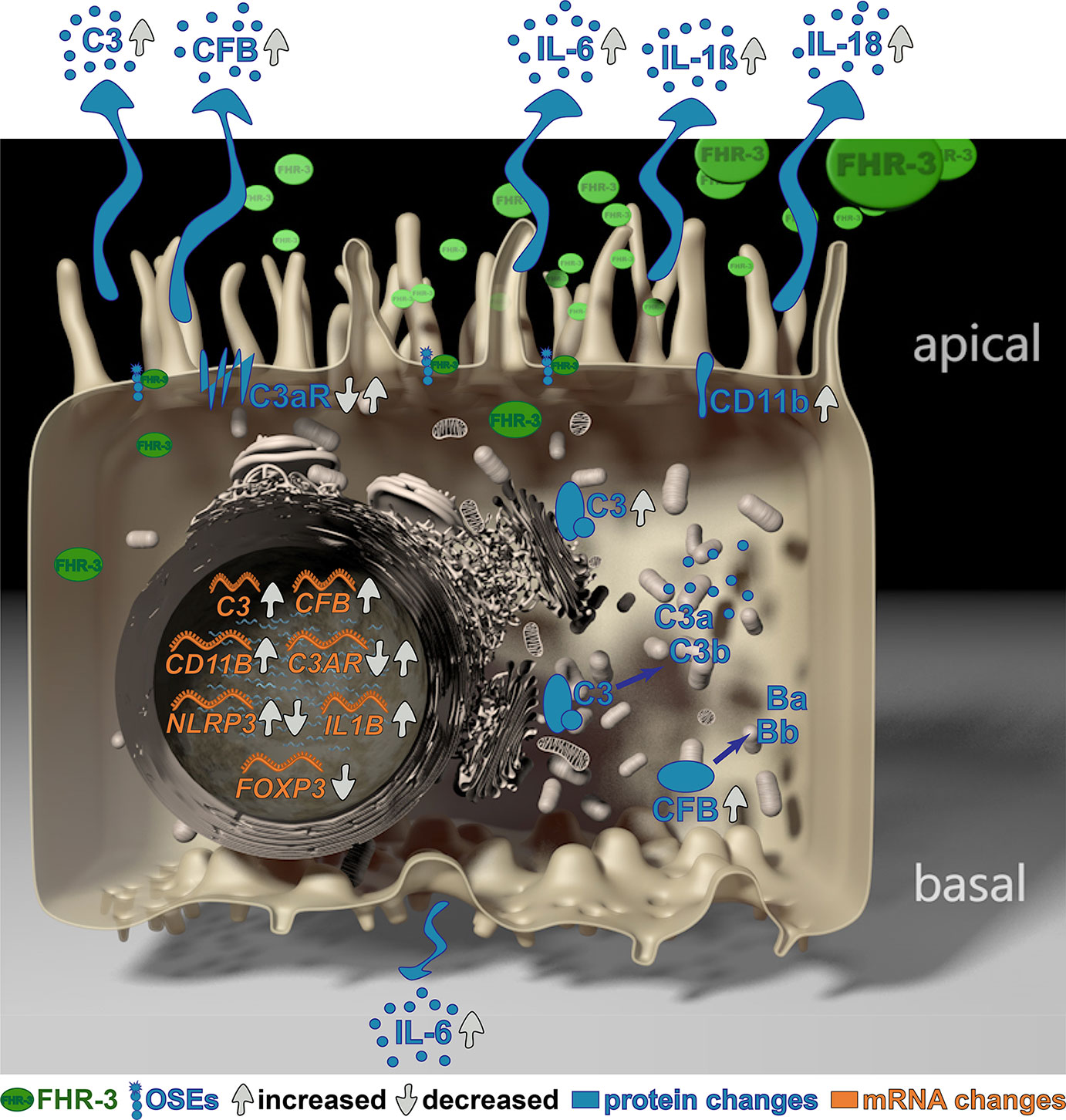
Graphical Abstract
Introduction
Studied for more than one century, but still not fully understood – the evolutionary ancient complement system is one of the first defense line of our body to protect from pathogens, cellular debris or dead cells. The complement system is tightly regulated to prevent dysregulation leading to progression of different degenerative diseases such as age-related macular degeneration (AMD) (1). One major group of soluble regulators are complement factor H (FH), its splice variant factor H-like protein 1 (FHL-1), and its related proteins 1 – 5 (FHR-1 – 5) (2–4). Single nucleotide polymorphisms (SNP) located in the complement factor H (CFH) gene locus have been described to be responsible for AMD pathology (5, 6). While the most relevant CFH SNP (Y402H) contributed to AMD progression, a joint deletion of the complement factor-H related 1 and 3 (ΔCFHR3/1) genes was protective for the disease (7, 8). FHR-1 and FHR-3 are mainly produced by hepatocytes and released into the systemic circulation. In the eye, a local expression could be reported for FHR-3 by retinal macrophages/microglia cells (9) and for FH and FHL-1 by retinal pigment epithelium (RPE) cells (10).
Retinal macrophages/microglia and RPE cells are important to maintain the physiological homeostasis of the neuronal tissue. The RPE forms the outer blood-retinal barrier isolating the retina from the systemic immune system. Besides absorption of light, recycling of photoreceptor outer segements and protection against photooxidation, the RPE supports the retina by secretion of various nutrients, which is precisely regulated. However, during AMD progression age-dependent disruption and loss of RPE cells occur (11, 12). This is accompanied by loss of RPE function, e.g. failure to degrade proteins, inflammaging or increased cellular oxidative stress (13, 14).
Oxidative stress can lead to structural modifications resulting in novel oxidation-specific epitopes (OSE) detected in RPE cells (15, 16). OSE can trigger inflammation in the retina, which was inhibited by FH binding (17, 18). FHR-1 and FHR-3 also attach to OSE on necrotic cells and compete with FH for binding (19). This interference of FHR-1 and FHR-3 prevents FH-mediated proteolysis of the central complement component C3 into iC3b and results in enhanced activity of the soluble complement system (20).
Besides its blood-related function, FH also acts on complement components in the cell, the so called “complosome” (21, 22). FH is internalized by stressed and apoptotic RPE cells (18, 21), resulting in reduced endogenous C3-cleavage and increased cell survival in stressed cells (18), but contrary increased C3b-fragments are detected in apoptotic RPE cells, facilitating RPE cell opsonization (21). A similar interaction was also described for FHR-1 on necrotic cells, including RPE cells, but a cellular incorporation of FHR-1 hadn’t been reported (20, 23). Cellular FHR-1 interaction increases complement activation as well as deposition of C3 and C4 fragments on cell surfaces (23, 24). Further, inflammasome reactivity is triggered on monocytes by FHR-1 binding but not upon FHR-3 interaction (20).
In RPE cells, inflammasome activation is so far mainly related to stress and complement receptor signaling (25–27), but not to FHR-binding. RPE cells autonomously produce and activate numerous complement components independently from blood-derived complement proteins (26, 27), which can than bind to complement receptors. This local complement expression and secretion by RPE cells is modulated by external stress (27–31). Stress-related activation of the inflammasome is associated with a pro-inflammatory secretion phenotype of RPE cells (25, 30, 32, 33), which in return correlates with complement activation (30).
However, a direct effect of FHR-3 on RPE cellular physiology and its non-canonical functions modulating the “complosome” has not been described so far. Knowing that FHR-3 is expressed by retinal macrophages/microglia cells in a degenerated human retina (9), we explored the functional role of apical FHR-3 on RPE cells in depth. Here, we describe a local function of FHR-3 as an exogenous danger molecule for RPE cells, sensing those cells for “complosome” activation independent of other extracellular complement sources and inducing inflammatory immune responses. This effect was attenuated by blocking FHR-3 using a specific chimerized monoclonal antibody RETC-2-ximab.
Materials and Methods
Human Material and Ethical Statements
The research complies with the human research act (HRA) stating that small quantities of bodily substances removed in the course of transplantation may be anonymized for research purposes without consent (HRA chapter 5, paragraph 38, Switzerland). Human primary retinal pigment epithelial cells (hpRPE) were prepared from two anonymized donor eyes as described below (34).
Complement-depleted human sera were purchased from Complement Technology. All human serum samples were stored at −80°C.
PCR for Genetic Analysis
DNA was isolated from human adult retinal pigment epithelium cells (ARPE-19 cells, American Type Culture Collection, #CRL-2302) lysates and sclera slices of donor eyes using ReliaPrep™ FFPE gDNA Miniprep System (Promega, Mannheim, Germany, #A2351). PCR amplification of relevant AMD-associated complement SNPs was performed using in-house generated primers (Supplementary Table 1) and the following cycle steps, according to the MIQE guidelines: denaturation (95°C, 1 min), annealing (60°C, 1 min), elongation (72°C, 1 min), 33 cycles. Afterwards, DNA-sequencing was performed by GeneArt (Thermo Fisher Scientific, Dreieich, Germany) using either forward or reverse in-house primers, respectively (Supplementary Table 1).
Cell Culture and Treatment
ARPE-19 cells (passage 38; passage 25 for Figures S1D–G) were cultivated in cell culture flasks with DMEM/F12 (Sigma Aldrich, Munich, Germany), 10% foetal calf serum (FCS; PanBiotech, Aidenbach, Germany) and 1% penicillin/streptomycin until they reached confluency of approx. 80% (37°C, 5% CO2). Human primary RPE (hpRPE) cells were harvested from the eyecup after enzymatic digestion with dispase (1 mg/ml) and DNAse (12.2 µg/ml) for 1 h at 37°C. hpRPE cells were centrifuged at 259 × g for 5 min at 4°C and cultivated in Dulbecco’s modified Eagle’s medium/Nutrient Mixture F-12 (DMEM/F-12 GlutaMax, Thermo Fisher Scientific, #31331-028) containing 5 % FCS (Thermo Fisher Scientific, #10500-064), 1 % Penicillin-Streptomycin (Thermo Fisher Scientific, #15070-063), 1 % N1 Medium Supplement (Merck, Darmstadt, Germany, #N6530), 10 mM MEM on-essential amino acids (Thermo Fisher Scientific, #11140-035), 0.25 mg/ml taurine (Merck, #T0625), 4.5 mg/ml glucose solution (Thermo Fisher Scientific, #G524940-01), 0.013 ng/ml triiodothyronine (Merck, #T2877), 0.02 µg/ml hydrocortisone (Merck, #H0888), 20 ng/ml human basic growth factor (hbFGF, R&D Systems, Minneapolis, MN, USA, #13256029), and 1 mg/ml human epidermal growth factor (hEGF, Thermo Fisher Scientific, #PHG0311) in laminin-coated Transwell® inserts under standard conditions (37°C, 5% CO2, 80% humidity). ARPE-19 cells were cultivated in laminin-coated Transwell® inserts for 4 – 6 weeks, as published previously (30).
Before treatment, cells were cultivated in FCS-reduced medium within 3 days (5% - 2.5% - 1.25% - 0%). Afterwards, polarized ARPE-19 cells and hpRPE cells were treated apically with serum-free medium containing either: a) 50 µg/ml (1 µM) FHR-3-strep (recombinant, in-house) (9), b) FHR-1-strep (recombinant, in-house), c) FH (native, Complement Technology, Tyler, TX, USA, #A137), d) Properdin (FP, native, Complement Technology, #A139, e) 50 µg/ml (1 µM) FHR-3 and 150 µg/ml (1 µM) anti-FHR-3 RETC-2-ximab (recombinant, in-house), or f) 50 µg/ml (1 µM) FHR-3 and 150 µg/ml (1 µM) anti-BSA control-ximab (recombinant, in-house), at different time points (see figure legends).
Immunofluorescence
After treatment, polarized ARPE-19 cells in Transwell® inserts were permeabilized (PBS/0.2% Tween20 (PBS-T), 45 min) and paraformaldehyde-fixated (4%, 20 min). Afterwards, unspecific binding was blocked (3% bovine serum albumin (BSA)/PBS-T, 1 h). Antigens were detected using specific primary antibodies (3% BSA/PBS-T, overnight): anti-C3 (Figures 3D, E, Supplementary Figure 5A, Abcam, Cambridge, UK, #ab181147), anti-C3a desArg (Figure 4C, Hycult, Uden, Netherlands, #HM2074), anti-FB (Figures 5D, E, Merck, #341272), anti-C3aR (Figure 6C, Antibodies-online, Aachen, Germany, #ABIN682213), anti-cluster of differentiation molecule 11B (CD11b, CR3 subunit) (Figure 7C, Biorbyt, Cambridge, UK, #orb19554), anti-FHR-3 (Figures 1A–C, RETC-2, in-house), anti-GM130 (Figure 3D, E, R&D Systems, #AF8199-SP) and anti-ZO-1 (Figures 1A, C and Supplementary Figure 5, Thermo Fisher Scientific, #61-7300). Fluorescence-conjugated secondary antibodies for detection were the following (3% BSA/PBS, 45 min): anti-mouse IgG-488 (Jackson ImmunoResearch, West Grove, PA, USA, #715-545-150), anti-mouse IgG-Cy3 (Jackson ImmunoResearch, #715-165-150), anti-goat IgG-Cy3 (Jackson ImmunoResearch, #305-035-003), anti-rabbit IgG-488 (Jackson ImmunoResearch, #711-545-152) and GAPDH-HRP (Cell Signaling Technology, Beverly, MA, USA, #3683). The fluorochrome HOECHST 33342 (Thermo Fisher Scientific, #H3570) was used to stain DNA and acti-stain™ 488 phalloidin (Figures 5D, E, Cytoskeleton, Denver, CO, USA, #PHDG1-A) detected specifically actin filaments in ARPE-19 cells. Cells were covered with fluorescence mounting medium (Dako, Agilent Technologies, Boeblingen, Germany, #S302380-2). Images were taken with a VisiScope CSU-X1 Confocal System (Visitron Systems, Puchheim, Germany) and a high-resolution sCMOS camera, and further processed with Adobe Photoshop CC 2019.
FHR-3 labelling for internalization studies was performed using pHrodo™ Red Microscale Labeling Kit (Figure 1D, Thermo Fisher Scientific, #P35363) according to the manufacturer’s protocol. Labelled FHR-3 (FHR-3-pHrodo) was incubated apically on ARPE-19 cells for 60 min, cells were then fixed with paraformaldehyde (4%, 20 min). Internalized FHR-3 was detected immunohistologically by means of a Leica AF6000LX fluorescence microscope, equipped with a Leica DFC350 FX digital camera. The images were visualized by 3D modeling using Leica deconvolution software module, and further processed with Adobe Photoshop CC 2019.
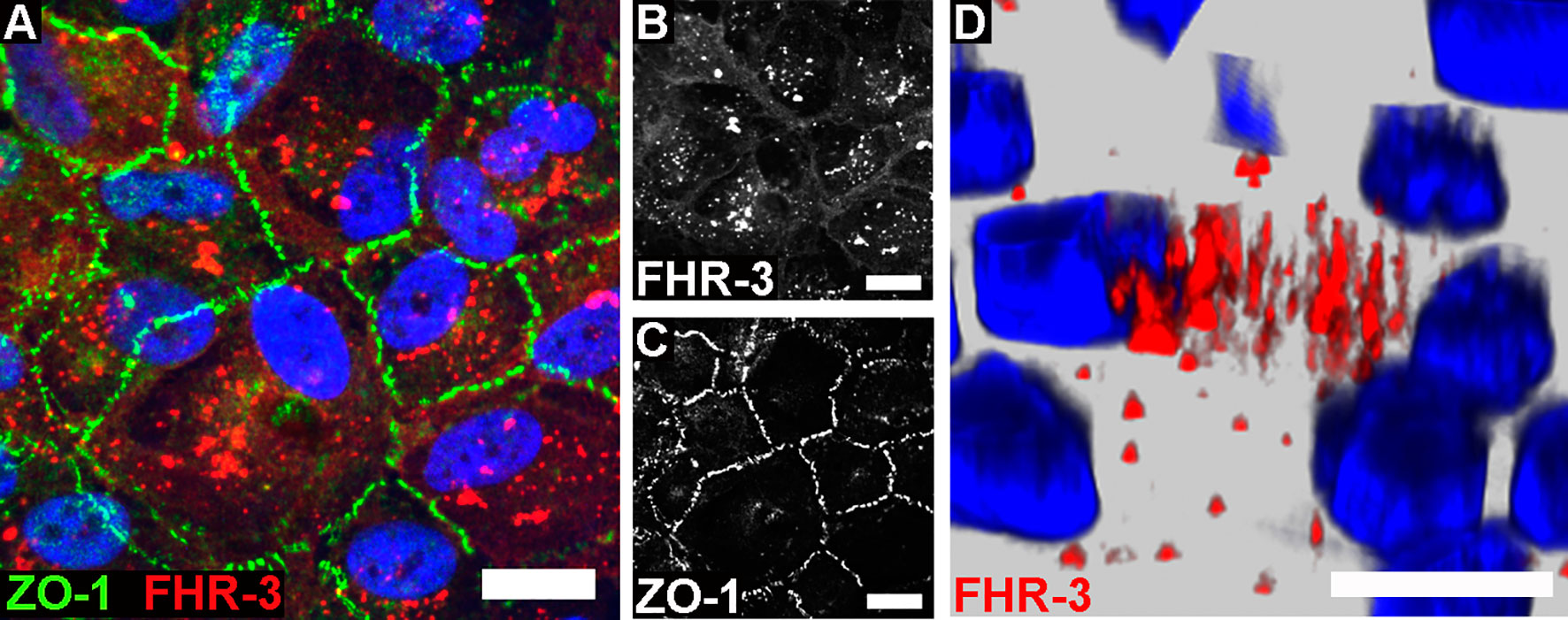
Figure 1 FHR-3 was internalized by ARPE-19 cells. Polarized, differentiated ARPE-19 cells were apically treated with FHR-3. (A) FHR-3 was specifically detected after 2 h incubation time by anti-FHR-3 antibody RETC-2 [red, (B)], cell-cell connections were labelled with anti-ZO-1 antibody [green, (C)]. Magnification 200x. (D) FHR-3-pHrodo (red) was specifically detected inside high-passage ARPE-19 cells. Magnification 400x. Scale bars 40 μm. Cell nuclei were stained with Hoechst.
Transepithelial Resistance and Cellular Capacitance
Transepithelial resistance (TER), a measure of cell monolayer barrier function, and cellular capacitance (CCl), an indicator of the expression of membrane folding such as microvilli, of polarized ARPE-19 cells were recorded online using the established cellZscope device (nanoAnalytics, Münster, Germany), as described previously (30).
RT-qPCR
RNA was isolated using a NucleoSpin® RNA/Protein kit (Macherey-Nagel, Düren, Germany). Sample quality and purity were ensured by randomly testing using QIAxcel® RNA analyses (Qiagen, Hilden, Germany). Purified mRNA was transcribed into cDNA with a QuantiTect®Reverse Transcription Kit (Qiagen, #205313). Transcripts of complement components, receptors, and inflammation-associated markers were analyzed using a Rotor-Gene SYBR®Green PCR Kit either with QuantiTect Primer Assays (Qiagen, Supplementary Table 1) or in-house-designed primer pairs (Metabion, Planegg, Germany, Supplementary Table 1) in a Rotor Gene Q 2plex cycler (Qiagen), using the following conditions: hold (95°C, 5 min), cycling (95°C, 5 sec; 60°C, 10 sec; 72°C, 20 sec; 40 cycles). Data were normalized to GAPDH housekeeper expression, analysed by using the 2-ΔΔCT method. Values were depicted on a linear scale using log2-transformed scores to equally visualize increases and decreases in expression levels.
Synthesis of 2-(ω-Carboxyethyl) Pyrrole Bovine Serum Albumin
2-(w-carboxyethyl) pyrrole (CEP)-BSA was prepared using a method by Lu et al. (35), which was shortened by one step using succinic anhydride together with the Grignard reagent of 2-(2-bromoethyl)-1,3-dioxane to get directly 6-(1,3-dioxan-2-yl)-4-oxohexanoic acid in 57% yield.
The pyrrole content in CEP-BSA was determined by Ehrlich assay using the Ehrlich reagent 4-(dimethylamino)benzaldehyde and 3-(1-methyl-1H-pyrrol-2-yl)propanoic acid (Sigma Aldrich) as the pyrrole standard measuring the absorbance of the formed pyrrole adduct at 540 nm.
ELISA for Oxidative Stress Epitopes Interaction
MaxiSorp microtiter plates (Nalgene Nunc, Rochester, NY, USA) were coated either with CEP-BSA, malondialdehyde (MDA)-BSA (Hölzel Diagnostika, Köln, Germany, #20P-MD-BS102) or malondialdehyde-acetaldehyde (MAA)-BSA (10 μg/ml, PBS, overnight, 4°C). Blocking was performed with PBS-T (1 h). CEP-BSA plates (Figures 2A–C) were incubated with FHR-3 (200 nM), FH (6.4 nM), or with anti-FHR-3 antibody RETC-2 (666 nM). MDA-BSA plates (Figures 2D–F) were incubated with FHR-3 (700 nM), FH (64.5 nM), or with anti-FHR-3 antibody RETC-2 (2333 nM). MAA-BSA plates (Figures 2G–I) were incubated with FHR-3 (7 nM), FH (0.64 nM), or with anti-FHR-3 antibody RETC-2 (233 nM). All proteins were diluted in PBS and incubated for 1 h. FHR-3 and anti-FHR-3 antibody RETC-2 were preincubated for 1 h. For the standard curves antigen serial dilutions (FHR-3 1.8 – 4000 nM, FH 0.8 – 416 nM) were incubated (PBS, 1 h). Binding was detected either with mouse anti-FH (R&D Systems, #AF4999, PBS, 1 h) and anti-mouse IgG-Fcγ-POD (Jackson ImmunoResearch, #115-035-164, PBS, 30 min) or StrepMAB-HRP (IBA Lifesciences, Goettingen, Germany, #2-1509-001, PBS, 30 min) to detect the FHR-3-strep. The signal was developed with 3,3’,5,5’-Tetramethylbenzidine (TMB, Seramun Diagnostica, Heidesee, Germany, #S-004-4), and absorption was determined at 450 nm.
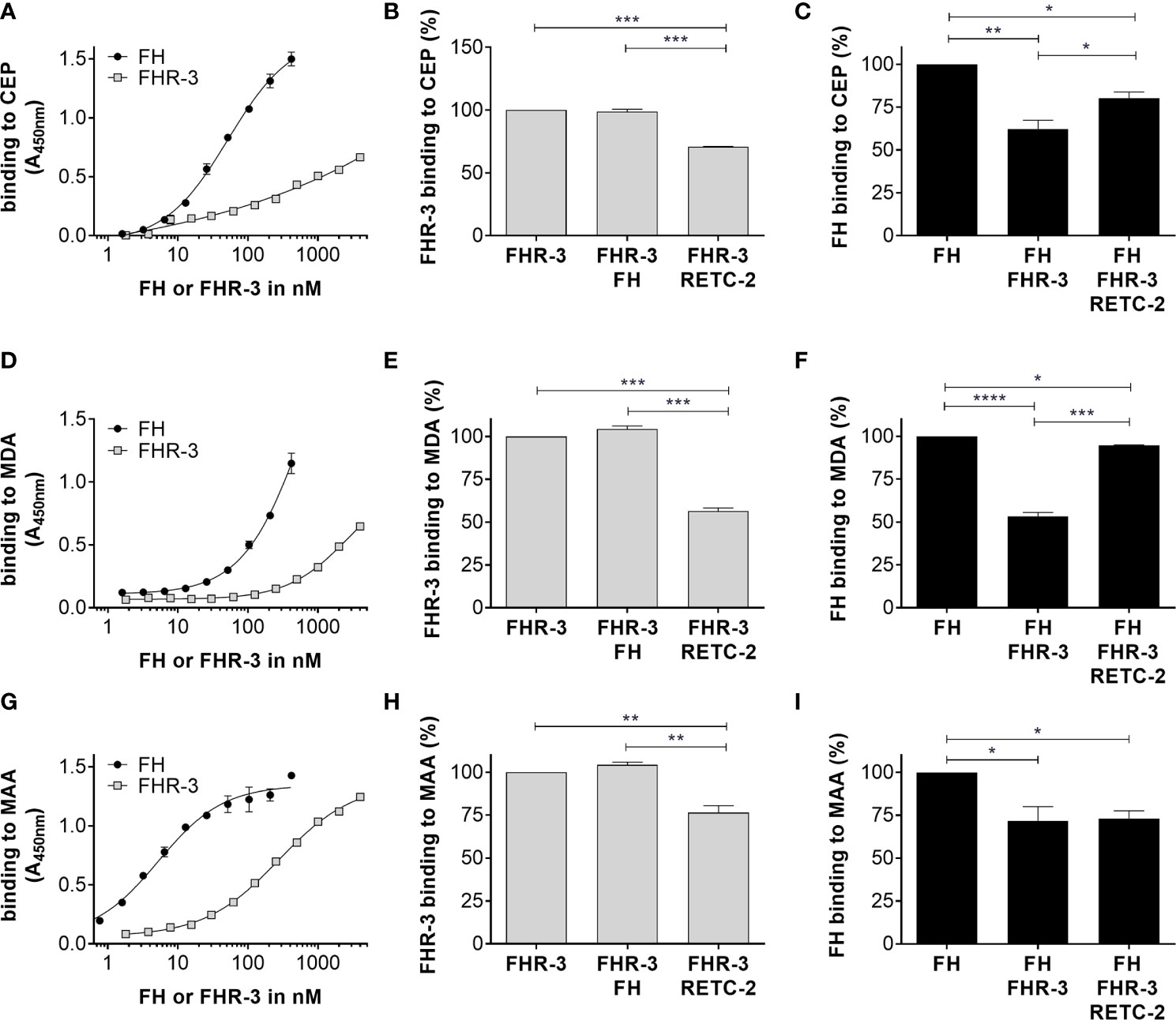
Figure 2 FHR-3 and FH interacted with OSEs and RETC-2 reduced this competitive binding. All three lipid peroxidation products (A) CEP, (D) MDA, and (G) MAA interacted concentration dependently with FHR-3 and FH. FH (black) showed a higher binding strength than FHR-3 (grey) to the OSE. Interaction of FHR-3 with (B) CEP, (E) MDA, or (H) MAA could not be reduced by additional incubation with FH, but RETC-2 significantly reduced the binding of FHR-3 to all three peroxidation products. Interaction of FH to (C) CEP, (F) MDA, and (I) MAA was significantly reduced by additional incubation with FHR-3, and anti-FHR-3 antibody RETC-2 was able to reverse this inhibitory effect completely (F, MDA), partially (C, CEP), or not at all (I, MAA). ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05 (one-way ANOVA with Dunnett’s multiple comparisons test, n = 3).
Expression of recombinant FHR-3 and FHR-1
FHR-3 with a Strep-tag II was expressed in HEK293 cells (American Type Culture Collection) as previously described (10).
FHR-1 was transiently expressed in HEK293 cells. For this, the CFHR1 gene was cloned into expression vector pEXPR-IBA103 containing a c-terminal Strep-tag II (IBA) by using specific in-house-designed primer pairs (Metabion, Supplementary Table 1). The generated construct pEXPR-CFHR1 was transiently inserted into HEK293 cells (Thermo Fisher Scientific) with TransIT-LT1 Transfection Reagent (Mirus Bio, Madison, WI, USA, #MIR2305), according to the manufacturer’s protocol. FHR-1 with Strep-tag II was purified from HEK293 supernatants using Strep-Tactin Sepharose columns (IBA). After gradient elution of FHR-1, the recombinant protein was concentrated by vacuum centrifugation. Protein purity was detected with Coomassie staining and Western blot using a specific anti-FHR-1 antibody (R&D Systems, #MAB4247). Strep-tagged FHR-1 was used as a control protein to exclude an off-side effect of the Strep-tag on FHR-3-treated RPE cell inflammation and complement expression.
Protein Secretion Assays
FHR-3 and properdin levels in cell culture supernatants were determined using sandwich ELISA, as described previously (9, 36). Complement secretion levels of ARPE-19 cell supernatants were determined using MILLIPLEX MAP Human Complement Panel 1 and 2 (Merck, #HCMP1MAG-19K, HCMP2MAG-19K). Cytokine concentrations were determined according to the protocol of a custom ProcartaPlex® multiplex immunoassay kit (Thermo Fisher Scientific, #PPX-05). The readout of the multiplex assays was performed using a Magpix instrument (Luminex, Austin, TX, USA). Vascular endothelial growth factor (VEGF)-α concentrations were determined using a human VEGF Quantikine ELISA Kit (R&D systems).
Western Blot
ARPE-19 cell lysates were dissolved in RIPA buffer (Sigma-Aldrich, #R0278) with protease and phosphatase inhibitors (1:100, Sigma-Aldrich, #P8340). Samples were diluted in reducing ROTI®Load 1 (Carl Roth, Karlsruhe, Germany, #K929.1) and denatured (95°C, 10 min). Following sample separation in a 12% SDS-PAGE, proteins were transferred onto an activated polyvinylidene difluoride membrane using a wet-blotting system. Membranes were blocked with 5% BSA/TBS-T (1 h) and incubated with specific primary antibodies (5% BSA/TBS-T, overnight): anti-C3b-α (Figures 3C, 4A, B, Progen, Heidelberg, Germany, #61019), anti-FB (Figure 5C, Merck, #341272), anti-C3aR (Figure 6B, Antibodies-online, #ABIN682213), anti-CD11b (Figure 7B, Biorbyt, #orb19554), anti-GAPDH. Detection was performed using peroxidase-conjugated anti-species antibodies (TBS, 1 h): anti-mouse IgG-Fcγ-POD (Jackson ImmunoResearch, #115-035-164), anti-goat IgG-Fcγ-POD (Jackson ImmunoResearch, #305-035-003), anti-rabbit IgG-Fcγ-POD (Jackson ImmunoResearch, #111-035-003), anti-GAPDH-HRP (Cell Signaling Technology, #3683). Visualization was performed by WesternSure PREMIUM Chemiluminescent Substrate (LI-COR, Bad Homburg, Germany) in a Fluor Chem FC2 Imaging System (Alpha Innotech, San Leandro, CA, USA). Afterwards, Western blots were quantified against a GAPDH housekeeping protein and calculated using Image Studio Lite (LI-COR).
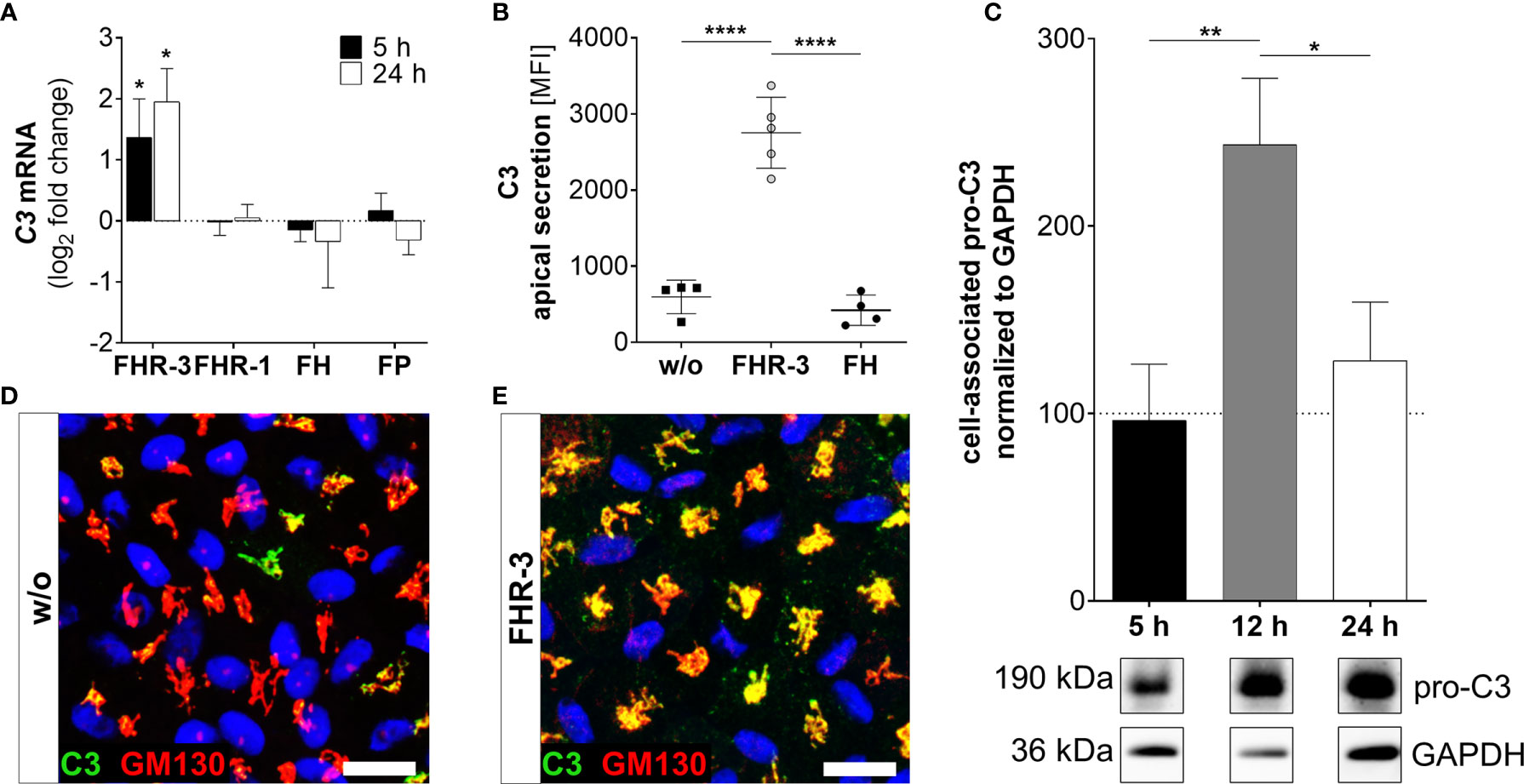
Figure 3 FHR-3 increased C3 expression and secretion in ARPE-19 cells. (A) C3 mRNA expression was increased 5 h and 24 h after apical treatment of ARPE-19 cells with FHR-3, but not after incubation with FHR-1, FH or FP, respectively. This effect could be confirmed at the protein level showing an FHR-3-dependent increase (B) in C3 protein secretion after 24 h, (C) elevated pro-C3 protein (190 kDa) in ARPE-19 cell lysates in Western blots after 12 h, and (D, E) increased intracellular C3 protein levels by immunofluorescence using anti-C3 (green) and anti-GM130 (red, cis-Golgi marker) antibodies after 12 h treatment. C3 was co-localized with the cis-Golgi (yellow). Scale bars 40 µm. w/o untreated control [(A, C) dotted line]. Mean with standard deviation is shown. Full Western blot in Supplementary Figure 1K. ****p < 0.0001, **p < 0.01, *p < 0.05. (A) Wilcoxon matched-pairs signed rank test (n = 3); (B, C) Ordinary one-way ANOVA, Turkey’s multiple comparisons test (n = 3).
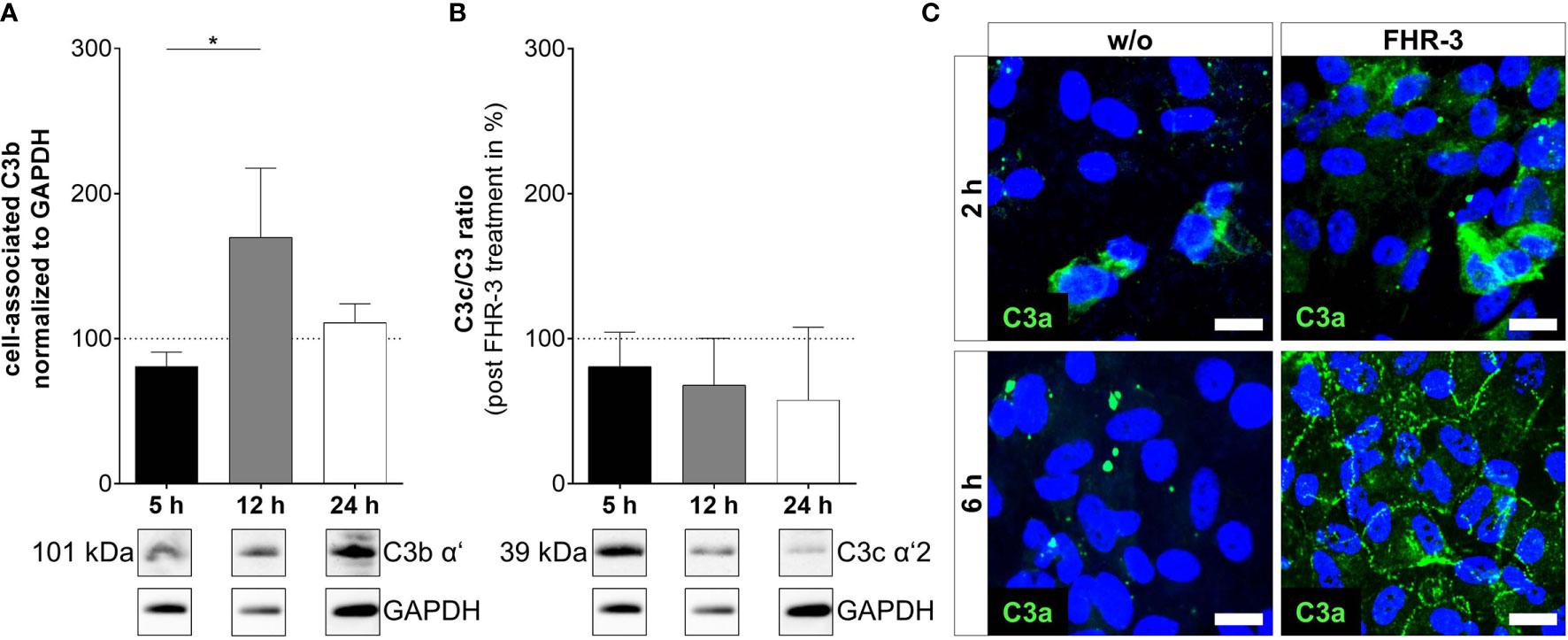
Figure 4 C3 cleavage products were changed in ARPE-19 cells after FHR-3 incubation. (A) Increased C3b levels (101 kDa) 12 h after FHR-3 treatment and (B) a time-dependent reduction of C3c fragments (39 kDa) in ratio to pro-C3 were detected in Western blots of ARPE-19 cell lysates. Supplementary Figure 1K shows full Western blots. (C) Anaphylatoxin C3a was detected by immunofluorescence using a specific anti-C3a antibody (green). C3a increased time-dependently from 2 h to 6 h after FHR-3 treatment and was translocated from the cytoplasm (upper right panel) to the cell membrane (lower right panel). Scale bars 40 µm. w/o untreated control (dotted line). Mean with standard deviation is shown. *p < 0.05. (A, B) Ordinary one-way ANOVA, Turkey’s multiple comparisons test (n = 3).
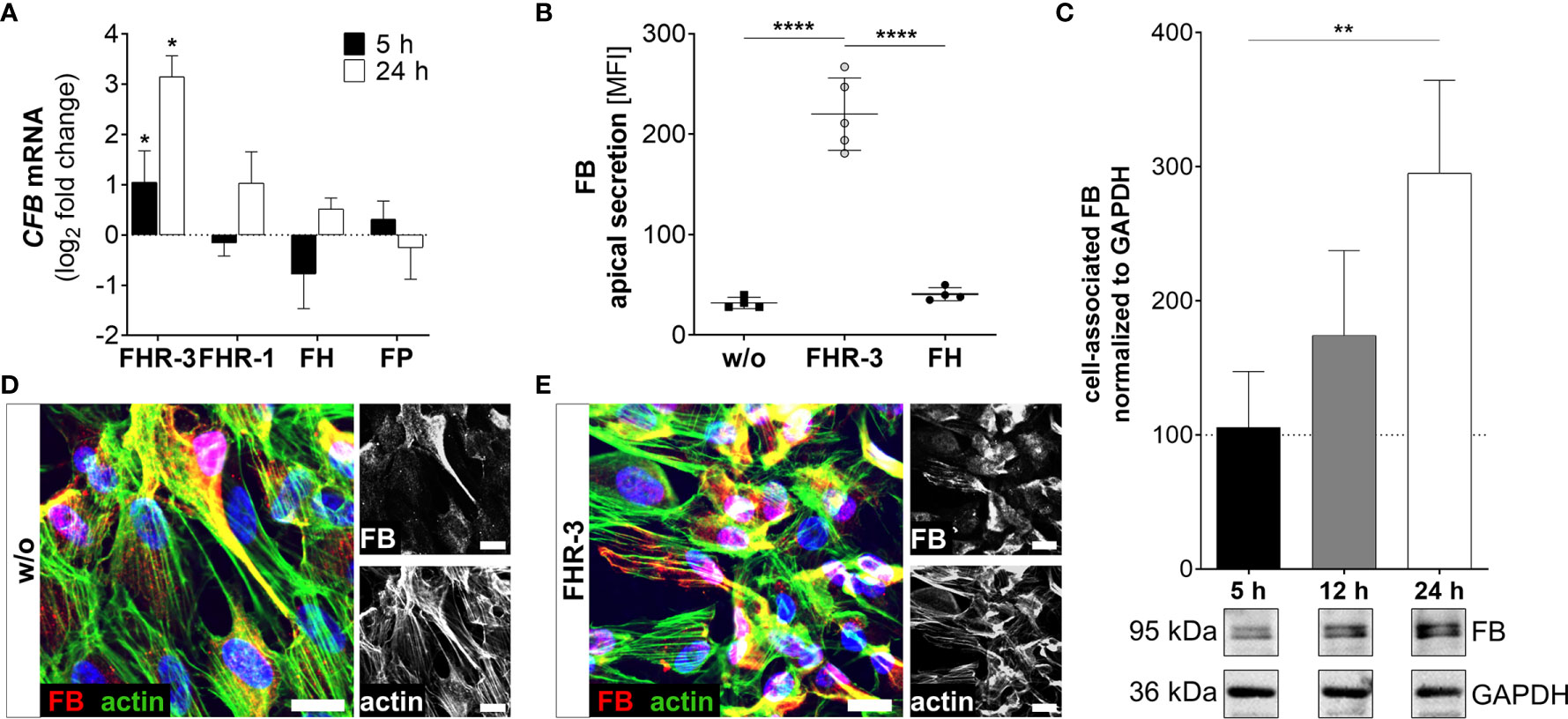
Figure 5 FHR-3 increased FB expression and secretion in ARPE-19 cells. (A) CFB mRNA increased after apical FHR-3 treatment of ARPE-19 cells, but not following incubation with FHR-1, FH or FP. This effect could be confirmed at the protein level: (B) Apical FB secretion was increased 24 h after FHR-3 incubation. (C) Western blots of ARPE-19 cell lysates detected a time-dependent increase in FB levels (95 kDa) 24 h after FHR-3 treatment. Supplementary Figure 1L shows full Western blots. (D, E) Increased FB protein levels were detected by immunofluorescence using specific anti-FB (red) and anti-actin (green) antibodies 12 h after FHR-3 treatment. FB was co-localized partly with actin stress fibers (yellow). Scale bars 40 µm. (A–C) w/o untreated control (dotted line). (A–C) Mean with standard deviation is shown. ****p < 0.0001, **p < 0.01, *p < 0.05. (A) Wilcoxon matched-pairs signed rank test (n = 3); (B, C) Ordinary one-way ANOVA, Turkey’s multiple comparisons test (n = 3).
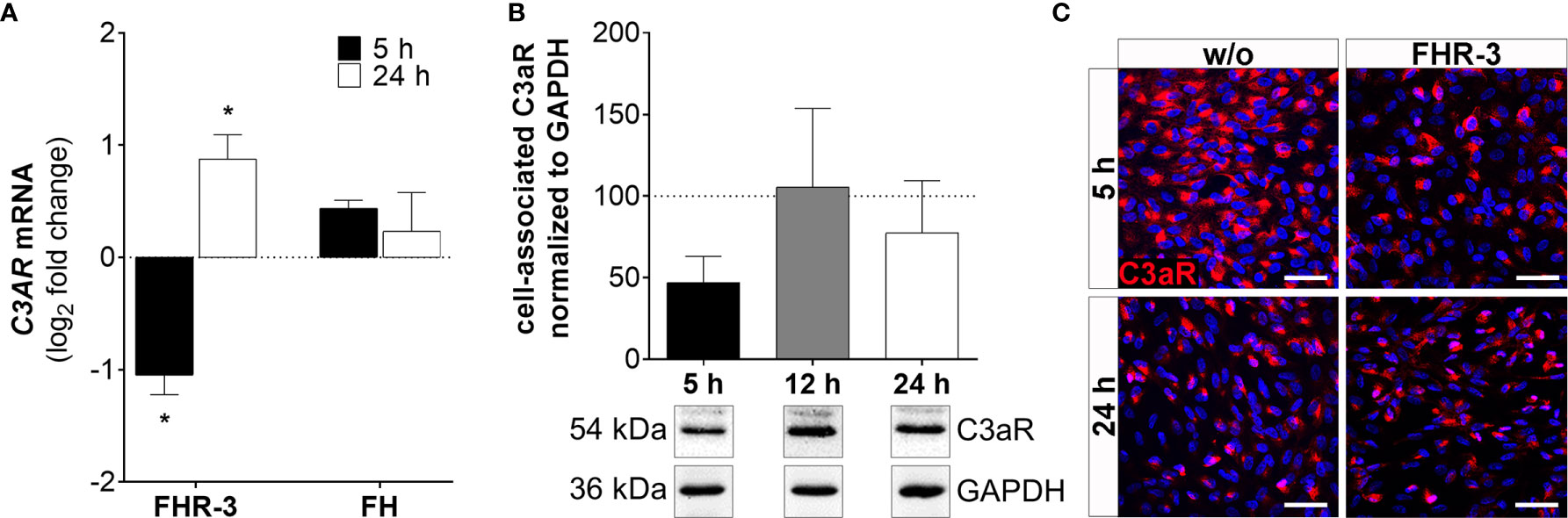
Figure 6 C3aR expression was time-dependently regulated in FHR-3 stressed ARPE-19 cells. (A) C3AR mRNA expression was decreased after 5 h and increased after 24 h FHR-3 incubation. This effect could be confirmed on protein level: (B) Western blots of ARPE-19 cell lysates showed a tendency for decreased C3aR levels (54 kDa) 5 h after FHR-3 treatment. Supplementary Figure 1M shows full Western blots. (C) Decreased C3aR protein levels were detected by immunofluorescence using a specific anti-C3aR antibody (red) after 5 h FHR-3 incubation (upper panels), whereas no differences between FHR-3 stressed and unstressed controls were observed after 24 h (lower panels). Scale bars 40 µm. w/o untreated control (dotted line). Mean with standard deviation is shown. *p < 0.05. (A) Wilcoxon matched-pairs signed rank test (n = 3); (B) Ordinary one-way ANOVA, Turkey’s multiple comparisons test (n = 3).
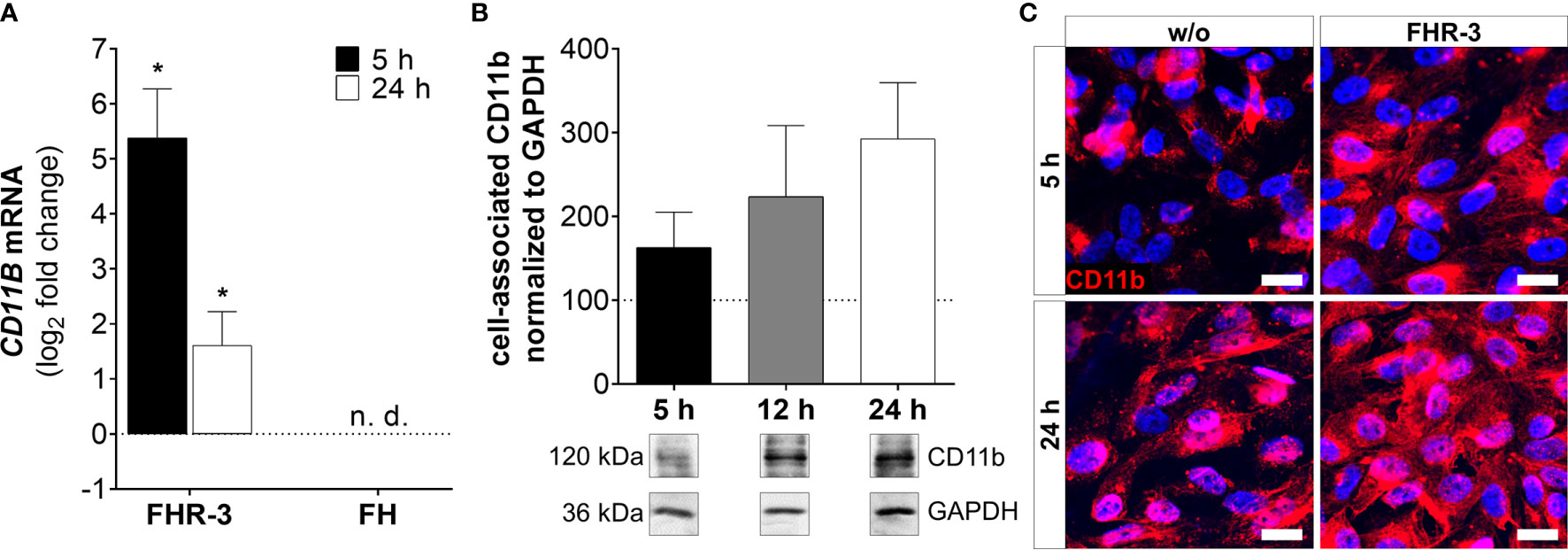
Figure 7 CD11b expression was highly elevated in FHR-3 treated ARPE-19 cells. (A) CD11B mRNA expression was highly elevated after 5 h and 24 h FHR-3 incubation. No expression of CD11B mRNA could be detected when cells were treated with FH. The effect of FHR-3 could be confirmed at the protein level: (B) Western blots of ARPE-19 cell lysates showed a time-dependent increase in CD11b levels (120 kDa) from 5 h – 24 h after FHR-3 incubation. Supplementary Figure 1N shows full Western blots. (C) Elevated CD11b protein levels were detected by immunofluorescence using a specific anti-CD11b antibody (red) after 5 h and 24 h of FHR-3 incubation (right panels). Scale bars 40 µm. w/o untreated control (dotted line). n.d. not detected. Mean with standard deviation is shown. *p < 0.05. (A) Wilcoxon matched-pairs signed rank test (n = 3); (B) Ordinary one-way ANOVA, Turkey’s multiple comparisons test (n = 3).
Chimerization of Anti-FHR-3 Antibody RETC-2
Variable regions (heavy and light chain) of murine RETC-2 (anti-FHR-3) and murine control antibody (anti-BSA) were determined as described previously (9) and cloned into plasmids containing either human IgG4 heavy chain (pFUSE-CHIg-hG4e1, In vivogen, Toulouse, France, #pfuse-hchg4e1) or human IgG kappa light chain (pFUSE2-CLIg-hk, In vivo gen, #pfuse2-hclk), according to the manufacturer’s protocol. The generated constructs for the heavy and light chains were transiently co-transfected into HEK293 cells (Thermo Fisher Scientific, using IgG-free FCS in Medium) using a ratio of 3 : 2 (light chain construct : heavy chain construct) with TransIT-LT1 Transfection Reagent (Mirus), according to the manufacturer’s protocol. Cell supernatants were tested for antibody amount and for specific FHR-3 binding using ELISA and Western blots. Chimerized antibodies were purified using Protein-A columns HiTrap® MabSelect™ SuRe™ (Merck, #GE11-0034-94) and named as follows: RETC-2-ximab, control-ximab.
Statistics
Statistics were performed using GraphPad Prism 8 (GraphPad Software Inc., San Diego, CA, USA). All data are expressed as mean ± standard deviation (SD) unless stated otherwise. Detailed information about specific n-values, implemented statistical analyses and coding of significance levels are documented in each figure and figure legend, respectively.
Results
We previously reported that FHR-3 is co-localized with activated macrophages/microglia cells in an aged and inflamed retina, but not detectable in a healthy retina (9). Due to our additional results, showing that RPE cells in response to stress increased the expression of complement components and pro-inflammatory factors (30), we proposed that FHR-3 could be a stress factor for the RPE in the aged retina promoting retinal degeneration (9). Consequently, we investigated here the cell-specific complement and inflammation-associated response of RPE cells exposed to FHR-3.
To exclude a genotype-primed reactivity ARPE-19 cells and human primary RPE cells (hpRPE) were used in this study. We determined the most common AMD-associated SNPs within genes of the complement pathway in these cells (37). Homogenous AMD-risk SNPs could not be detected in the examined RPE cells, instead heterozygous SNPs were present in the CFH and C3 gene of ARPE-19 and CFH, C2/CFB, CFI and ARMS gene of hpRPE cells (Supplementary Figure 1A).
We verified the epithelial phenotype of the used ARPE-19 cells, passaged for 38 times and cultivated under in vivo-like conditions, by staining tight junction protein zonula occludens 1 (ZO-1). A polarized monolayer could be detected for untreated and FHR-3 treated cells, showing that FHR-3 had no effect on stable cell-cell contacts. We also excluded that ARPE-19 cell passaging had any influence on FHR-3 dependent tight junction formation by comparing ZO-1 stainings in passage number 38 (P38) with 25 (P25) (Supplementary Figures 1D–G). Transepithelial resistance and cellular capacitance of the polarized ARPE-19 cells were measured between 0.5 – 72 h of FHR-3-treatment. FHR-3 had no impact on cell barrier function and on cell membrane folding (Supplementary Figure 1B). These characterizations resulted in a specific RPE phenotype, with a slight shift to mesenchymal characteristics established by mRNA expression analysis of epithelial-mesenchymal transition (EMT) markers vimentin (VIM), α‐smooth muscle actin (ACTA2) and collagen type 1 (COL1A1) (Supplementary Figures 1H–J). FHR-3 treatment increased VIM expression in ARPE-19 cells P38 and P25 compared to untreated controls (Supplementary Figure 1H), whereas ACTA2 and COL1A1 were only raised in ARPE-19 cells P25 (Supplementary Figures 1I, J) indicating an early EMT caused by FHR-3 and already existing mesenchymal characteristics in high-passage ARPE-19 cells (P38).
Due to the number of passages and the slight EMT, which is typical for aged human RPE cells (38), the investigated ARPE-19 cells at P38 were used in this study.
FHR-3 Was Internalized by ARPE-19 Cells
Previous reports described an interaction of proteins of the FH-protein family with damaged cells: FH was internalized by apoptotic ARPE-19 cells (21), FHR-1 and FHR-5 bound to necrotic ARPE-19 cells (23), and FHR-1 and FHR-3 interacted with a necrotic-type endothelial cell line (19, 20). In this study, we showed that FHR-3 was bound to and internalized by viable ARPE-19 cells (Figure 1 and Supplementary Figure 1C), knowing that CFHR3 mRNA is not expressed in these cells (Supplementary Figure 3). Interaction of FHR-3 with ARPE-19 cells was confirmed by immunofluorescence resulting in FHR-3-positive ARPE-19 cells following FHR-3 incubation (Figures 1A, B). Further, in the supernatant of the ARPE-19 cells supplemented with FHR-3 only 30% of the added FHR-3 remained in the apical and 5% in the basal supernatant compared to the added complement control protein properdin (FP), which was stable to 92% in the apical and 3% in the basal supernatant after incubation with ARPE-19 cells (Supplementary Figure 1C). To investigate whether the complement regulator was internalized, we labelled FHR-3 with pH-sensitive dye pHrodo (FHR-3-pHrodo). This dye is non-fluorescent outside the cell, but fluoresces in acidic, cellular compartments like endosomes, phagosomes or lysosomes. Added FHR-3-pHrodo was detected inside high-passage ARPE-19 cells indicating a phagocytosis or receptor-mediated endocytosis (Figure 1D). This shift in fluorescence activity could not be detected when cells were incubated with FH-pHrodo concluding that FH was not internalized in viable ARPE-19 cells.
Just recently, it was shown that FHR-3 binds to malondialdehyde (MDA)-epitopes (19). These OSE are present on the surface of stressed ARPE-19 cells (16). We determined as well specific binding of FHR-3 and FH either to OSE 2-(ω-carboxyethyl)pyrrole (CEP), MDA or malondialdehyde-acetaldehyde (MAA) (Figures 2A, D, G). We detected a competitive binding of FHR-3 and FH to CEP, MDA and MAA. Interaction of FHR-3 and oxidative stress epitopes was not changed by additional incubation of FH (Figures 2B, E, H), whereas a reduced FH interaction with CEP (38%), MDA (47%) and MAA (28%) could be observed when FHR-3 was added as a competitor (Figures 2C, F, I). Our previously published anti-FHR-3 antibody RETC-2 (9) decreased binding of FHR-3 to CEP (29%), MDA (44%) and MAA (24%) (Figures 2B, E, H), and prevented the competitive effect of FHR-3 with FH for the interaction with CEP (47%) and MDA (87%). This impact could not be detected for MAA (Figures 2C, F, I).
Here, we showed FHR-3 internalization by viable, high-passage ARPE-19 cells (Figure 1), and determined peroxidation products CEP, MDA and MAA as potential ligands. FHR-3 bound to OSE and prevented their interaction with FH, which was reversed by anti-FHR-3 antibody RETC-2 (9) (Figure 2).
FHR-3 Increased Endogenous Complement Activation
Complement components are locally expressed by different retinal cell types and by ARPE-19 cells (27, 30, 31, 39). Here, we showed that the complement regulator FHR-3 enhanced complement expression and secretion of RPE cells (Figures 3–5 and Supplementary Figure 2A). We demonstrated an increase of C3 mRNA expression in polarized high-passage ARPE-19 cells and hpRPE cells after FHR-3 treatment, whereas no expression change was shown for FHR-1, FH or FP incubation (Figure 3A, Supplementary Figure 2A). On protein level, an increased C3 secretion after 24 h (Figure 3B) and enhanced cell-associated precursor form of C3 protein (pro-C3, 190 kDa) detection in cell lysates after 12 h and 24 h FHR-3 incubation compared to untreated cells were examined (Figure 3C and Supplementary Figure 1K). We visualized accumulated intracellular C3, co-localized with Golgi complex, which seems to be mainly pro-C3, increased in 12 h FHR-3-treated ARPE-19 cells (Figures 3D, E).
In line with our previous published data, where we reported C3 activation fragments in ARPE-19 cells under oxidative stress (30), we assume an indirect involvement of FHR-3 in C3 cleavage in ARPE-19 cells due to increase of C3 protein concentration (Figure 4). However, we did not detect a direct contact of FHR-3 and C3 in ARPE-19 cells (Supplementary Figure 5), nor an increase in cathepsin L (CTSL) expression, which cleaves intracellular C3 (Supplementary Figure 3) (40). We detected raised levels of cell-associated C3b (101 kDa) after 12 h of FHR-3 treatment (Figure 4A). However, C3c as a marker for inactivated C3 was time-dependently reduced (5 h – 24 h) when ARPE-19 cells were incubated with FHR-3 (Figure 4B). Anaphylatoxin C3a was increased in FHR-3-treated cells and showed a translocation from the cytoplasm to the cell membrane after FHR-3 exposure (Figure 4C).
Increased detection of FB was also a consequence of FHR-3 addition to ARPE-19 cells and hpRPE cells (Figure 5 and Supplementary Figure 2B). CFB transcripts were highly elevated after 5 h and 24 h of FHR-3 incubation and no expression changes were shown for FHR-1, FH or FP treatment (Figure 5A and Supplementary Figure 2B). Enhanced FB protein secretion after 24 h (Figure 5B) and a time-dependent increase in cell-associated FB protein expression in cell lysates from 5 h to 24 h of FHR-3 treatment were determined (Figure 5C). FB cleavage products Bb and Ba were also detected in ARPE-19 cells and raised in FHR-3-treated cells (Supplementary Figure 1L). Using immunofluorescence, we showed accumulated intracellular FB, partly co-localized with actin stress fibers in FHR-3 treated ARPE-19 cells (Figures 5D, E).
Our data indicated a cell-associated complement-activating effect of FHR-3 by enhancing C3 and FB expression as well as secretion and by anaphylatoxin C3a increase in immortal ARPE-19 cells (Figures 3–5 and Supplementary Figure 4) as well as cultivated, post-mitotic, hpRPE cells (Supplementary Figures 2A, B).
FHR-3 Altered Complement Receptor Expression of ARPE-19 Cells
ARPE-19 cells express a variety of complement receptors (30, 41). It has recently been reported, that oxidatively stressed ARPE-19 cells increased expression of complement receptors CR3 and C5aR1 (30). Here, we showed that FHR-3 modified complement receptor C3aR (Figure 6 and Supplementary Figure 2C) and CD11b (Figure 7 and Supplementary Figure 2D) expression on high-passage ARPE-19 cells and hpRPE cells, independently from any systemic complement.
C3AR mRNA expression was time-dependently changed with decreased transcripts after 5 h (Figure 6 and Supplementary Figure 2C) and elevated expression after 24 h of FHR-3 exposure (Figure 6A). This was partly in accordance with protein data showing decreased cell-associated C3aR levels in Western blots (Figure 6B) and immunohistochemically (Figure 6C) 5 h after FHR-3 incubation. An increase in C3aR protein expression 24 h after treatment, as shown at the mRNA level, could not be verified. This might be explained by storage of C3AR RNA into RNA granules, either stress granules or processing bodies (42).
ARPE-19 cells express the α-chain CD11b of the integrin complement receptor CR3 (30). We detected very low mRNA expression in untreated-, and no expression in FH-treated ARPE-19 cells. When cells were incubated with FHR-3, CD11B expression increased significantly in ARPE-19 and hpRPE cells, respectively (Figure 7A and Supplementary Figure 2D). Western blot analyses revealed a time-dependent upregulation of CD11b from 5 h – 24 h after FHR-3 incubation (Figure 7B). Immunostainings of FHR-3 treated and untreated cells confirmed CD11b accumulation after 5 h and 24 h (Figure 7C). We did not observe a change in C5aR1 mRNA transcript levels following FHR-3 treatment, and C5aR2 mRNA transcripts were not detected in ARPE-19 cells (Supplementary Figure 3).
These results revealed an FHR-3 triggered alteration of complement receptor C3aR and CD11b expression in immortal, high-passage ARPE-19 cells (Figures 6, 7 and Supplementary Figure 4) as well as cultivated, post-mitotic, hpRPE cells (Supplementary Figures 2C, D).
FHR-3 Induced Pro-Inflammatory Markers in ARPE-19 Cells
NLRP3 inflammasome activation is considered as an additional hallmark for the development of AMD (43). It has already been shown that anaphylatoxins C3a and C5a, and oxidative stress lead to the priming of NLRP3 and secretion of pro-inflammatory cytokines in ARPE-19 cells (25, 30, 44). Here, we describe that FHR-3, a potential risk factor of AMD progression, induced inflammasome-associated pro-inflammation in ARPE-19 cells (Figures 8A–D). NLRP3 mRNA expression was elevated 5 h and slightly decreased 24 h after FHR-3 incubation, whereas treatment with FHR-1, FH or FP did not show any expression changes (Figure 8A). Accordingly, transcripts of IL1B were also upregulated 5 h and 24 h after FHR-3 incubation (Figure 8B). In line with this, determination of cytokine secretion levels of FHR-3-treated ARPE-19 cells revealed an upregulation of apical secretion of IL-1ß (Figure 8C) and IL-18 (Figure 8D) compared to untreated cells (Figures 8C, D, dotted line).
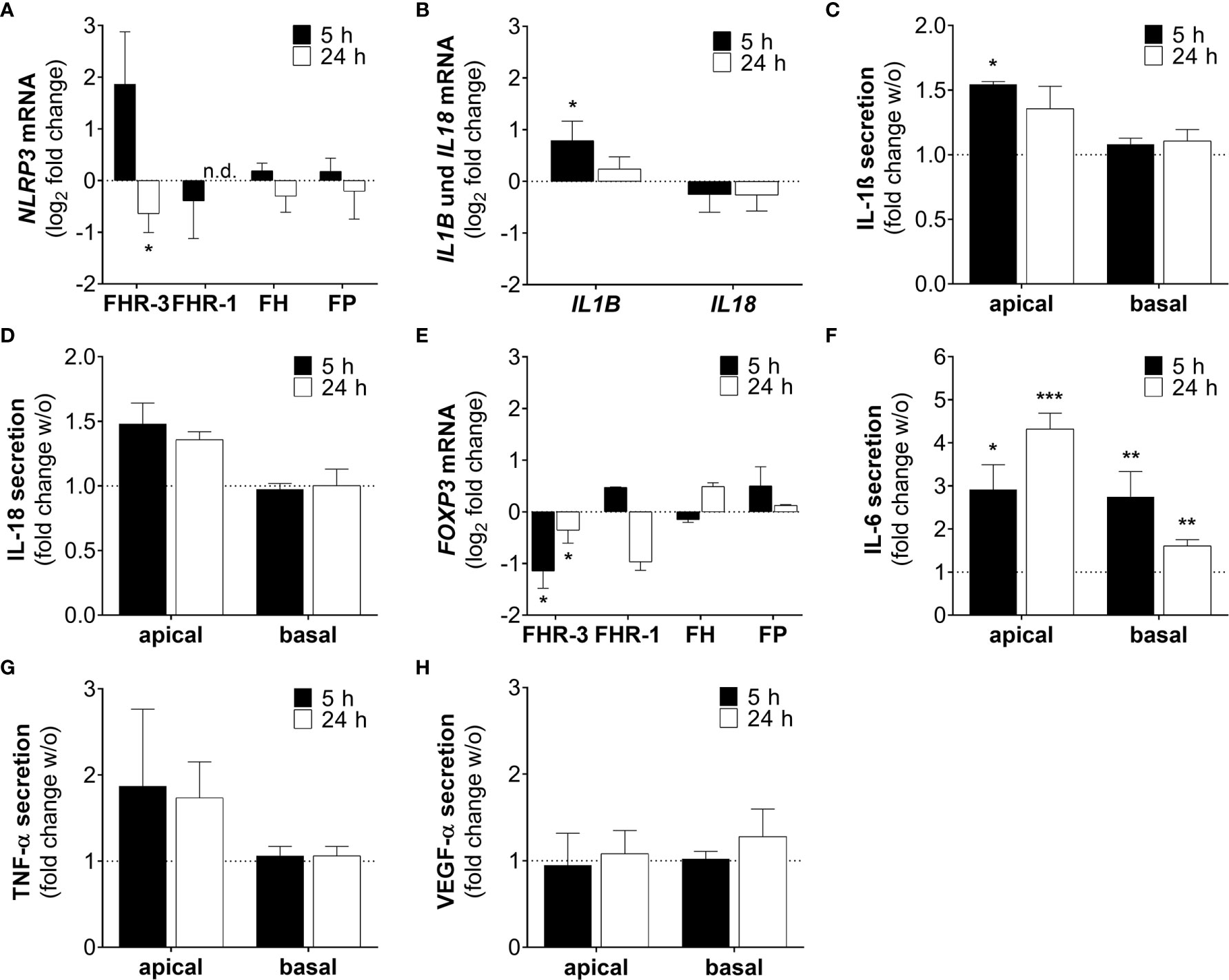
Figure 8 FHR-3 induced an ARPE-19 cell dependent pro-inflammatory microenvironment. (A) NLRP3 mRNA expression in ARPE-19 cells was either increased or decreased after 5 h or 24 h FHR-3 treatment, respectively. This was in contrast to no expression changes following FHR-1, FH or FP incubation. (B) Transcripts of IL1B were significantly elevated, whereas IL18 mRNA expression was slightly decreased after FHR-3 treatment. (C, D) Apical secretion levels of (C) IL-1ß and (D) IL-18 were increased in ARPE-19 cells after FHR-3 stress. (E) FOXP3 mRNA expression was reduced 5 h and 24 h after FHR-3 incubation. (F) Secretion of IL-6 protein was significantly raised after FHR-3 incubation, both on the apical and basal cell site. (G) ARPE-19 cell secretion of TNF-α protein was on trend increased after FHR-3 incubation on the apical site compared to w/o. (H) VEGF-α was secreted by ARPE-19 cells, but showed no differences between FHR-3 treated and untreated cells. w/o untreated control (dotted line). n.d. not detected. Mean with standard deviation is shown. ***p < 0.001, **p < 0.01, *p < 0.05. (A, B, E) Wilcoxon matched-pairs signed rank test (n = 3); (C, D, F–H) Unpaired t-test (n = 5).
Inflammatory cellular micro-environments are shaped by further key players, e.g. forkhead box P3 (FOXP3), an inflammation-associated transcription factor, which triggers secretion of anti-inflammatory cytokines in regulatory T-cells. Expression of FOXP3 was detected recently in ARPE-19 cells (30, 45, 46). We determined a time-dependent significant decrease in FOXP3 mRNA expression, after FHR-3 incubation (Figure 8E), indicating a pro-inflammatory effect of FHR-3.
Additionally, the concentration of the pro-inflammatory cytokine IL-6 was elevated in both, apical and basal supernatants of FHR-3-incubated ARPE-19 cells (Figure 8F). A tendency to higher secretion levels of tumor necrosis factor (TNF)-α could be also detected in apical supernatants of ARPE-19 cells incubated with FHR-3 (Figure 8G). However, a regulation of pro-angiogenic marker vascular endothelial growth factor (VEGF)-α, which is important for proper RPE function, could not be determined after FHR-3 incubation (Figure 8H).
In sum, these results proposed a pro-inflammatory role of FHR-3 on aged RPE cells, independent from blood-derived complement components, which may have a so far unknown impact on AMD progression (Supplementary Figure 4).
RETC-2-ximab Diminished The Inflammatory Effect of FHR-3
In our previous studies we generated a highly specific monoclonal mouse antibody (mAb) against human FHR-3, RETC-2. We showed that RETC-2 inhibits binding of FHR-3 to C3b and regained interaction of FH to C3b (9). Similar results could be observed regarding FHR-3 and FH binding to OSEs (CEP, MDA, MAA), as described afore (Figure 2). To further investigate a therapeutic potential of RETC-2, chimerization was performed to replace complement-activating mouse regions of the mAb. The chimerized anti-FHR-3 mAb, called RETC-2-ximab, was tested in in vitro studies with polarized high-passage ARPE-19 cells, which were treated apically for 24 h with FHR-3, FHR-3 and RETC-2-ximab or with FHR-3 and an antibody isotype control (control-ximab, specific for BSA) (Figure 9). Gene expression of C3 decreased by 27% (Figure 9A), CFB was reduced significantly by 48% (Figure 9B), respectively, and C3AR mRNA expression was decreased by 21% (Figure 9C) after combined treatment with FHR-3/RETC-2-ximab compared to FHR-3 alone or with the FHR-3/control-ximab in high-passage ARPE-19 cells.
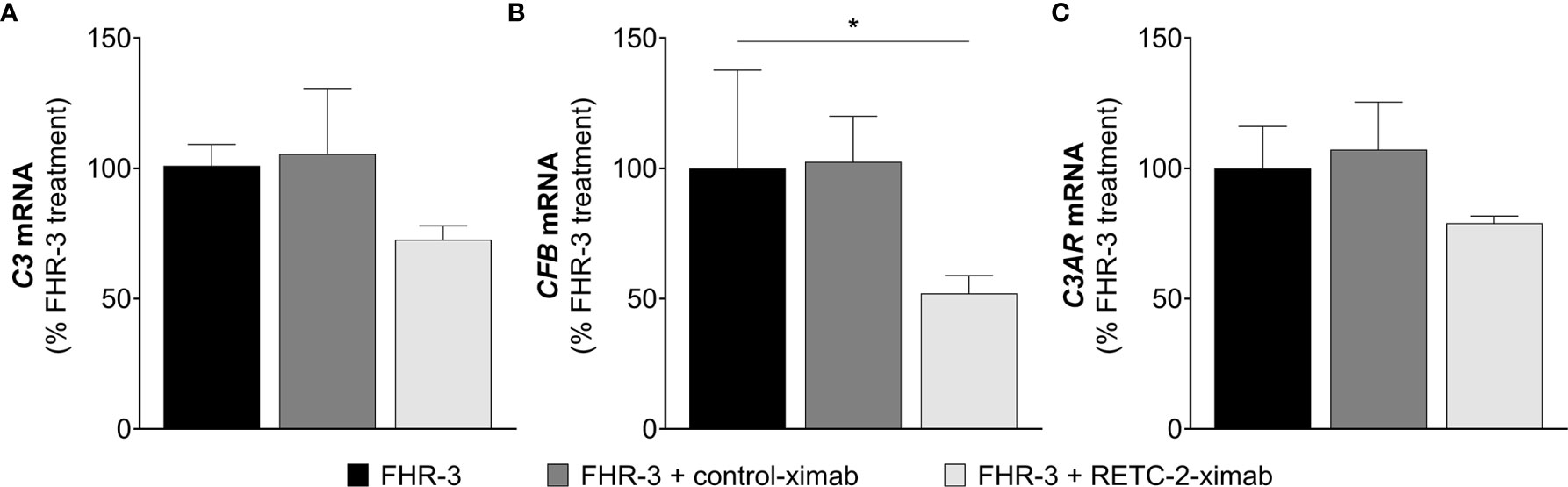
Figure 9 RETC-2-ximab slightly ameliorated the effect of FHR-3 on ARPE-19 cells. ARPE-19 cells were treated with either FHR-3 (black), FHR-3 and control-ximab (anti-BSA, dark grey) or FHR-3 and RETC-2-ximab (anti-FHR-3, light grey). (A) C3 mRNA expression was reduced by 27%, (B) CFB mRNA expression was reduced by 48%, and (C) C3AR mRNA expression was reduced by 21% when incubated with FHR-3 and RETC-2-ximab compared to FHR-3 alone. Shown is the relative mRNA expression to FHR-3 treated cells in %. Mean with standard deviation is shown. *p < 0.05. Unpaired t-test (n = 2).
These promising results could offer a potential recovery of local complement homeostasis and a reduced progression of retinal degeneration using RETC-2-ximab in the future.
Discussion
It has been known for 15 years, that the deletion of the genes for CFHR3 and CFHR1 (ΔCFHR3/1) is associated with a protective effect for the development of AMD (7, 47). However, the local biological function of FHR-3 and FHR-1 proteins is still poorly understood. We discovered recently, that FHR-3 – not expressed in RPE cells – is produced by macrophages/microglia within an aged, inflamed retina (9). In this study, we focussed on the intraocular role of FHR-3 on RPE cells and did not consider whether FHR-3 could interact basally with the RPE to examine a systemic effect, as Cipriani et al. recently described for FHR-4 (48).
Here, we demonstrated a complement activating and pro-inflammatory effect on human RPE cells mediated by FHR-3, which could be ameliorated by a chimeric anti-FHR-3 antibody.
Endocytosis of FHR-3 by Viable ARPE-19 Cells
Our data revealed an incorporation of FHR-3 by high-passage viable RPE cells. So far, internalization of other FH-family proteins has been solely described for apoptotic cells. Martin et al. examined a specific FH uptake by apoptotic rendered Jurkat T- and ARPE-19 cells, whereas neither binding nor uptake could be confirmed by living cells (21). FHR-1 and FHR-5 have also been reported to bind to necrotic RPE and endothelial cells, but an uptake was not described (20, 23).
FHR-3 uptake could be linked to OSE and potentially unknown co-receptors on aged RPE cells as potential ligands for FHR-3-dependent membrane invagination. Joseph et al. showed that oxidatively stressed RPE cells express lipid peroxidation product MDA on their cell surface (16). In accordance to Alic et al. we showed a specific binding of FHR-3 to MDA-epitopes and a competitive interaction with FH to these OSE (19). We also observed the same interaction of FHR-3 with CEP, a peroxidation product serving as biomarker for AMD (49). It is known that FH binds to OSE and thereby inhibits pro-inflammation in the aging eye (17, 18), indicating that FHR-3-FH-OSE interaction may impact the risk for AMD progression. Just recently, Irmscher et al. detected a binding of FHR-1 to MDA-epitopes (20) and Rudnick et al. showed an interaction of FHR-5 with lipid peroxidation products (50), both indicated an enhanced complement activity and inflammation in endothelial cells and monocytes. To what extend lipid peroxidation may be sufficient for cell membrane invagination and consequent uptake of FHR-3, rather than FH in viable RPE cells – even this pathway of lipid-mediated endocytosis is poorly understood (51) – needs to be further investigated.
FHR-3, a Danger Associated Molecule Within the Eye?
Beyond the classical function of the complement system as part of the humoral immune system, which has been known for more than 100 years, more and more non-canonical functions and the “complosome” have been assigned in the last decade, including the influence on cell metabolism, cell development and regeneration (22, 52, 53). In the past years, rising data indicate that FHR proteins have complement activating properties (20, 21, 23, 24, 48, 50). While FHR-1, FHR-3, FHR-4 and FHR-5 compete systemically with FH for C3b binding, less is known about their non-canonical functions as cellular modulators (23, 48, 54, 55). Here, we treated serum-free, in vivo-like cultivated ARPE-19 and hpRPE cells apically with FHR-3, FHR-1, FH or properdin, respectively, to investigate the cell-associated complement and inflammatory response independent of any external complement sources.
We showed that FHR-3 is an inflammatory stimulator and may serve as a damage-associated sensing molecule for RPE cells (56). Just recently, we reported complement expression in hpRPE cells (27, 31), ARPE-19 cells (30) and murine RPE (39). Here, we described that FHR-3 enhanced complement activation in cultured RPE cells. C3 cleavage was indicated by increased C3b protein detection and accumulation of C3a in ARPE-19 cells specifically following FHR-3 treatment, but not with FHR-1, FH or properdin incubation. Based on our results we suggest FHR-3 as a modulator of cellular C3/FB protein concentration mediated by an unknown ligand-receptor signal transduction. Contrary, Martin et al. showed that apoptotic ARPE-19 cells – not cultivated under in vivo-like conditions – internalized FH leading to increased cleavage and deposition of endogenous C3 (21). Previous studies investigating oxidative stress in ARPE-19 cells also reported increased C3 accumulation and secretion of C3 and C3a (30, 57).
We also showed a time-dependent redistribution of C3a to the cell membrane in FHR-3 treated ARPE-19 cells. C3a, especially via C3aR signaling, is known to disrupt RPE function and promote AMD progression (58). In a previous study, C3a overexpression leaded to formation of sub-RPE deposits which are associated with early-stage AMD (59). Local C3a effects could be also reported in other cells: Liszewski et al. described that intracellular lysosomal C3a led to the activation of the C3aR, which was essential for the survival of T cells (40). In endothelial cells, C3a overexpression disrupted barrier integrity via C3aR-signaling (60).
Our data revealed FHR-3 as a cellular stressor for RPE cells, which could balance the physiological and inflammatory state of the outer retinal-blood barrier.
Asgari et al. described that C3a-mediated C3aR signaling in monocytes caused initiation of the NLRP3 inflammasome and secretion of the pro-inflammatory cytokine IL-1ß (61). Further, the C3 fragment receptor CR3, has been also associated with NLRP3 inflammasome activation in human primary RPE cells (62). Besides recently shown for ARPE-19 (30) and hpRPE cells (27), CR3 expression has been mainly associated with macrophages (63) so far.
Here, we showed that FHR-3 regulated time-dependently C3aR and CR3 expression in RPE cells. Long-term FHR-3 incubation increased complement receptor C3AR and CD11b transcript levels in ARPE-19 cells.
FHR-3 does not only possess complement-activating properties, but also triggers pro-inflammatory potentials in RPE cells. Supplementation of ARPE-19 cells with FHR-3 increased the mRNA expression of NLRP3 and IL1B and, subsequently, enhanced the secretion of pro-inflammatory cytokines IL-1ß and IL-18. Irmscher et al. described an FHR-1 triggered induction of NLRP3 inflammasome in monocytes (20). Inflammasome priming in RPE cells was shown to be influenced by addition of extracellular anaphylatoxins and oxidative stress so far (25). We added FHR-3 to this list, potentially mediating NLRP3-priming by cell-autonomous C3a. However, we recently demonstrated that exogenous added properdin, the main positive regulator of the complement system, depicted an opposed effect and showed anti-inflammatory properties (27).
In line with these results, we also observed that FHR-3 increased secretion of the pro-inflammatory cytokine IL-6 by ARPE-19 cells. IL-6 plays an important role during intraocular inflammation. Previous studies demonstrated an increased IL-6 release by oxidatively stressed (64) and C3a-stimulated RPE cells (65).
FOXP3, an inflammation-associated transcription factor, promotes the secretion of anti-inflammatory cytokines in regulatory T cells (45). Busch et al. demonstrated that FOXP3 is expressed in ARPE-19 cells and activated by extracellular addition of anaphylatoxins C3a and C5a (46). Recent studies determined increased FOXP3 mRNA expression in oxidatively stressed ARPE-19 cells (30). In the present study, FOXP3 mRNA was decreased in FHR-3 stimulated ARPE-19 cells, which may be consistent with the pro-inflammatory effect of FHR-3 on RPE cells. However, the exact role of FOXP3 in RPE cells is not known so far.
Related to these pro-inflammatory events triggered by FHR-3, we chimerized our anti-FHR-3 antibody RETC-2 and hypothesized that RETC-2-ximab might serve as a new therapeutic approach for treatment of degenerated eye diseases such as AMD. We demonstrated that RETC-2-ximab slightly mitigated the effect of FHR-3 on high-passage ARPE-19 cells. These promising data needs to be further investigated to elucidate the potential of RETC-2-ximab restoring local complement homeostasis. Just recently, it has been shown that a local complement suppression can be an effective tool for treating geographic atrophy, a late-stage AMD. In a phase 2 trial, intravitreal injection of the C3 inhibitor Pegcetacoplan significantly reduced the geographic atrophy rate over 12 months (66).
Here, we demonstrated FHR-3 – but not FHR-1, FH or properdin – as a complement promoting and pro-inflammatory protein on human RPE cells. Our in vitro studies, using ARPE-19 cells and hpRPE cells, unveiled a putative local, intraocular function of FHR-3. In addition, we chimerized our highly specific monoclonal antibody against FHR-3 (9) – RETC-2-ximab – and showed promising inhibitory potential.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics Statement
The research complies with the human research act (HRA) stating that small quantities of bodily substances removed in the course of transplantation may be anonymized for research purposes without consent (HRA chapter 5, paragraph 38, Switzerland). Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
Author Contributions
Conceptualization: NS and DP. Data curation: NS and DP. Formal analysis: NS. Funding acquisition: NS and DP. Investigation: NS, AR, DMO, SA, VE, and DP. Methodology: NS, AR, SA, VE, and DP. Project administration: NS and DP. Supervision: HJ and DP. Visualization: NS and DP. Writing – original draft: NS. Writing – review and editing: NS, SA, VE and DP. All authors have read and agreed to the published version of the manuscript.
Funding
This project was supported by the Bright Focus Foundation (Grant Reference ID: M2015186 to DP), Dr. Werner Jackstädt-Stiftung (DP), European Union’s Horizon 2020 research and innovation programme (SciFiMed, grant agreement No 899163) (DP) and by the Maloch Stiftung (NS).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Renate Foeckler, Elfriede Eckert, Andrea Dannullis and Stephanie Lötscher for excellent technical support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.769242/full#supplementary-material
Abbreviations
ACTA2, α‐smooth muscle actin; AMD, age-related macular degeneration; ARPE-19, human adult retinal pigment epithelium cells; BSA, bovine serum albumin; CCl, cellular capacitance; CD11b, cluster of differentiation molecule 11B (CR3 subunit); CEP, ω−carboxyethyl)pyrrole; COL1A1, collagen type I, alpha 1; CR3, complement receptor 3; EMT, epithelial-mesenchymal transition; FH, factor H; FHL-1, factor-H like protein 1; FHR-1 – 5, factor H-related proteins 1 – 5; FOXP3, forkhead box P3; FP, properdin; hpRPE, primary human RPE cells; MAA, malondialdehyde-acetaldehyde; MDA, malondialdehyde; NLRP3, NLR family pyrin domain containing 3; OSE, oxidation-specific epitopes; RETC-2, REgensburg Therapy Complement 2 (anti-FHR-3 antibody); RETC-2-ximab, chimerized REgensburg Therapy Complement 2 (anti-FHR-3 antibody); RPE, retinal pigment epithelium; SNP, single nucleotide polymorphisms; TER, Transepithelial Resistance; TNF, tumor necrosis factor; VIM, vimentin; ZO-1, zonula occludens 1.
References
1. Sjoberg AP, Trouw LA, Blom AM. Complement Activation and Inhibition: A Delicate Balance. Trends Immunol (2009) 30:83–90. doi: 10.1016/j.it.2008.11.003
2. Medjeral-Thomas N, Pickering MC. The Complement Factor H-Related Proteins. Immunol Rev (2016) 274(1):191–201. doi: 10.1111/imr.12477
3. Sánchez-Corral P, Pouw RB, López-Trascasa M, Józsi M. Self-Damage Caused by Dysregulation of the Complement Alternative Pathway: Relevance of the Factor H Protein Family. Front Immunol (2018) 9:1607. doi: 10.3389/fimmu.2018.01607
4. Cserhalmi M, Papp A, Brandus B, Uzonyi B, Józsi M. Regulation of Regulators: Role of the Complement Factor H-Related Proteins. Semin Immunol (2019) 45:101341. doi: 10.1016/j.smim.2019.101341
5. Klein RJ, Zeiss C, Chew EY, Tsai J-Y, Sackler RS, Haynes C, et al. Complement Factor H Polymorphism in Age-Related Macular Degeneration. Science (2005) 308(5720):385–9. doi: 10.1126/science.1109557
6. Hageman GS, Anderson DH, Johnson LV, Hancox LS, Taiber AJ, Hardisty LI, et al. A Common Haplotype in the Complement Regulatory Gene Factor H (HF1/CFH) Predisposes Individuals to Age-Related Macular Degeneration. Proc Natl Acad Sci USA (2005) 102(20):7227–32. doi: 10.1073/pnas.0501536102
7. Spencer KL, Hauser M, Olson LM, Schmidt S, Scott WK, Gallins P, et al. Deletion of CFHR3 and CFHR1 Genes in Age-Related Macular Degeneration. Hum Mol Genet (2008) 17(7):971–7. doi: 10.1093/hmg/ddm369
8. Zouache MA, Bennion A, Hageman JL, Pappas C, Richards BT, Hageman GS. Macular Retinal Thickness Differs Markedly in Age-Related Macular Degeneration Driven by Risk Polymorphisms on Chromosomes 1 and 10. Sci Rep (2020) 10:21093. doi: 10.1038/s41598-020-78059-x
9. Schäfer N, Grosche A, Reinders J, Hauck SM, Pouw RB, Kuijpers TW, et al. Complement Regulator FHR-3 Is Elevated Either Locally or Systemically in a Selection of Autoimmune Diseases. Front Immunol (2016) 7:1–16. doi: 10.3389/fimmu.2016.00542
10. Clark SJ, Schmidt CQ, White AM, Hakobyan S, Morgan BP, Bishop PN. Identification of Factor H-Like Protein 1 as the Predominant Complement Regulator in Bruch's Membrane: Implications for Age-Related Macular Degeneration. J Immunol (Baltimore Md 1950) (2014) 193(10):4962–70. doi: 10.4049/jimmunol.1401613
11. Strauss O. The Retinal Pigment Epithelium in Visual Function. Physiol Rev (2005) 85(3):845–81. doi: 10.1152/physrev.00021.2004
12. Lakkaraju A, Umapathy A, Tan LX, Daniele L, Philp NJ, Boesze-Battaglia K, et al. The Cell Biology of the Retinal Pigment Epithelium. Prog Retinal Eye Res (2020) 2020:100846. doi: 10.1016/j.preteyeres.2020.100846
13. Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The Hallmarks of Aging. Cell (2013) 153:1194–217. doi: 10.1016/j.cell.2013.05.039
14. Kaarniranta K, Kajdanek J, Morawiec J, Pawlowska E, Blasiak J. PGC-1alpha Protects RPE Cells of the Aging Retina Against Oxidative Stress-Induced Degeneration Through the Regulation of Senescence and Mitochondrial Quality Control. The Significance for AMD Pathogenesis. Int J Mol Sci (2018) 19:2317. doi: 10.3390/ijms19082317
15. Schutt F, Bergmann M, Holz FG, Kopitz J. Proteins Modified by Malondialdehyde, 4-Hydroxynonenal, or Advanced Glycation End Products in Lipofuscin of Human Retinal Pigment Epithelium. Invest Opthalmol Visual Sci (2003) 44(8):3663–. doi: 10.1167/iovs.03-0172
16. Joseph K, Kulik L, Coughlin B, Kunchithapautham K, Bandyopadhyay M, Thiel S, et al. Oxidative Stress Sensitizes Retinal Pigmented Epithelial (RPE) Cells to Complement-Mediated Injury in a Natural Antibody-, Lectin Pathway-, and Phospholipid Epitope-Dependent Manner. J Biol Chem (2013) 288(18):12753–65. doi: 10.1074/jbc.M112.421891
17. Weismann D, Hartvigsen K, Lauer N, Bennett KL, Scholl HP, Charbel Issa P, et al. Complement Factor H Binds Malondialdehyde Epitopes and Protects From Oxidative Stress. Nature (2011) 478(7367):76–81. doi: 10.1038/nature10449
18. Borras C, Canonica J, Jorieux S, Abache T, El Sanharawi M, Klein C, et al. CFH Exerts Anti-Oxidant Effects on Retinal Pigment Epithelial Cells Independently From Protecting Against Membrane Attack Complex. Sci Rep (2019) 9(1):13873. doi: 10.1038/s41598-019-50420-9
19. Alic L, Papac-Milicevic N, Czamara D, Rudnick RB, Ozsvar-Kozma M, Hartmann A, et al. A Genome-Wide Association Study Identifies Key Modulators of Complement Factor H Binding to Malondialdehyde-Epitopes. Proc Natl Acad Sci (2020) 117(18):9942–51. doi: 10.1073/pnas.1913970117
20. Irmscher S, Brix SR, Zipfel SLH, Halder LD, Mutlutürk S, Wulf S, et al. Serum FHR1 Binding to Necrotic-Type Cells Activates Monocytic Inflammasome and Marks Necrotic Sites in Vasculopathies. Nat Commun (2019) 10(1):2961. doi: 10.1038/s41467-019-10766-0
21. Martin M, Leffler J, Smoląg KI, Mytych J, Björk A, Chaves LD, et al. Factor H Uptake Regulates Intracellular C3 Activation During Apoptosis and Decreases the Inflammatory Potential of Nucleosomes. Cell Death Differ (2016) 23(5):903–11. doi: 10.1038/cdd.2015.164
22. Arbore G, Kemper C, Kolev M. Intracellular Complement – The Complosome – in Immune Cell Regulation. Mol Immunol (2017) 89:2–9. doi: 10.1016/j.molimm.2017.05.012
23. Kárpáti É, Papp A, Schneider AE, Hajnal D, Cserhalmi M, Csincsi ÁI, et al. Interaction of the Factor H Family Proteins FHR-1 and FHR-5 With DNA and Dead Cells: Implications for the Regulation of Complement Activation and Opsonization. Front Immunol (2020) 11:1297. doi: 10.3389/fimmu.2020.01297
24. Csincsi ÁI, Szabó Z, Bánlaki Z, Uzonyi B, Cserhalmi M, Kárpáti É, et al. FHR-1 Binds to C-Reactive Protein and Enhances Rather Than Inhibits Complement Activation. J Immunol (Baltimore Md 1950) (2017) 199(1):292–303. doi: 10.4049/jimmunol.1600483
25. Brandstetter C, Holz FG, Krohne TU. Complement Component C5a Primes Retinal Pigment Epithelial Cells for Inflammasome Activation by Lipofuscin-Mediated Photooxidative Damage. J Biol Chem (2015) 290(52):31189–98. doi: 10.1074/jbc.M115.671180
26. Wang L, Cano M, Datta S, Wei H, Ebrahimi KB, Gorashi Y, et al. Pentraxin 3 Recruits Complement Factor H to Protect Against Oxidative Stress-Induced Complement and Inflammasome Overactivation. J Pathol (2016) 240(4):495–506. doi: 10.1002/path.4811
27. Schäfer N, Wolf HN, Enzbrenner A, Schikora J, Reichenthaler M, Enzmann V, et al. Properdin Modulates Complement Component Production in Stressed Human Primary Retinal Pigment Epithelium Cells. Antioxidants (2020) 9(9):793–. doi: 10.3390/antiox9090793
28. Fernandez-Godino R, Bujakowska KM, Pierce EA. Changes in Extracellular Matrix Cause RPE Cells to Make Basal Deposits and Activate the Alternative Complement Pathway. Hum Mol Genet (2018) 27(1):147–59. doi: 10.1093/hmg/ddx392
29. Sugita S, Makabe K, Fujii S, Takahashi M. Detection of Complement Activators in Immune Attack Eyes After iPS-Derived Retinal Pigment Epithelial Cell Transplantation. Invest Ophthalmol Visual Sci (2018) 59(10):4198–209. doi: 10.1167/iovs.18-24769
30. Trakkides T-O, Schäfer N, Reichenthaler M, Kühn K, Brandwijk RJMGE, Toonen EJM, et al. Oxidative Stress Increases Endogenous Complement-Dependent Inflammatory and Angiogenic Responses in Retinal Pigment Epithelial Cells Independently of Exogenous Complement Sources. Antioxidants (2019) 8(11):548–. doi: 10.3390/antiox8110548
31. Pauly D, Agarwal D, Dana N, Schafer N, Biber J, Wunderlich KA, et al. Cell-Type-Specific Complement Expression in the Healthy and Diseased Retina. Cell Rep (2019) 29(9):2835–48.e4. doi: 10.1016/j.celrep.2019.10.084
32. Brandstetter C, Mohr LKM, Latz E, Holz FG, Krohne TU. Light Induces NLRP3 Inflammasome Activation in Retinal Pigment Epithelial Cells via Lipofuscin-Mediated Photooxidative Damage. J Mol Med (Berlin Germany) (2015) 93(8):905–16. doi: 10.1007/s00109-015-1275-1
33. Mohr LKM, Hoffmann AV, Brandstetter C, Holz FG, Krohne TU. Effects of Inflammasome Activation on Secretion of Inflammatory Cytokines and Vascular Endothelial Growth Factor by Retinal Pigment Epithelial Cells. Invest Opthalmol Visual Sci (2015) 56(11):6404. doi: 10.1167/iovs.15-16898
34. Blenkinsop TA, Salero E, Stern JH, Temple S. The Culture and Maintenance of Functional Retinal Pigment Epithelial Monolayers From Adult Human Eye. Methods Mol Biol (2012) 945:45–65. doi: 10.1007/978-1-62703-125-7_4
35. Lu L, Gu X, Hong L, Laird J, Jaffe K, Choi J, et al. Synthesis and Structural Characterization of Carboxyethylpyrrole-Modified Proteins: Mediators of Age-Related Macular Degeneration. Bioorganic Medicinal Chem (2009) 17(21):7548–61. doi: 10.1016/j.bmc.2009.09.009
36. Pauly D, Nagel BM, Reinders J, Killian T, Wulf M, Ackermann S, et al. A Novel Antibody Against Human Properdin Inhibits the Alternative Complement System and Specifically Detects Properdin From Blood Samples. PLoS One (2014) 9(5):e96371. doi: 10.1371/journal.pone.0096371
37. Fritsche LG, Igl W, Bailey JNC, Grassmann F, Sengupta S, Bragg-Gresham JL, et al. A Large Genome-Wide Association Study of Age-Related Macular Degeneration Highlights Contributions of Rare and Common Variants. Nat Genet (2016) 48(2):134–43. doi: 10.1038/ng.3448
38. Zhou M, Geathers JS, Grillo SL, Weber SR, Wang W, Zhao Y, et al. Role of Epithelial-Mesenchymal Transition in Retinal Pigment Epithelium Dysfunction. Front Cell Dev Biol (2020) 8:501. doi: 10.3389/fcell.2020.00501
39. Schäfer N, Grosche A, Schmitt SI, Braunger BM, Pauly D. Complement Components Showed a Time-Dependent Local Expression Pattern in Constant and Acute White Light-Induced Photoreceptor Damage. Front Mol Neurosci (2017) 10:197. doi: 10.3389/fnmol.2017.00197
40. Liszewski MK, Kolev M, Le Friec G, Leung M, Bertram Paula G, Fara Antonella F, et al. Intracellular Complement Activation Sustains T Cell Homeostasis and Mediates Effector Differentiation. Immunity (2013) 39(6):1143–57. doi: 10.1016/j.immuni.2013.10.018
41. Thurman JM, Renner B, Kunchithapautham K, Ferreira VP, Pangburn MK, Ablonczy Z, et al. Oxidative Stress Renders Retinal Pigment Epithelial Cells Susceptible to Complement-Mediated Injury. J Biol Chem (2009) 284(25):16939–47. doi: 10.1074/jbc.M808166200
42. Hwang YE, Baek YM, Baek A, Kim D-E. Oxidative Stress Causes Alu RNA Accumulation via PIWIL4 Sequestration Into Stress Granules. BMB Rep (2019) 52(3):196–201. doi: 10.5483/BMBRep.2019.52.3.146
43. Gao J, Liu RT, Cao S, Cui JZ, Wang A, To E, et al. NLRP3 Inflammasome: Activation and Regulation in Age-Related Macular Degeneration. Mediators Inflammation (2015) 2015:690243. doi: 10.1155/2015/690243
44. Brandstetter C, Patt J, Holz FG, Krohne TU. Inflammasome Priming Increases Retinal Pigment Epithelial Cell Susceptibility to Lipofuscin Phototoxicity by Changing the Cell Death Mechanism From Apoptosis to Pyroptosis. J Photochemistry Photobiol B: Biol (2016) 161:177–83. doi: 10.1016/j.jphotobiol.2016.05.018
45. Le Friec G, Köhl J, Kemper C. A Complement a Day Keeps the Fox(p3) Away. Nat Immunol (2013) 14(2):110–2. doi: 10.1038/ni.2515
46. Busch C, Annamalai B, Abdusalamova K, Reichhart N, Huber C, Lin Y, et al. Anaphylatoxins Activate Ca2+, Akt/PI3-Kinase, and FOXO1/FoxP3 in the Retinal Pigment Epithelium. Front Immunol (2017) 8:703. doi: 10.3389/fimmu.2017.00703
47. Hageman GS, Hancox LS, Taiber AJ, Gehrs KM, Anderson DH, Johnson LV, et al. Extended Haplotypes in the Complement Factor H (CFH) and CFH-Related (CFHR) Family of Genes Protect Against Age-Related Macular Degeneration: Characterization, Ethnic Distribution and Evolutionary Implications. Ann Med (2006) 38(8):592–604. doi: 10.1080/07853890601097030
48. Cipriani V, Lorés-Motta L, He F, Fathalla D, Tilakaratna V, McHarg S, et al. Increased Circulating Levels of Factor H-Related Protein 4 Are Strongly Associated With Age-Related Macular Degeneration. Nat Commun (2020) 11(1):778. doi: 10.1038/s41467-020-14499-3
49. Gu X, Meer SG, Miyagi M, Rayborn ME, Hollyfield JG, Crabb JW, et al. Carboxyethylpyrrole Protein Adducts and Autoantibodies, Biomarkers for Age-Related Macular Degeneration. J Biol Chem (2003) 278:42027–35. doi: 10.1074/jbc.M305460200
50. Rudnick RB, Chen Q, Stea ED, Hartmann A, Papac-Milicevic N, Person F, et al. FHR5 Binds to Laminins, Uses Separate C3b and Surface-Binding Sites, and Activates Complement on Malondialdehyde-Acetaldehyde Surfaces. J Immunol (2018) 200(7):2280–90. doi: 10.4049/jimmunol.1701641
51. Ewers H, Helenius A. Lipid-Mediated Endocytosis. Cold Spring Harbor Perspect Biol (2011) 3(8):1–14. doi: 10.1101/cshperspect.a004721
52. Mastellos DC, DeAngelis RA, Lambris JD. Complement-Triggered Pathways Orchestrate Regenerative Responses Throughout Phylogenesis. Semin Immunol (2013) 25(1):29–38. doi: 10.1016/j.smim.2013.04.002
53. Kolev M, Dimeloe S, Le Friec G, Navarini A, Arbore G, Povoleri Giovanni A, et al. Complement Regulates Nutrient Influx and Metabolic Reprogramming During Th1 Cell Responses. Immunity (2015) 42(6):1033–47. doi: 10.1016/j.immuni.2015.05.024
54. Goicoechea de Jorge E, Caesar JJE, Malik TH, Patel M, Colledge M, Johnson S, et al. Dimerization of Complement Factor H-Related Proteins Modulates Complement Activation In Vivo. Proc Natl Acad Sci USA (2013) 110(12):4685–90. doi: 10.1073/pnas.1219260110
55. Tortajada A, Uzonyi B, Jorge EGD. Factor H-Related Proteins Determine Complement-Activating Surfaces. Trends Immunol (2015) 36: (6):374–84. doi: 10.1016/j.it.2015.04.008
56. Schaefer L. Complexity of Danger: The Diverse Nature of Damage-Associated Molecular Patterns. J Biol Chem (2014) 289:35237–45. doi: 10.1074/jbc.R114.619304
57. Kunchithapautham K, Atkinson C, Rohrer B. Smoke Exposure Causes Endoplasmic Reticulum Stress and Lipid Accumulation in Retinal Pigment Epithelium Through Oxidative Stress and Complement Activation. J Biol Chem (2014) 289(21):14534–46. doi: 10.1074/jbc.M114.564674
58. Rohrer B. Anaphylatoxin Signaling in Retinal Pigment and Choroidal Endothelial Cells: Characteristics and Relevance to Age-Related Macular Degeneration. Adv Exp Med Biol (2018) 1074:45–51. doi: 10.1007/978-3-319-75402-4_6
59. Fernandez-Godino R, Pierce EA. C3a Triggers Formation of Sub-Retinal Pigment Epithelium Deposits via the Ubiquitin Proteasome Pathway. Sci Rep (2018) 8(1):9679. doi: 10.1038/s41598-018-28143-0
60. Propson NE, Roy ER, Litvinchuk A, Köhl J, Zheng H. Endothelial C3a Receptor Mediates Vascular Inflammation and BBB Permeability During Aging. J Clin Invest (2020) 131(1):e140966. doi: 10.1172/JCI140966
61. Asgari E, Friec GL, Yamamoto H, Perucha E, Sacks SS, Cook HT, et al. C3a Modulates IL-1 B Secretion in Human Monocytes by Regulating ATP Ef Fl Ux and Subsequent NLRP3 in Fl Ammasome Activation. Blood (2013) 122(20):3473–82. doi: 10.1182/blood-2013-05-502229
62. Bian Z-M, Field MG, Elner SG, Kahlenberg JM, Elner VM. Distinct CD40L Receptors Mediate Inflammasome Activation and Secretion of IL-1β and MCP-1 in Cultured Human Retinal Pigment Epithelial Cells. Exp Eye Res (2018) 170:29–39. doi: 10.1016/j.exer.2018.02.014
63. Wagner C, Hänsch GM, Stegmaier S, Denefleh B, Hug F, Schoels M. The Complement Receptor 3, CR3 (CD11b/CD18), on T Lymphocytes: Activation-Dependent Up-Regulation and Regulatory Function. Eur J Immunol (2001) 31(4):1173–80. doi: 10.1002/1521-4141(200104)31:4<1173::AID-IMMU1173>3.0.CO;2-9
64. Wu WC, Hu DN, Gao HX, Chen M, Wang D, Rosen R, et al. Subtoxic Levels Hydrogen Peroxide-Induced Production of Interleukin-6 by Retinal Pigment Epithelial Cells. Mol Vision (2010) 16:1864–73.
65. Fernandez-Godino R, Garland DL, Pierce EA. A Local Complement Response by RPE Causes Early-Stage Macular Degeneration. Hum Mol Genet (2015) 24(19):5555–69. doi: 10.1093/hmg/ddv287
Keywords: AMD, complement activation, complosome, FHR-3, inflammation, oxidative stress epitopes, RETC-2, RPE cells FHR-3 alters RPE cell complosome
Citation: Schäfer N, Rasras A, Ormenisan DM, Amslinger S, Enzmann V, Jägle H and Pauly D (2021) Complement Factor H-Related 3 Enhanced Inflammation and Complement Activation in Human RPE Cells. Front. Immunol. 12:769242. doi: 10.3389/fimmu.2021.769242
Received: 01 September 2021; Accepted: 18 October 2021;
Published: 08 November 2021.
Edited by:
Nicole Thielens, UMR5075 Institut de Biologie Structurale (IBS), FranceReviewed by:
Lubka T. Roumenina, U1138 Centre de Recherche des Cordeliers (CRC) (INSERM), FranceJózsef Dobó, Hungarian Academy of Sciences (MTA), Hungary
Copyright © 2021 Schäfer, Rasras, Ormenisan, Amslinger, Enzmann, Jägle and Pauly. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Diana Pauly, ZGlhbmEucGF1bHlAdW5pLW1hcmJ1cmcuZGU=