 Xiaoting Liao
Xiaoting Liao Weikang Zhang2†
Weikang Zhang2†- 1Department of Anesthesiology, Guangxi Key Laboratory of Basic Research on Perioperative Organ Function Injury & Control, and Guangxi Medical Engineering Research Center of Tissue Injury and Repair, Guangxi Medical University Cancer Hospital, Nanning, China
- 2Department of Anesthesiology, The Cancer Hospital of the University of Chinese Academy of Sciences (Zhejiang Cancer Hospital), Hangzhou, China
Ventilator-induced lung injury (VILI) is one of the most common complications of mechanical ventilation and can severely affect health. VILI appears to involve excessive inflammatory responses, but its pathogenesis has not yet been clarified. Since interleukin-17 (IL-17) plays a critical role in the immune system and the development of infectious and inflammatory diseases, we investigated here whether it plays a role in VILI. In a mouse model of VILI, mechanical ventilation with high tidal volume promoted the accumulation of lung neutrophils, leading to increased IL-17 levels in the lung, which in turn upregulated macrophage chemoattractant protein-1 via p38 mitogen-activated protein kinase. Depletion of neutrophils decreases the production IL-17 in mice and inhibition of IL-17 significantly reduced HTV-induced lung injury and inflammatory response. These results were confirmed in vitro using RAW264.7 macrophage cultures. Our results suggest that IL-17 plays a pro-inflammatory role in VILI and could serve as a new target for its treatment.
Introduction
Mechanical ventilation is widely used to support patients with acute respiratory failure and other severe diseases. However, it is associated with ventilator-induced lung injury (VILI), a syndrome that can cause or exacerbate lung injury through volu-, baro- and biotrauma (1–3). VILI can lead to overwhelming inflammatory responses, such as activation of the innate immune system and release of inflammatory molecules, which increase alveolar edema and pulmonary vascular permeability (4). We previously found that alveolar macrophages (AM) played an important role in promoting inflammation in the occurrence and development of VILI-mediated lung inflammation (5). VILI-induced inflammation and the release of inflammatory factors appear to involve phosphorylation of p38 mitogen-activated protein kinase (MAPK) (6), but many details of the processes behind VILI remain unclear.
Interleukin (IL)-17, a pro-inflammatory cytokine that was first detected in human peripheral blood (7, 8), contributes to inflammatory, tumor, and autoimmune diseases (9), as it can induce the production of chemokines and inflammatory molecules such as tumor necrosis factor-α, thereby activating innate immune responses and recruiting immunecells (10). Increased IL-17 levels in the serum, sputum, and broncho alveolar lavage fluid (BALF) of asthmatic patients positively correlate with disease severity (11–13). Moreover, IL-17 was found to aggravate lung inflammation and reduce lung function during H1N1 influenza virus infection and Klebsiella pneumoniae-induced pneumonia (14, 15).
The p38 MAPK, a member of the MAPK superfamily, is activated by various pro-inflammatory and stressful stimuli, mediating the release of inflammatory cytokines involved in the regulation of various diseases (16–19). Macrophage chemoattractant protein-1 (MCP-1), also known as C–C chemokine ligand 2 (CCL2), is an important downstream molecule of p38 MAPK activation that modulates the recruitment of inflammatory cells into damaged organs and tissues (20). MCP-1 may also exacerbate lung injury in patients with polycystic kidney disease and acute respiratory distress syndrome (21, 22).
In this study, we aimed to determine whether IL-17 plays a role in VILI. In particular, we asked whether IL-17 influences VILI by activating p38 MAPK/MCP-1 signaling.
Materials and Methods
Animals
Male C57BL/6 mice aged 6–8 weeks with a body weight of 22 ± 5 g were purchased from the Experimental Animal Center of Guangxi Medical University. All mice were raised at 20–25°C and 30–70% relative humidity and provided with protein-containing feed and purified water. The study was approved by the Experimental Animal Committee of Guangxi Medical University, and all animal experiments were performed in accordance with local and international ethical guidelines. Age- and weight-matched controls were used in all experiments.
Mouse Model of VILI
Before tracheal incubation, all mice were fasted for 8h and left without water for 4 h. Mice were then anesthetized by intraperitoneal injection of sodium pentobarbital (60 mg/kg); they were administered in 1/3 of the first dose to maintain anesthesia every 45 min. A sterile 20G intravenous trocar was used for tracheal intubation at a depth of 1.2 ± 0.5 cm. The catheter was fixed in the trachea to prevent one-lung ventilation and was connected to a small-animal ventilator (catalog no. SAR-1000, CWE) for mechanical ventilation with air. Non-ventilated mice were used as controls, while mechanically ventilated mice were ventilated with either normal tidal volume (NTV, 7 mL/kg) or high tidal volume (HTV, 20 mL/kg).
To explore the source of IL-17 in the mice treated with HTV, the mice were injected intraperitoneally with anti-Ly6G antibody (400 µg; BP0075, BioXCell) one day before ventilation to deplete neutrophils in vivo. To assess the role of IL-17 in the p38 MAPK/MCP-1 pathway after HTV ventilation, mice were randomly divided into four groups (H1-4) depending on whether ventilation lasted 1, 2, 3, or 4 h. One day before ventilation, the mice were injected intraperitoneally with anti-IL-17 antibody (400 µg; BE0173, BioXCell), and at 30 min before anesthesia 200 µg anti-IL-17 antibody was administered through intratracheal instillation. Recombinant mouse IL-17 (rmIL-17; 0.5 μg, catalog no. 576006, Biolegend) was administered through intratracheal instillation 30 min before anesthesia. HTV-ventilated mice administered with saline served as controls.
Cell Strains
RAW264.7 mononuclear macrophage leukemia cells were purchased from the American Type Culture Collection. Cells were grown in Dulbecco’s Modified Eagle’s medium (DMEM; catalog no. C11995500BT, Gibco), and the optimal infection conditions and multiplicity of infection were determined. In the optimized procedure, a 500 μL suspension of RAW246.7 cells and 20 μL of HitransG A infection-enhancing solution were added to each well of a 24-cell well plate. After cells reached 50% confluence, they were co-cultured with 20 μL of HitransG P infection-enhancing solution and lentivirus infection solution. After 16 h, the medium was replaced with fresh DMEM medium. At 72 h after transfection, the transfection efficiency was determined by fluorescence microscopy. Transfected cells were selected using 2 μg/mL puromycin, which was reduced to 1 μg/mL for further screening.
To clarify the mechanism of IL-17 in vitro, wild-type RAW264.7 cells were stimulated with rmIL-17(100 ng/mL) and cultured for 1, 2, 3, 4, or 5 h. Non-stimulated wild-type RAW264.7 cells served as controls. RAW264.7 cells transfected with negative-control lentivirus and stimulated with rmIL-17 for 4 h are referred to below as the CN group. RAW264.7 cells transfected with lentivirus encoding short hairpin RNA (shRNA) against p38 MAPK (Shanghai Jikai Gene Technology, Shanghai, China) and cultured with rmIL-17 for 4 h are referred to below as the shRNA group.
Inflammatory Response
After the animal model was established, all mice were sacrificed by carotid artery bleeding, and their lungs were excised and weighed immediately (wet weight) and again after drying in an oven at 60°C for 72 h (dry weight). All operations were conducted carefully to minimize the risk of inflammatory activation.
The total protein concentration in BALF was estimated using the bicinchoninic acid (BCA) assay (P0012S, Beyotime) to assess pulmonary permeability. The levels of IL-17 and MCP-1were assayed in BALF and culture medium were detected using the ELISA kits (CSB-E04608m, Cusabio Biotech and EK0568, Boster) according to the manufacturers’ instructions.
Histopathological Analysis
The pathological damage in mouse lungs after HTV ventilation was assessed after hematoxylin–eosin (H&E) staining. Briefly, the collected mouse lungs were fixed with 3.7% paraformaldehyde, embedded in paraffin, and cut into 5-μm sections. After dewaxing, the sections were stained and the tissue morphology was observed with a microscope. The degree of lung injury was evaluated by applying a four-point scale (0, no injury; 1, minor injury; 2, moderate injury; 3, severe injury) to each of the following parameters (6, 23, 24): alveolar congestion, hemorrhage, inflammatory cell infiltration, and alveolar wall thickening. These individual scores were summed to give the total score of lung damage.
Immunoblot Analysis
Lung tissues and RAW264.7 cells were lysed in RIPA lysis buffer (P0013B,Beyotime) supplemented with protease inhibitor (ST505, Beyotime) and phosphatase inhibitor (P1050, Beyotime), and the protein concentration in each sample was determined using the BCA assay. Samples were denatured, fractionated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and transferred to polyvinylidene difluoride membranes. The non-specific membrane binding sites were blocked with 5% bovine serum albumin (BSA) for 1 h. Membranes were then incubated at 4°C overnight with primary antibodies (all diluted 1:1000) against the following proteins: IL-17 (ab79056, Abcam), MCP-1 (catalog no. 2029, CST),p38 MAPK (catalog no. 9212, CST), and phosphorylated (p)-p38 MAPK (catalog no. 4511, CST). Blots were washed several times with PBS buffer, then incubated for 1 h with goat anti-rabbit IgG secondary antibody (diluted 1:15000; ab96899, Abcam). The bands were visualized using a fluorescent scanner. As an internal reference, GAPDH was immunostained using an antibody (1:1000; catalog no. 5174, CST).
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
Total RNA was isolated from lungs and RAW264.7 cells using the MiniBEST Universal RNA Extraction kit (catalog no. 9767, TaKaRa Bio, Otsu) following the manufacturer’s instructions. After determining RNA quality and quantity by spectrophotometry, cDNA was synthesized using the PrimeScript™ RT Master Mix kit(RR036A, TaKaRa) and amplified with the SYBR® Premix Ex Taq™ II(TliRNaseH Plus) kit(RR820A, TaKaRa). The following primers were used: GAPDH forward, 5′-TGTGTCCGTCGTGGATCTGA-3′; GAPDH reverse,5′-TTGCTGTTGAAGTCGCAGGAG-3′; MCP-1 forward, 5′-CAGGTCCCTGTCATGCTTCT-3′; MCP-1 reverse, 5′-GTGGGGCGTTAACTGCATCT-3′; p38 forward, 5′-CTGTCGAGACCGTTTCAGTCCA-3′; p38 reverse,5′-GTGTGAACACATCCAACAGACCAA-3′. The relative expression of GAPDH, MCP-1, and p38 was determined using the 2−ΔΔCt method and normalized to expression of GAPDH.
Flow Cytometry
Flow cytometry was used to detect the accumulation of neutrophils in the lung tissues of mice and the expression of IL-17 in neutrophils, Th17 cells, and T cells. Lung sections were treated with 200 U/mL Type I collagenase (950 μL, 17018029, Gibco), DNaseII(20 U,4942078001, Merck), and FBS (50 μL,10099141, Gibco)to prepare single-cell suspensions. Red blood cell lysis solution (1mL) was then added to each cell suspension to remove red blood cells and adjust the density to 5 × 106 mL−1. Cells were labelled using antibodies against Ly6G (catalog no.REA256, Miltenyi Biotec), CD4 (catalog no. GK1.5, Miltenyi Biotec), and CD90 (catalog no. 105201, Biolegend). After washing with PBST buffer, cells were ruptured and intracellular proteins were labelled for 20 min using anti-IL-17 antibody (catalog no. 560522, BD Horizon) and analyzed at 4 °C using a flow cytometer.
Immunofluorescence
Immunofluorescence was used to assess the expression of MCP-1 in RAW264.7 cells; the expression of IL-17 (catalog no.14-7179-82, Thermo Fisher Scientific), CD4 (catalog no.100401, Biolegend), CD90 (catalog no.105201, Biolegend), and Ly6G (catalog no.127601, Biolegend) in mouse lungs; and the source of IL-17 in mouse lung tissues. All sections were fixed with 3.7% paraformaldehyde, ruptured with 0.2% Triton X-100, and blocked with BSA and goat serum. After incubation with the above- mentioned primary antibodies at 4°C overnight, samples were incubated with goat anti-rabbit IgG secondary antibody for 1 h. Nuclei were stained with 4′,6-diamidino-2-phenylindole, then samples were observed using a confocal laser scanning microscope.
Statistical Analysis
Statistical analysis was performed using SPSS 22.0 software (IBM, USA). All data were reported as mean ± standard deviation (SD). Differences in parametric data that showed a normal distribution were assessed for significance using one-way analysis of variance and the Bonferroni test. Differences associated with a two-tailed P < 0.05 were considered statistically significant.
Results
HTV Ventilation Upregulates IL-17 in Lung Tissues
To assess the effect of HTV ventilation on IL-17 expression, we determined the levels of IL-17 in non-ventilated, NTV, and HTV ventilated mice. The IL-17 levels in lungs were significantly higher in the HTV group than in the control and NTV groups (Figures 1A–C). Compared with CON group, the concentration of IL-17 in BALF also increased in HTV group (Figure 1D). These results suggest that IL-17 is closely associated with the development of VILI.
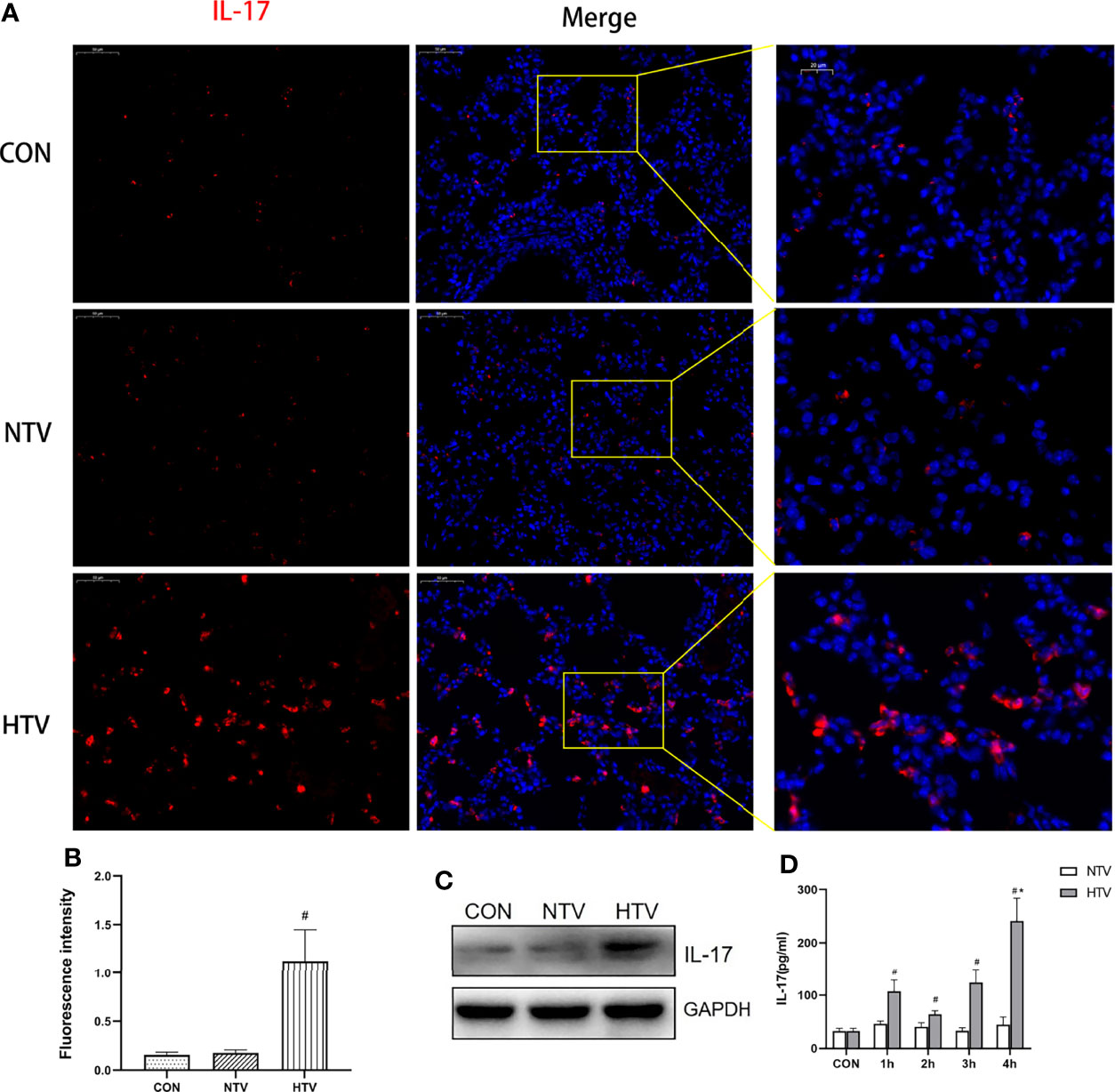
Figure 1 Ventilation with high tidal volume (HTV) up regulates interleukin (IL)-17 in the lungs. (A–C) IL-17 expression in the lung tissue of mice ventilated for 4 h with normal tidal volume (NTV, 7 mL/kg) or high tidal volume (HTV, 20 mL/kg). (D) Concentration of IL-17 in bronchoalveolar lavage fluid (BALF) of mice ventilated with NTV and HTV for 1 h, 2 h, 3 h, or 4 h. CON, non-ventilated mice. #P < 0.05 vs. CON group; *P < 0.05 vs. H1, H2, H3, or H4 group.
IL-17 Is Mainly Produced by Lung Neutrophils After HTV Ventilation
IL-17 is expressed mainly by Th17 cells and neutrophils (25, 26). Flow cytometry showed no significant difference in the numbers of CD4+IL-17+ cells among control, NTV and HTV groups (Figure 2A). IL-17 in the HTV group did not co-localize with the Th17 cell surface marker CD4 or the T cell surface marker CD90 (Figure 2B), suggesting that IL-17 is not produced by Th17 cells and T cells during VILI. In contrast, the number of Ly6G+neutrophils was significantly higher in the HTV group than in the other two groups (Figure 3A). Immunofluorescence shows that IL-17 co-localizes with the neutrophils surface marker Ly6G (Figure 3B). Furthermore, when neutrophils were depleted in mice (Figures 3C, F), compared with the HTV group, the levels of IL-17 in lung tissue (Figure 3D), BALF (Figure 3E) and plasma (Figure 3G) were significantly reduced in the HTV+anti-Ly6G group. These results indicate that IL-17 is produced mainly by Ly6G+neutrophils that are recruited to the lungs at the onset of VILI.
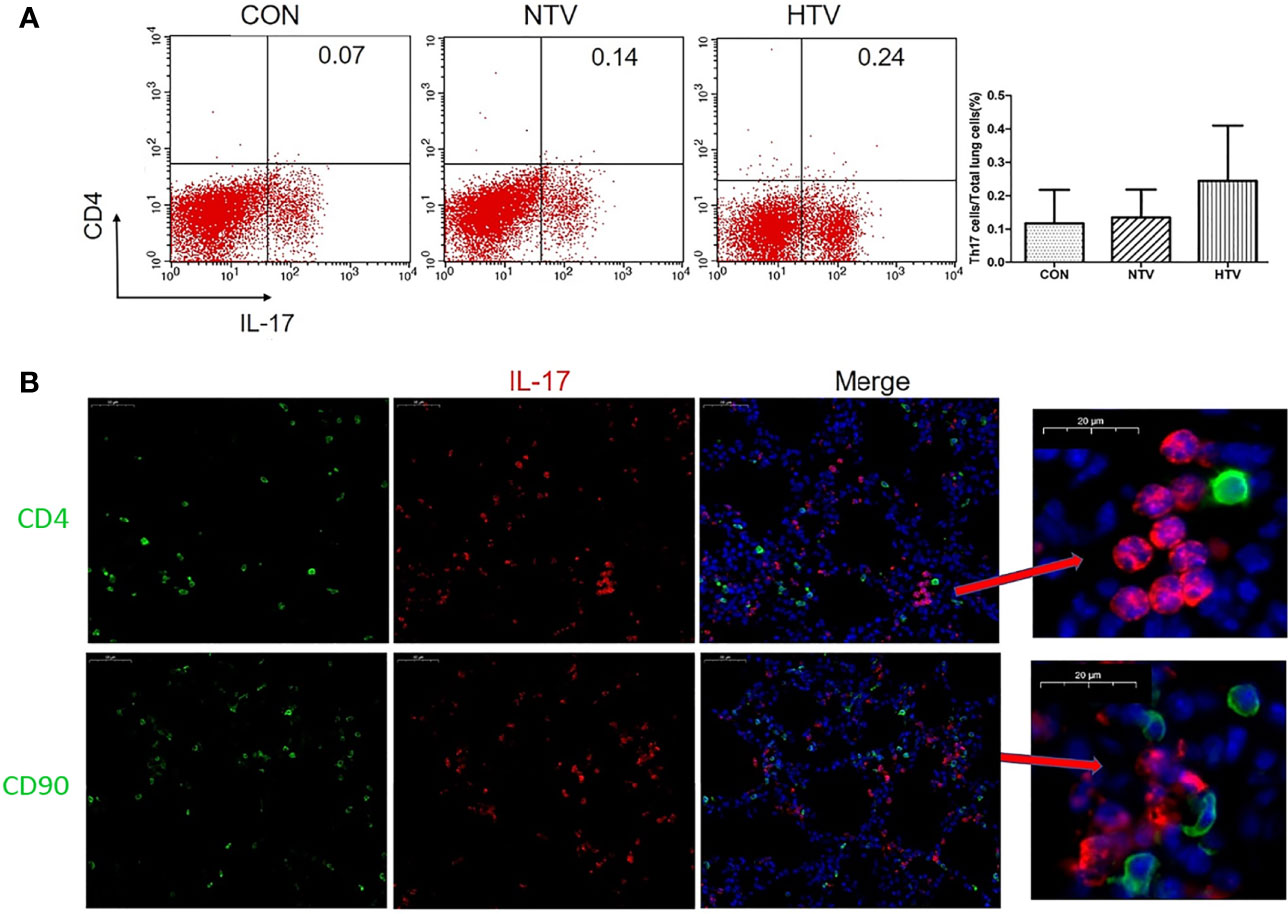
Figure 2 Interleukin (IL)-17 does not originate from Th17 cells and T cells after high tidal volume (HTV) ventilation. (A) Number of CD4+IL-17+ cells in mouse lung tissue. (B) Co-localization of IL-17 with CD4 and CD90 in the lung tissues. Data are shown as mean ± SD (n = 6). CON, non-ventilated mice; NTV, ventilated for 4 h with normal tidal volume; HTV, ventilated for 4 h with high tidal volume.
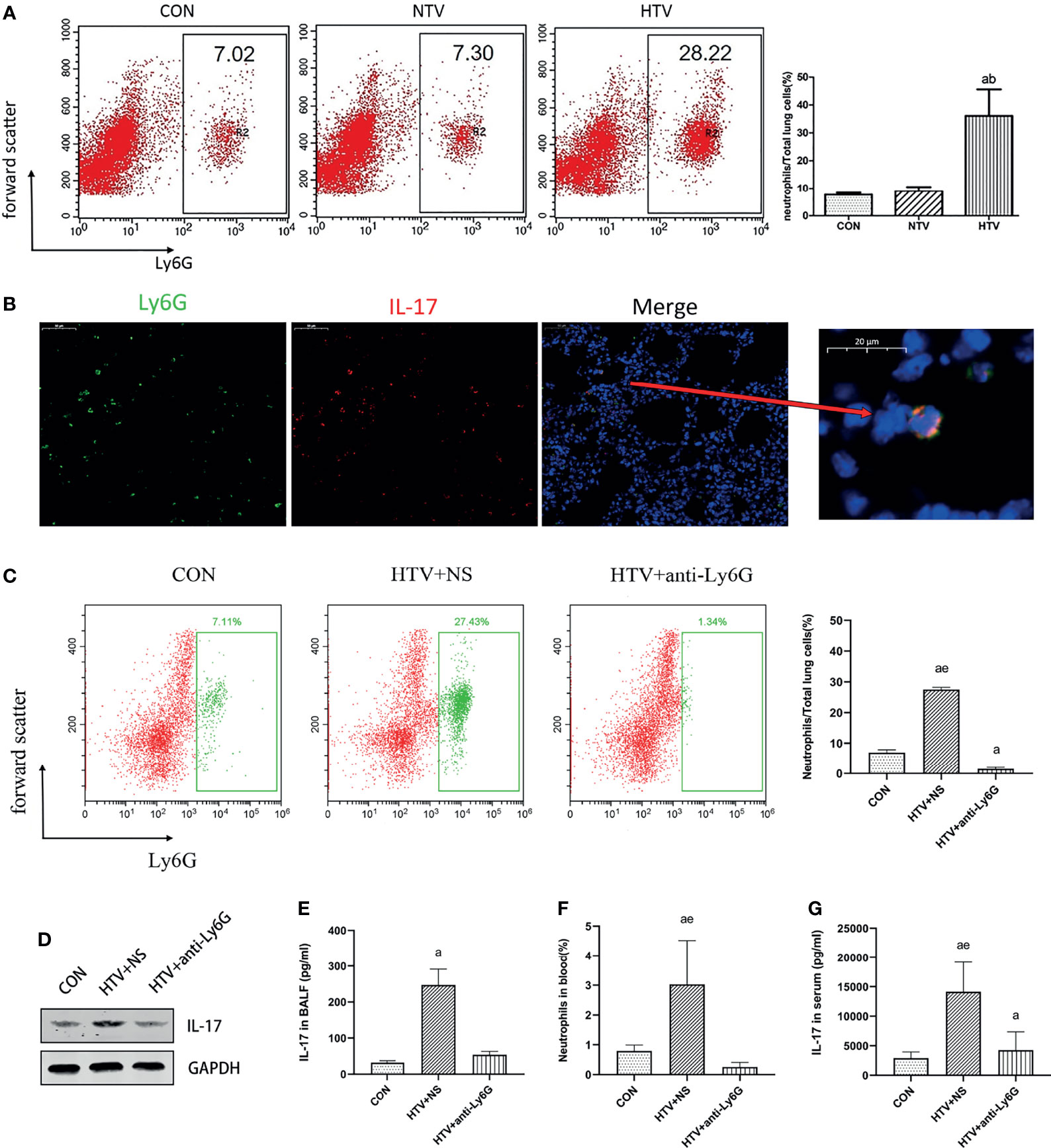
Figure 3 Interleukin (IL)-17 is produced by lung neutrophils after HTV. (A) Aggregation of neutrophils in mouse lung tissue. (B) Co-localization of IL-17 with Ly6G in the lung tissues. (C) Neutrophils in mouse lung cells. (D) IL-17 levels in mouse lung tissues. (E) Concentration of IL-17 in BALF. (F) Neutrophils in blood of mice. (G) Concentration of IL-17 in serum. Data are shown as mean ± SD (n = 6). aP < 0.05 vs. CON group, bP < 0.05 vs. NTV group, eP < 0.05 vs. HTV+anti-Ly6G group. CON, non-ventilated mice; HTV+NS, intraperitoneal injection with normal saline (NS) and ventilated for 4 h with high tidal volume; HTV+anti-Ly6G, intraperitoneal injection with anti-Ly6G antibody and ventilated for 4 h with high tidal volume.
IL-17 Inhibition Attenuates VILI-Induced Inflammation
The wet weight to dry weight ratio (W/D) of mouse lungs reflects the degree of lung edema, as the fluid that accumulates in the lung interstitium, alveolar cavity, and small bronchi increases lung tissue wet weight. Moreover, lung inflammation leads to the secretion of several inflammatory molecules in the alveolar cavity. We took advantage of these phenomena to assess whether inhibiting the function of IL-17 using an anti-IL-17 antibody (Figure 4A) would reduce pulmonary edema and total protein levels in BALF. Indeed, treating the HTV group with anti-IL-17 antibody led to significantly lower pulmonary edema and total BALF protein (Figures 4B, C), as well as milder alveolar congestion, alveolar interstitial thickening, and neutrophil infiltration (Figure 4D). These results suggest that blocking IL-17 can mitigate inflammation caused by VILI.
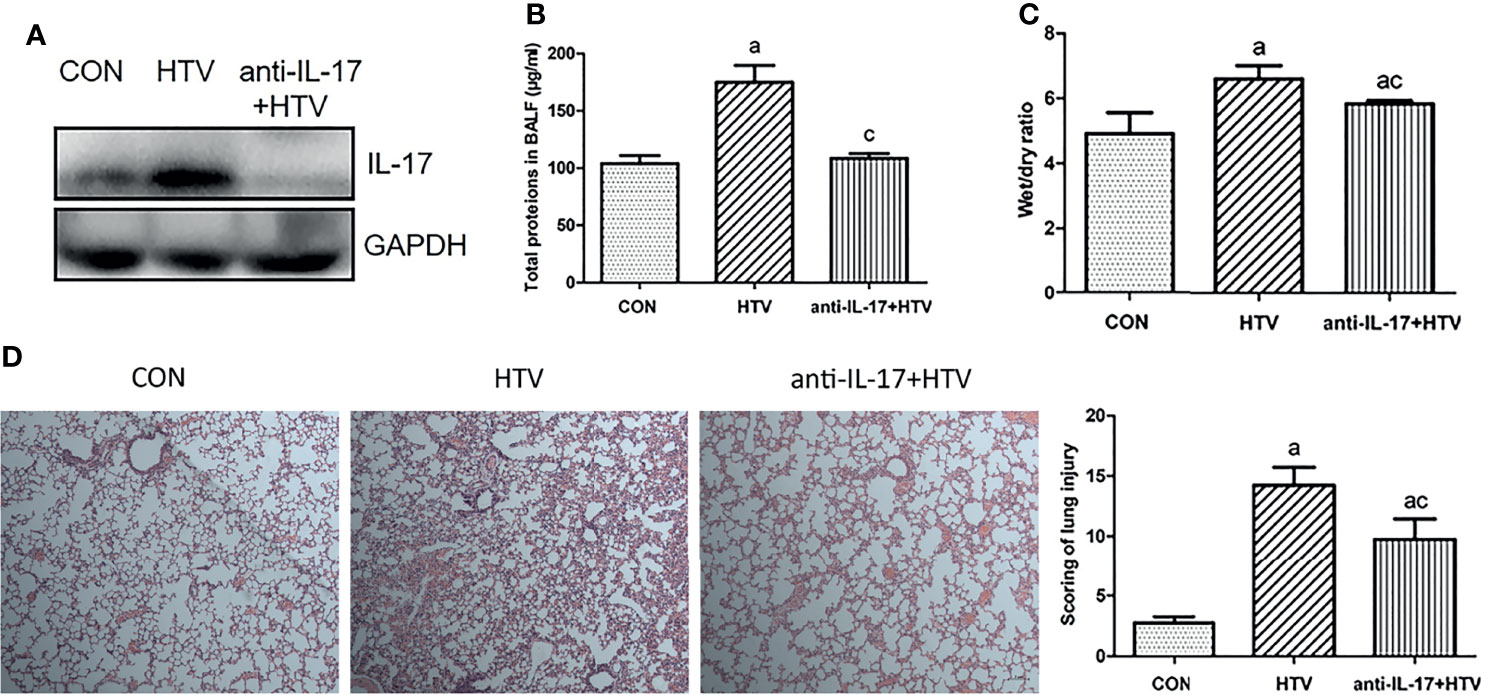
Figure 4 Inhibition of interleukin (IL)-17 attenuates ventilator-induced pulmonary edema. (A) IL-17 levels in mouse lung tissues. (B) Total protein concentration in bronchoalveolar lavage fluid (BALF) of mice. (C) Wet weight to dry weight (W/D) ratio of mouse lungs. (D) Histopathological analysis and degree of lung injury after ventilation with high tidal volume (HTV). Magnification, 400×. Data are shown as mean ± SD (n = 6). aP < 0.05 vs. CON group, cP < 0.05 vs. HTV group. CON, non-ventilated mice; HTV, ventilated for 4 h with high tidal volume; anti-IL-17+HTV, treated with anti-IL-17 antibody and ventilated for 4 h with high tidal volume.
rmIL-17 Upregulates MCP-1 in RAW264.7 Cells via the p38 MAPK Pathway
The p38 MAPK, as a classic downstream molecule of TLR4, has been associated with the pathogenesis of VILI (6). RAW264.7 cells were stimulated with rmIL-17 to examine whether IL-17 may interact with p38 MAPK. RT-qPCR analysis revealed that rmIL-17 significantly upregulatedMCP-1 mRNA expression (Figures 5A, B), which p38 MAPK knockdown partially reversed (Figures 5C–F). Western blot and RT-qPCR analysis also showed that rmIL-17 promoted the phosphorylation of p38 MAPK and MCP-1, without affecting total levelsofp38-MAPK (Figures 5A, E). These results suggest that IL-17 upregulates the expression ofMCP-1 by promoting the phosphorylation of p38 MAPK in lung macrophages.
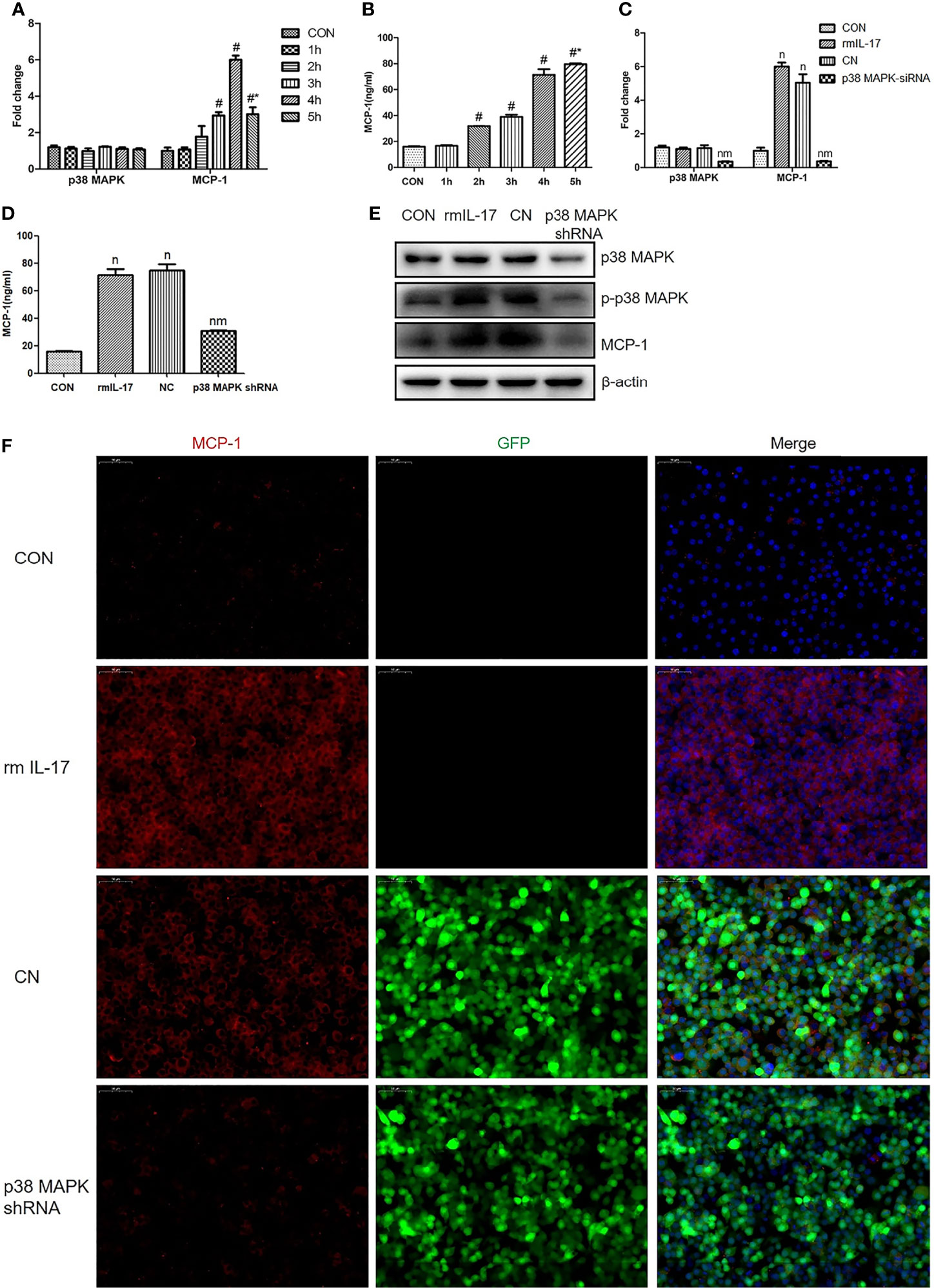
Figure 5 Recombinant mouse interleukin-17 (rmIL-17) upregulates MCP-1 in RAW264.7 cells via the p38 MAPK pathway. (A, C) Levels of mRNAs encoding p38 and MCP-1 in RAW264.7 cells. (B, D) Concentration of MCP-1 in medium from RAW264.7 cultures. (E) Levels of p38, phosphorylated (p)-p38, and MCP-1 in RAW264.7 cells. (F) Expression of MCP-1 in RAW264.7 cells, as determined by immunofluorescence. Data are shown as mean ± SD (n = 4). #P < 0.05 vs. CON group, *P < 0.05 vs. wild-type RAW264.7 cells cultured with rmIL-17, nP < 0.05 vs. CON group, mP < 0.05 vs. CN group. CON, wild-type RAW264.7 cells; 1h, 2h, 3h, 4h, wild-type RAW264.7 cells stimulated with rmIL-17 (100 ng/mL) and cultured for 1, 2, 3, 4, or 5 h; rmIL-17, wild-type RAW264.7 cells cultured with rmIL-17 (100 ng/mL); CN, negative lentivirus-transfected RAW264.7 cells stimulated with rmIL-17 (100 ng/mL) for 4 h; p38 MAPK shRNA, RAW264.7 cells transfected with lentivirus encoding short hairpin RNA against p38 MAPK and cultured with rmIL-17 (100 ng/mL) for 4 h.
IL-17 Upregulates MCP-1 in Mouse Lung Tissues via the p38 MAPK Pathway
To explore whether the observed effects of IL-17 on the p38 MAPK/MCP-1 pathway in vitro also occur in vivo, HTV ventilated mice were injected with anti-IL-17 antibody orrmIL-17 before ventilation. Anti-IL-17 antibody significantly reduced MCP-1 and p-p38 MAPK expression (Figures 6A–C), whereas rmIL-17 exerted the opposite effects (Figures 6D, E). At the same time, neither treatment affected the total level of p38 MAPK in lung or BALF (Figures 6C, E). These results confirm that IL-17 can upregulate MCP-1 by promoting the phosphorylation of p38 MAPK.
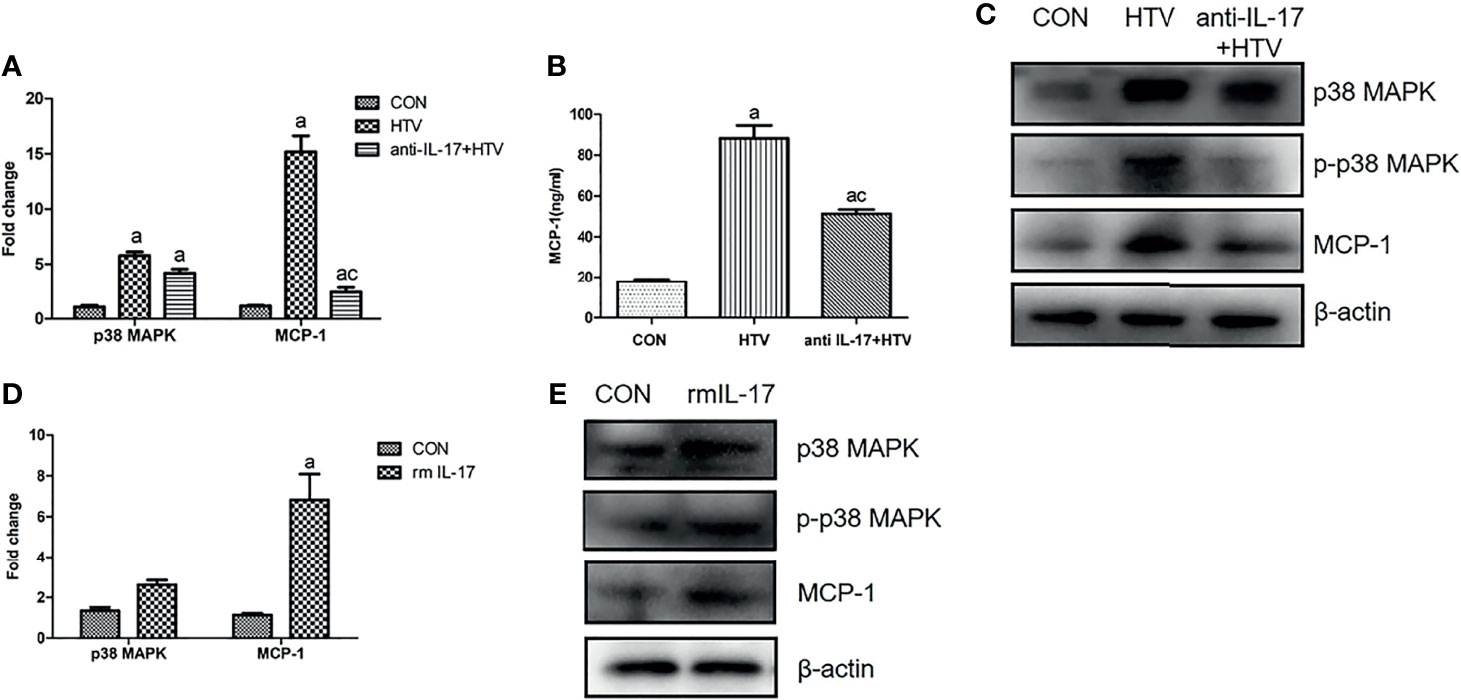
Figure 6 Interleukin (IL)-17 upregulates MCP-1 in mouse lung tissues via the p38 MAPK pathway. (A, D) Levels of mRNAs encoding p38 and MCP-1 in mouse lung tissues. (B) Concentration of MCP-1 in bronchoalveolar lavage fluid of mice. (C, E) Expression of p38 MAPK, phosphorylated (p)-p38 MAPK, and MCP-1 in mouse lung tissues. Data are shown as mean ± SD (n = 6). aP < 0.05 vs. CON group, cP < 0.05 vs. HTV. CON, non-ventilated mice; HTV, ventilated for 4 h with high tidal volume; anti-IL-17+HTV, treated with anti-IL-17 antibody and ventilated for 4 h with high tidal volume; rmIL-17+HTV, treated with rmIL-17 and ventilated for 4 h with high tidal volume.
Discussion
VILI is a pathophysiological process that leads to overwhelming inflammatory responses when alveolar macrophages, neutrophils, alveolar epithelial cells, and endothelial cells migrate to the lungs (27–29) and when pro-inflammatory cytokines are expressed. Current treatment methods for VILI are mainly based on regulating the tidal volume, oxygen concentration, and positive end-expiratory pressure of the ventilator (1, 30, 31). However, these therapies generally show low efficacy, highlighting the need to clarify the underlying mechanism of VILI in order to identify more effective targets.
In this study, we explored the role of IL-17 and the p38 MAPK/MCP-1 pathway in the pathogenesis of VILI. In our experiments in vitro and in vivo, HTV ventilation significantly increased the number of lung neutrophils, which secreted large amounts of IL-17, and this cytokine in turn upregulatedMCP-1 via the p38 MAPK pathway. Inhibition of IL-17 significantly reduced lung injury caused by HTV mechanical ventilation, suggesting that IL-17 may be a promising new target for the treatment of VILI-induced inflammation.
IL-17 receptors contain a conserved SEFIR domain, which binds to Act1 to activate downstream signal cascades (32, 33). Downstream targets of IL-17 include the NF-κB signaling pathway (34, 35), three MAPK pathways (JNK, ERK, and p38), and the phosphoinositide kinase pathway (36–38). Our findings about the role of IL-17 in VILI extend the list of conditions in which the cytokine can contribute to excessive inflammation, which include bacterial and fungal infections (39, 40). In fact, in certain cases of bacterial infection, higher levels of IL-17 may be beneficial to eliminate pathogens (41–43). Thus, whether elevated IL-17 levels lead to injury or benefit may depend on the cells secreting the cytokine and on other factors.
Inflammatory cells infiltrate into affected tissues, where they trigger local immune responses. Neutrophils, a type of polymorphonuclear leukocytes, are the first inflammatory cells recruited to inflammatory sites, where they help eliminate pathogens through multiple mechanisms (44–46). Although neutrophils are short-lived, they can be activated several times during inflammation (47, 48) and their chemotaxis is elevated in the lungs of patients with bacterial pneumonia or chronic bronchitis (49, 50). Here, we found a significantly high number of lung neutrophils in HTV ventilated mice, and flow cytometry revealed that lung neutrophils rather than Th17 cells are the main source of IL-17 during VILI. Therefore, we speculate that lung neutrophils may promote pneumonia response after HTV ventilation by promoting inflammation on their own, as well as by producing IL-17 to trigger inflammatory responses in other cell types. Future studies should clarify the role of neutrophils in VILI.
Although IL-17 participates in VILI through the p38 MAPK/MCP-1 pathway, there are still some limitations in this research that deserve improvement. First of all, HTV ventilation may cause respiratory alkalosis, we should continue to monitor and control the PCO2 and pH. However we had some difficulties in the monitoring methods mentioned above. Another limitation was there are some other factors that affect the severity of lung injury besides tidal volume. To reduce the influence of these factors, we should continuously monitor hemodynamics, PEEP or lung recruitment.
Conclusion
Our study suggests that IL-17 produced by lung neutrophils can act via the p38 MAPK/MCP-1 pathway to drive pulmonary inflammatory responses in VILI. Inhibition of IL-17 can effectively relieve VILI, suggesting that IL-17 may serve as a new target for attenuating lung inflammation induced by HTV ventilation.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics Statement
The animal study was reviewed and approved by The Animal Care & Welfare Committee of Guangxi Medical University.
Author Contributions
LP designed and directed the overall study. XL wrote the manuscript and carried out experiments. WZ wrote the manuscript and guided the experiments. HD, RJ, and MY carried out the experiments. WG and SP collected and analyzed data. All authors read and approved the final manuscript.
Funding
This work was supported by grants from the Natural Science Foundation of Guangxi (Grant No.2018GXNSFDA138007), the National Natural Science Foundation of China (Grant No. 81970078) and Innovation Project of Guangxi Graduate Education (Grant No.YCSW2021130).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.768813/full#supplementary-material
Supplementary Figure 1 | Lung neutrophils increase after HTV. Neutrophils in mouse lung cells. Data are shown as mean ± SD (n = 6). *P < 0.05 vs. CON group, #P < 0.05 vs. H0.5h group. CON, non-ventilated mice; H0.5h, H0.75h, H1h, H4h, ventilated for 0.5h, 0.75h, 1h and 4 h with high tidal volume.
References
1. Nieman GF, Satalin J, Andrews P, Aiash H, Habashi NM, Gatto LA. Personalizing Mechanical Ventilation According to Physiologic Parameters to Stabilize Alveoli and Minimize Ventilator Induced Lung Injury (VILI). Intensive Care Med Exp (2017) 5(1):8. doi: 10.1186/s40635-017-0121-x
2. Bates JHT, Smith BJ. Ventilator-Induced Lung Injury and Lung Mechanics. Ann Transl Med (2018) 6(19):378. doi: 10.21037/atm.2018.06.29
3. Wu Y, Kharge AB, Perlman CE. Lung Ventilation Injures Areas With Discrete Alveolar Flooding, in a Surface Tension-Dependent Fashion. J Appl Physiol (1985) (2014) 117(7):788–96. doi: 10.1152/japplphysiol.00569.2014
4. Seah AS, Grant KA, Aliyeva M, Allen GB, Bates JH. Quantifying the Roles of Tidal Volume and PEEP in the Pathogenesis of Ventilator-Induced Lung Injury. Ann BioMed Eng (2011) 39(5):1505–16. doi: 10.1007/s10439-010-0237-6
5. Huang C, Pan L, Lin F, Dai H, Fu R. Monoclonal Antibody Against Toll-Like Receptor 4 Attenuates Ventilator-Induced Lung Injury in Rats by Inhibiting MyD88- and NF-κb-Dependent Signaling. Int J Mol Med (2017) 39(3):693–700. doi: 10.3892/ijmm.2017.2873
6. Zhang W, Dai H, Lin F, Zhao C, Wang X, Zhang S, et al. Ly-6C(High) Inflammatory-Monocyte Recruitment Is Regulated by P38 MAPK/MCP-1 Activation and Promotes Ventilator-Induced Lung Injury. Int Immunopharmacol (2020) 78:106015. doi: 10.1016/j.intimp.2019.106015
7. Rouvier E, Luciani MF, Mattéi MG, Denizot F, Golstein P. CTLA-8, Cloned From an Activated T Cell, Bearing AU-Rich Messenger RNA Instability Sequences, and Homologous to a Herpesvirus Saimiri Gene. J Immunol (1993) 150(12):5445–56.
8. Yao Z, Fanslow WC, Seldin MF, Rousseau AM, Painter SL, Comeau MR, et al. Herpesvirus Saimiri Encodes a New Cytokine, IL-17, Which Binds to a Novel Cytokine Receptor. Immunity (1995) 3(6):811–21. doi: 10.1016/1074-7613(95)90070-5
9. Wang W, Fan YQ, Lv Z, Yao XJ, Wang W, Huang KW, et al. Interleukin-25 Promotes Basic Fibroblast Growth Factor Expression by Human Endothelial Cells Through Interaction With IL-17RB, But Not IL-17ra. Clin Exp Allergy (2012) 42(11):1604–14. doi: 10.1111/j.1365-2222.2012.04062.x
10. Friedrich M, Diegelmann J, Schauber J, Auernhammer CJ, Brand S. Intestinal Neuroendocrine Cells and Goblet Cells Are Mediators of IL-17A-Amplified Epithelial IL-17C Production in Human Inflammatory Bowel Disease. Mucosal Immunol (2015) 8(4):943–58. doi: 10.1038/mi.2014.124
11. Ramakrishnan RK, Al Heialy S, Hamid Q. Role of IL-17 in Asthma Pathogenesis and Its Implications for the Clinic. Expert Rev Respir Med (2019) 13(11):1057–68. doi: 10.1080/17476348.2019.1666002
12. Östling J, van Geest M, Schofield JPR, Jevnikar Z, Wilson S, Ward J, et al. IL-17-High Asthma With Features of a Psoriasis Immunophenotype. J Allergy Clin Immunol (2019) 144(5):1198–213. doi: 10.1016/j.jaci.2019.03.027
13. Al-Kufaidy R, Vazquez-Tello A, BaHammam AS, Al-Muhsen S, Hamid Q, Halwani R. IL-17 Enhances the Migration of B Cells During Asthma by Inducing CXCL13 Chemokine Production in Structural Lung Cells. J Allergy Clin Immunol (2017) 139(2):696–9.e5. doi: 10.1016/j.jaci.2016.07.037
14. Chuammitri P, Wongsawan K, Pringproa K, Thanawongnuwech R. Interleukin 17 (IL-17) Manipulates Mouse Bone Marrow- Derived Neutrophils in Response to Acute Lung Inflammation. Comp Immunol Microbiol Infect Dis (2019) 67:101356. doi: 10.1016/j.cimid.2019.101356
15. Xiong H, Keith JW, Samilo DW, Carter RA, Leiner IM, Pamer EG. Innate Lymphocyte/Ly6C(hi) Monocyte Crosstalk Promotes Klebsiella Pneumoniae Clearance. Cell (2016) 165(3):679–89. doi: 10.1016/j.cell.2016.03.017
16. Keszler M, Sant'Anna G. Mechanical Ventilation and Bronchopulmonary Dysplasia. Clin Perinatol (2015) 42(4):781–96. doi: 10.1016/j.clp.2015.08.006
17. Sur S, Nakanishi H, Steele R, Ray RB. Depletion of PCAT-1 in Head and Neck Cancer Cells Inhibits Tumor Growth and Induces Apoptosis by Modulating C-Myc-AKT1-P38 MAPK Signalling Pathways. BMC Cancer (2019) 19(1):354. doi: 10.1186/s12885-019-5562-z
18. Feng PC, Ke XF, Kuang HL, Pan LL, Ye Q, Wu JB. BMP2 Secretion From Hepatocellular Carcinoma Cell HepG2 Enhances Angiogenesis and Tumor Growth in Endothelial Cells via Activation of the MAPK/p38 Signaling Pathway. Stem Cell Res Ther (2019) 10(1):237. doi: 10.1186/s13287-019-1301-2
19. He F, Xiao Z, Yao H, Li S, Feng M, Wang W, et al. The Protective Role of microRNA-21 Against Coxsackievirus B3 Infection Through Targeting the MAP2K3/P38 MAPK Signaling Pathway. J Transl Med (2019) 17(1):335. doi: 10.1186/s12967-019-2077-y
20. Eltzschig HK, Eckle T. Ischemia and Reperfusion–From Mechanism to Translation. Nat Med (2011) 17(11):1391–401. doi: 10.1038/nm.2507
21. Salah SM, Meisenheimer JD, Rao R, Peda JD, Wallace DP, Foster D, et al. MCP-1 Promotes Detrimental Cardiac Physiology, Pulmonary Edema, and Death in the Cpk Model of Polycystic Kidney Disease. Am J Physiol Renal Physiol (2019) 317(2):F343–60. doi: 10.1152/ajprenal.00240.2018
22. He L, Ding Y, Zhang Q, Che X, He Y, Shen H, et al. Expression of Elevated Levels of Pro-Inflammatory Cytokines in SARS-CoV-Infected ACE2+ Cells in SARS Patients: Relation to the Acute Lung Injury and Pathogenesis of SARS. J Pathol (2006) 210(3):288–97. doi: 10.1002/path.2067
23. Belperio JA, Keane MP, Burdick MD, Londhe V, Xue YY, Li K, et al. Critical Role for CXCR2 and CXCR2 Ligands During the Pathogenesis of Ventilator-Induced Lung Injury. J Clin Invest (2002) 110(11):1703–16. doi: 10.1172/JCI0215849
24. Dai H, Pan L, Lin F, Ge W, Li W, He S. Mechanical Ventilation Modulates Toll-Like Receptors 2, 4, and 9 on Alveolar Macrophages in a Ventilator-Induced Lung Injury Model. J Thorac Dis (2015) 7(4):616–24. doi: 10.3978/j.issn.2072-1439.2015.02.10
25. Xu S, Cao X. Interleukin-17 and Its Expanding Biological Functions. Cell Mol Immunol (2010) 7(3):164–74. doi: 10.1038/cmi.2010.21
26. Kudo M, Melton AC, Chen C, Engler MB, Huang KE, Ren X, et al. IL-17A Produced by αβ T Cells Drives Airway Hyper-Responsiveness in Mice and Enhances Mouse and Human Airway Smooth Muscle Contraction. Nat Med (2012) 18(4):547–54. doi: 10.1038/nm.2684
27. Grommes J, Soehnlein O. Contribution of Neutrophils to Acute Lung Injury. Mol Med (2011) 17(3-4):293–307. doi: 10.2119/molmed.2010.00138
28. Dolinay T, Kim YS, Howrylak J, Hunninghake GM, An CH, Fredenburgh L, et al. Inflammasome-Regulated Cytokines Are Critical Mediators of Acute Lung Injury. Am J Respir Crit Care Med (2012) 185(11):1225–34. doi: 10.1164/rccm.201201-0003OC
29. Tremblay LN, Slutsky AS. Ventilator-Induced Lung Injury: From the Bench to the Bedside. Intensive Care Med (2006) 32(1):24–33. doi: 10.1007/s00134-005-2817-8
30. Curley GF, Laffey JG, Zhang H, Slutsky AS. Biotrauma and Ventilator-Induced Lung Injury: Clinical Implications. Chest (2016) 150(5):1109–17. doi: 10.1016/j.chest.2016.07.019
31. Cressoni M, Gotti M, Chiurazzi C, Massari D, Algieri I, Amini M, et al. Mechanical Power and Development of Ventilator-Induced Lung Injury. Anesthesiology (2016) 124(5):1100–8. doi: 10.1097/ALN.0000000000001056
32. Mellett M, Atzei P, Bergin R, Horgan A, Floss T, Wurst W, et al. Orphan Receptor IL-17RD Regulates Toll-Like Receptor Signalling via SEFIR/TIR Interactions. Nat Commun (2015) 6:6669. doi: 10.1038/ncomms7669
33. Novatchkova M, Leibbrandt A, Werzowa J, Neubüser A, Eisenhaber F. The STIR-Domain Superfamily in Signal Transduction, Development and Immunity. Trends Biochem Sci (2003) 28(5):226–9. doi: 10.1016/S0968-0004(03)00067-7
34. Wang B, Zhao CH, Sun G, Zhang ZW, Qian BM, Zhu YF, et al. IL-17 Induces the Proliferation and Migration of Glioma Cells Through the Activation of PI3K/Akt1/NF-κb-P65. Cancer Lett (2019) 447:93–104. doi: 10.1016/j.canlet.2019.01.008
35. Herjan T, Hong L, Bubenik J, Bulek K, Qian W, Liu C, et al. IL-17-Receptor-Associated Adaptor Act1 Directly Stabilizes mRNAs to Mediate IL-17 Inflammatory Signaling. Nat Immunol (2018) 19(4):354–65. doi: 10.1038/s41590-018-0071-9
36. Ning W, Choi AM, Li C. Carbon Monoxide Inhibits IL-17-Induced IL-6 Production Through the MAPK Pathway in Human Pulmonary Epithelial Cells. Am J Physiol Lung Cell Mol Physiol (2005) 289(2):L268–73. doi: 10.1152/ajplung.00168.2004
37. Li JK, Nie L, Zhao YP, Zhang YQ, Wang X, Wang SS, et al. IL-17 Mediates Inflammatory Reactions via P38/C-Fos and JNK/c-Jun Activation in an AP-1-Dependent Manner in Human Nucleus Pulposus Cells. J Transl Med (2016) 14:77. doi: 10.1186/s12967-016-0833-9
38. Song Y, Yang M, Zhang H, Sun Y, Tao Y, Li H, et al. IL-17 Affects the Progression, Metastasis, and Recurrence of Laryngeal Cancer via the Inhibition of Apoptosis Through Activation of the PI3K/AKT/FAS/FASL Pathways. J Immunol Res (2020) 2020:2953191. doi: 10.21203/rs.2.12638/v1
39. Miyazaki Y, Hamano S, Wang S, Shimanoe Y, Iwakura Y, Yoshida H. IL-17 Is Necessary for Host Protection Against Acute-Phase Trypanosoma Cruzi Infection. J Immunol (2010) 185(2):1150–7. doi: 10.4049/jimmunol.0900047
40. Ritchie ND, Ritchie R, Bayes HK, Mitchell TJ, Evans TJ. IL-17 Can be Protective or Deleterious in Murine Pneumococcal Pneumonia. PloS Pathog (2018) 14(5):e1007099. doi: 10.1371/journal.ppat.1007099
41. Wolf L, Sapich S, Honecker A, Jungnickel C, Seiler F, Bischoff M, et al. IL-17A-Mediated Expression of Epithelial IL-17C Promotes Inflammation During Acute Pseudomonas Aeruginosa Pneumonia. Am J Physiol Lung Cell Mol Physiol (2016) 311(5):L1015–22. doi: 10.1152/ajplung.00158.2016
42. Ramirez-Carrozzi V, Sambandam A, Luis E, Lin Z, Jeet S, Lesch J, et al. IL-17C Regulates the Innate Immune Function of Epithelial Cells in an Autocrine Manner. Nat Immunol (2011) 12(12):1159–66. doi: 10.1038/ni.2156
43. Kusagaya H, Fujisawa T, Yamanaka K, Mori K, Hashimoto D, Enomoto N, et al. Toll-Like Receptor-Mediated Airway IL-17C Enhances Epithelial Host Defense in an Autocrine/Paracrine Manner. Am J Respir Cell Mol Biol (2014) 50(1):30–9. doi: 10.1165/rcmb.2013-0130OC
44. Sadik CD, Kim ND, Luster AD. Neutrophils Cascading Their Way to Inflammation. Trends Immunol (2011) 32(10):452–60. doi: 10.1016/j.it.2011.06.008
45. Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. Neutrophil Function: From Mechanisms to Disease. Annu Rev Immunol (2012) 30:459–89. doi: 10.1146/annurev-immunol-020711-074942
46. Soehnlein O, Lindbom L. Phagocyte Partnership During the Onset and Resolution of Inflammation. Nat Rev Immunol (2010) 10(6):427–39. doi: 10.1038/nri2779
47. Colotta F, Re F, Polentarutti N, Sozzani S, Mantovani A. Modulation of Granulocyte Survival and Programmed Cell Death by Cytokines and Bacterial Products. Blood (1992) 80(8):2012–20. doi: 10.1182/blood.V80.8.2012.2012
48. Kim MH, Granick JL, Kwok C, Walker NJ, Borjesson DL, Curry FR, et al. Neutrophil Survival and C-Kit(+)-Progenitor Proliferation in Staphylococcus Aureus-Infected Skin Wounds Promote Resolution. Blood (2011) 117(12):3343–52. doi: 10.1182/blood-2010-07-296970
49. Hopkins H, Stull T, Von Essen SG, Robbins RA, Rennard SI. Neutrophil Chemotactic Factors in Bacterial Pneumonia. Chest (1989) 95(5):1021–7. doi: 10.1378/chest.95.5.1021
Keywords: ventilator-induced lung injury, IL-17, inflammatory response, p38 MAPK, MCP-1
Citation: Liao X, Zhang W, Dai H, Jing R, Ye M, Ge W, Pei S and Pan L (2021) Neutrophil-Derived IL-17 Promotes Ventilator-Induced Lung Injury via p38 MAPK/MCP-1 Pathway Activation. Front. Immunol. 12:768813. doi: 10.3389/fimmu.2021.768813
Received: 01 September 2021; Accepted: 29 November 2021;
Published: 15 December 2021.
Edited by:
Heiko Mühl, Goethe University Frankfurt, GermanyReviewed by:
Ulrich Steinhoff, University of Marburg, GermanyMichael Wilson, Imperial College London, United Kingdom
Copyright © 2021 Liao, Zhang, Dai, Jing, Ye, Ge, Pei and Pan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Linghui Pan, cGFubGluZ2h1aUBneG11LmVkdS5jbg==
†These authors have contributed equally to this work and share first authorship