 Jonathan W. Wallace
Jonathan W. Wallace David A. Constant
David A. Constant Timothy J. Nice
Timothy J. Nice- Department of Molecular Microbiology and Immunology, Oregon Health & Science University, Portland, OR, United States
Interferon λ (IFN-λ) is critical for host viral defense at mucosal surfaces and stimulates immunomodulatory signals, acting on epithelial cells and few other cell types due to restricted IFN-λ receptor expression. Epithelial cells of the intestine play a critical role in the pathogenesis of Inflammatory Bowel Disease (IBD), and the related type II interferons (IFN-γ) have been extensively studied in the context of IBD. However, a role for IFN-λ in IBD onset and progression remains unclear. Recent investigations of IFN-λ in IBD are beginning to uncover complex and sometimes opposing actions, including pro-healing roles in colonic epithelial tissues and potentiation of epithelial cell death in the small intestine. Additionally, IFN-λ has been shown to act through non-epithelial cell types, such as neutrophils, to protect against excessive inflammation. In most cases IFN-λ demonstrates an ability to coordinate the host antiviral response without inducing collateral hyperinflammation, suggesting that IFN-λ signaling pathways could be a therapeutic target in IBD. This mini review discusses existing data on the role of IFN-λ in the pathogenesis of inflammatory bowel disease, current gaps in the research, and therapeutic potential of modulating the IFN-λ-stimulated response.
Introduction
Inflammatory bowel disease (IBD) is the collective term for Ulcerative Colitis (UC) and Crohn’s Disease (CD). IBD has historically been more prevalent in North America, Europe and Oceania, but worldwide incidence is rising, especially in developing nations (1). The Centers for Disease Control recently estimated 3.1 million diagnosed adults in the United States alone (2). Overall prevalence of IBD among Medicare fee-for-service beneficiaries is slightly increasing and the largest increases in incidence are seen in non-Hispanic black persons. IBD and its associated comorbidities can be disabling and patients are often vulnerable to decreasing quality of life (3) as well as significant financial toxicity due to the high cost of therapy (4). Therefore, there remains significant interest in furthering understanding of the mechanisms behind initiation and progression of IBD.
The intestinal epithelium plays a critical role in the pathogenesis of IBD (5), so fundamental understanding of intestinal epithelial cell (IEC) contributions to disease has been of major interest in the field for decades. In the absence of disease, IECs maintain intestinal homeostasis by providing a physical barrier and forming the interface between enteric microbes and the host immune system (6, 7). Disruption of epithelial homeostasis through environmental or infectious triggers can perturb this otherwise peaceful relationship between commensal microbes and the immune system. For example, epithelial barrier breakdown allows excessive exposure to luminal products that promote chronic inflammation and collateral tissue damage and ulceration seen in IBD (8, 9). The chronic inflammatory immune response of IBD results in increased levels of cytokines including tumor necrosis factor alpha (TNFα) and interferons (IFNs).
IFNs are named for their important role in interfering with viral replication (10, 11), but have also been implicated in autoinflammatory diseases (12, 13). There are three types of IFNs, each utilizing its own distinctive receptor. Type I IFN consists of at least 17 functional subtypes, including IFN-α subtypes and IFN-β (simplified herein as IFN-α/β) (14). Type II IFN (or IFN-γ) is a single signaling protein that has been of particular interest in studies of IBD. Type III IFN is the most recently identified IFN type and consists of 3-4 IFN-λ subtypes in humans. IFN-λ responses have many similarities with IFN-α/β, but IFN-λ is more restricted in expression of its receptor and physiological functions (15). The receptor for IFN-λ is primarily localized to epithelial cells and IFN-λ has been shown to play a unique and important role in protection of intestinal epithelial cells from enteric viral infections (16–18). Recently, evidence is emerging about a role for IFN-λ in the pathogenesis of IBD, serving as the impetus for this review. In the following sections, we will introduce IFN-λ signaling, discuss protective actions of IFN-λ, and consider contexts where its protective role may not be preserved (Figure 1).
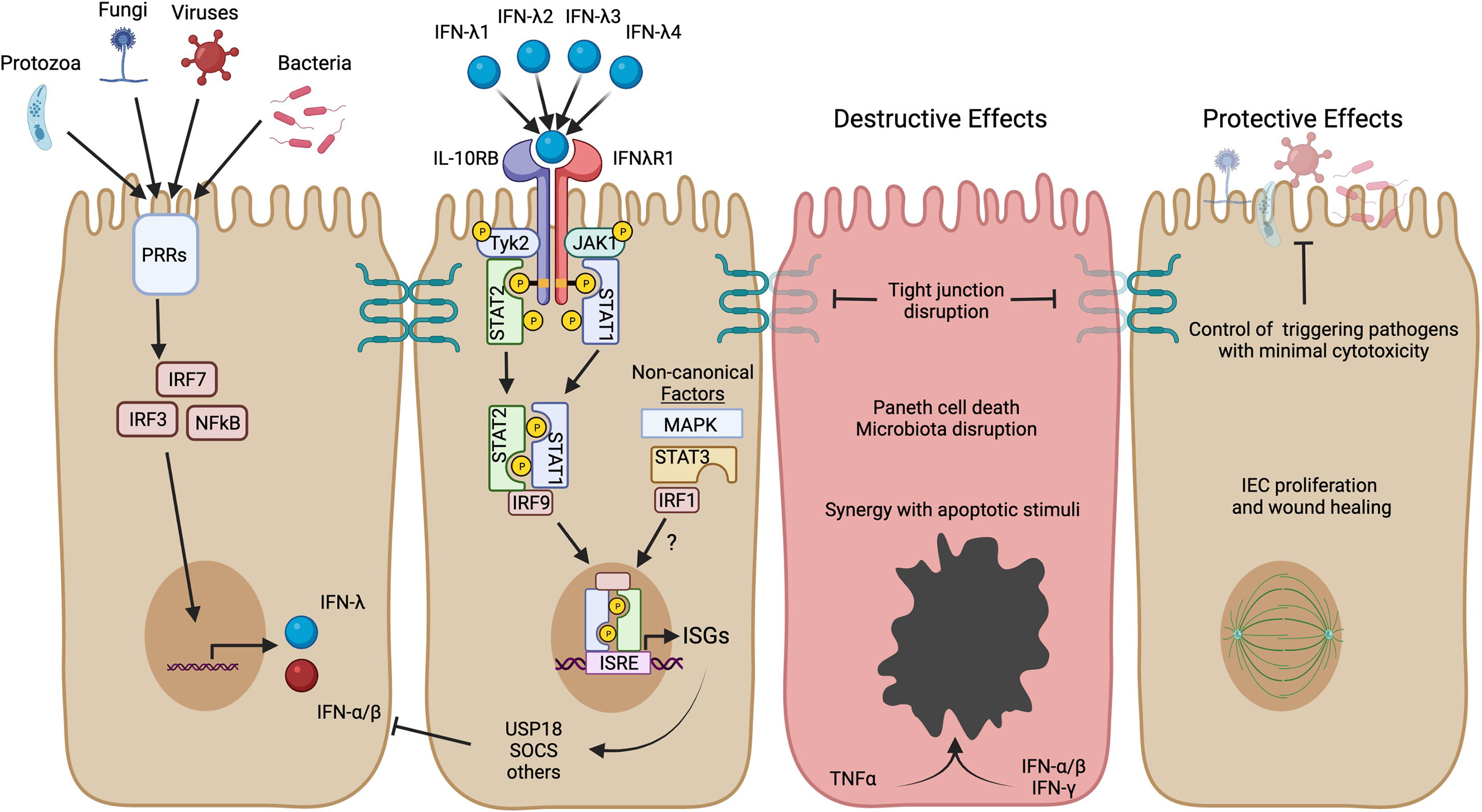
Figure 1 IFN-λ induction, signaling, and potential roles in IBD. Production of IFN-λ can be stimulated by microbial activation of pattern recognition receptors (PRRs) and stimulation of downstream transcription factors (IRF3, IRF7, NFκB). These same transcription factors can also stimulate IFN-α/β, but IECs are able to preferentially produce IFN-λ (19). The four subtypes of human IFN-λ bind to the heterodimeric IFN-λ receptor to activate JAK/STAT signaling and transcription of ISGs. IFN-λ activates the canonical heterotrimeric transcription factor ISGF3 (STAT1, STAT2, and IRF9) to upregulate a core set of ISGs, including direct-acting antiviral effectors and mediators of negative feedback such as USP18 and SOCS genes. Other non-canonical factors such as MAPK, STAT3, and IRF1 can further influence the magnitude and scope of ISGs expressed and may have distinct roles in the context of inflammatory disease. The figure highlights possible detrimental and protective roles for IFN-λ in the context of IBD based on human organoid studies and mouse models of intestinal inflammation (see text for more detail). Created in BioRender.com.
IFN-λ Signaling
Three human IFN-λs (originally named IL-28A, IL-28B and IL-29; later renamed IFN-λ2, IFN-λ3, IFN-λ1, respectively) were discovered in 2003 (20, 21) and a fourth IFN-λ gene (IFN-λ4) was discovered in 2013 (22). The four IFN-λ genes in humans may have non-overlapping roles in UC and CD because studies in other contexts revealed that subtypes of IFN-λ are not functionally equivalent. For example, IFN-λ4 is thought of as having an anti-inflammatory effect due to observations that it is not induced strongly by viral infections like the other IFN-λ subtypes (23) and a disruptive SNP in IFNL4 is associated with clearance of Hepatitis C (22, 24). Comparative transcriptomic studies in hepatocytes suggested minimal differences between IFN-λ1-3 (25), but differing receptor affinity or receptor abundance could significantly impact the scope of a downstream response (26–28). Additionally, differences in the transcription factor usage within the promoter for IFN-λ1 relative to promoter for IFN-λ2 or IFN-λ3 (29) may influence subtype-specific gene expression. Mouse models have been essential to our basic understanding of IFN biology, but it is notable that mice only have two functional IFN-λ genes (IFN-λ2 and IFN-λ3), with IFN-λ1 being a pseudogene and IFN-λ4 being absent (30). Potential differences between subtypes are important considerations for interpretation of results from mouse models and will be interesting to consider in future studies of a role in human disease.
The IFN-λ receptor (IFNLR) is a heterodimeric complex composed of IL10RB and IFNLR1 subunits. IFNLR1 provides specificity for IFN-λ while IL10RB is shared by other IL-10-family cytokines with anti-inflammatory and wound healing functions (IL-10, IL-22, and IL-26) (20, 21, 27). Canonically, the IFNLR1 subunit is associated with Janus-kinase 1 (JAK1) and the IL10RB subunit is associated with the related tyrosine kinase 2 (TYK2) (31). Upon IFN-λ binding to IFNLR, JAK1 and TYK2 phosphorylate signal transducer and activator of transcription 1 (STAT1) and STAT2, which combine with interferon regulatory factor 9 (IRF9) to form a heterotrimeric transcription factor (interferon-stimulated gene factor 3; ISGF3). ISGF3 activates transcription of hundreds of interferon-stimulated genes (ISGs) through binding IFN-stimulated response elements (ISREs) in promoter regions (10).
In the context of complex inflammatory disease such as IBD, multiple cytokine pathways and transcription factors may act in concert (32), with synergy or antagonism in the downstream responses. For IFN family cytokines, homologous interferon regulatory factors (IRF1-IRF9) play combinatorial roles in defining the transcription of IFN-λ, IFN-α/β, and ISGs (33, 34). For example, increased expression of IRF1 can potentiate inflammatory responses to IFN (26) and increased IRF1 has been associated with IBD by expression studies and genome-wide association studies (GWAS) (35, 36). Additionally, JAK and STAT homologs (4 JAKs and 7 STATs) are responsible for an immense complexity of signaling function for a diverse array of cytokines. The complexity relates to cell type specificity, duration, and intensity of STAT signaling (37). The contributions of JAK/STAT signaling to IBD is an active area of study in the field, and is critical to understand in relation to use of existing JAK/STAT inhibitors as therapeutics.
It is clear that the IFN-STAT axis contributes to CD, and recent studies suggest non-redundant contributions of STAT1 and STAT2 in a mouse model of disease (38). STAT3 is also a known gene susceptibility locus for IBD (39, 40), and can be a target of IFN-λ receptor activation in addition to the canonical STAT1/STAT2 response (20, 41). STAT3 was phosphorylated to a low extent by IFN-λ3, but not by IFN-λ1 in one study of the Huh-7 hepatocyte cell line (42), further suggesting the potential for subtype-specific differences. Induction of STAT3 by IFN-λ or IFN-α/β subtypes could be increased in the context of disease and potentiate other proinflammatory cytokines that utilize STAT3 such as IL-6. The role of STAT3 in IBD is likely to be complex with different functions in distinct cell types (43). For example, STAT3 can drive pro-inflammatory effects in Th-17 lymphocytes through IL-23, but STAT3 in IECs has been associated with tissue healing through IL-22 (44). Furthermore, STAT3 can negatively regulate STAT1 signaling via sequestration of STAT1 monomers (45, 46). Thus, it is important to consider the contribution of non-canonical signals such as STAT3 when studying the role of IFN-λ in disease.
In addition to the JAK/STAT pathway, IFN-λ also stimulates mitogen-activated protein kinase (MAPK) pathways (47). In contrast to IFN-λ, IFN-α/β-stimulated antiviral protection of IECs does not depend on MAPK (48). In addition to epithelial cells, IFN-λ was found to activate the MAPK pathway in human fibroblasts, which may promote tissue healing via upregulation of collagens (49). However, this pro-healing effect may not persist during chronic inflammation where fibrosis is a major complication. MAPK signaling may also be stimulated downstream of TNFα to reduce cell proliferation and increase apoptosis (50), and it would be of interest to study the interactions with IFN-λ in this process. Future studies may further our understanding of the potential for combinatorial MAPK stimulation to be either beneficial in restoring homeostasis or maladaptive in the chronic inflammatory landscape of IBD.
Evidence for a Protective Role of IFN-λ in IBD
Biopsies from IBD patients show increased expression of both IFN-λ and IFNLR in the intestinal mucosa, with no discernable differences between CD and UC (51). However, these associations do not indicate whether IFN-λ plays a protective or detrimental role. In fact, distinct roles may manifest in different anatomical locations exemplified by one study showing overexpression of IFN-λ contributes to Paneth cell death in the mouse small intestine (52) and a separate study showing that IFN-λ improves mucosal healing in colonic tissue using a knockout mouse model (51). In this section we will consider the evidence for protective roles of IFN-λ, with evidence for detrimental roles considered in the next section.
IFNLR knockout mice exhibited increased tissue inflammation following dextran sulfate sodium (DSS)-induced colitis relative to WT controls, suggesting that induction of IFN-λ could be tissue protective in this acute inflammation model (51). The authors show that the IFN-λ response promotes mucosal healing and increases IEC proliferation, with associated increases in expression of specific ISGs such as USP18, which has been shown to desensitize cells to the IFN-α/β response (53, 54). Additional studies in IFNLR knockout mice have corroborated findings that IFNLR promotes mucosal healing in the colon. When IFNLR was knocked out alone or in IFNLR/IFNAR double knockouts, DSS colitis susceptibility increased compared with controls (55, 56). In double receptor knockouts, slowed recovery from DSS colitis was associated with impaired IEC proliferation and loss of goblet cells with mucin granules (55). Increased infiltration of neutrophils and CD169+ macrophages were also observed in double receptor knockouts, but interestingly, larger increases occurred in mice deficient in only the IFNLR receptor. Bone marrow chimeras demonstrated that IFN-α/β exerts its effects primarily haematopoietically whereas IFN-λ exerts its effect primarily from within the colonic epithelium (55). However, separate studies have implicated IFN-λ in protection from intestinal inflammation by acting haematopoietically. IFN-λ was found to suppress neutrophil generation of reactive oxygen species in a DSS colitis mouse model (57). Other recent studies of intestinal inflammation associated with graft versus host disease reported a partial, protective role for hematopoietic expression of IFN-λ receptor that is distinct from promoting epithelial cell proliferation and mucosal healing (58). Taken together, these studies point to protective mechanisms for IFN-λ in promoting epithelial healing during acute colonic inflammation and suggest additional protective roles of non-epithelial cell types.
There are also indications of a protective role for IFN pathway factors. IRF3 and IRF7 are important for induction of IFN-λ, IFN-α/β, and ISGs; experiments in IRF3/IRF7 double knockout mice with DSS colitis showed greater tissue inflammation relative to controls (51), suggesting a mucosal protective effect of IFN responses in colonic tissues. Toll-like receptor 3 (TLR3) is important in antiviral immunity and can induce IFN-λ as well as IFN-α/β. While the specific patterns of TLR expression by IECs remains a controversial area, one study reported that TLR3 is downregulated in CD but not UC (59). Additionally, polyinosinic-polycytidylic acid (poly-I:C, TLR3 agonist) treatment was shown to protect mice from tissue damage in DSS colitis in a TLR3-dependant manner (60, 61). However, TLR3 stimulation also increases expression of proinflammatory chemokine CXCL10 which is increased in active IBD (62), and thought to promote pathology. Thus, the roles of IFN-λ may be more complex when combined with other IFN family members.
In addition to direct effects on inflamed tissues, IFN-λ may be indirectly protective against IBD via defense against pathogenic microbes that could trigger IBD symptom onset or exacerbate flares (63). Norovirus is of particular interest because murine norovirus infection synergizes with deficiency in Atg16L1 (autophagy- and CD-associated gene) to increase risk of DSS-triggered colitis (64). This interactive phenotype was ameliorated by anti-TNFα, anti-IFN-γ, or antibiotics, implicating similar pathways influential in human IBD. Mouse norovirus infection of IECs is controlled by IFN-λ (17, 65), which would indirectly block inflammation triggered by this virus infection. Furthermore, IFN-λ stimulates antiviral gene expression in IECs, with limited stimulation of proapoptotic genes, thereby minimizing epithelial damage during an inflammatory immune response (66). In addition to antiviral defense, IFN-λ restricts fungal infections such as Aspergillus fumigatus (67), protozoal infections such as Cryptosporidium parvum (68), and is produced upon TLR stimulation by bacterial products (69). Defense against Aspergillus is particularly notable because increased colonization is observed in areas of inflamed UC tissue (70). Aspergillus has also been observed to be overabundant in fecal samples of patients with CD compared with healthy subjects (71). In addition to pathogens, it is interesting to speculate that IFN-λ responses may influence the host response to the non-pathogenic microbiota, but definitive studies in this area are still needed. In sum, the anti-pathogen activities of IFN-λ are anticipated to indirectly limit infectious triggers of IBD.
Evidence for a Destructive Role of IFN-λ in IBD
Changes to the structure and expression of tight junction proteins is known to occur in both CD and UC (72–76). Studies of IEC organoids indicate that barrier permeability is increased by IFN-λ exposure without an increase in apoptosis, but rather associated with disruption of junctional proteins E-cadherin and ZO-1 (77). The barrier disruption caused by IFN-λ in organoids is eliminated by pretreatment with the clinically-relevant JAK1 inhibitor filgotinib (77), consistent with a requirement for this IFNLR1-associated kinase. Disruption of E-cadherin is especially notable because it has been associated with barrier function breakdown in CD (78). Even though IFN-λ may have some disruptive properties, IFN-γ stimulation of IEC organoids more potently disrupted barrier integrity (77), suggesting it may be a more prominent mediator of pathology in IBD. Additionally, IFN-γ inhibits expression of SLC26A3 (79), a chloride bicarbonate ion exchanger which is known to normally protect against TNFα–induced barrier destruction (80). Lower levels of SLC26A3 are associated with intestinal epithelial barrier dysfunction via modulation of junctional proteins (81). It is likely that the different IFN types may synergize or inhibit each other in their effect on junctional proteins, an area for future study.
Paneth cell death is an important factor in CD as it causes dysregulation of microbial control at epithelial barriers due to loss of secreted antimicrobial peptides. A recent study associated elevated IFN-λ with increased Paneth cell death in CD compared to controls (52). A mouse model of elevated IFN-λ also revealed decreased Paneth cell numbers and epithelial apoptosis that was restored by STAT1-deficiency (52). Additionally, STAT1 (but not STAT2) deficiency partly restores Paneth cell numbers in the Caspase-8-deficient mouse model of intestinal inflammation (38). A cytotoxic effect may be even stronger for other IFN types because organoid studies have shown increased IEC death following treatment with IFN-β or IFN-γ (66, 77). Even so, the effects of IFN-λ on death of Paneth cells or other IEC types may be amplified by signaling factors such as IRF1. IRF1 was shown to have increased expression in the epithelium of IBD patients (36), and its expression amplifies inflammatory chemokine secretion and cytotoxic responses to TNFα (26, 36). Additionally, IFN-stimulated effects may be further amplified by deficiency in autophagy, as indicated by discovery of a role for autophagy gene ATG16L1 in preventing TNFα-driven necroptosis of Paneth cells (82). In sum, excessive death of IECs is a critical driver of intestinal mucosa disruption during IBD and further study is needed regarding the role that IFN stimulation plays in combination with other signals such as TNFα or autophagy.
Therapeutic Potential of the IFN-λ Pathway
Polyethylene glycol (PEG)-conjugated IFN-λ therapy has been examined for viral infections, including Hepatitis C, Hepatitis B, and recently SARS-CoV-2. PEG-IFN-λ has not been tested as a therapy for IBD. However, IFN-α/β has been tested in multiple trials, but there was no improvement compared to placebo and generally low tolerability with increased withdrawal due to adverse events (83). These results are consistent with the generally low tolerability of therapy. Although mouse models of UC show evidence of a pro-healing role for IFN-λ, any potential for PEG-IFN-λ therapy would need to identify a critical balance between its helpful and detrimental roles such as its potential to cause Paneth cell death. On the other hand, IFN-λ neutralization might be an option based on potential for detrimental effects in CD. However, there has been more interest in neutralization of IFN-γ because of its more robust role in driving IBD pathology. Although neutralization of IFN-γ has shown efficacy in mouse models (64), anti-IFN-γ (fontolizumab) trials for CD showed poor clinical efficacy and were discontinued (84), but a combined inhibition across IFN types or together with anti-TNFα may be worthy of consideration.
The JAK/STAT pathway is an attractive therapeutic option based on the potential synergistic effects across IFNs and other cytokine networks, and has currently approved inhibitors and several investigational products. JAK inhibitors may have differential efficacy in IBD subsets due to evidence for differential regulation in CD vs UC (39). Tofacitinib, an inhibitor of JAK1-3, was found to be efficacious in UC trials (85), but not in trials for treatment of CD (86). In contrast, a selective inhibitor of JAK1 (filglotinib) was found to increase remission of CD (87), and other trials are underway with a different JAK1 selective agent, upadacitinib (88). Due to the central role of JAK1 in the IFN response, the necessity of JAK1 selective inhibition in CD is supportive of a pathogenic role for IFN signaling in this disease subset.
In addition to therapeutic modulation of IFN signaling, IFN-λ or ISGs may be useful biomarkers to predict responsiveness to anti-TNFα treatments. The heterogeneity in therapeutic responsiveness and the significant systemic adverse effects associated with anti-TNFα therapy have compelled an extensive search for predictive biomarkers (89). Although anti-TNFα is among the best available treatment options, approximately one third of patients do not respond (90). ISG signatures prior to treatment have been shown to be lower in peripheral blood of patients who responded to anti-TNFα therapy (91), suggesting that increased IFN response can limit therapeutic efficacy. IFN-λ is elevated in serum during active CD, but showed significant heterogeneity among patients (52, 77). Thus, it would be interesting to determine whether IFN-λ, other IFN types, or specific sets of ISGs are superior biomarkers for prediction of anti-TNFα responsiveness.
Conclusion
IFN-λ is critical for immunological function and barrier integrity at mucosal surfaces. Control of pathogenic microbes at the intestinal epithelium without inducing overly robust inflammatory responses is a well-established normal function of IFN-λ. Additionally, there is evidence that IFN-λ promotes healing in colonic epithelial tissues and via ISG expression in IECs themselves as well as via regulation of other inflammatory cell types such as neutrophils. However, a maladaptive role might be assumed in the setting of chronic inflammation. In particular, studies of Paneth cells in human subjects and mouse models suggest that IFN-λ takes a detrimental rather than protective role in the small intestine in CD. Finally, IFN-λ as a biomarker for prediction of response to anti-TNFα is an intriguing area for investigation, and continued consideration of JAK inhibitors that act across IFN types will be important.
Author Contributions
JW wrote the initial draft and all authors revised, edited, and approved the final manuscript. All authors contributed to the article and approved the submitted version.
Funding
DC was supported by NIH grant T32-AI007472 and the Medical Research Foundation of Oregon (OHSU). TN was supported by NIH grant R01-AI130055. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ng SC, Shi HY, Hamidi N, Underwood FE, Tang W, Benchimol EI, et al. Worldwide Incidence and Prevalence of Inflammatory Bowel Disease in the 21st Century: A Systematic Review of Population-Based Studies. Lancet (2017) 390:2769–78. doi: 10.1016/S0140-6736(17)32448-0
2. Dahlhamer JM, Zammitti EP, Ward BW, Wheaton AG, Croft JB. Prevalence of Inflammatory Bowel Disease Among Adults Aged ≥18 Years — United States, 2015. MMWR Morb Mortal Wkly Rep (2016) 65:1166–9. doi: 10.15585/mmwr.mm6542a3
3. Knowles SR, Graff LA, Wilding H, Hewitt C, Keefer L, Mikocka-Walus A. Quality of Life in Inflammatory Bowel Disease: A Systematic Review and Meta-Analyses-Part I. Inflamm Bowel Dis (2018) 24:742–51. doi: 10.1093/ibd/izx100
4. Park KT, Ehrlich OG, Allen JI, Meadows P, Szigethy EM, Henrichsen K, et al. The Cost of Inflammatory Bowel Disease: An Initiative From the Crohn’s & Colitis Foundation. Inflammation Bowel Dis (2020) 26:1–10. doi: 10.1093/ibd/izz104
5. Okamoto R, Watanabe M. Role of Epithelial Cells in the Pathogenesis and Treatment of Inflammatory Bowel Disease. J Gastroenterol (2016) 51:11–21. doi: 10.1007/s00535-015-1098-4
6. Peterson LW, Artis D. Intestinal Epithelial Cells: Regulators of Barrier Function and Immune Homeostasis. Nat Rev Immunol (2014) 14:141–53. doi: 10.1038/nri3608
7. Parikh K, Antanaviciute A, Fawkner-Corbett D, Jagielowicz M, Aulicino A, Lagerholm C, et al. Colonic Epithelial Cell Diversity in Health and Inflammatory Bowel Disease. Nature (2019) 567:49–55. doi: 10.1038/s41586-019-0992-y
8. Mankertz J, Schulzke JD. Altered Permeability in Inflammatory Bowel Disease: Pathophysiology and Clinical Implications. Curr Opin Gastroenterol (2007) 23:379–83. doi: 10.1097/MOG.0b013e32816aa392
9. Muehler A, Slizgi JR, Kohlhof H, Groeppel M, Peelen E, Vitt D. Clinical Relevance of Intestinal Barrier Dysfunction in Common Gastrointestinal Diseases. World J Gastrointest Pathophysiol (2020) 11:114–30. doi: 10.4291/wjgp.v11.i6.114
10. Mesev EV, LeDesma RA, Ploss A. Decoding Type I and III Interferon Signalling During Viral Infection. Nat Microbiol (2019) 4:914–24. doi: 10.1038/s41564-019-0421-x
11. Negishi H, Taniguchi T, Yanai H. The Interferon (IFN) Class of Cytokines and the IFN Regulatory Factor (IRF) Transcription Factor Family. Cold Spring Harb Perspect Biol (2018) 10:a028423. doi: 10.1101/cshperspect.a028423
12. Barrat FJ, Crow MK, Ivashkiv LB. Interferon Target-Gene Expression and Epigenomic Signatures in Health and Disease. Nat Immunol (2019) 20:1574–83. doi: 10.1038/s41590-019-0466-2
13. Uggenti C, Lepelley A, Crow YJ. Self-Awareness: Nucleic Acid–Driven Inflammation and the Type I Interferonopathies. Annu Rev Immunol (2019) 37:247–67. doi: 10.1146/annurev-immunol-042718-041257
14. Wittling MC, Cahalan SR, Levenson EA, Rabin RL. Shared and Unique Features of Human Interferon-Beta and Interferon-Alpha Subtypes. Front Immunol (2021) 11:605673. doi: 10.3389/fimmu.2020.605673
15. Kotenko SV, Rivera A, Parker D, Durbin JE. Type III IFNs: Beyond Antiviral Protection. Semin Immunol (2019) 43:101303. doi: 10.1016/j.smim.2019.101303
16. Lee S, Baldridge MT. Interferon-Lambda: A Potent Regulator of Intestinal Viral Infections. Front Immunol (2017) 8:749. doi: 10.3389/fimmu.2017.00749
17. Baldridge MT, Lee S, Brown JJ, McAllister N, Urbanek K, Dermody TS, et al. Expression of Ifnlr1 on Intestinal Epithelial Cells Is Critical to the Antiviral Effects of Interferon Lambda Against Norovirus and Reovirus. J Virol (2017) 91:e02079–16. doi: 10.1128/JVI.02079-16
18. Stanifer ML, Guo C, Doldan P, Boulant S. Importance of Type I and III Interferons at Respiratory and Intestinal Barrier Surfaces. Front Immunol (2020) 11:608645. doi: 10.3389/fimmu.2020.608645
19. Odendall C, Dixit E, Stavru F, Bierne H, Franz KM, Durbin AF, et al. Diverse Intracellular Pathogens Activate Type III Interferon Expression From Peroxisomes. Nat Immunol (2014) 15:717–28. doi: 10.1038/ni.2915
20. Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK, et al. IFN-λs Mediate Antiviral Protection Through a Distinct Class II Cytokine Receptor Complex. Nat Immunol (2003) 4:69–77. doi: 10.1038/ni875
21. Sheppard P, Kindsvogel W, Xu W, Henderson K, Schlutsmeyer S, Whitmore TE, et al. IL-28, IL-29 and Their Class II Cytokine Receptor IL-28r. Nat Immunol (2003) 4:63–8. doi: 10.1038/ni873
22. Prokunina-Olsson L, Muchmore B, Tang W, Pfeiffer RM, Park H, Dickensheets H, et al. A Variant Upstream of IFNL3 (IL28B) Creating a New Interferon Gene IFNL4 Is Associated With Impaired Clearance of Hepatitis C Virus. Nat Genet (2013) 45:164–71. doi: 10.1038/ng.2521
23. Hong M, Schwerk J, Lim C, Kell A, Jarret A, Pangallo J, et al. Interferon Lambda 4 Expression is Suppressed by the Host During Viral Infection. J Exp Med (2016) 213:2539–52. doi: 10.1084/jem.20160437
24. Zhou H, Mohlenberg M, Terczynska-Dyla E, Winther KG, Hansen NH, Vad-Nielsen J, et al. The IFNL4 Gene Is a Noncanonical Interferon Gene With a Unique But Evolutionarily Conserved Regulation. J Virol (2020) 94:e01535–19. doi: 10.1128/JVI.01535-19
25. Bolen CR, Ding S, Robek MD, Kleinstein SH. Dynamic Expression Profiling of Type I and Type III Interferon-Stimulated Hepatocytes Reveals a Stable Hierarchy of Gene Expression. Hepatology (2014) 59:1262–72. doi: 10.1002/hep.26657
26. Forero A, Ozarkar S, Li H, Lee CH, Hemann EA, Nadjsombati MS, et al. Differential Activation of the Transcription Factor IRF1 Underlies the Distinct Immune Responses Elicited by Type I and Type III Interferons. Immunity (2019) 51:451–64.e6. doi: 10.1016/j.immuni.2019.07.007
27. Mendoza JL, Schneider WM, Hoffmann H-H, Vercauteren K, Jude KM, Xiong A, et al. The IFN-λ-IFN-λr1-IL-10rβ Complex Reveals Structural Features Underlying Type III IFN Functional Plasticity. Immunity (2017) 46:379–92. doi: 10.1016/j.immuni.2017.02.017
28. Pervolaraki K, Rastgou Talemi S, Albrecht D, Bormann F, Bamford C, Mendoza JL, et al. Differential Induction of Interferon Stimulated Genes Between Type I and Type III Interferons Is Independent of Interferon Receptor Abundance. PloS Pathog (2018) 14:e1007420. doi: 10.1371/journal.ppat.1007420
29. Osterlund PI, Pietilä TE, Veckman V, Kotenko SV, Julkunen I. IFN Regulatory Factor Family Members Differentially Regulate the Expression of Type III IFN (IFN-Lambda) Genes. J Immunol (2007) 179:3434–42. doi: 179/6/3434 [pii
30. Lazear HM, Nice TJ, Diamond MS. Interferon-λ: Immune Functions at Barrier Surfaces and Beyond. Immunity (2015) 43:15–28. doi: 10.1016/j.immuni.2015.07.001
31. Kotenko SV, Langer JA. Full House: 12 Receptors for 27 Cytokines. Int Immunopharmacol (2004) 4:593–608. doi: 10.1016/j.intimp.2004.01.003
32. Friedrich M, Pohin M, Powrie F. Cytokine Networks in the Pathophysiology of Inflammatory Bowel Disease. Immunity (2019) 50:992–1006. doi: 10.1016/j.immuni.2019.03.017
33. Taniguchi T, Ogasawara K, Takaoka A, Tanaka N. IRF Family of Transcription Factors as Regulators of Host Defense. Annu Rev Immunol (2001) 19:623–55. doi: 10.1146/annurev.immunol.19.1.623
34. Zhao GN, Jiang DS, Li H. Interferon Regulatory Factors: At the Crossroads of Immunity, Metabolism, and Disease. Biochim Biophys Acta (2015) 1852:365–78. doi: 10.1016/j.bbadis.2014.04.030
35. Brandt M, Kim-Hellmuth S, Ziosi M, Gokden A, Wolman A, Lam N, et al. An Autoimmune Disease Risk Variant: A Trans Master Regulatory Effect Mediated by IRF1 Under Immune Stimulation? PloS Genet (2021) 17:e1009684. doi: 10.1371/journal.pgen.1009684
36. Tang R, Yang G, Zhang S, Wu C, Chen M. Opposite Effects of Interferon Regulatory Factor 1 and Osteopontin on the Apoptosis of Epithelial Cells Induced by TNF-α in Inflammatory Bowel Disease. Inflamm Bowel Dis (2014) 20:1950–61. doi: 10.1097/MIB.0000000000000192
37. Villarino AV, Kanno Y, Ferdinand JR, O’Shea JJ. Mechanisms of Jak/STAT Signaling in Immunity and Disease. J Immunol (2015) 194:21–7. doi: 10.4049/jimmunol.1401867
38. Stolzer I, Dressel A, Chiriac MT, Neurath MF, Günther C. An IFN-STAT Axis Augments Tissue Damage and Inflammation in a Mouse Model of Crohn’s Disease. Front Med (2021) 8:644244. doi: 10.3389/fmed.2021.644244
39. Cordes F, Foell D, Ding JN, Varga G, Bettenworth D. Differential Regulation of JAK/STAT-Signaling in Patients With Ulcerative Colitis and Crohn’s Disease. World J Gastroenterol (2020) 26:4055–75. doi: 10.3748/wjg.v26.i28.4055
40. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host–microbe Interactions Have Shaped the Genetic Architecture of Inflammatory Bowel Disease. Nature (2012) 491:119–24. doi: 10.1038/nature11582
41. Dumoutier L, Lejeune D, Hor S, Fickenscher H, Renauld JC. Cloning of a New Type II Cytokine Receptor Activating Signal Transducer and Activator of Transcription (STAT)1, STAT2 and STAT3. Biochem J (2003) 370:391–6. doi: 10.1042/BJ20021935
42. Diegelmann J, Beigel F, Zitzmann K, Kaul A, Goke B, Auernhammer CJ, et al. Comparative Analysis of the Lambda-Interferons IL-28A and IL-29 Regarding Their Transcriptome and Their Antiviral Properties Against Hepatitis C Virus. PloS One (2010) 5:e15200. doi: 10.1371/journal.pone.0015200
43. Kasembeli MM, Bharadwaj U, Robinson P, Tweardy DJ. Contribution of STAT3 to Inflammatory and Fibrotic Diseases and Prospects for its Targeting for Treatment. Int J Mol Sci (2018) 19:2299. doi: 10.3390/ijms19082299
44. Li Y, de Haar C, Peppelenbosch MP, van der Woude CJ. New Insights Into the Role of STAT3 in IBD. Inflamm Bowel Dis (2012) 18:1177–83. doi: 10.1002/ibd.21884
45. Ho HH, Ivashkiv LB. Role of STAT3 in Type I Interferon Responses. Negative Regulation of STAT1-Dependent Inflammatory Gene Activation. J Biol Chem (2006) 281:14111–8. doi: 10.1074/jbc.M511797200
46. Tsai MH, Pai LM, Lee CK. Fine-Tuning of Type I Interferon Response by STAT3. Front Immunol (2019) 10:1448. doi: 10.3389/fimmu.2019.01448
47. Zhou Z, Hamming OJ, Ank N, Paludan SR, Nielsen AL, Hartmann R. Type III Interferon (IFN) Induces a Type I IFN-Like Response in a Restricted Subset of Cells Through Signaling Pathways Involving Both the Jak-STAT Pathway and the Mitogen-Activated Protein Kinases. J Virol (2007) 81:7749–58. doi: 10.1128/JVI.02438-06
48. Pervolaraki K, Stanifer ML, Münchau S, Renn LA, Albrecht D, Kurzhals S, et al. Type I and Type III Interferons Display Different Dependency on Mitogen-Activated Protein Kinases to Mount an Antiviral State in the Human Gut. Front Immunol (2017) 8:459. doi: 10.3389/fimmu.2017.00459
49. Alase AA, El-Sherbiny YM, Vital EM, Tobin DJ, Turner NA, Wittmann M. IFNlambda Stimulates MxA Production in Human Dermal Fibroblasts via a MAPK-Dependent STAT1-Independent Mechanism. J Invest Dermatol (2015) 135:2935–43. doi: 10.1038/jid.2015.317
50. Yuan Y, Ding D, Zhang N, Xia Z, Wang J, Yang H, et al. TNF-Alpha Induces Autophagy Through ERK1/2 Pathway to Regulate Apoptosis in Neonatal Necrotizing Enterocolitis Model Cells IEC-6. Cell Cycle (2018) 17:1390–402. doi: 10.1080/15384101.2018.1482150
51. Chiriac MT, Buchen B, Wandersee A, Hundorfean G, Günther C, Bourjau Y, et al. Activation of Epithelial Signal Transducer and Activator of Transcription 1 by Interleukin 28 Controls Mucosal Healing in Mice With Colitis and Is Increased in Mucosa of Patients With Inflammatory Bowel Disease. Gastroenterology (2017) 153:123–38.e8. doi: 10.1053/j.gastro.2017.03.015
52. Günther C, Ruder B, Stolzer I, Dorner H, He G-W, Chiriac MT, et al. Interferon Lambda Promotes Paneth Cell Death Via STAT1 Signaling in Mice and Is Increased in Inflamed Ileal Tissues of Patients With Crohn’s Disease. Gastroenterology (2019) 157:1310–22.e13. doi: 10.1053/j.gastro.2019.07.031
53. Malakhova OA, Kim KII, Luo J-K, Zou W, Kumar KGS, SY F, et al. UBP43 is a Novel Regulator of Interferon Signaling Independent of its ISG15 Isopeptidase Activity. EMBO J (2006) 25:2358–67. doi: 10.1038/sj.emboj.7601149
54. Basters A, Knobeloch KP, Fritz G. USP18 - A Multifunctional Component in the Interferon Response. Biosci Rep (2018) 38:BSR20180250. doi: 10.1042/BSR20180250
55. McElrath C, Espinosa V, Lin JD, Peng J, Sridhar R, Dutta O, et al. Critical Role of Interferons in Gastrointestinal Injury Repair. Nat Commun (2021) 12:2624. doi: 10.1038/s41467-021-22928-0
56. Rauch I, Rosebrock F, Hainzl E, Heider S, Majoros A, Wienerroither S, et al. Noncanonical Effects of IRF9 in Intestinal Inflammation: More Than Type I and Type III Interferons. Mol Cell Biol (2015) 35:2332–43. doi: 10.1128/MCB.01498-14
57. Broggi A, Tan Y, Granucci F, Zanoni I. IFN-λ Suppresses Intestinal Inflammation by Non-Translational Regulation of Neutrophil Function. Nat Immunol (2017) 18:1084–93. doi: 10.1038/ni.3821
58. Henden AS, Koyama M, Robb RJ, Forero A, Kuns RD, Chang K, et al. IFN-λ Therapy Prevents Severe Gastrointestinal Graft-Versus-Host Disease. Blood (2021) 138:722–37. doi: 10.1182/blood.2020006375
59. Cario E, Podolsky DK. Differential Alteration in Intestinal Epithelial Cell Expression of Toll-Like Receptor 3 (TLR3) and TLR4 in Inflammatory Bowel Disease. Infect Immun (2000) 68:7010–7. doi: 10.1128/IAI.68.12.7010-7017.2000
60. Vijay-Kumar M, Wu H, Aitken J, Kolachala VL, Neish AS, Sitaraman SV, et al. Activation of Toll-Like Receptor 3 Protects Against DSS-Induced Acute Colitis. Inflammation Bowel Dis (2007) 13:856–64. doi: 10.1002/ibd.20142
61. Zhao HW, Yue YH, Han H, Chen XL, Lu YG, Zheng JM, et al. Effect of Toll-Like Receptor 3 Agonist Poly I:C on Intestinal Mucosa and Epithelial Barrier Function in Mouse Models of Acute Colitis. World J Gastroenterol (2017) 23:999–1009. doi: 10.3748/wjg.v23.i6.999
62. Ostvik AE, Granlund AV, Bugge M, Nilsen NJ, Torp SH, Waldum HL, et al. Enhanced Expression of CXCL10 in Inflammatory Bowel Disease: Potential Role of Mucosal Toll-Like Receptor 3 Stimulation. Inflamm Bowel Dis (2013) 19:265–74. doi: 10.1002/ibd.23034
63. Iliev ID, Cadwell K. Effects of Intestinal Fungi and Viruses on Immune Responses and Inflammatory Bowel Diseases. Gastroenterology (2021) 160:1050–66. doi: 10.1053/j.gastro.2020.06.100
64. Cadwell K, Patel KK, Maloney NS, Liu T-C, Ng ACY, Storer CE, et al. Virus-Plus-Susceptibility Gene Interaction Determines Crohn’s Disease Gene Atg16L1 Phenotypes in Intestine. Cell (2010) 141:1135–45. doi: 10.1016/j.cell.2010.05.009
65. Nice TJ, Baldridge MT, McCune BT, Norman JM, Lazear HM, Artyomov M, et al. Interferon-λ Cures Persistent Murine Norovirus Infection in the Absence of Adaptive Immunity. Science (2015) 347:269–73. doi: 10.1126/science.1258100
66. Van Winkle JA, Constant DA, Li L, Nice TJ. Selective Interferon Responses of Intestinal Epithelial Cells Minimize Tumor Necrosis Factor Alpha Cytotoxicity. J Virol (2020) 94:1–16. doi: 10.1128/JVI.00603-20
67. Espinosa V, Dutta O, McElrath C, Du P, Chang YJ, Cicciarelli B, et al. Type III Interferon is a Critical Regulator of Innate Antifungal Immunity. Sci Immunol (2017) 2:eaan5357. doi: 10.1126/sciimmunol.aan5357
68. Ferguson SH, Foster DM, Sherry B, Magness ST, Nielsen DM, Gookin JL. Interferon-Lambda3 Promotes Epithelial Defense and Barrier Function Against Cryptosporidium Parvum Infection. Cell Mol Gastroenterol Hepatol (2019) 8:1–20. doi: 10.1016/j.jcmgh.2019.02.007
69. Odendall C, Voak AA, Kagan JC. Type III IFNs Are Commonly Induced by Bacteria-Sensing TLRs and Reinforce Epithelial Barriers During Infection. J Immunol (2017) 199:3270–9. doi: 10.4049/jimmunol.1700250
70. Qiu X, Ma J, Jiao C, Mao X, Zhao X, Lu M, et al. Alterations in the Mucosa-Associated Fungal Microbiota in Patients With Ulcerative Colitis. Oncotarget (2017) 8:107577–88. doi: 10.18632/oncotarget.22534
71. Li Q, Wang C, Tang C, He Q, Li N, Li J. Dysbiosis of Gut Fungal Microbiota is Associated With Mucosal Inflammation in Crohn’s Disease. J Clin Gastroenterol (2014) 48:513–23. doi: 10.1097/MCG.0000000000000035
72. Das P, Goswami P, Das TK, Nag T, Sreenivas V, Ahuja V, et al. Comparative Tight Junction Protein Expressions in Colonic Crohn’s Disease, Ulcerative Colitis, and Tuberculosis: A New Perspective. Virchows Arch (2012) 460:261–70. doi: 10.1007/s00428-012-1195-1
73. Heller F, Florian P, Bojarski C, Richter J, Christ M, Hillenbrand B, et al. Interleukin-13 Is the Key Effector Th2 Cytokine in Ulcerative Colitis That Affects Epithelial Tight Junctions, Apoptosis, and Cell Restitution. Gastroenterology (2005) 129:550–64. doi: 10.1016/j.gastro.2005.05.002
74. Landy J, Ronde E, English N, Clark SK, Hart AL, Knight SC, et al. Tight Junctions in Inflammatory Bowel Diseases and Inflammatory Bowel Disease Associated Colorectal Cancer. World J Gastroenterol (2016) 22:3117–26. doi: 10.3748/wjg.v22.i11.3117
75. Schmitz H, Barmeyer C, Fromm M, Runkel N, Foss HD, Bentzel CJ, et al. Altered Tight Junction Structure Contributes to the Impaired Epithelial Barrier Function in Ulcerative Colitis. Gastroenterology (1999) 116:301–9. doi: 10.1016/s0016-5085(99)70126-5
76. Zeissig S, Burgel N, Gunzel D, Richter J, Mankertz J, Wahnschaffe U, et al. Changes in Expression and Distribution of Claudin 2, 5 and 8 Lead to Discontinuous Tight Junctions and Barrier Dysfunction in Active Crohn’s Disease. Gut (2007) 56:61–72. doi: 10.1136/gut.2006.094375
77. Xu P, Becker H, Elizalde M, Pierik M, Masclee A, Jonkers D. Interleukin-28A Induces Epithelial Barrier Dysfunction in CD Patient-Derived Intestinal Organoids. Am J Physiol Gastrointest Liver Physiol (2021) 320:G689–99. doi: 10.1152/ajpgi.00064.2020
78. Muise AM, Walters TD, Glowacka WK, Griffiths AM, Ngan BY, Lan H, et al. Polymorphisms in E-Cadherin (CDH1) Result in a Mis-Localised Cytoplasmic Protein That is Associated With Crohn’s Disease. Gut (2009) 58:1121–7. doi: 10.1136/gut.2008.175117
79. Saksena S, Singla A, Goyal S, Katyal S, Bansal N, Gill RK, et al. Mechanisms of Transcriptional Modulation of the Human Anion Exchanger SLC26A3 Gene Expression by IFN-{Gamma}. Am J Physiol Gastrointest Liver Physiol (2010) 298:G159–66. doi: 10.1152/ajpgi.00374.2009
80. Ding X, Li D, Li M, Wang H, He Q, Wang Y, et al. SLC26A3 (DRA) Prevents TNF-Alpha-Induced Barrier Dysfunction and Dextran Sulfate Sodium-Induced Acute Colitis. Lab Investig (2018) 98:462–76. doi: 10.1038/s41374-017-0005-4
81. Kumar A, Priyamvada S, Ge Y, Jayawardena D, Singhal M, Anbazhagan AN, et al. A Novel Role of SLC26A3 in the Maintenance of Intestinal Epithelial Barrier Integrity. Gastroenterology (2021) 160:1240–55.e3. doi: 10.1053/j.gastro.2020.11.008
82. Matsuzawa-Ishimoto Y, Shono Y, Gomez LE, Hubbard-Lucey VM, Cammer M, Neil J, et al. Autophagy Protein ATG16L1 Prevents Necroptosis in the Intestinal Epithelium. J Exp Med (2017) 214:3687–705. doi: 10.1084/jem.20170558
83. Wang Y, MacDonald JK, Benchimol EI, Griffiths AM, Steinhart AH, Panaccione R, et al. Type I Interferons for Induction of Remission in Ulcerative Colitis. Cochrane Database Syst Rev (2015) 9:CD006790. doi: 10.1002/14651858.CD006790.pub3
84. Reinisch W, de Villiers W, Bene L, Simon L, Racz I, Katz S, et al. Fontolizumab in Moderate to Severe Crohn’s Disease: A Phase 2, Randomized, Double-Blind, Placebo-Controlled, Multiple-Dose Study. Inflamm Bowel Dis (2010) 16:233–42. doi: 10.1002/ibd.21038
85. Sandborn WJ, Su C, Panes J. Tofacitinib as Induction and Maintenance Therapy for Ulcerative Colitis. N Engl J Med (2017) 377:496–7. doi: 10.1056/NEJMc1707500
86. Panes J, Sandborn WJ, Schreiber S, Sands BE, Vermeire S, D’Haens G, et al. Tofacitinib for Induction and Maintenance Therapy of Crohn’s Disease: Results of Two Phase IIb Randomised Placebo-Controlled Trials. Gut (2017) 66:1049–59. doi: 10.1136/gutjnl-2016-312735
87. Vermeire S, Schreiber S, Petryka R, Kuehbacher T, Hebuterne X, Roblin X, et al. Clinical Remission in Patients With Moderate-to-Severe Crohn’s Disease Treated With Filgotinib (the FITZROY Study): Results From a Phase 2, Double-Blind, Randomised, Placebo-Controlled Trial. Lancet (2017) 389:266–75. doi: 10.1016/S0140-6736(16)32537-5
88. Ma C, Jairath V, Vande Casteele N. Pharmacology, Efficacy and Safety of JAK Inhibitors in Crohn’s Disease. Best Pr Res Clin Gastroenterol (2019) 38–39:101606. doi: 10.1016/j.bpg.2019.03.002
89. Atreya R, Neurath MF, Siegmund B. Personalizing Treatment in IBD: Hype or Reality in 2020? Can We Predict Response to Anti-TNF? Front Med (2020) 7:517. doi: 10.3389/fmed.2020.00517
90. Gisbert JP, Panes J. Loss of Response and Requirement of Infliximab Dose Intensification in Crohn’s Disease: A Review. Am J Gastroenterol (2009) 104:760–7. doi: 10.1038/ajg.2008.88
Keywords: interferon lambda (IFN-λ), intestinal epithelial cell (IEC), inflammatory bowel disease (IBD), ulcerative colitis (UC), Crohn’s disease (CD)
Citation: Wallace JW, Constant DA and Nice TJ (2021) Interferon Lambda in the Pathogenesis of Inflammatory Bowel Diseases. Front. Immunol. 12:767505. doi: 10.3389/fimmu.2021.767505
Received: 30 August 2021; Accepted: 23 September 2021;
Published: 12 October 2021.
Edited by:
Deanna Santer, University of Manitoba, CanadaReviewed by:
Karen L. Edelblum, The State University of New Jersey, United StatesAna Maria Gamero, Temple University, United States
Copyright © 2021 Wallace, Constant and Nice. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Timothy J. Nice, bmljZUBvaHN1LmVkdQ==