 Richard Felix Kraus*†
Richard Felix Kraus*† Michael Andreas Gruber†
Michael Andreas Gruber†- Department of Anesthesiology, University Medical Center Regensburg, Regensburg, Germany
Neutrophils (polymorphonuclear cells; PMNs) form a first line of defense against pathogens and are therefore an important component of the innate immune response. As a result of poorly controlled activation, however, PMNs can also mediate tissue damage in numerous diseases, often by increasing tissue inflammation and injury. According to current knowledge, PMNs are not only part of the pathogenesis of infectious and autoimmune diseases but also of conditions with disturbed tissue homeostasis such as trauma and shock. Scientific advances in the past two decades have changed the role of neutrophils from that of solely immune defense cells to cells that are responsible for the general integrity of the body, even in the absence of pathogens. To better understand PMN function in the human organism, our review outlines the role of PMNs within the innate immune system. This review provides an overview of the migration of PMNs from the vascular compartment to the target tissue as well as their chemotactic processes and illuminates crucial neutrophil immune properties at the site of the lesion. The review is focused on the formation of chemotactic gradients in interaction with the extracellular matrix (ECM) and the influence of the ECM on PMN function. In addition, our review summarizes current knowledge about the phenomenon of bidirectional and reverse PMN migration, neutrophil microtubules, and the microtubule organizing center in PMN migration. As a conclusive feature, we review and discuss new findings about neutrophil behavior in cancer environment and tumor tissue.
1 The Role of Neutrophils in Non-Specific Immune Defense
Granulocytes are an important component of the innate immune system. The three types of granulocytes eosinophils, basophils, and neutrophils are distinguished by their histological, morphological, and immunological properties. Each of the three types matures in the bone marrow (1). The view that PMNs only have a life span of a few hours to fewer than 3 days after maturation has recently been challenged. Crucial aspects of the neutrophil life cycle, namely their life span in different tissues and different inflammatory states, are still considered not yet fully defined (1, 2).
Making up 50–70% of all circulating leukocytes, neutrophil granulocytes (neutrophils, polymorphonuclear cells; PMNs) are the most mobile and abundant cellular component of the innate immune system of the human body. They act as an important first line of defense within the innate immune response (see below) (3, 4). Neutrophils have a diameter of 10–12 µm, and their nucleus is usually lobed into three to four segments. Therefore, PMNs are also referred to as being polymorphonuclear. Their granules are very small (<1 µm) and have a pinkish to lilac color when exposed to Pappenheim staining (see Figure 1) (5).
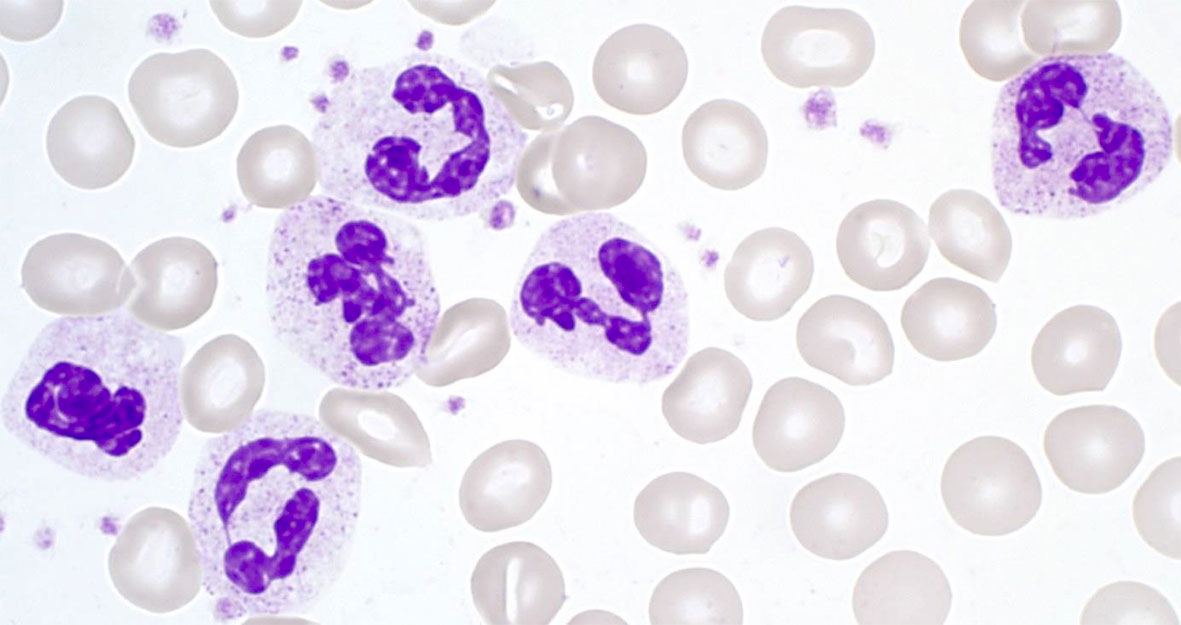
Figure 1 Segmented neutrophil granulocytes in Pappenheim‐stained blood cell smears (graphic provided by the laboratory for Paediatric Oncology and Haematology at the University Medical Centre Regensburg).
The neutrophil life cycle begins with the granulopoiesis in the bone marrow and is illustrated in Figure 2. Every day, approximately 1011 PMNs are generated in the hematopoietic strands interspersed in the venous sinuses of the bone marrow in the human body. Granulocyte differentiation is regulated by the coordinated expression of myeloid key transcription factors (6, 7). The amount of PMNs released and renewed daily constitutes about 1% of all nucleated cells (approximately 1013) of the human body (8, 9). If PMNs did not have any crucial role within the human immune system, such an enormous effort would probably have become obsolete long ago in the history of evolution.
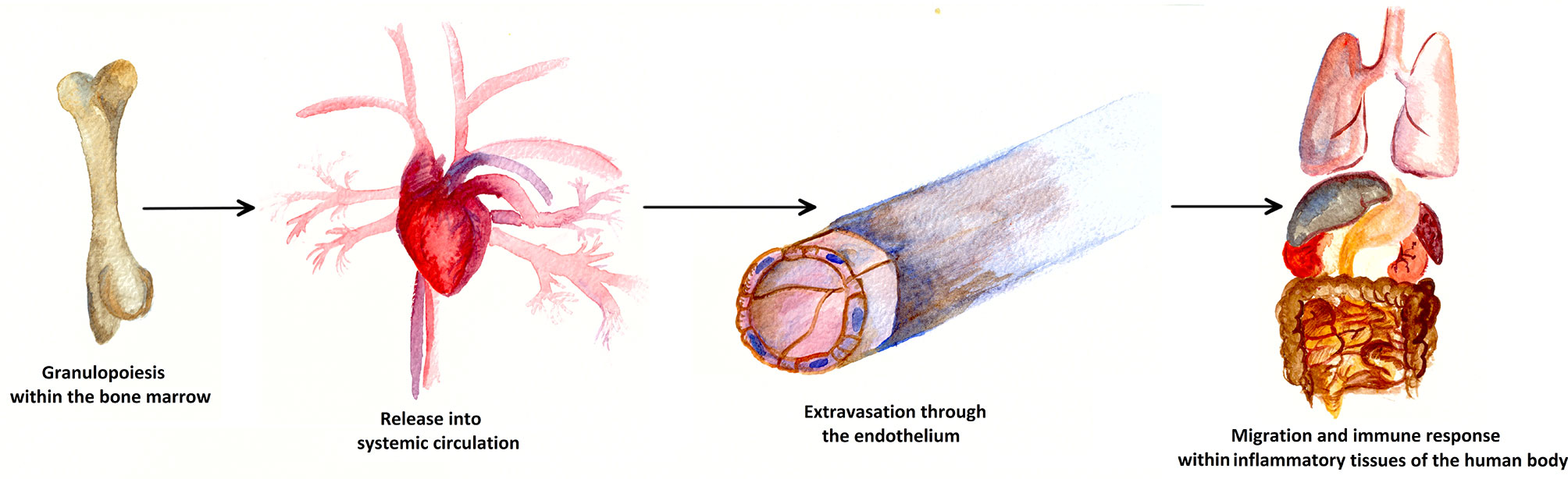
Figure 2 Life cycle of a neutrophil cell. Approximately 1011 PMNs are generated in the bone marrow via granulopoiesis every day. Attracted by cytokines, PMNs are consecutively released into the blood stream and thus into systemic circulation. At the sites of inflammation, PMNs leave the blood vessels through the endothelium, a process known as extravasation. In inflammatory human tissue, PMNs migrate along chemotactic gradients in the interstitium and perform specific neutrophil immune functions as a first line defense of the innate immune system.
To leave the bone marrow, mature neutrophils have to migrate through the sinusoidal endothelium separating the hematopoietic compartment from the blood stream. Thereby, neutrophils seem to migrate across the bone marrow endothelium through tight-fitting pores in the transcellular rather than the paracellular pathway (7, 10, 11). For this process, endothelial penetrability is of importance, whereby endothelial homoeostasis and, as a consequence, cell release from the bone marrow are regulated and decisively influenced by vascular endothelial cadherin (12).
PMN release is stimulated by gradients across the sinus wall of bone marrow sinusoids generated by the production of mediators (such as the macrophage inflammatory protein-2 [MIP-2], granulocyte-colony stimulating factor [G-CSF], and c-x-c motive chemokine 1 [CXCL1]) (7, 10). The precise mechanism by which inflammation leads to circulating neutrophilia is not yet fully understood. Nevertheless, acute mobilization of neutrophils from the bone marrow may require the coordinated, yet unambiguous, actions of G-CSF and CXC chemokines, whereby G-CSF disrupts the retention mechanisms in the bone marrow (such as the CXCR4-stromal-derived-factor-1 axis) (7, 13). Besides the participation in the release of mature PMNs from the bone marrow, G-CSF is the principal regulator of physiological granulopoiesis. Effects include commitment of progenitor cells to the myeloid lineage, proliferation of granulocytic precursors, and reduction in transit time across the granulocytic compartment (7, 14, 15). Because of this proliferation and the reduction in transit time, G−CSF is nowadays also used therapeutically for the pretreatment of granulocyte donors (16).
After their release from the bone marrow, PMNs circulate in the blood stream under physiological conditions for fewer than 24 hours. The short life span and high production rate of PMNs require the equivalent elimination of PMNs from circulation to maintain homeostasis (6). Therefore, a certain part of circulating PMNs undergo constitutive apoptosis. Apoptotic PMNs are sorted out in the bone marrow, liver, and spleen and subsequently eliminated by efferocytosis (17, 18).
Besides PMNs circulating in the blood (circulating pool), there are two or maybe three reservoirs in which PMNs are resting and can be released on demand.
First, the bone marrow contains a reserve of less mature (band-shaped) and mature PMNs, which are retained in the reserve by life-sustaining cytokines of the bone marrow (bone marrow pool). The prevailing idea in the literature is that neutrophils receive complex anti-apoptotic signals in the bone marrow, which are less present or even absent in the bloodstream. These signals may include G−CSF and GM-CSF, although GM-CSF has a much smaller effect on mouse neutrophils than G-CSF (19–21). Additionally, mesenchymal stem cells (MSC) of the bone marrow are likely to protect neutrophils from apoptosis, maintaining their effector functions and preventing the disproportionate activation of the oxidative metabolism. Thereby, the key MSC-derived soluble factor IL-6 has been shown to be responsible for neutrophil protection from apoptosis (22). Moreover, SerpinB1 (an inhibitor of neutrophil serine proteases NE, CG, and PR-3) seems to be essential for maintaining a healthy PMN bone marrow pool by preserving anti-apoptotic signals (23).
Second, some PMNs are not intravascularly located in the main blood stream but adhere loosely to the endothelium of venous blood vessels (marginated pool). By recruiting this reserve, the number of neutrophils in the blood stream can be rapidly increased (5, 7). Such marginated pools can be found in the bone marrow, liver, spleen, and, as currently discussed, also in the lungs (7).
The size of individual marginated pools is considered to be the product of the mean intravascular transit time through the organ (i.e. the mean time it takes for neutrophils to transmigrate the capillary bed) and its blood flow (7). Peters et al. and Ussov et al. quantified the mean neutrophil intravascular transit time for the bone marrow (10 min), spleen (10 min), and liver (2 min) (24, 25). Although the size of the marginated pulmonary pool is predominantly determined by the mean pulmonary transit time (approximately 3–6 min), exact specification is challenging and controversial (7, 26–30). It has been estimated that 49% of the total blood granulocyte pool resides in the circulating pool, whereas the remaining 51% of the pool is attributed to PMNs of the marginated pool (7, 31).
Until recently, medical education books, such as Janeway’s Immunobiology, stated that PMNs are not present in healthy tissue in contrast to other phagocytizing cells (32). However, very recent studies (reviewed in (4, 7)) have demonstrated that PMNs are also physiologically present in the interstitium of the organs in which marginated pools are observed, namely in the bone marrow, liver, spleen, and lungs (4). As reported recently, PMNs may also be found, albeit to a smaller degree, in uninfected lymph nodes, the intestine, white adipose tissue, the skin, and in skeletal muscles (33, 34). Nevertheless, the question how and why PMNs are physiologically concentrated in these tissues remains unanswered. On the one hand, it is possible that these organs are further reservoirs that can rapidly supply PMNs in case of emergency. On the other hand, organ-resident PMNs may patrol through the above-mentioned organs, searching for damaged tissue and micro-organisms (4). To develop this idea further, PMNs detecting a pathogen would be able to rapidly and directly activate the adaptive immune system by physical contact and communication with other organ resident immune cells. In line with this theory, Puga et al. showed that splenic PMNs are able to directly activate B cells even under physiological conditions (30, 35). Moreover, the interaction of organ-resident pulmonary PMNs with B cells appears to play an important role in regulating the immune response (30, 36).
For this purpose, PMNs seem to be endowed with important non-immune regulatory functions when migrating through healthy tissues (34). As shown by Doerschuk et al. and Downey et al., one mechanism for restricting the presence of PMNs in pulmonary capillaries is the need for neutrophils to deform (27, 28).
Currently, the question of why PMNs are present in healthy tissue has not been fully clarified yet. So far, it is not known which tissues are actually involved, which dynamic processes take place, and—maybe more decisively—to what extent infiltrating PMNs contribute to healthy tissue homeostasis (4, 34).
Besides the controversially discussed physiological presence of PMNs in tissue, PMNs migrate very quickly, either as a consequence of infections or sterile tissue damage, from peripheral blood into peripheral tissues to fight a lesion there (37). This process is called extravasation (38). In an acute inflammatory reaction, granulopoiesis in the bone marrow increases, and a large number of PMNs accumulate very rapidly at the site of infection or the lesion. In the process, the life span of the circulating PMNs also becomes significantly extended (3).
Inflammatory reactions are modulated by inflammatory mediators, which are released by sensitive leukocytes (such as macrophages, dendritic cells, or mast cells) in the tissue when pathogens or disturbed tissue homeostasis are detected. Mediators may also be released by endothelial cells, by epithelial cells, by fibroblasts or by PMNs themselves upon activation (4, 37, 39, 40).
The most important mediators are chemokines, peptides, and eicosanoids. Chemokines are a large group of chemotactic cytokines, which are divided into four groups according to the arrangement of the two N-terminal cysteine residues designated CXC, CC, C, and CX3C, depending on the spacing of the conserved cytokines (“X” stands for an amino acid). CXC chemokines mainly target neutrophils and lymphocytes, whereas CC chemokines target a variety of cell types including macrophages, eosinophils, basophils, and dendritic cells (see Table 1) (84, 85).
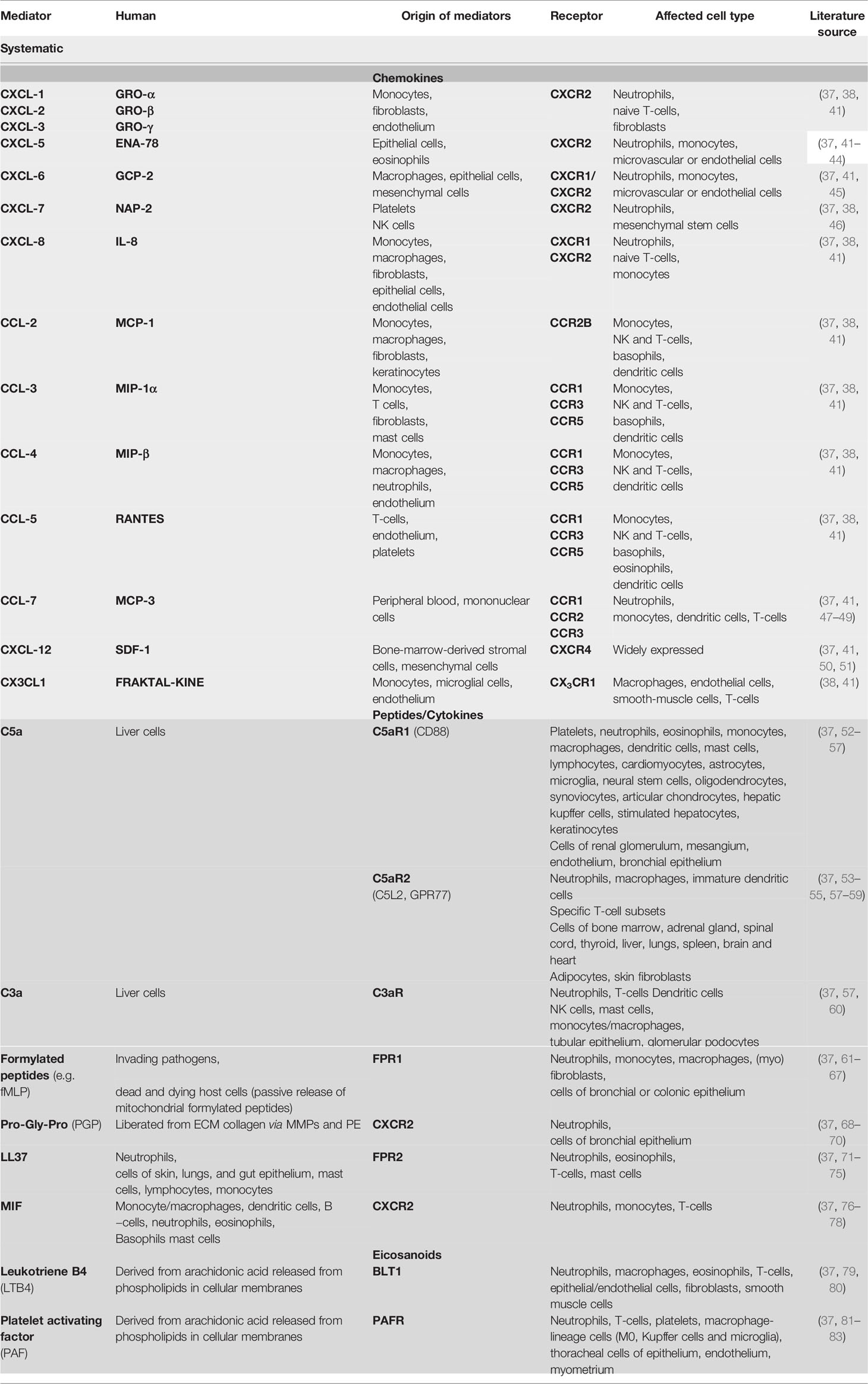
Table 1 Important inflammatory mediators and associated cell types..
Sensitive leukocytes are activated by certain surface structures found on pathogens (pathogen-associated molecular patterns, PAMPs) or endogenously released from cells through inflammasome activation or passively after cell damage (damage-associated molecular patterns, DAMPs) (86, 87). PAMPs and DAMPs can be recognized by sensitive leukocytes via certain receptors (pattern recognition receptors, PRR). PRR-mediated activation of sensitive leukocytes induce the release of proinflammatory mediators, such as IL-1β, IL-6, TNF−α, and other specific neutrophil-active chemo-attractants (see chapter 5) (37, 88).
These mediators trigger the recruitment of leukocytes to inflammatory tissues, regulate cell death in inflammatory tissues, induce the production of acute-phase proteins, and modify vascular endothelial permeability (86). In the specific case of PMNs, mobilization from the bone marrow into the blood stream is regulated by the mediators leukotriene B4, active complement component C5 (C5a), and the interleukin C-X-C motif chemokine-ligand 8 (CXCL8, formerly also called IL-8), which are released by the mechanisms described above. Furthermore, these mediators direct PMNs to the lesion site via chemotactic gradients (for details on chemotaxis, see chapter 5) and finally induce them to leave the blood and migrate into the surrounding tissue (extravasation) (38, 89).
2 The Process of Extravasation
The recruitment of PMN requires adhesion to and subsequent transmigration through vascular walls (see Figure 3) (90). In most tissues, PMNs leave the vascular system through postcapillary venules (91). Only in the lungs does the extravasation process occur through capillaries (91).
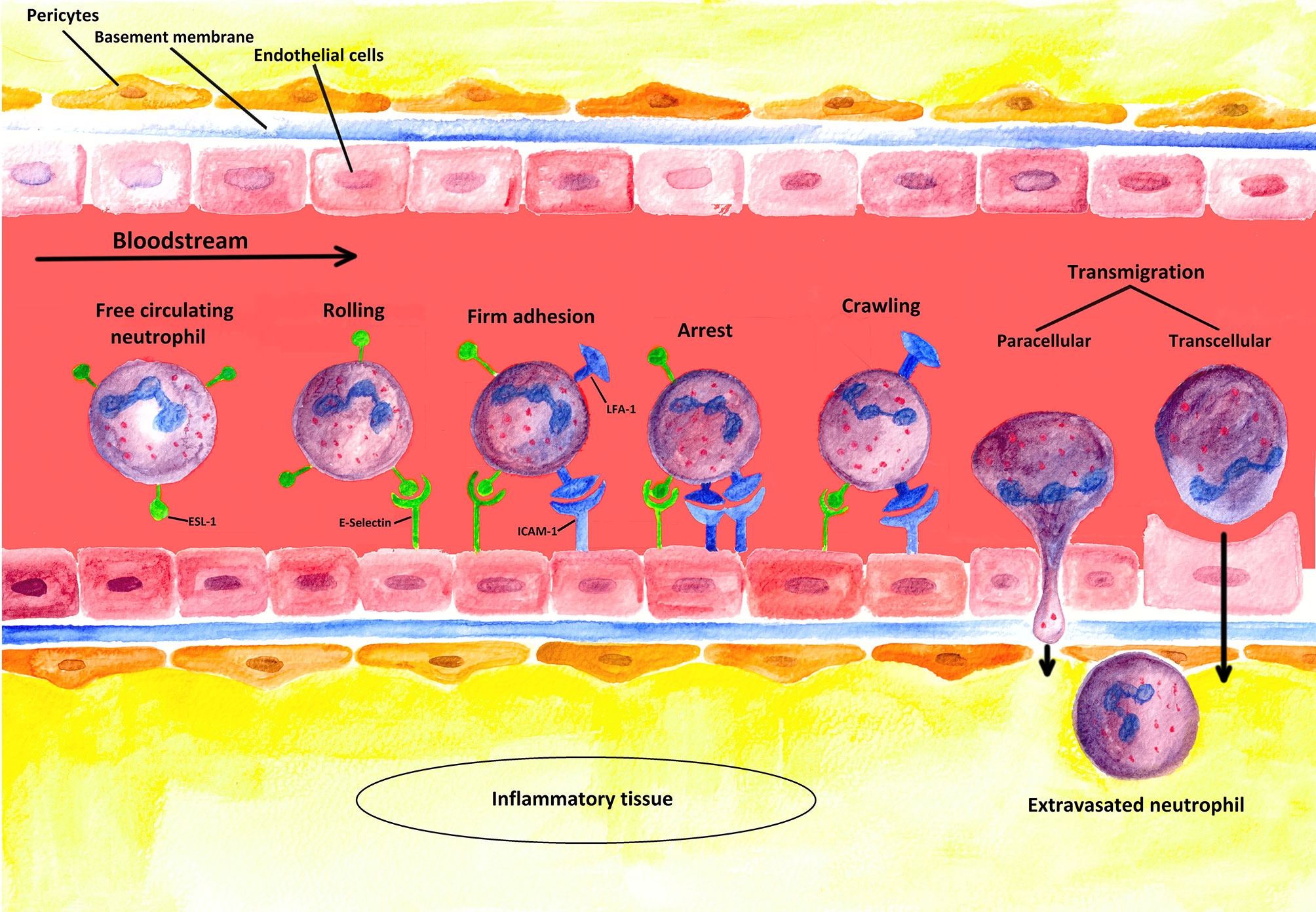
Figure 3 Schematic illustration of the extravasation process: PMNs leaving a blood vessel through the endothelium. The first step of the multi-stage process is the weak binding of PMNs to the endothelium due to interactions between selectins induced on endothelial cells and their corresponding ligands on the PMNs. In this figure, the process is illustrated for E-selectin and its ligand ESL-1 [containing sialyl-Lewisx-unit (s-Lex)]. However, such binding is not strong enough to resist the shear forces of the blood flow, so that new bondages are continuously formed and released again (rolling). Stronger interactions are only induced, however, when a chemokine (such as CXCL-8) binds to its specific receptor (not shown) on the neutrophil cell, which triggers the activation of the integrins LFA-1 and CR-3 (Mac-1) (firm adhesion). To induce the expression of adhesion molecules [such as ICAM-1 (ligand of LFA-1)] on the endothelium, inflammation-specific cytokines such as TNF-α are additionally required. Strong binding between ICAMs and integrins terminates rolling (arrest) and allows PMNs to squeeze between the endothelial cells (paracellular transmigration); yet, a transcellular way of transmigration is also possible as described in the literature. The neutrophil cell then crosses the basement membrane with the help of matrix metalloproteinases (like MMP-9), which are expressed on the neutrophil cell surface. Finally, the extravasated PMN migrates along a concentration gradient of chemokines secreted by cells at the sites of infection in the interstitium (4, 32).
The initial action of PMN extravasation is the activation and upregulation of adhesion molecules in the endothelium situated in close proximity to inflammatory tissue (37). As described, such activation can be mediator-induced. However, the endothelium itself can also recognize PAMPs and DAMPs via its own PRRs (4). The decisive point is that adhesion molecules are upregulated in both ways, mediator-induced and endothelium-induced. This process is crucial for initiating the recruitment of neutrophils (89).
The most important adhesion molecules for the recruitment process are P- and E-selectins. P−selectins are physiologically stored in the Weibel-Palade bodies in dormant endothelial cells and in α-Granula in platelets. When activated, P−selectins can be immediately relocated to the apical cell membrane. E-selectins, however, are de novo synthesized, and appear on the endothelial surface within 90 min (4, 92). Having reached the endothelial surface, the selectins bind to the adhesive ligands present on the PMNs (89). Both selectins bind to the sialyl-LewisX unit, an oligosaccharide present on the cell surface protein of circulating PMNs (38). E−selectin preferentially binds to E-selectin ligand-1 (ESL-1), whereas P-selectin mainly binds to P-selectin glycoprotein ligand-1 (PSGL−1, CD162). Both ligands have sialyl-LewisX units (93).
Owing to the Fåhraeus-Lindqvist effect, cellular components are usually located in the center of small blood vessels, in which flow velocity is at its maximum. In inflammation foci, blood vessels are dilated. The resulting lower flow velocity enables PMNs to interact more easily with the endothelial surface by the mechanism just described. As a consequence, PMNs that are freely circulating in the blood stream become attached to the endothelial surface. This first interaction of P- and E-selectins with their ligands (see above), however, cannot anchor the cells against the shear forces of the blood stream. Subsequently, the cells “roll” by reversible binding along the endothelium by constantly making and breaking contact with the endothelium and the cells (for details see chapter 3) (38, 89, 94, 95).
As a next step, G-protein-coupled receptors on the “rolling” granulocytes bind to PMN attractants secreted on the apical membrane (for details, see chapter 3 and 5) (89). By this binding, an “inside-out signal” is transmitted to the PMNs, which causes conformational changes in the PMN surface proteins termed β2-integrins. Of particular importance here are the two continuously expressed β2-integrins lymphocyte function-associated antigen 1 (LFA-1) and macrophage antigen 1 (MAC-1; alternative name: complement receptor 3, CR3) (4).
LFA-1 und MAC-1 interact with the intercellular adhesion molecules ICAM-1 and ICAM−2 on the endothelial surface (4). Usually, LFA-1 und MAC−1 bind their ligands only weakly (38). Due to the conformational change, however, LFA-1 und MAC-1 bind very firmly to the ICAMs, causing the end of the “rolling” and firm adhesion of the PMNs to the endothelium (“arrest”) (38).
Besides G-protein signaling, the conformational activation of LFA-1 required for neutrophil arrest can be induced by selectin engagement (96). On the one hand, E-selectin binding to its ligands on PMNs supports slow rolling and facilitates activation of high-affinity β2-integrins. Thereby, bond formation with ICAMs leads to PMN arrest on inflamed endothelium (97–101). On the other hand, a study published by Morikis et al. in 2017 showed the important role of L−selectin in transitioning neutrophils from rolling to arrest, which led to a paradigm shift in understanding mechanosignaling of human PMNs during recruitment (97, 102). E-selectin ligation on L-selectin and PSGL-1 receptors induces their redistribution into membrane clusters (97, 98). The PSGL-1/L-selectin complex signals through Src family kinases, ITAM domain–containing adaptor proteins, and other kinases, which ultimately results in LFA-1 activation (96).
It is noteworthy that, in neutrophil arrest, G-protein-coupled and selectin-mediated outside-in signaling can effectively amplify the number of high-affinity β2-integrins (103). Thus, cooperation of both signaling ways is temporally required for regulating the number and affinity state of β2-integrins (97).
After neutrophil “arrest”, PMNs actively “crawl” to suitable endothelial passageways (“crawling”) (4). Such “crawling” is based on the firm binding of MAC−1 to ICAM-1 (104). These bindings maintain adhesion to the endothelial surface at all times, thus enabling the PMNs to “crawl” perpendicularly along the endothelium or against the blood flow under the shear conditions of the blood stream until they reach the preferential site of transmigration or a passageway (4, 104, 105).
To finally leave the blood vessels, PMNs must first pass through the endothelium (transmigration). Suitable passageways are located at the cell-to-cell junctions between endothelial cells. Of particular importance during paracellular transmigration through the endothelium is the binding of the integrins LFA-1 und MAC-1 to the cellular adhesion molecules ICAM−1 and ICAM−2 or to the vascular cell adhesion protein 1 (VCAM−1). However, other adhesive interactions involving junctional proteins such as the platelet endothelial cell adhesion molecule-1 (PECAM1; alternative name CD31) are also important. All these interactions finally allow PMNs to force their way through the endothelium (4, 38).
However, neutrophil recruitment does not follow this classical cascade in every organ and may sometimes require organ-specific mechanisms (4, 106, 107). For instance, neutrophils recruited into an inflamed liver appear to lack rolling and to adhere directly due to the interaction of CD44 on PMNs and hyaluronan (HA) on liver sinusoidal endothelium (106–109). In the lungs, PMNs mainly exit vessels at the alveolar capillary level; thus, PMN rolling is unlikely and seems to be replaced by mechanical sequestration because cytokine-induced cytoskeleton-dependent PMN stiffening due to F-actin polymerization provokes dramatic slowing of PMNs within narrow-caliber capillaries (110). Moreover, neutrophil recruitment in the brain seems to depend on the presence of platelets adhering to the endothelium and building a “bridge” between the endothelium and the PMNs (4, 111). Table 2 shows receptors and corresponding ligands that are important for the neutrophil extravasation process.
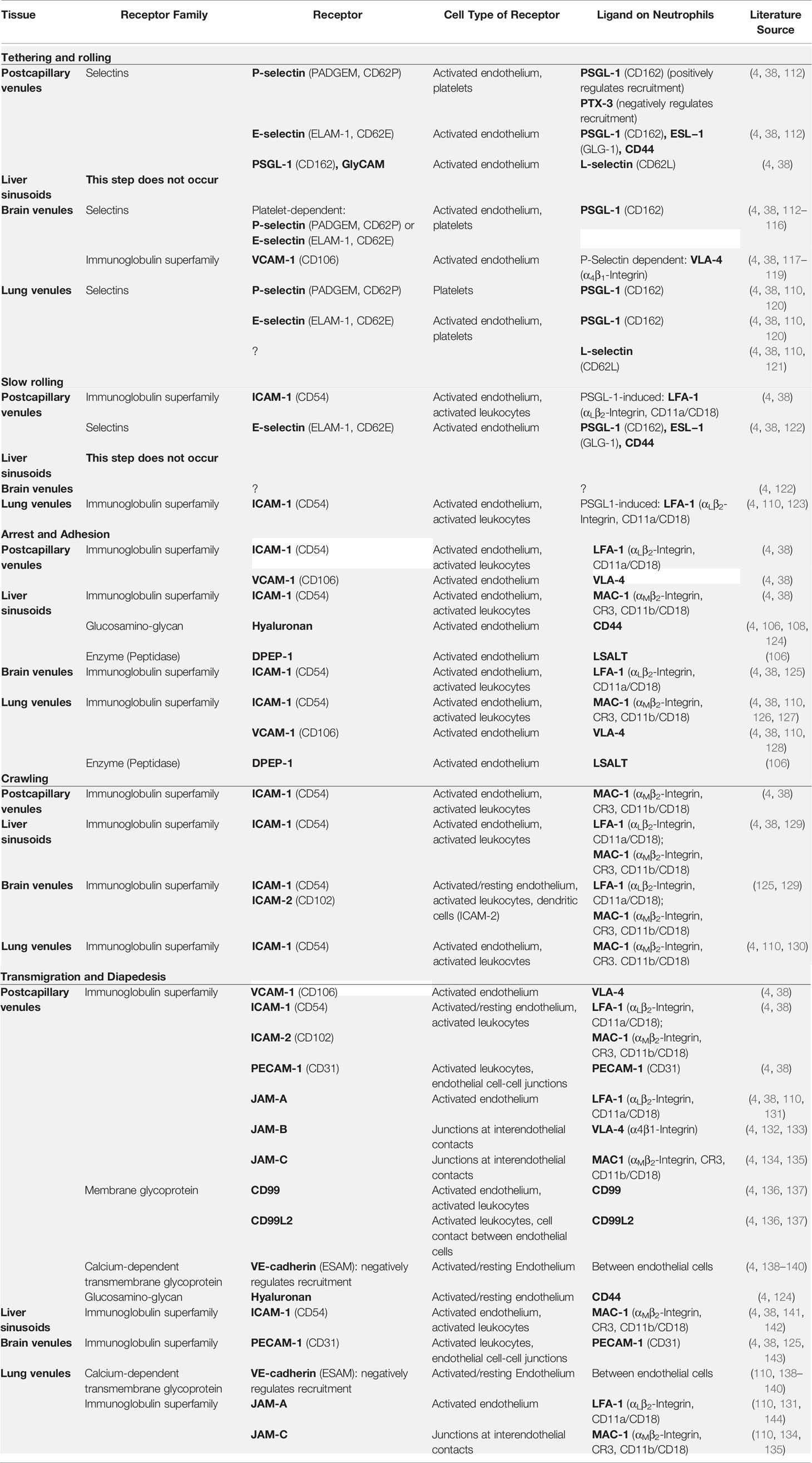
Table 2 Important receptors and corresponding ligands involving neutrophil adhesion and signaling..
However, PMNs can also get past the endothelial layer on the transcellular pathway through endothelial cells. To what extent the endothelium actively participates in this transmigration has not yet been fully elucidated. The current assumption is that—at this point in the transendothelial migration process—, the endothelium actively participates by inducing actin-rich structures that surround transmigrating leukocytes, which extend dorsally in some cases (146, 147).
During extravasation, PMNs penetrate the vascular endothelial lining, which requires opening of the endothelial barrier. Remarkably, this process does not necessarily cause any plasma leakage. The questions how endothelial cells form transmigration areas through which PMNs can migrate and what mechanisms are behind the ability of the endothelium to prevent leakage and maintain integrity while numerous leukocytes are penetrating are still under investigation. Platelets docking to von Willebrand factor seem to be essential for closing endothelial gaps induced by transmigrating neutrophils through stimulating the angiopoietin receptor Tie-2 (148–150). Nevertheless, paracellular and transcellular pathways do coexist, but current data are contradictory in terms of which pathway is preferred by PMNs (151).
When PMNs reach the end of the endothelial cell layer, they must overcome the endothelial basement membrane, a passage termed diapedesis (38). The basement membrane is a continuous structure consisting of proteins of the extracellular matrix (ECM proteins) such as collagen (mainly collagen IV) and laminin. Neutrophils possess specific proteases with enzymatic activity against ECM proteins, which include matrix metalloproteases such as MMP-9 and serine proteases such as neutrophil elastase. Although one may easily conclude that PMNs “cut” their way through the basement membrane, this process has not been conclusively proven yet. Even if histological examination did not show any rupture of the basement membrane in inflammatory tissue, it is nevertheless currently assumed that PMNs preferentially migrate through areas of the basement membrane that have a low content of ECM molecules (<60% as compared to otherwise dense areas). Thereby, MMPs seem to provide assistance in the process (4).
After overcoming the basement membrane, PMNs subsequently migrate through the pericytic region before reaching the interstitium. Pericytes are cells that wrap around endothelial cells, thus forming an interface between the circulating blood and the interstitial space. Interestingly, gaps in pericytic regions overlap with regions with lower basement membrane density. In the extravasation process, PMNs are therefore assumed to choose the path of least resistance when migrating to the interstitium (4).
3 Neutrophil Endothelial Adhesion and Rolling Mechanisms
In contrast to most leucocytes, which—for the most part—are only able to roll along the walls of venules at low shear stress, neutrophils have the ability to roll at a 10-fold higher shear stress level (152). Although the mechanisms are not yet completely understood, four potential mechanisms have been identified that enable neutrophils to roll at high shear stress of the bloodstream: cell flattening, catch bond behavior, membrane tethers, and slings (see Figure 4) (153).
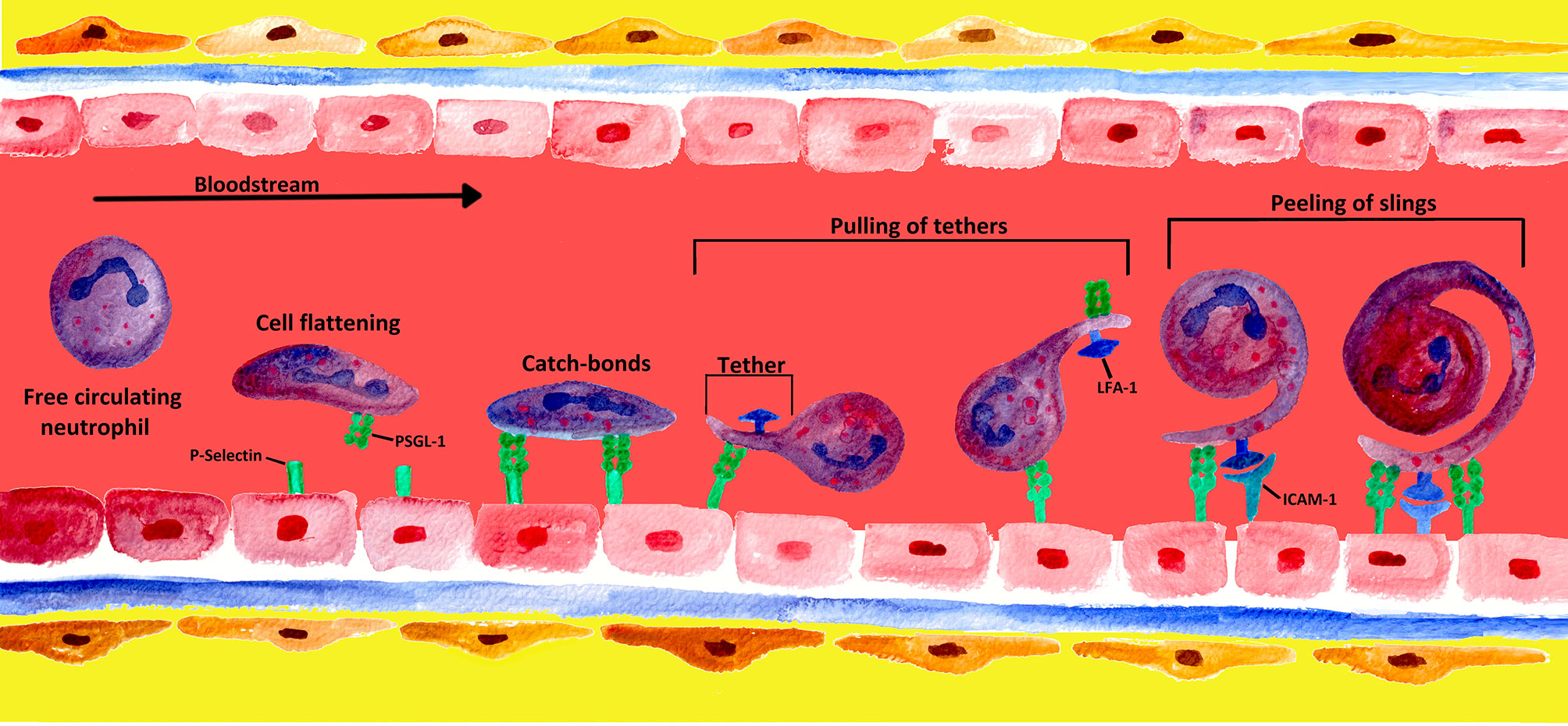
Figure 4 Mechanisms of formation and engagement of tethers and slings by rolling neutrophils. Rolling neutrophils experience high shear stress in the blood stream and have to overcome tensile stretch due to rolling. When PMNs converge into a blood vessel wall, the shear stress of the blood leads to cell flattening. PMNs limit stress forces during the rolling process by adhesive bonds generated at the front and disrupted at the rear of the PMN (4). At low detachment forces, these adhesive P-selectin-PSGL-1 bonds behave like catch bonds. With increasing force, the bonds become stronger, and long membrane bonds called tethers are created at the rear of the PMN (153, 154). The tethers bind to endothelial P−selectin via PSGL-1, forming temporary anchorage points that are subsequently disconnected from the endothelium by the pulling of tethers (4, 155). Once the tethers break at the rear of the rolling PMN, they swing forward and wrap around the cell as a sling, thereby decelerating the PMN. On slings, multiple patches along the whole projection are formed via the binding of PSGL-1 to the endothelium. This sequential attachment and pulling apart is referred to as the “step-wise peeling of slings”. The final deceleration and arrest of the cell results from the interaction of neutrophil LFA-1 with endothelial ICAM-2, leading to an even tighter wrapping of the sling around the cell body (4, 123).
At high shear stress in post-capillary venules, rolling neutrophils deform into a tear drop shape and undergo flattening. This process is the result of elongation in the flow direction imposed by the hydrodynamic drag acting on the rolling cell. On the one hand, this process decreases cell height and subsequently reduces the hydrodynamic drag experienced by the rolling cell. On the other hand, as the cell flattens, the contact area or cell footprint on the vessel wall is increased, raising the probability of P-selectin-PSGL-1 bond formation (153, 156–159).
Neutrophils rolling along the vascular endothelium are mainly a result of rapid formation and dissociation of P-selectin-PSGL-1 bonds at the center and rear of the rolling cell, which balance the hydrodynamic drag of the blood stream (152). P-selectin-PSGL-1 bonds behave like catch bonds at small detachment forces and thus become stronger with increasing force (153, 154).
Nevertheless, PSGL-1 does not only bind to P-selectin but is also one of the major ligands of L-selectin (CD62L) (160, 161). Unlike E- and P-selectin, which are expressed on activated endothelium (see chapter 2), L-selectin is the only selectin constitutively expressed at the tips of microvilli on PMNs (162, 163). L-selectin undergoes split second changes in bond lifetime with its ligand (most likely PSGL-1 with sialyl-LewisX unit) under flow conditions, classified into catch and slip bonds (102). Interestingly, L-selectin on human neutrophils is loaded itself with sialyl-LewisX, whereby it can be recognized by E-selectin (164). Therefore, L−selectin is one of the first neutrophil adhesion molecules to be in contact with the endothelium under flow conditions (165).
During early adhesion, initial contact between the calcium dependent (C-type) lectin domain (CTLD) of neutrophil L-selectin and its ligand exerts low tenacity, which starts at the leading edge of the cell (slip bond). At low shear stress (<0.3 dyn/cm2), slip bonds usually last less than a second, and their lifetimes are shortened by force (102, 154, 166). As shear stress rises up to an optimum level (∼1.0 dyn/cm2), the tenacity between the CTLD and its ligand increases to unfold. Increasing force prolongs bond lifetimes (catch bonds), now located at the trailing end of the cell. Above the optimum level of shear stress, force shortens bond lifetimes (slip bonds); when the tenacity exceeds the limit for catch bonds, bond lifetime decreases, and CTLD and its ligand separate again (102, 154, 166, 167). Under conditions of abundant ligand availability, a new catch bond will form at the new leading edge to repeat the process, culminating in classic cell rolling behavior (102).
In contrast to E- and P-selectin, L-selectin is rapidly cleaved from the cell surface in response to cellular activation, inflammatory stimuli, and mechanical force, a process termed “shedding” (162, 168, 169). Although many issues regarding the physiologic role of shedding remain unanswered, this process seems to play a key role in regulating neutrophil rolling and adhesion dynamics mediated by L-selectin-ligand interactions (168–171).
In summary, increasing force leads to triphasic (slip-catch-slip) behavior of selectin-ligand interactions and lifetimes (154, 166, 167, 172–174). In vitro and in silico studies have shown that such catch-behaving bonds stabilize neutrophil rolling at low shear stresses of less than 1 dyn/cm2 (160, 175–178). Nevertheless, no study has yet analyzed the role of catch-behaving bonds in facilitating neutrophil rolling at shear stresses higher than 6 dyn/cm2 (153).
Moreover, neutrophils rolling at high shear stresses form membrane tethers, which can be longer than the cell diameter and promote the survival of P-selectin-PSGL-1 bonds (153). Membrane tethers are nano-tubes extruded from the lipid bilayer membranes of blood cells. These tethers are formed when a microvillus on the surface of a neutrophil is pulled with force that is increased over time (179). Sundd et al. showed that such long membrane tethers, attached to the substrate via highly strained P-selectin-PSGL-1 bonds, contribute to the catch-bond behavior of the system (159, 180). Such bonds tend to increase their lifetime in response to the pulling force, thus allowing the tethers to stay attached to the P-selectin substrate for a longer time and to grow in length (153).
As mentioned above, blood flow imposes a hydrodynamic drag on the rolling cell, enabling the cell to move forward and to also rotate like a ball along the vessel wall. To continue rolling, the cell needs to at least partially balance both the forward and the rotating components of the hydrodynamic drag (180). Membrane tethers in cooperation with the catch bond phenomenon extend under pulling force and appear as “slings” at the front of the rolling cells (153, 180).
According to Sundd et al., neutrophil rolling at shear stresses of 6–10 dyn/cm2 is facilitated by slings, which are cell-autonomous adhesive structures extended at the front of rolling neutrophils. As the cell rolls over the sling laid in front of it, the sling starts to wrap around the rolling cell, which undergoes a step-wise peeling process at the rear of the cell due to the tandem failure of PSGL-1 patches under the hydrodynamic drag (180). When a PSGL-1 patch on a peeling sling fails, the cell tries to jump forward, but only for a short distance until the next patch downstream of the first patch on the same sling becomes loadbearing (180). This step-wise peeling distinguishes a tether from a sling because unlike the failure of a sling, failure of a tether is catastrophic as there are no other bonds available that can keep the tether attached to the substrate; thus, the cell accelerates forward (153).
The patchy distribution of PSGL-1 along each sling provides a unique adhesive substrate once the cell rolls over the sling. As each PSGL-1 patch fails, a new patch is already lined up that now becomes loadbearing. This step-wise peeling makes slings even more effective than tethers in slowing down rolling neutrophils (180).
Unlike PSGL-1, LFA-1 is expressed all over the neutrophil surface and the entire length of the slings (180). Although, LFA-1-ICAM-1 bonds have been shown to behave as catch-like bonds at small bonding forces, there is no report of such behavior of LFA-1-ICAM-2 bonds (181). Besides stabilizing, rolling slings are unique structures that also enable rolling neutrophils to present LFA-1 to their ligand ICAM-2. However, catch-like LFA-1-ICAM-2 interactions will probably result in even tighter wrapping of slings at smaller bonding forces compared to slip bonds. Eventually, the long tethers detach from the substrate and transform into slings, which stabilize rolling by undergoing a step-wise peeling process (180). As rolling progresses, the number of slings increases, which may explain the well-known phenomenon of rolling to become more stable over time (180, 182).
Interestingly, circulating PMNs can not only tether and role on the endothelium but also on adherent leukocytes. This process is termed “secondary tethering and rolling” and is enabled by interaction of the sialyl-LewisX unit of PSGL-1 with L-selectin. Secondary tethering extends PMN recruitment when endothelial cell-derived ligands are already masked by adherent leukocytes (151, 165, 173, 183). By promoting primary tethering and rolling, PSGL−1/L-selectin may contribute to chronic inflammation (165, 184–186).
Taken together, catch bonds, long tethers, cell flattening, and slings act together and contribute to the forces balancing the hydrodynamic drag, which may explain why neutrophils can roll even at very high shear stress as observed in acute inflammation in vivo (153, 180). How the synergy between the four mechanisms leads to stable rolling and whether catch-behaving bonds are responsible for formation of long tethers and slings are topics that need to be investigated further (153).
4 Migration in the Interstitium of the Target Tissue
After completing transendothelial migration, PMNs reach their target tissue. Once arrived at their target, PMNs migrate through the inflammatory interstitium along a chemokine gradient (see chapter 5) to reach their final destination (187). Depending on the site of the damage, PMNs can encounter very different tissues, such as fibrillar networks, cell-rich environments of an organ parenchyma, or lymphatic tissues (188). To be able to migrate through different tissues, PMNs preferentially use an amoeboid mode of locomotion, which is characterized by smooth and fast migration (189).
Crucial for this mode of locomotion is active cell body deformation. In PMNs, the intracellular forces of such deformation are almost exclusively generated by the actin-myosin cytoskeleton and characterized by the alternation of an intracellular network extension by actin polymerization followed by network contraction through actin-myosin. On the one hand, this contractility generates hydrostatic pressure on the rear side, which compresses cytoplasmic material and pushes it forward. On the other hand, adhesions at the rear edge of the cells are released (188). To move the cell, the cytoskeletal forces must be transferred to the ECM. The transfer can be integrin-mediated by the weak interaction of adhesion molecules, whereby, in contrast to the dominant participation of β2-integrins in the extravasation process, the interstitial migration process seems to be mainly associated with the activation of β1−integrins (91, 190, 191).
In addition, Nourshargh et al. described that, when integrin receptors are missing or unable to bind to the substrate, PMNs could also physically interact with the extracellular environment and thus achieve force transmission. The authors postulated that the possibility to use both modes of locomotion, i.e. integrin-dependent and integrin-independent locomotion, enables PMNs to migrate through a wide variety of different interstitial tissues (188).
Accordingly, Wolf et al. considered every single migration step in the interstitium as adaptive in response to cell-intrinsic signals and extracellular chemical and mechanical signals (regulation of adhesion, cytoskeletal dynamics, proteolysis, forming of the cell body, or geometry of the ECM) (192).
Friedl et al. summarized cell migration within the interstitial tissue as a complex mechano-chemical process that requires the interaction of key processes of the signaling, cytoskeletal, membrane, and adhesive systems (193).
5 Current Knowledge and Controversies of the Influence of the Extra-Cellular Matrix on the Function of Neutrophils
The mode of locomotion in the interstitium significantly differs from the mode of locomotion during extravasation. In the latter, cells remain firmly integrated into a tissue context by cell-to-cell or cell-to-ECM adhesion (188). The amoeboid mode of locomotion in the interstitium, however, is characterized by the absence of such strong adhesive interactions (194). Although intravascular events and transmigration through the endothelium have been comprehensively examined in numerous studies, comparatively relatively little interest has been paid to the steps after the extravasation cascade. As a consequence, the mechanisms regulating the passage through the interstitium are less well characterized (190). Cell-matrix interaction is not completely clarified (188), and little is known about the adhesive interactions determining the motility of migrating leukocytes in the interstitium (190).
The question whether and to what extent PMNs use extracellular conditions as guidance structures is not finally answered (188). Furthermore, the literature contains different and sometimes contradictory information as to whether the composition of the ECM influences the functions of granulocytes: Nourshargh et al. reported in their review that the amoeboid locomotion of leukocytes and thus also that of PMNs is independent of the composition of the extracellular environment (188). In contrast, in reference to a study by van Goethem et al. in which the migration of macrophages was influenced by ECM conditions, Jennings et al. postulated that PMNs adapt their mode of locomotion to the composition of the ECM. Jennings et al. assumed that the ECM environment encountered by PMNs determines the input of β1- or β2-integrins as well as the actin polymerization and the myosin-II-driven forces of the locomotion behavior (195).
Burns et al. gave an overview of how the adhesive properties of different ECM elements influence both the direction and speed of leukocyte movement. In addition, the authors described integrin-mediated adherence to the ECM as being very important for PMN locomotion towards inflammatory sites (91).
Relying on previous studies by Cox and Huttenlocher (196), Lindbom et al. endorsed the concept that repeated cycles of temporary adhesion to and detachment from matrix structures are necessary for the effective motility of PMNs. Integrins seem to be crucial here in so far that they establish contact with matrix molecules, thus enabling locomotion by acting as an anchor for the filaments of the cytoskeleton. Of course, there is also integrin-independent PMN migration (196), but such adhesion-independent mechanisms are far from being able to achieve significant effectiveness of the locomotor system under physiological conditions (190, 196). Furthermore, Lindbom et al. stated that the chemotaxis of granulocytes is also influenced by the relative frequency of matrix proteins within the tissue. Conditions in the extracellular environment can increase the binding strength between integrins and their ligands, thus antagonizing motility (190). Kuntz et al. assumed that migration must depend on adhesion to the ECM (197).
The previous findings on the influence of the ECM on PMN migration can be summarized insofar that the migration patterns determined by the ECM seem to be more modulated rather than strictly determined (188). The idea that the ECM can have a structural function by serving as a barrier or scaffold for cells infiltrating inflamed tissue is per se easy to imagine. Besides the influence on the migration of immune cells, Sorokin et al. also reported an influence of the ECM on the inflammation of tissues. As mentioned above, by binding chemokines in a spatially structured and regulated manner, the ECM can integrate and deliver multiple complex signals to leukocytes, which influences their behavior in inflammatory tissues (198). In line with this fact, we observed in a recent study that the ECM does not only impact neutrophil migration but also neutrophil immunological functions, such as ROS production, MPO release, and NETosis (199). Moreover, chemokines in inflammatory tissues increase both the tissue turnover and protease secretion of tissue-resident cells (198). Several publications—inter alia by Houghton et al. and by Ospelt and Gay—suggested that such aberrantly expressed ECM molecules can influence the activation, differentiation, and survival of immune cells (200, 201). Gaggar et al. described that the release of MMP8 and MMP9 by PMNs during an inflammation breaks down collagen into bioactive ECM fragments, which in turn have chemotactic activity (202). Weathington et al. provided evidence of such chemotactic activity in an in vivo lung inflammation model, thus proving the physiological relevance of ECM influence on inflammation and in particular on the function of PMNs (203). Nissen et al. demonstrated that, in the presence of bioactive fragments of collagen, PMNs produce less ROS and reduce their interstitial velocity of migration in vitro. Above all, Nissen et al. asserted in an in vivo asthma model that the same bioactive fragments selectively inhibit the accumulation of PMNs in lung interstitium, thereby proving the (patho−)physiological relevance of ECM influence on inflammation and especially on PMN functionality (204).
In the last 5 years, increased interest in the interaction of neutrophils with their surrounding ECM has advanced in vivo research in this matter. Both the presence of ECM and the interplay between neutrophils and their ECM are now considered as a vital mechanistic aspect of inflammation. There is mounting evidence of complex interactions between ECM macromolecules and PMN (reviewed recently by Zhu et al.). We now know that the close relationship between ECM and PMNs plays an important role in the progression of various diseases in humans (205). Recent studies for example demonstrated an important role for ECM in fighting against infectious diseases by mediating an antifungal response of PMNs (205–207). Moreover, experimental studies have shown that released NETs cleave fibronectin via NE and MMP-9 to further degrade ECM in alveoli, thereby promoting the development of bronchopulmonary dysplasia (205, 208). In addition, the interactions of PMN with ECM play a fundamental role in inflammatory conditions of many organs like myocardial injury and pulmonary diseases (205, 209, 210). Furthermore, there is a growing evidence that neutrophil invasion into tumor ECM is associated with cancer progression and subsequent metastatic dissemination (for details see 12 Neutrophil Behavior in Cancer Environment and Tumor Tissue) (205, 211–214). Nevertheless, ECM-neutrophil interactions do have the potential for treatment options of PMN-associated diseases. However, gaps remain in understanding the regulatory role of ECM in determining neutrophil function. Hence, future studies are required to fill the gaps and decover underlying mechanisms, which could be used to treat patients with PMN associated diseases (205).
6 Chemotactic Signal Transduction of Migration
To be able to perform their functions adequately, PMNs must know the exact location of the lesion focus. Therefore, the targeted guidance of PMNs from the reservoirs through the vascular system to the affected tissue is of crucial importance (89). For this effective response, PMNs can detect extracellular chemotactic concentration gradients and move up the gradients towards higher concentrations. This process is referred to as chemotaxis (215, 216).
Neutrophil chemotaxis is characterized by three different processes: gradient detection, polarization, and cell motility (217). PMNs have receptors for chemokines and chemo-attractants, such as the endogenous molecules C5a, LTB4, and CXCL8 released in the course of an inflammatory response, but also for exogenous molecules such as the peptide N−formylmethionine-leucyl-phenylalanine (fMLP) released by bacteria. The receptors are linked to G-protein-receptor signaling pathways, which provide “outside-in” signals. These signals induce PMNs to undergo polarization of their cell form. This process results in the formation of a front end (“leading edge”) and a rear end (“uropod”). At the same time, neutrophil integrins are activated for targeted cell (trans)migration (see chapter 4), enabling PMNs to move intra- and extravascularly with their “front edge” in the direction of the higher concentration of the gradient (88).
However, the exact mechanism underlying the navigation in the complex lymphoid or inflammatory target tissues is not yet fully understood (88). Early studies by Foxman et al. assumed that chemotactic migration is based on a multi-stage process (218). An advanced model of this step-by-step migration developed by Heit et al. described the hierarchy of chemo-attractants. PMNs prioritize chemotactic signals by distinguishing “intermediary” (LTB4, CXCL8, and PAF) and “end-target” chemo-attractants (fMLP and C5a) with significantly different intracellular signaling pathways. Thus, PMNs are able to avoid “distraction” in a complex environment of chemo-attractants and move to the lesion site in a targeted manner (219).
7 Formation of Chemotactic Gradients in Interaction With the Extracellular Matrix
The original concept of the chemotaxis of cells was described as directional migration heading for a concentration gradient of soluble chemo-attractants. Later, the gradient of chemo-attractants was found to be generally determined by the binding and immobilization of these chemical signals to a substrate. The concept of haptotaxis was introduced, denoting directional cell movement induced by a gradient of structure-bound adhesion sites or signal molecules (216, 220–222).
Within the vascular compartment, chemo-attractants are immobilized by glycosaminoglycans (GAGs) or heparan sulfate, mainly on the luminal membrane of endothelial cells (223–227). Outside vessels, chemo-attractants can bind to the ECM, thus directing the migration of neutrophils to lesion foci (216). On their migration path, PMNs come into contact with two different basic forms of the ECM: On the one hand, with basement membranes consisting of thin networks of tightly interconnected glycoproteins, and, on the other hand, by meeting loose fibril-like interstitial matrices after transmigration (198). As described above, the basement membrane consists of the four main components collagen IV, laminin, nidogen, and heparan sulfate as well as of the proteoglycan perlecan. With the exception of the CNS, the interstitial matrix in most tissues is composed of fibrillae that mainly contain collagen of types I, III, V, and XI (198). In addition, specialized ECM structures exist that combine the properties of both basement membrane and interstitial matrix. These structures form the reticular fiber network of the secondary lymphatic organs and share properties with the provisional matrix formed at injury sites (198). The negative charge of many ECM molecules, in particular of proteoglycans, and the large surface they occupy in the tissue offers a large potential for interactions with other charged molecules such as chemokines (198). Thus, chemo-attractants can bind to the proteoglycans of the ECM, thereby directing the migration of neutrophils to lesion foci (216).
8 The Role of Microtubules and the Microtubule Organizing Center in the Migration of Neutrophils
As postmitotic cells, neutrophils are not able to undergo mitosis. To run their function in host defense, PMNs are not reliant on the mitotic machinery. The advantage of this minor microtubule architecture in combination with the segmented nucleus may enable high cellular flexibility, which facilitates PMNs to migrate more rapidly than other leukocytes and to infiltrate many different and even dense tissues because of their high morphological dynamics (228). Microtubules (MTs) are known to be substantially involved in intracellular transport. However, the role of MTs in chemotactic PMN migration and neutrophil effector functions is far from being resolved (229).
Whereas resting PMNs contain few MTs, which are gathered in the Microtubule Organizing Center (MTOC, Centrosome) behind the neutrophil multilobular nucleus, PMNs prolongate their MTs within minutes in response to in vitro stimulation with chemotactic peptides (such as CXCL-1 or fMLP), as confirmed by Yadav et al. in 2019 (229). Anderson et al. did not report any changes in the number of MTs per neutrophil granulocyte after in vitro chemotactic stimulation but a significant increase in the average length of MTs. Thereby, MTs in the direction of migration were lengthened, whereas MTs perpendicular to the direction of migration were shortened (230).
To establish and maintain the necessary cell polarity for amoeboid locomotion (see above), small, rapidly moving cells (as PMNs are) perform actin- and myosin II-dependent reorientation of the MTs array toward their uropod. According to a theory by Eddy et al., such reorientation and compacting accumulation of the MTs into the uropod could make cells more streamlined. Thus, polarization seems to be alleviated, and cell motility could be maximized to two- or three-dimensional matrices. To further elaborate this theory, reorientation of MTs could supply positional information, which would serve to reinforce cell polarity during migration (231).
The change between spontaneous neutrophil locomotion (chemokinesis) and chemotaxis does not seem to involve any changes in the collocation of the microtubule cytoskeleton itself (228). In most migrating cells—including PMNs—, cell polarity is rather characterized by the position of the nuclear-centrosome-axis (NC-axis) in relation to the front-back-axis of the cell. According to Luxton et al., in case of chemotaxing neutrophils in contrast to mesenchymal cells, this NC-axis is oriented in posterior direction. Thus, the neutrophil nucleus is located directly at the leading edge of the cell, and the MTOC is arranged behind the nucleus (232).
Hence, amoeboid PMN migration in vitro seems to be consequently characterized by the fact that the nucleus is located ahead and in front of the MTOC in the direction of PMN migration (“nucleus-first-configuration”) (233). Chiplonkar et al. reported that in resting PMNs, the MTOC takes a “predefined” apical location, and only upon chemotactic stimulation in vitro do they translocate to a newly defined basal location, if microtubules are intact (234). Anderson et al. reported that MTs spread almost exclusively from a single MTOC after stimulation. In contrast, Schliwa et al. described the transient separation of the centrosome into two single centrioles surrounded by an aster of MTs after PMN stimulation. Schliwa et al. further explicated that 10% of the cells with separated centrosome had a third centriole free aster consisting of microtubules with compactly accumulated seeds (230, 235).
To be able to choose the path of least resistance during migration (see above), PMNs ‘palpate’ their immediate environment. Renkawitz et al. accounted the nucleus-first-configuration as a type of “measure instrument”, possibly enabling the differentiation of the extent of PMNs surrounding pores. In this way, the bulkiest part of the neutrophil, the nucleus, is used as a mechanical gauge and acts as a selector of the migratory direction (233).
In the context of the observed close proximity between nucleus and MTOC and the proof that polarization is determined by the MTOC in other cellular systems, Renkawitz et al. made this assumption as a conclusion of their migration experiments. In these in vitro experiments, the MTOC predominantly located itself in between nuclear lobes when cells moved through straight channels. At narrow pores (“decision points”), the neutrophil nucleus unfolded; initially, one nuclear lobe passed preferably the largest pore, before the MTOC and the other nuclear lobes ultimately followed. The MTOC quasi specified the nuclear lobe, resulting in the choice of direction (233). Renkawitz et al. also made the observation that after the nucleus and the attached MTOC had completely overcome the largest pore, cytoplasmatic protuberances located in smaller pores were retightened. This step was coordinated by dynamic MTs, whereby migrating cells loose integrity and fragments in fluid cytoplasmatic pieces in the case of MT rupture (233).
In 2019, a study by Yadav et al. showed that drug-controlled suppression of MT polymerization, which in turn was triggered by chemotactic peptides (for instance, CXCL1 and fMLP), inhibited neutrophil chemotactic migration in vitro. This suppression disabled CXCL1- and fMLP-triggered elastase-dependent neutrophil traverse through collagen I hurdles (229). Interestingly, CXCL1-regulated transendothelial migration did not depend on MT polymerization in vitro, since the break of existing or de novo generated MTs did neither impair protrusion not squeezing through IL-1β stimulated endothelium in vitro (229).
Despite the in vitro findings described above, we still do not know how microtubules regulate PMN migration in vivo (236). In contrast to in vitro studies, an in vivo zebrafish study by Yoo et al. showed the MTOC in migrating PMNs in front of the nucleus. MT depolymerization inhibited the activity of polarized Phosphoinositol-3-Kinase (PI3K) at the leading cell edge and activated fast PI3K-independent motility. MTs seem to exert their effects on neutrophil polarity and motility in vivo, at least partly, via negative regulation of both Rho- and Rac-activity (236).
In view of the discrepancies between the in vitro und in vivo findings, the current state of research is as follows: de novo chemoattractant-triggered MT polymerization seems to be the key to neutrophil chemotaxis and elastase-dependent infiltration into tissue but does not seem to be responsible for chemotactic overcoming of the inflammatory endothelial barrier (229).
To conclude, further experiments are required to uncover the discrepancies between in vitro and in vivo insights and to gain better knowledge about the true role of MTs in neutrophil chemotactic migration and host defense.
9 Bidirectional and Reverse Migration of Neutrophils
Perseverance of neutrophils in tissues may result in tissue damage and chronic inflammation as outlined in chapter 11. Therefore, PMNs must be cleared away from the injury site after fulfilling their duty, whereby such neutrophil clearance from affected tissues is crucial to induce a pro-resolution cascade (as reviewed in (237, 238)). Until a few years ago, the predominating dogma of neutrophil clearance after recruitment to tissue was that PMNs undergo apoptosis before they are cleared by macrophages via efferocytosis (see chapter 1) (239, 240).
However, a number of questions regarding neutrophil clearance remain undetermined. In various models of sterile inflammation, PMNs infiltrated tissues and disappeared long before the presence of monocytes. Furthermore, in these models, depletion of monocytes or macrophages did not compromise neutrophil removal (237, 241).
An often underappreciated or perhaps ignored issue in the past was whether transmigrated leukocytes can leave inflammatory sites and perhaps even return across the endothelium and re-enter circulation (239). The mechanisms of unidirectional migration of neutrophils through the endothelium into tissues have been extensively investigated; migration from tissues back in the opposite direction, however, has attracted the attention of the scientific world only in recent years (90, 242). In the past two decades, several notable studies have shown that PMNs are able to undergo bi-directional movement and can move in the direction opposite to the direction that was to be expected, a process termed “reverse migration” (243–245). Figure 5 gives a graphical overview of the different forms of neutrophil migration.
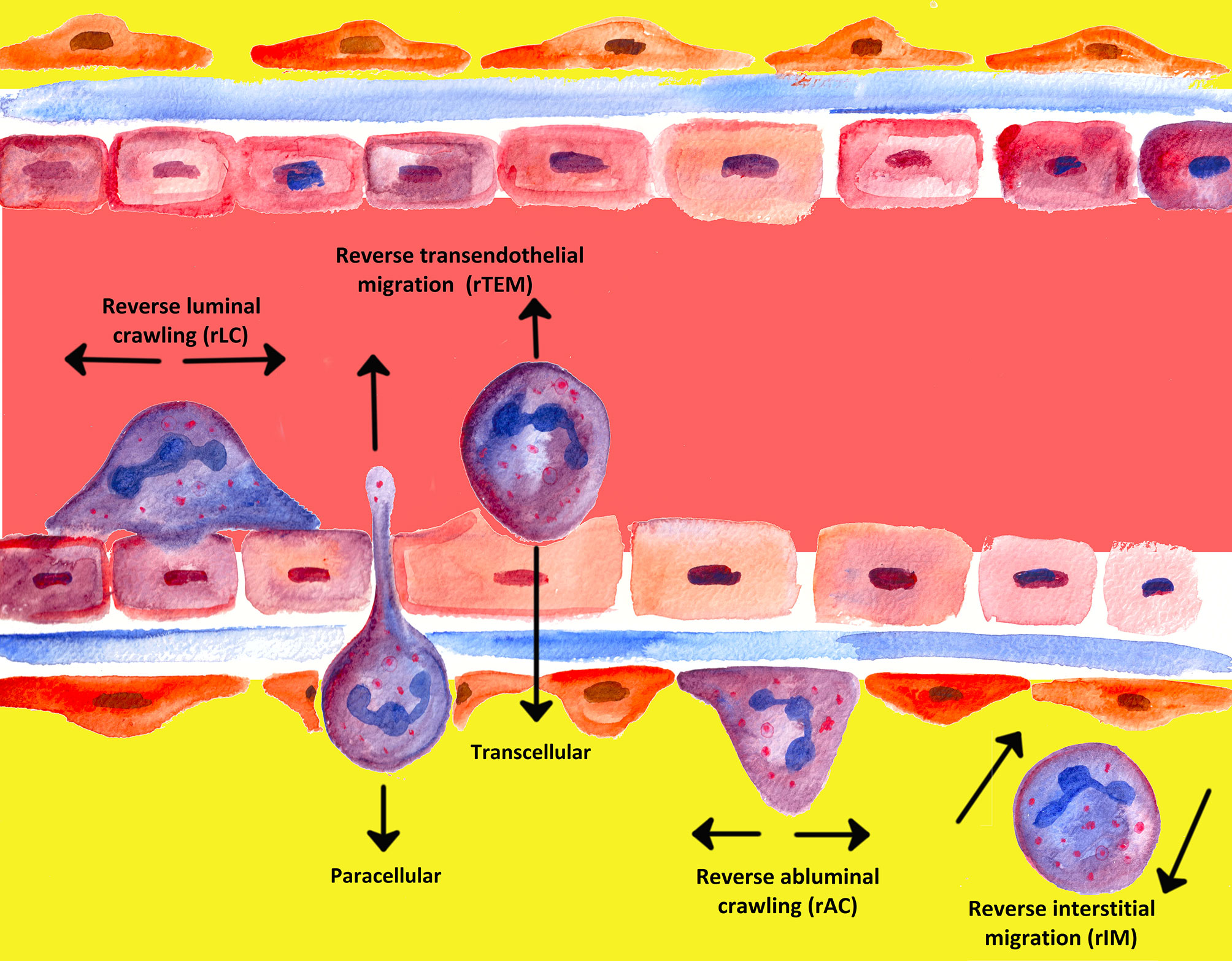
Figure 5 Overview of the different types of migration known for PMNs so far.
In 1997, Hughes et al. were the first to report in a rat model of glomerular capillary injury that neutrophils can migrate bi-directionally during inflammation (242, 246). Since then, various types of neutrophil reverse migration have been described (243, 245). As outlined by Nourshargh et al., a number of different modes of reverse migration are assumed to (co−)exist, each with its own signal mechanisms and subsequent cell effects (239, 245).
Meanwhile, different denotations have been introduced in the literature for various phenomena of reverse migration, depending on the site. Neutrophil transmigration through endothelial layers in abluminal to luminal direction was denoted as reverse transendothelial migration (rTEM), whereby migration of neutrophils away from inflammatory foci in interstitial tissues was termed reverse interstitial migration (rIM). Besides, reverse abluminal crawling (rAC) constitutes reverse migration of neutrophils in pericyte layers (104, 243–245, 247–249).
The first in vivo evidence of reverse transmigration was provided by Mathias et al. in 2006, who demonstrated neutrophils migrating away from a wound back to the vasculature (237, 248). The authors used a genetically engineered zebrafish model, in which neutrophils could be observed by real-time visualization within larvae (239, 248). In the zebrafish model, as many as 80% of PMNs recruited to the injured site migrated back towards the vasculature, whereby some of the cells went even back to circulation; merely 3% of invading neutrophils underwent apoptosis at the site of injury (239, 250, 251). Nevertheless, in zebrafish models, the outcome of PMNs returning to the endothelium has not been fully clarified yet (239).
In 2006, bidirectional movement of human neutrophils through endothelial monolayers was detected by Buckley et al. (252). The authors also described PMNs that displayed reverse transmigration had an altered cell surface phenotype, in which they expressed high levels of ICAM-1 and downregulated expression of the chemokine receptor CXCR1. In this context, it is striking that patients with systemic inflammation show increased levels of this PMN population (ICAM-1high/CXCR1low) in peripheral blood (239, 252).
In 2011, neutrophil reverse transmigration was also live-imaged in ischemia/reperfusion injury in mouse models (244, 253). Conducting confocal intravital microscopy in mice, Woodfin et al. observed that nearly 10% of transendothelial migration events were reversely migrating PMNs. This finding differed considerably from observations in in vivo zebrafish experiments, in which almost all wound responsive neutrophils had migrated reversely (244, 248, 254).
Tharp et al. found that increased levels of cytokine-induced neutrophil chemoattractant-1 [CINC-1, a rat orthologue of human CXCL-8] ultimately result in PMN movement in opposite direction towards venular walls, implicating the concentration of chemo-attractants in one of the major determinants for rTEM regulation (242, 255). Indeed, the process of rTEM seems to depend on the capability of PMNs degrading the junctional adhesion molecule C (JAM-C) by proteolysis (237, 244). As shown by Bradfield et al., JAM-C regulates the unidirectional migration of leucocytes and is ubiquitously expressed on endothelial cells (133, 256). Due to the fact that blockade or genetic deletion of endothelial JAM-C increased neutrophil rTEM, JAM-C was considered an important regulator of rTEM by Woodfin et al. and Zindel et al. (244, 257).
Furthermore, Colom et al. showed that neutrophil elastase was essential for promoting TEM by degrading JAM-C in mice (237, 258). Moreover, LTB4 also seems to influence the regulation of rTEM via JAM-C because the application of LTB4, which was observed to enhance the degradation of JAM-C between endothelial cells, increased rTEM in mice. Conversely, in mice pretreated with an LTB4 receptor antagonist, JAM-C expression persisted and neutrophil transmigration decreased (239, 258).
In 2014, Tauzin et al. described the interaction of macrophages with PMN-stimulated neutrophil reverse migration via redox-Src family kinase (SFK) signaling, which mediates migration in neutrophils in response to oxidative stress as a redox sensing element (239, 242, 250). Thus, SFK signaling may remove invaded neutrophils to help mitigate neutrophil-mediated inflammation of wounds in zebrafish (242). Neutrophils have been shown to not necessarily require contact with macrophages or monocytes to set up reverse migration. Nevertheless, in the absence of macrophages, the number of recruited neutrophils undergoing reverse migration was significantly decreased (239).
Another factor influencing neutrophil clearance from the site of injury is (de-)stabilization of hypoxia-inducible factor-1a (HIF-1a). Elks et al. demonstrated delayed neutrophil clearance as a consequence of genetic or pharmacologic stabilization of HIF-1a activity. Furthermore, HIF-1a supported inflammation by decelerating neutrophil apoptosis through inhibiting prolyl hydroxylase activity. Burn et al. did not view this decrease in reverse migration as a consequence of overall reduced migration but rather as a change in directionality. This view lead to the suggestion that signaling pathways exist that normally drive PMNs away from the site of initial recruitment (239, 251).
In 2017, Wang et al. described a neutrophil reverse migration cascade from the interstitium backwards using a model of focal hepatic sterile injury (237, 238, 257). The authors observed that PMNs initially performed important repair functions in the interstitial space before migrating back to the bloodstream, whereby PMNs at the injury border showed directional movement away from the lesion (237, 238). After PMNs had entered the bloodstream, they stopped in the lung capillaries, in which CXCR4 was upregulated, which in turn enabled the PMNs to ultimately return to the bone marrow. This process was followed by neutrophil apoptosis and clearance (237, 238, 257). Interestingly, mice deficient in cathepsin C (and thereby unable to activate several proteases) showed normal numbers of neutrophils migrating to the site of injury but fewer neutrophils leaving the lesion, which disrupted the normal revascularization process (237, 238).
Strikingly, CXCR4high neutrophils (“aged neutrophils”) performed reinforced NET formation under inflammatory conditions as asserted by Zhang et al. (259). Moreover, rTEM neutrophils (with phenotype ICAM-1high) showed enhanced ability to produce ROS, which in turn is required for NET production (242, 252, 260). These observations led to the assumption that rTEM neutrophils tend to exhibit exceeding NET formation, which—apart from killing invading pathogens—may have negative effects such as tissue injury or disproportionate coagulation during inflammation (242, 260–262).
In 2019, a study on patients with acute ischemic stroke by Weisenburger-Lile et al. determined an increased percentage of neutrophils with a reverse transendothelial migration (ICAM-1highCXCR1low) phenotype and continuous basal hyperactivation of circulating neutrophils. Importantly, these neutrophil alterations were associated with the clinical severity of the stroke (263). Moreover, Lohri et al. showed that medical interventions can also affect human rTEM neutrophils: After adjuvant chemotherapy in patients with breast cancer, the number of reverse transmigrating (ICAM-1high/CXCR1low) human neutrophils had decreased significantly (264). These studies highlight not only the diversity of diseases and treatments affecting human rTEM neutrophils but also contribute to the in vivo importance of reversely migrating neutrophils and outline the desideratum for a better understanding of proceedings involving reverse neutrophil migration.
On the one hand, reverse migration of neutrophils leads to PMN removing from the lesion site and resolution of local inflammation. On the other hand, reversely migrating neutrophils that re-enter the bloodstream may disperse into different parts of the body by circulation. Taking this hypothesis further, reversely migrating PMNs may transmigrate into other—initially non-inflammatory—organs again, thus contributing to accessory organ injuries and systemic inflammation (242).
Indeed, Yoo et al. observed in a zebrafish model that reversely transmigrating PMNs tended to distribute in tissues throughout the body (242, 250). Similarly, Woodfin et al. found PMNs with phenotype ICAM-1high within pulmonary vasculature after lower-limb ischemic/reperfusion injury in mice. Because of a significant association between the frequency of ICAM1high neutrophils in pulmonary vasculature of ischemic/reperfusion stimulated mice and the extent of lung inflammation, Woodfin et al. assumed an association of neutrophil rTEM with inflammation in a second organ (242, 244). According to Colom et al., increased JAM-C levels in plasma (as an indirect marker of neutrophil rTEM) correlated significantly with consecutive severity of multiple organ failure in trauma patients (242, 258). Based on these observations, Colom et al. stated that tissue-experienced neutrophils returning to circulation may contribute to propagated systemic inflammation (213, 250, 258).
However, the idea that reverse PMN transmigration promote systemic inflammation after an episode of localized tissue inflammation is controversial. Downregulation of the chemokine receptor CXCR1 (CXCR1low-phenotype) and thus the inability of reversely transmigrating PMNs to transmigrate again across inflamed endothelium make it seem unlikely that such PMNs have the capability to reinfiltrate tissue at inflammatory sites (242, 252). Moreover, it is hardly possible to identify rTEM neutrophils by means of upregulated ICAM-1 because ICAM−1 is also upregulated after long-term PMN stimulation by bacterial lipopolysaccharide or cytokines such as TNF- α, as shown by Wang et al. (242, 265).
Previous research mostly focused on the process of reverse migration as a whole (sometimes by reason of investigation methods); in the past few years, however, more attention has been paid to distinguish the different sections of reverse neutrophil migration. Recently, the importance of distinguishing rTEM from reverse interstitial migration (rIM) has been underlined by Nourshargh et al. (245). In contrast to rTEM, rIM constitutes a relatively new field of investigation, which describes movements away from the foci of inflammation within tissues, whereby rIM does not necessarily involve re-entry into circulation via the endothelium (239, 245).
So far, a possible connection between these two modes of reverse locomotion has not been examined. The two modes may exist as two separated and autonomous phenomena. Yet, rTEM may also be the continuation of rIM so that PMNs moving from inflammatory foci within tissues (rIM) are able to undergo rTEM after rIM (239). Besides, another purpose of rIM may be the transport of captured antigens to lymph nodes for the initiation of adaptive immune responses as contemplated by Nicolás-Ávila et al. and Maletto et al. (213, 266).
In conclusion, many questions in the field of neutrophil reverse migration remain unanswered. Although recent studies have indicated that neutrophil reverse migration can be physiological as well as pathological, the true (patho-)physiological role of neutrophil reverse migration has not yet been fully elucidated (239, 243). The question which mechanisms and signals are required for establishing reverse migration versus apoptosis also needs to be further investigated (237). Nonetheless, it is noteworthy at this point that both, reverse migration and efferocytosis of PMNs, are not mutually exclusive; in fact, both processes may be necessary for appropriate resolution of tissue inflammation (257).
Even though a few phenotypic markers of rTEM neutrophils have been identified, the specific molecular mechanisms underlying neutrophil reverse migration are far from being completely understood (242). Ultimately, it is indispensable for neutrophils to be removed from the lesion site either by apoptosis or by reverse migration, since the failure to remove neutrophils may lead to disrepair and chronic inflammation (237).
10 The Immune Effects of Neutrophils at the Site of Action
After arriving at the sites of action, PMNs have different first line immune defense strategies at their disposal (see Figure 6). First, PMNs ensue a form of receptor-mediated endocytosis termed phagocytosis (267). The two main targets of elimination by phagocytosis are foreign particles (pathogens) and “altered self cells”, whereby the term “altered self cell” typically corresponds to apoptotic and necrotic (host-)cells (267–269). In wounds, PMNs also remove dead tissue by phagocytosis, thus preparing the wound for the formation and deposition of new tissue (270). PMNs are professional phagocytes. A single PMN can kill up to 50 individual bacteria. Moreover, neutrophil phagocytosis is a rapid process, which can be completed in just a few seconds (267, 271).
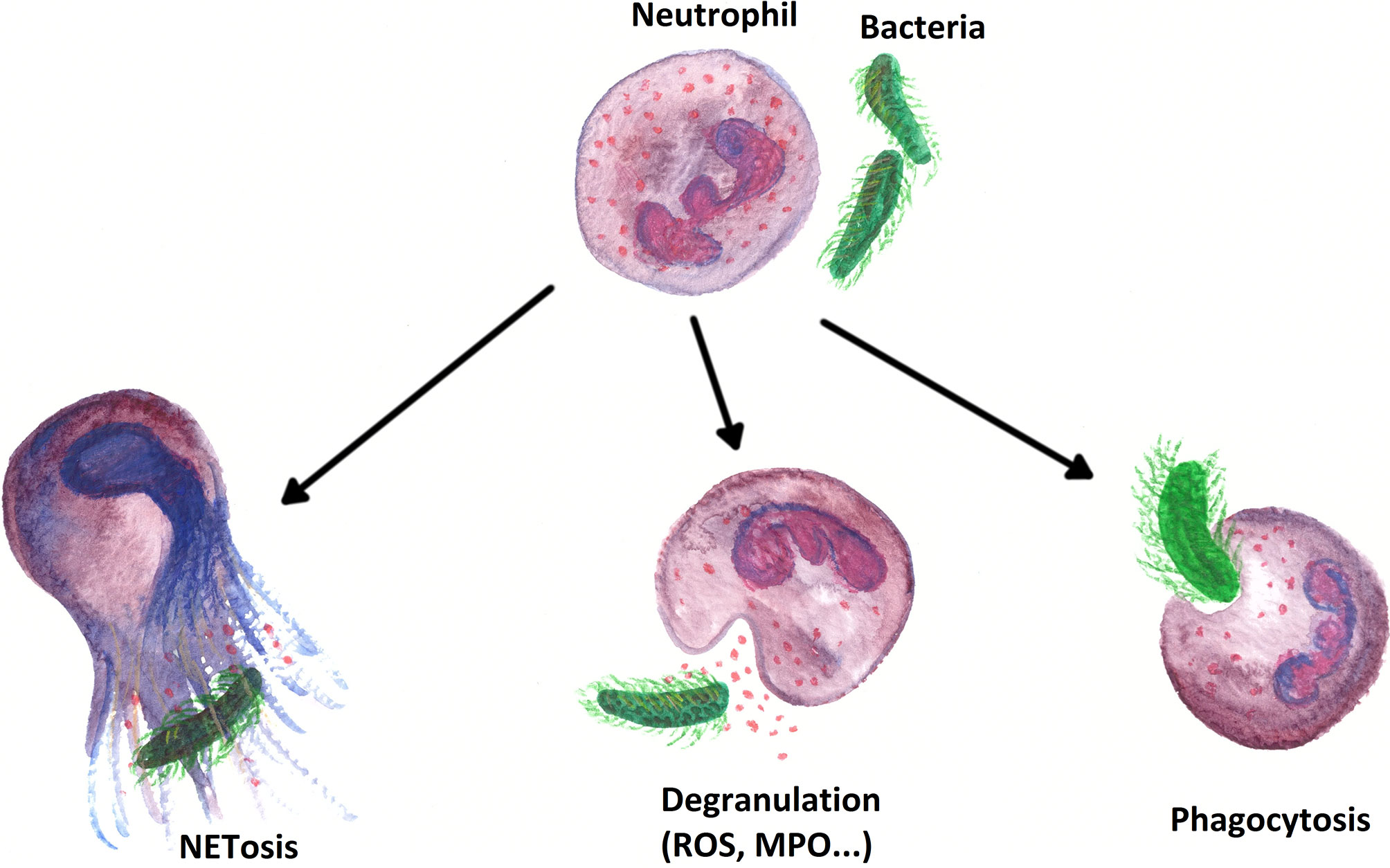
Figure 6 Overview of the most important immune effects PMNs perform within the first line defense of the innate immune system.
Neutrophil phagocytosis involves a diversity of receptors and starts with the recognition of the target, namely the binding of a phagocytic receptor to its correspondent ligand (267, 269). Receptors on PMN surfaces are capable of recognizing phagocytic determinants that are intrinsic to pathogens (i.e., PAMPs) and classified by the C-type lectins Dectin-1 (which binds to β-glucan) and Dectin-2 (which is able to bind to a variety of ligands on the surface of mycobacteria, fungi, and even cancer cells) (267, 272–274). Receptors that detect eat-me-signals of “altered self cells” bind directly to phosphatidylserine (PS) or PS-binding bridging proteins, altered sugars (recognized by lectins), or thrombospondin (267, 275).
Although recognition of PAMPs can trigger phagocytosis, microbial engulfment is at its optimum when targets are “marked” as foreign cells by being coated with distinct serum components that can be detected by effective phagocytic receptors. This process of “labeling” certain microorganisms by antibodies and the complement system are known as opsonization (267). The most important opsonins in serum are immunoglobulins and certain components of the complement cascade (reviewed in (267)); opsonins are recognized by both Fc receptors (FcRs) and complement receptors (CRs) (reviewed in (276)) (267, 277).
The binding of multivalent ligands to the surface of the target leads to the clustering of receptors on the PMN and—after various intermediate steps—to the recruitment of GTPases of the Rho family (278, 279).
The following signal cascade results in the actin-dependent formation of a phagocytic cup and the elongation of pseudopodia around the ligand. Finally, the target is ingested into a vacuole—the phagosome—that is completely internalized into the neutrophil cell. The phagosome undergoes extensive remodeling to increase its hostile mechanisms against pathogenic particles. This process is known as maturation, by which internalized particles are moved into a series of soaring acidified membrane-bound structures, culminating in particle degradation and elimination of the ingested microorganisms (267, 280). Although some bacteria have developed strategies to survive phagocytosis, it should be mentioned at this point that phagocytosis does not inevitably lead to the destruction of all microbes. Staphylococcus aureus, for example, impedes phagocytosis on itself by complement inhibitors but can also escape intracellular destruction by enzymes such as superoxide dismutase (281, 282). Nevertheless, neutrophil phagocytosis is an effective first line defense within the innate immune system.
Video 1 was provided by Franz Reichelt (Laboratory of anesthesiology, University Medical Center Regensburg) and shows the process of phagocytosis as described above by means of in vitro phagocytosis of Escherichia coli (stained red) by PMN. The experimental assay shown is a chemotactic experiment according to Doblinger et al.; in this experiment, an fMLP-chemotactic gradient was built in a 3D collagen matrix, in which PMNs were embedded, enabling them to move and mediate their immune effects along the gradient (283).
In addition to the phagocytosis of pathogens, PMNs use two fundamentally different mechanisms for the defense against infectious pathogens: oxygen-dependent and oxygen-independent mechanisms (284, 285).
As oxygen-dependent mechanism, the formation of reactive oxygen species (ROS) should be mentioned in particular (285). In the context of a process termed “respiratory burst reaction”, phagocytizing PMNs show a strong increase in their oxygen consumption. This increase is caused by the NADPH-dependent production of superoxide anions , which are the trigger that leads to the formation of ROS, i.e. to the formation of hydrogen peroxide (H2O2), hydroxyl radical (OH•), and hypochlorous acid (HOCl). These acids contribute to the destruction of bacteria (286, 287). In Video 2, the process of ROS production is illustrated showing fluorescence images of an in vitro chemotaxis experiment with human PMNs (199): Human PMNs were embedded in a type I collagen matrix and exposed to an fMLP gradient. ROS production was visualized using 1,2,3-dihydrorhodamine (DHR). The red glowing signal around the cells indicates an ongoing ROS production in the videos.
As oxygen-independent mechanism, the degranulation of histologically visible granules is of importance because of its release of lytic enzymes and bactericidal peptides (284). Cytoplasmic granules are characteristic for neutrophils (which belong to the granulocyte family) and instrumental in microbicidal response. These granules can be subdivided into three dissimilar classes based on the contents of their matrix and their integral membrane proteins: azurophilic (primary) granules, specific (secondary) granules, and gelatinase (tertiary) granules (267, 281). Primary granules contain antimicrobial substances, such as lytic enzymes and antimicrobial peptides, and include defensins and myeloperoxidase (MPO). Secondary granules contain phagocytic receptors (e.g., Fc receptors and CRs; see above) and the NADPH oxidase complex (see above). Tertiary granules contain receptors and enzymes that degrade ECM to facilitate the extravasation process and the migration of neutrophils to the site of inflammation (see The Process of Extravasation) (267, 288). Taken together, degranulation results in the release of lytic enzymes and bactericidal peptides, procuring an effective host defense against microbial pathogens.
However, one granule component that plays a special role in oxygen-independent defense mechanisms is the enzyme myeloperoxidase (MPO). In the presence of H2O2 and chloride anions (Cl-), MPO catalyzes the formation of reactive oxygen intermediates including HOCl, which destroys cell membranes and cell walls. Besides the antimicrobial effect of the MPO/HOCl system, MPO has proved to be a local mediator of tissue damage and the resulting inflammation in various inflammatory diseases (289). Video 3 shows the release of neutrophil MPO in an chemotaxis experiment, where PMN were embedded in a type I collagen matrix and exposed to an fMLP-gradient. In this experiment MPO was made visible by ANTI-MPO-APC anti-body staining, so that the green signal in the video near the cells indicates just released MPO (199).
In 2004, Brinkmann et al. (290) described another, previously unknown ability of PMNs: At the end of a cytolytic process, the nucleus of PMNs is released as a net-like DNA structure into the extracellular space (284). These neutrophil extracellular traps (NETs) have histones and cationic peptides on their surface (284). Once released, NETs can surround, immobilize, and finally kill both bacteria and fungi. The phenomenon of NET release mainly occurs in inflammation foci and is referred to as NETosis (290). In Video 4 NETosis was visualized in an in-vitro-chemotaxis experiment with human PMNs (199). The PMNs were embedded in a type III collagen Matrix and exposed to an fMLP gradient. NETosis was visualized was assessed with 4´,6-diamidino-2-phenylindole. In the beginning, PMNs migrate along an fMLP gradient within the matrix, whereby they produce ROS (visualized by DHR red signal). With increasing experimental time, neutrophil migration stopped and the cells underwent NETosis. Thereby, the blue signal in the videos indicates PMNs undergoing NETosis.
Three models of NETosis have been described so far.
First, the best described model is suicidal NETosis with a duration of 2–4 h (291). NETosis begins with the activation of neutrophils through the recognition of stimuli (PMA or fMLP, among others), leading PMNs to stimulate the NADPH oxidase complex through protein kinase C (PKC), Raf, MERK, and MAPK/ERK signaling (292, 293). Furthermore, the activation of peptidyl arginine deiminase 4 (PAD4)-dependent citrullination of histones induces the decondensation of chromatin (292, 294–296). Suicidal NETosis is dependent on ROS for the disintegration of the nuclear membrane and for histone citrullination by PAD4 (295–297). Suicidal NETosis also depends on elastase and MPO transport from granules to the nucleus (298). Ultimately, pores in the ruptured plasma membrane allow the liberation of NETs, leading to cell death and the loss of viable cell functions (292, 294, 299–301).
The second model is vital NETosis, during which PMNs release NETs without destructing the plasma membrane or the nucleus. This type of NETosis lasts about 5–60 min and consists of the release of nuclear DNA through nuclear shell growth and vesicle release, decondensation of the nucleus, and nuclear shell disruption (291, 302–304). Vital NETosis is promoted by activation of TLRs and complement receptor for C3 protein (292, 305–307). Moreover, interaction between platelet glycoprotein Ib with β2-integrin may induce NET formation by activating ERK, PI3K, and Src kinases (292, 308). Neutrophils undergoing vital NETosis are still able to run phagocytosis with preservation of chemotaxis (294, 309, 310), allowing the coexistence of NET forming and conventional host defense (292, 304).
Finally, a third type of NETosis was described by Yousefi et al. in 2009. In this subtype of vital NETosis, which is dependent on ROS, mitochondrial DNA is released instead of nuclear DNA. After recognition of complement C5a or lipopolysaccharide (LPS), mitochondrial NETs are released from 80% of neutrophils within 15 min (292, 311).
Even though great progress has already been made in this field in the past decade, the cellular mechanisms that mediate neutrophil NET release are still not fully explored (304).
The chronological sequence of neutrophil immune effects (migration, ROS production, MPO release and NETosis) is recognized in the current scientific literature and reflects the fact that ROS und MPO are released through degranulation first, whereas NETosis occurs last (260, 283, 312). While Figures 7A, B illustrates this sequence of immune function using images, Video 5 shows the processes using film techniques. In both media an in vitro chemotaxis experiment (like those in Videos 2–4) is presented in which PMNs were embedded in a matrix of type I collagen and exposed to an fMLP gradient. First, PMNs migrate along the gradient, whereby they produce ROS (red signal). With increasing experimental duration, migration decreases and the cells undergo NETosis one after another.
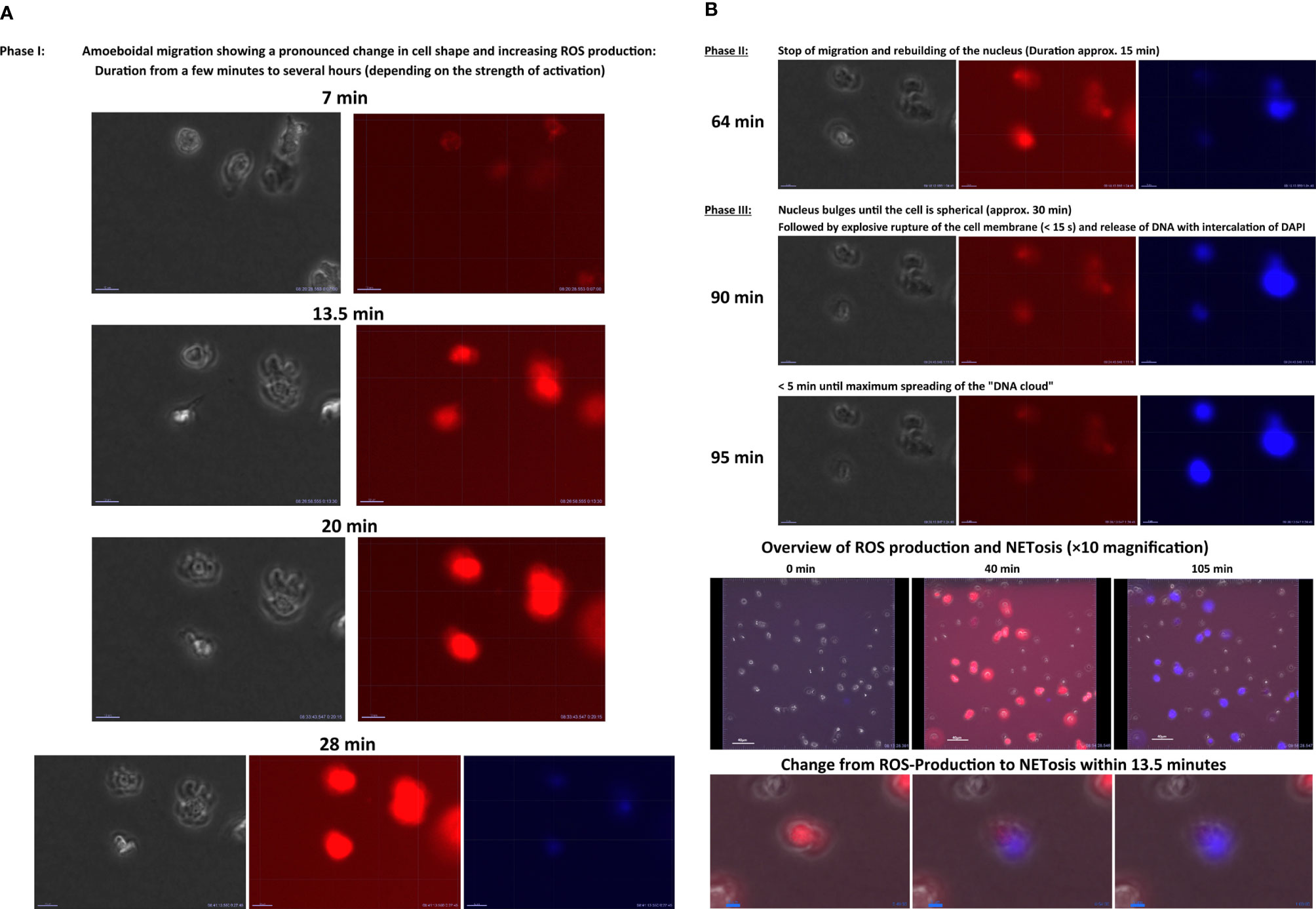
Figure 7 (A, B) Graphical presentation of the chronological sequence of the neutrophil immune effects ROS production and NETosis. Fluorescence images of an in vitro chemotaxis experiment with human PMNs: The cells were embedded in a type I collagen matrix and exposed to an fMLP gradient. ROS production was visualized using dihydrorhodamine 123 (red). NETosis was assessed with 4´,6-diamidino-2-phenylindole (DAPI, blue). The time points in the headlines of the images refer to the time of first gel contact. Overview of the sequence of neutrophil immune effects ROS production and NETosis in in-vitro-chemotaxix experiment (x40 magnification).
The relationship between neutrophil phagocytosis and NETosis is still controversially discussed. Some literature reports suggest that the two immune effects cannot coexist within one cell at the same time. Some researchers, such as Manfredi et al. and Branzk. et al., postulated that—after phagocytosis—PMNs can no longer perform NETosis and, conversely, PMNs perform NETosis if they are unable to phagocytose the pathogen (for example because of its size) (313, 314). This theory is based on results indicating that the presence of MPO and neutrophil elastase in the cytosol is a prerequisite of NETosis. These enzymes would be captured during phagocytosis and phagosome maturation; therefore, they would not be available in the cytosol and thus at the nucleus for NETosis any longer (314–316). Manfredi et al. concluded that neutrophils encountering a pathogen make an irrevocable decision to either phagocytose or to form NETs (313).
However, there are doubts if the only option for neutrophils is truly dichotomous and not a consecutive process from phagocyosis to NETosis. This assumption is supported by a publication by Ullah et al. showing pneumococcal-induced autophagocytosis as a promoter of NETosis (317). Furthermore, Pelletier et al. demonstrated by means of flow cytometry that PMNs, which have already phagocytosed subsequently went into NETosis (318). A matter of interest in this context is the observation that after the phagocytosis of certain bacteria with evolved mechanisms for escaping intracellular death (such as E. coli or S. aureus), PMNs released the previously internalized bacteria just before neutrophil cell dead was induced (319–323).
The above process is shown in Video 6, which was provided by Franz Reichelt (Laboratory of anesthesiology, UMC Regensburg). The experimental setup in Video 6 is equal to that in Video 1. The video shows a neutrophil cell that phagocytosed 6–8 individual bacteria of E. coli in vitro. Having obviously already committed to the defense function of phagocytosis, the neutrophil cell releases the bacteria and still undergoes NETosis afterwards. As the bacteria are shown to actively move in the video, they appear to be alive after their release. Thus, the bacteria seem to be still able to replicate and be capable of causing harm to the host (321, 323).
The inconsistencies just reflect the need for further elucidative work in the area of granulocyte functionality in contact with bacteria.
In this context, we introduce another (rather unknown) neutrophil immune function. In the presence of bacteria, PMNs develop dynamic, thin, and very long membrane tubules that are able to catch pathogens. These protrusions of the cytoskeleton and the cell membrane are referred to as tubulovesicular extensions (TVE), protrusions, or cytonemes (324, 325). Galkina et al. showed that cytonemes are able to bind and aggregate bacteria at a distance by telescopic exocytosis to release bactericidal molecules directly at the bound pathogen but not in their own vicinity (326, 327). The outstanding length of cytonemes—which can reach several cell diameters in length—allows PMNs to secrete targeted aggressive bactericides over a long distance without diluting or injuring the surrounding tissues (328). Cytonemes participate in neutrophil migration in an actin-dependent manner but seem to be dispensable for cell locomotion (325, 329).
Furthermore, cytonemes have been shown to execute long-range adhesion and binding objects for phagocytosis, such as serum-opsonized zymosan particles and erythrocytes (155).
In addition, Kornberg et al. suggested in 2014 that the main task of cytonemes was intercellular communication by means of receptor-ligand-interactions. By allowing signals to be transmitted from a source cell to target cells in a selective manner over a range of distances, cytonemes have recently emerged as a means of communication between cells in a highly specific manner (330–333). In recent years, the influence on the formation of cytonemes has been investigated. Cytonemes are assumed to be influenced by certain microbial substances such as the alkaloid staurosporine, Adenosine-A3 receptors agonists, and the presence of nitric oxide, which also seems to play a crucial role in the regulation of cytonemes (155, 334, 335).
Although or just because they are occasionally rather difficult to see with light microscopy due to their extremely small diameter, cytonemes are a wonderful example of the fascinating range of functions and peculiarities of PMNs.
Video 7 (provided by Franz Reichelt, UMC Regensburg) shows the formation of neutrophil cytonemes in an in vitro chemotaxis experiment, in which PMNs were embedded in a type I collagen matrix, exposed to an fMLP gradient, and placed next to E. coli. Phase-contrast exposure shows some kind of cell filament formation with long, thin protrusions suddenly spreading from the cell followed by their prompt regress in the middle of the screen. The process takes about 10 minutes and includes protrusions up to 110 µm in length. In this video, it is also worth to pay attention to the cell touched by the protrusions, which is followed by NETosis as indicated by blue DAPI signaling. In addition, it looks like cytonemes are also spreading from this second cell.
To sum up, cell migration, phagocytosis, oxidative burst, degranulation, and NETosis are some of the most important functional responses that enable PMNs to fulfil their tasks in immune defense (336).
11 Importance of a Balanced Immune Response of Neutrophils
The importance of a functioning PMN immune response can be above all seen in the severe courses of disease in which a disordered PMN immune response can lead to life-threatening infections, such as chronic granulomatosis, leukocyte adhesion deficiency, or all forms of neutropenia (337–339).
However, the mechanisms used by PMNs to kill microorganisms also have the potential to injure healthy tissue. Thus, excessive PMN response has a negative effect on the course of certain inflammatory diseases, such as acute respiratory distress syndrome (ARDS), cerebral apoplexy, acute coronary syndrome, or sepsis (340–342). Hereunto, it is important to point out that neutrophil NETs could have problematic effects under certain conditions, as most recently shown by studies of SARS-CoV-2 (343, 344). Furthermore, a number of autoimmune diseases are not directly caused by malfunctioning PMNs but indirectly by the significant contribution of PMNs to the pathogenesis of these diseases (345). Thus, an important influence, inter alia, on the autoimmune diseases systemic lupus erythematodes (SLE), rheumatoid arthritis (RA), or pyoderma gangrenosum (PG) is attributed to dysregulated PMN immune response (312, 346, 347). PMNs even appear to be involved in the pathogenesis of degenerative CNS diseases such as Alzheimer’s disease or multiple sclerosis (348–350). In the context of dysregulated PMN migration, it is worth mentioning that, in Alzheimer’s disease, LFA-1-mediated neutrophil transmigration through the blood-brain-barrier (BBB) may promote neutrophil inflammation within the brain together with amyloid deposits, leading to far-flung neutrophil-dependent CNS damage (348, 350). Besides autoimmune and degenerative diseases, neutrophil defense mechanisms also seem to have a destructive effect on the integrity of the BBB in infectious diseases. Thus, diseases with lesion sites primarily outside the CNS may suddenly involve the CNS, as observed in the cerebral manifestation of malaria (350, 351).
The important role of neutrophils in innate immunity, together with their tendency to cause tissue damage, requires the balanced and strict control of PMN activity (7).
12 Neutrophil Behavior in Cancer Environment and Tumor Tissue
Cancer is a chronic disease, which critically relies on the interplay of tumor cells with their supporting environment. Cancer presents with inflammation, and inflammatory response is an important factor for the development of tumors (213, 352). Compared to other immune cells, neutrophils have traditionally received little attention in this field, partly because their limited lifespan seems to tergiversate with the chronic nature of cancer. Experimental evidence generated in the past decade, however, supports a causal role for neutrophils in malignant transformation, tumor progression, antitumoral immunity, and angiogenesis (213, 353, 354).
It becomes more and more evident that tumor-associated neutrophils (TANs) and their myeloid precursors (peripheral neutrophils and granulocytic Myeloid Derived Suppressor Cells [G-MDSCs]) in bone marrow, spleen, and blood have an important role in cancer biology (353, 355). Although it is unlikely that immune suppression is their only biological function (as noted by Coffelt et al.), the term G-MDSC is used to indicate the immunosuppressive pro-tumoral properties of this heterogeneous group of cells of myeloid origin, including neutrophils (212, 356).
A transcriptome study by Fridlender et al. showed that TANs are not “tissue-based G−MDSCs” modulated by the tumor micro-environment (TME) but are a different population of neutrophils from both bone marrow-derived neutrophils and G-MDSCs (353, 355). The spleen is known as the site of TAN precursor localization, from which they physically relocate to tumor stroma, whereby CXCL8 (IL-8) is mainly responsible for the recruitment of TANs (353, 357). The make-up of the myeloid compartment in tumor stroma seems to be determined by the TME rather than by the anatomic site of tumor development or tumor-derived circulating factors (358).
Extravasation from blood into a tumor is a regulated multistep process involving a series of coordinated interactions between PMNs and endothelial cells. This process is partly different from non-tumorous situations. A cytokine-endothelium cross-talk is the first step in the intratumoral accumulation of PMNs (359, 360). Some pro-inflammatory mediators or other factors directly secreted by tumor cells or elicited as downstream mediators by the released cytokine increase the endothelial expression of several leukocyte adhesion and activation molecules. IL-1β and TNF-α induce or up-regulate the expression of endothelial-leukocyte adhesion molecule 1 (ELAM-1), P-selectin, ICAM-1, and vascular cell adhesion molecule-1 (VCAM-1) in endothelial cells, whereas Interferon-γ (IFN- γ) mainly promotes ICAM-1 expression (359, 361–364).
Integrin-mediated adhesion leads to the extravasation of PMNs, which are highly attracted to the tumor site by the macrophage inflammatory protein 2 (MIP-2) binding to the CXCR1 or CXCR2 counter receptor of PMNs. MIP-2 expression was associated with marked recruitment of PMNs, whose accumulation was enhanced by the further release of MIP-2 produced by PMNs themselves in response to the stimulation by TNF-α in the TME (359, 365).
PMNs also accumulate in tumor stroma when IL-10 is present in the TME (359, 366, 367). IL-10 is typically regarded as an anti-inflammatory mediator because it inhibits the release of other interleukins and chemokines (359, 368–370). Furthermore, a distinct adhesion pathway, mediated by CD11b/CD18 up-regulation on activated PMNs, enables these cells to adhere to the vascular endothelium, thus creating a subjacent micro-environment. The subsequent accumulation of neutrophil effector molecules at local concentrations is sufficient to cause endothelial damage and matrix degradation (359, 371).
The role of TANs in tumor progression or eradication and metastasis has been controversially discussed in the literature [reviewed in (253)]. TANs have pro-tumorous properties but may also act as antitumor effector cells (372–374). In the early 2000s, this contradictory role of neutrophils in both tumor suppression and tumor promotion was re-evaluated in terms of the characterization of different types of TANs with polarized N1 (anti-tumorous) or N2 (pro-tumorigenic) phenotypes (356, 373). The contradictory evidence can be partly explained by the high plasticity of neutrophils in response to primary tumors. After the migration into tumor tissues, neutrophils specialize under the direct influence of factors secreted by tumor cells and acquire various phenotypes and functions. This process seem to be controlled by TGFβ in tumor proximity (356, 375). The description of TAN subtypes N1 and N2 illustrates how the TME can influence the phenotype of these cells (356).
Under the influence of TGFβ in the TME, TANs polarize to N2 cells, which are characterized by an expression profile that promotes tumor angiogenesis and metastasis and inhibits antitumor immune response (372, 376–379). In the context of TME, secreted ROS, RNS, and proteases may lead to oxidative damage, thus inducing genetic damage or signaling in pre-tumoral cells, which subsequently results in boosted tumorigenesis (213, 372). During tumor progression, N2 cells become predominantly pro-tumorigenic: The transfer of neutrophil elastase (NE) by N2 cells activates proliferation within tumor cells. The liberation of arginase-1 (ARG-1) suppresses CD8+ T-cell and NK cell responses, and the release of MMP9 activates the vascular endothelial growth factor A (VEGFA) and fibroblast growth factor (FGF2), which support angiogenesis (213). Furthermore, N2 neutrophils are characterized by the high expression of CCL2 and CCL5 chemokines and the ability to inhibit effector T-cell functions (356, 373).
Under TGFβ-inhibiting conditions, neutrophils acquire an antitumor N1 phenotype, which promotes tumor death and inhibits tumor growth (359, 373, 380, 381). N1 neutrophils can be identified by hypersegmented nuclei, increased expression of intercellular adhesion molecule (ICAM) and TNF-α, and the ability to activate CD8+ T lymphocytes (356, 372). It is still unclear whether the adhesion of PMNs to tumor cells is necessary to cause injury. However, the ultrastructural studies conducted during the growth and rejection phases of several tumors engineered to release cytokines have shown PMNs to be in close contact with damaged tumor cells (359, 382, 383).
Interestingly, N1 and N2 neutrophils were shown to control the activation status of CD8+ T−cells. This interplay seemed to be reciprocal because activated CD8+ T-cells also controlled the activation and migration of neutrophils to the TME (372, 384).
A reverse reprogramming effect has been shown to be exerted by interferons (IFN); IFN-γ and the granulocyte macrophage colony-stimulating factor (GM-CSF) induce an anti-tumoral phenotype in human neutrophils; such neutrophils are capable of cross-presenting antigens, which triggers and augments T-cell responses (213, 385).
Clinical evidence also indicates a negative association between the number of TANs and the prognosis for many types of cancer including malignant melanoma, renal carcinoma, colorectal cancer, gastric or pancreatic ductal carcinoma, hepatocellular carcinoma, intrahepatic cholangiocarcinoma, and head and neck cancer (386). Templeton et al. demonstrated a significant correlation between circulating neutrophil counts (respective neutrophil-to-lymphocyte ratios) and the overall survival of patients with solid and hematological tumors (387). Up to now, the neutrophil/lymphocyte ratio is being used as a prognostic factor in colorectal and non-small-cell lung cancers (356, 388, 389). Tumor infiltration by MPO expressing neutrophils was shown to be an independent prognostic biomarker with a favorable prognosis in human breast cancer (390).
Besides prognostic issues, the secretion of MPO by TANs is also important for the recruitment of monocytes and macrophages and the activation of platelets in tumor settings (356, 391). In turn, the combination of platelet count and neutrophil to lymphocyte ratio is considered to be a useful predictor of postoperative survival in patients with colorectal cancer, which shows the close interconnectedness of the different myeolic cells in tumor processes (392).
Although NETosis has also been found in non-microorganism-induced inflammatory environments such as tumors, the precise details of the connection between NETosis and tumor processes are not yet known (393, 394). The limited data available do not provide sufficient evidence to conclusively demonstrate whether TANs actually produce NETs and to outline what signaling pathways are involved in NETosis in the TME. Despite the available knowledge about the relationship between the deposition of NETs and the recruitment of MPO-rich population of neutrophils in tumors, there does not seem to be enough evidence to prove the existence of TAN-specific NETosis (353, 393–395).
The contradictory role of neutrophils in tumor events remains the subject of intense research. However, the detailed mechanisms of the immunosuppressive function of TANs and their exact role in tumor progression are still largely unknown (396). What we do know is that in the absence of pro-inflammatory cytokines such as IL-1β, TNF-α, and GM-CSF, paracrine IL-10 debilitates the early influx of PMNs and permits initial tumor formation by transitorily paralyzing a prompt non-specific antitumor response (359, 366, 397). Recently, a new hypothesis regarding the immunosuppressive ability of TANs has been presented in the literature by Hiramatsu et al., who investigated the mechanisms behind the immunosuppressive ability of TANs in gastric cancer. The authors observed that neutrophils incubated with tumor-tissue-culture supernatants (TTCS) of gastric tumor cells showed upregulation of programmed cell death ligand 1 (PDL−1) expression, a decreased ratio of apoptotic cells, decreased expression of human leukocyte antigen DR (HLA−DR), and diminished levels of H2O2. Subsequently, Hiramatsu et al. found that neutrophils in non-inflammatory tumor tissue inhibit the proliferation of CD4+ T-cells and may form a local immunosuppressive environment through the PD−1/PDL−1 pathway (396).
The programmed cell death protein 1 (PD-1) and PDL-1 is a negative immune checkpoint pathway that inhibits immune responses, whereby upregulation of PD-1 in exhausted T-cells enables cancer cells to evade immune responses (398). Blockade of the PD-1/PD-L1 pathway has been shown to invert immunosuppression and to rehabilitate the function of T-cells in tumor tissues. Currently, immune checkpoint inhibitors are one of the most encouraging cancer immunotherapy strategies (399).
To sum up, the role of TANs is not yet fully clarified. A better understanding of the mechanisms by which PMNs interact with the specific immune system in tumor defense and act to enhance or inhibit tumor growth is essential to provide novel approaches for cancer treatment by promoting antitumor immune responses based on stimulation of neutrophil antitumor capabilities (356, 399).
13 Summary
To contribute to a better understanding of the role of neutrophils in the human organism, this review summarized current knowledge about PMN chemotaxis and bidirectional migration and PMN interaction with ECM. We considered the role of neutrophil microtubules in migration and discussed neutrophil behavior in the context of cancer environment and tumor tissue. Despite recent successes in elucidating newly discovered neutrophil properties and functions, many processes are not yet fully clarified and require further research. The aspiration of future studies should therefore be to mimic physiological conditions as closely as possible by refining existing models or by creating new assays. In this way, neutrophil key mechanisms along with signaling pathways can be investigated, enabling the development of effective treatment methods (88).
Author Contributions
The corresponding author RK gathered the information, did the literature research, summarized the findings, wrote the first, the following drafts and the endversion of the manuscript. RK also created the tables, edited the video material and took care for the visual and added all changes that occurred during the process of writing. MG had the idea for this review, corrected the drafts, gave suggestions about the topics and did the professional correction of the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.767175/full#supplementary-material
Glossary

References
1. Murphy K, Weaver C. Principles of Innate Immunity. In: Murphy K, Weaver C, editors. Janeway´s Immunobiology. New York, London: GARLAND Science (2017). p. 1–11.
2. Hidalgo A, Chilvers ER, Summers C, Koenderman L. The Neutrophil Life Cycle. Trends Immunol (2019) 40(7):584–97. doi: 10.1016/j.it.2019.04.013
3. Quinn MT, DeLeo FR. Preface. In: Quinn MT, DeLeo FR, editors. Neutrophil Methods and Protocols. Totowa, New Jersey: Humana Press (2014). p. vii–viii.
4. Kolaczkowska E, Kubes P. Neutrophil Recruitment and Function in Health and Inflammation. Nat Rev Immunol (2013) 13(3):159–75. doi: 10.1038/nri3399
5. Lüllmann-Rauch R, Paulsen F. Blut Und Blutbildung. In: Lüllmann-Rauch R, Paulsen F, editors. Taschenlehrbuch Histologie. Stuttgart: Thieme (2012). p. 280–303.
6. McCracken JM, Allen L-AH. Regulation of Human Neutrophil Apoptosis and Lifespan in Health and Disease. J Cell Death (2014) 7:15–23. doi: 10.4137/JCD.S11038
7. Summers C, Rankin SM, Condliffe AM, Singh N, Peters AM, Chilvers ER. Neutrophil Kinetics in Health and Disease. Trends Immunol (2010) 31(8):318–24. doi: 10.1016/j.it.2010.05.006
8. Sender R, Fuchs S, Milo R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PloS Biol (2016) 14(8):e1002533. doi: 10.1371/journal.pbio.1002533
9. Bianconi E, Piovesan A, Facchin F, Beraudi A, Casadei R, Frabetti F, et al. An Estimation of the Number of Cells in the Human Body. Ann Hum Biol (2013) 40(6):463–71. doi: 10.3109/03014460.2013.807878
10. Burdon PC, Martin C, Rankin SM. Migration Across the Sinusoidal Endothelium Regulates Neutrophil Mobilization in Response to ELR + CXC Chemokines. Br J Haematol (2008) 142(1):100–8. doi: 10.1111/j.1365-2141.2008.07018.x
11. Weiss L. Transmural Cellular Passage in Vascular Sinuses of Rat Bone Marrow. Blood (1970) 36(2):189–208. doi: 10.1182/blood.V36.2.189.189
12. Rademakers T, Goedhart M, Hoogenboezem M, García Ponce A, van Rijssel J, Samus M, et al. Hematopoietic Stem and Progenitor Cells Use Podosomes to Transcellularly Cross the Bone Marrow Endothelium. Haematologica (2020) 105(12):2746–56. doi: 10.3324/haematol.2018.196329
13. Wengner AM, Pitchford SC, Furze RC, Rankin SM. The Coordinated Action of G-CSF and ELR + CXC Chemokines in Neutrophil Mobilization During Acute Inflammation. Blood (2008) 111(1):42–9. doi: 10.1182/blood-2007-07-099648
14. Richards MK, Liu F, Iwasaki H, Akashi K, Link DC. Pivotal Role of Granulocyte Colony-Stimulating Factor in the Development of Progenitors in the Common Myeloid Pathway. Blood (2003) 102(10):3562–8. doi: 10.1182/blood-2003-02-0593
15. Lord BI, Bronchud MH, Owens S, Chang J, Howell A, Souza L, et al. The Kinetics of Human Granulopoiesis Following Treatment With Granulocyte Colony-Stimulating Factor. vivo Proc Natl Acad Sci USA (1989) 86(23):9499–503. doi: 10.1073/pnas.86.23.9499
16. Moog R. Donor Tolerance and Results of Stimulation With G-CSF Alone or in Combination With Dexamethasone for the Collection of Granulocytes. J Clin Apher (2004) 19(3):115–8. doi: 10.1002/jca.20013
17. Saverymuttu SH, Peters AM, Keshavarzian A, Reavy HJ, Lavender JP. The Kinetics of 111Indium Distribution Following Injection of 111Indium Labelled Autologous Granulocytes in Man. Br J Haematol (1985) 61(4):675–85. doi: 10.1111/j.1365-2141.1985.tb02882.x
18. Greenlee-Wacker MC. Clearance of Apoptotic Neutrophils and Resolution of Inflammation. Immunol Rev (2016) 273(1):357–70. doi: 10.1111/imr.12453
19. Boxio R, Bossenmeyer-Pourié C, Steinckwich N, Dournon C, Nüße O. Mouse Bone Marrow Contains Large Numbers of Functionally Competent Neutrophils. J Leukoc Biol (2004) 75(4):604–11. doi: 10.1189/jlb.0703340
20. Lord BI, Molineux G, Pojda Z, Souza LM, Mermod J-J, Dexter TM. Myeloid Cell Kinetics in Mice Treated With Recombinant Interleukin-3, Granulocyte Colony-Stimulating Factor (CSF), or Granulocyte-Macrophage CSF In Vivo. Blood (1991) 77(10):2154–9. doi: 10.1182/blood.V77.10.2154.2154
21. Basu S, Hodgson G, Katz M, Dunn AR. Evaluation of Role of G-CSF in the Production, Survival, and Release of Neutrophils From Bone Marrow Into Circulation. Blood (2002) 100(3):854–61. doi: 10.1182/blood.v100.3.854
22. Raffaghello L, Bianchi G, Bertolotto M, Montecucco F, Busca A, Dallegri F, et al. Human Mesenchymal Stem Cells Inhibit Neutrophil Apoptosis: A Model for Neutrophil Preservation in the Bone Marrow Niche. Stem Cells (2008) 26(1):151–62. doi: 10.1634/stemcells.2007-0416
23. Benarafa C, LeCuyer TE, Baumann M, Stolley JM, Cremona TP, Remold-O’Donnell E. SerpinB1 Protects the Mature Neutrophil Reserve in the Bone Marrow. J Leukoc Biol (2011) 90(1):21–9. doi: 10.1189/jlb.0810461
24. Peters AM, Saverymuttu SH, Keshavarzian A, Bell RN, Lavender JP. Splenic Pooling of Granulocytes. Clin Sci (Lond) (1985) 68(3):283–9. doi: 10.1042/cs0680283
25. Ussov WY, Aktolun C, Myers MJ, Jamar F, Peters AM. Granulocyte Margination in Bone Marrow: Comparison With Margination in the Spleen and Liver. Scand J Clin Lab Invest (1995) 55(1):87–96. doi: 10.3109/00365519509075382
26. Peters AM. Just How Big is the Pulmonary Granulocyte Pool? Clin Sci (Lond) (1998) 94(1):7–19. doi: 10.1042/cs0940007
27. Doerschuk CM, Allard MF, Martin BA, MacKenzie A, Autor AP, Hogg JC. Marginated Pool of Neutrophils in Rabbit Lungs. J Appl Physiol (1987) 63(5):1806–15. doi: 10.1152/jappl.1987.63.5.1806
28. Downey GP, Doherty DE, Schwab B, Elson EL, Henson PM, Worthen GS. Retention of Leukocytes in Capillaries: Role of Cell Size and Deformability. J Appl Physiol (1990) 69(5):1767–78. doi: 10.1152/jappl.1990.69.5.1767
29. Motosugi H, Graham L, Noblitt TW, Doyle NA, Quinlan WM, Li Y, et al. Changes in Neutrophil Actin and Shape During Sequestration Induced by Complement Fragments in Rabbits. Am J Pathol (1996) 149(3):963–73.
30. Granton E, Kim JH, Podstawka J, Yipp BG. The Lung Microvasculature Is a Functional Immune Niche. Trends Immunol (2018) 39(11):890–9. doi: 10.1016/j.it.2018.09.002
31. Athens JW, Haab OP, Raab SO, Mauer AM, Ashenbrucker H, Cartwright GE, et al. Leukokinetic Studies. IV. The Total Blood, Circulating and Marginal Granulocyte Pools and the Granulocyte Turnover Rate in Normal Subjects. J Clin Invest (1961) 40(6):989–95. doi: 10.1172/jci104338
32. Murphy K, Weaver C. Pattern Recognition by Cells of the Innate Immune System. In: Murphy K, Weaver C, editors. Janeway´s Immunobiology. New York, London: GARLAND Science (2017). p. 77–106.
33. Lok LS, Dennison TW, Mahbubani KM, Saeb-Parsy K, Chilvers ER, Clatworthy MR. Phenotypically Distinct Neutrophils Patrol Uninfected Human and Mouse Lymph Nodes. Proc Natl Acad Sci USA (2019) 116(38):19083–9. doi: 10.1073/pnas.1905054116
34. Casanova-Acebes M, Nicolás-Ávila JA, Li JL, García-Silva S, Balachander A, Rubio-Ponce A, et al. Neutrophils Instruct Homeostatic and Pathological States in Naive Tissues. J Exp Med (2018) 215(11):2778–95. doi: 10.1084/jem.20181468
35. Puga I, Cols M, Barra CM, He B, Cassis L, Gentile M, et al. B Cell-Helper Neutrophils Stimulate the Diversification and Production of Immunoglobulin in the Marginal Zone of the Spleen. Nat Immunol (2011) 13(2):170–80. doi: 10.1038/ni.2194
36. Kim JH, Podstawka J, Lou Y, Li L, Lee EK, Divangahi M, et al. Aged Polymorphonuclear Leukocytes Cause Fibrotic Interstitial Lung Disease in the Absence of Regulation by B Cells. Nat Immunol (2018) 19(2):192–201. doi: 10.1038/s41590-017-0030-x
37. Sadik CD, Kim ND, Luster AD. Neutrophils Cascading Their Way to Inflammation. Trends Immunol (2011) 32(10):452–60. doi: 10.1016/j.it.2011.06.008
38. Murphy K, Weaver C. Induced Innate Responses to Infection. In: Murphy K, Weaver C, editors. Janeway´s Immunobiology. New York, London: GARLAND Science (2017). p. 107–33.
39. Mahapatro M, Erkert L, Becker C. Cytokine-Mediated Crosstalk Between Immune Cells and Epithelial Cells in the Gut. Cells (2021) 10(1):111. doi: 10.3390/cells10010111
40. Mahmoudi S, Mancini E, Xu L, Moore A, Jahanbani F, Hebestreit K, et al. Heterogeneity in Old Fibroblasts is Linked to Variability in Reprogramming and Wound Healing. Nature (2019) 574(7779):553–8. doi: 10.1038/s41586-019-1658-5
41. Charo IF, Ransohoff RM. The Many Roles of Chemokines and Chemokine Receptors in Inflammation. N Engl J Med (2006) 354(6):610–21. doi: 10.1056/NEJMra052723
42. Persson T, Monsef N, Andersson P, Bjartell A, Malm J, Calafat J, et al. Expression of the Neutrophil-Activating CXC Chemokine ENA-78/CXCL5 by Human Eosinophils. Clin Exp Allergy (2003) 33(4):531–7. doi: 10.1046/j.1365-2222.2003.01609.x
43. Bisset LR, Schmid-Grendelmeier P. Chemokines and Their Receptors in the Pathogenesis of Allergic Asthma: Progress and Perspective. Curr Opin Pulm Med (2005) 11(1):35–42. doi: 10.1097/01.mcp.0000144502.50149.e0
44. Chang MS, McNinch J, Basu R, Simonet S. Cloning and Characterization of the Human Neutrophil-Activating Peptide (ENA-78) Gene. J Biol Chem (1994) 269(41):25277–82. doi: 10.1016/S0021-9258(18)47243-2
45. Yücel Ç, Fırat Oğuz E, Er S, Balamir İ, Turhan T, Tez M. Diagnostic Value of GCP-2/CXCL-6 and Hs-CRP in the Diagnosis of Acute Appendicitis. Turkish J Trauma Emergency Surg (2020) 26(2):191–6. doi: 10.14744/tjtes.2019.26270
46. Almeida CR, Caires HR, Vasconcelos DP, Barbosa MA. NAP-2 Secreted by Human NK Cells Can Stimulate Mesenchymal Stem/Stromal Cell Recruitment. Stem Cell Rep (2016) 6(4):466–73. doi: 10.1016/j.stemcr.2016.02.012
47. Proost P, Wuyts A, van Damme J. Human Monocyte Chemotactic Proteins-2 and -3: Structural and Functional Comparison With MCP-1. J Leukoc Biol (1996) 59(1):67–74. doi: 10.1002/jlb.59.1.67
48. van Damme J, Proost P, Lenaerts JP, Opdenakker G. Structural and Functional Identification of Two Human, Tumor-Derived Monocyte Chemotactic Proteins (MCP-2 and MCP-3) Belonging to the Chemokine Family. J Exp Med (1992) 176(1):59–65. doi: 10.1084/jem.176.1.59
49. Menten P, Proost P, Struyf S, van Coillie E, Put W, Lenaerts J-P, et al. Differential Induction of Monocyte Chemotactic Protein-3 in Mononuclear Leukocytes and Fibroblasts by Interferon-α / β and Interferon-γ Reveals MCP-3 Heterogeneity. Eur J Immunol (1999) 29(2):678–85. doi: 10.1002/(SICI)1521-4141(199902)29:02<678:AID-IMMU678>3.0.CO;2-J
50. Nagasawa T, Hirota S, Tachibana K, Takakura N, Nishikawa S, Kitamura Y, et al. Defects of B-Cell Lymphopoiesis and Bone-Marrow Myelopoiesis in Mice Lacking the CXC Chemokine PBSF/SDF-1. Nature (1996) 382(6592):635–8. doi: 10.1038/382635a0
51. Tachibana K, Hirota S, Iizasa H, Yoshida H, Kawabata K, Kataoka Y, et al. The Chemokine Receptor CXCR4 is Essential for Vascularization of the Gastrointestinal Tract. Nature (1998) 393(6685):591–4. doi: 10.1038/31261
52. Nording H, Baron L, Haberthür D, Emschermann F, Mezger M, Sauter M, et al. The C5a/C5a Receptor 1 Axis Controls Tissue Neovascularization Through CXCL4 Release From Platelets. Nat Commun (2021) 12(1):3352. doi: 10.1038/s41467-021-23499-w
53. Giorgio C, Zippoli M, Cocchiaro P, Castelli V, Varrassi G, Aramini A, et al. Emerging Role of C5 Complement Pathway in Peripheral Neuropathies: Current Treatments and Future Perspectives. Biomedicines (2021) 9(4):399. doi: 10.3390/biomedicines9040399
54. Guo R-F, Ward PA. Role of C5a in Inflammatory Responses. Annu Rev Immunol (2005) 23:821–52. doi: 10.1146/annurev.immunol.23.021704.115835
55. Lee H, Whitfeld PL, Mackay CR. Receptors for Complement C5a. The Importance of C5aR and the Enigmatic Role of C5L2. Immunol Cell Biol (2008) 86(2):153–60. doi: 10.1038/sj.icb.7100166
56. Fayyazi A, Scheel O, Werfel T, Schweyer S, Oppermann M, Götze O, et al. The C5a Receptor is Expressed in Normal Renal Proximal Tubular But Not in Normal Pulmonary or Hepatic Epithelial Cells. Immunology (2000) 99(1):38–45. doi: 10.1046/j.1365-2567.2000.00911.x
57. Kolev M, Le Friec G, Kemper C. Complement–tapping Into New Sites and Effector Systems. Nat Rev Immunol (2014) 14(12):811–20. doi: 10.1038/nri3761
58. Ohno M, Hirata T, Enomoto M, Araki T, Ishimaru H, Takahashi TA. A Putative Chemoattractant Receptor, C5L2, is Expressed in Granulocyte and Immature Dendritic Cells, But Not in Mature Dendritic Cells. Mol Immunol (2000) 37(8):407–12. doi: 10.1016/S0161-5890(00)00067-5
59. Li XX, Lee JD, Kemper C, Woodruff TM. The Complement Receptor C5aR2: A Powerful Modulator of Innate and Adaptive Immunity. J Immunol (2019) 202(12):3339–48. doi: 10.4049/jimmunol.1900371
60. Gao S, Cui Z, Zhao M-H. The Complement C3a and C3a Receptor Pathway in Kidney Diseases. Front Immunol (2020) 11:1875. doi: 10.3389/fimmu.2020.01875
61. Leslie J, Millar BJ, Del Carpio Pons A, Burgoyne RA, Frost JD, Barksby BS, et al. FPR-1 is an Important Regulator of Neutrophil Recruitment and a Tissue-Specific Driver of Pulmonary Fibrosis. JCI Insight (2020) 5(4):e125937. doi: 10.1172/jci.insight.125937
62. Rabiet M-J, Huet E, Boulay F. Human Mitochondria-Derived N-Formylated Peptides are Novel Agonists Equally Active on FPR and FPRL1, While Listeria Monocytogenes-Derived Peptides Preferentially Activate FPR. Eur J Immunol (2005) 35(8):2486–95. doi: 10.1002/eji.200526338
63. Crouser ED, Shao G, Julian MW, Macre JE, Shadel GS, Tridandapani S, et al. Monocyte Activation by Necrotic Cells is Promoted by Mitochondrial Proteins and Formyl Peptide Receptors. Crit Care Med (2009) 37(6):2000–9. doi: 10.1097/CCM.0b013e3181a001ae
64. Alam A, Leoni G, Wentworth CC, Kwal JM, Wu H, Ardita CS, et al. Redox Signaling Regulates Commensal-Mediated Mucosal Homeostasis and Restitution and Requires Formyl Peptide Receptor 1. Mucosal Immunol (2014) 7(3):645–55. doi: 10.1038/mi.2013.84
65. Shao G, Julian MW, Bao S, McCullers MK, Lai J-P, Knoell DL, et al. Formyl Peptide Receptor Ligands Promote Wound Closure in Lung Epithelial Cells. Am J Respir Cell Mol Biol (2011) 44(3):264–9. doi: 10.1165/rcmb.2010-0246RC
66. VanCompernolle SE, Clark KL, Rummel KA, Todd SC. Expression and Function of Formyl Peptide Receptors on Human Fibroblast Cells. J Immunol (2003) 171(4):2050–6. doi: 10.4049/jimmunol.171.4.2050
67. Rossi FW, Napolitano F, Pesapane A, Mascolo M, Staibano S, Matucci-Cerinic M, et al. Upregulation of the N-Formyl Peptide Receptors in Scleroderma Fibroblasts Fosters the Switch to Myofibroblasts. J Immunol (2015) 194(11):5161–73. doi: 10.4049/jimmunol.1402819
68. Patel DF, Peiró T, Shoemark A, Akthar S, Walker SA, Grabiec AM, et al. An Extracellular Matrix Fragment Drives Epithelial Remodeling and Airway Hyperresponsiveness. Sci Transl Med (2018) 10(455):eaaq0693. doi: 10.1126/scitranslmed.aaq0693
69. Patel DF, Snelgrove RJ. The Multifaceted Roles of the Matrikine Pro-Gly-Pro in Pulmonary Health and Disease. Eur Respir Rev (2018) 27(148):180017. doi: 10.1183/16000617.0017-2018
70. Turnbull AR, Pyle CJ, Patel DF, Jackson PL, Hilliard TN, Regamey N, et al. Abnormal Pro-Gly-Pro Pathway and Airway Neutrophilia in Pediatric Cystic Fibrosis. J Cyst Fibros (2020) 19(1):40–8. doi: 10.1016/j.jcf.2019.05.017
71. Agerberth B, Charo J, Werr J, Olsson B, Idali F, Lindbom L, et al. The Human Antimicrobial and Chemotactic Peptides LL-37 and α-Defensins are Expressed by Specific Lymphocyte and Monocyte Populations. Blood (2000) 96(9):3086–93. doi: 10.1182/blood.V96.9.3086
72. Sun J, Dahlén B, Agerberth B, Haeggström JZ. The Antimicrobial Peptide LL-37 Induces Synthesis and Release of Cysteinyl Leukotrienes From Human Eosinophils–Implications for Asthma. Allergy (2013) 68(3):304–11. doi: 10.1111/all.12087
73. Di Nardo A, Vitiello A, Gallo RL. Cutting Edge: Mast Cell Antimicrobial Activity is Mediated by Expression of Cathelicidin Antimicrobial Peptide. J Immunol (2003) 170(5):2274–8. doi: 10.4049/jimmunol.170.5.2274
74. Kai-Larsen Y, Agerberth B. The Role of the Multifunctional Peptide LL-37 in Host Defense. Front Biosci (2008) 13:3760–7. doi: 10.2741/2964
75. Tripathi S, Wang G, White M, Rynkiewicz M, Seaton B, Hartshorn K. Identifying the Critical Domain of LL-37 Involved in Mediating Neutrophil Activation in the Presence of Influenza Virus: Functional and Structural Analysis. PloS One (2015) 10(8):e0133454. doi: 10.1371/journal.pone.0133454
76. Calandra T, Roger T. Macrophage Migration Inhibitory Factor: A Regulator of Innate Immunity. Nat Rev Immunol (2003) 3(10):791–800. doi: 10.1038/nri1200
77. Bernhagen J, Krohn R, Lue H, Gregory JL, Zernecke A, Koenen RR, et al. MIF is a Noncognate Ligand of CXC Chemokine Receptors in Inflammatory and Atherogenic Cell Recruitment. Nat Med (2007) 13(5):587–96. doi: 10.1038/nm1567
78. Krammer C, Kontos C, Dewor M, Hille K, Dalla Volta B, El Bounkari O, et al. A MIF-Derived Cyclopeptide That Inhibits MIF Binding and Atherogenic Signaling via the Chemokine Receptor Cxcr2. Chembiochem (2021) 22(6):1012–9. doi: 10.1002/cbic.202000574
79. He R, Chen Y, Cai Q. The Role of the LTB4-BLT1 Axis in Health and Disease. Pharmacol Res (2020) 158:104857. doi: 10.1016/j.phrs.2020.104857
80. Peters-Golden M, Henderson WR. Leukotrienes. N Engl J Med (2007) 357(18):1841–54. doi: 10.1056/NEJMra071371
81. Ishii S, Shimizu T. Platelet-Activating Factor (PAF) Receptor and Genetically Engineered PAF Receptor Mutant Mice. Prog Lipid Res (2000) 39(1):41–82. doi: 10.1016/S0163-7827(99)00016-8
82. Honda Z-I, Ishii S, Shimizu T. Platelet-Activating Factor Receptor. J Biochem (2002) 131(6):773–9. doi: 10.1093/oxfordjournals.jbchem.a003164
83. Midgley A, Barakat D, Braitch M, Nichols C, Nebozhyn M, Edwards LJ, et al. PAF-R on Activated T Cells: Role in the IL-23/Th17 Pathway and Relevance to Multiple Sclerosis. Immunobiology (2021) 226(1):152023. doi: 10.1016/j.imbio.2020.152023
84. Mostafa GA, Al-Ayadhi LY. The Possible Link Between Elevated Serum Levels of Epithelial Cell-Derived Neutrophil-Activating Peptide-78 (ENA-78/CXCL5) and Autoimmunity in Autistic Children. Behav Brain Funct (2015) 11:11. doi: 10.1186/s12993-015-0056-x
85. Zlotnik A, Yoshie O. Chemokines. Immunity (2000) 12(2):121–7. doi: 10.1016/s1074-7613(00)80165-x
86. Takeuchi O, Akira S. Pattern Recognition Receptors and Inflammation. Cell (2010) 140(6):805–20. doi: 10.1016/j.cell.2010.01.022
87. Denning N-L, Aziz M, Gurien SD, Wang P. DAMPs and NETs in Sepsis. Front Immunol (2019) 10:2536. doi: 10.3389/fimmu.2019.02536
88. Williams MR, Azcutia V, Newton G, Alcaide P, Luscinskas FW. Emerging Mechanisms of Neutrophil Recruitment Across Endothelium. Trends Immunol (2011) 32(10):461–9. doi: 10.1016/j.it.2011.06.009
89. Phillipson M, Kubes P. The Neutrophil in Vascular Inflammation. Nat Med (2011) 17(11):1381–90. doi: 10.1038/nm.2514
90. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the Site of Inflammation: The Leukocyte Adhesion Cascade Updated. Nat Rev Immunol (2007) 7(9):678–89. doi: 10.1038/nri2156
91. Burns AR, Smith CW, Walker DC. Unique Structural Features That Influence Neutrophil Emigration Into the Lung. Physiol Rev (2003) 83(2):309–36. doi: 10.1152/physrev.00023.2002
92. McCormack JJ, Lopes da Silva M, Ferraro F, Patella F, Cutler DF. Weibel-Palade Bodies at a Glance. J Cell Sci (2017) 130(21):3611–7. doi: 10.1242/jcs.208033
93. Kannagi R, Izawa M, Koike T, Miyazaki K, Kimura N. Carbohydrate-Mediated Cell Adhesion in Cancer Metastasis and Angiogenesis. Cancer Sci (2004) 95(5):377–84. doi: 10.1111/j.1349-7006.2004.tb03219.x
94. Chebbi R. Dynamics of Blood Flow: Modeling of Fåhraeus and Fåhraeus-Lindqvist Effects Using a Shear-Induced Red Blood Cell Migration Model. J Biol Phys (2018) 44(4):591–603. doi: 10.1007/s10867-018-9508-5
95. Ascolese M, Farina A, Fasano A. The Fåhræus-Lindqvist Effect in Small Blood Vessels: How Does it Help the Heart? J Biol Phys (2019) 45(4):379–94. doi: 10.1007/s10867-019-09534-4
96. Stadtmann A, Germena G, Block H, Boras M, Rossaint J, Sundd P, et al. The PSGL-1-L-Selectin Signaling Complex Regulates Neutrophil Adhesion Under Flow. J Exp Med (2013) 210(11):2171–80. doi: 10.1084/jem.20130664
97. Morikis VA, Chase S, Wun T, Chaikof EL, Magnani JL, Simon SI. Selectin Catch-Bonds Mechanotransduce Integrin Activation and Neutrophil Arrest on Inflamed Endothelium Under Shear Flow. Blood (2017) 130(19):2101–10. doi: 10.1182/blood-2017-05-783027
98. Smolen JE, Petersen TK, Koch C, O’Keefe SJ, Hanlon WA, Seo S, et al. L-Selectin Signaling of Neutrophil Adhesion and Degranulation Involves P38 Mitogen-Activated Protein Kinase. J Biol Chem (2000) 275(21):15876–84. doi: 10.1074/jbc.M906232199
99. Green CE, Pearson DN, Camphausen RT, Staunton DE, Simon SI. Shear-Dependent Capping of L-Selectin and P-Selectin Glycoprotein Ligand 1 by E-Selectin Signals Activation of High-Avidity Beta2-Integrin on Neutrophils. J Immunol (2004) 172(12):7780–90. doi: 10.4049/jimmunol.172.12.7780
100. Green CE, Pearson DN, Christensen NB, Simon SI. Topographic Requirements and Dynamics of Signaling via L-Selectin on Neutrophils. Am J Physiol Cell Physiol (2003) 284(3):C705–17. doi: 10.1152/ajpcell.00331.2002
101. Steeber DA, Engel P, Miller AS, Sheetz MP, Tedder TF. Ligation of L-Selectin Through Conserved Regions Within the Lectin Domain Activates Signal Transduction Pathways and Integrin Function in Human, Mouse, and Rat Leukocytes. J Immunol (1997) 159(2):952–63.
102. Ivetic A. A Head-to-Tail View of L-Selectin and its Impact on Neutrophil Behaviour. Cell Tissue Res (2018) 371(3):437–53. doi: 10.1007/s00441-017-2774-x
103. Schaff UY, Yamayoshi I, Tse T, Griffin D, Kibathi L, Simon SI. Calcium Flux in Neutrophils Synchronizes Beta2 Integrin Adhesive and Signaling Events That Guide Inflammatory Recruitment. Ann BioMed Eng (2008) 36(4):632–46. doi: 10.1007/s10439-008-9453-8
104. Phillipson M, Heit B, Colarusso P, Liu L, Ballantyne CM, Kubes P. Intraluminal Crawling of Neutrophils to Emigration Sites: A Molecularly Distinct Process From Adhesion in the Recruitment Cascade. J Exp Med (2006) 203(12):2569–75. doi: 10.1084/jem.20060925
105. Phillipson M, Heit B, Parsons SA, Petri B, Mullaly SC, Colarusso P, et al. Vav1 is Essential for Mechanotactic Crawling and Migration of Neutrophils Out of the Inflamed Microvasculature. J Immunol (2009) 182(11):6870–8. doi: 10.4049/jimmunol.0803414
106. Choudhury SR, Babes L, Rahn JJ, Ahn B-Y, Goring K-AR, King JC, et al. Dipeptidase-1 Is an Adhesion Receptor for Neutrophil Recruitment in Lungs and Liver. Cell (2019) 178(5):1205–1221.e17. doi: 10.1016/j.cell.2019.07.017
107. Lee W-Y, Kubes P. Leukocyte Adhesion in the Liver: Distinct Adhesion Paradigm From Other Organs. J Hepatol (2008) 48(3):504–12. doi: 10.1016/j.jhep.2007.12.005
108. McDonald B, McAvoy EF, Lam F, Gill V, de La Motte C, Savani RC, et al. Interaction of CD44 and Hyaluronan is the Dominant Mechanism for Neutrophil Sequestration in Inflamed Liver Sinusoids. J Exp Med (2008) 205(4):915–27. doi: 10.1084/jem.20071765
109. Wong J, Johnston B, Lee SS, Bullard DC, Smith CW, Beaudet AL, et al. A Minimal Role for Selectins in the Recruitment of Leukocytes Into the Inflamed Liver Microvasculature. J Clin Invest (1997) 99(11):2782–90. doi: 10.1172/JCI119468
110. Lin W-C, Fessler MB. Regulatory Mechanisms of Neutrophil Migration From the Circulation to the Airspace. Cell Mol Life Sci (2021) 78(9):4095–124. doi: 10.1007/s00018-021-03768-z
111. Carvalho-Tavares J, Hickey MJ, Hutchison J, Michaud J, Sutcliffe IT, Kubes P. A Role for Platelets and Endothelial Selectins in Tumor Necrosis Factor-Alpha-Induced Leukocyte Recruitment in the Brain Microvasculature. Circ Res (2000) 87(12):1141–8. doi: 10.1161/01.RES.87.12.1141
112. Zarbock A, Ley K, McEver RP, Hidalgo A. Leukocyte Ligands for Endothelial Selectins: Specialized Glycoconjugates That Mediate Rolling and Signaling Under Flow. Blood (2011) 118(26):6743–51. doi: 10.1182/blood-2011-07-343566
113. Zhou H, Andonegui G, Wong CH, Kubes P. Role of Endothelial TLR4 for Neutrophil Recruitment Into Central Nervous System Microvessels in Systemic Inflammation. J Immunol (2009) 183(8):5244–50. doi: 10.4049/jimmunol.0901309
114. Wang H, Knight JS, Hodgin JB, Wang J, Guo C, Kleiman K, et al. Psgl-1 Deficiency is Protective Against Stroke in a Murine Model of Lupus. Sci Rep (2016) 6:28997. doi: 10.1038/srep28997
115. Tsai N-W, Chang W-N, Shaw C-F, Jan C-R, Huang C-R, Chen S-D, et al. The Value of Leukocyte Adhesion Molecules in Patients After Ischemic Stroke. J Neurol (2009) 256(8):1296–302. doi: 10.1007/s00415-009-5117-3
116. Tao L, Changfu W, Linyun L, Bing M, Xiaohui H. Correlations of Platelet-Leukocyte Aggregates With P-Selectin S290N and P-Selectin Glycoprotein Ligand-1 M62I Genetic Polymorphisms in Patients With Acute Ischemic Stroke. J Neurol Sci (2016) 367:95–100. doi: 10.1016/j.jns.2016.05.046
117. Pietronigro E, Zenaro E, Della Bianca V, Dusi S, Terrabuio E, Iannoto G, et al. Blockade of α4 Integrins Reduces Leukocyte-Endothelial Interactions in Cerebral Vessels and Improves Memory in a Mouse Model of Alzheimer’s Disease. Sci Rep (2019) 9(1):12055. doi: 10.1038/s41598-019-48538-x
118. Neumann J, Riek-Burchardt M, Herz J, Doeppner TR, König R, Hütten H, et al. Very-Late-Antigen-4 (VLA-4)-Mediated Brain Invasion by Neutrophils Leads to Interactions With Microglia, Increased Ischemic Injury and Impaired Behavior in Experimental Stroke. Acta Neuropathol (2015) 129(2):259–77. doi: 10.1007/s00401-014-1355-2
119. Fabene PF, Navarro Mora G, Martinello M, Rossi B, Merigo F, Ottoboni L, et al. A Role for Leukocyte-Endothelial Adhesion Mechanisms in Epilepsy. Nat Med (2008) 14(12):1377–83. doi: 10.1038/nm.1878
120. Kornerup KN, Salmon GP, Pitchford SC, Liu WL, Page CP. Circulating Platelet-Neutrophil Complexes are Important for Subsequent Neutrophil Activation and Migration. J Appl Physiol (1985) (2010) 109(3):758–67. doi: 10.1152/japplphysiol.01086.2009
121. Cappenberg A, Margraf A, Thomas K, Bardel B, McCreedy DA, van Marck V, et al. L-Selectin Shedding Affects Bacterial Clearance in the Lung: A New Regulatory Pathway for Integrin Outside-in Signaling. Blood (2019) 134(17):1445–57. doi: 10.1182/blood.2019000685
122. Katayama Y, Hidalgo A, Chang J, Peired A, Frenette PS. CD44 is a Physiological E-Selectin Ligand on Neutrophils. J Exp Med (2005) 201(8):1183–9. doi: 10.1084/jem.20042014
123. Begandt D, Thome S, Sperandio M, Walzog B. How Neutrophils Resist Shear Stress at Blood Vessel Walls: Molecular Mechanisms, Subcellular Structures, and Cell-Cell Interactions. J Leukoc Biol (2017) 102(3):699–709. doi: 10.1189/jlb.3MR0117-026RR
124. Khan AI, Kerfoot SM, Heit B, Liu L, Andonegui G, Ruffell B, et al. Role of CD44 and Hyaluronan in Neutrophil Recruitment. J Immunol (2004) 173(12):7594–601. doi: 10.4049/jimmunol.173.12.7594
125. Gorina R, Lyck R, Vestweber D, Engelhardt B. β2 Integrin-Mediated Crawling on Endothelial ICAM-1 and ICAM-2 is a Prerequisite for Transcellular Neutrophil Diapedesis Across the Inflamed Blood-Brain Barrier. J Immunol (2014) 192(1):324–37. doi: 10.4049/jimmunol.1300858
126. Burns AR, Takei F, Doerschuk CM. Quantitation of ICAM-1 Expression in Mouse Lung During Pneumonia. J Immunol (1994) 153(7):3189–98.
127. Moreland JG, Fuhrman RM, Pruessner JA, Schwartz DA. CD11b and Intercellular Adhesion Molecule-1 are Involved in Pulmonary Neutrophil Recruitment in Lipopolysaccharide-Induced Airway Disease. Am J Respir Cell Mol Biol (2002) 27(4):474–80. doi: 10.1165/rcmb.4694
128. Tasaka S, Richer SE, Mizgerd JP, Doerschuk CM. Very Late Antigen-4 in CD18-Independent Neutrophil Emigration During Acute Bacterial Pneumonia in Mice. Am J Respir Crit Care Med (2002) 166(1):53–60. doi: 10.1164/rccm.2105034
129. Tong C-F, Zhang Y, Lü S-Q, Li N, Gong Y-X, Yang H, et al. Binding of Intercellular Adhesion Molecule 1 to β2-Integrin Regulates Distinct Cell Adhesion Processes on Hepatic and Cerebral Endothelium. Am J Physiol Cell Physiol (2018) 315(3):C409–21. doi: 10.1152/ajpcell.00083.2017
130. Phillipson M, Kaur J, Colarusso P, Ballantyne CM, Kubes P. Endothelial Domes Encapsulate Adherent Neutrophils and Minimize Increases in Vascular Permeability in Paracellular and Transcellular Emigration. PloS One (2008) 3(2):e1649. doi: 10.1371/journal.pone.0001649
131. Ostermann G, Weber KS, Zernecke A, Schröder A, Weber C. JAM-1 is a Ligand of the Beta(2) Integrin LFA-1 Involved in Transendothelial Migration of Leukocytes. Nat Immunol (2002) 3(2):151–8. doi: 10.1038/ni755
132. Lamagna C, Meda P, Mandicourt G, Brown J, Gilbert RJ, Jones EY, et al. Dual Interaction of JAM-C With JAM-B and Alpha(M)beta2 Integrin: Function in Junctional Complexes and Leukocyte Adhesion. Mol Biol Cell (2005) 16(10):4992–5003. doi: 10.1091/mbc.e05-04-0310
133. Bradfield PF, Scheiermann C, Nourshargh S, Ody C, Luscinskas FW, Rainger GE, et al. JAM-C Regulates Unidirectional Monocyte Transendothelial Migration in Inflammation. Blood (2007) 110(7):2545–55. doi: 10.1182/blood-2007-03-078733
134. Chavakis T, Keiper T, Matz-Westphal R, Hersemeyer K, Sachs UJ, Nawroth PP, et al. The Junctional Adhesion Molecule-C Promotes Neutrophil Transendothelial Migration In Vitro and In Vivo. J Biol Chem (2004) 279(53):55602–8. doi: 10.1074/jbc.M404676200
135. Aurrand-Lions M, Lamagna C, Dangerfield JP, Wang S, Herrera P, Nourshargh S, et al. Junctional Adhesion Molecule-C Regulates the Early Influx of Leukocytes Into Tissues During Inflammation. J Immunol (2005) 174(10):6406–15. doi: 10.4049/jimmunol.174.10.6406
136. Bixel MG, Li H, Petri B, Khandoga AG, Khandoga A, Zarbock A, et al. CD99 and CD99L2 Act at the Same Site as, But Independently of, PECAM-1 During Leukocyte Diapedesis. Blood (2010) 116(7):1172–84. doi: 10.1182/blood-2009-12-256388
137. Schenkel AR, Dufour EM, Chew TW, Sorg E, Muller WA. The Murine CD99-Related Molecule CD99-Like 2 (CD99L2) is an Adhesion Molecule Involved in the Inflammatory Response. Cell Commun Adhes (2007) 14(5):227–37. doi: 10.1080/15419060701755966
138. Lampugnani MG, Dejana E, Giampietro C. Vascular Endothelial (VE)-Cadherin, Endothelial Adherens Junctions, and Vascular Disease. Cold Spring Harb Perspect Biol (2018) 10(10):a029322. doi: 10.1101/cshperspect.a029322
139. Broermann A, Winderlich M, Block H, Frye M, Rossaint J, Zarbock A, et al. Dissociation of VE-PTP From VE-Cadherin is Required for Leukocyte Extravasation and for VEGF-Induced Vascular Permeability In Vivo. J Exp Med (2011) 208(12):2393–401. doi: 10.1084/jem.20110525
140. Schulte D, Küppers V, Dartsch N, Broermann A, Li H, Zarbock A, et al. Stabilizing the VE-Cadherin-Catenin Complex Blocks Leukocyte Extravasation and Vascular Permeability. EMBO J (2011) 30(20):4157–70. doi: 10.1038/emboj.2011.304
141. Zhou Z, Xu M-J, Cai Y, Wang W, Jiang JX, Varga ZV, et al. Neutrophil-Hepatic Stellate Cell Interactions Promote Fibrosis in Experimental Steatohepatitis. Cell Mol Gastroenterol Hepatol (2018) 5(3):399–413. doi: 10.1016/j.jcmgh.2018.01.003
142. Bartneck M, Wang J. Therapeutic Targeting of Neutrophil Granulocytes in Inflammatory Liver Disease. Front Immunol (2019) 10:2257. doi: 10.3389/fimmu.2019.02257
143. Winneberger J, Schöls S, Lessmann K, Rández-Garbayo J, Bauer AT, Mohamud Yusuf A, et al. Platelet Endothelial Cell Adhesion Molecule-1 is a Gatekeeper of Neutrophil Transendothelial Migration in Ischemic Stroke. Brain Behav Immun (2021) 93:277–87. doi: 10.1016/j.bbi.2020.12.026
144. Lakshmi SP, Reddy AT, Naik MU, Naik UP, Reddy RC. Effects of JAM-A Deficiency or Blocking Antibodies on Neutrophil Migration and Lung Injury in a Murine Model of ALI. Am J Physiol Lung Cell Mol Physiol (2012) 303(9):L758–66. doi: 10.1152/ajplung.00107.2012
145. Maas SL, Soehnlein O, Viola JR. Organ-Specific Mechanisms of Transendothelial Neutrophil Migration in the Lung, Liver, Kidney, and Aorta. Front Immunol (2018) 9:2739. doi: 10.3389/fimmu.2018.02739
146. Schoppmeyer R, van Buul JD. The Diapedesis Synapse: Dynamic Leukocyte-Endothelium Interactions. Curr Opin Physiol (2021) 19:1–9. doi: 10.1016/j.cophys.2020.06.003
147. Feng D, Nagy JA, Pyne K, Dvorak HF, Dvorak AM. Neutrophils Emigrate From Venules by a Transendothelial Cell Pathway in Response to FMLP. J Exp Med (1998) 187(6):903–15. doi: 10.1084/jem.187.6.903
148. Carman CV, Springer TA. A Transmigratory Cup in Leukocyte Diapedesis Both Through Individual Vascular Endothelial Cells and Between Them. J Cell Biol (2004) 167(2):377–88. doi: 10.1083/jcb.200404129
149. van Buul JD. Why Vessels do Not Leak When Leukocytes Migrate Out. Blood (2020) 136(5):531–2. doi: 10.1182/blood.2020006568
150. Braun LJ, Stegmeyer RI, Schäfer K, Volkery S, Currie SM, Kempe B, et al. Platelets Docking to VWF Prevent Leaks During Leukocyte Extravasation by Stimulating Tie-2. Blood (2020) 136(5):627–39. doi: 10.1182/blood.2019003442
151. Petri B, Phillipson M, Kubes P. The Physiology of Leukocyte Recruitment: An In Vivo Perspective. J Immunol (2008) 180(10):6439–46. doi: 10.4049/jimmunol.180.10.6439
152. Sundd P, Pospieszalska MK, Cheung LS-L, Konstantopoulos K, Ley K. Biomechanics of Leukocyte Rolling. Biorheology (2011) 48(1):1–35. doi: 10.3233/BIR-2011-0579
153. Sundd P, Pospieszalska MK, Ley K. Neutrophil Rolling at High Shear: Flattening, Catch Bond Behavior, Tethers and Slings. Mol Immunol (2013) 55(1):59–69. doi: 10.1016/j.molimm.2012.10.025
154. Marshall BT, Long M, Piper JW, Yago T, McEver RP, Zhu C. Direct Observation of Catch Bonds Involving Cell-Adhesion Molecules. Nature (2003) 423(6936):190–3. doi: 10.1038/nature01605
155. Galkina SI, Molotkovsky JG, Ullrich V, Sud’ina GF. Scanning Electron Microscopy Study of Neutrophil Membrane Tubulovesicular Extensions (Cytonemes) and Their Role in Anchoring, Aggregation and Phagocytosis. The Effect of Nitric Oxide. Exp Cell Res (2005) 304(2):620–9. doi: 10.1016/j.yexcr.2004.12.005
156. Dong C, Cao J, Struble EJ, Lipowsky HH. Mechanics of Leukocyte Deformation and Adhesion to Endothelium in Shear Flow. Ann Biomed Eng (1999) 27(3):298–312. doi: 10.1114/1.143
157. Firrell JC, Lipowsky HH. Leukocyte Margination and Deformation in Mesenteric Venules of Rat. Am J Physiol (1989) 256(6 Pt 2):H1667–74. doi: 10.1152/ajpheart.1989.256.6.H1667
158. Damiano ER, Westheider J, Tözeren A, Ley K. Variation in the Velocity, Deformation, and Adhesion Energy Density of Leukocytes Rolling Within Venules. Circ Res (1996) 79(6):1122–30. doi: 10.1161/01.res.79.6.1122
159. Sundd P, Gutierrez E, Pospieszalska MK, Zhang H, Groisman A, Ley K. Quantitative Dynamic Footprinting Microscopy Reveals Mechanisms of Neutrophil Rolling. Nat Methods (2010) 7(10):821–4. doi: 10.1038/nmeth.1508
160. Yago T, Wu J, Wey CD, Klopocki AG, Zhu C, McEver RP. Catch Bonds Govern Adhesion Through L-Selectin at Threshold Shear. J Cell Biol (2004) 166(6):913–23. doi: 10.1083/jcb.200403144
161. McEver RP. Selectins: Lectins That Initiate Cell Adhesion Under Flow. Curr Opin Cell Biol (2002) 14(5):581–6. doi: 10.1016/s0955-0674(02)00367-8
162. Lee D, Schultz JB, Knauf PA, King MR. Mechanical Shedding of L-Selectin From the Neutrophil Surface During Rolling on Sialyl Lewis X Under Flow. J Biol Chem (2007) 282(7):4812–20. doi: 10.1074/jbc.M609994200
163. Bruehl RE, Springer TA, Bainton DF. Quantitation of L-Selectin Distribution on Human Leukocyte Microvilli by Immunogold Labeling and Electron Microscopy. J Histochem Cytochem (1996) 44(8):835–44. doi: 10.1177/44.8.8756756
164. Zöllner O, Lenter MC, Blanks JE, Borges E, Steegmaier M, Zerwes HG, et al. L-Selectin From Human, But Not From Mouse Neutrophils Binds Directly to E-Selectin. J Cell Biol (1997) 136(3):707–16. doi: 10.1083/jcb.136.3.707
165. Ivetic A. Signals Regulating L-Selectin-Dependent Leucocyte Adhesion and Transmigration. Int J Biochem Cell Biol (2013) 45(3):550–5. doi: 10.1016/j.biocel.2012.12.023
166. McEver RP. Selectins: Initiators of Leucocyte Adhesion and Signalling at the Vascular Wall. Cardiovasc Res (2015) 107(3):331–9. doi: 10.1093/cvr/cvv154
167. Sarangapani KK, Yago T, Klopocki AG, Lawrence MB, Fieger CB, Rosen SD, et al. Low Force Decelerates L-Selectin Dissociation From P-Selectin Glycoprotein Ligand-1 and Endoglycan. J Biol Chem (2004) 279(3):2291–8. doi: 10.1074/jbc.M310396200
168. Hafezi-Moghadam A, Ley K. Relevance of L-Selectin Shedding for Leukocyte Rolling In Vivo. J Exp Med (1999) 189(6):939–48. doi: 10.1084/jem.189.6.939
169. Hafezi-Moghadam A, Thomas KL, Prorock AJ, Huo Y, Ley K. L-Selectin Shedding Regulates Leukocyte Recruitment. J Exp Med (2001) 193(7):863–72. doi: 10.1084/jem.193.7.863
170. Peng S, Chen S-B, Li L-D, Tong C-F, Li N, Lü S-Q, et al. Impact of Real-Time Shedding on Binding Kinetics of Membrane-Remaining L-Selectin to PSGL-1. Am J Physiol Cell Physiol (2019) 316(5):C678–89. doi: 10.1152/ajpcell.00212.2018
171. Allport JR, Ding HT, Ager A, Steeber DA, Tedder TF, Luscinskas FW. L-Selectin Shedding Does Not Regulate Human Neutrophil Attachment, Rolling, or Transmigration Across Human Vascular Endothelium In Vitro. J Immunol (1997) 158(9):4365–72.
172. Ivetic A, Hoskins Green HL, Hart SJ. L-Selectin: A Major Regulator of Leukocyte Adhesion, Migration and Signaling. Front Immunol (2019) 10:1068. doi: 10.3389/fimmu.2019.01068
173. McEver RP, Zhu C. Rolling Cell Adhesion. Annu Rev Cell Dev Biol (2010) 26:363–96. doi: 10.1146/annurev.cellbio.042308.113238
174. Wayman AM, Chen W, McEver RP, Zhu C. Triphasic Force Dependence of E-Selectin/Ligand Dissociation Governs Cell Rolling Under Flow. Biophys J (2010) 99(4):1166–74. doi: 10.1016/j.bpj.2010.05.040
175. Lou J, Yago T, Klopocki AG, Mehta P, Chen W, Zarnitsyna VI, et al. Flow-Enhanced Adhesion Regulated by a Selectin Interdomain Hinge. J Cell Biol (2006) 174(7):1107–17. doi: 10.1083/jcb.200606056
176. Zhu C, Yago T, Lou J, Zarnitsyna VI, McEver RP. Mechanisms for Flow-Enhanced Cell Adhesion. Ann BioMed Eng (2008) 36(4):604–21. doi: 10.1007/s10439-008-9464-5
177. Beste MT, Hammer DA. Selectin Catch–Slip Kinetics Encode Shear Threshold Adhesive Behavior of Rolling Leukocytes. PNAS (2008) 105(52):20716–21. doi: 10.1073/pnas.0808213105
178. Caputo KE, Lee D, King MR, Hammer DA. Adhesive Dynamics Simulations of the Shear Threshold Effect for Leukocytes. Biophys J (2007) 92(3):787–97. doi: 10.1529/biophysj.106.082321
179. Waugh RE, Hochmuth RM. Mechanical Equilibrium of Thick, Hollow, Liquid Membrane Cylinders. Biophys J (1987) 52(3):391–400. doi: 10.1016/S0006-3495(87)83227-7
180. Sundd P, Gutierrez E, Koltsova EK, Kuwano Y, Fukuda S, Pospieszalska MK, et al. ’Slings’ Enable Neutrophil Rolling at High Shear. Nature (2012) 488(7411):399–403. doi: 10.1038/nature11248
181. Chen W, Lou J, Zhu C. Forcing Switch From Short- to Intermediate- and Long-Lived States of the αa Domain Generates LFA-1/ICAM-1 Catch Bonds. J Biol Chem (2010) 285(46):35967–78. doi: 10.1074/jbc.M110.155770
182. Ramachandran V, Williams M, Yago T, Schmidtke DW, McEver RP. Dynamic Alterations of Membrane Tethers Stabilize Leukocyte Rolling on P-Selectin. Proc Natl Acad Sci USA (2004) 101(37):13519–24. doi: 10.1073/pnas.0403608101
183. Kunkel EJ, Chomas JE, Ley K. Role of Primary and Secondary Capture for Leukocyte Accumulation In Vivo. Circ Res (1998) 82(1):30–8. doi: 10.1161/01.res.82.1.30
184. Eriksson EE, Xie X, Werr J, Thoren P, Lindbom L. Importance of Primary Capture and L-Selectin-Dependent Secondary Capture in Leukocyte Accumulation in Inflammation and Atherosclerosis In Vivo. J Exp Med (2001) 194(2):205–18. doi: 10.1084/jem.194.2.205
185. Rivera-Nieves J, Burcin TL, Olson TS, Morris MA, McDuffie M, Cominelli F, et al. Critical Role of Endothelial P-Selectin Glycoprotein Ligand 1 in Chronic Murine Ileitis. J Exp Med (2006) 203(4):907–17. doi: 10.1084/jem.20052530
186. Eriksson EE. Intravital Microscopy on Atherosclerosis in Apolipoprotein E-Deficient Mice Establishes Microvessels as Major Entry Pathways for Leukocytes to Advanced Lesions. Circulation (2011) 124(19):2129–38. doi: 10.1161/CIRCULATIONAHA.111.030627
187. Salvermoser M, Begandt D, Alon R, Walzog B. Nuclear Deformation During Neutrophil Migration at Sites of Inflammation. Front Immunol (2018) 9:2680. doi: 10.3389/fimmu.2018.02680
188. Nourshargh S, Hordijk PL, Sixt M. Breaching Multiple Barriers: Leukocyte Motility Through Venular Walls and the Interstitium. Nat Rev Mol Cell Biol (2010) 11(5):366–78. doi: 10.1038/nrm2889
189. Huttenlocher A, Horwitz AR. Integrins in Cell Migration. Cold Spring Harb Perspect Biol (2011) 3(9):a005074. doi: 10.1101/cshperspect.a005074
190. Lindbom L, Werr J. Integrin-Dependent Neutrophil Migration in Extravascular Tissue. Semin Immunol (2002) 14(2):115–21. doi: 10.1006/smim.2001.0348
191. Sixt M, Hallmann R, Wendler O, Scharffetter-Kochanek K, Sorokin LM. Cell Adhesion and Migration Properties of β2-Integrin Negative Polymorphonuclear Granulocytes on Defined Extracellular Matrix Molecules. J Biol Chem (2001) 276(22):18878–87. doi: 10.1074/jbc.M010898200
192. Wolf K, Te Lindert M, Krause M, Alexander S, Te Riet J, Willis AL, et al. Physical Limits of Cell Migration: Control by ECM Space and Nuclear Deformation and Tuning by Proteolysis and Traction Force. J Cell Biol (2013) 201(7):1069–84. doi: 10.1083/jcb.201210152
193. Friedl P, Borgmann S, Bröcker EB. Amoeboid Leukocyte Crawling Through Extracellular Matrix: Lessons From the Dictyostelium Paradigm of Cell Movement. J Leukoc Biol (2001) 70(4):491–509. doi: 10.1189/jlb.70.4.491
194. van Goethem E, Poincloux R, Gauffre F, Maridonneau-Parini I, Le Cabec V. Matrix Architecture Dictates Three-Dimensional Migration Modes of Human Macrophages: Differential Involvement of Proteases and Podosome-Like Structures. J Immunol (2010) 184(2):1049–61. doi: 10.4049/jimmunol.0902223
195. Jennings RT, Knaus UG. Neutrophil Migration Through Extracellular Matrix. Methods Mol Biol (2014) 1124:209–18. doi: 10.1007/978-1-62703-845-4_13
196. Cox EA, Huttenlocher A. Regulation of Integrin-Mediated Adhesion During Cell Migration. Microsc Res Tech (1998) 43(5):412–9. doi: 10.1002/(SICI)1097-0029(19981201)43:5<412:AID-JEMT7>3.0.CO;2-F
197. Kuntz RM, Saltzman WM. Neutrophil Motility in Extracellular Matrix Gels: Mesh Size and Adhesion Affect Speed of Migration. Biophys J (1997) 72(3):1472–80. doi: 10.1016/S0006-3495(97)78793-9
198. Sorokin L. The Impact of the Extracellular Matrix on Inflammation. Nat Rev Immunol (2010) 10(10):712–23. doi: 10.1038/nri2852
199. Kraus RF, Gruber MA, Kieninger M. The Influence of Extracellular Tissue on Neutrophil Function and its Possible Linkage to Inflammatory Diseases. Immun Inflammation Dis (2021) 9(4):1237–51. doi: 10.1002/iid3.472
200. Houghton AM, Quintero PA, Perkins DL, Kobayashi DK, Kelley DG, Marconcini LA, et al. Elastin Fragments Drive Disease Progression in a Murine Model of Emphysema. J Clin Invest (2006) 116(3):753–9. doi: 10.1172/JCI25617
201. Ospelt C, Gay S. TLRs and Chronic Inflammation. Int J Biochem Cell Biol (2010) 42(4):495–505. doi: 10.1016/j.biocel.2009.10.010
202. Gaggar A, Jackson PL, Noerager BD, O’Reilly PJ, McQuaid DB, Rowe SM, et al. A Novel Proteolytic Cascade Generates an Extracellular Matrix-Derived Chemoattractant in Chronic Neutrophilic Inflammation. J Immunol (2008) 180(8):5662–9. doi: 10.4049/jimmunol.180.8.5662
203. Weathington NM, van Houwelingen AH, Noerager BD, Jackson PL, Kraneveld AD, Galin FS, et al. A Novel Peptide CXCR Ligand Derived From Extracellular Matrix Degradation During Airway Inflammation. Nat Med (2006) 12(3):317–23. doi: 10.1038/nm1361
204. Nissen G, Hollaender H, Tang FS, Wegmann M, Lunding L, Vock C, et al. Tumstatin Fragment Selectively Inhibits Neutrophil Infiltration in Experimental Asthma Exacerbation. Clin Exp Allergy (2018) 48(11):1483–93. doi: 10.1111/cea.13236
205. Zhu Y, Huang Y, Ji Q, Fu S, Gu J, Tai N, et al. Interplay Between Extracellular Matrix and Neutrophils in Diseases. J Immunol Res (2021) 2021(4):1–11. doi: 10.1155/2021/8243378
206. Byrd AS, O’Brien XM, Johnson CM, Lavigne LM, Reichner JS. An Extracellular Matrix–Based Mechanism of Rapid Neutrophil Extracellular Trap Formation in Response to Candida Albicans. J Immunol (2013) 190(8):4136–48. doi: 10.4049/jimmunol.1202671
207. Johnson CJ, Cabezas-Olcoz J, Kernien JF, Wang SX, Beebe DJ, Huttenlocher A, et al. The Extracellular Matrix of Candida Albicans Biofilms Impairs Formation of Neutrophil Extracellular Traps. PloS Pathog (2016) 12(9):e1005884. doi: 10.1371/journal.ppat.1005884
208. Xu J, Mao X, Jin R, Yin J, Lu K, Guo Y, et al. Neutrophil Extracellular Traps Degrade Fibronectin in a Rat Model of Bronchopulmonary Dysplasia Induced by Perinatal Exposure to Lipopolysaccharide. J Cell Mol Med (2020) 24(24):14645–9. doi: 10.1111/jcmm.15842
209. Frangogiannis NG. The Extracellular Matrix in Myocardial Injury, Repair, and Remodeling. J Clin Invest (2017) 127(5):1600–12. doi: 10.1172/JCI87491
210. O’Dwyer DN, Gurczynski SJ, Moore BB. Pulmonary Immunity and Extracellular Matrix Interactions. Matrix Biol (2018) 73(3):122–34. doi: 10.1016/j.matbio.2018.04.003
211. Liang W, Ferrara N. The Complex Role of Neutrophils in Tumor Angiogenesis and Metastasis. Cancer Immunol Res (2016) 4(2):83–91. doi: 10.1158/2326-6066.CIR-15-0313
212. Coffelt SB, Wellenstein MD, de Visser KE. Neutrophils in Cancer: Neutral No More. Nat Rev Cancer (2016) 16(7):431–46. doi: 10.1038/nrc.2016.52
213. Nicolás-Ávila JÁ, Adrover JM, Hidalgo A. Neutrophils in Homeostasis, Immunity, and Cancer. Immunity (2017) 46(1):15–28. doi: 10.1016/j.immuni.2016.12.012
214. Winkler J, Abisoye-Ogunniyan A, Metcalf KJ, Werb Z. Concepts of Extracellular Matrix Remodelling in Tumour Progression and Metastasis. Nat Commun (2020) 11(1):5120. doi: 10.1016/j.jconrel.2017.01.034
215. Moghe PV, Nelson RD, Tranquillo RT. Cytokine-Stimulated Chemotaxis of Human Neutrophils in a 3-D Conjoined Fibrin Gel Assay. J Immunol Methods (1995) 180(2):193–211. doi: 10.1016/0022-1759(94)00314-M
216. Petri B, Sanz M-J. Neutrophil Chemotaxis. Cell Tissue Res (2018) 371(3):425–36. doi: 10.1007/s00441-017-2776-8
217. Pablo I, Peter D. Navigating Through Models of Chemotaxis. Curr Opin Cell Biol (2008) 20(1):35–40. doi: 10.1016/j.ceb.2007.11.011
218. Foxman EF, Campbell JJ, Butcher EC. Multistep Navigation and the Combinatorial Control of Leukocyte Chemotaxis. J Cell Biol (1997) 139(5):1349–60. doi: 10.1083/jcb.139.5.1349
219. Heit B, Tavener S, Raharjo E, Kubes P. An Intracellular Signaling Hierarchy Determines Direction of Migration in Opposing Chemotactic Gradients. J Cell Biol (2002) 159(1):91–102. doi: 10.1083/jcb.200202114
220. Rot A. Endothelial Cell Binding of NAP-1/IL-8: Role in Neutrophil Emigration. Immunol Today (1992) 13(8):291–4. doi: 10.1016/0167-5699(92)90039-A
221. Rot A. Binding of Neutrophil Attractant/Activation Protein-1 (Interleukin 8) to Resident Dermal Cells. Cytokine (1992) 4(5):347–52. doi: 10.1016/1043-4666(92)90077-5
222. Rot A. Neutrophil Attractant/Activation Protein-1 (Interleukin-8) Induces In Vitro Neutrophil Migration by Haptotactic Mechanism. Eur J Immunol (1993) 23(1):303–6. doi: 10.1002/eji.1830230150
223. Handel TM, Johnson Z, Crown SE, Lau EK, Proudfoot AE. Regulation of Protein Function by Glycosaminoglycans – as Exemplified by Chemokines. Annu Rev Biochem (2005) 74:385–410. doi: 10.1146/annurev.biochem.72.121801.161747
224. Wang L, Fuster M, Sriramarao P, Esko JD. Endothelial Heparan Sulfate Deficiency Impairs L-Selectin- and Chemokine-Mediated Neutrophil Trafficking During Inflammatory Responses. Nat Immunol (2005) 6(9):902–10. doi: 10.1038/ni1233
225. Middleton J, Patterson AM, Gardner L, Schmutz C, Ashton BA. Leukocyte Extravasation: Chemokine Transport and Presentation by the Endothelium. Blood (2002) 100(12):3853–60. doi: 10.1182/blood.V100.12.3853
226. Graham GJ, Handel TM, Proudfoot AE. Leukocyte Adhesion: Reconceptualizing Chemokine Presentation by Glycosaminoglycans. Trends Immunol (2019) 40(6):472–81. doi: 10.1016/j.it.2019.03.009
227. Parish CR. Heparan Sulfate and Inflammation. Nat Immunol (2005) 6(9):861–2. doi: 10.1038/ni0905-861
228. Entschladen F, Gunzer M, Scheuffele CM, Niggemann B, Zänker KS. T Lymphocytes and Neutrophil Granulocytes Differ in Regulatory Signaling and Migratory Dynamics With Regard to Spontaneous Locomotion and Chemotaxis. Cell Immunol (2000) 199(2):104–14. doi: 10.1006/cimm.1999.1605
229. Yadav SK, Stojkov D, Feigelson SW, Roncato F, Simon H-U, Yousefi S, et al. Chemokine-Triggered Microtubule Polymerization Promotes Neutrophil Chemotaxis and Invasion But Not Transendothelial Migration. J Leukoc Biol (2019) 105(4):755–66. doi: 10.1002/JLB.3A1118-437RR
230. Anderson DC, Wible LJ, Hughes BJ, Smith CW, Brinkley BR. Cytoplasmic Microtubules in Polymorphonuclear Leukocytes: Effects of Chemotactic Stimulation and Colchicine. Cell (1982) 31(3 Pt 2):719–29. doi: 10.1016/0092-8674(82)90326-9
231. Eddy RJ, Pierini LM, Maxfield FR. Microtubule Asymmetry During Neutrophil Polarization and Migration. Mol Biol Cell (2002) 13(12):4470–83. doi: 10.1091/mbc.e02-04-0241
232. Luxton GG, Gundersen GG. Orientation and Function of the Nuclear–Centrosomal Axis During Cell Migration. Curr Opin Cell Biol (2011) 23(5):579–88. doi: 10.1016/j.ceb.2011.08.001
233. Renkawitz Jörg, Kopf A, Stopp J, de Vries I, Driscoll MK, Merrin J, et al. Nuclear Positioning Facilitates Amoeboid Migration Along the Path of Least Resistance. Nature (2019) 568(7753):546–50. doi: 10.1038/s41586-019-1087-5
234. Chiplonkar JM, Vandré DD, Robinson JM. Stimulus-Dependent Relocation of the Microtubule Organizing Center in Human Polymorphonuclear Leukocytes. J Cell Sci (1992) 102(Pt 4):723–8. doi: 10.1242/jcs.102.4.723
235. Schliwa M, Pryzwansky KB, Euteneuer U. Centrosome Splitting in Neutrophils: An Unusual Phenomenon Related to Cell Activation and Motility. Cell (1982) 31(3 Pt 2):705–17. doi: 10.1016/0092-8674(82)90325-7
236. Yoo SaK, Lam P-y, Eichelberg MR, Zasadil L, Bement WM, Huttenlocher A. The Role of Microtubules in Neutrophil Polarity and Migration in Live Zebrafish. J Cell Sci (2012) 125(23):5702–10. doi: 10.1242/jcs.108324
237. Peiseler M, Kubes P. More Friend Than Foe: The Emerging Role of Neutrophils in Tissue Repair. J Clin Invest (2019) 129(7):2629–39. doi: 10.1172/JCI124616
238. Wang J. Neutrophils in Tissue Injury and Repair. Cell Tissue Res (2018) 371(3):531–9. doi: 10.1007/s00441-017-2785-7
239. Burn T, Alvarez JI. Reverse Transendothelial Cell Migration in Inflammation: To Help or to Hinder? Cell Mol Life Sci (2017) 74(10):1871–81. doi: 10.1007/s00018-016-2444-2
240. Bratton DL, Henson PM. Neutrophil Clearance: When the Party is Over, Clean-Up Begins. Trends Immunol (2011) 32(8):350–7. doi: 10.1016/j.it.2011.04.009
241. Wang J, Hossain M, Thanabalasuriar A, Gunzer M, Cynthia M, Paul K. Visualizing the Function and Fate of Neutrophils in Sterile Injury and Repair. Science (2017) 358(6359):111–6. doi: 10.1126/science.aam9690
242. Hirano Y, Aziz M, Wang P. Role of Reverse Transendothelial Migration of Neutrophils in Inflammation. Biol Chem (2016) 397(6):497–506. doi: 10.1515/hsz-2015-0309
243. Hyun Y-M, Hong C-W. Deep Insight Into Neutrophil Trafficking in Various Organs. J Leukoc Biol (2017) 102(3):617–29. doi: 10.1189/jlb.1RU1216-521R
244. Woodfin A, Voisin M-B, Beyrau M, Colom B, Caille D, Diapouli F-M, et al. The Junctional Adhesion Molecule JAM-C Regulates Polarized Transendothelial Migration of Neutrophils In Vivo. Nat Immunol (2011) 12(8):761–9. doi: 10.1038/ni.2062
245. Nourshargh S, Renshaw SA, Imhof BA. Reverse Migration of Neutrophils: Where, When, How, and Why? Trends Immunol (2016) 37(5):273–86. doi: 10.1016/j.it.2016.03.006
246. Hughes J, Johnson RJ, Mooney A, Hugo C, Gordon K, Savill J. Neutrophil Fate in Experimental Glomerular Capillary Injury in the Rat. Emigration Exceeds in Situ Clearance by Apoptosis. Am J Pathol (1997) 150(1):223–34.
247. Kienle K, Lämmermann T. Neutrophil Swarming: An Essential Process of the Neutrophil Tissue Response. Immunol Rev (2016) 273(1):76–93. doi: 10.1111/imr.12458
248. Mathias JR, Perrin BJ, Liu T-X, Kanki J, Look AT, Huttenlocher A. Resolution of Inflammation by Retrograde Chemotaxis of Neutrophils in Transgenic Zebrafish. J Leukoc Biol (2006) 80(6):1281–8. doi: 10.1189/jlb.0506346
249. Tauzin S, Starnes TW, Becker FB, Lam P-Y, Huttenlocher A. Redox and Src Family Kinase Signaling Control Leukocyte Wound Attraction and Neutrophil Reverse Migration. J Cell Biol (2014) 207(5):589–98. doi: 10.1083/jcb.201408090
250. Yoo SK, Huttenlocher A. Spatiotemporal Photolabeling of Neutrophil Trafficking During Inflammation in Live Zebrafish. J Leukoc Biol (2011) 89(5):661–7. doi: 10.1189/jlb.1010567
251. Elks PM, van Eeden FJ, Dixon G, Wang X, Reyes-Aldasoro CC, Ingham PW, et al. Activation of Hypoxia-Inducible Factor-1α (Hif-1α) Delays Inflammation Resolution by Reducing Neutrophil Apoptosis and Reverse Migration in a Zebrafish Inflammation Model. Blood (2011) 118(3):712–22. doi: 10.1182/blood-2010-12-324186
252. Buckley CD, Ross EA, McGettrick HM, Osborne CE, Haworth O, Schmutz C, et al. Identification of a Phenotypically and Functionally Distinct Population of Long-Lived Neutrophils in a Model of Reverse Endothelial Migration. J Leukoc Biol (2006) 79(2):303–11. doi: 10.1189/jlb.0905496
253. Powell DR, Huttenlocher A. Neutrophils in the Tumor Microenvironment. Trends Immunol (2016) 37(1):41–52. doi: 10.1016/j.it.2015.11.008
254. Langereis JD. Neutrophil Integrin Affinity Regulation in Adhesion, Migration, and Bacterial Clearance. Cell Adh Migr (2013) 7(6):476–81. doi: 10.4161/cam.27293
255. Tharp WG, Yadav R, Irimia D, Upadhyaya A, Samadani A, Hurtado O, et al. Neutrophil Chemorepulsion in Defined Interleukin-8 Gradients In Vitro and In Vivo. J Leukoc Biol (2006) 79(3):539–54. doi: 10.1189/jlb.0905516
256. Weber C, Fraemohs L, Dejana E. The Role of Junctional Adhesion Molecules in Vascular Inflammation. Nat Rev Immunol (2007) 7(6):467–77. doi: 10.1038/nri2096
257. Zindel J, Kubes P. DAMPs, PAMPs, and LAMPs in Immunity and Sterile Inflammation. Annu Rev Pathol (2020) 15:493–518. doi: 10.1146/annurev-pathmechdis-012419-032847
258. Colom B, Bodkin JV, Beyrau M, Woodfin A, Ody C, Rourke C, et al. Leukotriene B4-Neutrophil Elastase Axis Drives Neutrophil Reverse Transendothelial Cell Migration In Vivo. Immunity (2015) 42(6):1075–86. doi: 10.1016/j.immuni.2015.05.010
259. Zhang D, Chen G, Manwani D, Mortha A, Xu C, Faith JJ, et al. Neutrophil Ageing is Regulated by the Microbiome. Nature (2015) 525(7570):528–32. doi: 10.1038/nature15367
260. Brinkmann V, Zychlinsky A. Neutrophil Extracellular Traps: Is Immunity the Second Function of Chromatin? J Cell Biol (2012) 198(5):773–83. doi: 10.1083/jcb.201203170
261. Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, et al. Extracellular Histones Promote Thrombin Generation Through Platelet-Dependent Mechanisms: Involvement of Platelet TLR2 and TLR4. Blood (2011) 118(7):1952–61. doi: 10.1182/blood-2011-03-343061
262. Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD, et al. Extracellular DNA Traps Promote Thrombosis. PNAS (2010) 107(36):15880–5. doi: 10.1073/pnas.1005743107
263. Weisenburger-Lile D, Dong Y, Yger M, Weisenburger G, Polara GF, Chaigneau T, et al. Harmful Neutrophil Subsets in Patients With Ischemic Stroke: Association With Disease Severity. Neurol Neuroimmunol Neuroinflamm (2019) 6(4):e571. doi: 10.1212/NXI.0000000000000571
264. Lohri C, Schaltegger CS, van den Broek M, Wenger RH, Ruegg C, Fink D, et al. Neutrophil Expression of ICAM1, CXCR1, and VEGFR1 in Patients With Breast Cancer Before and After Adjuvant Chemotherapy. Anticancer Res (2014) 34(9):4693–9.
265. Wang JH, Sexton DM, Redmond HP, Watson RW, Croke DT, Bouchier-Hayes D. Intercellular Adhesion Molecule-1 (ICAM-1) is Expressed on Human Neutrophils and is Essential for Neutrophil Adherence and Aggregation. Shock (1997) 8(5):357–61. doi: 10.1097/00024382-199711000-00007
266. Maletto BA, Ropolo AS, Alignani DO, Liscovsky MV, Ranocchia RP, Moron VG, et al. Presence of Neutrophil-Bearing Antigen in Lymphoid Organs of Immune Mice. Blood (2006) 108(9):3094–102. doi: 10.1182/blood-2006-04-016659
267. Lim JJ, Grinstein S, Roth Z. Diversity and Versatility of Phagocytosis: Roles in Innate Immunity, Tissue Remodeling, and Homeostasis. Front Cell Infect Microbiol (2017) 7:191. doi: 10.3389/fcimb.2017.00191
268. Brown GC, Neher JJ. Eaten Alive!: Cell Death by Primary Phagocytosis: ‘Phagoptosis’. Trends Biochem Sci (2012) 37(8):325–32. doi: 10.1016/j.tibs.2012.05.002
269. Gordon S. Phagocytosis: An Immunobiologic Process. Immunity (2016) 44(3):463–75. doi: 10.1016/j.immuni.2016.02.026
270. Hart J. Inflammation: Its Role in the Healing of Acute Wounds. J Wound Care (2002) 11(6):205–9. doi: 10.12968/jowc.2002.11.6.26411
271. Segal AW, Dorling J, Coade S. Kinetics of Fusion of the Cytoplasmic Granules With Phagocytic Vacuoles in Human Polymorphonuclear Leukocytes. Biochemical and Morphological Studies. J Cell Biol (1980) 85(1):42–59. doi: 10.1083/jcb.85.1.42
272. Kerscher B, Willment JA, Brown GD. The Dectin-2 Family of C-Type Lectin-Like Receptors: An Update. Int Immunol (2013) 25(5):271–7. doi: 10.1093/intimm/dxt006
273. Kimura Y, Inoue A, Hangai S, Saijo S, Negishi H, Nishio J, et al. The Innate Immune Receptor Dectin-2 Mediates the Phagocytosis of Cancer Cells by Kupffer Cells for the Suppression of Liver Metastasis. PNAS (2016) 113(49):14097–102. doi: 10.1073/pnas.1617903113
274. Kennedy AD, Willment JA, Dorward DW, Williams DL, Brown GD, DeLeo FR. Dectin-1 Promotes Fungicidal Activity of Human Neutrophils. Eur J Immunol (2007) 37(2):467–78. doi: 10.1002/eji.200636653
275. Hochreiter-Hufford A, Ravichandran KS. Clearing the Dead: Apoptotic Cell Sensing, Recognition, Engulfment, and Digestion. Cold Spring Harb Perspect Biol (2013) 5(1):a008748–a008748. doi: 10.1101/cshperspect.a008748
276. Nordenfelt P, Tapper H. Phagosome Dynamics During Phagocytosis by Neutrophils. J Leukoc Biol (2011) 90(2):271–84. doi: 10.1189/jlb.0810457
277. Hoffmann JJ. Neutrophil CD64: A Diagnostic Marker for Infection and Sepsis. Clin Chem Lab Med (2009) 47(8):903–16. doi: 10.1515/CCLM.2009.224
278. Tollis S, Dart AE, Tzircotis G, Endres RG. The Zipper Mechanism in Phagocytosis: Energetic Requirements and Variability in Phagocytic Cup Shape. BMC Syst Biol (2010) 4:149. doi: 10.1186/1752-0509-4-149
279. Swanson JA, Hoppe AD. The Coordination of Signaling During Fc Receptor-Mediated Phagocytosis. J Leukoc Biol (2004) 76(6):1093–103. doi: 10.1189/jlb.0804439
280. Kinchen JM, Ravichandran KS. Phagosome Maturation: Going Through the Acid Test. Nat Rev Mol Cell Biol (2008) 9(10):781–95. doi: 10.1038/nrm2515
281. Teng T-S, Ji A-L, Ji X-Y, Li Y-Z. Neutrophils and Immunity: From Bactericidal Action to Being Conquered. J Immunol Res (2017) 2017:9671604. doi: 10.1155/2017/9671604
282. Thammavongsa V, Kim HK, Missiakas D, Schneewind O. Staphylococcal Manipulation of Host Immune Responses. Nat Rev Microbiol (2015) 13(9):529–43. doi: 10.1038/nrmicro3521
283. Doblinger N, Bredthauer A, Mohrez M, Hähnel V, Graf B, Gruber M, et al. Impact of Hydroxyethyl Starch and Modified Fluid Gelatin on Granulocyte Phenotype and Function. Transfusion (2019) 59(6):2121–30. doi: 10.1111/trf.15279
285. Weckmann M, Becker T, Nissen G, Pech M, Kopp MV. SiMA: A Simplified Migration Assay for Analyzing Neutrophil Migration. Cytometry A (2017) 91(7):675–85. doi: 10.1002/cyto.a.23114
286. Hampton MB, Kettle AJ, Winterbourn CC. Inside the Neutrophil Phagosome: Oxidants, Myeloperoxidase, and Bacterial Killing. Blood (1998) 92(9):3007–17. doi: 10.1182/blood.V92.9.3007.421k47_3007_3017
287. El-Benna J, Hurtado-Nedelec M, Marzaioli V, Marie J-C, Gougerot-Pocidalo M-A, Dang PM-C. Priming of the Neutrophil Respiratory Burst: Role in Host Defense and Inflammation. Immunol Rev (2016) 273(1):180–93. doi: 10.1111/imr.12447
288. Faurschou M, Borregaard N. Neutrophil Granules and Secretory Vesicles in Inflammation. Microbes Infect (2003) 5(14):1317–27. doi: 10.1016/j.micinf.2003.09.008
289. Aratani Y. Myeloperoxidase: Its Role for Host Defense, Inflammation, and Neutrophil Function. Arch Biochem Biophys (2018) 640:47–52. doi: 10.1016/j.abb.2018.01.004
290. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil Extracellular Traps Kill Bacteria. Science (2004) 303(5663):1532–5. doi: 10.1126/science.1092385
291. Pilsczek FH, Salina D, Poon KK, Fahey C, Yipp BG, Sibley CD, et al. A Novel Mechanism of Rapid Nuclear Neutrophil Extracellular Trap Formation in Response to Staphylococcus Aureus. J Immunol (2010) 185(12):7413–25. doi: 10.4049/jimmunol.1000675
292. Delgado-Rizo V, Martínez-Guzmán MA, Iñiguez-Gutierrez L, García-Orozco A, Alvarado-Navarro A, Fafutis-Morris M. Neutrophil Extracellular Traps and Its Implications in Inflammation: An Overview. Front Immunol (2017) 8:81. doi: 10.3389/fimmu.2017.00081
293. Al-Khafaji AB, Tohme S, Yazdani HO, Miller D, Huang H, Tsung A. Superoxide Induces Neutrophil Extracellular Trap Formation in a TLR-4 and NOX-Dependent Mechanism. Mol Med (2016) 22:621–31. doi: 10.2119/molmed.2016.00054
294. Masuda S, Nakazawa D, Shida H, Miyoshi A, Kusunoki Y, Tomaru U, et al. NETosis Markers: Quest for Specific, Objective, and Quantitative Markers. Clin Chim Acta (2016) 459:89–93. doi: 10.1016/j.cca.2016.05.029
295. Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y. PAD4 is Essential for Antibacterial Innate Immunity Mediated by Neutrophil Extracellular Traps. J Exp Med (2010) 207(9):1853–62. doi: 10.1084/jem.20100239
296. Lewis HD, Liddle J, Coote JE, Atkinson SJ, Barker MD, Bax BD, et al. Inhibition of PAD4 Activity is Sufficient to Disrupt Mouse and Human NET Formation. Nat Chem Biol (2015) 11(3):189–91. doi: 10.1038/nchembio.1735
297. Neeli I, Khan SN, Radic M. Histone Deimination as a Response to Inflammatory Stimuli in Neutrophils. J Immunol (2008) 180(3):1895–902. doi: 10.4049/jimmunol.180.3.1895
298. Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil Elastase and Myeloperoxidase Regulate the Formation of Neutrophil Extracellular Traps. J Cell Biol (2010) 191(3):677–91. doi: 10.1083/jcb.201006052
299. Yang H, Biermann MH, Brauner JM, Liu Y, Zhao Y, Herrmann M. New Insights Into Neutrophil Extracellular Traps: Mechanisms of Formation and Role in Inflammation. Front Immunol (2016) 7:302. doi: 10.3389/fimmu.2016.00302
300. Brinkmann V, Zychlinsky A. Beneficial Suicide: Why Neutrophils Die to Make NETs. Nat Rev Microbiol (2007) 5(8):577–82. doi: 10.1038/nrmicro1710
301. Neeli I, Dwivedi N, Khan S, Radic M. Regulation of Extracellular Chromatin Release From Neutrophils. J Innate Immun (2009) 1(3):194–201. doi: 10.1159/000206974
302. Branitzki-Heinemann K, Möllerherm H, Völlger L, Husein DM, de Buhr N, Blodkamp S, et al. Formation of Neutrophil Extracellular Traps Under Low Oxygen Level. Front Immunol (2016) 7:518. doi: 10.3389/fimmu.2016.00518
303. Douda DN, Khan MA, Grasemann H, Palaniyar N. SK3 Channel and Mitochondrial ROS Mediate NADPH Oxidase-Independent NETosis Induced by Calcium Influx. Proc Natl Acad Sci USA (2015) 112(9):2817–22. doi: 10.1073/pnas.1414055112
304. Yipp BG, Kubes P. NETosis: How Vital is it? Blood (2013) 122(16):2784–94. doi: 10.1182/blood-2013-04-457671
305. Byrd AS, O’Brien XM, Johnson CM, Lavigne LM, Reichner JS. An Extracellular Matrix-Based Mechanism of Rapid Neutrophil Extracellular Trap Formation in Response to Candida Albicans. J Immunol (2013) 190(8):4136–48. doi: 10.4049/jimmunol.1202671
306. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 Activates Neutrophil Extracellular Traps to Ensnare Bacteria in Septic Blood. Nat Med (2007) 13(4):463–9. doi: 10.1038/nm1565
307. Yipp BG, Petri B, Salina D, Jenne CN, Scott BN, Zbytnuik LD, et al. Infection-Induced NETosis is a Dynamic Process Involving Neutrophil Multitasking. vivo Nat Med (2012) 18(9):1386–93. doi: 10.1038/nm.2847
308. Carestia A, Kaufman T, Rivadeneyra L, Landoni VI, Pozner RG, Negrotto S, et al. Mediators and Molecular Pathways Involved in the Regulation of Neutrophil Extracellular Trap Formation Mediated by Activated Platelets. J Leukoc Biol (2016) 99(1):153–62. doi: 10.1189/jlb.3A0415-161R
309. Lominadze G, Powell DW, Luerman GC, Link AJ, Ward RA, McLeish KR. Proteomic Analysis of Human Neutrophil Granules. Mol Cell Proteomics (2005) 4(10):1503–21. doi: 10.1074/mcp.M500143-MCP200
311. Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU. Viable Neutrophils Release Mitochondrial DNA to Form Neutrophil Extracellular Traps. Cell Death Differ (2009) 16(11):1438–44. doi: 10.1038/cdd.2009.96
312. Gupta S, Kaplan MJ. The Role of Neutrophils and NETosis in Autoimmune and Renal Diseases. nrneph (2016) 12(7):402–13. doi: 10.1038/nrneph.2016.71
313. Manfredi AA, Ramirez GA, Rovere-Querini P, Maugeri N. The Neutrophil’s Choice: Phagocytose vs Make Neutrophil Extracellular Traps. Front Immunol (2018) 9:288. doi: 10.3389/fimmu.2018.00288
314. Branzk N, Lubojemska A, Hardison SE, Wang Q, Gutierrez MG, Brown GD, et al. Neutrophils Sense Microbe Size and Selectively Release Neutrophil Extracellular Traps in Response to Large Pathogens. Nat Immunol (2014) 15(11):1017–25. doi: 10.1038/ni.2987
315. Metzler KD, Fuchs TA, Nauseef WM, Reumaux D, Roesler J, Schulze I, et al. Myeloperoxidase is Required for Neutrophil Extracellular Trap Formation: Implications for Innate Immunity. Blood (2011) 117(3):953–9. doi: 10.1182/blood-2010-06-290171
316. Parker H, Dragunow M, Hampton MB, Kettle AJ, Winterbourn CC. Requirements for NADPH Oxidase and Myeloperoxidase in Neutrophil Extracellular Trap Formation Differ Depending on the Stimulus. J Leukoc Biol (2012) 92(4):841–9. doi: 10.1189/jlb.1211601
317. Ullah I, Ritchie ND, Evans TJ. The Interrelationship Between Phagocytosis, Autophagy and Formation of Neutrophil Extracellular Traps Following Infection of Human Neutrophils by Streptococcus Pneumoniae. Innate Immun (2017) 23(5):413–23. doi: 10.1177/1753425917704299
318. Pelletier MG, Szymczak K, Barbeau AM, Prata GN, O’Fallon KS, Gaines P. Characterization of Neutrophils and Macrophages From Ex Vivo-Cultured Murine Bone Marrow for Morphologic Maturation and Functional Responses by Imaging Flow Cytometry. Methods (2017) 112:124–46. doi: 10.1016/j.ymeth.2016.09.005
319. Fexby S, Bjarnsholt T, Jensen PØ, Roos V, Høiby N, Givskov M, et al. Biological Trojan Horse: Antigen 43 Provides Specific Bacterial Uptake and Survival in Human Neutrophils. Infect Immun (2007) 75(1):30–4. doi: 10.1128/IAI.01117-06
320. DuMont AL, Yoong P, Surewaard BG, Benson MA, Nijland R, van Strijp JA, et al. Staphylococcus Aureus Elaborates Leukocidin AB to Mediate Escape From Within Human Neutrophils. Infect Immun (2013) 81(5):1830–41. doi: 10.1128/IAI.00095-13
321. Surewaard BG, de Haas CJ, Vervoort F, Rigby KM, DeLeo FR, Otto M, et al. Staphylococcal Alpha-Phenol Soluble Modulins Contribute to Neutrophil Lysis After Phagocytosis. Cell Microbiol (2013) 15(8):1427–37. doi: 10.1111/cmi.12130
322. Genestier A-L, Michallet M-C, Prévost G, Bellot G, Chalabreysse L, Peyrol S, et al. Staphylococcus Aureus Panton-Valentine Leukocidin Directly Targets Mitochondria and Induces Bax-Independent Apoptosis of Human Neutrophils. J Clin Invest (2005) 115(11):3117–27. doi: 10.1172/JCI22684
323. Nazareth H, Genagon SA, Russo TA. Extraintestinal Pathogenic Escherichia Coli Survives Within Neutrophils. Infect Immun (2007) 75(6):2776–85. doi: 10.1128/IAI.01095-06
324. Galkina SI, Fedorova NV, Stadnichuk VI, Sud’ina GF. Membrane Tubulovesicular Extensions (Cytonemes). Cell Adh Migr (2013) 7(2):174–86. doi: 10.4161/cam.23130
325. Leithner A, Eichner A, Müller J, Reversat A, Brown M, Schwarz J, et al. Diversified Actin Protrusions Promote Environmental Exploration But are Dispensable for Locomotion of Leukocytes. Nat Cell Biol (2016) 18(11):1253–9. doi: 10.1038/ncb3426
326. Galkina SI, Romanova JM, Stadnichuk VI, Molotkovsky JG, Sud’ina GF, Klein T. Nitric Oxide-Induced Membrane Tubulovesicular Extensions (Cytonemes) of Human Neutrophils Catch and Hold Salmonella Enterica Serovar Typhimurium at a Distance From the Cell Surface. FEMS Immunol Med Microbiol (2009) 56(2):162–71. doi: 10.1111/j.1574-695X.2009.00560.x
327. Galkina SI, Sud’ina GF, Ullrich V. Inhibition of Neutrophil Spreading During Adhesion to Fibronectin Reveals Formation of Long Tubulovesicular Cell Extensions (Cytonemes). Exp Cell Res (2001) 266(2):222–8. doi: 10.1006/excr.2001.5227
328. Galkina SI, Fedorova NV, Serebryakova MV, Romanova JM, Golyshev SA, Stadnichuk VI, et al. Proteome Analysis Identified Human Neutrophil Membrane Tubulovesicular Extensions (Cytonemes, Membrane Tethers) as Bactericide Trafficking. Biochim Biophys Acta (BBA) - Gen Subj (2012) 1820(11):1705–14. doi: 10.1016/j.bbagen.2012.06.016
329. Galkina SI, Fedorova NV, Serebryakova MV, Arifulin EA, Stadnichuk VI, Gaponova TV, et al. Inhibition of the GTPase Dynamin or Actin Depolymerisation Initiates Outward Plasma Membrane Tubulation/Vesiculation (Cytoneme Formation) in Neutrophils. Biol Cell (2015) 107(5):144–58. doi: 10.1111/boc.201400063
330. Buszczak M, Inaba M, Yamashita YM. Signaling by Cellular Protrusions: Keeping the Conversation Private. Trends Cell Biol (2016) 26(7):526–34. doi: 10.1016/j.tcb.2016.03.003
331. Kornberg TB. The Contrasting Roles of Primary Cilia and Cytonemes in Hh Signaling. Dev Biol (2014) 394(1):1–5. doi: 10.1016/j.ydbio.2014.07.015
332. Kornberg TB, Roy S. Cytonemes as Specialized Signaling Filopodia. Development (2014) 141(4):729–36. doi: 10.1242/dev.086223
333. Fairchild CL, Barna M. Specialized Filopodia: At the ’Tip’ of Morphogen Transport and Vertebrate Tissue Patterning. Curr Opin Genet Dev (2014) 27:67–73. doi: 10.1016/j.gde.2014.03.013
334. Galkina SI, Stadnichuk VI, Molotkovsky JG, Romanova JM, Sud’ina GF, Klein T. Microbial Alkaloid Staurosporine Induces Formation of Nanometer-Wide Membrane Tubular Extensions (Cytonemes, Membrane Tethers) in Human Neutrophils. Cell Adh Migr (2014) 4(1):32–8. doi: 10.4161/cam.4.1.10314
335. Corriden R, Self T, Akong-Moore K, Nizet V, Kellam B, Briddon SJ, et al. Adenosine-A 3 Receptors in Neutrophil Microdomains Promote the Formation of Bacteria-Tethering Cytonemes. EMBO Rep (2013) 14(8):726–32. doi: 10.1038/embor.2013.89
336. Chen Y, Junger WG. Measurement of Oxidative Burst in Neutrophils. Methods Mol Biol (2012) 844:115–24. doi: 10.1007/978-1-61779-527-5_8
337. Arnold DE. Heimall JR. A Review of Chronic Granulomatous Disease. Adv Ther (2017) 34(12):2543–57. doi: 10.1007/s12325-017-0636-2
338. van de Vijver E, van den Berg TK, Kuijpers TW. Leukocyte Adhesion Deficiencies. Hematol Oncol Clin North Am (2013) 27(1):S. 101–116, viii. doi: 10.1016/j.hoc.2012.10.001
339. Spoor J, Farajifarda H, Rezaeia N. Congenital Neutropenia and Primary Immunodeficiency Diseases. Crit Rev Oncology/Hematology (2019) 133:149–62. doi: 10.1016/j.critrevonc.2018.10.003
340. Segel GB, Halterman MW, Lichtman MA. The Paradox of the Neutrophil’s Role in Tissue Injury. J Leukoc Biol (2011) 89(3):359–72. doi: 10.1189/jlb.0910538
341. Shen X-F, Cao K, Jiang J-P, Guan W-X, Du J-F. Neutrophil Dysregulation During Sepsis: An Overview and Update. J Cell Mol Med (2017) 21(9):1687–97. doi: 10.1111/jcmm.13112
342. Chen C, Huang T, Zhai X, Ma Y, Xie L, Lu B, et al. Targeting Neutrophils as a Novel Therapeutic Strategy After Stroke. J Cereb Blood Flow Metab (2021) 41(9):2150–61. doi: 10.1177/0271678X211000137
343. Veras FP, Pontelli MC, Silva CM, Toller-Kawahisa JE, de Lima M, Nascimento DC, et al. SARS-CoV-2-Triggered Neutrophil Extracellular Traps Mediate COVID-19 Pathology. J Exp Med (2020) 217(12):e20201129. doi: 10.1084/jem.20201129
344. Leppkes M, Knopf J, Naschberger E, Lindemann A, Singh J, Herrmann I, et al. Vascular Occlusion by Neutrophil Extracellular Traps in COVID-19. EBioMedicine (2020) 58(102925):1–9. doi: 10.1016/j.ebiom.2020.102925
345. Stefan B. Chemisches Signal Und Biologische Antwort: Modulation Der Generierung Reaktiver Sauerstoffverbindungen Aus Neutrophilen Granulozyten. Leipzig: Akademische Verlagsanstalt Engelsdorf (2000).
346. Bardoel BW, Kenny EF, Sollberger G, Zychlinsky A. The Balancing Act of Neutrophils. Cell Host Microbe (2014) 15(5):526–36. doi: 10.1016/j.chom.2014.04.011
347. Alavi A, French LE, Davis MD, Brassard A, Kirsner RS. Pyoderma Gangrenosum: An Update on Pathophysiology, Diagnosis and Treatment. Am J Clin Dermatol (2017) 18(3):355–72. doi: 10.1007/s40257-017-0251-7
348. Zenaro E, Pietronigro E, Della Bianca V, Piacentino G, Marongiu L, Budui S, et al. Neutrophils Promote Alzheimer’s Disease-Like Pathology and Cognitive Decline via LFA-1 Integrin. Nat Med (2015) 21(8):880–6. doi: 10.1038/nm.3913
349. Woodberry T, Bouffler S, Wilson A, Buckland R, Brüstle A. The Emerging Role of Neutrophil Granulocytes in Multiple Sclerosis. JCM (2018) 7(12):511. doi: 10.3390/jcm7120511
350. Manda-Handzlik A, Demkow U. The Brain Entangled: The Contribution of Neutrophil Extracellular Traps to the Diseases of the Central Nervous System. Cells (2019) 8(12):1477. doi: 10.3390/cells8121477
351. Boeltz S, Muñoz LE, Fuchs TA, Herrmann M. Neutrophil Extracellular Traps Open the Pandora’s Box in Severe Malaria. Front Immunol (2017) 8:874. doi: 10.3389/fimmu.2017.00874
352. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-Related Inflammation. Nature (2008) 454(7203):436–44. doi: 10.1038/nature07205
353. Kim J, Bae J-S. Tumor-Associated Macrophages and Neutrophils in Tumor Microenvironment. Mediators Inflammation (2016) 2016. doi: 10.1155/2016/6058147
354. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the Activation and Regulation of Innate and Adaptive Immunity. Nat Rev Immunol (2011) 11(8):519–31. doi: 10.1038/nri3024
355. Fridlender ZG, Sun J, Mishalian I, Singhal S, Cheng G, Kapoor V, et al. Transcriptomic Analysis Comparing Tumor-Associated Neutrophils With Granulocytic Myeloid-Derived Suppressor Cells and Normal Neutrophils. PloS One (2012) 7(2):e31524. doi: 10.1371/journal.pone.0031524
356. Perobelli SM, Galvani RG, Gonçalves-Silva T, Xavier CR, Nóbrega A, Bonomo A. Plasticity of Neutrophils Reveals Modulatory Capacity. Braz J Med Biol Res (2015) 48(8):665–75. doi: 10.1590/1414-431X20154524
357. Cortez-Retamozo V, Etzrodt M, Newton A, Rauch PJ, Chudnovskiy A, Berger C, et al. Origins of Tumor-Associated Macrophages and Neutrophils. Proc Natl Acad Sci USA (2012) 109(7):2491–6. doi: 10.1073/pnas.1113744109
358. Elpek KG, Cremasco V, Shen H, Harvey CJ, Wucherpfennig KW, Goldstein DR, et al. The Tumor Microenvironment Shapes Lineage, Transcriptional, and Functional Diversity of Infiltrating Myeloid Cells. Cancer Immunol Res (2014) 2(7):655–67. doi: 10.1158/2326-6066.CIR-13-0209
359. Di Carlo E, Forni G, Lollini P, Colombo MP, Modesti A, Musiani P. The Intriguing Role of Polymorphonuclear Neutrophils in Antitumor Reactions. Blood (2001) 97(2):339–45. doi: 10.1182/blood.V97.2.339
360. Springer TA. Traffic Signals on Endothelium for Lymphocyte Recirculation and Leukocyte Emigration. Annu Rev Physiol (1995) 57:827–72. doi: 10.1146/annurev.ph.57.030195.004143
361. Carlos TM, Harlan JM. Leukocyte-Endothelial Adhesion Molecules. Blood (1994) 84(7):2068–101. doi: 10.1182/blood.V84.7.2068.bloodjournal8472068
362. Bevilacqua MP, Stengelin S, Gimbrone MA, Seed B. Endothelial Leukocyte Adhesion Molecule 1: An Inducible Receptor for Neutrophils Related to Complement Regulatory Proteins and Lectins. Science (1989) 243(4895):1160–5. doi: 10.1126/science.2466335
363. Pober JS, Bevilacqua MP, Mendrick DL, Lapierre LA, Fiers W, Gimbrone MA. Two Distinct Monokines, Interleukin 1 and Tumor Necrosis Factor, Each Independently Induce Biosynthesis and Transient Expression of the Same Antigen on the Surface of Cultured Human Vascular Endothelial Cells. J Immunol (1986) 136(5):1680–7.
364. Dustin ML, Rothlein R, Bhan AK, Dinarello CA, Springer TA. Induction by IL 1 and Interferon-Gamma: Tissue Distribution, Biochemistry, and Function of a Natural Adherence Molecule (ICAM-1). J Immunol (1986) 137(1):245–54.
365. Mantovani A. The Chemokine System: Redundancy for Robust Outputs. Immunol Today (1999) 20(6):254–7. doi: 10.1016/s0167-5699(99)01469-3
366. Di Carlo E, Coletti A, Modesti A, Giovarelli M, Forni G, Musiani P. Local Release of Interleukin-10 by Transfected Mouse Adenocarcinoma Cells Exhibits Pro- and Anti-Inflammatory Activity and Results in a Delayed Tumor Rejection. Eur Cytokine Netw (1998) 9(1):61–8.
367. Vora M, Romero LI, Karasek MA. Interleukin-10 Induces E-Selectin on Small and Large Blood Vessel Endothelial Cells. J Exp Med (1996) 184(3):821–9. doi: 10.1084/jem.184.3.821
368. De Waal Malefyt R, Yssel H, Roncarolo MG, Spits H, De Vries JE. Interleukin-10. Curr Opin Immunol (1992) 4(3):314–20. doi: 10.1016/0952-7915(92)90082-P
369. Berkman N, John M, Roesems G, Jose PJ, Barnes PJ, Chung KF. Inhibition of Macrophage Inflammatory Protein-1 Alpha Expression by IL-10. Differential Sensitivities in Human Blood Monocytes and Alveolar Macrophages. J Immunol (1995) 155(9):4412–8.
370. Kasama T, Strieter RM, Lukacs NW, Burdick MD, Kunkel SL. Regulation of Neutrophil-Derived Chemokine Expression by IL-10. J Immunol (1994) 152(7):3559–69.
371. Vedder NB, Fouty BW, Winn RK, Harlan JM, Rice CL. Role of Neutrophils in Generalized Reperfusion Injury Associated With Resuscitation From Shock. Surgery (1989) 106(3):509–16.
372. Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limón P. The Polarization of Immune Cells in the Tumour Environment by TGFbeta. Nat Rev Immunol (2010) 10(8):554–67. doi: 10.1038/nri2808
373. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of Tumor-Associated Neutrophil Phenotype by TGF-Beta: "N1" Versus "N2" TAN. Cancer Cell (2009) 16(3):183–94. doi: 10.1016/j.ccr.2009.06.017
374. Piccard H, Muschel RJ, Opdenakker G. On the Dual Roles and Polarized Phenotypes of Neutrophils in Tumor Development and Progression. Crit Rev Oncology/Hematology (2012) 82(3):296–309. doi: 10.1016/j.critrevonc.2011.06.004
375. Kobayashi Y. The Role of Chemokines in Neutrophil Biology. Front Biosci (2008) 13:2400–7. doi: 10.2741/2853
376. Nozawa H, Chiu C, Hanahan D. Infiltrating Neutrophils Mediate the Initial Angiogenic Switch in a Mouse Model of Multistage Carcinogenesis. Proc Natl Acad Sci USA (2006) 103(33):12493–8. doi: 10.1073/pnas.0601807103
377. Pekarek LA, Starr BA, Toledano AY, Schreiber H. Inhibition of Tumor Growth by Elimination of Granulocytes. J Exp Med (1995) 181(1):435–40. doi: 10.1084/jem.181.1.435
378. Tazawa H, Okada F, Kobayashi T, Tada M, Mori Y, Une Y, et al. Infiltration of Neutrophils Is Required for Acquisition of Metastatic Phenotype of Benign Murine Fibrosarcoma Cells. Am J Pathol (2003) 163(6):2221–32. doi: 10.1016/S0002-9440(10)63580-8
379. Schmielau J, Finn OJ. Activated Granulocytes and Granulocyte-Derived Hydrogen Peroxide are the Underlying Mechanism of Suppression of T-Cell Function in Advanced Cancer Patients. Cancer Res (2001) 61(12):4756–60.
380. Colombo MP, Modesti A, Parmiani G, Forni G. Local Cytokine Availability Elicits Tumor Rejection and Systemic Immunity Through Granulocyte-T-Lymphocyte Cross-Talk. Cancer Res (1992) 52(18):4853–7.
381. Hicks AM, Riedlinger G, Willingham MC, Alexander-Miller MA, von Kap-Herr C, Pettenati MJ, et al. Transferable Anticancer Innate Immunity in Spontaneous Regression/Complete Resistance Mice. Proc Natl Acad Sci USA (2006) 103(20):7753–8. doi: 10.1073/pnas.0602382103
382. Colombo MP, Lombardi L, Stoppacciaro A, Melani C, Parenza M, Bottazzi B, et al. Granulocyte Colony-Stimulating Factor (G-CSF) Gene Transduction in Murine Adenocarcinoma Drives Neutrophil-Mediated Tumor Inhibition In Vivo. Neutrophils Discriminate Between G-CSF-Producing and G-CSF-Nonproducing Tumor Cells. J Immunol (1992) 149(1):113–9.
383. Musiani P, Allione A, Modica A, Lollini PL, Giovarelli M, Cavallo F, et al. Role of Neutrophils and Lymphocytes in Inhibition of a Mouse Mammary Adenocarcinoma Engineered to Release IL-2, IL-4, IL-7, IL-10, IFN-Alpha, IFN-Gamma, and TNF-Alpha. Lab Invest J Tech Methods Pathol (1996) 74(1):146–57.
384. Stoppacciaro A, Melani C, Parenza M, Mastracchio A, Bassi C, Baroni C, et al. Regression of an Established Tumor Genetically Modified to Release Granulocyte Colony-Stimulating Factor Requires Granulocyte-T Cell Cooperation and T Cell-Produced Interferon Gamma. J Exp Med (1993) 178(1):151–61. doi: 10.1084/jem.178.1.151
385. Singhal S, Bhojnagarwala PS, O’Brien S, Moon EK, Garfall AL, Rao AS, et al. Origin and Role of a Subset of Tumor-Associated Neutrophils With Antigen-Presenting Cell Features in Early-Stage Human Lung Cancer. Cancer Cell (2016) 30(1):120–35. doi: 10.1016/j.ccell.2016.06.001
386. Shen M, Hu P, Donskov F, Wang G, Liu Q, Du J. Tumor-Associated Neutrophils as a New Prognostic Factor in Cancer: A Systematic Review and Meta-Analysis. PloS One (2014) 9(6):e98259. doi: 10.1371/journal.pone.0098259
387. Templeton AJ, McNamara MG, Šeruga B, Vera-Badillo FE, Aneja P, Ocaña A, et al. Prognostic Role of Neutrophil-To-Lymphocyte Ratio in Solid Tumors: A Systematic Review and Meta-Analysis. J Natl Cancer Inst (2014) 106(6):dju124. doi: 10.1093/jnci/dju124
388. Walsh SR, Cook EJ, Goulder F, Justin TA, Keeling NJ. Neutrophil-Lymphocyte Ratio as a Prognostic Factor in Colorectal Cancer. J Surg Oncol (2005) 91(3):181–4. doi: 10.1002/jso.20329
389. Sarraf KM, Belcher E, Raevsky E, Nicholson AG, Goldstraw P, Lim E. Neutrophil/lymphocyte Ratio and its Association With Survival After Complete Resection in non-Small Cell Lung Cancer. J Thorac Cardiovasc Surg (2009) 137(2):425–8. doi: 10.1016/j.jtcvs.2008.05.046
390. Zeindler J, Angehrn F, Droeser R, Däster S, Piscuoglio S, Ng CK, et al. Infiltration by Myeloperoxidase-Positive Neutrophils is an Independent Prognostic Factor in Breast Cancer. Breast Cancer Res Treat (2019) 177(3):581–9. doi: 10.1007/s10549-019-05336-3
391. Kolarova H, Klinke A, Kremserova S, Adam M, Pekarova M, Baldus S, et al. Myeloperoxidase Induces the Priming of Platelets. Free Radic Biol Med (2013) 61:357–69. doi: 10.1016/j.freeradbiomed.2013.04.014
392. Ishizuka M, Nagata H, Takagi K, Iwasaki Y, Kubota K. Combination of Platelet Count and Neutrophil to Lymphocyte Ratio is a Useful Predictor of Postoperative Survival in Patients With Colorectal Cancer. Br J Cancer (2013) 109(2):401–7. doi: 10.1038/bjc.2013.350
393. Cools-Lartigue J, Spicer J, McDonald B, Gowing S, Chow S, Giannias B, et al. Neutrophil Extracellular Traps Sequester Circulating Tumor Cells and Promote Metastasis. J Clin Invest (2013) 123(8):3446–58. doi: 10.1172/JCI67484
394. Sangaletti S, Tripodo C, Vitali C, Portararo P, Guarnotta C, Casalini P, et al. Defective Stromal Remodeling and Neutrophil Extracellular Traps in Lymphoid Tissues Favor the Transition From Autoimmunity to Lymphoma. Cancer Discovery (2014) 4(1):110–29. doi: 10.1158/2159-8290.CD-13-0276
395. Berger-Achituv S, Brinkmann V, Abed UA, Kühn LI, Ben-Ezra J, Elhasid R, et al. A Proposed Role for Neutrophil Extracellular Traps in Cancer Immunoediting. Front Immunol (2013) 4:48. doi: 10.3389/fimmu.2013.00048
396. Hiramatsu S, Tanaka H, Nishimura J, Yamakoshi Y, Sakimura C, Tamura T, et al. Gastric Cancer Cells Alter the Immunosuppressive Function of Neutrophils. Oncol Rep (2020) 43(1):251–9. doi: 10.3892/or.2019.7410
397. Giovarelli M, Musiani P, Modesti A, Dellabona P, Casorati G, Allione A, et al. Local Release of IL-10 by Transfected Mouse Mammary Adenocarcinoma Cells Does Not Suppress But Enhances Antitumor Reaction and Elicits a Strong Cytotoxic Lymphocyte and Antibody-Dependent Immune Memory. J Immunol (1995) 155(6):3112–23.
398. Zak KM, Grudnik P, Magiera K, Dömling A, Dubin G, Holak TA. Structural Biology of the Immune Checkpoint Receptor PD-1 and Its Ligands PD-L1/PD-L2. Structure (2017) 25(8):1163–74. doi: 10.1016/j.str.2017.06.011
Keywords: microtubule organization center, NEtosis, tumor association, neutrophil (PMN) function, neutrophil extravasation, extracellular matrix (ECM), chemotactic gradients, bidirectional (trans)migration
Citation: Kraus RF and Gruber MA (2021) Neutrophils—From Bone Marrow to First-Line Defense of the Innate Immune System. Front. Immunol. 12:767175. doi: 10.3389/fimmu.2021.767175
Received: 30 August 2021; Accepted: 03 December 2021;
Published: 23 December 2021.
Edited by:
Deirdre R. Coombe, Curtin University, AustraliaReviewed by:
Benyamin Rosental, Ben-Gurion University of the Negev, IsraelCharles W. Frevert, University of Washington Tacoma, United States
Copyright © 2021 Kraus and Gruber. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Richard Felix Kraus, UmljaGFyZC1GZWxpeC5LcmF1c0BzdHVkLnVuaS1yZWdlbnNidXJnLmRl
†ORCID: Richard Felix Kraus, orcid.org/0000-0001-5280-6530
Michael Andreas Gruber, orcid.org/0000-0002-5509-9760