 Patrick Haubruck
Patrick Haubruck Marlene Magalhaes Pinto3
Marlene Magalhaes Pinto3 Christopher B. Little
Christopher B. Little Rebecca Gentek
Rebecca Gentek- 1Centre for Orthopaedics, Trauma Surgery and Spinal Cord Injury, Trauma and Reconstructive Surgery, Heidelberg University Hospital, Heidelberg, Germany
- 2Raymond Purves Bone and Joint Research Laboratory, Kolling Institute, Institute of Bone and Joint Research, Faculty of Medicine and Health University of Sydney, Royal North Shore Hospital, St. Leonards, NSW, Australia
- 3Centre for Inflammation Research & Centre for Reproductive Health, University of Edinburgh, Edinburgh, United Kingdom
- 4Clinic of Orthopaedics and Trauma Surgery, University Clinic of Schleswig-Holstein, Kiel, Germany
Synovial joints are complex structures that enable normal locomotion. Following injury, they undergo a series of changes, including a prevalent inflammatory response. This increases the risk for development of osteoarthritis (OA), the most common joint disorder. In healthy joints, macrophages are the predominant immune cells. They regulate bone turnover, constantly scavenge debris from the joint cavity and, together with synovial fibroblasts, form a protective barrier. Macrophages thus work in concert with the non-hematopoietic stroma. In turn, the stroma provides a scaffold as well as molecular signals for macrophage survival and functional imprinting: “a macrophage niche”. These intricate cellular interactions are susceptible to perturbations like those induced by joint injury. With this review, we explore how the concepts of local tissue niches apply to synovial joints. We introduce the joint micro-anatomy and cellular players, and discuss their potential interactions in healthy joints, with an emphasis on molecular cues underlying their crosstalk and relevance to joint functionality. We then consider how these interactions are perturbed by joint injury and how they may contribute to OA pathogenesis. We conclude by discussing how understanding these changes might help identify novel therapeutic avenues with the potential of restoring joint function and reducing post-traumatic OA risk.
Introduction
Osteoarthritis (OA) is the most common joint disorder (1). Its prevalence is expected to increase further (2) due to rising societal levels of ageing, obesity and injury, key risk factors for OA. While the disease commonly affects knees, hips, hands and feet, OA of the knee accounts for more than 80% of the disease burden (1, 3). The knee is particularly susceptible to injury, with approximately 40% of patients that suffer a traumatic knee injury developing so-called “post-traumatic” (pt)OA, and surgical reconstruction and restoration of joint biomechanics insufficient to prevent its development (4). Treatment options for OA are very limited, and there is a particular need for effective preventive and disease modifying drugs (DMD). This is highlighted by clinical data showing comparable disease burden at diagnosis but significantly higher burden 6 months later in OA compared to rheumatoid arthritis (RA) patients (5). Owing to this paucity of treatment options and the high and rising prevalence, OA contributes substantially to the global burden of disease. In a 2015 survey, OA was identified as the second most prevalent cause for years lived with disability (2), highlighting the impact OA has on both individuals and society (2).
Although the name osteoarthritis implies an inherent inflammatory process (6), it was historically believed that OA had purely biomechanical causes (7). Indeed, OA was regarded a disease of the elderly, inevitably caused by years of “wear and tear”. Breaking with this previously held view, we now know that OA development involves a complex active biological response with local interaction between joint tissues and their resident cells, and these with systemic mediators. This includes an inflammatory response (8) that is accompanied by complex metabolic changes, which contribute to cartilage degradation and activation of bone remodeling (9). Although innate immune cells, and monocytes and macrophages in particular have been implicated (7, 10, 11), the exact nature of the inflammatory response in OA, its underlying mechanisms and its relative contribution to onset or progression of structural pathology and symptoms remain incompletely resolved (12).
Much like OA etiology, our understanding of the complex development and functional heterogeneity of macrophages and monocytes as well as their interactions within local tissue “niches” has dramatically changed in recent years. It is now firmly established that the long-held paradigm of discrete, polarized monocyte and macrophage activation states is an oversimplification of what in reality is a spectrum of cell states. Likewise, it is now recognized that macrophages established from fetal progenitors can persist in adult tissues, and that many macrophages self-maintain independently of monocytes (13–16). Lastly, we are beginning to appreciate that macrophages engage in bidirectional crosstalk with other cell types within their local niches, interactions that are of mutual benefit and implicate macrophages as gatekeepers of tissue function (17, 18). While much remains to be learned and confirmed, these concepts developed in other organs and tissues appear to also apply to macrophages in joints (19, 20). Indeed, macrophages found in the healthy synovium are predominantly monocyte-independent, and they protect and contribute to joint homeostasis in several ways, including barrier formation, clearance of debris and even lubrication (20). Under inflammatory conditions, such as may occur with joint injury however, monocytes are recruited to the affected joint and can differentiate into macrophages, which retain a more inflammatory phenotype (21). These joint macrophage populations thus not only differ in their origins, but also exert distinct functions (22, 23).
Modifying the developmental, functional and in situ dynamics of joint macrophages and monocytes might therefore represent an attractive avenue for novel therapeutic approaches in OA. This may be particularly relevant in ptOA, where causal initiation and subsequent temporal changes in monocytes, macrophages and their activation with disease onset and progression may be targeted. This review aims to explore this notion, with a focus on the synovial rather than osseous joint tissue niche. We will summarize experimental and clinical studies on macrophages and monocytes in healthy and diseased joints and interpret these in the context of current paradigms of myeloid biology. Our emphasis in this review is on joint injury and ptOA, as this represents the major OA phenotype studied in pre-clinical research, and as noted above, it has the most well demarcated disease stages and thus potentially the broadest therapeutic opportunity. In doing so, we hope to bridge persisting gaps between bench and bedside and highlight research questions with the potential to pave the way towards better treatment options for ptOA, but also other OA phenotypes more broadly.
Macro- and Microanatomy of the Knee Joint
Synovial joints provide critical motion segments that allow low friction movement between rigid (osseous) skeletal components. They enable diverse and essential bio-mechanical functions ranging from fine movements of arms, hands and fingers through to walking, running and jumping. The knee represents an anatomically complex example of a joint (Figure 1) that enables locomotion in a variety of terrains, while minimizing muscular energy requirements and absorbing and redistributing forces that originate from the contact between the walking surface and the foot (24). Its main osseous components are the femur, tibia and patella, that articulate in two locations: the tibiofemoral and patellofemoral joints. The menisci, two C-shaped fibro-cartilaginous structures, absorb and distribute load between the femoral and tibial surfaces. Together with a multitude of extra- and intra-articular ligaments and the fibrous joint capsule, the menisci also provide stability in flexion/extension and rotation, enabling the unique biomechanical function of the knee (25). As in all joints, the osseous surfaces in the knee are covered by hyaline cartilage, a sparsely cellular, deformable connective tissue matrix with key components of collagen type II and highly hydrated proteoglycans. Cartilage is heterogenous and can be broadly divided into three zones based on depth from the surface. These have distinct composition, biomechanical properties and functions (26). Chondrocytes make up about 2% of the articular cartilage volume (27) and are responsible for the maintenance and repair of the cartilage extracellular matrix. They are highly specialized cells derived from mesenchymal stem cells that have limited potential for replication in situ, but can react to a plethora of mechanical and molecular stimuli (26). The knee also harbors several adipose tissues. These are located intra-articularly and extra-synovially, and include the infrapatellar fat pad, which can be considered a highly specialized compartment in the sublining interstitial tissue [in humans known as Hoffa’s fat pad (28)]. Beyond filling the space in the joint cavity and absorbing shock, adipose tissues also secrete cytokines and adipokines (29, 30) and are therefore potent immune-modulators. It is also believed that the infrapatellar fat pad engages in intimate crosstalk with the synovial membrane, a specialized connective tissue that lines the inner surface of the joint capsule (31). The synovial membrane consists of three layers: The intimal lining layer is found closest to the joint cavity and consists mainly of macrophages (“type A cells”) and fibroblasts (“type B cells”) that show low degrees of cell division (31). Beneath this is the vascularized subintimal layer, also referred to as sublining interstitial tissue, and finally a fibrous stromal layer forming the joint capsule. The synovial membrane maintains joint homeostasis by providing lubrication and nutrition to the cartilage. It also forms a semi-permeable protective barrier that controls the molecular traffic in and out of the joint (32) and renders the synovial cavity relatively immune-privileged (20). Because of its critical role in joint homeostasis, this review will largely focus on monocyte and macrophage biology of the synovial membrane, including its sublining interstitial layer.
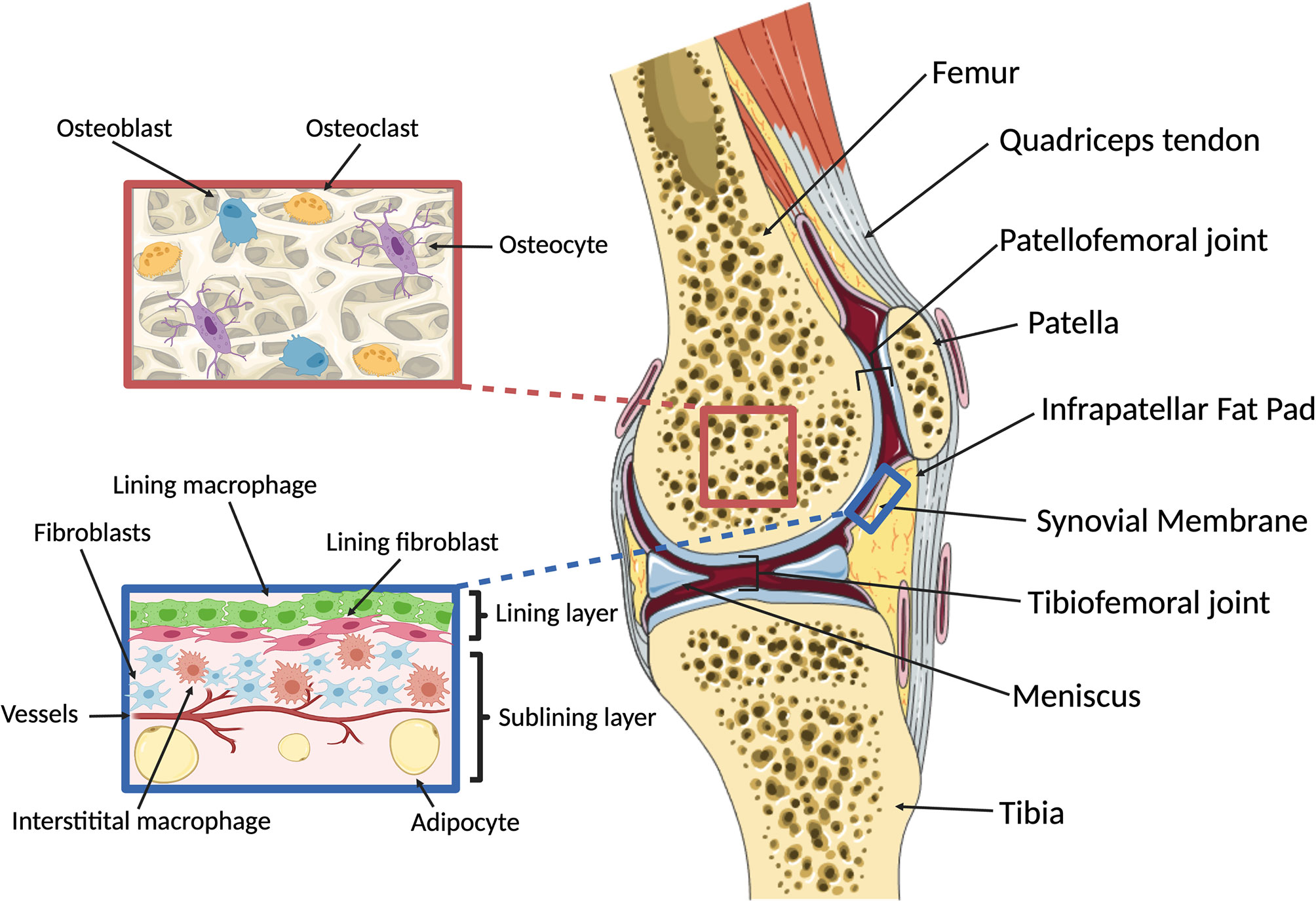
Figure 1 Overview of knee macro- and microanatomy. (Right) Sagittal cut through a human knee. The femur and tibia articulate in the tibiofemoral joint, with two fibrocartilaginous menisci serving to provide rotational and anterior-posterior stability and load distribution. The patella is a hypomochlion (pivot point) for the quadriceps tendon that articulates with the femur in the patellofemoral joint. The intra-articular components of the osseous structures are covered by cartilage, enabling low-friction bearing. Joint function and stability are maintained by ligaments and the joint capsule. The inner surface of the joint capsule is lined with the synovial membrane, which is accompanied by adipose tissues located intra-articularly and extra-synovially, including the infrapatellar fat pad. (Left, top) Microanatomy of subchondral bone. The cellular components of bone include osteoblasts, osteoclasts and osteocytes that dynamically respond to changes in mechanical loading and potentially communicate with the overlying cartilage via soluble signals. (Left, bottom) Microanatomy of the synovial membrane. The synovium comprises three layers: the intimal lining layer which consists of macrophages and fibroblasts that together form a semi-permeable protective barrier; the vascularized subintimal or sublining layer which contains interstitial macrophages and fibroblasts as well as adipocytes; and an outer fibrous stromal layer forming the joint capsule (not shown). Created with BioRender.com and smart.servier.com.
A Revised View of Monocyte and Macrophage Biology
Many historically held views of monocyte and macrophage biology have been overhauled in recent times, including their phenotypic and functional heterogeneity, developmental dynamics as well as their crosstalk and functional interdependence with other cell types in the same tissue microenvironment.
Monocyte and Macrophage Development
It was previously believed that the key (if not sole) function of monocytes was to produce macrophages, and that in turn, all macrophages found in peripheral tissues originate exclusively from monocytes (33). Elegant studies exploiting genetic fate mapping have since shown that most tissue-resident macrophages are in fact of fetal origin and self-maintain in adult tissues independently of bone marrow (BM)-derived monocytes (33). Indeed, macrophages colonize tissues concomitantly with their development in what appears to be a demand-driven way. They are generated from successive, but overlapping waves of hematopoietic progenitors produced at distinct anatomical sites (34). The majority of fetal macrophages originate from erythro-myeloid progenitors (EMP) produced in the extra-embryonic yolk sac (15, 16). EMP are fetal-restricted progenitors that differentiate into macrophages either directly or via fetal liver intermediates, but as an uncommitted entity do not persist into adulthood.
This new paradigm of predominantly fetal origins of tissue macrophages notwithstanding, monocytes can still complement tissue phagocyte compartments on demand (33). While this applies to some tissues at homeostasis (e.g. skin and gastro-intestinal tract), it is particularly true and important in inflammatory conditions. Importantly, in both scenarios, monocytes themselves have a number of key effector functions (35).
Monocytes differentiate from BM hematopoietic stem cells (HSC) in a strictly hierarchical, tree-like maturation process (35). They share a common progenitor with dendritic cells (DCs) known as “monocyte-macrophage DC progenitor” (MDP) (36, 37), which gives rise to a monocyte-committed intermediate, designated the “common monocyte progenitor” (cMoP) (38). The downstream “transitional pre-monocytes” (TpMos) (38, 39) are believed to be the final intermediate stage in monocyte differentiation (39). They are capable of rapid proliferation and express high levels of C-X-C motif chemokine Receptors (CXCR) 4, which anchors them to the BM. Based on differential expression of Lymphocyte antigen 6C (Ly6C), CX3CR1 and C-Chemokine Receptor type 2 (CCR2) in mice (40) or Cluster of Differentiation (CD)14 and CD16 in humans (37), mature monocytes can be broadly classified into classical (mice: Ly6Chigh CX3CR1low CCR2high; humans: CD14high CD16-) and non-classical monocytes (mice: Ly6Clow CX3CR1high CCR2low; humans: CD14low CD16+) (35, 41–44). This binary classification is now widely established (45) and has more recently been backed up by extensive high-dimensional studies, the latter also revealing a previously underappreciated heterogeneity (46). A third monocyte population with an intermediate phenotype is exclusive to humans (CD14+ CD16+) (35). Classical and non-classical monocytes differ in a number of features, including their relative abundance and the regulatory mechanisms governing their retention in and egress from the BM (47). The mature monocyte compartment in the BM is vastly predominated by Ly6Chigh monocytes, which downregulate CXCR4 (48, 49) and highly express CCR2 (50, 51), collectively enabling their egress from the BM. Ly6Clow monocytes, on the other hand, only express very low levels of CCR2, and while still under investigation, Sphingosine-1-Phosphate Receptor 5 (S1PR5) signaling has been implicated in orchestrating their BM egress (52).
Once released into the blood stream, classical Ly6Chigh monocytes have a relatively short half-life lasting for a mere 20-24 hours in mice (53–55), whereas their non-classical counterparts are slightly longer-lived with a half-life of around 2 days in mice and 7 days in humans (53). The two populations are also developmentally connected: lineage tracing indicating that Ly6Clow monocytes originate from aging Ly6Chigh monocytes (41, 53), a gradual conversion that is dependent on Nuclear Receptor subfamily 4 group A member 1 (NR4A1) signaling (56) and involves direct cellular contact with endothelial cells and Notch signaling (57, 58). Similar mechanisms appear to be at play in human monocytes (59, 60). At homeostasis, Ly6Clow monocytes do not normally extravasate but instead patrol the luminal side of the endothelium (61). They roll along the vascular endothelium, independent of the direction of the blood flow, via CX3CR1, β2 integrin (58, 62) and interactions between Lymphocyte Function-associated Antigen-1 (LFA-1) and IntraCellular Adhesion Molecule 1 (ICAM1) and ICAM2 (58, 62). They have thus been considered the “tissue-resident” macrophages of blood vessels. In non-homeostatic conditions, Ly6Clow monocytes are thought to promote resolution of inflammation, however, they can also contribute to autoimmunity and chronic inflammatory diseases (58), as we will discuss further below. Intriguingly, experiments using bleomycin-induced lung fibrosis in mice identified an alternative pathway to Ly6Clow monocytes, consisting of a separate progenitor referred to as a “Segregated-nucleus-containing atypical Monocyte (SatM)”, whose production depends on the transcription factor C/EBPβ (63). Whether this pathway is relevant to other pathologies remains to be determined.
Unlike their non-classical counterparts, Ly6Chigh monocytes do traffic into peripheral tissues even at steady state (64). In tissues that (partially) rely on homeostatic renewal from the BM, such as the skin and gastro-intestinal tract (65, 66), the majority of recruited Ly6Chigh monocytes gradually differentiate into macrophages, a process phenotypically characterized as a “monocyte waterfall” (67). These macrophages are functionally imprinted in response to local cues that superpose tissue-specific identity onto a transcriptional core lineage program (68, 69). Provided monocytes encounter a homeostatic environment and are allowed sufficient time in the tissue, monocyte-derived macrophages are phenotypically, transcriptomically and epigenetically indistinguishable from pre-existing tissue-resident macrophages (70, 71). However, this is not the case following inflammation or other insults resulting in perturbed homeostasis, which might have important functional implications. Indeed, different and sometimes even opposing roles have been reported for developmentally distinct macrophages in conditions like cancer (72–74) and stroke (75), and this might also be the case in joint pathology, as we will discuss below.
Monocyte Effector Functions
In addition to representing an “on-demand” source for macrophages, monocytes also have important effector functions in their own right. Indeed, a fraction of classical monocytes recruited at steady state maintains their monocytic phenotype with minimal transcriptional changes (76). In the parenchyma of non-lymphoid organs like the skin, lung, and heart (43, 66), they contribute to immune surveillance. During sterile inflammatory responses, as would occur following closed traumatic knee injury, Ly6Chigh and Ly6Clow monocytes are recruited in a highly orchestrated manner facilitated by differential chemokine release. Under such conditions, Ly6Clow monocytes have primarily been attributed beneficial, anti-inflammatory roles. In the ischemic heart and kidney for example, deficiency in Ly6Clow monocytes results in higher inflammatory levels and impaired restoration of organ function (77–79). In line with this, Ly6Clow monocytes predominantly produce anti-inflammatory mediators like Interleukin (IL)-10 (80, 81) as well as Vascular Endothelial Growth Factor (VEGF) and other pro-angiogenic factors (82), as observed during spinal cord injury and myocardial infarction (82), respectively.
Somewhat contradictory evidence exists regarding the role of Ly6Chigh monocytes. Historically, these classical monocytes have been recognized as potent pro-inflammatory effector cells. Indeed, CCR2 knockout mice, which are largely deficient in classical monocytes in the periphery, show decreased levels of IL-1β and Tumor Necrosis Factor (TNF)-α and an increase in the anti-inflammatory cytokines IL-4, IL-5 and IL-13 at the site of inflammation during cerebral ischemia (83). Ly6Chigh monocytes also show high levels of reactive oxygen species, TNF-α and IL-6 (84) in the context of liver ischemia reperfusion injury (84). In line with this pro-inflammatory phenotype, Ly6Chigh monocytes mediate tissue damage in the ischemic liver as well as the heart following myocardial infarction, and contribute to progression of atherosclerosis (84–87). At the same time however, Ly6Chigh monocytes have also been implicated in regression of atherosclerosis (88), although this may be attributable to anti-inflammatory effects of monocyte-derived macrophages, rather than a true monocyte effector function. Nonetheless, these findings collectively suggest that instead of being globally pro- and anti-inflammatory, classical and non-classical monocytes differentially shape the local inflammatory response via the tailored production of cytokines and other cellular mediators. Although similarities exist between tissues and insults, their exact trafficking patterns and effector functions appear to be context-dependent, and therefore need to be delineated specifically in the homeostatic, injured and OA joint.
Tissue Adaptation and Activation of Monocytes and Macrophages
Monocytes and macrophages dynamically respond to a variety of cues in their microenvironment, which shape their local tissue adaptation and activation state. Consequently, although they share a lineage-defining core transcriptomic signature, macrophages in different tissues are transcriptionally, phenotypically and functionally very diverse (70, 71, 89). The core macrophage program is initiated in committed fetal progenitors or BM-derived monocytes and driven by lineage-determining transcription factors (68–71).
Acquisition of tissue-specific identity and function is subsequently orchestrated by additional transcription factors in response to signals present in the local microenvironment (69). In the spleen, for example, heme from senescent red blood cells induces expression of the transcription factor SPI-C, which in turn activates a transcriptional program inducing differentiation of red pulp macrophages (90). Experimental data from adoptive transfer experiments demonstrate that exposure to different environments partially, though not fully, rewires the tissue-specific identity of macrophages, indicating a limited degree of plasticity even under such non-physiological conditions (70, 91).
At the same time, the activation state of terminally differentiated macrophages can vary as a function of microenvironmental signals, in particular cytokines. Historically, it has been thought that macrophages polarize into either classically activated, pro-inflammatory (“M1”) or alternatively activated, anti-inflammatory (“M2”) (92, 93) subtypes in response to cytokines associated respectively with type I or type II immunity (94, 95). However, it is now abundantly clear that this strict dichotomy is a drastic oversimplification of real-life in vivo physiology. Rather, these opposing polarization states represent extremes (94, 95) of a much wider and more fluid spectrum of activation states (96, 97). Understanding the different activation states of macrophages and monocytes in OA and the signals that drive them will be paramount in delineating their respective contribution to disease pathogenesis. Since circulating monocytes represent a modifiable source, they – and their relationship with macrophages found in the joints – are of particular translational relevance.
Macrophage Niches
The intricate developmental dynamics between monocytes and macrophages and their adaptation to tissue-derived signals illustrate that these cell types actively engage with each other and their immediate environment or “niche”. Research into such niches represents a current focus in the field of myeloid cell biology. The niche concept postulates that macrophages are not only functionally imprinted by tissue-specific cues, but that their niches also provide them with a physical scaffold for anchoring and survival factors (17, 18). In turn, macrophages support appropriate functioning of their cellular partners. They thus form mutually beneficial cellular circuits (18) with their niches. In line with this, organ function is heavily impaired in mice lacking numerous tissue-resident macrophages owing to genetic deficiency in Colony Stimulating Factor (CSF)1, a key macrophage survival signal, or its receptor (98–100). Niches consist of macrophages and other, often non-hematopoietic stromal cell types, as well as the extracellular matrix surrounding them, and they can also “call” monocytes for replenishment. In the liver, for example, hepatocytes, endothelial and stellate cells together provide numerous signals to resident Kupffer cells and incoming monocytes, including CSF1, IL-34, CCL2 and Notch ligands (101), whereas in the red pulp of the spleen, macrophages depend on CSF1 produced by fibroblasts (102). In return, macrophages help facilitate tissue-specific functions and homeostasis. Beyond their role in immune surveillance and protective immunity, macrophages have been implicated in diverse physiological processes ranging from haemoglobin recycling, intestinal motility, surfactant degradation in the lung, to cardiac conduction (64, 102–109). The circuits underlying some of these less-traditional macrophage effects are starting to be deciphered. For example, macrophages located in the interstitial space of the testis produce cholesterol, which stimulates steroidogenesis in Leydig cells (110–112).
Whilst their cellular partners, signaling circuitry and functions remain incompletely understood, it is highly conceivable that distinct macrophage niches also exist in the joint. In the following, we will thus discuss how the current concepts of monocyte and macrophage biology in other tissues and organs reviewed above, apply to synovial joints, with particular emphasis on molecular and cellular mechanisms bearing potential for translational exploitation in OA. By interpreting the dynamics between these pleiotropic cell types and their functions within their potential joint-associated niches, we aim to provide an integrative view of their contribution to joint health and disease.
Monocytes and Macrophages in Joint Homeostasis
Bone and Adipose Tissue
The bone-resident macrophages are known as osteoclasts, peculiar large and multinucleated cells whose primary function is bone resorption. They are essential for skeleton remodeling and maintenance of the hematopoietic environment in the BM. Consequently, defects in osteoclasts cause osteopetrosis and hematopoietic failure, while their overactivation leads to osteoporosis. Osteoclasts allow for homeostatic bone turnover in joint-associated subchondral bone in response to loading. Osteoclasts first colonize the ossification centers of developing bones in the fetus from EMP, where they form long-lived syncytia that are maintained throughout life by low-grade fusion with incoming monocytes (113, 114). Adding to this complexity, elegant recent intra-vital imaging has shown that osteoclasts do not necessarily undergo apoptosis following activation and bone resorption, but instead, can fission into daughter cells termed “osteomorphs” (115). These can be recycled by fusion with osteoclasts but remain transcriptionally distinct from both osteoclasts and other macrophages.
As in many other tissues, adipose tissue-resident macrophages are developmentally and functionally heterogeneous. In the healthy adipose tissue of lean mice and likely humans (116, 117), monocyte-derived macrophages co-exist with long lived, fetal yolk sac EMP-derived macrophages and regulate appropriate development of adipose tissues and lipid storage during homeostasis (116, 118). Of note, it is currently unclear whether the macrophage compartments within joint-specific adipose tissues, such as the infra-patellar fat pad, are developmentally and functionally equivalent to those in more commonly studied adipose depots, such as the subcutaneous or inguinal fat. The infrapatellar fat is highly vascularized and innervated, and thus more reminiscent of visceral than subcutaneous fat (Reviewed in Urban and Little 2018) (30). It is also interesting to note that although generally considered a type of white adipose tissue, the infrapatellar fat may not always behave like other adipose tissues, for example in conditions of obesity. Although the infrapatellar fat pad increases in volume, vascularization and adipocyte size in response to obesity like other adipose tissues, it may be more protected from obesity-induced inflammation (119–121). This suggests that infrapatellar fat may show features of both white and brown adipose tissue in response to obesity, and distinct responses to other white adipose deposits have also been observed in OA (122). In end-stage knee OA patients the infrapatellar fat pad had significantly less macrophages, toll-like receptor 4 expression and fibrosis compared with other peri-synovial adipose tissue. In these same patients both adipose tissues had increases in adipocyte size and haematopoietic and M2 macrophage cell infiltration correlated with body mass index. This complex interplay between systemic and local joint factors related to post-traumatic OA, and how these affect and are affected by infrapatellar fat pad macrophage polarization, has been demonstrated in mouse models (123). The infrapatellar fat has been implicated as a major player in sustaining and perpetuating inflammation in OA (29). While macrophage deregulation has been associated with pathological changes in other adipose depots, those in the infrapatellar fat can contribute directly or indirectly to OA pathogenesis and future research is needed to better characterize which macrophage features it shares with other adipose tissues and which are unique.
Synovial Membrane and Interstitial Connective Tissue
At homeostasis, macrophages are virtually the only immune cells in the synovial membrane (124, 125), and whilst the underlying interstitial connective tissue does harbor other lineages like mast cells and lymphocytes, macrophages predominate by far (126). Importantly, both the steady state synovial membrane and interstitium are largely devoid of monocytes. Healthy synovial tissue contains three populations of macrophages that are dynamically interconnected: lining macrophages gradually turn over from proliferating MHCII+ macrophages found in the sub-lining connective tissue, which also generate a second population of interstitial macrophages characterized by expression of Hypoxia-Induced Mitogenic Factor (Resistin-like alpha; RELMa) (22). The exact sources from which synovial macrophages are originally established during development remain to be determined with appropriate additional fate mapping systems. However, chimeras and parabiosis have now firmly established that all three populations receive minimal if any monocyte input in the adult steady state (22).
Despite their developmental interdependence, the distinct populations of synovial macrophages are phenotypically and functionally highly specialized. In addition to being a source of other synovial macrophages, MHCII+ sub-lining macrophages are particularly well-equipped for antigen presentation, while the RELMa+ population shows a regulatory phenotype and abundantly expresses scavenger receptors like CD206 and CD163 (22). Lining macrophages protect joint functionality and the immune privilege of the joint space through a multitude of mechanisms: they act as sentinels for molecular and cellular changes in the joint cavity (124) and facilitate clearance of cartilage and bone debris, highly immunogenic and hence dangerous signals that are constantly shed into the synovial fluid due to mechanical shear stress. In both mice and humans, lining macrophages express high levels of scavenger receptors, in particular Triggering Receptor Expressed on Myeloid Cells 2 (TREM2) and CD163 and are highly phagocytic and anti-inflammatory (22, 124, 127–129). Lining macrophages also actively participate in production of extracellular matrix (ECM) components and synovial fluid (22). Finally, sophisticated genetic and imaging approaches recently revealed that reminiscent of epithelial cells, lining macrophages form tight junctions with one another and thereby constitute a structural and immunological barrier (22). This barrier limits immune cell trafficking across the synovial membrane and thereby protects the avascular cavity from systemic threats. Conversely, it shelters the synovial connective tissue from immunogenic stimuli present in the joint space. Collectively, these features make synovial macrophages key regulators of joint homeostasis.
Potential Macrophage Niches and Signals in Healthy Joints
The exact cellular interactions and molecular signals comprising macrophage niches in healthy joint tissues remain to be deciphered with state-of-the-art approaches, however, fibroblasts are likely key players. This is the case for the spleen and peritoneal cavity (102, 130) and may also be particularly true for the synovial lining, where in the absence of a basement membrane they are in intimate contact with lining macrophages. Synovial fibroblasts and macrophages have been characterized individually in great detail over the last several years (22, 131, 132), and their potential interplay has been discussed in excellent recent reviews (133–135). Fibroblasts are ideally suited to provide anchorage to macrophages, and they are also a recognized source of key macrophage survival factors, such as CSF1 (Figure 2). Synovial lining macrophages are lacking in CSF1-deficient osteopetrotic (“op/op”) mice (99) demonstrating their CSF1-dependence, at least during development. Intriguingly, systemic administration of CSF1 does not restore synovial macrophages, whereas transgenic overexpression of the full-length transmembrane protein does, suggesting they depend on the membrane-bound isoform of the growth factor and thus, local sources (99, 136). Whilst synovial lining macrophages express the receptor for CSF1 receptor (CSF1R) at steady state (22), it is currently unclear whether they also rely on CSF1 for their homeostatic maintenance and turnover.
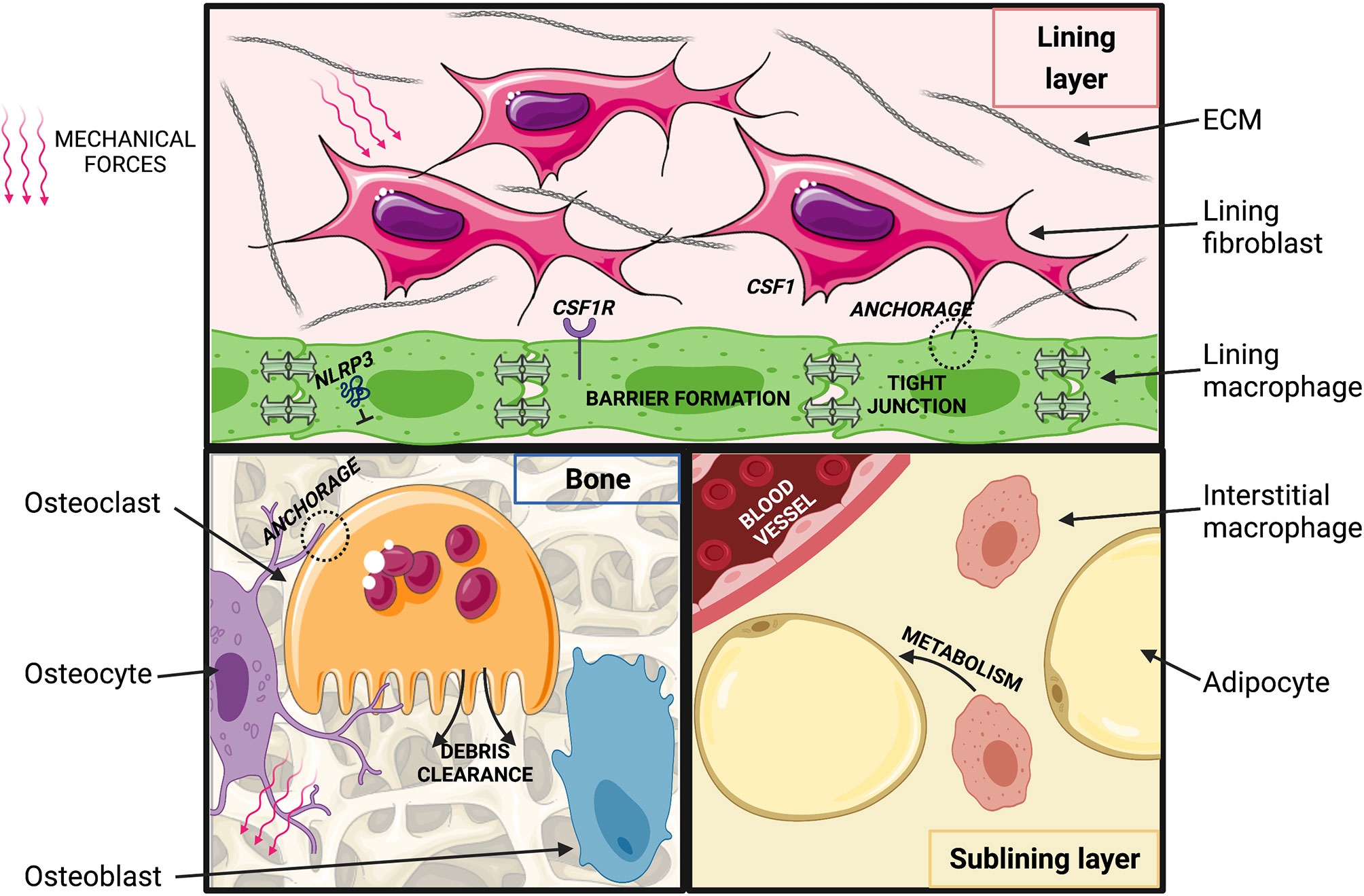
Figure 2 Putative macrophage niches in the healthy joint. (Top) Synovial lining: macrophages are connected via tight junctions and are in close contact with fibroblasts. Fibroblasts may provide CSF1 and anchorage to macrophages, which may be imprinted by exposure to ECM degradation products. Movement-induced cyclic stretch may inhibit NLRP3 inflammasome activation. (Bottom, left) Osteoblasts lining the bone surface synthesize bone matrix in response to soluble mediators released by osteocytes that sense changes in mechanical loading and bone deformation. Osteoclasts resorb bone and thereby regulate balanced homeostatic bone turnover (“modelling”) in response to anchorage and soluble signals from osteocytes and osteoblasts. (Bottom, right) In the synovial sub-lining, self-maintained interstitial macrophages may regulate adipose tissue metabolism and act as a reservoir to replenish synovial lining macrophages. Created with BioRender.com and smart.servier.com.
Fibroblasts could also act to bring macrophages in proximity of tissue-specific cues that imprint their functional identity, although this process may be orchestrated by additional stromal cell types in the joint, such as adipocytes, endothelial cells and chondrocytes. Such complexity is seen in the liver, where incoming monocytes are functionally imprinted by a triad of hepatocytes, endothelial cells and stellate cells (101). Joint tissues are constantly exposed to shear stress and tensile forces that dynamically change with variations in joint loading. Mechanical stimuli are amongst the candidate cues that could play a particularly important role in instructing the specific identity of macrophages in joint tissues. Macrophages are in principle capable of sensing mechanical forces. Human BM-derived macrophages for example respond to substrates with different stiffness with changes in their shape, migration and proliferation (137). Mechanotransduction can also directly modulate their inflammatory cytokine production (138, 139). This latter effect is dependent on the NLRP3 (NOD-, LRR- and pyrin domain containing 3) inflammasome and can also involve signaling through TRPV1 and 4 (Transient receptor potential vanilloid-type 1 and 4) cation channels, which have been implicated in ptOA pathophysiology in mice (140–142). Transduction of mechanical signals through TRPV4 has also been implicated in the formation of multinucleated giant cells, inflammatory and destructive multinucleated macrophages (143). In addition to mechanical stress, normal ECM turnover products represent candidate signals that could imprint joint macrophage identities. Indeed, synovial lining macrophages appear to be highly phagocytic and constantly scavenge cartilage debris from the joint cavity (144–146). Joint biomechanics are altered during OA pathogenesis, and joint-tissue ECM degradation products more prevalent than at homeostasis, thus these pathways likely also impact macrophage identities and functions in arthritic joints.
Monocyte and Macrophage Functions in the Injured and Osteo-Arthritic Joint
Macrophage Functions in ptOA Pathogenesis
In addition to self-maintained lining and interstitial sub-lining macrophages already present at steady state, the arthritic synovium contains inflammatory monocyte-derived macrophages (147, 148). Similar changes also occur in other tissues within the joint (e.g. subchondral bone), regional (e.g. lymph node) and in remote tissues (e.g. spleen, peripheral blood). The necessity to delineate the specific roles of these distinct macrophage populations is highlighted by discrepant findings on the consequences of macrophage depletion depending on the experimental approach. Although the precise contribution of the various populations remains to be shown, systemic depletion of macrophages in mice in which apoptosis is induced in cells expressing CSF1R exacerbates experimental ptOA, whereas local clodronate liposome-mediate depletion within the joint is beneficial (149).
In RA, the respective functions of the distinct macrophage subsets have now been well explored, and macrophages originating from recruited monocytes appear to have overall disease-promoting functions (150, 151). Similarly, the majority of studies on OA-affected joints have identified inflammatory, monocyte-derived macrophages as the main culprit in promoting and sustaining inflammation (124, 152). These cells produce pro-inflammatory cytokines and release additional signaling molecules associated with tissue-injury, which can attract lymphocytes that further propagate inflammation. However, exploiting these findings therapeutically is currently hindered by a lack of detailed understanding of the exact interplay between monocyte-derived macrophages and different types of lymphocytes, and how these change in the distinct stages of ptOA pathogenesis. Monocyte-derived macrophages also participate in cartilage destruction via production of IL-1β and TNF-α, which suppress synthesis of the ECM components aggrecan and collagen by chondrocytes and upregulate expression of catabolic enzymes like ADAMTS-4 and MMP-13 (153–155). Soluble matrix degradation products in turn can activate resident synovial macrophages via Toll-Like Receptors (TLRs) and other pattern-recognition receptors (156). As this example illustrates, different macrophage populations in the joint can be functionally interlinked.
Another effector by which macrophages might contribute to ptOA pathogenesis is B cell Activating Factor (BAFF), a member of the TNF superfamily. BAFF is a crucial B cell survival factor, but also exerts co-stimulatory effects on T cell activation via upregulation of B-cell lymphoma 2 (BCL-2) (157). Furthermore, BAFF promotes T-helper-cell (Th)1 and suppresses Th2 responses (158), and drives Th17 differentiation via Il-6 signaling (87, 158–161). BAFF levels are elevated in serum and synovial fluid from RA patients (162), and BAFF appears to have a pathogenic role in RA (163, 164). During established RA, BAFF promotes pro-inflammatory polarization of CD4+ T cells, DC maturation as well as proliferation of inflammatory fibroblasts (163). In the inflamed joint, macrophages (165) are the main source of BAFF, although it is unclear if these are monocyte-derived or resident macrophages, or both. This compelling evidence led to the development of BAFF antagonists as DMDs for RA, which are currently being tested in early phase clinical trials (163). Whether BAFF production is also a mechanism by which macrophages contribute to pathogenesis of OA has not been determined but elevated BAFF levels have been detected in OA synovial fluid (166).
Unlike their monocyte-derived counterparts, and some controversy notwithstanding, self-maintained resident synovial macrophages have largely been attributed protective roles in arthritis. The barrier generated by synovial lining macrophages is disrupted in both RA patients and experimental RA (22). In mice, this occurs rapidly upon induction of serum transfer-mediated arthritis, and thus constitutes an early event in disease development. In this model, barrier breakdown occurs following phagocytosis of immune complexes containing auto-antibodies, which activate lining macrophages and induce structural joint pathologic changes. Consequently, depletion of lining macrophages or specific disruption of their tight junctions worsens experimental RA. In turn, drug-mediated stabilization of tight junctions protects mice from RA (22), a finding that is translationally promising. Whilst the role of synovial lining macrophages has not yet been addressed specifically in OA pathogenesis, it is worthwhile noting that targeting lining macrophages or tight junctions not only exacerbates RA but may also result in spontaneous inflammation in the joint cavity in otherwise healthy animals (20, 22). With respect to ptOA, one could thus envision a scenario in which following injury, mechanical disruption of the synovial lining macrophage barrier enables rapid influx of inflammatory cells and hence, transition to the inflammatory phase of OA pathogenesis. Unlike in RA however, this barrier breach might be transient in nature, since the barrier appears more intact in patients with established OA compared to RA (22). This might be due to differences between immune complex-mediated and mechanical barrier-breakdown and could contribute to the, often considerable, lag phase between joint injury and ptOA onset.
Monocyte Functions in Joint Pathogenesis
Monocytes are critical players in OA pathogenesis, both as effector cells and a source of additional macrophages. As described earlier, it is widely accepted that Ly6Chigh and Ly6Clow monocytes can differentiate into macrophages with distinct polarization profiles in response to the cytokine milieu encountered in the tissue microenvironment. Reflecting this complexity, the overall impact of classical and non-classical monocytes on joint disease pathogenesis remains unclear. On the one hand, adoptive transfer of Ly6Clow monocytes following pan monocyte depletion increases the development of serum-transfer induced arthritis (58, 167, 168). In this model, Ly6Clow monocytes are actively recruited to the joint, where they are critical for the initiation of sterile joint inflammation and differentiate into inflammatory macrophages (169). On the other hand, Ly6Clow monocytes were also found to limit excessive inflammation in arthritic mice via enhanced recruitment of regulatory T-cells (Tregs) (58, 170). This seemingly contradictory evidence regarding the role of Ly6Clow monocytes in RA underscores the need for further studies that improve the understanding of the complex role of monocytes in inflammatory arthritis, and similar considerations apply to OA. The diverse roles of monocytes and macrophages in ptOA pathogenesis will be discussed in more detail in the following section, focusing on molecular and cellular factors shaping their respective functions.
Signals and Cellular Interactions Shaping Monocytes and Macrophages in ptOA
Depending on severity, joint injury can induce marked mechanical, anatomical and immunological changes, initially resulting in recruitment of monocytes and other inflammatory cells. Pathological changes persist throughout ptOA development and in established disease, and impact both incoming monocytes and previously resident macrophages. This section discusses how the perturbed joint tissue environment might affect monocytes and macrophages (Figure 3). An overview of murine and human monocytes and macrophages found in the synovial tissue during homeostasis, rheumatoid arthritis and in as far as known osteoarthritis, can be found in Table 1.
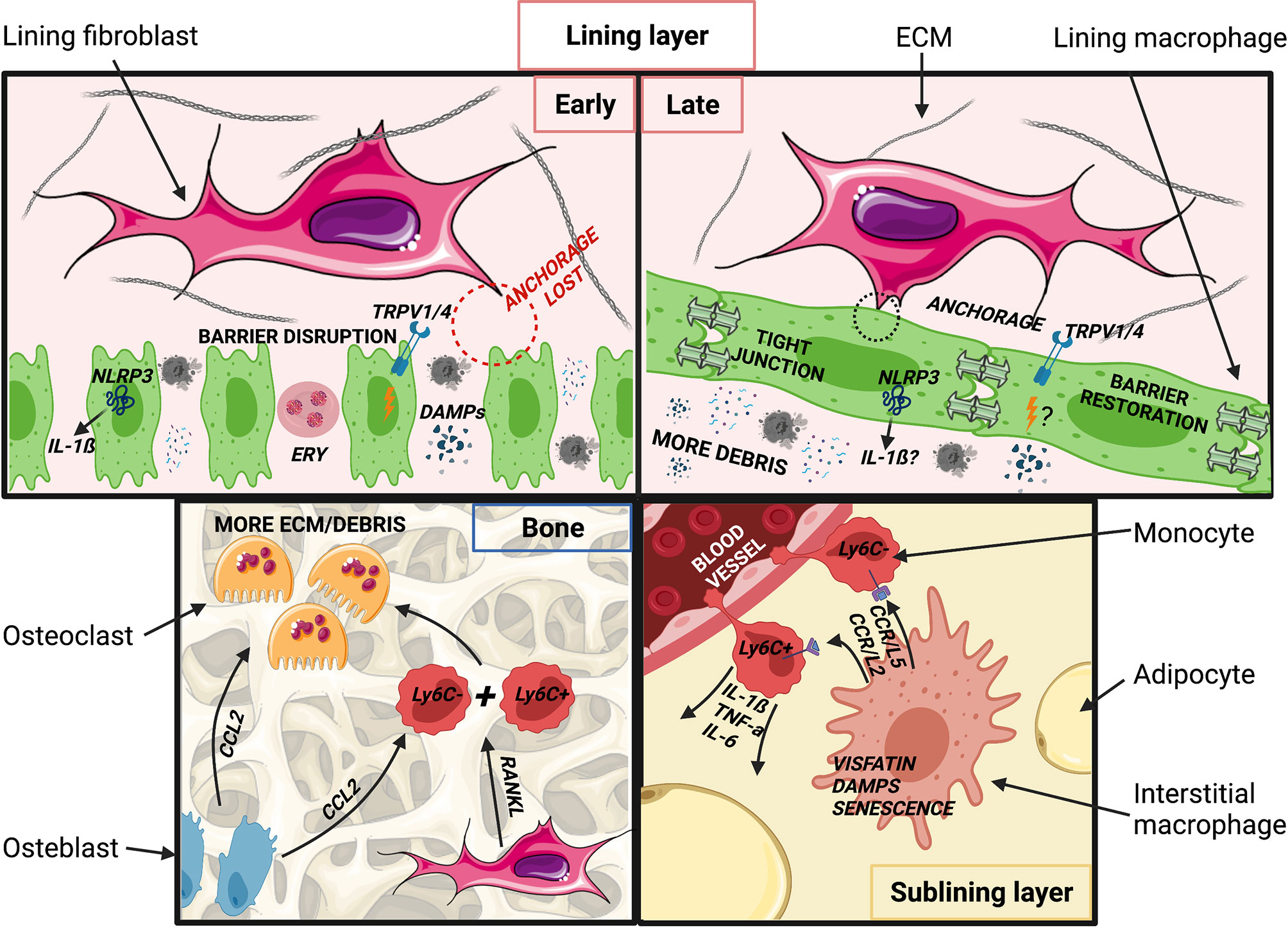
Figure 3 Putative changes in joint tissues after injury and during post-traumatic OA development. (Top, left) Acutely following injury, synovial lining macrophages are spatially re-orientated and the barrier is disrupted. DAMPs, PAMPs and catabolic enzymes are released into the synovial cavity by chondrocytes and damaged tissues (ligament, meniscus). Extra-vascular erythrocytes and associated free heme from blood vessel injury may pathologically imprint synovial macrophages. Barrier disruption may impede cyclic stretching of lining macrophages, resulting in NLRP3 inflammasome activation and increased IL-1ß production, known to promote of OA. Altered mechanics may also promote joint inflammation through TRPV1/4 cation channels. (Top right) At later stages of ptOA pathogenesis, the synovial lining layer may be restored. Levels of IL-1ß remain elevated, though involvement of the NLRP3 inflammasome is unclear. TRPV1 activation may continue to promote OA pathogenesis, although likely via signals other than or in addition to mechanical stimuli. Cellular debris, DAMPs and PAMPs remain abundant in the synovial cavity and thus potentially imprint pathological macrophage phenotypes. (Bottom, left) Increased numbers and activation of osteoclasts contribute to accelerated bone turnover and remodeling in the arthritic joint. Osteoclastogenesis may be promoted by CCL2 produced by activated osteoblasts and inflammatory cells, potentially resulting in recruitment and fusion of Ly6Chigh and Ly6Clow monocytes, a process that may be further stimulated by RANKL produced by lining fibroblasts. (Bottom, right) In the sublining layer, exposure to ECM degradation products may stimulate interstitial macrophages to produce CCL2 and CCL5, leading to recruitment of Ly6Chigh and Ly6Clow monocytes. Ly6Chigh monocytes produce IL-1ß, TNF-α and IL-6, potentially in response to the adipokine visfatin, a TLR4 receptor agonist, which also induces changes in the subchondral bone. Ly6Clow monocytes may supply the interstitial macrophage pool, but these macrophages may retain higher baseline NF-κB and IL-1ß activity than those in healthy joints. Created with BioRender.com and smart.servier.com.
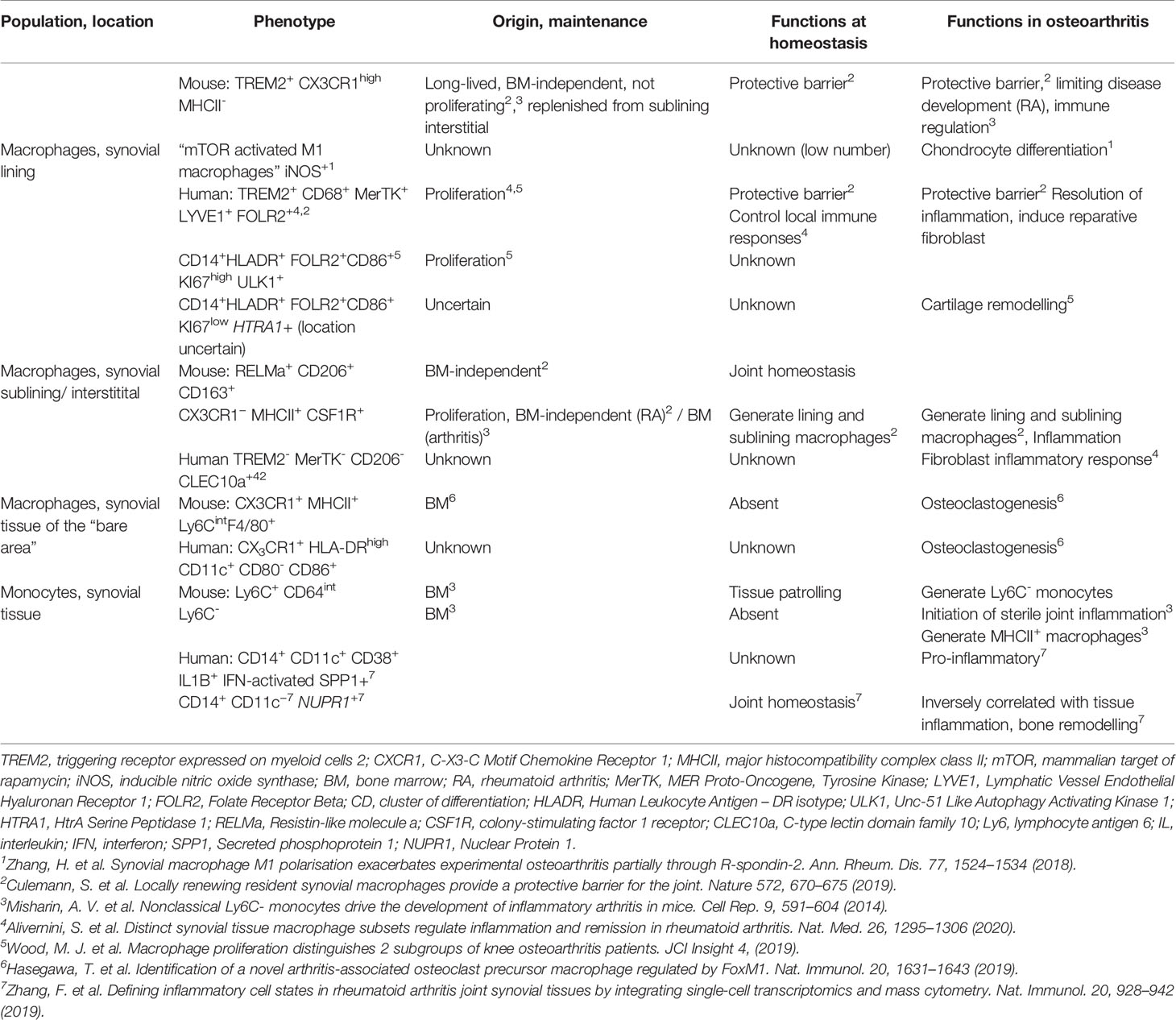
Table 1 Markers, origin and putative function of monocytes and synovial macrophage subsets in mouse and human.
Perturbations Following Joint Injury
Joint injury triggers a series of complex mechano-biological and immunological changes, which can be broadly separated into three successive phases. Immediately after injury, mechanical perturbation effects predominate. These are direct results of the injury (9, 171) and may include tissue disruption (e.g., subchondral bone (micro)fractures, ligament tearing), collagenous matrix disruption and cartilage swelling, blood-vessel injury and hemarthrosis i.e. the presence of blood in the synovial cavity. This immediate joint-tissue injury is followed by an acute inflammatory phase (172), which is characterized by abundant cell death and pro-inflammatory signaling involving both innate and adaptive lineages (173). The nature and duration of this inflammatory response have been identified as major determinants for the risk of developing ptOA post injury (9, 172, 173). While appropriate control and resolution of inflammation is essential for normal wound-healing and might prevent ptOA development, perpetuated inflammation leads to the chronic and final phase of OA pathogenesis, which is defined by fluctuating low-level synovitis (174) and continuous tissue remodeling processes that ultimately lead to destruction of the cartilage and joint failure (175). Delayed or failed resolution of inflammation can be due to a permanently disrupted equilibrium of pro- and anti-inflammatory factors (171) or inadequate post-injury inflammation control (172). In addition, biomechanical factors such as instability or recurring joint injuries (176) can result in continuous re-triggering of acute mechano-biological responses, which initiate an inflammatory vicious circle. These considerations identify the acute inflammatory phase as a potential target for ptOA DMDs and highlight the need for a better understanding of its cellular and molecular regulators.
Mechanisms Underlying Monocyte Recruitment During OA Pathogenesis
The exact nature of the inflammatory response subsequent to joint injury is still under investigation, and differences might exist depending on the type of injury and/or tissues injured, as we will discuss below. Overall, however, a growing body of evidence implicates chemokines and their receptors in monocyte recruitment during OA pathogenesis. As reported for other pathologies (177), classical and non-classical monocytes differentially depend on CCL2/CCR2 or CCL5/CCR5 and CX3CL1/CX3CR1. CCL2 (also known as MCP-1) is a key regulator of Ly6Chigh monocyte egress from the BM and their recruitment to peripheral tissues (178). Following joint injury, release of ECM degradation products and complement factors temporally induces CCL2 production by chondrocytes, resident synovial macrophages (179) and endothelial cells (180). This occurs via a positive feedback loop, stimulated by increased IL-1β and TNF-α expression in synovial macrophages (181) and fibroblasts (182). In keeping with this, clinical studies have reported elevated levels of CCL2 in synovial fluid immediately after traumatic joint injury (183) and subsequent to meniscal tears (184), and concentrations correlate with severity of OA (185, 186). Its expression is also significantly elevated in the serum of OA patients (187), and CCL2 might also affect other cells relevant to OA pathogenesis. In chondrocytes, for example, CCL2 increases expression of the catabolic enzymes MMP3 and MMP13 (179) and inhibits proliferation and enhances apoptosis. CCL2 might thus promote OA pathogenesis via attracting monocytes to the joint, but also by directly promoting cartilage destruction (179).
Unlike CCL2, which is found mainly in the intimal lining of the synovium, CCL5 [also known as RANTES (Regulated on Activation, Normal T Cell Expressed and Secreted)] is distributed more diffusely throughout the synovial tissue (188). Conflicting evidence exists regarding the role of CCL5 and its receptor CCR5, which possess strong chemo-attractive properties for Ly6Clow monocytes. In line with a disease-promoting role of Ly6Clow monocytes, CCR5-/- mice were initially reported to exhibit reduced cartilage destruction (189), however, a recent study by Raghu and colleagues found that neither deficiency in CCL5, nor CCR5 protects mice from ptOA (21). This was further corroborated by a clinical study of synovial biopsy specimens, which found significantly higher levels of CCL5 in RA compared to OA patients, whilst expression of all other chemokines and receptors is comparable (188). This may suggest that the inflammatory responses underlying these different arthritides have some unique molecular signatures or phenotypes. Considering its role in attracting Ly6Clow monocytes, which are known to promote pathogenesis of inflammatory arthritis, it seems surprising that depletion of CCR5 has no protective function in ptOA (21). However, CX3CL1/CX3CR1 signaling also participates in recruitment of non-classical monocytes, and elevated levels of CX3CL1 have been found in peripheral blood (190) and synovial fluid (186) of OA patients. Functional experiments revealed that in addition to its chemo-attractive properties, CX3CL1 also stimulates inflammation specifically at the early stages of OA (190). Finally, soluble CX3CL1 induces production of the pro-inflammatory cytokines IL-1β, IL-6 and TNF-α in recently recruited monocytes (191). Taken together these data indicate that the CX3CL1/CX3CR1 axis predominates in recruitment and pro-inflammatory activation of Ly6Clow monocytes in the context of OA initiation. Ly6Chigh monocytes recruited and activated via CCL2/CCR2, on the other hand, might help sustain inflammation at later stages (192). Temporal changes in chemokine expression and associated monocyte sub-population recruitment and accumulation, may in part explain the recently described loss with time post-injury, of an initially protective/anabolic effect of injured synovium on chondrocytes (193).
Osteoclasts have been implicated in progressive joint destruction. Osteoclastogenesis is controlled by RANK (Receptor Activator of Nuclear factor Kappa B (NF-κB) (194). Its ligand (RANKL) is expressed by fibroblast-like synoviocytes and Th17 cells, and expression is regulated by pro-inflammatory cytokines secreted by monocytes and macrophages (IL-1β, IL-6, IL-17 and TNF-α) (195). A particularly interesting mechanism by which monocytes could contribute to increased osteoclastogenesis in the context of arthritis was proposed by Hirose et al. They postulated that CCL2 secreted by osteoblasts leads to fusion of Ly6Chigh with Ly6Clow monocytes stimulated by RANKL, resulting in mature, multinucleated osteoclasts (195). In addition to osteoblasts, activated inflammatory cells produce large amounts of CCL2 and this might explain the accelerated osteoclastogenesis and joint destruction in an inflammatory setting (195).
Signals Governing Monocyte Differentiation, Activation, and Functions in OA
Following their recruitment to the tissue, monocyte functions can be shaped by a variety of signals present in the non-homeostatic joint. Calcium binding proteins can act as damage- or pathogen-associated molecular patterns (DAMPs and PAMPs, respectively) and have multi-faceted effects on OA pathogenesis. Damage to the cartilage leads to increased levels of S100A8 and its binding partner S100A9 specifically in synovial pro-inflammatory macrophages, but not fibroblasts (196). Secretion of these factors induces the production of pro-inflammatory cytokines in these macrophages in an autocrine manner (196). In addition, in a murine collagenase-induced OA model, which has a more inflammatory phenotype than surgically-induced disease, release of S100A8/9 elicits influx of Ly6Chigh monocytes via upregulation of CCL2 (197) and increased egress of Ly6Chigh monocytes from the BM (197). In turn, activated monocytes are a major source for S100A8/9, which might thus constitute a positive feedback loop. Finally, S100A8/9 might also be derived from chondrocytes and directly contribute to cartilage disruption in OA by inducing production of ADAMTS-4 and -5, MMP-1, -3, -9 and -13 and the pro-inflammatory cytokines IL-6, IL-8 and CCL2 in chondrocytes in a TLR4-dependent manner (198, 199). Interestingly, chondrocyte-derived S100A8/9 may play a predominant role in the acute post-injury phase, as expression and protein levels in cartilage decrease with post-traumatic OA disease progression while levels are maintained in immune-mediate inflammatory arthropathy (199). S100A8/9 thus fuel the initial pro-inflammatory microenvironment in the joint, provide chemotactic cues for Ly6Chigh monocytes and exert direct catabolic effects within the cartilage, processes which are further amplified by several feedback loops. Another class of OA-associated DAMPs are basic calcium phosphate (BCP) crystals. Whilst in vivo data on their relevance to OA are currently lacking, in vitro exposure of monocyte-derived macrophages to BCP crystals leads to a classically activated, pro-inflammatory phenotype, a bioenergetic switch towards glycolysis and increased expression of S100A8 (200). Promisingly, both BCP-induced phenotypic polarization and S100A8 expression are inhibited by a glycolytic inhibitor (2DG) indicating that metabolic reprogramming might be underlying these effects (200).
The Janus Kinase/Signal Transducer and Activator of Transcription (JAK/STAT), Mitogen-Activated Protein Kinase (MAPK) and NF-κB pathways are involved in differentiation of monocytes into macrophages and their functional polarization. The latter depends on interferon regulatory factors (IRFs) (201). IRF5 is a also downstream target of Granulocyte-Macrophage Colony-Stimulating Factor Receptor (GM-CSFR), and plays a critical role in pro-inflammatory macrophage polarization (202). A recent clinical study investigated the role of IRF5 in OA and found it to be overexpressed in synovial macrophages, but not circulating monocytes (203). However, exposure to synovial fluid from OA patients induced expression of IRF5 and IL-12 (via the individual subunits IL-12p35 and IL-12p40) in monocytes, in turn making them potent inducers of a Th1 response characterized by expression of IFN-γ and Tbx21 in co-cultured naïve CD4+ T-cells (203). This suggests that patient synovial fluid contains soluble factors capable of inducing IRF5 in monocytes, thus contributing towards a Th1 inflammatory response.
TLR 4 is a receptor for PAMPs and DAMPs expressed on monocytes that plays an important role in the activation of innate immunity (204), and has been implicated in the inflammatory reaction associated with OA (205). The adipokine visfatin was recently identified as a TLR4 receptor agonist capable of evoking inflammatory responses (206). In addition, visfatin stimulates production of IL-1β, TNF-α and IL-6 by monocytes (207), and the resulting inflammatory environment displays higher levels of circulating visfatin, thus constituting a positive feedback loop (208, 209). Visfatin is also involved in inter-tissue joint communication underlying changes in the subchondral bone (210). These have long been described in OA, but the exact mechanisms of this remodeling and pathways of its activation has remained elusive. Emerging evidence now points towards direct communication between the subchondral bone and cartilage via diffusion (211). Laiguillon et al. found that visfatin is produced in cartilage, synovium and subchondral bone and exerts an enzymatic effector function selectively inducing a pro-inflammatory phenotype in chondrocytes, osteoblasts and synoviocytes, characterized by increased secretion of IL-6 and CCL2 (210, 212–214). Because of its contribution to the inflammatory response and tissue remodeling, inhibition of visfatin might be a promising DMD approach.
The gene expression profiles of monocytes and monocyte-derived macrophages, and hence, their functional polarization, might also be shaped by microRNAs expressed in response to environmental stimuli. A recent study identified miR-155 as a potential genomic switch in monocyte-derived macrophages generated in vitro, which regulates their inflammatory profile (215). Intriguingly, this phenomenon is partially reversed by treatment with monoclonal anti-TNF antibodies, but not a soluble TNF receptor (Etanercept) (215). MiRNAs expressed by monocytes and their macrophage progeny, such as miR-155, might therefore represent promising candidate DMD targets.
While we have focused primarily on joint injury and ptOA, monocytes and macrophages in the arthritic joint might also be affected by age and cellular senescence, as has been demonstrated for RA. In this context, an elegant mouse study from Misharin et al. is of note, where the role of different monocyte subsets in RA pathogenesis using serum-transfer induced arthritis was investigated. Ly6Clow monocytes are recruited to the joint and initially develop into classically activated macrophages, but the macrophage compartment gradually undergoes a switch towards a more alternatively activated phenotype (169). The initial highly pro-inflammatory nature of the Ly6Clow monocytes could be caused by a senescence-associated secretory phenotype, which is associated with high baseline NF-κB and IL-1α activity (216). In addition, accumulation of Ly6Clow monocytes is found in the elderly. These findings suggest that senescence might correlate with increased numbers and pro-inflammatory skewing of Ly6Clow monocytes, which might further exaggerate the inflammatory response unfolding during RA progression. Whether similar mechanisms might be at play in ptOA requires evaluation, but it is interesting to note the accelerated disease progression, increased expression of inflammatory genes and inhibitory effects of MIF ablation following medial meniscal destabilization in older versus younger mice (217, 218).
Immunogenic and Imprinting Signals in the Injured Joint
As introduced, the synovial membrane plays a key role in maintaining joint homeostasis as it guarantees the relative immune privilege of the synovial cavity. However, while the joint space itself is not vascularized, the synovial membrane also features a vascular net located just below the intima. This comprises capillaries, venules, arterioles and lymphatics (219) through which systemic and local inflammatory stimuli can be sensed (125, 220). Hemarthrosis and cartilage damage are direct consequences of joint trauma, which affect the joint not only macroscopically, but also on a cellular and molecular level (176). Of note, the presence of blood in synovial fluid is an independent predictor or poorer 2-year outcome following joint injury (221). It is tempting to speculate that heme could be a cellular cue shaping macrophages in the early stages of ptOA pathogenesis, but unlike in the homeostatic spleen, it may instruct more inflammatory cell states (222). Moreover, hemarthrosis directly activates the complement system, leading to production of complement anaphylatoxins (C3a and C5a) and formation of the membrane attack complex (223). Intriguingly, genetic deficiency for individual components of the complement system in mice leads to either attenuated [C5 and C6 (224)] or aggravated [CD59, also known as protectin (224)] ptOA joint damage following injury.
As discussed above, mechanical forces may directly shape macrophage functions in the homeostatic joint. It is therefore plausible that the mechanical changes following joint injury, impact on resident and recruited macrophages. This appears to be the case at least in experimental RA, where the extent of mechanical loading determines the local distribution of inflammation and degree of damage (225). Mechanical damage to the cartilage also leads to substantial ECM degradation. Collagen fibers fail to contract (226, 227) and ECM degradation is further enhanced by the lack of maintenance and repair (228) following chondrocyte death. ECM-derived tissue fragments are widely recognized as pro-inflammatory and immunogenic (229, 230). These fragments and the complement anaphylatoxin C5a can act as effective chemo-attractants for innate and adaptive immune cells (231, 232) and directly activate macrophages via NF-kB signaling (233). Released cartilage destruction products such as matrilin-3 (229, 234), tenascin-C (235), fragmented biglycan (236) and fibronectin (237) can also potently activate resident synovial macrophages. Finally, cartilage and other joint tissue degradation can induce release of additional DAMPs (238) capable of activating innate immune cells via TLR2 and 4 (239) and NF-kB signaling. The mechano-biological damage induced by injury thus generates an inflammatory microenvironment in the joint space, which is characterized by an increase in soluble inflammatory mediators and chemo-attractants that might induce transition to the acute inflammatory phase, and importantly shape subsequent responses of both recruited monocytes and resident macrophages.
T Cell-Mediated Monocyte and Macrophage Activation in OA?
Other immune cells may provide signals amplifying the effects of monocytes and macrophages. In the context of joint injury, recruitment and activation of lymphocytes have traditionally been thought of as secondary events that follow monocyte influx and changes in macrophages (11). However, T cells are found in synovium at higher levels in early versus late OA (240) and might actively contribute to monocyte and macrophage activation via co-stimulatory pathways. One such pathway relies on interactions between CD40, a member of the TNF receptor family found primarily on antigen-presenting cells and monocytes, and its ligand CD40L (CD154), which is almost exclusively expressed by activated CD4+ T cells (241, 242). CD40/CD40L interactions elicit a broad pro-inflammatory response (243) that involves B cell differentiation (244) and macrophage activation. In turn, activated macrophages and other antigen-presenting cells enhance immunoglobulin antigen affinity (241), activate cytotoxic T cells and promote a Th1 immune response (245). This co-stimulatory pathway therefore has the potential to initiate a powerful amplification loop that propagates joint inflammation. In keeping with this notion, exaggerated CD40/CD40L signaling contributes to autoimmunity (246), including RA. Multiple studies have shown overexpression of both CD40L (247–249) and CD40 (246) in RA, and levels of CD40L are associated with disease activity (248) and perpetuation (247). Based on these findings, biological treatments targeting this axis in RA have been developed, which are currently undergoing early phase clinical trials (250). Despite differences in the pathogenesis and mechanisms involved in the development of RA and OA, shared elements in the underlying inflammatory response seem plausible (251). The effects of targeting the CD40/CD40L axis in OA remain to be determined, however we have found that CD40L mRNA levels are elevated exclusively in the synovium immediately after ACL rupture and during early onset of OA development (252). These preliminary findings support the notion that CD40/CD40L may be an early driver of T cell-mediated synovial macrophage activation and warrant future research into the CD40/CD40L axis specifically in ptOA.
Additional Candidate Pathways and Mechanisms Leading to Macrophage Dysregulation During OA Pathogenesis
In addition to the factors discussed above, obesity is a well-established risk factor contributing to OA development (253), through increased mechanical loading but also via dysregulated secretion of adipokines and other metabolic factors (254). In mice, high-fat diet (HFD) results in elevated leptin-induced levels of lysophosphatidylcholine (lysoPC), which in turn increases MMP13 production by chondrocytes. As a consequence, obese mice show an earlier onset and progressive course of spontaneous OA (254). Direct links between obesity and OA have also been shown in mouse models of ptOA. HFD was associated with inflammation in the infrapatellar fat pad, characterized by macrophage crown-like structures, which may have a priming effect on the fat pad leading to a metabolic state of progressive OA following injury (123). HFD was also shown to aggravate inflammation of the synovial membrane post-injury, which was marked by increased macrophage infiltration (255). These findings are in line with the notion that obesity contributes to aberrant macrophage activation in OA pathogenesis. Intriguingly, these detrimental effects of HFD on OA persisted even after a normal diet was resumed (254), indicating long-lasting effects and potential windows or particular susceptibility. Some of these effects may even be programmed in early life and transmitted across generations. Indeed, increased higher susceptibility to experimental ptOA has been reported in the first and second generation offspring of mice fed a HFD during breeding (256). Immune cells have been implicated as mediators of such programming and transgenerational effects of obesity in offspring (256), and epigenetic dysregulation has been postulated as a central mechanism.
While some epigenetic modifications are stable and passed on across generations, others are more dynamic and responsive to environmental stimuli (257). These are believed to play a significant role in OA development. Of the studies that have investigated epigenetic changes in OA development, most have focused on epigenetic mechanisms modulating chondrocyte biology and inflammatory mediators (258). Evidence for epigenetic modifications of macrophage remains scarce in the context of OA. In principle, epigenetic processes govern various aspects of macrophage biology, including their development, differentiation, and activation, as well as the specification of their tissue identity (259–261). For example, active DNA demethylation occurs during monocyte to macrophage differentiation in vitro (262) and the identity of tissue-resident macrophages is shaped by unique enhancer landscapes in response to microenvironmental cues (70, 71). They also activate genes governing embryonic stem cell-like self-renewal through macrophage-specific enhancers (263). Fully differentiated macrophages are maintained in a “balanced” state through a combination of activating (such as PU.1, H3K4me1 and open chromatin) (264) and repressive (such as H3K9me3, H3K27me3 and H4K20me3) (265) epigenetic marks and regulators (262). These repressive marks are removed upon stimulation of macrophages through TLR, and specifically TLR4, ultimately resulting in the production of inflammatory cytokines such as IL-1β, CXCL10, IL-6 and TNF (262). TLR4 signaling has also been implicated in low-grade inflammation mediated by plasma proteins present in the synovial fluid of OA patients (266). Whether epigenetic changes in macrophages contribute to this remains to be formally shown, however.
Activation via TLR4 also initiates metabolic reprogramming of macrophages, and distinct metabolic states have been linked to functional differences in macrophage subsets. For example, metabolic reprogramming towards increased glycolysis promotes pro-inflammatory polarization (267). In OA, increased glucose uptake correlates with disease progression, and the hypoxic environment in the OA synovium may enhance osteoclastogenesis (267, 268). Osteoclastogenesis appears to also be promoted by metabolic syndrome through NF-κB activation and advanced glycation end products (269). A bioenergetic switch towards glycolysis is also induced in macrophages by basic calcium phosphate crystals, which are specifically found in OA (220), further supporting the notion that macrophages may undergo metabolic reprogramming during OA pathogenesis.
Finally, epigenetic and immunometabolic changes are also hallmarks of “trained immunity”. This recently coined concept (270) acknowledges that innate immune cells, including macrophages, show increased responsiveness to secondary stimuli following “training” by primary exposures. Whilst trained immunity has not specifically been studied in the context of OA, many of the cellular signals that impact on macrophages during OA pathogenesis – or even preceding disease onset - could mediate long-term effects through inducing this type of innate memory in macrophages. Thus, obesity/HFD may be primary exposures that train heightened or specific OA inducing immune responses to a secondary stimulus such as injury (123, 256). It is interesting to speculate that this may also be relevant in the context of prior even minor joint injuries increasing the risk and/or severity of ptOA following a critical/destabilizing injury such as anterior cruciate ligament (ACL) rupture (see Blaker et al., 2021 and references therein) (271).
Harnessing Monocyte and Macrophage Biology for OA Risk Stratification, Diagnosis, and Therapy?
It is now well recognized that ptOA development features an early inflammatory response. This involves systemic processes resulting in monocyte recruitment, as well as a local disbalance within the immune “niches” of the affected joint, whose immune privilege is therefore compromised. This recognition has several implications that in the future could be exploited for prognostic, diagnostic and therapeutic benefit, examples of which we discuss below.
Monocytes and Macrophages as Biomarkers for OA
To this day, OA diagnosis largely depends on clinical presentation/symptoms and conventional imaging methods like x-ray, computerized tomography (CT) scans or magnetic resonance imaging (MRI) (272). Diagnostic biomarkers are currently missing, as are reliable predictive markers. Access to synovial fluid and hence, the search for useful biomarkers, is limited by the invasive nature of the acquisition procedure. Nonetheless, advances have been made recently in the search for cellular and molecular biomarkers with diagnostic and/or predictive potential (221, 273), using synovial fluid where available, or peripheral blood, which can be more readily obtained.
On the cellular level, a growing body of data implicates monocytes and monocyte-derived macrophages in OA pathogenesis, as discussed in this review. Several recent clinical studies therefore investigated the prognostic value of peripheral immune cell ratios. While the exact ratios differ between studies, monocytes represent a common denominator. In particular, the neutrophil to monocyte ratio is independently and inversely associated with OA severity as classified using the Kellgren-Lawrence scale (274). Similarly, the monocyte to lymphocyte ratio reliably predicts OA progression (275). MicroRNA analysis of peripheral blood mononuclear cells (PBMCs) from OA patients showed elevated expression of miRNA-146a and miRNA-155 (276), which influence inflammatory cell signaling via the NF-κB pathway (277, 278). Moreover, transcriptomic analysis of PBMCs from OA patients identified more than 1000 differently expressed genes, pathway analysis of which implicated inhibition of chondrocyte differentiation, increased osteoclastogenesis and MAPK activation (279). These data collectively indicate that peripheral blood monocytes of OA patients differ from healthy controls both quantitatively and qualitatively. It is tempting to speculate that specific OA-primed inflammatory monocytes exist in the peripheral blood during disease progression, and even potentially prior to onset. This notion is corroborated by data from Loukov and colleagues, who demonstrated that following in vitro exposure to DAMPs, peripheral blood monocytes from women with knee OA produced higher levels of the pro-inflammatory cytokines IL-1β and TNF-α than monocytes from healthy controls (280). The same group also demonstrated significantly higher levels of CD14 expression on monocytes of OA patients, further implicating non-classical activated monocytes.
Within the synovial fluid of knee OA patients, monocytes and macrophages constitute the second most abundant cell population after T cells, and a large proportion of these are CD16-, thus further implicating non-classical monocytes (281). Liu et al. investigated the relative abundance of phenotypically distinct macrophages in synovial fluid of knee OA patients and found an increased ratio of “classically” compared to “alternatively” activated macrophages (282). This ratio further correlated with disease severity, suggesting that despite the limitations of this simplistic dichotomy, such analyses can yield clinically relevant data.
A wealth of experimental and clinical studies has analysed inflammatory parameters and markers in synovial fluid for their potential to serve as biomarkers. These studies have shown that levels of CCL2, IL-6 and IL-8 accurately distinguish OA from normal joints (283–285) and inflammatory markers can even predict the outcome of ACL reconstruction. Similarly, the presence and severity of synovitis following meniscal injury are associated with the risk of progressive cartilage damage, even if inflammation subsequently resolves (286). Elevated levels of several additional synovial fluid biomarkers associate not only with radiographic OA severity (sVCAM-1, sICAM-1, TIMP-1 and VEGF) and OA symptoms (VEGF, MMP-3, TIMP-1, sVCAM-1, sICAM-1 and MCP-1) but are also highly correlated with levels of neutrophil elastase (287). This highlights a potential role for neutrophil activation in the onset of OA. These initial findings were further corroborated by a recent study indicating that expression levels of TGF-ß1 and elastase were associated with radiographic severity scores and predictive of knee OA progression (288).
Based on such findings, Jayadev et al. used a novel machine learning approach to develop a “cytokine fingerprint” for end-stage OA. Using a panel of eight biomarkers (PIIANP, TIMP-1, ADAMTS-4, CCL2, IP-10 and TGF-β3), this model distinguishes between OA, knee injury and inflammatory knee arthritis (i.e. RA or psoriatic arthritis) with almost 100% efficacy (289). Interestingly, knee/hip arthroplasty further increases the levels of angiogenic and pro-inflammatory cytokines, but leaves anti-inflammatory cytokines unaffected, suggesting underlying changes specifically in pro-inflammatory pathways, which might be further exacerbated with surgical treatment (290).
In summary, biomarker research has both leveraged and fueled the notion that monocytes contribute to OA pathogenesis, and that OA-primed non-classical monocytes might exist. Although this progress is encouraging, the majority of recently identified biomarkers are associated with disease progression, rather than onset. There remains thus a pressing, unmet clinical need for biomarkers instrumental in diagnosis and stratification of individuals at risk of developing ptOA following knee injury.
Disease Modifying Drugs for OA: Where Are We on the Clinical and Pre-Clinical Level?
To target the early inflammatory phase that follows joint injury and initiates OA pathogenesis, adjuvant DMDs are urgently needed. A variety of agents with the potential to serve as DMDs are currently being tested, but unfortunately, with limited success (291). Some studies on local inhibition of IL-1 in animal models showed promising results. In particular after closed articular fracture, intra-articular injection of an antagonist for IL-1 receptor (IL-1Ra) reduces post-traumatic OA, cartilage degeneration and synovitis (292–294). However, the clinical efficacy of this approach remains controversial. While an early clinical study found that intra-articular administration of IL-1Ra performed within 1 month of severe knee injury led to reduced knee pain and improved function over a 2-week follow-up period (295), other clinical studies using either IL-1Ra (Anakira) or a dual variable domain immunoglobulin that simultaneously inhibits IL-1α and IL-1β (Lutikizumab) have yielded no benefit in OA patients compared to placebo controls (291). Interestingly, retrospective secondary analysis of a large-scale clinical trial of a monoclonal antibody targeting IL-1β for cardiovascular disease found reduced incidence of hip and knee replacement in patients with high C-reactive protein, suggesting a potential effect on OA progression in an inflammatory setting (296).
It is noteworthy, that the OA-specific studies of DMD candidates described above were tested mostly in patients with advanced OA with the intention of reducing established clinical and radiological disease progression. In line with the current difficulties in risk stratification and early diagnosis outlined above, to date, no drugs have been advanced to the clinical stage that target the inflammatory response at the onset or early stages of disease. Experimentally, however, attempts to advance in this direction have been made recently. One study explored the role of incretin hormone receptors in vitro. It found that an analogue for the human Glucagon-Like Peptide-1 (GLP-1), liraglutide, reduced production of reactive oxygen species, IL-6 and CCL2, reduced collagen and aggrecan degradation and inhibited inflammation via deactivation of NF-kB signaling (297). Similarly, administration of dexamethasone, rapamycin or BMP-7 results in a more anti-inflammatory macrophage phenotype in vitro. Whilst these findings await in vivo confirmation, these drugs might hold potential of modulating synovial inflammation in patients (298).
One of the few in vivo studies investigating the impact of anti-inflammatory therapy immediately after joint injury utilized a porcine model. Here, the authors induced OA via transection of the anterior cruciate ligament (ACL) and immediately provided corticosteroids by intraarticular injection, which resulted in mitigated collagen degradation, reduced monocyte recruitment and a less inflammatory macrophage profile/phenotype (299). Our group has investigated the respective effects of intraarticular administration of hyaluronan or BM-derived mesenchymal stem cells in a murine OA model. We found that hyaluronan therapy increased anti-fibrotic macrophages and decreased pain sensitization, while treatment with MSCs did not impact pain, but led to long-term chondroprotection (300). Along similar lines, another recent study demonstrated that administration of Alpha defensin-1 renders macrophages less inflammatory and attenuates OA in a surgical model (301). Collectively, these data suggest that specific anti-inflammatory treatment immediately after knee injury might represent a promising future therapeutic approach, and thus, justify additional experimental and ultimately clinical studies.
Concluding remarks, Open Questions, and Future Directions
Distinct Inflammatory Responses in Different Joint Injuries?
Post-traumatic OA accounts for nearly 12% of all cases of symptomatic OA (302) and a recent longitudinal cohort study showed that the risk of developing OA is almost sixfold increased by knee injury at a young age (303). Further stratification of these data revealed distinct risks for different injury types: ACL injury, meniscal tears and articular fractures of the tibia (risk difference (RD) of 19.5%, 10.5% and 6.6%, respectively) were associated with the highest risk (303). In trying to identify possible reasons for these differences, the simplest explanation of variable biomechanical aberration is insufficient, as an abundance of data has shown that restoration of biomechanics alone does not prevent ptOA development (4). An alternative hypothesis is that phenotypic differences exist in the pathogenesis of ptOA depending on the type of tissue affected by injury. Thus, metabolic and immunobiological differences may determine the individual risk of developing ptOA. Although comprehensive studies investigating the tissue- and phenotype-specific immune response after joint injury are missing, existing evidence supports an immunological role for the meniscus (304), which engages in a pro-inflammatory crosstalk with the synovium in OA (305). Furthermore, synovial fluid, cartilage tissue and isolated cartilage cells display distinct pro-inflammatory cytokine profiles depending on the type of pathology, further supporting the notion that a phenotype-specific cytokine topography exists in the joint (306). Future studies should therefore be designed to examine the inflammatory reaction associated with different types of joint injury.
Could Restoring Joint Immune Homeostasis Hold the Key to OA?
Owing to decades of research, we have come to understand that OA cannot simply be attributed to “wear and tear” resulting from biomechanical changes. Rather, OA results from a complex biological response of different cells in multiple joint tissues, with “inflammation” playing a crucial role this process. Monocytes and macrophages are emerging as key players in the inflammatory process associated with OA. Circulating monocytes are particularly attractive as druggable targets, but selective targeting of tissue-resident macrophages might be equally feasible, for example using antibody-conjugated lipid nanoparticles (72). It is tempting to speculate that dysregulated dynamics between monocytes, macrophages and other cell types they cross-react with in the joint not only fuels pathology, but in fact represents an underlying cause of OA. To harness these intricate cellular interactions for diagnostic and therapeutic purposes, we need to further improve our understanding of monocyte and macrophage biology in both healthy and arthritic joints. Deciphering their developmental and functional dynamics harbours the potential of one day being able to restore synovial immune homeostasis and thus, finally provide a causative treatment for this debilitating disease.
Can Findings From ptOA Be Translated to Other OA Phenotypes?
A wide range of small and large animal models for OA have been developed, and these have recently been reviewed (307). Far and away the most commonly used models are those induced by joint injury. They share a comparably rapid onset and highly standardized disease progression, and allow investigation of underlying molecular and cellular mechanisms, as well as evaluation of potential treatments at different stages of disease progression. However, the translation of findings made in these models to clinical settings has remained challenging (308). One explanation is the current discrepancy between preclinical research predominantly using ptOA models and clinical studies, the majority of which investigate late-stage “primary OA” which occurs in the absence of prior trauma or disease (309). There is emerging evidence that the pathophysiologic mechanisms of structural and symptomatic OA differs depending on the key initiating factors or disease phenotype (310, 311). How different the complex cellular inflammatory response is in different OA phenotypes remains to be resolved. In a first attempt to overcome this issue, findings from preclinical ptOA studies should be tested in preclinical models of primary OA, such as spontaneous age-associated and metabolic/obesity-induced disease. Whilst the associated immune response may be different in strength and spatio-temporal patterns, it is likely that at least some aspects of monocyte and macrophage biology relevant to ptOA apply to other OA phenotypes, such as their roles in ECM degradation and chondrocyte death, which are universal OA disease features. Likewise, some molecular signals identified in ptOA models as regulators of macrophage activation and polarization, such as specific cytokines and chemokines, and are also present in multiple OA phenotypes (312).
Open Questions and Future Directions for Research
- Does the initial inflammatory response following joint injury pathologically imprint monocytes, macrophages and their stromal niche in the joint?
- Do disease-specific, “imprinted” populations of monocytes and macrophages emerge prior to disease onset? Do they mediate (or: propagate) OA pathogenesis?
- Are monocyte and macrophage dynamics permanently altered following joint injury?
- Can these populations be targeted therapeutically?
- Is there an optimal ratio between pro- and anti-inflammatory monocyte/macrophage subsets that mitigates the risk of OA after joint injury?
- Can a threshold be determined that governs the future direction of either resolution of inflammation and restoration of joint function or ongoing inflammation that contributes towards development of OA?
- Do similar considerations also apply to non-traumatic forms of OA?
Addressing these questions will provide the critical scientific understanding necessary to improve diagnosis, risk prognosis, and underpin development of specific targeted therapies to prevent OA onset and/or slow its progression following joint injury. Recognizing that the targets may differ depending on injury and change with time and being able to identify these therapeutic stages/windows, will be key to providing effective individualized patient management.
Author Contributions
Conception and design of study: PH, CL, BM, and RG. Acquisition of data: PH, RG, CL, and MP. Drafting of article: PH, RG, MP, CL, and BM. Revising it critically for important intellectual content: PH, RG, MP, CL, and BM. All authors have approved the final version of the manuscript.
Funding
The authors acknowledge salary support from the following agencies: Raymond E Purves Foundation PhD Scholarship (PH), University of Edinburgh and the Kennedy Trust for Rheumatology Research (RG, MP). The funding bodies had no input into the design, drafting, editing or content of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to acknowledge the general contribution of colleagues, institutions, and agencies that have aided the research efforts of the authors in areas related to the topic of this review.
References
1. Spitaels D, Mamouris P, Vaes B, Smeets M, Luyten F, Hermens R, et al. Epidemiology of Knee Osteoarthritis in General Practice: A Registry-Based Study. BMJ Open (2020) 10(1):e031734. doi: 10.1136/bmjopen-2019-031734
2. Kloppenburg M, Berenbaum F. Osteoarthritis Year in Review 2019: Epidemiology and Therapy. Osteoarthritis Cartilage (2020) 28(3):242–8. doi: 10.1016/j.joca.2020.01.002
3. Vos T, Flaxman AD, Naghavi M, Lozano R, Michaud C, Ezzati M, et al. Years Lived With Disability (YLDs) for 1160 Sequelae of 289 Diseases and Injuries 1990-2010: A Systematic Analysis for the Global Burden of Disease Study 2010. Lancet (2012) 380(9859):2163–96. doi: 10.1016/S0140-6736(12)61729-2
4. Barton KI, Shekarforoush M, Heard BJ, Sevick JL, Vakil P, Atarod M, et al. Use of Pre-Clinical Surgically Induced Models to Understand Biomechanical and Biological Consequences of PTOA Development. J Orthop Res (2017) 35(3):454–65. doi: 10.1002/jor.23322
5. Chua JR, Jamal S, Riad M, Castrejon I, Malfait AM, Block JA, et al. Disease Burden in Osteoarthritis Is Similar to That of Rheumatoid Arthritis at Initial Rheumatology Visit and Significantly Greater Six Months Later. Arthritis Rheumatol (2019) 71(8):1276–84. doi: 10.1002/art.40869
6. Dequeker J, Luyten FP. The History of Osteoarthritis-Osteoarthrosis. Ann Rheum Dis (2008) 67(1):5–10. doi: 10.1136/ard.2007.079764
7. Orlowsky EW, Kraus VB. The Role of Innate Immunity in Osteoarthritis: When Our First Line of Defense Goes on the Offensive. J Rheumatol (2015) 42(3):363–71. doi: 10.3899/jrheum.140382
8. Sokolove J, Lepus CM. Role of Inflammation in the Pathogenesis of Osteoarthritis: Latest Findings and Interpretations. Ther Adv Musculoskelet Dis (2013) 5(2):77–94. doi: 10.1177/1759720X12467868
9. Punzi L, Galozzi P, Luisetto R, Favero M, Ramonda R, Oliviero F, et al. Post-Traumatic Arthritis: Overview on Pathogenic Mechanisms and Role of Inflammation. RMD Open (2016) 2(2):e000279. doi: 10.1136/rmdopen-2016-000279
10. Berenbaum F. Osteoarthritis as an Inflammatory Disease (Osteoarthritis is Not Osteoarthrosis!). Osteoarthritis Cartilage (2013) 21(1):16–21. doi: 10.1016/j.joca.2012.11.012
11. Woodell-May JE, Sommerfeld SD. Role of Inflammation and the Immune System in the Progression of Osteoarthritis. J Orthop Res (2020) 38(2):253–7. doi: 10.1002/jor.24457
12. Mora JC, Przkora R, Cruz-Almeida Y. Knee Osteoarthritis: Pathophysiology and Current Treatment Modalities. J Pain Res (2018) 11:2189–96. doi: 10.2147/JPR.S154002
13. Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, et al. A Lineage of Myeloid Cells Independent of Myb and Hematopoietic Stem Cells. Science (2012) 336(6077):86–90. doi: 10.1126/science.1219179
14. Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, et al. Tissue-Resident Macrophages Self-Maintain Locally Throughout Adult Life With Minimal Contribution From Circulating Monocytes. Immunity (2013) 38(4):792–804. doi: 10.1016/j.immuni.2013.04.004
15. Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-Resident Macrophages Originate From Yolk-Sac-Derived Erythro-Myeloid Progenitors. Nature (2015) 518(7540):547–51. doi: 10.1038/nature13989
16. Hoeffel G, Chen J, Lavin Y, Low D, Almeida FF, See P, et al. C-Myb(+) Erythro-Myeloid Progenitor-Derived Fetal Monocytes Give Rise to Adult Tissue-Resident Macrophages. Immunity (2015) 42(4):665–78. doi: 10.1016/j.immuni.2015.03.011
17. Guilliams M, Scott CL. Does Niche Competition Determine the Origin of Tissue-Resident Macrophages? Nat Rev Immunol (2017) 17(7):451–60. doi: 10.1038/nri.2017.42
18. Guilliams M, Thierry GR, Bonnardel J, Bajenoff M. Establishment and Maintenance of the Macrophage Niche. Immunity (2020) 52(3):434–51. doi: 10.1016/j.immuni.2020.02.015
19. Ginhoux F, Guilliams M. Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity (2016) 44(3):439–49. doi: 10.1016/j.immuni.2016.02.024
20. Culemann S, Gruneboom A, Kronke G. Origin and Function of Synovial Macrophage Subsets During Inflammatory Joint Disease. Adv Immunol (2019) 143:75–98. doi: 10.1016/bs.ai.2019.08.006
21. Raghu H, Lepus CM, Wang Q, Wong HH, Lingampalli N, Oliviero F, et al. CCL2/CCR2, But Not CCL5/CCR5, Mediates Monocyte Recruitment, Inflammation and Cartilage Destruction in Osteoarthritis. Ann Rheum Dis (2017) 76(5):914–22. doi: 10.1136/annrheumdis-2016-210426
22. Culemann S, Gruneboom A, Nicolas-Avila JA, Weidner D, Lammle KF, Rothe T, et al. Locally Renewing Resident Synovial Macrophages Provide a Protective Barrier for the Joint. Nature (2019) 572(7771):670–5. doi: 10.1038/s41586-019-1471-1
23. Boersema GSA, Utomo L, Bayon Y, Kops N, van der Harst E, Lange JF, et al. Monocyte Subsets in Blood Correlate With Obesity Related Response of Macrophages to Biomaterials. Vitro Biomater (2016) 109:32–9. doi: 10.1016/j.biomaterials.2016.09.009
24. Masouros SD, Bull AMJ. Amis AA.(i) Biomechanics of the Knee Joint. Orthopaedics Trauma (2010) 24(2):84–91. doi: 10.1016/j.mporth.2010.03.005
25. Blackburn TA, Craig E. Knee Anatomy: A Brief Review. Phys Ther (1980) 60(12):1556–60. doi: 10.1093/ptj/60.12.1556
26. Sophia Fox AJ, Bedi A, Rodeo SA. The Basic Science of Articular Cartilage: Structure, Composition, and Function. Sports Health (2009) 1(6):461–8. doi: 10.1177/1941738109350438
27. Alford JW, Cole BJ. Cartilage Restoration, Part 1: Basic Science, Historical Perspective, Patient Evaluation, and Treatment Options. Am J Sports Med (2005) 33(2):295–306. doi: 10.1177/0363546504273510
28. Hoffa A. The Influence Of The Adipose Tissue With Regard To The Pathology Of The Knee Joint. J Am Med Assoc (1904) XLIII(12):795–6. doi: 10.1001/jama.1904.92500120002h
29. Jiang LF, Fang JH, Wu LD. Role of Infrapatellar Fat Pad in Pathological Process of Knee Osteoarthritis: Future Applications in Treatment. World J Clin cases (2019) 7(16):2134–42. doi: 10.12998/wjcc.v7.i16.2134
30. Urban H, Little CB. The Role of Fat and Inflammation in the Pathogenesis and Management of Osteoarthritis. Rheumatol (Oxford) (2018) 57(suppl_4):iv10–21. doi: 10.1093/rheumatology/kex399
31. Kung MS, Markantonis J, Nelson SD, Campbell P. The Synovial Lining and Synovial Fluid Properties After Joint Arthroplasty. Lubricants (2015) 3(2):394–412. doi: 10.3390/lubricants3020394
32. Scanzello CR, Goldring SR. The Role of Synovitis in Osteoarthritis Pathogenesis. Bone (2012) 51(2):249–57. doi: 10.1016/j.bone.2012.02.012
33. Varol C, Mildner A, Jung S. Macrophages: Development and Tissue Specialization. Annu Rev Immunol (2015) 33:643–75. doi: 10.1146/annurev-immunol-032414-112220
34. Mass E, Gentek R. Fetal-Derived Immune Cells at the Roots of Lifelong Pathophysiology. Front Cell Dev Biol (2021) 9:648313. doi: 10.3389/fcell.2021.648313
35. Guilliams M, Mildner A, Yona S. Developmental and Functional Heterogeneity of Monocytes. Immunity (2018) 49(4):595–613. doi: 10.1016/j.immuni.2018.10.005
36. Fogg DK, Sibon C, Miled C, Jung S, Aucouturier P, Littman DR, et al. A Clonogenic Bone Marrow Progenitor Specific for Macrophages and Dendritic Cells. Science (2006) 311(5757):83–7. doi: 10.1126/science.1117729
37. Auffray C, Fogg DK, Narni-Mancinelli E, Senechal B, Trouillet C, Saederup N, et al. CX3CR1+ CD115+ CD135+ Common Macrophage/DC Precursors and the Role of CX3CR1 in Their Response to Inflammation. J Exp Med (2009) 206(3):595–606. doi: 10.1084/jem.20081385
38. Hettinger J, Richards DM, Hansson J, Barra MM, Joschko AC, Krijgsveld J, et al. Origin of Monocytes and Macrophages in a Committed Progenitor. Nat Immunol (2013) 14(8):821–30. doi: 10.1038/ni.2638
39. Chong SZ, Evrard M, Devi S, Chen J, Lim JY, See P, et al. CXCR4 Identifies Transitional Bone Marrow Premonocytes That Replenish the Mature Monocyte Pool for Peripheral Responses. J Exp Med (2016) 213(11):2293–314. doi: 10.1084/jem.20160800
40. Geissmann F, Jung S, Littman DR. Blood Monocytes Consist of Two Principal Subsets With Distinct Migratory Properties. Immunity (2003) 19(1):71–82. doi: 10.1016/s1074-7613(03)00174-2
41. Teh YC, Ding JL, Ng LG, Chong SZ. Capturing the Fantastic Voyage of Monocytes Through Time and Space. Front Immunol (2019) 10:834. doi: 10.3389/fimmu.2019.00834
42. Ginhoux F, Jung S. Monocytes and Macrophages: Developmental Pathways and Tissue Homeostasis. Nat Rev Immunol (2014) 14(6):392–404. doi: 10.1038/nri3671
43. Jakubzick C, Gautier EL, Gibbings SL, Sojka DK, Schlitzer A, Johnson TE, et al. Minimal Differentiation of Classical Monocytes as They Survey Steady-State Tissues and Transport Antigen to Lymph Nodes. Immunity (2013) 39(3):599–610. doi: 10.1016/j.immuni.2013.08.007
44. Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, et al. Analysis of Fractalkine Receptor CX(3)CR1 Function by Targeted Deletion and Green Fluorescent Protein Reporter Gene Insertion. Mol Cell Biol (2000) 20(11):4106–14. doi: 10.1128/mcb.20.11.4106-4114.2000
45. Kratofil RM, Kubes P, Deniset JF. Monocyte Conversion During Inflammation and Injury. Arterioscler Thromb Vasc Biol (2017) 37(1):35–42. doi: 10.1161/ATVBAHA.116.308198
46. Zhao M, Tuo H, Wang S, Zhao L. The Roles of Monocyte and Monocyte-Derived Macrophages in Common Brain Disorders. BioMed Res Int (2020) 2020:9396021. doi: 10.1155/2020/9396021
47. Carlin LM, Stamatiades EG, Auffray C, Hanna RN, Glover L, Vizcay-Barrena G, et al. Nr4a1-Dependent Ly6C(low) Monocytes Monitor Endothelial Cells and Orchestrate Their Disposal. Cell (2013) 153(2):362–75. doi: 10.1016/j.cell.2013.03.010
48. Jung H, Mithal DS, Park JE, Miller RJ. Localized CCR2 Activation in the Bone Marrow Niche Mobilizes Monocytes by Desensitizing Cxcr4. PloS One (2015) 10(6):e0128387. doi: 10.1371/journal.pone.0128387
49. Liu Q, Li Z, Gao JL, Wan W, Ganesan S, McDermott DH, et al. CXCR4 Antagonist AMD3100 Redistributes Leukocytes From Primary Immune Organs to Secondary Immune Organs, Lung, and Blood in Mice. Eur J Immunol (2015) 45(6):1855–67. doi: 10.1002/eji.201445245
50. Tsou CL, Peters W, Si Y, Slaymaker S, Aslanian AM, Weisberg SP, et al. Critical Roles for CCR2 and MCP-3 in Monocyte Mobilization From Bone Marrow and Recruitment to Inflammatory Sites. J Clin Invest (2007) 117(4):902–9. doi: 10.1172/JCI29919
51. Serbina NV, Pamer EG. Monocyte Emigration From Bone Marrow During Bacterial Infection Requires Signals Mediated by Chemokine Receptor CCR2. Nat Immunol (2006) 7(3):311–7. doi: 10.1038/ni1309
52. Debien E, Mayol K, Biajoux V, Daussy C, De Aguero MG, Taillardet M, et al. S1PR5 is Pivotal for the Homeostasis of Patrolling Monocytes. Eur J Immunol (2013) 43(6):1667–75. doi: 10.1002/eji.201343312
53. Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate Mapping Reveals Origins and Dynamics of Monocytes and Tissue Macrophages Under Homeostasis. Immunity (2013) 38(1):79–91. doi: 10.1016/j.immuni.2012.12.001
54. Scott CL, Zheng F, De Baetselier P, Martens L, Saeys Y, De Prijck S, et al. Bone Marrow-Derived Monocytes Give Rise to Self-Renewing and Fully Differentiated Kupffer Cells. Nat Commun (2016) 7:10321. doi: 10.1038/ncomms10321
55. Varol C, Landsman L, Fogg DK, Greenshtein L, Gildor B, Margalit R, et al. Monocytes Give Rise to Mucosal, But Not Splenic, Conventional Dendritic Cells. J Exp Med (2007) 204(1):171–80. doi: 10.1084/jem.20061011
56. Hanna RN, Carlin LM, Hubbeling HG, Nackiewicz D, Green AM, Punt JA, et al. The Transcription Factor NR4A1 (Nur77) Controls Bone Marrow Differentiation and the Survival of Ly6C- Monocytes. Nat Immunol (2011) 12(8):778–85. doi: 10.1038/ni.2063
57. Gamrekelashvili J, Giagnorio R, Jussofie J, Soehnlein O, Duchene J, Briseno CG, et al. Regulation of Monocyte Cell Fate by Blood Vessels Mediated by Notch Signalling. Nat Commun (2016) 7:12597. doi: 10.1038/ncomms12597
58. Narasimhan PB, Marcovecchio P, Hamers AAJ, Hedrick CC. Nonclassical Monocytes in Health and Disease. Annu Rev Immunol (2019) 37:439–56. doi: 10.1146/annurev-immunol-042617-053119
59. Schmidl C, Renner K, Peter K, Eder R, Lassmann T, Balwierz PJ, et al. Transcription and Enhancer Profiling in Human Monocyte Subsets. Blood (2014) 123(17):e90–9. doi: 10.1182/blood-2013-02-484188
60. Buenrostro JD, Corces MR, Lareau CA, Wu B, Schep AN, Aryee MJ, et al. Integrated Single-Cell Analysis Maps the Continuous Regulatory Landscape of Human Hematopoietic Differentiation. Cell (2018) 173(6):1535–48 e16. doi: 10.1016/j.cell.2018.03.074
61. Quintar A, McArdle S, Wolf D, Marki A, Ehinger E, Vassallo M, et al. Endothelial Protective Monocyte Patrolling in Large Arteries Intensified by Western Diet and Atherosclerosis. Circ Res (2017) 120(11):1789–99. doi: 10.1161/CIRCRESAHA.117.310739
62. Thomas G, Tacke R, Hedrick CC, Hanna RN. Nonclassical Patrolling Monocyte Function in the Vasculature. Arterioscler Thromb Vasc Biol (2015) 35(6):1306–16. doi: 10.1161/ATVBAHA.114.304650
63. Satoh T, Nakagawa K, Sugihara F, Kuwahara R, Ashihara M, Yamane F, et al. Identification of an Atypical Monocyte and Committed Progenitor Involved in Fibrosis. Nature (2017) 541(7635):96–101. doi: 10.1038/nature20611
64. Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, et al. Embryonic and Adult-Derived Resident Cardiac Macrophages are Maintained Through Distinct Mechanisms at Steady State and During Inflammation. Immunity (2014) 40(1):91–104. doi: 10.1016/j.immuni.2013.11.019
65. Bain CC, Bravo-Blas A, Scott CL, Perdiguero EG, Geissmann F, Henri S, et al. Constant Replenishment From Circulating Monocytes Maintains the Macrophage Pool in the Intestine of Adult Mice. Nat Immunol (2014) 15(10):929–37. doi: 10.1038/ni.2967
66. Tamoutounour S, Guilliams M, Montanana Sanchis F, Liu H, Terhorst D, Malosse C, et al. Origins and Functional Specialization of Macrophages and of Conventional and Monocyte-Derived Dendritic Cells in Mouse Skin. Immunity (2013) 39(5):925–38. doi: 10.1016/j.immuni.2013.10.004
67. Tamoutounour S, Henri S, Lelouard H, de Bovis B, de Haar C, van der Woude CJ, et al. CD64 Distinguishes Macrophages From Dendritic Cells in the Gut and Reveals the Th1-Inducing Role of Mesenteric Lymph Node Macrophages During Colitis. Eur J Immunol (2012) 42(12):3150–66. doi: 10.1002/eji.201242847
68. Mass E, Ballesteros I, Farlik M, Halbritter F, Gunther P, Crozet L, et al. Specification of Tissue-Resident Macrophages During Organogenesis. Science (2016) 353(6304):353–85. doi: 10.1126/science.aaf4238
69. T'Jonck W, Guilliams M, Bonnardel J. Niche Signals and Transcription Factors Involved in Tissue-Resident Macrophage Development. Cell Immunol (2018) 330:43–53. doi: 10.1016/j.cellimm.2018.02.005
70. Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, et al. Tissue-Resident Macrophage Enhancer Landscapes are Shaped by the Local Microenvironment. Cell (2014) 159(6):1312–26. doi: 10.1016/j.cell.2014.11.018
71. Gosselin D, Link VM, Romanoski CE, Fonseca GJ, Eichenfield DZ, Spann NJ, et al. Environment Drives Selection and Function of Enhancers Controlling Tissue-Specific Macrophage Identities. Cell (2014) 159(6):1327–40. doi: 10.1016/j.cell.2014.11.023
72. Etzerodt A, Tsalkitzi K, Maniecki M, Damsky W, Delfini M, Baudoin E, et al. Specific Targeting of CD163(+) TAMs Mobilizes Inflammatory Monocytes and Promotes T Cell-Mediated Tumor Regression. J Exp Med (2019) 216(10):2394–411. doi: 10.1084/jem.20182124
73. Zhu Y, Herndon JM, Sojka DK, Kim KW, Knolhoff BL, Zuo C, et al. Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate From Embryonic Hematopoiesis and Promote Tumor Progression. Immunity (2017) 47(3):597. doi: 10.1016/j.immuni.2017.08.018
74. Loyher PL, Hamon P, Laviron M, Meghraoui-Kheddar A, Goncalves E, Deng Z, et al. Macrophages of Distinct Origins Contribute to Tumor Development in the Lung. J Exp Med (2018) 215(10):2536–53. doi: 10.1084/jem.20180534
75. Werner Y, Mass E, Ashok Kumar P, Ulas T, Handler K, Horne A, et al. Cxcr4 Distinguishes HSC-Derived Monocytes From Microglia and Reveals Monocyte Immune Responses to Experimental Stroke. Nat Neurosci (2020) 23(3):351–62. doi: 10.1038/s41593-020-0585-y
76. Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, et al. Identification of Splenic Reservoir Monocytes and Their Deployment to Inflammatory Sites. Science (2009) 325(5940):612–6. doi: 10.1126/science.1175202
77. Li L, Huang L, Sung SS, Vergis AL, Rosin DL, Rose CE Jr., et al. The Chemokine Receptors CCR2 and CX3CR1 Mediate Monocyte/Macrophage Trafficking in Kidney Ischemia-Reperfusion Injury. Kidney Int (2008) 74(12):1526–37. doi: 10.1038/ki.2008.500
78. Huen SC, Cantley LG. Macrophages in Renal Injury and Repair. Annu Rev Physiol (2017) 79:449–69. doi: 10.1146/annurev-physiol-022516-034219
79. Furuichi K, Wada T, Iwata Y, Kitagawa K, Kobayashi K, Hashimoto H, et al. CCR2 Signaling Contributes to Ischemia-Reperfusion Injury in Kidney. J Am Soc Nephrol (2003) 14(10):2503–15. doi: 10.1097/01.asn.0000089563.63641.a8
80. Shechter R, Raposo C, London A, Sagi I, Schwartz M. The Glial Scar-Monocyte Interplay: A Pivotal Resolution Phase in Spinal Cord Repair. PloS One (2011) 6(12):e27969. doi: 10.1371/journal.pone.0027969
81. Shechter R, London A, Varol C, Raposo C, Cusimano M, Yovel G, et al. Infiltrating Blood-Derived Macrophages are Vital Cells Playing an Anti-Inflammatory Role in Recovery From Spinal Cord Injury in Mice. PloS Med (2009) 6(7):e1000113. doi: 10.1371/journal.pmed.1000113
82. Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, et al. The Healing Myocardium Sequentially Mobilizes Two Monocyte Subsets With Divergent and Complementary Functions. J Exp Med (2007) 204(12):3037–47. doi: 10.1084/jem.20070885
83. Dimitrijevic OB, Stamatovic SM, Keep RF, Andjelkovic AV. Absence of the Chemokine Receptor CCR2 Protects Against Cerebral Ischemia/Reperfusion Injury in Mice. Stroke (2007) 38(4):1345–53. doi: 10.1161/01.STR.0000259709.16654.8f
84. Bamboat ZM, Ocuin LM, Balachandran VP, Obaid H, Plitas G, DeMatteo RP. Conventional DCs Reduce Liver Ischemia/Reperfusion Injury in Mice. Via IL-10 Secretion J Clin Invest (2010) 120(2):559–69. doi: 10.1172/JCI40008
85. Spahn JH, Kreisel D. Monocytes in Sterile Inflammation: Recruitment and Functional Consequences. Arch Immunol Ther Exp (Warsz) (2014) 62(3):187–94. doi: 10.1007/s00005-013-0267-5
86. Robbins CS, Chudnovskiy A, Rauch PJ, Figueiredo JL, Iwamoto Y, Gorbatov R, et al. Extramedullary Hematopoiesis Generates Ly-6C(High) Monocytes That Infiltrate Atherosclerotic Lesions. Circulation (2012) 125(2):364–74. doi: 10.1161/CIRCULATIONAHA.111.061986
87. Zhou X, Liu XL, Ji WJ, Liu JX, Guo ZZ, Ren D, et al. The Kinetics of Circulating Monocyte Subsets and Monocyte-Platelet Aggregates in the Acute Phase of ST-Elevation Myocardial Infarction: Associations With 2-Year Cardiovascular Events. Med (Baltimore) (2016) 95(18):e3466. doi: 10.1097/MD.0000000000003466
88. Rahman K, Vengrenyuk Y, Ramsey SA, Vila NR, Girgis NM, Liu J, et al. Inflammatory Ly6Chi Monocytes and Their Conversion to M2 Macrophages Drive Atherosclerosis Regression. J Clin Invest (2017) 127(8):2904–15. doi: 10.1172/JCI75005
89. Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, et al. Gene-Expression Profiles and Transcriptional Regulatory Pathways That Underlie the Identity and Diversity of Mouse Tissue Macrophages. Nat Immunol (2012) 13(11):1118–28. doi: 10.1038/ni.2419
90. Haldar M, Kohyama M, So AY, Kc W, Wu X, Briseno CG, et al. Heme-Mediated SPI-C Induction Promotes Monocyte Differentiation Into Iron-Recycling Macrophages. Cell (2014) 156(6):1223–34. doi: 10.1016/j.cell.2014.01.069
91. van de Laar L, Saelens W, De Prijck S, Martens L, Scott CL, Van Isterdael G, et al. Yolk Sac Macrophages, Fetal Liver, and Adult Monocytes Can Colonize an Empty Niche and Develop Into Functional Tissue-Resident Macrophages. Immunity (2016) 44(4):755–68. doi: 10.1016/j.immuni.2016.02.017
92. Gordon S, Martinez FO. Alternative Activation of Macrophages: Mechanism and Functions. Immunity (2010) 32(5):593–604. doi: 10.1016/j.immuni.2010.05.007
93. Sica A, Mantovani A. Macrophage Plasticity and Polarization: In Vivo Veritas. J Clin Invest (2012) 122(3):787–95. doi: 10.1172/JCI59643
94. Hume DA. The Many Alternative Faces of Macrophage Activation. Front Immunol (2015) 6:370. doi: 10.3389/fimmu.2015.00370
95. Murray PJ. Macrophage Polarization. Annu Rev Physiol (2017) 79:541–66. doi: 10.1146/annurev-physiol-022516-034339
96. Martinez FO, Gordon S. The M1 and M2 Paradigm of Macrophage Activation: Time for Reassessment. F1000Prime Rep (2014) 6:13. doi: 10.12703/P6-13
97. Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, et al. Transcriptome-Based Network Analysis Reveals a Spectrum Model of Human Macrophage Activation. Immunity (2014) 40(2):274–88. doi: 10.1016/j.immuni.2014.01.006
98. Wiktor-Jedrzejczak W, Bartocci A, Ferrante AW Jr., Ahmed-Ansari A, Sell KW, Pollard JW, et al. Total Absence of Colony-Stimulating Factor 1 in the Macrophage-Deficient Osteopetrotic (Op/Op) Mouse. Proc Natl Acad Sci U.S.A. (1990) 87(12):4828–32. doi: 10.1073/pnas.87.12.4828
99. Cecchini MG, Dominguez MG, Mocci S, Wetterwald A, Felix R, Fleisch H, et al. Role of Colony Stimulating Factor-1 in the Establishment and Regulation of Tissue Macrophages During Postnatal Development of the Mouse. Development (1994) 120(6):1357–72. doi: 10.1242/dev.120.6.1357
100. Dai XM, Ryan GR, Hapel AJ, Dominguez MG, Russell RG, Kapp S, et al. Targeted Disruption of the Mouse Colony-Stimulating Factor 1 Receptor Gene Results in Osteopetrosis, Mononuclear Phagocyte Deficiency, Increased Primitive Progenitor Cell Frequencies, and Reproductive Defects. Blood (2002) 99(1):111–20. doi: 10.1182/blood.v99.1.111
101. Bonnardel J, T'Jonck W, Gaublomme D, Browaeys R, Scott CL, Martens L, et al. Stellate Cells, Hepatocytes, and Endothelial Cells Imprint the Kupffer Cell Identity on Monocytes Colonizing the Liver Macrophage Niche. Immunity (2019) 51(4):638–54 e9. doi: 10.1016/j.immuni.2019.08.017
102. Bellomo A, Mondor I, Spinelli L, Lagueyrie M, Stewart BJ, Brouilly N, et al. Reticular Fibroblasts Expressing the Transcription Factor WT1 Define a Stromal Niche That Maintains and Replenishes Splenic Red Pulp Macrophages. Immunity (2020) 53(1):127–42 e7. doi: 10.1016/j.immuni.2020.06.008
103. Mortha A, Chudnovskiy A, Hashimoto D, Bogunovic M, Spencer SP, Belkaid Y, et al. Microbiota-Dependent Crosstalk Between Macrophages and ILC3 Promotes Intestinal Homeostasis. Science (2014) 343(6178):1249288. doi: 10.1126/science.1249288
104. Kana V, Desland FA, Casanova-Acebes M, Ayata P, Badimon A, Nabel E, et al. CSF-1 Controls Cerebellar Microglia and is Required for Motor Function and Social Interaction. J Exp Med (2019) 216(10):2265–81. doi: 10.1084/jem.20182037
105. Muller PA, Koscso B, Rajani GM, Stevanovic K, Berres ML, Hashimoto D, et al. Crosstalk Between Muscularis Macrophages and Enteric Neurons Regulates Gastrointestinal Motility. Cell (2014) 158(2):300–13. doi: 10.1016/j.cell.2014.04.050
106. Yoshida M, Ikegami M, Reed JA, Chroneos ZC, Whitsett JA. GM-CSF Regulates Protein and Lipid Catabolism by Alveolar Macrophages. Am J Physiol Lung Cell Mol Physiol (2001) 280(3):L379–86. doi: 10.1152/ajplung.2001.280.3.L379
107. Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, et al. Alveolar Macrophages Develop From Fetal Monocytes That Differentiate Into Long-Lived Cells in the First Week of Life via GM-CSF. J Exp Med (2013) 210(10):1977–92. doi: 10.1084/jem.20131199
108. De Schepper S, Verheijden S, Aguilera-Lizarraga J, Viola MF, Boesmans W, Stakenborg N, et al. Self-Maintaining Gut Macrophages Are Essential for Intestinal Homeostasis. Cell (2018) 175(2):400–15.e13. doi: 10.1016/j.cell.2018.07.048
109. Hulsmans M, Clauss S, Xiao L, Aguirre AD, King KR, Hanley A, et al. Macrophages Facilitate Electrical Conduction in the Heart. Cell (2017) 169(3):510–22 e20. doi: 10.1016/j.cell.2017.03.050
110. Bergh A, Damber JE, van Rooijen N. Liposome-Mediated Macrophage Depletion: An Experimental Approach to Study the Role of Testicular Macrophages in the Rat. J Endocrinol (1993) 136(3):407–13. doi: 10.1677/joe.0.1360407
111. Hutson JC, Garner CW, Doris PA. Purification and Characterization of a Lipophilic Factor From Testicular Macrophages That Stimulates Testosterone Production by Leydig Cells. J Androl (1996) 17(5):502–8.
112. Lukyanenko YO, Chen JJ, Hutson JC. Production of 25-Hydroxycholesterol by Testicular Macrophages and its Effects on Leydig Cells. Biol Reprod (2001) 64(3):790–6. doi: 10.1095/biolreprod64.3.790
113. Yahara Y, Barrientos T, Tang YJ, Puviindran V, Nadesan P, Zhang H, et al. Erythromyeloid Progenitors Give Rise to a Population of Osteoclasts That Contribute to Bone Homeostasis and Repair. Nat Cell Biol (2020) 22(1):49–59. doi: 10.1038/s41556-019-0437-8
114. Jacome-Galarza CE, Percin GI, Muller JT, Mass E, Lazarov T, Eitler J, et al. Developmental Origin, Functional Maintenance and Genetic Rescue of Osteoclasts. Nature (2019) 568(7753):541–5. doi: 10.1038/s41586-019-1105-7
115. McDonald MM, Khoo WH, Ng PY, Xiao Y, Zamerli J, Thatcher P, et al. Osteoclasts Recycle via Osteomorphs During RANKL-Stimulated Bone Resorption. Cell (2021) 184(5):1330–47 e13. doi: 10.1016/j.cell.2021.02.002
116. Cox N, Geissmann F. Macrophage Ontogeny in the Control of Adipose Tissue Biology. Curr Opin Immunol (2020) 62:1–8. doi: 10.1016/j.coi.2019.08.002
117. Zeyda M, Farmer D, Todoric J, Aszmann O, Speiser M, Gyori G, et al. Human Adipose Tissue Macrophages are of an Anti-Inflammatory Phenotype But Capable of Excessive Pro-Inflammatory Mediator Production. Int J Obes (Lond) (2007) 31(9):1420–8. doi: 10.1038/sj.ijo.0803632
118. Cox N, Crozet L, Holtman IR, Loyher PL, Lazarov T, White JB, et al. Diet-Regulated Production of PDGFcc by Macrophages Controls Energy Storage. bioRxiv (2021) 373(6550):eabe9383. doi: 10.1126/science.abe9383
119. Barboza E, Hudson J, Chang WP, Kovats S, Towner RA, Silasi-Mansat R, et al. Profibrotic Infrapatellar Fat Pad Remodeling Without M1 Macrophage Polarization Precedes Knee Osteoarthritis in Mice With Diet-Induced Obesity. Arthritis Rheumatol (2017) 69(6):1221–32. doi: 10.1002/art.40056
120. Fujisaka S, Usui I, Bukhari A, Ikutani M, Oya T, Kanatani Y, et al. Regulatory Mechanisms for Adipose Tissue M1 and M2 Macrophages in Diet-Induced Obese Mice. Diabetes (2009) 58(11):2574–82. doi: 10.2337/db08-1475
121. Iwata M, Ochi H, Hara Y, Tagawa M, Koga D, Okawa A, et al. Initial Responses of Articular Tissues in a Murine High-Fat Diet-Induced Osteoarthritis Model: Pivotal Role of the IPFP as a Cytokine Fountain. PloS One (2013) 8(4):e60706. doi: 10.1371/journal.pone.0060706
122. Harasymowicz NS, Clement ND, Azfer A, Burnett R, Salter DM, Simpson A. Regional Differences Between Perisynovial and Infrapatellar Adipose Tissue Depots and Their Response to Class II and Class III Obesity in Patients With Osteoarthritis. Arthritis Rheumatol (2017) 69(7):1396–406. doi: 10.1002/art.40102
123. Warmink K, Kozijn AE, Bobeldijk I, Stoop R, Weinans H, Korthagen NM. High-Fat Feeding Primes the Mouse Knee Joint to Develop Osteoarthritis and Pathologic Infrapatellar Fat Pad Changes After Surgically Induced Injury. Osteoarthritis Cartilage (2020) 28(5):593–602. doi: 10.1016/j.joca.2020.03.008
124. Kurowska-Stolarska M, Alivernini S. Synovial Tissue Macrophages: Friend or Foe? RMD Open (2017) 3(2):e000527. doi: 10.1136/rmdopen-2017-000527
125. de Lange-Brokaar BJE, Ioan-Facsinay A, van Osch GJVM, Zuurmond AM, Schoones J, Toes REM, et al. Synovial Inflammation, Immune Cells and Their Cytokines in Osteoarthritis: A Review. Osteoarthritis Cartilage (2012) 20(12):1484–99. doi: 10.1016/j.joca.2012.08.027
126. Buckley CD, Ospelt C, Gay S, Midwood KS. Location, Location, Location: How the Tissue Microenvironment Affects Inflammation in RA. Nat Rev Rheumatol (2021) 17(4):195–212. doi: 10.1038/s41584-020-00570-2
127. Ambarus CA, Noordenbos T, de Hair MJ, Tak PP, Baeten DL. Intimal Lining Layer Macrophages But Not Synovial Sublining Macrophages Display an IL-10 Polarized-Like Phenotype in Chronic Synovitis. Arthritis Res Ther (2012) 14(2):R74. doi: 10.1186/ar3796
128. Fonseca JE, Edwards JC, Blades S, Goulding NJ. Macrophage Subpopulations in Rheumatoid Synovium: Reduced CD163 Expression in CD4+ T Lymphocyte-Rich Microenvironments. Arthritis Rheum (2002) 46(5):1210–6. doi: 10.1002/art.10207
129. Smith MD, Barg E, Weedon H, Papengelis V, Smeets T, Tak PP, et al. Microarchitecture and Protective Mechanisms in Synovial Tissue From Clinically and Arthroscopically Normal Knee Joints. Ann Rheum Dis (2003) 62(4):303–7. doi: 10.1136/ard.62.4.303
130. Buechler MB, Kim KW, Onufer EJ, Williams JW, Little CC, Dominguez CX, et al. A Stromal Niche Defined by Expression of the Transcription Factor WT1 Mediates Programming and Homeostasis of Cavity-Resident Macrophages. Immunity (2019) 51(1):119–30 e5. doi: 10.1016/j.immuni.2019.05.010
131. Alivernini S, MacDonald L, Elmesmari A, Finlay S, Tolusso B, Gigante MR, et al. Distinct Synovial Tissue Macrophage Subsets Regulate Inflammation and Remission in Rheumatoid Arthritis. Nat Med (2020) 26(8):1295–306. doi: 10.1038/s41591-020-0939-8
132. Croft AP, Campos J, Jansen K, Turner JD, Marshall J, Attar M, et al. Distinct Fibroblast Subsets Drive Inflammation and Damage in Arthritis. Nature (2019) 570(7760):246–51. doi: 10.1038/s41586-019-1263-7
133. Schuster R, Rockel JS, Kapoor M, Hinz B. The Inflammatory Speech of Fibroblasts. Immunol Rev (2021) 302:126–46. doi: 10.1111/imr.12971
134. Hannemann N, Apparailly F, Courties G. Synovial Macrophages: From Ordinary Eaters to Extraordinary Multitaskers. Trends Immunol (2021) 42(5):368–71. doi: 10.1016/j.it.2021.03.002
135. Franklin RA. Fibroblasts and Macrophages: Collaborators in Tissue Homeostasis. Immunol Rev (2021) 302:86–103. doi: 10.1111/imr.12989
136. Ryan GR, Dai X-M, Dominguez MG, Tong W, Chuan F, Chisholm O, et al. Rescue of the Colony-Stimulating Factor 1 (CSF-1)–Nullizygous Mouse (Csf1op/Csf1op) Phenotype With a CSF-1 Transgene and Identification of Sites of Local CSF-1 Synthesis. Blood (2001) 98(1):74–84. doi: 10.1182/blood.V98.1.74
137. Adlerz KM, Aranda-Espinoza H, Hayenga HN. Substrate Elasticity Regulates the Behavior of Human Monocyte-Derived Macrophages. Eur Biophys J (2016) 45(4):301–9. doi: 10.1007/s00249-015-1096-8
138. Escolano JC, Taubenberger AV, Abuhattum S, Schweitzer C, Farrukh A, Del Campo A, et al. Compliant Substrates Enhance Macrophage Cytokine Release and NLRP3 Inflammasome Formation During Their Pro-Inflammatory Response. Front Cell Dev Biol (2021) 9:639815. doi: 10.3389/fcell.2021.639815
139. Maruyama K, Nemoto E, Yamada S. Mechanical Regulation of Macrophage Function - Cyclic Tensile Force Inhibits NLRP3 Inflammasome-Dependent IL-1beta Secretion in Murine Macrophages. Inflamm Regener (2019) 39:3. doi: 10.1186/s41232-019-0092-2
140. O’Conor CJ, Ramalingam S, Zelenski NA, Benefield HC, Rigo I, Little D, et al. Cartilage-Specific Knockout of the Mechanosensory Ion Channel TRPV4 Decreases Age-Related Osteoarthritis. Sci Rep (2016) 6(1):29053. doi: 10.1038/srep29053
141. Lv Z, Xu X, Sun Z, Yang YX, Guo H, Li J, et al. TRPV1 Alleviates Osteoarthritis by Inhibiting M1 Macrophage Polarization via Ca2+/CaMKII/Nrf2 Signaling Pathway. Cell Death Dis (2021) 12(6):504. doi: 10.1038/s41419-021-03792-8
142. Michalick L, Kuebler WM. Trpv4—A Missing Link Between Mechanosensation and Immunity. Front Immunol (2020) 11:413(413). doi: 10.3389/fimmu.2020.00413
143. Arya RK, Goswami R, Rahaman SO. Mechanotransduction via a TRPV4-Rac1 Signaling Axis Plays a Role in Multinucleated Giant Cell Formation. J Biol Chem (2020) 296:100–29. doi: 10.1074/jbc.RA120.014597
144. Bhattaram P, Chandrasekharan U. The Joint Synovium: A Critical Determinant of Articular Cartilage Fate in Inflammatory Joint Diseases. Semin Cell Dev Biol (2017) 62:86–93. doi: 10.1016/j.semcdb.2016.05.009
145. Kennedy A, Fearon U, Veale DJ, Godson C. Macrophages in Synovial Inflammation. Front Immunol (2011) 2:52. doi: 10.3389/fimmu.2011.00052
146. van Lent PL, Licht R, Dijkman H, Holthuysen AE, Berden JH, van den Berg WB. Uptake of Apoptotic Leukocytes by Synovial Lining Macrophages Inhibits Immune Complex-Mediated Arthritis. J Leukoc Biol (2001) 70(5):708–14.
147. Goldring MB, Otero M. Inflammation in Osteoarthritis. Curr Opin Rheumatol (2011) 23(5):471–8. doi: 10.1097/BOR.0b013e328349c2b1
148. Mills CD, Kincaid K, Alt JM, Heilman MJ. Hill AM. M-1/M-2 Macrophages and the Th1/Th2 Paradigm. J Immunol (2000) 164(12):6166–73. doi: 10.4049/jimmunol.164.12.6166
149. Wu CL, McNeill J, Goon K, Little D, Kimmerling K, Huebner J, et al. Conditional Macrophage Depletion Increases Inflammation and Does Not Inhibit the Development of Osteoarthritis in Obese Macrophage Fas-Induced Apoptosis-Transgenic Mice. Arthritis Rheumatol (2017) 69(9):1772–83. doi: 10.1002/art.40161
150. Tu J, Wang X, Gong X, Hong W, Han D, Fang Y, et al. Synovial Macrophages in Rheumatoid Arthritis: The Past, Present, and Future. Mediators Inflammation (2020) 2020:1583647–. doi: 10.1155/2020/1583647
151. Udalova IA, Mantovani A, Feldmann M. Macrophage Heterogeneity in the Context of Rheumatoid Arthritis. Nat Rev Rheumatol (2016) 12(8):472–85. doi: 10.1038/nrrheum.2016.91
152. Shi C, Pamer EG. Monocyte Recruitment During Infection and Inflammation. Nat Rev Immunol (2011) 11(11):762–74. doi: 10.1038/nri3070
153. Fernandes JC, Martel-Pelletier J, Pelletier JP. The Role of Cytokines in Osteoarthritis Pathophysiology. Biorheology (2002) 39(1-2):237–46.
154. Schlaak JF, Pfers I, Meyer Zum Buschenfelde KH, Marker-Hermann E. Different Cytokine Profiles in the Synovial Fluid of Patients With Osteoarthritis, Rheumatoid Arthritis and Seronegative Spondylarthropathies. Clin Exp Rheumatol (1996) 14(2):155–62.
155. Blom AB, van der Kraan PM, van den Berg WB. Cytokine Targeting in Osteoarthritis. Curr Drug Targets (2007) 8(2):283–92. doi: 10.2174/138945007779940179
156. Cui J, Chen Y, Wang HY, Wang RF. Mechanisms and Pathways of Innate Immune Activation and Regulation in Health and Cancer. Hum Vaccin Immunother (2014) 10(11):3270–85. doi: 10.4161/21645515.2014.979640
157. Huard B, Schneider P, Mauri D, Tschopp J, French LE. T Cell Costimulation by the TNF Ligand BAFF. J Immunol (2001) 167(11):6225–31. doi: 10.4049/jimmunol.167.11.6225
158. Chen M, Lin X, Liu Y, Li Q, Deng Y, Liu Z, et al. The Function of BAFF on T Helper Cells in Autoimmunity. Cytokine Growth Factor Rev (2014) 25(3):301–5. doi: 10.1016/j.cytogfr.2013.12.011
159. Lai Kwan Lam Q, King Hung Ko O, Zheng BJ, Lu L. Local BAFF Gene Silencing Suppresses Th17-Cell Generation and Ameliorates Autoimmune Arthritis. Proc Natl Acad Sci U.S.A. (2008) 105(39):14993–8. doi: 10.1073/pnas.0806044105
160. Zheng SG, Wang J, Horwitz DA. Cutting Edge: Foxp3+CD4+CD25+ Regulatory T Cells Induced by IL-2 and TGF-Beta are Resistant to Th17 Conversion by IL-6. J Immunol (2008) 180(11):7112–6. doi: 10.4049/jimmunol.180.11.7112
161. Serada S, Fujimoto M, Mihara M, Koike N, Ohsugi Y, Nomura S, et al. IL-6 Blockade Inhibits the Induction of Myelin Antigen-Specific Th17 Cells and Th1 Cells in Experimental Autoimmune Encephalomyelitis. Proc Natl Acad Sci USA (2008) 105(26):9041–6. doi: 10.1073/pnas.0802218105
162. Bosello S, Youinou P, Daridon C, Tolusso B, Bendaoud B, Pietrapertosa D, et al. Concentrations of BAFF Correlate With Autoantibody Levels, Clinical Disease Activity, and Response to Treatment in Early Rheumatoid Arthritis. J Rheumatol (2008) 35(7):1256–64.
163. Wei F, Chang Y, Wei W. The Role of BAFF in the Progression of Rheumatoid Arthritis. Cytokine (2015) 76(2):537–44. doi: 10.1016/j.cyto.2015.07.014
164. Weyand CM. Immunopathologic Aspects of Rheumatoid Arthritis: Who is the Conductor and Who Plays the Immunologic Instrument? J Rheumatol Suppl (2007) 79:9–14.
165. Seyler TM, Park YW, Takemura S, Bram RJ, Kurtin PJ, Goronzy JJ, et al. BLyS and APRIL in Rheumatoid Arthritis. J Clin Invest (2005) 115(11):3083–92. doi: 10.1172/JCI25265
166. Yang L, Chen Z, Guo H, Wang Z, Sun K, Yang X, et al. Extensive Cytokine Analysis in Synovial Fluid of Osteoarthritis Patients. Cytokine (2021) 143:155546. doi: 10.1016/j.cyto.2021.155546
167. Kouskoff V, Korganow AS, Duchatelle V, Degott C, Benoist C, Mathis D. Organ-Specific Disease Provoked by Systemic Autoimmunity. Cell (1996) 87(5):811–22. doi: 10.1016/s0092-8674(00)81989-3
168. Korganow AS, Ji H, Mangialaio S, Duchatelle V, Pelanda R, Martin T, et al. From Systemic T Cell Self-Reactivity to Organ-Specific Autoimmune Disease via Immunoglobulins. Immunity (1999) 10(4):451–61. doi: 10.1016/s1074-7613(00)80045-x
169. Misharin AV, Cuda CM, Saber R, Turner JD, Gierut AK, Haines GK 3rd, et al. Nonclassical Ly6C(-) Monocytes Drive the Development of Inflammatory Arthritis in Mice. Cell Rep (2014) 9(2):591–604. doi: 10.1016/j.celrep.2014.09.032
170. Brunet A, LeBel M, Egarnes B, Paquet-Bouchard C, Lessard AJ, Brown JP, et al. NR4A1-Dependent Ly6C(low) Monocytes Contribute to Reducing Joint Inflammation in Arthritic Mice Through Treg Cells. Eur J Immunol (2016) 46(12):2789–800. doi: 10.1002/eji.201646406
171. Anderson DD, Chubinskaya S, Guilak F, Martin JA, Oegema TR, Olson SA, et al. Post-Traumatic Osteoarthritis: Improved Understanding and Opportunities for Early Intervention. J Orthop Res (2011) 29(6):802–9. doi: 10.1002/jor.21359
172. Lieberthal J, Sambamurthy N, Scanzello CR. Inflammation in Joint Injury and Post-Traumatic Osteoarthritis. Osteoarthritis Cartilage (2015) 23(11):1825–34. doi: 10.1016/j.joca.2015.08.015
173. Kandahari AM, Yang X, Dighe AS, Pan D, Cui Q. Recognition of Immune Response for the Early Diagnosis and Treatment of Osteoarthritis. J Immunol Res (2015) 2015:192415. doi: 10.1155/2015/192415
174. Jackson MT, Moradi B, Zaki S, Smith MM, McCracken S, Smith SM, et al. Depletion of Protease-Activated Receptor 2 But Not Protease-Activated Receptor 1 may Confer Protection Against Osteoarthritis in Mice Through Extracartilaginous Mechanisms. Arthritis Rheumatol (2014) 66(12):3337–48. doi: 10.1002/art.38876
175. Chen L, Zheng JJY, Li G, Yuan J, Ebert JR, Li H, et al. Pathogenesis and Clinical Management of Obesity-Related Knee Osteoarthritis: Impact of Mechanical Loading. J Orthop Translat (2020) 24:66–75. doi: 10.1016/j.jot.2020.05.001
176. Lotz MK, Kraus VB. New Developments in Osteoarthritis. Posttraumatic Osteoarthritis: Pathogenesis and Pharmacological Treatment Options. Arthritis Res Ther (2010) 12(3):211. doi: 10.1186/ar3046
177. Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, et al. Monocyte Subsets Differentially Employ CCR2, CCR5, and CX3CR1 to Accumulate Within Atherosclerotic Plaques. J Clin Invest (2007) 117(1):185–94. doi: 10.1172/JCI28549
178. Conti P, DiGioacchino M. MCP-1 and RANTES are Mediators of Acute and Chronic Inflammation. Allergy Asthma Proc (2001) 22(3):133–7. doi: 10.2500/108854101778148737
179. Yuan GH, Masuko-Hongo K, Sakata M, Tsuruha J, Onuma H, Nakamura H, et al. The Role of C-C Chemokines and Their Receptors in Osteoarthritis. Arthritis Rheum (2001) 44(5):1056–70. doi: 10.1002/1529-0131(200105)44:5<1056::AID-ANR186>3.0.CO;2-U
180. Tong X, Zeng H, Gu P, Wang K, Zhang H, Lin X. Monocyte Chemoattractant Protein1 Promotes the Proliferation, Migration and Differentiation Potential of Fibroblastlike Synoviocytes via the PI3K/P38 Cellular Signaling Pathway. Mol Med Rep (2020) 21(3):1623–32. doi: 10.3892/mmr.2020.10969
181. Tu J, Hong W, Guo Y, Zhang P, Fang Y, Wang X, et al. Ontogeny of Synovial Macrophages and the Roles of Synovial Macrophages From Different Origins in Arthritis. Front Immunol (2019) 10:1146. doi: 10.3389/fimmu.2019.01146
182. Nadiv O, Beer Y, Goldberg M, Agar G, Loos M, Katz Y. Decreased Induction of IL-1beta in Fibroblast-Like Synoviocytes: A Possible Regulatory Mechanism Maintaining Joint Homeostasis. Mol Immunol (2007) 44(12):3147–54. doi: 10.1016/j.molimm.2007.02.001
183. Watt FE, Paterson E, Freidin A, Kenny M, Judge A, Saklatvala J, et al. Acute Molecular Changes in Synovial Fluid Following Human Knee Injury: Association With Early Clinical Outcomes. Arthritis Rheumatol (2016) 68(9):2129–40. doi: 10.1002/art.39677
184. Scanzello CR, McKeon B, Swaim BH, DiCarlo E, Asomugha EU, Kanda V, et al. Synovial Inflammation in Patients Undergoing Arthroscopic Meniscectomy: Molecular Characterization and Relationship to Symptoms. Arthritis Rheum (2011) 63(2):391–400. doi: 10.1002/art.30137
185. Li L, Jiang BE. Serum and Synovial Fluid Chemokine Ligand 2/Monocyte Chemoattractant Protein 1 Concentrations Correlates With Symptomatic Severity in Patients With Knee Osteoarthritis. Ann Clin Biochem (2015) 52(Pt 2):276–82. doi: 10.1177/0004563214545117
186. Endres M, Andreas K, Kalwitz G, Freymann U, Neumann K, Ringe J, et al. Chemokine Profile of Synovial Fluid From Normal, Osteoarthritis and Rheumatoid Arthritis Patients: CCL25, CXCL10 and XCL1 Recruit Human Subchondral Mesenchymal Progenitor Cells. Osteoarthritis Cartilage (2010) 18(11):1458–66. doi: 10.1016/j.joca.2010.08.003
187. Ni F, Zhang Y, Peng X, Li J. Correlation Between Osteoarthritis and Monocyte Chemotactic Protein-1 Expression: A Meta-Analysis. J Orthop Surg Res (2020) 15(1):516. doi: 10.1186/s13018-020-02045-2
188. Haringman JJ, Smeets TJ, Reinders-Blankert P, Tak PP. Chemokine and Chemokine Receptor Expression in Paired Peripheral Blood Mononuclear Cells and Synovial Tissue of Patients With Rheumatoid Arthritis, Osteoarthritis, and Reactive Arthritis. Ann Rheum Dis (2006) 65(3):294–300. doi: 10.1136/ard.2005.037176
189. Takebe K, Rai MF, Schmidt EJ, Sandell LJ. The Chemokine Receptor CCR5 Plays a Role in Post-Traumatic Cartilage Loss in Mice, But Does Not Affect Synovium and Bone. Osteoarthritis Cartilage (2015) 23(3):454–61. doi: 10.1016/j.joca.2014.12.002
190. Wojdasiewicz P, Poniatowski LA, Kotela A, Deszczynski J, Kotela I, Szukiewicz D. The Chemokine CX3CL1 (Fractalkine) and its Receptor CX3CR1: Occurrence and Potential Role in Osteoarthritis. Arch Immunol Ther Exp (Warsz) (2014) 62(5):395–403. doi: 10.1007/s00005-014-0275-0
191. Yano R, Yamamura M, Sunahori K, Takasugi K, Yamana J, Kawashima M, et al. Recruitment of CD16+ Monocytes Into Synovial Tissues is Mediated by Fractalkine and CX3CR1 in Rheumatoid Arthritis Patients. Acta Med Okayama (2007) 61(2):89–98. doi: 10.18926/AMO/32882
192. Yarmola EG, Shah YY, Lakes EH, Pacheco YC, Xie DF, Dobson J, et al. Use of Magnetic Capture to Identify Elevated Levels of CCL2 Following Intra-Articular Injection of Monoiodoacetate in Rats. Connect Tissue Res (2020) 61(5):485–97. doi: 10.1080/03008207.2019.1620223
193. Lai-Zhao Y, Pitchers KK, Appleton CT. Transient Anabolic Effects of Synovium in Early Post-Traumatic Osteoarthritis: A Novel Ex Vivo Joint Tissue Co-Culture System for Investigating Synovium-Chondrocyte Interactions. Osteoarthritis Cartilage (2021) 29(7):1060–70. doi: 10.1016/j.joca.2021.03.010
194. Takayanagi H. New Developments in Osteoimmunology. Nat Rev Rheumatol (2012) 8(11):684–9. doi: 10.1038/nrrheum.2012.167
195. Hirose S, Lin Q, Ohtsuji M, Nishimura H, Verbeek JS. Monocyte Subsets Involved in the Development of Systemic Lupus Erythematosus and Rheumatoid Arthritis. Int Immunol (2019) 31(11):687–96. doi: 10.1093/intimm/dxz036
196. van den Bosch MH, Blom AB, Schelbergen RF, Koenders MI, van de Loo FA, van den Berg WB, et al. Alarmin S100A9 Induces Proinflammatory and Catabolic Effects Predominantly in the M1 Macrophages of Human Osteoarthritic Synovium. J Rheumatol (2016) 43(10):1874–84. doi: 10.3899/jrheum.160270
197. Cremers NAJ, van den Bosch MHJ, van Dalen S, Di Ceglie I, Ascone G, van de Loo F, et al. S100A8/A9 Increases the Mobilization of Pro-Inflammatory Ly6C(high) Monocytes to the Synovium During Experimental Osteoarthritis. Arthritis Res Ther (2017) 19(1):217. doi: 10.1186/s13075-017-1426-6
198. Schelbergen RF, Blom AB, van den Bosch MH, Sloetjes A, Abdollahi-Roodsaz S, Schreurs BW, et al. Alarmins S100A8 and S100A9 Elicit a Catabolic Effect in Human Osteoarthritic Chondrocytes That is Dependent on Toll-Like Receptor 4. Arthritis Rheum (2012) 64(5):1477–87. doi: 10.1002/art.33495
199. Zreiqat H, Belluoccio D, Smith MM, Wilson R, Rowley LA, Jones K, et al. S100A8 and S100A9 in Experimental Osteoarthritis. Arthritis Res Ther (2010) 12(1):R16. doi: 10.1186/ar2917
200. Mahon OR, Kelly DJ, McCarthy GM, Dunne A. Osteoarthritis-Associated Basic Calcium Phosphate Crystals Alter Immune Cell Metabolism and Promote M1 Macrophage Polarization. Osteoarthritis Cartilage (2020) 28(5):603–12. doi: 10.1016/j.joca.2019.10.010
201. Schneider A, Weier M, Herderschee J, Perreau M, Calandra T, Roger T, et al. IRF5 Is a Key Regulator of Macrophage Response to Lipopolysaccharide in Newborns. Front Immunol (2018) 9:1597. doi: 10.3389/fimmu.2018.01597
202. Krausgruber T, Blazek K, Smallie T, Alzabin S, Lockstone H, Sahgal N, et al. IRF5 Promotes Inflammatory Macrophage Polarization and TH1-TH17 Responses. Nat Immunol (2011) 12(3):231–8. doi: 10.1038/ni.1990
203. Ni Z, Zhao X, Dai X, Zhao L, Xia J. The Role of Interferon Regulatory Factor 5 in Macrophage Inflammation During Osteoarthritis. Inflammation (2019) 42(5):1821–9. doi: 10.1007/s10753-019-01044-8
204. Alonso-Perez A, Franco-Trepat E, Guillan-Fresco M, Jorge-Mora A, Lopez V, Pino J, et al. Role of Toll-Like Receptor 4 on Osteoblast Metabolism and Function. Front Physiol (2018) 9:504. doi: 10.3389/fphys.2018.00504
205. Kalaitzoglou E, Lopes EBP, Fu Y, Herron JC, Flaming JM, Donovan EL, et al. TLR4 Promotes and DAP12 Limits Obesity-Induced Osteoarthritis in Aged Female Mice. JBMR Plus (2019) 3(4):e10079. doi: 10.1002/jbm4.10079
206. Franco-Trepat E, Guillan-Fresco M, Alonso-Perez A, Jorge-Mora A, Francisco V, Gualillo O, et al. Visfatin Connection: Present and Future in Osteoarthritis and Osteoporosis. J Clin Med (2019) 8(8). doi: 10.3390/jcm8081178
207. Moschen AR, Kaser A, Enrich B, Mosheimer B, Theurl M, Niederegger H, et al. Visfatin, an Adipocytokine With Proinflammatory and Immunomodulating Properties. J Immunol (2007) 178(3):1748–58. doi: 10.4049/jimmunol.178.3.1748
208. Dahl TB, Yndestad A, Skjelland M, Oie E, Dahl A, Michelsen A, et al. Increased Expression of Visfatin in Macrophages of Human Unstable Carotid and Coronary Atherosclerosis: Possible Role in Inflammation and Plaque Destabilization. Circulation (2007) 115(8):972–80. doi: 10.1161/CIRCULATIONAHA.106.665893
209. Hong EH, Yun HS, Kim J, Um HD, Lee KH, Kang CM, et al. Nicotinamide Phosphoribosyltransferase is Essential for Interleukin-1beta-Mediated Dedifferentiation of Articular Chondrocytes via SIRT1 and Extracellular Signal-Regulated Kinase (ERK) Complex Signaling. J Biol Chem (2011) 286(32):28619–31. doi: 10.1074/jbc.M111.219832
210. Laiguillon MC, Houard X, Bougault C, Gosset M, Nourissat G, Sautet A, et al. Expression and Function of Visfatin (Nampt), an Adipokine-Enzyme Involved in Inflammatory Pathways of Osteoarthritis. Arthritis Res Ther (2014) 16(1):R38. doi: 10.1186/ar4467
211. Mahjoub M, Berenbaum F, Houard X. Why Subchondral Bone in Osteoarthritis? The Importance of the Cartilage Bone Interface in Osteoarthritis. Osteoporos Int (2012) 23 Suppl 8:S841–6. doi: 10.1007/s00198-012-2161-0
212. Brentano F, Schorr O, Ospelt C, Stanczyk J, Gay RE, Gay S, et al. Pre-B Cell Colony-Enhancing Factor/Visfatin, a New Marker of Inflammation in Rheumatoid Arthritis With Proinflammatory and Matrix-Degrading Activities. Arthritis Rheum (2007) 56(9):2829–39. doi: 10.1002/art.22833
213. Meier FM, Frommer KW, Peters MA, Brentano F, Lefevre S, Schroder D, et al. Visfatin/pre-B-Cell Colony-Enhancing Factor (PBEF), a Proinflammatory and Cell Motility-Changing Factor in Rheumatoid Arthritis. J Biol Chem (2012) 287(34):28378–85. doi: 10.1074/jbc.M111.312884
214. Evans L, Williams AS, Hayes AJ, Jones SA, Nowell M. Suppression of Leukocyte Infiltration and Cartilage Degradation by Selective Inhibition of Pre-B Cell Colony-Enhancing Factor/Visfatin/Nicotinamide Phosphoribosyltransferase: Apo866-Mediated Therapy in Human Fibroblasts and Murine Collagen-Induced Arthritis. Arthritis Rheum (2011) 63(7):1866–77. doi: 10.1002/art.30338
215. Paoletti A, Rohmer J, Ly B, Pascaud J, Riviere E, Seror R, et al. Monocyte/Macrophage Abnormalities Specific to Rheumatoid Arthritis Are Linked to miR-155 and Are Differentially Modulated by Different TNF Inhibitors. J Immunol (2019) 203(7):1766–75. doi: 10.4049/jimmunol.1900386
216. Ong SM, Hadadi E, Dang TM, Yeap WH, Tan CT, Ng TP, et al. The Pro-Inflammatory Phenotype of the Human non-Classical Monocyte Subset is Attributed to Senescence. Cell Death Dis (2018) 9(3):266. doi: 10.1038/s41419-018-0327-1
217. Loeser RF, Olex AL, McNulty MA, Carlson CS, Callahan MF, Ferguson CM, et al. Microarray Analysis Reveals Age-Related Differences in Gene Expression During the Development of Osteoarthritis in Mice. Arthritis Rheum (2012) 64(3):705–17. doi: 10.1002/art.33388
218. Rowe MA, Harper LR, McNulty MA, Lau AG, Carlson CS, Leng L, et al. Reduced Osteoarthritis Severity in Aged Mice With Deletion of Macrophage Migration Inhibitory Factor. Arthritis Rheumatol (2017) 69(2):352–61. doi: 10.1002/art.39844
219. Smith MD. The Normal Synovium. Open Rheumatol J (2011) 5:100–6. doi: 10.2174/1874312901105010100
220. Klein-Wieringa IR, de Lange-Brokaar BJ, Yusuf E, Andersen SN, Kwekkeboom JC, Kroon HM, et al. Inflammatory Cells in Patients With Endstage Knee Osteoarthritis: A Comparison Between the Synovium and the Infrapatellar Fat Pad. J Rheumatol (2016) 43(4):771–8. doi: 10.3899/jrheum.151068
221. Garriga C, Goff M, Paterson E, Hrusecka R, Hamid B, Alderson J, et al. Clinical and Molecular Associations With Outcomes at 2 Years After Acute Knee Injury: A Longitudinal Study in the Knee Injury Cohort at the Kennedy (KICK). Lancet Rheumatol (2021) 3:648–58. doi: 10.1016/S2665-9913(21)00116-8
222. Pradhan P, Vijayan V, Gueler F, Immenschuh S. Interplay of Heme With Macrophages in Homeostasis and Inflammation. Int J Mol Sci (2020) 21(3). doi: 10.3390/ijms21030740
223. Struglics A, Okroj M, Sward P, Frobell R, Saxne T, Lohmander LS, et al. The Complement System is Activated in Synovial Fluid From Subjects With Knee Injury and From Patients With Osteoarthritis. Arthritis Res Ther (2016) 18(1):223. doi: 10.1186/s13075-016-1123-x
224. Wang Q, Rozelle AL, Lepus CM, Scanzello CR, Song JJ, Larsen DM, et al. Identification of a Central Role for Complement in Osteoarthritis. Nat Med (2011) 17(12):1674–9. doi: 10.1038/nm.2543
225. Cambre I, Gaublomme D, Burssens A, Jacques P, Schryvers N, De Muynck A, et al. Mechanical Strain Determines the Site-Specific Localization of Inflammation and Tissue Damage in Arthritis. Nat Commun (2018) 9(1):4613. doi: 10.1038/s41467-018-06933-4
226. Otsuki S, Brinson DC, Creighton L, Kinoshita M, Sah RL, D'Lima D, et al. The Effect of Glycosaminoglycan Loss on Chondrocyte Viability: A Study on Porcine Cartilage Explants. Arthritis Rheum (2008) 58(4):1076–85. doi: 10.1002/art.23381
227. Quinn TM, Grodzinsky AJ, Hunziker EB, Sandy JD. Effects of Injurious Compression on Matrix Turnover Around Individual Cells in Calf Articular Cartilage Explants. J Orthop Res (1998) 16(4):490–9. doi: 10.1002/jor.1100160415
228. D'Lima DD, Hashimoto S, Chen PC, Colwell CW Jr., Lotz MK. Impact of Mechanical Trauma on Matrix and Cells. Clin Orthop Relat Res (2001) 391 Suppl):S90–9. doi: 10.1097/00003086-200110001-00009
229. Maldonado M, Nam J. The Role of Changes in Extracellular Matrix of Cartilage in the Presence of Inflammation on the Pathology of Osteoarthritis. BioMed Res Int (2013) 2013:284873. doi: 10.1155/2013/284873
230. Adair-Kirk TL, Senior RM. Fragments of Extracellular Matrix as Mediators of Inflammation. Int J Biochem Cell Biol (2008) 40(6-7):1101–10. doi: 10.1016/j.biocel.2007.12.005
231. Senior RM, Hinek A, Griffin GL, Pipoly DJ, Crouch EC, Mecham RP. Neutrophils Show Chemotaxis to Type IV Collagen and its 7S Domain and Contain a 67 kD Type IV Collagen Binding Protein With Lectin Properties. Am J Respir Cell Mol Biol (1989) 1(6):479–87. doi: 10.1165/ajrcmb/1.6.479
232. Duca L, Blanchevoye C, Cantarelli B, Ghoneim C, Dedieu S, Delacoux F, et al. The Elastin Receptor Complex Transduces Signals Through the Catalytic Activity of its Neu-1 Subunit. J Biol Chem (2007) 282(17):12484–91. doi: 10.1074/jbc.M609505200
233. Noble PW, McKee CM, Cowman M, Shin HS. Hyaluronan Fragments Activate an NF-Kappa B/I-Kappa B Alpha Autoregulatory Loop in Murine Macrophages. J Exp Med (1996) 183(5):2373–8. doi: 10.1084/jem.183.5.2373
234. Bonnans C, Chou J, Werb Z. Remodelling the Extracellular Matrix in Development and Disease. Nat Rev Mol Cell Biol (2014) 15(12):786–801. doi: 10.1038/nrm3904
235. Patel L, Sun W, Glasson SS, Morris EA, Flannery CR, Chockalingam PS. Tenascin-C Induces Inflammatory Mediators and Matrix Degradation in Osteoarthritic Cartilage. BMC Musculoskelet Disord (2011) 12:164. doi: 10.1186/1471-2474-12-164
236. Schaefer L, Babelova A, Kiss E, Hausser HJ, Baliova M, Krzyzankova M, et al. The Matrix Component Biglycan is Proinflammatory and Signals Through Toll-Like Receptors 4 and 2 in Macrophages. J Clin Invest (2005) 115(8):2223–33. doi: 10.1172/JCI23755
237. Fan L, Wang Q, Liu R, Zong M, He D, Zhang H, et al. Citrullinated Fibronectin Inhibits Apoptosis and Promotes the Secretion of Pro-Inflammatory Cytokines in Fibroblast-Like Synoviocytes in Rheumatoid Arthritis. Arthritis Res Ther (2012) 14(6):R266. doi: 10.1186/ar4112
238. Newton K, Dixit VM. Signaling in Innate Immunity and Inflammation. Cold Spring Harb Perspect Biol (2012) 4(3). doi: 10.1101/cshperspect.a006049
239. Kim HA, Cho ML, Choi HY, Yoon CS, Jhun JY, Oh HJ, et al. The Catabolic Pathway Mediated by Toll-Like Receptors in Human Osteoarthritic Chondrocytes. Arthritis Rheum (2006) 54(7):2152–63. doi: 10.1002/art.21951
240. Benito MJ, Veale DJ, FitzGerald O, van den Berg WB, Bresnihan B. Synovial Tissue Inflammation in Early and Late Osteoarthritis. Ann Rheum Dis (2005) 64(9):1263–7. doi: 10.1136/ard.2004.025270
241. Elgueta R, Benson MJ, de Vries VC, Wasiuk A, Guo Y, Noelle RJ. Molecular Mechanism and Function of CD40/CD40L Engagement in the Immune System. Immunol Rev (2009) 229(1):152–72. doi: 10.1111/j.1600-065X.2009.00782.x
242. Graf D, Muller S, Korthauer U, van Kooten C, Weise C, Kroczek RA. A Soluble Form of TRAP (CD40 Ligand) is Rapidly Released After T Cell Activation. Eur J Immunol (1995) 25(6):1749–54. doi: 10.1002/eji.1830250639
243. Zirlik A, Maier C, Gerdes N, MacFarlane L, Soosairajah J, Bavendiek U, et al. CD40 Ligand Mediates Inflammation Independently of CD40 by Interaction With Mac-1. Circulation (2007) 115(12):1571–80. doi: 10.1161/CIRCULATIONAHA.106.683201
244. Perazzio SF, Soeiro-Pereira PV, Dos Santos VC, de Brito MV, Salu B, Oliva MLV, et al. Soluble CD40L is Associated With Increased Oxidative Burst and Neutrophil Extracellular Trap Release in Behcet's Disease. Arthritis Res Ther (2017) 19(1):235. doi: 10.1186/s13075-017-1443-5
245. Soong RS, Song L, Trieu J, Lee SY, He L, Tsai YC, et al. Direct T Cell Activation via CD40 Ligand Generates High Avidity CD8+ T Cells Capable of Breaking Immunological Tolerance for the Control of Tumors. PloS One (2014) 9(3):e93162. doi: 10.1371/journal.pone.0093162
246. Guo Y, Walsh AM, Fearon U, Smith MD, Wechalekar MD, Yin X, et al. CD40L-Dependent Pathway Is Active at Various Stages of Rheumatoid Arthritis Disease Progression. J Immunol (2017) 198(11):4490–501. doi: 10.4049/jimmunol.1601988
247. MacDonald KP, Nishioka Y, Lipsky PE, Thomas R. Functional CD40 Ligand is Expressed by T Cells in Rheumatoid Arthritis. J Clin Invest (1997) 100(9):2404–14. doi: 10.1172/JCI119781
248. Roman-Fernandez IV, Garcia-Chagollan M, Cerpa-Cruz S, Jave-Suarez LF, Palafox-Sanchez CA, Garcia-Arellano S, et al. Assessment of CD40 and CD40L Expression in Rheumatoid Arthritis Patients, Association With Clinical Features and DAS28. Clin Exp Med (2019) 19(4):427–37. doi: 10.1007/s10238-019-00568-5
249. Liu MF, Chao SC, Wang CR, Lei HY. Expression of CD40 and CD40 Ligand Among Cell Populations Within Rheumatoid Synovial Compartment. Autoimmunity (2001) 34(2):107–13. doi: 10.3109/08916930109001958
250. Lai JH, Luo SF, Ho LJ. Targeting the CD40-CD154 Signaling Pathway for Treatment of Autoimmune Arthritis. Cells (2019) 8(8). doi: 10.3390/cells8080927
251. Gierut A, Perlman H, Pope RM. Innate Immunity and Rheumatoid Arthritis. Rheum Dis Clin North Am (2010) 36(2):271–96. doi: 10.1016/j.rdc.2010.03.004
252. Haubruck PSC, Liu Y, Moradi B, Blaker C, Clarke E, Little CB. Evaluating the Role and Origin of Pro-Inflammatory Macrophage Subsets as Part of the Cellular Immune Response During the Onset and Development of Posttraumatic Osteoarthritis in Mice. In: ORS 2021 Annual Meeting (2020). Virtual.
253. Pottie P, Presle N, Terlain B, Netter P, Mainard D, Berenbaum F. Obesity and Osteoarthritis: More Complex Than Predicted! Ann Rheum Dis (2006) 65(11):1403–5. doi: 10.1136/ard.2006.061994
254. Datta P, Zhang Y, Parousis A, Sharma A, Rossomacha E, Endisha H, et al. High-Fat Diet-Induced Acceleration of Osteoarthritis is Associated With a Distinct and Sustained Plasma Metabolite Signature. Sci Rep (2017) 7(1):8205. doi: 10.1038/s41598-017-07963-6
255. Larrañaga-Vera A, Lamuedra A, Pérez-Baos S, Prieto-Potin I, Peña L, Herrero-Beaumont G, et al. Increased Synovial Lipodystrophy Induced by High Fat Diet Aggravates Synovitis in Experimental Osteoarthritis. Arthritis Res Ther (2017) 19(1):264. doi: 10.1186/s13075-017-1473-z
256. Harasymowicz NS, Choi Y-R, Wu C-L, Iannucci L, Tang R, Guilak F. Intergenerational Transmission of Diet-Induced Obesity, Metabolic Imbalance, and Osteoarthritis in Mice. Arthritis Rheumatol (2020) 72(4):632–44. doi: 10.1002/art.41147
257. Shen J, Abu-Amer Y, O'Keefe RJ, McAlinden A. Inflammation and Epigenetic Regulation in Osteoarthritis. Connective Tissue Res (2017) 58(1):49–63. doi: 10.1080/03008207.2016.1208655
258. Raman S, FitzGerald U, Murphy JM. Interplay of Inflammatory Mediators With Epigenetics and Cartilage Modifications in Osteoarthritis. Front Bioeng Biotechnol (2018) 6:22. doi: 10.3389/fbioe.2018.00022
259. Hoeksema MA, de Winther MP. Epigenetic Regulation of Monocyte and Macrophage Function. Antioxid Redox Signal (2016) 25(14):758–74. doi: 10.1089/ars.2016.6695
260. Kapellos TS, Iqbal AJ. Epigenetic Control of Macrophage Polarisation and Soluble Mediator Gene Expression During Inflammation. Mediators Inflammation (2016) 2016:6591703. doi: 10.1155/2016/6591703
261. Logie C, Stunnenberg HG. Epigenetic Memory: A Macrophage Perspective. Semin Immunol (2016) 28(4):359–67. doi: 10.1016/j.smim.2016.06.003
262. Chen S, Yang J, Wei Y, Wei X. Epigenetic Regulation of Macrophages: From Homeostasis Maintenance to Host Defense. Cell Mol Immunol (2020) 17(1):36–49. doi: 10.1038/s41423-019-0315-0
263. Soucie EL, Weng Z, Geirsdóttir L, Molawi K, Maurizio J, Fenouil R, et al. Lineage-Specific Enhancers Activate Self-Renewal Genes in Macrophages and Embryonic Stem Cells. Science (2016) 351(6274):aad5510. doi: 10.1126/science.aad5510
264. Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple Combinations of Lineage-Determining Transcription Factors Prime Cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol Cell (2010) 38(4):576–89. doi: 10.1016/j.molcel.2010.05.004
265. Kruidenier L, Chung CW, Cheng Z, Liddle J, Che K, Joberty G, et al. A Selective Jumonji H3K27 Demethylase Inhibitor Modulates the Proinflammatory Macrophage Response. Nature (2012) 488(7411):404–8. doi: 10.1038/nature11262
266. Sohn DH, Sokolove J, Sharpe O, Erhart JC, Chandra PE, Lahey LJ, et al. Plasma Proteins Present in Osteoarthritic Synovial Fluid can Stimulate Cytokine Production via Toll-Like Receptor 4. Arthritis Res Ther (2012) 14(1):R7–R. doi: 10.1186/ar3555
267. Rhoads JP, Major AS, Rathmell JC. Fine Tuning of Immunometabolism for the Treatment of Rheumatic Diseases. Nat Rev Rheumatol (2017) 13(5):313–20. doi: 10.1038/nrrheum.2017.54
268. Hong YH, Kong EJ. (18f)Fluoro-Deoxy-D-Glucose Uptake of Knee Joints in the Aspect of Age-Related Osteoarthritis: A Case-Control Study. BMC Musculoskelet Disord (2013) 14:141. doi: 10.1186/1471-2474-14-141
269. Courties A, Gualillo O, Berenbaum F, Sellam J. Metabolic Stress-Induced Joint Inflammation and Osteoarthritis. Osteoarthritis Cartilage (2015) 23(11):1955–65. doi: 10.1016/j.joca.2015.05.016
270. Netea MG, Domínguez-Andrés J, Barreiro LB, Chavakis T, Divangahi M, Fuchs E, et al. Defining Trained Immunity and its Role in Health and Disease. Nat Rev Immunol (2020) 20(6):375–88. doi: 10.1038/s41577-020-0285-6
271. Blaker CL, Zaki S, Little CB, Clarke EC. Long-Term Effect of a Single Subcritical Knee Injury: Increasing the Risk of Anterior Cruciate Ligament Rupture and Osteoarthritis. Am J Sports Med (2021) 49(2):391–403. doi: 10.1177/0363546520977505
272. Wang X, Oo WM, Linklater JM. What is the Role of Imaging in the Clinical Diagnosis of Osteoarthritis and Disease Management? Rheumatol (Oxford) (2018) 57(suppl_4):iv51–60. doi: 10.1093/rheumatology/kex501
273. Glyn-Jones S, Palmer AJ, Agricola R, Price AJ, Vincent TL, Weinans H, et al. Osteoarthritis. Lancet (2015) 386(9991):376–87. doi: 10.1016/S0140-6736(14)60802-3
274. Shi J, Zhao W, Ying H, Du J, Chen J, Chen S, et al. The Relationship of Platelet to Lymphocyte Ratio and Neutrophil to Monocyte Ratio to Radiographic Grades of Knee Osteoarthritis. Z Rheumatol (2018) 77(6):533–7. doi: 10.1007/s00393-017-0348-7
275. Gao K, Zhu W, Liu W, Ma D, Li H, Yu W, et al. Diagnostic Value of the Blood Monocyte-Lymphocyte Ratio in Knee Osteoarthritis. J Int Med Res (2019) 47(9):4413–21. doi: 10.1177/0300060519860686
276. Soyocak A, Kurt H, Ozgen M, Turgut Cosan D, Colak E, Gunes HV. miRNA-146a, miRNA-155 and JNK Expression Levels in Peripheral Blood Mononuclear Cells According to Grade of Knee Osteoarthritis. Gene (2017) 627:207–11. doi: 10.1016/j.gene.2017.06.027
277. Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-Dependent Induction of microRNA miR-146, an Inhibitor Targeted to Signaling Proteins of Innate Immune Responses. Proc Natl Acad Sci U.S.A. (2006) 103(33):12481–6. doi: 10.1073/pnas.0605298103
278. Ceppi M, Pereira PM, Dunand-Sauthier I, Barras E, Reith W, Santos MA, et al. MicroRNA-155 Modulates the Interleukin-1 Signaling Pathway in Activated Human Monocyte-Derived Dendritic Cells. Proc Natl Acad Sci U.S.A. (2009) 106(8):2735–40. doi: 10.1073/pnas.0811073106
279. Shi T, Shen X, Gao G. Gene Expression Profiles of Peripheral Blood Monocytes in Osteoarthritis and Analysis of Differentially Expressed Genes. BioMed Res Int (2019) 2019:4291689. doi: 10.1155/2019/4291689
280. Loukov D, Karampatos S, Maly MR, Bowdish DME. Monocyte Activation is Elevated in Women With Knee-Osteoarthritis and Associated With Inflammation, BMI and Pain. Osteoarthritis Cartilage (2018) 26(2):255–63. doi: 10.1016/j.joca.2017.10.018
281. Kriegova E, Manukyan G, Mikulkova Z, Gabcova G, Kudelka M, Gajdos P, et al. Gender-Related Differences Observed Among Immune Cells in Synovial Fluid in Knee Osteoarthritis. Osteoarthritis Cartilage (2018) 26(9):1247–56. doi: 10.1016/j.joca.2018.04.016
282. Liu B, Zhang M, Zhao J, Zheng M, Yang H. Imbalance of M1/M2 Macrophages is Linked to Severity Level of Knee Osteoarthritis. Exp Ther Med (2018) 16(6):5009–14. doi: 10.3892/etm.2018.6852
283. Allen PI, Conzemius MG, Evans RB, Kiefer K. Correlation Between Synovial Fluid Cytokine Concentrations and Limb Function in Normal Dogs and in Dogs With Lameness From Spontaneous Osteoarthritis. Vet Surg (2019) 48(5):770–9. doi: 10.1111/vsu.13212
284. Monibi F, Roller BL, Stoker A, Garner B, Bal S, Cook JL. Identification of Synovial Fluid Biomarkers for Knee Osteoarthritis and Correlation With Radiographic Assessment. J Knee Surg (2016) 29(3):242–7. doi: 10.1055/s-0035-1549022
285. Cuellar VG, Cuellar JM, Kirsch T, Strauss EJ. Correlation of Synovial Fluid Biomarkers With Cartilage Pathology and Associated Outcomes in Knee Arthroscopy. Arthroscopy (2016) 32(3):475–85. doi: 10.1016/j.arthro.2015.08.033
286. MacFarlane LA, Yang H, Collins JE, Jarraya M, Guermazi A, Mandl LA, et al. Association of Changes in Effusion-Synovitis With Progression of Cartilage Damage Over Eighteen Months in Patients With Osteoarthritis and Meniscal Tear. Arthritis Rheumatol (2019) 71(1):73–81. doi: 10.1002/art.40660
287. Haraden CA, Huebner JL, Hsueh MF, Li YJ, Kraus VB. Synovial Fluid Biomarkers Associated With Osteoarthritis Severity Reflect Macrophage and Neutrophil Related Inflammation. Arthritis Res Ther (2019) 21(1):146. doi: 10.1186/s13075-019-1923-x
288. Hsueh MF, Zhang X, Wellman SS, Bolognesi MP, Kraus VB. Synergistic Roles of Macrophages and Neutrophils in Osteoarthritis Progression. Arthritis Rheumatol (2021) 73(1):89–99. doi: 10.1002/art.41486
289. Jayadev C, Hulley P, Swales C, Snelling S, Collins G, Taylor P, et al. Synovial Fluid Fingerprinting in End-Stage Knee Osteoarthritis: A Novel Biomarker Concept. Bone Joint Res (2020) 9(9):623–32. doi: 10.1302/2046-3758.99.BJR-2019-0192.R1
290. Shih L, Guler N, Syed D, Hopkinson W, McComas KN, Walborn A, et al. Postoperative Changes in the Systemic Inflammatory Milieu in Older Surgical Patients. Clin Appl Thromb Hemost (2018) 24(4):583–8. doi: 10.1177/1076029617747412
291. Ghouri A, Conaghan PG. Update on Novel Pharmacological Therapies for Osteoarthritis. Ther Adv Musculoskelet Dis (2019) 11:1759720X19864492. doi: 10.1177/1759720X19864492
292. Furman BD, Mangiapani DS, Zeitler E, Bailey KN, Horne PH, Huebner JL, et al. Targeting Pro-Inflammatory Cytokines Following Joint Injury: Acute Intra-Articular Inhibition of Interleukin-1 Following Knee Injury Prevents Post-Traumatic Arthritis. Arthritis Res Ther (2014) 16(3):R134. doi: 10.1186/ar4591
293. Kimmerling KA, Furman BD, Mangiapani DS, Moverman MA, Sinclair SM, Huebner JL, et al. Sustained Intra-Articular Delivery of IL-1RA From a Thermally-Responsive Elastin-Like Polypeptide as a Therapy for Post-Traumatic Arthritis. Eur Cell Mater (2015) 29:124–39; discussion 39-40. doi: 10.22203/ecm.v029a10
294. Olson SA, Furman BD, Kraus VB, Huebner JL, Guilak F. Therapeutic Opportunities to Prevent Post-Traumatic Arthritis: Lessons From the Natural History of Arthritis After Articular Fracture. J Orthop Res (2015) 33(9):1266–77. doi: 10.1002/jor.22940
295. Kraus VB, Birmingham J, Stabler TV, Feng S, Taylor DC, Moorman CT, et al. 3rdEffects of Intraarticular IL1-Ra for Acute Anterior Cruciate Ligament Knee Injury: A Randomized Controlled Pilot Trial (NCT00332254). Osteoarthritis Cartilage (2012) 20(4):271–8. doi: 10.1016/j.joca.2011.12.009
296. Schieker M, Conaghan PG, Mindeholm L, Praestgaard J, Solomon DH, Scotti C, et al. Effects of Interleukin-1β Inhibition on Incident Hip and Knee Replacement : Exploratory Analyses From a Randomized, Double-Blind, Placebo-Controlled Trial. Ann Intern Med (2020) 173(7):509–15. doi: 10.7326/m20-0527
297. Mei J, Sun J, Wu J, Zheng X. Liraglutide Suppresses TNF-Alpha-Induced Degradation of Extracellular Matrix in Human Chondrocytes: A Therapeutic Implication in Osteoarthritis. Am J Transl Res (2019) 11(8):4800–8.
298. Utomo L, van Osch GJ, Bayon Y, Verhaar JA, Bastiaansen-Jenniskens YM. Guiding Synovial Inflammation by Macrophage Phenotype Modulation: An In Vitro Study Towards a Therapy for Osteoarthritis. Osteoarthritis Cartilage (2016) 24(9):1629–38. doi: 10.1016/j.joca.2016.04.013
299. Sieker JT, Ayturk UM, Proffen BL, Weissenberger MH, Kiapour AM, Murray MM. Immediate Administration of Intraarticular Triamcinolone Acetonide After Joint Injury Modulates Molecular Outcomes Associated With Early Synovitis. Arthritis Rheumatol (2016) 68(7):1637–47. doi: 10.1002/art.39631
300. Shu CC, Zaki S, Ravi V, Schiavinato A, Smith MM, Little CB. The Relationship Between Synovial Inflammation, Structural Pathology, and Pain in Post-Traumatic Osteoarthritis: Differential Effect of Stem Cell and Hyaluronan Treatment. Arthritis Res Ther (2020) 22(1):29. doi: 10.1186/s13075-020-2117-2
301. Xie JW, Wang Y, Xiao K, Xu H, Luo ZY, Li L, et al. Alpha Defensin-1 Attenuates Surgically Induced Osteoarthritis in Association With Promoting M1 to M2 Macrophage Polarization. Osteoarthritis Cartilage (2021) 4. doi: 10.1016/j.joca.2021.04.006
302. Thomas AC, Hubbard-Turner T, Wikstrom EA, Palmieri-Smith RM. Epidemiology of Posttraumatic Osteoarthritis. J Athl Train (2017) 52(6):491–6. doi: 10.4085/1062-6050-51.5.08
303. Snoeker B, Turkiewicz A, Magnusson K, Frobell R, Yu D, Peat G, et al. Risk of Knee Osteoarthritis After Different Types of Knee Injuries in Young Adults: A Population-Based Cohort Study. Br J Sports Med (2020) 54(12):725–30. doi: 10.1136/bjsports-2019-100959
304. Cook AE, Cook JL, Stoker AM. Metabolic Responses of Meniscus to IL-1beta. J Knee Surg (2018) 31(9):834–40. doi: 10.1055/s-0037-1615821
305. Favero M, Belluzzi E, Trisolino G, Goldring MB, Goldring SR, Cigolotti A, et al. Inflammatory Molecules Produced by Meniscus and Synovium in Early and End-Stage Osteoarthritis: A Coculture Study. J Cell Physiol (2019) 234(7):11176–87. doi: 10.1002/jcp.27766
306. Tsuchida AI, Beekhuizen M, t Hart MC, Radstake TR, Dhert WJ, Saris DB, et al. Cytokine Profiles in the Joint Depend on Pathology, But are Different Between Synovial Fluid, Cartilage Tissue and Cultured Chondrocytes. Arthritis Res Ther (2014) 16(5):441. doi: 10.1186/s13075-014-0441-0
307. Zaki S, Blaker CL, Little CB. OA Foundations - Experimental Models of Osteoarthritis. Osteoarthritis Cartilage (2021) 3. doi: 10.1016/j.joca.2021.03.024
308. Cucchiarini M, de Girolamo L, Filardo G, Oliveira JM, Orth P, Pape D, et al. Basic Science of Osteoarthritis. J Exp Orthopaedics (2016) 3(1):22. doi: 10.1186/s40634-016-0060-6
309. Little CB, Hunter DJ. Post-Traumatic Osteoarthritis: From Mouse Models to Clinical Trials. Nat Rev Rheumatol (2013) 9(8):485–97. doi: 10.1038/nrrheum.2013.72
310. Zaki S, Smith MM, Little CB. Pathology-Pain Relationships in Different Osteoarthritis Animal Model Phenotypes: It Matters What You Measure, When You Measure, and How You Got There. Osteoarthritis Cartilage (2021) 29(10):1448–61. doi: 10.1016/j.joca.2021.03.023
311. Zaki S, Smith MM, Smith SM, Little CB. Differential Patterns of Pathology in and Interaction Between Joint Tissues in Long-Term Osteoarthritis With Different Initiating Causes: Phenotype Matters. Osteoarthritis Cartilage (2020) 28(7):953–65. doi: 10.1016/j.joca.2020.04.009
Keywords: osteoarthritis, monocyte - macrophage, inflammation, niche, native immune functions, synovitis, immunomodulation
Citation: Haubruck P, Pinto MM, Moradi B, Little CB and Gentek R (2021) Monocytes, Macrophages, and Their Potential Niches in Synovial Joints – Therapeutic Targets in Post-Traumatic Osteoarthritis? Front. Immunol. 12:763702. doi: 10.3389/fimmu.2021.763702
Received: 24 August 2021; Accepted: 18 October 2021;
Published: 04 November 2021.
Edited by:
Matthew William Grol, Western University, CanadaReviewed by:
Oreste Gualillo, Servicio Gallego de Salud, SpainKyuho Kang, Chungbuk National University, South Korea
Copyright © 2021 Haubruck, Pinto, Moradi, Little and Gentek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christopher B. Little, Q2hyaXN0b3BoZXIubGl0dGxlQHN5ZG5leS5lZHUuYXU=
†These authors have contributed equally to this work