 Hua Jiang
Hua Jiang Denggang Fu
Denggang Fu Alan Bidgoli
Alan Bidgoli Sophie Paczesny
Sophie Paczesny- Department of Microbiology and Immunology and Pediatrics, Medical University of South Carolina, Charleston, SC, United States
Allogeneic hematopoietic cell transplantation (allo-HCT) is an essential therapeutic modality for patients with hematological malignancies and other blood disorders. Unfortunately, acute graft-versus-host disease (aGVHD) remains a major source of morbidity and mortality following allo-HCT, which limits its use in a broader spectrum of patients. Chronic graft-versus-host disease (cGVHD) also remains the most common long-term complication of allo-HCT, occurring in reportedly 30-70% of patients surviving more than 100 days. Chronic GVHD is also the leading cause of non-relapse mortality (NRM) occurring more than 2 years after HCT for malignant disease. Graft versus tumor (GVT) is a major component of the overall beneficial effects of allogeneic HCT in the treatment of hematological malignancies. Better understanding of GVHD pathogenesis is important to identify new therapeutic targets for GVHD prevention and therapy. Emerging data suggest opposing roles for different T cell subsets, e.g., IFN-γ producing CD4+ and CD8+ T cells (Th1 and Tc1), IL-4 producing T cells (Th2 and Tc2), IL-17 producing T cells (Th17 and Tc17), IL-9 producing T cells (Th9 and Tc9), IL-22 producing T cells (Th22), T follicular helper cells (Tfh), regulatory T-cells (Treg) and tissue resident memory T cells (Trm) in GVHD and GVT etiology. In this review, we first summarize the general description of the cytokine signals that promote the differentiation of T cell subsets and the roles of these T cell subsets in the pathogenesis of GVHD. Next, we extensively explore preclinical findings of T cell subsets in both GVHD/GVT animal models and humans. Finally, we address recent findings about the roles of T-cell subsets in clinical GVHD and current strategies to modulate T-cell differentiation for treating and preventing GVHD in patients. Further exploring and outlining the immune biology of T-cell differentiation in GVHD that will provide more therapeutic options for maintaining success of allo-HCT.
Introduction
Allogeneic hematopoietic cell transplant (allo-HCT) is a remarkably successful immunotherapy in large part due to the graft-versus-tumor (GVT) effect. Unfortunately, GVT is tethered to the pathogenesis of acute graft versus host disease (aGVHD). The detailed pathogenesis of acute GVHD (aGVHD) has recently been reviewed in depth (1, 2). Overall, T cells are indispensable mediators of aGVHD pathogenesis since GVHD rarely develops after syngeneic or T cell-depleted transplants (3–7). Both aGVHD and GVT have been found to be initiated by antigen presenting cells (APCs) derived from the donor and from host activating donor T cells (8, 9). Such activation leads to the release of inflammatory cytokines, with subsequent proliferation of alloreactive T cells, resulting in host damage and further inflammation. Around 15-20% of hematopoietic cell transplant patients develop severe refractory GVHD leading to mortality (10, 11). Chronic GVHD (cGVHD) pathogenesis is a complex process involving both B and T cells (12). The process was reviewed recently in detail (1). Essentially, crosstalk between B and T cells leading to the proliferation of germinal centers allowing the production of allo-reactive antibodies appears to be the overlying process of the disease. The mainstays of GVHD prevention include anti-thymocyte globulin, calcineurin inhibitors and post-transplant cyclophosphamide, and first line therapies include corticosteroids in addition to calcineurin inhibitors. However, treatment with these drugs negatively affect desirable GVT (13). In addition, steroid-refractory GVHD (SR-aGVHD) patients have dismal outcomes, thus representing an urgent need for developing new treatment strategies in the field of transplant medicine (14). That said, recent breakthroughs have been made including the positive result of the randomized phase III clinical trial evaluating ruxolitinib versus best available treatment (BAT) in SR-aGVHD (15). Similarly, positive results were seen in SR-cGVHD comparing ruxolitinib versus BAT (16). The central role of T cells in the pathogenesis of GVHD has also led to extensive studies in manipulating T cell populations to reduce GVHD severity. Specific T cell subsets have been found to either exacerbate or alleviate GVHD/GVT, a finding that is currently being exploited in novel treatment options in preclinical and/or clinical studies.
T Cells Inducers of GVHD
T cells differentiation is initiated when naïve T cells are stimulated by antigens in the presence of MHC molecules under a particular milieu of cytokines their corresponding signaling pathways to develop into different T cell subsets that acquire specialized effector cell phenotypes (17). As shown in Figure 1, these T cell subsets are characterized by the production of signature cytokines and expression of specific transcription factors (TFs). The specific cytokines and TFs are activated by signal transducer and activator of transcription (STAT) family members to confer specialized functions to the T cell subsets. These cytokines and TFs that regulate T cells differentiation may have effects on the development of multiple T cell subsets. For example, interleukin 6 (IL-6) is essential for T follicular helper (Tfh) and T helper type 17 (Th17) differentiation through the STAT5 signaling pathway (18). Different T cell subsets have been involved in several inflammatory diseases (19, 20), and may allow the development of novel treatment strategies (21).
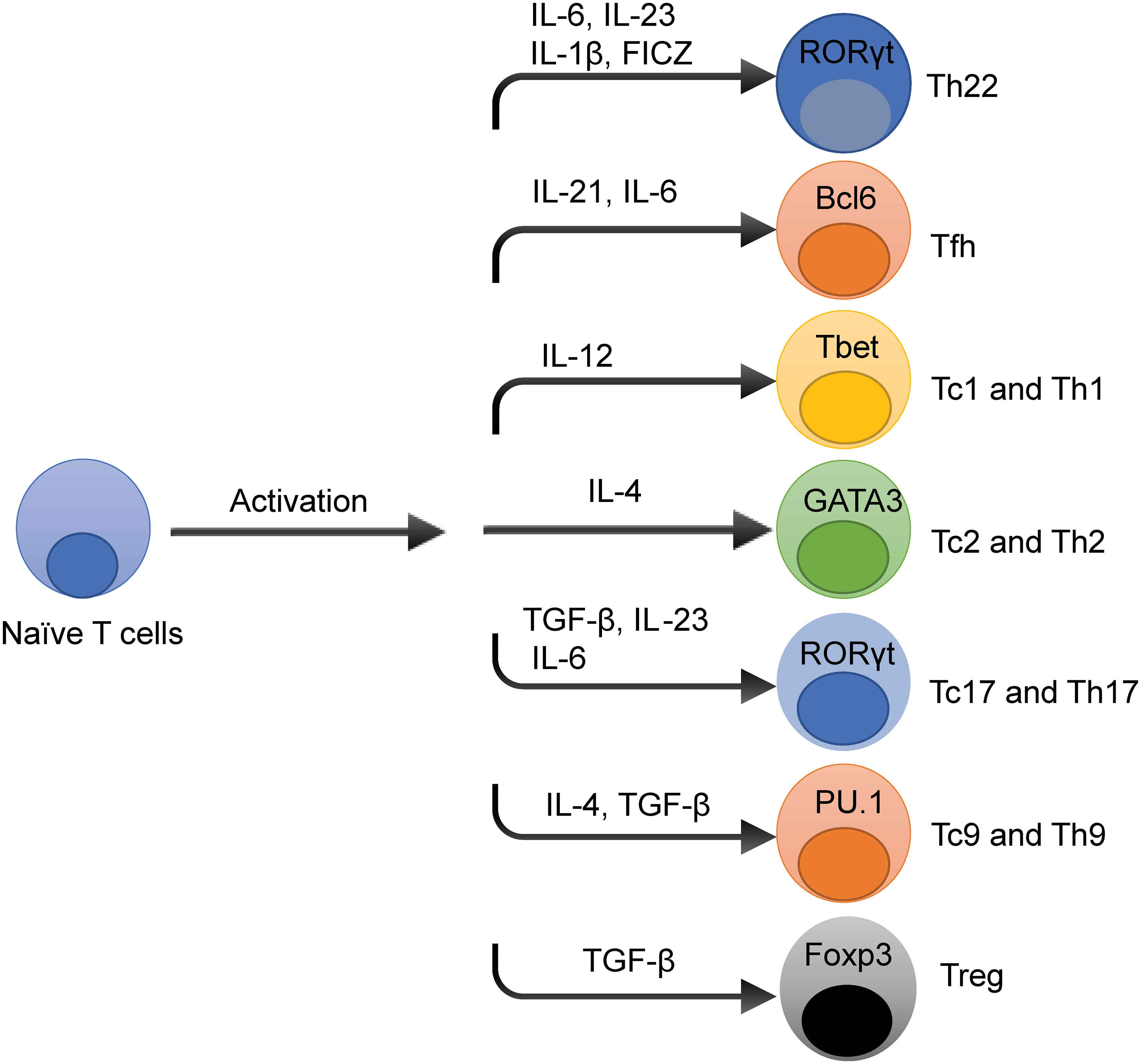
Figure 1 Overview of T Cells Differentiation Pathways. The cytokine and transcription factors (TFs) niche dictates T cell differentiation in spite of the stimulation of T cell receptor signaling pathways. The prototypical cytokines and TFs that regulate each T cell subset differentiation fate are depicted. These cytokines and TFs that influence T cell differentiation have effects on the development of multiple T cell subsets, such as interleukin-6 (IL-6) is essential for T helper type 22 (Th22), T follicular helper (Tfh), and T helper 17 (Th17) cell development.
Figure 2 summarizes T cells subsets demonstrated or putative roles in GVHD/GVT. The gut and other tissues are damaged during irradiation and/or chemotherapy, leading to the release of various DAMPs/PAMPs, and inflammatory cytokines (22). These DAMPs, PAMPs, and cytokines activate both host and donor antigen-presenting cells (APCs), which then activate the donor T cells. The APCs are also secreting various cytokines that promotes T cell differentiation toward different T cell subsets including T helper type 1 (Th1), T helper type 2 (Th2), T helper type 17 (Th17), T helper type 9 (Th9), and regulatory T cells (Tregs). Activated T cells are able to secrete various pro-inflammatory cytokines including IFNγ, IL-17, IL-22 leading to cytolysis of cells in target tissues, mainly in the gut, liver, and skin, which can be alleviated by anti-inflammatory cytokine produced by Th2, Th9 and Treg cells, such as IL-33-producing Th9 (23).
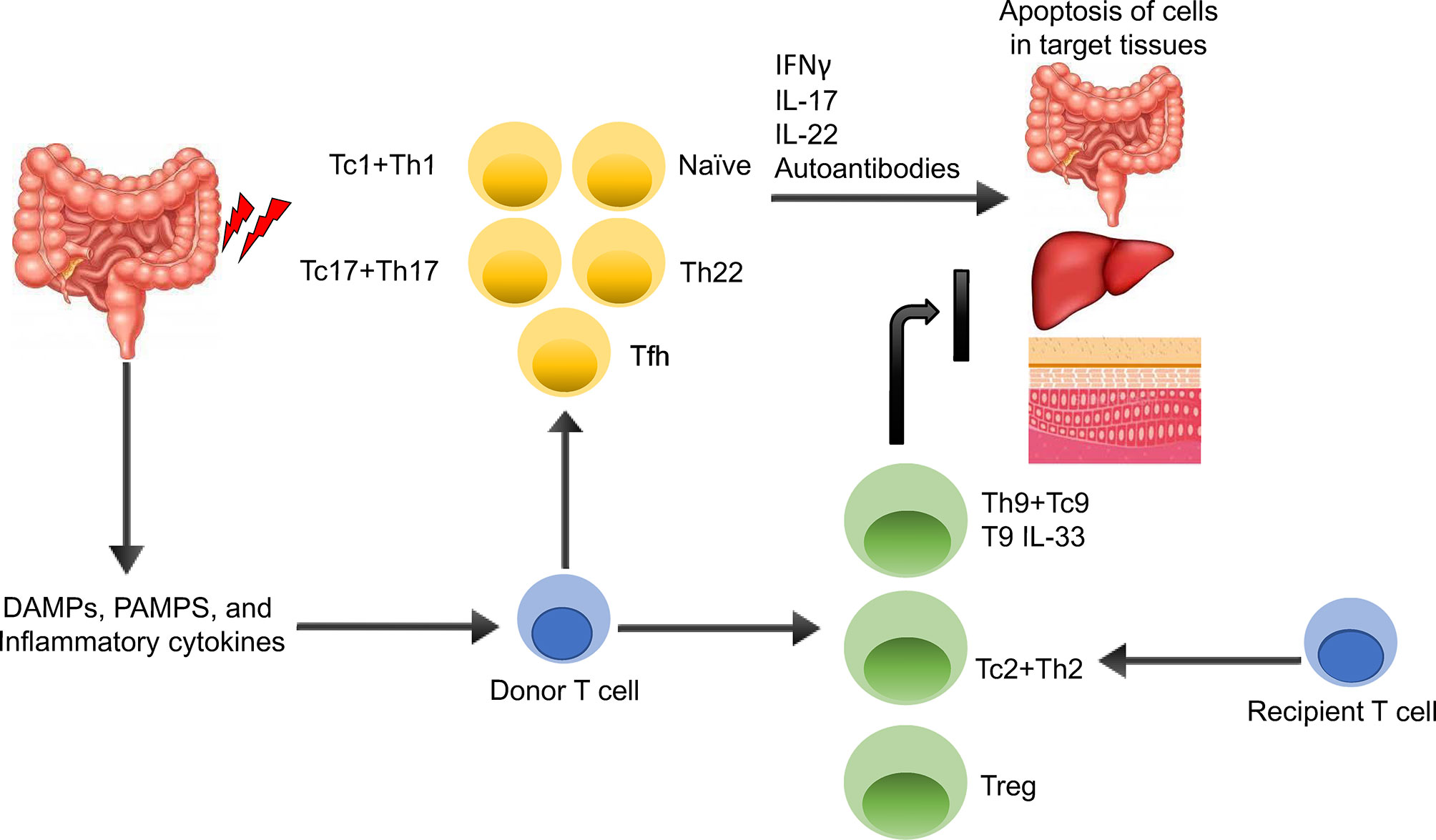
Figure 2 Overview of GVHD Pathogenesis. The gut and other issues are damaged during irradiation or chemotherapy, leading to the release of various DAMPs, PAMPs, and inflammatory cytokines. These DAMPs, PAMPs, and cytokines activate both host and donor antigen-presenting cells (APCs), which then activate the donor T cells. The APCs are also secreting various cytokines that promotes T cell differentiation toward different T cell subsets including T helper type 1 (Th1), T helper type 2 (Th2), T helper type 17 (Th17), T helper type 9 (Th9), and regulatory T cells (Tregs). Activated Th1 and Th17 T cells are able to secrete various pro-inflammatory cytokines including IFNγ, IL-17, IL-22 leading to apoptosis of cells in target tissues, mainly in the gut, liver, and skin, which can be alleviated by anti-inflammatory cytokine producing Th2, Th9 and Treg cells, such as IL-33-producing Th9.
Naïve T Cells
T cell depletion previous to HCT and the use of T cell-depleting antibodies have been used resulting in a significant reduction in GVHD (24). Anti-T-lymphocyte Globulin (ATG) has been used to prevent GVHD in the conditioning regimen but also as treatment while the patient’s response is still unsatisfactory. In the steroid-refractory GVHD therapeutic setting, the response rate is reported to be 24%-41% using ATG and overall survival is poor (25). A recent phase 3 clinical trial aimed to take advantage of the benefits of T-cell depletion with respect to GVHD by using an anti-CD25 antibody (inolimomab) versus ATG, which found no difference in overall survival (26). Unfortunately, the experimental group suffered from issues with infection and relapse, common to T cell-depletion strategies leading to mortality in both arms of the study. The differences between naïve T cells and memory T cells have being investigated to determine which specific subsets of T cells were particularly inductive of pathological immune responses. Naïve T cells (TN) are CD45RA+CD62L+ antigen inexperienced cells with a diverse TCR repertoire (27, 28). Preclinical studies have supported the role of naïve T cells (TN) in inducing GVHD as opposed to central memory T cells (TCM) (29, 30). In allogeneic mouse models of HCT, it was determined that TN caused more severe GVHD compared to that of TCM and effector memory T cells (TEM) cells in isolation (29–35). It was also found in in vitro studies that CD8+ TN were 5-20 times more likely to be specific for a minor histocompatibility antigen than TM (36), supporting the role of this subset in the pathogenesis of GVHD disease. Concurrently, grafts in mice performed with memory cells retained GVT activity when challenged with malignancy (29, 34, 37). A recent phase II clinical trial applied these findings to humans. Naïve T cell depletion was used to reduce GVHD in acute leukemia patients (38). Naïve T cells were depleted from peripheral blood stem cells (PBSCs) using an iron-dextran bead conjugated to a monoclonal anti-CD45RA antibody. Thirty-five patients with acute leukemia or advanced myelodysplastic syndrome received TN-depleted HCT after myeloablative conditioning with 50 days of tacrolimus as immunosuppression. Durable engraftment was achieved in 34 out of 35 patients. Acute GVHD was not reduced in this trial. However, chronic GVHD, a secondary endpoint, was very low (9%) over the 2.5-year median follow up time. This method had other advantages when compared to historical controls, such as faster immune reconstitution compared to T-Cell Depleted (TCD) HCT, a 2-year disease free survival of 70% compared to 50% in TCD and 65% in T-cell-replete grafts, and a 28% relapse rate compared with 60% in TCD and 37% in T-cell replete grafts. These findings require confirmation in future randomized clinical trials.
Th1 and Tc1
The early phase of aGVHD pathogenesis is predominantly mediated by Th1/Tc1 cells, with hyperproliferation and high cytotoxicity driving disease. These cells arise in response to the transplant-conditioning-induced cytokine storm and resultant release of DAMPs and PAMPs, and they can often be targeted by standard immunosuppression regimens, which focus on the inhibition of proliferation and NFAT-driven T cell signaling (39). CD4+ (Th) and CD8+ (Tc) T cells are stimulated to differentiate into the Th1/Tc1 together type 1 subtype when they receive T Cell Receptor (TCR) stimulation from their specific antigen, here allo-antigen, as well as co-stimulation from a variety of different pathways. IL-12, in its activation of STAT4 has been found to be a critical component of the Th1 pathway (40, 41). In the same way, CD8+ T cells are encouraged to differentiate into Tc1 cells by TCR activation, co-stimulation, and the cytokines IL-2 and IL-12 (42–44). Interferon gamma (IFNγ), a primary mediator of inflammation and tissue damage, is a primary product of activated Th1 cells (45). The defining Th1 transcription factor is T-box expressed in T cells (T-bet or Tbx21) (46). Similar to Th1’s, Tc1 cells also depend on T-bet as a transcription factor and are also induced by concurrent expression of eomesodermin (Eomes) (47).
Allogeneic donor Th1 and Tc1 cells have been shown in multiple experiments to induce both GVT and GVHD in mouse models and is classically thought to be the main propagator of GVHD. The IFNγ secreted by donor Th1 cells have been found to both encourage further Th1 cell differentiation and direct damage to the gut mucosa (48). Preclinical models have shown elevation of Th1-derived cytokines including tumor necrosis factor (TNF) and IFNγ in association with GVHD. The cytokines have also been implicated directly in target organ damage (48–51). T-bet is a crucial regulator of Th1 differentiation and IFNγ production, and T-bet-/- T cells alleviate GVHD after adoptive transfer in both major and minor MHC mismatched mouse model (52). Blockade of Th1 and Th17 differentiation by targeting T-bet and RORgammat in mice ameliorates GVHD while surprisingly not decreasing GVT activity (53). As a transcription factor, a promising strategy is to target its downstream effectors for preventing GVHD instead of directly inhibiting T-bet. In human patients suffering from acute GVHD, Th1 cytokines are found in pathologic lesions, supporting the clinical relevance of this subset in the pathogenesis of GVHD (54, 55).
The Janus Kinases (JAKs) family members (JAK-1, -2, -3, Tyrosine kinase 2) and its downstream regulators signal transducers and activators of transcription (STAT) are crucial in the pathogenesis of GVHD (56). Different JAK inhibitors, such as JAK1/2-inhibitor ruxolitinib and JAK1-inhibitor itacitinib, have been developed and applied to prevent or treat aGVHD and cGVHD with different clinical indications. JAK1/2 antagonists can suppress Th1 and Th17 cell function, activation of antigen presenting cells (APCs), MHC expression and co-stimulatory signals through inhibition of STAT1 and STAT3 signaling pathways (57). The regulatory T cell function is retained by reserving IL-2-JAK3-STAT5 signaling pathway followed JAK1/2 inhibition. In 2019, ruxolitinib (Jakafi), a JAK1/2 inhibitor, was approved by the U.S. Food and Drug Administration to treat steroid-refractory aGVHD in adults and children age 12 years and older based on the randomized phase III trial (15). It was also recently approved for steroid-refractory cGVHD in the same population based on the randomized phase III trial (16). In recently published preclinical work, the JAK1/2-inhibitor baricitinib has shown to prevent GVHD by increasing Tregs via the JAK3 pathway (58).
Th17 and Tc17
Th17 cells are the other major subtype of inflammatory T cell implicated in the pathogenesis of GVHD. Cytokines TFG-β1 and IL-6 ± IL-23 direct the differentiation of Th17 cells (59–63). The cells are defined by their expression of IL-17 and lack of expression of IL-4 and IFNγ (64, 65). Retinoic acid receptor-related orphan receptor gamma (RORγt) is the main transcription factor of the Th17 lineage (66). The role Th17 in the pathogenesis of aGVHD is complicated, but overall aGVHD appears to be primarily a Th1 but not Th17 process (67) while cGVHD is both a Th1 and Th17 process (68). Another Th17-like cytokine, IL-21 has also been shown in many preclinical models to induce aGVHD, either via knockout of IL-21 or inhibition of the IL-21 receptor (68–72). The role of Th17 cells have also been investigated in mouse models by way of their transcription factors. Multiple studies have found that the absence of both RORγt and Tbet greatly diminished the severity of aGVHD. In addition, absence of critical Th17 transcription factors led to a significant decrease in the frequency of IL-17A and TNF in subjects’ serum and pathogenic lesions (73). A third method to investigate the role of the Th17 subset in the pathogenesis of GVHD has been to target the cytokines that produced Th17. While likely not specific to Th17, IL-6 inhibition in mouse models was shown to significantly decrease aGVHD severity (74, 75). In addition, blockade of IL-23 was found to diminish aGVHD severity (76–78). Finally, allogeneic donor Th17 cells have been shown to be capable of inducing lethal GVHD in isolation, but they have also been shown to be unnecessary in doing so, as the Th1/Tc1 subset is also sufficient to do so in isolation (79, 80).
In patients suffering from aGVHD, the frequency of Th17 cells in peripheral blood was increased along with the frequency of IL-17 (81). As time progresses after transplantation, Th17/Tc17 cells may become a major driving force of GVHD, secreting proinflammatory cytokines, providing a cellular reservoir for effector alloimmune cells, and supporting the Tfh-driven immune response that characterizes cGVHD (39). Indeed, Th17 cells have been even more heavily implicated in cGVHD in humans. They have been found to be present in increased frequency in the blood of cGVHD patients (81) and mixed Th1/Th17 cells were found in histological examination of cGVHD skin lesions (82). In addition, CD146 and CCR5+CD146+ CD4 T cells are present in increased frequencies in humans suffering from aGVHD and cGVHD, and these cells have been shown to be skewed toward a mixed Th1/Th17 phenotype (83, 84). In a murine model experiment, the potential application of RORγt inhibition has been studied with TMP778. Treatment resulted in a significant decrease in the observed pathology, like a group treated with an anti-IL-17 antibody (84). Furthermore, KD025, was explored in a murine model of cGVHD, which demonstrated a significant reduction in the symptoms of disease. The same study also demonstrated that KD025 inhibition decreased the production of IL-21, IL-17, and IFNγ in the PBMCs of patients suffering from GVHD (85). To follow up on these findings, a phase II clinical trial investigating ROCK2 inhibition with belumosudil (KD025) in the treatment of SR-cGVHD (NCT02841995) showed overall response rates (ORR) (95% CI) with belumosudil 200 mg once daily, 200 mg twice daily, and 400 mg once daily of 65% (38% to 86%), 69% (41% to 89%), and 62% (38% to 82%), respectively. Responses were clinically meaningful, with a median duration of response of 35 weeks, and were associated with quality-of-life improvements and corticosteroid (CS) dose reductions (86). Furthermore, the ROCKstar study showed that belumosudil showed responses for cGVHD after 2 or more prior lines of therapy (87). Based on these findings, belumosudil was recently FDA approved for patients 12 years and older who have received 2 or more prior lines of therapy (88).
Th22
Recently defined as a separate lineage from Th17 cells, Th22 cells were first described in the context of epidermis-infiltrating cells in individuals with inflammatory skin conditions that produced IL-22 and TNFα without producing IFNγ, IL-4, or IL-17 (89). Th22 cells have been shown to develop under the influence of IL-6, IL-23, IL-1β, and 6-formylindolo[3,2-B] carbazole (FICZ) in vitro, along with the tyrosine kinase inhibitor galunsertib. However, ideal conditions for differentiation of Th22 in vitro and in vivo have yet to be determined (90). RORγt has been established as the critical transcription factor for Th22 differentiation, while Tbet is an inhibitory transcription factor for this lineage (90). In contrast to the relatively well-established roles of Th1 and Th17 cells in the pathogenesis of GVHD, the role of Th22 cells and their trademark cytokine, IL-22, remains controversial. In murine models of aGVHD, approximately half the cytokine IL-22 was derived from Th22 cells (91). However, IL-22 has been associated with a protective effect on intestinal stem cells in an experiment that showed recipient deficiency in IL-22 led to more severe immune-mediated damage in the intestine (92). Simultaneously, it was demonstrated in a murine allo-HCT model that deficiency of IL-22 in donor T cells led to diminished aGVHD severity without inhibiting GVT (93). In line with this latter finding, exogenous injection of IL-22 into a murine model after allo-transplant was associated with increased aGVHD severity secondary to Th1 and Tc1 cell expansion, while diminishing Treg levels (94). However, the tissue protective functions of IL-22 can be decoupled from pro-inflammatory actions through structure-based design (95). Based on these findings, a study of IL-22 IgG2-Fc (F-652) along with corticosteroids for subjects with grade II-IV lower gastro-intestinal (GI) aGVHD has been conducted (NCT02406651). Preliminary results of the multicenter prospective phase 2 study showed the combination with corticosteroids was well tolerated and met primary efficacy endpoint (96). Based on these preliminary results, Genentech has sponsored an ongoing clinical trial investigating the use of IL-22Fc in addition to standard therapy for prophylaxis of aGVHD in patients undergoing allogeneic HCT (NCT04539470). Altogether, the action of IL-22 appears to depend on its source and location with donor IL-22 leading to increased aGVHD.
Tfh
Tfh cell differentiation is a multi-step process that is initiated by dendritic cell priming of a naïve CD4+ cell (97). IL-6 is key to this priming process, and its signaling will increase the key transcription factor B-cell lymphoma 6 (Bcl6) in the maturing cell (98–101). Tfh secrete IL-21 as its lead cytokine (102). IL-2 acts as an inhibitor of the Tfh pathway (103, 104). While Tfh cells have not yet been investigated for their roles in aGVHD, donor Tfh cells have been shown to induce cGVHD via their secretion of IL-21. This cytokine leads to the proliferation of germinal centers, differentiation of plasma cells, and the production of auto-antibodies characteristic of cGVHD (105–107). Patients with active cGVHD had a significantly lower frequency of circulating Tfh compared with patients without cGVHD which was associated with higher CXCL13 plasma levels suggesting increased homing of Tfh to secondary lymphoid organs. Further, cTfh were skewed toward a Th2/Th17 phenotype in turn promoting B-cell immunoglobulin secretion and maturation (106).
Trm
It was previously thought that T cells were exclusively found in the blood and secondary lymphoid organs at steady state. Recent observations suggest that the majority of memory T cells reside in human peripheral tissues, primarily located in the skin, gut, liver and lung. Increasing studies unraveled that tissue resident memory T (Trm) cells, representing a lineage of memory T cells, are thought to be contributors in the pathogenesis of GVHD. The Trm cells can be identified by specific markers like CD69 (108). Contribution from host T cells has been recognized recently. Pretransplant conditioning which typically consists of chemoimmunotherapeutic drugs and/or total body irradiation were thought to eliminate host T cells and therefore not play a role in GVHD, but new studies indicate that host T cells resident in peripheral tissues are highly resistant to depletion, even after high-intensity conditioning (109). In humans, host-derived Trm cells have been found in patients’ skin lesions before and after allo-HCT and showed distinct transcriptomic program with RUNX3 and galectin-3 as the phenotypic signatures for these cells as compared to blood T cells (110). Similarly, host T cells were found in all skin and colon from patients with aGVHD after allo-HCT. A subset of host-derived Trm cells is highly proliferative and can be directly activated by donor-derived monocytes. These Trm cells promote the development of GVHD through production of proinflammatory cytokines such as IFNγ and IL-17 (109). Skin Trm cells are HCT conditioning resistant and can be maintained during a long period of time with replenishing T cells rapidly acquiring Trm phenotype. The role of Trm cells in other GVHD target organs is also being explored in preclinical models as well as additional functional roles. For example, murine PSGL1loCD4+ T cells from GVHD target tissues enhance B cell differentiation into plasma cells and production of autoantibodies via their PD-1 interaction with PD-L2 on B cells. Similar evidence was found from humanized GVHD target tissues. In addition, human PSGL1loCD4+ T cells were apposed with memory B cells in the liver tissues of humanized mice and cGVHD patients (111). By creating three spatiotemporal T cell compartments in non-human primates, development of pathogenic Trm into donor CD8+ T cells after allo-HCT was observed. Results showed that by day 8 after transplant, donor T cells infiltrated into the GI tract and exhibit Trm hallmarks. The T cells displayed highly activated and cytotoxic phenotype driven by IL-15 and IL-21 signaling (112).
Regulatory T Cells in GVHD
Th2
Th2 cells mainly produce IL-4, IL-5, IL-10, and IL-13 (113). GATA-binding protein 3 (GATA3) was found to be the master transcription factor for Th2 cells (114). Tc2 cells overlap with Th2 cells in many ways, including their cytokine profile and transcription factor. However, they express both less IL-4 and GATA3 than Th2 cells (115, 116). While we placed Th2 cells under the “regulatory” section due to their protective role when adoptively transferred (117, 118), their overall role is still controversial as they have also been shown to be involved in the pathogenesis of GVHD of the skin and lungs at later stages (48, 119). IL-10-producing Th2 subset has been associated with decreased GVHD in animal models. Also, the natural protective effect of Th2 cells on the gut may prove beneficial for preventing severe gut GVHD (120), the most lethal location of the disease. Additionally, Th2 and Tc2 cells have been described to mediate significantly less severe GVHD compared to Th1 and Tc1 cells after adoptive cell transfer (117, 118). However, these cells concurrently have little to no ability to kill malignant leukemia cells in vivo (117, 118). No conclusive results can be drawn for an association between Th2/IL-4 and cGVHD.
Rapamycin resistant T cells (Trapa) hold promise in preventing GVHD in adoptive cell transfer. Trapa cells have the advantage of being more robust in vivo due to their increased frequency of the T central memory phenotype (Tcm). Rapamycin resistant T cells also have the advantage of proliferating to a greater degree compared to rapamycin sensitive cells once removed from rapamycin (121). These qualities have been exploited in both preclinical and clinical studies. Ex vivo murine Trapa cells polarized with IL-4 toward a Th2 phenotype differentiated into the Th2-type cell and was more effective at preventing GVHD and graft rejection than control Th2 cells (122). Rapamycin-resistance in T cells has also been shown to support Treg cell populations in vivo in the setting of transplant, denoting another potential avenue of rapamycin and rapamycin resistance to combat GVHD (123). A phase II clinical trial investigated Th2-skewed Trapa cells used as donor leukocyte infusion (DLI) after allo-HCT. Trapa cells showed a mix of Th2 and Th1 phenotype and cumulative incidence probability of aGVHD was 20% and 40% at days 100 and 180 post-HCT, respectively. Safety was demonstrated, as none of the patients experienced transplant-related mortality (124). However, there are no phase III Trapa DLI clinical trials in process.
Th9
Th9 cells were shown to be a subset of CD4 cells unique from Th2 cells due to their significant IL-9 production and minimal IL-4 production (125). Characterization of this subset continued as transforming growth factor-beta was found to induce IL-9 expression in Th2 cells (126). A concurrent study similarly found that IL-4 along with TGF-beta led to an IL-9+ IL-10+ Foxp3- phenotype (127). Eventually, PU.1 was deciphered to be a defining transcription factor of this unique subset (128). Recently, it was described that CD8+ cells could also differentiate into this IL-9-producing subset, representing Tc9 cells (129). Unlike the relatively straightforward role of Tregs, the function of Th9 and Tc9 cells in the context of immunomodulation is complicated, as they have been implicated in both pro-inflammatory and anti-inflammatory actions. It was suggested that Th9 cells may prevent GVHD in an experiment that showed in a murine allogeneic model, mice treated with a co-transfer of rapamycin resistant Th9 cells showed decreased donor CD8+ cell engraftment and decreased donor IFN-γ production (130). In addition, two studies suggested the importance of IL-9 specifically to immune-mediated limitation of tumor growth (131, 132).
Aside from the decreased IFN-γ with Th9 cell transfer, it was postulated that this subset may decrease GVHD through their expression of membrane-bound Stimulation-2 (ST2), the IL-33 receptor (23, 133). The IL-33/ST2 pathway has been shown to induce type 2 cytokine production, which is implicated in both supporting tissue repair and maladaptive allergic responses (134). Elevated levels of soluble ST2 (sST2), the decoy receptor, was found to be a risk factor for severe GVHD (135). Using an anti-sST2 antibody GVHD severity could be reduced. This treatment also simultaneously maintained membrane-bound ST2 expression on T cells, increasing the ratio of ST2 to sST2, as well as maintaining GVT. This combination of findings pointed to the inverse relationship between the two related receptors (136).
It has previously been described that Th2 cells express ST2 (137, 138) and that its ST2 expression is increased upon exposure to TGF-beta and IL-33. Further, it was found that IL-33 and TGF-beta treatment increased the expression of IL-9 by Th2 cells (139). With these existing data, it was hypothesized that cells polarized under Th9/Tc9 conditions with the addition of IL-33 would exhibit even greater anti-GVHD effect with maintenance of GVT (23). Indeed, this was found to be the case. Furthermore, supporting evidence was found for the mechanism of the T9IL-33 subset’s mechanism of GVHD prevention in that this subset expressed significantly more amphiregulin (AREG) on its surface than other subsets. Further research on the new T9 cell subset found that cholesterol blockade in Th9 cells with beta-cyclodextrin led to significantly increased IL-9 production as well as increased tumor killing in both a melanoma model and a metastatic lung tumor model (140). The combination of IL-33 with an anti-cholesterol agent to further enhance the desirable phenotypic characteristics of this subset is an exciting potential avenue of research that could be applied in the near future to combat GVHD while maintaining GVT. Of note, like aGVHD, sST2 is elevated in patients with cGVHD (141). However, the role of the ST2/IL-33 pathway in preclinical model of cGVHD is still under study.
Amphiregulin (AREG)-Expressing T Cell
AREG, a member of Epidermal Growth Factor (EGF) family, binds to EGF receptor and promote the proliferation of normal and malignant epithelial cells, fibroblasts and keratinocytes. Deficiency of AREG in mice showed slower clearance of helminth parasite, Trichuris muris, which was driven by Th2-biased responses (142). Recent study has revealed that IL-33 via its receptor ST2 enhances the production of AREG from ST2hi memory T helper 2 (Th2) subset, and directly involved in the reprogramming eosinophils to an inflammatory state with a boost production of osteopontin, a key profibrotic immunomodulatory protein which hence contribute to establishing of lung fibrosis (143). As described above, T9IL-33 surface expressed amphiregulin (AREG) contributes to its GVHD prevention. Furthermore, AREG was found to be essential to the anti-GVHD effect of T9IL-33 cells co-cultured with allogeneic colonic epithelial cells, as AREG blockade significantly increased epithelial cell damage. It was also found that AREG did not cause suppression of effector T cell subsets, explaining the ability of T9IL-33 cells to simultaneously inhibit GVHD and maintain GVT (23). In further support of AREG’s lack of suppressive activity, AREG was previously found to be inconsequential in the suppressive activity of Tregs via genetic ablation (144). Another recent work has revealed that ex vivo IL-33-stimulated Tregs (termed as TregIL-33) expressed higher AREG and displayed stronger immunosuppression. Adoptive transfer of TregIL-33 led to a marked improvement of GVHD prevention compared to either naïve control Tregs or IL-23/IL-17-stimulated TregIL-33. Consistently, blocking AREG with neutralizing antibody in vivo abolished the immunosuppression function of TregIL-33, which collectively suggest a critical role for AREG in IL-33/Treg-mediated GVHD control (145).
Classical Regulatory T Cells (Tregs)
In vivo, Tregs have been shown to develop under the influence of IL-2, IL-15, and TGF-β with FOXP3 as the most critical transcription factor (146–149). Tregs have an extensive experimental history with respect to GVHD, as recently reviewed (150). As predicted by their inherent biology of immunomodulation and self-tolerance, Treg populations have been shown to be decreased during GVHD, allowing for alloreactive T cells to exert their effect (151). Tregs that are FOXP3 negative are known as Type 1 regulatory (Tr1) cells (152). Tr1 cells were characterized as being generated due to alloantigen stimulation by a recipient dendritic cell as well as being stimulated by IL-27. The source of IL-27 is mainly donor macrophages in the context of allo-HCT. In conjunction with this finding, it was determined in this work that IL-6 inhibition increases the proliferation of Tr1 by increasing T cell sensitivity to IL-27 (153). Supporting the inhibitory role of Tregs in the pathogenesis of aGVHD, Tr1 deficiency has been found to exacerbate aGVHD in mouse models (153).
Naturally occurring Tregs (nTregs) have been studied for over a decade in mouse models investigating their ability to prevent aGVHD with nTreg transfer (154, 155). Recipient Treg populations have been expanded before allo-transplant in mice using tumor necrosis factor receptor-2 agonists, leading to prolonged survival and decreased aGVHD (156). Chimeric antigen receptor therapy has been applied to Tregs as well. An alloantigen (HLA-A2) specific CAR was created and applied to Tregs, thus creating an alloantigen-specific human Treg phenotype. In murine models, these CAR-T cells demonstrated superior xenogeneic GVHD prevention caused by HLA-A2+ T cells compared to Treg cells expressing an irrelevant CAR (157).
In human studies, Treg cells have become the front-runner in the use of cell transfer to treat GVHD. Naturally occurring Tregs (nTregs) hold significant promise as a therapy, but nTreg use in clinic has been hindered by a limited amount of Tregs in the peripheral blood (1-2%) (158, 159) and contamination of nTregs with CD25+ T-effector or T memory cells (159–161). However, good-manufacturing practice, large-scale ex-vivo expansion of Tregs has been demonstrated (159). And despite these limitations, human clinical trials using Tregs have shown promising results. A phase one and dose escalation study with umbilical cord-derived nTreg cells in the prevention of GVHD was encouraging for this style of therapy in the future (162, 163). Another trial investigated the effect of early infusion of freshly sorted Tregs followed by conventional T cells (Tcons) on immune reconstitution and GVHD after haplo-identical HCT. Results of this trial showed promise for GVHD prevention, immune reconstitution, preserved GVT, and resistance to opportunistic infections (164). It is worth noticing that the role of Tregs in cGVHD pathogenesis is controversial (165). Both donor and recipient derived Tregs are known to use TGFβ as the effector of suppression in several models. In contrast to its protective role in aGVHD, Treg-produced TGFβ may exacerbate cGVHD since TGFβ can result in fibrosis of organs such as the skin and lung (120).
In the ALT-TEN trial, patients underwent haplo-identical T-cell depleted HCT combined with IL-10 pretreated T cells. The IL-10 treated cells contained Tr1 cells and T memory cells. The results demonstrated the feasibility of using Tr1 cells as a treatment for immune-mediated disorders such as aGVHD (166). The inhibitory role of IL-6 on Treg and Tr1 expansion has been explored in a phase I/II clinical trial as a potential therapeutic target for aGVHD. Anti-IL-6 tocilizumab was used in a single dose before allo-matched HCT, which showed low incidence of aGVHD with treatment, and called for further study of this method in GVHD prophylaxis (167). However, a more recent randomized phase 3 trial evaluating the addition of tocilizumab to cyclosporin and methotrexate for aGVHD prophylaxis, did not show statistically significant reduction in grade II-IV aGVHD or long-term survival (168).
T Cells Inducers of GVT
Donor grafts-derived allogeneic immune cells, particularly the T cells, recognize and eradicate leukemic cells via GVT reactivity, which hence could harness the power for high-risk hematological malignancies such as acute myeloid leukemia (AML) and multiple myeloma (MM). However, the normal tissues of the recipient will also be recognized and attacked by these cells also attack host normal tissues by GVHD (169). Separation of GVT reactivity from GVHD reaction is a necessary step for improving allo-HCT outcomes. Previous study indicated that Th9 cells, a unique subset of CD4+ T cell that produce the pleiotropic cytokine IL-9 and boost antitumor immune responses in vivo via CD8+ CTL-mediated antitumor immunity (131). Further study revealed that IL-9–produced CD8+ T (Tc9) cells generated various cytokines and showed less cytolytic activity in vitro but surprisingly elicited enhanced antitumor responses against advanced tumors in OT-I/B16-OVA and Pmel-1/B16 melanoma models (170). As proof of principle of better antitumoral activity, human chimeric antigen receptor (CAR) T cells polarized and expanded under a Th9-culture condition (T9 CAR-T) showed enhanced antitumor activity against established tumors compared to IL2-polarized (T1) cells. T9 CAR-T cells secrete IL9 but little IFN-γ, express central memory phenotype and lower levels of exhaustion markers and display robust proliferative capacity (171). In allo-HCT settings, T9 cells activated with IL-33 during in vitro differentiation boosted their ST2 expression and IL-9 production. Adoptive cell transfer (ACT) of IL-33 activated T9 cells (T9IL-33) decreased GVHD severity and increased GVT activity via two distinct mechanisms: decrease of fatal immunity by amphiregulin expression and increase of antileukemic activity via CD8α expression (23).
Novel GVHD Treatments Based on Fundamental T Cell Biology
Cellular Therapy
Recent reviews have summarized the scope of cellular therapies to treat GVHD (172, 173). Table 1 list potential T-cells based cellular therapies at different stages of development. Cellular therapies are likely to expand their scope in patients with diverse diseases (174), although delivery of such “live” drugs are not easily scalable (175). Fortunately, in parallel strikes have been made in GVHD treatment with classical drugs as summarized below and in Table 2.

Table 1 Summary of Cellular Therapies for GVHD based on T cells subsets.

Table 2 Summary of recent novel small molecule treatments for GVHD.
Small Molecules Inhibitors
ITK Inhibitors
Ibrutinib is an Interleukin-2-inducible T-cell kinase (ITK) and Bruton tyrosine kinase (BTK) inhibitor that hinders the survival of reactive T-cells, and B cells, respectively (176, 177). In mice, transplant of bone marrow deficient in ITK and BTK showed the importance of these molecules in the pathogenesis of cGVHD, as the transplanted mice did not experience cGVHD (25). Concurrently, mice treated with ibrutinib experienced less severe cGVHD (178). In a phase I/II study for patients with SR-cGVHD, ibrutinib was shown to significantly improve symptoms in most patients, as well as decrease the frequency of chemotactic and fibrotic factors in patients’ blood (179). A significant number of adverse events (AEs) including grade ≥ 3 infectious complications were seen; however the safety profile was deemed acceptable as the AEs were similar to those observed in cGVHD patients treated with concomitant steroids (179). These studies led to the first ever drug in cGVHD to obtain the FDA breakthrough denomination.
JAK Inhibitors
Janus kinases (JAKs) are tyrosine kinases that mediate cytokine-signaling in T cells, propagating survival and differentiation signals (180). The activation of a JAK leads to phosphorylation of signal transducers and activators of transcription (STATs) (181). JAK signaling has also been associated with dendritic cell function, thus amplifying this pathway’s potential importance in GVHD (182, 183).
In mouse models, JAK1/2 blockade with ruxolitinib has displayed decreased IFNy Receptor (IFNyR) receptor signaling, leading to reduced severity of GVHD and preserved GVT (184, 185). In addition, JAK1/2 inhibition in mouse models led to increased frequency of Tregs and decreased frequency of inflammatory cytokines in association with the decreased severity of aGVHD (181).
Following up on findings in murine models, a preliminary trial of 6 human patients with SR-GVHD treated with ruxolitinib showed an improvement in symptoms and similar reduction of the frequency of inflammatory cytokines in peripheral blood (181). In addition, a large multicenter retrospective survey of patients who had received ruxolitinib for steroid-refractory GVHD suggested that ruxolitinib had significant efficacy (186). The results of the phase III randomized clinical trials have recently been published with an overall response of 62% in the ruxolitinib group vs. 39% in the control group (P<0.001) in steroid-refractory aGVHD (15), and an overall response of 50% in the ruxolitinib group vs. 26% in the control group (P<0.001) in steroid refractory cGVHD (16), respectively. Ruxolitinib is now the second drug to get the FDA breakthrough denomination for both cGVHD and aGVHD.
A specific blockade of JAK1 was explored in a phase I trial with itacitinib (INCB039110), which showed responses rates of 64.7% and 88.3% for steroid refractory and treatment naïve disease, respectively (187). Similar AEs were seen with this drug as with ruxolininib, including cytopenia and CMV reactivations. However, itacitinib missed the mark in phase III when given in combination with corticosteroids in patients with treatment-naïve aGVHD.
RORγt Inhibitors
TMP778
As mentioned above, one of the RORγt transcription factor small molecule inhibitors, TMP778, has showed promise in a GVHD murine model similar to an anti-IL-17 antibody (84). However, global inhibition of a transcription factor is generally too toxic to implement in clinic ad alternative have been found such as ROCK2 inhibitors upstream of transcription factors.
ROCK2 Inhibitors
Belumosudil (KD025) is a serine-threonine kinase inhibiting ROCK2 that rebalances the immune system in GVHD by downregulating pro-inflammatory Th17 cells and increasing Tregs, also acting on JAK2/JAK3 and STAT3 (85). Further, ROCK2 is an intracellular integrator of profibrotic signals. Excellent responses were seen in the phase II clinical trials (86) (87) and belumosudil was FDA approved for cGVHD patients who are received 2 prior lines of therapy as mentioned above (88).
Anti-Cytokines
Anti- IL6
The addition of a humanized anti-IL-6R mAb (Tocilizumab) to standard GVHD prophylaxis has shown in promise in reducing the incidence of aGVHD in a prospective phase I/II clinical study (167). The phase III double-blinded study of the addition of Tocilizumab vs. Placebo to cyclosporin/methotrexate GVHD Prophylaxis after HLA-Matched allo-HCT failed to meet the primary endpoint (168).
Conjugated Antibodies (Ab)
Anti-CD30 Ab
Higher frequency of CD30+ CD8 T cells, plasma soluble CD30, and CD30+ lymphocytes have been demonstrated in the intestinal lesions of aGVHD patients (188). This led to the proposal of using the anti-CD30 monomethyl auristatin E (MMAE) conjugate for use in GVHD. A phase I trial for patients with SR-aGVHD showed significant toxicity associated with this drug, including neutropenic sepsis leading to death along with other grade III toxicities of headache, hypoxia, ileus, and elevated bilirubin (189).
Anti-Integrins Abs
Blockade of alpha4beta7 in the gut has been used effectively for inflammatory bowel disease as it disallows effector T cells from being trafficked to the area of inflammation. Natalizumab is one such example that has been used for autoimmune diseases such as Crohn’s, however, its lack of specificity gives it the associated risk of progressive multifocal leukoencephalopathy (PML) (190). Two phase II clinical trials are currently investigating the effectiveness of natalizumab for GVHD (NCT02176031 and NCT02133924) (14). Vedolizumab represents an example of a monoclonal antibody that is specific to the gut, as it inhibits alpha4beta7 integrin’s interaction with MAdCAM-1 and therefore carries significantly lower risk for the serious adverse outcome of PML (191–193). Similarly, this integrin has been shown to be important in the pathogenesis of intestinal GVHD (184). A recent case series of 6 patients explored the use of vedolizumab for the treatment of steroid-refractory intestinal GVHD. Patients treated with vedolizumab almost universally achieved remarkable improvement in gastrointestinal GVHD symptoms, in some cases having symptoms reduced from grade IV to grade I or absent (194). However, a phase II clinical trial, NCT02993783, to evaluate the safety and efficacy of this treatment was recently terminated due to lack of efficacy. Indeed, ORR at day 28 was 50% in patients treated at 300 mg (n = 8) and 22% in patients treated at 600 mg (n = 9); 12% and 0% of patients, respectively, achieved complete response (CR). Thus, higher the dose the less efficacious vedolizumab was. Due to its mechanism of action of blocking T cell migration to the intestine, it is likely that there cannot be an effect when GVHD is already full-blown and T cells in the gut. However, it is possible that preventing effector T cells migration to the GI tract may be beneficial at early stages of GVHD or as GVHD prophylaxis.
Conclusion
Acute and chronic GVHD remain severe and common complications of hematopoietic stem cell transplant. Prevention and treatment of these diseases remain a critical frontier in transplant medicine. New understandings of T cell biology have led to novel treatments with a variety of targets and fundamental mechanisms. The plethora of recent human clinical trials as well as the exciting preclinical experiments have suggested the real possibility of a significant breakthrough for HSCT patients in the near future.
Author Contributions
HJ, DF, and SP conceived and wrote the manuscript. All authors contributed to critical revisions of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The National Cancer Institute (R01 CA168814, and U01 CA232491), and the National Heart Lung and Blood Institute R21-HL139934 to SP, and Medical University of South Carolina Smartstate funds for the Sally Abney Rose Endowment to SP.
Conflict of Interest
SP holds a patent on “Biomarkers and assays to detect chronic graft versus host disease” (U.S. Patent #10,571,478 B2).
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to acknowledge our funding sources including: The National Cancer Institute (R01 CA168814, and U01 CA232491), and the National Heart Lung and Blood Institute R21-HL139934 to SP, and Medical University of South Carolina Smartstate funds for the Sally Abney Rose Endowment to SP.
References
1. Magenau J, Runaas L, Reddy P. Advances in Understanding the Pathogenesis of Graft-Versus-Host Disease. Br J Haematol (2016) 173(2):190–205. doi: 10.1111/bjh.13959
2. Zeiser R, Socie G, Blazar BR. Pathogenesis of Acute Graft-Versus-Host Disease: From Intestinal Microbiota Alterations to Donor T Cell Activation. Br J Haematol (2016) 175(2):191–207. doi: 10.1111/bjh.14295
3. Koyama M, Kuns RD, Olver SD, Raffelt NC, Wilson YA, Don AL, et al. Recipient Nonhematopoietic Antigen-Presenting Cells Are Sufficient to Induce Lethal Acute Graft-Versus-Host Disease. Nat Med (2011) 18(1):135–42. doi: 10.1038/nm.2597
4. Matte CC, Liu J, Cormier J, Anderson BE, Athanasiadis I, Jain D, et al. Donor APCs Are Required for Maximal GVHD But Not for GVL. Nat Med (2004) 10(9):987–92. doi: 10.1038/nm1089
5. Reddy P, Maeda Y, Liu C, Krijanovski OI, Korngold R, Ferrara JL. A Crucial Role for Antigen-Presenting Cells and Alloantigen Expression in Graft-Versus-Leukemia Responses. Nat Med (2005) 11(11):1244–9. doi: 10.1038/nm1309
6. Shlomchik WD, Couzens MS, Tang CB, McNiff J, Robert ME, Liu J, et al. Prevention of Graft Versus Host Disease by Inactivation of Host Antigen-Presenting Cells. Science (1999) 285(5426):412–5. doi: 10.1126/science.285.5426.412
7. Toubai T, Tawara I, Sun Y, Liu C, Nieves E, Evers R, et al. Induction of Acute GVHD by Sex-Mismatched H-Y Antigens in the Absence of Functional Radiosensitive Host Hematopoietic-Derived Antigen-Presenting Cells. Blood (2012) 119(16):3844–53. doi: 10.1182/blood-2011-10-384057
8. Chakraverty R, Sykes M. The Role of Antigen-Presenting Cells in Triggering Graft-Versus-Host Disease and Graft-Versus-Leukemia. Blood (2007) 110(1):9–17. doi: 10.1182/blood-2006-12-022038
9. Xia G, Truitt RL, Johnson BD. Graft-Versus-Leukemia and Graft-Versus-Host Reactions After Donor Lymphocyte Infusion Are Initiated by Host-Type Antigen-Presenting Cells and Regulated by Regulatory T Cells in Early and Long-Term Chimeras. Biol Blood Marrow Transplant (2006) 12(4):397–407. doi: 10.1016/j.bbmt.2005.11.519
10. Blazar BR, Murphy WJ, Abedi M. Advances in Graft-Versus-Host Disease Biology and Therapy. Nat Rev Immunol (2012) 12(6):443–58. doi: 10.1038/nri3212
11. Anasetti C, Logan BR, Lee SJ, Waller EK, Weisdorf DJ, Wingard JR, et al. Peripheral-Blood Stem Cells Versus Bone Marrow From Unrelated Donors. N Engl J Med (2012) 367(16):1487–96. doi: 10.1056/NEJMoa1203517
12. MacDonald KP, Hill GR, Blazar BR. Chronic Graft-Versus-Host Disease: Biological Insights From Preclinical and Clinical Studies. Blood (2017) 129(1):13–21. doi: 10.1182/blood-2016-06-686618
13. Koyama M, Hill GR. Alloantigen Presentation and Graft-Versus-Host Disease: Fuel for the Fire. Blood (2016) 127(24):2963–70. doi: 10.1182/blood-2016-02-697250
14. Hill L, Alousi A, Kebriaei P, Mehta R, Rezvani K, Shpall E. New and Emerging Therapies for Acute and Chronic Graft Versus Host Disease. Ther Adv Hematol (2018) 9(1):21–46. doi: 10.1177/2040620717741860
15. Zeiser R, von Bubnoff N, Butler J, Mohty M, Niederwieser D, Or R, et al. Ruxolitinib for Glucocorticoid-Refractory Acute Graft-Versus-Host Disease. N Engl J Med (2020) 382(19):1800–10. doi: 10.1056/NEJMoa1917635
16. Zeiser R, Polverelli N, Ram R, Hashmi SK, Chakraverty R, Middeke JM, et al. Ruxolitinib for Glucocorticoid-Refractory Chronic Graft-Versus-Host Disease. N Engl J Med (2021) 385(3):228–38. doi: 10.1056/NEJMoa2033122
17. Kaplan MH, Hufford MM, Olson MR. The Development and In Vivo Function of T Helper 9 Cells. Nat Rev Immunol (2015) 15(5):295–307. doi: 10.1038/nri3824
18. Dienz O, Rincon M. The Effects of IL-6 on CD4 T Cell Responses. Clin Immunol (2009) 130(1):27–33. doi: 10.1016/j.clim.2008.08.018
19. Imam T, Park S, Kaplan MH, Olson MR. Effector T Helper Cell Subsets in Inflammatory Bowel Diseases. Front Immunol (2018) 9:1212. doi: 10.3389/fimmu.2018.01212
20. Hirahara K, Nakayama T. CD4+ T-Cell Subsets in Inflammatory Diseases: Beyond the Th1/Th2 Paradigm. Int Immunol (2016) 28(4):163–71. doi: 10.1093/intimm/dxw006
21. Busch DH, Frassle SP, Sommermeyer D, Buchholz VR, Riddell SR. Role of Memory T Cell Subsets for Adoptive Immunotherapy. Semin Immunol (2016) 28(1):28–34. doi: 10.1016/j.smim.2016.02.001
22. Ramadan A, Paczesny S. Various Forms of Tissue Damage and Danger Signals Following Hematopoietic Stem-Cell Transplantation. Front Immunol (2015) 6:14. doi: 10.3389/fimmu.2015.00014
23. Ramadan A, Griesenauer B, Adom D, Kapur R, Hanenberg H, Liu C, et al. Specifically Differentiated T Cell Subset Promotes Tumor Immunity Over Fatal Immunity. J Exp Med (2017) 214(12):3577–96. doi: 10.1084/jem.20170041
24. Saad A, Lamb LS. Ex Vivo T-Cell Depletion in Allogeneic Hematopoietic Stem Cell Transplant: Past, Present and Future. Bone Marrow Transplant (2017) 52(9):1241–8. doi: 10.1038/bmt.2017.22
25. Arai S, Margolis J, Zahurak M, Anders V, Vogelsang GB. Poor Outcome in Steroid-Refractory Graft-Versus-Host Disease With Antithymocyte Globulin Treatment. Biol Blood Marrow Transplant (2002) 8(3):155–60. doi: 10.1053/bbmt.2002.v8.pm11939605
26. Socie G, Vigouroux S, Yakoub-Agha I, Bay JO, Furst S, Bilger K, et al. A Phase 3 Randomized Trial Comparing Inolimomab vs Usual Care in Steroid-Resistant Acute GVHD. Blood (2017) 129(5):643–9. doi: 10.1182/blood-2016-09-738625
27. Arstila TP, Casrouge A, Baron V, Even J, Kanellopoulos J, Kourilsky P. A Direct Estimate of the Human Alphabeta T Cell Receptor Diversity. Science (1999) 286(5441):958–61. doi: 10.1126/science.286.5441.958
28. Robins HS, Campregher PV, Srivastava SK, Wacher A, Turtle CJ, Kahsai O, et al. Comprehensive Assessment of T-Cell Receptor Beta-Chain Diversity in Alphabeta T Cells. Blood (2009) 114(19):4099–107. doi: 10.1182/blood-2009-04-217604
29. Anderson BE, McNiff J, Yan J, Doyle H, Mamula M, Shlomchik MJ, et al. Memory CD4+ T Cells do Not Induce Graft-Versus-Host Disease. J Clin Invest (2003) 112(1):101–8. doi: 10.1172/JCI17601
30. Chen BJ, Cui X, Sempowski GD, Liu C, Chao NJ. Transfer of Allogeneic CD62L- Memory T Cells Without Graft-Versus-Host Disease. Blood (2004) 103(4):1534–41. doi: 10.1182/blood-2003-08-2987
31. Zhang Y, Joe G, Zhu J, Carroll R, Levine B, Hexner E, et al. Dendritic Cell-Activated CD44hiCD8+ T Cells Are Defective in Mediating Acute Graft-Versus-Host Disease But Retain Graft-Versus-Leukemia Activity. Blood (2004) 103(10):3970–8. doi: 10.1182/blood-2003-09-3135
32. Dutt S, Tseng D, Ermann J, George TI, Liu YP, Davis CR, et al. Naive and Memory T Cells Induce Different Types of Graft-Versus-Host Disease. J Immunol (2007) 179(10):6547–54. doi: 10.4049/jimmunol.179.10.6547
33. Chen BJ, Deoliveira D, Cui X, Le NT, Son J, Whitesides JF, et al. Inability of Memory T Cells to Induce Graft-Versus-Host Disease is a Result of an Abortive Alloresponse. Blood (2007) 109(7):3115–23. doi: 10.1182/blood-2006-04-016410
34. Zheng H, Matte-Martone C, Li H, Anderson BE, Venketesan S, Sheng Tan H, et al. Effector Memory CD4+ T Cells Mediate Graft-Versus-Leukemia Without Inducing Graft-Versus-Host Disease. Blood (2008) 111(4):2476–84. doi: 10.1182/blood-2007-08-109678
35. Zheng H, Matte-Martone C, Jain D, McNiff J, Shlomchik WD. Central Memory CD8+ T Cells Induce Graft-Versus-Host Disease and Mediate Graft-Versus-Leukemia. J Immunol (2009) 182(10):5938–48. doi: 10.4049/jimmunol.0802212
36. Bleakley M, Otterud BE, Richardt JL, Mollerup AD, Hudecek M, Nishida T, et al. Leukemia-Associated Minor Histocompatibility Antigen Discovery Using T-Cell Clones Isolated by In Vitro Stimulation of Naive CD8+ T Cells. Blood (2010) 115(23):4923–33. doi: 10.1182/blood-2009-12-260539
37. Li N, Matte-Martone C, Zheng H, Cui W, Venkatesan S, Tan HS, et al. Memory T Cells From Minor Histocompatibility Antigen-Vaccinated and Virus-Immune Donors Improve GVL and Immune Reconstitution. Blood (2011) 118(22):5965–76. doi: 10.1182/blood-2011-07-367011
38. Bleakley M, Heimfeld S, Loeb KR, Jones LA, Chaney C, Seropian S, et al. Outcomes of Acute Leukemia Patients Transplanted With Naive T Cell-Depleted Stem Cell Grafts. J Clin Invest (2015) 125(7):2677–89. doi: 10.1172/JCI81229
39. Hill GR, Betts BC, Tkachev V, Kean LS, Blazar BR. Current Concepts and Advances in Graft-Versus-Host Disease Immunology. Annu Rev Immunol (2021) 39:19–49. doi: 10.1146/annurev-immunol-102119-073227
40. Kaplan MH, Sun YL, Hoey T, Grusby MJ. Impaired IL-12 Responses and Enhanced Development of Th2 Cells in Stat4-Deficient Mice. Nature (1996) 382(6587):174–7. doi: 10.1038/382174a0
41. Magram J, Sfarra J, Connaughton S, Faherty D, Warrier R, Carvajal D, et al. IL-12-Deficient Mice are Defective But Not Devoid of Type 1 Cytokine Responses. Ann N Y Acad Sci (1996) 795:60–70. doi: 10.1111/j.1749-6632.1996.tb52655.x
42. Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, et al. Inflammation Directs Memory Precursor and Short-Lived Effector CD8(+) T Cell Fates via the Graded Expression of T-Bet Transcription Factor. Immunity (2007) 27(2):281–95. doi: 10.1016/j.immuni.2007.07.010
43. Kalia V, Sarkar S, Subramaniam S, Haining WN, Smith KA, Ahmed R. Prolonged Interleukin-2Ralpha Expression on Virus-Specific CD8+ T Cells Favors Terminal-Effector Differentiation In Vivo. Immunity (2010) 32(1):91–103. doi: 10.1016/j.immuni.2009.11.010
44. Pipkin ME, Sacks JA, Cruz-Guilloty F, Lichtenheld MG, Bevan MJ, Rao A. Interleukin-2 and Inflammation Induce Distinct Transcriptional Programs That Promote the Differentiation of Effector Cytolytic T Cells. Immunity (2010) 32(1):79–90. doi: 10.1016/j.immuni.2009.11.012
45. Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two Types of Murine Helper T Cell Clone. I. Definition According to Profiles of Lymphokine Activities and Secreted Proteins. J Immunol (1986) 136(7):2348–57.
46. Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A Novel Transcription Factor, T-Bet, Directs Th1 Lineage Commitment. Cell (2000) 100(6):655–69. doi: 10.1016/S0092-8674(00)80702-3
47. Glimcher LH, Townsend MJ, Sullivan BM, Lord GM. Recent Developments in the Transcriptional Regulation of Cytolytic Effector Cells. Nat Rev Immunol (2004) 4(11):900–11. doi: 10.1038/nri1490
48. Burman AC, Banovic T, Kuns RD, Clouston AD, Stanley AC, Morris ES, et al. IFNgamma Differentially Controls the Development of Idiopathic Pneumonia Syndrome and GVHD of the Gastrointestinal Tract. Blood (2007) 110(3):1064–72. doi: 10.1182/blood-2006-12-063982
49. Hill GR, Crawford JM, Cooke KR, Brinson YS, Pan L, Ferrara JL. Total Body Irradiation and Acute Graft-Versus-Host Disease: The Role of Gastrointestinal Damage and Inflammatory Cytokines. Blood (1997) 90(8):3204–13. doi: 10.1182/blood.V90.8.3204
50. Hill GR, Ferrara JL. The Primacy of the Gastrointestinal Tract as a Target Organ of Acute Graft-Versus-Host Disease: Rationale for the Use of Cytokine Shields in Allogeneic Bone Marrow Transplantation. Blood (2000) 95(9):2754–9. doi: 10.1182/blood.V95.9.2754.009k25_2754_2759
51. Robb RJ, Hill GR. The Interferon-Dependent Orchestration of Innate and Adaptive Immunity After Transplantation. Blood (2012) 119(23):5351–8. doi: 10.1182/blood-2012-02-368076
52. Fu J, Wang D, Yu Y, Heinrichs J, Wu Y, Schutt S, et al. T-Bet is Critical for the Development of Acute Graft-Versus-Host Disease Through Controlling T Cell Differentiation and Function. J Immunol (2015) 194(1):388–97. doi: 10.4049/jimmunol.1401618
53. Yu Y, Wang D, Liu C, Kaosaard K, Semple K, Anasetti C, et al. Prevention of GVHD While Sparing GVL Effect by Targeting Th1 and Th17 Transcription Factor T-Bet and RORgammat in Mice. Blood (2011) 118(18):5011–20. doi: 10.1182/blood-2011-03-340315
54. Krenger W, Ferrara JL. Graft-Versus-Host Disease and the Th1/Th2 Paradigm. Immunol Res (1996) 15(1):50–73. doi: 10.1007/BF02918284
55. Morris ES, Hill GR. Advances in the Understanding of Acute Graft-Versus-Host Disease. Br J Haematol (2007) 137(1):3–19. doi: 10.1111/j.1365-2141.2007.06510.x
56. Schroeder MA, Choi J, Staser K, DiPersio JF. The Role of Janus Kinase Signaling in Graft-Versus-Host Disease and Graft Versus Leukemia. Biol Blood Marrow Transplant (2018) 24(6):1125–34. doi: 10.1016/j.bbmt.2017.12.797
57. Mannina D, Kroger N. Janus Kinase Inhibition for Graft-Versus-Host Disease: Current Status and Future Prospects. Drugs (2019) 79(14):1499–509. doi: 10.1007/s40265-019-01174-1
58. Kim S, Ashami K, Lim S, Staser K, Vij K, Santhanam S, et al. Baricitinib Prevents GvHD by Increasing Tregs via JAK3 and Treats Established GvHD by Promoting Intestinal Tissue Repair via EGFR. Leukemia (2021) 38:42 Z. doi: 10.1038/s41375-021-01360-9
59. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the Context of an Inflammatory Cytokine Milieu Supports De Novo Differentiation of IL-17-Producing T Cells. Immunity (2006) 24(2):179–89. doi: 10.1016/j.immuni.2006.01.001
60. Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming Growth Factor-Beta Induces Development of the T(H)17 Lineage. Nature (2006) 441(7090):231–4. doi: 10.1038/nature04754
61. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal Developmental Pathways for the Generation of Pathogenic Effector TH17 and Regulatory T Cells. Nature (2006) 441(7090):235–8. doi: 10.1038/nature04753
62. Yang L, Anderson DE, Baecher-Allan C, Hastings WD, Bettelli E, Oukka M, et al. IL-21 and TGF-Beta are Required for Differentiation of Human T(H)17 Cells. Nature (2008) 454(7202):350–2. doi: 10.1038/nature07021
63. Huber M, Heink S, Grothe H, Guralnik A, Reinhard K, Elflein K, et al. A Th17-Like Developmental Process Leads to CD8(+) Tc17 Cells With Reduced Cytotoxic Activity. Eur J Immunol (2009) 39(7):1716–25. doi: 10.1002/eji.200939412
64. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A Distinct Lineage of CD4 T Cells Regulates Tissue Inflammation by Producing Interleukin 17. Nat Immunol (2005) 6(11):1133–41. doi: 10.1038/ni1261
65. Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-Producing CD4+ Effector T Cells Develop via a Lineage Distinct From the T Helper Type 1 and 2 Lineages. Nat Immunol (2005) 6(11):1123–32. doi: 10.1038/ni1254
66. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The Orphan Nuclear Receptor RORgammat Directs the Differentiation Program of Proinflammatory IL-17+ T Helper Cells. Cell (2006) 126(6):1121–33. doi: 10.1016/j.cell.2006.07.035
67. Murphy KM, Reiner SL. The Lineage Decisions of Helper T Cells. Nat Rev Immunol (2002) 2(12):933–44. doi: 10.1038/nri954
68. Hill GR, Olver SD, Kuns RD, Varelias A, Raffelt NC, Don AL, et al. Stem Cell Mobilization With G-CSF Induces Type 17 Differentiation and Promotes Scleroderma. Blood (2010) 116(5):819–28. doi: 10.1182/blood-2009-11-256495
69. Meguro A, Ozaki K, Hatanaka K, Oh I, Sudo K, Ohmori T, et al. Lack of IL-21 Signal Attenuates Graft-Versus-Leukemia Effect in the Absence of CD8 T-Cells. Bone Marrow Transplant (2011) 46(12):1557–65. doi: 10.1038/bmt.2010.342
70. Meguro A, Ozaki K, Oh I, Hatanaka K, Matsu H, Tatara R, et al. IL-21 is Critical for GVHD in a Mouse Model. Bone Marrow Transplant (2010) 45(4):723–9. doi: 10.1038/bmt.2009.223
71. Bucher C, Koch L, Vogtenhuber C, Goren E, Munger M, Panoskaltsis-Mortari A, et al. IL-21 Blockade Reduces Graft-Versus-Host Disease Mortality by Supporting Inducible T Regulatory Cell Generation. Blood (2009) 114(26):5375–84. doi: 10.1182/blood-2009-05-221135
72. Hanash AM, Kappel LW, Yim NL, Nejat RA, Goldberg GL, Smith OM, et al. Abrogation of Donor T-Cell IL-21 Signaling Leads to Tissue-Specific Modulation of Immunity and Separation of GVHD From GVL. Blood (2011) 118(2):446–55. doi: 10.1182/blood-2010-07-294785
73. Fulton LM, Carlson MJ, Coghill JM, Ott LE, West ML, Panoskaltsis-Mortari A, et al. Attenuation of Acute Graft-Versus-Host Disease in the Absence of the Transcription Factor RORgammat. J Immunol (2012) 189(4):1765–72. doi: 10.4049/jimmunol.1200858
74. Chen X, Das R, Komorowski R, Beres A, Hessner MJ, Mihara M, et al. Blockade of Interleukin-6 Signaling Augments Regulatory T-Cell Reconstitution and Attenuates the Severity of Graft-Versus-Host Disease. Blood (2009) 114(4):891–900. doi: 10.1182/blood-2009-01-197178
75. Tawara I, Koyama M, Liu C, Toubai T, Thomas D, Evers R, et al. Interleukin-6 Modulates Graft-Versus-Host Responses After Experimental Allogeneic Bone Marrow Transplantation. Clin Cancer Res (2011) 17(1):77–88. doi: 10.1158/1078-0432.CCR-10-1198
76. Das R, Chen X, Komorowski R, Hessner MJ, Drobyski WR. Interleukin-23 Secretion by Donor Antigen-Presenting Cells is Critical for Organ-Specific Pathology in Graft-Versus-Host Disease. Blood (2009) 113(10):2352–62. doi: 10.1182/blood-2008-08-175448
77. Thompson JS, Chu Y, Glass JF, Brown SA. Absence of IL-23p19 in Donor Allogeneic Cells Reduces Mortality From Acute GVHD. Bone Marrow Transplant (2010) 45(4):712–22. doi: 10.1038/bmt.2009.215
78. Das R, Komorowski R, Hessner MJ, Subramanian H, Huettner CS, Cua D, et al. Blockade of Interleukin-23 Signaling Results in Targeted Protection of the Colon and Allows for Separation of Graft-Versus-Host and Graft-Versus-Leukemia Responses. Blood (2010) 115(25):5249–58. doi: 10.1182/blood-2009-11-255422
79. Carlson MJ, West ML, Coghill JM, Panoskaltsis-Mortari A, Blazar BR, Serody JS. In Vitro-Differentiated TH17 Cells Mediate Lethal Acute Graft-Versus-Host Disease With Severe Cutaneous and Pulmonary Pathologic Manifestations. Blood (2009) 113(6):1365–74. doi: 10.1182/blood-2008-06-162420
80. Iclozan C, Yu Y, Liu C, Liang Y, Yi T, Anasetti C, et al. T Helper17 Cells are Sufficient But Not Necessary to Induce Acute Graft-Versus-Host Disease. Biol Blood Marrow Transplant (2010) 16(2):170–8. doi: 10.1016/j.bbmt.2009.09.023
81. Dander E, Balduzzi A, Zappa G, Lucchini G, Perseghin P, Andre V, et al. Interleukin-17-Producing T-Helper Cells as New Potential Player Mediating Graft-Versus-Host Disease in Patients Undergoing Allogeneic Stem-Cell Transplantation. Transplantation (2009) 88(11):1261–72. doi: 10.1097/TP.0b013e3181bc267e
82. Bruggen MC, Klein I, Greinix H, Bauer W, Kuzmina Z, Rabitsch W, et al. Diverse T-Cell Responses Characterize the Different Manifestations of Cutaneous Graft-Versus-Host Disease. Blood (2014) 123(2):290–9. doi: 10.1182/blood-2013-07-514372
83. Li W, Liu L, Gomez A, Zhang Q, Zhang J, Ramadan A, et al. A Novel Th17-Prone CD146+CCR5+ T-Cell Population As an Early Marker of Intestinal Graft-Versus-Host Disease. Blood (2014) 124(21):3. doi: 10.1182/blood.V124.21.3.3
84. Forcade E, Paz K, Flynn R, Griesenauer B, Amet T, Li W, et al. An Activated Th17-Prone T Cell Subset Involved in Chronic Graft-Versus-Host Disease Sensitive to Pharmacological Inhibition. JCI Insight (2017) 2(12):e92111. doi: 10.1172/jci.insight.92111
85. Flynn R, Paz K, Du J, Reichenbach DK, Taylor PA, Panoskaltsis-Mortari A, et al. Targeted Rho-Associated Kinase 2 Inhibition Suppresses Murine and Human Chronic GVHD Through a Stat3-Dependent Mechanism. Blood (2016) 127(17):2144–54. doi: 10.1182/blood-2015-10-678706
86. Jagasia M, Lazaryan A, Bachier CR, Salhotra A, Weisdorf DJ, Zoghi B, et al. ROCK2 Inhibition With Belumosudil (KD025) for the Treatment of Chronic Graft-Versus-Host Disease. J Clin Oncol Off J Am Soc Clin Oncol (2021) 39(17):1888–98. doi: 10.1200/JCO.20.02754
87. Cutler CS, Lee SJ, Arai S, Rotta M, Zoghi B, Lazaryan A, et al. Belumosudil for Chronic Graft-Versus-Host Disease (cGVHD) After 2 or More Prior Lines of Therapy: The ROCKstar Study. Blood (2021). doi: 10.1182/blood.2021012021
88. FDA. FDA Approves Belumosudil for Chronic Graft-Versus-Host Disease, in: FDA/CEDR Resources Page. Food and Drug Administration. Available at: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-belumosudil-chronic-graft-versus-host-disease (Accessed September 9, 2021).
89. Eyerich S, Eyerich K, Pennino D, Carbone T, Nasorri F, Pallotta S, et al. Th22 Cells Represent a Distinct Human T Cell Subset Involved in Epidermal Immunity and Remodeling. J Clin Invest (2009) 119(12):3573–85. doi: 10.1172/JCI40202
90. Plank MW, Kaiko GE, Maltby S, Weaver J, Tay HL, Shen W, et al. Th22 Cells Form a Distinct Th Lineage From Th17 Cells In Vitro With Unique Transcriptional Properties and Tbet-Dependent Th1 Plasticity. J Immunol (2017) 198(5):2182–90. doi: 10.4049/jimmunol.1601480
91. Zhao K, Zhao D, Huang D, Song X, Chen C, Pan B, et al. The Identification and Characteristics of IL-22-Producing T Cells in Acute Graft-Versus-Host Disease Following Allogeneic Bone Marrow Transplantation. Immunobiology (2013) 218(12):1505–13. doi: 10.1016/j.imbio.2013.05.005
92. Hanash AM, Dudakov JA, Hua G, O’Connor MH, Young LF, Singer NV, et al. Interleukin-22 Protects Intestinal Stem Cells From Immune-Mediated Tissue Damage and Regulates Sensitivity to Graft Versus Host Disease. Immunity (2012) 37(2):339–50. doi: 10.1016/j.immuni.2012.05.028
93. Couturier M, Lamarthée B, Arbez J, Renauld JC, Bossard C, Malard F, et al. IL-22 Deficiency in Donor T Cells Attenuates Murine Acute Graft-Versus-Host Disease Mortality While Sparing the Graft-Versus-Leukemia Effect. Leukemia (2013) 27:1527. doi: 10.1038/leu.2013.39
94. Zhao K, Zhao D, Huang D, Yin L, Chen C, Pan B, et al. Interleukin-22 Aggravates Murine Acute Graft-Versus-Host Disease by Expanding Effector T Cell and Reducing Regulatory T Cell. J Interferon Cytokine Res (2014) 34(9):707–15. doi: 10.1089/jir.2013.0099
95. Saxton RA, Henneberg LT, Calafiore M, Su L, Jude KM, Hanash AM, et al. The Tissue Protective Functions of Interleukin-22 can be Decoupled From Pro-Inflammatory Actions Through Structure-Based Design. Immunity (2021) 54(4):660–72 e9. doi: 10.1016/j.immuni.2021.03.008
96. Ponce D, Alousi A, Nakamura R, Sandu K, Barker J, Shia J, et al. A Phase 2 Study of F-652, a Novel Tissue-Targeted Recombinant Human Interleukin-22 (IL-22) Dimer, for Treatment of Newly Diagnosed Acute Gvhd of the Lower GI Tract. Biol Blood Marrow Transplant (2021) 26(3):S51 – S2. doi: 10.1016/j.bbmt.2019.12.124
97. Goenka R, Barnett LG, Silver JS, O’Neill PJ, Hunter CA, Cancro MP, et al. Cutting Edge: Dendritic Cell-Restricted Antigen Presentation Initiates the Follicular Helper T Cell Program But Cannot Complete Ultimate Effector Differentiation. J Immunol (2011) 187(3):1091–5. doi: 10.4049/jimmunol.1100853
98. Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD, et al. Bcl6 Mediates the Development of T Follicular Helper Cells. Science (2009) 325(5943):1001–5. doi: 10.1126/science.1176676
99. Choi YS, Kageyama R, Eto D, Escobar TC, Johnston RJ, Monticelli L, et al. ICOS Receptor Instructs T Follicular Helper Cell Versus Effector Cell Differentiation via Induction of the Transcriptional Repressor Bcl6. Immunity (2011) 34(6):932–46. doi: 10.1016/j.immuni.2011.03.023
100. Choi YS, Eto D, Yang JA, Lao C, Crotty S. Cutting Edge: STAT1 is Required for IL-6-Mediated Bcl6 Induction for Early Follicular Helper Cell Differentiation. J Immunol (2013) 190(7):3049–53. doi: 10.4049/jimmunol.1203032
101. Pepper M, Pagan AJ, Igyarto BZ, Taylor JJ, Jenkins MK. Opposing Signals From the Bcl6 Transcription Factor and the Interleukin-2 Receptor Generate T Helper 1 Central and Effector Memory Cells. Immunity (2011) 35(4):583–95. doi: 10.1016/j.immuni.2011.09.009
102. Fang D, Cui K, Mao K, Hu G, Li R, Zheng M, et al. Transient T-Bet Expression Functionally Specifies a Distinct T Follicular Helper Subset. J Exp Med (2018) 215(11):2705–14. doi: 10.1084/jem.20180927
103. Ballesteros-Tato A, Leon B, Graf BA, Moquin A, Adams PS, Lund FE, et al. Interleukin-2 Inhibits Germinal Center Formation by Limiting T Follicular Helper Cell Differentiation. Immunity (2012) 36(5):847–56. doi: 10.1016/j.immuni.2012.02.012
104. Johnston RJ, Choi YS, Diamond JA, Yang JA, Crotty S. STAT5 is a Potent Negative Regulator of TFH Cell Differentiation. J Exp Med (2012) 209(2):243–50. doi: 10.1084/jem.20111174
105. Nguyen V, Luzina I, Rus H, Tegla C, Chen C, Rus V. IL-21 Promotes Lupus-Like Disease in Chronic Graft-Versus-Host Disease Through Both CD4 T Cell- and B Cell-Intrinsic Mechanisms. J Immunol (2012) 189(2):1081–93. doi: 10.4049/jimmunol.1200318
106. Forcade E, Kim HT, Cutler C, Wang K, Alho AC, Nikiforow S, et al. Circulating T Follicular Helper Cells With Increased Function During Chronic Graft-Versus-Host Disease. Blood (2016) 127(20):2489–97. doi: 10.1182/blood-2015-12-688895
107. Kamihara YFE, Koreth J, Liu H, Kubo T, Whangbo J, Hirakawa M, et al. Blood. Atlanta, GA: American Society of Hematology (2017).
108. Kumar BV, Ma W, Miron M, Granot T, Guyer RS, Carpenter DJ, et al. Human Tissue-Resident Memory T Cells Are Defined by Core Transcriptional and Functional Signatures in Lymphoid and Mucosal Sites. Cell Rep (2017) 20(12):2921–34. doi: 10.1016/j.celrep.2017.08.078
109. Divito SJ, Aasebo AT, Matos TR, Hsieh PC, Collin M, Elco CP, et al. Peripheral Host T Cells Survive Hematopoietic Stem Cell Transplantation and Promote Graft-Versus-Host Disease. J Clin Invest (2020) 130(9):4624–36. doi: 10.1172/JCI129965
110. Strobl J, Pandey RV, Krausgruber T, Bayer N, Kleissl L, Reininger B, et al. Long-Term Skin-Resident Memory T Cells Proliferate in Situ and are Involved in Human Graft-Versus-Host Disease. Sci Transl Med (2020) 12(570):eabb7028. doi: 10.1126/scitranslmed.abb7028
111. Kong X, Zeng D, Wu X, Wang B, Yang S, Song Q, et al. Tissue-Resident PSGL1loCD4+ T Cells Promote B Cell Differentiation and Chronic Graft-Versus-Host Disease-Associated Autoimmunity. J Clin Invest (2021) 131(1):e135468. doi: 10.1172/JCI135468
112. Tkachev V, Kaminski J, Potter EL, Furlan SN, Yu A, Hunt DJ, et al. Spatiotemporal Single-Cell Profiling Reveals That Invasive and Tissue-Resident Memory Donor CD8(+) T Cells Drive Gastrointestinal Acute Graft-Versus-Host Disease. Sci Transl Med (2021) 13(576):eabc0227. doi: 10.1126/scitranslmed.abc0227
113. Nurieva RI, Chung Y. Understanding the Development and Function of T Follicular Helper Cells. Cell Mol Immunol (2010) 7(3):190–7. doi: 10.1038/cmi.2010.24
114. Zheng W, Flavell RA. The Transcription Factor GATA-3 is Necessary and Sufficient for Th2 Cytokine Gene Expression in CD4 T Cells. Cell (1997) 89(4):587–96. doi: 10.1016/S0092-8674(00)80240-8
115. Croft M, Carter L, Swain SL, Dutton RW. Generation of Polarized Antigen-Specific CD8 Effector Populations: Reciprocal Action of Interleukin (IL)-4 and IL-12 in Promoting Type 2 Versus Type 1 Cytokine Profiles. J Exp Med (1994) 180(5):1715–28. doi: 10.1084/jem.180.5.1715
116. Omori M, Yamashita M, Inami M, Ukai-Tadenuma M, Kimura M, Nigo Y, et al. CD8 T Cell-Specific Downregulation of Histone Hyperacetylation and Gene Activation of the IL-4 Gene Locus by ROG, Repressor of GATA. Immunity (2003) 19(2):281–94. doi: 10.1016/S1074-7613(03)00210-3
117. Jung U, Foley JE, Erdmann AA, Eckhaus MA, Fowler DH. CD3/CD28-Costimulated T1 and T2 Subsets: Differential In Vivo Allosensitization Generates Distinct GVT and GVHD Effects. Blood (2003) 102(9):3439–46. doi: 10.1182/blood-2002-12-3936
118. Tawara I, Maeda Y, Sun Y, Lowler KP, Liu C, Toubai T, et al. Combined Th2 Cytokine Deficiency in Donor T Cells Aggravates Experimental Acute Graft-vs-Host Disease. Exp Hematol (2008) 36(8):988–96. doi: 10.1016/j.exphem.2008.02.010
119. Yi T, Chen Y, Wang L, Du G, Huang D, Zhao D, et al. Reciprocal Differentiation and Tissue-Specific Pathogenesis of Th1, Th2, and Th17 Cells in Graft-Versus-Host Disease. Blood (2009) 114(14):3101–12. doi: 10.1182/blood-2009-05-219402
120. Henden AS, Hill GR. Cytokines in Graft-Versus-Host Disease. J Immunol (2015) 194(10):4604–12. doi: 10.4049/jimmunol.1500117
121. Jung U, Foley JE, Erdmann AA, Toda Y, Borenstein T, Mariotti J, et al. Ex Vivo Rapamycin Generates Th1/Tc1 or Th2/Tc2 Effector T Cells With Enhanced In Vivo Function and Differential Sensitivity to Post-Transplant Rapamycin Therapy. Biol Blood Marrow Transplant (2006) 12(9):905–18. doi: 10.1016/j.bbmt.2006.05.014
122. Mariotti J, Foley J, Ryan K, Buxhoeveden N, Kapoor V, Amarnath S, et al. Graft Rejection as a Th1-Type Process Amenable to Regulation by Donor Th2-Type Cells Through an Interleukin-4/STAT6 Pathway. Blood (2008) 112(12):4765–75. doi: 10.1182/blood-2008-05-154278
123. Shin HJ, Baker J, Leveson-Gower DB, Smith AT, Sega EI, Negrin RS. Rapamycin and IL-2 Reduce Lethal Acute Graft-Versus-Host Disease Associated With Increased Expansion of Donor Type CD4+CD25+Foxp3+ Regulatory T Cells. Blood (2011) 118(8):2342–50. doi: 10.1182/blood-2010-10-313684
124. Fowler DH, Mossoba ME, Steinberg SM, Halverson DC, Stroncek D, Khuu HM, et al. Phase 2 Clinical Trial of Rapamycin-Resistant Donor CD4+ Th2/Th1 (T-Rapa) Cells After Low-Intensity Allogeneic Hematopoietic Cell Transplantation. Blood (2013) 121(15):2864–74. doi: 10.1182/blood-2012-08-446872
125. Chang HC, Zhang S, Thieu VT, Slee RB, Bruns HA, Laribee RN, et al. PU.1 Expression Delineates Heterogeneity in Primary Th2 Cells. Immunity (2005) 22(6):693–703. doi: 10.1016/j.immuni.2005.03.016
126. Veldhoen M, Uyttenhove C, van Snick J, Helmby H, Westendorf A, Buer J, et al. Transforming Growth Factor-Beta ’Reprograms’ the Differentiation of T Helper 2 Cells and Promotes an Interleukin 9-Producing Subset. Nat Immunol (2008) 9(12):1341–6. doi: 10.1038/ni.1659
127. Dardalhon V, Awasthi A, Kwon H, Galileos G, Gao W, Sobel RA, et al. IL-4 Inhibits TGF-Beta-Induced Foxp3+ T Cells and, Together With TGF-Beta, Generates IL-9+ IL-10+ Foxp3(-) Effector T Cells. Nat Immunol (2008) 9(12):1347–55. doi: 10.1038/ni.1677
128. Chang HC, Sehra S, Goswami R, Yao W, Yu Q, Stritesky GL, et al. The Transcription Factor PU.1 Is Required for the Development of IL-9-Producing T Cells and Allergic Inflammation. Nat Immunol (2010) 11(6):527–34. doi: 10.1038/ni.1867
129. Visekruna A, Ritter J, Scholz T, Campos L, Guralnik A, Poncette L, et al. Tc9 Cells, a New Subset of CD8(+) T Cells, Support Th2-Mediated Airway Inflammation. Eur J Immunol (2013) 43(3):606–18. doi: 10.1002/eji.201242825
130. Mangus CW, Massey PR, Fowler DH, Amarnath S. Rapamycin Resistant Murine Th9 Cells Have a Stable In Vivo Phenotype and Inhibit Graft-Versus-Host Reactivity. PloS One (2013) 8(8):e72305. doi: 10.1371/journal.pone.0072305
131. Lu Y, Hong S, Li H, Park J, Hong B, Wang L, et al. Th9 Cells Promote Antitumor Immune Responses In Vivo. J Clin Invest (2012) 122(11):4160–71. doi: 10.1172/JCI65459
132. Purwar R, Schlapbach C, Xiao S, Kang HS, Elyaman W, Jiang X, et al. Robust Tumor Immunity to Melanoma Mediated by Interleukin-9-Producing T Cells. Nat Med (2012) 18(8):1248–53. doi: 10.1038/nm.2856
133. Griesenauer B, Paczesny S. The ST2/IL-33 Axis in Immune Cells During Inflammatory Diseases. Front Immunol (2017) 8:475. doi: 10.3389/fimmu.2017.00475
134. Garlanda C, Dinarello CA, Mantovani A. The Interleukin-1 Family: Back to the Future. Immunity (2013) 39(6):1003–18. doi: 10.1016/j.immuni.2013.11.010
135. Vander Lugt MT, Braun TM, Hanash S, Ritz J, Ho VT, Antin JH, et al. ST2 as a Marker for Risk of Therapy-Resistant Graft-Versus-Host Disease and Death. N Engl J Med (2013) 369(6):529–39. doi: 10.1056/NEJMoa1213299
136. Zhang J, Ramadan AM, Griesenauer B, Li W, Turner MJ, Liu C, et al. ST2 Blockade Reduces Sst2-Producing T Cells While Maintaining Protective Mst2-Expressing T Cells During Graft-Versus-Host Disease. Sci Transl Med (2015) 7(308):308ra160. doi: 10.1126/scitranslmed.aab0166
137. Lohning M, Stroehmann A, Coyle AJ, Grogan JL, Lin S, Gutierrez-Ramos JC, et al. T1/ST2 is Preferentially Expressed on Murine Th2 Cells, Independent of Interleukin 4, Interleukin 5, and Interleukin 10, and Important for Th2 Effector Function. Proc Natl Acad Sci USA (1998) 95(12):6930–5. doi: 10.1073/pnas.95.12.6930
138. Xu D, Chan WL, Leung BP, Huang F, Wheeler R, Piedrafita D, et al. Selective Expression of a Stable Cell Surface Molecule on Type 2 But Not Type 1 Helper T Cells. J Exp Med (1998) 187(5):787–94. doi: 10.1084/jem.187.5.787
139. Blom L, Poulsen BC, Jensen BM, Hansen A, Poulsen LK. IL-33 Induces IL-9 Production in Human CD4+ T Cells and Basophils. PloS One (2011) 6(7):e21695. doi: 10.1371/journal.pone.0021695
140. Ma X, Bi E, Huang C, Lu Y, Xue G, Guo X, et al. Cholesterol Negatively Regulates IL-9-Producing CD8(+) T Cell Differentiation and Antitumor Activity. J Exp Med (2018) 215(6):1555–69. doi: 10.1084/jem.20171576
141. Yu J, Storer BE, Kushekhar K, Abu Zaid M, Zhang Q, Gafken PR, et al. Biomarker Panel for Chronic Graft-Versus-Host Disease. J Clin Oncol Off J Am Soc Clin Oncol (2016) 34(22):2583–90. doi: 10.1200/JCO.2015.65.9615
142. Zaiss DM, Yang L, Shah PR, Kobie JJ, Urban JF, Mosmann TR. Amphiregulin, a TH2 Cytokine Enhancing Resistance to Nematodes. Science (2006) 314(5806):1746. doi: 10.1126/science.1133715
143. Morimoto Y, Hirahara K, Kiuchi M, Wada T, Ichikawa T, Kanno T, et al. Amphiregulin-Producing Pathogenic Memory T Helper 2 Cells Instruct Eosinophils to Secrete Osteopontin and Facilitate Airway Fibrosis. Immunity (2018) 49(1):134–50 e6. doi: 10.1016/j.immuni.2018.04.023
144. Arpaia N, Green JA, Moltedo B, Arvey A, Hemmers S, Yuan S, et al. A Distinct Function of Regulatory T Cells in Tissue Protection. Cell (2015) 162(5):1078–89. doi: 10.1016/j.cell.2015.08.021
145. Yang J, Ramadan A, Reichenbach DK, Loschi M, Zhang J, Griesenauer B, et al. Rorc Restrains the Potency of ST2+ Regulatory T Cells in Ameliorating Intestinal Graft-Versus-Host Disease. JCI Insight (2019) 4(5):e122014. doi: 10.1172/jci.insight.122014
146. Burchill MA, Yang J, Vogtenhuber C, Blazar BR, Farrar MA. IL-2 Receptor Beta-Dependent STAT5 Activation Is Required for the Development of Foxp3+ Regulatory T Cells. J Immunol (2007) 178(1):280–90. doi: 10.4049/jimmunol.178.1.280
147. Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A Function for Interleukin 2 in Foxp3-Expressing Regulatory T Cells. Nat Immunol (2005) 6(11):1142–51. doi: 10.1038/ni1263
148. Marshall D, Sinclair C, Tung S, Seddon B. Differential Requirement for IL-2 and IL-15 During Bifurcated Development of Thymic Regulatory T Cells. J Immunol (2014) 193(11):5525–33. doi: 10.4049/jimmunol.1402144
149. Liu Y, Zhang P, Li J, Kulkarni AB, Perruche S, Chen W. A Critical Function for TGF-Beta Signaling in the Development of Natural CD4+CD25+Foxp3+ Regulatory T Cells. Nat Immunol (2008) 9(6):632–40. doi: 10.1038/ni.1607
150. Zeiser R, Blazar BR. Acute Graft-Versus-Host Disease - Biologic Process, Prevention, and Therapy. N Engl J Med (2017) 377(22):2167–79. doi: 10.1056/NEJMra1609337
151. Chen X, Vodanovic-Jankovic S, Johnson B, Keller M, Komorowski R, Drobyski WR. Absence of Regulatory T-Cell Control of TH1 and TH17 Cells Is Responsible for the Autoimmune-Mediated Pathology in Chronic Graft-Versus-Host Disease. Blood (2007) 110(10):3804–13. doi: 10.1182/blood-2007-05-091074
152. Zeng H, Zhang R, Jin B, Chen L. Type 1 Regulatory T Cells: A New Mechanism of Peripheral Immune Tolerance. Cell Mol Immunol (2015) 12(5):566–71. doi: 10.1038/cmi.2015.44
153. Zhang P, Lee JS, Gartlan KH, Schuster IS, Comerford I, Varelias A, et al. Eomesodermin Promotes the Development of Type 1 Regulatory T (TR1) Cells. Sci Immunol (2017) 2(10):eaah7152. doi: 10.1126/sciimmunol.aah7152
154. Edinger M, Hoffmann P, Ermann J, Drago K, Fathman CG, Strober S, et al. CD4+CD25+ Regulatory T Cells Preserve Graft-Versus-Tumor Activity While Inhibiting Graft-Versus-Host Disease After Bone Marrow Transplantation. Nat Med (2003) 9(9):1144–50. doi: 10.1038/nm915
155. Trenado A, Charlotte F, Fisson S, Yagello M, Klatzmann D, Salomon BL, et al. Recipient-Type Specific CD4+CD25+ Regulatory T Cells Favor Immune Reconstitution and Control Graft-Versus-Host Disease While Maintaining Graft-Versus-Leukemia. J Clin Invest (2003) 112(11):1688–96. doi: 10.1172/JCI17702
156. Chopra M, Biehl M, Steinfatt T, Brandl A, Kums J, Amich J, et al. Exogenous TNFR2 Activation Protects From Acute GvHD via Host T Reg Cell Expansion. J Exp Med (2016) 213(9):1881–900. doi: 10.1084/jem.20151563
157. MacDonald KG, Hoeppli RE, Huang Q, Gillies J, Luciani DS, Orban PC, et al. Alloantigen-Specific Regulatory T Cells Generated With a Chimeric Antigen Receptor. J Clin Invest (2016) 126(4):1413–24. doi: 10.1172/JCI82771
158. Baecher-Allan C, Brown JA, Freeman GJ, Hafler DA. CD4+CD25high Regulatory Cells in Human Peripheral Blood. J Immunol (2001) 167(3):1245–53. doi: 10.4049/jimmunol.167.3.1245
159. Hippen KL, Merkel SC, Schirm DK, Sieben CM, Sumstad D, Kadidlo DM, et al. Massive Ex Vivo Expansion of Human Natural Regulatory T Cells (T(regs)) With Minimal Loss of In Vivo Functional Activity. Sci Transl Med (2011) 3(83):83ra41. doi: 10.1126/scitranslmed.3001809
160. June CH, Blazar BR. Clinical Application of Expanded CD4+25+ Cells. Semin Immunol (2006) 18(2):78–88. doi: 10.1016/j.smim.2006.01.006
161. Godfrey WR, Ge YG, Spoden DJ, Levine BL, June CH, Blazar BR, et al. In Vitro-Expanded Human CD4(+)CD25(+) T-Regulatory Cells can Markedly Inhibit Allogeneic Dendritic Cell-Stimulated MLR Cultures. Blood (2004) 104(2):453–61. doi: 10.1182/blood-2004-01-0151
162. Brunstein CG, Miller JS, Cao Q, McKenna DH, Hippen KL, Curtsinger J, et al. Infusion of Ex Vivo Expanded T Regulatory Cells in Adults Transplanted With Umbilical Cord Blood: Safety Profile and Detection Kinetics. Blood (2011) 117(3):1061–70. doi: 10.1182/blood-2010-07-293795
163. Brunstein CG, Miller JS, McKenna DH, Hippen KL, DeFor TE, Sumstad D, et al. Umbilical Cord Blood-Derived T Regulatory Cells to Prevent GVHD: Kinetics, Toxicity Profile, and Clinical Effect. Blood (2016) 127(8):1044–51. doi: 10.1182/blood-2015-06-653667
164. Di Ianni M, Falzetti F, Carotti A, Terenzi A, Castellino F, Bonifacio E, et al. Tregs Prevent GVHD and Promote Immune Reconstitution in HLA-Haploidentical Transplantation. Blood (2011) 117(14):3921–8. doi: 10.1182/blood-2010-10-311894
165. Clark FJ, Gregg R, Piper K, Dunnion D, Freeman L, Griffiths M, et al. Chronic Graft-Versus-Host Disease is Associated With Increased Numbers of Peripheral Blood CD4+CD25high Regulatory T Cells. Blood (2004) 103(6):2410–6. doi: 10.1182/blood-2003-06-2073
166. Bacchetta R, Lucarelli B, Sartirana C, Gregori S, Lupo Stanghellini MT, Miqueu P, et al. Immunological Outcome in Haploidentical-HSC Transplanted Patients Treated With IL-10-Anergized Donor T Cells. Front Immunol (2014) 5:16. doi: 10.3389/fimmu.2014.00016
167. Kennedy GA, Varelias A, Vuckovic S, Le Texier L, Gartlan KH, Zhang P, et al. Addition of Interleukin-6 Inhibition With Tocilizumab to Standard Graft-Versus-Host Disease Prophylaxis After Allogeneic Stem-Cell Transplantation: A Phase 1/2 Trial. Lancet Oncol (2014) 15(13):1451–9. doi: 10.1016/S1470-2045(14)71017-4
168. Kennedy GA, Tey SK, Buizen L, Varelias A, Gartlan KH, Curley C, et al. A Phase 3 Double-Blind Study of the Addition of Tocilizumab vs Placebo to Cyclosporin/Methotrexate GVHD Prophylaxis. Blood (2021) 137(14):1970–9. doi: 10.1182/blood.2020009050
169. van den Brink MR, Burakoff SJ. Cytolytic Pathways in Haematopoietic Stem-Cell Transplantation. Nat Rev Immunol (2002) 2(4):273–81. doi: 10.1038/nri775
170. Lu Y, Hong B, Li H, Zheng Y, Zhang M, Wang S, et al. Tumor-Specific IL-9-Producing CD8+ Tc9 Cells are Superior Effector Than Type-I Cytotoxic Tc1 Cells for Adoptive Immunotherapy of Cancers. Proc Natl Acad Sci USA (2014) 111(6):2265–70. doi: 10.1073/pnas.1317431111
171. Liu L, Bi E, Ma X, Xiong W, Qian J, Ye L, et al. Enhanced CAR-T Activity Against Established Tumors by Polarizing Human T Cells to Secrete Interleukin-9. Nat Commun (2020) 11(1):5902. doi: 10.1038/s41467-020-19672-2
172. Voermans C, Hazenberg MD. Cellular Therapies for Graft-Versus-Host Disease: A Tale of Tissue Repair and Tolerance. Blood (2020) 136(4):410–7. doi: 10.1182/blood.2019000951
173. Blazar BR, MacDonald KPA, Hill GR. Immune Regulatory Cell Infusion for Graft-Versus-Host Disease Prevention and Therapy. Blood (2018) 131(24):2651–60. doi: 10.1182/blood-2017-11-785865
174. Weber EW, Maus MV, Mackall CL. The Emerging Landscape of Immune Cell Therapies. Cell (2020) 181(1):46–62. doi: 10.1016/j.cell.2020.03.001
175. Elverum K, Whitman M. Delivering Cellular and Gene Therapies to Patients: Solutions for Realizing the Potential of the Next Generation of Medicine. Gene Ther (2020) 27(12):537–44. doi: 10.1038/s41434-019-0074-7
176. Shinners NP, Carlesso G, Castro I, Hoek KL, Corn RA, Woodland RT, et al. Bruton’s Tyrosine Kinase Mediates NF-Kappa B Activation and B Cell Survival by B Cell-Activating Factor Receptor of the TNF-R Family. J Immunol (2007) 179(6):3872–80. doi: 10.4049/jimmunol.179.6.3872
177. Dubovsky JA, Beckwith KA, Natarajan G, Woyach JA, Jaglowski S, Zhong Y, et al. Ibrutinib is an Irreversible Molecular Inhibitor of ITK Driving a Th1-Selective Pressure in T Lymphocytes. Blood (2013) 122(15):2539–49. doi: 10.1182/blood-2013-06-507947
178. Dubovsky JA, Flynn R, Du J, Harrington BK, Zhong Y, Kaffenberger B, et al. Ibrutinib Treatment Ameliorates Murine Chronic Graft-Versus-Host Disease. J Clin Invest (2014) 124(11):4867–76. doi: 10.1172/JCI75328
179. Miklos D, Cutler CS, Arora M, Waller EK, Jagasia M, Pusic I, et al. Ibrutinib for Chronic Graft-Versus-Host Disease After Failure of Prior Therapy. Blood (2017) 130(21):2243–50. doi: 10.1182/blood-2017-07-793786
180. Yamaoka K, Saharinen P, Pesu M, Holt VE 3rd, Silvennoinen O, O’Shea JJ. The Janus Kinases (Jaks). Genome Biol (2004) 5(12):253. doi: 10.1186/gb-2004-5-12-253
181. Spoerl S, Mathew NR, Bscheider M, Schmitt-Graeff A, Chen S, Mueller T, et al. Activity of Therapeutic JAK 1/2 Blockade in Graft-Versus-Host Disease. Blood (2014) 123(24):3832–42. doi: 10.1182/blood-2013-12-543736
182. Heine A, Held SA, Daecke SN, Wallner S, Yajnanarayana SP, Kurts C, et al. The JAK-Inhibitor Ruxolitinib Impairs Dendritic Cell Function In Vitro and In Vivo. Blood (2013) 122(7):1192–202. doi: 10.1182/blood-2013-03-484642
183. Betts BC, Abdel-Wahab O, Curran SA, St Angelo ET, Koppikar P, Heller G, et al. Janus Kinase-2 Inhibition Induces Durable Tolerance to Alloantigen by Human Dendritic Cell-Stimulated T Cells Yet Preserves Immunity to Recall Antigen. Blood (2011) 118(19):5330–9. doi: 10.1182/blood-2011-06-363408
184. Choi J, Ziga ED, Ritchey J, Collins L, Prior JL, Cooper ML, et al. IFNgammaR Signaling Mediates Alloreactive T-Cell Trafficking and GVHD. Blood (2012) 120(19):4093–103. doi: 10.1182/blood-2012-01-403196
185. Choi J, Cooper ML, Alahmari B, Ritchey J, Collins L, Holt M, et al. Pharmacologic Blockade of JAK1/JAK2 Reduces GvHD and Preserves the Graft-Versus-Leukemia Effect. PloS One (2014) 9(10):e109799. doi: 10.1371/journal.pone.0109799
186. Zeiser R, Burchert A, Lengerke C, Verbeek M, Maas-Bauer K, Metzelder SK, et al. Ruxolitinib in Corticosteroid-Refractory Graft-Versus-Host Disease After Allogeneic Stem Cell Transplantation: A Multicenter Survey. Leukemia (2015) 29(10):2062–8. doi: 10.1038/leu.2015.212
187. Schroeder MA, Khoury HJ, Jagasia M, Ali H, Schiller GJ, Staser K, et al. A Phase 1 Trial of Itacitinib, a Selective JAK1 Inhibitor, in Patients With Acute Graft-Versus-Host Disease. Blood Adv (2020) 4(8):1656–69. doi: 10.1182/bloodadvances.2019001043
188. Chen YB, McDonough S, Hasserjian R, Chen H, Coughlin E, Illiano C, et al. Expression of CD30 in Patients With Acute Graft-Versus-Host Disease. Blood (2012) 120(3):691–6. doi: 10.1182/blood-2012-03-415422
189. Chen YB, Perales MA, Li S, Kempner M, Reynolds C, Brown J, et al. Phase 1 Multicenter Trial of Brentuximab Vedotin for Steroid-Refractory Acute Graft-Versus-Host Disease. Blood (2017) 129(24):3256–61. doi: 10.1182/blood-2017-03-772210
190. Schwab N, Schneider-Hohendorf T, Melzer N, Cutter G, Wiendl H. Natalizumab-Associated PML: Challenges With Incidence, Resulting Risk, and Risk Stratification. Neurology (2017) 88(12):1197–205. doi: 10.1212/WNL.0000000000003739
191. Lam MC, Bressler B. Vedolizumab for Ulcerative Colitis and Crohn’s Disease: Results and Implications of GEMINI Studies. Immunotherapy (2014) 6(9):963–71. doi: 10.2217/imt.14.66
192. Milch C, Wyant T, Xu J, Parikh A, Kent W, Fox I, et al. Vedolizumab, a Monoclonal Antibody to the Gut Homing Alpha4beta7 Integrin, Does Not Affect Cerebrospinal Fluid T-Lymphocyte Immunophenotype. J Neuroimmunol (2013) 264(1-2):123–6. doi: 10.1016/j.jneuroim.2013.08.011
193. Soler D, Chapman T, Yang LL, Wyant T, Egan R, Fedyk ER. The Binding Specificity and Selective Antagonism of Vedolizumab, an Anti-Alpha4beta7 Integrin Therapeutic Antibody in Development for Inflammatory Bowel Diseases. J Pharmacol Exp Ther (2009) 330(3):864–75. doi: 10.1124/jpet.109.153973
Keywords: graft versus host disease, graft versus tumor, tissue resident memory t cell, cell therapy, t cells subsets
Citation: Jiang H, Fu D, Bidgoli A and Paczesny S (2021) T Cell Subsets in Graft Versus Host Disease and Graft Versus Tumor. Front. Immunol. 12:761448. doi: 10.3389/fimmu.2021.761448
Received: 19 August 2021; Accepted: 13 September 2021;
Published: 05 October 2021.
Edited by:
Robert Zeiser, University of Freiburg, GermanyReviewed by:
Lazaros Lekakis, University of Miami, United StatesFlorent Malard, Sorbonne Universités, France
Copyright © 2021 Jiang, Fu, Bidgoli and Paczesny. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sophie Paczesny, cGFjemVzbnNAbXVzYy5lZHU=
†These authors have contributed equally to this work